
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 17 June 2022
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.925880
This article is part of the Research Topic Ion Channel Structure and Drug Discovery Accelerated by Cryo-EM View all 11 articles
 Adam C. Oken1
Adam C. Oken1 Ipsita Krishnamurthy1Jonathan C. Savage1Nicolas E. Lisi1Michael H. Godsey1
Ipsita Krishnamurthy1Jonathan C. Savage1Nicolas E. Lisi1Michael H. Godsey1 Steven E. Mansoor1,2*
Steven E. Mansoor1,2*Extracellular ATP is a critical signaling molecule that is found in a wide range of concentrations across cellular environments. The family of nonselective cation channels that sense extracellular ATP, termed P2X receptors (P2XRs), is composed of seven subtypes (P2X1-P2X7) that assemble as functional homotrimeric and heterotrimeric ion channels. Each P2XR is activated by a distinct concentration of extracellular ATP, spanning from high nanomolar to low millimolar. P2XRs are implicated in a variety of physiological and pathophysiological processes in the cardiovascular, immune, and central nervous systems, corresponding to the spatiotemporal expression, regulation, and activation of each subtype. The therapeutic potential of P2XRs is an emerging area of research in which structural biology has seemingly exceeded medicinal chemistry, as there are several published P2XR structures but currently no FDA-approved drugs targeting these ion channels. Cryogenic electron microscopy is ideally suited to facilitate structure-based drug design for P2XRs by revealing and characterizing novel ligand-binding sites. This review covers structural elements in P2XRs including the extracellular orthosteric ATP-binding site, extracellular allosteric modulator sites, channel pore, and cytoplasmic substructures, with an emphasis on potential therapeutic ligand development.
Membrane proteins that recognize extracellular purine nucleotides, termed purinergic receptors, are grouped into three unique families: G-protein coupled P1 receptors that recognize adenosine, G-protein coupled P2Y receptors that recognize ADP and ATP, and ligand-gated P2X receptor (P2XR) ion channels that recognize ATP exclusively (Burnstock, 1976; van Calker et al., 1979; Londos et al., 1980; Webb et al., 1993; Brake et al., 1994; Valera et al., 1994). The seven P2XR subtypes, denoted P2X1-P2X7, are trimeric non-selective cation channels that are activated by distinct extracellular concentrations of ATP, a key signaling molecule released from cells in a broad range of physiological and pathophysiological states, from low concentrations during homeostasis to high concentrations in chronic inflammation or ischemia (Burnstock, 1972; Roman and Fitz, 1999; Taylor et al., 1999; Burnstock, 2004). Individual P2XR subtypes are activated by a wide range of distinct extracellular ATP (eATP) concentrations, from high nanomolar to low millimolar (Jarvis and Khakh, 2009; Illes et al., 2021). The binding of eATP to the receptor induces a conformational change, opening the ion channel and facilitating the influx of Na+ and Ca2+ ions and the efflux of K+ ions (Samways et al., 2014). The resultant net inward current following P2XR activation plays a significant role in downstream signaling and cellular function.
The functional and clinical significance of P2XR subtypes is related to both their individualized affinity to eATP and the cell types in which they are expressed. These include, but are not limited to, platelets (P2X1), smooth muscle cells (P2X1-7, predominantly P2X1), sensory neurons (P2X1-7, predominantly P2X3), epithelial cells (P2X4, P2X5, P2X6, P2X7), and immune cells (P2X4 and P2X7) (Burnstock and Kennedy, 2011). Accordingly, P2XRs are implicated in an array of pathological conditions. For instance, in animal models of Alzheimer’s disease (AD), pharmacologic inhibition or genetic depletion of P2X7 significantly improves the symptoms and neuropathology of AD (Francistiova et al., 2020). Several other examples of P2XR-implicated pathological conditions include: platelet aggregation (P2X1), hearing loss (P2X2), asthma (P2X3), vascular inflammation (P2X7), and cancer (P2X7) (Kamei et al., 2005; Baroni et al., 2007; Burnstock and Kennedy, 2011; Furlan-Freguia et al., 2011; Mahaut-Smith et al., 2011; North and Jarvis, 2013; Yan et al., 2013; Burnstock and Knight, 2018; Lara et al., 2020; Illes et al., 2021). As a result, P2XRs are an active area of therapeutic research with several subtype-specific antagonists currently in clinical trials to treat persistent cough (clinicaltrials.gov: NCT02502097), rheumatoid arthritis (clinicaltrials.gov: NCT00628095), and depression (clinicaltrials.gov: NCT04116606), among others (Kamei et al., 2005; Pfizer, 2008; Keystone et al., 2012; Abdulqawi et al., 2015; Afferent Pharmaceuticals, 2015; CCTU-Core et al., 2019; Richards et al., 2019).
P2XRs assemble as both homotrimeric and heterotrimeric channels. The known heterotrimeric assemblies include: P2X1/2, P2X1/4, P2X1/5, P2X2/3, P2X2/6, P2X4/5, P2X4/6, and P2X4/7 (Murrell-Lagnado and Qureshi, 2008). Very little is known about heterotrimeric composition, stoichiometry, and function. This is compounded by the increased pharmacologic complexity of heterotrimeric receptors, which poses an additional challenge to therapeutic targeting of P2XR activity. For example, heterotrimeric P2X2/3 has a ligand dose-response profile that is distinct from either homotrimeric P2X2 or P2X3 (Lewis et al., 1995). Currently, there are published structures for only three of seven homotrimeric subtypes and none for heterotrimeric receptors. The first P2XR structure, solved by X-ray crystallography, characterized the apo (unbound) closed state, defined the overall architecture of this receptor family, and confirmed three-fold symmetry for the homotrimeric assembly (Kawate et al., 2009). Subsequent crystallographic studies have identified the extracellular orthosteric ATP-binding site, revealed two distinct extracellular allosteric binding sites, and defined the molecular mechanisms of P2XR gating (Hattori and Gouaux, 2012; Karasawa and Kawate, 2016; Mansoor et al., 2016; Kasuya et al., 2017; Wang et al., 2018). While these crystallographic studies established a foundational understanding of P2XR structure and function, truncation of the cytoplasmic N- and C- termini (necessary for crystallization) limited the scope of our understanding of P2XR biology. The application of single-particle cryogenic electron microscopy (cryo-EM) to rat P2X7 overcame these limitations and provided visualization of the first full-length, wild-type P2XR, revealing novel features in the cytoplasmic domain that are essential for receptor function (McCarthy et al., 2019).
The “resolution revolution” in cryo-EM is advancing the ability of researchers to solve high-resolution protein structures (Kuhlbrandt, 2014). Compared to X-ray crystallography, cryo-EM requires less protein, tolerates more sample heterogeneity, and allows for the study of proteins in more native-like membrane environments, all of which are challenges in studying membrane protein structure (Su et al., 2021). Moreover, cryo-EM structures of wild-type proteins and protein complexes are consistently reaching sufficient resolutions to discover novel ligands and identify post-translational modifications (McCarthy et al., 2019; Flores et al., 2020; Nakane et al., 2020; Rahman et al., 2020). In turn, these new high-resolution structures of therapeutically relevant proteins have accelerated structure-based drug design (SBDD) efforts, which are significantly more efficient and generate higher-specificity ligands when compared to classical in vitro based assays (Lionta et al., 2014; Batool et al., 2019; Ballante et al., 2021; Ford et al., 2021; Lees et al., 2021). Cryo-EM is well suited to visualize novel receptor structures and receptor-ligand interactions for use in SBDD techniques, ultimately leading to more potent therapeutic compounds with fewer and less severe side effects for patients.
Careful examination of the available P2XR structures provides insights into the molecular pharmacology of this therapeutically relevant receptor family. This review highlights both known and potentially targetable sites within P2XRs that are critical to modulate receptor function.
The first P2XR structure to be solved, the apo closed state of a truncated zebrafish P2X4 (zfP2X4), defined the architecture of the extracellular and transmembrane regions for this receptor family (Kawate et al., 2009). Within each protomer of a trimeric receptor, the extracellular and transmembrane domains were imagined to resemble a breaching dolphin (Figure 1A) (Kawate et al., 2009). This overall architecture was confirmed to be conserved for apo closed states of the human P2X3 (hP2X3), giant panda P2X7 (pdP2X7), and rat P2X7 (rP2X7) receptors in successive structural studies (Karasawa and Kawate, 2016; Mansoor et al., 2016; McCarthy et al., 2019). Comparing the molecular architecture of several P2XRs reveals conserved sites that are targetable across this receptor family and highlights the structural differences across subtypes.
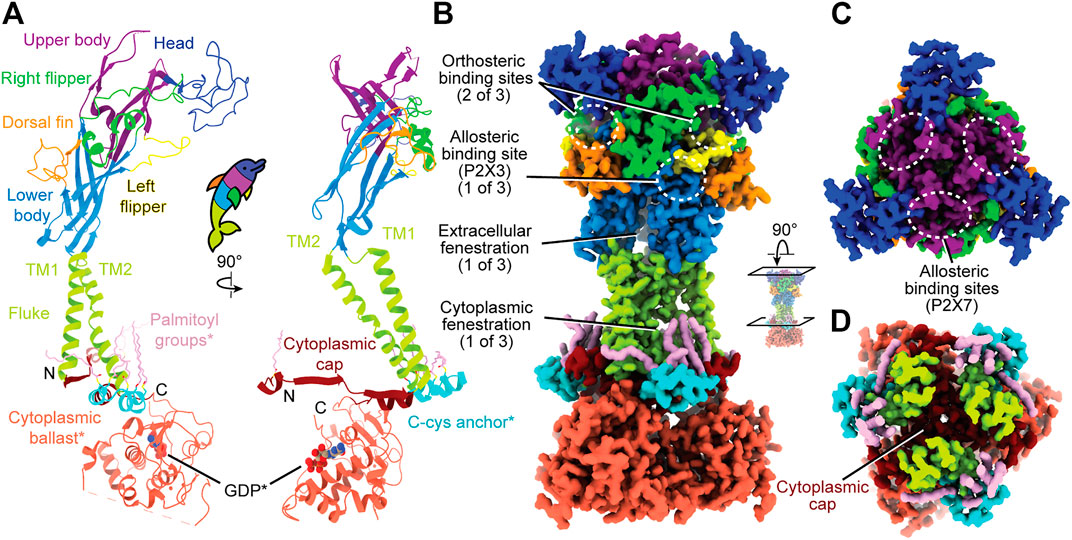
FIGURE 1. Overall, dolphin-resembling architecture of P2XRs exemplified by rP2X7 (PDB: 6U9V). (A) Left: Ribbon representation of one subunit of rP2X7 colored by domain architecture. Right: Orthogonal view colored by domain architecture. (B) Surface representation of the biological trimeric assembly of rP2X7 colored by domain architecture, viewed parallel to the membrane and with select structural elements labeled. The orthosteric ATP-binding site and the extracellular and cytoplasmic fenestrations are structurally conserved amongst P2XRs. The allosteric site found in hP2X3 is mapped to its respective spatial position on the presented P2X7 model. This allosteric site has not been confirmed to exist in P2X7, but the structural similarity amongst P2XRs allows visualization of its relative position. (C) Top-down view of the trimeric assembly of the rP2X7 extracellular domain colored by domain architecture, with denoted symmetry-related allosteric site confirmed in P2X7. This view highlights the three identical, symmetry-related, allosteric binding sites found on top of the extracellular domain of homotrimeric P2X7. (D) Biological trimeric assembly of rP2X7, colored by domain architecture, viewed perpendicular to the membrane from the center of the transmembrane domain into the cytoplasm. *Denotes structural elements found only in the P2X7 subtype.
The extracellular domain, which has been likened to a dolphin’s head and body, is composed of thirteen β-strands and four α-helices (Figure 1A). The β-sheets of the lower and upper body compose the central backbone of the extracellular domain of all P2XRs, with peripheral α-helices and loops variable between subtypes. Tucked within the loops, β-sheets, and α-helices of the extracellular domain are several ligand-binding pockets that modulate P2XR function. The orthosteric ATP-binding site, defined as the location of endogenous ligand (eATP) binding, has been structurally characterized for three P2XR subtypes: hP2X3, zfP2X4, and rP2X7 (Mansoor et al., 2016; Kawate et al., 2009; Hattori and Gouaux, 2012; McCarthy et al., 2019). This site is found in all P2XRs at an interface formed by the head, left flipper, and upper body of one protomer and the lower body of the neighboring protomer (Figures 1A,B). Thus, a functional trimeric P2XR has three symmetry-related orthosteric binding sites. However, it is generally accepted that the occupation of all three of these binding sites by ATP is not necessary to activate the channel (Friel and Bean, 1988; Coddou et al., 2011; Kawate, 2017).
In addition, extracellular allosteric binding sites—distinct from the orthosteric binding site—have been structurally confirmed in hP2X3 and pdP2X7 and are predicted in other P2XRs (Figures 1B,C) (Ase et al., 2019; Bidula et al., 2022; Karasawa and Kawate, 2016; Obrecht et al., 2019; Wang et al., 2018). One structurally confirmed allosteric site in hP2X3 is found towards the base of the extracellular domain, formed by the left flipper of one subunit and the lower body and dorsal fin of another (Figure 1B) (Wang et al., 2018). Due to symmetry, there are three such allosteric sites in homotrimeric hP2X3 (Figure 1B). Another allosteric site, structurally confirmed in pdP2X7, is located on top of the extracellular domain and is formed by the upper body between neighboring protomers (Figure 1C) (Karasawa and Kawate, 2016). Similarly, due to symmetry, there are three such allosteric sites in homotrimeric pdP2X7 (Figure 1C). Ligand binding at allosteric sites modulates protein activity through structural rearrangements, resulting in either inhibited (negative allostery) or enhanced (positive allostery) orthosteric ligand binding. Negative allosteric modulators are non-competitive antagonists as they do not compete with orthosteric ligand binding. The orthosteric and two distinct allosteric sites are critical targets to consider when developing potential therapeutic ligands that modulate P2XR activity.
The transmembrane (TM) domain, the dolphin’s tail, is composed of two α-helices (the peripheral TM1 and internal, pore-lining TM2) that span the membrane, connecting the extracellular and cytoplasmic domains (Figures 1A,B). Together, the six TM helices of the trimeric receptor form the pore and undergo distinct conformational rearrangements upon ATP binding (Li et al., 2010). The three conformational states of the gating cycle include the apo closed state, the ATP-bound open state, and the ATP-bound desensitized (closed) state (Mansoor et al., 2016). Desensitization refers to progressive pore closure during sustained agonist binding, which has been thoroughly reviewed for P2XRs (Werner et al., 1996; North, 2002; Kawate, 2017; Mansoor, 2022, in press). The rearrangements of the pore during the gating cycle are described in the helical recoil model of receptor desensitization, first proposed from the published structures of hP2X3 (Mansoor et al., 2016). The P2X7 subtype is the only P2XR that does not undergo desensitization. Currently, there are no structures characterizing P2XR pore-modulating ligands, but such a class of antagonists would have therapeutic potential for modulating P2XR function.
Not much is known about the structure of cytoplasmic domains for the majority of P2XR subtypes. However, the hP2X3 ATP-bound open state structure and the full-length rP2X7 structures highlight aspects of intracellular elements (Mansoor et al., 2016; McCarthy et al., 2019). The first segment of the cytoplasmic domain was elucidated by the hP2X3 ATP-bound open state structure, which revealed the cytoplasmic ends of the TM helices are flanked by a substructure termed the “cytoplasmic cap” (Figures 1A,D) (Mansoor et al., 2016). The cytoplasmic cap is a product of domain swapping, composed of intertwining β-strands from all three protomers knitted together to form β-sheets that run parallel to the membrane. The stability of the cytoplasmic cap plays a pivotal role in the gating cycle of distinct subtypes by setting the rate of desensitization (Mansoor et al., 2016). Two additional cytoplasmic structural features unique to P2X7 were revealed by cryo-EM, a cysteine rich domain termed the “C-cys anchor“ and a ∼200 residue C-terminal domain termed the “cytoplasmic ballast” (Figures 1A,B) (McCarthy et al., 2019). Further elucidation of P2XR cytoplasmic structural diversity by cryo-EM will provide invaluable information about receptor biology.
The array of P2XR structures to date have supplemented our understanding of P2XR function and defined orthosteric and allosteric ligand binding, the path of ion flow through the pore, and the helical recoil model of desensitization. The recent discoveries of the previously unknown cytoplasmic elements of rP2X7, facilitated by cryo-EM, have raised new questions regarding its metabotropic signaling properties (Surprenant et al., 1996; Wilson et al., 2002; Cheewatrakoolpong et al., 2005; Adinolfi et al., 2010; Costa-Junior et al., 2011; McCarthy et al., 2019). The nuances of the structural elements contained within the P2XR architecture are discussed below, focusing on potential strategies to target them therapeutically with ligands.
Protein sequence alignments of human P2XRs reveal these receptors share ∼35–53% sequence identity between any two subtypes (Figure 2). P2XRs vary in length by up to 207 amino acids per protomer, with P2X7 having the longest sequence and P2X4 having the shortest (595 and 388 amino acids per protomer, respectively). The most significant variations between P2XR sequences lay in their cytoplasmic termini, which are known to play a critical role in receptor desensitization, trafficking, and signaling (Brandle et al., 1997; Koshimizu et al., 1999; Boue-Grabot et al., 2000; North, 2002; Chaumont et al., 2004; Jarvis and Khakh, 2009; Hausmann et al., 2014; Mansoor et al., 2016; Sattler and Benndorf, 2022). The N-termini vary in length by up to 18 amino acids, with P2X2 having the longest N-terminus at 20 residues before the first α-helix of the cytoplasmic cap and P2X3 having the shortest at only two residues. The C-termini vary in length by up to 191 amino acids after the last β-strand in the cytoplasmic cap, with P2X7 having the longest C-terminus at 203 residues and P2X4 having the shortest at 12 residues. Despite significant sequence similarity between P2XRs, subtype-specific sequence differences confer distinct receptor functionality. As our interests lie in human specific drug design, unless stated otherwise, all residues discussed will be from the register of human orthologs.

FIGURE 2. Protein sequence alignment of the seven human P2XR subtypes, P2X1-P2X7. Amino acids are shown in grayscale by level of sequence conservation across human subtypes as calculated with Alscript (Barton, 1993) (white text on black background denotes full sequence conservation). Residues with direct ligand interactions are highlighted, with the orthosteric ATP-binding site residues in yellow and the residues of the two distinct allosteric sites in pink and blue. These residues are highlighted only for subtypes for which there are published structures. Secondary structure (SS) is denoted in the top row and is colored by domain architecture corresponding to the color scheme in Figure 1. The break at the end of the sequence alignment condenses the divergent cytoplasmic domains. The ancestral difference in TM2 of hP2X5, as described in the text, is indicated with lower case letters. The sequence alignment was generated in Clustal Omega and the figure created using Aline (Bond and Schuttelkopf, 2009; Sievers and Higgins, 2018). Sequences were obtained from the UniProt database with accession numbers P51575, Q9UBL9, P56373, Q99571, Q93086, O15547, and Q99572 for hP2X1-hP2X7, respectively.
There is no published structure of P2X5 but this subtype is known to be expressed in humans predominantly as a non-functional isoform (Kotnis et al., 2010). A single-nucleotide polymorphism leads to the canonical isoform which lacks 22 residues (328–349) encoded by exon 10, including the N-terminal (outer leaflet) portion of TM2 (Figure 2) (Le et al., 1997). Without a significant portion of TM2, this isoform is prone to subunit aggregation (Duckwitz et al., 2006). Restoration of the amino acids encoded by exon 10 is found to re-establish P2X5 function, producing strong currents in response to ATP (Bo et al., 2003). While, in humans, the allele encoding the non-functional protein is predominant, alleles encoding full-length, functional P2X5 have also been reported (Kotnis et al., 2010). Notably, the full-length, functional P2X5 isoform predominates in other species. Structures of the truncated and full-length isoforms might provide insight into the evolution of this P2XR subtype and the structural basis for the corresponding pharmacological effects.
While the sequence of hP2X6 appears to be quite similar to other P2XRs, this subtype lacks nine residues that compose the main portion of the left flipper, a key element in the activation of P2XRs (Figure 2) (Wang et al., 2017; Zhao et al., 2014; Li et al., 2010; Kawate, 2017; Jiang et al., 2012). These missing residues might explain why P2X6 homotrimers do not produce currents in response to ATP (Soto et al., 1996; Le et al., 1998; King et al., 2000). However, all residues that are known to coordinate ATP in other P2X subtypes are conserved within hP2X6 (Figure 2). This suggests that while ATP may be able to bind in the orthosteric binding site of hP2X6, the lack of a functional left flipper may be the cause of this subtype’s inability to transition to an ATP-bound open state. The functional significance of hP2X6 may be found in its ability to form heterotrimers, thereby expanding the pharmacological complexity of purinergic signaling (Le et al., 1998; King et al., 2000; Antonio et al., 2014).
Easily seen in the sequence alignment, P2X7 has several features that are specific to this subtype, including the C-cys anchor (residues 360–377) and its uniquely large cytoplasmic domain, referred to as the cytoplasmic ballast (Figure 2) (McCarthy et al., 2019). Post-translational palmitoylation of residues on the C-cys anchor explains the distinct ability of P2X7 to remain open without undergoing desensitization (McCarthy et al., 2019). Mapping the unique features of P2X7 from sequence to structure demonstrates the importance of resolving the cytoplasmic domains for each of the other P2XRs.
It is crucial to elucidate how ATP interacts with the orthosteric binding site in order to understand the mechanism and modulation of P2XR activation. One approach to inhibit P2XR function is to develop competitive antagonists that target the orthosteric ATP-binding site (Figure 1B). The residues that compose the orthosteric pocket are positively charged, hydrophilic, and highly conserved across all receptor subtypes (Figures 2, 3A,B) (Chataigneau et al., 2013). Similarity of the orthosteric site across P2XRs might impede the development of subtype-selective competitive antagonists. However, structural studies can provide crucial insights into the subtle differences in molecular pharmacology between P2XR subtypes at the orthosteric site.
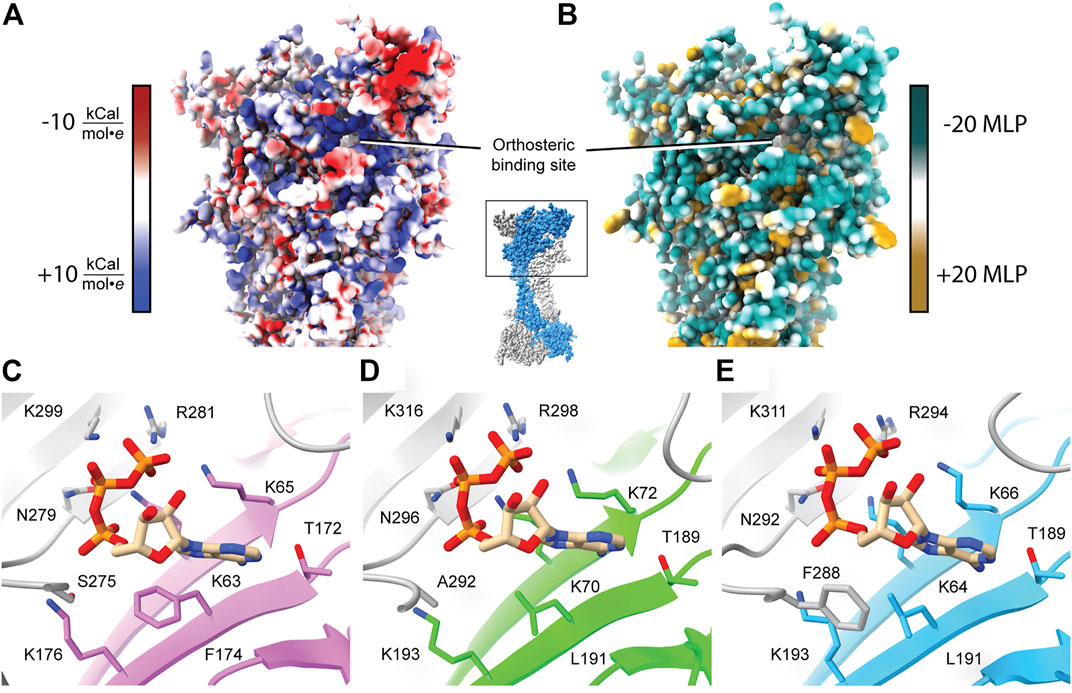
FIGURE 3. Characteristics of the orthosteric binding site of hP2X3, zfP2X4, and rP2X7. (A) Electrostatic surface rendering of rP2X7 (PDB: 6U9V) with blue and red coloring representing positive and negative electrostatic potential, respectively, highlighting the electropositive nature of the P2XR orthosteric ATP-binding site. (B) Hydrophobic surface rendering of rP2X7 (PDB: 6U9V) with hydrophobic regions colored brown and hydrophilic regions colored turquoise, each defined by positive and negative molecular lipophilicity potential (MLP), respectively (Laguerre et al., 1997). This rendering highlights the hydrophilic nature of the orthosteric ATP-binding pocket. (C–E) Conserved residues involved in the coordination of ATP in the orthosteric binding pocket across structurally characterized P2XRs. Panels are annotated with ortholog-specific residue numbering. Orthosteric site of (C) the ATP-bound open state hP2X3 structure (light purple, PDB: 5SVK), (D) the ATP-bound open state zfP2X4 structure (green, PDB: 4DW1), and (E) the ATP-bound open state rP2X7 structure (blue, PDB: 6U9W). All bound ATP molecules occupy a nearly identical U-shaped pose. The MLP and electrostatic surface potential were calculated in ChimeraX (Pettersen et al., 2021).
The published structures of hP2X3, zfP2X4, and rP2X7 receptors in an ATP-bound open state define the orthosteric binding site (Figure 3). A close look at these structures reveals that ATP occupies a virtually identical U-shaped pose and the principal residues responsible for ATP coordination are highly conserved: four lysines, a threonine, an asparagine, and an arginine (Figures 2, 3C–E) (Hattori and Gouaux, 2012; Mansoor et al., 2016; McCarthy et al., 2019). Despite this conservation, the molecular pharmacology of P2XR activation by ATP is remarkably variable across subtypes. For example, P2X7 requires ∼200-fold higher concentration of eATP than P2X3 for activation (Jarvis and Khakh, 2009; Illes et al., 2021). A comparison of the ATP-bound open state structures of hP2X3, zfP2X4, and rP2X7 reveals there are differences in only two of the residues that directly coordinate ATP. The first difference is a hydrophobic residue that interacts with the adenosine base (F174, L191, and L191 in hP2X3, zfP2X4, and rP2X7, respectively) (Figures 2, 3). The second variable residue is on a loop within the left flipper which differs between a serine (S275) in hP2X3 that interacts with the α-phosphate of ATP, an alanine (A292) in zfP2X4 that makes no contacts to ATP, or a phenylalanine (F288) in rP2X7 that interacts with C5 on the ribose of ATP. These minor variations in the principal residues of the orthosteric binding site between subtypes seem unlikely to fully explain the dramatic differences in ATP sensitivity, suggesting there are other factors at play.
A kinetic limitation to ATP-binding may be one such factor contributing to differential ATP sensitivities between P2XR subtypes. Solvent accessibility to the orthosteric binding site in the apo closed state structures of hP2X3, zfP2X4, and rP2X7 are strikingly different (Figure 4) (Stank et al., 2016; McCarthy et al., 2019). The surface-accessible volume of the orthosteric binding site in rP2X7 is approximately 44% smaller than in hP2X3 and 32% smaller than in zfP2X4 (Figure 4) (Tian et al., 2018). The effects of variable accessibility can perhaps be explained by small side-chain fluctuations and backbone or interdomain vibrational movements, referred to as “pocket breathing” (Ferrari et al., 2003; Stank et al., 2016). Local protein dynamics and flexibility would affect the kinetics of ligand binding by restricting pocket accessibility, thereby requiring higher concentrations of ATP to activate P2X7. The identity of the coordinating residues and the accessibility of the orthosteric site are valuable insights gained from structural analysis, both of which are necessary considerations during the design of subtype-selective antagonists.
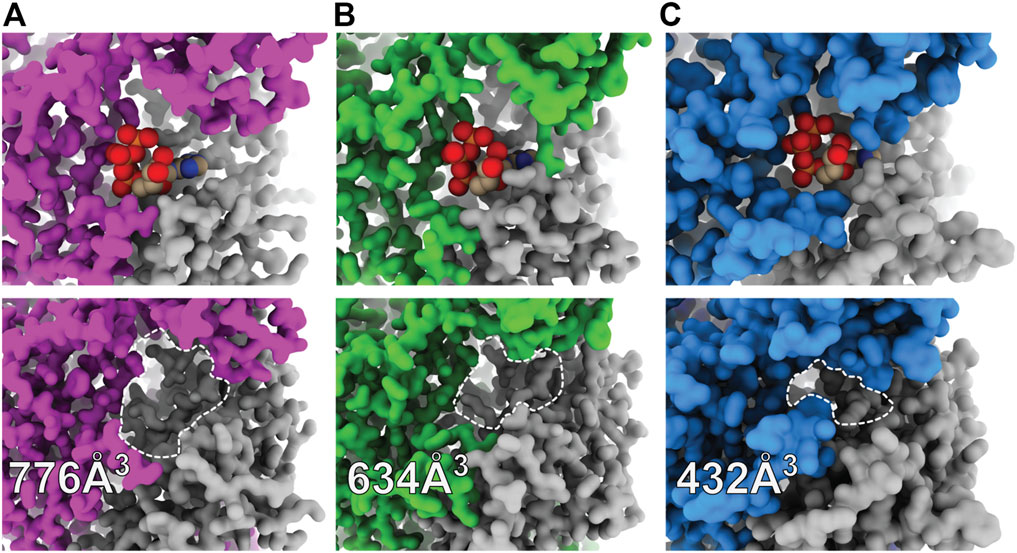
FIGURE 4. Surface representations of orthosteric ATP-binding pockets highlight differential ligand accessibility between P2XR subtypes. (A) ATP-bound open (top) and apo closed (bottom) states of hP2X3 (light purple, PDBs: 5SVK and 5SVJ) reveal the highly accessible pocket. (B) ATP-bound open (top) and apo closed (bottom) states of zfP2X4 (green, PDBs: 4DW1 and 4DW0) reveal a slightly less accessible pocket. (C) ATP-bound open (top) and apo closed (bottom) states of rP2X7 (blue, PDBs: 6U9W and 6U9V) reveal the least accessible pocket. The approximate binding pockets are outlined with dashed lines for visual clarity. Reported solvent accessible pocket volumes of the orthosteric ATP-binding site in the apo states of hP2X3, zfP2X4, and rP2X7 were calculated with CASTp (using a 2.0 Å probe radius) to be 776, 634, and 432 Å3, respectively (Tian et al., 2018).
The effect that divalent cations have on the activity of select P2XRs is another consideration for the difference in ATP sensitivities between subtypes. The presence of divalent cations in the extracellular environment shifts the activation requirements for P2X2, P2X4, and P2X7 to higher concentrations of ATP (Garcia-Guzman et al., 1997; Virginio et al., 1997; Li et al., 2013). In P2X2, the affinity and efficacy of MgATP2- is lower than that of free ATP, suggesting Mg2+ acts as an inhibitor and constrains receptor function. In contrast, MgATP2- effectively agonizes P2X1 and P2X3 at similar concentrations to free ATP (Li et al., 2013). The crystal structures of the apo closed state and ATP-bound open state of hP2X3 confirm that Mg2+ binds in two distinct conformations at an acidic chamber near the orthosteric binding site (Mansoor et al., 2016; Li et al., 2019). Functional data for hP2X3 suggest that Mg2+ slows ATP release and receptor recovery from desensitization (Li et al., 2019). However, direct equilibrium binding data demonstrate the presence of Mg2+ does not influence the affinity of hP2X3 for ATP (Mansoor et al., 2016). The subtype-specific effects of divalent cations on P2XR activation and gating need to be investigated further.
The information gained from the few known competitive antagonists that target P2XRs provides crucial insight into receptor modulation (Wolf et al., 2011; North and Jarvis, 2013; Muller, 2015; Muller and Namasivayam, 2021; Illes et al., 2021). The two structures of competitive antagonists bound to hP2X3 (TNP-ATP and A-317491) reveal these high-affinity antagonists bind at the orthosteric binding site in a Y-shaped pose, distinct from the U-shaped pose of ATP (Figures 5A,B) (Mansoor et al., 2016). This shift in ligand pose allows for deeper penetration into the binding pocket and is postulated to prevent the conformational changes necessary for channel opening (Mansoor et al., 2016). Residues D158 and F174 (hP2X3) are theorized to confer high specificity of TNP-ATP for hP2X3 and hP2X1, as this pair of key residues is only conserved in these two P2XR subtypes (Figure 2) (Mansoor et al., 2016). TNP-ATP occupies a different, extended conformational pose in the orthosteric binding site of ckP2X7, distinct from both the U-shaped pose of ATP in rP2X7 and Y-shaped pose of TNP-ATP in hP2X3 (Figures 5B,C) (Kasuya et al., 2017). The phosphate groups of TNP-ATP in ckP2X7 are stabilized in this extended conformation by hydrogen bond interactions from residues K236 and K298. Notably, while TNP-ATP inhibits P2X1 and P2X3 with low nanomolar affinity, hundreds of micromolar of TNP-ATP are required to antagonize P2X7 (Virginio et al., 1998). This drastic difference in apparent affinity and the distinct binding poses of TNP-ATP between hP2X3 and ckP2X7 may be correlated. The multitude of poses occupied by ATP, TNP-ATP, and A-317491 reveal the targetable space within the orthosteric pocket, providing knowledge that should be used in development of general and subtype-specific competitive antagonists. These structures reveal the importance of subtype-specific residues and pocket accessibility as critical factors for consideration in SBDD.
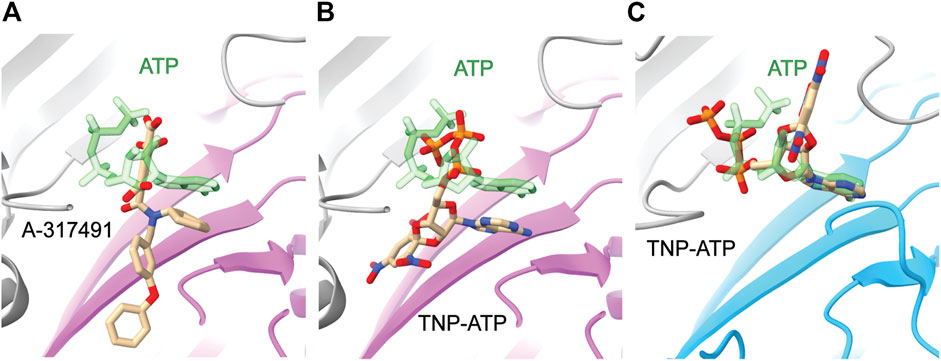
FIGURE 5. Comparative ligand poses of competitive antagonist-bound closed state and ATP-bound open state P2XR structures. (A) The Y-shaped pose of competitive antagonist A-317491 (tan) bound to hP2X3 contrasts with the U-shaped pose of ATP (green) bound to hP2X3 (purple). Superimposed PDBs: 5SVR and 5SVK. (B) The Y-shaped pose of competitive antagonist TNP-ATP (tan) bound to hP2X3 contrasts with the U-shaped pose of ATP (green) bound to hP2X3 (purple). Superimposed PDBs: 5SVQ and 5SVK. (C) The extended pose of competitive antagonist TNP-ATP (tan) bound to ckP2X7 contrasts with the U-shaped pose of ATP (green) bound to rP2X7 (blue). Superimposed PDBs: 5XW6 and 6U9W.
Identification of allosteric binding sites in P2XRs is crucial for the development of novel therapeutic ligands (Muller, 2015; Muller and Namasivayam, 2021). These sites bind secondary ligands non-competitively, promoting structural conformations that enhance or suppress orthosteric ligand binding. From a therapeutic perspective, allosteric modulators are advantageous as they often have fewer side effects compared to orthosteric ligands due to higher receptor specificity, resulting in less disruption of other functional pathways (Coddou et al., 2011; Nussinov and Tsai, 2012; Wenthur et al., 2014; Muller, 2015; Changeux and Christopoulos, 2016; Wang et al., 2018). This is particularly important when targeting a protein whose orthosteric ligand is ATP, a common substrate for proteins involved in metabolism and neurotransmission. Until recently, all known allosteric modulators against P2XRs were non-natural synthetic ligands. Current data now indicate bilirubin is an endogenous ligand that acts as a negative allosteric modulator of P2X7, suggesting the possibility for native cellular mechanisms of P2XR allosteric regulation (Zhao et al., 2021). It would be intriguing to discover other endogenous ligands that modulate P2XRs, determine their role in physiological or pathophysiological states, and adapt their chemical scaffold for the development of novel ligands. In support of the proposed therapeutic value of the P2XR allosteric sites, there are numerous small-molecule antagonists of P2XRs in various phases of clinical trials (Kamei et al., 2005; Pfizer, 2008; Keystone et al., 2012; Abdulqawi et al., 2015; Afferent Pharmaceuticals, 2015; CCTU-Core et al., 2019; Richards et al., 2019).
There are currently two distinct allosteric binding sites within P2XRs that have been confirmed with high-resolution structures, one near the orthosteric binding site and another on the top of the extracellular domain, visualized in hP2X3 and pdP2X7, respectively (Figures 1B,C) (Karasawa and Kawate, 2016; Wang et al., 2018). Electrophysiological and biochemical studies additionally show allosteric sites exist in P2X1 and P2X4 (Muller, 2015; Ase et al., 2019; Obrecht et al., 2019; Illes et al., 2021; Muller and Namasivayam, 2021; Bidula et al., 2022). Sequence analysis further suggests the remaining P2XRs also contain allosteric sites (Figure 2). Since each distinct allosteric site exists at the interface of two protomers, it is unclear to what extent similar allosteric sites exist within heterotrimeric receptors where the interface between subtypes might be markedly different. Examination of the available P2XR structures in complex with allosteric modulators provides pivotal insights into the regulation of P2XRs.
One of the visualized allosteric sites in P2XRs was revealed in the structure of hP2X3 bound to the nanomolar-affinity negative allosteric modulator gefapixant (Wang et al., 2018). This site is composed of residues at a protomer interface formed by the left flipper of one subunit and the lower body and dorsal fin of another (Figures 1A,B, 6A,B). This allosteric site is located on the opposite side of the left flipper as the orthosteric binding site. Movement of the left flipper is an essential step in the transition to an ATP-bound open state (Wang et al., 2017; Zhao et al., 2014; Li et al., 2010; Kawate, 2017; Jiang et al., 2012). Negative allosteric modulators that bind this site, including gefapixant, restrict left flipper mobility and thereby inhibit channel opening. Analysis of the gefapixant-bound hP2X3 structure indicates that residue K176, which is fully conserved across all human P2XRs, is of particular importance (Figure 2). It is the only residue in common between this allosteric site and the orthosteric ATP-binding site, interacting with the two oxygens of the sulfonyl nitrene moiety of gefapixant or with an oxygen on the α-phosphate of ATP (Figures 3C, 6B). More biochemical experimentation is required to determine if both ligands can occupy their respective binding sites simultaneously. Several other residues interacting with gefapixant in hP2X3 are of note; N190 is fully conserved and L191, V238, and L265 display hydrophobicity across all P2XR subtypes (Figures 2, 6B). Interestingly, all residues in this allosteric site (except V61 and S267) are conserved between hP2X3 and hP2X1, suggesting this allosteric site is present and quite similar in both subtypes. To support this idea, the ligand aurintricarboxylic acid (ATA) is a high-affinity negative allosteric modulator of hP2X1 and hP2X3 (proposed to bind the same site as gefapixant), but does not effectively antagonize other subtypes (Obrecht et al., 2019). The characterization of this allosteric site within P2X3 (and potentially P2X1) provides a structural template for the development of P2XR subtype-specific antagonists to this novel site.
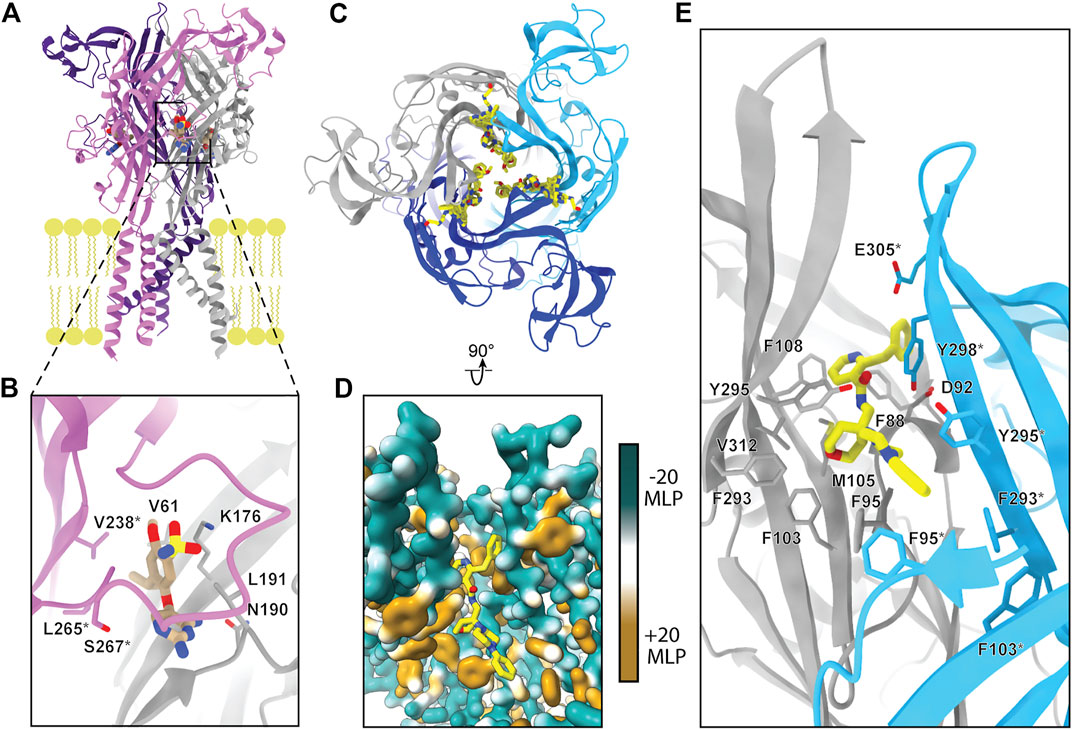
FIGURE 6. The two known allosteric sites of P2XRs. (A) Ribbon representation of the negative allosteric modulator gefapixant (tan) bound to hP2X3 (PDB: 5YVE). (B) Magnified view of panel A showing gefapixant interacting with residues at the interface of the left flipper of one protomer (purple) and lower body of another (grey) in hP2X3. (C) Top-down view of five superimposed negative allosteric modulators (yellow) bound to a ribbon representation of pdP2X7, highlighting the nearly identical binding mode of the chemically distinct ligands (PDBs: 5U1W, 5U1X, 5U1Y, 5U1V, and 5U1U). (D) Magnified and rotated view of panel C showing one of the five negative allosteric modulators (JNJ-47965567) bound to the hydrophobic pocket of pdP2X7 (PDB: 5U1X). Hydrophobic surface rendering of pdP2X7 with hydrophobic regions colored brown and hydrophilic regions colored turquoise, each defined by positive and negative molecular lipophilicity potential (MLP), respectively (Laguerre et al., 1997). (E) Ribbon representation of panel D with negative allosteric modulator JNJ-47965567 bound to pdP2X7 highlighting interacting residues (PDB: 5U1X). *Demarcating residues in the purple and blue protomers in panel B and in panel E, respectively. The MLP was calculated in ChimeraX (Pettersen et al., 2021).
Crystallographic structures of five unique negative allosteric modulators bound to pdP2X7 provide a view of another allosteric site present in P2XRs (Karasawa and Kawate, 2016). Located on the top of the extracellular domain, ligands bound at this allosteric site prevent a conformational rearrangement required for all P2XRs to undergo transition to an ATP-bound open state (Figures 1C, 6C) (Karasawa and Kawate, 2016; Kawate, 2017). This binding pocket is generally hydrophobic and the entrance is lined by charged residues (Figures 6D,E). The five negative allosteric modulators structurally characterized in complex with pdP2X7, despite varying in size, share a common scaffold that generally complements the properties of the pocket, with a mostly hydrophobic tail and a hydrophilic head connected through a narrow, nitrogenous linker (Figure 6D) (Karasawa and Kawate, 2016). The residues at this allosteric binding pocket of pdP2X7 are similar or conserved in hP2X7 and across other human P2XRs, suggesting this site may be present in other subtypes. In support of this, the proposed negative allosteric modulator 5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro [3,2-e]-1,4-diazepin-2-one (5-BDBD) is reported to bind to P2X4 at the equivalent allosteric site (Bidula et al., 2022). Altogether, the structures of allosteric modulators bound to pdP2X7 and the biochemical experiments on P2X4 suggest this site is a promising target for the development of novel high-affinity and subtype-specific P2XR antagonists by SBDD.
As the primary function of P2XRs is to serve as non-selective cation channels in response to activation by eATP, another approach to inhibit ion channel function is by obstructing the ion path through the protein. The pore through the transmembrane region of P2XRs is formed by a bundle of TM helices in a structure similar to those found in acid-sensing ion channels (ASICs) and epithelial Na+ channels (ENaCs) (Noreng et al., 2018; Jasti et al., 2007; Baconguis et al., 2014). Ions flow through lateral, phospholipid-lined fenestrations into vestibules on both sides of the TM helices (Figures 1, 7, 8) (Samways et al., 2011; Samways et al., 2014). To validate the ion flow path, hP2X3 was crystallized in the presence of CsCl and probed for the location of Cs+ ions by anomalous signal (Mansoor et al., 2016). Glutamate E46 was found to coordinate a Cs+ ion at the entrance to each of the three extracellular fenestrations, indicating this residue similarly coordinates Na+ ions under physiological conditions (Figure 7B). The residue at this position is a glutamate or aspartate in four of the human subtypes (P2X1, P2X3, P2X4, and P2X7), but a glutamine in hP2X2 and a lysine in hP2X5 and hP2X6 (Figure 2). In hP2X2, hP2X5, and hP2X6, this ion-coordinating role in the fenestrations is likely fulfilled by a nearby acidic residue on an alternate protomer (E69, D57, or E66, respectively). This conserved charge at the lateral extracellular fenestrations could be targeted by designing ligands that prevent ion entry, regardless of whether the pore is open or closed. Such a ligand would need to selectively bind and block each extracellular fenestration, which are large surface domains with substantial void space in the ATP-bound open state (Figures 1B, 7A). This may be one instance where P2XR modulation is best achieved by a larger biologic therapeutic, such as an antibody or an aptamer (Nelson et al., 2010; Zhou and Rossi, 2017). These structured protein and nucleic acid affinity reagents are ideally suited for targeting large extracellular epitopes, where accessing small binding pockets is not necessary and diffusion across the cell membrane is not desired.
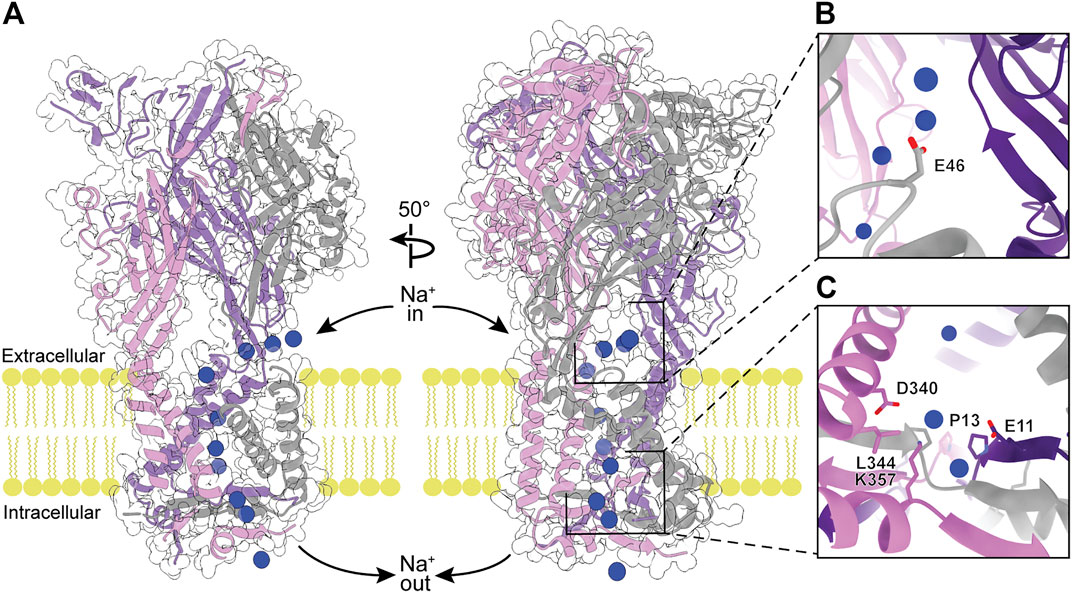
FIGURE 7. The modeled flow of Na+ ions through the ATP-bound open state hP2X3 structure (PDB: 5SVK). (A) Molecular dynamic simulations show Na+ ions (blue spheres) enter the receptor through the extracellular fenestrations, pass through the extracellular vestibule and the pore, and enter the cell via the cytoplasmic fenestrations (Mansoor et al., 2016). Left: View highlighting the flow of Na+ ions through the pore. Right: 50o rotated view highlighting the flow of Na+ ions through extracellular and cytoplasmic fenestrations. (B) Magnified view of Na+-coordinating residue (E46) within the extracellular fenestration. (C) Magnified view of key residues in the cytoplasmic fenestration. Mutations T13P/S15V/V16I were needed to slow the rate of desensitization and capture the ATP-bound open state hP2X3 crystal structure (Hausmann et al., 2014).
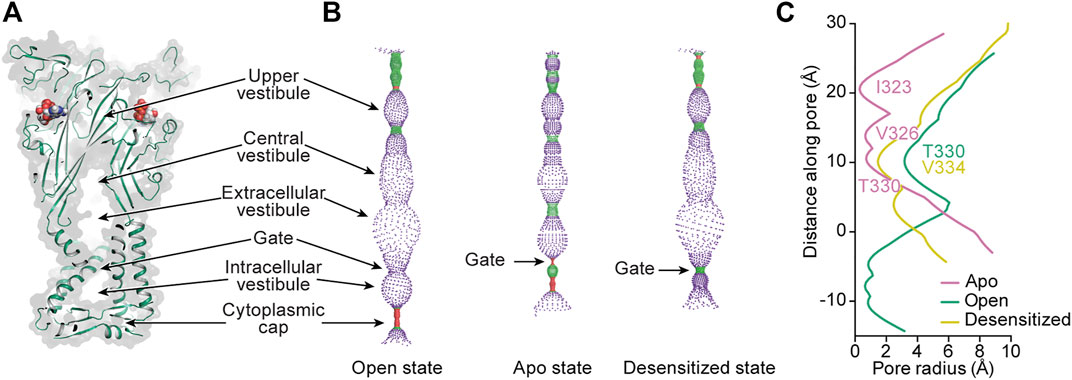
FIGURE 8. Mapping the interior space of the hP2X3 pore in different conformational states. (A) A coronal section of a surface representation of the ATP-bound open state of hP2X3 (PDB: 5SVK) reveals the upper, central, extracellular, and intracellular vestibules.(B) Pore-lining surface along the axis of hP2X3 for the ATP-bound open, apo closed, and ATP-bound desensitized states (PDBs: 5SVK, 5SVJ, and 5SVL, respectively), calculated by the program HOLE: red <1.15 Å radius, green between 1.15–2.30 Å radius, and purple >2.30 Å radius (Smart et al., 1996). (C) Plot of pore radius as a function of distance along the pore axis for the ATP-bound open, apo closed, and ATP-bound desensitized states of hP2X3. The positions of the residues making up the narrowest radius in each conformational state are labeled. The Cα position of I341 in hP2X3 is set as zero. This figure was from adapted from Mansoor et al. (2016).
Ions travel from one of the extracellular fenestrations to the extracellular/central vestibule lined by the first β-strand after TM1 and the last β-strand before TM2 (Figures 1A, 8A,B). Through molecular modeling and mutagenesis studies, this large vestibule has been found to be a binding site for ginsenoside (“steroid-like” dammarane triterpenoid glycosides) allosteric modulators of hP2X7 (Bidula et al., 2019). Simulations suggest these molecules bind in the upper part of this vestibule (central vestibule), making key interactions to the β-strand immediately preceding TM2. While no structures of ginsenoside molecules bound to P2XRs exist at this time, this site may prove to be an attractive target for new allosteric modulators due to its location near the transmembrane pore. The upper vestibule is also the location of a Zn2+-binding site seen in the tick P2X structure (AmP2X) (Kasuya et al., 2016). Of the human subtypes, only hP2X4 has residues (Q94, E95) similar to the Zn2+-coordinating residues (E105, E106) of AmP2X (Figure 2). The activity of hP2X4 has been shown to be modulated by Zn2+, an interesting consideration for development of P2X4-specific ligands (Garcia-Guzman et al., 1997).
From the extracellular/central vestibule, ions cross the membrane through a channel (the pore) at the symmetry axis formed by a set of six TM helices consisting of two antiparallel helices from each protomer (Figures 1, 8A). In each protomer, the N-terminal α-helix (TM1) is oriented from cytosolic to extracellular and TM2 is oriented oppositely, forming a short section of antiparallel coiled-coil. The interior of the bundle is formed by TM2 from each protomer. The pore of the apo closed state is gated towards the extracellular side of the transmembrane region, with minimum radii of 0.3 Å at symmetry-related I323 in hP2X3 and 0.1 Å at S339 in rP2X7 (Figures 8B,C). Interestingly, these residues are on different turns of TM2 in the superimposed structures, showing there is some variability in the placement of the extracellular gate between subtypes. Upon the binding of ATP, movement of the three TM2 helices opens the channel to a pore radius large enough for passage of partially hydrated sodium ions (minimum radii of 3.2 Å in hP2X3 and 2.5 Å in rP2X7) (Degrève et al., 1996). Desensitization of P2X3, and presumably other P2XRs (except P2X7 which does not desensitize), occurs through another movement of the TM2 helices, whereby disassembly of the cytoplasmic cap leads to a new gate near the center of the transmembrane region at V334 in hP2X3 (radius of 1.5 Å). This new gate is too narrow to allow passage of hydrated sodium ions (Figures 8B,C) (Mansoor et al., 2016).
ASICs and ENaCs have known pore-blocking molecules such as amiloride, which binds near the extracellular end of the TM2 helices (PDB: 4NTX) (Kleyman and Cragoe, 1988; Baconguis et al., 2014). Considering the structural similarities between ASICs, ENaCs, and P2XRs, it is conceivable that amiloride derivatives or other molecules might also be found to target this region in P2XRs. Deeper areas of the pore, elucidated by cryo-EM, could also prove to be attractive drug targets despite being less accessible to extracellular molecules.
At the cytosolic end of the P2XR pore, the cytoplasmic cap forces the ions to egress laterally through the three cytoplasmic fenestrations (Figures 7A,C) (Mansoor et al., 2016). These fenestrations are roughly triangular in shape, with two sides formed by the ends of TM1 and TM2 from adjacent protomers and the third (distal) side formed by part of the cytoplasmic cap. The distal sides of the fenestrations are lined with charged side chains from each of the protomers, providing a favorable path for ions to exit the pore and enter the cell. The residues D340 and K357 form a salt bridge in the cytoplasmic fenestration of hP2X3 (Mansoor et al., 2016). Of note, these two residues are absolutely conserved across human subtypes while there is diversity in the other residues of the fenestrations (Figures 2, 7C). As in the extracellular fenestrations, these conserved charges may be useful in designing drugs to target the cytosolic fenestrations.
The P2XR ion path contains several locations that may prove to be attractive drug targets, including the extracellular and cytoplasmic fenestrations, the extracellular vestibule, and the transmembrane portion of the pore. The existing P2XR structures provide starting points for SBDD. Structures of the other human P2XRs would prove extremely useful in designing ligands that effectively and specifically target the pore.
P2XR C-terminal domains are divergent in sequence and size and their impacts on receptor function remain poorly understood. These domains dramatically vary from approximately 3%–34% of total protomer length, corresponding to 12 residues per protomer for hP2X4 and 203 residues per protomer for hP2X7, respectively (Figure 2). Unfortunately, the full C-terminal domains were not present in any of the constructs used to obtain crystal structures (Mansoor et al., 2016; Hattori and Gouaux, 2012; Kawate et al., 2009; Karasawa and Kawate, 2016; Kasuya et al., 2017; Wang et al., 2018). The recent full-length cryo-EM structures of rP2X7 contain the only structurally characterized P2XR cytoplasmic domain beyond the cytoplasmic cap, including the subtype-unique C-cys anchor and cytoplasmic ballast (Figure 9) (McCarthy et al., 2019).
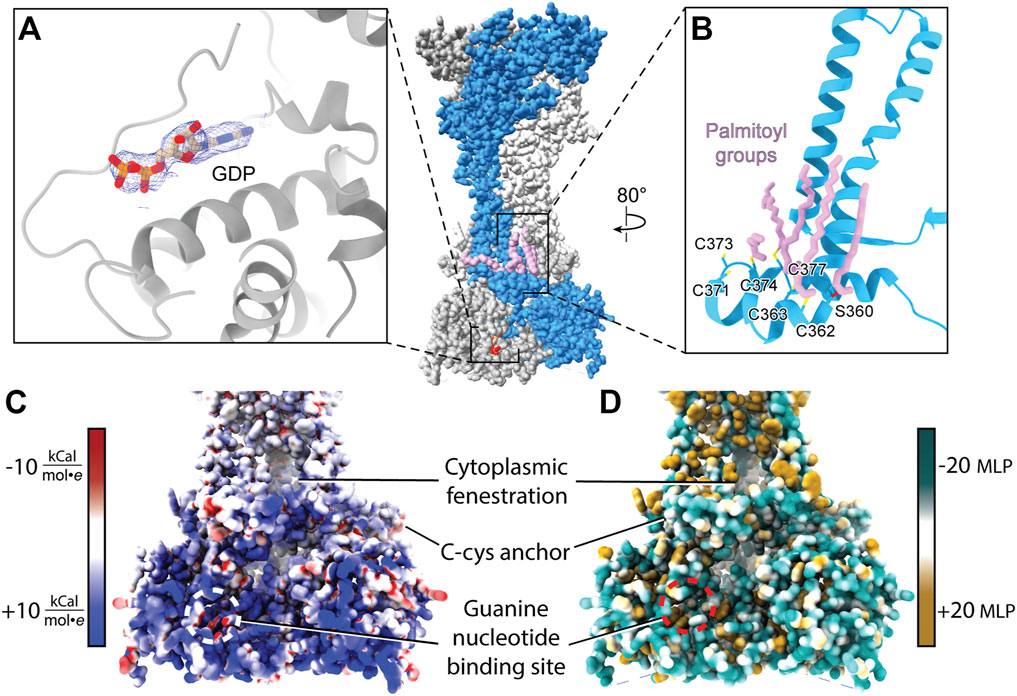
FIGURE 9. Attributes of the cytoplasmic domain of rP2X7 (PDB: 6U9V). (A) GDP bound to the cytoplasmic ballast of rP2X7 with its cryo-EM density shown in blue mesh (EMDB: 20702). (B) The C-cys anchor of rP2X7 contains palmitoyl groups (light purple) that extend into the membrane and prevent desensitization. (C) Electrostatic surface rendering of the rP2X7 cytoplasmic domain (PDB: 6U9V) with blue and red coloring representing positive and negative electrostatic potential, respectively, highlighting the electropositive nature of the cytoplasmic ballast. (D) Hydrophobic surface rendering of the rP2X7 cytoplasmic domain with hydrophobic regions colored brown and hydrophilic regions colored turquoise, each defined by positive and negative molecular lipophilicity potential (MLP), respectively, highlighting the hydrophilic nature of the cytoplasmic ballast (Laguerre et al., 1997). The location of the guanine nucleotide-binding site is outlined in white and red in panel C and in panel D, respectively. The MLP and electrostatic surface potential were calculated in ChimeraX (Pettersen et al., 2021).
Within each protomer of P2X7 is a C-cys anchor which contains six cysteines and one serine that can be palmitoylated (Figure 9B). The palmitoyl moieties extend into the inner leaflet of the membrane, preventing the helical recoil movement of TM2 that would otherwise result in channel desensitization (McCarthy et al., 2019). When the C-cys anchor is removed or mutated to prevent palmitoylation, rP2X7 desensitizes similarly to each of the other P2XR subtypes (McCarthy et al., 2019). While the role of the C-cys anchor in the gating cycle has been characterized, details surrounding post-translational modifications to this element remain unclear. For example, it is currently unknown in what stage of folding or trafficking the palmitoyl groups are added, nor which specific palmitoyl acyl transferase (PAT) enzyme or adapter protein is responsible for this post-translational modification. PATs generally target specific substrates and are localized to the ER, the Golgi, the plasma membrane, and endosomes, providing precise spatial regulation to their activity (Charollais and Van Der Goot, 2009). Modifications to the palmitoylation of P2X7 have a dramatic effect on receptor gating and would presumably alter downstream signaling (Allsopp and Evans, 2015). As a result, modulating C-cys anchor palmitoylation poses as an interesting therapeutic avenue to pursue, despite the significant challenges. As with any drug that has an intracellular target, the membrane is a barrier to delivery (Aungst, 1993; Di and Kerns, 2003; Naylor et al., 2018). Given that many other proteins are also palmitoylated, selectively preventing P2X7 palmitoylation would require determining and specifically targeting the correct PAT(s) at the appropriate point in post-translational processing. A greater understanding of the basic biology of P2X7 maturation is necessary before such a pharmacological strategy can be attempted.
The discovery of a nanomolar-affinity guanine nucleotide-binding site in the cytoplasmic ballast of rP2X7, the residues of which are conserved in hP2X7, is quite intriguing (Figure 9A). While only GDP is visualized in the apo closed and ATP-bound open state rP2X7 structures following purification, both GTP and GDP were shown to bind with equally high affinity (McCarthy et al., 2019). The pose of GDP is such that the ribose and nucleobase are positioned internally and the phosphate tail is on the periphery of the cytoplasmic ballast and easily accessible to the intracellular environment and potential accessory proteins (Figure 9A). The guanine nucleotide-binding site is electropositive, hydrophilic, and exposed, making it an accessible docking site for protein-protein interactions (Figures 9C,D). It would be interesting if P2X7, like G-protein coupled receptors (GPCRs), has associated guanine nucleotide exchange factors (GEFs) that facilitate GDP release (Cherfils and Zeghouf, 2013). The presence of the cytoplasmic ballast is necessary for the metabotropic properties of P2X7, including the release of cytokines as well as activation of various lipases and kinases (El-Moatassim and Dubyak, 1992; Humphreys and Dubyak, 1996; Surprenant et al., 1996; Wilson et al., 2002; Cheewatrakoolpong et al., 2005; Costa-Junior et al., 2011; Kopp et al., 2019). While there is no direct data regarding a role of the guanine nucleotide-binding site in P2X7 signaling, it is intriguing to imagine the metabotropic signaling properties of the cytoplasmic ballast are dependent on guanine nucleotide binding. If, similar to GPCRs, guanine nucleotide binding proves to be critical for P2X7 signaling, then guanine nucleotide analog ligands (such as abacavir, acyclovir, and entecavir) may be applied to study and therapeutically target P2X7-mediated signaling (Seley-Radtke and Yates, 2018). As with targeting the orthosteric ATP-binding site, targeting intracellular guanine nucleotide-binding sites must be done carefully as GTP and GDP are crucial metabolic and signaling molecules for numerous biological processes. Altogether, cryo-EM facilitated the study of full-length rP2X7 and thus revealed the cytoplasmic domain in its entirety and the therapeutic potential it holds. With the unique cytoplasmic domains of the remaining six P2XRs yet to be structurally characterized, there are more avenues for novel subtype-specific therapeutic development to be discovered.
The recent structures of full-length rP2X7 facilitated key discoveries that were exclusively enabled by cryo-EM. The guanine nucleotide-binding site was identified only after the cryo-EM map revealed an unexplained non-protein density of sufficient quality to accurately predict ligand identity without a priori knowledge of its existence (Figure 9A) (McCarthy et al., 2019). The identity of the guanine nucleotide was subsequently validated to be GDP by mass spectrometry. The cryo-EM reconstructions similarly revealed the precise location of the palmitoylated residues in the C-cys anchor (including the unexpected palmitoylation of a serine residue), a task that proved to be challenging by other methods (Gonnord et al., 2009). Altogether, the discoveries empowered by cryo-EM have unveiled novel elements that will further our understanding of the biological functions of P2X7.
There remain four homotrimeric P2XR subtypes without published structures: P2X1, P2X2, P2X5, and P2X6. Given that these receptors are implicated in a wide range of physiological and pathophysiological states, it is crucial to understand the molecular pharmacology of each receptor subtype (Burnstock and Kennedy, 2011; North and Jarvis, 2013; Burnstock and Knight, 2018; Lara et al., 2020). Obtaining cryo-EM structures of these receptors will not only reveal their subtype-specific features but also facilitate SBDD to improve ligand selectivity. It is important to note that while homology modeling and machine-learning programs like AlphaFold are improving, there is no replacement for the certainty afforded by empirically determined ligand-bound or protein-protein complexed structures (Jumper et al., 2021). To this point, there are also many known P2XR ligands for which we do not know how (or even where) they bind receptors. These ligands dramatically vary in size, shape, and chemical identity—even between ligands known to be selective for a specific receptor subtype—suggesting there are additional uncharacterized allosteric sites present in P2XRs. Structures of each receptor subtype in complex with these different ligands will help define the molecular pharmacology of P2XRs and confirm the locations of novel allosteric sites.
To date, the published P2XR structures are predominantly of non-human orthologs. While these structures are helpful and enable more accurate homology models, it is known that P2XR orthologs have unique pharmacological features which can significantly impact drug development. For example, human, mouse, and rat P2X7 dramatically differ in response to known allosteric modulators. The negative allosteric modulator AZ11645373 is a high-affinity antagonist for hP2X7, yet is ∼100-fold less effective against mouse P2X7 (mP2X7) and >500-fold less effective against rP2X7 (Stokes et al., 2006; Michel et al., 2009). There are also negative allosteric modulators such as A-438079 and A-740003 that antagonize all three of these P2X7 orthologs with similar efficacy (Donnelly-Roberts et al., 2009). Therefore, using cryo-EM to understand the structural basis for the pharmacologic variability across P2XR orthologs is crucial for the development of novel therapeutics that effectively target human P2XRs.
The feasibility of SBDD has dramatically increased with access to more advanced computational tools and the availability of structures for therapeutically relevant membrane proteins, largely due to the cryo-EM resolution revolution (Kuhlbrandt, 2014; Lionta et al., 2014; Lees et al., 2021). With the information gained from the currently published P2XR structures, SBDD can facilitate the discovery of ligands that target this receptor family (Figure 10). Molecular docking and in silico screening can probe any of the targetable sites within P2XRs for potential “hit” compounds (Ballante et al., 2021). Then, structure-based modeling can be used to optimize chemical groups, adding or substituting different moieties to improve affinity and specificity. Once computationally optimized, a ligand can be synthesized, evaluated in vitro, and (if validated) structurally solved in complex with its target receptor to high-resolution. This process will be repeated iteratively until a threshold of affinity and specificity is reached (Figure 10). Even though the use of cryo-EM has led to a dramatic number of new structures, the application of SBDD driven by this structural method is still an emerging area of research. Some recent successful cases that exemplify this approach include ligands that specifically target the TRPV5 channel, µ-opioid receptor, and the 80S ribosome (Wong et al., 2017; Hughes et al., 2019; Wang et al., 2022). Cryo-EM is currently the best method to solve ligand-bound P2XR structures and empower the use of SBDD.
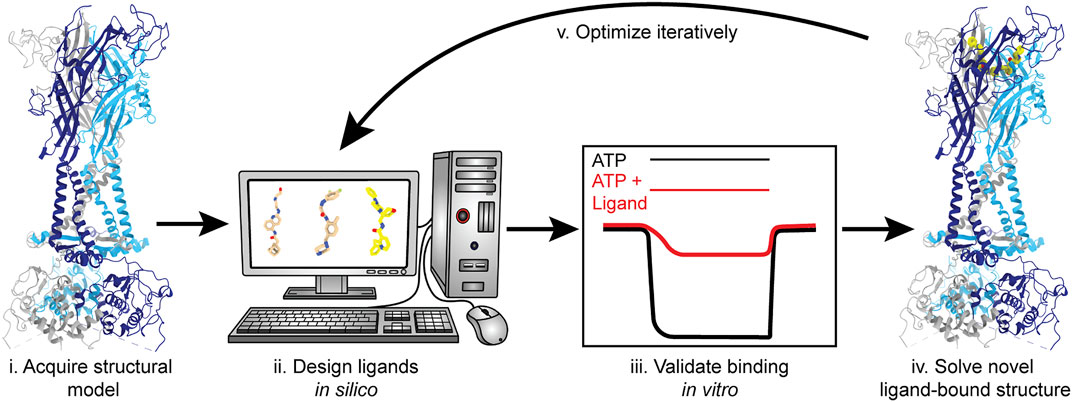
FIGURE 10. Structure-based drug design (SBDD) workflow using P2XRs as an example. SBDD includes five broad steps. (i) Solve receptor structure to high-resolution or use a reliable theoretical model to identify and define ligand-binding sites; (ii) Use molecular docking and modeling software (in silico) to identify and optimize small molecules capable of occupying a binding pocket; (iii) Chemically synthesize the most promising in silico “hits” and validate binding and selectivity by biochemical and electrophysiological methods; (iv) Solve the structure of the ligand-bound receptor complex empirically; and (v) Iterate steps (ii–iv) until ligand(s) with satisfactory properties are obtained and fully characterized. Representative structures generated from PDBs: 6U9V (for protein backbone) and 5U1X (for the bound ligand).
The substantial body of work characterizing P2XR structures has provided invaluable information on the biology and molecular pharmacology for this receptor family, however there remain many unanswered questions. Little is known about the structure and function of P2XR cytoplasmic domains and even less about heterotrimeric P2XRs. While the structural determination of the P2X7 cytoplasmic domain provided valuable insights, unexpected findings raised even more questions about its biological function. Most importantly, we have yet to fully understand how to selectively modulate the seven homotrimeric P2XRs or how effective such modulators would be against heterotrimeric P2XRs, considering their pharmacological complexity. The recent advancements in cryo-EM make this technique ideally suited to study P2XRs, including novel receptor structures and ligand-bound receptor complexes. While P2XRs are implicated in a host of physiological and pathophysiological conditions found within the cardiovascular, central nervous, and immune systems, there are no FDA-approved drugs targeting this receptor family. The success of antiplatelet drugs which antagonize the related P2Y GPCR family exemplifies the therapeutic value of targeting purinergic signaling (Kamran et al., 2021). To this point, there remains substantial untapped therapeutic potential for P2XR modulation. Using the currently published structures as a roadmap, we have discussed the known targetable sites and postulated potential areas for SBDD targeting P2XRs. Continued structural investigation will advance our understanding of P2XR biology and reveal novel approaches for receptor modulation and therapeutic intervention.
SM and AO outlined the manuscript. AO, IK, MG, and JS wrote the manuscript. AO, NL, and MG made the figures. All authors edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We want to thank David L. Farrens for reading the manuscript and providing constructive feedback.
Abdulqawi, R., Dockry, R., Holt, K., Layton, G., Mccarthy, B. G., Ford, A. P., et al. (2015). P2x3 Receptor Antagonist (Af-219) in Refractory Chronic Cough: A Randomised, Double-Blind, Placebo-Controlled Phase 2 Study. Lancet 385, 1198–1205. doi:10.1016/S0140-6736(14)61255-1
Adinolfi, E., Cirillo, M., Woltersdorf, R., Falzoni, S., Chiozzi, P., Pellegatti, P., et al. (2010). Trophic Activity of A Naturally Occurring Truncated Isoform of the P2x7 Receptor. Faseb J. 24, 3393–3404. doi:10.1096/fj.09-153601
Afferent Pharmaceuticals, I. (2015). A Study of Gefapixant (Af-219/Mk-7264) in Participants with Idiopathic Pulmonary Fibrosis (Ipf) with Persistent Cough (Mk-7264-016). Available at: https://Clinicaltrials.Gov/Show/Nct02502097.
Allsopp, R. C., and Evans, R. J. (2015). Contribution of the Juxtatransmembrane Intracellular Regions to the Time Course and Permeation of Atp-Gated P2x7 Receptor Ion Channels. J. Biol. Chem. 290, 14556–14566. doi:10.1074/jbc.M115.642033
Antonio, L. S., Stewart, A. P., Varanda, W. A., and Edwardson, J. M. (2014). Identification of P2x2/P2x4/P2x6 Heterotrimeric Receptors Using Atomic Force Microscopy (Afm) Imaging. Febs Lett. 588, 2125–2128. doi:10.1016/j.febslet.2014.04.048
Ase, A. R., Therrien, É., and Séguéla, P. (2019). An Allosteric Inhibitory Site Conserved in the Ectodomain of P2x Receptor Channels. Front. Cell Neurosci. 13, 121. doi:10.3389/fncel.2019.00121
Aungst, B. J. (1993). Novel Formulation Strategies for Improving Oral Bioavailability of Drugs with Poor Membrane Permeation or Presystemic Metabolism. J. Pharm. Sci. 82, 979–987. doi:10.1002/jps.2600821008
Baconguis, I., Bohlen, C. J., Goehring, A., Julius, D., and Gouaux, E. (2014). X-Ray Structure of Acid-Sensing Ion Channel 1-Snake Toxin Complex Reveals Open State of A Na(+)-Selective Channel. Cell 156, 717–729. doi:10.1016/j.cell.2014.01.011
Ballante, F., Kooistra, A. J., Kampen, S., De Graaf, C., and Carlsson, J. (2021). Structure-Based Virtual Screening for Ligands of G Protein-Coupled Receptors: What Can Molecular Docking Do for You? Pharmacol. Rev. 73, 527–565. doi:10.1124/pharmrev.120.000246
Baroni, M., Pizzirani, C., Pinotti, M., Ferrari, D., Adinolfi, E., Calzavarini, S., et al. (2007). Stimulation of P2 (P2x7) Receptors in Human Dendritic Cells Induces the Release of Tissue Factor-Bearing Microparticles. Faseb J. 21, 1926–1933. doi:10.1096/fj.06-7238com
Barton, G. J. (1993). ALSCRIPT: A Tool to Format Multiple Sequence Alignments. Prot. Eng. 6 (1), 37–40. doi:10.1093/protein/6.1.37
Batool, M., Ahmad, B., and Choi, S. (2019). A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 20. doi:10.3390/ijms20112783
Bidula, S., Nadzirin, I. B., Cominetti, M., Hickey, H., Cullum, S. A., Searcey, M., et al. (2022). Structural Basis of the Negative Allosteric Modulation of 5-Bdbd at Human P2x4 Receptors. Mol. Pharmacol. 101, 33–44. doi:10.1124/molpharm.121.000402
Bidula, S. M., Cromer, B. A., Walpole, S., Angulo, J., and Stokes, L. (2019). Mapping A Novel Positive Allosteric Modulator Binding Site in the Central Vestibule Region of Human P2x7. Sci. Rep. 9, 3231. doi:10.1038/s41598-019-39771-5
Bo, X., Jiang, L. H., Wilson, H. L., Kim, M., Burnstock, G., Surprenant, A., et al. (2003). Pharmacological and Biophysical Properties of the Human P2x5 Receptor. Mol. Pharmacol. 63, 1407–1416. doi:10.1124/mol.63.6.1407
Bond, C. S., and Schüttelkopf, A. W. (2009). Aline: A Wysiwyg Protein-Sequence Alignment Editor for Publication-Quality Alignments. Acta Crystallogr. D. Biol. Crystallogr. 65, 510–512. doi:10.1107/S0907444909007835
Boué-Grabot, E., Archambault, V., and Séguéla, P. (2000). A Protein Kinase C Site Highly Conserved in P2x Subunits Controls the Desensitization Kinetics of P2x(2) Atp-Gated Channels. J. Biol. Chem. 275, 10190–10195. doi:10.1074/jbc.275.14.10190
Brake, A. J., Wagenbach, M. J., and Julius, D. (1994). New Structural Motif for Ligand-Gated Ion Channels Defined by an Ionotropic Atp Receptor. Nature 371, 519–523. doi:10.1038/371519a0
Brändle, U., Spielmanns, P., Osteroth, R., Sim, J., Surprenant, A., Buell, G., et al. (1997). Desensitization of the P2x(2) Receptor Controlled by Alternative Splicing. Febs Lett. 404, 294–298. doi:10.1016/s0014-5793(97)00128-2
Burnstock, G. (1976). Do Some Nerve Cells Release More Than One Transmitter? Neuroscience 1, 239–248. doi:10.1016/0306-4522(76)90054-3
Burnstock, G. (2004). Introduction: P2 Receptors. Curr. Top. Med. Chem. 4, 793–803. doi:10.2174/1568026043451014
Burnstock, G., and Kennedy, C. (2011). P2x Receptors in Health and Disease. Adv. Pharmacol. 61, 333–372. doi:10.1016/B978-0-12-385526-8.00011-4
Burnstock, G., and Knight, G. E. (2018). The Potential of P2x7 Receptors as A Therapeutic Target, Including Inflammation and Tumour Progression. Purinergic Signal. 14, 1–18. doi:10.1007/s11302-017-9593-0
CCTU-Core Pharmaceuticals, J.Cambridgeshire and Peterborough NHS Foundation Trust (2019). Antidepressant Trial with P2x7 Antagonist Jnj-54175446. Available at: https://Clinicaltrials.Gov/Show/Nct04116606.
Changeux, J. P., and Christopoulos, A. (2016). Allosteric Modulation as A Unifying Mechanism for Receptor Function and Regulation. Cell 166, 1084–1102. doi:10.1016/j.cell.2016.08.015
Charollais, J., and Van Der Goot, F. G. (2009). Palmitoylation of Membrane Proteins (Review). Mol. Membr. Biol. 26, 55–66. doi:10.1080/09687680802620369
Chataigneau, T., Lemoine, D., and Grutter, T. (2013). Exploring the Atp-Binding Site of P2x Receptors. Front. Cell Neurosci. 7, 273. doi:10.3389/fncel.2013.00273
Chaumont, S., Jiang, L. H., Penna, A., North, R. A., and Rassendren, F. (2004). Identification of A Trafficking Motif Involved in the Stabilization and Polarization of P2x Receptors. J. Biol. Chem. 279, 29628–29638. doi:10.1074/jbc.M403940200
Cheewatrakoolpong, B., Gilchrest, H., Anthes, J. C., and Greenfeder, S. (2005). Identification and Characterization of Splice Variants of the Human P2x7 Atp Channel. Biochem. Biophys. Res. Commun. 332, 17–27. doi:10.1016/j.bbrc.2005.04.087
Cherfils, J., and Zeghouf, M. (2013). Regulation of Small Gtpases by Gefs, Gaps, and Gdis. Physiol. Rev. 93, 269–309. doi:10.1152/physrev.00003.2012
Coddou, C., Stojilkovic, S. S., and Huidobro-Toro, J. P. (2011). Allosteric Modulation of Atp-Gated P2x Receptor Channels. Rev. Neurosci. 22, 335–354. doi:10.1515/RNS.2011.014
Costa-Junior, H. M., Sarmento Vieira, F., and Coutinho-Silva, R. (2011). C Terminus of the P2x7 Receptor: Treasure Hunting. Purinergic Signal. 7, 7–19. doi:10.1007/s11302-011-9215-1
Degrève, L., Vechi, S. M., and Junior, C. Q. (1996). The Hydration Structure of the Na+ and K+ Ions and the Selectivity of Their Ionic Channels. Biochimica Biophysica Acta (Bba) - Bioenergetics 1274, 149–156.
Di, L., and Kerns, E. H. (2003). Profiling Drug-like Properties in Discovery Research. Curr. Opin. Chem. Biol. 7, 402–408. doi:10.1016/s1367-5931(03)00055-3
Donnelly-Roberts, D. L., Namovic, M. T., Han, P., and Jarvis, M. F. (2009). Mammalian P2x7 Receptor Pharmacology: Comparison of Recombinant Mouse, Rat and Human P2x7 Receptors. Br. J. Pharmacol. 157, 1203–1214. doi:10.1111/j.1476-5381.2009.00233.x
Duckwitz, W., Hausmann, R., Aschrafi, A., and Schmalzing, G. (2006). P2x5 Subunit Assembly Requires Scaffolding by the Second Transmembrane Domain and A Conserved Aspartate. J. Biol. Chem. 281, 39561–39572. doi:10.1074/jbc.M606113200
El-Moatassim, C., and Dubyak, G. R. (1992). A Novel Pathway for the Activation of Phospholipase D by P2z Purinergic Receptors in Bac1.2f5 Macrophages. J. Biol. Chem. 267, 23664–23673. doi:10.1016/s0021-9258(18)35890-3
Ferrari, S., Costi, P. M., and Wade, R. C. (2003). Inhibitor Specificity via Protein Dynamics: Insights from the Design of Antibacterial Agents Targeted against Thymidylate Synthase. Chem. Biol. 10, 1183–1193. doi:10.1016/j.chembiol.2003.11.012
Flores, J. A., Haddad, B. G., Dolan, K. A., Myers, J. B., Yoshioka, C. C., Copperman, J., et al. (2020). Connexin-46/50 in a Dynamic Lipid Environment Resolved by CryoEM at 1.9 Å. Nat. Commun. 11, 4331. doi:10.1038/s41467-020-18120-5
Ford, A. P., Dillon, M. P., Kitt, M. M., and Gever, J. R. (2021). The Discovery and Development of Gefapixant. Auton. Neurosci. 235, 102859. doi:10.1016/j.autneu.2021.102859
Francistiová, L., Bianchi, C., Di Lauro, C., Sebastián-Serrano, Á., de Diego-García, L., Kobolák, J., et al. (2020). The Role of P2x7 Receptor in Alzheimer's Disease. Front. Mol. Neurosci. 13, 94. doi:10.3389/fnmol.2020.00094
Friel, D. D., and Bean, B. P. (1988). Two Atp-Activated Conductances in Bullfrog Atrial Cells. J. Gen. Physiol. 91, 1–27. doi:10.1085/jgp.91.1.1
Furlan-Freguia, C., Marchese, P., Gruber, A., Ruggeri, Z. M., and Ruf, W. (2011). P2x7 Receptor Signaling Contributes to Tissue Factor-dependent Thrombosis in Mice. J. Clin. Invest. 121, 2932–2944. doi:10.1172/JCI46129
Garcia-Guzman, M., Soto, F., Gomez-Hernandez, J. M., Lund, P. E., and Stühmer, W. (1997). Characterization of Recombinant Human P2x4 Receptor Reveals Pharmacological Differences to the Rat Homologue. Mol. Pharmacol. 51, 109–118. doi:10.1124/mol.51.1.109
Gonnord, P., Delarasse, C., Auger, R., Benihoud, K., Prigent, M., Cuif, M. H., et al. (2009). Palmitoylation of the P2x7 Receptor, an Atp-Gated Channel, Controls its Expression and Association with Lipid Rafts. Faseb J. 23, 795–805. doi:10.1096/fj.08-114637
Hattori, M., and Gouaux, E. (2012). Molecular Mechanism of Atp Binding and Ion Channel Activation in P2x Receptors. Nature 485, 207–212. doi:10.1038/nature11010
Hausmann, R., Bahrenberg, G., Kuhlmann, D., Schumacher, M., Braam, U., Bieler, D., et al. (2014). A Hydrophobic Residue in Position 15 of the Rp2x3 Receptor Slows Desensitization and Reveals Properties Beneficial for Pharmacological Analysis and High-Throughput Screening. Neuropharmacology 79, 603–615. doi:10.1016/j.neuropharm.2014.01.010
Hughes, T. E., Del Rosario, J. S., Kapoor, A., Yazici, A. T., Yudin, Y., Fluck, E. C., et al. (2019). Structure-Based Characterization of Novel Trpv5 Inhibitors. Elife 8. doi:10.7554/eLife.49572
Humphreys, B. D., and Dubyak, G. R. (1996). Induction of the P2z/P2x7 Nucleotide Receptor and Associated Phospholipase D Activity by Lipopolysaccharide and Ifn-Gamma in the Human Thp-1 Monocytic Cell Line. J. Immunol. 157, 5627–5637.
Illes, P., Müller, C. E., Jacobson, K. A., Grutter, T., Nicke, A., Fountain, S. J., et al. (2021). Update of P2x Receptor Properties and Their Pharmacology: Iuphar Review 30. Br. J. Pharmacol. 178, 489–514. doi:10.1111/bph.15299
Jarvis, M. F., and Khakh, B. S. (2009). Atp-Gated P2x Cation-Channels. Neuropharmacology 56, 208–215. doi:10.1016/j.neuropharm.2008.06.067
Jasti, J., Furukawa, H., Gonzales, E. B., and Gouaux, E. (2007). Structure of Acid-Sensing Ion Channel 1 at 1.9 A Resolution and Low Ph. Nature 449, 316–323. doi:10.1038/nature06163
Jiang, R., Taly, A., Lemoine, D., Martz, A., Cunrath, O., and Grutter, T. (2012). Tightening of the Atp-Binding Sites Induces the Opening of P2x Receptor Channels. Embo J. 31, 2134–2143. doi:10.1038/emboj.2012.75
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly Accurate Protein Structure Prediction with Alphafold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kamei, J., Takahashi, Y., Yoshikawa, Y., and Saitoh, A. (2005). Involvement of P2x Receptor Subtypes in Atp-Induced Enhancement of the Cough Reflex Sensitivity. Eur. J. Pharmacol. 528, 158–161. doi:10.1016/j.ejphar.2005.10.030
Kamran, H., Jneid, H., Kayani, W. T., Virani, S. S., Levine, G. N., Nambi, V., et al. (2021). Oral Antiplatelet Therapy after Acute Coronary Syndrome: A Review. Jama 325, 1545–1555. doi:10.1001/jama.2021.0716
Karasawa, A., and Kawate, T. (2016). Structural Basis for Subtype-specific Inhibition of the P2x7 Receptor. Elife 5. doi:10.7554/eLife.22153
Kasuya, G., Fujiwara, Y., Takemoto, M., Dohmae, N., Nakada-Nakura, Y., Ishitani, R., et al. (2016). Structural Insights into Divalent Cation Modulations of Atp-Gated P2x Receptor Channels. Cell Rep. 14, 932–944. doi:10.1016/j.celrep.2015.12.087
Kasuya, G., Yamaura, T., Ma, X. B., Nakamura, R., Takemoto, M., Nagumo, H., et al. (2017). Structural Insights into the Competitive Inhibition of the Atp-Gated P2x Receptor Channel. Nat. Commun. 8, 876. doi:10.1038/s41467-017-00887-9
Kawate, T., Michel, J. C., Birdsong, W. T., and Gouaux, E. (2009). Crystal Structure of the Atp-Gated P2x(4) Ion Channel in the Closed State. Nature 460, 592–598. doi:10.1038/nature08198
Kawate, T. (2017). P2x Receptor Activation. Adv. Exp. Med. Biol. 1051, 55–69. doi:10.1007/5584_2017_55
Keystone, E. C., Wang, M. M., Layton, M., Hollis, S., Mcinnes, I. B., and Team, D. C. S. (2012). Clinical Evaluation of the Efficacy of the P2x7 Purinergic Receptor Antagonist Azd9056 on the Signs and Symptoms of Rheumatoid Arthritis in Patients with Active Disease Despite Treatment with Methotrexate or Sulphasalazine. Ann. Rheum. Dis. 71, 1630–1635. doi:10.1136/annrheumdis-2011-143578
King, B. F., Townsend-Nicholson, A., Wildman, S. S., Thomas, T., Spyer, K. M., and Burnstock, G. (2000). Coexpression of Rat P2x2 and P2x6 Subunits in Xenopus Oocytes. J. Neurosci. 20, 4871–4877. doi:10.1523/jneurosci.20-13-04871.2000
Kleyman, T. R., and Cragoe, E. J. (1988). Amiloride and its Analogs as Tools in the Study of Ion Transport. J. Membr. Biol. 105, 1–21. doi:10.1007/BF01871102
Kopp, R., Krautloher, A., Ramírez-Fernández, A., and Nicke, A. (2019). P2x7 Interactions and Signaling - Making Head or Tail of it. Front. Mol. Neurosci. 12, 183. doi:10.3389/fnmol.2019.00183
Koshimizu, T., Koshimizu, M., and Stojilkovic, S. S. (1999). Contributions of the C-Terminal Domain to the Control of P2x Receptor Desensitization. J. Biol. Chem. 274, 37651–37657. doi:10.1074/jbc.274.53.37651
Kotnis, S., Bingham, B., Vasilyev, D. V., Miller, S. W., Bai, Y., Yeola, S., et al. (2010). Genetic and Functional Analysis of Human P2x5 Reveals A Distinct Pattern of Exon 10 Polymorphism with Predominant Expression of the Nonfunctional Receptor Isoform. Mol. Pharmacol. 77, 953–960. doi:10.1124/mol.110.063636
Kühlbrandt, W. (2014). Biochemistry. The Resolution Revolution. Science 343, 1443–1444. doi:10.1126/science.1251652
Laguerre, M., Saux, M., Dubost, J., and Carpy, A. (1997). Mlpp: A Progarm for the Calculation of Molecular Lipophilicity Potential in Proteins. Pharm. Pharmacol. Commun. 3 (5-6), 217–222.
Lara, R., Adinolfi, E., Harwood, C. A., Philpott, M., Barden, J. A., Di Virgilio, F., et al. (2020). P2x7 in Cancer: From Molecular Mechanisms to Therapeutics. Front. Pharmacol. 11, 793. doi:10.3389/fphar.2020.00793
Lê, K. T., Babinski, K., and Séguéla, P. (1998). Central P2x4 and P2x6 Channel Subunits Coassemble into A Novel Heteromeric Atp Receptor. J. Neurosci. 18, 7152–7159. doi:10.1523/jneurosci.18-18-07152.1998
Lê, K. T., Paquet, M., Nouel, D., Babinski, K., and Séguéla, P. (1997). Primary Structure and Expression of A Naturally Truncated Human P2x Atp Receptor Subunit from Brain and Immune System. Febs Lett. 418, 195–199. doi:10.1016/s0014-5793(97)01380-x
Lees, J. A., Dias, J. M., and Han, S. (2021). Applications of Cryo-Em in Small Molecule and Biologics Drug Design. Biochem. Soc. Trans. 49, 2627–2638. doi:10.1042/BST20210444
Lewis, C., Neidhart, S., Holy, C., North, R. A., Buell, G., and Surprenant, A. (1995). Coexpression of P2x2 and P2x3 Receptor Subunits Can Account for Atp-Gated Currents in Sensory Neurons. Nature 377, 432–435. doi:10.1038/377432a0
Li, M., Kawate, T., Silberberg, S. D., and Swartz, K. J. (2010). Pore-Opening Mechanism in Trimeric P2x Receptor Channels. Nat. Commun. 1, 44. doi:10.1038/ncomms1048
Li, M., Silberberg, S. D., and Swartz, K. J. (2013). Subtype-Specific Control of P2x Receptor Channel Signaling by Atp and Mg2+. Proc. Natl. Acad. Sci. U. S. A. 110, E3455–E3463. doi:10.1073/pnas.1308088110
Li, M., Wang, Y., Banerjee, R., Marinelli, F., Silberberg, S., Faraldo-Gómez, J. D., et al. (2019). Molecular Mechanisms of Human P2x3 Receptor Channel Activation and Modulation by Divalent Cation Bound Atp. Elife 8. doi:10.7554/eLife.47060
Lionta, E., Spyrou, G., Vassilatis, D. K., and Cournia, Z. (2014). Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 14, 1923–1938. doi:10.2174/1568026614666140929124445
Londos, C., Cooper, D. M., and Wolff, J. (1980). Subclasses of External Adenosine Receptors. Proc. Natl. Acad. Sci. U. S. A. 77, 2551–2554. doi:10.1073/pnas.77.5.2551
Mahaut-Smith, M. P., Jones, S., and Evans, R. J. (2011). The P2x1 Receptor and Platelet Function. Purinergic Signal 7, 341–356. doi:10.1007/s11302-011-9224-0
Mansoor, S. E., Lü, W., Oosterheert, W., Shekhar, M., Tajkhorshid, E., and Gouaux, E. (2016). X-Ray Structures Define Human P2x(3) Receptor Gating Cycle and Antagonist Action. Nature 538, 66–71. doi:10.1038/nature19367
Mansoor, S. E. (2022). “How Structural Biology Has Directly Impacted Our Understanding of P2x Receptor Function and Gating,” in The P2x7 Receptor. Editor A. Nicke. 1 Ed. (New York: Humana). in Press.
McCarthy, A. E., Yoshioka, C., and Mansoor, S. E. (2019). Full-Length P2x7 Structures Reveal How Palmitoylation Prevents Channel Desensitization. Cell 179, 659–e13. doi:10.1016/j.cell.2019.09.017
Michel, A. D., Ng, S. W., Roman, S., Clay, W. C., Dean, D. K., and Walter, D. S. (2009). Mechanism of Action of Species-Selective P2x(7) Receptor Antagonists. Br. J. Pharmacol. 156, 1312–1325. doi:10.1111/j.1476-5381.2009.00135.x
Müller, C. E. (2015). Medicinal Chemistry of P2x Receptors: Allosteric Modulators. Curr. Med. Chem. 22, 929–941. doi:10.2174/0929867322666141210155610
Müller, C. E., and Namasivayam, V. (2021). Recommended Tool Compounds and Drugs for Blocking P2x and P2y Receptors. Purinergic Signal 17, 633–648. doi:10.1007/s11302-021-09813-7
Murrell-Lagnado, R. D., and Qureshi, O. S. (2008). Assembly and Trafficking of P2x Purinergic Receptors (Review). Mol. Membr. Biol. 25, 321–331. doi:10.1080/09687680802050385
Nakane, T., Kotecha, A., Sente, A., Mcmullan, G., Masiulis, S., Brown, P. M. G. E., et al. (2020). Single-Particle Cryo-Em at Atomic Resolution. Nature 587, 152–156. doi:10.1038/s41586-020-2829-0
Naylor, M. R., Ly, A. M., Handford, M. J., Ramos, D. P., Pye, C. R., Furukawa, A., et al. (2018). Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem. 61, 11169–11182. doi:10.1021/acs.jmedchem.8b01259
Nelson, A. L., Dhimolea, E., and Reichert, J. M. (2010). Development Trends for Human Monoclonal Antibody Therapeutics. Nat. Rev. Drug Discov. 9, 767–774. doi:10.1038/nrd3229
Noreng, S., Bharadwaj, A., Posert, R., Yoshioka, C., and Baconguis, I. (2018). Structure of the Human Epithelial Sodium Channel by Cryo-Electron Microscopy. Elife 7. doi:10.7554/eLife.39340
North, R. A., and Jarvis, M. F. (2013). P2x Receptors as Drug Targets. Mol. Pharmacol. 83, 759–769. doi:10.1124/mol.112.083758
North, R. A. (2002). Molecular Physiology of P2x Receptors. Physiol. Rev. 82, 1013–1067. doi:10.1152/physrev.00015.2002
Nussinov, R., and Tsai, C. J. (2012). The Different Ways through Which Specificity Works in Orthosteric and Allosteric Drugs. Curr. Pharm. Des. 18, 1311–1316. doi:10.2174/138161212799436377
Obrecht, A. S., Urban, N., Schaefer, M., Röse, A., Kless, A., Meents, J. E., et al. (2019). Identification of Aurintricarboxylic Acid as A Potent Allosteric Antagonist of P2x1 and P2x3 Receptors. Neuropharmacology 158, 107749. doi:10.1016/j.neuropharm.2019.107749
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). Ucsf Chimerax: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 30, 70–82. doi:10.1002/pro.3943
Pfizer (2008). Study of Ce-224,535 A Twice Daily Pill to Control Rheumatoid Arthritis in Patients Who Have Not Totally Improved with Methotrexate. Available at: https://Clinicaltrials.Gov/Show/Nct00628095.
Rahman, M. M., Teng, J., Worrell, B. T., Noviello, C. M., Lee, M., Karlin, A., et al. (2020). Structure of the Native Muscle-type Nicotinic Receptor and Inhibition by Snake Venom Toxins. Neuron 106, 952–e5. doi:10.1016/j.neuron.2020.03.012
Richards, D., Gever, J. R., Ford, A. P., and Fountain, S. J. (2019). Action of Mk-7264 (Gefapixant) at Human P2x3 and P2x2/3 Receptors and In Vivo Efficacy in Models of Sensitisation. Br. J. Pharmacol. 176, 2279–2291. doi:10.1111/bph.14677
Roman, R. M., and Fitz, J. G. (1999). Emerging Roles of Purinergic Signaling in Gastrointestinal Epithelial Secretion and Hepatobiliary Function. Gastroenterology 116, 964–979. doi:10.1016/s0016-5085(99)70081-8
Samways, D. S., Khakh, B. S., Dutertre, S., and Egan, T. M. (2011). Preferential Use of Unobstructed Lateral Portals as the Access Route to the Pore of Human Atp-Gated Ion Channels (P2x Receptors). Proc. Natl. Acad. Sci. U. S. A. 108, 13800–13805. doi:10.1073/pnas.1017550108
Samways, D. S., Li, Z., and Egan, T. M. (2014). Principles and Properties of Ion Flow in P2x Receptors. Front. Cell Neurosci. 8, 6. doi:10.3389/fncel.2014.00006
Sattler, C., and Benndorf, K. (2022). Enlightening Activation Gating in P2x Receptors. Purinergic Signal. doi:10.1007/s11302-022-09850-w
Seley-Radtke, K. L., and Yates, M. K. (2018). The Evolution of Nucleoside Analogue Antivirals: A Review for Chemists and Non-chemists. Part 1: Early Structural Modifications to the Nucleoside Scaffold. Antivir. Res. 154, 66–86. doi:10.1016/j.antiviral.2018.04.004
Sievers, F., and Higgins, D. G. (2018). Clustal Omega for Making Accurate Alignments of Many Protein Sequences. Protein Sci. 27, 135–145. doi:10.1002/pro.3290
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A., and Sansom, M. S. (1996). Hole: A Program for the Analysis of the Pore Dimensions of Ion Channel Structural Models. J. Mol. Graph 14, 354376–376. doi:10.1016/s0263-7855(97)00009-x
Soto, F., Garcia-Guzman, M., Karschin, C., and Stühmer, W. (1996). Cloning and Tissue Distribution of A Novel P2x Receptor from Rat Brain. Biochem. Biophys. Res. Commun. 223, 456–460. doi:10.1006/bbrc.1996.0915
Stank, A., Kokh, D. B., Fuller, J. C., and Wade, R. C. (2016). Protein Binding Pocket Dynamics. Acc. Chem. Res. 49, 809–815. doi:10.1021/acs.accounts.5b00516
Stokes, L., Jiang, L. H., Alcaraz, L., Bent, J., Bowers, K., Fagura, M., et al. (2006). Characterization of A Selective and Potent Antagonist of Human P2x(7) Receptors, Az11645373. Br. J. Pharmacol. 149, 880–887. doi:10.1038/sj.bjp.0706933
Su, C. C., Lyu, M., Morgan, C. E., Bolla, J. R., Robinson, C. V., and Yu, E. W. (2021). A 'Build and Retrieve' Methodology to Simultaneously Solve Cryo-Em Structures of Membrane Proteins. Nat. Methods 18, 69–75. doi:10.1038/s41592-020-01021-2
Surprenant, A., Rassendren, F., Kawashima, E., North, R. A., and Buell, G. (1996). The Cytolytic P2z Receptor for Extracellular Atp Identified as A P2x Receptor (P2x7). Science 272, 735–738. doi:10.1126/science.272.5262.735
Taylor, A. L., Schwiebert, L. M., Smith, J. J., King, C., Jones, J. R., Sorscher, E. J., et al. (1999). Epithelial P2x Purinergic Receptor Channel Expression and Function. J. Clin. Invest. 104, 875–884. doi:10.1172/JCI7270
Tian, W., Chen, C., Lei, X., Zhao, J., and Liang, J. (2018). Castp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 46, W363–W367. doi:10.1093/nar/gky473
Valera, S., Hussy, N., Evans, R. J., Adami, N., North, R. A., Surprenant, A., et al. (1994). A New Class of Ligand-Gated Ion Channel Defined by P2x Receptor for Extracellular Atp. Nature 371, 516–519. doi:10.1038/371516a0
van Calker, D., Müller, M., and Hamprecht, B. (1979). Adenosine Regulates via Two Different Types of Receptors, the Accumulation of Cyclic Amp in Cultured Brain Cells. J. Neurochem. 33, 999–1005. doi:10.1111/j.1471-4159.1979.tb05236.x
Virginio, C., Church, D., North, R. A., and Surprenant, A. (1997). Effects of Divalent Cations, Protons and Calmidazolium at the Rat P2x7 Receptor. Neuropharmacology 36, 1285–1294. doi:10.1016/s0028-3908(97)00141-x
Virginio, C., Robertson, G., Surprenant, A., and North, R. A. (1998). Trinitrophenyl-Substituted Nucleotides Are Potent Antagonists Selective for P2x1, P2x3, and Heteromeric P2x2/3 Receptors. Mol. Pharmacol. 53, 969–973.
Wang, H., Hetzer, F., Huang, W., Qu, Q., Meyerowitz, J., Kaindl, J., et al. (2022). Structure-Based Evolution of G Protein-Biased μ-Opioid Receptor Agonists. Angew. Chem. Int. Ed. Engl. 6, E202200269. doi:10.1002/anie.202200269
Wang, J., Sun, L. F., Cui, W. W., Zhao, W. S., Ma, X. F., Li, B., et al. (2017). Intersubunit Physical Couplings Fostered by the Left Flipper Domain Facilitate Channel Opening of P2x4 Receptors. J. Biol. Chem. 292, 7619–7635. doi:10.1074/jbc.M116.771121
Wang, J., Wang, Y., Cui, W. W., Huang, Y., Yang, Y., Liu, Y., et al. (2018). Druggable Negative Allosteric Site of P2x3 Receptors. Proc. Natl. Acad. Sci. U. S. A. 115, 4939–4944. doi:10.1073/pnas.1800907115
Webb, T. E., Simon, J., Krishek, B. J., Bateson, A. N., Smart, T. G., King, B. F., et al. (1993). Cloning and Functional Expression of A Brain G-Protein-Coupled Atp Receptor. Febs Lett. 324, 219–225. doi:10.1016/0014-5793(93)81397-i
Wenthur, C. J., Gentry, P. R., Mathews, T. P., and Lindsley, C. W. (2014). Drugs for Allosteric Sites on Receptors. Annu. Rev. Pharmacol. Toxicol. 54, 165–184. doi:10.1146/annurev-pharmtox-010611-134525
Werner, P., Seward, E. P., Buell, G. N., and North, R. A. (1996). Domains of P2x Receptors Involved in Desensitization. Proc. Natl. Acad. Sci. U. S. A. 93, 15485–15490. doi:10.1073/pnas.93.26.15485
Wilson, H. L., Wilson, S. A., Surprenant, A., and North, R. A. (2002). Epithelial Membrane Proteins Induce Membrane Blebbing and Interact with the P2x7 Receptor C Terminus. J. Biol. Chem. 277, 34017–34023. doi:10.1074/jbc.M205120200
Wolf, C., Rosefort, C., Fallah, G., Kassack, M. U., Hamacher, A., Bodnar, M., et al. (2011). Molecular Determinants of Potent P2x2 Antagonism Identified by Functional Analysis, Mutagenesis, and Homology Docking. Mol. Pharmacol. 79, 649–661. doi:10.1124/mol.110.068700
Wong, W., Bai, X. C., Sleebs, B. E., Triglia, T., Brown, A., Thompson, J. K., et al. (2017). Mefloquine Targets the Plasmodium Falciparum 80s Ribosome to Inhibit Protein Synthesis. Nat. Microbiol. 2, 17031. doi:10.1038/nmicrobiol.2017.31
Yan, D., Zhu, Y., Walsh, T., Xie, D., Yuan, H., Sirmaci, A., et al. (2013). Mutation of the Atp-Gated P2x(2) Receptor Leads to Progressive Hearing Loss and Increased Susceptibility to Noise. Proc. Natl. Acad. Sci. U. S. A. 110, 2228–2233. doi:10.1073/pnas.1222285110
Zhao, W. S., Wang, J., Ma, X. J., Yang, Y., Liu, Y., Huang, L. D., et al. (2014). Relative Motions between Left Flipper and Dorsal Fin Domains Favour P2x4 Receptor Activation. Nat. Commun. 5, 4189. doi:10.1038/ncomms5189
Zhao, Y., Chen, X., He, C., Gao, G., Chen, Z., and Du, J. (2021). Discovery of Bilirubin as Novel P2x7r Antagonist with Anti-tumor Activity. Bioorg Med. Chem. Lett. 51, 128361. doi:10.1016/j.bmcl.2021.128361
Keywords: purinergic receptors, P2X, ion channels, cryo-EM, X-ray crystallography, antagonist, allosteric modulator, structure-based drug design
Citation: Oken AC, Krishnamurthy I, Savage JC, Lisi NE, Godsey MH and Mansoor SE (2022) Molecular Pharmacology of P2X Receptors: Exploring Druggable Domains Revealed by Structural Biology. Front. Pharmacol. 13:925880. doi: 10.3389/fphar.2022.925880
Received: 22 April 2022; Accepted: 10 May 2022;
Published: 17 June 2022.
Edited by:
Shujia Zhu, Shanghai Institute for Biological Sciences (CAS), ChinaReviewed by:
Ye Yu, China Pharmaceutical University, ChinaCopyright © 2022 Oken, Krishnamurthy, Savage, Lisi, Godsey and Mansoor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven E. Mansoor, bWFuc29vcnNAb2hzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.