 Priya Bisht
Priya Bisht V. Udaya Kumar
V. Udaya Kumar Ruchi Pandey
Ruchi Pandey Ravichandiran Velayutham1
Ravichandiran Velayutham1 Nitesh Kumar
Nitesh Kumar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 06 July 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.939570
This article is part of the Research Topic Emerging Talents in Frontiers in Pharmacology: Experimental Pharmacology and Drug Discovery 2022 View all 4 articles
Glioblastoma multiform is the most aggressive primary type of brain tumor, representing 54% of all gliomas. The average life span for glioblastoma multiform is around 14–15 months instead of treatment. The current treatment for glioblastoma multiform includes surgical removal of the tumor followed by radiation therapy and temozolomide chemotherapy for 6.5 months, followed by another 6 months of maintenance therapy with temozolomide chemotherapy (5 days every month). However, resistance to temozolomide is frequently one of the limiting factors in effective treatment. Poly (ADP-ribose) polymerase (PARP) inhibitors have recently been investigated as sensitizing drugs to enhance temozolomide potency. However, clinical use of PARP inhibitors in glioblastoma multiform is difficult due to a number of factors such as limited blood–brain barrier penetration of PARP inhibitors, inducing resistance due to frequent use of PARP inhibitors, and overlapping hematologic toxicities of PARP inhibitors when co-administered with glioblastoma multiform standard treatment (radiation therapy and temozolomide). This review elucidates the role of PARP inhibitors in temozolomide resistance, multiple factors that make development of these PARP inhibitor drugs challenging, and the strategies such as the development of targeted drug therapies and combination therapy to combat the resistance of PARP inhibitors that can be adopted to overcome these challenges.
Glioma is defined as one of the primary brain tumors that are categorized based on their origin cells such as astrocytic tumors (anaplastic astrocytoma or glioblastoma multiform), oligodendrogliomas, ependymomas, and mixed gliomas. They are among the most prevalent types of tumors present in the central nervous system (CNS), accounting for over 80% of overall primary brain tumors which are malignant (Hanif et al., 2017). Glioblastoma multiform (GBM)/glioblastoma is one such type of glioma which is highly prevalent and accounts for 54% of all gliomas, among which malignant primary brain as well as CNS tumors comprise 45.2%, while 16% is carried by primary brain as well as CNS tumors. Additionally, it is an aggressive primary malignant brain tumor, which has been categorized as grade IV type by the World Health Organization (WHO). The average life span for GBM is around 14–15 months after diagnosis (Louis et al., 2007; Ghosh et al., 2018). GBM has a global incidence of 0.59–3.69 per 100,000 live births, with a male-adjusted incidence rate of 3.97 cases per 100,000 and a female-adjusted incidence rate of 2.53 cases per 100,000 (Ghosh et al., 2018). GBM most frequently occurs in the hemispheres of the cerebrum, among which 95% of GBM originates in the supratentorial area, although hardly a small percentage of GBM arises in the cerebellum, brainstem, or spinal cord (Nakada et al., 2011). GBM is divided into two main subtypes: primary and secondary, and the term was first coined in Antwerp in 1940 by the German neuropathologist Hans Joachim Sherer (Kleihues and Ohgaki, 1999). Primary GBM is the most common subtype with 80% of cases and manifests later in life at the median age of 62 years, while secondary GBM manifests earlier in life at the median age of 45 years (Kleihues and Ohgaki, 1999; Ohgaki and Kleihues, 2009; Ohgaki et al., 2004; Watanabe et al., 2009). Primary GBM is associated with overexpression of epidermal growth factor receptor (EGFR) as well as mouse double minute 2 (MDM2) gene along with deletion of p16, loss of heterozygosity (LOH) of chromosome 10q holding PTEN (phosphatase and tensin homolog), and telomerase reverse transcriptase (TERT) promoter mutation while secondary class of GBM originates from low-grade astrocytoma or oligodendrogliomas and often contain mutations in tumor protein 53 (TP53), ATRX, and isocitrate dehydrogenase 1/2 (IDH1/2) or overexpression of LOH of 19q, retinoblastoma (RB), platelet-derived growth factor A, and platelet-derived growth receptor alpha (PDGFA/PDGFRa) (Davis, 2016; Hanif et al., 2017). A rare subtype of GBM named GBM-0 is also added by WHO with an oligodendroglioma feature, which is characterized as GBM with regions resembling anaplastic oligodendroglioma, having GBM-like characteristics and necrosis but without microvascular proliferation (Louis et al., 2007). The current therapy available for the treatment of GBM includes clinical removal of the tumor adjuvant to radiation therapy (RT) with temozolomide (TMZ) administration. TMZ is a first-line chemotherapeutic agent widely used in the treatment of GBM; however, resistance to TMZ is frequently the limiting factor in effective treatment (Gupta et al., 2019; Singh et al., 2021). The principal pathway involved in the induction of resistance toward TMZ is through the activation of multiple DNA repair pathways (MGMT-O6-methylguanine DNA methyltransferase, MMR-mismatch repair, and BMR-base excision repair). However, many other pathways also contribute to developing resistance to TMZ which includes hyperactivation of DNA repair pathways, aberrant signaling pathways, epigenetic modifications, autophagy, extracellular vesicle production, and microRNAs (Singh et al., 2021; Tomar et al., 2021). Poly (ADP-ribose) polymerase (PARP) inhibitors have been recently investigated as sensitizing drugs to enhance TMZ potency. PARP is a class of enzyme that participates in the BER pathway and is also involved in the MGMT pathway by physically interacting with and ultimately PARylates MGMT as a response to TMZ chemotherapy to eliminate adducts of O6-methylguanine (O6-MetG) from the damaged segment of DNA. Second, PARP works as a sensor, triggering the BER response pathways. PARP inhibitor drugs block binding of PARP-MGMT or PARylation of MGMT, reducing MGMT function and preventing O6-MetG repair. As a result, the MGMT function is reduced, resulting in TMZ sensitization and providing a rationale to sensitize (Wu et al., 2021). PARP inhibitor drugs have been investigated in GBM patients in a number of clinical trials. However, multiple factors make the clinical development of these inhibitors challenging. This review elucidates PARP and the relevance of PARP inhibitor drugs in the treatment of TMZ resistance in GBM. Furthermore, the challenges that may arise during the clinical trials of PARP inhibitors as well as the strategies to overcome these hurdles are highlighted.
TMZ is a type of prodrug which is an imidazotetrazine analog of an anticancer alkylating agent, dacarbazine. This is commonly used as a chemotherapeutic agent under the brand name Temodar (Moody and Wheelhouse, 2014). TMZ is lipophilic in nature, and it can penetrate the BBB and be taken orally (Lee, 2016). It is a DNA alkylating agent that causes arrest at the G2/M phase of the cell cycle and subsequently induces apoptosis (Alonso et al., 2007). At physiological pH, TMZ drug is being transformed into 5-(3-methyltriazen-1-yl) imidazole-4-carboxamide (MTIC), a form of an active metabolite which is again degraded in 5-aminoimidazole-4-carboxamide (AIC) or methylhydrazine. The cytotoxic response of TMZ relies mainly on its DNA methylation efficacy, which takes place at the N7 or O6 guanine position or at the O3 position of adenine within the genomic DNA. Methylation at O6 guanine causes the addition of thymine rather than cytosine in front of methylguanine during the upcoming DNA replication process which further triggers the death of tumor cells (Lee, 2016). TMZ has been shown to be effective against human malignancies such as astrocytomas and melanomas (Middleton et al., 2000; Quirt et al., 2007; Yung et al., 1999; Hart et al., 2013). In 1999, TMZ was the first approved medicament used for recently diagnosed glioblastoma treatment as well as in refractory anaplastic astrocytoma in young patients by the United States Food and Drug Administration (USFDA). Patients who are newly diagnosed with adult GBM, when received concurrent Temodar and radiation, had shown a higher overall survival count than those who received radiation only (12.1/14.6 month’s average survival) (Cohen et al., 2005). Therefore, the currently available treatment for GBM is removal via surgery and subsequently by RT with adjuvant TMZ. The first-line chemotherapeutic agent TMZ is used for the treatment of GBM (Gupta et al., 2019; Singh et al., 2021). Unfortunately, due to the resistance, only around 50% of patients respond to TMZ (Lee, 2016). The principal pathway involved in the development of resistance of TMZ is through activation of various DNA repair pathways (MGMT, MMR, and BMR). However, many other pathways also play a vital role in TMZ resistance which includes hyperactivation of DNA repair pathways, aberrant signaling pathways, epigenetic modifications, autophagy, extracellular vesicle production, and microRNAs (Singh et al., 2021; Tomar et al., 2021). Recently, poly (ADP-ribose) polymerase (PARP) inhibitor drugs are being investigated as sensitizing drugs to enhance TMZ potency (Wu et al., 2021).
PARPs are a new class of enzymes that use β-NAD+ as a substrate to catalyze transmission of ADP-ribose on target proteins (i.e., poly ADP-ribosylation) (Amé et al., 2004; d'AMOURS et al., 1999; Lal and Snape, 2021). The process of attaching ADP-ribose to target proteins through PARP is known as PARsylation (Lal and Snape, 2021). PARPs are implied in a variety of processes at the molecular level, such as transcription, recombination, replication, DNA repair, and chromatin structure modulation (Morales et al., 2014). The PARP family comprises 18 members, encoded by a distinct gene and has a fixed catalytic domain. Some members of the PARP family such as PARP1 or PARP2 are widely recognized for their roles in DNA repair mechanism (Amé et al., 2004). PARP is a member of the BER complex, which includes XRCC1 protein, DNA ligase, and the DNA polymerase beta, and is involved in the BER-mediated pathway in response to single-stranded DNA breaks (SSBs) (Caldecott et al., 1996). In cell-free conditions, it has been found that the PARP enzyme in an unmodified form, attaches strongly to DNA strand breaks and, subsequently, auto-poly ADP-ribosylation is released, allowing repair enzyme exposure to access the damaged DNA (d'AMOURS et al., 1999; Satoh and Lindahl, 1992). Both PARP1 and PARP2 act as well as share similar partners in the SSR repair mechanism and BER processes (Amé et al., 2004; Schreiber et al., 2002). PARP1 has been found to play a functional response in nucleotide excision repair (NER), as NER functions are diminished when the PARP1 enzyme is being inhibited (Flohr et al., 2003). PARP is also involved in the MGMT pathway by physically interacting with it and ultimately PARylates MGMT as an effect of TMZ treatment to eliminate the O6-MetG complex in double-stranded damaged DNA, independent of the BER pathway (Wu et al., 2021). BER, NER, and MGMT are important pathways for repairing damaged DNA induced by alkylating and chemotherapeutic drugs. (15,103) Figure 1 elaborates on the role of PARP inhibiting drugs in TMZ resistance.
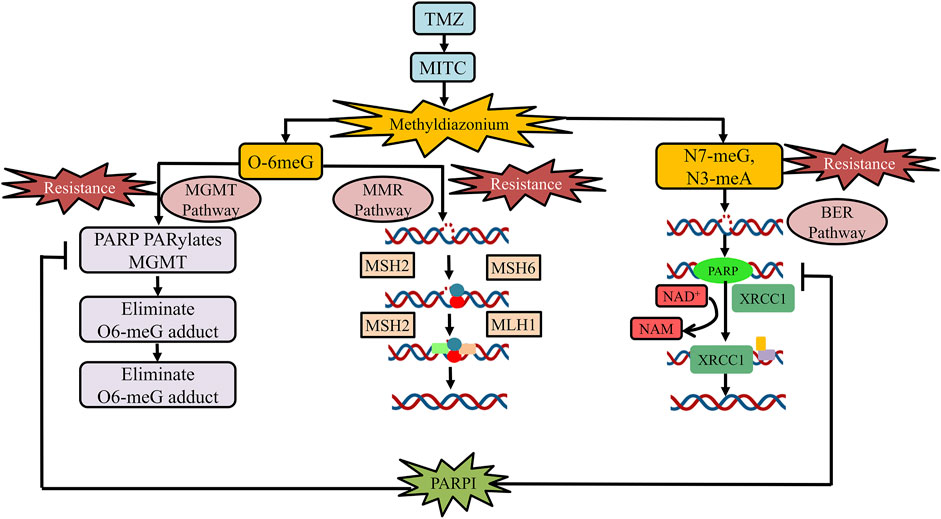
FIGURE 1. Role of PARP inhibitor drugs in TMZ resistance. At physiological pH, TMZ will be converted into MTIC and this MTIC is further hydrolyzed to methylhydrazine. The cytotoxicity of TMZ drug depends on the methylating/alkylating ability of methylhydrazine at the N7 or O6 positions of guanines or the O3 position of adenines in genomic DNA. Methylation at O6 position of guanine causes the addition of thymine rather than cytosine nucleotide opposite to methylguanine during the subsequent process of DNA replication, which triggers the death of tumor cells. The principal pathway involved in TMZ resistance development is the activation of DNA repair pathways (MGMT, MMR, and BMR). PARP is a class of enzyme that is being involved in the MGMT pathway by physically interacting with and PARylates MGMT as an effect of TMZ treatment to eliminate adducts of O6-MetG present in the damaged DNA strand. PARP inhibitors usually reduce the binding of PARP-MGMT along with PARylation as well as silencing of MGMT expression for O6-MetG repair. Hence, diminishing MGMT activity and rendering sensitization to TMZ. In the MMR pathway, the O6-meG:C pair gets mismatched after the first round of replication and results in O6-meG:T formation in the progeny DNA. The MMR system repairs the mismatch of O6-meG:T, by detaching the thymine-containing patch of newly generated strand, utilizing the number of complex proteins such as MSH2, MSH6, MLH1, and PMS2, respectively. Furthermore, PARP is a part of the BER complex that comprises XRCC1 protein, DNA ligase, and the DNA polymerase beta and is involved in BER in response to SSBs. PARP inhibitors reduced PARP binding in the BER complex, thus, reducing BER function to repair SSBs. Abbreviation: PARPI, PARP inhibitor; TMZ, temozolomide; MGMT, O6-methylguanine-DNA methyltransferase; MTIC, 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide; MMR, mismatch repair; O6-MetG, O6-methylguanine; BMR, base excision repair; N7-meG, N7 methylguanine; N3meA, N3 methyl alanine; O6-meG:C, O6-methyl guanine:cytosine; O6-meG:T, O6-methylguanine:thymine; SSBs, single-stranded DNA breaks.
The PARP inhibitors are promising therapeutic agents involved in the treatment of different types of cancers. They exclusively produce synthetic toxicity in cancerous cells through homologous recombination deficiencies (HRDs), and one of the most prominent mechanisms is via mutations in the BRCA1/2 genes present in cancer cells. Current clinical studies suggest that PARP inhibitors can play a beneficial role in cancer therapy irrespective of BRCA1/2 or HRD status (Kim et al., 2021). Initially classified as an HR deficient cancer treatment, olaparib has been approved by FDA for the treatment of serous ovarian cancer as well as breast cancer having a mutation in BRCA1 or BRCA2 germline (Min and Im, 2020). Currently, the FDA has approved four PARP inhibiting drugs (olaparib, rucaparib, talazoparib, and niraparib) to ensure the treatment of breast carcinoma (having detrimental BRCA mutation) along with advanced ovarian cancer (Kim et al., 2021; Min and Im, 2020). The FDA, in May 2020, has also approved rucaparib and olaparib for young patients who are diagnosed with metastatic castration-resistant prostate cancer (mCRPC), having deleterious or suspected deleterious germline or mutation in somatic homologous recombination repair (HRR) gene (Maughan and Antonarakis, 2021). Along with these studies, olaparib has also been approved as a maintenance drug, used in germline BRCA1/2 mutant advanced PDAC (pancreatic ductal adenocarcinoma) by the United States FDA as a phase III Pancreatic Cancer Olaparib Ongoing (POLO) trials have revealed that the administration of olaparib (as a maintenance treatment) magnified the progression-free survival (PFS) rate when compared to a placebo (Chi et al., 2021; Zhu et al., 2020). The role of multiple PARP inhibitors has also been evaluated in gastric cancer (GC) conditions as mutated homologous DNA recombination genes such as BRC1/2, PALB2, ATM, RAD51C, and ARID1A carry somatic HRD (Sahasrabudhe et al., 2017). Therefore, various PARP inhibitors either in the form of mono (olaparib, talazoparib, pamiparib, rucaparib, and niraparib) or combinational therapy (olaparib + paclitaxel, olaparib + ramucirumab, and ceralasertib + olaparib) have gone under multiple phases of clinical trials. Also, FDA has approved ramucirumab in the treatment of gastroesophageal union adenocarcinoma or GC (Wang et al., 2021). In addition to these findings, a contrary role of PARP inhibitors has been reported in AML (acute myeloid leukemia)/MDS (myelodysplastic syndrome) conditions as microsatellite instability was linked to the reduced expression of HR repair genes (Kontandreopoulou et al., 2021). A phase I clinical trial data have shown a tolerable response produced by talazoparib (a PARP inhibitor), with decitabine, a DNA methyltransferase inhibitor when used against relapsed/refractory AML (Baer et al., 2022). On the other hand, a review of randomized controlled trials has reported that the use of different PARP inhibiting agents such as olaparib, niraparib, rucaparib, veliparib, and talazoparib significantly enhances the risk of AML/MDS in comparison to placebo (Kontandreopoulou et al., 2021). Also, mutation of the IDH1/2 gene in primary AML cells manifested HR abnormalities as well as a reduction in the expression of ATM, making AML cells PARP inhibitors susceptible. IDH1/2 inhibitors guard the cells from PARP inhibitors because DNA damage is reduced due to restoration of ATM expression. As a result, in IDH1/2 mutant AML, the use of PARP inhibitors with IDH1/2 inhibitors should be prevented (Molenaar et al., 2018). Notably, tumors without mutated HR-related genes (such as small-cell lung malignancies) have shown some susceptibility toward PARP inhibitors, potentially because of enhanced replication stress caused by RB1 mutations (Dias et al., 2021).
In addition to other cancers, PARP inhibitors have demonstrated a significant response in various preclinical and clinical trials of glioma. Multiple studies investigated the efficacy of PARP inhibitors as they potentiate the anticancer activity of chemotherapeutic drugs and radiation when used simultaneously (Lal and Snape, 2021). Examples of actively acting PARP inhibitors against GBM, with their preclinical significance, are summarized in Table 1 while Table 2 comprises the PARP inhibitors which are effective against GBM, as well as their clinical status.
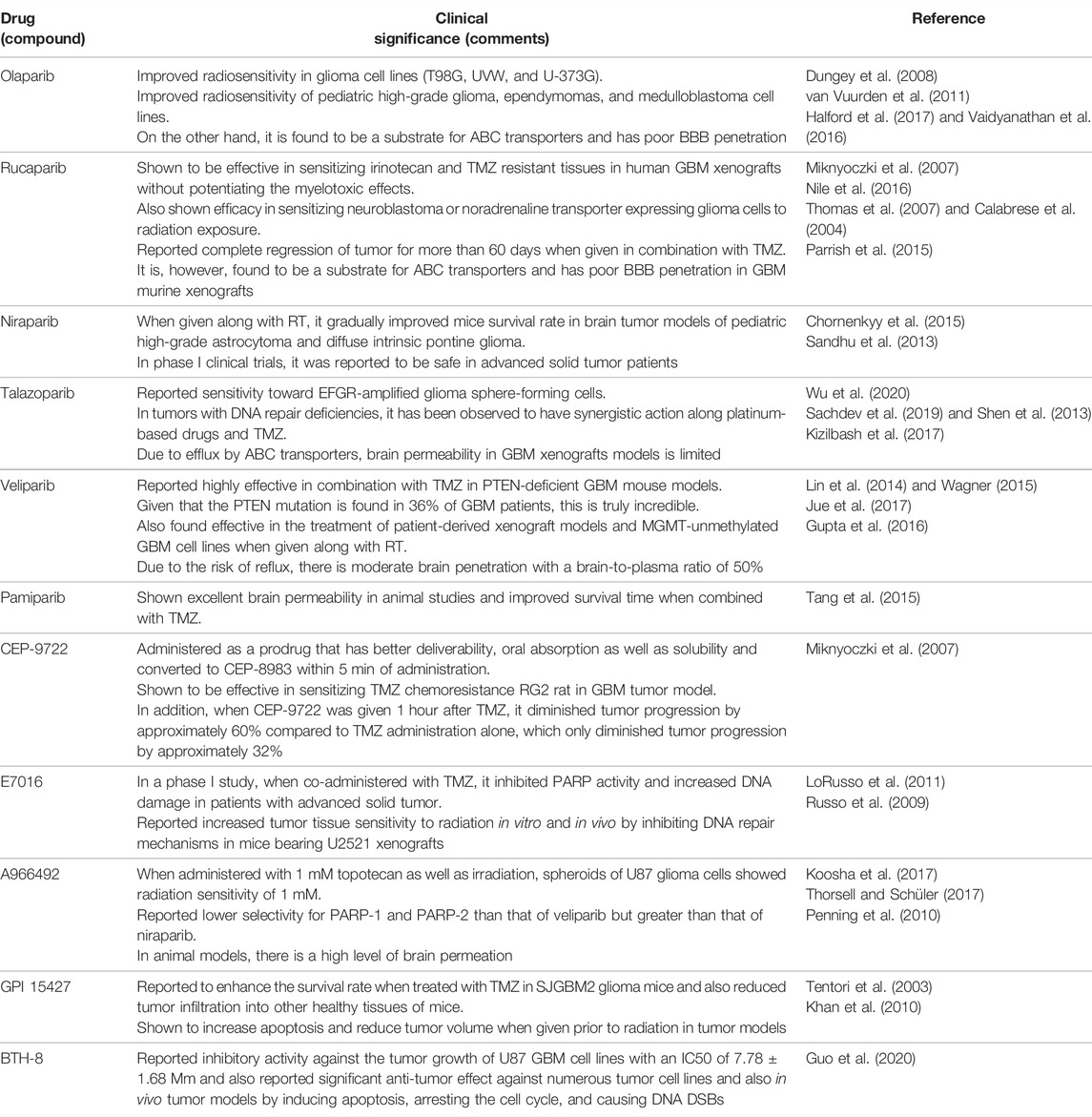
TABLE 1. Examples of actively acting PARP inhibitors against GBM, with their preclinical significance.
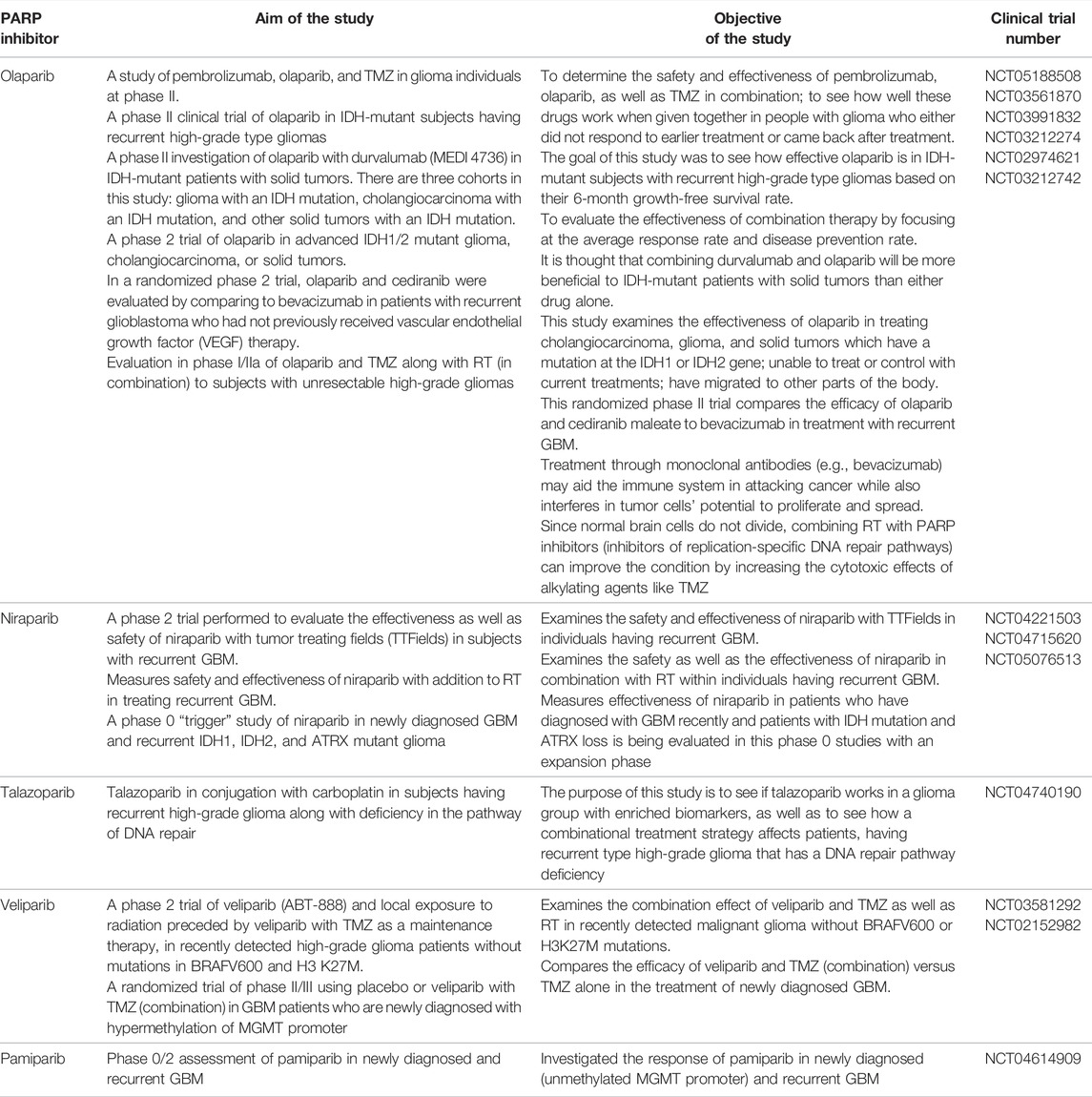
TABLE 2. PARP inhibitor drugs which are under investigation in clinical trials and are active against GBM.
In 2008, a preclinical study by F. A. Dungey et al. showed that olaparib improves the radiosensitivity in glioma cell lines (T98G, UVW, and U-373G). The findings also reveal that olaparib improves radiosensitivity in a replication-dependent way which was increased by fractionation (Dungey et al., 2008). In addition, van Vuurden et al. reported a preclinical study in 2011 that showed olaparib improved radiosensitivity in pediatric high-grade glioma, ependymomas, and medulloblastoma cell lines. Also, gene expression profiling of pediatric high-grade glioma, ependymoma, and medulloblastoma showed that high expression of PARP1 has been linked to a poor prognosis (van Vuurden et al., 2011). Furthermore, a preclinical study by Miknyoczki et al. in 2007 explored another PAPP-1 inhibitor rucaparib which was reported effective in sensitizing irinotecan and TMZ resistant tissues in human GBM xenografts without potentiating the myelotoxic effects (Miknyoczki et al., 2007). A combination study of PARP inhibitors was performed by Nile et al. in 2016. This combined preclinical study elucidated the efficacy of olaparib and rucaparib in sensitizing neuroblastoma or noradrenaline transporter expressing glioma cells to radiation exposure. Rucaparib and olaparib were found to be equally potent inhibitors of PARP-1 functioning. PARP-1 inhibitors were used in combination with X-rays, and DNA damage was gradually increased 10-fold 2 h following irradiation (Nile et al., 2016). Another study of rucaparib by Calabrese et al. reported complete regression of tumor for more than 60 days when given in combination with TMZ. Rucaparib was found to be retained in xenograft tumors where PARP-1 activity was inhibited up to 70% for at least 4 h (Calabrese et al., 2004). Another PARP-1 inhibitor, niraparib, has been shown to be effective in both in vitro and in vivo models. Reduced rate of DNA damage repair, formation of the colony, and relative cell count were observed in an in vitro model before RT. pHGA cells were pretreated with a niraparib sublethal dose of 1 mol/L, and when given prior RT, in vivo niraparib (50 mg/kg) reduced PARP1 activity and increased mice survival rate in orthotopic xenograft model of pHGA (Chornenkyy et al., 2015). Another PARP-1 inhibitor, talazoparib, increased DNA damage and PARP–DNA trapping, enhancing cytotoxicity against EGFR-amplified glioma sphere-forming cells, Additionally, it reduced tumor growth significantly in EGFR-amplified subcutaneous models but not in nonamplified models (Wu et al., 2020). Shen et al. reported that talazoparib specifically targeted tumor cells with BRCA2, PTEN, or BRCA1 gene mutations with 20- to 200-fold better efficacy than current PARP1/2 inhibitors. Talazoparib showed a significant anticancer effect in vivo against xenografted tumors with DNA repair deficiencies caused by BRCA mutations and PTEN loss and was found to be sensitive to well-tolerated doses of talazoparib. Additionally, when talazoparib was administered in combination with TMZ, SN38, or platinum-based drugs, it had additive or synergistic antitumor effects (Shen et al., 2013). PARP-1 inhibitor veliparib was found to be highly effective in combination with TMZ in PTEN-deficient GBM mouse models. Given that the PTEN mutation is found in 36% of GBM patients, this is truly incredible (Lin et al., 2014). In addition, Jue et al. found that combining veliparib and RT inhibits the formation of the colony and increases apoptosis in most patient-derived cell lines. Furthermore, in a PDX model of MGMT unmethylated GBM, treatment with veliparib in a dose of 12.5 mg/kg in combination with complete brain RT (4 Gy) caused apoptosis and reduced cell proliferation. When compared to RT alone or veliparib alone, PDX treated with the combined treatment had significantly higher survival rates (Jue et al., 2017). In a study by Tang et al., pamiparib showed excellent brain permeability in animal studies and improved survival time when combined with TMZ. The in vitro combined effect of pamiparib and TMZ in 7 small-cell lung cancer (SCLC) and 8 GBM cell lines were investigated in this study. Pamiparib showed a significantly greater synergistic effect with TMZ in the majority of those cell lines. In addition, pamiparib showed considerable brain infiltration in C57 mice. To study the combination of pamiparib and TMZ on SCLC in the brain, mice models with established H209 xenografts (intracranial) were used. PARylation in brain or tumor tissues were significantly inhibited 4 h after 3 mg/kg single unit oral dose pamiparib. In this intracranial model, the addition of pamiparib dramatically increased animal survival compared to TMZ alone (Tang et al., 2015).
A phase II clinical study of pembrolizumab, olaparib, and TMZ is now underway to determine the safety and effectiveness of these drugs when given in combination in glioma patients who failed to respond to earlier treatment or relapse. (NCT05188508) Another phase II clinical trial of olaparib in IDH-mutant subjects having recurrent high-grade type gliomas is completed, and this study elucidated the treatment efficacy of olaparib in IDH-mutant subjects with recurrent high-grade type gliomas based on their 6-month growth-free survival rate. (NCT03561870) A phase II investigation of the combination of olaparib and durvalumab (MEDI 4736) in IDH-mutant patients with solid tumors is nearing completion at the University Health Network in Toronto. There are three cohorts in this study: glioma with an IDH mutation, cholangiocarcinoma with an IDH mutation, and other solid tumors with an IDH mutation. Researchers hypothesized that combining durvalumab and olaparib will be more beneficial to IDH-mutant patients with solid tumors than either drug alone. (NCT03991832) Another phase 2 trial of olaparib in advanced IDH1/2 mutant glioma, cholangiocarcinoma, or solid tumors is also under completion. This study examines the effectiveness of olaparib in treating cholangiocarcinoma, glioma, and solid tumors which have mutations in the IDH1 or IDH2 gene; unable to treat or control with current treatments; have migrated to other parts of the body. (NCT03212274) In a randomized phase 2 trial, olaparib and cediranib combination was evaluated by comparing it to bevacizumab in patients with recurrent GBM who had not previously received vascular endothelial growth factor (VEGF) therapy. This randomized trial compares the efficacy of olaparib and cediranib maleate to bevacizumab in treatment with recurrent GBM. Treatment through monoclonal antibodies (e.g., bevacizumab) may aid the immune system in attacking cancer while also interfering with tumor cells’ potential to proliferate and spread. (NCT02974621) Evaluation in phase I/IIa of olaparib and TMZ along with RT (in combination) to subjects with unresectable high-grade gliomas. Scientists hypothesized that since normal brain cells do not divide, combining RT with PARP inhibitors (inhibitors of replication-specific DNA repair pathways) can improve the condition by increasing the cytotoxic effects of alkylating agents like TMZ. (NCT03212742) Niraparib has been established as an effective drug to treat ovarian cancer, and multiple clinical studies have reported an increased progression-free survival rate post therapy with niraparib. A phase II randomized trial with an estimated enrolment of N = 30 patients aged > 22 years has been hypothesized to evaluate the efficacy and safety of niraparib as primary outcomes and estimation of overall survival and progression-free survival as secondary outcomes [NCT04715620]. A phase I multicentered study by Kurzrock et al. has evaluated the tolerability and efficacy of niraparib and TMZ in individuals (N = 19) with recurrent GBM and melanoma, thrombocytopenia, progression of neoplasm, and leukopenia as major reported adverse events. At 40 mg, niraparib has shown antitumor activity (Kurzrock et al., 2014). Phase II study was executed to study efficacy and toxicity of veliparib in patients N = 66 with diffuse intrinsic pontine glioma. Veliparib 25 mg/m2 was added to the therapy along with chemotherapy. Grade 3 nervous system disorder, maculopapular rash, and hemorrhage inside the tumor were found to be major toxic effects, and there was no significant survival benefit compared to the control group (Baxter et al., 2020). A phase I tolerability study by Baxter et al. explored veliparib as maintenance therapy in combination with TMZ in pontine glioma patients N = 18. Veliparib 62 mg/m2 was well tolerated in the study population (Baxter et al., 2020). Ptirowski et al. (2019) concluded pamiparib 60 mg in combination with TMZ and radiation therapy was very well tolerated in GBM patients, with nausea and thrombocytopenia as major reported adverse events related to treatment. Recently, in 2021, the University of Hong Kong has initiated a study (NCT04740190) to explore the treatment efficacy of talazoparib and carboplatin combination in severe grade glioma patients.
IDH1/2 gene mutation was first recognized in the AML as well as glioma and afterward in numerous other cancers. GBM, low-grade glioma (Yan et al., 2009), cholangiocarcinoma (Jiao et al., 2013), acute myeloid leukemia (AML) (Mardis et al., 2009), chondrosarcoma, and melanoma (Amary et al., 2011; Krauthammer et al., 2012) have all been linked to IDH1 mutations. According to Parsons et al. (2008), almost 12% of patients suffering from GBM have IDH1 gene mutation i.e., R132H type. Although other variants such as R132C, R132L, and R132S at codon 132 are also found (Parsons et al., 2008). The 2q33 position of the chromosome carries the IDH1 gene which encodes for the enzyme IDH1 (Balss et al., 2008). Various studies have reported spontaneous mutations in the NADP+-dependent IDH1 gene in glioma along with mutation in the IDH2 gene which is located on 15q26.1 chromosomal position with a frequent mutation at R172K (sometimes also at R172K, R172W, and R172M) of R172 (Horbinski, 2013; Yan et al., 2009). Also, as per WHO classification, GBM has been classified into two main types. One is IDH-wt (wild type) GBMs which comprise primary or de novo GBMs while another is IDH-mut (mutation) GBMs which are secondary or progressive GBMs (Kaminska et al., 2019). Therefore, within glioma patients, a mutation in the IDH1/2 gene promotes the growth as well as the progression of glioma. The conversion of isocitrate to α-ketoglutarate (αKG) is being catalyzed by these enzymes, i.e., IDH1/2 to produce nicotinamide adenine dinucleotide phosphate (NADPH), and due to neomorphic mutations, an oncometabolite, i.e., 2-hydroxyglutarate (2HG) is being generated. A study has previously reported that the accumulation of 2HG inhibits the DNA repair process mediated by the HR process which results in the condition known as “BRCAness,” which offers sensitivity to PARP inhibitors (Dang et al., 2009). On the basis of these results, a multicenter study has been conducted in which patients suffering from IDH1/2 mutated gliomas were administered with a monotherapy olaparib and has shown tolerable response (Fanucci et al., 2022). Also, as per preliminary findings, olaparib has been found to be safe as well as effective in IDH1/2-mutant mesenchymal sarcoma condition (Eder et al., 2021). Another in vitro research has evaluated the effect of PARP inhibitors in combination with ATR inhibiting molecules in IDH1/2-mutant cells and suggested that this combination therapy elevates the premature mitotic entry in the presence of IDH1/2 mutations (Sule et al., 2021). Various clinical studies are still ongoing in different phases in which PARP inhibitors are used in IDH mutated glioma patients. Niraparib (NCT05076513) is being evaluated in recurrent IDH-mutant glioma (cohort B) study, while many combinational drugs like pamiparib/TMZ (NCT03914742) and radiotherapy along with carboplatin/talazoparib (NCT04740190) are studied in high-grade glioma with PTEN-mutant, IDH-mutant, and BRCAness signatures (Sim et al., 2022). Therefore, PARP inhibitors are one of the promising as well as desperately needed domains to be explored more in the field of neuro-oncology research.
Clinical development of PARP inhibitors against glioblastoma is quite difficult because of multiple factors (Figure 2). Some of them are enlisted as:
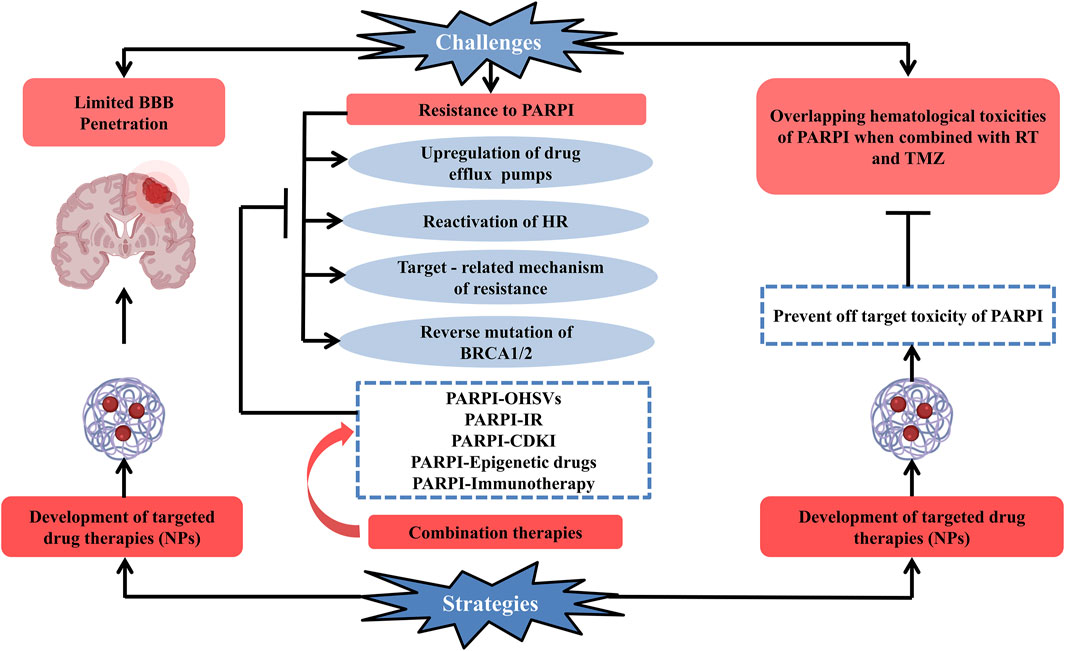
FIGURE 2. Schematic representation of current challenges along with the possible strategies which can be opted for the efficient clinical development of PARP enzyme inhibitors used for the treatment of glioblastoma. Development of PARP enzyme inhibitors for glioblastoma is clinically challenging due to multiple factors: limited blood–brain barrier penetration of PARP inhibitor drugs and development of resistance toward PARP inhibitors (upregulation of drug efflux pumps, reactivation of HR, targeted-related mechanism of resistance, and reverse mutation of BRCA1/2), overlapping hematologic toxicities of PARP inhibitor drugs when conjugated with glioblastoma’s standard treatment (radiation therapy + temozolomide). One of the promising strategies to deal with the challenges is the development of targeted drug therapies and combination therapy (PARPI-IR, PARPI-OHSVs, PARPI-CDKI, PARPI-epigenetic drugs, and PARPI-immunotherapy) to combat the resistance of PARP inhibitors to overcome these factors. Abbreviations: PARPI, PARP inhibitor; OHSVs, oncolytic herpes simplex viruses; IR, ionizing radiation; CDKI, cyclin-dependent kinase; NPs, nanoparticles.
The targeted delivery of therapeutically active compounds into the CNS is a significant challenge in the treatment of most neurological diseases. The BBB is one of the most complex and well-protected neurovascular unit composed of tight junctions between endothelial cells and brain capillary, which restrict paracellular diffusion and protects the brain from harmful chemicals and toxins by separating it from peripheral circulation (Agarwal et al., 2012; Gupta et al., 2019). Thus, it limits the exclusion of many anticancer drugs including PARP inhibitors and undermines their effectiveness (Agarwal et al., 2012; Saran et al., 2018). In GBM, glioma cells can infiltrate normal brain tissue some centimeters apart from the tumor, evading surgical resection and so being secured by a relatively intact BBB (Berens and Giese, 1999; Pitz et al., 2011). Even though the BBB may be damaged at or around the tumor core, it stays intact in locations distant from the core, limiting anticancer drug delivery in these areas (Levin et al., 1980). This implies that in several GBM patients, restricted drug delivery to invasive tumor cells could play a crucial role in therapeutic resistance. This type of failure in drug delivery needs more exploration in preclinical models. Since some preclinical models are not developed in the brain such as the flank model, and some involve mice with well-circumscribed brain tumors obtained from implanted cell lines that demonstrate a leaky BBB without any significant invasion (Blakeley et al., 2009). Preclinical data involving GBM animal models imply that the efficiency of PARP inhibitors in GBM could be restricted because of limited transport over the BBB membrane and heterogeneous glioma response (Gupta et al., 2014; Kizilbash et al., 2017; Parrish et al., 2015; Gupta et al., 2016). Significant attempts had been made to acknowledge the brain pharmacokinetics of PARP inhibitors, and many PARP inhibitors, particularly the trapping agents like rucaparib and talazoparib, exhibit efflux liabilities around the BBB, and thus a complete absence of sensitizing response is observed in orthotopic models of tumor in spite of great activity in heterotopic cancer models (Hopkins et al., 2015; Parrish et al., 2015). These results support the hypothesis that targeted delivery of drugs within the healthy brain or orthotopically implanted tumor can simulate their potency in the GBM. Talazoparib, a PARP inhibitor, enhances the potency of temozolomide in a variety of tumor types. However, when the pharmacokinetic parameters of talazoparib were compared to another PARP in normal rodents, a low ratio of brain-plasma concentration of talazoparib (approximately 0.02) was observed, indicating that it lacks effectiveness in the orthotopic type of glioma models (Parrish et al., 2015; Kizilbash et al., 2017). Alternative PARP enzyme trapping compound bearing efflux liability as well as limited distribution around BBB is olaparib (Halford et al., 2017; Vaidyanathan et al., 2016). Despite the reality that olaparib was found to penetrate the center of GBM tumors in the phase I clinical trial in individuals with repetitive GBM (Halford et al., 2017), these findings must be considered with caution since GBM cells infiltrate tissues far away from the margins. Rucaparib in combination with TMZ was found to be extremely effective in a study performed in short explant cultures (in vitro) obtained from GBM12, and the same combination (when dosed for 5 days in a consecutive manner every 28 days for three cycles) significantly prolonged the tumor regrowth duration by at least 40% in heterotopic xenografts. However, a PK/PD investigation following a unit dose revealed that rucaparib deposition within the brain is related to elevate over left PARP enzymatic activity (Parrish et al., 2015). Conversely, veliparib seems to have a significantly greater ratio of brain-plasma concentration (around 0.47) rather than rucaparib and talazoparib in spite of its efflux liability toward MDR1 as well as BCRP (Gupta et al., 2016; Li et al., 2011). In addition, orthotopic GBM models are sensitized better from veliparib rather than rucaparib and talazoparib comparatively, although being much less powerful with respect to PARP trapping (Gupta et al., 2016). This premise sheds light on the importance of factors like penetrability across the BBB and efflux liability in orthotopic models of glioma which has a role to improve efficacy. These findings highlight the significance of neuronal pharmacokinetic parameters in preclinical models for the successful clinical development of innovative GBM therapies.
Although PARP inhibitor drugs impart a positive response initially, many subjects acquire resistance to them, resulting in disease recurrence. The emergence of resistance to PARP inhibitor drugs can occur via five basic mechanisms: overexpression of drug efflux pumps, target-related mechanism for resistance, restoration of BRCA1 or BRCA2 gene activity, BRCA1-independent restoration of HR, and restoration of fork stability.
Overexpression of ABCB1 type transporters (also known as P-glycoprotein), which are mainly responsible for the efflux of drugs, has been reported among the earliest reasons for the generation of resistance to PARP inhibitor drugs. ABCB1 is a type of ATP-binding cassette (ABC) transporter, which has been shown to confer resistance to a variety of chemotherapeutic and many other drugs by limiting their accumulation intracellularly. The development of resistance against PARP inhibitor is being mediated by ABCB1, and it was first reported in BRCA1/2-deficient breast cancer mice models (Rottenberg et al., 2008). Patients who have received chemotherapy before starting PARP inhibitor treatment are more susceptible to this resistance mechanism, as ABCB1 transporters have been shown to be upregulated in tumors that are formerly exposed to chemotherapies because of chromosomal translocations that occur during treatment through paclitaxel anticancer agent (Marzolini et al., 2004). Interestingly, resistance may be overcome by ABCB1 inhibitors (verapamil and elacridar), which can be used against drug resistance mediated by ABCB1, in ovarian cancer treated by olaparib and paclitaxel (Vaidyanathan et al., 2016). Clinical trials with ABCB1 inhibitors have shown unsatisfactory results, and recent research indicated toward ABCB1 is crucial for effective immunological response (Chen et al., 2020). Further investigation is required to clarify the significance of ABCB1 gene screening in developing therapy regimens.
Each available PARP inhibitor drug competes with coenzyme NAD+ to target the PARP enzymes at its catalytic domain site. Resistance could thus develop from PARP1 mutations that decide whether it decreases the efficacy of the PARP inhibitor compound or conserve primary in-built activities of the PARP enzymes when bound to the PARP inhibitor. In vitro results indicate that the development of resistance to PARP inhibitors, associated with point mutations is present not only in the enzyme’s catalytic region but also present in domains through which PARP1 is being trapped on the DNA strand (Pettitt et al., 2018; Pettitt et al., 2013). Confirming these findings, in a PARP inhibitor-resistant ovarian tumor, a mutated PARP1 was found that does not alter the mobilization of the PARP1 enzyme toward the damaged locus of DNA but prevents the trapping of PARP1 (Pettitt et al., 2018). However, mutations in PARP1 exclusively develop resistance in cells which are HR-proficient and with hypomorphic BRCA1 mutation in cells and synthetic lethal effect are produced which causes loss of both PARP1 and BRCA1 in response to BRCA1 activity at the residual level. Another enzyme poly (ADP-ribose) glycohydrolase (PARG) is an important factor that was found to play a pivotal role in the generation of resistance toward PARP inhibitors. This enzyme separates the PAR chain from the targeted protein. For example, in genetically modified models of BRCA1/2-deficient mice for breast carcinoma, depletion of PARG leads to the development of resistance to PARP inhibitor drugs. Significantly, in these models, PARG lacking cells treated to inhibit PARP are predicted to maintain enough PARylation at desired targeted proteins so that they can initiate the signaling cascade for DNA damage as well as minimize PARP1 protein trapping onto DNA strand due to remaining PARP activity. Despite the lack of clinical evidence, two small cohort studies reported that PARG negative areas accounting for 10% or more of the overall tumor mass which have been found in a significant proportion of tumors present in females suffering from triple-negative breast cancers (76.8%) or ovarian cancer (78.4%), for which PARP inhibitors can be used as a therapeutic approach for both (Gogola et al., 2018).
One of the clinically proven mechanisms for the development of resistance toward PARP inhibiting drugs is reverse mutation and modification at the epigenetic level, which causes the repetitive expression of a BRCA1/2 protein and leads to hypomorphic variations. After long-term treatment with PARP inhibitors or cisplatin, the mutation in reversion of protein-truncating BRCA1 or BRCA2 gene was first observed in an in vitro study performed in BRCA2 mutated gene in pancreatic as well as ovarian cancerous cell lines (Edwards et al., 2008; Sakai et al., 2008). Patient-derived xenograft (PDX) models of mutated BRCA1 gene or BRCA1 methylated gene in TNBC disclosed resistant after the exposure to PARP inhibiting drugs due to intragenic removal which reinstate the reading sequence of mutant BRCA1, and absence in BRCA1 gene promoter hypermethylation as well as de-novo gene fusions that results in upregulated expression of epigenetically silenced gene BRCA1 (Ter Brugge et al., 2016). Over many recent years, multiple investigations had identified that reversions of BRCA1/2 at the genetic level are a cause of PARP inhibitor resistance during ovarian (Edwards et al., 2008; Kondrashova et al., 2017; Weigelt et al., 2017; Barber et al., 2013; Norquist et al., 2011; Domchek, 2017; Lin et al., 2019), prostate (Quigley et al., 2017; Goodall et al., 2017), breast (Weigelt et al., 2017; Barber et al., 2013; Afghahi et al., 2017), and pancreatic cancers (Pishvaian et al., 2017). Interestingly, reversions linked with PARP inhibitor resistance are not only reported in BRCA1 and BRCA2 but have also been reported in some HR-related genes including PALB2 and RAD51C/1D (Kondrashova et al., 2017; Goodall et al., 2017). Due to the challenges of detecting reversion mutation and the limited usage of PARP inhibitors in clinical trials, data gathered from massive-scale research to assess the prevalence of BRCA1/2 gene reactivation in cancer patients having PARP inhibitor-resistant tumors are missing even now.
A mutation in the NHEJ (non-homologous end-joining) pathway results in the resurgence of homologous recombination (HR) in mutant cells of BRCA1/2. BRCA1 induces strand exchange through its interaction with BRCA2 and PALB2 in normal cells. Additionally, it antagonizes 53BP1 and impairs the NHEJ pathway (Isono et al., 2017). 53BP1 is an essential element of NHEJ machinery and imparts a crucial response in repairing DNA as well as checkpoint control (Bunting et al., 2010; Mirman and de Lange, 2020; Zimmermann and De Lange, 2014). It stimulates NHEJ through decreasing DNA end-resection process that is essential for HR. Furthermore, it interacts with a few genes such as RIF1, REV7 and SHLD1, SHLD2, and SHLD3 (shieldin complex) whose resulting complex known as 53BP1-RIF1-REV7-shieldin axis that prevents incision (Xu et al., 2015; Zimmermann et al., 2013; Noordermeer et al., 2018). The depletion of any of the components in this complex has been implicated in PARP inhibitor resistance in cells that are BRCA1 deficient but notably not in cells that lack the BRCA2 gene (Bouwman et al., 2010). It is hypothesized that the suppression of the shieldin axis, i.e., 53BP1-RIF1-REV7-axis, enables resection at the end and then HR to take place in an independent manner in BRCA1 while in a dependent manner in RNF168. RNF168 belongs to the E3 ubiquitin ligase category enzyme which can activate HR without any need for BRCA1 requirement by interacting directly with PALB2. Dynein light chain 1 (DYNLL1) is yet another molecule which has been associated to develop PARP inhibitor resistance, like 53BP1, acts by blocking the end resection of DNA in normal cells and promotes NHEJ. Although it has been reported that DYNLL1, which is a coding gene for protein, interacts with 53BP1 to limit terminal resection or that DYNLL1 gene which acts by blocking resection machinery elements like MRE11 (Lo et al., 2005; Becker et al., 2018; He et al., 2018), the actual relation between 53BP1 axis and DYNLL1 remains unknown. Similarly, inhibition of 53BP1 and inactivation of the DYNLL1 gene are linked to PARP inhibitor resistance (He et al., 2018). The deprivation of ERCC6L2, which is an additional NHEJ component, has been reported to reestablish DNA end-resection, leading to incomplete (half) HR repair and acquiring resistance toward PARP inhibiting compounds in cells lacking the BRCA1 gene (He et al., 2018; Francica et al., 2020). Furthermore, upregulation of factors that promotes HR and inhibits NHEJ, including TIRR134 (Drané et al., 2017), TRIP13 (Clairmont et al., 2020), and miRNA-622 (Choi et al., 2016), have been reported to protect HR plus diminish the sensitivity toward PARP inhibiting compounds in BRCA1 gene-deficient cells. Such results support the hypothesis which states that the induction of resistance to PARP inhibitor drugs develops from the failure of DNA end protection within functionally active BRCA1 diminished cells. Importantly, the foregoing data indicates that in the HR pathway mediated by RAD51, BRCA2 protein is critical for the pathway while BRCA1 protein is somewhat required for the last few steps in the same pathway.
Generation of resistance to PARP inhibitor drugs from reactivation of fork stability is frequent in both BRCA1- and BRCA2-deficient cells. As stated formerly, BRCA1/2 is needed not just for HR but, moreover, to maintain the stability as well as to provide protection to the replication forks during replicative strain. MRE11 or MUS81 are the two nucleases which are needed for the formation of halted replication forks. Uncontrolled excision of unprotected halted forks by MRE11 causes collapse of the fork and results in elevated instability at the genomic level in BRCA1/2-deficient cells (Schlacher et al., 2011; Schlacher et al., 2012; Ray Chaudhuri et al., 2016; Ying et al., 2012). In view of this fact, loss of protein, i.e. PTIP, a complex of MLL3/4 as well as nucleosome remodeling element CHD4 impairs MRE11 attachment in obstructed forks; resulting in protection of fork that leads to PARP inhibitor resistance in cells deficient in BRCA1/2 (Ray Chaudhuri et al., 2016; Guillemette et al., 2015). SMARCAL1, a chromatin remodeling complex depletion, reduces the sensitivity of tumor cells deficient in BRCA1/2 and causes PARP inhibitor resistance, however, this effect appears to be cell-type specific (Taglialatela et al., 2017; Kolinjivadi et al., 2017). Another protein associated with replication fork protection is RADX; reduction of this protein in cells deficient in BRCA2 restores protection of replication fork and reduces the toxic effects of PARP inhibitor compounds (Dungrawala et al., 2017). Also, preventing MUS81 gathering by inhibiting the methyltransferase activity of EZH2 induces protection of fork and partially PARP inhibitor resistance, especially in cells deficient in BRCA2 (Rondinelli et al., 2017). Unfortunately, there is conflicting evidence on the involvement of MUS81, with studies claiming that this nuclease either disrupts (Dungrawala et al., 2017; Rondinelli et al., 2017) or protects (Lai et al., 2017; Lemaçon et al., 2017) the unprotected forks; consequently, whether the cytotoxic response of PARP inhibiting drugs is affected by MUS81 within BRCA1 and BRCA2 diminished cells seems unknown (Lemaçon et al., 2017). Interestingly, loss of PTIP, RADX, or EZH2 does not improve the functioning of HR in cells having low BRCA1/2, implying that reactivation of protection of replication fork is a major aspect for induction of resistance toward PARP inhibitor compounds (Ray Chaudhuri et al., 2016; Dungrawala et al., 2017; Rondinelli et al., 2017). Another replication stress factor is Schlafen 11 (SLFN11) whose depletion lowers the cytotoxic response of PARP inhibiting compounds in BRCA1/2 proficient as well as BRCA2 diminished cells (Murai et al., 2016). Lastly, and perhaps most notably, PARP1 is required to facilitate MRE11 localization to a stalled replication fork. PARP1 deficiency causes synthetic toxicity in BRCA1/2 diminished cells; however, PARP1 repression prior to BRCA1or BRCA2 depletion re-establishes the integrity within the stalled forks and increases cell viability, widely through decreasing MRE11 localization at the replication fork (Ray Chaudhuri et al., 2016; Ding et al., 2016). Considering the multifaceted function of PARP1 at the replication fork, more research is needed to clarify how it may alter the outcomes of PARP inhibitor-based combination therapy.
The acceptability of veliparib in conjugation with the standard treatment of newly diagnosed GBM (RT and TMZ) was investigated in a phase 1 trial. The study showed that a daily 10 mg dose (twice a day) of veliparib when administered orally, with concurrent RT/TMZ was intolerable in individuals suffering from GBM due to hematological toxicity (Kleinberg et al., 2013). A randomized study under phase 1/2 clinical assessment investigated a combination along with TMZ for treating persistent GBM with TMZ resistance. The myelosuppression with grade 3/4 was reported in approximately 20% of patients, with a usual progression-free survival (PFS) for 2 months (Robins et al., 2016). PARP inhibitors in addition to TMZ have also been studied in neuro-oncology and in the pediatric population. A phase I study of veliparib in conjugation with TMZ in the juvenile population suffering from recurrent brain tumors has been reported. Dose-dependent myelosuppression was observed in children (Su et al., 2014). Furthermore, in the phase I concentration escalation study, using olaparib in addition to TMZ within relapsed GBM patients was performed in the US. Pharmacokinetics study of orally bioavailable olaparib against tumor was studied after four doses using tumor resection. Olaparib penetrated tumors, with a mean concentration of 588 nM in the tumor core and 500 nM in the tumor tissues. When a daily dose of 150 mg of olaparib was administered for 1–3 days weekly in conjunction with 75 mg/m2 of TMZ, the response was well tolerated. Totally, 45% of the evaluable subjects were progression free following 6 months. However, 24 of the 35 evaluable patients reported adverse effects ≥3. The most frequent symptom was lymphopenia (51%), followed by neutropenia (26%), thrombocytopenia (17%), anemia (14%), and fatigue (Halford et al., 2017). This finding prompted us to conduct a clinical trial in GBM patients who are newly diagnosed and treated with olaparib in adjuvant to RT or RT + TMZ depending on MGMT status stratification (phase 1, PARADIGM-2). In this PARADIGM-2 study, within a single trial protocol, patients were allocated into two respective groups parallelly. Patients who are allocated within parallel 1 of the study (MGMT methylated) were administered olaparib in combination with radical RT (60 Gy in 30 fractions for 6 weeks) with concomitant TMZ chemotherapy (75 mg/m2 each day throughout RT), followed by 4 weeks of oral olaparib, subsequently followed by TMZ at its standard effective dose for six cycles and schedule commencing 4 weeks post radiotherapy. Patients assigned to parallel 2 of the study (MGMT unmethylated) received olaparib with radical RT in combination (60 Gy in 30 fractions for 6 weeks), and a further 4 weeks of oral olaparib. Hematological toxicities such as neutropenia grade 4 (lasting for ≥5 days), febrile neutropenia of grade ≥3 (absolute neutrophil count <1.0 × 109/L with ≥38.5°C fever), and thrombocytopenia of grade 4 (lasts for ≥5 days) linked with heavy bleeding or requiring platelet transfusion reported during the treatment period of olaparib in both MGMT methylated and methylated patients (Fulton et al., 2018). A newer generation of PARP inhibitor BGB-290 (pamiparib) is now also being studied in clinical trials. A phase 1b/2 assessment was conducted in patients having first-line or recurrent/refractory GBM to evaluate the tolerability and safety as well as the effectiveness of BGB-290 with RT + TMZ (in conjugation). In a dose-escalation/phase 1b study, pamiparib was conjugated with RT (arm A) and RT plus TMZ (arm B) in recently detected unmethylated GBM patients while arm C of the test has pamiparib in conjugation with TMZ for methylated or unmethylated recurrent/refractory GBM patients. The dose expansion or phase 2 study enrolled up to four cohorts: recently detected unmethylated GBM patients in arm A as well as arm B, and cohorts of patients bearing recurrent/refractory GBM divided via MGMT status (unmethylated/methylated) within arm C. Patients of arms A or B have been treated till their RT is completed, while patients in arm C could undergo treatment if there are no safety concerns or tumor growth. BGB-290 at 60 mg BID in combination with RT/TMZ was well tolerated in recently diagnosed or recurrent/refractory GBM patients. However, one dose-limiting adverse event (grade 3 type febrile neutropenia) was observed in patients of arm B. Although, treatment-associated adverse effects (≥10%) were arm A experiences nausea (around 23 percent/2 percent); B reported a decrease in WBC count (approximately 11 percent/11 percent); C (none) (Piotrowski et al., 2019).
The development of nanoscale drug delivery vehicles including nanoparticles, liposomes, dendrimers, nanomicelles, polymeric micelles, liposomes, and dendrimers has completely transformed new drug delivery approaches. These nanotherapeutic techniques are widely implemented in clinics for the betterment of the efficacy of active small-molecule inhibitors due to their remarkable cancer-targeting efficacy and sustained release. PARP inhibitor drugs are small artificial molecules that have proven to be one of the most effective new approaches to treating carcinoma cells with alterations in repair genes of DNA. PARP inhibitor efficacy has been greatly enhanced due to the advancement of nanotherapeutic-based delivery of drugs. Nanoparticles could especially accumulate inside the tumor and cancer cell’s leaky vasculature, releasing the cytotoxic compound inside the microenvironment of the tumor cell. But on the other hand, nanoparticles will not be able to diffuse through the non-cancerous body tissues as well as organs, so their toxic response is minimal to non-existent. Targeting PARP inhibitors to treat cancer frequently results in toxicity; thus, a delivery carrier is required to encapsulate them (Sargazi et al., 2021). Several nanovehicles have now been used to transmit PARP inhibitor compounds in various cancerous cells (Mensah et al., 2019; Baldwin et al., 2018b; Di Zhang et al., 2019; Patel et al., 2017; Baldwin et al., 2018a). In a study, a nanoformulation of olaparib in conjugation with platinum was developed, and its activity was illustrated in the cell line of ovarian cancer. The ADME as well as bioavailability profiles were improved by the nanoformulation. They improved the therapeutic response by inhibiting cell proliferation (Baldwin et al., 2018b). In a different study, lipid-based nanoformulation was developed to deliver olaparib. The lipospheres are created through melt dispersion along with nanosuspension via wet processing or evaporation of the solvent. The ADME profiles or hematological adverse response of olaparib nanosuspension and lipospheres were compared. The nanoparticles had a high bioavailability and no toxicity in the tissues (Pathade et al., 2019). In another study, polymeric vehicles were used to develop drug carriers. Drug nanocrystals, pectin, and bioadhesive hydrogel were encapsulated with polylactic acid-polyethylene glycol and sprayed in the brain’s parenchyma to treat GBM. The nanoparticles gelled at the calcium concentration inside the brain. Both olaparib as well as etoposide were packed in the polymeric carrier synergistically, and for spray-drying purposes, pluronic F127 was used. Over the course of 120 h, the drug was released. The nanoparticles had aggregated in the brain according to fluorescent imaging results. A pharmacological platform for malignant brain cancers was introduced by the sprayable hydrogel (McCrorie et al., 2020). These simplistic but novel strategic approaches can boost the activity of PARP inhibitors.
One of the promising strategies to deal with the resistance of PARP inhibitors in tumor cell is combination therapy.
Oncolytic herpes simplex viruses (oHSVs) are compounds synthesized through genetic modification to specifically destroy cancer cells because of their unique property of reproducing as well as propagating inside the malignant cells except for the normal ones. The FDA has approved oHSVs for the treatment of recurrent melanoma. They actively participate in DNA damage repair (DDR) manipulation (Kohlhapp and Kaufman, 2016). MG18L, a recently discovered activity of oHSV, has been shown to deteriorate RAD51 as well as sensitize GBM stem cells toward PARP inhibitors, by killing cells in an artificial lethal like manner both in vitro as well as in vivo. A combined effect of two, i.e., olaparib and MG181, significantly improves the condition in PARP inhibitor sensitive cells along with resistant GBM stem cells derived from tumor cells. Fused treatment (olaparib + MG181) not just combats PARP inhibitor resistance but it also broadens the application to malignancies along with HR-proficient. Especially, when compared to traditional treatments, oHSVs only destroy tumor cells by not affecting normal cells, and this implies that they could have minimal side effects (Ning et al., 2017).
BRCA1 requires localization of the nucleus in order to participate in homologous recombination-mediated repair of DNA (Wang et al., 2010). Ionizing radiation can cause BRCA1 to be exported to the cytoplasm from the nucleus, increasing the sensitivity of PARP inhibitors in the wild-type BRCA1 and homologous recombination proficient tumor cells (Jiang et al., 2011; Yang et al., 2012). The combination therapy can be used only in patients with p53 wild-type because its synthetic lethality is p53-dependent (Sizemore et al., 2018). Furthermore, it was reported that tumors mutated due to the BRCA1 gene developed resistance as a result of BRCA1-independent homologous recombination restoration which can be sensitized to radiotherapy (Barazas et al., 2019). Based on preclinical findings, multiple clinical experiments were performed in order to optimize the potency of PARP inhibitors-ionizing radiation combination. A phase-1 concentration escalation study (open-labelled) in brain metastasis individuals to evaluate the combination of veliparib with whole-brain radiation therapy and found better results (Mehta et al., 2015). Furthermore, two phase-I trials concluded that the PARP inhibitors-ionizing radiation combination was well-tolerated and produced decent results (Reiss et al., 2015; Czito et al., 2017). More studies are currently underway on the basis of encouraging preliminary safety and efficacy outcomes.
The end resection of DNA is dependent on the activity of cyclin-dependent kinases. Numerous studies have found that CDKs play prime roles in PARP inhibitor resistance (Tomimatsu et al., 2014; Ali et al., 2014; Joshi et al., 2014; Johnson et al., 2016; Ning et al., 2019; Militello et al., 2019). Dinaciclib, a CDK inhibitor, re-sensitized triple-negative breast cancer cells that had developed niraparib resistance. Furthermore, the combination of dinaciclib and niraparib was extremely effective in pancreatic, lung, colon, prostate, and ovarian cancer (Carey et al., 2018). Cyclin-dependent kinase-12 has recently gained more interest in PARP inhibitor resistance owing to its inactivating somatic alterations that have been found in various cancers. Several studies have shown that CDK12 mutation or deficiency causes cancer cells to be more sensitive to PARP inhibitors (Ali et al., 2014). In addition to this, cyclin-dependent kinase-12 inhibitors have reversed de novo and developed PARP inhibitor resistance in breast cancer cells mutated due to the BRCA1 gene (Johnson et al., 2016).
Jiao et al. have concluded that PD-L1 expression was upregulated by PARP inhibitor in cell lines of breast cancer by inactivated GSK3, resulting in impaired anticancer immunity. In addition, the anti-PD-L1 therapy and PARP inhibitor combination show higher therapeutic effectiveness (Jiao et al., 2017). Independent of BRCA1/2 mutation, PARP inhibitor-mediated immune response modulation accords to the therapeutic value. According to recent findings, PARP inhibitors promote the buildup of cytosolic fragments of DNA as a result of unsolved DNA lesions, further activate the DNA-sensing cGAS-STING pathway which results in stimulating type I interferon production to further stimulate anti-tumor immunity which is independent of BRCA status (Shen et al., 2019).
As stated previously, epigenetic modifications were linked to PARP inhibitor sensitivity (Kondrashova et al., 2018; Guo et al., 2018; Fukumoto et al., 2019). The two notable mechanisms of post-translational gene expression regulation are histone acetylation and deacetylation (Audia and Campbell, 2016). Studies concluded that treatment of cancer cells with histone deacetylation (HDAC) inhibitor and PARP inhibitor had a synergistic effect because of the induction of HDAC inhibitor on homologous recombination deficiencies, which further sensitized the cancer cells to PARP inhibitor (Jasek et al., 2014; Baldan et al., 2015; Ha et al., 2014; Min et al., 2015). Numerous mechanisms have been discovered. Initially, it was found that HDAC inhibitor reduced the DNA repair gene expression of BRCA1, RAD21, CHK1, and RAD51 via the transcription factor E2F1 (Kachhap et al., 2010). Additionally, HDAC inhibitors inhibited HSP90 deacetylation, leading to the degradation of its substrate RAD52, CHK1, BRCA1, and ATR (Kim et al., 2017). Eventually, recent research has elucidated that acetylation inhibited DNA damage-induced chromatin PARylation and that HDAC inhibitor therapy significantly enhances PARP1 trapping at DSB sites in chromatin (Robert et al., 2016; Abbotts et al., 2019). Besides that, the DNA methyltransferase (DNMT) inhibitors in low doses stimulated BRCAnes by decreased regulation of key homologous recombination gene expression. The DNMTi and PARP inhibitor combination increased cytotoxicity by enhancing the PARP “trapping” upon double-stranded break sites independent of BRCA mutation (Pulliam et al., 2018; Muvarak et al., 2016). However, the data to evaluate the clinical effectiveness remain unavailable and need further research.
Since TMZ is the first-line drug that is effective in the treatment of GBM, resistance to it is the most serious problem in the context. PARP inhibitors have recently been used to overcome TMZ resistance. However, there are a number of factors that make PARP inhibitors' clinical development challenging. The limited BBB penetration, resistance to PARP inhibitors, and hematological toxicities that occur when PARP inhibitors are used in combination with TMZ/IR are three of the most significant challenges in PARP inhibitors’ clinical development. As a result, this review concludes that challenges can be overcome if good strategies are implemented. For example, if targeted delivery is used, the drug can penetrate the BBB and act. The prevention of off-target toxicity is the second advantage of targeted delivery. Third, we can overcome PARPI resistance by using combination therapies. The use of targeted delivery and combination therapies is one step toward achieving the goal of successful clinical development of PARP inhibitors in GBM.
PB wrote the first draft of the manuscript and worked on the coding of figures. VK contributed to the conception and design of the study. RP contributed to the conception and design of the study. RV read the manuscript and approved the final manuscript. NK read the manuscript and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbotts, R., Topper, M. J., Biondi, C., Fontaine, D., Goswami, R., Stojanovic, L., et al. (2019). DNA Methyltransferase Inhibitors Induce a BRCAness Phenotype that Sensitizes NSCLC to PARP Inhibitor and Ionizing Radiation. Proc. Natl. Acad. Sci. U. S. A. 116 (45), 22609–22618. doi:10.1073/pnas.1903765116
Afghahi, A., Timms, K. M., Vinayak, S., Jensen, K. C., Kurian, A. W., Carlson, R. W., et al. (2017). Tumor BRCA1 Reversion Mutation Arising during Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin. Cancer Res. 23 (13), 3365–3370. doi:10.1158/1078-0432.CCR-16-2174
Agarwal, S., Mittapalli, R. K., Zellmer, D. M., Gallardo, J. L., Donelson, R., Seiler, C., et al. (2012). Active Efflux of Dasatinib from the Brain Limits Efficacy against Murine Glioblastoma: Broad Implications for the Clinical Use of Molecularly Targeted Agents. Mol. Cancer Ther. 11 (10), 2183–2192. doi:10.1158/1535-7163.MCT-12-0552
Ali, S. M., Hawryluk, M. J., Wang, K., Chmielecki, J., Palmer, G. A., Garcia, L., et al. (2014). Abstract A28: Clinical Next-Generation Sequencing (NGS) Reveals Genomic Alterations (GAs) to Guide Targeted Therapy in Advanced Neuroblastoma Patients. Cancer Res. 74. doi:10.1158/1538-7445.PEDCAN-A28
Alonso, M. M., Gomez-Manzano, C., Bekele, B. N., Yung, W. K., and Fueyo, J. (2007). Adenovirus-based Strategies Overcome Temozolomide Resistance by Silencing the O6-Methylguanine-DNA Methyltransferase Promoter. Cancer Res. 67 (24), 11499–11504. doi:10.1158/0008-5472.CAN-07-5312
Amary, M. F., Bacsi, K., Maggiani, F., Damato, S., Halai, D., Berisha, F., et al. (2011). IDH1 and IDH2 Mutations Are Frequent Events in Central Chondrosarcoma and Central and Periosteal Chondromas but Not in Other Mesenchymal Tumours. J. Pathol. 224 (3), 334–343. doi:10.1002/path.2913
Amé, J. C., Spenlehauer, C., and de Murcia, G. (2004). The PARP Superfamily. Bioessays 26 (8), 882–893.
Audia, J. E., and Campbell, R. M. (2016). Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 8 (4), a019521. doi:10.1101/cshperspect.a019521
Baer, M. R., Kogan, A. A., Bentzen, S. M., Mi, T., Lapidus, R. G., Duong, V. H., et al. (2022). Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/refractory Acute Myeloid Leukemia. Clin. Cancer Res. 28 (7), 1313–1322. doi:10.1158/1078-0432.CCR-21-3729
Baldan, F., Mio, C., Allegri, L., Puppin, C., Russo, D., Filetti, S., et al. (2015). Synergy between HDAC and PARP Inhibitors on Proliferation of a Human Anaplastic Thyroid Cancer-Derived Cell Line. Int. J. Endocrinol. doi:10.1155/2015/978371
Baldwin, P., Ohman, A. W., Tangutoori, S., Dinulescu, D. M., and Sridhar, S. (2018a). Intraperitoneal Delivery of NanoOlaparib for Disseminated Late-Stage Cancer Treatment. Int. J. Nanomedicine 13, 8063–8074. doi:10.2147/IJN.S186881
Baldwin, P., Tangutoori, S., and Sridhar, S. (2018b). In Vitro analysis of PARP Inhibitor Nanoformulations. Int. J. Nanomedicine 13, 59–61. doi:10.2147/IJN.S124992
Balss, J., Meyer, J., Mueller, W., Korshunov, A., Hartmann, C., and von Deimling, A. (2008). Analysis of the IDH1 Codon 132 Mutation in Brain Tumors. Acta Neuropathol. 116 (6), 597–602. doi:10.1007/s00401-008-0455-2
Barazas, M., Gasparini, A., Huang, Y., Küçükosmanoğlu, A., Annunziato, S., Bouwman, P., et al. (2019). Radiosensitivity Is an Acquired Vulnerability of PARPi-Resistant BRCA1-Deficient Tumors. Cancer Res. 79 (3), 452–460. doi:10.1158/0008-5472.CAN-18-2077
Barber, L. J., Sandhu, S., Chen, L., Campbell, J., Kozarewa, I., Fenwick, K., et al. (2013). Secondary Mutations in BRCA2 Associated with Clinical Resistance to a PARP Inhibitor. J. Pathol. 229 (3), 422–429. doi:10.1002/path.4140
Baxter, P. A., Su, J. M., Onar-Thomas, A., Billups, C. A., Li, X. N., Poussaint, T. Y., et al. (2020). A Phase I/II Study of Veliparib (ABT-888) with Radiation and Temozolomide in Newly Diagnosed Diffuse Pontine Glioma: a Pediatric Brain Tumor Consortium Study. Neuro Oncol. 22 (6), 875–885. doi:10.1093/neuonc/noaa016
Becker, J. R., Cuella-Martin, R., Barazas, M., Liu, R., Oliveira, C., Oliver, A. W., et al. (2018). The ASCIZ-DYNLL1 axis Promotes 53BP1-dependent Non-homologous End Joining and PARP Inhibitor Sensitivity. Nat. Commun. 9 (1), 5406–5412. doi:10.1038/s41467-018-07855-x
Berens, M. E., and Giese, A. (1999). "...those Left behind." Biology and Oncology of Invasive Glioma cellsBiology and Oncology of Invasive Glioma Cells. Neoplasia 1 (3), 208–219. doi:10.1038/sj.neo.7900034
Blakeley, J. O., Olson, J., Grossman, S. A., He, X., Weingart, J., and Supko, J. G. (2009). Effect of Blood Brain Barrier Permeability in Recurrent High Grade Gliomas on the Intratumoral Pharmacokinetics of Methotrexate: a Microdialysis Study. J. Neurooncol 91 (1), 51–58. doi:10.1007/s11060-008-9678-2
Bouwman, P., Aly, A., Escandell, J. M., Pieterse, M., Bartkova, J., Van Der Gulden, H., et al. (2010). 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struct. Mol. Biol. 17 (6), 688–695. doi:10.1038/nsmb.1831
Bunting, S. F., Callén, E., Wong, N., Chen, H. T., Polato, F., Gunn, A., et al. (2010). 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 141 (2), 243–254. doi:10.1016/j.cell.2010.03.012
Calabrese, C. R., Almassy, R., Barton, S., Batey, M. A., Calvert, A. H., Canan-Koch, S., et al. (2004). Anticancer Chemosensitization and Radiosensitization by the Novel poly(ADP-Ribose) Polymerase-1 Inhibitor AG14361. J. Natl. Cancer Inst. 96 (1), 56–67. doi:10.1093/jnci/djh005
Caldecott, K. W., Aoufouchi, S., Johnson, P., and Shall, S. (1996). XRCC1 Polypeptide Interacts with DNA Polymerase and Possibly Poly (ADP-Ribose) Polymerase, and DNA Ligase III Is a Novel Molecular 'Nick-Sensor' In Vitro. Nucleic acids Res. 24 (22), 4387–4394. doi:10.1093/nar/24.22.4387
Carey, J. P. W., Karakas, C., Bui, T., Chen, X., Vijayaraghavan, S., Zhao, Y., et al. (2018). Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 78 (3), 742–757. doi:10.1158/0008-5472.CAN-17-1494
Chaudhuri, A. R., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J. E., et al. (2016). Erratum: Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 539 (7612), 456–387. doi:10.1038/nature19826
Chen, M. L., Sun, A., Cao, W., Eliason, A., Mendez, K. M., Getzler, A. J., et al. (2020). Physiological Expression and Function of the MDR1 Transporter in Cytotoxic T Lymphocytes. J. Exp. Med. 217 (5). doi:10.1084/jem.20191388
Chi, J., Chung, S. Y., Parakrama, R., Fayyaz, F., Jose, J., and Saif, M. W. (2021). The Role of PARP Inhibitors in BRCA Mutated Pancreatic Cancer. Ther. Adv. Gastroenterol. 14, 17562848211014818. doi:10.1177/17562848211014818
Choi, Y. E., Meghani, K., Brault, M. E., Leclerc, L., He, Y. J., Day, T. A., et al. (2016). Platinum and PARP Inhibitor Resistance Due to Overexpression of microRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 14 (3), 429–439. doi:10.1016/j.celrep.2015.12.046
Chornenkyy, Y., Agnihotri, S., Yu, M., Buczkowicz, P., Rakopoulos, P., Golbourn, B., et al. (2015). Poly-ADP-ribose Polymerase as a Therapeutic Target in Pediatric Diffuse Intrinsic Pontine Glioma and Pediatric High-Grade Astrocytoma. Mol. Cancer Ther. 14 (11), 2560–2568. doi:10.1158/1535-7163.MCT-15-0282
Clairmont, C. S., Sarangi, P., Ponnienselvan, K., Galli, L. D., Csete, I., Moreau, L., et al. (2020). TRIP13 Regulates DNA Repair Pathway Choice through REV7 Conformational Change. Nat. Cell Biol. 22 (1), 87–96. doi:10.1038/s41556-019-0442-y
Cohen, M. H., Johnson, J. R., and Pazdur, R. (2005). Food and Drug Administration Drug Approval Summary: Temozolomide Plus Radiation Therapy for the Treatment of Newly Diagnosed Glioblastoma Multiforme. Clin. Cancer Res. 11 (19), 6767–6771. doi:10.1158/1078-0432.CCR-05-0722
Czito, B. G., Deming, D. A., Jameson, G. S., Mulcahy, M. F., Vaghefi, H., Dudley, M. W., et al. (2017). Safety and Tolerability of Veliparib Combined with Capecitabine Plus Radiotherapy in Patients with Locally Advanced Rectal Cancer: a Phase 1b Study. Lancet Gastroenterol. Hepatol. 2 (6), 418–426. doi:10.1016/S2468-1253(17)30012-2
d'Amours, D., Desnoyers, S., d'Silva, I., and Poirier, G. G. (1999). Poly(ADP-ribosyl)ation Reactions in the Regulation of Nuclear Functions. Biochem. J. 342 ( Pt 2) (2), 249–268. doi:10.1042/bj3420249
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 Mutations Produce 2-hydroxyglutarate. Nature 462 (7274), 739–744. doi:10.1038/nature08617
Davis, M. E. (2016). Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 20 (5), S2–S8. doi:10.1188/16.CJON.S1.2-8
Dias, M. P., Moser, S. C., Ganesan, S., and Jonkers, J. (2021). Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 18 (12), 773–791. doi:10.1038/s41571-021-00532-x
Ding, X., Ray Chaudhuri, A., Callen, E., Pang, Y., Biswas, K., Klarmann, K. D., et al. (2016). Synthetic Viability by BRCA2 and PARP1/ARTD1 Deficiencies. Nat. Commun. 7 (1), 1–12. doi:10.1038/ncomms12425
Domchek, S. M. (2017). Reversion Mutations with Clinical Use of PARP Inhibitors: Many Genes, Many Versions. Cancer Discov. 7 (9), 937–939. doi:10.1158/2159-8290.CD-17-0734
Drané, P., Brault, M. E., Cui, G., Meghani, K., Chaubey, S., Detappe, A., et al. (2017). TIRR Regulates 53BP1 by Masking its Histone Methyl-Lysine Binding Function. Nature 543 (7644), 211–216. doi:10.1038/nature21358
Dungey, F. A., Löser, D. A., and Chalmers, A. J. (2008). Replication-dependent Radiosensitization of Human Glioma Cells by Inhibition of poly(ADP-Ribose) Polymerase: Mechanisms and Therapeutic Potential. Int. J. Radiat. Oncol. Biol. Phys. 72 (4), 1188–1197. doi:10.1016/j.ijrobp.2008.07.031
Dungrawala, H., Bhat, K. P., Le Meur, R., Chazin, W. J., Ding, X., Sharan, S. K., et al. (2017). RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 67 (3), 374e375–e5. doi:10.1016/j.molcel.2017.06.023
Eder, J. P., Doroshow, D. B., Do, K. T., Keedy, V. L., Sklar, J. S., Glazer, P., et al. (2021). Clinical Efficacy of Olaparib in IDH1/IDH2-Mutant Mesenchymal Sarcomas. JCO Precis. Oncol. 5, 466–472. doi:10.1200/PO.20.00247
Edwards, S. L., Brough, R., Lord, C. J., Natrajan, R., Vatcheva, R., Levine, D. A., et al. (2008). Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature 451 (7182), 1111–1115. doi:10.1038/nature06548
Fanucci, K., Pilat, M. J. P., Shah, R., Boerner, S. A., Li, J., Durecki, D. E., et al. (2022). Multicenter Phase 2 Trial of the PARP Inhibitor (PARPi) Olaparib in Recurrent IDH1 and IDH2-Mutant Contrast-Enhancing Glioma. Am. Soc. Clin. Oncol. 40, 2035. doi:10.1200/JCO.2022.40.16_suppl.2035
Flohr, C., Bürkle, A., Radicella, J. P., and Epe, B. (2003). Poly(ADP-ribosyl)ation Accelerates DNA Repair in a Pathway Dependent on Cockayne Syndrome B Protein. Nucleic Acids Res. 31 (18), 5332–5337. doi:10.1093/nar/gkg715
Francica, P., Mutlu, M., Blomen, V. A., Oliveira, C., Nowicka, Z., Trenner, A., et al. (2020). Functional Radiogenetic Profiling Implicates ERCC6L2 in Non-homologous End Joining. Cell Rep. 32 (8), 108068. doi:10.1016/j.celrep.2020.108068
Fukumoto, T., Zhu, H., Nacarelli, T., Karakashev, S., Fatkhutdinov, N., Wu, S., et al. (2019). N6-methylation of Adenosine of FZD10 mRNA Contributes to PARP Inhibitor Resistance. Cancer Res. 79 (11), 2812–2820. doi:10.1158/0008-5472.CAN-18-3592
Fulton, B., Short, S. C., James, A., Nowicki, S., McBain, C., Jefferies, S., et al. (2018). PARADIGM-2: Two Parallel Phase I Studies of Olaparib and Radiotherapy or Olaparib and Radiotherapy Plus Temozolomide in Patients with Newly Diagnosed Glioblastoma, with Treatment Stratified by MGMT Status. Clin. Transl. Radiat. Oncol. 8, 12–16. doi:10.1016/j.ctro.2017.11.003
Ghosh, D., Nandi, S., and Bhattacharjee, S. (2018). Combination Therapy to Checkmate Glioblastoma: Clinical Challenges and Advances. Clin. Transl. Med. 7 (1), 33–12. doi:10.1186/s40169-018-0211-8
Gogola, E., Duarte, A. A., de Ruiter, J. R., Wiegant, W. W., Schmid, J. A., de Bruijn, R., et al. (2018). Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer cell 33 (6), 1078–e12. doi:10.1016/j.ccell.2018.05.008
Goodall, J., Mateo, J., Yuan, W., Mossop, H., Porta, N., Miranda, S., et al. (2017). Circulating Cell-free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 7 (9), 1006–1017. doi:10.1158/2159-8290.CD-17-0261
Guillemette, S., Serra, R. W., Peng, M., Hayes, J. A., Konstantinopoulos, P. A., Green, M. R., et al. (2015). Resistance to Therapy in BRCA2 Mutant Cells Due to Loss of the Nucleosome Remodeling Factor CHD4. Genes Dev. 29 (5), 489–494. doi:10.1101/gad.256214.114
Guo, C., Zhang, F., Wu, X., Yu, X., Wu, X., Shi, D., et al. (2020). BTH-8, a Novel Poly (ADP-Ribose) Polymerase-1 (PARP-1) Inhibitor, Causes DNA Double-Strand Breaks and Exhibits Anticancer Activities In Vitro and In Vivo. Int. J. Biol. Macromol. 150, 238–245. doi:10.1016/j.ijbiomac.2020.02.069
Guo, X., Bai, Y., Zhao, M., Zhou, M., Shen, Q., Yun, C. H., et al. (2018). Acetylation of 53BP1 Dictates the DNA Double Strand Break Repair Pathway. Nucleic Acids Res. 46 (2), 689–703. doi:10.1093/nar/gkx1208
Gupta, S. K., Kizilbash, S. H., Carlson, B. L., Mladek, A. C., Boakye-Agyeman, F., Bakken, K. K., et al. (2016). Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 108 (5), djv369. doi:10.1093/jnci/djv369
Gupta, S. K., Mladek, A. C., Carlson, B. L., Boakye-Agyeman, F., Bakken, K. K., Kizilbash, S. H., et al. (2014). Discordant In Vitro and In Vivo Chemopotentiating Effects of the PARP Inhibitor Veliparib in Temozolomide-Sensitive versus -resistant Glioblastoma Multiforme Xenografts. Clin. Cancer Res. 20 (14), 3730–3741. doi:10.1158/1078-0432.CCR-13-3446
Gupta, S. K., Smith, E. J., Mladek, A. C., Tian, S., Decker, P. A., Kizilbash, S. H., et al. (2019). PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front. Oncol. 8, 670. doi:10.3389/fonc.2018.00670
Ha, K., Fiskus, W., Choi, D. S., Bhaskara, S., Cerchietti, L., Devaraj, S. G., et al. (2014). Histone Deacetylase Inhibitor Treatment Induces 'BRCAness' and Synergistic Lethality with PARP Inhibitor and Cisplatin against Human Triple Negative Breast Cancer Cells. Oncotarget 5 (14), 5637–5650. doi:10.18632/oncotarget.2154
Halford, S. E., Cruickshank, G., Dunn, L., Erridge, S., Godfrey, L., Herbert, C., et al. (2017). Results of the OPARATIC Trial: A Phase I Dose Escalation Study of Olaparib in Combination with Temozolomide (TMZ) in Patients with Relapsed Glioblastoma (GBM). Am. Soc. Clin. Oncol. 35, 2022. doi:10.1200/JCO.2017.35.15_suppl.2022
Hanif, F., Muzaffar, K., Perveen, K., Malhi, S. M., and Simjee, ShU. (2017). Glioblastoma Multiforme: a Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac J. Cancer Prev. 18 (1), 3–9. doi:10.22034/APJCP.2017.18.1.3
Hart, M. G., Garside, R., Rogers, G., Stein, K., and Grant, R. (2013). Temozolomide for High Grade Glioma. Cochrane Database Syst. Rev. (4), CD007415. doi:10.1002/14651858.CD007415.pub2
He, Y. J., Meghani, K., Caron, M. C., Yang, C., Ronato, D. A., Bian, J., et al. (2018). DYNLL1 Binds to MRE11 to Limit DNA End Resection in BRCA1-Deficient Cells. Nature 563 (7732), 522–526. doi:10.1038/s41586-018-0670-5
Hopkins, T. A., Shi, Y., Rodriguez, L. E., Solomon, L. R., Donawho, C. K., DiGiammarino, E. L., et al. (2015). Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 13 (11), 1465–1477. doi:10.1158/1541-7786.MCR-15-0191-T
Horbinski, C. (2013). What Do We Know about IDH1/2 Mutations So Far, and How Do We Use it? Acta Neuropathol. 125 (5), 621–636. doi:10.1007/s00401-013-1106-9
Isono, M., Niimi, A., Oike, T., Hagiwara, Y., Sato, H., Sekine, R., et al. (2017). BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 18 (2), 520–532. doi:10.1016/j.celrep.2016.12.042
Jasek, E., Gajda, M., Lis, G. J., Jasińska, M., and Litwin, J. A. (2014). Combinatorial Effects of PARP Inhibitor PJ34 and Histone Deacetylase Inhibitor Vorinostat on Leukemia Cell Lines. Anticancer Res. 34 (4), 1849–1856.
Jiang, J., Yang, E. S., Jiang, G., Nowsheen, S., Wang, H., Wang, T., et al. (2011). p53-dependent BRCA1 Nuclear Export Controls Cellular Susceptibility to DNA Damage. Cancer Res. 71 (16), 5546–5557. doi:10.1158/0008-5472.CAN-10-3423
Jiao, S., Xia, W., Yamaguchi, H., Wei, Y., Chen, M. K., Hsu, J. M., et al. (2017). PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 23 (14), 3711–3720. doi:10.1158/1078-0432.CCR-16-3215
Jiao, Y., Pawlik, T. M., Anders, R. A., Selaru, F. M., Streppel, M. M., Lucas, D. J., et al. (2013). Exome Sequencing Identifies Frequent Inactivating Mutations in BAP1, ARID1A and PBRM1 in Intrahepatic Cholangiocarcinomas. Nat. Genet. 45 (12), 1470–1473. doi:10.1038/ng.2813
Johnson, S. F., Cruz, C., Greifenberg, A. K., Dust, S., Stover, D. G., Chi, D., et al. (2016). CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 17 (9), 2367–2381. doi:10.1016/j.celrep.2016.10.077
Joshi, P. M., Sutor, S. L., Huntoon, C. J., and Karnitz, L. M. (2014). Ovarian Cancer-Associated Mutations Disable Catalytic Activity of CDK12, a Kinase that Promotes Homologous Recombination Repair and Resistance to Cisplatin and poly(ADP-Ribose) Polymerase Inhibitors. J. Biol. Chem. 289 (13), 9247–9253. doi:10.1074/jbc.M114.551143
Jue, T. R., Nozue, K., Lester, A. J., Joshi, S., Schroder, L. B., Whittaker, S. P., et al. (2017). Veliparib in Combination with Radiotherapy for the Treatment of MGMT Unmethylated Glioblastoma. J. Transl. Med. 15 (1), 61–68. doi:10.1186/s12967-017-1164-1
Kachhap, S. K., Rosmus, N., Collis, S. J., Kortenhorst, M. S., Wissing, M. D., Hedayati, M., et al. (2010). Downregulation of Homologous Recombination DNA Repair Genes by HDAC Inhibition in Prostate Cancer Is Mediated through the E2F1 Transcription Factor. PloS one 5 (6), e11208. doi:10.1371/journal.pone.0011208
Kaminska, B., Czapski, B., Guzik, R., Król, S. K., and Gielniewski, B. (2019). Consequences of IDH1/2 Mutations in Gliomas and an Assessment of Inhibitors Targeting Mutated IDH Proteins. Molecules 24 (5), 968. doi:10.3390/molecules24050968
Khan, K., Araki, K., Wang, D., Li, G., Li, X., Zhang, J., et al. (2010). Head and Neck Cancer Radiosensitization by the Novel poly(ADP-Ribose) Polymerase Inhibitor GPI-15427. Head. Neck 32 (3), 381–391. doi:10.1002/hed.21195
Kim, D.-S., Camacho, C. V., and Kraus, W. L. (2021). Alternate Therapeutic Pathways for PARP Inhibitors and Potential Mechanisms of Resistance. Exp. Mol. Med. 53 (1), 42–51. doi:10.1038/s12276-021-00557-3
Kim, Y., Kim, A., Sharip, A., Sharip, A., Jiang, J., Yang, Q., et al. (2017). Reverse the Resistance to PARP Inhibitors. Int. J. Biol. Sci. 13 (2), 198–208. doi:10.7150/ijbs.17240
Kizilbash, S. H., Gupta, S. K., Chang, K., Kawashima, R., Parrish, K. E., Carlson, B. L., et al. (2017). Restricted Delivery of Talazoparib across the Blood-Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer Ther. 16 (12), 2735–2746. doi:10.1158/1535-7163.MCT-17-0365
Kleihues, P., and Ohgaki, H. (1999). Primary and Secondary Glioblastomas: from Concept to Clinical Diagnosis. Neuro Oncol. 1 (1), 44–51. doi:10.1093/neuonc/1.1.44
Kleinberg, L., Supko, J. G., Mikkelsen, T., Blakeley, J. O. N., Stevens, G., Ye, X., et al. (2013). Phase I Adult Brain Tumor Consortium (ABTC) Trial of ABT-888 (Veliparib), Temozolomide (TMZ), and Radiotherapy (RT) for Newly Diagnosed Glioblastoma Multiforme (GBM) Including Pharmacokinetic (PK) Data. Am. Soc. Clin. Oncol. 31, 2065. doi:10.1200/jco.2013.31.15_suppl.2065
Kohlhapp, F. J., and Kaufman, H. L. (2016). Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin. Cancer Res. 22 (5), 1048–1054. doi:10.1158/1078-0432.CCR-15-2667
Kolinjivadi, A. M., Sannino, V., De Antoni, A., Zadorozhny, K., Kilkenny, M., Técher, H., et al. (2017). Smarcal1-mediated Fork Reversal Triggers Mre11-dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 67 (5), 867–e7. e867. doi:10.1016/j.molcel.2017.07.001
Kondrashova, O., Nguyen, M., Shield-Artin, K., Tinker, A. V., Teng, N. N. H., Harrell, M. I., et al. (2017). Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 7 (9), 984–998. doi:10.1158/2159-8290.CD-17-0419
Kondrashova, O., Topp, M., Nesic, K., Lieschke, E., Ho, G. Y., Harrell, M. I., et al. (2018). Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat. Commun. 9 (1), 1–16. doi:10.1038/s41467-018-05564-z
Kontandreopoulou, C. N., Diamantopoulos, P. T., Tiblalexi, D., Giannakopoulou, N., and Viniou, N. A. (2021). PARP1 as a Therapeutic Target in Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood Adv. 5 (22), 4794–4805. doi:10.1182/bloodadvances.2021004638
Koosha, F., Neshasteh-Riz, A., Takavar, A., Eyvazzadeh, N., Mazaheri, Z., Eynali, S., et al. (2017). The Combination of A-966492 and Topotecan for Effective Radiosensitization on Glioblastoma Spheroids. Biochem. Biophys. Res. Commun. 491 (4), 1092–1097. doi:10.1016/j.bbrc.2017.08.018
Krauthammer, M., Kong, Y., Ha, B. H., Evans, P., Bacchiocchi, A., McCusker, J. P., et al. (2012). Exome Sequencing Identifies Recurrent Somatic RAC1 Mutations in Melanoma. Nat. Genet. 44 (9), 1006–1014. doi:10.1038/ng.2359
Kurzrock, R., Galanis, E., Johnson, D. R., Kansra, V., Wilcoxen, K., Mcclure, T., et al. (2014). A Phase I Study of Niraparib in Combination with Temozolomide (TMZ) in Patients with Advanced Cancer. Am. Soc. Clin. Oncol. 31. doi:10.1200/jco.2013.31.15_suppl.2065
Lai, X., Broderick, R., Bergoglio, V., Zimmer, J., Badie, S., Niedzwiedz, W., et al. (2017). MUS81 Nuclease Activity Is Essential for Replication Stress Tolerance and Chromosome Segregation in BRCA2-Deficient Cells. Nat. Commun. 8 (1), 1–13. doi:10.1038/ncomms15983
Lal, S., and Snape, T. J. (2021). A Therapeutic Update on PARP Inhibitors: Implications in the Treatment of Glioma. Drug Discov. Today 26 (2), 532–541. doi:10.1016/j.drudis.2020.10.029
Lee, S. Y. (2016). Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 3 (3), 198–210. doi:10.1016/j.gendis.2016.04.007
Lemaçon, D., Jackson, J., Quinet, A., Brickner, J. R., Li, S., Yazinski, S., et al. (2017). MRE11 and EXO1 Nucleases Degrade Reversed Forks and Elicit MUS81-dependent Fork Rescue in BRCA2-Deficient Cells. Nat. Commun. 8 (1), 1–12. doi:10.1038/s41467-017-01180-5
Levin, V. A., Patlak, C. S., and Landahl, H. D. (1980). Heuristic Modeling of Drug Delivery to Malignant Brain Tumors. J. Pharmacokinet. Biopharm. 8 (3), 257–296. doi:10.1007/BF01059646
Li, X., Delzer, J., Voorman, R., de Morais, S. M., and Lao, Y. (2011). Disposition and Drug-Drug Interaction Potential of Veliparib (ABT-888), a Novel and Potent Inhibitor of poly(ADP-Ribose) Polymerase. Drug Metab. Dispos. 39 (7), 1161–1169. doi:10.1124/dmd.110.037820
Lin, F., de Gooijer, M. C., Roig, E. M., Buil, L. C., Christner, S. M., Beumer, J. H., et al. (2014). ABCB1, ABCG2, and PTEN Determine the Response of Glioblastoma to Temozolomide and ABT-888 Therapy. Clin. Cancer Res. 20 (10), 2703–2713. doi:10.1158/1078-0432.CCR-14-0084
Lin, K. K., Harrell, M. I., Oza, A. M., Oaknin, A., Ray-Coquard, I., Tinker, A. V., et al. (2019). BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 9 (2), 210–219. doi:10.1158/2159-8290.CD-18-0715
Lo, K. W., Kan, H. M., Chan, L. N., Xu, W. G., Wang, K. P., Wu, Z., et al. (2005). The 8-kDa Dynein Light Chain Binds to P53-Binding Protein 1 and Mediates DNA Damage-Induced P53 Nuclear Accumulation. J. Biol. Chem. 280 (9), 8172–8179. doi:10.1074/jbc.M411408200
LoRusso, P., Ji, J. J., Li, J., Heilbrun, L. K., Shapiro, G., Sausville, E. A., et al. (2011). Phase I Study of the Safety, Pharmacokinetics (PK), and Pharmacodynamics (PD) of the poly(ADP-Ribose) Polymerase (PARP) Inhibitor Veliparib (ABT-888; V) in Combination with Irinotecan (CPT-11; Ir) in Patients (Pts) with Advanced Solid Tumors. Jco 29 (15_Suppl. l), 3000. doi:10.1200/jco.2011.29.15_suppl.3000
Louis, D. N., Ohgaki, H., Wiestler, O. D., Cavenee, W. K., Burger, P. C., Jouvet, A., et al. (2007). The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 114 (2), 97–109. doi:10.1007/s00401-007-0243-4
Mardis, E. R., Ding, L., Dooling, D. J., Larson, D. E., McLellan, M. D., Chen, K., et al. (2009). Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 361 (11), 1058–1066. doi:10.1056/NEJMoa0903840
Marzolini, C., Paus, E., Buclin, T., and Kim, R. B. (2004). Polymorphisms in Human MDR1 (P-Glycoprotein): Recent Advances and Clinical Relevance. Clin. Pharmacol. Ther. 75 (1), 13–33. doi:10.1016/j.clpt.2003.09.012
Maughan, B. L., and Antonarakis, E. S. (2021). Olaparib and Rucaparib for the Treatment of DNA Repair-Deficient Metastatic Castration-Resistant Prostate Cancer. Expert Opin. Pharmacother. 22 (12), 1625–1632. doi:10.1080/14656566.2021.1912015
McCrorie, P., Mistry, J., Taresco, V., Lovato, T., Fay, M., Ward, I., et al. (2020). Etoposide and Olaparib Polymer-Coated Nanoparticles within a Bioadhesive Sprayable Hydrogel for Post-surgical Localised Delivery to Brain Tumours. Eur. J. Pharm. Biopharm. 157, 108–120. doi:10.1016/j.ejpb.2020.10.005
Mehta, M. P., Wang, D., Wang, F., Kleinberg, L., Brade, A., Robins, H. I., et al. (2015). Veliparib in Combination with Whole Brain Radiation Therapy in Patients with Brain Metastases: Results of a Phase 1 Study. J. Neurooncol 122 (2), 409–417. doi:10.1007/s11060-015-1733-1
Mensah, L. B., Morton, S. W., Li, J., Xiao, H., Quadir, M. A., Elias, K. M., et al. (2019). Layer-by-layer Nanoparticles for Novel Delivery of Cisplatin and PARP Inhibitors for Platinum-Based Drug Resistance Therapy in Ovarian Cancer. Bioeng. Transl. Med. 4 (2), e10131. doi:10.1002/btm2.10131
Middleton, M. R., Grob, J. J., Aaronson, N., Fierlbeck, G., Tilgen, W., Seiter, S., et al. (2000). Randomized Phase III Study of Temozolomide versus Dacarbazine in the Treatment of Patients with Advanced Metastatic Malignant Melanoma. J. Clin. Oncol. 18 (1), 158–166. doi:10.1200/JCO.2000.18.1.158
Miknyoczki, S., Chang, H., Grobelny, J., Pritchard, S., Worrell, C., McGann, N., et al. (2007). The Selective poly(ADP-Ribose) Polymerase-1(2) Inhibitor, CEP-8983, Increases the Sensitivity of Chemoresistant Tumor Cells to Temozolomide and Irinotecan but Does Not Potentiate Myelotoxicity. Mol. Cancer Ther. 6 (8), 2290–2302. doi:10.1158/1535-7163.MCT-07-0062
Militello, A. M., Zielli, T., Boggiani, D., Michiara, M., Naldi, N., Bortesi, B., et al. (2019). Mechanism of Action and Clinical Efficacy of CDK4/6 Inhibitors in BRCA-Mutated, Estrogen Receptor-Positive Breast Cancers: Case Report and Literature Review. Front. Oncol. 9, 759. doi:10.3389/fonc.2019.00759
Min, A., Im, S. A., Kim, D. K., Song, S. H., Kim, H. J., Lee, K. H., et al. (2015). Histone Deacetylase Inhibitor, Suberoylanilide Hydroxamic Acid (SAHA), Enhances Anti-tumor Effects of the Poly (ADP-Ribose) Polymerase (PARP) Inhibitor Olaparib in Triple-Negative Breast Cancer Cells. Breast Cancer Res. 17 (1), 33–13. doi:10.1186/s13058-015-0534-y
Min, A., and Im, S. A. (2020). PARP Inhibitors as Therapeutics: beyond Modulation of PARylation. Cancers (Basel) 12 (2), 394. doi:10.3390/cancers12020394
Mirman, Z., and de Lange, T. (2020). 53BP1: a DSB Escort. Genes Dev. 34 (1-2), 7–23. doi:10.1101/gad.333237.119
Molenaar, R. J., Radivoyevitch, T., Nagata, Y., Khurshed, M., Przychodzen, B., Makishima, H., et al. (2018). IDH1/2 Mutations Sensitize Acute Myeloid Leukemia to PARP Inhibition and This Is Reversed by IDH1/2-Mutant Inhibitors. Clin. Cancer Res. 24 (7), 1705–1715. doi:10.1158/1078-0432.CCR-17-2796
Moody, C. L., and Wheelhouse, R. T. (2014). The Medicinal Chemistry of Imidazotetrazine Prodrugs. Pharm. (Basel) 7 (7), 797–838. doi:10.3390/ph7070797
Morales, J., Li, L., Fattah, F. J., Dong, Y., Bey, E. A., Patel, M., et al. (2014). Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 24 (1), 15–28. doi:10.1615/critreveukaryotgeneexpr.2013006875
Murai, J., Feng, Y., Yu, G. K., Ru, Y., Tang, S. W., Shen, Y., et al. (2016). Resistance to PARP Inhibitors by SLFN11 Inactivation Can Be Overcome by ATR Inhibition. Oncotarget 7 (47), 76534–76550. doi:10.18632/oncotarget.12266
Muvarak, N. E., Chowdhury, K., Xia, L., Robert, C., Choi, E. Y., Cai, Y., et al. (2016). Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents - A Potential Therapy for Cancer. Cancer cell 30 (4), 637–650. doi:10.1016/j.ccell.2016.09.002
Nakada, M., Kita, D., Watanabe, T., Hayashi, Y., Teng, L., Pyko, I. V., et al. (2011). Aberrant Signaling Pathways in Glioma. Cancers (Basel) 3 (3), 3242–3278. doi:10.3390/cancers3033242
Nile, D. L., Rae, C., Hyndman, I. J., Gaze, M. N., and Mairs, R. J. (2016). An Evaluation In Vitro of PARP-1 Inhibitors, Rucaparib and Olaparib, as Radiosensitisers for the Treatment of Neuroblastoma. BMC cancer 16 (1), 1–13. doi:10.1186/s12885-016-2656-8
Ning, J., Wakimoto, H., Peters, C., Martuza, R. L., and Rabkin, S. D. (2017). Rad51 Degradation: Role in Oncolytic Virus-Poly(ADP-Ribose) Polymerase Inhibitor Combination Therapy in Glioblastoma. J. Natl. Cancer Inst. 109 (3), 1–13. doi:10.1093/jnci/djw229
Ning, J. F., Stanciu, M., Humphrey, M. R., Gorham, J., Wakimoto, H., Nishihara, R., et al. (2019). Myc Targeted CDK18 Promotes ATR and Homologous Recombination to Mediate PARP Inhibitor Resistance in Glioblastoma. Nat. Commun. 10 (1), 2910–2918. doi:10.1038/s41467-019-10993-5
Noordermeer, S. M., Adam, S., Setiaputra, D., Barazas, M., Pettitt, S. J., Ling, A. K., et al. (2018). The Shieldin Complex Mediates 53BP1-dependent DNA Repair. Nature 560 (7716), 117–121. doi:10.1038/s41586-018-0340-7
Norquist, B., Wurz, K. A., Pennil, C. C., Garcia, R., Gross, J., Sakai, W., et al. (2011). Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J. Clin. Oncol. 29 (22), 3008–3015. doi:10.1200/JCO.2010.34.2980
Ohgaki, H., Dessen, P., Jourde, B., Horstmann, S., Nishikawa, T., Di Patre, P. L., et al. (2004). Genetic Pathways to Glioblastoma: a Population-Based Study. Cancer Res. 64 (19), 6892–6899. doi:10.1158/0008-5472.CAN-04-1337
Ohgaki, H., and Kleihues, P. (2009). Genetic Alterations and Signaling Pathways in the Evolution of Gliomas. Cancer Sci. 100 (12), 2235–2241. doi:10.1111/j.1349-7006.2009.01308.x
Parrish, K. E., Cen, L., Murray, J., Calligaris, D., Kizilbash, S., Mittapalli, R. K., et al. (2015). Efficacy of PARP Inhibitor Rucaparib in Orthotopic Glioblastoma Xenografts Is Limited by Ineffective Drug Penetration into the Central Nervous System. Mol. Cancer Ther. 14 (12), 2735–2743. doi:10.1158/1535-7163.MCT-15-0553
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C., Leary, R. J., Angenendt, P., et al. (2008). An Integrated Genomic Analysis of Human Glioblastoma Multiforme. science 321 (5897), 1807–1812. doi:10.1126/science.1164382
Patel, P., Misra, S., Rodriguez, N. S., Vulugundam, G., Oh, A. L., Senyuk, V., et al. (2017). Combined Nanoparticle Delivery of PARP and DNA-PK Inhibition for Multiple Myeloma. Blood 130, 1809.
Pathade, A. D., Kommineni, N., Bulbake, U., Thummar, M. M., Samanthula, G., and Khan, W. (2019). Preparation and Comparison of Oral Bioavailability for Different Nano-Formulations of Olaparib. AAPS PharmSciTech 20 (7), 1–13. doi:10.1208/s12249-019-1468-y
Penning, T. D., Zhu, G. D., Gong, J., Thomas, S., Gandhi, V. B., Liu, X., et al. (2010). Optimization of Phenyl-Substituted Benzimidazole Carboxamide poly(ADP-Ribose) Polymerase Inhibitors: Identification of (S)-2-(2-fluoro-4-(pyrrolidin-2-yl)phenyl)-1H-benzimidazole-4-carboxamide (A-966492), a Highly Potent and Efficacious Inhibitor. J. Med. Chem. 53 (8), 3142–3153. doi:10.1021/jm901775y
Pettitt, S. J., Krastev, D. B., Brandsma, I., Dréan, A., Song, F., Aleksandrov, R., et al. (2018). Genome-wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 9 (1), 1–14. doi:10.1038/s41467-018-03917-2
Pettitt, S. J., Rehman, F. L., Bajrami, I., Brough, R., Wallberg, F., Kozarewa, I., et al. (2013). A Genetic Screen Using the PiggyBac Transposon in Haploid Cells Identifies Parp1 as a Mediator of Olaparib Toxicity. PloS one 8 (4), e61520. doi:10.1371/journal.pone.0061520
Piotrowski, A., Puduvalli, V., Wen, P., Campian, J., Colman, H., Pearlman, M., et al. (2019). ACTR-39. Pamiparib in Combination with Radiation Therapy (RT) And/or Temozolomide (TMZ) in Patients with Newly Diagnosed or Recurrent/refractory (R/R) Glioblastoma (GBM); Phase 1b/2 Study Update. Neuro-Oncology 21 (Suppl. ment_6), vi21–vi22. doi:10.1093/neuonc/noz175.081
Pishvaian, M. J., Biankin, A. V., Bailey, P., Chang, D. K., Laheru, D., Wolfgang, C. L., et al. (2017). BRCA2 Secondary Mutation-Mediated Resistance to Platinum and PARP Inhibitor-Based Therapy in Pancreatic Cancer. Br. J. Cancer 116 (8), 1021–1026. doi:10.1038/bjc.2017.40
Pitz, M. W., Desai, A., Grossman, S. A., and Blakeley, J. O. (2011). Tissue Concentration of Systemically Administered Antineoplastic Agents in Human Brain Tumors. J. Neurooncol 104 (3), 629–638. doi:10.1007/s11060-011-0564-y
Pulliam, N., Fang, F., Ozes, A. R., Tang, J., Adewuyi, A., Keer, H., et al. (2018). An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 24 (13), 3163–3175. doi:10.1158/1078-0432.CCR-18-0204
Quigley, D., Alumkal, J. J., Wyatt, A. W., Kothari, V., Foye, A., Lloyd, P., et al. (2017). Analysis of Circulating Cell-free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 7 (9), 999–1005. doi:10.1158/2159-8290.CD-17-0146
Quirt, I., Verma, S., Petrella, T., Bak, K., and Charette, M. (2007). Temozolomide for the Treatment of Metastatic Melanoma: a Systematic Review. Oncologist 12 (9), 1114–1123. doi:10.1634/theoncologist.12-9-1114
Reiss, K. A., Herman, J. M., Zahurak, M., Brade, A., Dawson, L. A., Scardina, A., et al. (2015). A Phase I Study of Veliparib (ABT-888) in Combination with Low-Dose Fractionated Whole Abdominal Radiation Therapy in Patients with Advanced Solid Malignancies and Peritoneal Carcinomatosis. Clin. Cancer Res. 21 (1), 68–76. doi:10.1158/1078-0432.CCR-14-1552
Robert, C., Nagaria, P. K., Pawar, N., Adewuyi, A., Gojo, I., Meyers, D. J., et al. (2016). Histone Deacetylase Inhibitors Decrease NHEJ Both by Acetylation of Repair Factors and Trapping of PARP1 at DNA Double-Strand Breaks in Chromatin. Leuk. Res. 45, 14–23. doi:10.1016/j.leukres.2016.03.007
Robins, H. I., Zhang, P., Gilbert, M. R., Chakravarti, A., de Groot, J. F., Grimm, S. A., et al. (2016). A Randomized Phase I/II Study of ABT-888 in Combination with Temozolomide in Recurrent Temozolomide Resistant Glioblastoma: an NRG Oncology RTOG Group Study. J. Neurooncol 126 (2), 309–316. doi:10.1007/s11060-015-1966-z
Rondinelli, B., Gogola, E., Yücel, H., Duarte, A. A., van de Ven, M., van der Sluijs, R., et al. (2017). EZH2 Promotes Degradation of Stalled Replication Forks by Recruiting MUS81 through Histone H3 Trimethylation. Nat. Cell Biol. 19 (11), 1371–1378. doi:10.1038/ncb3626
Rottenberg, S., Jaspers, J. E., Kersbergen, A., van der Burg, E., Nygren, A. O., Zander, S. A., et al. (2008). High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination with Platinum Drugs. Proc. Natl. Acad. Sci. U. S. A. 105 (44), 17079–17084. doi:10.1073/pnas.0806092105
Russo, A. L., Kwon, H. C., Burgan, W. E., Carter, D., Beam, K., Weizheng, X., et al. (2009). In Vitro and In Vivo Radiosensitization of Glioblastoma Cells by the Poly (ADP-Ribose) Polymerase Inhibitor E7016. Clin. Cancer Res. 15 (2), 607–612. doi:10.1158/1078-0432.CCR-08-2079
Sachdev, E., Tabatabai, R., Roy, V., Rimel, B. J., and Mita, M. M. (2019). PARP Inhibition in Cancer: an Update on Clinical Development. Target Oncol. 14 (6), 657–679. doi:10.1007/s11523-019-00680-2
Sahasrabudhe, R., Lott, P., Bohorquez, M., Toal, T., Estrada, A. P., Suarez, J. J., et al. (2017). Germline Mutations in PALB2, BRCA1, and RAD51C, Which Regulate DNA Recombination Repair, in Patients with Gastric Cancer. Gastroenterology 152 (5), 983–e6. e986. doi:10.1053/j.gastro.2016.12.010
Sakai, W., Swisher, E. M., Karlan, B. Y., Agarwal, M. K., Higgins, J., Friedman, C., et al. (2008). Secondary Mutations as a Mechanism of Cisplatin Resistance in BRCA2-Mutated Cancers. Nature 451 (7182), 1116–1120. doi:10.1038/nature06633
Sandhu, S. K., Schelman, W. R., Wilding, G., Moreno, V., Baird, R. D., Miranda, S., et al. (2013). The poly(ADP-Ribose) Polymerase Inhibitor Niraparib (MK4827) in BRCA Mutation Carriers and Patients with Sporadic Cancer: a Phase 1 Dose-Escalation Trial. Lancet Oncol. 14 (9), 882–892. doi:10.1016/S1470-2045(13)70240-7
Saran, F., James, A., McBain, C., Jefferies, S., Harris, F., Cseh, A., et al. (2018). ACTR-38. A Phase I Trial of Afatinib and Radiotherapy (Rt) with or without Temozolomide (Tmz) in Patients with Newly Diagnosed Glioblastoma (GBM). Neuro-Oncology 20 (Suppl. 6), vi20. doi:10.1093/neuonc/noy148.071
Sargazi, S., Mukhtar, M., Rahdar, A., Barani, M., Pandey, S., and Díez-Pascual, A. M. (2021). Active Targeted Nanoparticles for Delivery of Poly(ADP-Ribose) Polymerase (PARP) Inhibitors: A Preliminary Review. Int. J. Mol. Sci. 22 (19), 10319. doi:10.3390/ijms221910319
Satoh, M. S., and Lindahl, T. (1992). Role of poly(ADP-Ribose) Formation in DNA Repair. Nature 356 (6367), 356–358. doi:10.1038/356356a0
Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H., and Jasin, M. (2011). Double-strand Break Repair-independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 145 (4), 529–542. doi:10.1016/j.cell.2011.03.041
Schlacher, K., Wu, H., and Jasin, M. (2012). A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-Brca1/2. Cancer cell 22 (1), 106–116. doi:10.1016/j.ccr.2012.05.015
Schreiber, V., Amé, J. C., Dollé, P., Schultz, I., Rinaldi, B., Fraulob, V., et al. (2002). Poly(ADP-ribose) Polymerase-2 (PARP-2) Is Required for Efficient Base Excision DNA Repair in Association with PARP-1 and XRCC1. J. Biol. Chem. 277 (25), 23028–23036. doi:10.1074/jbc.M202390200
Shen, J., Zhao, W., Ju, Z., Wang, L., Peng, Y., Labrie, M., et al. (2019). PARPi Triggers the STING-dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 79 (2), 311–319. doi:10.1158/0008-5472.CAN-18-1003
Shen, Y., Rehman, F. L., Feng, Y., Boshuizen, J., Bajrami, I., Elliott, R., et al. (2013). BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 19 (18), 5003–5015. doi:10.1158/1078-0432.CCR-13-1391
Sim, H. W., Galanis, E., and Khasraw, M. (2022). PARP Inhibitors in Glioma: A Review of Therapeutic Opportunities. Cancers (Basel) 14 (4), 1003. doi:10.3390/cancers14041003
Singh, N., Miner, A., Hennis, L., and Mittal, S. (2021). Mechanisms of Temozolomide Resistance in Glioblastoma-A Comprehensive Review. Cancer drug Resist. (Alhambra, Calif.) 4, 17.
Sizemore, S. T., Mohammad, R., Sizemore, G. M., Nowsheen, S., Yu, H., Ostrowski, M. C., et al. (2018). Synthetic Lethality of PARP Inhibition and Ionizing Radiation Is P53-dependent. Mol. Cancer Res. 16 (7), 1092–1102. doi:10.1158/1541-7786.MCR-18-0106
Su, J. M., Thompson, P., Adesina, A., Li, X. N., Kilburn, L., Onar-Thomas, A., et al. (2014). A Phase I Trial of Veliparib (ABT-888) and Temozolomide in Children with Recurrent CNS Tumors: a Pediatric Brain Tumor Consortium Report. Neuro Oncol. 16 (12), 1661–1668. doi:10.1093/neuonc/nou103
Sule, A., Van Doorn, J., Sundaram, R. K., Ganesa, S., Vasquez, J. C., and Bindra, R. S. (2021). Targeting IDH1/2 Mutant Cancers with Combinations of ATR and PARP Inhibitors. Nar. cancer 3 (2), zcab018. doi:10.1093/narcan/zcab018
Taglialatela, A., Alvarez, S., Leuzzi, G., Sannino, V., Ranjha, L., Huang, J. W., et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 68 (2), 414–e8. e418. doi:10.1016/j.molcel.2017.09.036
Tang, Z., Jiang, B., Shi, Z., Gong, W., Liu, Y., Wang, X., et al. (2015). BGB-290, a Novel PARP Inhibitor with Unique Brain Penetration Ability, Demonstrated Strong Synergism with Temozolomide in Subcutaneous and Intracranial Xenograft Models. Cancer Res. 75. doi:10.1158/1538-7445.AM2015-1651
Tentori, L., Leonetti, C., Scarsella, M., D'Amati, G., Vergati, M., Portarena, I., et al. (2003). Systemic Administration of GPI 15427, a Novel poly(ADP-Ribose) Polymerase-1 Inhibitor, Increases the Antitumor Activity of Temozolomide against Intracranial Melanoma, Glioma, Lymphoma. Clin. Cancer Res. 9 (14), 5370–5379.
Ter Brugge, P., Kristel, P., Van Der Burg, E., Boon, U., De Maaker, M., Lips, E., et al. (2016). Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 108 (11). doi:10.1093/jnci/djw148
Thomas, H. D., Calabrese, C. R., Batey, M. A., Canan, S., Hostomsky, Z., Kyle, S., et al. (2007). Preclinical Selection of a Novel poly(ADP-Ribose) Polymerase Inhibitor for Clinical Trial. Mol. Cancer Ther. 6 (3), 945–956. doi:10.1158/1535-7163.MCT-06-0552
Thorsell, A.-G., and Schüler, H. (2017). Selectivity Profile of the Poly (ADP-Ribose) Polymerase (PARP) Inhibitor, A-966492. bioRxiv, 119818.
Tomar, M. S., Kumar, A., Srivastava, C., and Shrivastava, A. (2021). Elucidating the Mechanisms of Temozolomide Resistance in Gliomas and the Strategies to Overcome the Resistance. Biochimica Biophysica Acta (BBA) - Rev. Cancer 1876 (2), 188616. doi:10.1016/j.bbcan.2021.188616
Tomimatsu, N., Mukherjee, B., Catherine Hardebeck, M., Ilcheva, M., Vanessa Camacho, C., Louise Harris, J., et al. (2014). Phosphorylation of EXO1 by CDKs 1 and 2 Regulates DNA End Resection and Repair Pathway Choice. Nat. Commun. 5 (1), 1–10. doi:10.1038/ncomms4561
Vaidyanathan, A., Sawers, L., Gannon, A. L., Chakravarty, P., Scott, A. L., Bray, S. E., et al. (2016). ABCB1 (MDR1) Induction Defines a Common Resistance Mechanism in Paclitaxel- and Olaparib-Resistant Ovarian Cancer Cells. Br. J. Cancer 115 (4), 431–441. doi:10.1038/bjc.2016.203
van Vuurden, D. G., Hulleman, E., Meijer, O. L., Wedekind, L. E., Kool, M., Witt, H., et al. (2011). PARP Inhibition Sensitizes Childhood High Grade Glioma, Medulloblastoma and Ependymoma to Radiation. Oncotarget 2 (12), 984–996. doi:10.18632/oncotarget.362
Wagner, L. M. (2015). Profile of Veliparib and its Potential in the Treatment of Solid Tumors. Onco Targets Ther. 8, 1931–1939. doi:10.2147/OTT.S69935
Wang, H., Yang, E. S., Jiang, J., Nowsheen, S., and Xia, F. (2010). DNA Damage-Induced Cytotoxicity Is Dissociated from BRCA1's DNA Repair Function but Is Dependent on its Cytosolic Accumulation. Cancer Res. 70 (15), 6258–6267. doi:10.1158/0008-5472.CAN-09-4713
Wang, Y., Zheng, K., Huang, Y., Xiong, H., Su, J., Chen, R., et al. (2021). PARP Inhibitors in Gastric Cancer: Beacon of Hope. J. Exp. Clin. Cancer Res. 40 (1), 1–13. doi:10.1186/s13046-021-02005-6
Watanabe, T., Nobusawa, S., Kleihues, P., and Ohgaki, H. (2009). IDH1 Mutations Are Early Events in the Development of Astrocytomas and Oligodendrogliomas. Am. J. Pathol. 174 (4), 1149–1153. doi:10.2353/ajpath.2009.080958
Weigelt, B., Comino-Méndez, I., de Bruijn, I., Tian, L., Meisel, J. L., García-Murillas, I., et al. (2017). Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 23 (21), 6708–6720. doi:10.1158/1078-0432.CCR-17-0544
Wu, S., Gao, F., Zheng, S., Zhang, C., Martinez-Ledesma, E., Ezhilarasan, R., et al. (2020). EGFR Amplification Induces Increased DNA Damage Response and Renders Selective Sensitivity to Talazoparib (PARP Inhibitor) in Glioblastoma. Clin. Cancer Res. 26 (6), 1395–1407. doi:10.1158/1078-0432.CCR-19-2549
Wu, S., Li, X., Gao, F., de Groot, J. F., Koul, D., and Yung, W. K. A. (2021). PARP-mediated PARylation of MGMT Is Critical to Promote Repair of Temozolomide-Induced O6-Methylguanine DNA Damage in Glioblastoma. Neuro Oncol. 23 (6), 920–931. doi:10.1093/neuonc/noab003
Xu, G., Chapman, J. R., Brandsma, I., Yuan, J., Mistrik, M., Bouwman, P., et al. (2015). REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature 521 (7553), 541–544. doi:10.1038/nature14328
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 360 (8), 765–773. doi:10.1056/NEJMoa0808710
Yang, E. S., Nowsheen, S., Rahman, M. A., Cook, R. S., and Xia, F. (2012). Targeting BRCA1 Localization to Augment Breast Tumor Sensitivity to poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 72 (21), 5547–5555. doi:10.1158/0008-5472.CAN-12-0934
Ying, S., Hamdy, F. C., and Helleday, T. (2012). Mre11-dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1. Cancer Res. 72 (11), 2814–2821. doi:10.1158/0008-5472.CAN-11-3417
Yung, W. K., Prados, M. D., Yaya-Tur, R., Rosenfeld, S. S., Brada, M., Friedman, H. S., et al. (1999). Multicenter Phase II Trial of Temozolomide in Patients with Anaplastic Astrocytoma or Anaplastic Oligoastrocytoma at First Relapse. Temodal Brain Tumor Group. J. Clin. Oncol. 17 (9), 2762–2771. doi:10.1200/JCO.1999.17.9.2762
Zhang, D., Baldwin, P., Leal, A. S., Carapellucci, S., Sridhar, S., and Liby, K. T. (2019). A Nano-Liposome Formulation of the PARP Inhibitor Talazoparib Enhances Treatment Efficacy and Modulates Immune Cell Populations in Mammary Tumors of BRCA-Deficient Mice. Theranostics 9 (21), 6224–6238. doi:10.7150/thno.36281
Zhu, H., Wei, M., Xu, J., Hua, J., Liang, C., Meng, Q., et al. (2020). PARP Inhibitors in Pancreatic Cancer: Molecular Mechanisms and Clinical Applications. Mol. Cancer 19 (1), 49–15. doi:10.1186/s12943-020-01167-9
Zimmermann, M., and De Lange, T. (2014). 53BP1: Pro Choice in DNA Repair. Trends Cell Biol. 24 (2), 108–117. doi:10.1016/j.tcb.2013.09.003
Keywords: GBM, TMZ, PARP inhibitors, PARP inhibitor resistance, hematological toxicity, targeted delivery, combination therapy
Citation: Bisht P, Kumar VU, Pandey R, Velayutham R and Kumar N (2022) Role of PARP Inhibitors in Glioblastoma and Perceiving Challenges as Well as Strategies for Successful Clinical Development. Front. Pharmacol. 13:939570. doi: 10.3389/fphar.2022.939570
Received: 09 May 2022; Accepted: 10 June 2022;
Published: 06 July 2022.
Edited by:
Syed Nasir Abbas Bukhari, Al Jouf University, Saudi ArabiaReviewed by:
Ciler Celik-Ozenci, Koç University, TurkeyCopyright © 2022 Bisht, Kumar, Pandey, Velayutham and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nitesh Kumar, bml0ZXNoa3VtYXIwNEBuaXBlcmhhamlwdXIuYWMuaW4=, bml0ZXNoa3VtYXI0M0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.