 Xiangyun Tian1
Xiangyun Tian1 Junjie Zhang
Junjie Zhang Wei Fu
Wei Fu Ruibin Su
Ruibin Su- 1State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Key Laboratory of Neuropsychopharmacology, Beijing Institute of Pharmacology and Toxicology, Beijing, China
- 2Department of Medicinal Chemistry, School of Pharmacy, Fudan University, Shanghai, China
- 3Nanjing University of Chinese Medicine, Nanjing, China
- 4School of Pharmacy, Yantai University, Yantai, China
G protein–coupled receptors can signal through both G proteins and ß-arrestin2. For the µ-opioid receptor (MOR), early experimental evidence from a single study suggested that G protein signaling mediates analgesia and sedation, whereas ß-arrestin signaling mediates respiratory depression and constipation. Then, receptor mutations were used to clarify which residues interact with ligands to selectively regulate signals in a ligand-specific manner. However, there is no systematic study on how to determine these residues and clarify the molecular mechanism of their influence on signal pathways. We have therefore used molecular docking to predict the amino acid sites that affect the binding of ligands and MOR. Then, the corresponding sites were mutated to determine the effect of the structural determinant of MOR on Gi/o protein and ß-arrestin pathways. The pharmacological and animal behavioral experiments in combination with molecular dynamics simulations were used to elucidate the molecular mechanism of key residues governing the signaling. Without affecting ligand binding to MOR, MORY7.43A attenuated the activation of both Gi/o protein and ß-arrestin signaling pathways stimulated by fentanyl, whereas it did not change these two pathways stimulated by morphine. Likewise, the activation peak time of extracellular regulated protein kinases was significantly prolonged at MORY7.43A compared with that at MORwildtype stimulated by fentanyl, but there was no difference stimulated by morphine. In addition, MORY7.43A significantly enhanced analgesia by fentanyl but not by morphine in the mice behavioral experiment. Furthermore, the molecular dynamics simulations showed that H6 moves toward the cellular membrane. H6 of the fentanyl–Y7.43A system moved outward more than that in the morphine–Y7.43A system. Y7.43 mutation disrupted hydrophobic interactions between W6.48 and Y7.43 in the fentanyl–Y7.43A system but not in the morphine–Y7.43A system. Our results have disclosed novel mechanisms of Y7.43 mutation affecting MOR signaling pathways. Y7.43 mutation reduced the activation of the Gi/o protein pathway and blocked the ß-arrestin2 recruitment, increased the H6 outward movement of MOR, and disrupted hydrophobic interactions. This may be responsible for the enhanced fentanyl analgesia. These findings are conducive to designing new drugs from the perspective of ligand and receptor binding, and Y7.43 is also expected to be a key site to structure optimization of synthesized compounds.
Introduction
Opioids, such as morphine and the synthetic opioid fentanyl, are the mainstay analgesics for the treatment of acute and chronic pain in the clinic (Frank and Pollack, 2017; Eidson and Murphy, 2019). Opioid receptors (ORs) are the main target of opioids, and they are divided into three classical subtypes: µ-opioid receptor (MOR), δ-opioid receptor (DOR), and κ-opioid receptor (KOR) (Stein, 2016). They are widely distributed in the central and peripheral nervous systems (Kieffer et al., 1992; Chen et al., 1993; Furst et al., 2020). MOR is predominantly coupled to the Gi/o protein family, which transduces signals by inhibiting the production of cyclic adenosine 5′-monophosphate (cAMP) by adenylyl cyclase (AC) through Gα-subunits and activating G protein–coupled inwardly rectifying potassium channels through Gβγ subunits, among other effectors. MOR signaling is regulated by the phosphorylation of intracellular C-terminal serine and threonine residues, which stabilizes the recruitment of ß-arrestin2, which leads to receptor internalization and additional kinase-driven signaling events. Upon the activation, there are two main downstream signaling pathways of MOR, namely, the G protein–dependent signaling pathway and the ß-arrestin2–mediated signaling pathway (Schmid et al., 2017), which are not only mechanically independent but also pharmacologically separated (Galandrin and Bouvier, 2006). The discovery of ligands that preferentially stimulate G protein or ß-arrestin2 signaling has led to the concept of a “biased agonist.”
It was reported that the G protein–dependent signaling pathway mediates pharmacological effects, such as analgesia and sedation, whereas the ß-arrestin–dependent signaling pathway mediates respiratory depression (Kelly, 2013). At present, the original suggestion that ß-arrestin2 signaling plays a key role in opioid-induced respiratory depression has been questioned (Kliewer et al., 2020; Bachmutsky et al., 2021). Even so, there is still a growing interest in the development of functionally selective G protein–coupled receptor (GPCR) ligands facilitated by hopes of producing more effective drugs with reduced side effects (Wacker et al., 2017). In the development of new opioid analgesic drugs, a milestone was a study published in 2005 on ß-arrestin2 KO mice that reported enhanced morphine analgesia with greatly diminished respiratory depression and constipation (Raehal et al., 2005). The first representative MOR ligand with G protein bias (oliceridine; brand name Olinvyk, previously TRV130) was obtained through a series of methods involving biological screening, analysis of structure–activity relationships, and structural modification (Chen et al., 2013; Soergel et al., 2014). However, recent clinical studies have failed to demonstrate a significantly broader therapeutic window in humans, when compared with the administration of morphine (Hertz, 2018). All the same, it is approved by the Food and Drug Administration for the treatment of moderate to severe acute pain (Tan and Habib, 2021). Then, TRV734, a close analog of TRV130, is an orally bioavailable G protein–biased MOR agonist developed by Trevena and currently in phase I trials (James et al., 2020; De Neve et al., 2021). The data suggested that TRV734 was safe and well tolerated. These studies add valuable information to the increasingly discussed concept of biased agonism at MOR.
Recent studies of the mechanism of opioids (DeWire et al., 2013) indicate that the mechanism of functional selectivity of ORs involves a wide range of interactions. Hothersall et al. (2017) found that the MORW7.35A downregulates ß-arrestin biased signaling of the ligand DAMGO, whereas the MORY7.43F exhibits abrogated ß-arrestin signaling. Using residue mutation experiments, Sun et al. (2017) confirmed that DORW6.48 significantly affects the ß-arrestin pathway but not G protein signaling. In the structure-based optimized synthesis of PZM21, Manglik et al. (2016) found that the disruption of hydrogen bond interactions between the urea and D3.32, Y7.43, and Q2.60 would result in a loss between 30- and 230-fold potency of the compound. For other GPCRs, residues S3.36 and Y7.43 of 5-HT2C impacted ligand-binding pocket structure via hydrogen bond formation (Canal et al., 2011). These results suggest that the mechanism of functional selectivity and signal transduction of ORs is highly complex.
It remains elusive how the agonist binding influences receptor conformational dynamics and then affects the downstream signal pathway of the receptor. To detect the structural determinant controlling the effector coupling in MOR, receptor mutagenesis experiments were conducted here. It was demonstrated to be an effective approach to identifying the molecular switch that controls the signal transduction in a ligand-specific manner (Tschammer et al., 2011; Bock et al., 2012; Fowler et al., 2012; Fenalti et al., 2014; Wootten et al., 2016). It is important to note that evidence from a β2-adrenergic receptor mutation has indicated the residues in the ligand-binding pocket that can mediate agonist-selective effector coupling, thus supporting the hypothesis that qualitative differences in receptor signaling can be regulated at the level of ligand/receptor interactions (Woo et al., 2014). Likewise, mutations in MOR have been increasingly studied in recent years (Hothersall et al., 2017). However, few studies were conducted on how to determine the key residues in MOR that affect the downstream signaling pathways of the receptor. In this study, we first predict the key ligand-binding sites of MOR via molecular docking simulation, investigate the impact of site mutations on signaling pathways, and finally clarify the signal transduction mechanism via molecular dynamics (MD) simulation.
Materials and methods
Plasmids, antibodies, and chemicals
To construct MOR and Y7.43A mutant plasmids, full-length hOPRM1 cDNA was subcloned into the Tag-lite® pT8-SNAP vector (Cisbio, Codolet, France). The hOPRM1 cDNA sequence was obtained from the NCBI website. The Vazyme® Fast Mutagenesis Kit V2 (Vazyme Biotech Co., Ltd., Nanjing, China) was used to introduce the Y7.43A site mutation to the MOR recombinant plasmid using the following primers: F 5′-CTCTAGGTgctACAAACAGCTGCCTCAACCCA-3′ and R 5′-GTTTGTagcACCTAGAGCAATGCAGAAGTGCC-3′. The nucleotide sequences of mutant MOR were confirmed through DNA sequencing and sequence alignment.
Antibodies recognizing phosphorylated p44/42 MAPK (p-ERK1/2), ERK1/2, and ß-arrestin2 were purchased from Cell Signaling Technology (Frankfurt, HE, Germany). Polyclonal rabbit MOR antibody was purchased from Sigma-Aldrich (Darmstadt, Germany). GAPDH monoclonal antibody was purchased from Abcam (Cambridge, United Kingdom). HRP-coupled secondary antimouse and antirabbit antibodies were obtained from Jackson ImmunoResearch (Philadelphia, PA, United States).
Morphine, 7,8-Didehydro-4,5-epoxy-17-methyl-(5α,6α)-morphinan-3,6-diol hydrochloride, item no. 20020201 was purchased from Qinghai Pharmaceutical Factory (Xining, Qinghai, China). Fentanyl, N-Phenyl-N-[1-(2-phenylethyl) piperidin-4-yl] propanamide 2-hydroxypropane-1,2,3-tricarboxylate (1:1) was synthesized by the Military Medical Research Institute (Beijing, China). Forskolin was purchased from Sigma-Aldrich (Darmstadt, Germany). Naltrindole hydrochloride was purchased from Tocris Bioscience (Bristol, United Kingdom).
Molecular modeling and docking
The Homo sapiens active MOR was built by homology modeling with the X-ray structure of Mus musculus active MOR (PDB code: 5C1M), using Discovery Studio 3.5. Align Sequences module was utilized to align Homo sapiens MOR and murine MOR sequences. All water molecules in the X-ray structure were retained. The Ramachandran plot was used to evaluate the validity of the homology models.
The fentanyl and morphine were docked into the 3D structure of Homo sapiens active MOR. Also, DAMGO was involved in docking as a reference agonist. The Induce Fit Docking module of the Schrödinger 3.5 software was used to dock them into the binding site of the active MOR. The two systems were subjected to Monte Carlo Multiple Minimum conformational searches using the OPLS_2005 force field, and the optimal conformations were chosen as binding conformations.
Molecular dynamics simulations
The MD simulations were conducted using Gromacs 5.1.2 package. We built six systems for MD simulations: 1) MOR without ligand (Apo); 2) fentanyl-bound active MOR; 3) morphine-bound active MOR; 4) Y7.43A mutant MOR without ligand (Apo-Y7.43A); 5) fentanyl-bound active Y7.43A mutant MOR; and 6) morphine-bound active Y7.43A mutant MOR. All systems were embedded into the hydrated, equilibrated palmitoyloleoylphosphatidylcholine bilayer using the CHARMM-GUI interface. Sodium and chloride ions were added to neutralize systems and adjusted NaCl concentration to approximately 0.15 mol/l. All systems were minimized and gradually equilibrated in an NPT ensemble at 310 K and 1 bar. The MD simulations were conducted for 100 ns. All analyses of MD trajectories were conducted using tools implemented in the GROMACS 5.1.2 package.
Homogeneous time-resolved fluorescence competitive binding assay
Homogeneous time-resolved fluorescence (HTRF) competitive binding assay was conducted according to the Tag-lite® binding assay recommended protocol (Cisbio, Codolet, France). HEK293T cells were cultured in a DMEM medium (GIBCO, Grand Island, United States) containing 10% fetal bovine serum at 37°C with 5% CO2. They were grown on 60 mm dishes and transiently transfected with 4 μg WT/Y7.43A mutant MOR plasmid using Lipofectamine® 3,000 (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. All cells were labeled in batch with 700 μl/dish SNAP-Lumi4-Tb and suspended in Tag-lite® labeling buffer (TLB), and then, cells were dispensed in a 384 well small volume white microplate (Thermo Scientific Nunc, Waltham, MA, United States) at a density of 5,000 cells/well.
To determine the saturation binding constant Kd of the fluorescent ligand, which is a naltrexone derivative labeled with a red-emitting HTRF fluorescent probe (Cisbio, Codolet, France), cells were incubated with a series of concentrations (final concentration 0.1–100 nM) of the fluorescent ligand in TLB. Nonspecific binding signal wells were incubated with 100 nM Naltrindole hydrochloride. For competition binding experiments, cells were added fentanyl or morphine (final concentration 10−14 M–10−4 M) in competition against the 8 nM fluorescent ligand. All samples were mixed with a final volume of 20 μl and incubated at room temperature for 3 h. After incubation, HTRF signals were measured using Envision Multilabel Reader (PerkinElmer, Waltham, MA, United States) after excitation at 337 nm at both 620 and 665 nm emission, and the HTRF signal was calculated as a two-wavelength signal ratio: [intensity (665 nm)/intensity (620 nm)].
Time-resolved cyclic adenosine 5′-monophosphate measurement assay
Measurement of ligand-induced cAMP production in HEK293T cells transiently expressing MORwildtype/MORY7.43A was performed using the GloSensor cAMP biosensor (Promega, Madison, WI, United States) according to the manufacturer’s protocol. In a word, HEK293T cells were grown on 60 mm dishes and transiently cotransfected with 2 μg MORwildtype/1.58 μg MORY7.43A plasmid and 1.4 μg pGloSensor-22 F cAMP plasmid (Promega, E2301). After 8 h of transient transfection, the cells were digested by trypsin and distributed evenly into the white 96-well poly-D-lysine-coated plate (Thermo Fisher Scientific Nunc, Roskilde, Denmark). After 24 h, cell media were replaced by 90 ml of fresh full DMEM with 2% v/v GloSensor cAMP Reagent (Promega, E1290) and incubated for 120 min at 37°C. Firefly luciferase activity was measured using Envision Multilabel Reader. After initial measurement of the baseline signal for 15 min, cells were stimulated with 10 μl of 10× drug solution of the MOR agonist fentanyl or morphine (final concentration 10−12 M–10−4 M) for 10 min. Then, 1 μl of forskolin (final concentration 10 μM) solution was added for 15 min, and the real-time chemiluminescent signal was measured at once. The inhibition of cAMP production was calculated using the baseline signal, drug response signal, and forskolin signal. Then, sigmoidal dose–response curves with variable slope (three-parameter logistic regression) were plotted. Results are from three independent experiments.
Time-resolved ß-arrestin2 recruitment assay
Coupling between MOR and ß-arrestin2 was monitored using a NanoBiT protein:protein interaction assay (Promega, M2014). MORwildtype and MORY7.43A were cloned into pBiT2.1-N [TK/SmBiT] vector and ß-arrestin2 into pBiT1.1-C [TK/LgBiT] vector. For the negative control, the MOR-SmBiT construct was substituted with the NanoBiT negative control vector (HaloTag-SmBiT). The positive control consisted of SmBiT-PRKACA and LgBiT-PRKAR2A vectors; 2.4 μg WT/Y7.43A-SmBiT together with 0.8 μg LgBiT-β-arrestin2 were used to transiently transfect HEK293T cells in 60 mm dish using Lipofectamine 3,000 according to the manufacturer’s protocol. After 8 h, the cells were digested by trypsin and distributed evenly into the white 96-well poly-D-lysine-coated plate. The next day, 30 min prior to the assay, the culture medium was substituted for 100 μl/well serum-free Opti-MEM medium (GIBCO, Grand Island, United States). The nanoluciferase activity was measured using Envision Multilabel Reader. After the initial measurement of the background signal, 25 μl/well Nano-Glo® Live Cell Assay System (Promega, N2013) was added and cells were equilibrated and basal luciferase activity was measured for 15 min. Then, cells were stimulated with 10 μl of 13.5× drug solution of the MOR agonist fentanyl or morphine (final concentration 10−12 M–10−5 M), and a chemiluminescent signal was measured for 15 min (5 min intervals). The ß-arrestin2 recruitment was calculated using the baseline signal and the drug response signal. Then, sigmoidal dose–response curves with variable slope (three-parameter logistic regression) were plotted. Results are from three independent experiments.
Quantitative real-time polymerase chain reaction analysis
The transfected samples for the ß-arrestin2 recruitment assay were seeded in six-well plates. The next day, RNA was extracted using TRIzol (Sigma-Aldrich, St. Louis, MO, United States). cDNA was synthesized using the RevertAid first strand cDNA synthesis kit (Thermo Fisher Scientific Baltics UAB, Vilnius, Lithuania). Relative MORwildtype and MORY7.43A expressions were determined on 250 ng cDNA using SYBR Green quantitative polymerase chain reaction (PCR) reagents (Thermo Fisher Scientific Baltics UAB, Vilnius, Lithuania), normalized to GAPDH expression levels. The forward primer of MORwildtype/MORY7.43A was GCCCTTCCAGAGTGTGAATTAC, the reverse primer of MORwildtype/MORY7.43A was GTGCAGAGGGTGAATATGCTG, the forward primer of GAPDH was AGGTCATCCCAGAGCTGAACG, and the reverse primer of GAPDH was TCAGATGCCTGCTTCACCAC.
Western blot analysis
After 24 h of transient transfection of MORwildtype and MORY7.43A plasmids into HEK 293T cells, the transfected cells were starved in serum-free DMEM medium for 2 h. Then, the cells were exposed to 10 μM fentanyl and morphine for an indicated time. Immediately following stimulation, all cells were lysed in an ice-cold RIPA lysis buffer (50 mM pH 7.4 Tris, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail and phosphatase inhibitor (Roche, Basel, Switzerland). Protein samples were resolved by 10% SDS-PAGE and transferred onto PVDF membranes (Millipore, Carrigtwohill, Co. Cork, Ireland). After the membranes were blocked with 5% milk, they were incubated overnight with primary antibodies against p-ERK1/2, ERK1/2, MOR, and GAPDH at 4°C. The membranes were washed with TBST and incubated with the secondary antibody for 1 h at room temperature. The immune complexes were detected using western blot detection reagents (Millipore, Burlington, MA, United States).
Mice
MOR knockout mouse strain B6.129S2-Oprm1tm1Kff/J (MOR−/−, RRID: IMSR_JAX:007,559) was obtained from The Jackson Laboratory. Mice were genotyped via PCR analysis of genomic tail-biopsy DNA using the following primers: 5′-GCCAGAGGCCACTTGTGTAG-3′, 5′-ATCTTCACCCTCTGCACCAT-3′, and 5′-TGCTGGGCTCAAGCTTTAAT-3’. JAX™C57BL/6J (RRID: IMSR_JAX:000,664) mice from The Jackson Laboratory were used as wild-type control. Animals were housed four to six per cage under a 12 h light–dark cycle (lights on at 8 a.m.) and ventilated at a controlled temperature with ad libitum access to food and water. In hot plate experiments, we used female mice aged 8–16 weeks because the genitals of male mice touched the hot plate, which can produce an allergic reaction. Each animal was used only once for each dose and each drug tested. Studies were conducted in parallel such that age-matched mice received the same drug treatment at the same time. All experimental protocols were approved by and conducted in accordance with the Animal Care and Use Committee of the Beijing Institute of Pharmacology & Toxicology (Beijing, China).
AAV package, intracerebroventricular injection, and expression in central nervous system
The AAV (BrainVTA Co., Ltd., Wuhan, Hubei, China) that overexpressed MORwildtype/MORY7.43A was rAAV2-EF1α-EGFP-WPRE-hGH polyA, AAV-PHP.eB vector containing MORwildtype/MORY7.43A insert of mice.
The heads of mice were firmly fixed by the thumb and index finger. A 25 µl tip microsyringe was used to slowly inject AAVs (5 µl) into the right cerebral lateral ventricle (at coordinates −1.0 mm mediolateral, −0.5 mm anteroposterior from Bregma; −2.0 mm dorsal–ventral from the skull) (Craft et al., 2005).
Twenty-one days following virus injection, mice were sacrificed through transcardial perfusion of deeply anesthetized mice using intraperitoneal-injected 1% sodium pentobarbital. Mice were perfused with 0.9% saline followed by 4% paraformaldehyde in 0.1 M PO4, after which whole-brain dissections were performed. Brains were postfixed overnight in 4% paraformaldehyde in 0.1 M PO4 followed by incubation for a minimum of 48 h in 30% sucrose in 0.1 M PO4 (Kofoed et al., 2021). Fifty-micrometer sections were cut on a sliding microtome with a freezing stage (Leica Biosystems, Buffalo Grove, IL, United States) (Rincon et al., 2018). Sections were collected in a 24-well plate filled with antifreeze (PBS: glycerol: ethylene glycol = 5:2:3) and stored in a refrigerator at −20°C and were scanned using Olympus VS.120.
Hot plate test
Opiate effects on paw withdrawal latencies were assessed as the time to response (licking hind paw, quick pedaling, and jumping (s)) after placement on a hot plate maintained at 55 ± 0.5°C (Shao et al., 2020). To avoid tissue damage, we used a 60 s cutoff. The hot plate test was conducted 15 min (fentanyl) or 30 min (morphine) after drug administration and expressed as the percent maximum possible effect (%MPE), calculated as follows: %MPE = [(drug response latency−basal response latency)/(60 s−basal response latency)] ×100. For the analgesic model, after testing basal response time, mice were subcutaneously (s.c.) injected with fentanyl (0.1 mg/kg) or morphine (10 mg/kg). The time courses for antinociception were determined at various time points after drug treatment.
Statistical analysis
Data are presented as mean ± SEM of at least three individual experiments. For HTRF competitive binding assay, concentration–response curves were fit to one-site binding models provided in GraphPad Prism software to determine Kd and Ki. For EC50 values of concentration–response curves, the best-fit line was generated following nonlinear regression analysis, as described in each section above. The results of the HTRF competitive binding assay, cAMP measurement assay, and ß-arrestin2 recruitment assay were evaluated via an unpaired two-tailed t-test. For the hot plate test experiment, data were analyzed using two-way ANOVA, followed by Bonferroni’s post hoc tests. In all the experiments, a value of p < 0.05 was considered significant and all calculations were performed using GraphPad Prism 8.0 software.
Results
Y7.43 site was involved in the interaction of fentanyl and morphine with µ-opioid receptor
The 3D model of Homo sapiens active OR was obtained by homology modeling with the X-ray structure of Mus musculus active MOR (PDB code: 5C1M) and is shown in Figure 1A. The sequence identity and sequence similarity between active Homo sapiens MOR and murine MOR were 95.2% and 99.6%, respectively. In the Ramachandran plot, 96.7% amino acids were located in the most favored regions and 3.3% amino acids were located in additional allowed regions (Figure 1B). The snake diagram of MOR with residue Y7.43 highlighted is shown in Figure 1C.
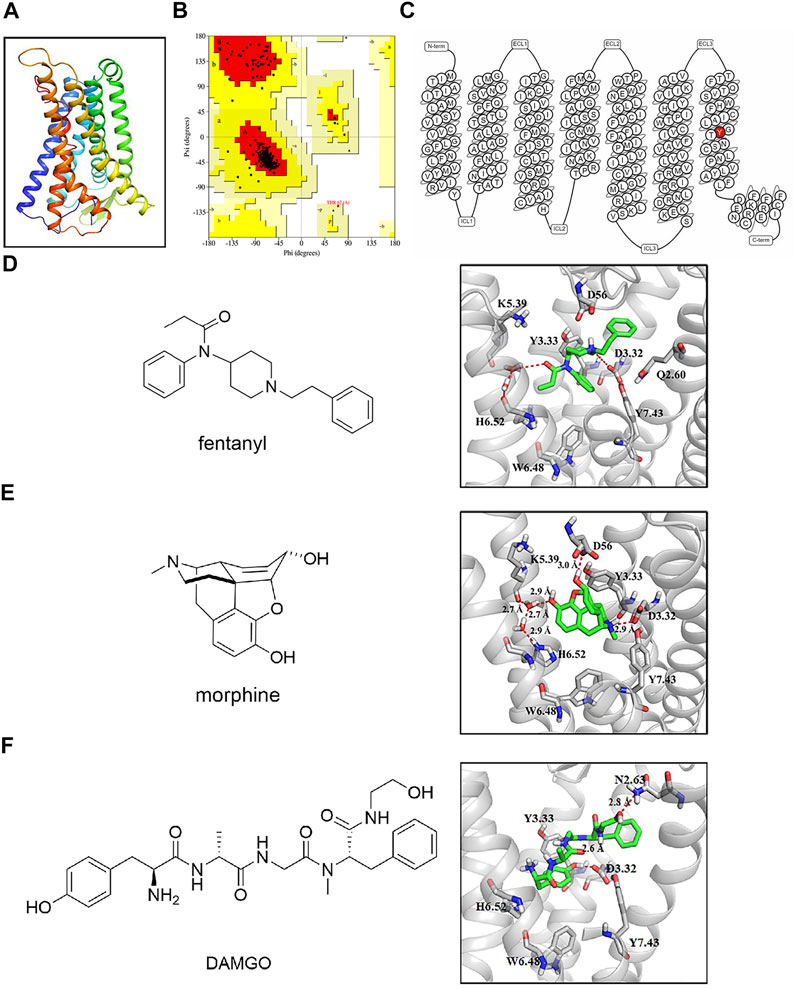
FIGURE 1. Binding mode of agonists with active µ-opioid receptor (MOR). (A) three-dimensional structure model of Homo sapiens MOR. (B) Ramachandran plot of active MOR model from active Mus musculus MOR. (C) snake diagram of MOR with residue Y7.43 highlighted in magenta. Image created from www.gpcrdb.org. (D–F) binding mode of agonists with active MOR. Active MOR is shown in gray ribbons; residues are shown as a gray ball–stick model. Hydrogen bond is shown as a dashed line. Agonists are shown as a green ball–stick model.
The molecular docking diagram showed residues of MOR that interact with agonists. The most reasonable conformation of fentanyl in MOR was selected in our docking process based on the following criteria: 1) the lowest binding free energy and 2) the most frequently binding pose. The docking pose of fentanyl was subsequently optimized through MD simulation. It is consistent with the message-address concept: the protonated amine of the piperidine ring in fentanyl formed electrostatic interaction with the acidic D3.32 (message site), and the n-alkyl phenyl formed extensive hydrophobic contacts with transmembrane helices (TM) 2/3 (address site). The result was consistent with de Waal’s studies (de Waal et al., 2020). Also, the DAMGO participates in the docking process as a reference agonist. As shown in the binding mode with MOR in Figures 1D–F, the protonated nitrogen of three compounds engaged in a strong salt bridge with the carboxyl oxygen atom of D3.32, and the oxygen of amide of fentanyl and the phenolic hydroxyl of morphine participated in the hydrogen-bonding network with H6.52 via two water molecules. In addition, D3.32 forms the bidentate hydrogen bonds with Y7.43. As a critical site, D3.32 formed conserved ionic bonds with the protonated ligands. This interaction was observed in structures of the MOR, DOR, KOR, and nociceptin receptor bound to ligands of different scaffolds (Manglik et al., 2016). Therefore, Y7.43 was selected for a mutation to study the mechanism of activating signaling pathways.
Y7.43 site mutation did not change the affinity of ligands in binding with µ-opioid receptor
The binding affinity of fentanyl and morphine in membranes from cells expressing MORwildtype/MORY7.43A was estimated using a competitive TR-FRET (time-resolved fluorescence resonance energy transfer)–based assay (Perez-Aguilar et al., 2013), and derived pKi values are compiled in Table 1. From these experiments, the Kd of fluorescent ligand was estimated as (13.81 ± 2.69) and (35.49 ± 2.97) nM, respectively, at MORwildtype and MORY7.43A. In addition, we determined the expression of MORwildtype and MORY7.43A. There was no statistical difference in both whole cell and cell membrane via western blot and fluorescence labeling, respectively (Supplementary Figures S1A,B). In conclusion, the relative affinity of morphine at MORY7.43A was 101% of MORwildtype pKi, and the pKi value of fentanyl was 91% of MORwildtype pKi.
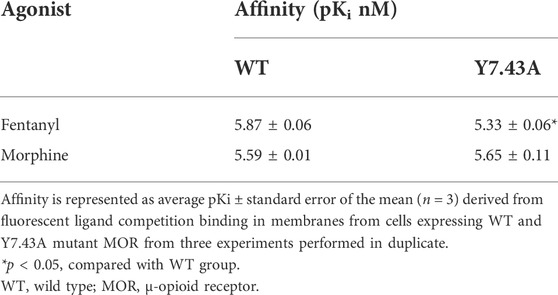
TABLE 1. Binding affinity of the agonists of wild type and Y7.43A mutant µ-opioid receptor
Y7.43 site mutation reduced the activation of µ-opioid receptor–mediated Gi/o protein–dependent signaling stimulated by fentanyl
To explore the effect of Y7.43 mutation on the activity of the Gi/o protein pathway downstream of MOR, we set up a GloSensor cAMP bioassay by transiently transfecting the plasmid pGloSensor-22F™ into HEK293T cells (Gilissen et al., 2015). The cAMP production was detected by this bioassay. The results showed that EC50 of fentanyl was (546.67 ± 22.78) nM for MORY7.43A, which is much higher than that observed for MORwildtype (0.53 ± 0.15) nM. By contrast, the EC50 of morphine was (291.13 ± 181.44) nM for MORY7.43A, with no significant difference between the MORY7.43A and MORwildtype (119.94 ± 52.02) nM (Figures 2D, E, and Table 2). Next, we determined the expression of MORwildtype and MORY7.43A. There was no statistical difference in both whole cell and cell membrane based on western blot and fluorescence labeling, respectively (Supplementary Figures S1C,D), indicating that the MORY7.43A could weaken the activity of the Gi/o protein pathway stimulated by fentanyl.
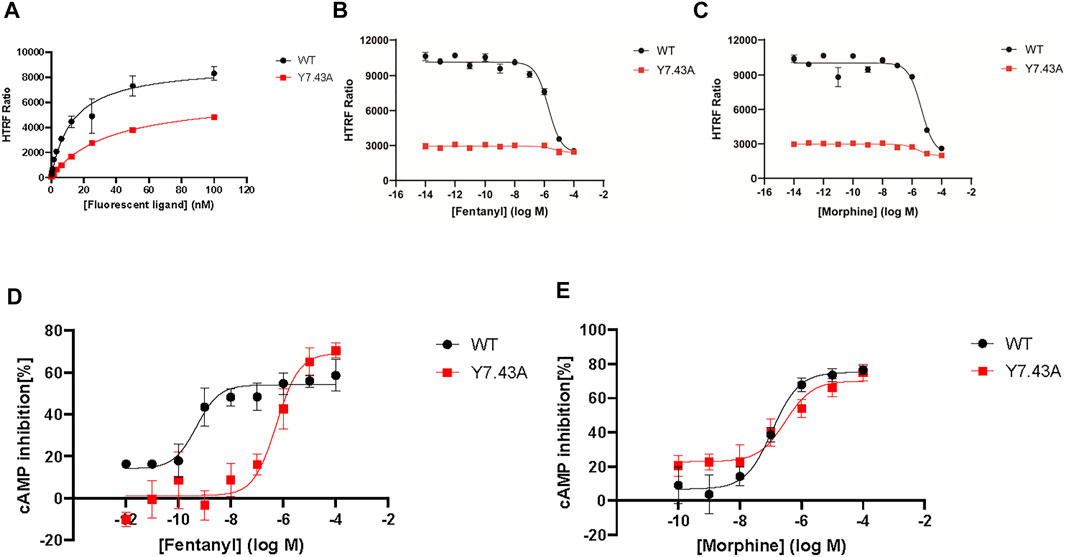
FIGURE 2. Effect of fentanyl and morphine on 10-μM forskolin-stimulated cyclic adenosine 5′-monophosphate (cAMP) accumulation in wild type (WT) and Y7.43A mutant µ-opioid receptor (MOR). (A) fluorescent ligand saturation binding of WT and Y.43A mutant MOR. (B) homogeneous time-resolved fluorescence (HTRF) competition binding of fluorescent ligand to fentanyl (WT: pKi = 5.87 ± 0.06 nM; Y7.43A: pKi = 5.33 ± 0.06 nM). (C) HTRF competition binding of fluorescent ligand to morphine (WT: pKi = 5.59 ± 0.01 nM; Y7.43A: pKi = 5.65 ± 0.11 nM). (D,E) dose–response curves for fentanyl- and morphine-induced cAMP production for WT (fentanyl: EC50 = 0.53 ± 0.15 nM, Emax = 54.18 ± 1.86%; morphine: EC50 = 119.94 ± 52.02 nM, Emax = 75.28 ± 3.09%) and Y7.43A mutant MOR transfected cells (fentanyl: EC50 = 546.67 ± 22.78 nM, Emax = 69.00 ± 0.58%; morphine: EC50 = 291.13 ± 181.44 nM, Emax = 69.86 ± 3.93% compared with WT). Data represented as mean ± standard error of the mean from three independent experiments; the statistical significance of differences between WT and Y7.43A mutant MOR analyzed using an unpaired two-tailed t-test.
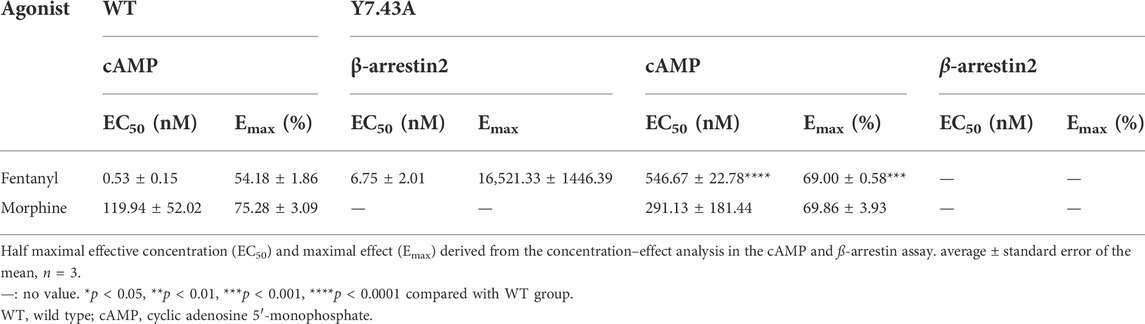
TABLE 2. Activation parameters of test agonists to stimulate µ-opioid receptor–mediated Gi/o protein and ß-arrestin signaling pathways
Y7.43 site mutation blocked ß-arrestin2 recruitment to µ-opioid receptor and affected downstream signaling
To further explore Y7.43 site mutation effects on the ß-arrestin–dependent signaling pathway, we focused on the interaction between MOR and ß-arrestin2 using NanoBiT assay. The results showed that MORY7.43A markedly inhibited the ß-arrestin2 recruitment induced by fentanyl (Figure 3A), whereas MORY7.43A did not change the status of ß-arrestin2 induced by morphine. That is, MORwildtype and MORY7.43A failed to recruit ß-arrestin2 (Figure 3B). We also confirmed that the MORwildtype and MORY7.43A were successfully expressed using quantitative real-time PCR analysis (Figure 3C). ERK plays a crucial role in the regulation of MOR signaling and opioid analgesia (Ruan et al., 2019; Zhang et al., 2020). MOR-mediated ERK1/2 activation is involved not only in G protein–dependent signaling but also in ß-arrestin–dependent signaling (Rozenfeld and Devi, 2007). Therefore, we monitored the magnitude and duration of ERK1/2 phosphorylation stimulated by fentanyl and morphine. For MORwildtype and MORY7.43A induced by morphine, the activation peaks of ERK1/2 were similar (Figures 3D,E). Combined with the similar degree of Gi/o protein signaling pathway activation, it was suggested that morphine-mediated ERK1/2 phosphorylation was G protein–dependent rather than relying on ß-arrestin. The results also showed that the peak activation of MORwildtype ERK1/2 by fentanyl was at 3 min time point, whereas MORY7.43A prolonged the activation peak time that appeared at 15 min. Combined with the fact that MORY7.43A significantly reduced the activity of the Gi/o protein pathway and inhibited the ß-arrestin2 recruitment induced by fentanyl, this indicates that there is a relationship between ERK1/2 activation and ß-arrestin2 downstream of MOR stimulated by fentanyl. These results demonstrated that Y7.43 may be an important amino acid site affecting fentanyl-mediated ß-arrestin2 recruitment and ERK1/2 activation downstream of MOR.
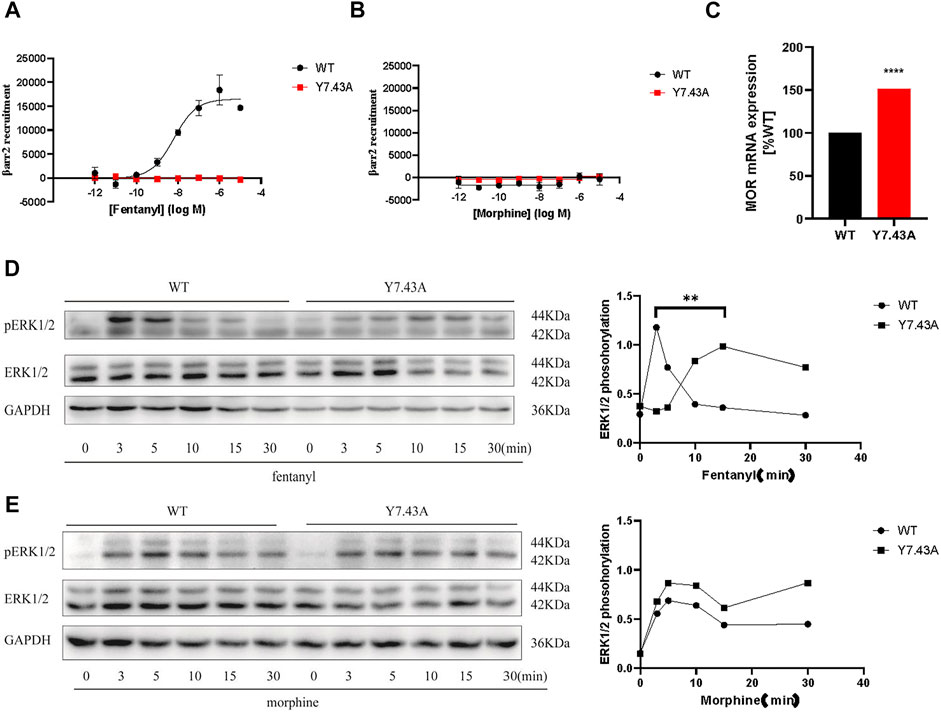
FIGURE 3. Effect of ß-arrestin2 recruitment stimulated by fentanyl and morphine in wild type (WT) and Y7.43A mutant µ-opioid receptor (MOR). (A,B) dose–response curves for fentanyl- and morphine-induced ß-arrestin2 recruitment for WT (fentanyl: EC50 = 6.75 ± 2.01 nM, Emax = 16,521.33 ± 1446.39) and Y7.43A mutant MOR transfected cells. (C) receptor cell surface expression via quantitative real-time polymerase chain reaction assay. Data represented as mean ± standard error of the mean from three independent experiments; the statistical significance of differences between WT and Y7.43A mutant MOR analyzed using an unpaired two-tailed t-test (p**** <0.0001). (D,E) representative western blots and quantification of ERK1/2 phosphorylation by WT and Y7.43A mutant MOR before (−) and after (+) stimulation by (D) fentanyl and (E) morphine in a time-course experiment; GAPDH used as a loading control; the quantification of ERK1/2 phosphorylation refers to the pERK/ERK ratio. * indicates statistically significant differences at the peak time point of ERK1/2 activation compared with WT.
µ-opioid receptorY7.43A enhanced fentanyl analgesia
To study the Y7.43 site mutation for analgesia in vivo, we conducted the hot plate test in mice expressing MORY7.43A. We created suitable mutant mouse models, which were injected with MORwildtype/MORY7.43A AAV in the right cerebral lateral ventricle of MOR KO mice. The absence of MOR protein expression was confirmed by the mice-tail genotyping (Figure 4A). We also confirmed that MOR KO mice injected with fentanyl and morphine had the completely opposite analgesic effect to WT mice given the same dose, whereas WT and KO mice showed no difference in basic response to hot plate (WT, 11.30 ± 1.48 s, n = 8; KO, 11.33 ± 2.39, n = 9; Figures 4B–D).
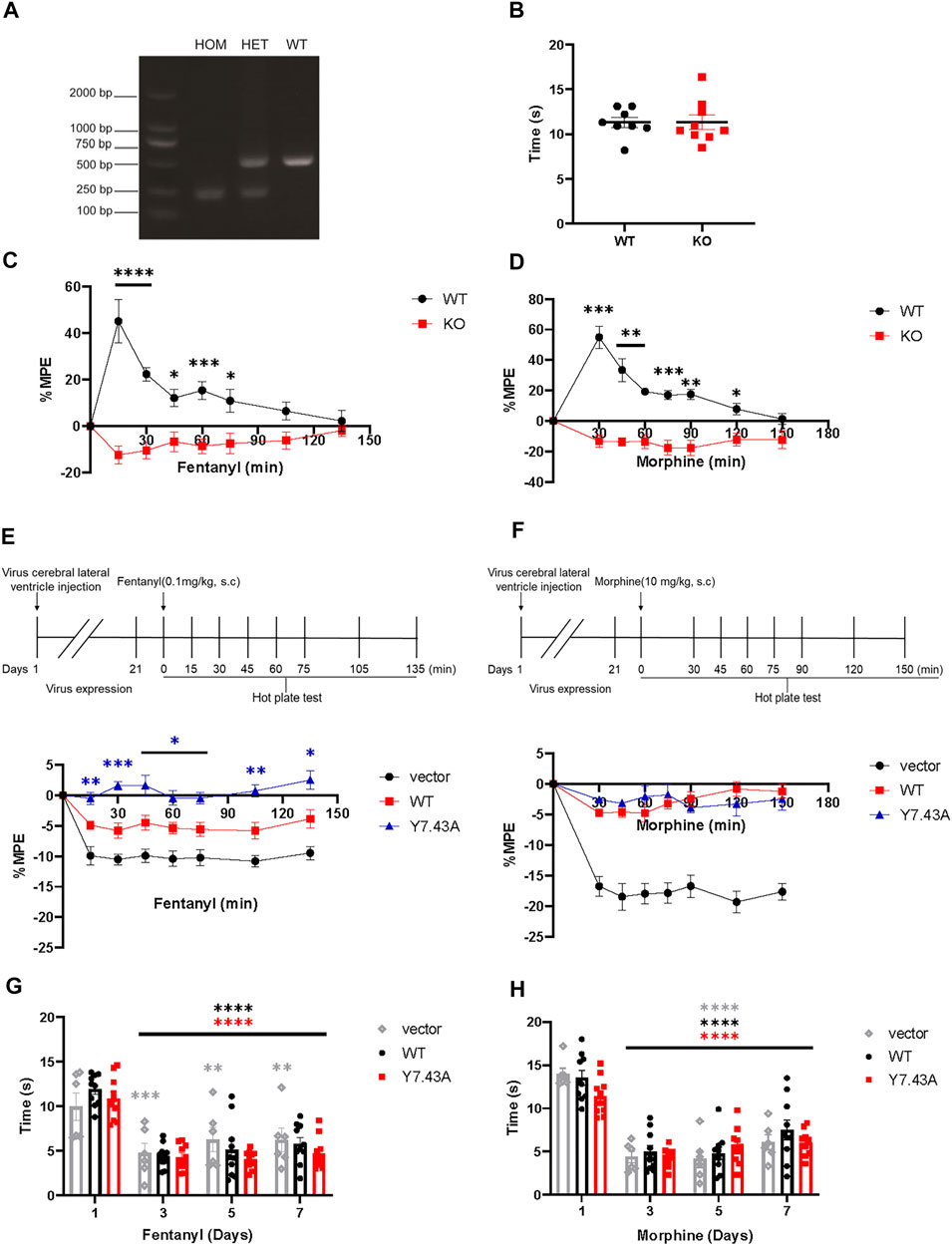
FIGURE 4. Results of hot plate test. (A,B) verification of µ-opioid receptor (MOR) KO mice. (A) mice were genotyped via polymerase chain reaction analysis of genomic tail-biopsy DNA using the following primers: 5′-GCCAGAGGCCACTTGTGTAG-3′, 5′-ATCTTCACCCTCTGCACCAT-3′, and 5′-TGCTGGGCTCAAGCTTTAAT-3′. (B) baseline of pain threshold of MOR KO or JAX™C57BL/6J wild type (WT) control mice (WT, 11.30 ± 1.48 s, n = 8; KO, 11.33 ± 2.39 s, n = 9). (C,D) antinociceptive effect of MOR KO mice given 0.1 mg/kg fentanyl (C) and 10 mg/kg morphine (D) was completely opposite to WT mice. Significant differences of analgesia between MOR KO and WT groups are indicated by asterisks: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (n = 7). Two-way analysis of variance (ANOVA) with Bonferroni post hoc test; MOR KO mice aged 8–16 weeks were injected with AAV-wildtype and AAV-Y7.43A allowed 21 days for virus expression. (E,F) acute antinociceptive response measured in the mice hot plate test 15 min (fentanyl) or 30 min (morphine) after drug administration. Nociceptive latencies were defined by licking, stomping, or jumping and are reported as percent maximum possible effect with a 60 s cutoff. Analgesic time course of acutely administered fentanyl (E), (Fgenotype (2, 27) = 59.79, p < 0.0001; Ftime (5.310, 131.2) = 11.22, p < 0.0001) or morphine (F), (Fgenotype (2, 27) = 71.41, p < 0.0001; Ftime (5.151, 131.7) = 20.58, p < 0.0001) was repeated after fentanyl and morphine administration, respectively, for the indicated times. Significant differences of analgesia between WT and Y7.43A groups are indicated by asterisks: *p < 0.05, **p < 0.01, ***p < 0.001 (n = 10). Two-way ANOVA with Bonferroni post hoc test. (G,H) administration drugs for 7 consecutive days, the change of basal pain threshold in MOR KO mice injected with AAV-vector, AAV-WT, and AAV-Y7.43A. Mice were given fentanyl and morphine twice daily (8 a.m. and 6 p.m.) for 7 days, and then, baseline pain thresholds were measured before drug administration on the first, third, fifth, and seventh days. Basal pain thresholds on the third, fifth, and seventh days were significantly lower than on the first day (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10). Two-way ANOVA with Bonferroni post hoc test.
After 21 days of AAV injection, MORwildtype and MORY7.43A were expressed in the whole brain of mice (Supplementary Figure S2), indicating the mouse models were built successfully. MORwildtype partially rescued the fentanyl and morphine analgesia compared with the vector AAV group. The MORY7.43A group significantly increased the %MPE of fentanyl compared with MORwildtype, but there was no difference in the development of morphine analgesia (Figures 4E,F). Altogether, these observations revealed that the Y7.43 site was associated with fentanyl analgesia.
Activation mechanism of µ-opioid receptor by fentanyl and morphine
The available results suggested that Y7.43 is a key residue for differential changes of downstream signal pathways of MOR stimulated by fentanyl and morphine. MORY7.43A enhanced analgesia of fentanyl but not morphine. To investigate why Y7.43 had varying impacts on different types of MOR agonists, we used MD simulations to investigate the mechanism of fentanyl and morphine activating MOR-mediated signaling.
The whole simulation process lasted for 100 ns, and all the systems remained stable (Supplementary Figure S2). In the fentanyl–receptor system, Y7.43 site mutation disrupted hydrophobic interactions between W6.48 and Y7.43 (Figures 5A,B). In terms of morphine–WT and morphine–Y7.43A systems, the hydrophobic interactions have been maintained (Figures 5C,D). In addition, the conserved P5.50-I3.40-F6.44 triad, an important activation switch that stayed at the bottom of the binding pocket of the agonist, rearranged after the binding of the agonist with MOR (Huang et al., 2015; Huang et al., 2021). As shown in Figure 6, F6.44 moved outwardly toward the cellular membrane (intracellular view), leading to the 7.3 and 8.4 Å outward movement of H6 in the fentanyl–WT and fentanyl–Y7.43A systems, respectively. This result suggested that the Y7.43 site mutation slightly increased the outward migration of H6 compared with WT. In the morphine–receptor activated system, H6 moved outward 6.3 Å after site mutation, which was less than 8.9 Å in the WT system (intracellular view). Besides, in the fentanyl–WT and fentanyl–Y7.43A systems, we also found the degree of I3.40 flipping was consistent compared to the apo state, while it varied in the two activated states of morphine binding with a receptor. In summary, the binding of fentanyl with MORY7.43A broke up hydrophobic interactions and rearranged the conserved P5.50-I3.40-F6.44 triad. The rearrangement of the conserved P5.50-I3.40-F6.44 triad induced the outward movement of H6, which ultimately may lead to a differential impact on signal pathways by agonists stimulating the receptor.
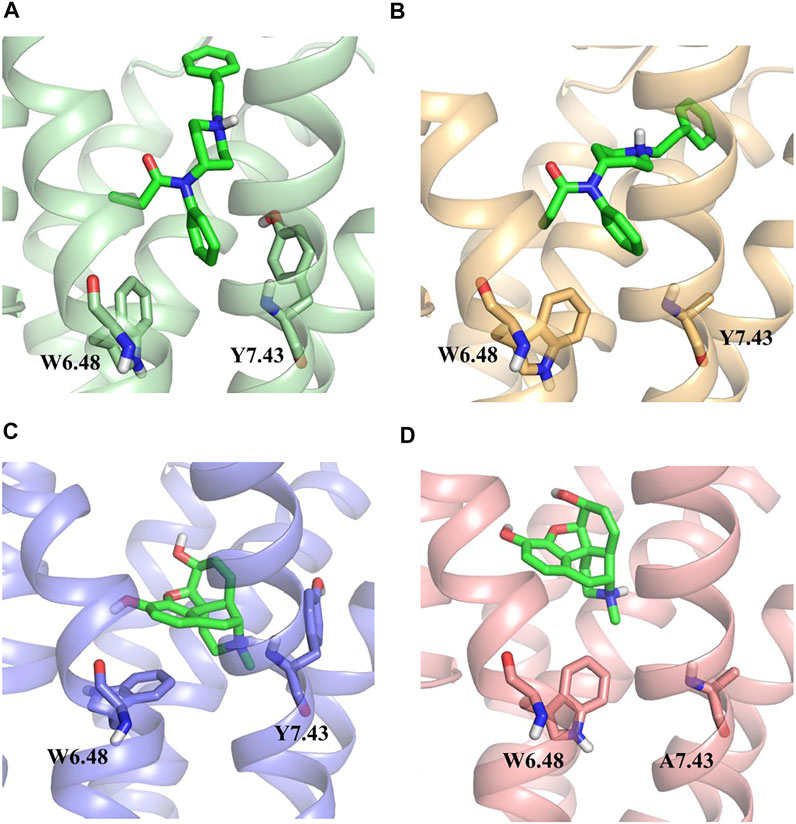
FIGURE 5. Hydrophobic interactions between W6.48 and Y7.43 during simulations. (A,B) hydrophobic interactions between W6.48 and Y7.43 during the binding process of fentanyl with µ-opioid receptor (MOR). Residues are shown in the ball–stick model; wild type (WT), green ribbon; Y7.43A, yellow ribbon. (C,D) hydrophobic interactions between W6.48 and Y7.43 during the binding process of morphine with MOR. Residues are shown in the ball–stick model; WT, light blue ribbon; Y7.43A, pink ribbon.
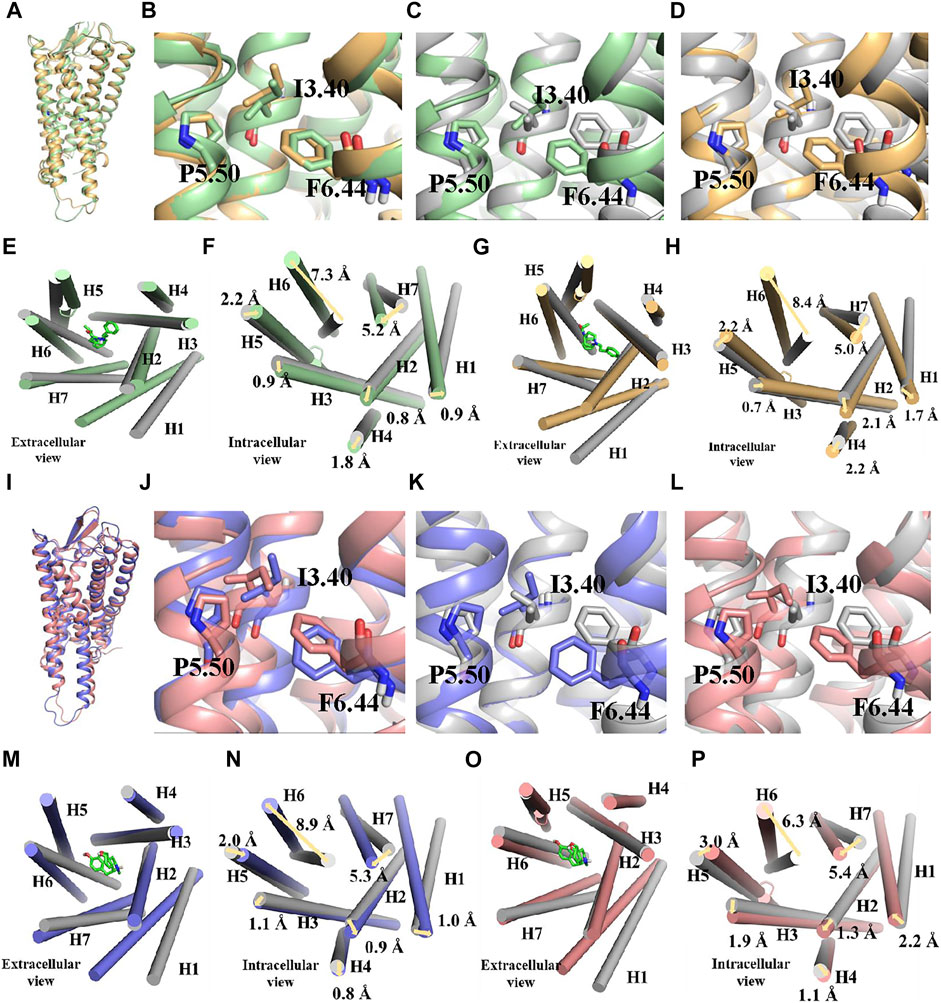
FIGURE 6. Conserved P5.50-I3.40-F6.44 triad rearrangement and transmembrane helix change during simulations. (A–H) binding poses and changes of transmembrane helix during the binding process of fentanyl with µ-opioid receptor (MOR). Active wild type (WT), green; active Y7.43A, yellow; apo state, gray; they are shown in ribbon and cylindrical shape in binding pose and transmembrane helix, respectively. Residues are shown in the ball–stick model. (I–P) binding poses and changes of transmembrane helix during the binding process of morphine with MOR. Active WT, blue; active Y7.43A, pink; apo state, gray; they are shown in ribbon and cylindrical shape in binding pose and transmembrane helix, respectively. Residues are shown in the ball–stick model.
Discussion
In our studies, we first predicted the key amino acid residues playing an important role in agonists binding to the receptor by molecular docking. We then constructed MORY7.43A to characterize the binding affinities and functional activities of fentanyl and morphine. The conventional hot plate test was used to determine the analgesic effect in vivo when the Y7.43 site was mutated. It showed that MORY7.43A reduced the activation of Gi/o protein–dependent signaling, blocked ß-arrestin2 recruitment, and altered the kinetics of ERK1/2 phosphorylation induced by fentanyl. That is, MORY7.43A delayed the occurrence of maximum ERK1/2 phosphorylation induced by fentanyl, compared with the MORwildtype group. It is worth noting that we observed that MORY7.43A mice exhibited significantly greater analgesic potency of fentanyl than MORwildtype. Moreover, compared with WT, Y7.43 site mutation broke up the hydrophobic interactions and promoted the outward movement of H6 in MOR stimulated by fentanyl but not morphine. Taken together, we show for the first time that Y7.43 site mutation reduced Gi/o protein signaling and blocked ß-arrestin2 recruitment, which may engage the enhanced analgesia induced by fentanyl.
Functional activity at MOR was characterized using cell-based assays designed to measure Gi/o protein signaling and ß-arrestin2 recruitment. In general, it is necessary to measure the affinity of MOR and ligands in determining the functional activity of MOR. In our research, we used HTRF competitive binding assay to determine the affinity of drugs in binding with MOR. Figure 2A shows the affinity of fluorescent ligand binding with MOR, and the Kd value was obtained from this curve. Figures 2B,C reflects the ability of drugs to compete with fluorescent ligand to bind to MOR, and the IC50 of drugs was obtained from this curve. The Ki value was calculated as the Cheng–Prusoff equation:
Recent research has clarified how and where a ligand should bind with a receptor to cause adequate signaling activation. Y7.43 of MOR TM7 forms a ligand-binding pocket with W7.35 as highlighted by recent receptor crystallography data (Manglik et al., 2012; Huang et al., 2015) and previous mutagenesis and docking studies (Subramanian et al., 2000; Metzger et al., 2001). Molecular docking studies found that Y7.43 engaged in a hydrogen bond with D3.32. The interaction between D3.32 residue and the protonated nitrogen of fentanyl and morphine combined with hydrogen bond orients the molecules. These interactions have been proposed to be important for ligand-specific binding with MOR. In our results, receptor mutation functioned differently by fentanyl and morphine in signal transduction. MORY7.43A did not change both cAMP and ß-arrestin2 signaling response by morphine, as it was similar to MORwildtype. In complete contrast, decreased cAMP activity and no ß-arrestin2 activity were discovered at MORY7.43A by fentanyl.
In addition, unremitting efforts are continuously put into research on the agonist-GPCR binding mode. In MD simulations, we also observed three other indexes except for hydrophobic interaction and P5.50-I3.40-F6.44 triad rearrangement, which were mentioned in the result section. The first index is a three to seven lock, which was formed by the hydrogen bond between D3.32 and Y7.43, and plays a key role in the activation of MOR (Cheng et al., 2018). As shown in Supplementary Figure S4, the hydrogen bonds were unstable and broken in fentanyl–WT and morphine–WT systems, respectively, and they disappeared because of the Y7.43 mutation in the fentanyl/morphine–Y7.43A system. Then, we monitored the ionic interaction between D3.32 and protonated nitrogen of agonists (Supplementary Figure S5), which facilitates the binding of an agonist with MOR (Pasternak and Pan, 2013; Huang et al., 2021). The third index is the hydrogen bond of T6.34-R3.50, which regulates conformation, and may stabilize the receptor in an inactive state (Jones et al., 1995; Lu et al., 1997; Rasmussen et al., 1999; Alewijnse et al., 2000; Scheer et al., 2000; Ohyama et al., 2002; Schneider et al., 2010; Manglik et al., 2012). As shown in Supplementary Figure S6, the hydrogen bonds were always broken during all stimulations in four systems. Therefore, the above three monitored indexes showed no difference in the binding process between agonists and receptors. Therefore, the hydrophobic interaction and the outward movement in H6 were significant clues in activating different signaling transduction mediated by fentanyl and morphine.
β-arrestin was previously thought to be the regulatory factor involved in endocytosis and desensitization of receptors (Luttrell and Lefkowitz, 2002). In studies spanning more than a decade, researchers have shown that the carboxyl-terminal phosphorylated MOR stimulated by agonists recruits ß-arrestin2, which may drive many of the pharmacological and unwanted side effects of MOR activation (Raehal et al., 2011; Bu et al., 2015). However, the molecular mechanisms initiating these effects remained unresolved. In our results, the ß-arrestin assay has revealed that Y7.43 site mutation was sufficient to abolish ß-arrestin2 recruitment. Fentanyl-enhanced analgesia was observed in MOR KO mice with rescued MORY7.43A, this could be because of the lack of ß-arrestin2 recruitment as the ß-arrestin assay showed. This is consistent with the conclusion that carboxyl-terminal phosphorylation site S375 mutation aimed at reducing ß-arrestin2 recruitment to MOR increased the analgesic potency of fentanyl but not morphine (Kliewer et al., 2019). Besides, ß-arrestin2 recruitment is also thought to be initiated by the phosphorylation of Tyrosine residues on the carboxyl-terminal tail following receptor activation. Next, we will focus on the relationship between Y7.43 and carboxy-terminal phosphorylation of MOR.
Morphine and fentanyl, as MOR agonists, were tested for analgesic potency at fixed doses. For the control JAX™C57BL/6J group, we found that fentanyl-induced analgesic rate peaked at 15 min after administration and disappeared after 135 min, compared with 30 and 150 min for morphine, respectively (Figures 4C,D). Fentanyl has a rapid onset and then rapid decay, whereas morphine has the opposite, which aligns with previous research in clinical profiles and animal studies (Gillis et al., 2020). It is of interest that MOR KO mice injected with AAV were sensitized when they were treated with fentanyl and morphine, which was completely different from MOR KO mice. Sensitivity and mania are expressed as jumping quickly in a hot plate apparatus and running circles in a cage. One plausible explanation for this difference is the stimulation of the AAV injection, and another possible explanation is the opioid-induced hyperalgesia (OIH) development in mice given repeated doses of fentanyl and morphine. Figures 4G,H show that basal pain thresholds were significantly decreased on days 3, 5, and 7 compared with that on day 1. Consistent with the existing literature, central sensitization is characterized by increased spontaneous activity and reduced thresholds (LaMotte et al., 1982; Coderre et al., 1993). Psychological factors, including anxiety and amplification of pain, could be modulating factors in the development of OIH (Lee and Yeomans, 2014).
Biased agonism has been proposed as a means to separate desirable from adverse drug responses downstream of MOR (Kenakin, 2011; Rankovic et al., 2016). The most accepted method for detection and quantification of biased agonism is the comparison of “transduction coefficients” based on the Black and Leff operational model of agonism (Black and Leff, 1983; Kenakin and Christopoulos, 2013). However, the contribution of the degree of bias to the improved separation of therapeutic benefit from side effects is unclear. A recent report has developed a novel idea that bias factor correlates with therapeutic window, which can be used to predict safer opioid analgesics (Schmid et al., 2017). In essence, the bias factor defines the extent of difference in relative agonist activity between two signaling assays (the bias factor is
In summary, we indeed first elucidate the novel mechanisms of Y7.43 mutation affecting MOR signaling pathways. Further, functional selectivity in fentanyl and morphine can engage different mechanisms to induce analgesia. Through a better understanding of the molecular mechanisms of ligands specific binding with MOR, it is anticipated that we can bridge the knowledge gap on how chemical information encoded within the receptor regions’ controlled-effector coupling is decoded into the effect mediated by signaling pathways through the specific interactions it makes within the receptor binding site. Thus, by rationally designing novel compounds to interact predictably with MORY7.43, we might improve the chance of successfully designing new analgesics with fewer side effects and stronger analgesic ability.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
This animal study was reviewed and approved by the Animal Care and Use Committee of the Beijing Institute of Pharmacology and Toxicology.
Author contributions
BT and RS conceived the study and revised the manuscript; XT designed and performed the experiments, collected and analyzed the data, and wrote the original draft; JZ performed molecular docking and molecular dynamics simulations experiments and analyzed the data; SW and XL assisted with animal experiments; HG participated in the discussion; and YS and WF contributed advice. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (no. 81773709).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.919325/full#supplementary-material
References
Alewijnse, A. E., Timmerman, H., Jacobs, E. H., Smit, M. J., Roovers, E., Cotecchia, S., et al. (2000). The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol. Pharmacol. 57 (5), 890–898.
Bachmutsky, I., Wei, X. P., Durand, A., and Yackle, K. (2021). ß-arrestin 2 germline knockout does not attenuate opioid respiratory depression. Elife 10, e62552. doi:10.7554/eLife.62552
Black, J. W., and Leff, P. (1983). Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 220 (1219), 141–162. doi:10.1098/rspb.1983.0093
Bock, A., Merten, N., Schrage, R., Dallanoce, C., Batz, J., Klockner, J., et al. (2012). The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 3, 1044. doi:10.1038/ncomms2028
Bu, H., Liu, X., Tian, X., Yang, H., and Gao, F. (2015). Enhancement of morphine analgesia and prevention of morphine tolerance by downregulation of beta-arrestin 2 with antigene RNAs in mice. Int. J. Neurosci. 125 (1), 56–65. doi:10.3109/00207454.2014.896913
Canal, C. E., Cordova-Sintjago, T. C., Villa, N. Y., Fang, L. J., and Booth, R. G. (2011). Drug discovery targeting human 5-HT(2C) receptors: residues S3.36 and Y7.43 impact ligand-binding pocket structure via hydrogen bond formation. Eur. J. Pharmacol. 673 (1-3), 1–12. doi:10.1016/j.ejphar.2011.10.006
Chen, X. T., Pitis, P., Liu, G., Yuan, C., Gotchev, D., Cowan, C. L., et al. (2013). Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan- 9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem. 56 (20), 8019–8031. doi:10.1021/jm4010829
Chen, Y., Mestek, A., Liu, J., Hurley, J. A., and Yu, L. (1993). Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol. Pharmacol. 44 (1), 8–12.
Cheng, J. X., Cheng, T., Li, W. H., Liu, G. X., Zhu, W. L., and Tang, Y. (2018). Computational insights into the G-protein-biased activation and inactivation mechanisms of the mu opioid receptor. Acta Pharmacol. Sin. 39 (1), 154–164. doi:10.1038/aps.2017.158
Coderre, T. J., Katz, J., Vaccarino, A. L., and Melzack, R. (1993). Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain 52 (3), 259–285. doi:10.1016/0304-3959(93)90161-H
Craft, J. M., Watterson, D. M., Hirsch, E., and Van Eldik, L. J. (2005). Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J. Neuroinflammation 2, 15. doi:10.1186/1742-2094-2-15
De Neve, J., Barlow, T. M. A., Tourwe, D., Bihel, F., Simonin, F., and Ballet, S. (2021). Comprehensive overview of biased pharmacology at the opioid receptors: biased ligands and bias factors. RSC Med. Chem. 12 (6), 828–870. doi:10.1039/d1md00041a
de Waal, P. W., Shi, J., You, E., Wang, X., Melcher, K., Jiang, Y., et al. (2020). Molecular mechanisms of fentanyl mediated beta-arrestin biased signaling. PLoS Comput. Biol. 16 (4), e1007394. doi:10.1371/journal.pcbi.1007394
DeWire, S. M., Yamashita, D. S., Rominger, D. H., Liu, G., Cowan, C. L., Graczyk, T. M., et al. (2013). A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 344 (3), 708–717. doi:10.1124/jpet.112.201616
Eidson, L. N., and Murphy, A. Z. (2019). Inflammatory mediators of opioid tolerance: implications for dependency and addiction. Peptides 115, 51–58. doi:10.1016/j.peptides.2019.01.003
Fenalti, G., Giguere, P. M., Katritch, V., Huang, X. P., Thompson, A. A., Cherezov, V., et al. (2014). Molecular control of delta-opioid receptor signalling. Nature 506 (7487), 191–196. doi:10.1038/nature12944
Fowler, J. C., Bhattacharya, S., Urban, J. D., Vaidehi, N., and Mailman, R. B. (2012). Receptor conformations involved in dopamine D(2L) receptor functional selectivity induced by selected transmembrane-5 serine mutations. Mol. Pharmacol. 81 (6), 820–831. doi:10.1124/mol.111.075457
Frank, R. G., and Pollack, H. A. (2017). Addressing the fentanyl threat to public health. N. Engl. J. Med. 376 (7), 605–607. doi:10.1056/NEJMp1615145
Furst, S., Zadori, Z. S., Zador, F., Kiraly, K., Balogh, M., Laszlo, S. B., et al. (2020). On the role of peripheral sensory and gut mu opioid receptors: Peripheral analgesia and tolerance. Molecules 25 (11), 2473. doi:10.3390/molecules25112473
Galandrin, S., and Bouvier, M. (2006). Distinct signaling profiles of beta1 and beta2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol. Pharmacol. 70 (5), 1575–1584. doi:10.1124/mol.106.026716
Gilissen, J., Geubelle, P., Dupuis, N., Laschet, C., Pirotte, B., and Hanson, J. (2015). Forskolin-free cAMP assay for Gi-coupled receptors. Biochem. Pharmacol. 98 (3), 381–391. doi:10.1016/j.bcp.2015.09.010
Gillis, A., Gondin, A. B., Kliewer, A., Sanchez, J., Lim, H. D., Alamein, C., et al. (2020). Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 13 (625), eaaz3140. doi:10.1126/scisignal.aaz3140
Hertz, S. (2018). Anesthetic and analgesic drug products advisory committee (AADPAC). FDA Briefing Document.
Hothersall, J. D., Torella, R., Humphreys, S., Hooley, M., Brown, A., McMurray, G., et al. (2017). Residues W320 and Y328 within the binding site of the mu-opioid receptor influence opiate ligand bias. Neuropharmacology 118, 46–58. doi:10.1016/j.neuropharm.2017.03.007
Huang, H., Li, X., Guo, W., Zhu, C., Qian, Y., Shen, Q., et al. (2021). Design, synthesis, and structure-activity relationship exploration of alkyl/phenylalkyl piperidine analogues as novel highly potent and selective mu opioid receptor agonists. ACS Chem. Neurosci. 12 (2), 285–299. doi:10.1021/acschemneuro.0c00487
Huang, W., Manglik, A., Venkatakrishnan, A. J., Laeremans, T., Feinberg, E. N., Sanborn, A. L., et al. (2015). Structural insights into µ-opioid receptor activation. Nature 524 (7565), 315–321. doi:10.1038/nature14886
James, I. E., Skobieranda, F., Soergel, D. G., Ramos, K. A., Ruff, D., and Fossler, M. J. (2020). A first-in-human clinical study with TRV734, an orally bioavailable G-protein-biased ligand at the mu-opioid receptor. Clin. Pharmacol. Drug Dev. 9 (2), 256–266. doi:10.1002/cpdd.721
Jones, P. G., Curtis, C. A., and Hulme, E. C. (1995). The function of a highly-conserved arginine residue in activation of the muscarinic M1 receptor. Eur. J. Pharmacol. 288 (3), 251–257. doi:10.1016/0922-4106(95)90036-5
Kelly, E. (2013). Efficacy and ligand bias at the mu-opioid receptor. Br. J. Pharmacol. 169 (7), 1430–1446. doi:10.1111/bph.12222
Kenakin, T., and Christopoulos, A. (2013). Measurements of ligand bias and functional affinity. Nat. Rev. Drug Discov. 12 (6), 483. doi:10.1038/nrd3954-c2
Kenakin, T. (2011). Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 336 (2), 296–302. doi:10.1124/jpet.110.173948
Kenakin, T., Watson, C., Muniz-Medina, V., Christopoulos, A., and Novick, S. (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3 (3), 193–203. doi:10.1021/cn200111m
Kieffer, B. L., Befort, K., Gaveriaux-Ruff, C., and Hirth, C. G. (1992). The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. U. S. A. 89 (24), 12048–12052. doi:10.1073/pnas.89.24.12048
Kliewer, A., Gillis, A., Hill, R., Schmiedel, F., Bailey, C., Kelly, E., et al. (2020). Morphine-induced respiratory depression is independent of beta-arrestin2 signalling. Br. J. Pharmacol. 177 (13), 2923–2931. doi:10.1111/bph.15004
Kliewer, A., Schmiedel, F., Sianati, S., Bailey, A., Bateman, J. T., Levitt, E. S., et al. (2019). Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 10 (1), 367. doi:10.1038/s41467-018-08162-1
Kofoed, R. H., Heinen, S., Silburt, J., Dubey, S., Dibia, C. L., Maes, M., et al. (2021). Transgene distribution and immune response after ultrasound delivery of rAAV9 and PHP.B to the brain in a mouse model of amyloidosis. Mol. Ther. Methods Clin. Dev. 23, 390–405. doi:10.1016/j.omtm.2021.10.001
LaMotte, R. H., Thalhammer, J. G., Torebjork, H. E., and Robinson, C. J. (1982). Peripheral neural mechanisms of cutaneous hyperalgesia following mild injury by heat. J. Neurosci. 2 (6), 765–781. doi:10.1523/jneurosci.02-06-00765.1982
Lee, H. J., and Yeomans, D. C. (2014). Opioid induced hyperalgesia in anesthetic settings. Korean J. Anesthesiol. 67 (5), 299–304. doi:10.4097/kjae.2014.67.5.299
Lu, Z. L., Curtis, C. A., Jones, P. G., Pavia, J., and Hulme, E. C. (1997). The role of the aspartate-arginine-tyrosine triad in the m1 muscarinic receptor: mutations of aspartate 122 and tyrosine 124 decrease receptor expression but do not abolish signaling. Mol. Pharmacol. 51 (2), 234–241. doi:10.1124/mol.51.2.234
Luttrell, L. M., and Lefkowitz, R. J. (2002). The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115 (3), 455–465. doi:10.1242/jcs.115.3.455
Manglik, A., Kruse, A. C., Kobilka, T. S., Thian, F. S., Mathiesen, J. M., Sunahara, R. K., et al. (2012). Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 485 (7398), 321–326. doi:10.1038/nature10954
Manglik, A., Lin, H., Aryal, D. K., McCorvy, J. D., Dengler, D., Corder, G., et al. (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature 537 (7619), 185–190. doi:10.1038/nature19112
Metzger, T. G., Paterlini, M. G., Ferguson, D. M., and Portoghese, P. S. (2001). Investigation of the selectivity of oxymorphone- and naltrexone-derived ligands via site-directed mutagenesis of opioid receptors: exploring the "address" recognition locus. J. Med. Chem. 44 (6), 857–862. doi:10.1021/jm000381r
Ohyama, K., Yamano, Y., Sano, T., Nakagomi, Y., Wada, M., and Inagami, T. (2002). Role of the conserved DRY motif on G protein activation of rat angiotensin II receptor type 1A. Biochem. Biophys. Res. Commun. 292 (2), 362–367. doi:10.1006/bbrc.2002.6670
Pasternak, G. W., and Pan, Y. X. (2013). Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev. 65 (4), 1257–1317. doi:10.1124/pr.112.007138
Perez-Aguilar, J. M., Xi, J., Matsunaga, F., Cui, X., Selling, B., Saven, J. G., et al. (2013). A computationally designed water-soluble variant of a G-protein-coupled receptor: the human mu opioid receptor. PLoS One 8 (6), e66009. doi:10.1371/journal.pone.0066009
Raehal, K. M., Schmid, C. L., Groer, C. E., and Bohn, L. M. (2011). Functional selectivity at the mu-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 63 (4), 1001–1019. doi:10.1124/pr.111.004598
Raehal, K. M., Walker, J. K., and Bohn, L. M. (2005). Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 314 (3), 1195–1201. doi:10.1124/jpet.105.087254
Rankovic, Z., Brust, T. F., and Bohn, L. M. (2016). Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 26 (2), 241–250. doi:10.1016/j.bmcl.2015.12.024
Rasmussen, S. G., Jensen, A. D., Liapakis, G., Ghanouni, P., Javitch, J. A., and Gether, U. (1999). Mutation of a highly conserved aspartic acid in the beta2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol. Pharmacol. 56 (1), 175–184. doi:10.1124/mol.56.1.175
Rincon, M. Y., de Vin, F., Duque, S. I., Fripont, S., Castaldo, S. A., Bouhuijzen-Wenger, J., et al. (2018). Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene Ther. 25 (2), 83–92. doi:10.1038/s41434-018-0005-z
Rozenfeld, R., and Devi, L. A. (2007). Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 21 (10), 2455–2465. doi:10.1096/fj.06-7793com
Ruan, J. P., Chen, L., and Ma, Z. L. (2019). Activation of spinal Extacellular Signal-Regulated Kinases and c-jun N-terminal kinase signaling pathways contributes to morphine-induced acute and chronic hyperalgesia in mice. J. Cell. Biochem. 120 (9), 15045–15056. doi:10.1002/jcb.28766
Scheer, A., Costa, T., Fanelli, F., De Benedetti, P. G., Mhaouty-Kodja, S., Abuin, L., et al. (2000). Mutational analysis of the highly conserved arginine within the glu/asp-arg-tyr motif of the alpha(1b)-adrenergic receptor: effects on receptor isomerization and activation. Mol. Pharmacol. 57 (2), 219–231.
Schmid, C. L., Kennedy, N. M., Ross, N. C., Lovell, K. M., Yue, Z., Morgenweck, J., et al. (2017). Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 171 (5), 1165–1175. e13. doi:10.1016/j.cell.2017.10.035
Schneider, E. H., Schnell, D., Strasser, A., Dove, S., and Seifert, R. (2010). Impact of the DRY motif and the missing "ionic lock" on constitutive activity and G-protein coupling of the human histamine H4 receptor. J. Pharmacol. Exp. Ther. 333 (2), 382–392. doi:10.1124/jpet.109.163220
Shao, S., Sun, Y., Zhang, Y., Tian, X., Li, Y., Tan, B., et al. (2020). A20 enhances mu-opioid receptor function by inhibiting beta-arrestin2 recruitment. Biochem. Biophys. Res. Commun. 528 (1), 127–133. doi:10.1016/j.bbrc.2020.05.073
Soergel, D. G., Subach, R. A., Sadler, B., Connell, J., Marion, A. S., Cowan, C. L., et al. (2014). First clinical experience with TRV130: pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 54 (3), 351–357. doi:10.1002/jcph.207
Stein, C. (2016). Opioid receptors. Annu. Rev. Med. 67 (1), 433–451. doi:10.1146/annurev-med-062613-093100
Subramanian, G., Paterlini, M. G., Portoghese, P. S., and Ferguson, D. M. (2000). Molecular docking reveals a novel binding site model for fentanyl at the mu-opioid receptor. J. Med. Chem. 43 (3), 381–391. doi:10.1021/jm9903702
Sun, X., Laroche, G., Wang, X., Agren, H., Bowman, G. R., Giguere, P. M., et al. (2017). Propagation of the allosteric modulation induced by sodium in the delta-opioid receptor. Chemistry 23 (19), 4615–4624. doi:10.1002/chem.201605575
Tan, H. S., and Habib, A. S. (2021). Oliceridine: A novel drug for the management of moderate to severe acute pain - a review of current evidence. J. Pain Res. 14, 969–979. doi:10.2147/JPR.S278279
Tschammer, N., Bollinger, S., Kenakin, T., and Gmeiner, P. (2011). Histidine 6.55 is a major determinant of ligand-biased signaling in dopamine D2L receptor. Mol. Pharmacol. 79 (3), 575–585. doi:10.1124/mol.110.068106
Wacker, D., Stevens, R. C., and Roth, B. L. (2017). How ligands illuminate GPCR molecular pharmacology. Cell 170 (3), 414–427. doi:10.1016/j.cell.2017.07.009
Woo, A. Y., Jozwiak, K., Toll, L., Tanga, M. J., Kozocas, J. A., Jimenez, L., et al. (2014). Tyrosine 308 is necessary for ligand-directed Gs protein-biased signaling of β2-adrenoceptor. J. Biol. Chem. 289 (28), 19351–19363. doi:10.1074/jbc.M114.558882
Wootten, D., Reynolds, C. A., Koole, C., Smith, K. J., Mobarec, J. C., Simms, J., et al. (2016). A hydrogen-bonded polar network in the core of the glucagon-like peptide-1 receptor is a fulcrum for biased agonism: lessons from class B crystal structures. Mol. Pharmacol. 89 (3), 335–347. doi:10.1124/mol.115.101246
Keywords: μ-opioid receptor, molecular docking, cAMP, β-arrestin2 recruitment, signaling pathway, analgesia, molecular dynamics simulations
Citation: Tian X, Zhang J, Wang S, Gao H, Sun Y, Liu X, Fu W, Tan B and Su R (2022) Tyrosine 7.43 is important for mu-opioid receptor downstream signaling pathways activated by fentanyl. Front. Pharmacol. 13:919325. doi: 10.3389/fphar.2022.919325
Received: 13 April 2022; Accepted: 01 August 2022;
Published: 02 September 2022.
Edited by:
Samuel D. Banister, Psylo, AustraliaReviewed by:
Christophe Stove, Ghent University, BelgiumTaka-Aki Koshimizu, Jichi Medical University, Japan
Deepak K. Saini, Indian Institute of Science (IISc), India
Copyright © 2022 Tian, Zhang, Wang, Gao, Sun, Liu, Fu, Tan and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Tan, dGFuYm9AYm1pLmFjLmNu, NDY5NDM2MDk0QDE2My5jb20=; Ruibin Su, c3VyYkBibWkuYWMuY24=, cnVpYmluc3VAMTI2LmNvbQ==