 Congwu Du1*
Congwu Du1* Yueming Hua1†
Yueming Hua1† Kevin Clare1†
Kevin Clare1† Kicheon Park1†
Kicheon Park1† Craig P. Allen1
Craig P. Allen1 Nora D. Volkow2*
Nora D. Volkow2* Xiu-Ti Hu3*
Xiu-Ti Hu3* Yingtian Pan1
Yingtian Pan1- 1Department of Biomedical Engineering, Stony Brook University, New York, NY, United States
- 2National Institute on Drug Abuse, Bethesda, MD, United States
- 3Department of Microbial Pathogens and Immunity, Rush University Medical Center, Chicago, IL, United States
Individuals with substance use disorder are at a higher risk of contracting HIV and progress more rapidly to AIDS as drugs of abuse, such as cocaine, potentiate the neurotoxic effects of HIV-associated proteins including, but not limited to, HIV-1 trans-activator of transcription (Tat) and the envelope protein Gp120. Neurotoxicity and neurodegeneration are hallmarks of HIV-1-associated neurocognitive disorders (HANDs), which are hypothesized to occur secondary to excitotoxicity from NMDA-induced neuronal calcium dysregulation, which could be targeted with NMDA antagonist drugs. Multiple studies have examined how Gp120 affects calcium influx and how cocaine potentiates this influx; however, they mostly focused on single cells and did not analyze effects in neuronal and vascular brain networks. Here, we utilize a custom multi-wavelength imaging platform to simultaneously study the neuronal activity (detected using genetically encoded Ca2+ indicator, GcaMP6f, expressed in neurons) and hemodynamic changes (measured by total hemoglobin and oxygenated hemoglobin within the tissue) in the prefrontal cortex (PFC) of HIV-1 Tg rats in response to cocaine and evaluate the effects of the selective NMDA antagonist drug memantine on cocaine and HIV neurotoxicity compared to those of non-HIV-1 Tg animals (controls). Our results show that memantine improved cocaine-induced deficit in cerebral blood volume while also attenuating an abnormal increase of the neuronal calcium influx and influx duration in both control rats and HIV-1 Tg rats. Cocaine-induced neuronal and hemodynamic dysregulations were significantly greater in HIV-1 Tg rats than in control rats. With memantine pretreatment, HIV-1 Tg rats showed attenuated cocaine’s effects on neuronal and hemodynamic responses, with responses similar to those observed in control rats. These imaging results document an enhancement of neuronal Ca2+ influx, hypoxemia, and ischemia with cocaine in the PFC of HIV-1 Tg rats that were attenuated by memantine pretreatment. Thus, the potential utility of memantine in the treatment of HAND and of cocaine-induced neurotoxicity deserves further investigation.
Introduction
Drug addiction places individuals at a higher risk for HIV infection and also for rapid progression to neuroHIV (Larrat and Zierler, 1993; Kral et al., 1998; Vignoles et al., 2006). Specifically, cocaine can accelerate the progression of HIV/AIDS as it enhances viral replication and impairs immune responses, targeting macrophages and lymphocytes (Peterson et al., 1990; Peterson et al., 1991). Cocaine might also accelerate the development of HIV-1-associated neurocognitive disorder (HAND), which can manifest as mild cognitive and motor impairment up to dementia and encephalitis (Larrat and Zierler, 1993; Antinori et al., 2007; McArthur et al., 2010). A hallmark of HAND is neuronal degeneration, which is hypothesized to occur due to excitotoxicity from the dysregulation of calcium homeostasis mediated by overactive NMDA receptors and L-type Ca2+ channels (Lipton et al., 1991; New et al., 1997; Hu, 2016). The neurotoxic HIV-1 proteins trans-activator of transcription (Tat) and Gp120 are implicated in mediating the dysfunction of NMDA receptors, thus contributing to the abnormally enhanced calcium influx (Nath et al., 1996; Bansal et al., 2000; Nath et al., 2000). Cocaine acts synergistically with HIV-1 Tat and Gp120, potentiating further excessive calcium influx and neurotoxicity (Koutsilieri et al., 1997; Yao et al., 2009). This sudden and prolonged increase in neuronal calcium ([Ca2+]in) leads to mitochondrial dysfunction, increased reactive oxygen species, and caspase activation (Aksenov et al., 2006; Armada-Moreira et al., 2020). Clinically, patients with HAND have shown attenuated cerebral blood flow including in prefrontal cortex (PFC) regions that might reflect neurotoxicity-related decreases in neuronal activity (Schielke et al., 1990).
Like HAND, preclinical models of cocaine use disorders (CUDs) showed that cocaine alone led to neuronal calcium dysregulation in brain tissues as well as hypoxemia in the PFC (Allen et al., 2019; Du et al., 2021). In combination with the dysregulation of voltage-gated L-type Ca2+ channels (Nasif et al., 2005a; Nasif et al., 2005b; Ford et al., 2009; Napier et al., 2014; Wayman et al., 2015; Wayman et al., 2016), NMDA receptors are also pivotal in the development of addictive behaviors and neural adaptations that are mediated by neuronal Ca2+ dysregulation. Conditional knockout of these receptors leads to the loss of condition place preference with cocaine administration, while the application of an NMDA antagonist inhibits neuronal spine adaptions in the striatum (Agatsuma et al., 2010; Ren et al., 2010).
Multiple studies have revealed how HIV proteins such as Tat and Gp120 disturb neuronal calcium influx as well as how cocaine potentiates this influx and vice versa (Peterson et al., 1991; Yao et al., 2009; Buch et al., 2011; Wayman et al., 2012; Napier et al., 2014; Wayman et al., 2015; Wayman et al., 2016). Moreover, HIV-1 Tat also facilitates dopamine (DA) neurotransmission in the mesocorticolimbic systems by inhibiting the activity of DA transporters (DATs) (Zhu et al., 2018; Quizon et al., 2021), which is also the target of cocaine’s effects, and could also promote the cocaine-induced dysregulation of intracellular calcium homeostasis. However, many of these studies have been limited to looking at single cells either in vivo or in vitro and did not analyze the concurrent pathogenesis of hemodynamics, especially in the brain regions that regulate neurocognition and addiction. To study the impact of HIV-1 and its associated proteins on the brain’s reward pathways, we utilized a rodent model of neuroHIV that expresses the HIV-1-associated proteins Tat, Gp120, and several others without a viral load. This rodent model is used in conjunction with a custom-built multi-spectral imaging modality with high spatiotemporal resolution to define the alterations in the prefrontal neuronal and hemodynamic function following exposure to cocaine. Additionally, we also sought to understand how memantine, a selective NMDA antagonist, may modify the PFC’s response to cocaine in both wild-type (non-Tg) and HIV-1 Tg rats. To our knowledge, this is the first neuroimaging study that demonstrates the dysfunction of neuronal activity (i.e., excessive Ca2+ influx; [Ca2+]in) and hemodynamics simultaneously in a brain region regulating neurocognition and addiction in the context of neuroHIV and cocaine use.
Methods
Animals and Experimental Design
Experimental protocols utilizing animals were approved by the Stony Brook Institutional Animal Care and Use committee. F344 adult male (wild-type) were acquired from Charles River Lab, and HIV-1 Tg rats were acquired from the University of Maryland, the creator of this neuroHIV rat model (F344 background). The rats were single-housed in a 12-h light/dark cycle and were given ad libitum access to food and water. To avoid possible interference of sex hormones on cerebral vasculature and neuronal excitability, we choose to only study male rats, but future studies will evaluate female animals. Animals were divided into experimental groups, as outlined in Table 1.
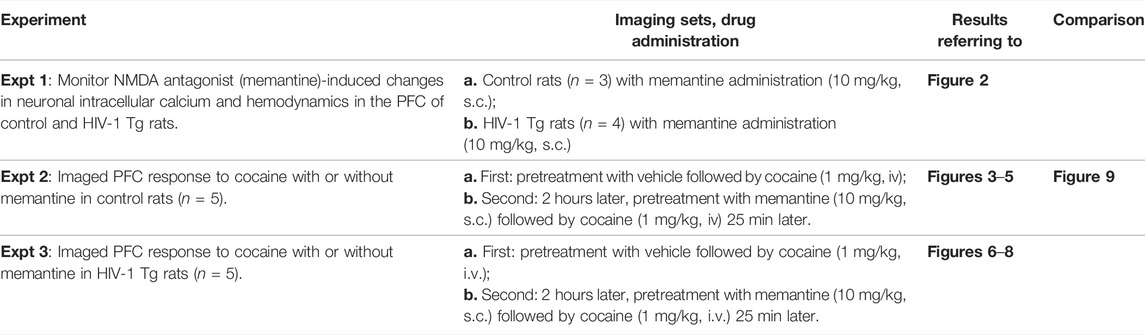
TABLE 1. Animal groups, experimental design, and outcome referring to/comparison.
Infusion of GCaMP6f Into Rat PFC
The genetically encoded calcium indicator GCaMP6f was delivered into the PFC of rats to study neuronal activity. Briefly, rats were anesthetized with isoflurane and mounted onto a stereotaxic frame. An incision was made along the scalp, and a hole was drilled into the skull over the PFC area (A/P: +3 mm; M/L:+0.8 mm). A total of 1 μl of AAV5. Syn.gCaMP6f.WPRE.SV40 (Addgene) was infused at two depths (D/V: −1.4mm; D/V: −1.0 mm) at a rate of 0.2 μl/min. Following the infusion of 0.5 μl, a 20-min break occurred to allow for viral diffusion through the tissue. After the surgery, rats were returned to their home cage and observed until awake. Flunixin was given immediately post-operatively and every 12 h thereafter for 3 days. Post-operative recovery was evaluated based on the level of activity as well as food and water intake. Imaging of PFC took place 3–4 weeks after viral delivery. Ex vivo imaging was conducted at the end of experiments to confirm the location of GCaMP6f expression (Figure 1B0).
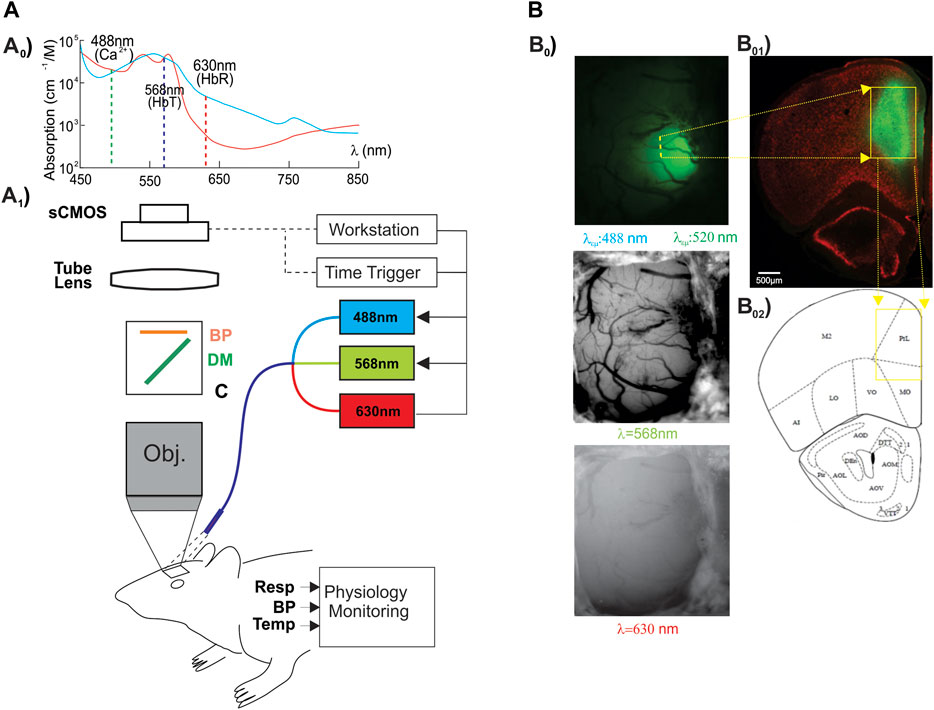
FIGURE 1. Experimental paradigm and image acquisition. (Ao) Hemoglobin (Hb) absorbance spectra. The vertical lines illustrate the center wavelengths of Ca2+ fluorescent excitation (λex = 488 nm), total hemoglobin (HbT, sensitive at λ1 = 568 nm), and deoxygenated-Hb (HbR, sensitive at λ2 = 630 nm). (A1) Schematic illustrating the custom multi-wavelength imaging platform (MIP) for the simultaneous capture of neuronal and hemodynamic activities. (B0) Representative in vivo images acquired by the MWIP of the GCaMP6f-expressed Ca2+ fluorescence, HbT, and HbR with ex vivo confirmation of the GCaMP6f expression in the rat’s PFC (B01). B02: rat brain atlas, showing the location of the GCaMP6f expression in PFC, as shown in a yellow cube (Paxinos and Watson, 1997).
Surgical Preparation for Imaging
Three to four weeks after viral infusion, rats were anesthetized with isoflurane and placed on a ventilator via intubation with a 16-gauge IV catheter into the trachea. Venous and arterial accesses were obtained through the insertion of catheters into the left lower limb femoral artery and vein (0.58 mm ID, 0.99 mm OD, Scientific Commodities Inc.). The arterial line was used to monitor blood pressure, whereas the venous line was used for drug delivery. The rat was then placed in a stereotaxic frame (Kopf 900), and a 4 × 6 mm2 craniotomy was performed above the PFC (A/P: 1–5 mm, M/l: −3 to 3 mm). The dura was carefully removed, and 1.25% agarose gel was placed onto the brain surface. A glass coverslip was placed over the agarose gel and secured into place using superglue. During experimentation, the animal’s physiology was recorded including the mean arterial pressure (mean = 82.28, SE = 1.88), body temperature (∼37–38°C), and respiration (∼40–45 breaths per minute) (small Animal Monitoring and Gating System, model 1025L, SA Instruments Inc.).
Drug Preparation
The NIDA drug supply program provided the cocaine–HCL used in this experiment. Cocaine solutions were freshly made for each experiment by diluting the drug in saline. Memantine (MEM) was purchased from Sigma-Aldrich (St. Louis, MO) and was dissolved in saline (10 mg/ml). For vehicle, saline was utilized.
Imaging of PFC Through Optical Window
Using a custom-built multi-modality imaging platform (MIP) utilizing previously described methods by our group (Allen et al., 2019; Yuan et al., 2011; Chen et al., 2016), in vivo images of the PFC were acquired. As shown in Figure 1, the simultaneous acquisition of [Ca2+]i, HbR, and HbO2 was achieved by the time-sharing approach (Yuan et al., 2011). The light source cycled three LEDs at three different wavelengths (λ1 = 568 nm, λ2 = 630 nm, and λexcitation = 488 nm; Spectra Light Engine, Lumicor; Figure 1A0) at a rate of 1 Hz to illuminate the brain tissue through the cranial window implanted over the PFC (Figure 1A1). Images were acquired by a cMOS camera (pixel size: 6.5 μm; Zyla 4.3, Andor) using modified Solis software (version 4.26, Andor). Figure 1B0 demonstrates the representative multi-wavelength images obtained from the PFC of a rat in vivo. Total hemoglobin (HbT) was calculated based on the changes in HbR and HbO2 (Eq. 1).
To determine the effects of memantine on baseline Ca2+ and hemodynamics, both HIV-1 Tg and control F344 were given 10 mg/kg s.c. of memantine (Table 1: experiment 1). These animals were first imaged for 5–10 min to acquire a baseline followed by SC injection of memantine (MEM) with an additional 20 min of recording.
Animals in experiments 2 and 3 underwent two sets of imaging with at least 90 min between each cocaine infusion (set a: VEH pretreatment followed by cocaine challenge; set b: memantine (MEM) pretreatment followed by cocaine challenge). Rats were first (in set a) pre-treated with VEH (saline, 1 ml/kg) followed by a 10-min baseline acquisition. Cocaine was infused (1 mg/kg i.v.) over 1 min followed by an additional 60 min of imaging. For the second set of images (in set b), rats were pre-treated with 10 mg/kg s.c. memantine. A baseline was acquired over 10 min followed by the administration of acute cocaine challenge over 1 min (1 mg/kg i.v.) with an additional 60-min imaging.
Processing Acquired Images
To quantify changes in Ca2+ and hemodynamics in response to cocaine challenge, we selected five regions of interest (ROI) for each animal (as illustrated in Figure 3: yellow circles). From full-field images, we selected the hemisphere in which GCaMP6f was injected and expressed (Supplementary Figures S2, S3). ROIs were then positioned in regions along the periphery of the GCaMP6f expression and were placed to avoid vasculature, as described in our previous publications (Allen et al., 2019; Du et al., 2020; Du et al., 2021; Park et al., 2022). This ROI positioning criterion was used to minimize the risk of GCaMP6f overexposure following cocaine administration as well as to prevent confounding effects of hemodynamic changes on the measured [Ca2+]i fluorescent signal. The same ROIs are used for all three image channels to ensure that the quantification of [Ca2+]i, HbT, and HbO2 was conducted in the same location of the cortex.
Hemodynamic changes (i.e., Δ[HbR] and Δ[HbO2]) were calculated using a modified Beer–Lambert law, as outlined in Eq. 1:
Through the MIP, images from illumination with λ1 and λ2 were captured and the reflectance matrices were determined (Rλ1(t) and Rλ2 (t)). Molar excitation coefficients for HbO2 and HbR at the given wavelengths are represented by ελ1HbO2, ελ1HbR, ελ2HbO2, and ελ2HbR and Lλ1 (t) and Lλ2 (t) are the path lengths of light propagation. When the sum of Δ[HbR] and Δ[HbO2] is calculated, the changes in total hemoglobin (Δ[HbT]) can be determined and used as a correlate for total blood volume in the cerebral cortex (Yuan et al., 2011).
Changes in intracellular Ca2+ levels were calculated based on the relative change in fluorescent intensity when compared to those in baseline (Δ[Ca2+]i). Data are represented as the percent change relative to the baseline. To correct for possible absorption in emitted light due to changes in hemodynamics, the fluorescent intensity in the ROIs was normalized to the intensity from areas of non-fluorescence.
Statistical Analysis
Mean and standard error of the mean were calculated using Microsoft Excel. Statistics were carried out using SigmaStat software (Systat Software INC.). Two-way ANOVAs were used to compare the difference between groups at each time point. Either one-way or two-way ANOVAs with a Bonferroni post hoc test were used for comparison of curve parameters (i.e., peak). p<0.05 was considered statistically significant. Raw data and p-values are summarized in Supplementary Tables S1–S4.
Results
Memantine (NMDA Antagonist) Activates Neuronal [Ca2+]i, But Has No Effect on Tissue Oxygenation in the PFC of Control and HIV-1 Tg Rats
To determine whether memantine would affect neuronal [Ca2+]i and hemodynamics (e.g., Δ[HbT], Δ[HbO2]) in the PFC, we administered memantine (10 mg/kg, s.c.) into control (F344, n = 3) and HIV-1 Tg (n = 4) animals. Neuronal Δ[Ca2+]i fluorescence, tissue oxygen hemoglobin (Δ[HbO2]), and total blood volume (Δ[HbT]) in PFC were detected for 15–20 min (Supplementary Figure S1). Figure 2 summarizes the changes in these three parameters for control and HIV- 1 Tg rats. Figure 2A shows that MEM induced a slight increase in the mean neuronal Δ[Ca2+]i to 1.40 ± 0.23 (%/minute, F (1,4) = 29.391, p = 0.006) and 0.81 ± 0.16 (%/minute, F (1,6) = 19.126, p = 0.005) in PFCs of control and HIV-1 Tg rats, respectively. These changes did not differ between CTL and HIV-1 Tg animals (F (1,5) = 4.756, p = 0.081). The mean blood volumes tended to decrease, but these effects were not significant neither in HIV-1 Tg animals (∆[HbT] = -0.062 ± 0.018 (%/min), F (1,6) = 1.28, p = 0.061) nor in controls (∆[HbT] = -0.64 ± 0.33 (%/min), F (1,4) = 3.144, p = 0.15). Oxygenated hemoglobin did not change in control (∆[HbO2] = −1.15 ± 0.37 (%/min), F (1,4) = 7.608 p = 0.051) or HIV-1 Tg (∆[HbO2] = -2.87 ± 1.52 (%/min), F (1,6) = 3,648, p = 0.105) animals. None of the responses to memantine differed significantly between the two groups.
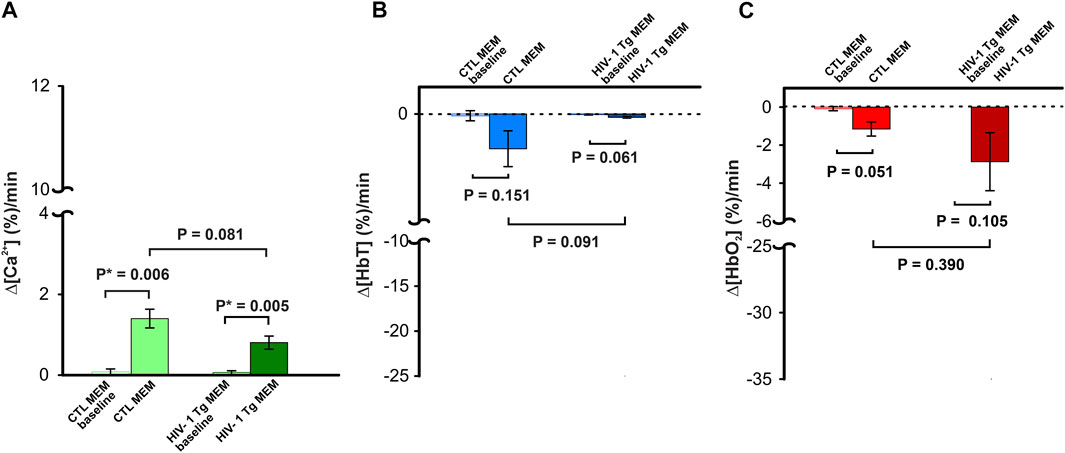
FIGURE 2. Neuronal Δ[Ca2+]i and brain hemodynamic responses to memantine between control and HIV-1 Tg rats. Memantine increased (A) neuronal Δ[Ca2+]i activity but did not change (B) blood volume (Δ[HbT]) nor (C) oxygenated hemoglobin (Δ[HbO2]) in PFC, and these responses did not differ between controls and HIV-1 Tg rats.
Memantine Shortened Cocaine-Induced Neuronal [Ca2+]i Increases and Reduced Tissue Oxygenation and Total Blood Volumes in Drug-Naïve Animals
Panel B0 of Figure 1 shows a representative image of GCaMP6f neuronal expression in the left PFC, where the virus was infused. Fluorescence was primarily expressed in the prelimbic cortex (PrL), which is implicated in cocaine intake (Chen et al., 2013), and reinstatement (Kalivas and McFarland, 2003). To compare cocaine’s effect on PFC with and without NMDA blockade by memantine in the same animal, we administered cocaine twice, first following the vehicle and then after MEM pretreatment (expt 2, Table 1). To minimize the effects of remaining cocaine from the first injection on the response of the second cocaine injection, we waited 2 h between infusions (Du et al., 2021).
Figure 3AVeh and 3AMEM include representative images of the GCaMP6f neuronal expression before and after MEM infusion, respectively. Figure 3Aveh (t = 0 min) and 3Aveh (t = 20 min) show the ratio images of GCaMP6f [Ca2+]in fluorescence at baseline and 20 min after cocaine without MEM pretreatment, whereas Figure 3AMEM(t = 0 min) and 3AMEM(t = 20 min) are ratio images of Ca2+ fluorescence at baseline and 20 min after cocaine with MEM pretreatment. These images demonstrate that neuronal [Ca2+]i fluorescence increases after cocaine with either vehicle or MEM pretreatment. Figure 3B shows the mean time courses of cocaine-induced neuronal [Ca2+]i changes in the PFC (n = 5) with VEH or MEM-pretreatment, indicating that cocaine triggered an influx in neuronal [Ca2+]i in the PFC with different recovery profiles when given after vehicle (cocaine only) or after cocaine with MEM pretreatment (10 mg/kg, s.c.). It illustrates a long-lasting [Ca2+]i increase in response to cocaine in VEH animals (i.e., >60 min after cocaine challenge). However, with MEM pretreatment, cocaine-induced Δ[Ca2+]i increase recovered at 45–60 min. A two-way ANOVA showed no significant difference between these two-time courses from baseline to 40 min after cocaine (F (13, 84) = 0.706, p = 0.753). However, a significant difference was observed from 40 min until the end of recording at 60 min post cocaine (p = 0.04). The average rate of Ca2+ influx did not differ following memantine pretreatment (F (1, 6) = 1.268, p = 0.303; Figure 3C). The peak neuronal ∆[Ca2+]i fluorescence increase induced by cocaine corresponded to 4.46 ± 0.92% for vehicle and did not differ from 4.41 ± 0.78% induced with MEM pretreatment (F (1, 6) = 0.00188, p = 0.967) (Figure 3D).
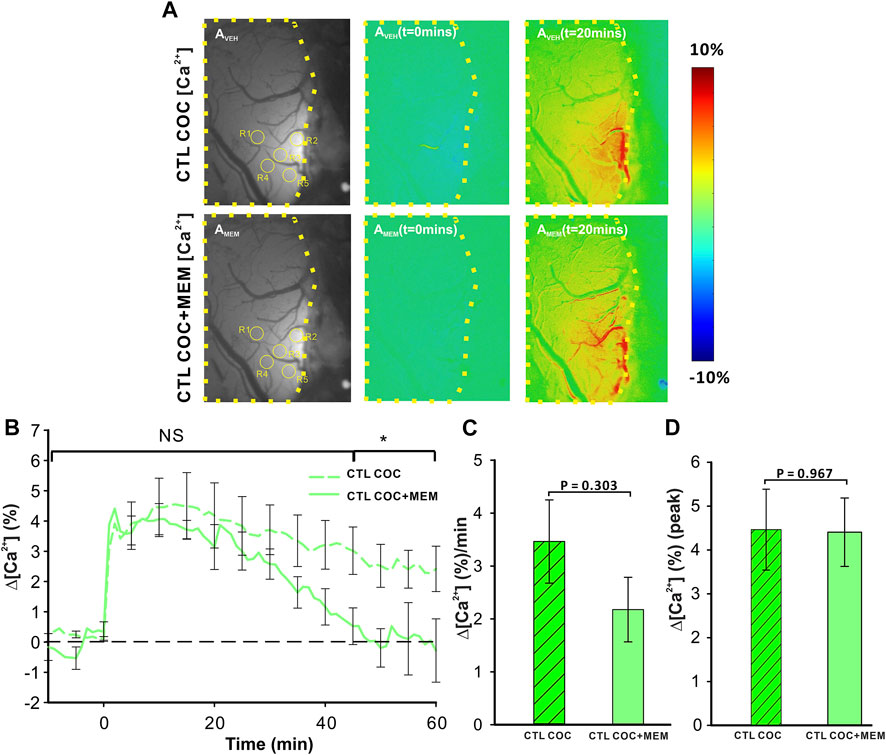
FIGURE 3. NMDA antagonist (memantine) shortens the duration of [Ca2+]i influx in control animals. (A) Representative images demonstrating the [Ca2+]i fluorescence changes in PFC following cocaine administration (1 mg/kg, i.v.) in vehicle pretreatment (upper panels) and memantine pretreatment (lower panel) rats at t = 20 min relative to the baseline (t = 0 min). Region of brain exposed through the cranial window outlined in yellow, with regions of interest (ROI) for quantification marked with yellow circles. (B) Time course of the mean cocaine-induced Δ[Ca2+]i response revealing a reduction in the duration of the Ca2+ influx following memantine pretreatment. (C) Total cocaine-induced Ca2+ influx was reduced following memantine pretreatment. (D) The presence of memantine did not affect the peak Ca2+ influx. Graphed values represent the mean and standard error. NS: not significant *: p<0.05, significant difference.
Acute cocaine reduced cerebral blood volume as measured by changes in ∆[HbT] (Figure 4). Figure 4A displays grayscale images with representative images of PFC in a drug-naïve animal at baseline and after MEM pretreatment. Ratio images of PFC before (t = 0 min) and after (t = 20 min) cocaine injection in VEH (upper panels) or MEM pretreatment (lower panel) indicate a decrease in ∆[HbT] in PFC after cocaine (1 mg/kg, i.v.). Comparison between CTL-Coc and MEM-Coc with a two-way ANOVA showed no significant difference within 5 min after cocaine (p>0.05) followed by a significant difference from t = 10–60 min post cocaine (F(13, 84) = 5.47, p<0.001). Cocaine’s reduction of ∆[HbT] was persistent (i.e., >60 min) after vehicle but returned to baseline after 58.83 ± 14.83 min in the MEM group (Figure 4B). We also compared that cocaine-induced changes in ∆[HbT] integrated over 60 min, which showed significant differences between the vehicle and MEM corresponding to −7.0 ± 0.78% and −1.87 ± 0.31%, respectively (F(1, 6) = 23.075, p = 0.003). Figure 4D illustrates the mean peak changes in ∆[HbT] following cocaine, showing no differences between the vehicle and MEM pretreatment (F(1, 6) = 3.887, p = 0.096).
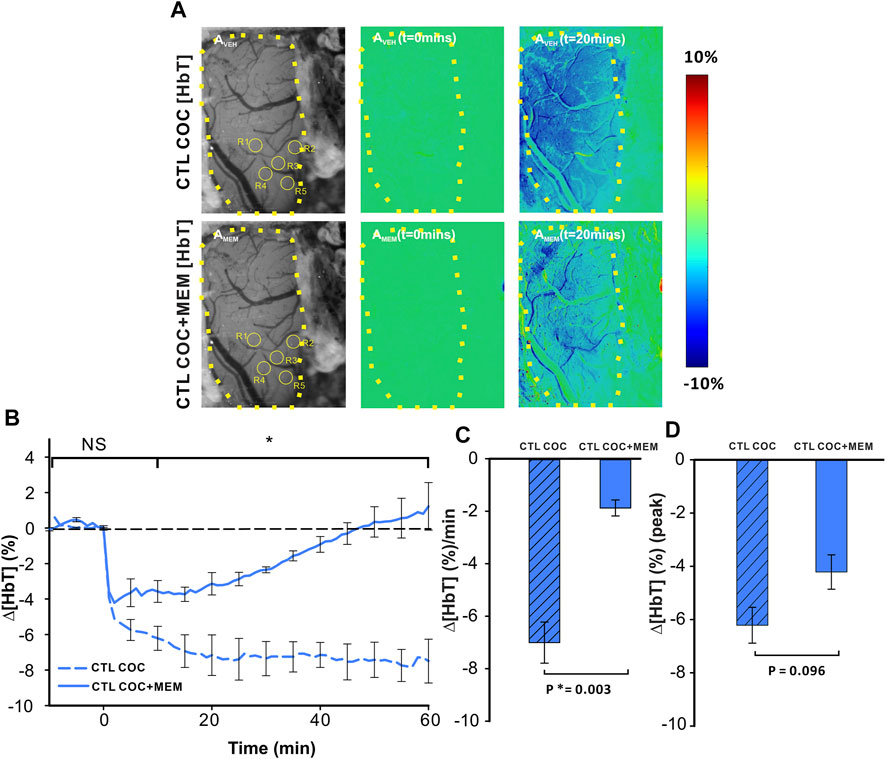
FIGURE 4. Memantine attenuates cocaine-induced Δ[HbT] decrease in the PFC of control animals. (A) Ratio imaging demonstrating the spatiotemporal cocaine-induced reduction in total hemoglobin (Δ[HbT]) in the rat PFC with vehicle (upper panels) and memantine (lower panels) pretreatments. The area encompassed by dashed yellow lines represents the region of brain visualized with the yellow circles indicting the location of ROI for quantification in AVEH and AMEM. (B) Exposure to memantine prior to cocaine improved cocaine-induced Δ[HbT] reduction as well as shortened the duration of Δ[HbT] response to cocaine. (C) Pretreatment with memantine significantly reduced the average Δ[HbT] reduction rate in the PFC. (D) Maximal Δ[HbT] decrease (i.e., peak) in response to cocaine did not differ significantly for vehicle and memantine. Graphed values represent the mean and standard error. NS: not significant. *: p<0.05, significant difference.
Cocaine decreased ∆[HbO2] in the PFC after vehicle with MEM pretreatment, attenuating the amplitude and duration of the cocaine-induced decrease (Figure 5). Specifically, for VEH, cocaine induced a persistent ∆[HbO2] decrease, whereas with MEM pretreatment, the decrease from cocaine returned to baseline at 38 min (t = 1–38 min; p < 0.001, t = 38 min; p = 0.909) (Figure 5B). Panels AVEH (upper panels in Figure 5A) and AMEM (lower panels in Figure 5A) show representative images of the surface of the PFC with ROI selections and ratio images to illustrate the percent change in [HbO2] concentration at t = 0 min and t = 20 min following cocaine infusion (vehicle and MEM pretreatment, respectively). A two-way ANOVA of Δ[HbO2] of VEH and MEM showed a significant interaction between pretreatment and time (F(13, 84) = 4.493, p<0.001). Point-by-point comparison between these two-time courses showed no differences within 5 min after cocaine between VEH and MEM pretreatment (p>0.05) followed by significant differences between them from t = 10–60 min (p < 0.05; Figure 5B). Cocaine-induced mean ∆[HbO2] change showed a −6.12 ± 0.76 (%/min) reduction with vehicle that was significantly attenuated to −1.23 ± 0.49 (%/min) with MEM (F(1, 6) = 21.023, p = 0.004; Figure 5C). The peak cocaine-induced decrease in ∆[HbO2] did not differ significantly between vehicle and MEM (−7.63 ± 0.84% with vehicle and −5.95 ± 0.64% with MEM, (F(1, 6) = 1.937, p = 0.213; Figure 5D).
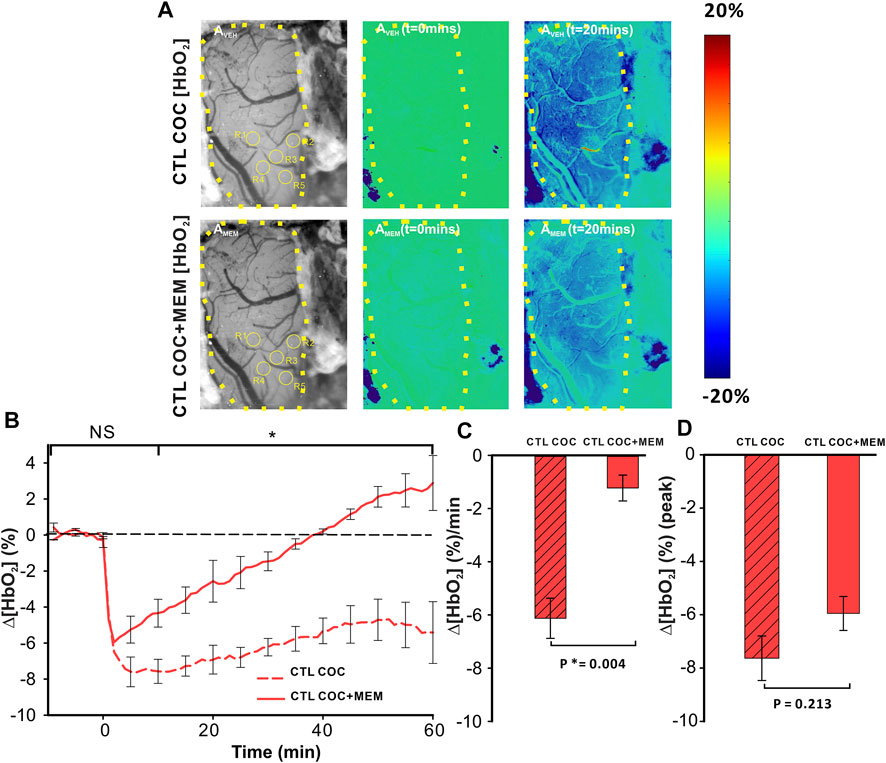
FIGURE 5. Reduction of Δ[HbO2] in the PFC of control animals induced by cocaine is attenuated following memantine pretreatment. (A) In vivo imaging illustrating the time- and treatment-dependent reduction in oxygenated hemoglobin (HbO2) following cocaine exposure in control rats pretreated with vehicle (upper panels) or memantine (lower panel). (B) Comparison of the time course for tissue oxygenation showed a faster recovery from cocaine-induced reduction in Δ[HbO2] after memantine pretreatment. (C) Quantification of the total change in Δ[HbO2] indicated that memantine pretreatment significantly improved Δ[HbO2] decreases in response to cocaine even though it (D) did not significantly change the peak reduction in Δ[HbO2]. Graphed values represent the mean and standard error. NS: not significant. *: p < 0.05, significant difference.
Memantine Depressed Cocaine-Induced Persistent Neuronal [Ca2+]i Activity and Reduced Hypoxia in HIV-1 Tg Animals
Cocaine triggered significant increases in neuronal Δ[Ca2+]i in PFC of HIV-1 Tg animals that differed when given after vehicle or after MEM pretreatment (Figure 6). Figure 6AVEH-HIV and Figure 6AMEM-HIV show the representative images of GCaMP6f neuronal expression in the PFC of a HIV-1 Tg rat with and without MEM. Figure 6AVEH (t = 0 min) and AVEH (t = 20 min) show the ratio images before and 20 min after cocaine (1 mg/kg, i.v.) for vehicle pretreatment and Figure 6AMEM-HIV(t = 0 min) and AMEM-HIV(t = 20 min) for MEM pretreatment. A two-way ANOVA showed a significant time effect ([F(13, 84) = 1.701, p = 0.072]). Comparing the responses to cocaine at each time point showed no significant difference in Δ[Ca2+]i increases from baseline to 30 min (p>0.05) between VEH and MEM pretreatment. This is followed by a significant difference after 35 min (p35-60<0.05; Figure 6B). The cocaine-induced mean Δ[Ca2+]i increase was 7.96 ± 1.66 (%/min) with vehicle, which was significantly reduced to 3.01 ± 0.57 (%/min) with MEM (F(1, 7) = 6.451, p = 0.039; Figure 6C). Also, the peaks of ∆[Ca2+]i in responses to cocaine differed significantly, corresponding to 6.71 ± 0.86% and 3.32 ± 0.63% for vehicle and MEM, respectively (F(1, 7) = 9.102, p = 0.019) (Figure 6D).
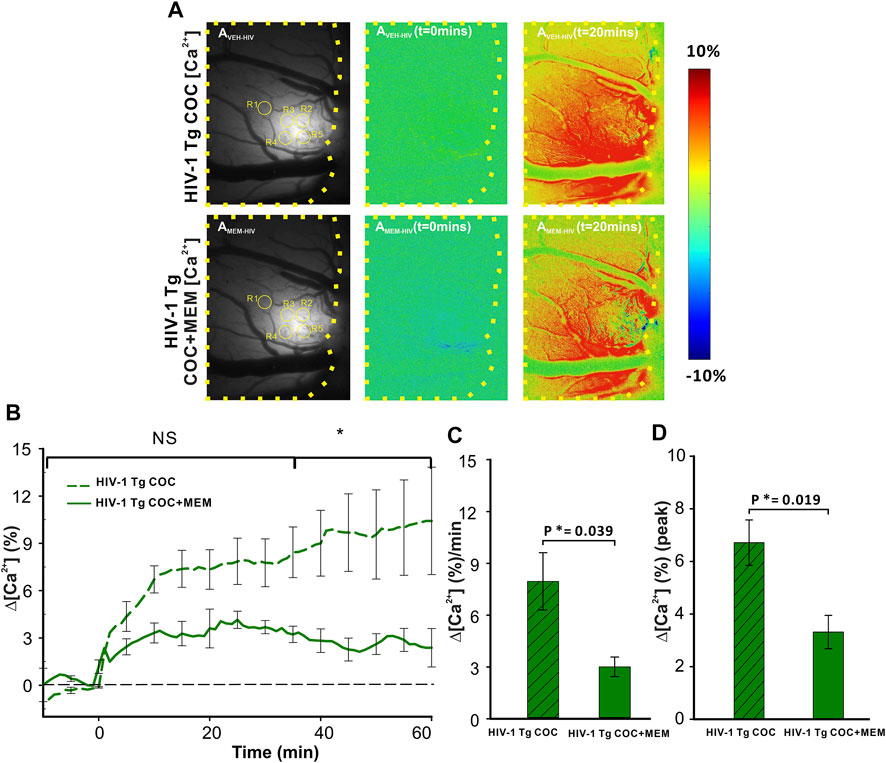
FIGURE 6. Attenuated neuronal Δ[Ca2+]i response to cocaine with memantine pretreatment in HIV-1 Tg rats. (A) Representative images of the [Ca2+]i fluorescent dynamics in the PFC, illustrating how the response varies spatiotemporally in the PFC of HIV-1 Tg rats. Upper panels represent the response in vehicle-pretreated animals, whereas the lower panel shows the PFC response to cocaine in memantine-pretreated HIV-1 Tg rats. (B) Time courses of neuronal Δ[Ca2+]i fluorescent changes in both the vehicle and memantine pretreated rats, including the attenuation of cocaine-induced neuronal Δ[Ca2+]i with memantine pretreatment. (C) Mean Δ[Ca2+]i change rate and (D) peak of the Δ[Ca2+]i signals were significantly reduced in memantine-pretreated rats. Values in graph represent the mean and standard error. The region of the PFC encircled by a yellow line represents the brain region of interest. ROIs quantified are contained within yellow circles in AVEH-HIV and AMEM-HIV. NS: not significant. *: p<0.05, significant difference.
Cocaine decreased Δ[HbT] in the PFC of HIV-1 Tg animals with both vehicle and MEM pretreatments (Figure 7A). A two-way ANOVA for the comparison between CTL-Coc and MEM-Coc showed a significant interaction of pretreatment and time ([F(13, 84) = 1.236, p = 0.27]). Cocaine’s reduction of ∆[HbT] was persistent with vehicle but recovered at 58.78 ± 26.91 min with MEM pretreatment (Figure 7B). The mean ∆[HbT] was −15.49 ± 3.2 (%/min) and −4.04 ± 3.01 (%/min) for vehicle and MEM pretreatment, respectively (F(1, 7) = 6.418, p = 0.039). There was no significant difference in the cocaine-induced peak of ∆[HbT] between vehicle (−14.83 ± 2.41%) and MEM (−6.95 ± 2.3%) (F(1, 7) = 4.306, p = 0.077; Figure 7D).
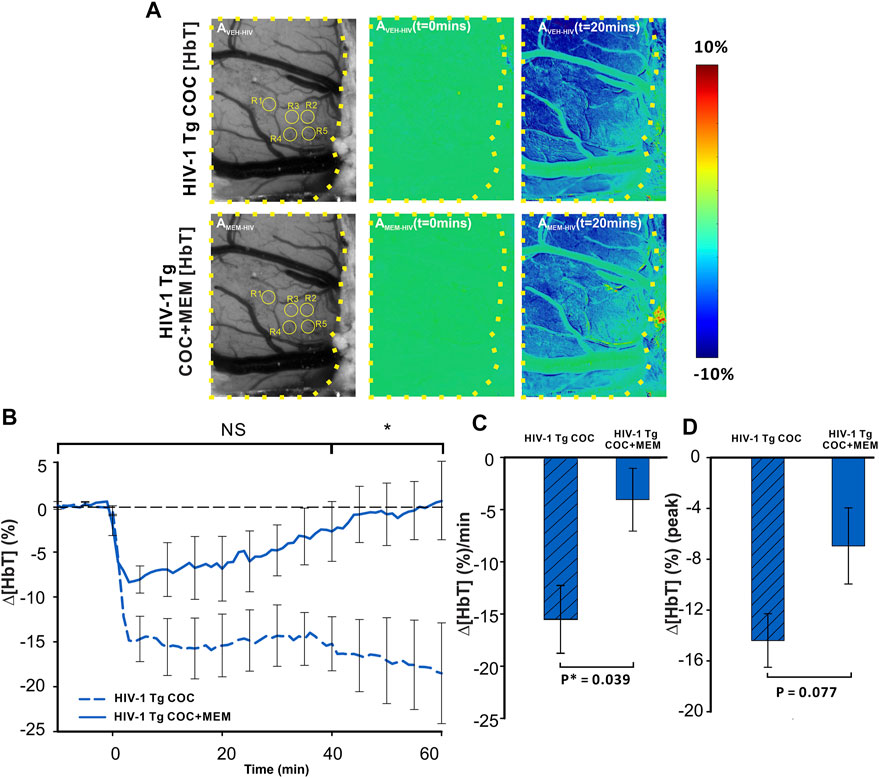
FIGURE 7. Memantine improves cocaine-induced decrease in Δ[HbT] in HIV-1 Tg rats. (A) Spatiotemporal changes of Δ[HbT] in the PFC of HIV-1 Tg rats in response to cocaine with either pretreatment with vehicle (upper panel) or with memantine (lower panel). (B) Time course of Δ[HbT] response to cocaine in PFC, illustrating that HIV-1 Tg rats pretreated with memantine returned to baseline within 60 min but not with vehicle pretreatment. (C) Memantine attenuated cocaine-induced reductions in Δ[HbT] and (D) peak of Δ[HbT] changes. Graphed values represent the mean with standard error. ROIs for quantification are encircled in yellow. The brain region of interest is encompassed by a yellow dashed line. NS: not significant. *: p<0.05, significant difference.
Cocaine reduced Δ[HbO2] in the PFC of HIV-1 Tg animals with vehicle or MEM pretreatment (Figure 8). Similar to memantine’s attenuation of the reduction of cerebral blood volume (∆[HbT]; Figure 7), oxygenated hemoglobin in PFC was also reduced with MEM pretreatment (Figure 8). With vehicle, cocaine decreased Δ[HbO2] to −33.97 ± 2.3% at t = 4 min and with memantine to −23.48 ± 4.0% (F(1, 7) = 5.714, p = 0.048; Figure 8D). Additionally, cocaine induced a persistent decrease in ∆[HbO2] over 60 min with vehicle, whereas with MEM pretreatment, the decrease recovered at 34.93 ± 12 min followed by an overshoot (Figure 8B). The mean changes in cocaine-induced ∆[HbO2] were significantly reduced from −18.7 ± 1.6 (%/min) with vehicle to −1.98 ± 5.3 (%/min) with MEM pretreatment (F(1, 7) = 11.066, p = 0.013; Figure 8C).
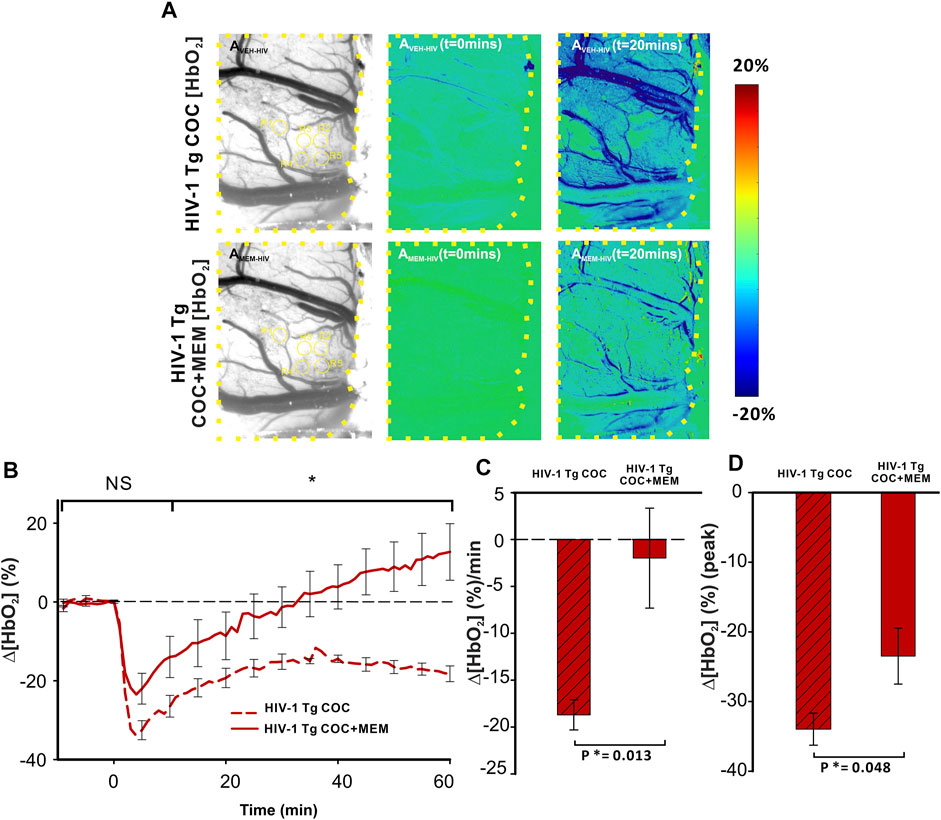
FIGURE 8. Cocaine-induced decrease in oxygenated hemoglobin improved with memantine in HIV-1 Tg rats. (A) Ratio imaging of HbO2 in the PFC of HIV-1 Tg rats following exposure to cocaine after pretreatment with either vehicle (upper panel) or memantine (lower panel). Areas encompassed within the dashed yellow lines represent the brain region of interest. ROIs used for quantification are in yellow circles. (B) Memantine administration prior to cocaine improved the cocaine-induced reduction in Δ[HbO2] returning to baseline but remained decreased in the PFC of animals with vehicle pretreatment. (C,D) Total cocaine-induced reduction in Δ[HbO2] and peak was significantly improved when HIV-1 Tg rats were pretreated with the NMDA antagonist. NS: not significant. *: p<0.05, significant difference.
Cocaine-Induced Neuronal [Ca2+]i and Hemodynamic Dysregulations in the PFC Between HIV-1 Tg Rats and Controls, With Memantine Reducing Cocaine’s Effect on Neural and Hemodynamic Changes in PFCs of HIV-Tg Rats
Figure 9 summarizes the comparison of cocaine’s effects on neuronal [Ca2+]i and hemodynamic changes in PFC between control and HIV-1 Tg animals with vehicle and memantine pretreatment. As shown in Figure 9A, the time course of ∆[Ca2+]i in the PFC of saline pretreated HIV-1 Tg animals showed a persistent increase over 60 min, whereas in controls, it reached a peak at ∼10 min after cocaine followed by a slow recovery. HIV-1 Tg rats pretreated with memantine displayed a similar neuronal Δ[Ca2+]i response to cocaine as control rats (from t = −5 min to t = 45 min, p-5 to 45>0.05, green shaded area) (Figure 9D). However, the ∆[Ca2+]i fluorescence in controls returned to baseline at t = 47.88 ± 10.53 min but not in HIV-1 Tg animals. A two-way ANOVA examining the interaction of drug (pretreatment with saline v memantine) and animal (control v HIV-1 Tg) for ∆[Ca2+]i found no significant interaction between memantine treatment and animals (F(16,1) = 2.409, p = 0.145). The significant main effects were found for both memantine (p = 0.02) and animal (p = 0.042) on ∆[Ca2+]i. Post hoc analysis revealed that the mean neuronal ∆[Ca2+]i was significantly higher in HIV-1 Tg rats pretreated with saline (∆[Ca2+]i = 7.96 ± 1.6 (%/min), n = 5) than in controls (∆[Ca2+]i = 3.46 ± 0.8 (%/min), n = 5, p = 0.01; Figure 9G). The mean rate of accumulated ∆[Ca2+]i did not differ between controls (∆[Ca2+]i = 2.18 ± 0.6 (%/min)) and HIV-1 Tg rats (3.01 ± 0.57 (%/min), p = 0.654). Pretreatment of HIV-1 Tg rats with memantine significantly reduced the accumulated Δ[Ca2+]i following cocaine exposure (p = 0.008; Figure 9G).
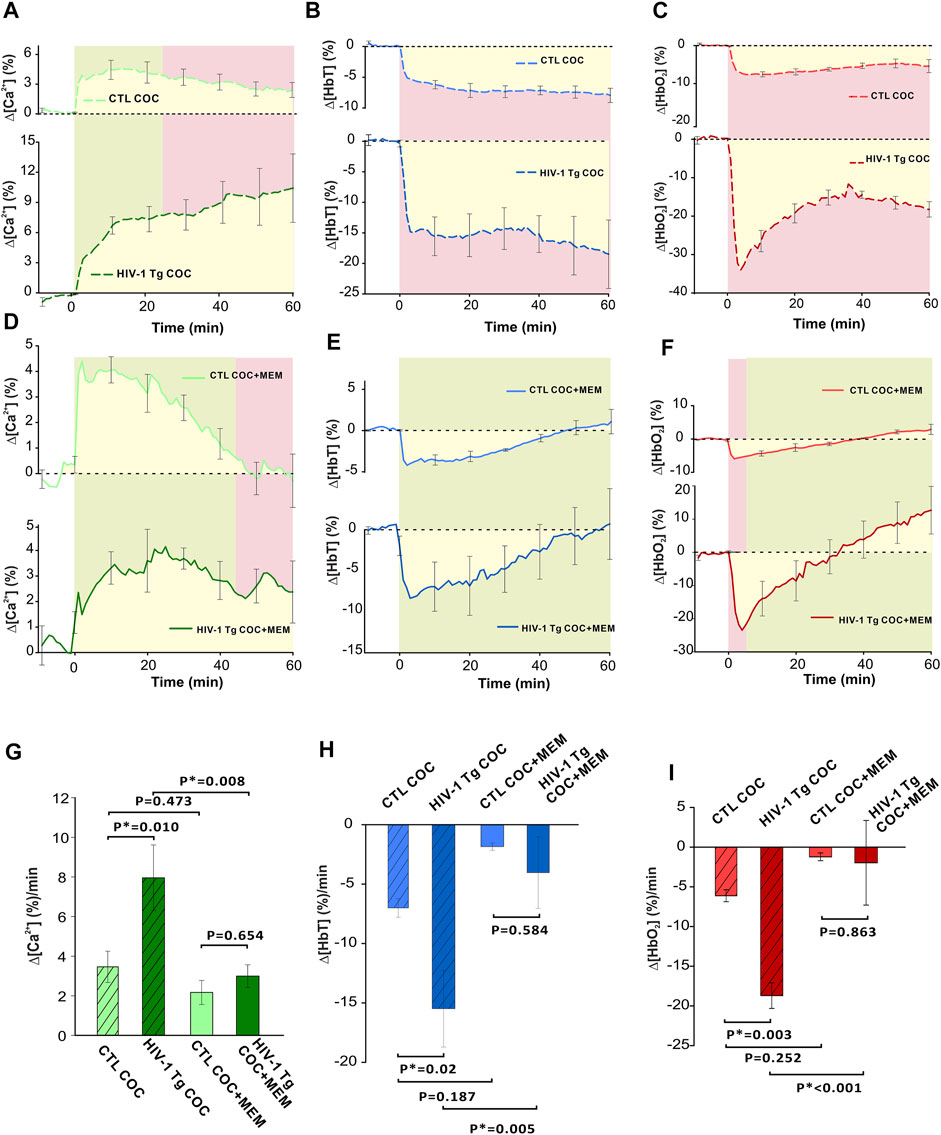
FIGURE 9. Comparison of cocaine-induced changes between control and transgenic HIV-1 Tg animals with saline or memantine pretreatment. (A) Time course of neuronal response to cocaine in HIV-1 Tg and control (F344 non-Tg) rats showing the variation in response to cocaine (1 mg/kg, i.v.). In control rats, Δ[Ca2+]i peak was followed by a slow decay, whereas HIV-1 Tg rats display a long, persistent increase in neuronal Δ[Ca2+]i. (B) HIV-1 Tg rats had a greater reduction in total hemoglobin (Δ[HbT]) as well as (C) a rapid decrease in oxygenated hemoglobin (Δ[HbO2]) followed by a slight recovery from -30% to -20% that remained decreased over 60 min. (D) Memantine-pretreated HIV-1 Tg rats had a similar Δ[Ca2+]i response to cocaine as control animals. However, control animals returned to baseline where HIV-1 Tg animals did not. (E) Time courses of control and HIV-1 Tg rat responses to cocaine showed similar temporal profiles in Δ[HbT] reductions. (F) Cocaine-induced Δ[HbO2] peak reduction was larger in HIV-1 Tg than in control rats. However, they returned to baseline at the same time. (G–I) HIV-1 Tg rats accumulated more intracellular neuronal Ca2+ and demonstrated significantly greater reductions in total and oxygenated hemoglobin. These enhanced responses were significantly reduced following memantine pretreatment in HIV-1 Tg. Green shaded area: no significant difference between curves. Red shaded area denotes significant differences between curves. Yellow area indicates the area quantified for the average rate of change for Δ[Ca2+]i, Δ[HbT], and Δ[HbO2]. *: p < 0.05, significant difference.
In addition, the total cerebral volume in the PFC of HIV-1 Tg animals pretreated with saline decreased to −15% within t = 5 min after cocaine and remained reduced over the time of recording (60 min; Figure 9B); whereas in controls, Δ[HbT] decreased to −5% at t = 5 min and also remained reduced until the end of recording (Figure 9B). The time courses of hemodynamic response to cocaine for Δ[HbT] for memantine pretreated control and HIV-1 Tg rats showed no significant differences (Figure 9E). A two-way ANOVA of ∆[HbT] found only a simple main effect of drug pretreatment of the total cerebral blood volume (p = 0.006). The mean amplitude of Δ[HbT] was 2-fold larger in saline pretreated HIV-1 Tg PFC (−15.49 ± 3.2 (%/min)) than in controls (−7.0 ± 0.78 (%/min), p = 0.02). Cocaine-induced mean Δ[HbT] reductions in control and HIV-1 Tg animals were −1.87 ± 0.3 (%/min) and −4.04 ± 3.01 (%/min), respectively, and were not significantly different (p = 0.584; Figure 9H). Similar to ∆[Ca2+]i, a reduction in the total blood volume was attenuated in HIV-1 Tg rats pretreated with memantine when compared to saline pretreatment (p = 0.005).
HIV-1 Tg rats pretreated with saline had a rapid decrease in oxygenated hemoglobin followed by a slight recovery from −30% to −20% that remained over 60 min (Figure 9C). Cocaine-associated reduction in Δ[HbO2] that peaked at t = 4 min (Figure 9F) was larger in HIV-1 Tg rats (−23.48 ± 4.02 (%)) than in controls (−5.95 ± 0.64 (%), p = 0.015), but the returned time to baseline was similar (control: 38.33 ± 1.67 min and HIV-1Tg: 34.93 ± 12 min, p = 0.821; Figure 9F). A two-way ANOVA of ∆[HbO2] found no significant interaction between pretreatment and animal (F16,1) = 4.556, p = 0.052). Only the simple main effect of pretreatment was found to be significant (p = 0.002). The mean reduction in Δ[HbO2] was greater for HIV-1 Tg rats (−18.7 ± 1.6 (%/min)) than for controls (−6.12 ± 0.76 (%/min), p = 0.003; Figure 9I). The mean reduction in Δ[HbO2] following memantine pretreatment did not differ between the two groups (p = 0.863; Figure 9I). Memantine pretreatment of HIV-1Tg rats significantly attenuated Δ[HbO2] reduction (p<0.001).
Conclusion and Discussion
HAND is likely due in part to the HIV-induced neurotoxicity associated with HIV reservoirs in the CNS, which can be exacerbated with cocaine exposure and vice versa. To our knowledge, this is the first study that looks at the effects of cocaine on the brain of a rodent model of neuroHIV that used cutting-edge imaging approach to simultaneously demonstrate the neuronal and hemodynamics responses to cocaine in the context of neuroHIV. Here, we investigated neuroHIV-associated synergistic potentiation of cocaine-induced neuronal and hemodynamic dysregulation in the PFC of rats, as well as the ability of NMDAR blockade (by memantine) to attenuate these deleterious effects. In wild-type (non-Tg) rats, memantine significantly shortened the duration of cocaine-induced neuronal calcium increase and attenuated the cocaine-associated reduction in brain tissue oxygenation and total blood volume. Memantine pretreatment of HIV-1 Tg rats also depressed the amplitude of the neuronal calcium increase and its long persistence while attenuating the reductions in tissue oxygenation and total blood volume from cocaine. When compared to non-Tg rats, HIV-1 Tg rats were significantly more reactive to cocaine-induced dysregulation of neuronal activity and hemodynamics. Pretreatment of both non-Tg and HIV-1 Tg rats with memantine significantly reduced the differences in cocaine response between them, suggesting that NMDAR blockade may blunt neuroHIV-associated PFC neuronal hyper-reactivity that is enhanced further by cocaine. Importantly, this result is similar to findings in our earlier studies, showing that the blockade of voltage-gated L-type calcium channels (L-channels) significantly reduces cocaine- and/or neuroHIV-induced PFC neuronal calcium and hemodynamic dysregulation (Nasif et al., 2005b; Ford et al., 2009; Khodr et al., 2016; Wayman et al., 2016; Du et al., 2021).
Consistent with our previous findings, when drug-naïve rats were exposed to cocaine, there was a rapid increase in calcium concentration in neurons in PFC (Allen et al., 2019; Du et al., 2021), especially in glutamatergic pyramidal neurons (Nasif et al., 2005a; Nasif et al., 2005b; Ford et al., 2009; Napier et al., 2014; Wayman et al., 2015; Wayman et al., 2016). Excessive calcium influx could be one of the triggers for excitotoxicity that contributes to dysregulation, injury, and ultimately loss of cortical neurons in mice and rats treated chronically with cocaine (Olney et al., 1991; George et al., 2008; Clare et al., 2021). Various mechanisms through which calcium influx induces excitotoxicity have been suggested, including NMDA receptor overactivation (Dodt et al., 1993; Colwell and Levine, 1996). In NDMA-mediated excitotoxicity, the sustained calcium influx stimulates intracellular cascades that ultimately lead to the activation of proteases and nucleases. Increased cytosolic positive charge (Ca2+) from the calcium influx is initially buffered by mitochondria. However, mitochondria eventually fail to regulate [Ca2+]in, leading to abnormal membrane depolarization and failure of ATP creation, decreased anti-oxidate production, and release of caspases furthering neuronal death (Armada-Moreira et al., 2020).
Rats in the context of neuroHIV displayed a significantly elevated PFC neuronal calcium influx, and this was further enhanced when exposed to cocaine as compared to control rats (∼2-fold). Previous studies into understanding the mechanism through which HIV exacerbates calcium influx point to HIV proteins including Tat and the envelope protein Gp120 as major contributors (Buch et al., 2011; Hu, 2016). Tat has been linked to upregulating tumor necrosis factor alpha, which enhances calcium influx in rat hippocampal pyramidal neurons and leads to the promotion of the cAMP/PKA pathway, which can increase the phosphorylation of NMDA receptors and calcium influx (Zidovetzki et al., 1998; Jara et al., 2007). Moreover, we also demonstrate the dysfunction of voltage-gated Ca2+ channels (VGCCs) induced by Tat. For instance, Tat not only significantly enhances calcium influx through VGCCs (Napier et al., 2014) but also increases the expression of L-channel VGCCs in mPFC pyramidal neurons (Wayman et al., 2012). In addition, in conjunction with Tat, Gp120 has been shown to work synergistically with Tat, potentiating its calcium influx even when levels of Tat were subtoxic (Nath et al., 2000). Astrocytes, which clear 90% of extracellular glutamate, are also impacted by Tat, which inhibits their ability to remove extracellular glutamate, the main ligand of NMDA receptors (Kojima et al., 1999; Zhou et al., 2004). Collectively, all of these changes to normal cellular physiology likely contribute to Ca2+-induced neuronal excitotoxicity and ultimately to the mechanism of HAND.
Pretreatment with memantine not only significantly attenuated calcium influx in this rat model of neuroHIV but also attenuated the influx in non-Tg control rats, and this was associated with a return of intracellular calcium back to baseline levels within 60 min. When we compared the calcium transients of the memantine-pretreated control rats to the memantine-pretreated HIV-1 Tg rats, we found that there was no significant difference between the curves. This suggests that the blockade of overactive NMDARs was able to diminish the HIV-induced abnormal increase of neuronal calcium influx. Taken together, our novel findings support that, in combination with the selective blockade of overactive L-type calcium channels, antagonizing hyperactive NMDARs by memantine has a potential to be used clinically to decrease neuronal excitotoxicity in both HIV(+) and HIV (-) cocaine users, with the possibility of substantially more benefit to those individuals who are HIV(+) (Khodr et al., 2016). Given that previous clinical trial studies failed to significantly decrease neurocognitive deficits in patients with HAND, likely due to specific blockade of NMDARs (Schifitto et al., 2007) or L-channels (Navia et al., 1998), the combined treatment with selective NMDA antagonist and L-type calcium channel blocker may over time lead to reducing cocaine-induced excitotoxicity and limiting neuronal degeneration in the brain regions that regulate neurocognition. Further work is required to validate this novel concept and hypothesis (Hu, 2016; Du et al., 2021).
The ability of memantine to treat drug addiction remains understudied. Preclinically, memantine produces different results depending on the species. For example, pretreatment with the NMDA antagonist in rats led to a reduction in drug intake in a self-administering mouse model, whereas it increased drug intake in self-administering monkeys (Blokhina et al., 2005; Newman and Beardsley, 2006). On the other hand, clinically, memantine alone has shown no benefit in the treatment of cocaine use disorders as it does not reduce craving or cocaine consumption (Collins et al., 2006; Bisaga et al., 2010). However, these studies were limited in duration (range: 47 days to 12 weeks) and failed to assess additional complications of chronic cocaine use such as its impact on neurotoxicity. Furthermore, there was no combined L-channel blockade in these studies, and this could be a potential but very crucial reason.
Besides cocaine- and neuroHIV-induced functional alterations in the PFC per se, PFC neuronal and hemodynamic dysregulation demonstrated in the present study might also be affected by the dysfunction of the mesocorticolimbic dopamine (DA) system. Previous studies have reported that similar to cocaine, Tat also significantly inhibits the function of DA transporters (DATs), which abnormally decreases DA reuptake, promotes DA neurotransmission, and prolongs/enhances the effects of DA on postsynaptic brain regions (Zhu et al., 2018; Quizon et al., 2021). Additionally, in HIV-1 Tg rats that express Tat and several other HIV proteins, we previously revealed dysfunction in glutamatergic PFC pyramidal neurons (Nasif et al., 2005a; Nasif et al., 2005b; Ford et al., 2009; Wayman et al., 2012; Napier et al., 2014; Wayman et al., 2015; Wayman et al., 2016; Allen et al., 2019; Du et al., 2021) and GABAergic (dorsal/ventral) medium spiny striatal neurons (Chen et al., 2018), which are both influenced by DA as they are targets of DA terminals. The consequential effects of such DAT dysfunction may abnormally increase the functional activity/expression of L-channels and/or NMDARs, thereby jointly causing neuronal and hemodynamic dysregulation in the brain regions that regulate neurocognition. In combination with such alterations in these neurons per se, these neuronal dysfunctions associated with and in response to changes in DA neurotransmission may also contribute to the mechanism of HAND, as well as that underling the comorbidity of HAND and cocaine use disorders.
Both the naïve wild-type and HIV transgenic rats displayed significant hypoxemia (Δ[HbO2]) and reduction in cerebral blood volume (Δ[HbT]) following acute cocaine. While the decreases in both of these parameters is consistent with our previous reports as well as with clinical findings, the HIV 1-Tg rat reduction was more severe (2-fold) (Volkow et al., 1988; Chen et al., 2016; Allen et al., 2019; Du et al., 2020). Pretreatments with memantine in HIV 1-Tg rats abolished this substantial hypoxia, as demonstrated by the time-course imaging and quantification of oxygenated hemoglobin and total blood volume, which showed no significant differences with controls. This may be due to maintained neurovascular coupling as both blood volume and oxygenation return to baseline at the same time as neuronal activity.
Attenuation of blood flow in the PFC, a critical brain region that regulates neurocognition, has been observed in both chronic cocaine usage and HAND (Maini et al., 1990; Schielke et al., 1990; Allen et al., 2019). Referred to as hypofrontality, this reduction in blood flow might underlie decreases in higher-order cognitive functions such as learning and advanced planning (Volkow and Morales, 2015). Studies involving the use of memantine to treat diseases with reduced blood volume, such as stroke and Alzheimer’s disease, have demonstrated success in modifying hemodynamics and disease progression. In patients with Alzheimer’s disease, the addition of 20 mg/kg of memantine to donepezil (acetylcholinesterase inhibitor) significantly improved the mini mental status examination and clinical global impression improvement scale after 24 weeks. This increase in function relative to control was associated with more flow in the frontal gyri (Araki et al., 2014). Moreover, in animal models of stroke, memantine reduced the infarcted area and penumbra development (López-Valdés et al., 2021). These studies suggest that memantine may have a role in treating drug addiction and HAND by modifying the pathogenesis of the cerebral vasculature. Consistent with this hypothesis, an open-label study of 40 mg/kg of memantine that treated patients for 32 weeks found an improvement in their neurocognitive function (Zhao et al., 2010). Whether this hypothesized decrease in ischemia is due to NMDA antagonist’s effect on the brain’s vasculature directly or is a consequence of decreased overactivation of the neurons leading to decreased flow demands remains unclear and requires further investigation. However, attenuating ischemia and hypofrontality through NDMA antagonist such as memantine may slow the onset and progression of cognitive decline.
Although the present study provides insight into the pathophysiology of HIV and cocaine-induced neurotoxicity, there are some limitations. This study is limited by its small sample size and use of only male rats. Despite the limited sample, cocaine induced large, repeatable results in both the wild-type and HIV-1 Tg rats, which allowed us to document significant effects of HIV and blockade of overactive NMDARs on cocaine-induced excessive neuronal calcium influx and hemodynamics. We selectively choose to study these dysfunctional alterations in male rats as sex hormones can affect the cerebral vasculature (Duckles and Krause, 2007). However, studies continue to demonstrate that sexes respond to cocaine differently (Clare et al., 2021). Therefore, future studies should investigate the impact of sex on cocaine-induced changes in HIV. Lastly, to obtain these results, all animals were anesthetized during experimentation. It is possible that our outcomes could have been influenced by interactions between the anesthetic and the compounds used. Also, our animals were individually housed, and further studies are needed to determine the generalizability of our findings to animals reared in group housing.
In summary, in the present study, we have demonstrated the use of a custom imaging modality to acquire simultaneous information regarding the pathophysiology of neuronal and hemodynamic responses in the brain of a rat model of neuroHIV exposed to cocaine. Moreover, our novel findings also indicated that the blockade of overactive NMDARs with memantine attenuated the exaggerated calcium influx and hypoxia seen in the context of neuroHIV, either with or without concurrent exposure to cocaine. Thus, this research provides new evidence to support our perspective that, in combination with the appropriate blockade of overactivation/overexpression of L-type calcium channels, antagonism of overactive NMDARs in hyperactive PFC pyramidal neurons may be a useful and effective novel therapeutic approach for treating neuroAIDS-associated or cocaine-induced neurotoxicity, as well as the comorbidity of HAND and cocaine use disorders.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Stony Brook University Institutional Animal Care and Use committee.
Author Contributions
The research was designed by CD, NDV, X-TH, and YP; KP and CA carried out the experiments; KP, YH, and CA processed and analyzed the data; and KC, CD, NDV, X-TH, and YP contributed significantly to data interpretation, discussing the results, and writing the article.
Funding
This work was supported, in part, by the National Institutes of Health (NIH) grants 2R01 DA029718 (CD), RF1DA048808 (YP and CD), R21DA042597 (CD), R01NS084817, and R01DA044552 (X-TH) and NIH’s Intramural Program of NIAAA (NDV).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.895006/full#supplementary-material
References
Agatsuma, S., Dang, M. T., Li, Y., and Hiroi, N. (2010). N-methyl-D-aspartic Acid Receptors on Striatal Neurons Are Essential for Cocaine Cue Reactivity in Mice. Biol. Psychiatry 67, 778–780. doi:10.1016/j.biopsych.2009.12.023
Aksenov, M. Y., Aksenova, M. V., Nath, A., Ray, P. D., Mactutus, C. F., and Booze, R. M. (2006). Cocaine-mediated Enhancement of Tat Toxicity in Rat Hippocampal Cell Cultures: The Role of Oxidative Stress and D1 Dopamine Receptor. Neurotoxicology 27, 217–228. doi:10.1016/j.neuro.2005.10.003
Allen, C. P., Park, K., Li, A., Volkow, N. D., Koob, G. F., Pan, Y., et al. (2019). Enhanced Neuronal and Blunted Hemodynamic Reactivity to Cocaine in the Prefrontal Cortex Following Extended Cocaine Access: Optical Imaging Study in Anesthetized Rats. Addict. Biol. 24, 485–497. doi:10.1111/adb.12615
Antinori, A., Arendt, G., Becker, J. T., Brew, B. J., Byrd, D. A., Cherner, M., et al. (2007). Updated Research Nosology for HIV-Associated Neurocognitive Disorders. Neurology 69, 1789–1799. doi:10.1212/01.WNL.0000287431.88658.8b
Araki, T., Wake, R., Miyaoka, T., Kawakami, K., Nagahama, M., Furuya, M., et al. (2014). The Effects of Combine Treatment of Memantine and Donepezil on Alzheimer's Disease Patients and its Relationship with Cerebral Blood Flow in the Prefrontal Area. Int. J. Geriatr. Psychiatry 29, 881–889. doi:10.1002/gps.4074
Armada-Moreira, A., Gomes, J. I., Pina, C. C., Savchak, O. K., Gonçalves-Ribeiro, J., Rei, N., et al. (2020). Going the Extra (Synaptic) Mile: Excitotoxicity as the Road toward Neurodegenerative Diseases. Front. Cell Neurosci. 14, 90. doi:10.3389/fncel.2020.00090
Bansal, A. K., Mactutus, C. F., Nath, A., Maragos, W., Hauser, K. F., and Booze, R. M. (2000). Neurotoxicity of HIV-1 Proteins Gp120 and Tat in the Rat Striatum. Brain Res. 879, 42–49. doi:10.1016/s0006-8993(00)02725-6
Bisaga, A., Aharonovich, E., Cheng, W. Y., Levin, F. R., Mariani, J. J., Raby, W. N., et al. (2010). A Placebo-Controlled Trial of Memantine for Cocaine Dependence with High-Value Voucher Incentives during a Pre-randomization Lead-In Period. Drug Alcohol Depend. 111, 97–104. doi:10.1016/j.drugalcdep.2010.04.006
Blokhina, E. A., Kashkin, V. A., Zvartau, E. E., Danysz, W., and Bespalov, A. Y. (2005). Effects of Nicotinic and NMDA Receptor Channel Blockers on Intravenous Cocaine and Nicotine Self-Administration in Mice. Eur. Neuropsychopharmacol. 15, 219–225. doi:10.1016/j.euroneuro.2004.07.005
Buch, S., Yao, H., Guo, M., Mori, T., Su, T. P., and Wang, J. (2011). Cocaine and HIV-1 Interplay: Molecular Mechanisms of Action and Addiction. J. Neuroimmune Pharmacol. 6, 503–515. doi:10.1007/s11481-011-9297-0
Chen, B. T., Yau, H. J., Hatch, C., Kusumoto-Yoshida, I., Cho, S. L., Hopf, F. W., et al. (2013). Rescuing Cocaine-Induced Prefrontal Cortex Hypoactivity Prevents Compulsive Cocaine Seeking. Nature 496, 359–362. doi:10.1038/nature12024
Chen, L., Khodr, C., Al-Harthi, L., and Hu, X. T. (2018). Hyperactivity of Medium Spiny Neurons in the Dorsal Striatum Is Associated with Unique Ca2+ and K+ Channel Dysfunction in 12-Month-Old HIV-1 Tg Rats. J. Neuroimmune Pharmacol. 13, S15. doi:10.1007/s11481-018-9786-5
Chen, W., Park, K., Volkow, N., Pan, Y., and Du, C. (2016). Cocaine-Induced Abnormal Cerebral Hemodynamic Responses to Forepaw Stimulation Assessed by Integrated Multi-Wavelength Spectroimaging and Laser Speckle Contrast Imaging. IEEE J. Sel. Top. Quantum Electron 22, 6802608. doi:10.1109/JSTQE.2015.2503319
Clare, K., Pan, C., Kim, G., Park, K., Zhao, J., Volkow, N. D., et al. (2021). Cocaine Reduces the Neuronal Population while Upregulating Dopamine D2-Receptor-Expressing Neurons in Brain Reward Regions: Sex-Effects. Front. Pharmacol. 12, 624127. doi:10.3389/fphar.2021.624127
Collins, E. D., Vosburg, S. K., Ward, A. S., Haney, M., and Foltin, R. W. (2006). Memantine Increases Cardiovascular but Not Behavioral Effects of Cocaine in Methadone-Maintained Humans. Pharmacol. Biochem. Behav. 83, 47–55. doi:10.1016/j.pbb.2005.12.003
Colwell, C. S., and Levine, M. S. (1996). Glutamate Receptor-Induced Toxicity in Neostriatal Cells. Brain Res. 724, 205–212. doi:10.1016/0006-8993(96)00323-x
Dodt, H. U., Hager, G., and Zieglgänsberger, W. (1993). Direct Observation of Neurotoxicity in Brain Slices with Infrared Videomicroscopy. J. Neurosci. Methods 50, 165–171. doi:10.1016/0165-0270(93)90005-c
Du, C., Park, K., Allen, C. P., Hu, X. T., Volkow, N. D., and Pan, Y. (2021). Ca2+ Channel Blockade Reduces Cocaine's Vasoconstriction and Neurotoxicity in the Prefrontal Cortex. Transl. Psychiatry 11, 459. doi:10.1038/s41398-021-01573-7
Du, C., Volkow, N. D., You, J., Park, K., Allen, C. P., Koob, G. F., et al. (2020). Cocaine-induced Ischemia in Prefrontal Cortex Is Associated with Escalation of Cocaine Intake in Rodents. Mol. Psychiatry 25, 1759–1776. doi:10.1038/s41380-018-0261-8
Duckles, S. P., and Krause, D. N. (2007). Cerebrovascular Effects of Oestrogen: Multiplicity of Action. Clin. Exp. Pharmacol. Physiol. 34, 801–808. doi:10.1111/j.1440-1681.2007.04683.x
Ford, K. A., Wolf, M. E., and Hu, X. T. (2009). Plasticity of L-type Ca2+ Channels after Cocaine Withdrawal. Synapse 63, 690–697. doi:10.1002/syn.20651
George, O., Mandyam, C. D., Wee, S., and Koob, G. F. (2008). Extended Access to Cocaine Self-Administration Produces Long-Lasting Prefrontal Cortex-dependent Working Memory Impairments. Neuropsychopharmacology 33, 2474–2482. doi:10.1038/sj.npp.1301626
Hu, X. T. (2016). HIV-1 Tat-Mediated Calcium Dysregulation and Neuronal Dysfunction in Vulnerable Brain Regions. Curr. Drug Targets 17, 4–14. doi:10.2174/1389450116666150531162212
Jara, J. H., Singh, B. B., Floden, A. M., and Combs, C. K. (2007). Tumor Necrosis Factor Alpha Stimulates NMDA Receptor Activity in Mouse Cortical Neurons Resulting in ERK-dependent Death. J. Neurochem. 100, 1407–1420. doi:10.1111/j.1471-4159.2006.04330.x
Kalivas, P. W., and McFarland, K. (2003). Brain Circuitry and the Reinstatement of Cocaine-Seeking Behavior. Psychopharmacol. Berl. 168, 44–56. doi:10.1007/s00213-003-1393-2
Khodr, C. E., Chen, L., Dave, S., Al-Harthi, L., and Hu, X. T. (2016). Combined Chronic Blockade of Hyper-Active L-type Calcium Channels and NMDA Receptors Ameliorates HIV-1 Associated Hyper-Excitability of mPFC Pyramidal Neurons. Neurobiol. Dis. 94, 85–94. doi:10.1016/j.nbd.2016.06.008
Kojima, S., Nakamura, T., Nidaira, T., Nakamura, K., Ooashi, N., Ito, E., et al. (1999). Optical Detection of Synaptically Induced Glutamate Transport in Hippocampal Slices. J. Neurosci. 19, 2580–2588. doi:10.1523/jneurosci.19-07-02580.1999
Koutsilieri, E., Götz, M. E., Sopper, S., Sauer, U., Demuth, M., ter Meulen, V., et al. (1997). Regulation of Glutathione and Cell Toxicity Following Exposure to Neurotropic Substances and Human Immunodeficiency Virus-1 In Vitro. J. Neurovirol 3, 342–349. doi:10.3109/13550289709030748
Kral, A. H., Bluthenthal, R. N., Booth, R. E., and Watters, J. K. (1998). HIV Seroprevalence Among Street-Recruited Injection Drug and Crack Cocaine Users in 16 US Municipalities. Am. J. Public Health 88, 108–113. doi:10.2105/ajph.88.1.108
Larrat, E. P., and Zierler, S. (1993). Entangled Epidemics: Cocaine Use and HIV Disease. J. Psychoact. Drugs 25, 207–221. doi:10.1080/02791072.1993.10472272
Lipton, S. A., Sucher, N. J., Kaiser, P. K., and Dreyer, E. B. (1991). Synergistic Effects of HIV Coat Protein and NMDA Receptor-Mediated Neurotoxicity. Neuron 7, 111–118. doi:10.1016/0896-6273(91)90079-f
López-Valdés, H., Martínez-Coria, H., Arrieta-Cruz, I., and Cruz, M.-E. (2021). Physiopathology of Ischemic Stroke and its Modulation Using Memantine: Evidence from Preclinical Stroke. Neural Regen. Res. 16, 433. doi:10.4103/1673-5374.293129
Maini, C. L., Pigorini, F., Pau, F. M., Volpini, V., Galgani, S., Rosci, M. A., et al. (1990). Cortical Cerebral Blood Flow in HIV-1-Related Dementia Complex. Nucl. Med. Commun. 11, 639–648. doi:10.1097/00006231-199009000-00007
McArthur, J. C., Steiner, J., Sacktor, N., and Nath, A. (2010). Human Immunodeficiency Virus-Associated Neurocognitive Disorders: Mind the Gap. Ann. Neurol. 67, 699–714. doi:10.1002/ana.22053
Napier, T. C., Chen, L., Kashanchi, F., and Hu, X. T. (2014). Repeated Cocaine Treatment Enhances HIV-1 Tat-Induced Cortical Excitability via Over-activation of L-type Calcium Channels. J. Neuroimmune Pharmacol. 9, 354–368. doi:10.1007/s11481-014-9524-6
Nasif, F. J., Hu, X. T., and White, F. J. (2005). Repeated Cocaine Administration Increases Voltage-Sensitive Calcium Currents in Response to Membrane Depolarization in Medial Prefrontal Cortex Pyramidal Neurons. J. Neurosci. 25, 3674–3679. doi:10.1523/JNEUROSCI.0010-05.2005
Nasif, F. J., Sidiropoulou, K., Hu, X.-T., and White, F. J. (2005). Repeated Cocaine Administration Increases Membrane Excitability of Pyramidal Neurons in the Rat Medial Prefrontal Cortex. J. Pharmacol. Exp. Ther. 312, 1305–1313. doi:10.1124/jpet.104.075184
Nath, A., Haughey, N. J., Jones, M., Anderson, C., Bell, J. E., and Geiger, J. D. (2000). Synergistic Neurotoxicity by Human Immunodeficiency Virus Proteins Tat and Gp120: Protection by Memantine. Ann. Neurol. 47, 186–194. doi:10.1002/1531-8249(200002)47:2<186::aid-ana8>3.0.co;2-3
Nath, A., Psooy, K., Martin, C., Knudsen, B., Magnuson, D. S., Haughey, N., et al. (1996). Identification of a Human Immunodeficiency Virus Type 1 Tat Epitope that Is Neuroexcitatory and Neurotoxic. J. Virol. 70, 1475–1480. doi:10.1128/JVI.70.3.1475-1480.1996
Navia, B. A., Dafni, U., Simpson, D., Tucker, T., Singer, E., McArthur, J. C., et al. (1998). A Phase I/II Trial of Nimodipine for HIV-Related Neurologic Complications. Neurology 51, 221–228. doi:10.1212/wnl.51.1.221
New, D. R., Ma, M., Epstein, L. G., Nath, A., and Gelbard, H. A. (1997). Human Immunodeficiency Virus Type 1 Tat Protein Induces Death by Apoptosis in Primary Human Neuron Cultures. J. Neurovirol 3, 168–173. doi:10.3109/13550289709015806
Newman, J. L., and Beardsley, P. M. (2006). Effects of Memantine, Haloperidol, and Cocaine on Primary and Conditioned Reinforcement Associated with Cocaine in Rhesus Monkeys. Psychopharmacol. Berl. 185, 142–149. doi:10.1007/s00213-005-0282-2
Olney, J. W., Labruyere, J., Wang, G., Wozniak, D. F., Price, M. T., and Sesma, M. A. (1991). NMDA Antagonist Neurotoxicity: Mechanism and Prevention. Science 254, 1515–1518. doi:10.1126/science.1835799
Park, K., Clare, K., Volkow, N. D., Pan, Y., and Du, C. (2022). Cocaine's Effects on the Reactivity of the Medial Prefrontal Cortex to Ventral Tegmental Area Stimulation: Optical Imaging Study in Mice. Addiction. doi:10.1111/add.15869
Paxinos, G., and Watson, C. (1997). “The Rat Brain,” in Stereotaxic Coordinates (San Diego: Academic Press).
Peterson, P. K., Gekker, G., Chao, C. C., Schut, R., Molitor, T. W., and Balfour, H. H. (1991). Cocaine Potentiates HIV-1 Replication in Human Peripheral Blood Mononuclear Cell Cocultures. Involvement of Transforming Growth Factor-Beta. J. Immunol. 146, 81–84.
Peterson, P. K., Sharp, B. M., Gekker, G., Portoghese, P. S., Sannerud, K., and Balfour, H. H. (1990). Morphine Promotes the Growth of HIV-1 in Human Peripheral Blood Mononuclear Cell Cocultures. AIDS 4, 869–873. doi:10.1097/00002030-199009000-00006
Quizon, P. M., Yuan, Y., Zhu, Y., Zhou, Y., Strauss, M. J., Sun, W. L., et al. (2021). Mutations of Human DopamineTransporter at Tyrosine88, Aspartic Acid206, and Histidine547 Influence Basal and HIV-1 Tat-Inhibited Dopamine Transport. J. Neuroimmune Pharmacol. 16, 854–869. doi:10.1007/s11481-021-09984-5
Ren, Z., Sun, W. L., Jiao, H., Zhang, D., Kong, H., Wang, X., et al. (2010). Dopamine D1 and N-Methyl-D-Aspartate Receptors and Extracellular Signal-Regulated Kinase Mediate Neuronal Morphological Changes Induced by Repeated Cocaine Administration. Neuroscience 168, 48–60. doi:10.1016/j.neuroscience.2010.03.034
Schielke, E., Tatsch, K., Pfister, H. W., Trenkwalder, C., Leinsinger, G., Kirsch, C. M., et al. (1990). Reduced Cerebral Blood Flow in Early Stages of Human Immunodeficiency Virus Infection. Arch. Neurol. 47, 1342–1345. doi:10.1001/archneur.1990.00530120088015
Schifitto, G., Navia, B. A., Yiannoutsos, C. T., Marra, C. M., Chang, L., Ernst, T., et al. (2007). Memantine and HIV-Associated Cognitive Impairment: A Neuropsychological and Proton Magnetic Resonance Spectroscopy Study. AIDS 21, 1877–1886. doi:10.1097/QAD.0b013e32813384e8
Vignoles, M., Avila, M. M., Osimani, M. L., Pando, M. d. L. A., Rossi, D., Sheppard, H., et al. (2006). HIV Seroincidence Estimates Among At-Risk Populations in Buenos Aires and Montevideo: Use of the Serologic Testing Algorithm for Recent HIV Seroconversion. J. Acquir Immune Defic. Syndr. 42, 494–500. doi:10.1097/01.qai.0000221678.06822.8b
Volkow, N. D., and Morales, M. (2015). The Brain on Drugs: From Reward to Addiction. Cell 162, 712–725. doi:10.1016/j.cell.2015.07.046
Volkow, N. D., Mullani, N., Gould, K. L., Adler, S., and Krajewski, K. (1988). Cerebral Blood Flow in Chronic Cocaine Users: A Study with Positron Emission Tomography. Br. J. Psychiatry 152, 641–648. doi:10.1192/bjp.152.5.641
Wayman, W. N., Chen, L., Napier, T. C., and Hu, X. T. (2015). Cocaine Self-Administration Enhances Excitatory Responses of Pyramidal Neurons in the Rat Medial Prefrontal Cortex to Human Immunodeficiency Virus-1 Tat. Eur. J. Neurosci. 41, 1195–1206. doi:10.1111/ejn.12853
Wayman, W. N., Dodiya, H. B., Persons, A. L., Kashanchi, F., Kordower, J. H., Hu, X. T., et al. (2012). Enduring Cortical Alterations after a Single Iin-Vvivo Treatment of HIV-1 Tat. Neuroreport 23, 825–829. doi:10.1097/WNR.0b013e3283578050
Wayman, W. N., Chen, L., Hu, X.-T., and Napier, T. C. (2016). HIV-1 Transgenic Rat Prefrontal Cortex Hyper-Excitability Is Enhanced by Cocaine Self-Administration. Neuropsychopharmacol 41, 1965–1973. doi:10.1038/npp.2015.366
Yao, H., Allen, J. E., Zhu, X., Callen, S., and Buch, S. (2009). Cocaine and Human Immunodeficiency Virus Type 1 Gp120 Mediate Neurotoxicity through Overlapping Signaling Pathways. J. Neurovirol 15, 164–175. doi:10.1080/13550280902755375
Yuan, Z., Luo, Z., Volkow, N. D., Pan, Y., and Du, C. (2011). Imaging Separation of Neuronal from Vascular Effects of Cocaine on Rat Cortical Brain In Vivo. Neuroimage 54, 1130–1139. doi:10.1016/j.neuroimage.2010.08.045
Zhao, Y., Navia, B. A., Marra, C. M., Singer, E. J., Chang, L., Berger, J., et al. (2010). Memantine for AIDS Dementia Complex: Open-Label Report of ACTG 301. HIV Clin. Trials 11, 59–67. doi:10.1310/hct1101-59
Zhou, B. Y., Liu, Y., Kim, Bo., Xiao, Y., and He, J. J. (2004). Astrocyte Activation and Dysfunction and Neuron Death by HIV-1 Tat Expression in Astrocytes. Mol. Cell Neurosci. 27, 296–305. doi:10.1016/j.mcn.2004.07.003
Zhu, J., Ananthan, S., and Zhan, C. G. (2018). The Role of Human Dopamine Transporter in NeuroAIDS. Pharmacol. Ther. 183, 78–89. doi:10.1016/j.pharmthera.2017.10.007
Zidovetzki, R., Wang, J. L., Chen, P., Jeyaseelan, R., and Hofman, F. (1998). Human Immunodeficiency Virus Tat Protein Induces Interleukin 6 mRNA Expression in Human Brain Endothelial Cells via Protein Kinase C- and cAMP-dependent Protein Kinase Pathways. AIDS Res. Hum. Retroviruses 14, 825–833. doi:10.1089/aid.1998.14.825
Keywords: NMDA antagonist, addiction, neuroHIV, imaging, prefrontal cortex
Citation: Du C, Hua Y, Clare K, Park K, Allen CP, Volkow ND, Hu X-T and Pan Y (2022) Memantine Attenuates Cocaine and neuroHIV Neurotoxicity in the Medial Prefrontal Cortex. Front. Pharmacol. 13:895006. doi: 10.3389/fphar.2022.895006
Received: 12 March 2022; Accepted: 20 April 2022;
Published: 25 May 2022.
Edited by:
Li Zhang, National Institutes of Health (NIH), United StatesReviewed by:
Sylvia Fitting, University of North Carolina, United StatesIrma Elisa Cisneros, University of Texas Medical Branch, United States
Copyright © 2022 Du, Hua, Clare, Park, Allen, Volkow, Hu and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Congwu Du, Q29uZ3d1LkR1QHN0b255YnJvb2suZWR1; Nora D. Volkow, bnZvbGtvd0BuaWRhLm5paC5nb3Y=; Xiu-Ti Hu, eGl1LXRpX2h1QHJ1c2guZWR1
†These authors have contributed equally to this work