 Yanzhao Dong1†
Yanzhao Dong1† Haiying Zhou1†
Haiying Zhou1† Ahmad Alhaskawi1
Ahmad Alhaskawi1 Zewei Wang2
Zewei Wang2 Jingtian Lai2
Jingtian Lai2 Sohaib Hasan Abdullah Ezzi2
Sohaib Hasan Abdullah Ezzi2 Vishnu Goutham Kota2Mohamed Hasan Abdulla Hasan Abdulla2Zhenyu Sun1
Vishnu Goutham Kota2Mohamed Hasan Abdulla Hasan Abdulla2Zhenyu Sun1 Hui Lu1,3*
Hui Lu1,3*- 1Department of Orthopedics, B Department of Rehabilitation Medicine, the First Affiliated Hospital, Zhejiang University, Hangzhou, China
- 2Zhejiang University School of Medicine, Hangzhou, China
- 3Alibaba-Zhejiang University Joint Research Center of Future Digital Healthcare, Zhejiang University, Hangzhou, China
Bone fracture healing is a complex process involving various signaling pathways. It remains an unsolved issue the fast and optimal management of complex or multiple fractures in the field of orthopedics and rehabilitation. Bone fracture healing is largely a four-stage process, including initial hematoma formation, intramembrane ossification, chondrogenesis, and endochondral ossification followed by further bone remodeling. Many studies have reported the involvement of immune cells and cytokines in fracture healing. On the other hand, the Tumor Necrosis Factor (TNF) family and TNF receptor superfamily (TNFRSF) play a pivotal role in many physiological processes. The functions of the TNF family and TNFRSF in immune processes, tissue homeostasis, and cell differentiation have been extensively studied by many groups, and treatments targeting specific TNFRSF members are in progress. In terms of bone fracture management, it has been discovered that several members of TNFRSF have very distinct functions in different stages of fracture healing, including TNFR1, TNFR2, and receptor activator of nuclear factor kappa-B (RANK) pathways. More specifically, TNFR1 is associated with osteoclastogenesis and TNFR2 is associated with osteogenic differentiation, while RANK is in association with bone remodeling. In this review, we will discuss and summarize the involvement of members of TNFRSF including TNFR1, TNFR2, and Receptor activator of nuclear factor kappa-B (RANK) pathways in different stages of fracture healing and bone remodeling and the current treatment trend involving TNFRSF agonists and antagonists.
1 Background
General background of TNFRSF signaling pathways and clinical application of TNFRSF agonists and antagonists, an overview of the bone regeneration process following bone fracture, and the future perspectives of application of TNFRSF agonists and antagonists in the treatment of bone fracture. TNF was first identified as early as 4 decades ago as a product of lymphocytes that exerts lytic effects against certain cell types, tumor cells in particular. Later in 1975, Carswell et al. chemically and genetically described this cytokine as “tumor necrotizing factors”, which was later called tumor necrosis factor (Granger et al., 1969; Carswell et al., 1975). Using large-scale sequencing, a series of similar proteins were discovered, and collectively they were categorized as TNF superfamily (TNFSF), their receptors, therefore, are categorized as TNF receptor superfamily (TNFRSF) (Idriss and Naismith, 2000).
1.1 TNFRSF signaling pathways
TNFRSF is an important category of receptors for cytokines that provides crucial communication signals between various cell types during development and homeostasis, especially in the skin, bones, and lymphoid organs, and functions to maintain organ homeostasis and initiates tissue responses (Locksley et al., 2001). TNFRSF is further categorized into three groups according to different structures and functions, which are respectively characterized by death domains (DD) or death receptors, TNF receptor-associated factor (TRAF)-interacting receptors, and soluble or membrane-anchored TNFRSF receptors that act as decoy receptors of the death and TRAF-interacting receptors (Lang et al., 2016). Depending on different physiological processes, the consequences of signal transduction by TNF receptors include cell apoptosis, proliferation, or differentiation. During bone fracture healing, three members of TNFRSF are mainly involved, respectively TNFR1, TNFR2, and Osteoprotegerin (OPG), a decoy receptor for Receptor activator of nuclear factor-kappa-Β (RANK) that regulates the stimulation of RANK via competing for RANKL. (Figure 1).
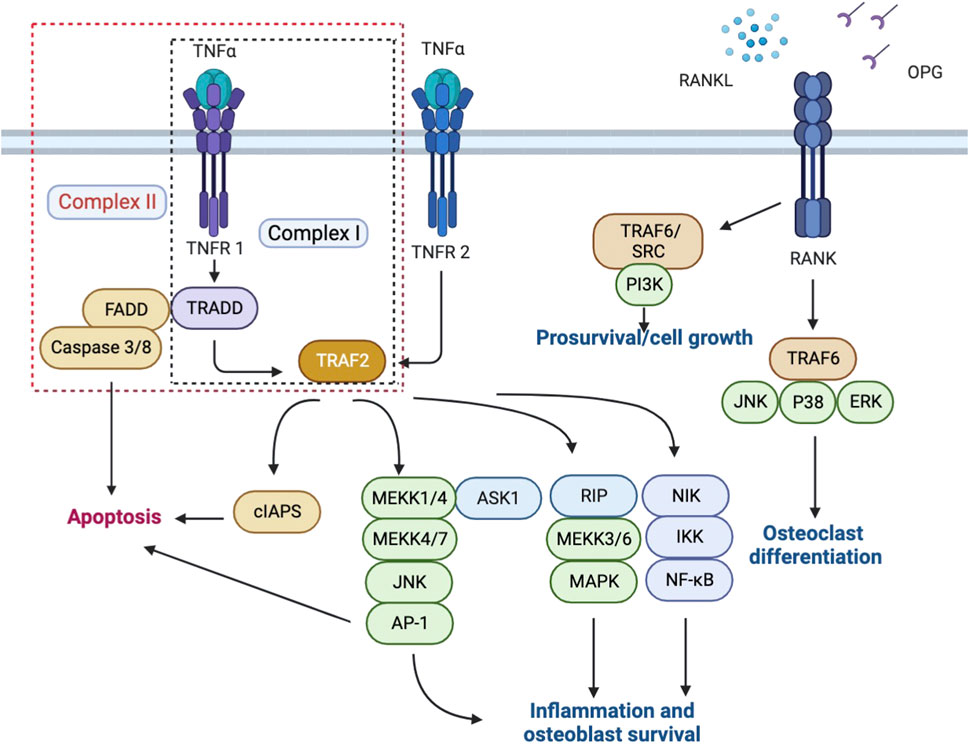
FIGURE 1. TNFR1, TNFR2 and RANK/RANKL/OPG intracellular signaling pathways.
1.2 TNFR1, TNFR2, and RANK
TNFα has long been identified as one of the most potent pro-inflammatory cytokines and a major driver of cell apoptosis and necroptosis in vitro. The anti-inflammatory effect of TNFR agonists and antagonists in the treatment of rheumatic diseases has been associated with their effect on blocking TNF from binding to TNFR1 and TNFR2.
TNFR1, also referred to as tumor necrosis factor receptor superfamily member 1A (TNFRSF1A), exerts its functions as a ubiquitous member receptor for TNFα. In bone, TNFR1 is expressed by both osteoblasts and osteoclasts (Bu et al., 2003). Once bound to TNFα, TNFR1 activates the mitogen-activated protein kinase (MAPKs) and canonical NF-kB pathways, and collectively led to the upregulation of pro-inflammatory genes transcription and results in inflammation. On the other hand, upon binding with TNFα, TNFR1 could also exert a cytotoxic effect via sequential assembly of a membrane-bound primary signaling complex (Complex I) that activates the formation of a secondary cytoplasmic complex (Complex II) and leads to cell death (Hsu et al., 1996; Micheau and Tschopp, 2003). However, it has now been increasingly clear that only when certain cell death checkpoints are activated would TNFR1 activation result in apoptosis or necroptosis, and in most cell types, TNFR1 activation instead triggers a pro-survival response. (Ting and Bertrand, 2016). This is achieved via the adaptor molecule TNFR1-associated death domain protein (TRADD), through which different signaling complexes could trigger either cell death or cell survival depending on the cellular context. (Brenner et al., 2015). TNFR2, or TNFRSF1B, is widely expressed by the immune, neuronal and epithelial cells, and has a high affinity for the membrane-bound form of TNFα, which leads to activation of the NF-kB and MAPKs pathways. (Al-Lamki and Mayadas, 2015; Mancusi et al., 2019). Apart from TNFR1, TNFR2 is the only membrane receptor that could bind to TNFα and exert signaling functions. Differences between TNFR1 and TNFR2 majorly lie in their intracellular structures; for instance, compared to TNFR2, TNFR1 contains an intracellular death domain (DD), which allows interaction between TNFR1 and DD-containing protein and evokes not only pro-inflammatory responses but also cytotoxic signaling. (Medler and Wajant, 2019). While TNFR2 plays a protective role upon interaction with TNF Receptor Associated Factor 2 (TRAF2) in many vital organs, the role of TNFR2 in the bone fracture healing process is more complicated and remains to be summarized. The polymorphisms in TNFRSF1B are often observed in patients suffering from rheumatic diseases, which seem to result in altered binding kinetics between TNFα and TNFR2 and eventually inhibition of the downstream NF-kB pathways, suggesting a protective role of TNFR2 signaling. (Yang et al., 2018).
RANK (TNFRSF11), RANKL, and osteoprotegerin (OPG) consist the signaling axis RANK/RANKL/OPG pathway, which plays a pivotal role in bone turnover in the context of inflammation. (Soos et al., 2022). Once activated by pro-inflammatory cytokines including TNF and interleukin, the RANKL-dependent pathways subsequently induce osteoclastogenesis, osteoclast differentiation, and osteoclast activation. (Schett and Gravallese, 2012; van den Berg and Miossec, 2009). In particular, upon binding with TNF, the expression level of RANKL by osteoblasts, B cells, and T cells is increased, which indirectly enhances osteoclastogenesis. (Lam et al., 2000; Schett and Gravallese, 2012). RANK/RANKL/OPG pathway has also been identified as an apoptosis regulator that maintains bone mass by regulating chondroclast differentiation, a crucial step during endochondral ossification. (Vu et al., 1998).
1.3 Current application of TNFRSF agonists and antagonists
Identified as inflammatory mediators, a growing body of evidence suggests that TNFRSF has the potential to be integrated into the new generation of biotherapy for the treatment of immune disease. (Croft et al., 2012). Currently, several anti-TNF drugs have been approved for clinical use to counteract the pro-inflammatory effect of TNF, which include infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), and etanercept (Enbrel). (Monaco et al., 2015). While these medications prove effective in the treatment of immune disorders including rheumatoid arthritis (RA) and inflammatory bowel disease (IBD), the side effects including opportunistic infections remains a problem for clinicians.
At the same time, the understanding of bone healing and reconstruction has advanced, and researchers nowadays hold the opinion that bone regeneration and reconstruction following a fracture are tightly regulated by both pro-inflammatory and anti-inflammatory cytokines, for example, the recruitment and activation of mesenchymal stem cells (MSCs) at the site of fracture to exert an anabolic effect on bone repair. (Liu et al., 2018). The future prospects of fracture treatment may, like many immune disorders, lie in local application of agonists and antagonists of TNFRSF at the site of fracture. (Table 1).
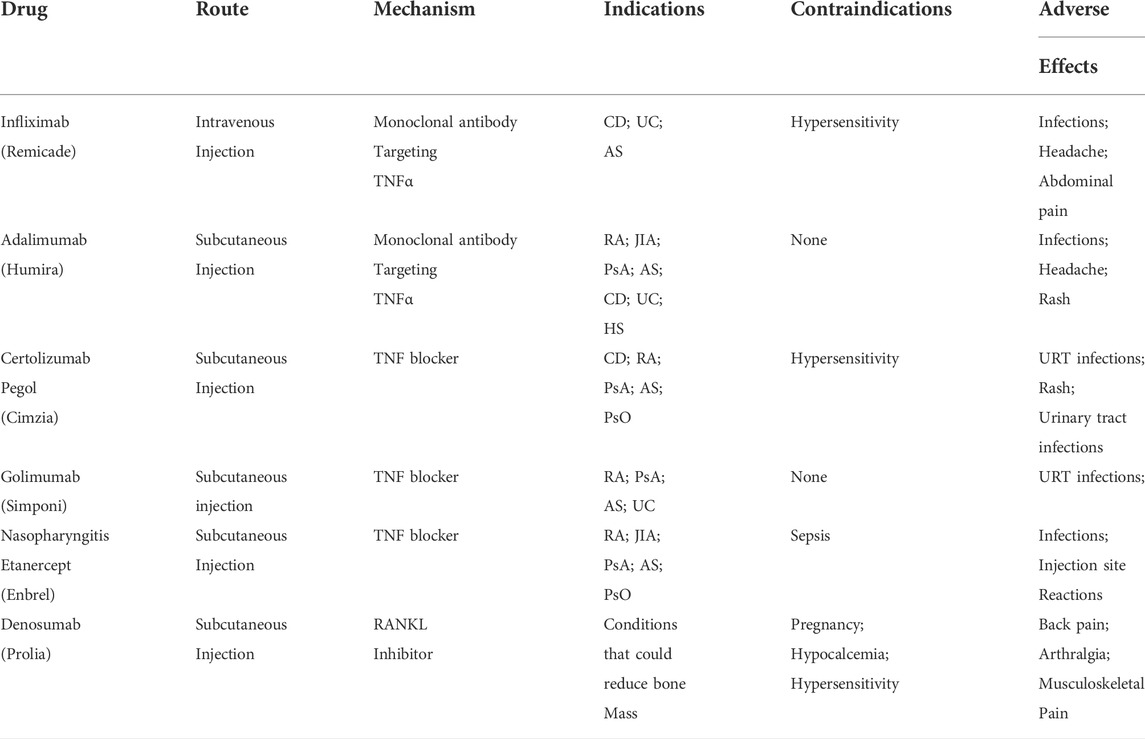
TABLE 1. Current medication options interfering with the TNFRSF signaling pathways. Abbreviation: CD - Crohn’s Disease; UC—Ulcerative Colitis; AS - Ankylosing Spondylitis; RA—Rheumatoid Arthritis; JIA - Juvenile Idiopathic Arthritis; PsA - Psoriatic Arthritis; HS - Hidradenitis Suppurativa; PsO - Plaque Psoriasis; URT—upper respiratory tract; (Information on Tumor Necrosis Factor, 2015; Medication Guide Prolia, 2022).
1.4 Bone fracture: burden, current treatment options, and future perspectives
Bone fracture is becoming a significant public health burden worldwide. In 2019, the total incidents of fracture reached 178 million in total, and the years lived with disability (YLDs) reached 25.8 million. (Collaborators, 2021). As an essential component of the musculoskeletal system, bone provides shape and support for the body and is subject to constant remodeling processes to maintain structural integrity and microarchitecture throughout life, and once fractured, these functions tend to be comprised, the quality of live lowered, and worse even, be lethal. (David and Schett, 2010). Promoting bone fracture healing in both time and quality is, therefore, an important measure to improve living standards.
Bone fracture healing is a multi-stage process that majorly involves two types of cells: osteoblasts and osteoclasts. Fracture healing can be divided into four stages, which are hematoma formation, intramembranous ossification, chondrogenesis, and endochondral ossification, each characterized by specific cellular events and extracellular matrix formation. (Bolander, 1992; Karnes et al., 2015). The healed bone will then undergo remodeling to improve the biological and physiological functions.
Current treatment methods for bone fracture vary depending on the sites and types. These methods generally include conservative treatment characterized by traction and external fixation, where the fracture is less severe and cause no blood flow disorder and nerve injuries, and surgical treatment followed by internal fixation, where circumstances are worse and influence the vessels and nerves. All methods, however, require several weeks to months for bone regeneration and functional rehabilitation to improve prognosis. The fixation process could be agonizing for patients, and clinicians are eager to find a new adjuvant therapy to facilitate fracture healing and lessen the timespan. As observed in the past, fracture healing is tightly regulated and the regulators including TNFRSF could be assimilated into the treatment.
While many studies have investigated the process of bone fracture healing, the involvement of signaling via TNFRSF in this process remains to be summarized. Therefore in this review, we seek to highlight processes where novel therapeutic interventions in TNFRSF signaling to enhance bone fracture healing are possible.
2 TNFR1, TNFR2 and RANK/RANKL/osteoprotegerin in bone fracture healing
2.1 TNFR1 signaling, osteoclastogenesis and fracture healing
To further discuss the roles TNFR1 plays in fracture healing, it is essential to understand two main stages of fracture healing, intramembranous and endochondral ossification. Intramembranous ossification occurs adjacent to the fracture site, where osteoblasts synthesize new bone tissue without cartilaginous intermediates. Endochondral ossification, by contrast, is the process of calcification in previously formed cartilages and continues until all cartilages are replaced by bone. While these processes are well-understood, many groups seek to study the roles TNFRSF plays in fracture healing on this basis. Lukić et al. (2005) found that while TNFR1 signaling does not affect intramembranous ossification, it is involved in endochondral ossification in adult mice. They then concluded that TNFR1 functions as a negative regulator during new tissue formation in endochondral ossification but not intramembranous ossification. (Lukic et al., 2005). In comparison, Gerstenfeld et al, (2001); Gerstenfeld et al., 2003)demonstrated that in TNFR1/2 double knockout mice, intramembranous ossification is also impaired, which indicates that TNFR2 could potentially play an imperative role in intramembranous ossification. To investigate whether TNFR1 regulates the formation and survival of osteoclasts and osteoblasts, Hiroki et al. (2010) used lipopolysaccharides (LPS) to induce bone loss in wild-type and TNFR1 knockout mice. (Ochi et al., 2010). The research group concluded that TNFR1 is indispensable in osteoclastogenesis and that TNFR1 is necessary to induce osteoclast precursors. More importantly, it is the first study to demonstrate that TNFR1 has an anti-apoptotic effect in LPS-induced inflammation in vivo. Furthermore, the study administrated osteoprotegerin (OPG) in mice with LPS-induced bone loss and determined that RANK/RANKL/OPG signaling affects osteoclast differentiation rather than the osteoclast precursor population.
In terms of disease management, although elevated TNFα levels have been identified in patients suffering from autoimmune and degenerative diseases, including Rheumatoid Arthritis (RA), and Irritable Bowel Disease, it is essential for maintaining homeostasis and fighting infections. While anti-TNF therapeutics have succeeded in the treatment of these autoimmune diseases, the severe side effects, including opportunistic infections, have limited further clinical application. (Monaco et al., 2015). To avoid side-effects caused by TNFα inhibition, Kontermann et al, (2008) developed a humanized TNFR1-specific antagonist designated as IZI-06.1 (Atrosab), an IgG1 molecule derived from mice immunized with human TNFR1. Concurrently as the molecule act as a potent TNFR1 antagonist, TNFR2 and other TNF-mediated immunity showed no significant signs of inhibition. In 2019, following this study, Richter et al, (2019) generated a monovalent anti-TNFR1 antibody fragment (Fab 13.7) and fused the variable heavy and light chains to the N-termini of the newly-formed heterodimerizing Fc chains. The constructed protein Atrosimab displays improved pharmacokinetic properties in the presence of anti-human IgG antibodies, retains strong binding to TNFR1, and shows a potent inhibition effect. The development of Atrosab and Atrosimab sheds light on the possibility that TNFR1 antagonists targeted to shorten the inflammation process and enhance new tissue formation during fracture healing be applied clinically without the risk of causing decreased immunity and opportunistic infections.
To sum up, TNFR1 plays a pivotal role in osteoclastogenesis, functioning as a negative regulator in new tissue formation in bone fracture healing. TNFR1 antagonists, including Atrosimab and Atrosab, have seen tremendous advances in affinity to TNFR1 and potency as antagonists. The future perspective regarding bone fracture healing could assimilate the current understanding of TNFR1 signaling pathways and test the safety of these medications in phase 3 clinical trials while exploring other possible methods to deter TNFR1 signaling pathways and achieve faster recovery of bone fracture (Figure 2).

FIGURE 2. Organization of the bone remodeling units. MSCs: Mesenchymal stromal/stem cells.
2.2 TNFR2, osteoclastogenesis, and osteogenic differentiation
TNFR2 expression, in comparison with the ubiquitous expression of TNFR1, is primarily on immune cells. (Medler and Wajant, 2019). Although signaling via TNFR1 and outcomes are well characterized, much less is known about TNFR2. (Sabio and Davis, 2014). Nagano et al. (2011) designed a study where they used subcutaneous injections of TNFα onto calvariae to induce bone resorption in both WT and TNFR2-deficient mice and observed the magnitude of bone resorption lacunae in each group to determine whether TNFR2 plays a protective role in the bone resorption process. (Nagano et al., 2011). The result showed that compared to mTNFα, hTNFα/Cholesterol-bearing pullulan (CHP) nanogel could significantly reduce bone mineral density (BMD), indicating that signaling via TNFR1 is essential for bone resorption induction and that signaling via both TNFR1 and TNFR2 could not induce bone resorption lacunae. To further rule out the possibility that TNFR2 plays no role in bone resorption, the research group used TNFR2-deficient mice and found that both hTNFα/CHP and mTNFα/CHP could significantly reduce BMD compared to the control group. These results, in summary, suggest that TNFR2 could have a suppressive effect against TNFR1-induced osteoclastogenesis in mice, which is the primary cause of bone resorption in chronic inflammatory diseases, and that TNFR2 agonists could be a potential therapeutic method in the treatment of bone fractures.
With progressing studies into the effect of TNFR2 on osteoclastogenesis, other research groups seek to find whether the TNFR2 signaling pathway has a role in osteogenic differentiation. In 2020, Y Zhang et al. led a study on EphB4/TNFR2/ERK/MAPK signaling. (Zhang et al., 2020). Previous studies showed that low concentrations of TNFα and TNFR2 could upregulate EphB4 expression and promote osteogenic differentiation of osteoblast precursor cells. However, the roles of TNFR2 signaling and EphB4 in the osteogenic differentiation process and whether there is any crosstalk between these signaling pathways remain elucidated. Previous studies showed that TNFα could significantly enhance EphB4 expression. (Wang et al., 2017). Zhang et al, (2020) used lentivirus-mediated shRNA to knockdown TNFR2 expression level in MC3T3-E1 osteoblasts, which showed an approximately 70% reduction in TNFR2 expression compared with the control group. RUNX 2 and bone sialoprotein (BSP) were selected as indicators downstream of TNFR2 in osteoblast differentiation. The research group first observed that when treated with TNFα, RUNX2 and BSP levels in TNFR2 knockdown osteoblasts were significantly lower than that of the control group. In contrast, the expression level of EphB4 showed no significant difference between TNFR2 knockdown osteoblasts and the control group, indicating that TNFR2 activation does not affect TNFα-induced EphB4 upregulation (Figure 3). On the other hand, when EphB4 forward signaling was suppressed, TNFα-induced TNFR2 expression was lowered, which indicates that TNFα-enhanced EphB4 expression could upregulate TNFR2 expression. In this study, p38, p-p38, ERK1/2, p-ERK1/2, JNK1+2 + 3, and p-JNK1+2 + 3 levels were monitored under the aforementioned conditions and the result showed that in TNFR2 knockdown osteoblasts, the pERK1/2 level was significantly decreased. Similarly, when EphB4 forward signaling was inhibited, pERK1/2 level was also significantly down-regulated. When MC3T3 osteoblasts were pretreated with ERK inhibitor U0216, the levels of RUNX2 and BSP showed a significant decrease. It could be concluded that EphB4, TNFR2, and ERK/MAPK signaling pathways comprises a signaling axis that partly mediates the osteogenic differentiation induced by TNFα.
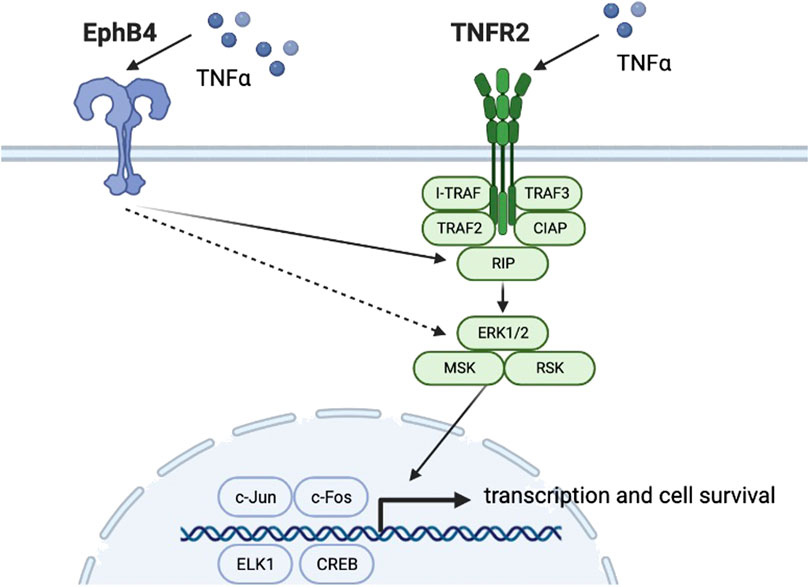
FIGURE 3. Schematic diagram of the EphB4, TNFR2 and ERK/MAPK signaling pathways.
Efforts to target TNFR2 and achieve therapeutic effects in degenerative diseases involving TNFR2 activation, including RA and Alzheimer’s disease have been made. (Fischer et al., 2020). In 2011, R Fischer et al, (2011) constructed a soluble, human TNFR2 agonist via genetic fusion of the trimerization domain of tenascin C to a TNFR2-selective single-chain TNF molecule designated as TNC-scTNF. TNC-scTNF could bind to TNFR2 and induce clustering of TNFR2 on a cellular level. Later in 2014, R Fischer et al, (2014) managed to apply this method to generate a mouse TNFR2-selective mouse TNC-scTNF and incorporated D221N and A223R gene in mouse TNF to improve specificity. Furthermore, Fisher et al. (2017) applied tetramerization on the agonist and obtain improved crosslinking activity. (Fischer et al., 2017).
In summary, the TNFR2 signaling pathway shows a protective effect in bone resorption via suppression of TNFR1-induced osteoclastogenesis, and TNFR2 is possibly a part of a complex signaling axis that mediates osteoblast differentiation. Studies have shown that TNFR2 agonism has therapeutic potential in treating degenerative diseases. Therefore, the potential therapeutic value of TNFR2-targeting agents should not be overlooked.
2.3 RANK, RANKL, and OPG in bone modeling and remodeling
Chondroclasts, as another important component in fracture healing, are less well-understood than their counterparts, such as osteoclasts and osteoblasts. Ota et al. (2009) led research using wild-type and OPG double-knockout tibial fracture mice model to estimate the effect of OPG on fracture healing. The results showed that in OPG double-knockout mice, fracture healing is accelerated in that both the turnover rate of cartilaginous callus and the chondroclasts at the chondroosseous junction are increased and led to the conclusion that OPG deficiency could clearly promote chondrocyte-dependent chondroclastogenesis, accelerate the cartilage replacement by bone tissue and promote fracture healing. (Ota et al., 2009). These findings provide insights for accelerating fracture healing via applying OPG agonists or competitive inhibitors for RANK adjacent to the fracture site and promoting cartilage replacement.
One major component of bone fracture healing, besides the classic four-stage histological progression, is bone remodeling, a continuous process that guarantees the dynamic balance between bone formation and bone resorption. (Takayanagi, 2007). The bone mass can differ enormously depending on different ages (Santos et al., 2017), hormone levels, and weight-bearing levels (Figure 4). The RANK/RANKL/OPG pathway is the pivotal regulator of this process, as this signaling pathway plays a vital role in regulating osteoblasts and osteoclasts. (Boyce and Xing, 2007). For patients suffering from bone fracture, one of the most effective methods following the treatment is to seek help from physical medicine or rehabilitation since studies have shown that exercise could facilitate bone modeling and remodeling. (Qi et al., 2016).

FIGURE 4. Interaction of exercise and RANKL/RANK/OPG biomolecular pathway.
Although the role of the RANK/RANKL/OPG signaling pathway in bone modeling and remodeling is researched by many groups, the scope of this matter remained at an individual level. To better understand whether the RANL/RANKL/OPG signaling pathway is associated with the risk of fracture in the general population, P. Tharabenjasin et al, (2022) conducted a systematic review in 2021 where they searched 13 articles available concerning OPG and fracture and examined four polymorphisms of genes encoding OPG. The result showed that OPG gene polymorphisms could reduce the risk of osteoporotic fracture in the senior population, especially in postmenopausal women over 60 years of age. This result shed light on the possible application of OPG gene sequencing in the population as early detection of possible osteoporosis or other diseases that could cause predisposition to bone fracture, followed by an early intervention to avoid such situations. The statistical result indicates the potential future application of RANK/RANKL/OPG is in the early detection and intervention of diseases or conditions that could cause bone fracture.
In fact, the attempt to target RANKL via artificial monoclonal antibody is not unprecedented. Raje et al. (2018) conducted a double-blinded, randomized phase-3 clinical study in adult patients suffering from newly diagnosed multiple myeloma (MM) with at least one lytic bone lesion. The research team selected denosumab, a monoclonal antibody targeting RANKL, to compare the safety and efficacy in preventing skeletal-related events caused by MM compared with zoledronic acid, an intravenous biphosphate. The study continued from May 2012 to March 2016, and a total of 1718 patients were included. At the endpoint, denosumab achieved non-inferiority compared with zoledronic acid, suggesting that denosumab could be an additional therapeutic option for MM patients suffering from skeletal implications. (Raje et al., 2018).
3 Conclusion
In the musculoskeletal system, TNFRSF members exert different functions and form a complex signaling network. TNFR1 has a crucial role in osteoblastogenesis and osteoclastogenesis, where it could either facilitate osteoblast differentiation and osteocyte survival or result in osteoblast apoptosis, while TNFR1 is also indispensable for osteoclast differentiation and activation. While the underlying mechanism that modulates osteoblastogenesis remains unclear, TNFRSF-targeted antagonists or other agents could selectively bind to TNFR1 and mediate osteoclasts formation, and therefore, facilitate endochondral ossification. This also applies to TNFR2-targeted agents, since TNFR2 has been discovered as a possible member of the EphB4 and ERK/MAPK signaling axis and that TNFR2 plays a vital role in osteogenic process and osteoclastogenesis. Similarly, for other members of TNFRSF, the therapeutic potential in bone fractures and other musculoskeletal diseases or events should not be ignored. In the RANK/RANKL/OPG pathway, monoclonal antibodies including denosumab have already emerged, confirming the therapeutic value of these targets (Figure 5.).
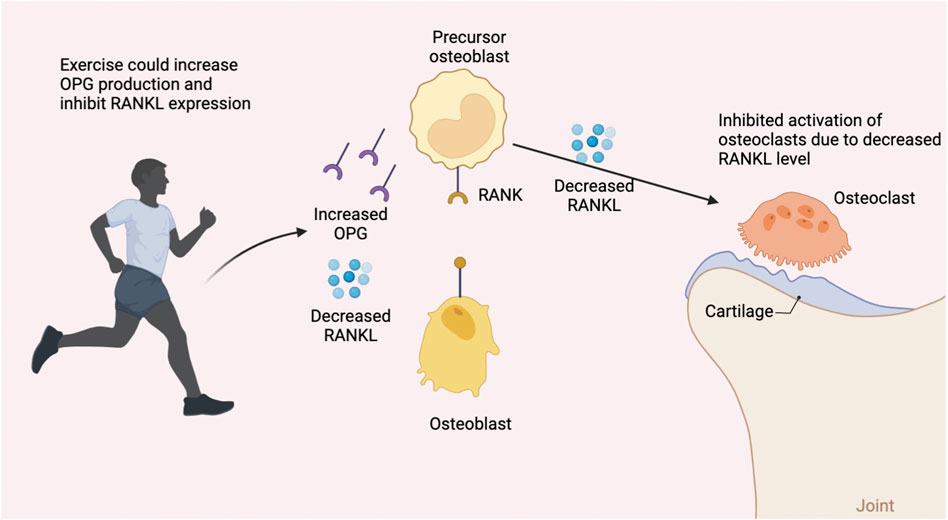
FIGURE 5. General overview of the four-stage fracture healing process and the respective signaling pathways and cytokines involved in initial inflammation, osteoblastogenesis, and osteoclastogenesis. As mentioned in 2.2, the Runx2 level is potentially associated with TNFR2 signaling. (Zhang et al., 2020) MSCs: mesenchymal stem cells; CFU-M: Colony forming unit M, namely monoblast. M-CSF: macrophage colony-stimulating factor.
For future perspectives on TNFRSF-targeted therapeutic regimens, one possible form of treatment is the implantation of hydrogel or other matrix loaded with medication at the fracture site, which could eliminate disseminated adverse effects and increase drug concentration to achieve a better therapeutic effect. In conclusion, TNFRSF has excellent therapeutic potential in treating and preventing skeletal-related diseases, including bone fractures. Indeed, the future application of such therapy may emerge in various forms, and this field remains in need of further investigation and experimentation.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
HL designed the study, SE and ZS performed data collection, ZW, JL and MA analyzed the results, HZ, YD and AA drafted the manuscript. The final paper has been reviewed and approved by the authors.
Funding
The study was funded by the Zhejiang Provincial Natural Science Foundation (grant number LS21H060001), Alibaba Youth Studio Project (grant number ZJU-032). The funding bodies had no role in the design of the study; in collection, analysis, and interpretation of data; and in drafting the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Lamki, R. S., and Mayadas, T. N. (2015). TNF receptors: Signaling pathways and contribution to renal dysfunction. Kidney Int. 87 (2), 281–296. doi:10.1038/ki.2014.285
Bolander, M. E. (1992). Regulation of fracture repair by growth factors. Proc. Soc. Exp. Biol. Med. 200 (2), 165–170. doi:10.3181/00379727-200-43410a
Boyce, B. F., and Xing, L. (2007). Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 9 (1), 12. doi:10.1186/ar2165
Brenner, D., Blaser, H., and Mak, T. W. (2015). Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 15 (6), 362–374. doi:10.1038/nri3834
Bu, R., Borysenko, C. W., Li, Y., Cao, L., Sabokbar, A., and Blair, H. C. (2003). Expression and function of TNF-family proteins and receptors in human osteoblasts. Bone 33 (5), 760–770. doi:10.1016/j.bone.2003.07.006
Carswell, E. A., Old, L. J., Kassel, R. L., Green, S., FioreN., , and Williamson, B. (1975). An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U. S. A. 72 (9), 3666–3670. doi:10.1073/pnas.72.9.3666
Collaborators, G. B. D. F. (2021). Global, regional, and national burden of bone fractures in 204 countries and territories, 1990-2019: A systematic analysis from the global burden of disease study 2019. Lancet. Healthy Longev. 2 (9), e580–e592. doi:10.1016/S2666-7568(21)00172-0
Croft, M., Duan, W., Choi, H., Eun, S. Y., Madireddi, S., and Mehta, A. (2012). TNF superfamily in inflammatory disease: Translating basic insights. Trends Immunol. 33 (3), 144–152. doi:10.1016/j.it.2011.10.004
David, J. P., and Schett, G. (2010). TNF and bone. Curr. Dir. Autoimmun. 11, 135–144. doi:10.1159/000289202
Fischer, R., Kontermann, R. E., and Pfizenmaier, K. (2020). Selective targeting of TNF receptors as a novel therapeutic approach. Front. Cell. Dev. Biol. 8, 401. doi:10.3389/fcell.2020.00401
Fischer, R., Maier, O., Siegemund, M., Wajant, H., Scheurich, P., and Pfizenmaier, K. (2011). A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS One 6 (11), e27621. doi:10.1371/journal.pone.0027621
Fischer, R., Marsal, J., Gutta, C., Eisler, S. A., Peters, N., Bethea, J. R., et al. (2017). Novel strategies to mimic transmembrane tumor necrosis factor-dependent activation of tumor necrosis factor receptor 2. Sci. Rep. 7 (1), 6607. doi:10.1038/s41598-017-06993-4
Fischer, R., Wajant, H., Kontermann, R., Pfizenmaier, K., and Maier, O. (2014). Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia 62 (2), 272–283. doi:10.1002/glia.22605
Gerstenfeld, L. C., Cho, T. J., Kon, T., Aizawa, T., Cruceta, J., Graves, B. D., et al. (2001). Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs 169 (3), 285–294. doi:10.1159/000047893
Gerstenfeld, L. C., Cho, T. J., Kon, T., Aizawa, T., TsAy, A., Fitch, J., et al. (2003). Impaired fracture healing in the absence of TNF-alpha signaling: The role of TNF-alpha in endochondral cartilage resorption. J. Bone Min. Res. 18 (9), 1584–1592. doi:10.1359/jbmr.2003.18.9.1584
Granger, G. A., Shacks, S. J., Williams, T. W., and Kolb, W. P. (1969). Lymphocyte in vitro cytotoxicity: Specific release of lymphotoxin-like materials from tuberculin-sensitive lymphoid cells. Nature 221 (5186), 1155–1157. doi:10.1038/2211155a0
Hsu, H., Shu, H. B., Pan, M. G., and Goeddel, D. V. (1996). TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 84 (2), 299–308. doi:10.1016/s0092-8674(00)80984-8
Idriss, H. T., and Naismith, J. H. (2000). TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 50 (3), 184–195. doi:10.1002/1097-0029(20000801)50:3<184::AID-JEMT2>3.0.CO;2-H
Information on tumor necrosis factor (TNF) blockers (marketed as remicade, enbrel, humira, cimzia, and simponi), in FDA drug safety communication. 2015, MD: U.S. Food and Drug Administration.
Karnes, J. M., Daffner, S. D., and Watkins, C. M. (2015). Multiple roles of tumor necrosis factor-alpha in fracture healing. Bone 78, 87–93. doi:10.1016/j.bone.2015.05.001
Kontermann, R. E., Munkel, S., Neumeyer, J., Muller, D., Branschadel, M., Scheurich, P., et al. (2008). A humanized tumor necrosis factor receptor 1 (TNFR1)-specific antagonistic antibody for selective inhibition of tumor necrosis factor (TNF) action. J. Immunother. 31 (3), 225–234. doi:10.1097/CJI.0b013e31816a88f9
Lam, J., TakeShita, S., Barker, J. E., Kanagawa, O., Ross, F. P., and Teitelbaum, S. L. (2000). TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 106 (12), 1481–1488. doi:10.1172/JCI11176
Lang, I., Fullsack, S., Wyzgol, A., Fick, A., Trebing, J., Arana, J. A. C., et al. (2016). Binding studies of TNF receptor superfamily (TNFRSF) receptors on intact cells. J. Biol. Chem. 291 (10), 5022–5037. doi:10.1074/jbc.M115.683946
Liu, H., Li, D., Zhang, Y., and Li, M. (2018). Inflammation, mesenchymal stem cells and bone regeneration. Histochem. Cell. Biol. 149 (4), 393–404. doi:10.1007/s00418-018-1643-3
Locksley, R. M., Killeen, N., and Lenardo, M. J. (2001). The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell. 104 (4), 487–501. doi:10.1016/s0092-8674(01)00237-9
Lukic, I. K., Grcevic, D., KovacicN., , KataVic, V., Ivcevic, S., KalajzIc, I., et al. (2005). Alteration of newly induced endochondral bone formation in adult mice without tumour necrosis factor receptor 1. Clin. Exp. Immunol. 139 (2), 236–244. doi:10.1111/j.1365-2249.2005.02680.x
Mancusi, A., Alvarez, M., Piccinelli, S., Velardi, A., and Pierini, A. (2019). TNFR2 signaling modulates immunity after allogeneic hematopoietic cell transplantation. Cytokine Growth Factor Rev. 47, 54–61. doi:10.1016/j.cytogfr.2019.05.001
Medication Guide Prolia Injection for subcutaneous use. 05/2022. CA: Amgen Inc. One Amgen Center Drive Thousand Oaks California.
Medler, J., and Wajant, H. (2019). Tumor necrosis factor receptor-2 (TNFR2): An overview of an emerging drug target. Expert Opin. Ther. Targets 23 (4), 295–307. doi:10.1080/14728222.2019.1586886
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 114 (2), 181–190. doi:10.1016/s0092-8674(03)00521-x
Monaco, C., Nanchahal, J., Taylor, P., and Feldmann, M. (2015). Anti-TNF therapy: Past, present and future. Int. Immunol. 27 (1), 55–62. doi:10.1093/intimm/dxu102
Nagano, K., Alles, N., Mian, A. H., Shimoda, A., Morimoto, N., Tamura, Y., et al. (2011). The tumor necrosis factor type 2 receptor plays a protective role in tumor necrosis factor-alpha-induced bone resorption lacunae on mouse calvariae. J. Bone Min. Metab. 29 (6), 671–681. doi:10.1007/s00774-011-0270-z
Ochi, H., Hara, Y., Tagawa, M., Shinomiya, K., and Asou, Y. (2010). The roles of TNFR1 in lipopolysaccharide-induced bone loss: Dual effects of TNFR1 on bone metabolism via osteoclastogenesis and osteoblast survival. J. Orthop. Res. 28 (5), 657–663. doi:10.1002/jor.21028
Ota, N., Takaishi, H., Kosaki, N., Takito, J., Yoda, M., Tohmonda, T., et al. (2009). Accelerated cartilage resorption by chondroclasts during bone fracture healing in osteoprotegerin-deficient mice. Endocrinology 150 (11), 4823–4834. doi:10.1210/en.2009-0452
Qi, Z., Liu, W., and Lu, J. (2016). The mechanisms underlying the beneficial effects of exercise on bone remodeling: Roles of bone-derived cytokines and microRNAs. Prog. Biophys. Mol. Biol. 122 (2), 131–139. doi:10.1016/j.pbiomolbio.2016.05.010
Raje, N., Terpos, E., Willenbacher, W., Shimizu, K., Garcia-Sanz, R., Durie, B., et al. (2018). Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet. Oncol. 19 (3), 370–381. doi:10.1016/S1470-2045(18)30072-X
Richter, F., Seifert, O., Herrmann, A., Pfizenmaier, K., and Kontermann, R. E. (2019). Improved monovalent TNF receptor 1-selective inhibitor with novel heterodimerizing Fc. MAbs 11 (4), 653–665. doi:10.1080/19420862.2019.1596512
Sabio, G., and Davis, R. J. (2014). TNF and MAP kinase signalling pathways. Semin. Immunol. 26 (3), 237–245. doi:10.1016/j.smim.2014.02.009
Santos, L., Elliott-Sale, K. J., and Sale, C. (2017). Exercise and bone health across the lifespan. Biogerontology 18 (6), 931–946. doi:10.1007/s10522-017-9732-6
Schett, G., and Gravallese, E. (2012). Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 8 (11), 656–664. doi:10.1038/nrrheum.2012.153
Soos, B., Szentpetery, A., Raterman, H. G., Lems, W. F., Bhattoa, H. P., and Szekanecz, Z. (2022). Effects of targeted therapies on bone in rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 18 (5), 249–257. doi:10.1038/s41584-022-00764-w
Takayanagi, H. (2007). Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 7 (4), 292–304. doi:10.1038/nri2062
Tharabenjasin, P., Pabalan, N., Jarjanazi, H., and Jinawath, N. (2022). Associations of osteoprotegerin (OPG) TNFRSF11B gene polymorphisms with risk of fractures in older adult populations: meta-analysis of genetic and genome-wide association studies. Osteoporos. Int. 33 (3), 563–575. doi:10.1007/s00198-021-06161-5
Ting, A. T., and Bertrand, M. J. M. (2016). More to life than NF-κB in TNFR1 signaling. Trends Immunol. 37 (8), 535–545. doi:10.1016/j.it.2016.06.002
van den Berg, W. B., and Miossec, P. (2009). IL-17 as a future therapeutic target for rheumatoid arthritis. Nat. Rev. Rheumatol. 5 (10), 549–553. doi:10.1038/nrrheum.2009.179
Vu, T. H., Shipley, J. M., BerGers, G., Berger, J. E., Helms, J. A., Hanahan, D., et al. (1998). MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 93 (3), 411–422. doi:10.1016/s0092-8674(00)81169-1
Wang, L., Zhang, J., Wang, C., Qi, Y., Du, M., Liu, W., et al. (2017). Low concentrations of TNF-alpha promote osteogenic differentiation via activation of the ephrinB2-EphB4 signalling pathway. Cell. Prolif. 50 (1), e12311. doi:10.1111/cpr.12311
Yang, S., Wang, J., Brand, D. D., and Zheng, S. G. (2018). Role of TNF-TNF receptor 2 signal in regulatory T cells and its therapeutic implications. Front. Immunol. 9, 784. doi:10.3389/fimmu.2018.00784
Keywords: fracture, TNFR1, TNFR2, RANK/RANKL/OPG, healing
Citation: Dong Y, Zhou H, Alhaskawi A, Wang Z, Lai J, Abdullah Ezzi SH, Kota VG, Abdulla Hasan Abdulla MH, Sun Z and Lu H (2022) Alterations in bone fracture healing associated with TNFRSF signaling pathways. Front. Pharmacol. 13:905535. doi: 10.3389/fphar.2022.905535
Received: 27 March 2022; Accepted: 03 October 2022;
Published: 17 October 2022.
Edited by:
James Jeiwen Chou, Harvard Medical School, United StatesReviewed by:
Linda Connelly, California University of Science and Medicine, United StatesZhifang Zhang, City of Hope National Medical Center, United States
Copyright © 2022 Dong, Zhou, Alhaskawi, Wang, Lai, Abdullah Ezzi, Kota, Abdulla Hasan Abdulla, Sun and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Lu, aHVpbHVAemp1LmVkdS5jbg==
†These authors have contributed equally to this work