 Chen Chen
Chen Chen Zehua Wang
Zehua Wang Yanru Qin
Yanru Qin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 22 July 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.935536
This article is part of the Research Topic New Anti-Cancer Strategies Targeting Epigenetic Modifications and Associated Metabolism Reprogramming View all 10 articles
Cancer cells undergo metabolic adaptations to sustain their growth and proliferation under several stress conditions thereby displaying metabolic plasticity. Epigenetic modification is known to occur at the DNA, histone, and RNA level, which can alter chromatin state. For almost a century, our focus in cancer biology is dominated by oncogenic mutations. Until recently, the connection between metabolism and epigenetics in a reciprocal manner was spotlighted. Explicitly, several metabolites serve as substrates and co-factors of epigenetic enzymes to carry out post-translational modifications of DNA and histone. Genetic mutations in metabolic enzymes facilitate the production of oncometabolites that ultimately impact epigenetics. Numerous evidences also indicate epigenome is sensitive to cancer metabolism. Conversely, epigenetic dysfunction is certified to alter metabolic enzymes leading to tumorigenesis. Further, the bidirectional relationship between epigenetics and metabolism can impact directly and indirectly on immune microenvironment, which might create a new avenue for drug discovery. Here we summarize the effects of metabolism reprogramming on epigenetic modification, and vice versa; and the latest advances in targeting metabolism-epigenetic crosstalk. We also discuss the principles linking cancer metabolism, epigenetics and immunity, and seek optimal immunotherapy-based combinations.
Cancer metabolism is based on the principle that cancer cells undergo metabolic adaptations to sustain their uncontrolled proliferation. Such adaptations render malignant cells to exhibit altered metabolism compared to the normal cells. In 1920s, Warburg firstly proposed (Kaye, 1998; Chinnaiyan et al., 2012) that cancer cells display enhanced glycolysis and increased secretion of lactate even with abundant oxygen supply. This phenomenon is termed as “Warburg effect” or aerobic glycolysis. Moreover, an emerging class of metabolic alterations enables tumor cells to take up available ample nutrients and utilize them to produce ATP, generate biosynthetic precursors for cell anabolism, and tolerate stresses related to malignancy, such as hypoxia and nutrient starvation (Owen et al., 2002; Koppenol et al., 2011; Lunt and Vander Heiden, 2011; Metallo et al., 2011; Mullen et al., 2011; Wise et al., 2011; Cantor and Sabatini, 2012; Ahn and Metallo, 2015). In this context, cancer metabolism provides a selective advantage during tumorigenesis. Metabolic reprogramming (Figure 1) is now recognized as a hallmark of cancer (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016), which could be intrinsically regulated by genotype and epigenotype, or extrinsically affected by tumor microenvironment (TME).
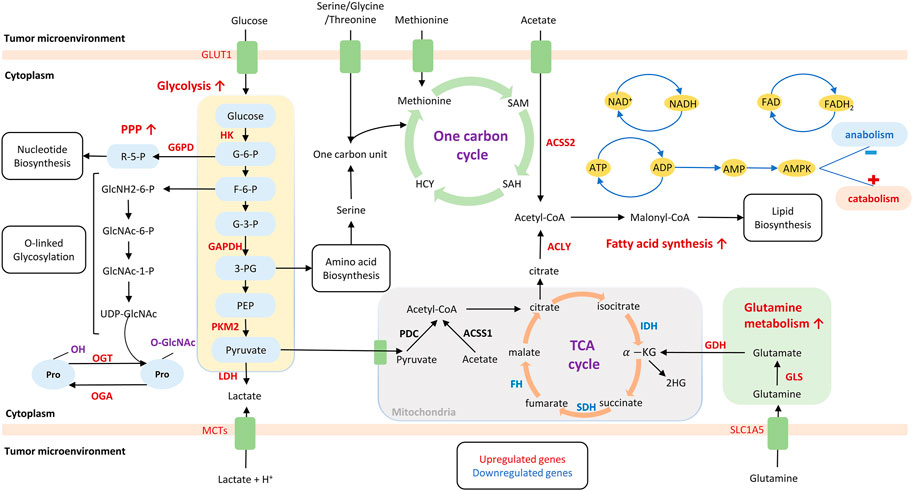
FIGURE 1. Metabolism reprogramming in cancer cells. Metabolism reprogramming is characterized by a class of altered pathway, including enhanced glycolysis with increased lactate production, and enhanced pentose phosphate pathway, fatty acid synthesis, and glutamine metabolism. These metabolic pathways support energy supply and macromolecule biosynthesis, such as nucleotides, amino acids, and lipids. Metabolites that are produced by altered metabolism have the potential to control signaling or epigenetic pathways by regulating reactive oxygen species, acetylation, and methylation. Upregulated genes or proteins are labels red, whereas downregulated genes or proteins are labeled blue. GLUT, glucose transporter; MCT, monocarboxylate transporter; SLC1A5, solute carrier family 1 member 5; TCA, Tricarboxylic acid cycle; G6PD, glucose-6-phosphate dehydrogenase; HK, hexokinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PKM, pyruvate kinase M 2; LDH, lactate dehydrogenase; ACSS2, Acyl-CoA short-chain synthetase-2; ACSS1: Acyl-CoA short-chain synthetase-1; ACLY: ATP citrate lyase; GLS, glutaminase; GDH, glutamate dehydrogenase; PDC: pyruvate dehydrogenase complex; FH, fumarate hydratase; SDH, succinate dehydrogenase; IDH1/2, isocitrate dehydrogenase 1/2; HCY, homocysteine; PPP, pentose phosphate pathway; ATP, adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; OGT, O-GlcNAc transferase; OGA, O-GlcNAcase.
Epigenetics was firstly established by Conrad Waddington in 1942 (Cairns et al., 2011), which refers to the study of modification in gene expression or cellular phenotype that occurs without changes in DNA nucleotide sequences (Possemato et al., 2011). The basic unit of chromatin organization is nucleosome, which is composed of DNA and histone octamer. Chromatin state is a dynamic event that controls gene transcription. Epigenetic modification of gene expression occurs at the DNA, histone, and RNA level. The most well-characterized examples are DNA methylation, histone methylation, acetylation, phosphorylation, ubiquitination, and microRNA-dependent gene silencing (Margueron and Reinberg, 2010). It is widely recognized that epigenetic dysfunction is a common feature of many cancers (Ribich et al., 2017). Numerous excellent reviews have summarized the biology fundamentals of chromatin-modified proteins (CMPs) (Tessarz and Kouzarides, 2014; Piunti and Shilatifard, 2016; Soshnev et al., 2016) and the therapeutic potentials to target CMPs in tumor (Pfister and Ashworth, 2017).
For almost a century, our focus in cancer is dominated by oncogenic mutations. Until recently, the connection between metabolism and epigenetics was emphasized in cancer biology. Metabolism reprogramming is known to affect epigenetic landscapes through different mechanisms. Conversely, epigenetic regulation contributes to altered metabolic activities. Hence, cancer metabolism and epigenetics are highly interwoven in a reciprocal manner. This great breakthrough has gained wide interest in targeting both altered metabolism and modified epigenetics. However, whether these two hallmarks synergistically attack tumor remains unknown. Noteworthy, such a complex relationship has the potential to affect immune system, such as trained immunity, T cell activation, macrophage activation. A novel strategy is to target epigenetics-metabolism axis in combination with immunotherapy, potentially boosting more potent antitumor responses.
In this review article, we firstly summarize the metabolic alterations that drive epigenetic changes in cancer, and vice versa. We next describe the therapeutic opportunities by targeting metabolism-epigenetic crosstalk. Further, we discuss the principles linking metabolism, epigenetics to immunity and introduce the rationale for novel immunotherapy-based combinations. Our aim is to introduce the fundamentals of connection between metabolism and epigenetics in cancer biology and discuss potential pharmacological strategies that can exploit the metabolism and epigenetics in malignancy.
Tumors are likely to harbor epigenetic changes driven by their cellular metabolism. There are several different mechanisms explaining the influx from metabolism to chromatin.
Epigenetic enzymes employ several metabolic intermediates as substrates or co-factors to carry out post-translational modifications of DNA and histone (Katada et al., 2012), which in turn influence metabolic gene expression. Examples of such metabolites include: SAM,

TABLE 1. Fundamental interface of metabolism and epigenetics.
DNA and histone methylation are respectively mediated by DNA methyltransferase (DNMT) enzymes and histone methyltransferase (HMT) enzymes (Varier and Timmers, 2011), both of which utilize S-Adenosyl-methionine (SAM) as a major methyl donor. Methylation is to transfer a methyl group from SAM to the receptor, and the remaining residue is S-adenosyl-homocysteine (SAH) that is inhibitory to methyltransferase. SAM is derived from one-carbon metabolism that plays integral roles in DNA synthesis and methylation reaction. The most studied metabolites, like glucose and glutamine, feed into the one-carbon cycle and increase the availability of SAM. Both global DNA hypomethylation and site-specific CpG hypermethylation are frequent epigenetic abnormities observed in cancer (Sandoval and Esteller, 2012), while histone methylation may activate or repress gene transcription (Vakoc et al., 2005; Berger, 2007; Bernstein et al., 2007). Therefore, SAM/SAH ratio directly affect the methylation status of chromatin.
Reversal of DNA and histone methylation is catalyzed by DNA and histone demethylase. Histone demethylation is regulated by two classes of enzymes: lysine-specific demethylase family (LSD1 and LSD2) (Fang et al., 2010) and JmjC-containing family, both of which are dependent on ferrous adenine dinucleotide (FAD). Also, JmjC family is ferrous ion-dependent oxygenase requiring
Histone acetylation is another important epigenetic modification that depends on histone acetyltransferase (HAT) and histone deacetylase (HDAC) (Shahbazian and Grunstein, 2007). Acetyl-CoA is a pivotal metabolite for energy production and anabolic process (Wellen and Thompson, 2012; Pietrocola et al., 2015). HAT transfers the acetyl moiety of acetyl-CoA to lysine residues of histone, while HDAC is responsible for removing the acetyl group to reverse histone acetylation. It is well-known histone acetylation can increase nucleosome mobility and activate transcription elongation (Racey and Byvoet, 1971; Cai et al., 2011). Previous study figured out, in yeast and mammalian cells, the glycolysis dynamically governs the acetyl-CoA quantity and correspondingly regulates HAT-dependent histone acetylation (Friis et al., 2009; Cai et al., 2011; Lee et al., 2014).
Histone deacetylation is catalyzed by two kinds of deacetylases: zinc-dependent and NAD+-dependent proteins. Deacetylation results in the tight wrapping of DNA by histone and hence promotes gene repression and silence (Imai et al., 2000; Finkel et al., 2009). Similarly, some metabolites function as antagonists that inhibit the activities of HDAC. For example, butyrate can robustly antagonize HDACs I, II and IV (Candido et al., 1978). Also, NAD+ is regarded as a catalytic co-factor for HDAC III to mediate histone deacetylation (Thakur and Chen, 2019). Further, evidence illustrated higher histone deacetylation levels are associated with poorer prognosis (Kurdistani, 2011).
Acetate has been implicated in driving histone acetylation and deacetylation. Recently, the role of acetate in the interaction between metabolism and epigenetics has been emphasized during tumorigenesis. Under hypoxia, cancer cells decrease the reliance on glucose and glutamate and inversely increase the demand of acetate as a substitute carbon source for lipid synthesis (Kamphorst et al., 2014). Consequently, acetate must be converted to acetyl-CoA either by ACSS1 in mitochondria or by ACSS2 in the cytoplasm or nucleus (Figure 1). There is already evidence that both acetate and acetyl-CoA facilitate tumor growth by histone acetylation in yeast (Cai et al., 2011). ACSS2, as the only known enzyme utilizing free acetate in nucleus (Moffett et al., 2020), could shape the epigenetic landscape via selective histone acetylation. More specifically, ACSS2 is translocated from cytoplasm to the nucleus supplying a local of acetyl-CoA (Chen et al., 2017), which contributes to all kinds of acetylation reactions in cell nuclei. One study indicated (Gao et al., 2016), under hypoxia condition, ACSS2 catalyzes the conversion of acetate to acetyl-CoA in the hepatoma carcinoma cells, facilitating the hyper-acetylation of histone K3K9, H3K27, and H3K56 and thereby upregulating the expression of lipogenic enzymes. This explains how acetate links metabolite levels to epigenetic regulation and gene transcription. Otherwise, ACSS2 acts to recycle acetate generated from HDAC-mediated deacetylation reactions under metabolic stresses, replenishing the cytoplasmic and nuclear storage and thus supporting chromatin remodeling events (Moffett et al., 2020).
Some kinase could be translocated to nucleus and straightly phosphorylate histone (Baek, 2011). For example, AMP-activated protein kinase (AMPK) acts as sensory signal of ATP/AMP ratio (Hardie, 2011). Conversion of ATP to AMP aids in anabolic process via AMPK-mediated pathway, whereas catabolism relies on the opposite switch from AMP to ATP. Owing to metabolic stress and low ATP/AMP ratio, AMPK is activated to phosphorylate histone H2B on serine 36 that triggers gene expression in favor of tumor survival (Bungard et al., 2010).
Protein glycosylation is carried by opposite actions of O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), respectively responsible for the addition and removal of O-GlcNAc from proteins. One of the most common features that cancer cells demonstrate is OGT overexpression leading to protein hyper-glycosylation (Pinho and Reis, 2015). Typically, O-GlcNAc is produced in Hexosamine biosynthetic pathway (HBP). In this pathway, glucose is firstly converted into glucose-6-P and then fructose-6-P. A series of metabolites, such as acetyl-CoA, UTP, glutamine, subsequently participate in the production of UDP-GlcNAc, the activated substrate for O-GlcNAcylation. Therefore, HBP integrated various metabolism pathways. Upregulation of HBP is associated with abnormal O-GlcNAcylation and more invasive behavior (Caldwell et al., 2010; Wellen et al., 2010; Itkonen et al., 2013; Onodera et al., 2014; Lucena et al., 2016). Recently, studies confirm that enhanced glycolysis aids in protein glycosylation (Wong et al., 2017). Moreover, OGT is associated with TETs to control O-GlcNAcylation of histone H2B for activation of gene transcription (Chen et al., 2013; Ito et al., 2014), while OGT is coordinated with EZH2 to modulate H3K27me3 for silence of tumor suppressor genes (Chu et al., 2014).
Taken together, either methylation or acetylation controls the activation and repression of gene transcription. This event is balanced by various epigenetic enzymes. The cellular metabolites, such as SAM/SAH, acetyl-CoA/CoA, NAD+/NADH, ATP/AMP ratio, commonly act as substrate or co-factors for these epigenetic-based enzymes (Table 2, Figure 2). Their fluctuating concentrations could regulate the epigenetic profile and affect gene transcription.
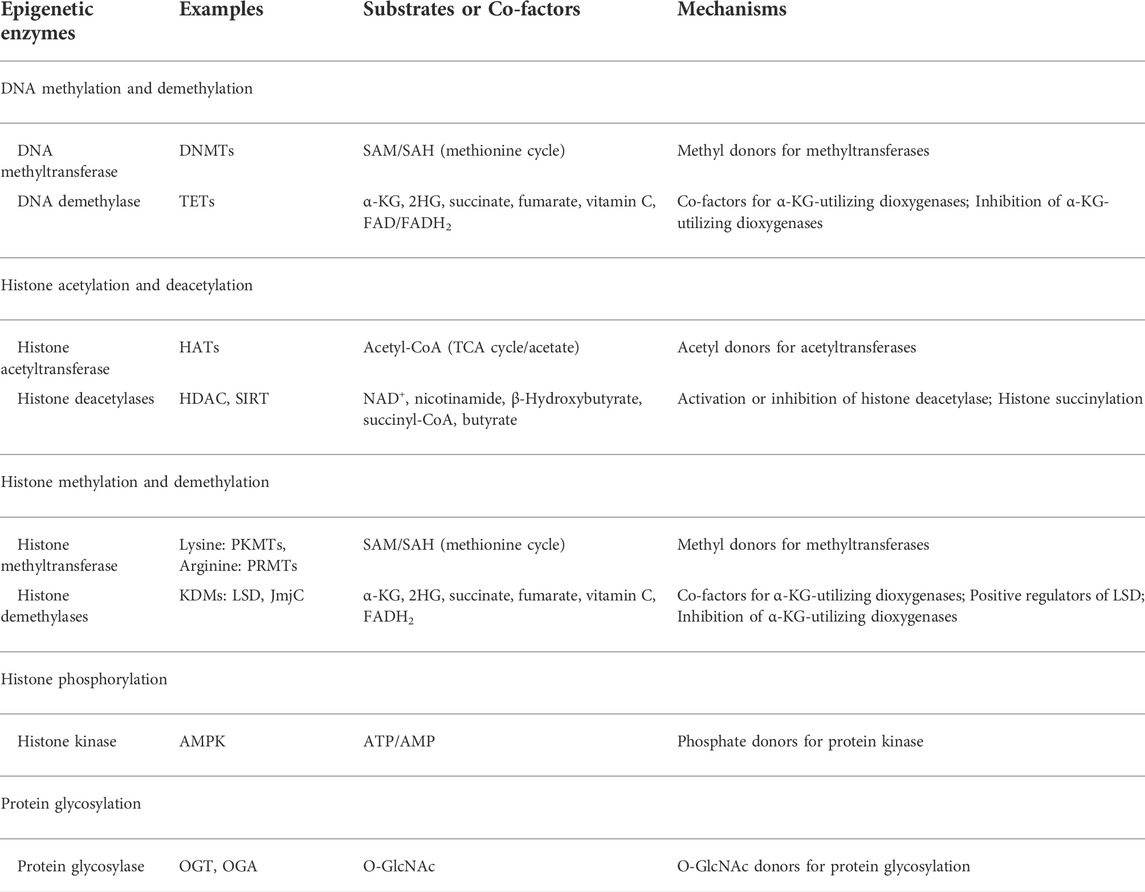
TABLE 2. Metabolites are either substrates or co-factors for epigenetic enzymes in cancer biology.
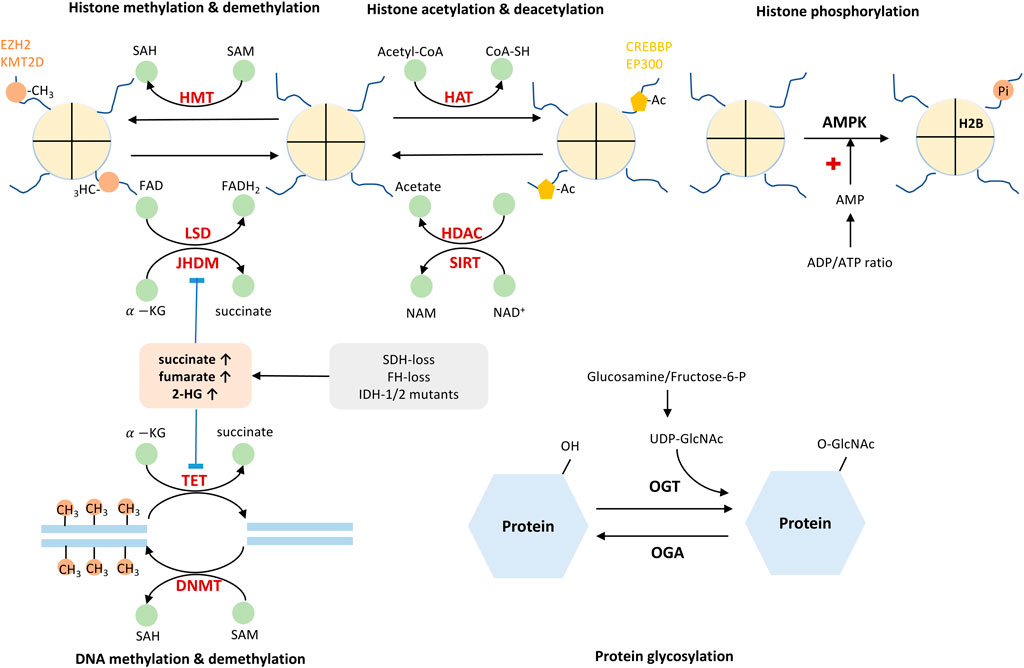
FIGURE 2. Cellular metabolites serve as co-factors or substrates for epigenetic enzymes. Addition or removal of epigenetic marks is catalyzed by epigenetic enzymes, of which process relies on several critical metabolites. SAH/SAM, NAD+/NADH, Acetyl-CoA/Co-A, ATP/ADP ratio act as important molecules or signals governing epigenetic modifications. In addition, Metabolites such as succinate, fumarate, 2-HG, and lactate could inhibit the activity of epigenetic enzymes. HMT, histone methyltransferase; LSD, lysine-specific histone demethylase; JHDM, Jumonji domain-containing histone demethylase; HAT, histone acetyltransferase; HDAC, histone deacetylase; SIRT, sirtuins; DNMT, DNA methyltransferase; TET, ten-eleven translocation methylcytosine dioxygenase; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine;
Mutations in metabolic enzymes subject the cells to tumorigenesis. Such changes facilitate the accumulation of metabolites that ultimately lead to epigenetic dysfunction (DeBerardinis and Chandel, 2016) and immunosuppression (Table 3).
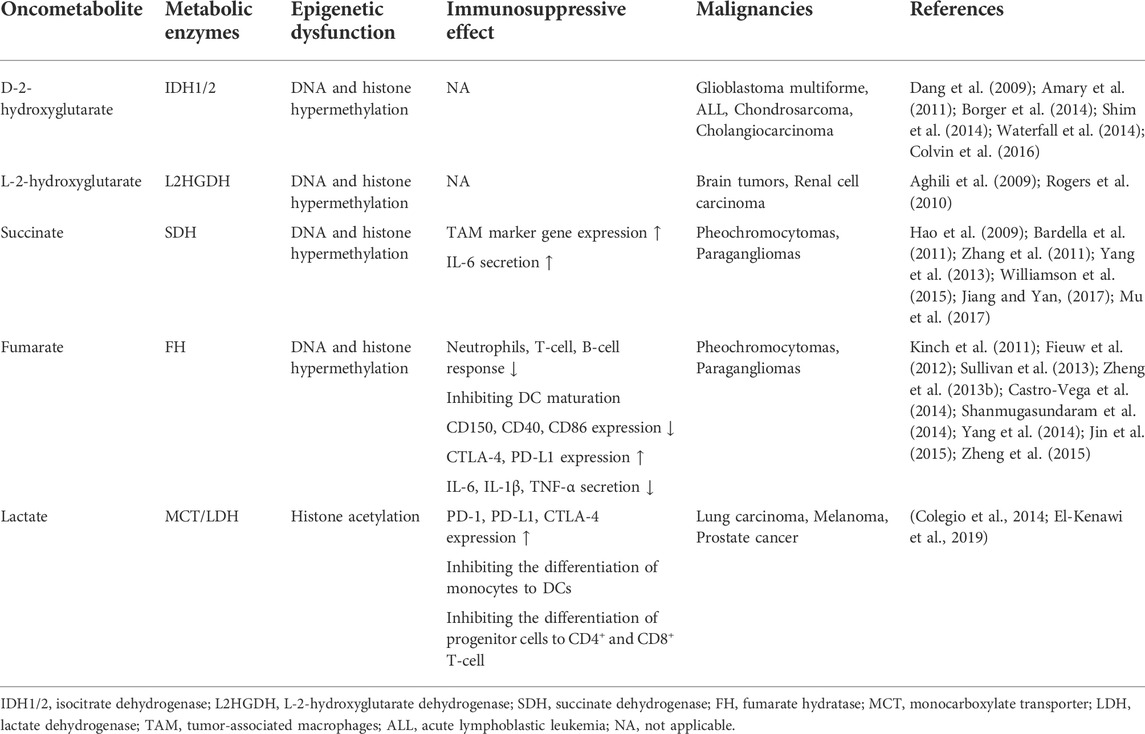
TABLE 3. The effect of oncometabolites on epigenetic dysfunction and immunosuppression.
One example is to generate oncometabolite. Oncometabolite refers to metabolites whose great quantity increases markedly in tumors compared with normal cells (Nowicki and Gottlieb, 2015). This new term is used to describe metabolites for which 1) there is a well-characterized mechanism connecting mutations in metabolic enzymes to accumulation of a certain metabolite; 2) there is convincing evidence for some metabolites as a predisposition to tumorigenesis. Oncometabolites are frequently associated with aberrant DNA damage and enable the tumor microenvironment (TME) more invasive. Currently, D-2-hydroxyglutarate (D2HG), L-2-hydroxyglutarate (L2HG), succinate, fumarate, and lactate are recognized oncometabolites.
The first emphasized oncometabolite is D2HG, a reduced form of the TCA cycle intermediate
This principle also applies to another two oncometabolites: succinate and fumarate (Yang et al., 2013). Mutational inactivation of succinate dehydrase (SDH) and fumarate hydratase (FH) respectively contributes to the stacking up of succinate and fumarate (Baysal et al., 2000; Tomlinson et al., 2002; Gottlieb and Tomlinson, 2005), both of which interfere with
To ensure adequate ATP supply, the malignant transformation is associated with an upregulated glycolysis (de Groof et al., 2009). Cancer cells upregulate glycolytic enzymes and metabolic transporters, which is connected with lactate overproduction. A new discovery considered lactate might have an effect on lysine residues of histone, acting in a similar way to acetylation and gene activation (Hou et al., 2019; Zhang et al., 2019). This phenomenon is based on the conversion of lactate to acetyl residues and thereby stimulates tumor angiogenesis. The accumulation of lactate also exerts an immunosuppressive effect on TME through inhibiting the differentiation and maturation of DC and T cell (Gottfried et al., 2006).
Cancer-specific mutations of metabolic enzymes with implications in epigenetic regulation have been reported. Phosphoglycerate dehydrogenase (PHGDH) is overexpressed in breast cancer and melanoma (Locasale et al., 2011; Possemato et al., 2011), directing the metabolism toward the serine biosynthesis pathway. Serine provides methyl donors to one-carbon metabolism, thereby affecting cellular epigenetics (Locasale, 2013). Conversely, PHGDH silence can downregulate serine synthesis leading to tumor growth suppression (Locasale et al., 2011; Possemato et al., 2011). Another example is proline dehydrogenase (PRODH) that catalyzes proline to produce pyrroline-5-carbonxylate (P5C), which is sequentially converted into glutamate and
As summarized, mutations in genes encoding metabolic enzymes have been recognized in caner, but they are rare. These lesions in genes related to metabolism constitute a new class of cancer-associated mutations that is able to subvert normal epigenetic regulation. It is tempting to speculate that these mutations provide the hope of identifying novel targets.
A number of metabolic enzymes are altered attributing to DNA methylation. Examples of such enzymes involve Fructose-1,6-bisphosphastase (FBP-1), fructose-1,6-bisphosphatase (FBP-2), glucose transporter 1 (GLUT-1), Hexokinase (HK2), and pyruvate kinase isozyme 2 (PKM-2).
As reported, promoter hypermethylation leads to the silence of FBP-1 and FBP-2 in gastric, colon, liver, and breast cancers (Kamphorst et al., 2014; Gao et al., 2016). Both FBP-1 and FBP-2 are rate-limiting enzymes for gluconeogenesis that antagonize glycolysis. Theoretically, the silence of FBP-1 or FBP-2 contributes to glycolytic phenotype, supporting macromolecular biosynthesis and energy production. DNA methylation also mediates the gene overexpression of GLUT-1 that transports glucose from tumor microenvironment to cytoplasm (Lopez-Serra et al., 2014). Oppositely, promoter hypomethylation results in the upregulation of HK2 in glioblastoma and hepatic carcinoma (Chen et al., 2011; Wolf et al., 2011) and the overexpression of PKM2 in multiple cancer types (Desai et al., 2014).
In brief, increased HK2 and PKM-2 levels promote enhanced glycolysis, while the silence of FBP-1 and FBP-2 limit gluconeogenesis. DNA methylation contributes to a higher glycolytic influx, which is beneficial to the proliferation of tumor cells.
Sirtuins (SIRTs), an enzyme catalyzing histone deacetylation, has been shown to function in cancer metabolism. Examples of epigenetic enzymes are SIRT6, SIRT7, and SIRT2.
NAD+-dependent SIRT6 optimizes energy homeostasis by regulating histone acetylation (Xiao et al., 2010). SIRT6 could directly repress glycolysis in the HIF1
SIRT7 could directly interacts with MYC that mediates the transcription of almost all the genes involved in glycolysis and glutaminolysis (Barber et al., 2012; Shin et al., 2013). SIRT7 selectively catalyzes H3K18 deacetylation that is a repressive mark (Wong et al., 2017). Hence, SIRT7 plays an opposite role in MYC-mediated metabolic reprogramming.
Compared to SIRT6/7, SIRT2 promotes cancer metabolism through stabilizing MYC (Liu et al., 2013). SIRT2 specifically deacetylases H4K16, resulting in decreased expression of ubiquitin-protein ligase NEDD4. NEDD4 serves as a negative regulator of MYC through ubiquitination and degradation (Wong et al., 2017). Consequently, SIRT2 facilitates MYC-dependent transcription and oncogenesis.
Targeting metabolic enzymes might be novel strategy for cancer therapy. LDH-A, a metabolic enzyme responsible for the conversion of pyruvate to lactate, was recognized as the first metabolic target of the oncogene MYC (Shim et al., 1997). Appealing evidence manifested genetic or pharmacologic ablation of LDH-A is able to dwindle MYC-driven tumors in the xenograft models (Fantin et al., 2006; Le et al., 2010). Inhibition of LDH-A could delay the progression of myeloid leukemia (Wang et al., 2014) and diminish NSCLC without systemic toxicity in genetically engineered mouse models (Xie et al., 2014). Hence, LDH-A is a promising target in MYC-mutant tumors. Another attractive target is the glycolytic protein Hexokinase (HK2). Many tumors express high levels of HK2. Specific inhibition of HK2 delays tumor progression in mouse models of NSCLC and breast cancer (Patra et al., 2013). Targeting HK2 might be efficacious in highly glycolytic tumors. Besides, PHGDH, an enzyme that functions in the de novo serine synthesis, is found to overexpress in human melanoma and breast cancers (Locasale et al., 2011; Possemato et al., 2011). Targeting PHGDH in the one-carbon metabolism has been shown to delay tumor progression, though more studies are needed to confirm it. Additionally, the concept of oncometabolite opened a new window for targeted therapy. Small molecules targeting IDH1/IDH2 demonstrate positive outcomes in ongoing clinical trials (Yen et al., 2017). Taken together, targeting metabolic enzyme holds great promise in the treatment of malignancy (Olivares et al., 2015).
Targeting metabolism pathways, such as glycolysis, glutamine metabolism, mitochondrial metabolism, and autophagy, provides new opportunities for drug discovery scheme. In the certain context, metabolites produced from these metabolic pathways are able to affect epigenome. For example, metformin, an anti-diabetic drug, has been spotlighted on mitochondrial-mediated metabolic activity emerging as a key target for cancer therapy (Weinberg and Chandel, 2015). Because diabetic patients treated with metformin not only control their blood glucose level but also improve survival rate if cancer was diagnosed already (Evans et al., 2005). Biguanide phenformin also displayed anti-tumor effect by inhibiting mitochondrial complex I (Birsoy et al., 2014). Another example is BPTES [bis-2-(5-phenylacetamido-1, 2, 4-thiadiazol-2-yl) ethyl sulfide], one inhibitor of glutaminase activity, is being explored for anti-cancer characteristics (Xiang et al., 2015). Autography offers amino acids that fuel TCA cycle. Autography inhibition is confirmed to decrease tumor progression without significant toxicity in the mouse models of NSCLC and pancreatic cancers (Son et al., 2013; Karsli-Uzunbas et al., 2014). An alternative approach is to target acetate metabolism. As discussed above, mitochondria conventionally provide acetyl-CoA to the normal cells, whereas cancer cells also utilize acetate to support cell survival under hypoxia or nutrient deprivation (Schug et al., 2015). ACCS2, a cytosolic enzyme that converts acetate to acetyl-CoA, is dispensable for acetate metabolism and holds great promise for cancer therapy. In models of hepatocellular carcinoma, genetic loss of ACSS2 is likely to reduce tumor burden (Comerford et al., 2014). Human glioblastoma is sensitive to inhibitors of ACSS2 as well (Mashimo et al., 2014).
Over the past decades, a few studies represent how advances of metabolic effects on epigenetics can be translated into potential therapies. One strategy is to reverse epigenetic dysfunction by targeting cancer metabolism (Table 4).
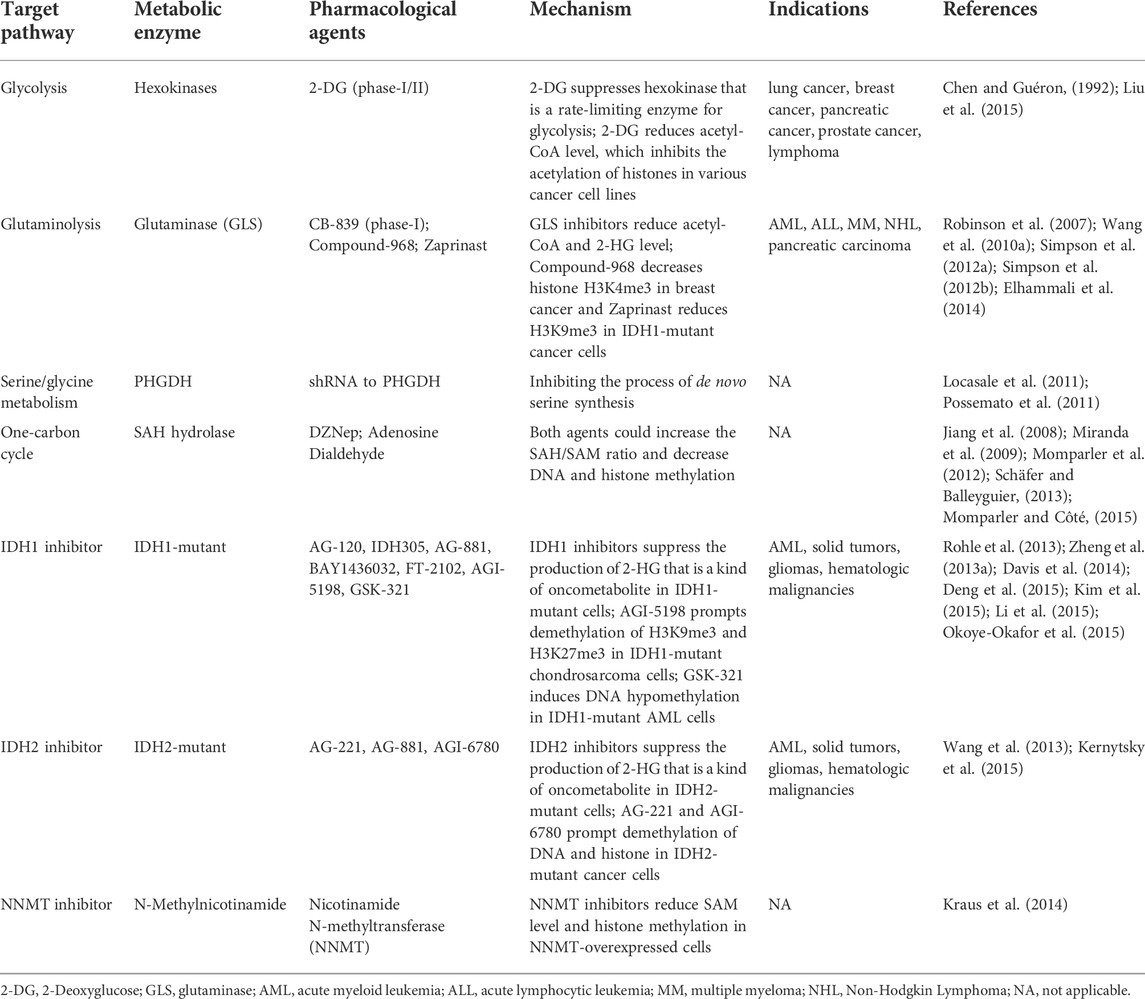
TABLE 4. Reversal of epigenetic dysfunction by targeting metabolism.
Glycolysis inhibitors could reverse global histone hyperacetylation. 2-Deoxyglucose (2-DG), a glucose analog, is a rate-limiting enzyme for glycolysis. The use of 2-DG inhibits acetyl-CoA levels, which rationally promotes histone deacetylation in multiple cancer cell lines. Hence, glycolysis inhibition represents a candidate target for regulating histone acetylation. Glutaminolysis produces
Instead, using epigenetic drugs could modulate metabolism rewiring as well (Table 5).
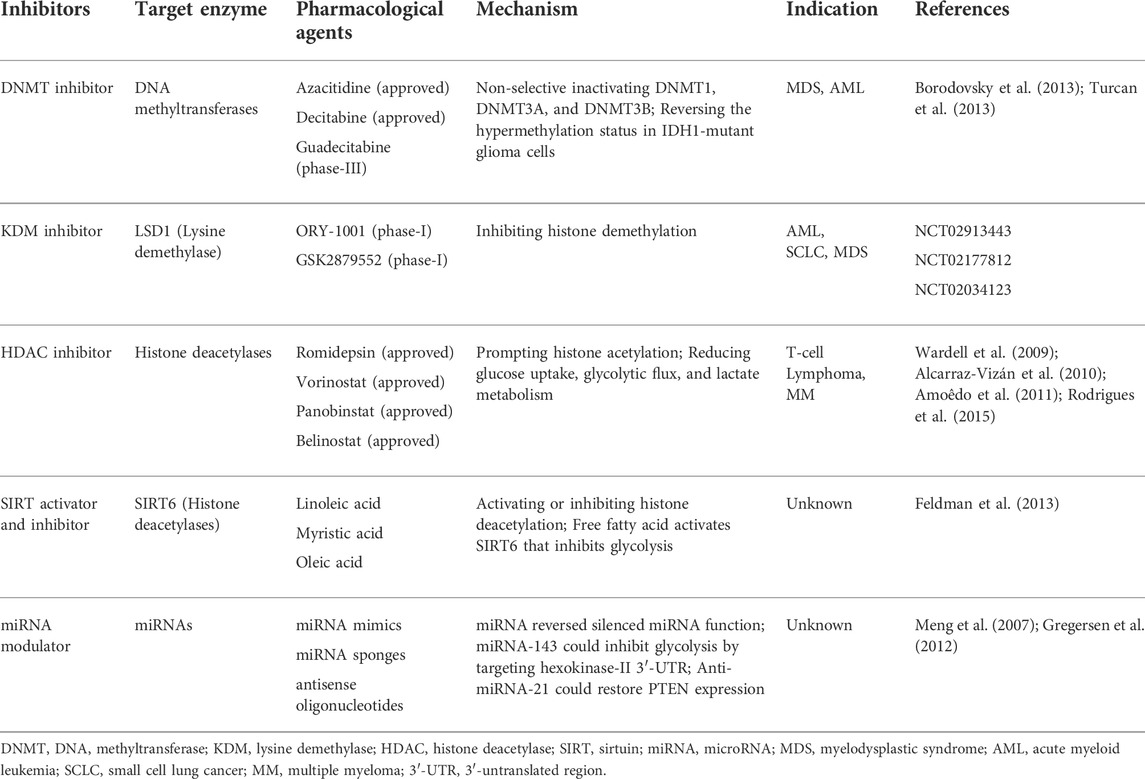
TABLE 5. Reversal of metabolism reprogramming by targeting epigenetics.
There are two kinds of DNMT inhibitors therapeutically targeting DNA methylation, respectively named 5-azacytidine and 5-aza-2′-deoxycytidine. Both of them have been approved by FDA to treat myelodysplastic syndrome (MDS). IDH 1/2-mutant tumors carrying DNA hypermethylation show a high sensitivity to DNMT inhibitor. In IDH 1-mutant glioma models, both of 5-azacytidine and 5-aza-2′-deoxycytidine induced tumor regression. When inducing the differentiation of IDH-mutant glioma cells, 5-aza-2′-deoxycytidine displayed a more potent efficacy than IDH inhibitors. Therefore, targeting epigenetics is a complementary approach to modulate the effect of oncometabolites in tumor. HDAC inhibitors could induce histone acetylation and reverse gene silence caused by HDACs. Growing evidence suggests HDAC inhibitors significantly suppressed glycolysis in various cancer types, such as lung cancer, breast cancer, and multiple myeloma. These findings manifest that inhibition of HDAC might reverse glycolytic phenotype. The modulation of SIRT activator and inhibitor holds promise as their regulatory roles in metabolism reprogramming. MiRNA-based therapeutics, such as miRNA-143, also inhibit glycolysis by targeting hexokinase-II 3′-UTR. More examples are summarized in Table 5.
Advancements in the area of cancer drug discovery have spotlighted on the inhibitors of metabolic pathways and cancer epigenetics. However, the efficacy of epigenetic inhibitors alone is not satisfactory, and this approach is usually prone to drug resistance (Zhang et al., 2020). Also, cancer cell could be drug-resistant to suppression of a particular metabolic pathway by upregulating compensatory pathways or expressing alternative isoforms. Further, inhibitions of metabolic enzymes might produce systemic toxicity owing to their physiological role in normal cells (Pearce et al., 2013; Ito and Suda, 2014; Erez and DeBerardinis, 2015). To achieve the purpose of less toxicity and potent efficiency, a rational strategy is to develop multiple drug combinations.
As an epigenetic regulator, enhancer of zeste homology (EZH2) inhibits gene transcription by trimethylation of histone H3K27 in cancer cells. Mounting evidence has suggested that EZH2 participated in the alteration of metabolic profiles in cancer through diverse pathways, covering glucose, lipid, amino acid metabolism. Meanwhile, metabolic activities also affect the stability and methyltransferase activity of EZH2, as some metabolites offer the donors for EZH2 post-translational modifications (Zhang et al., 2020). As a promising target, EZH2 inhibitors have been investigated in preclinical trials, but the effectiveness of EZH2 inhibitors alone is not satisfactory (De Raedt et al., 2011; Baude et al., 2014; Huang X. et al., 2018). Recently, researchers have found EZH2 inhibitor is able to weaken drug resistance caused by metabolic activities in tumor. Solid tumor is subject to hypoxia and glutamine deficiency because of the underdeveloped vascular system. Hypoxia induces a metabolic switch from oxidative to glycolytic metabolism, promoting the dedifferentiation of tumor cells and inducing resistance to radio- and chemotherapy. However, EZH2 inhibitors could directly block H3K27 methylation and consequently activate the transcription of pro-differentiation genes. Also, metabolic pathway is likely to downregulate EZH2 activity and thereby acts synergistically with EZH2 inhibitors (Zhang et al., 2020). More specifically, AMPK is activated in response to energy stress (glucose deficiency) and phosphorylates EZH2 (Cha et al., 2005). AKT-mediated phosphorylation of EZH2 suppresses trimethylation of lysine 27 in histone H3, facilitating the transcription of target genes to suppress tumor growth (Cha et al., 2005; Priebe et al., 2011; Gao et al., 2014; Kim and Yeom, 2018). Therefore, a combination of EZH2 inhibitors with metabolic regulators is a novel strategy to rescue the poor effectiveness of EZH2 inhibitor alone (Zhang et al., 2020). Briefly, epigenetic and metabolic alterations mediated by EZH2 are highly interlaced, demonstrating a synergistic effect in treating malignancy.
A model whereby linked metabolic-epigenetic programs reflects a new idea to target such an integrated axis. A study (McDonald et al., 2017) on the evolution of pancreatic ductal adenocarcinoma (PDAC) introduced an epigenetic mechanism that links glucose metabolism to distant metastasis. Remarkably, oxidative branch of the Pentose Phosphate Pathway (ox-PPP) was a driving force for epigenetic programming (histone H3K9 and DNA methylation) that enhanced tumorigenic fitness during the distant metastasis. Hence, targeting ox-PPP to reverse malignant epigenetic programs could be effective in metastatic PDAC. Another best-studied example is the use of AMPK activator metformin, which decreased EZHIP protein concentrations, elevated H3K27me3, inhibited TCA cycle, and suppressed tumor growth. Consequently, targeting integrated epigenetic-metabolic pathway shows hopeful therapeutic efficacy in mice models transplanted with PFA ependymomas (Panwalkar et al., 2021).
Oncogenic signal pathways also play important roles in novel combination therapy. A distinct work on melanoma demonstrated that reduced
Taken together, our understanding in targeting both altered metabolism and epigenetics remains at a very early stage. Whether these two hallmarks exert synergistic functions in tumor is less explored, though there are a few well-elaborated agents in ongoing clinical trials (Table 6).
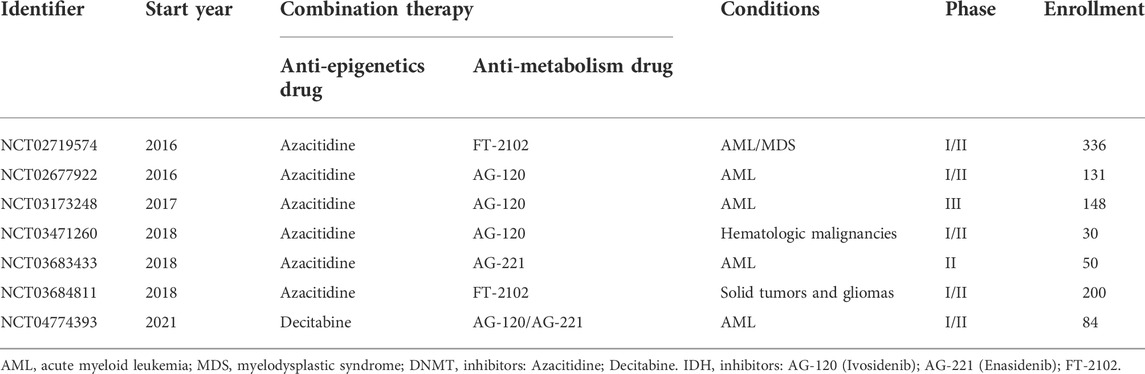
TABLE 6. Ongoing clinical trials of combined anti-epigenetic drugs and anti-metabolism drugs.
In the traditional viewpoint, immunological memory is a unique feature of the adaptive immune system (Netea et al., 2020a). However, “Trained immunity” is a relatively new term that refers to myeloid cells from the innate immune system also display memory capacity after pathogen exposure (Dominguez-Andres and Netea, 2019; Netea et al., 2020b; O'Neill and Netea, 2020). After the first stimuli, innate immune cells, such as macrophage and monocyte, are epigenetically programmed (Fanucchi et al., 2021). These epigenetic modifications unfold chromatin and expose promoter and enhancer regions controlling immune-associated genes, enabling them accessible to transcription factors (Klemm et al., 2019) and permitting cells to maintain a “trained” state after rechallenge (Saeed et al., 2014). Specifically, H3K4me3 frequently occurs on gene promoters; H3K4me1 and H3K27Ac accumulates on enhancers (Quintin et al., 2012; Novakovic et al., 2016). As such, upon the secondary stimulus, immune genes are more robustly transcribed (Fanucchi et al., 2021).
In addition, some metabolites act as substrates or co-factors for epigenetic enzymes, which alter chromatin state to cause transcriptional changes that are causal to trained immunity (Fanucchi et al., 2021). For example, acetyl-CoA mediates histone acetylation following immune stimuli (Wellen et al., 2009; Christ and Latz, 2019), while SAM level regulates DNA and histone methylation to control trained immunity (Mentch et al., 2015; Ji et al., 2019). On the contrary, NAD+ assist histone deacetylation to block trained immunity (Yeung et al., 2004; Zhong et al., 2010; Lo Sasso et al., 2014; Jia et al., 2018).
Apart from trained immunity, the crosstalk of metabolism and epigenetics has been reported in T cell (Bailis et al., 2019) and macrophage activation (Liu et al., 2017). A recent study has shown that both mitochondrial citrate export and malate-aspartate shuttle favor histone acetylation and influence the expression of specific genes involved in T cell activation (Bailis et al., 2019). Also, a research figured out
Cancer immunotherapy is rapidly developing in various research settings, including CAR-T cell therapy, immune checkpoint inhibitors, and adoptive transfer of tumor infiltrating lymphocytes (Rosenberg et al., 1988; Zhao et al., 2005; Robbins et al., 2011; Rosenberg et al., 2011; Rosenberg, 2012; Topalian et al., 2012; Maude et al., 2014). An innovative strategy is the combination of immunotherapy with either epigenetic inhibitors or metabolic inhibitors, or a triple combination of them.
Epigenetics and immunology are both fast-developing fields in cancer biology. Recent evidence provides unique opportunities to combine epigenetics-based drugs with immunotherapy (Zhang et al., 2020). Epigenetic-based drugs include four pan-HDAC inhibitors and two DNMT inhibitors approved by FDA before 2020 (Knutson et al., 2012; Yu et al., 2017). These agents are able to change the immunosuppressive tumor microenvironment and increased tumor-infiltrating lymphocytes (Yanagida et al., 2001; Wang L. et al., 2010; Li et al., 2013; Anwar et al., 2018), leading to enhanced tumor-associated antigen presentation, activation of DC cells, suppression of T cell exhaustion. Similar changes in TME are also observed in tumor tissues treated with other agents, such as inhibitors of KMT6A (EZH2), KDM1A (LSD1), PRMT5, and BET proteins (Hemmings and Restuccia, 2012; Kikuchi et al., 2015; Garcia and Shaw, 2017; Herzig and Shaw, 2018; Hoxhaj and Manning, 2020). Consequently, given that epigenetic drugs boosting antitumor immune response, immune checkpoint blockade therapies (ICBTs) and epigenetic-based inhibitors exert synergistic functions to sensitize less-immunogenic tumors and prevent both primary and acquired resistance (Zhang et al., 2020). There are numerous ongoing clinical trials summarized in Table 7.
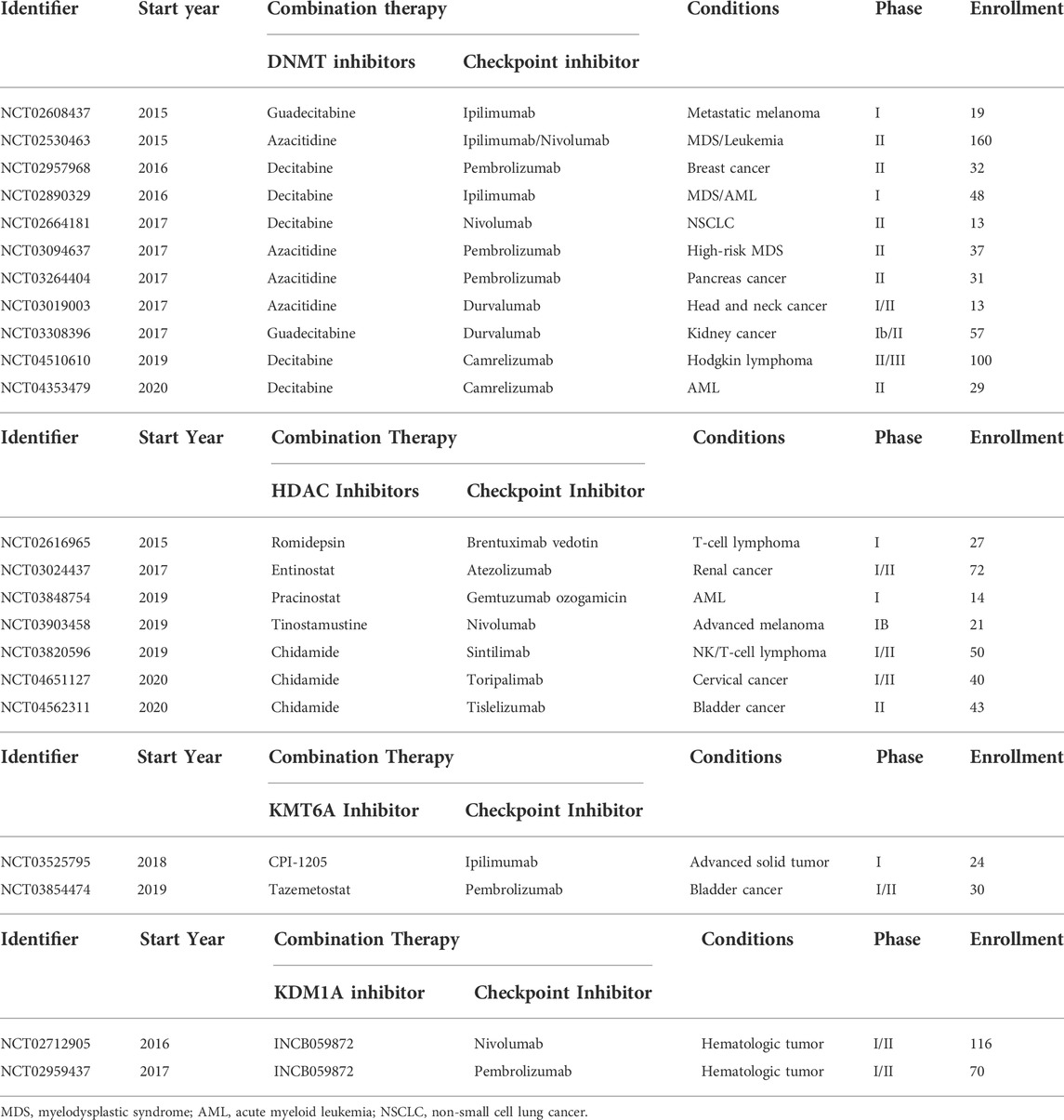
TABLE 7. Ongoing clinical trials of combined anti-epigenetic drugs and immune checkpoint inhibitors.
Metabolism can be modulated in vivo to govern anti-tumor T cell longevity and functionality, which determines the efficacy of immunotherapy (Chang and Pearce, 2016; O'Neill et al., 2016). The modulation of T cell metabolism is a promising strategy to enhance or suppress immune response (O'Sullivan and Pearce, 2015), as the characteristics of T cells are critical to determine clinical outcomes (Klebanoff et al., 2012). Several advances have been made in preclinical models. For example, when treating vascularized melanoma, limiting the ability of T cells engaged in glycolysis through suppression of hexokinase by 2-DG could ultimately leads to enhanced anti-tumor efficacy (Sukumar et al., 2013). Additionally, metabolic reprogramming occurs in other immune cells within tumor microenvironment, such as macrophages and dendritic cells (DCs). One research (Yan et al., 2021) put forward strategies to enhance cancer immunotherapy by manipulating metabolism reprogramming. For example, CB-839 is a glutaminase inhibitor that has been explored in numerous clinical trials with or without the combinations of immunotherapy (Cerezo and Rocchi, 2020). Acetyl-CoA acetyltransferase 1 (ACAT1) inhibitors could enhance the activity of CD8+ T cells and reduce the inflammatory response. Hence, ACAT1 might be a potential target to optimize immunotherapy (Yang et al., 2016; Huang L. H. et al., 2018; Bi et al., 2019). Indoleamine 2,3-dioxygenase (IDO) is responsible for the conversion of tryptophan to kynurenine in tumors. Blocking IDO can decrease Treg cells and preserve the functionality of T cells. Combination of IDO inhibitors (epacadostat) and immune checkpoint inhibitor (pembrolizumab) has been shown safe enough in clinical trials, though its efficacy needs further investigation (Prendergast et al., 2017; Komiya and Huang, 2018; Long et al., 2019). In summary, glutamine, acetyl-CoA acetyltransferase 1 (ATAC1), indoleamine 2,3-dioxygenase (IDO), lactate, and Toll-like receptors (TLRs) are likely to be considered as novel “metabolic checkpoints”, targeting of which could assist immune cells to achieve better anti-tumor effect.
Noteworthily, epigenetic, metabolism, and immune crosslink in germinal-cancer-derived B-cell lymphomas (GCB) uncover a rational triple combination therapy (Serganova et al., 2021). GCB lymphoma is significantly heterogenous based on genetic, epigenetic, and clinical characteristics. Epigenetic dysfunction, such as gain-of-function mutations of EZH2 and loss-of-function mutations of CREBP and EP300, disrupts the normal biological link between lymphoma cells and immune TME, and motivates immune evasion in GCB lymphoma. Also, lymphoma metabolism adaptions might aggravate immunosuppression, leading to poorly infiltrated effector T-cell. Considering the impacts of cancer metabolism on epigenetic modifier and immune microenvironment, triple combination therapy is a logic and feasible strategy for future treatment.
As reviewed, epigenetics and metabolism are highly interconnected in a reciprocal manner (Figure 3). Such a relationship is accentuated by the reversibility of both processes (Henikoff and Matzke, 1997). A major goal in exploring metabolism-dependent epigenetic modifications is the hope of identifying novel targets for cancer therapy. However, some aspects pertaining to metabolic-epigenetic axis in cancers remain poorly understood.
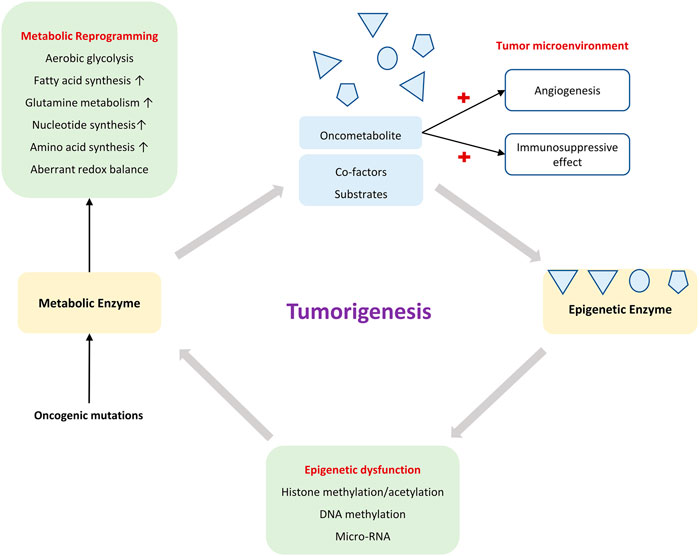
FIGURE 3. The crosstalk between metabolism and epigenetics in tumorigenesis.
Firstly, tumor heterogeneity is a major challenge that limits our understanding (Hensley et al., 2016). Inconsistent metabolic phenotypes were observed in various tumor tissues. Hence, tumor heterogeneity allows cancer cells to escape the deleterious attacks of inhibitors (Thakur and Chen, 2019). Secondly, the downstream factors mediating the tumorigenic activity of oncometabolites remains largely unknown. Thirdly, enzymatic parameters, such as Km, Vmax, and allosteric and inhibitory binding constants, constitute the basic element of the biochemistry (Reid et al., 2017). It is difficult to define physiological conditions in which the concentration dynamics of substrates and co-factors causally underlie an alteration of chromatin status. Discrepancies exist between artificial culture in vitro and physiological environment in vivo (Davidson et al., 2016). Another complexity is the precise input of metabolism into chromatin modifications, as both activation and suppression of histone marks need metabolites. For instance, how to predict the changes of SAM level establish the overall chromatin state and epigenetic phenotype. Additionally, though a bunch of metabolic enzymes function in nucleus have been identified, their individual contribution to epigenetic alterations was less defined. Robust experimental methods are needed to obtain accurate measurements of metabolites in specific cellular domain. Despite much interest in targeting both metabolism and epigenetics, poorly understood layers that whether these two hallmarks confer dependencies in tumors synergistically still exist.
In-depth connection between oncogenic signaling, metabolism, epigenetics, and immunity in cancer would facilitates effective designing of novel targeted drugs, which is the premise of precision medicine. It is anticipated that multiple combination therapies hold opportunities to improve care of cancer patients. Nevertheless, several outstanding challenges will be the major goal of future study.
YQ designed the study and reviewed the manuscript. CC and ZW participated in the study design and wrote the original draft of the manuscript. CC was mainly responsible for the design of tables and figures. All authors agreed to the submission of the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aghili, M., Zahedi, F., and Rafiee, E. (2009). Hydroxyglutaric aciduria and malignant brain tumor: A case report and literature review. J. Neurooncol. 91 (2), 233–236. doi:10.1007/s11060-008-9706-2
Ahn, C. S., and Metallo, C. M. (2015). Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 3 (1), 1. doi:10.1186/s40170-015-0128-2
Alcarraz-Vizán, G., Boren, J., Lee, W. N., and Cascante, M. (2010). Histone deacetylase inhibition results in a common metabolic profile associated with HT29 differentiation. Metabolomics 6 (2), 229–237. doi:10.1007/s11306-009-0192-0
Amary, M. F., Bacsi, K., Maggiani, F., Damato, S., Halai, D., Berisha, F., et al. (2011). IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 224 (3), 334–343. doi:10.1002/path.2913
Amoêdo, N. D., Rodrigues, M. F., Pezzuto, P., Galina, A., da Costa, R. M., de Almeida, F. C., et al. (2011). Energy metabolism in H460 lung cancer cells: Effects of histone deacetylase inhibitors. PLoS One 6 (7), e22264. doi:10.1371/journal.pone.0022264
Anwar, T., Arellano-Garcia, C., Ropa, J., Chen, Y. C., Kim, H. S., Yoon, E., et al. (2018). p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat. Commun. 9 (1), 2801. doi:10.1038/s41467-018-05078-8
Baek, S. H. (2011). When signaling kinases meet histones and histone modifiers in the nucleus. Mol. Cell. 42 (3), 274–284. doi:10.1016/j.molcel.2011.03.022
Bailis, W., Shyer, J. A., Zhao, J., Canaveras, J. C. G., Al Khazal, F. J., Qu, R., et al. (2019). Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571 (7765), 403–407. doi:10.1038/s41586-019-1311-3
Barber, M. F., Michishita-Kioi, E., Xi, Y., Tasselli, L., Kioi, M., Moqtaderi, Z., et al. (2012). SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487 (7405), 114–118. doi:10.1038/nature11043
Bardella, C., Pollard, P. J., and Tomlinson, I. (2011). SDH mutations in cancer. Biochim. Biophys. Acta 1807 (11), 1432–1443. doi:10.1016/j.bbabio.2011.07.003
Baude, A., Lindroth, A. M., and Plass, C. (2014). PRC2 loss amplifies Ras signaling in cancer. Nat. Genet. 46 (11), 1154–1155. doi:10.1038/ng.3124
Baysal, B. E., Ferrell, R. E., Willett-Brozick, J. E., Lawrence, E. C., Myssiorek, D., Bosch, A., et al. (2000). Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287 (5454), 848–851. doi:10.1126/science.287.5454.848
Berger, S. L. (2007). The complex language of chromatin regulation during transcription. Nature 447 (7143), 407–412. doi:10.1038/nature05915
Bernstein, B. E., Meissner, A., and Lander, E. S. (2007). The mammalian epigenome. Cell. 128 (4), 669–681. doi:10.1016/j.cell.2007.01.033
Bhutani, N., Burns, D. M., and Blau, H. M. (2011). DNA demethylation dynamics. Cell. 146 (6), 866–872. doi:10.1016/j.cell.2011.08.042
Bi, M., Qiao, X., Zhang, H., Wu, H., Gao, Z., Zhou, H., et al. (2019). Effect of inhibiting ACAT-1 expression on the growth and metastasis of Lewis lung carcinoma. Oncol. Lett. 18 (2), 1548–1556. doi:10.3892/ol.2019.10427
Birsoy, K., Possemato, R., Lorbeer, F. K., Bayraktar, E. C., Thiru, P., Yucel, B., et al. (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508 (7494), 108–112. doi:10.1038/nature13110
Borger, D. R., Goyal, L., Yau, T., Poon, R. T., Ancukiewicz, M., Deshpande, V., et al. (2014). Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin. Cancer Res. 20 (7), 1884–1890. doi:10.1158/1078-0432.Ccr-13-2649
Borodovsky, A., Salmasi, V., Turcan, S., Fabius, A. W., Baia, G. S., Eberhart, C. G., et al. (2013). 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 4 (10), 1737–1747. doi:10.18632/oncotarget.1408
Bungard, D., Fuerth, B. J., Zeng, P. Y., Faubert, B., Maas, N. L., Viollet, B., et al. (2010). Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science 329 (5996), 1201–1205. doi:10.1126/science.1191241
Cai, L., Sutter, B. M., Li, B., and Tu, B. P. (2011). Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell. 42 (4), 426–437. doi:10.1016/j.molcel.2011.05.004
Cairns, R. A., Harris, I. S., and Mak, T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11 (2), 85–95. doi:10.1038/nrc2981
Caldwell, S. A., Jackson, S. R., Shahriari, K. S., Lynch, T. P., Sethi, G., Walker, S., et al. (2010). Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 29 (19), 2831–2842. doi:10.1038/onc.2010.41
Candido, E. P., Reeves, R., and Davie, J. R. (1978). Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 14 (1), 105–113. doi:10.1016/0092-8674(78)90305-7
Cantor, J. R., and Sabatini, D. M. (2012). Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2 (10), 881–898. doi:10.1158/2159-8290.Cd-12-0345
Castro-Vega, L. J., Buffet, A., De Cubas, A. A., Cascón, A., Menara, M., Khalifa, E., et al. (2014). Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 23 (9), 2440–2446. doi:10.1093/hmg/ddt639
Cerezo, M., and Rocchi, S. (2020). Cancer cell metabolic reprogramming: A keystone for the response to immunotherapy. Cell. Death Dis. 11 (11), 964. doi:10.1038/s41419-020-03175-5
Cha, T. L., Zhou, B. P., Xia, W., Wu, Y., Yang, C. C., Chen, C. T., et al. (2005). Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310 (5746), 306–310. doi:10.1126/science.1118947
Chang, C. H., and Pearce, E. L. (2016). Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 17 (4), 364–368. doi:10.1038/ni.3415
Chen, M., Zhang, J., Li, N., Qian, Z., Zhu, M., Li, Q., et al. (2011). Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS One 6 (10), e25564. doi:10.1371/journal.pone.0025564
Chen, Q., Chen, Y., Bian, C., Fujiki, R., and Yu, X. (2013). TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493 (7433), 561–564. doi:10.1038/nature11742
Chen, R., Xu, M., Nagati, J., and Garcia, J. A. (2017). Coordinate regulation of stress signaling and epigenetic events by Acss2 and HIF-2 in cancer cells. PLoS One 12 (12), e0190241. doi:10.1371/journal.pone.0190241
Chen, W., and Guéron, M. (1992). The inhibition of bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate: Characterization by 31P NMR and metabolic implications. Biochimie 74 (9-10), 867–873. doi:10.1016/0300-9084(92)90070-u
Cheng, S. C., Quintin, J., Cramer, R. A., Shepardson, K. M., Saeed, S., Kumar, V., et al. (2014). mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345 (6204), 1250684. doi:10.1126/science.1250684
Chinnaiyan, P., Kensicki, E., Bloom, G., Prabhu, A., Sarcar, B., Kahali, S., et al. (2012). The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 72 (22), 5878–5888. doi:10.1158/0008-5472.Can-12-1572-t
Christ, A., and Latz, E. (2019). The Western lifestyle has lasting effects on metaflammation. Nat. Rev. Immunol. 19 (5), 267–268. doi:10.1038/s41577-019-0156-1
Chu, C. S., Lo, P. W., Yeh, Y. H., Hsu, P. H., Peng, S. H., Teng, Y. C., et al. (2014). O-GlcNAcylation regulates EZH2 protein stability and function. Proc. Natl. Acad. Sci. U. S. A. 111 (4), 1355–1360. doi:10.1073/pnas.1323226111
Colegio, O. R., Chu, N. Q., Szabo, A. L., Chu, T., Rhebergen, A. M., Jairam, V., et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513 (7519), 559–563. doi:10.1038/nature13490
Colvin, H., Nishida, N., Konno, M., Haraguchi, N., Takahashi, H., Nishimura, J., et al. (2016). Oncometabolite D-2-hydroxyglurate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci. Rep. 6, 36289. doi:10.1038/srep36289
Comerford, S. A., Huang, Z., Du, X., Wang, Y., Cai, L., Witkiewicz, A. K., et al. (2014). Acetate dependence of tumors. Cell. 159 (7), 1591–1602. doi:10.1016/j.cell.2014.11.020
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462 (7274), 739–744. doi:10.1038/nature08617
Davidson, S. M., Papagiannakopoulos, T., Olenchock, B. A., Heyman, J. E., Keibler, M. A., Luengo, A., et al. (2016). Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell. Metab. 23 (3), 517–528. doi:10.1016/j.cmet.2016.01.007
Davis, M. I., Gross, S., Shen, M., Straley, K. S., Pragani, R., Lea, W. A., et al. (2014). Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J. Biol. Chem. 289 (20), 13717–13725. doi:10.1074/jbc.M113.511030
de Groof, A. J., te Lindert, M. M., van Dommelen, M. M., Wu, M., Willemse, M., Smift, A. L., et al. (2009). Increased OXPHOS activity precedes rise in glycolytic rate in H-RasV12/E1A transformed fibroblasts that develop a Warburg phenotype. Mol. Cancer 8, 54. doi:10.1186/1476-4598-8-54
De Raedt, T., Walton, Z., Yecies, J. L., Li, D., Chen, Y., Malone, C. F., et al. (2011). Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 20 (3), 400–413. doi:10.1016/j.ccr.2011.08.014
DeBerardinis, R. J., and Chandel, N. S. (2016). Fundamentals of cancer metabolism. Sci. Adv. 2 (5), e1600200. doi:10.1126/sciadv.1600200
Deng, G., Shen, J., Yin, M., McManus, J., Mathieu, M., Gee, P., et al. (2015). Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 290 (2), 762–774. doi:10.1074/jbc.M114.608497
Desai, S., Ding, M., Wang, B., Lu, Z., Zhao, Q., Shaw, K., et al. (2014). Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget 5 (18), 8202–8210. doi:10.18632/oncotarget.1159
Dominguez-Andres, J., and Netea, M. G. (2019). Long-term reprogramming of the innate immune system. J. Leukoc. Biol. 105 (2), 329–338. doi:10.1002/jlb.Mr0318-104r
El-Kenawi, A., Gatenbee, C., Robertson-Tessi, M., Bravo, R., Dhillon, J., Balagurunathan, Y., et al. (2019). Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br. J. Cancer 121 (7), 556–566. doi:10.1038/s41416-019-0542-2
Elhammali, A., Ippolito, J. E., Collins, L., Crowley, J., Marasa, J., Piwnica-Worms, D., et al. (2014). A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer Discov. 4 (7), 828–839. doi:10.1158/2159-8290.Cd-13-0572
Erez, A., and DeBerardinis, R. J. (2015). Metabolic dysregulation in monogenic disorders and cancer - Finding method in madness. Nat. Rev. Cancer 15 (7), 440–448. doi:10.1038/nrc3949
Evans, J. M., Donnelly, L. A., Emslie-Smith, A. M., Alessi, D. R., and Morris, A. D. (2005). Metformin and reduced risk of cancer in diabetic patients. Bmj 330 (7503), 1304–1305. doi:10.1136/bmj.38415.708634.F7
Fang, R., Barbera, A. J., Xu, Y., Rutenberg, M., Leonor, T., Bi, Q., et al. (2010). Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell. 39 (2), 222–233. doi:10.1016/j.molcel.2010.07.008
Fantin, V. R., St-Pierre, J., and Leder, P. (2006). Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 9 (6), 425–434. doi:10.1016/j.ccr.2006.04.023
Fanucchi, S., Domínguez-Andrés, J., Joosten, L. A. B., Netea, M. G., and Mhlanga, M. M. (2021). The intersection of epigenetics and metabolism in trained immunity. Immunity 54 (1), 32–43. doi:10.1016/j.immuni.2020.10.011
Feldman, J. L., Baeza, J., and Denu, J. M. (2013). Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288 (43), 31350–31356. doi:10.1074/jbc.C113.511261
Fieuw, A., Kumps, C., Schramm, A., Pattyn, F., Menten, B., Antonacci, F., et al. (2012). Identification of a novel recurrent 1q42.2-1qter deletion in high risk MYCN single copy 11q deleted neuroblastomas. Int. J. Cancer 130 (11), 2599–2606. doi:10.1002/ijc.26317
Figueroa, M. E., Abdel-Wahab, O., Lu, C., Ward, P. S., Patel, J., Shih, A., et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 18 (6), 553–567. doi:10.1016/j.ccr.2010.11.015
Finkel, T., Deng, C. X., and Mostoslavsky, R. (2009). Recent progress in the biology and physiology of sirtuins. Nature 460 (7255), 587–591. doi:10.1038/nature08197
Friis, R. M., Wu, B. P., Reinke, S. N., Hockman, D. J., Sykes, B. D., Schultz, M. C., et al. (2009). A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Res. 37 (12), 3969–3980. doi:10.1093/nar/gkp270
Gao, M., Liang, J., Lu, Y., Guo, H., German, P., Bai, S., et al. (2014). Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene 33 (6), 745–755. doi:10.1038/onc.2013.2
Gao, X., Lin, S. H., Ren, F., Li, J. T., Chen, J. J., Yao, C. B., et al. (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 7, 11960. doi:10.1038/ncomms11960
Garcia, D., and Shaw, R. J. (2017). Ampk: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell. 66 (6), 789–800. doi:10.1016/j.molcel.2017.05.032
Gottfried, E., Kunz-Schughart, L. A., Ebner, S., Mueller-Klieser, W., Hoves, S., Andreesen, R., et al. (2006). Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107 (5), 2013–2021. doi:10.1182/blood-2005-05-1795
Gottlieb, E., and Tomlinson, I. P. (2005). Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 5 (11), 857–866. doi:10.1038/nrc1737
Gregersen, L. H., Jacobsen, A., Frankel, L. B., Wen, J., Krogh, A., Lund, A. H., et al. (2012). MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer 12, 232. doi:10.1186/1471-2407-12-232
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell. 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Hao, H. X., Khalimonchuk, O., Schraders, M., Dephoure, N., Bayley, J. P., Kunst, H., et al. (2009). SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325 (5944), 1139–1142. doi:10.1126/science.1175689
Hardie, D. G. (2011). Adenosine monophosphate-activated protein kinase: A central regulator of metabolism with roles in diabetes, cancer, and viral infection. Cold Spring Harb. Symp. Quant. Biol. 76, 155–164. doi:10.1101/sqb.2011.76.010819
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333 (6047), 1303–1307. doi:10.1126/science.1210944
Hemmings, B. A., and Restuccia, D. F. (2012). PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 4 (9), a011189. doi:10.1101/cshperspect.a011189
Henikoff, S., and Matzke, M. A. (1997). Exploring and explaining epigenetic effects. Trends Genet. 13 (8), 293–295. doi:10.1016/s0168-9525(97)01219-5
Hensley, C. T., Faubert, B., Yuan, Q., Lev-Cohain, N., Jin, E., Kim, J., et al. (2016). Metabolic heterogeneity in human lung tumors. Cell. 164 (4), 681–694. doi:10.1016/j.cell.2015.12.034
Herzig, S., and Shaw, R. J. (2018). Ampk: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 19 (2), 121–135. doi:10.1038/nrm.2017.95
Hou, X. M., Yuan, S. Q., Zhao, D., Liu, X. J., and Wu, X. A. (2019). LDH-A promotes malignant behavior via activation of epithelial-to-mesenchymal transition in lung adenocarcinoma. Biosci. Rep. 39 (1), BSR20181476. doi:10.1042/bsr20181476
Hoxhaj, G., and Manning, B. D. (2020). The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20 (2), 74–88. doi:10.1038/s41568-019-0216-7
Huang, L. H., Melton, E. M., Li, H., Sohn, P., Jung, D., Tsai, C. Y., et al. (2018a). Myeloid-specific Acat1 ablation attenuates inflammatory responses in macrophages, improves insulin sensitivity, and suppresses diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 315 (3), E340-E356. doi:10.1152/ajpendo.00174.2017
Huang, X., Yan, J., Zhang, M., Wang, Y., Chen, Y., Fu, X., et al. (2018b). Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 175 (1), 186–199. e119. doi:10.1016/j.cell.2018.08.058
Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403 (6771), 795–800. doi:10.1038/35001622
Ishii, M., Wen, H., Corsa, C. A., Liu, T., Coelho, A. L., Allen, R. M., et al. (2009). Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114 (15), 3244–3254. doi:10.1182/blood-2009-04-217620
Itkonen, H. M., Minner, S., Guldvik, I. J., Sandmann, M. J., Tsourlakis, M. C., Berge, V., et al. (2013). O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 73 (16), 5277–5287. doi:10.1158/0008-5472.Can-13-0549
Ito, K., and Suda, T. (2014). Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell. Biol. 15 (4), 243–256. doi:10.1038/nrm3772
Ito, R., Katsura, S., Shimada, H., Tsuchiya, H., Hada, M., Okumura, T., et al. (2014). TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes. cells. 19 (1), 52–65. doi:10.1111/gtc.12107
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333 (6047), 1300–1303. doi:10.1126/science.1210597
Ji, J., Xu, Y., Zheng, M., Luo, C., Lei, H., Qu, H., et al. (2019). Methionine attenuates lipopolysaccharide-induced inflammatory responses via DNA methylation in macrophages. ACS Omega 4 (1), 2331–2336. doi:10.1021/acsomega.8b03571
Jia, Y., Li, Z., Cai, W., Xiao, D., Han, S., Han, F., et al. (2018). SIRT1 regulates inflammation response of macrophages in sepsis mediated by long noncoding RNA. Biochim. Biophys. Acta. Mol. Basis Dis. 1864 (3), 784–792. doi:10.1016/j.bbadis.2017.12.029
Jiang, S., and Yan, W. (2017). Succinate in the cancer-immune cycle. Cancer Lett. 390, 45–47. doi:10.1016/j.canlet.2017.01.019
Jiang, X., Tan, J., Li, J., Kivimäe, S., Yang, X., Zhuang, L., et al. (2008). DACT3 is an epigenetic regulator of Wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell. 13 (6), 529–541. doi:10.1016/j.ccr.2008.04.019
Jin, L., Li, D., Alesi, G. N., Fan, J., Kang, H. B., Lu, Z., et al. (2015). Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 27 (2), 257–270. doi:10.1016/j.ccell.2014.12.006
Kamphorst, J. J., Chung, M. K., Fan, J., and Rabinowitz, J. D. (2014). Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2, 23. doi:10.1186/2049-3002-2-23
Karsli-Uzunbas, G., Guo, J. Y., Price, S., Teng, X., Laddha, S. V., Khor, S., et al. (2014). Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4 (8), 914–927. doi:10.1158/2159-8290.Cd-14-0363
Katada, S., Imhof, A., and Sassone-Corsi, P. (2012). Connecting threads: Epigenetics and metabolism. Cell. 148 (1-2), 24–28. doi:10.1016/j.cell.2012.01.001
Kaye, S. B. (1998). New antimetabolites in cancer chemotherapy and their clinical impact. Br. J. Cancer 78 (Suppl. 3Suppl 3), 1–7. doi:10.1038/bjc.1998.747
Kernytsky, A., Wang, F., Hansen, E., Schalm, S., Straley, K., Gliser, C., et al. (2015). IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood 125 (2), 296–303. doi:10.1182/blood-2013-10-533604
Kikuchi, J., Koyama, D., Wada, T., Izumi, T., Hofgaard, P. O., Bogen, B., et al. (2015). Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Investig. 125 (12), 4375–4390. doi:10.1172/jci80325
Kim, H. J., Choi, B. Y., and Keum, Y. S. (2015). Identification of a new selective chemical inhibitor of mutant isocitrate dehydrogenase-1. J. Cancer Prev. 20 (1), 78–83. doi:10.15430/jcp.2015.20.1.78
Kim, J. A., and Yeom, Y. I. (2018). Metabolic signaling to epigenetic alterations in cancer. Biomol. Ther. 26 (1), 69–80. doi:10.4062/biomolther.2017.185
Kinch, L., Grishin, N. V., and Brugarolas, J. (2011). Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell. 20 (4), 418–420. doi:10.1016/j.ccr.2011.10.005
Klebanoff, C. A., Gattinoni, L., and Restifo, N. P. (2012). Sorting through subsets: Which T-cell populations mediate highly effective adoptive immunotherapy? J. Immunother. 35 (9), 651–660. doi:10.1097/CJI.0b013e31827806e6
Klemm, S. L., Shipony, Z., and Greenleaf, W. J. (2019). Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20 (4), 207–220. doi:10.1038/s41576-018-0089-8
Klose, R. J., Kallin, E. M., and Zhang, Y. (2006). JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 7 (9), 715–727. doi:10.1038/nrg1945
Knutson, S. K., Wigle, T. J., Warholic, N. M., Sneeringer, C. J., Allain, C. J., Klaus, C. R., et al. (2012). A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8 (11), 890–896. doi:10.1038/nchembio.1084
Komiya, T., and Huang, C. H. (2018). Updates in the clinical development of epacadostat and other indoleamine 2, 3-dioxygenase 1 inhibitors (Ido1) for human cancers. Front. Oncol. 8, 423. doi:10.3389/fonc.2018.00423
Koppenol, W. H., Bounds, P. L., and Dang, C. V. (2011). Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11 (5), 325–337. doi:10.1038/nrc3038
Kottakis, F., Nicolay, B. N., Roumane, A., Karnik, R., Gu, H., Nagle, J. M., et al. (2016). LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539 (7629), 390–395. doi:10.1038/nature20132
Kraus, D., Yang, Q., Kong, D., Banks, A. S., Zhang, L., Rodgers, J. T., et al. (2014). Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 508 (7495), 258–262. doi:10.1038/nature13198
Kurdistani, S. K. (2011). Histone modifications in cancer biology and prognosis. Prog. Drug Res. 67, 91–106. doi:10.1007/978-3-7643-8989-5_5
Le, A., Cooper, C. R., Gouw, A. M., Dinavahi, R., Maitra, A., Deck, L. M., et al. (2010). Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U. S. A. 107 (5), 2037–2042. doi:10.1073/pnas.0914433107
Lee, J. V., Carrer, A., Shah, S., Snyder, N. W., Wei, S., Venneti, S., et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell. Metab. 20 (2), 306–319. doi:10.1016/j.cmet.2014.06.004
Li, K., Liu, C., Zhou, B., Bi, L., Huang, H., Lin, T., et al. (2013). Role of EZH2 in the growth of prostate cancer stem cells isolated from LNCaP cells. Int. J. Mol. Sci. 14 (6), 11981–11993. doi:10.3390/ijms140611981
Li, L., Paz, A. C., Wilky, B. A., Johnson, B., Galoian, K., Rosenberg, A., et al. (2015). Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS One 10 (9), e0133813. doi:10.1371/journal.pone.0133813
Liu, P. S., Wang, H., Li, X., Chao, T., Teav, T., Christen, S., et al. (2017). α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18 (9), 985–994. doi:10.1038/ni.3796
Liu, P. Y., Xu, N., Malyukova, A., Scarlett, C. J., Sun, Y. T., Zhang, X. D., et al. (2013). The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell. Death Differ. 20 (3), 503–514. doi:10.1038/cdd.2012.147
Liu, W., Zabirnyk, O., Wang, H., Shiao, Y. H., Nickerson, M. L., Khalil, S., et al. (2010). miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 29 (35), 4914–4924. doi:10.1038/onc.2010.237
Liu, X. S., Little, J. B., and Yuan, Z. M. (2015). Glycolytic metabolism influences global chromatin structure. Oncotarget 6 (6), 4214–4225. doi:10.18632/oncotarget.2929
Lo Sasso, G., Menzies, K. J., Mottis, A., Piersigilli, A., Perino, A., Yamamoto, H., et al. (2014). SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS One 9 (7), e103573. doi:10.1371/journal.pone.0103573
Locasale, J. W., Grassian, A. R., Melman, T., Lyssiotis, C. A., Mattaini, K. R., Bass, A. J., et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43 (9), 869–874. doi:10.1038/ng.890
Locasale, J. W. (2013). Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 13 (8), 572–583. doi:10.1038/nrc3557
Long, G. V., Dummer, R., Hamid, O., Gajewski, T. F., Caglevic, C., Dalle, S., et al. (2019). Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet. Oncol. 20 (8), 1083–1097. doi:10.1016/s1470-2045(19)30274-8
Lopez-Serra, P., Marcilla, M., Villanueva, A., Ramos-Fernandez, A., Palau, A., Leal, L., et al. (2014). A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 5, 3608. doi:10.1038/ncomms4608
Losman, J. A., and Kaelin, W. G. (2013). What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes. Dev. 27 (8), 836–852. doi:10.1101/gad.217406.113
Losman, J. A., Looper, R. E., Koivunen, P., Lee, S., Schneider, R. K., McMahon, C., et al. (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339 (6127), 1621–1625. doi:10.1126/science.1231677
Lucena, M. C., Carvalho-Cruz, P., Donadio, J. L., Oliveira, I. A., de Queiroz, R. M., Marinho-Carvalho, M. M., et al. (2016). Epithelial mesenchymal transition induces aberrant glycosylation through hexosamine biosynthetic pathway activation. J. Biol. Chem. 291 (25), 12917–12929. doi:10.1074/jbc.M116.729236
Lunt, S. Y., and Vander Heiden, M. G. (2011). Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell. Dev. Biol. 27, 441–464. doi:10.1146/annurev-cellbio-092910-154237
Margueron, R., and Reinberg, D. (2010). Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 11 (4), 285–296. doi:10.1038/nrg2752
Mashimo, T., Pichumani, K., Vemireddy, V., Hatanpaa, K. J., Singh, D. K., Sirasanagandla, S., et al. (2014). Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 159 (7), 1603–1614. doi:10.1016/j.cell.2014.11.025
Maude, S. L., Frey, N., Shaw, P. A., Aplenc, R., Barrett, D. M., Bunin, N. J., et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371 (16), 1507–1517. doi:10.1056/NEJMoa1407222
McDonald, O. G., Li, X., Saunders, T., Tryggvadottir, R., Mentch, S. J., Warmoes, M. O., et al. (2017). Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 49 (3), 367–376. doi:10.1038/ng.3753
Meng, F., Henson, R., Wehbe-Janek, H., Ghoshal, K., Jacob, S. T., Patel, T., et al. (2007). MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133 (2), 647–658. doi:10.1053/j.gastro.2007.05.022
Mentch, S. J., Mehrmohamadi, M., Huang, L., Liu, X., Gupta, D., Mattocks, D., et al. (2015). Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell. Metab. 22 (5), 861–873. doi:10.1016/j.cmet.2015.08.024
Metallo, C. M., Gameiro, P. A., Bell, E. L., Mattaini, K. R., Yang, J., Hiller, K., et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481 (7381), 380–384. doi:10.1038/nature10602
Miranda, T. B., Cortez, C. C., Yoo, C. B., Liang, G., Abe, M., Kelly, T. K., et al. (2009). DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 8 (6), 1579–1588. doi:10.1158/1535-7163.Mct-09-0013
Moffett, J. R., Puthillathu, N., Vengilote, R., Jaworski, D. M., and Namboodiri, A. M. (2020). Acetate revisited: A key biomolecule at the nexus of metabolism, epigenetics, and oncogenesis - Part 2: Acetate and ACSS2 in health and disease. Front. Physiol. 11, 580171. doi:10.3389/fphys.2020.580171
Momparler, R. L., and Côté, S. (2015). Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin. Investig. Drugs 24 (8), 1031–1043. doi:10.1517/13543784.2015.1051220
Momparler, R. L., Idaghdour, Y., Marquez, V. E., and Momparler, L. F. (2012). Synergistic antileukemic action of a combination of inhibitors of DNA methylation and histone methylation. Leuk. Res. 36 (8), 1049–1054. doi:10.1016/j.leukres.2012.03.001
Mu, X., Zhao, T., Xu, C., Shi, W., Geng, B., Shen, J., et al. (2017). Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 8 (8), 13174–13185. doi:10.18632/oncotarget.14485
Mullen, A. R., Wheaton, W. W., Jin, E. S., Chen, P. H., Sullivan, L. B., Cheng, T., et al. (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481 (7381), 385–388. doi:10.1038/nature10642
Netea, M. G., Domínguez-Andrés, J., Barreiro, L. B., Chavakis, T., Divangahi, M., Fuchs, E., et al. (2020a). Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20 (6), 375–388. doi:10.1038/s41577-020-0285-6
Netea, M. G., Giamarellos-Bourboulis, E. J., Domínguez-Andrés, J., Curtis, N., van Crevel, R., van de Veerdonk, F. L., et al. (2020b). Trained immunity: A tool for reducing susceptibility to and the severity of SARS-CoV-2 infection. Cell. 181 (5), 969–977. doi:10.1016/j.cell.2020.04.042
Novakovic, B., Habibi, E., Wang, S. Y., Arts, R. J. W., Davar, R., Megchelenbrink, W., et al. (2016). β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. 167 (5), 1354–1368. e1314. doi:10.1016/j.cell.2016.09.034
Nowicki, S., and Gottlieb, E. (2015). Oncometabolites: Tailoring our genes. Febs J. 282 (15), 2796–2805. doi:10.1111/febs.13295
O'Neill, L. A. J., and Netea, M. G. (2020). BCG-Induced trained immunity: Can it offer protection against COVID-19? Nat. Rev. Immunol. 20 (6), 335–337. doi:10.1038/s41577-020-0337-y
O'Neill, L. A., Kishton, R. J., and Rathmell, J. (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16 (9), 553–565. doi:10.1038/nri.2016.70
O'Sullivan, D., and Pearce, E. L. (2015). Targeting T cell metabolism for therapy. Trends Immunol. 36 (2), 71–80. doi:10.1016/j.it.2014.12.004
Okoye-Okafor, U. C., Bartholdy, B., Cartier, J., Gao, E. N., Pietrak, B., Rendina, A. R., et al. (2015). New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 11 (11), 878–886. doi:10.1038/nchembio.1930
Olivares, O., Däbritz, J. H. M., King, A., Gottlieb, E., and Halsey, C. (2015). Research into cancer metabolomics: Towards a clinical metamorphosis. Semin. Cell. Dev. Biol. 43, 52–64. doi:10.1016/j.semcdb.2015.09.008
Onodera, Y., Nam, J. M., and Bissell, M. J. (2014). Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Investig. 124 (1), 367–384. doi:10.1172/jci63146
Owen, O. E., Kalhan, S. C., and Hanson, R. W. (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277 (34), 30409–30412. doi:10.1074/jbc.R200006200
Pan, M., Reid, M. A., Lowman, X. H., Kulkarni, R. P., Tran, T. Q., Liu, X., et al. (2016). Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell. Biol. 18 (10), 1090–1101. doi:10.1038/ncb3410
Panwalkar, P., Tamrazi, B., Dang, D., Chung, C., Sweha, S., Natarajan, S. K., et al. (2021). Targeting integrated epigenetic and metabolic pathways in lethal childhood PFA ependymomas. Sci. Transl. Med. 13 (614), eabc0497. doi:10.1126/scitranslmed.abc0497
Patra, K. C., Wang, Q., Bhaskar, P. T., Miller, L., Wang, Z., Wheaton, W., et al. (2013). Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 24 (2), 213–228. doi:10.1016/j.ccr.2013.06.014
Pavlova, N. N., and Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell. Metab. 23 (1), 27–47. doi:10.1016/j.cmet.2015.12.006
Pearce, E. L., Poffenberger, M. C., Chang, C. H., and Jones, R. G. (2013). Fueling immunity: Insights into metabolism and lymphocyte function. Science 342 (6155), 1242454. doi:10.1126/science.1242454
Pfister, S. X., and Ashworth, A. (2017). Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 16 (4), 241–263. doi:10.1038/nrd.2016.256
Phang, J. M., Liu, W., and Hancock, C. (2013). Bridging epigenetics and metabolism: Role of non-essential amino acids. Epigenetics 8 (3), 231–236. doi:10.4161/epi.24042
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F., and Kroemer, G. (2015). Acetyl coenzyme A: A central metabolite and second messenger. Cell. Metab. 21 (6), 805–821. doi:10.1016/j.cmet.2015.05.014
Pinho, S. S., and Reis, C. A. (2015). Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 15 (9), 540–555. doi:10.1038/nrc3982
Piunti, A., and Shilatifard, A. (2016). Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352 (6290), aad9780. doi:10.1126/science.aad9780
Possemato, R., Marks, K. M., Shaul, Y. D., Pacold, M. E., Kim, D., Birsoy, K., et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476 (7360), 346–350. doi:10.1038/nature10350
Prendergast, G. C., Malachowski, W. P., DuHadaway, J. B., and Muller, A. J. (2017). Discovery of Ido1 inhibitors: From bench to bedside. Cancer Res. 77 (24), 6795–6811. doi:10.1158/0008-5472.Can-17-2285
Priebe, A., Tan, L., Wahl, H., Kueck, A., He, G., Kwok, R., et al. (2011). Glucose deprivation activates AMPK and induces cell death through modulation of Akt in ovarian cancer cells. Gynecol. Oncol. 122 (2), 389–395. doi:10.1016/j.ygyno.2011.04.024
Quintin, J., Saeed, S., Martens, J. H. A., Giamarellos-Bourboulis, E. J., Ifrim, D. C., Logie, C., et al. (2012). Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell. Host Microbe 12 (2), 223–232. doi:10.1016/j.chom.2012.06.006
Racey, L. A., and Byvoet, P. (1971). Histone acetyltransferase in chromatin. Evidence for in vitro enzymatic transfer of acetate from acetyl-coenzyme A to histones. Exp. Cell. Res. 64 (2), 366–370. doi:10.1016/0014-4827(71)90089-9
Reid, M. A., Dai, Z., and Locasale, J. W. (2017). The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell. Biol. 19 (11), 1298–1306. doi:10.1038/ncb3629
Ribich, S., Harvey, D., and Copeland, R. A. (2017). Drug discovery and chemical biology of cancer epigenetics. Cell. Chem. Biol. 24 (9), 1120–1147. doi:10.1016/j.chembiol.2017.08.020
Robbins, P. F., Morgan, R. A., Feldman, S. A., Yang, J. C., Sherry, R. M., Dudley, M. E., et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 29 (7), 917–924. doi:10.1200/jco.2010.32.2537
Robinson, M. M., McBryant, S. J., Tsukamoto, T., Rojas, C., Ferraris, D. V., Hamilton, S. K., et al. (2007). Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1, 2, 4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 406 (3), 407–414. doi:10.1042/bj20070039
Rodrigues, M. F., Carvalho, É., Pezzuto, P., Rumjanek, F. D., and Amoêdo, N. D. (2015). Reciprocal modulation of histone deacetylase inhibitors sodium butyrate and trichostatin A on the energy metabolism of breast cancer cells. J. Cell. Biochem. 116 (5), 797–808. doi:10.1002/jcb.25036
Rogers, R. E., Deberardinis, R. J., Klesse, L. J., Boriack, R. L., Margraf, L. R., Rakheja, D., et al. (2010). Wilms tumor in a child with L-2-hydroxyglutaric aciduria. Pediatr. Dev. Pathol. 13 (5), 408–411. doi:10.2350/09-12-0768-cr.1
Rohle, D., Popovici-Muller, J., Palaskas, N., Turcan, S., Grommes, C., Campos, C., et al. (2013). An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340 (6132), 626–630. doi:10.1126/science.1236062
Roichman, A., Elhanati, S., Aon, M. A., Abramovich, I., Di Francesco, A., Shahar, Y., et al. (2021). Restoration of energy homeostasis by SIRT6 extends healthy lifespan. Nat. Commun. 12 (1), 3208. doi:10.1038/s41467-021-23545-7
Rosenberg, S. A., Packard, B. S., Aebersold, P. M., Solomon, D., Topalian, S. L., Toy, S. T., et al. (1988). Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 319 (25), 1676–1680. doi:10.1056/nejm198812223192527
Rosenberg, S. A. (2012). Raising the bar: The curative potential of human cancer immunotherapy. Sci. Transl. Med. 4 (127), 127ps8. doi:10.1126/scitranslmed.3003634
Rosenberg, S. A., Yang, J. C., Sherry, R. M., Kammula, U. S., Hughes, M. S., Phan, G. Q., et al. (2011). Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17 (13), 4550–4557. doi:10.1158/1078-0432.Ccr-11-0116
Saeed, S., Quintin, J., Kerstens, H. H., Rao, N. A., Aghajanirefah, A., Matarese, F., et al. (2014). Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345 (6204), 1251086. doi:10.1126/science.1251086
Sandoval, J., and Esteller, M. (2012). Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 22 (1), 50–55. doi:10.1016/j.gde.2012.02.008
Satoh, T., Takeuchi, O., Vandenbon, A., Yasuda, K., Tanaka, Y., Kumagai, Y., et al. (2010). The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 11 (10), 936–944. doi:10.1038/ni.1920
Schäfer, F., Balleyguier, C., et al. (2013). Letter to editors regarding "breast elasticity: Principles, technique, results: An update and overview of commercially available software", C. Balleyguier et al. http://dx.doi.org/10.1016/j.ejrad.2012.03.001. Eur. J. Radiol. 82 (2), 385–386. doi:10.1016/j.ejrad.2012.03.00110.1016/j.ejrad.2012.11.016
Schug, Z. T., Peck, B., Jones, D. T., Zhang, Q., Grosskurth, S., Alam, I. S., et al. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 27 (1), 57–71. doi:10.1016/j.ccell.2014.12.002
Sciacovelli, M., Gonçalves, E., Johnson, T. I., Zecchini, V. R., da Costa, A. S., Gaude, E., et al. (2016). Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537 (7621), 544–547. doi:10.1038/nature19353
Sebastián, C., Zwaans, B. M., Silberman, D. M., Gymrek, M., Goren, A., Zhong, L., et al. (2012). The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 151 (6), 1185–1199. doi:10.1016/j.cell.2012.10.047
Serganova, I., Chakraborty, S., Yamshon, S., Isshiki, Y., Bucktrout, R., Melnick, A., et al. (2021). Epigenetic, metabolic, and immune crosstalk in germinal-center-derived B-cell lymphomas: Unveiling new vulnerabilities for rational combination therapies. Front. Cell. Dev. Biol. 9, 805195. doi:10.3389/fcell.2021.805195
Shahbazian, M. D., and Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. doi:10.1146/annurev.biochem.76.052705.162114
Shanmugasundaram, K., Nayak, B., Shim, E. H., Livi, C. B., Block, K., Sudarshan, S., et al. (2014). The oncometabolite fumarate promotes pseudohypoxia through noncanonical activation of NF-κB signaling. J. Biol. Chem. 289 (35), 24691–24699. doi:10.1074/jbc.M114.568162
Shi, Y. J., Matson, C., Lan, F., Iwase, S., Baba, T., Shi, Y., et al. (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell. 19 (6), 857–864. doi:10.1016/j.molcel.2005.08.027
Shim, E. H., Livi, C. B., Rakheja, D., Tan, J., Benson, D., Parekh, V., et al. (2014). L-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 4 (11), 1290–1298. doi:10.1158/2159-8290.Cd-13-0696
Shim, H., Dolde, C., Lewis, B. C., Wu, C. S., Dang, G., Jungmann, R. A., et al. (1997). c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. U. S. A. 94 (13), 6658–6663. doi:10.1073/pnas.94.13.6658
Shin, J., He, M., Liu, Y., Paredes, S., Villanova, L., Brown, K., et al. (2013). SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell. Rep. 5 (3), 654–665. doi:10.1016/j.celrep.2013.10.007
Simpson, N. E., Tryndyak, V. P., Beland, F. A., and Pogribny, I. P. (2012a). An in vitro investigation of metabolically sensitive biomarkers in breast cancer progression. Breast Cancer Res. Treat. 133 (3), 959–968. doi:10.1007/s10549-011-1871-x
Simpson, N. E., Tryndyak, V. P., Pogribna, M., Beland, F. A., and Pogribny, I. P. (2012b). Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics 7 (12), 1413–1420. doi:10.4161/epi.22713
Son, J., Lyssiotis, C. A., Ying, H., Wang, X., Hua, S., Ligorio, M., et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496 (7443), 101–105. doi:10.1038/nature12040
Soshnev, A. A., Josefowicz, S. Z., and Allis, C. D. (2016). Greater than the sum of parts: Complexity of the dynamic epigenome. Mol. Cell. 62 (5), 681–694. doi:10.1016/j.molcel.2016.05.004
Sowter, H. M., Raval, R. R., Moore, J. W., Ratcliffe, P. J., and Harris, A. L. (2003). Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 63 (19), 6130–6134.
Sukumar, M., Liu, J., Ji, Y., Subramanian, M., Crompton, J. G., Yu, Z., et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 123 (10), 4479–4488. doi:10.1172/jci69589
Sullivan, L. B., Martinez-Garcia, E., Nguyen, H., Mullen, A. R., Dufour, E., Sudarshan, S., et al. (2013). The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell. 51 (2), 236–248. doi:10.1016/j.molcel.2013.05.003
Tessarz, P., and Kouzarides, T. (2014). Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell. Biol. 15 (11), 703–708. doi:10.1038/nrm3890
Thakur, C., and Chen, F. (2019). Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 57, 52–58. doi:10.1016/j.semcancer.2019.06.006
Tomlinson, I. P., Alam, N. A., Rowan, A. J., Barclay, E., Jaeger, E. E., Kelsell, D., et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30 (4), 406–410. doi:10.1038/ng849
Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott, D. F., et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366 (26), 2443–2454. doi:10.1056/NEJMoa1200690
Turcan, S., Fabius, A. W., Borodovsky, A., Pedraza, A., Brennan, C., Huse, J., et al. (2013). Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 4 (10), 1729–1736. doi:10.18632/oncotarget.1412
Ulanovskaya, O. A., Zuhl, A. M., and Cravatt, B. F. (2013). NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 9 (5), 300–306. doi:10.1038/nchembio.1204
Vakoc, C. R., Mandat, S. A., Olenchock, B. A., and Blobel, G. A. (2005). Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell. 19 (3), 381–391. doi:10.1016/j.molcel.2005.06.011
Varier, R. A., and Timmers, H. T. (2011). Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Acta 1815 (1), 75–89. doi:10.1016/j.bbcan.2010.10.002
Vatrinet, R., Leone, G., De Luise, M., Girolimetti, G., Vidone, M., Gasparre, G., et al. (2017). The α-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. 5, 3. doi:10.1186/s40170-017-0165-0
Wang, F., Travins, J., DeLaBarre, B., Penard-Lacronique, V., Schalm, S., Hansen, E., et al. (2013). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340 (6132), 622–626. doi:10.1126/science.1234769
Wang, J. B., Erickson, J. W., Fuji, R., Ramachandran, S., Gao, P., Dinavahi, R., et al. (2010a). Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 18 (3), 207–219. doi:10.1016/j.ccr.2010.08.009
Wang, L., Jin, Q., Lee, J. E., Su, I. H., and Ge, K. (2010b). Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl. Acad. Sci. U. S. A. 107 (16), 7317–7322. doi:10.1073/pnas.1000031107
Wang, T., Yu, Q., Li, J., Hu, B., Zhao, Q., Ma, C., et al. (2017). O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat. Cell. Biol. 19 (7), 833–843. doi:10.1038/ncb3562
Wang, Y. H., Israelsen, W. J., Lee, D., Yu, V. W. C., Jeanson, N. T., Clish, C. B., et al. (2014). Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 158 (6), 1309–1323. doi:10.1016/j.cell.2014.07.048
Wang, Y. P., and Lei, Q. Y. (2018). Metabolic recoding of epigenetics in cancer. Cancer Commun. 38 (1), 25. doi:10.1186/s40880-018-0302-3
Wardell, S. E., Ilkayeva, O. R., Wieman, H. L., Frigo, D. E., Rathmell, J. C., Newgard, C. B., et al. (2009). Glucose metabolism as a target of histone deacetylase inhibitors. Mol. Endocrinol. 23 (3), 388–401. doi:10.1210/me.2008-0179
Waterfall, J. J., Killian, J. K., and Meltzer, P. S. (2014). The role of mutation of metabolism-related genes in genomic hypermethylation. Biochem. Biophys. Res. Commun. 455 (1-2), 16–23. doi:10.1016/j.bbrc.2014.08.003
Weinberg, S. E., and Chandel, N. S. (2015). Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 11 (1), 9–15. doi:10.1038/nchembio.1712
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., Thompson, C. B., et al. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324 (5930), 1076–1080. doi:10.1126/science.1164097
Wellen, K. E., Lu, C., Mancuso, A., Lemons, J. M., Ryczko, M., Dennis, J. W., et al. (2010). The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes. Dev. 24 (24), 2784–2799. doi:10.1101/gad.1985910
Wellen, K. E., and Thompson, C. B. (2012). A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell. Biol. 13 (4), 270–276. doi:10.1038/nrm3305
Williamson, S. R., Eble, J. N., Amin, M. B., Gupta, N. S., Smith, S. C., Sholl, L. M., et al. (2015). Succinate dehydrogenase-deficient renal cell carcinoma: Detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod. Pathol. 28 (1), 80–94. doi:10.1038/modpathol.2014.86
Wise, D. R., Ward, P. S., Shay, J. E., Cross, J. R., Gruber, J. J., Sachdeva, U. M., et al. (2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U. S. A. 108 (49), 19611–19616. doi:10.1073/pnas.1117773108
Wolf, A., Agnihotri, S., Munoz, D., and Guha, A. (2011). Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 44 (1), 84–91. doi:10.1016/j.nbd.2011.06.007
Wong, C. C., Qian, Y., and Yu, J. (2017). Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 36 (24), 3359–3374. doi:10.1038/onc.2016.485
Xiang, Y., Stine, Z. E., Xia, J., Lu, Y., O'Connor, R. S., Altman, B. J., et al. (2015). Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 125 (6), 2293–2306. doi:10.1172/jci75836
Xiao, C., Kim, H. S., Lahusen, T., Wang, R. H., Xu, X., Gavrilova, O., et al. (2010). SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 285 (47), 36776–36784. doi:10.1074/jbc.M110.168039
Xiao, M., Yang, H., Xu, W., Ma, S., Lin, H., Zhu, H., et al. (2012). Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes. Dev. 26 (12), 1326–1338. doi:10.1101/gad.191056.112
Xie, H., Hanai, J., Ren, J. G., Kats, L., Burgess, K., Bhargava, P., et al. (2014). Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell. Metab. 19 (5), 795–809. doi:10.1016/j.cmet.2014.03.003
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S. H., et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 19 (1), 17–30. doi:10.1016/j.ccr.2010.12.014
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360 (8), 765–773. doi:10.1056/NEJMoa0808710
Yan, L., Tan, Y., Chen, G., Fan, J., and Zhang, J. (2021). Harnessing metabolic reprogramming to improve cancer immunotherapy. Int. J. Mol. Sci. 22 (19), 10268. doi:10.3390/ijms221910268
Yanagida, O., Kanai, Y., Chairoungdua, A., Kim, D. K., Segawa, H., Nii, T., et al. (2001). Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 1514 (2), 291–302. doi:10.1016/s0005-2736(01)00384-4
Yang, M., Soga, T., and Pollard, P. J. (2013). Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 123 (9), 3652–3658. doi:10.1172/jci67228
Yang, M., Ternette, N., Su, H., Dabiri, R., Kessler, B. M., Adam, J., et al. (2014). The succinated proteome of FH-mutant tumours. Metabolites 4 (3), 640–654. doi:10.3390/metabo4030640
Yang, W., Bai, Y., Xiong, Y., Zhang, J., Chen, S., Zheng, X., et al. (2016). Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531 (7596), 651–655. doi:10.1038/nature17412
Yen, K., Travins, J., Wang, F., David, M. D., Artin, E., Straley, K., et al. (2017). AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 7 (5), 478–493. doi:10.1158/2159-8290.Cd-16-1034
Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones, D. R., Frye, R. A., et al. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 23 (12), 2369–2380. doi:10.1038/sj.emboj.7600244
Yu, T., Wang, Y., Hu, Q., Wu, W., Wu, Y., Wei, W., et al. (2017). The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget 8 (58), 98348–98359. doi:10.18632/oncotarget.21311
Zhang, D., Tang, Z., Huang, H., Zhou, G., Cui, C., Weng, Y., et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574 (7779), 575–580. doi:10.1038/s41586-019-1678-1
Zhang, T., Gong, Y., Meng, H., Li, C., and Xue, L. (2020). Symphony of epigenetic and metabolic regulation-interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin. Epigenetics 12 (1), 72. doi:10.1186/s13148-020-00862-0
Zhang, Z. G., and Qin, C. Y. (2014). Sirt6 suppresses hepatocellular carcinoma cell growth via inhibiting the extracellular signal-regulated kinase signaling pathway. Mol. Med. Rep. 9 (3), 882–888. doi:10.3892/mmr.2013.1879
Zhang, Z., Tan, M., Xie, Z., Dai, L., Chen, Y., Zhao, Y., et al. (2011). Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7 (1), 58–63. doi:10.1038/nchembio.495
Zhao, Y., Zheng, Z., Robbins, P. F., Khong, H. T., Rosenberg, S. A., Morgan, R. A., et al. (2005). Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J. Immunol. 174 (7), 4415–4423. doi:10.4049/jimmunol.174.7.4415
Zheng, B., Yao, Y., Liu, Z., Deng, L., Anglin, J. L., Jiang, H., et al. (2013a). Crystallographic investigation and selective inhibition of mutant isocitrate dehydrogenase. ACS Med. Chem. Lett. 4 (6), 542–546. doi:10.1021/ml400036z
Zheng, L., Cardaci, S., Jerby, L., MacKenzie, E. D., Sciacovelli, M., Johnson, T. I., et al. (2015). Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 6, 6001. doi:10.1038/ncomms7001
Zheng, L., MacKenzie, E. D., Karim, S. A., Hedley, A., Blyth, K., Kalna, G., et al. (2013b). Reversed argininosuccinate lyase activity in fumarate hydratase-deficient cancer cells. Cancer Metab. 1 (1), 12. doi:10.1186/2049-3002-1-12
Keywords: cancer metabolism, epigenetics, immunity, novel anti-cancer strategy, oncology
Citation: Chen C, Wang Z and Qin Y (2022) Connections between metabolism and epigenetics: mechanisms and novel anti-cancer strategy. Front. Pharmacol. 13:935536. doi: 10.3389/fphar.2022.935536
Received: 04 May 2022; Accepted: 29 June 2022;
Published: 22 July 2022.
Edited by:
Na Li, University of California, San Diego, United StatesReviewed by:
Xiawei Cheng, East China University of Science and Technology, ChinaCopyright © 2022 Chen, Wang and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanru Qin, eWFucnVxaW5AMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.