 Xi-xi Hou1†
Xi-xi Hou1† Xiao-qing Gong
Xiao-qing Gong Long-fei Mao
Long-fei Mao Ge Sun
Ge Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 10 August 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.940704
Erlotinib is a highly specific and reversible epidermal growth factor receptor tyrosine kinase inhibitor for the targeted therapy of non-small-cell lung cancer (NSCLC) However, the efficacy of erlotinib is limited because the development of drug resistance during chemotherapy. Indoleamine 2,3-dioxygenase-1 (IDO1) is a rate-limiting tryptophan catabolic enzyme that is activated in many human cancers. In this study, we designed a series of erlotinib-based 1,2,3-triazole compounds by combining erlotinib with phenyl or benzyl azide. Attentive FP prediction model was used to predict the bioactivity of those compounds. We discovered that most of the erlotinib-based 1,2,3-triazole compounds are capable of suppressing IDO1 activities in vitro experiments. Among them, compound 14b (IC50 = 0.59 ± 0.05 μM) had the strongest inhibitory effect on IDO1. In addition, compound 14b significantly inhibited tumor growth comparable to the antitumor activity of erlotinib and the IDO1 inhibitor epacadostat in murine tumor models.
Erlotinib (Figure 1, 1) is a standard EGFR inhibitor that was approved for sale by the FDA in 2004(Smith, 2005). As an orally administered small-molecule competitive reversible EGFR-TKI, erlotinib blocks the phosphorylation of EGFR by competing with mutant EGFR for ATP binding and inhibits activation of downstream signaling pathways (Mao et al., 2022). It was initially developed for the first-line treatment of advanced NSCLC caused by EGFR mutations in cases where conventional chemotherapy failed to provide an effective resolution (Hirsch et al., 2017). Compared with clinical treatment by conventional anticancer drugs, erlotinib treatment improves the median overall survival (OS) of patients from 4 months to >40 months and has better performance in the objective response rate (ORR), progression-free survival (PFS) and tolerance (Mathew et al., 2015; Wei et al., 2020). Although erlotinib significantly delays cancer progression in the targeted treatment of NSCLC, like the other two first-generation EGFR inhibitors, gefitinib (Cohen et al., 2003; Zhang et al., 2017), (Figure 1, 2) and icotinib(Shi et al., 2013; Mao et al., 2020) (Figure 1, 3), drug resistance starts to appear after about 9–14 months of application, and almost all tumors begin to re-grow(Ayyappan et al., 2013). The drug resistance problem has persisted with second-generation and third-generation EGFR inhibitors developed subsequently, such as afatinib (Figure 1, 4), dacomitinib (Figure 1, 5) and osimertinib (Figure 1, 6) (Jänne et al., 2015; Qin et al., 2016). Searching for an effective solution to the drug resistance problem of EGFR inhibitors, including erlotinib, remains the major effort of pharmaceutical research (Sun et al., 2022).
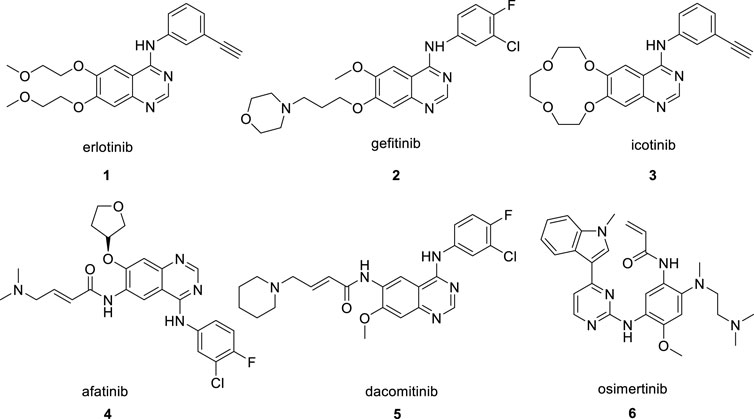
FIGURE 1. The reported EGFR inhibitors.
1,2,3-triazole structural building blocks exhibit a wide range of biological activities in marketed drugs, such as antimicrobial, antitumor (Deng et al., 2022), antitubercular, and antidiabetic effects (Maddili et al., 2018). In addition, abundant studies have shown various 1,2,3-triazole-containing hybrids, such as MMG-0358 (Figure 2, 7) (Röhrig et al., 2010) and Vertex-AT (Figure 2, 8) (Panda et al., 2019) also suppress IDO1 through structure-activity relationship and molecular docking studies. IDO1 is a promising cancer therapeutic target that over expressed in many human cancers (Theate et al., 2015). Especially in lung cancer, IDO1 is a driver of disease progression and metastasis, and is associated with poor prognosis (Smith et al., 2012; Wang et al., 2018). IDO1, which is expressed in multiple types of malignancies from the tumor microenvironment (TME), catalyzes the oxidation of tryptophan (TRP) to immunosupressive metabolite N-formylkynurenine. TRP depletion and catabolite production by IDO1 suppress the function of T effect or cells and natural killer (NK) cell, drive dendritic cells (DCs) (Eric et al., 2018) and macrophages toward an immunosuppressive phenotype, increase the activities of CD4+ T regulatory cells (Treg) and myeloid-derived suppressor cells (MDSCs), and sustain an immmnosuppressive environment in TME (Munn and Mellor, 2016; Schupp et al., 2019). Consequently, great efforts have been made to explore IDO1 inhibitors as the promising therapeutic candidate for cancer therapy. The clinical-stage IDO1 inhibitors such as EOS200271 (Figure 2, 9), BMS-986205 (Figure 2, 10), Epacadostat (Figure 2, 11) and Navoximod (Figure 2, 12), bind to IDO1 through different mechanisms (Platten et al., 2019).
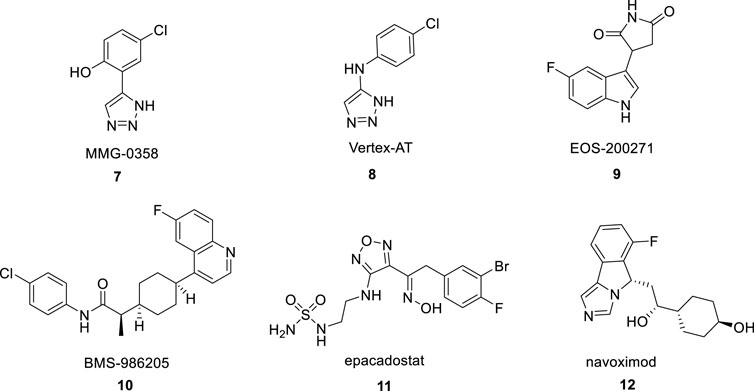
FIGURE 2. The reported IDO1 inhibitors.
The emerging evidence of IDO1 inhibitors in cancer immunotherapy provides ideas for the transformation of erlotinib. On the basis of erlotinib, Professor Yang Qing from Fudan University developed a compound with a triazole structure through click chemistry transformation (Figure 3, 13). The resulted compound displayed the inhibitory activity on the IDO1 enzyme (IC50 = 12.6 μM). Their study expands the application of the erlotinib structure in the development of tumor immunotherapy drugs. In our study, various substituents were introduced on the N atom of the 1-position of triazoles, including five benzyl derivatives substituted compounds (Figure 3, 14a-14e) and five phenyl derivatives substituted compounds (Figure 3, 14f-14j), with the hope to further improve the inhibitory activity against IDO1. In the current era of big data, as a new type of drug research and development technology, deep learning has been involved in multiple stages of drug research. To evaluate the biological activity of our designed molecules, we used the activity prediction function of the molecular representation model Attentive FP (Xiong et al., 2019) to predict the 10 synthesized compounds, so as to evaluate whether the molecules have the biological activity against IDO1. Attentive FP is a graph neural network model based on attention mechanism, which can be used for molecular property prediction, and it performs well in various property prediction results. Compound 13 was used as a control, the promising compounds as supported by Attentive FP were chosen for the further investigation of IDO inhibition, toxicity and tumor inhibitory property using in vitro and in vivo tumor models.
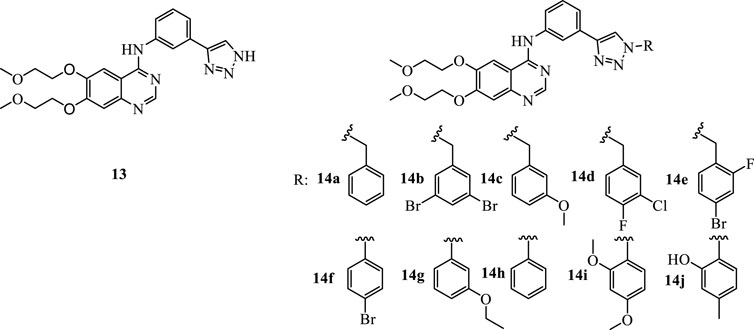
FIGURE 3. The structures of compound 13 and compounds 14a-14j.
The synthetic route for the preparation of the target compounds was showed in Figure 4. Copper(I)-catalysedazide–alkyne cycloaddition between compound 1 (erlotinib) and different azido compounds produced the target compounds 14a-14j (Table 1). The structures of all the target compounds were confirmed by nuclear magnetic resonance (1H NMR and 13C NMR).
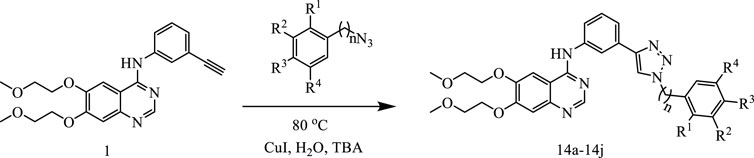
FIGURE 4. The reaction routes to erlotinib-1,2,3-triazole derivatives.
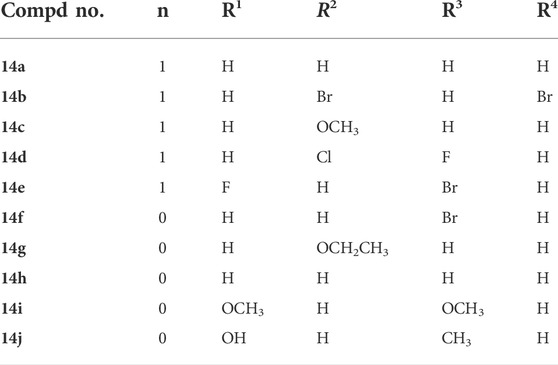
TABLE 1. The structures of erlotinib-1,2,3-triazole derivatives.
In the bioactivity prediction task of Attentive FP, the IDO1 dataset was randomly split according to the ratio of train, valid and test (8:1:1). The evaluation of the model obtained by training were shown in Figure 5. The AUC, F1-score, Precision and Recall are all above 0.8, and the IDO1 dataset showed promising results on Attentive FP. A series of compounds 14a-14j were designed based on the initial compound 13, and then the designed compounds were tested using the trained best model. The test results were shown in Figure 6. The probability of predicted bioactivity for compound 13 was at a medium level (0.580). The probability for compounds 14b, 14e and 14f were much higher than compound 13. Among them, compound 14b had the best bioactivity with a probability of 0.968. Compound 14e and compound 14f were at 0.931 and 0.874. The probability for Compounds 14d and 14j were at lower level compared to above group, while higher than compound 13. Compounds 14a, 14c, 14g, 14h and 14i were predicted to have weaker activity and activity levels were weaker than the compound 13. Based on this test result, we predict the designed compounds have bioactivities targeting IDO1, and justified further experimental studies in terms of synthesis and biological activity, especially compound 14b showed the best activity level.
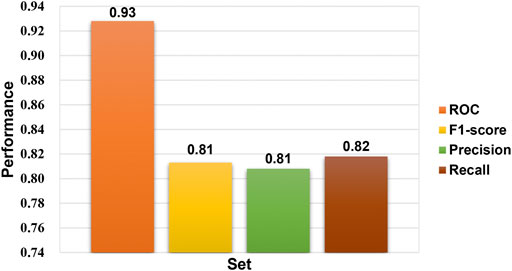
FIGURE 5. Performance of the Ido1 bioactivity model.
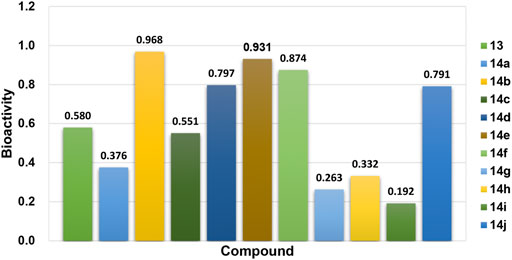
FIGURE 6. Bioactivity prediction results of the designed compounds.
To confirm the IDO1 inhibitory activities of the synthesized derivatives, all the compounds were screened using Hela cells as previously described (Malachowski et al., 2016; Qian et al., 2016). Cytotoxicity of compounds was performed by SHEE cell-based MTT assay (Xu et al., 2022). Epacadostat was chosen as a positive control with IC50 value of 90.7 nM, consistent with previously reported data (IC50 = 71.8 nM) (Nelp et al., 2018). The IDO1 inhibitory activities of synthesized compounds were shown in Table 2, indicating that most of the new compounds exhibited moderate to good IDO1 inhibitory activities (IC50 values <1.0 μM), particularly compound 14a (IC50 = 0.68 ± 0.42 μM) and compound 14b (IC50 = 0.59 ± 0.05 μM). The inhibition of most compounds on Hela cells exceeded 50 μM except for compound 14e (IC50 = 30.36 ± 3.61 μM). As a conclusion, these compounds could directly inhibit the activities of IDO1 enzyme without damaging Hela cells. Moreover, to test whether the inhibition of IDO1 by the compounds would produce cytotoxicity, we examined the growth inhibition of these compounds against SHEE cells. These results indicated that most compounds exhibited low inhibitory effects on SHEE cells. Therefore, we selected compound 14b to proceed for anti-tumor experiments in vivo considering its good IDO1 inhibitory activity and low cytotoxicity.
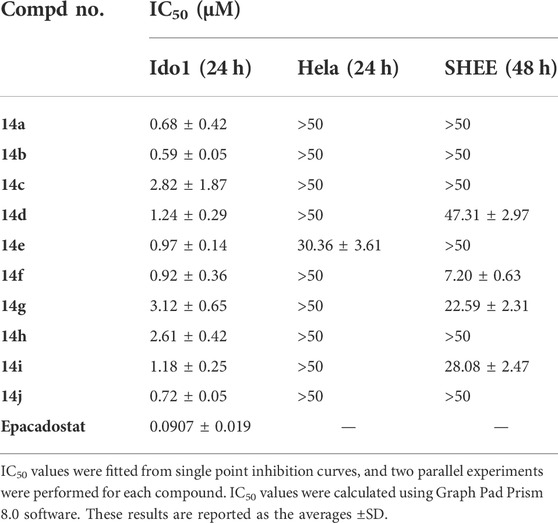
TABLE 2. IDO1 inhibitory activities of synthesized compounds.
Molecular docking is a computational modeling method that can explain the binding mechanism of IDO1 and compounds at the molecular level. Docking result depicted that the binding affinities of compounds 13 and 14b were both -9.0 kcal/mol. Figure 7 showed that compounds 13 and 14b were docked into the binding site of IDO1. The amino groups of Gly236 and Gly261 formed hydrogen bond with the oxygen of the ether chain of compound 13. The amino group of Gly261 formed a hydrogen bond with an oxygen of the ether chain of compound 14b. Both the quinazoline ring of compound 13 and the 1,3-dibromobenzyl group of compound 14b could form π-π stacking with Phe163. Additionally, the 1,3-dibromobenzyl of compound 14b was located in the hydrophobic pocket formed by Tyr126, Val130, Phe163, Ala264 and Gly261. The quinazoline ring of compound 14b was located in the active pocket formed by Phe226 and Arg231. According to Tojo et al., the interaction of ligand Amg-1 (Co-crystal ligand of 4PK5) with Phe226 and Arg231 is critical for the inhibitory activity of IDO1 (Tojo et al., 2014). The docking results indicated that compound 14b had better IDO1 inhibitory activity.
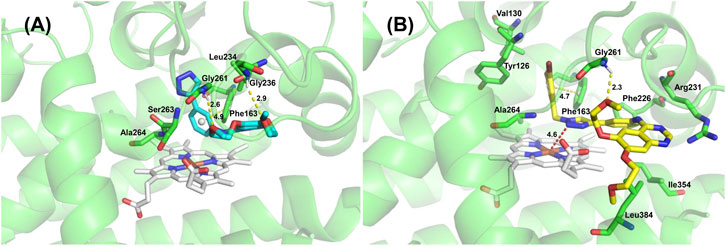
FIGURE 7. 3D binding modes of compound 13 (A) and compound 14b (B) in complex with IDO1.
The immunosuppressive effects of IDO1 promote resistance to cancer immune therapy. To better evaluate the role of IDO1 inhibitory activities of 14b in antitumor effects in vivo, we assessed the antitumor efficacy of 14b on an IDO1-overexpressing murine 4T1 breast model formed in immunocompetent BALB/c mice. The BALB/c mice harboring 4T1 tumors were intraperitoneally injected with 14b (25 mg/kg, qd), Erlotinib (25 mg/kg, qd) or well-known IDO1 inhibitor Epacadostat (25 mg/kg, bid) for 8 days, and then the tumors were harvested and weighed 14 days after inoculation (Figure 8A). As shown in Figure 8C, 14b and Epacadostat could efficiently suppress the 4T1 tumor growth (Figures 8B,D). Statistical analysis showed that 14b significantly inhibited tumor growth at day 8, 11 and 14 (p = 0.0359, p = 0.0431 and p = 0.0001) compared to NC group. While the results demonstrated that Erlotinib reduced the weight of the 4T1 tumors at day 14 post inoculation, the tumor volumes were not significantly changed compared to control group. Additionally, necrosis phenomenons such askaryolysis and karyorrhexis were frequently observed in tumor tissues exposed to 14b (Figure 8E).
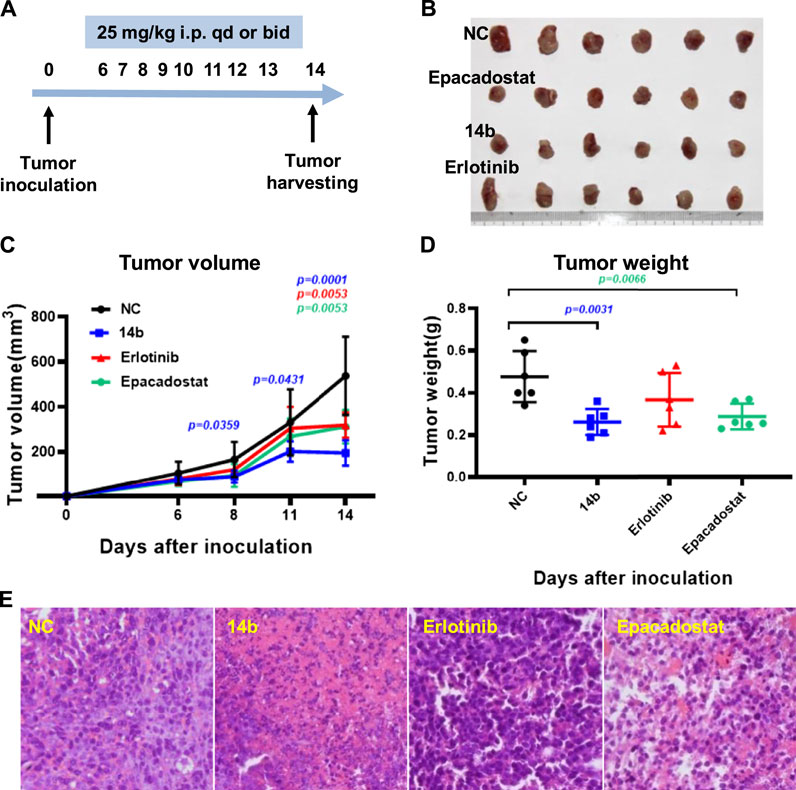
FIGURE 8. Effects of compound 14b on 4T1 allograft tumor growth in BALB/c mice. (A) Experimental procedure of the murine 4T1 breast model. (B) Growth morphology for the 4T1 tumors from the BALB/c mice after treatment with vehicle, compound 14b, Erlotinib and Epacadostat (n = 10, 9, 10 and 10, respectively). (C) Tumor volumes formed by 4T1 cells 14 days after inoculation (n = 6). (D) Tumor weight of 4T1 allograft were presented (n = 6). (E) Compound 14b-induced extensive necrosis within tumors detected by Hematoxylin and Eosin (H&E) staining.
To further explore the effects of 14b on antitumor immune response, cytokines associated with T cell functions within the 4T1 tumors were determined by qPCR. Interferon gamma (IFN-γ), Interleukin-2 (IL-2) and tumor necrosis factor (TNF-α) released by Th1 cells are typically associated with cytotoxic function, while transforming growth factor beta (TGF-β) and interleukin-10 (IL-10) driver T cells towards immune suppressive phenotypes in TME. Statistical analysis showed that 14b and Epacadostat significantly up regulated the RNA levels of IFN-γ within the tumors but the other cytokines were not affected obviously (Figure 9). These data indicated that 14b has emerged as a potential IDO1 inhibitor comparable to the clinical-stage IDO1 inhibitor. Besides, 14b increased the expression of IFN-γ in the TME, which was previously reported by other IDO1 inhibitors, and IFN-γ was associated with improved T cell function.
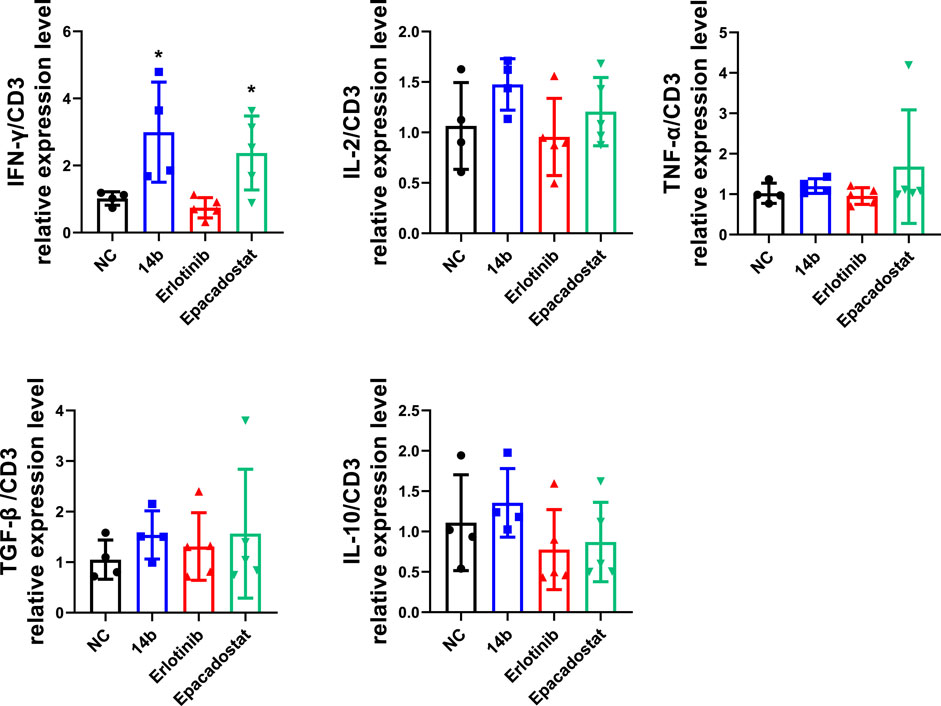
FIGURE 9. Compound 14b regulates T cell function-associated genes in TME. Cytokines associated with T cell functions including IFN-γ, IL-2, TNF-α, TGF-β and IL-10 within 4T1 tumors were analyzed by qPCR.
In conclusion, we designed and synthesized a series of erlotinib-based 1,2,3-triazole derivatives as potential IDO1 inhibitors. Attentive FP prediction results indicated that the inhibition of compound 14b against IDO1 was better than that of compound 13 (a reported IDO1 inhibitor). All designed compounds were evaluated by measuring the inhibitory activity of IDO1 using Hela cells and SHEE cells. Compound 14b (IC50 = 0.59 ± 0.05 μM) was found to have robust potency and low toxicity for IDO1 inhibition. In vivo antitumor studies shown that compound 14b could significantly inhibit the expression of IDO1 in mice with 4T1 tumor cells, as a result inhibited the tumor growth. Furthermore, the results of immune response experiments indicated that compound 14b could considerably up-regulate the RNA level of IFN-γ in tumors, thereby improving T cell function. These results warrant compound 14b for structural optimization and further study as a potential IDO1 inhibitor.
The target compounds were in-house synthesized. All reagents and solvents were purchased from Aladdin’s reagent or Sinopharm Group (China). 1H NMR spectra were acquired in DMSO-d6 solution with a Bruker600 or Bruker400 spectrometer. Hela cell line, DMEM medium and fetal bovine serum were purchased from ATCC(Virginia, United States).
Compound 1 (erlotinib, 1.0 mmol) and trinitrides (1.2 mmol) and cuprousiodide (0.1 mmol) were added to a mixed solvent (VH2O:V(CH3)3COH = 2:1, 30 ml).The reaction mixture was stirred for 10 h at 80°C. The mixture was extracted with DCM (dichloromethane, 20 ml × 3). The organic phase was washed successively with brine, then drying with Na2SO4 and desolventizing. The concentrate was purified by going through column chromatography to obtain the target compounds.
Compound 14a. Yield 69%. 1H NMR (600 MHz, DMSO-d6): δ9.56 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.27 (s, 1H), 7.92 (t, J1 = 6.0Hz, J2 = 12.0Hz, 2H), 7.57–7.56 (m, 1H), 7.46 (t, J1 = 6.0Hz, J2 = 6.0Hz, 1H),7.42–7.35 (m, 5H), 7.24 (s, 1H), 5.67 (s, 2H), 4.33–4.29 (m, 4H), 3.81–3.79 (m, 2H), 3.77–3.75 (m, 4H), 3.39 (s, 3H), 3.36 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 156.8, 154.1, 153.4, 148.6, 147.4, 147.1, 140.5, 136.5, 131.4, 129.5, 129.3, 128.7, 128.4, 122.3, 122.1, 120.8, 119.2, 109.4, 108.7, 103.7, 70.6, 70.5, 68.8, 68.5, 58.9, 58.8, 53.5. – MS (ESI): m/z (%) = 527 (100) [M + H]+.
Compound 14b. Yield 75%. 1H NMR (600 MHz, DMSO-d6): δ 9.58 (s, 1H), 8.72 (s, 1H), 8.49 (s, 1H), 8.28 (s, 1H), 7.92 (t, J1 = 12.0Hz, J2 = 6.0Hz, 2H), 7.86 (s, 1H), 7.64 (s, 2H), 7.57 (d, J = 6.0 Hz, 1H), 7.48–7.46 (m, 1H), 7.24 (s, 1H), 5.70 (s, 2H), 4.33–4.29 (m, 4H), 3.81–3.79 (m, 2H),3.77–3.75 (m, 2H), 3.39 (s, 3H), 3.36 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 156.8, 154.1, 153.4, 148.6, 147.5, 147.2, 140.8, 140.6, 133.7, 131.2, 130.7, 129.5, 123.2, 122.4, 120.8, 119.3, 109.4, 108.7, 103.7, 70.6, 70.5, 68.8, 68.5, 58.9, 58.8, 52.0. – MS (ESI): m/z (%) = 683 (100) [M + H]+.
Compound 14c. Yield 42%. 1H NMR (600 MHz, DMSO-d6): δ 9.61 (s, 1H), 8.66 (s, 1H), 8.27 (s, 1H), 8.01 (s, 1H), 7.91–7.90 (m, 1H), 7.58–7.56 (m, 1H), 7.47–7.45 (m, 1H), 7.33–7.32 (m, 2H), 6.97–6.92 (m, 3H), 5.63 (s, 2H), 4.33–4.27 (m, 4H), 3.80–3.76 (m, 4H), 3.76 (s, 3H), 3.39 (s, 3H), 3.37 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 159.9, 156.8, 154.0, 148.6, 147.1, 140.5, 137.9, 131.4, 130.5, 129.5, 122.4, 122.1, 120.9, 120.5, 119.3, 114.3, 114.0, 108.9, 103.8, 87.8, 70.6, 70.5, 68.9, 68.5, 58.9, 58.8, 55.6, 53.5, 22.6. – MS (ESI): m/z (%) = 557 (100) [M + H]+.
Compound 14d. Yield 87%. 1H NMR (400 MHz, DMSO-d6): 9.55 (s, 1H), 8.60 (s, 1H), 8.26 (s, 1H), 7.92 (t, J1 = 18.0Hz, J2 = 12.0Hz, 2H), 7.57 (t, J1 = 6.0Hz, J2 = 18.0Hz, 2H), 7.49–7.44 (m, 2H), 7.33–7.28 (m, 2H), 5.75 (s, 2H), 4.33–4.28 (m, 4H), 3.81–3.79 (m, 2H), 3.77–3.75 (m, 2H), 3.39 (s, 3H), 3.37 (s, 3H).13C NMR (100Hz, DMSO-d6): 163.5, 161.1, 153.7, 148.7, 146.9, 140.4, 134.3, 134.2, 133.0, 131.3, 130.0, 129.5, 122.4, 122.3, 120.9, 119.3, 117.6, 117.4, 115.5, 115.3, 70.5, 68.8, 68.5, 58.8, 50.7. – MS (ESI): m/z (%) = 579 (100) [M + H]+.
Compound 14e. Yield 62%. 1H NMR (400 MHz, DMSO-d6): δ 9.58 (s, 1H), 8.63 (s, 1H), 8.26 (s, 2H), 7.91 (s, 1H), 7.66–7.56 (m, 3H), 7.55–7.37 (m, 3H), 5.71 (s, 2H), 4.33–4.29 (m, 4H), 3.79–3.77 (m, 4H), 3.40 (s, 3H), 3.37 (s, 3H). 13C NMR (100Hz, DMSO-d6): 161.7, 159.2, 148.7, 147.0, 140.4, 132.8, 131.2, 129.5, 122.9, 122.7, 122.6, 122.4, 122.2, 120.9, 119.7, 119.4, 119.3, 70.5, 68.8, 68.5, 58.8, 47.2. – MS (ESI): m/z (%) = 623 (100) [M + H]+.
Compound 14f. Yield 58%. 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.37 (s, 1H), 8.51 (s, 1H), 8.38 (s, 1H), 7.98–7.94 (m, 4H), 7.87–7.85 (m, 2H), 7.67–7.65 (m, 1H), 7.54–7.52 (m, 1H), 7.25 (s, 1H), 4.34–4.32 (m, 2H),4.31–4.30 (m, 2H), 3.82–3.80 (m, 2H), 3.77–3.76 (m, 2H),3.39 (s, 3H), 3.37 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 156.9, 154.1, 153.4, 148.6, 148.0, 147.4, 140.6, 136.3, 133.3, 130.8, 129.6, 122.8, 122.4, 121.8, 121.0, 120.2, 119.5, 109.4, 108.7, 103.7, 70.6, 70.5, 68.8, 68.5, 58.9, 58.8. – MS (ESI): m/z (%) = 591 (100) [M + H]+.
Compound 14g. Yield 88%. 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.36 (s, 1H), 8.51 (s, 1H), 8.37 (s, 1H), 7.96–7.95 (m, 2H), 7.67–7.66 (m, 1H), 7.58–7.56 (m, 1H), 7.55–7.52 (m, 3H), 7.25 (s, 1H), 7.08–7.07 (m, 1H), 4.34–4.32 (m, 2H),4.31–4.30 (m, 2H), 4.16 (dd, J1 = 6.0Hz, J2 = 6.0Hz,2H), 3.82–3.80 (m, 2H), 3.77–3.76 (m, 2H), 3.39 (s, 3H), 3.37 (s, 3H), 1.40–1.38 (m, 3H). 13C NMR (150 MHz, DMSO-d6): 159.9, 156.9, 154.1, 153.4, 148.6, 147.8, 147.5, 140.6, 138.1, 131.4, 131.0, 129.6, 122.8, 121.0, 120.1, 119.5, 115.3, 112.3, 109.5, 108.7, 106.5, 103.7, 70.6, 70.5, 68.8, 68.5, 64.1, 58.9, 58.8, 15.0. – MS (ESI): m/z (%) = 557 (100) [M + H]+.
Compound 14h. Yield 71%. 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.34 (s, 1H), 8.50 (s, 1H), 8.38 (s, 1H), 8.00–7.94 (m, 4H), 7.68–7.64 (m, 3H), 7.54–7.51 (m, 2H), 7.24 (s, 1H), 4.34–4.32 (m, 2H),4.31–4.29 (m, 2H), 3.81–3.80 (m, 2H), 3.77–3.76 (m, 2H), 3.39 (s, 3H), 3.36 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 156.9, 154.1, 153.4, 148.6, 147.8, 147.5, 140.6, 137.1, 131.0, 130.4, 129.6, 129.2, 122.7, 121.0, 120.5, 120.2, 119.5, 109.4, 108.7, 103.7, 70.6, 70.5, 68.8, 68.5, 58.9, 58.8.—MS (ESI): m/z (%) = 513 (100) [M + H]+.
Compound 14i. Yield 48%. 1H NMR (400 MHz, DMSO-d6): δ 9.66 (s, 1H), 8.88 (s, 1H), 8.55 (s, 1H), 8.39 (s, 1H), 8.02–7.98 (m, 1H), 7.97–7.92 (m, 1H), 7.71–7.70 (m, 1H), 7.63–7.62 (m, 1H), 7.55–7.73 (m, 1H), 7.29 (s, 1H), 6.92–6.91 (m, 1H), 6.79–6.75 (m, 1H), 4.39–4.37 (m, 2H), 4.36–4.34 (m, 2H), 3.93 (s, 3H), 3.92 (s, 3H), 3.87–3.80 (m, 4H), 3.44 (s, 3H), 3.42 (s, 3H). 13C NMR (100 MHz, DMSO-d6): 161.7, 156.8, 154.0, 153.7, 153.4, 148.5, 146.5, 140.5, 131.3, 129.5, 127.5, 124.1, 122.5, 121.0, 119.6, 119.3, 108.6, 105.7, 103.6, 99.9, 70.6, 70.5, 68.8, 68.5, 58.9, 58.8, 56.7, 56.2.—MS (ESI): m/z (%) = 573 (100) [M + H]+.
Compound 14j. Yield 71%. 1H NMR (400 MHz, DMSO-d6): δ 9.66 (s, 1H), 8.98 (s, 1H), 8.55 (s, 1H), 8.40 (s, 1H), 8.00–7.98 (m, 2H), 7.75–7.71 (m, 2H), 7.63 (t, J = 6.0 Hz, 1H), 7.55 (t, J = 6.0 Hz, 1H), 7.41–7.40 (m, 1H), 7.29 (s, 1H), 7.24 (t, J = 6.0 Hz, 1H), 4.39–4.37 (m, 2H), 4.36–4.34 (m, 2H), 3.95 (s, 3H), 3.87–3.80 (m, 4H), 3.44 (s, 3H), 3.42 (s, 3H). 13C NMR (100 MHz, DMSO-d6): 156.9, 154.0, 153.4, 152.3, 148.5, 147.4, 146.7, 140.5, 131.4, 131.2, 129.5, 126.5, 126.2, 124.0, 122.6, 121.4, 121.0, 119.4, 113.5, 109.4, 108.6, 103.6, 70.6, 70.5, 68.9, 68.5, 58.9, 58.8, 56.6.—MS (ESI): m/z (%) = 543 (100) [M + H]+.
We used Attentive FP to predict bioactivities for the designed compounds. Firstly, we obtained the activity dataset of IDO1 from the ChEMBL database (1785 molecules), which used pIC50 (negative logarithm of IC50) as an indicator of activity. IDO1 activity dataset was split into positive and negative sets by setting a threshold of 6.0. Before training the model, we used k-means clustering and t-SNE dimensionality reduction to display the distribution of IDO1 activity data, as shown in Figure 10. In this work, IDO1 dataset was used to train the activity prediction classification model. The model used the Adam optimizer for gradient descent to maintain the best parameters of the model. After training for 800 epochs, the best model for the IDO1 target was output, and finally this model was used to test the activity probability of our designed compounds against IDO1. This model used CrossEntropyLoss to measure the cross entropy to calculate the loss function. The AUC, F1-score, Precision, Recall as performance evaluation indicators.
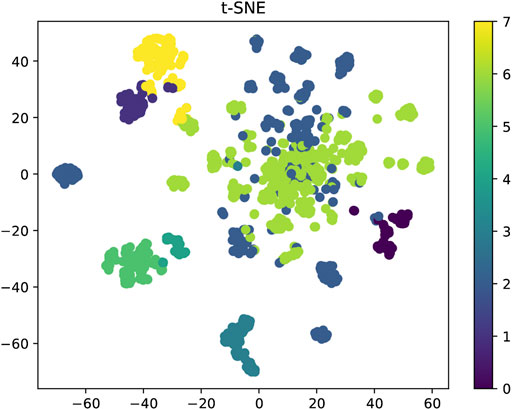
FIGURE 10. Distribution of IDO1 activity dataset based on pIC50.
To demonstrate the inhibitory effect of the designed compounds against IDO1, Hela cells were seeded at 50,000∼60,000 cells per well into 96-well plate in 100 μl of dulbecco’s modified eagle medium complete growth medium for 12∼18 h. The second day, 100 μl per well of diluted inhibitor was added at a final concentration of 100 ng/ml human IFNγ, and then incubated at 37°C, 5% CO2 for 18 h. The third day, 140 μl of medium was removed into a new 96-well plate and precipitate the protein with 10 μl of 6.1 N TCA (CCl3COOH) at 50°C for 30 min. The plate was centrifuged at 2,500 rpm for 10 min. Then 100 μl of supernatant per well was transferred to another 96-well plate and mixed with 100 μl of p-Dimethylaminobenzaldehyde in CH3COOH [2% (w/v)]. The plate was incubated at 25°C for 10 min, the yellow color derived from kynurenine was recorded by measuring absorbance at 480 nm using a microplate reader (PE, United States).
We used the Cell Counting Kit-8 (CCK-8, Meilunbio, Dalian, China) assay to evaluate whether compounds directly inhibit IDO1 in Hela cells. Make 1,000× compounds solution in DMSO. Add 1 μl 1,000× compounds to 49 μl growth medium to make 20× compounds. Dilute cell suspensions in growth medium to desired density and 95 μl were taken to 96-well plate. Add 5 µl 20× compounds into 96-well plate according to the plate map. Final DMSO concentration in each well was 0.1%. Then the cell was incubated at 37°C, 5% CO2 for 24 h. The next day, 20 μl CCK-8 solution was added into each well. After 2 h of incubation at 37°C, the plate was measured at 450 nm by an EnVision Multilabel Reader (PerkinElmer).
Cytoxicity of compounds against SHEE cells was evaluated by MTT assay. The SHEE cells were seeded in 96-well plates with densities of 2,200
In this study, we used AutoDockTools (ADT) 1.5.6 software to process protein and ligands, and then constructed the parameter files required for docking (Morris et al., 2009). The crystal structure of IDO1 was obtained from the RCSB PDB database for docking modeling (PDB code:4PK5). IDO1 Protein was prepared by removing water, adding polar hydrogens, calculating Gasteiger charges and assigning AD4 type. Ligands were constructed by ChemDraw and optimized by MMFF94 and MM2 force fields, and then the torsional degrees of freedom of the ligands were detected by ADT. AutoDock Vina was used to perform molecular docking operations (Trott and Olson, 2010). Protein-Ligand Interaction Profiler (PLIP) web tool and PyMol software were used to visualize the interactions of IDO1 and designed compounds (Adasme et al., 2021).
4T1 allograft were constructed by subcutaneous transplantation of cancer cells into the flanks of 4–6 weeks old BALB/c mice. Tumor volumes were measured using calipers and calculated as above, all mice were assigned randomly to four groups: one group was intraperitoneally injected with vehicle (5% DMSO, 30% PEG300 and 65% ddH2O; qd), one group was injected with compound 14b (25 mg/kg, qd), one group was injected with Erlotinib (25 mg/kg, qd) and one group received daily injections of Epacadostat (25 mg/kg, bid) in vehicle for eight consecutive days. Two weeks after inoculation, the mice were executed and the tumor tissues were harvested and weighed. Hematoxylin and Eosin (H&E) staining for the tumors was performed as we previously described (Xu et al., 2016). A piece of tumor was randomly selected for RNA extraction and RNA was converted to cDNA according to the manufacturer’s instructions. The primers for the target genes were shown in Table3 qPCR was performed to quantification of genes including IFN-γ, IL-2, TNF-α, TGF-β and IL-10. Statistical significance was evaluated by the Student’s t-test.
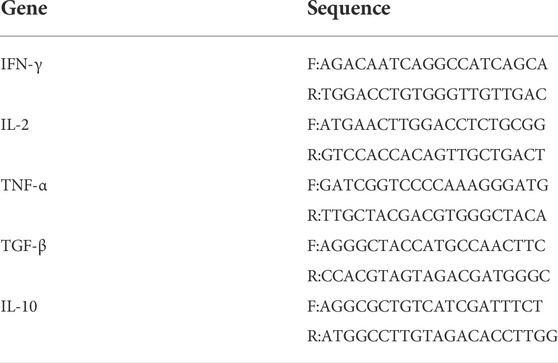
TABLE 3. Primer sequences of the target genes.
4T1 tumors cells were incubated in 12-well plates for 24 h, and then treated with 0.1% DMSO, various concentrations of either compound 14b for 16 h. Total RNA was extracted from 4T1 tumors cells tissue and cDNA was synthesized according to the manufacturer’s instructions. Quantitative analysis of IFN-γ, IL-2, TNF-α, TGF-β and IL-10 genes by qPCR, the primers of the genes were shown in Table 3 (Mao et al., 2020).
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by The Ethics Committee for the Care and Use of Laboratory Animals of Southern Medical University.
XH, XG and LM carried out the research and wrote the manuscript with support from GS and JY. XH completed the synthesis of compounds. XG completed the computational experiments. LM and GS carried out the biological activity experiments. GS and JY helped design and conceive the project.
The authors appreciate the financial support from the Scientific and Technological Project of Henan Province (No. 192102310142) and National Natural Science Foundation of China (NSFC 21672106). Computational resources were supported by Supercomputing Center of Lanzhou University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.940704/full#supplementary-material
Adasme, M. F., Linnemann, K. L., Bolz, S. N., Kaiser, F., Salentin, S., Haupt, V. J., et al. (2021). Plip 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 49 (W1), W530–W534. doi:10.1093/nar/gkab294
Ayyappan, S., Prabhakar, D., and Sharma, N. (2013). Epidermal growth factor receptor (EGFR)-targeted therapies in esophagogastric cancer. Anticancer Res. 33, 4139–4155. doi:10.1097/COC.0b013e31821dee4e
Cohen, M. H., Williams, G. A., Sridhara, R., Chen, G., and Pazdur, R. (2003). FDA drug approval summary: gefitinib (ZD1839) (iressa) tablets. Oncologist 8, 303–306. doi:10.1634/theoncologist.8-4-303
Deng, P., Sun, G., Zhao, J., Yao, K., Yuan, M., Peng, L., et al. (2022). Synthesis and antitumor activity of erlotinib derivatives linked with 1, 2, 3-triazole. Front. Pharmacol. 12, 793905. doi:10.3389/fphar.2021.793905
Fox, E., Oliver, T., Rowe, M., Thomas, S., Zakharia, Y., Gilman, P. B., et al. (2018). Indoximod: an immunometabolic adjuvant that empowers T cell activity in cancer. Front. Oncol. 8, 370. doi:10.3389/fonc.2018.00370
Hirsch, F. R., Scagliotti, G. V., Mulshine, J. L., Kwon, R., Curran, W. J., Wu, Y.-L., et al. (2017). Lung cancer: current therapies and new targeted treatments. Lancet 389, 299–311. doi:10.1016/S0140-6736(16)30958-8
Jänne, P. A., Yang, J. C.-H., Kim, D.-W., Planchard, D., Ohe, Y., Ramalingam, S. S., et al. (2015). AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 372, 1689–1699. doi:10.1056/NEJMoa1411817
Maddili, S. K., Katla, R., Kannekanti, V. K., Bejjanki, N. K., Tuniki, B., Zhou, C. H., et al. (2018). Molecular interaction of novel benzothiazolyl triazolium analogues with calf thymus DNA and HSA-their biological investigation as potent antimicrobial agents. Eur. J. Med. Chem. 150, 228–247. doi:10.1016/j.ejmech.2018.02.056
Malachowski, W. P., Winters, M., Duhadaway, J. B., Lewis-Ballester, A., Badir, S., Wai, J., et al. (2016). O-alkylhydroxylamines as rationally-designed mechanism-based inhibitors of indoleamine 2, 3-dioxygenase-1. Eur. J. Med. Chem. 108, 564–576. doi:10.1016/j.ejmech.2015.12.028
Mao, L. F., Wang, Z. Z., Wu, Q., Chen, X., Yang, J. X., Wang, X., et al. (2022). Design, synthesis, and antitumor activity of erlotinib derivatives. Front. Pharmacol. 13, 849364. doi:10.3389/fphar.2022.849364
Mao, L., Sun, G., Zhao, J., Xu, G., Yuan, M., Li, Y. M., et al. (2020). Design, synthesis and antitumor activity of icotinib derivatives. Bioorg. Chem. 105, 104421. doi:10.1016/j.bioorg.2020.104421
Mathew, M. P., Tan, E., Saeui, C. T., Bovonratwet, P., Liu, L., Bhattacharya, R., et al. (2015). Metabolic glycoengineering sensitizes drug-resistant pancreatic cancer cells to tyrosine kinase inhibitors erlotinib and gefitinib. Bioorg. Med. Chem. Lett. 25, 1223–1227. doi:10.1016/j.bmcl.2015.01.060
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30 (16), 2785–2791. doi:10.1002/jcc.21256
Munn, D. H., and Mellor, A. L. (2016). Ido in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 37, 193–207. doi:10.1016/j.it.2016.01.002
Nelp, M. T., Kates, P. A., Hunt, J. T., Newitt, J. A., Balog, A., Maley, D., et al. (2018). Immune-modulating enzyme indoleamine 2, 3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. U. S. A. 115, 3249–3254. doi:10.1073/pnas.1719190115
Panda, S., Pradhan, N., Chatterjee, S., Morla, S., Saha, A., Roy, A., et al. (2019). 4, 5-disubstituted 1, 2, 3-triazoles: effective inhibition of indoleamine 2, 3-dioxygenase 1 enzyme regulates T cell activity and mitigates tumor growth. Sci. Rep. 9, 18455. doi:10.1038/s41598-019-54963-9
Platten, M., Nollen, E. a. A., Rohrig, U. F., Fallarino, F., and Opitz, C. A. (2019). Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 18, 379–401. doi:10.1038/s41573-019-0016-5
Qian, S., He, T., Wang, W., He, Y., Zhang, M., Yang, L., et al. (2016). Discovery and preliminary structure-activity relationship of 1H-indazoles with promising indoleamine-2, 3-dioxygenase 1 (Ido1) inhibition properties. Bioorg. Med. Chem. 24, 6194–6205. doi:10.1016/j.bmc.2016.10.003
Qin, X., Li, Z., Yang, L., Liu, P., Hu, L., Zeng, C., et al. (2016). Discovery of new [1, 4]dioxino[2, 3-f]quinazoline-based inhibitors of EGFR including the T790M/L858R mutant. Bioorg. Med. Chem. 24, 2871–2881. doi:10.1016/j.bmc.2016.01.003
Röhrig, U. F., Awad, L., Grosdidier, A., Larrieu, P., Stroobant, V., Colau, D., et al. (2010). Rational design of indoleamine 2, 3-dioxygenase inhibitors. J. Med. Chem. 53 (3), 1172–1189. doi:10.1021/jm9014718
Schupp, J., Krebs, F. K., Zimmer, N., Trzeciak, E., Schuppan, D., Tuettenberg, A., et al. (2019). Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol. 343, 103713. doi:10.1016/j.cellimm.2017.10.013
Shi, Y., Zhang, L., Liu, X., Zhou, C., Zhang, L., Zhang, S., et al. (2013). Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial. Lancet. Oncol. 14, 953–961. doi:10.1016/S1470-2045(13)70355-3
Smith, C., Chang, M. Y., Parker, K. H., Beury, D. W., Duhadaway, J. B., Flick, H. E., et al. (2012). Ido is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2, 722–735. doi:10.1158/2159-8290.CD-12-0014
Smith, J. (2005). Erlotinib: small-molecule targeted therapy in the treatmentof non-small-cell lung cancer. Clin. Ther. 27 (10), 1513–1534. doi:10.1016/j.clinthera.2005.10.014
Sun, G., Mao, L., Deng, W., Xu, S., Zhao, J., Yang, J., et al. (2022). Discovery of a series of 1, 2, 3-triazole-containing erlotinib derivatives with potent anti-tumor activities against non-small cell lung cancer. Front. Chem. 9, 789030. doi:10.3389/fchem.2021.789030
Theate, I., Van Baren, N., Pilotte, L., Moulin, P., Larrieu, P., Renauld, J. C., et al. (2015). Extensive profiling of the expression of the indoleamine 2, 3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 3, 161–172. doi:10.1158/2326-6066.CIR-14-0137
Tojo, S., Kohno, T., Tanaka, T., Kamioka, S., Ota, Y., Ishii, T., et al. (2014). Crystal structures and structure-activity relationships of imidazothiazole derivatives as Ido1 inhibitors. ACS Med. Chem. Lett. 5 (10), 1119–1123. doi:10.1021/ml500247w
Trott, O., and Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31 (2), 455–461. doi:10.1002/jcc.21334
Wang, W., Huang, L., Jin, J. Y., Jolly, S., Zang, Y., Wu, H., et al. (2018). Ido immune status after chemoradiation may predict survival in lung cancer patients. Cancer Res. 78, 809–816. doi:10.1158/0008-5472.CAN-17-2995
Wei, W. T., Wang, L., Liang, J. X., Wang, J. F., and Zeng, J. (2020). LncRNA EIF3J-AS1 enhanced esophageal cancer invasion via regulating AKT1 expression through sponging miR-373-3p. Sci. Rep. 10, 13969. doi:10.1038/s41598-020-70886-2
Xiong, Z. P., Wang, D. Y., Liu, X. H., Zhong, F. S., Wang, X. Z., Li, X. T., et al. (2019). Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 63 (16), 8749–8760. doi:10.1021/acs.jmedchem.9b00959
Xu, G. Q., Gong, X. Q., Zhu, Y. Y., Yao, X. J., Peng, L. Z., Sun, G., et al. (2022). Novel 1, 2, 3-triazole erlotinib derivatives as potent Ido1 inhibitors: design, drug-target interactions prediction, synthesis, biological evaluation, molecular docking and adme properties studies. Front. Pharmacol. 13, 854965. doi:10.3389/fphar.2022.854965
Xu, S., Chen, W., Xie, Q., and Xu, Y. (2016). Obacunone activates the Nrf2-dependent antioxidant responses. Protein Cell 7, 684–688. doi:10.1007/s13238-016-0297-y
Keywords: erlotinib, 1,2,3-triazole, IDO1, Attentive FP, antitumor
Citation: Hou X-x, Gong X-q, Mao L-f, Sun G and Yang J-x (2022) Design, synthesis and biological evaluation of erlotinib-based IDO1 inhibitors. Front. Pharmacol. 13:940704. doi: 10.3389/fphar.2022.940704
Received: 23 May 2022; Accepted: 15 July 2022;
Published: 10 August 2022.
Edited by:
Yuanliang Yan, Central South University, ChinaReviewed by:
Andrea Angeli, University of Florence, ItalyCopyright © 2022 Hou, Gong, Mao, Sun and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ge Sun, c3VuZ2UyMDE0QDE2My5jb20=; Jian-xue Yang, RG9jeWp4MTk2OUAxMjYuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.