 Eiji Hishinuma1,2
Eiji Hishinuma1,2 Yoko Narita3
Yoko Narita3 Kai Obuchi3Akiko Ueda1
Kai Obuchi3Akiko Ueda1 Sakae Saito1,2Shu Tadaka2Kengo Kinoshita1,2,4
Sakae Saito1,2Shu Tadaka2Kengo Kinoshita1,2,4 Masamitsu Maekawa1,5Nariyasu Mano5
Masamitsu Maekawa1,5Nariyasu Mano5 Noriyasu Hirasawa1,3,5
Noriyasu Hirasawa1,3,5 Masahiro Hiratsuka1,3,2,5*
Masahiro Hiratsuka1,3,2,5*- 1Advanced Research Center for Innovations in Next-Generation Medicine, Tohoku University, Sendai, Japan
- 2Tohoku Medical Megabank Organization, Tohoku University, Sendai, Japan
- 3Laboratory of Pharmacotherapy of Life-Style Related Diseases, Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan
- 4Graduate School of Information Sciences, Tohoku University, Sendai, Japan
- 5Department of Pharmaceutical Sciences, Tohoku University Hospital, Sendai, Japan
Dihydropyrimidine dehydrogenase (DPD), encoded by the DPYD gene, is the rate-limiting enzyme in 5-fluorouracil (5-FU) degradation. In Caucasians, four DPYD risk variants are recognized to be responsible for interindividual variations in the development of 5-FU toxicity. However, these risk variants have not been identified in Asian populations. Recently, 41 DPYD allelic variants, including 15 novel single nucleotide variants, were identified in 3,554 Japanese individuals by analyzing their whole-genome sequences; however, the effects of these variants on DPD enzymatic activity remain unknown. In the present study, an in vitro analysis was performed on 41 DPD allelic variants and three DPD risk variants to elucidate the changes in enzymatic activity. Wild-type and 44 DPD-variant proteins were heterologously expressed in 293FT cells. DPD expression levels and dimerization of DPD were determined by immunoblotting after SDS-PAGE and blue native PAGE, respectively. The enzymatic activity of DPD was evaluated by quantification of dihydro-5-FU, a metabolite of 5-FU, using high-performance liquid chromatography-tandem mass spectrometry. Moreover, we used 3D simulation modeling to analyze the effect of amino acid substitutions on the conformation of DPD. Among the 41 DPD variants, seven exhibited drastically decreased intrinsic clearance (CLint) compared to the wild-type protein. Moreover, R353C and G926V exhibited no enzymatic activity, and the band patterns observed in the immunoblots after blue native PAGE indicated that DPD dimerization is required for its enzymatic activity. Our data suggest that these variants may contribute to the significant inter-individual variability observed in the pharmacokinetics and pharmacodynamics of 5-FU. In our study, nine DPD variants exhibited drastically decreased or no enzymatic activity due to dimerization inhibition or conformational changes in each domain. Especially, the rare DPYD variants, although at very low frequencies, may serve as important pharmacogenomic markers associated with the severe 5-FU toxicity in Japanese population.
Introduction
Dihydropyrimidine dehydrogenase (DPD; EC 1.3.1.2) is a rate-limiting enzyme involved in the degradation of the endogenous nucleobases uracil and thymine, and the anticancer drug 5-fluorouracil (5-FU) (Kikugawa et al., 1994; Gmeiner, 2021). 5-FU is phosphorylated and activated intracellularly, causing inhibition of DNA synthesis and RNA dysfunction (Wigmore et al., 2010). Although 5-FU is a key drug used to treat many solid tumors, including gastric, colorectal, and breast cancers, it causes severe side effects, such as neutropenia, thrombocytopenia, mucositis, diarrhea, and hand-foot syndrome, in 10%–30% of patients treated with 5-FU (Wigmore et al., 2010; Kobuchi and Ito, 2020). More than 80% of administered 5-FU is excreted through a three-step metabolic pathway involving DPD (Figure 1) (Heggie et al., 1987; Kunicka et al., 2016). First, DPD catalyzes the reduction of 5-FU to dihydro-5-FU (FUH2). Dihydropyrimidinase (DHP; EC 3.5.2.2) then catalyzes the hydrolysis of FUH2. Finally, the fluoro-β-ureidopropionic acid (FUPA) produced as a result of the previous steps is converted to fluoro-β-alanine by β-ureidopropionase (β-UP, EC 3.5.1.6). A loss of function of any of these enzymes can cause severe 5-FU toxicity; in particular, the loss of function of DPD, the rate-limiting enzyme, is an important factor (Botticelli et al., 2016; Vodenkova et al., 2020). DPD deficiency may result in increased anti-tumor effects or toxicity due to increased blood levels and accumulation of 5-FU (Beck et al., 1994; Botticelli et al., 2016).
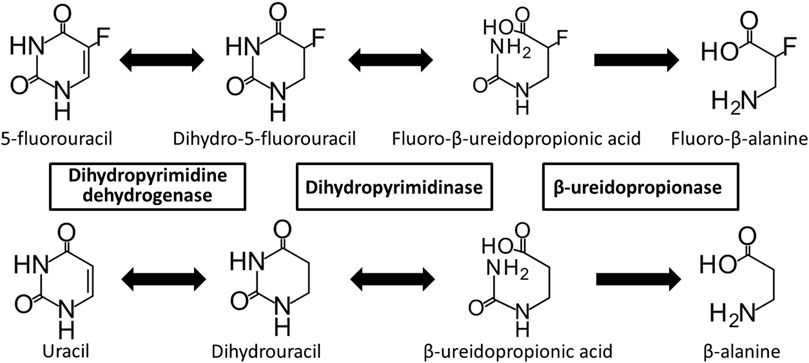
FIGURE 1. Metabolic pathway of 5-fluorouracil and uracil. 5-Fluorouracil and uracil are catabolized by dihydropyrimidine dehydrogenase, dihydropyrimidinase, and β-ureidopropionase.
DPD is expressed in almost all cells, but is particularly active in the liver and lymphocytes (van Kuilenburg et al., 2006). DPD is encoded by the DPYD gene, which is located on chromosome 1p21, comprises 23 exons, and consists of a 3,078 bp open-reading frame that encodes a polypeptide containing 1,025 amino acid residues (Lu et al., 1992). Single nucleotide variants (SNV), deletions, and insertions in DPYD can lead to changes in the amino acid sequence of DPD that alter its enzymatic activity. In addition, microdeletion and chromosomal instability in the 1p21 region of DPYD may cause DPD deficiency (Brečević et al., 2015), which is an autosomal recessive genetic disorder with clinical symptoms that vary widely and include seizures, mental developmental disorders, microcephaly, autism, and asymptomatic individuals (van Kuilenburg, 2004; van Kuilenburg et al., 2009). It is important to be able to predict the risk of developing toxicity because a DPD deficiency may only be detected after 5-FU chemotherapy leads to severe toxicity in asymptomatic patients (Ezzeldin and Diasio, 2004).
To date, more than 450 genetic variants have been identified in the DPYD gene (Sistonen et al., 2012; Thomas et al., 2016; Hishinuma et al., 2020). In Caucasians, four DPYD risk variants, including c.1905+1G>A (IVS14+1G>A, DPYD*2A), c.1129-5923C>G/hapB3, c.1679T>G (DPYD*13, p.I560S), and c.2846A>T (p.D949V), have been reported (Froehlich et al., 2015; Meulendijks et al., 2015). These variants are known to reduce DPD enzymatic activity due to splicing defects or amino acid substitutions (Meulendijks et al., 2015; Deenen et al., 2016). The Clinical Pharmacogenetics Implementation Consortium (CPIC) and Dutch Pharmacogenetics Working Group (DPWG) have established guidelines on 5-FU dosage adjustment based on DPYD genetic polymorphisms (Henricks et al., 2015; Amstutz et al., 2018; Lunenburg et al., 2020). However, there are considerable geographical-ethnicity differences in the variation and frequency of DPYD genetic polymorphisms, and these four DPYD risk variants have not been identified in Asian populations, including the Japanese population (van Kuilenburg, 2004; Maekawa et al., 2007; Yokoi et al., 2020). We previously reported enzymatic functional alterations in 21 DPD variants identified by whole-genome sequencing (WGS) of 1,070 Japanese individuals, suggesting that some DPD variants specific to the Japanese population may be responsible for the development of severe 5-FU toxicity (Hishinuma et al., 2018). To accurately predict this occurrence, a comprehensive functional analysis that includes rare DPYD genetic polymorphisms present in the Japanese population is necessary.
Recently, 15 novel DPYD variants were identified by WGS of 3,554 Japanese individuals conducted by the Tohoku University Tohoku Medical Megabank Organization (ToMMo), but any functional changes resulting from these variations remain unknown (Tadaka et al., 2021). Therefore, in this study, we characterized the enzymatic activity of wild-type DPD and 41 variants derived from DPYD genetic polymorphisms identified by ToMMo (Tables 1, 2) using recombinant proteins expressed in 293FT cells, and determined the kinetic parameters of these variants using a 5-FU reduction activity assay. In addition, three DPD risk variants (DPYD*2A, DPYD*13, and c.2846A>T) were analyzed. Our results indicate the potential for rare DPYD variants to be used as novel pharmacogenomic markers for predicting severe 5-FU toxicity in the Japanese population.
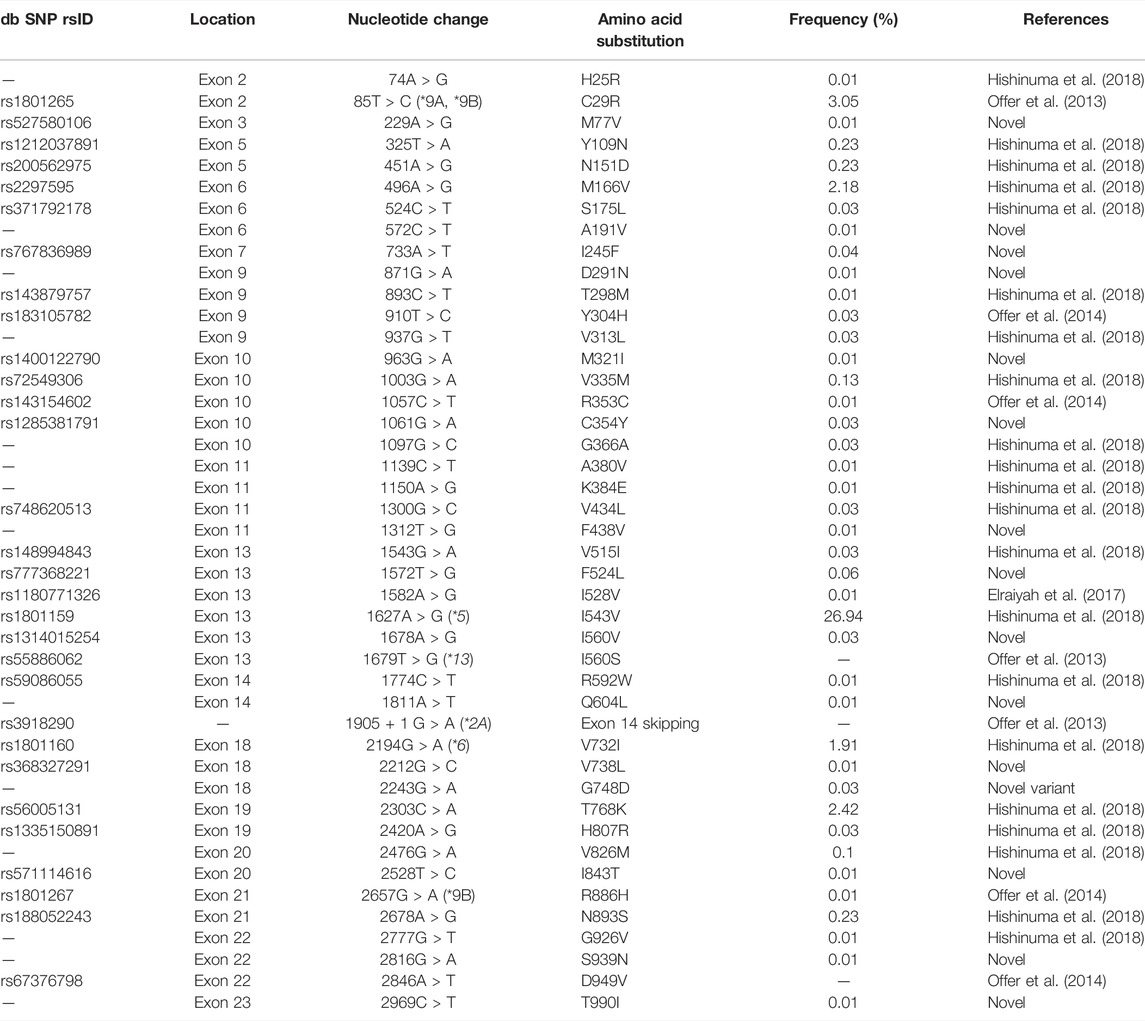
TABLE 1. DPYD variants identified in 3,554 Japanese subjects and three risk variants.
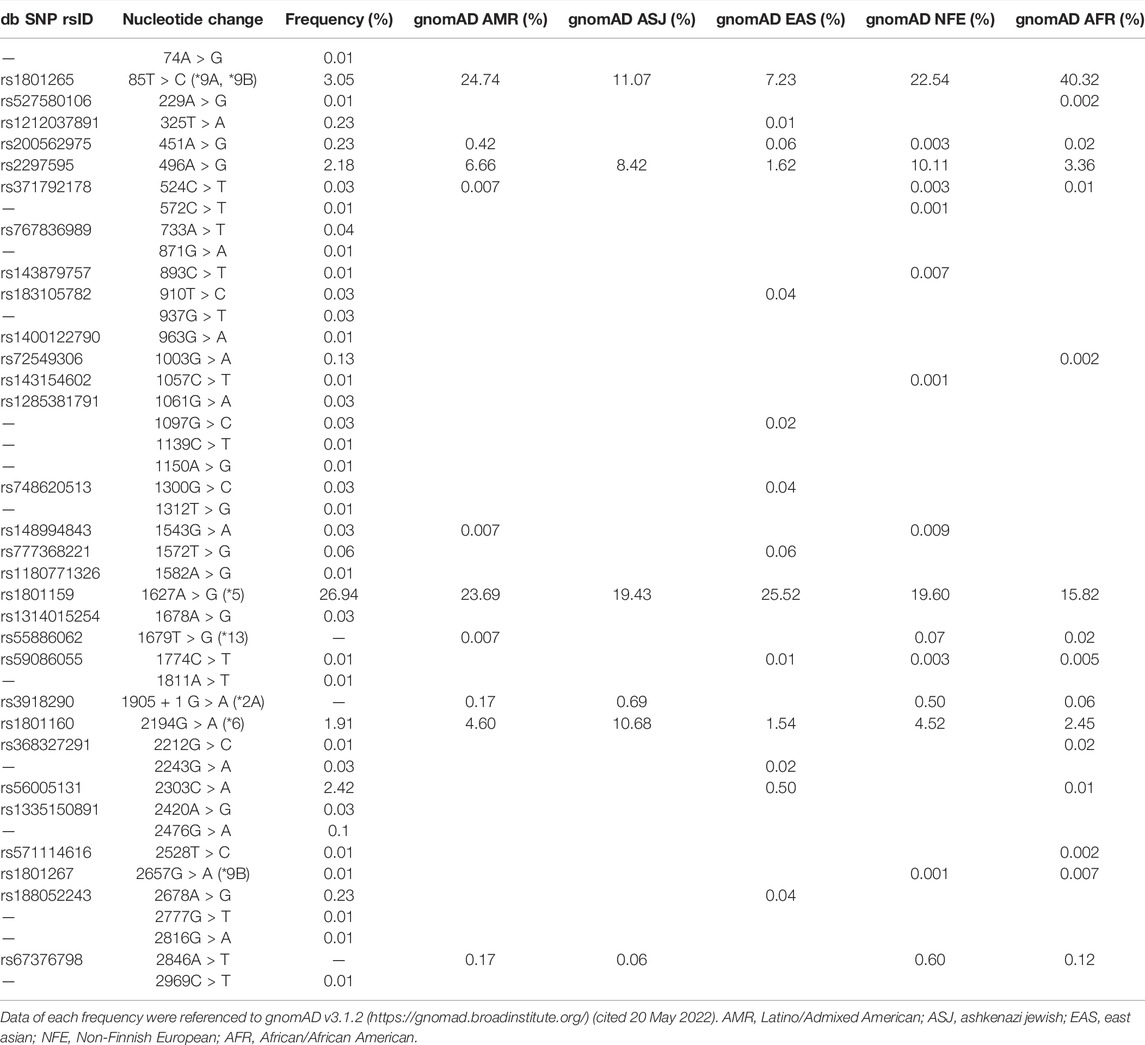
TABLE 2. Geographical-ethnicity differences in the variation and frequency of DPYD variants.
Materials and Methods
Chemicals and Reagents
Antibodies and reagents were purchased from the following commercial sources: cytosine-2,4-13C2,15N3 (TAIYO NIPPON SANSO, Tokyo, Japan); polyclonal anti-human DPD antibody (ABC451; Millipore, Tokyo, Japan); polyclonal anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase) antibody (G9545; Sigma-Aldrich, St. Louis, MO, United States). Other chemicals and reagents were the same as in previous reports (Hishinuma et al., 2018).
Sanger Sequencing of DPYD Gene
Genomic DNA was isolated from the whole blood of Japanese participants in a cohort study conducted by ToMMo (Kuriyama et al., 2016; Minegishi et al., 2019; Nagami et al., 2020). All study participants provided informed consent, and the study was approved by the Tohoku University Graduate School of Pharmaceutical Sciences Ethics Committee (permission number 14-08) and the ToMMo Ethics Committee (permission numbers 2017-4-26, 2017-4-58, and 2017-4-090). Sequence variations in the DPYD gene determined by WGS were confirmed using the polymerase chain reaction (PCR) and Sanger sequencing, according to previously described methods (Akai et al., 2015; Hiratsuka et al., 2015; Hishinuma et al., 2018). The primer pairs used to amplify the DPYD gene and Sanger sequencing results containing SNV are listed in Table 3.
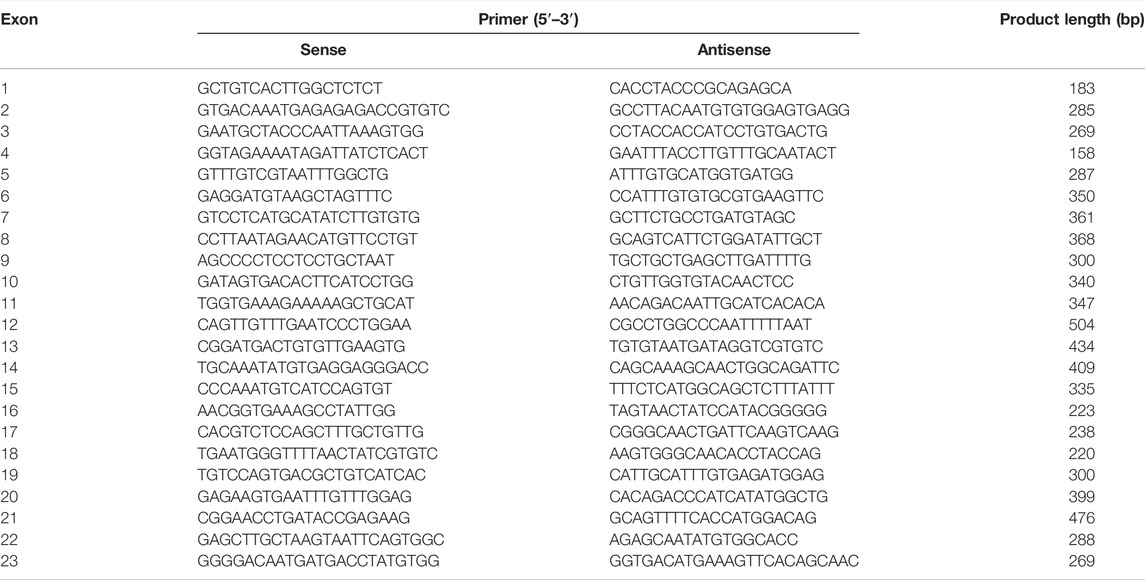
TABLE 3. Polymerase chain reaction primers used to amplify sequences of the human DPYD gene used in this study.
Construction of Expression Vectors
Plasmids containing wild-type human DPYD cDNA were constructed as previously described (Hishinuma et al., 2018). Constructs for 43 DPYD variants were generated using a QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, United States) according to the manufacturer’s instructions. DPYD*2A was deleted for exon 14 by inverse PCR using the KOD Plus Mutagenesis Kit (TOYOBO, Osaka, Japan). Wild-type and mutant DPYD cDNA were then inserted into pcDNA3.4 (Thermo Fisher Scientific, Waltham, MA, United States).
Dihydropyrimidine Dehydrogenase Variant Expression in 293FT Cells
Human embryonic kidney cell-derived 293FT cells were plated at a density of 2.0 × 106 cells/100-mm dish and transfected with plasmids carrying DPYD cDNA (7 μg each) 24 h later using polyethylenimine MAX reagent (Polysciences Inc., Warrington, PA, United States). Sodium sulfide (50 μM), ammonium ferric citrate (50 μM), flavin adenine dinucleotide (50 μM), and flavin mono nucleotide (50 μM) were added 18 h post-transfection. Following incubation for 30 h at 37°C, the cells were scraped off, centrifuged at 1,500 × g for 5 min, and resuspended in a homogenization buffer containing 10 mM Tris-HCl (pH 7.4), 1 mM ethylenediaminetetraacetic acid (EDTA), and 10% glycerol. Soluble fractions containing the DPD protein were prepared as previously described (Hishinuma et al., 2018).
Determination of Dihydropyrimidine Dehydrogenase Protein Expression Using Sodium Dodecyl Sulfate- Polyacrylamide Gel Electrophoresis and Immunoblotting
Proteins were separated by Sodium Dodecyl Sulfate (SDS)-Polyacrylamide Gel Electrophoresis (PAGE), then immunoblotting was performed as previously described (Hishinuma et al., 2018). Protein concentrations were quantified using the bicinchoninic acid assay, and 10 µg of soluble fraction per lane was applied. DPD was detected using a polyclonal anti-human DPD antibody (1:2,000) and goat anti-rabbit IgG (1:5,000). GAPDH antibody was used as a loading control (1:5,000), and goat anti-rabbit IgG (1:10,000) was used as the secondary antibody. Immunoblots were visualized using the SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific). Chemiluminescence was quantified using a ChemiDoc XRS+ with Image Lab Software (Bio-Rad Laboratories, Hercules, CA, United States).
Immunoblotting After Blue Native Polyacrylamide Gel Electrophoresis
Following blue native PAGE, immunoblotting was performed as previously described (Hishinuma et al., 2017; Hishinuma et al., 2018). DPD proteins were detected using a polyclonal anti-human DPD antibody (1:2,000) and goat anti-rabbit IgG (1:5,000). Immunoblots were visualized using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). NativeMarkTM Unstained Protein Standard (Thermo Fisher Scientific) was used as a molecular weight marker, and the proteins were visualized using Coomassie Brilliant Blue (CBB-R250) staining after electrophoresis.
5-FU Reduction Assays
5-FU reduction assay was performed as previously described, with several modifications (Hishinuma et al., 2018). 5-FU (0.1–20 μM) was added to the DPD variant protein (20 μg) and incubated at 37°C for 10 min. The reactions were terminated by adding acetonitrile containing 2 μM cytosine-2,4-13C2,15N3 as an internal standard.
After removal of proteins by centrifugation, the supernatant was vacuum dried at 40°C for 1.5 h and redissolved in 0.1% (v/v) formic acid solution. Sample solutions (5 μl) were injected into a high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) system, and FUH2 was measured using the positive ion detection mode. A standard curve was prepared using FUH2 standards in the range of 0.01–10 μM. Enzyme activity was normalized using the corresponding DPD expression levels quantified by immunoblotting after SDS-PAGE.
Data Analysis
Kinetic data were analyzed using SigmaPlot 12.5 Enzyme Kinetics Module (Systat Software Inc., Chicago, IL, United States); Michaelis constant (Km), maximum velocity (Vmax), and intrinsic clearance (CLint = Vmax/Km) values were determined using the average DPD expression values obtained in triplicate and are expressed as mean ± standard deviation. Statistical variance in protein expression and kinetic parameters was analyzed using Dunnett’s t-test, Dunnett’s T3 test, or the Kruskal-Wallis method (IBM SPSS Statistics Ver. 22, International Business Machines, Armonk, NY, United States). Statistical significance was set at p < 0.05.
3D Simulation Modeling of Dihydropyrimidine Dehydrogenase
A 3D simulation modeling analysis of DPD was performed as previously described (Hishinuma et al., 2018). Discovery Studio 4.5 was used for human DPD 3D imaging, and the Discovery Studio 2.5 (Accelrys, San Diego, CA, United States) CDOKER protocol was used to create docked DPD–5-FU structures.
Results
Confirmation of DPYD Exonic Single Nucleotide Variants Identified in 3,554 Japanese Individuals
A total of 41 DPYD allelic variants, including 15 novel variants in Japanese participants, were previously detecting using WGS (Tadaka et al., 2021). To confirm these alterations, Sanger sequencing was performed on DPYD variants according to previously described methods (Akai et al., 2015; Hiratsuka et al., 2015; Hishinuma et al., 2018). All exonic SNV and WGS results matched those obtained by Sanger sequencing (Table 1).
Wild-Type and Dihydropyrimidine Dehydrogenase Variant Expression Levels in 293FT Cells
DPD expression levels were determined by immunoblotting after SDS-PAGE with a polyclonal anti-human DPD antibody that recognizes all DPD variants (Figure 2A). The band for the DPYD*2A variant was detected at a lower molecular weight owing to skipping of exon 14. GAPDH expression levels were constant in all transfected 293FT cells. Endogenous DPD protein was not detected in the soluble fraction of 293FT cells.
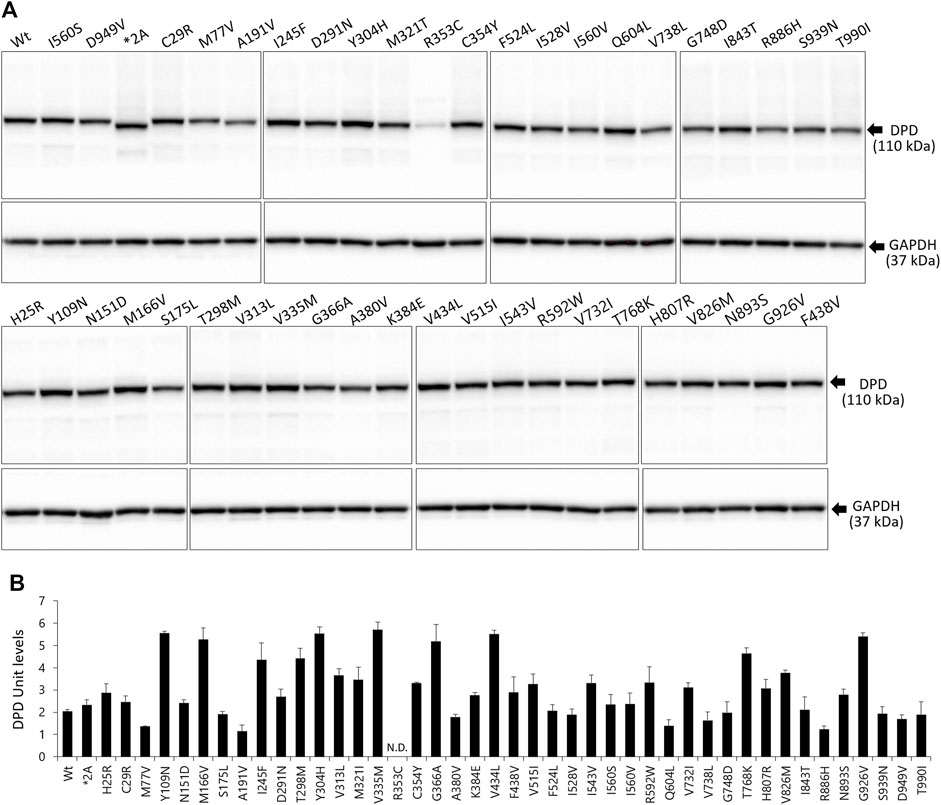
FIGURE 2. Expression levels of wild-type and variant DPD proteins. (A) DPD protein levels were determined by immunoblotting analysis following SDS-PAGE. (B) DPD units of proteins expressed in 293FT cells. Each bar represents the mean ± standard deviation of three independent assays.
The average levels of the wild-type and variant DPD proteins are shown in Figure 2B. We defined DPD expression levels equivalent to 10 μg of wild-type DPD as 1 DPD unit. The R353C variant was below the lower limit of quantification; therefore, the expression levels could not be determined. There were no significant differences in the expression levels of other variants, except for the R353C variant, compared to the wild-type.
Kinetics of 5-FU Reduction Mediated by Dihydropyrimidine Dehydrogenase
Kinetic parameters of 5-FU reduction were calculated for the wild-type and 44 DPD variants (Table 4). The nonlinear regression curves of the Michaelis-Menten equation are shown in Figure 3. Kinetic parameters could not be determined for R353C, DPYD*2A, and G926V, as no 5-FU reduction activity was detected.
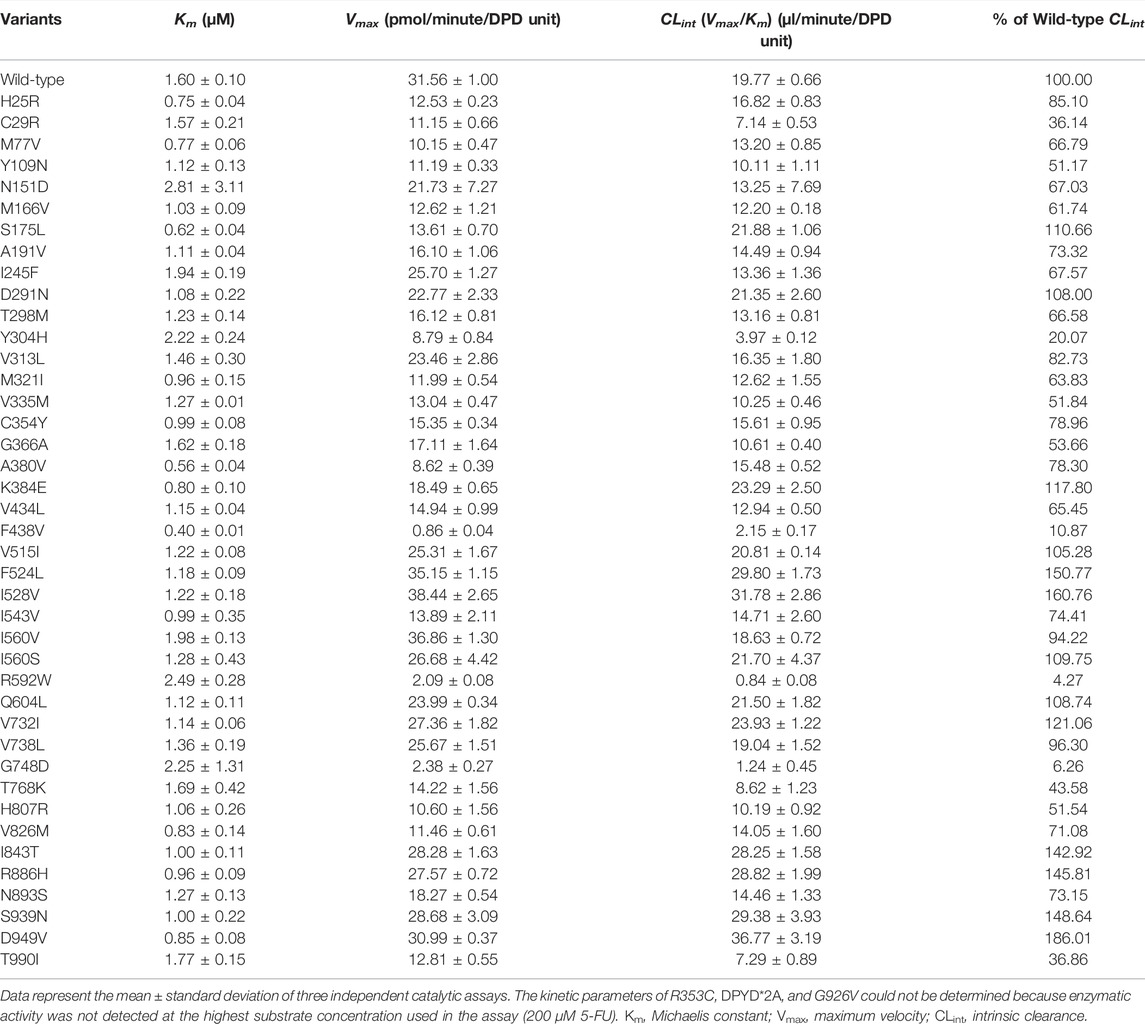
TABLE 4. Kinetic parameters of dihydropyrimidine dehydrogenase (DPD) variants in 5-FU metabolism.
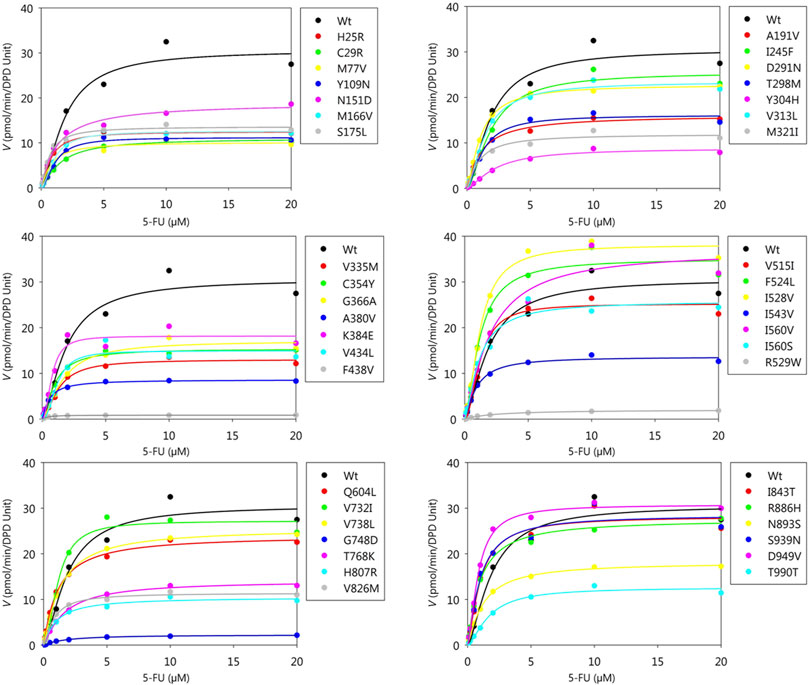
FIGURE 3. Michaelis-Menten curves of DPD variants. The kinetic parameters Km, Vmax, and CLint of 5-FU reduction were determined. The velocities for the reduction of 5-FU and 5-FU concentration are plotted on the horizontal and vertical axes, respectively.
The Km, Vmax, and CLint values for the reduction of 5-FU by wild-type DPD were 1.60 μM, 31.56 pmol/min/DPD unit, and 19.77 μl/min/DPD unit, respectively. None of the parameters showed significant differences because they did not follow a normal distribution; therefore, nonparametric tests were performed, but seven DPD variants (C29R, Y304H, F438V, R592W, G748D, T768K, and T990I) showed 50% or lower decrease in CLint compared to the wild-type. On the other hand, three variants (F524L, I528V, and D949V) showed 150% or higher increase in CLint compared to the wild-type.
Blue Native Polyacrylamide Gel Electrophoresis Analysis of Dihydropyrimidine Dehydrogenase Variants
Immunoblotting results after blue native PAGE are shown in Figure 4. The molecular weight of 43 variants, including wild-type DPD but excluding the DPYD*2A and G926V variants that lost enzymatic activity, was around 242 kDa, which corresponds to the DPD dimer. Six DPD variants (N151D, R353C, R592W, G748D, H807R, and T990I) showed a decreased band intensity compared to that of other variants. In addition, electrophoretic mobility was reduced in four variants (N151D, R592W, G748D, and H807R).
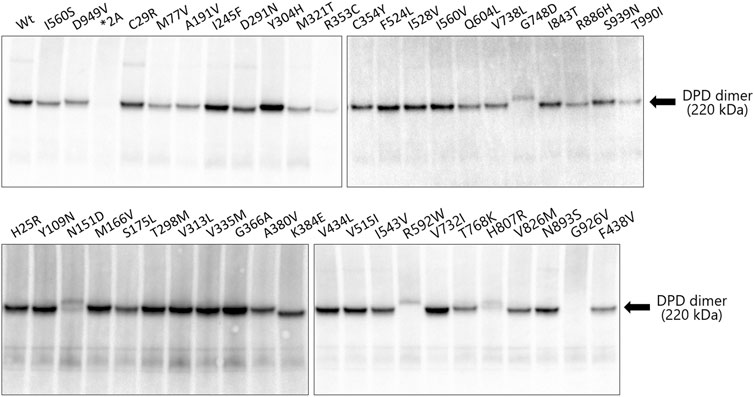
FIGURE 4. Immunoblotting analysis after blue native PAGE showing immunoreactive dihydropyrimidine dehydrogenase (DPD) variant proteins. Blue native PAGE was performed using Tris-glycine buffer and 5%–20% polyacrylamide gels; 9 μg of the soluble fraction of DPD variant proteins were loaded into each lane in triplicate. DPD variants were detected using polyclonal antibodies against human DPD.
3D Structural Modeling of Dihydropyrimidine Dehydrogenase
The crystal structure and five domains of human DPD created by homology modeling are shown in Figures 5A,B, respectively. Close-up views of the 3D crystal structure models of the C29R, Y304H, and F438V mutation sites are shown in Figures 6A–C, respectively. The R29 residue formed a hydrogen bond with N494. The H304 residue was bound to FAD via hydrophobic interactions with F309 and L310, and to NADPH via hydrogen bonds with S436 and A437. The V438 residue formed a direct hydrogen bond and hydrophobic interaction with NADPH. A close-up view of the R353C mutation site is shown in Figure 6D. The R353 residue was bound to FAD via hydrogen bonds with F234 and R235; however, substitution with C353 eliminated these interactions.
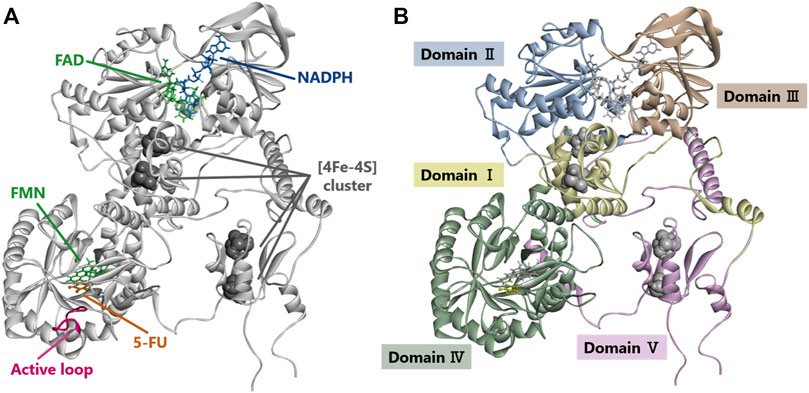
FIGURE 5. Dihydropyrimidine dehydrogenase (DPD) structural analysis. (A) Diagram showing overall structure of human DPD. (B) Diagram showing each domain of human DPD.
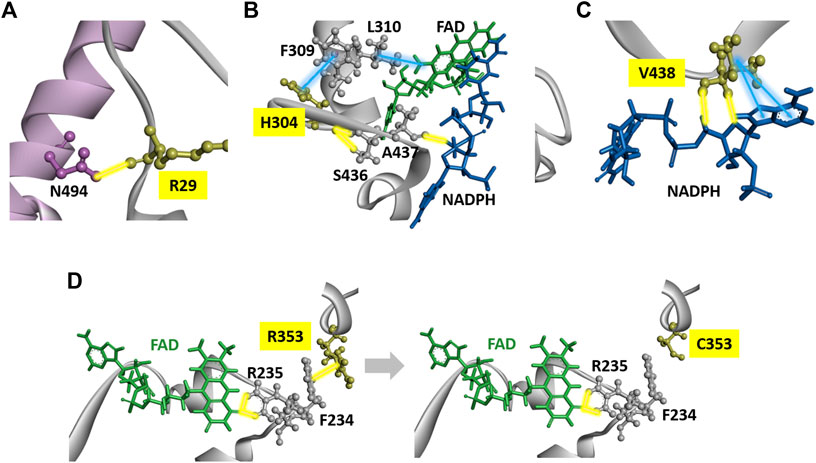
FIGURE 6. DPD structural analysis. (A) Diagram of a fragment of the crystal structures of the H25R variant. The R25 residues are shown in yellow. (B) Diagram of a fragment of the crystal structures of the H304Y variant. The Y304 residues are shown in yellow. (C) Diagram of a fragment of the crystal structures of the F438V variant. The F438 residues are shown in yellow. (D) Diagrams of a fragment of the crystal structures of wild-type DPD (left panel) and R353C (right panel). The R353 and C353 residues are shown in yellow. Yellow and blue lines indicate hydrogen bonds and hydrophobic interactions, respectively.
Discussion
DPD deficiencies can lead to the development of severe 5-FU toxicity; however, significant geographical-ethnicity differences exist with regards to polymorphisms of the DPYD gene (Table 2) (Lunenburg et al., 2016; Roberto et al., 2017). Therefore, it is important to identify genomic biomarkers for each ethnic group. In this study, we used in vitro assays to evaluate changes in the enzymatic function of DPD variants derived from 41 DPYD genetic polymorphisms identified by WGS in a Japanese population of 3,554 individuals along with three risk variants previously reported as predictive markers of 5-FU toxicity in Caucasians (Amstutz et al., 2018; Lunenburg et al., 2020). We previously demonstrated that the addition of four cofactors (iron, sulfur, FAD, and FMN) in the in vitro expression system of the DPD protein is crucial for the functional analysis of DPD variants (Hishinuma et al., 2018). Therefore, we transiently expressed 44 DPD variants in 293FT cells in the presence of these four cofactors, and evaluated the enzymatic activity of each DPD variant by determining the kinetic parameters of 5-FU reduction. The kinetic parameters of three variants (R353C, DPYD*2A, and G926V) could not be determined because no enzymatic activity was detected at the highest substrate concentration used in the assay. In addition, seven DPD variants (C29R, Y304H, F438V, R592W, G748D, T768K, and T990I) showed a CLint less than 50% of the wild-type DPD. DPYD*2A, which has been reported as a predictive marker of 5-FU toxicity in Caucasians, is known to lose enzymatic activity due to skipping of exon 14 (Van Kuilenburg et al., 1997; Nie et al., 2017). Consistent with our results, Offer et al. (2013) reported that recombinant DPD expressed in HEK293T/c17 cells also lost enzymatic activity. In contrast, two risk variants (I560S and D949V) showed increased enzymatic activity in the in vitro analysis of this study. In vivo and in vitro differences between these variants may be due to differences in expression conditions and cell lines, and may also be due to differential regulation of their expression by mRNA or other factors in vivo.
The DPD molecule is composed of five domains: domains I and V contain two (4Fe-4S) clusters of two molecules each, domain II contains the FAD binding site, and domain IV contains the FMN binding site. NADPH, which is required for reduction, binds to domain III to provide electrons, and the active site is located in domain IV (Lu et al., 1992; Dobritzsch et al., 2001; Schnackerz et al., 2004; Lohkamp et al., 2010). The amino acid sequences of these domains are highly conserved among various animal species, and the ability to form dimers plays an important role in the activity of DPD since it is activated by dimerization and the transporting of electrons through the (4Fe-4S) clusters (Dobritzsch et al., 2002; Lohkamp et al., 2010). To determine the effect of amino acid substitutions on DPD dimer formation we performed immunoblotting after blue native PAGE under non-denaturing conditions. A band of approximately 242 kDa, estimated to be a dimer, was detected in all DPD variants except DPYD*2A and G926V, for which a loss of enzymatic activity was observed. In addition, the resulting band of seven DPD variants (N151D, R353C, R592W, G748D, T768K, H807R, and T990I) had a decreased intensity compared to that of other variants, and these variants showed either decreased or no enzymatic activity. Furthermore, four variants (N151D, R592W, G748D, and H807R) showed a decrease in electrophoretic mobility that could be attributed to a change in the charge of the amino acids. These results suggest that these variants have reduced activity due to amino acid substitutions and changes in the charge of amino acids that prevent the dimerization of DPD. Three variants (R592W, T768K, and G926V) showed reduced enzymatic activity, similar to our previously reported analysis of DPD variants identified in 1,070 Japanese individuals (Hishinuma et al., 2018). In contrast, the C29R, Y304H, and F438V variants had a CLint less than 50% of the wild-type, although the intensity of their bands resulting from blue native PAGE were the same intensity as that of wild-type, and there was no clear correlation between the CLint value and dimerization ability in 5-FU reduction activity. It is possible that amino acid substitutions in these variants affected domain functions other than dimer formation.
The C29R variant has an allele frequency of 3.05% in the Japanese population, and is a common variant worldwide (Maekawa et al., 2007; Caudle et al., 2013). In this study, the C29R variant reduced the CLint of 5-FU reduction activity to 36% of wild-type DPD. Although the C29R variant did not show any alteration in side-chain binding or interaction in the 3D simulation analysis, this change occurs in domain I, and the substitution from cysteine to arginine might affect the transportation of electrons. Offer et al. (2013) reported that the C29R variant showed 13% higher activity than wild-type when expressed in HEK293T/c17 cells. In contrast, Kuilenburg et al. reported that the enzymatic activity of the C29R variant was reduced to approximately 80% of that of wild-type when expressed in HEK293 Flp-In cells (Kuilenburg et al., 2016). While there are reports of an increased incidence of grade 3–4 diarrhea in response to 5-FU treatment in patients carrying the C29R variant, there are also reports of no 5-FU toxicity developing in these individuals, and the relevance of this variant to the risk of developing 5-FU toxicity remains unclear (Joerger et al., 2015; Khushman et al., 2018; Maharjan et al., 2019).
In our study, the Y304H variant reduced the CLint of 5-FU reduction activity to 20% of that of wild-type DPD. Offer et al., 2013 reported that the enzymatic activity of the Y304H variant was reduced to approximately 80% of that of wild-type DPD in HEK293T/c17 cells (Offer et al., 2014). Y304 is in domain III and forms hydrophobic interactions or hydrogen bonds with F309 near FAD and S436 near NADPH. The F438V variant reduced the CLint of 5-FU reduction activity to 11% of that of wild-type DPD. F438 is also present in domain III and forms hydrophobic interactions and hydrogen bonds with NADPH. Although no changes in side chain binding or interactions were observed in the 3D simulation analysis of either variant, domain III binds NADPH, an important source of electrons, and it is assumed that the amino acid substitution affected the binding of NADPH, resulting in a decrease in enzymatic activity.
Offer et al., 2013 reported no enzymatic activity of the R353C variant when expressed in HEK293T/c17 cells (Offer et al., 2014). In the present study, the R353C variant also lost enzymatic activity. R353 is in domain III, and its electrostatic interaction with F234 was abolished by the substitution to C353. Since R235, located next to F234, forms a hydrogen bond with FAD, these results suggest that the substitution to C353 caused the loss of enzymatic activity due to a conformational change around FAD.
In conclusion, the wild-type and 41 DPD variants identified in a Japanese population of 3,554 individuals were expressed in 293FT cells, and their enzymatic activities were assessed in vitro. The results showed that nine DPD variants (C29R, Y304H, R353C, F438V, R592W, G748D, T768K, G926V, and T990I) reduced enzymatic activity to less than 50% of that of wild-type DPD. Therefore, we hypothesized that patients with these DPYD polymorphisms may develop severe toxicity due to elevated concentrations of 5-FU during chemotherapy. Except for C29R, these variants have been identified at very low frequencies and mostly in Asian populations, suggesting that several rare DPYD polymorphisms causing reduced DPD activity may be responsible for potential 5-FU toxicity in populations where the four risk variants have not been identified. Therefore, we suggest that rare DPYD polymorphism may be a useful predictive marker for 5-FU toxicity in the Japanese population. However, this information is insufficient to determine whether these variants affect the pharmacokinetics and pharmacodynamics of 5-FU in vivo. It is expected that large-scale clinical trials will be conducted in the future to analyze the correlation between the DPYD genotype and the blood levels of 5-FU and its derivatives in patients to evaluate its usefulness as a genetic biomarker for predicting 5-FU toxicity.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and approved by the Tohoku University Graduate School of Pharmaceutical Sciences Ethics Committee (permission number 14-08) and the Tohoku Medical Megabank Organization Ethics Committee (permission numbers 2017-4-26, 2017-4-58, and 2017-4-090).Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
Conceptualization, EH and MH; Data curation, EH and YN; Formal analysis, EH, YN, and MH; Funding acquisition, SS, ST, KK, and MH; Investigation, EH, YN, KO, AU, SS, ST, and MM; Methodology, EH, YN, MM, and MH; Project administration, MH; Resources, SS, KK, MM, NM, NH, and MH; Software, ST and KK; Supervision, KK, NM, NH, and MH; Validation, EH, YN, AU, and SS; Visualization, EH and YN; Writing—original draft, EH and YN; Writing—review and editing, MH.
Funding
MH was supported by grants from the Japan Agency for Medical Research and Development (AMED) (Grant No. JP19kk0305009), Takahashi Industrial and Economic Research Foundation, and Smoking Research Foundation. SS, ST, and KK were supported by grants from AMED (Grant Nos. JP17km0105001 and JP21tm0124005). This research was also supported in part by the Tohoku Medical Megabank Project: Promoting Public Utilization of Advanced Research Infrastructure, and the Sharing and Administrative Network for Research Equipment funded by the Ministry of Education, Culture, Sports, Science, and Technology (MEXT). ST and KK were supported by grants for the Facilitation of R&D Platform for AMED Genome Medicine Support from AMED (Grant No. JP16km0405001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akai, F., Hosono, H., Hirasawa, N., and Hiratsuka, M. (2015). Novel Single Nucleotide Polymorphisms of the Dihydropyrimidinase Gene (DPYS) in Japanese Individuals. Drug Metab. Pharmacokinet. 30 (1), 127–129. doi:10.1016/j.dmpk.2014.09.005
Amstutz, U., Henricks, L. M., Offer, S. M., Barbarino, J., Schellens, J. H. M., Swen, J. J., et al. (2018). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharmacol. Ther. 103 (2), 210–216. doi:10.1002/cpt.911
Beck, A., Etienne, M. C., Chéradame, S., Fischel, J. L., Formento, P., Renée, N., et al. (1994). A Role for Dihydropyrimidine Dehydrogenase and Thymidylate Synthase in Tumour Sensitivity to Fluorouracil. Eur. J. Cancer 30a (10), 1517–1522. doi:10.1016/0959-8049(94)00216-r
Botticelli, A., Borro, M., Onesti, C. E., Strigari, L., Gentile, G., Cerbelli, B., et al. (2016). Degradation Rate of 5-Fluorouracil in Metastatic Colorectal Cancer: A New Predictive Outcome Biomarker? PLoS One 11 (9), e0163105. doi:10.1371/journal.pone.0163105
Brečević, L., Rinčić, M., Krsnik, Ž., Sedmak, G., Hamid, A. B., Kosyakova, N., et al. (2015). Association of New Deletion/duplication Region at Chromosome 1p21 with Intellectual Disability, Severe Speech Deficit and Autism Spectrum Disorder-like Behavior: an All-In Approach to Solving the DPYD Enigma. Transl. Neurosci. 6 (1), 59–86. doi:10.1515/tnsci-2015-0007
Caudle, K. E., Thorn, C. F., Klein, T. E., Swen, J. J., McLeod, H. L., Diasio, R. B., et al. (2013). Clinical Pharmacogenetics Implementation Consortium Guidelines for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing. Clin. Pharmacol. Ther. 94 (6), 640–645. doi:10.1038/clpt.2013.172
Deenen, M. J., Meulendijks, D., Cats, A., Sechterberger, M. K., Severens, J. L., Boot, H., et al. (2016). Upfront Genotyping of DPYD*2A to Individualize Fluoropyrimidine Therapy: A Safety and Cost Analysis. J. Clin. Oncol. 34 (3), 227–234. doi:10.1200/JCO.2015.63.1325
Dobritzsch, D., Ricagno, S., Schneider, G., Schnackerz, K. D., and Lindqvist, Y. (2002). Crystal Structure of the Productive Ternary Complex of Dihydropyrimidine Dehydrogenase with NADPH and 5-iodouracil. Implications for Mechanism of Inhibition and Electron Transfer. J. Biol. Chem. 277 (15), 13155–13166. doi:10.1074/jbc.M111877200
Dobritzsch, D., Schneider, G., Schnackerz, K. D., and Lindqvist, Y. (2001). Crystal Structure of Dihydropyrimidine Dehydrogenase, a Major Determinant of the Pharmacokinetics of the Anti-cancer Drug 5-fluorouracil. EMBO J. 20 (4), 650–660. doi:10.1093/emboj/20.4.650
Elraiyah, T., Jerde, C. R., Shrestha, S., Wu, R., Nie, Q., Giama, N. H., et al. (2017). Novel Deleterious Dihydropyrimidine Dehydrogenase Variants May Contribute to 5-Fluorouracil Sensitivity in an East African Population. Clin. Pharmacol. Ther. 101 (3), 382–390. doi:10.1002/cpt.531
Ezzeldin, H., and Diasio, R. (2004). Dihydropyrimidine Dehydrogenase Deficiency, a Pharmacogenetic Syndrome Associated with Potentially Life-Threatening Toxicity Following 5-fluorouracil Administration. Clin. Colorectal Cancer 4 (3), 181–189. doi:10.3816/ccc.2004.n.018
Froehlich, T. K., Amstutz, U., Aebi, S., Joerger, M., and Largiadèr, C. R. (2015). Clinical Importance of Risk Variants in the Dihydropyrimidine Dehydrogenase Gene for the Prediction of Early-Onset Fluoropyrimidine Toxicity. Int. J. Cancer 136 (3), 730–739. doi:10.1002/ijc.29025
Gmeiner, W. H. (2021). A Narrative Review of Genetic Factors Affecting Fluoropyrimidine Toxicity. Precis. Cancer Med. 4, 38. doi:10.21037/pcm-21-17
Heggie, G. D., Sommadossi, J. P., Cross, D. S., Huster, W. J., and Diasio, R. B. (1987). Clinical Pharmacokinetics of 5-fluorouracil and its Metabolites in Plasma, Urine, and Bile. Cancer Res. 47 (8), 2203–2206.
Henricks, L. M., Lunenburg, C. A., Meulendijks, D., Gelderblom, H., Cats, A., Swen, J. J., et al. (2015). Translating DPYD Genotype into DPD Phenotype: Using the DPYD Gene Activity Score. Pharmacogenomics 16 (11), 1277–1286. doi:10.2217/pgs.15.70
Hiratsuka, M., Yamashita, H., Akai, F., Hosono, H., Hishinuma, E., Hirasawa, N., et al. (2015). Genetic Polymorphisms of Dihydropyrimidinase in a Japanese Patient with Capecitabine-Induced Toxicity. PLoS One 10 (4), e0124818. doi:10.1371/journal.pone.0124818
Hishinuma, E., Akai, F., Narita, Y., Maekawa, M., Yamaguchi, H., Mano, N., et al. (2017). Functional Characterization of 21 Allelic Variants of Dihydropyrimidinase. Biochem. Pharmacol. 143, 118–128. doi:10.1016/j.bcp.2017.06.121
Hishinuma, E., Gutiérrez Rico, E., and Hiratsuka, M. (2020). In Vitro Assessment of Fluoropyrimidine-Metabolizing Enzymes: Dihydropyrimidine Dehydrogenase, Dihydropyrimidinase, and β-Ureidopropionase. J. Clin. Med. 9 (8), 2342. doi:10.3390/jcm9082342
Hishinuma, E., Narita, Y., Saito, S., Maekawa, M., Akai, F., Nakanishi, Y., et al. (2018). Functional Characterization of 21 Allelic Variants of Dihydropyrimidine Dehydrogenase Identified in 1070 Japanese Individuals. Drug Metab. Dispos. 46 (8), 1083–1090. doi:10.1124/dmd.118.081737
Joerger, M., Huitema, A. D., Boot, H., Cats, A., Doodeman, V. D., Smits, P. H., et al. (2015). Germline TYMS Genotype Is Highly Predictive in Patients with Metastatic Gastrointestinal Malignancies Receiving Capecitabine-Based Chemotherapy. Cancer Chemother. Pharmacol. 75 (4), 763–772. doi:10.1007/s00280-015-2698-7
Khushman, M., Patel, G. K., Hosein, P. J., Laurini, J. A., Cameron, D., Clarkson, D. R., et al. (2018). Germline Pharmacogenomics of DPYD*9A (c.85T>C) Variant in Patients with Gastrointestinal Malignancies Treated with Fluoropyrimidines. J. Gastrointest. Oncol. 9 (3), 416–424. doi:10.21037/jgo.2018.02.03
Kikugawa, M., Kaneko, M., Fujimoto-Sakata, S., Maeda, M., Kawasaki, K., Takagi, T., et al. (1994). Purification, Characterization and Inhibition of Dihydropyrimidinase from Rat Liver. Eur. J. Biochem. 219 (1-2), 393–399. doi:10.1111/j.1432-1033.1994.tb19951.x
Kobuchi, S., and Ito, Y. (2020). Application of Pharmacometrics of 5-Fluorouracil to Personalized Medicine: A Tool for Predicting Pharmacokinetic-Pharmacodynamic/Toxicodynamic Responses. Anticancer Res. 40 (12), 6585–6597. doi:10.21873/anticanres.14683
Kuilenburg, A. B. P. V., Meijer, J., Tanck, M. W. T., Dobritzsch, D., Zoetekouw, L., Dekkers, L. L., et al. (2016). Phenotypic and Clinical Implications of Variants in the Dihydropyrimidine Dehydrogenase Gene. Biochim. Biophys. Acta 1862 (4), 754–762. doi:10.1016/j.bbadis.2016.01.009
Kunicka, T., Prochazka, P., Krus, I., Bendova, P., Protivova, M., Susova, S., et al. (2016). Molecular Profile of 5-fluorouracil Pathway Genes in Colorectal Carcinoma. BMC Cancer 16 (1), 795. doi:10.1186/s12885-016-2826-8
Kuriyama, S., Yaegashi, N., Nagami, F., Arai, T., Kawaguchi, Y., Osumi, N., et al. (2016). The Tohoku Medical Megabank Project: Design and Mission. J. Epidemiol. 26 (9), 493–511. doi:10.2188/jea.JE20150268
Lohkamp, B., Voevodskaya, N., Lindqvist, Y., and Dobritzsch, D. (2010). Insights into the Mechanism of Dihydropyrimidine Dehydrogenase from Site-Directed Mutagenesis Targeting the Active Site Loop and Redox Cofactor Coordination. Biochim. Biophys. Acta 1804 (12), 2198–2206. doi:10.1016/j.bbapap.2010.08.014
Lu, Z. H., Zhang, R., and Diasio, R. B. (1992). Purification and Characterization of Dihydropyrimidine Dehydrogenase from Human Liver. J. Biol. Chem. 267 (24), 17102–17109. doi:10.1016/s0021-9258(18)41899-6
Lunenburg, C. A. T. C., Henricks, L. M., Guchelaar, H. J., Swen, J. J., Deenen, M. J., Schellens, J. H. M., et al. (2016). Prospective DPYD Genotyping to Reduce the Risk of Fluoropyrimidine-Induced Severe Toxicity: Ready for Prime Time. Eur. J. Cancer 54, 40–48. doi:10.1016/j.ejca.2015.11.008
Lunenburg, C. A. T. C., van der Wouden, C. H., Nijenhuis, M., Crommentuijn-van Rhenen, M. H., de Boer-Veger, N. J., Buunk, A. M., et al. (2020). Dutch Pharmacogenetics Working Group (DPWG) Guideline for the Gene-Drug Interaction of DPYD and Fluoropyrimidines. Eur. J. Hum. Genet. 28 (4), 508–517. doi:10.1038/s41431-019-0540-0
Maekawa, K., Saeki, M., Saito, Y., Ozawa, S., Kurose, K., Kaniwa, N., et al. (2007). Genetic Variations and Haplotype Structures of the DPYD Gene Encoding Dihydropyrimidine Dehydrogenase in Japanese and Their Ethnic Differences. J. Hum. Genet. 52 (10), 804–819. doi:10.1007/s10038-007-0186-6
Maharjan, A. S., McMillin, G. A., Patel, G. K., Awan, S., Taylor, W. R., Pai, S., et al. (2019). The Prevalence of DPYD*9A(c.85T>C) Genotype and the Genotype-Phenotype Correlation in Patients with Gastrointestinal Malignancies Treated with Fluoropyrimidines: Updated Analysis. Clin. Colorectal Cancer 18 (3), e280–e286. doi:10.1016/j.clcc.2019.04.005
Meulendijks, D., Henricks, L. M., Sonke, G. S., Deenen, M. J., Froehlich, T. K., Amstutz, U., et al. (2015). Clinical Relevance of DPYD Variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as Predictors of Severe Fluoropyrimidine-Associated Toxicity: a Systematic Review and Meta-Analysis of Individual Patient Data. Lancet Oncol. 16 (16), 1639–1650. doi:10.1016/S1470-2045(15)00286-7
Minegishi, N., Nishijima, I., Nobukuni, T., Kudo, H., Ishida, N., Terakawa, T., et al. (2019). Biobank Establishment and Sample Management in the Tohoku Medical Megabank Project. Tohoku J. Exp. Med. 248 (1), 45–55. doi:10.1620/tjem.248.45
Nagami, F., Kuriki, M., Koreeda, S., Kageyama, M., Shimizu, O., Toda, S., et al. (2020). Public Relations and Communication Strategies in Construction of Large-Scale Cohorts and Biobank: Practice in the Tohoku Medical Megabank Project. Tohoku J. Exp. Med. 250 (4), 253–262. doi:10.1620/tjem.250.253
Nie, Q., Shrestha, S., Tapper, E. E., Trogstad-Isaacson, C. S., Bouchonville, K. J., Lee, A. M., et al. (2017). Quantitative Contribution of Rs75017182 to Dihydropyrimidine Dehydrogenase mRNA Splicing and Enzyme Activity. Clin. Pharmacol. Ther. 102 (4), 662–670. doi:10.1002/cpt.685
Offer, S. M., Fossum, C. C., Wegner, N. J., Stuflesser, A. J., Butterfield, G. L., and Diasio, R. B. (2014). Comparative Functional Analysis of DPYD Variants of Potential Clinical Relevance to Dihydropyrimidine Dehydrogenase Activity. Cancer Res. 74 (9), 2545–2554. doi:10.1158/0008-5472.CAN-13-2482
Offer, S. M., Wegner, N. J., Fossum, C., Wang, K., and Diasio, R. B. (2013). Phenotypic Profiling of DPYD Variations Relevant to 5-fluorouracil Sensitivity Using Real-Time Cellular Analysis and In Vitro Measurement of Enzyme Activity. Cancer Res. 73 (6), 1958–1968. doi:10.1158/0008-5472.CAN-12-3858
Roberto, M., Romiti, A., Botticelli, A., Mazzuca, F., Lionetto, L., Gentile, G., et al. (2017). Evaluation of 5-fluorouracil Degradation Rate and Pharmacogenetic Profiling to Predict Toxicity Following Adjuvant Capecitabine. Eur. J. Clin. Pharmacol. 73 (2), 157–164. doi:10.1007/s00228-016-2160-8
Schnackerz, K. D., Dobritzsch, D., Lindqvist, Y., and Cook, P. F. (2004). Dihydropyrimidine Dehydrogenase: a Flavoprotein with Four Iron-Sulfur Clusters. Biochim. Biophys. Acta 1701 (1-2), 61–74. doi:10.1016/j.bbapap.2004.06.009
Sistonen, J., Smith, C., Fu, Y. K., and Largiadèr, C. R. (2012). A New DPYD Genotyping Assay for Improving the Safety of 5-fluorouracil Therapy. Clin. Chim. Acta 414, 109–111. doi:10.1016/j.cca.2012.08.015
Tadaka, S., Hishinuma, E., Komaki, S., Motoike, I. N., Kawashima, J., Saigusa, D., et al. (2021). jMorp Updates in 2020: Large Enhancement of Multi-Omics Data Resources on the General Japanese Population. Nucleic Acids Res. 49 (D1), D536–d544. doi:10.1093/nar/gkaa1034
Thomas, F., Hennebelle, I., Delmas, C., Lochon, I., Dhelens, C., Garnier Tixidre, C., et al. (2016). Genotyping of a Family with a Novel Deleterious DPYD Mutation Supports the Pretherapeutic Screening of DPD Deficiency with Dihydrouracil/uracil Ratio. Clin. Pharmacol. Ther. 99 (2), 235–242. doi:10.1002/cpt.210
van Kuilenburg, A. B. (2004). Dihydropyrimidine Dehydrogenase and the Efficacy and Toxicity of 5-fluorouracil. Eur. J. Cancer 40 (7), 939–950. doi:10.1016/j.ejca.2003.12.004
van Kuilenburg, A. B., Meijer, J., Mul, A. N., Hennekam, R. C., Hoovers, J. M., de Die-Smulders, C. E., et al. (2009). Analysis of severely affected patients with dihydropyrimidine dehydrogenase deficiency reveals large intragenic rearrangements of DPYD and a De Novo interstitial deletion del(1)(p13.3p21.3). Hum. Genet. 125 (5-6), 581–590. doi:10.1007/s00439-009-0653-6
van Kuilenburg, A. B., van Lenthe, H., and van Gennip, A. H. (2006). Activity of Pyrimidine Degradation Enzymes in Normal Tissues. Nucleosides Nucleotides Nucleic Acids 25 (9-11), 1211–1214. doi:10.1080/15257770600894576
Van Kuilenburg, A. B., Vreken, P., Beex, L. V., Meinsma, R., Van Lenthe, H., De Abreu, R. A., et al. (1997). Heterozygosity for a Point Mutation in an Invariant Splice Donor Site of Dihydropyrimidine Dehydrogenase and Severe 5-fluorouracil Related Toxicity. Eur. J. Cancer 33 (13), 2258–2264. doi:10.1016/s0959-8049(97)00261-x
Vodenkova, S., Buchler, T., Cervena, K., Veskrnova, V., Vodicka, P., and au, V. (2020). 5-fluorouracil and Other Fluoropyrimidines in Colorectal Cancer: Past, Present and Future. Pharmacol. Ther. 206, 107447. doi:10.1016/j.pharmthera.2019.107447
Wigmore, P. M., Mustafa, S., El-Beltagy, M., Lyons, L., Umka, J., and Bennett, G. (2010). Effects of 5-FU. Adv. Exp. Med. Biol. 678, 157–164. doi:10.1007/978-1-4419-6306-2_20
Keywords: dihydropyrimidine dehydrogenase (DPD), DPYD, 5-fluorouracil, pharmacogenomics, genetic polymorphism
Citation: Hishinuma E, Narita Y, Obuchi K, Ueda A, Saito S, Tadaka S, Kinoshita K, Maekawa M, Mano N, Hirasawa N and Hiratsuka M (2022) Importance of Rare DPYD Genetic Polymorphisms for 5-Fluorouracil Therapy in the Japanese Population. Front. Pharmacol. 13:930470. doi: 10.3389/fphar.2022.930470
Received: 28 April 2022; Accepted: 30 May 2022;
Published: 15 June 2022.
Edited by:
Ryosuke Nakamura, National Institute of Health Sciences (NIHS), JapanReviewed by:
Haruhiko Sugimura, Hamamatsu University School of Medicine, JapanNuala Helsby, The University of Auckland, New Zealand
Copyright © 2022 Hishinuma, Narita, Obuchi, Ueda, Saito, Tadaka, Kinoshita, Maekawa, Mano, Hirasawa and Hiratsuka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masahiro Hiratsuka, bWFzYWhpcm8uaGlyYXRzdWthLmE4QHRvaG9rdS5hYy5qcA==