
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Pharmacol. , 02 June 2022
Sec. Translational Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.880448
This article is part of the Research Topic Lymphatic Delivery and Targeting of Drugs, Vaccines, and Imaging Agents View all 5 articles
 Jibriil P Ibrahim1
Jibriil P Ibrahim1 Shadabul Haque2Robert J Bischof3Andrew K Whittaker4
Shadabul Haque2Robert J Bischof3Andrew K Whittaker4 Michael R Whittaker2
Michael R Whittaker2 Lisa M Kaminskas1*
Lisa M Kaminskas1*Enhancing the delivery of therapeutic agents to the lung lymph, including drugs, transfection agents, vaccine antigens and vectors, has the potential to significantly improve the treatment and prevention of a range of lung-related illnesses. One way in which lymphatic delivery can be optimized is via the use of nanomaterial-based carriers, such as liposomes. After inhaled delivery however, there is conflicting information in the literature regarding whether nanomaterials can sufficiently access the lung lymphatics to have a therapeutic benefit, in large part due to a lack of reliable quantitative pharmacokinetic data. The aim of this work was to quantitatively evaluate the pulmonary lymphatic pharmacokinetics of a model nanomaterial-based drug delivery system (HSPC liposomes) in caudal mediastinal lymph duct cannulated sheep after nebulized administration to the lungs. Liposomes were labelled with 3H-phosphatidylcholine to facilitate evaluation of pharmacokinetics and biodistribution in biological samples. While nanomaterials administered to the lungs may access the lymphatics via direct absorption from the airways or after initial uptake by alveolar macrophages, only 0.3 and 0.001% of the 3H-lipid dose was recovered in lung lymph fluid and lymph cell pellets (containing immune cells) respectively over 5 days. This suggests limited lymphatic access of liposomes, despite apparent pulmonary bioavailability of the 3H-lipid being approximately 17%, likely a result of absorption of liberated 3H-lipid after breakdown of the liposome in the presence of lung surfactant. Similarly, biodistribution of 3H in the mediastinal lymph node was insignificant after 5 days. These data suggest that liposomes, that are normally absorbed via the lymphatics after interstitial administration, do not access the lung lymphatics after inhaled administration. Alternate approaches to maximize the lung lymphatic delivery of drugs and other therapeutics need to be identified.
The lung lymphatic system is responsible for regulating and optimizing lung fluid levels, (Weber et al., 2018), modulating local immune defenses (Bromley et al., 2005) and regulating the progression of some pulmonary diseases (Trevaskis et al., 2015). It is therefore now widely accepted as an important target for drug delivery applications, particularly for cancer chemotherapeutics and immunomodulators. To this end, nanomaterial-based drug delivery systems, such as liposomes, nanoparticles and dendrimers, have become important facilitators for improved drug access to the lymph after interstitial and, in some cases, intravenous administration (Kaminskas et al., 2009; Azzi et al., 2016; Ke et al., 2019; Marasini and Kaminskas, 2019; Siafaka et al., 2021; Zheng et al., 2021). After inhaled administration however, our understanding of lung lymphatic absorption and trafficking of nanomaterials is limited and research is sparse. Further, there is conflicting information in the literature regarding whether nanomaterials traffic towards the lung lymphatics after inhaled administration. This in part comes from a lack of quantitative lymphatic pharmacokinetic data and use of small animal models that have different pulmonary pharmacokinetics compared to humans and larger animals, particularly for macromolecules and nanomaterials (Choi et al., 2010; Ryan et al., 2016).
Using rodent models, previous papers have proposed that nanoparticles enter the lung lymphatics via non-cellular lymphatic draining or antigen-presenting cell (APC) mediated trafficking (Choi et al., 2010; Trevaskis et al., 2015). Optimal nanomaterial properties for lung lymphatic access after pulmonary administration, for example, have been reported to be smaller than 30 nm and exhibit a non-cationic charge (Choi et al., 2010). Lung lymphatic access of a 200 nm solid lipid nanoparticle however, has also been reported in rats after pulmonary administration (Videira et al., 2002; Choi et al., 2010). Liposomes (typically around 100–200 nm diameter) modified to mimic the bacterial wall have also been proposed to be efficiently absorbed via the lung lymph after pulmonary administration in pigs and allow radiometric visualization of the lung lymphatic network (Botelho et al., 2011; Liu et al., 2020). However, 10 nm dendrimers have been shown to exhibit poor absorption via the lung lymph after inhaled administration in sheep (Videira et al., 2012). Specifically, while a 22 kDa PEGylated dendrimer showed 16 and 9% pulmonary bioavailability in rats and sheep respectively after nebulized administration to the lungs, and 30% absorption via the lymph after interstitial administration, less than 0.5% of a nebulized dose was quantified in the lung lymph of sheep over 7 days (Ryan et al., 2016). This contradicted prior studies in rats and pigs that suggested good lymphatic absorption of inhaled nanomaterials. The latter study in sheep, however, quantified lung lymphatic pharmacokinetics by continuously collecting lung-derived lymph from the efferent caudal mediastinal lymph duct (CMLD) that collects the majority of lung-derived lymph before it enters the thoracic lymph duct (Chanana and Joel, 1985). The former studies in rats and pigs however, used non-quantitative imaging-based approaches.
While most nanomaterial structures are far from clinical translation as inhalable nanomedicines, there are currently over a dozen inhalable liposome-based formulations in the clinic and clinical trials for a range of diseases (Haque et al., 2016). Further, the inhaled delivery of liposome and non-liposome-based vaccines has the significant potential to enhance local mucosal immunity in the lungs compared to conventional interstitial injection. As an example, the PEGylated liposome-based Moderna and Pfizer Covid-19 vaccines would be expected to provide enhanced immunity and protection against Covid-19 after inhaled administration and may also minimize the risk of myocarditis compared to conventional intramuscular injection (Marasini et al., 2017; Abhyankar et al., 2021; Fanciullino et al., 2021; Li et al., 2021). It is therefore important to better understand the absorption and clearance mechanics of liposomes and other nanomaterials in the lungs to establish whether it is feasible to enhance the lung lymphatic delivery of drugs and other therapeutics via this route (Haque et al., 2016).
The objective of this work was therefore to quantify the lung lymphatic pharmacokinetics of 3H-labelled non-PEGylated conventional soy phosphatidylcholine (HSPC) liposomes using the established CMLD cannulated sheep model. While PEGylation can improve liposome stability in the lungs, lung lymphatic exposure was previously reported after inhaled administration of non-PEGylated nanoradioliposome (Botelho et al., 2011). Further, non-PEGylated liposomes dominate the inhalable nanomedicines pipeline and are more likely to be internalized by alveolar macrophages that can assist in trafficking the liposomes to the lymph, making them an appropriate investigational tool for this study.
Hydrogenated Soy Phosphatidylcholine (HSPC, Coatsome NC-21E) was purchased from Yushi-Sheihin (Singapore). 3H-dipalmitoyl phosphatidylcholine (ART0532) was from American Radiolabeled Chemicals (MO, United States). Cholesterol, Evans blue dye and urea assay kits (MAK006) were from Sigma-Aldrich (NSW, Australia). Bupivicaine, diazepam and heparin were each purchased from Clifford Hallam Healthcare (Vic, Australia). Saline (0.9% NaCl) was obtained from Baxter (NSW, Australia). Procaine penicillin, cephazolin and LethabarbTM were from Virbac (NSW, Australia). Transdermal Fentanyl patches were obtained from Janssen Pharmaceuticals (Beerse, Belgium). Isoflurane was purchased from Delvet (NSW, Australia). Thiopentone was from Troy Laboratories (NSW, Australia). Polyvinyl catheters (1.5 mm × 2.7 mm) and endotracheal tubes (Portex, 7–8mm i.d.) were purchased from Smiths Medical (Australia), while silastic tubing (0.63 mm × 1.19 mm) was purchased from Dow Corning (MI, United States). Soluene, Ultima GoldTM and scintillation vials were from PerkinElmer (MA, United States). All other reagents were AR grade and were used without further purification.
Liposomes were prepared via lipid film hydration as previously described (Haque et al., 2018). Briefly, HSPC (30 mg), cholesterol (8 mg) and 3H-dipalmitoyl phosphatidylcholine (250 µCi) were dissolved in chloroform (7 ml) and sonicated for 60 s using a VibraCell sonicator at 50% power (Fischer Scientific, Illkirich, France). This solution was then evaporated to dryness under reduced pressure using a rotary evaporator (Buchi Laabortechnik, Switzerland) at 60°C to form a dry, thin lipid film. Dried lipid films were then rehydrated in phosphate buffered saline (PBS pH 7.4) and agitated for 2 h at 40°C and re-sonicated for 60 s. After rehydration, the lipid solution was extruded using 7 passes through a mini-extruder set (Avanti Polar Lipids, Al, United States) using polycarbonate filters of pore diameter 200 nm, and 100 nm sequentially to form unilamellar liposomes. Unincorporated 3H radiolabel (as degraded phosphatidylcholine) was removed from the final liposome formulation by centrifuging liposomes for 5 min at 10,000 x g through a 3 kDa MWCO spin filter (Sigma Aldrich, Australia). Liposomes were then characterized for size, polydispersity and charge on a Malvern Zetasizer (Nano ZS, Malvern Instruments, Worcestershire, United Kingdom). Final characteristics were (as mean ± sd, n = 3) size: 161 ± 3 nm, PDI: 0.13 ± 0.03, charge: −2.47 ± 0.4 mV, specific radioactivity 6.5 uCi/mg lipid. Transmission electron cryomicroscopy (cryo-TEM) was also performed on non-radiolabeled liposomes and the image is shown in the supplementary information. Cryo-TEM confirmed the size, spherical and predominantly unilamellar nature of the liposomes.
To examine the stability of the liposomes during nebulization, non-radiolabeled liposomes were nebulized over 20 min in the PARI vibrating mesh nebulizer (sealed on either end of the chamber with parafilm to prevent venting) and analyzed via DLS. Nebulization led to a small increase in liposome size (30 nm) and PDI (0.16), but charge was not significantly affected (liposomes remained relatively uncharged). The results are shown in the supplementary information.
Female merino (ovis aries) sheep (approximately 28–35 kg, 1–2 years of age, n = 10) were sourced through the Monash Animal Research Platform, Monash University and acclimatized for 1 week in communal indoor pens. Sheep were housed under ambient conditions (20–22°C) on a 12 h light dark cycle with food and water provided ad libitum. Sheep were fasted for 12 h prior to surgery. Sheep remained in metabolism cages during post-surgical recovery and for the duration of the study. All experimental procedures were approved by the Monash University Animal Ethics Committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
The preparation, and surgical cannulation, of sheep under isoflurane anesthesia were conducted as described previously with some modification to antibiotics and analgesics used (details of pre- and post-surgical infection control and pain relief are reported in the supplementary information) (Enkhbaatar et al., 2003; Ryan et al., 2016; Kaminskas et al., 2020). Briefly, the right jugular vein was initially cannulated in all sheep to allow for saline (0.9% NaCl) administration during and after surgery (during the recovery phase—24–48 h post surgery), intravenous (IV) dosing and serial blood sample collection over the experimental phase (0–5 days post liposome administration). Sheep were then cannulated via the efferent CMLD via thoracotomy to continuously collect lung-derived lymph for pharmacokinetic analysis. All sheep underwent thoracotomy, but only sheep whose CMLD was well resolved underwent surgical cannulation of the CMLD (6 sheep). In all other sheep where cannulation of the CMLD was not possible, the rib cage was closed without further manipulation and these sheep were used as surgical “sham” controls that were subsequently used to evaluate intravenous pharmacokinetics for calculation of pulmonary bioavailability. Of the six sheep where the CMLD cannulation was attempted, four sheep were successfully cannulated.
The CMLD was cannulated with silastic tubing via a thoracotomy as previously described (Ryan et al., 2016). The cannula was exteriorized through a small chest incision to allow continuous collection of lymph into heparinized vessels. Sheep were then returned to individual metabolic cages to recover while on IV saline. Sheep were dosed with liposome 2–3 days after surgery.
Prior to liposome administration, samples of “blank” blood (5 ml), CMLD lymph, feces and urine were collected. Blood samples were collected via the jugular vein cannula into citrate-EDTA tubes, while lymph drained continuously into heparinized tubes.
CMLD-cannulated sheep (to be dosed via the lungs) were then transferred to a body sheath to restrain animals during dosing and collection of bronchoalveolar lavage fluid (BALF). Immediately before pulmonary dosing, a sample of pre-dose BALF was collected via a catheter placed in a lung lobe in CMLD-cannulated sheep using a fiber-optic endoscope to guide catheter placement as previously described (Ryan et al., 2016; Kaminskas et al., 2020). BALF samples were centrifuged at 4°C for 10 min (3,500 x g) to collect cell-free BALF and the cell pellet. Cell pellets were then mixed with 1 ml MQ water and frozen for storage and to disrupt the cell membranes.
Sheep were then administered 1 mg/kg liposome (as lipid, approximately 200 µCi 3H per sheep) in sterile saline via IV administration over 30 s (5 ml dose in non-CMLD cannulated sheep followed by 20 ml saline to flush through any remaining dose), or via nebulization (in 5 ml final volume in CMLD cannulated sheep). Nebulized administration was achieved by inserting a cuffed endotracheal tube into the trachea, via the nasal passage, and attaching the end of the tube to a respirator (Harvard Apparatus, MA, United States) to control breathing (20 breaths per minute, 1:2 IDE ratio). Liposome was then aerosolized and delivered to the lungs using a PARI eFlow® Inline vibrating mesh nebulizer (PARI, Gräfeling, Germany) until no dose remained (approximately 20 min). Immediately after the completion of aerosol dosing, post-dose BALF samples were collected from an alternate lung lobe to the pre-dose sample and processed as described above. Immediate post dose blood samples (5 ml) were also collected from all sheep (IV and pulmonary dosed), with further serial blood samples collected over the next 120 h. Once dosing was complete, all tubing used for nebulized administration, the nebulizing bulb and exhaled air filter were collected and rinsed with water to calculate the 3H dose not delivered and retained in the lungs. Pulmonary pharmacokinetics were then determined based on the actual dose delivered to the lungs, calculated as nominal 3H dose minus 3H dose recovered in nebulization equipment after completion of dosing as described previously (Ryan et al., 2016).
Plasma was isolated from blood via centrifugation at 3,500 x g for 10 min. Lymph was collected over post dose intervals 0–5 min, 5–30 min, 30–60 min, 1–2 h, 2–4 h, 4–6 h, 6–8 h, 8–12 h, 12–24 h, and daily thereafter. Lymph samples were centrifuged for 10 min at 3,500 x g to separate lymph fluid from the cell pellet (containing alveolar macrophages and other immune cells) that were subsequently processed as described above. Further BALF samples were also collected from pulmonary dosed sheep 24, 72 and 120 h after dosing to describe the rate of 3H-liposome clearance from the lungs and processed as described above. Feces and urine were collected continuously as described previously (Ryan et al., 2016).
After the last samples had been collected, sheep were euthanized via a bolus injection of Lethabarb (20 ml) via the jugular vein cannula. The lungs, liver, kidney, spleen and caudal mediastinal lymph node (CMLN) were then collected, weighed and tissue samples collected and stored at −20°C for further analysis.
The 3H content of biological samples were evaluated as previously described with some modification and details of the analysis described in the supplementary information (Thomson, 1999; Boyd et al., 2006; Ryan et al., 2016). After quantification of 3H content in biological samples, the mass of 3H-lipid from liposomes in each sample was determined using the specific activity of the liposomes.
Since BALF samples were diluted in saline, the actual concentration of 3H in BALF was determined using the urea correction method as described previously (Kaminskas et al., 2020). BALF data are represented as the % change in BALF concentration compared to the sample collected immediately after the completion of dosing.
Concentrations of 3H liposome have been expressed as ng/ml and were calculated based on the specific activity of the liposomes. This approach, for the purpose of pharmacokinetic calculations, assumes that 3H is entirely associated with intact liposomes, which is not always the case. Pharmacokinetic data therefore has to be evaluated with this caveat. This is an inherent issue in quantifying biodegradable nanomaterials with no intrinsic chromophore.
Non-compartmental pharmacokinetic parameters (including elimination rate constant, k; half life; area under the curve, AUC; apparent volume of distribution, Vz; clearance, Cl; maximum plasma concentration and time to maximum plasma concentration, Cmax and Tmax respectively) were calculated using PKSolver (Zhang et al., 2010). The fraction of the dose recovered in plasma was calculated by dividing AUC0-∞pulm by AUC0-∞IV. The cumulative proportion of the pulmonary dose recovered in lymph over time was calculated by determining the mass of 3H recovered in each sample and converting this to a % of the delivered dose. The % recovered at each time was added to the % recovered at all prior times to give the cumulative curve.
Statistical analysis was performed using GraphPad Prism, with statistical significance determined at a level of p < 0.05. Concentrations of plasma 3H between pulmonary and IV administration, and between plasma and lymph concentrations were compared via two-way ANOVA followed by Sidaks multiple comparison test. Differences organ biodistribution and excretion of 3H in urine and feces were compared between IV and pulmonary delivery via unpaired Students T-tests.
The plasma concentration-time profile of 3H-lipid (after dosing liposome) after IV and pulmonary administration in sheep are presented in Figure 1 and pharmacokinetic parameters are reported in Table 1. After IV dosing, plasma concentrations declined rapidly over the first hour to give a large apparent volume of distribution (Table 1), but showed slower elimination thereafter. The elimination half-life was calculated to be approximately 5 days (Table 1). Only approximately 20% of the dose was recovered in excreta, suggesting prolonged retention of the 3H-lipid in the body.
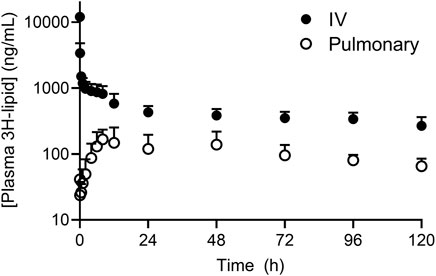
FIGURE 1. Plasma concentration-time profile of 3H-lipid after administration of radiolabeled (3H-phosphatidylcholine) liposomes to sheep via intravenous (black circles) or pulmonary (open circles) administration. Plasma concentrations were normalized to a dose of 1 mg/kg. Data represent mean ± SD (n = 4).
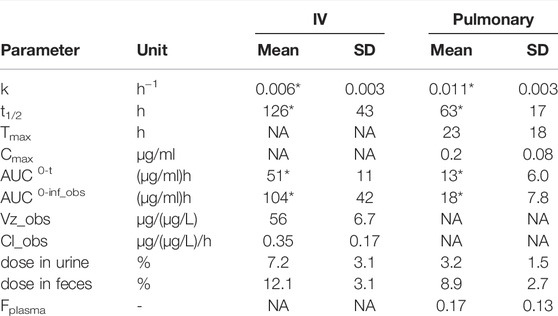
TABLE 1. Plasma pharmacokinetic parameters of 3H-liposomes after IV or pulmonary administration to sheep. Data are normalized to a dose of 1 mg/kg in all sheep and are presented as mean ± s.d. (n = 4). *Represents p <0.05 cf. IV via unpaired students T-test.
After pulmonary administration, plasma concentrations steadily increased with a Cmax of 200 ± 80 ng/ml at 23 h (Table 1). Thereafter, plasma concentrations decreased with an elimination half-life of 63 ± 17 h, approximately 2-fold more rapid than the elimination half-life after IV administration. The apparent bioavailability of the pulmonary 3H dose was 17 ± 13%. Approximately 3 ± 1.5% and 9 ± 2.7% of the 3H dose was respectively recovered in the urine and feces over 5 days after dosing. Cumulative excretion data over 5 days after pulmonary dosing are provided in the supplementary information and show appearance of radiolabel predominantly over the first 2 days after doing.
The rate of elimination of the 3H-lipid dose from BALF is shown in Figures 2A and is represented as % dose remaining in BALF compared to immediately post-dose. These data showed that 95 (± 4)% of the delivered 3H dose was eliminated from the BALF within the first 24 h. Thereafter, BALF concentrations steadily declined such that less than 1% of the dose initially deposited in the BALF remained after 5 days. The 3H content of the BALF cell pellet, which is comprised largely of alveolar macrophages, was separately analyzed and data are shown in Figure 2B. These data showed that the 3H content of the BALF cell pellet only declined by approximately 65% compared to immediate post dose, likely reflecting the slow uptake of liposomes by alveolar macrophages. Beyond 24 h though, a similar pattern of 3H elimination from the BALF cell pellet was apparent compared to cell-free BALF.
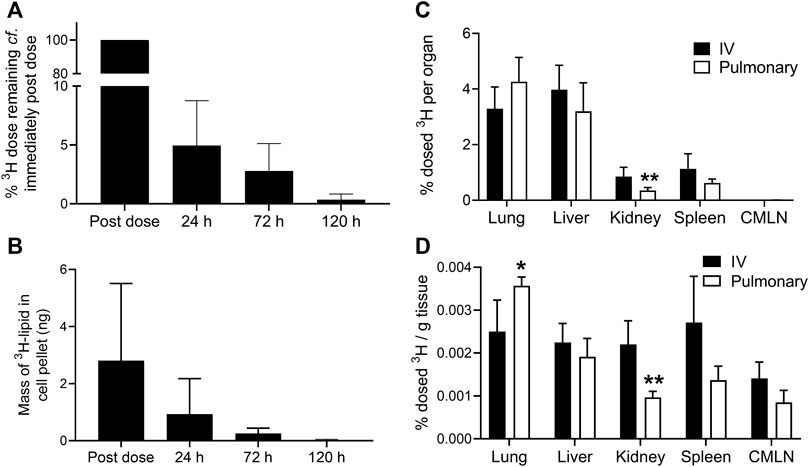
FIGURE 2. Biodistribution of 3H-lipid after IV or pulmonary administration of 3H-liposomes to sheep. (A) % Change in 3H-lipid concentration of cell-free BALF in sheep over time compared to immediately after the completion of pulmonary dosing. (B) Mass of 3H-lipid recovered in BALF cell pellets over time after pulmonary delivery. (C) Biodistribution of dosed 3H-lipid in whole organs and tissues 5 days after via intravenous or pulmonary dosing. (D) Mass normalized biodistribution of dosed 3H-lipid (% dose per gram of tissue) 5 days after IV or pulmonary dosing. Data represent mean ± SD (n = 4). * P<0.05, ** P<0.01 compared to IV dosing via two-way ANOVA with Sidaks Multiple Comparison test.
After 5 days, major organs and the CMLN were collected for biodistribution analysis (Figures 2C,D). Despite the low apparent bioavailability of pulmonary dosed 3H, no significant differences in absolute 3H-lipid biodistribution (p >0.05) were observed between IV and pulmonary dosed sheep, with the exception of kidneys (p <0.05). The 3H concentration of lungs was significantly higher in pulmonary dosed sheep, but only on a per gram basis. Less than 0.01% of the dose was recovered in the CMLN after 5 days and did not differ between pulmonary and IV dosed sheep.
The organ:plasma ratios (calculated from ng lipid/g of sample and reported in the supporting information) show higher concentrations of lipid in tissues at 5 days compared to plasma. Organ:plasma ratios were significantly higher in the pulmonary dosed group compared to the IV group for lungs (12.7 ± 2.2 vs. 3.4 ± 2.2 respectively) and liver (6.6 + 0.4 vs. 3.0 ± 1.5 respectively), but not in other organs.
The cumulative recovery of the 3H dose in cell-free CMLD lymph after pulmonary administration is shown in Figure 3A. Despite plasma and lymph concentrations reaching a peak around 6–23 h after pulmonary administration (Figure 3B), the cumulative recovery of 3H-lipid in lung lymph increased in a linear manner over 5 days and showed no evidence of plateauing by the last sample collected. Peak lymph concentrations were approximately 300 ng/ml and declined to approximately 180 ng/ml after 5 days. While concentrations of 3H in CMLD lymph after pulmonary dosing were approximately 2-fold higher compared to plasma obtained from the jugular vein, only 0.3 ± 0.02% of the delivered dose was recovered in CMLD lymph over 5 days.
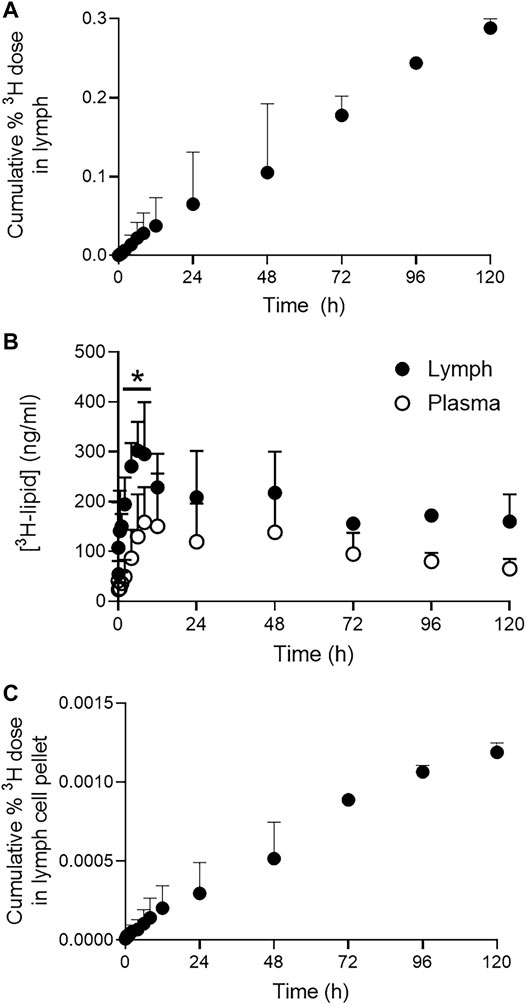
FIGURE 3. Pulmonary lymphatic pharmacokinetics of 3H-lipid in CMLD-cannulated sheep after nebulized administration of 3H-liposomes. (A) Cumulative recovery of dosed 3H-lipid in CMLD lymph after pulmonary administration. (B) Comparative concentrations of 3H-lipid in collected lymph (black circles) and plasma (open circles). Concentrations in plasma and lymph were normalized to a dose of 1 mg/kg. (C) Cumulative recovery of dosed 3H-lipid in lymph cell pellets after pulmonary administration. Data represents mean ± SD (n = 4). * P<0.05 between samples at each time point via two-way ANOVA with Sidaks Multiple Comparison test.
Since nanomaterials can access the lung lymph either passively via the interstitium, following initial passage through the pulmonary endothelium, or after uptake by local phagocytic cells (in particular, alveolar macrophages), the 3H content of lymph cell pellets were separately quantified. The cumulative recovery of the 3H dose in the lymph cell pellets reflected the linear profile apparent for the lymph fluid (Figure 3C). However, only 0.002% of the 3H dose was collectively recovered in the lymph cell pellets after 5 days compared to 0.3% via the lymph fluid.
The pulmonary lymphatic system is an important drug and vaccine target due to its involvement in disease progression and immune regulation (Pereira et al., 2015). There is therefore growing interest in developing approaches to enhance drug and vaccine access to the lung lymphatic system. In general, the pulmonary lymphatic system has a hybrid role, acting to regulate fluid levels within the lung to facilitate efficient gas exchange while also protecting the lungs via immune mediated pathways (Bromley et al., 2005; Weber et al., 2018). This is facilitated in part by the presence of lymphatic vessels primarily in the interalveolar septa in the deep lung and the large airways (Partanen et al., 2000; Weber et al., 2018). Together, this allows for a bi-directional flow of lymph; from the lung perimeter to the pleura and from the lung interior to the hilum, allowing the deeper lung lobes to clear faster than the upper lobes despite reduced lymphatic reach (Schraufnagel, 2010). However, the blood vascular supply of the lungs is significantly denser, particularly around the alveoli. This, together with the very tight cellular junctions in the lungs (Weber et al., 2018), particularly in the alveolar region, can limit the capacity of macromolecules and nanomaterials to access the lung lymphatics after pulmonary administration.
We have previously shown poor lymphatic absorption of a 10 nm PEGylated dendrimer following pulmonary administration, contradicting previously held assumptions that the lung lymphatic network mediates nanoparticle absorption (Ryan et al., 2016). In the present study, we again found that less than 0.5% of a pulmonary dose of HSPC liposome was absorbed into the lung lymphatic network. Given that pulmonary bioavailability was calculated to be approximately 17%, this suggests that less than 2% of the absorbed liposome dose was absorbed via the lymph. While the concentration of 3H-lipid in lung lymph was approximately 2-fold higher than in plasma, this does not contravene the suggestion of limited lymphatic availability. This is because the 3H content of lung lymph is derived primarily of lipid that has gained access to the lymph via absorption directly from the lungs, thereby representing “local” lymphatic concentrations. Plasma sampled from the jugular vein however, is systemic, not specifically lung derived, and 3H-lipid absorbed from the lungs via the blood is therefore subject to a more significant dilution effect compared to in CMLD lymph.
Despite this, given that 3H-lipid was still steadily being absorbed into lung lymph after 5 days, while less than 5% of the dose remained in the lungs at this point, the data suggest there is likely to be a degree of lymphatic recirculation occurring in the lungs. Specifically, since the plasma and lymph concentration profiles are very similar, it appears that a reasonable proportion of 3H-lipid absorbed via the blood is distributed back into the interstitium of the lungs and thorax, and made available for lymphatic reabsorption as suggested previously for PEGylated dendrimers (Kaminskas et al., 2009). Alternatively, 3H-lipid may be redistributed into lymph nodes via high endothelial venules (HEVs) that innervate lymph nodes and play a key role in lymphocyte transport (Ager, 2017). Nanoparticles have been shown to extravasate out of blood vessels in a size dependent manner in a microfluidic model and may enter the lymphatic circulation through HEVs, and this mechanism is now being explored as a possible method for optimizing intravenous drug delivery to lymph nodes (Kaminskas et al., 2009; Vu et al., 2019). Mouse model studies using nanoparticles engineered to target HEVs have already shown improved accumulation in lymph nodes after intravenous delivery (Azzi et al., 2016; Bahmani et al., 2018).
While lymphatic redistribution has typically been described for nanomaterials, this can also occur with lipids that bind to hydrophobic pockets in carrier proteins or chylomicrons, and as such, lipids have been developed as lymphatic targeting tools and drug delivery systems (Trevaskis et al., 2015; Han et al., 2016). To this end, the primary 3H species likely absorbed from the lungs into plasma and lymph was 3H-phosphatidylcholine or a metabolite, and not intact liposome. While liposomes have some capability to cross membranes, they are more likely to be adsorbed by cell membranes or, in the case of the lungs, be degraded by surfactants or oxidation/hydrolysis processes (Glukhova, 2020). This can be established by analyzing the 3H content of biological samples via size exclusion chromatography (SEC), as has been done previously in rats (Haque et al., 2018). However, due to the low level of radioactivity in plasma and lymph samples in this study, SEC analysis of samples was not possible. After IV and pulmonary administration of this liposome composition in rats, though, we formerly showed that the predominant 3H species in blood after IV administration was intact liposome. By 24 h however, and in all plasma samples examined after pulmonary administration, smaller species (including protein-associated 3H) dominated the SEC profiles, including a species that co-eluted with 3H-phosphatidylcholine (Haque et al., 2018). This likely also occurred here in sheep. Interestingly though, the elimination half-life of 3H in sheep after pulmonary administration was considerably shorter compared to after IV administration (2 vs. 5 days respectively). Further, while pulmonary bioavailability, was calculated to be only 17%, excreta and organ biodistribution data suggested higher bioavailability (> 50%, consistent with that found after pulmonary administration of this liposome composition in rats). Collectively, this points towards the possibility that the 3H species absorbed from the lungs and found in plasma after IV administration differed, resulting in the lower observed bioavailability. This may be due to differences in liposome and lipid erosion/biodegradation pathways between the lungs and blood.
The active uptake of non-PEGylated liposomes by alveolar macrophages and lymphatic immune cells may possibly explain the limited lymphatic exposure of liposomes after pulmonary administration (Haque et al., 2018). While the proportion of the 3H-lipid dose found in CMLD lymph fluid over 5 days was limited, as mentioned above, liposomes can also presumably traffic into the lung lymph after initial uptake by phagocytic cells, such as APCs or alveolar macrophages. However, only 0.002% of the pulmonary dose was quantified in total collected lymph cell pellets over 5 days. Further, only 7% of the 3H label quantified in total BALF (fluid and cell pellet) was identified in the cell pellets (data not shown) and 1.5% of the dose was found in the CMLD after 5 days. This collectively suggested that non-PEGylated, and non-targeted liposomes are not significantly transported into lung lymph by APCs or macrophages over 5 days, despite the fact they are avidly internalized by these cells (Haque et al., 2018; Haque et al., 2019). This was interesting given that alveolar macrophages have previously been reported to transport nanoparticles to lung draining lymph nodes (Kirby et al., 2009). Attempts to expand on this discovery, however, have been disappointing, with one mouse study showing only dendritic cells are capable of mediating transport of nanoparticles into draining lung lymph nodes (Hardy et al., 2013). It is worth noting, however, that the cellular composition of lymph fluid was not measured in this study.
In conclusion, this study quantitatively demonstrates that inhaled HSPC liposomes are poorly absorbed via the lung lymphatics. While previous studies have shown the potential for lung lymphatic access after pulmonary administration in rodents, or after inhaled administration of nanoradioliposomes that reflect the bacterial cell wall in pigs, we found less than 0.5% of the pulmonary dose was absorbed into the lung lymphatic network. Further, lymphatic exposure of 3H was likely a result of a combination of lymphatic absorption of products of liposome degradation and lipid metabolism, and lymphatic redistribution from the systemic circulation. This highlights the need to look for alternative approaches to enhance the delivery of drugs and other therapeutic agents towards the lung lymph after inhaled administration. While it is feasible to target nanomaterials to the lung lymphatics by relying upon lymphatic redistribution after IV administration, this approach results in higher systemic exposure and related off-target effects.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Monash University Animal Ethics Committee.
LK was the lead investigator and responsible for formulating the study hypothesis and design. JI, SH, and MW oversaw liposome production. Sheep experiments were conducted by JI, RB, and LK. Data analysis was conducted by JI, JI, and LK wrote the manuscript with editing from SH, RB, and MW formulation. JI, SH, RB, and LK were involved in collection and/or analysis of samples from sheep. All authors contributed to the manuscript draft.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Cryo-TEM image was generated at the Bio21 Institute Ian Holmes Imaging Center for the cryo EM imaging.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.880448/full#supplementary-material
Abhyankar, M. M., Mann, B. J., Sturek, J. M., Brovero, S., Moreau, G. B., Sengar, A., et al. (2021). Development of COVID-19 Vaccine Using a Dual Toll-like Receptor Ligand Liposome Adjuvant. NPJ vaccines 6 (1), 137. doi:10.1038/s41541-021-00399-0
Ager, A. (2017). High Endothelial Venules and Other Blood Vessels: Critical Regulators of Lymphoid Organ Development and Function. Front. Immunol. 8, 45. doi:10.3389/fimmu.2017.00045
Azzi, J., Yin, Q., Uehara, M., Ohori, S., Tang, L., Cai, K., et al. (2016). Targeted Delivery of Immunomodulators to Lymph Nodes. Cell Rep. 15 (6), 1202–1213. doi:10.1016/j.celrep.2016.04.007
Bahmani, B., Uehara, M., Jiang, L., Ordikhani, F., Banouni, N., Ichimura, T., et al. (2018). Targeted Delivery of Immune Therapeutics to Lymph Nodes Prolongs Cardiac Allograft Survival. J. Clin. Invest. 128 (11), 4770–4786. doi:10.1172/JCI120923
Botelho, M. F., De Lima, J. J., Dormehl, I. C., Baganha, M. F., Marques, M. A., Gomes, C. M., et al. (2011). Visualization of Deep Lung Lymphatic Network Using Radioliposomes. Rev. Port. Pneumol. 17 (3), 124–130. doi:10.1016/j.rppneu.2011.03.003
Boyd, B. J., Kaminskas, L. M., Karellas, P., Krippner, G., Lessene, R., and Porter, C. J. (2006). Cationic Poly-L-Lysine Dendrimers: Pharmacokinetics, Biodistribution, and Evidence for Metabolism and Bioresorption after Intravenous Administration to Rats. Mol. Pharm. 3 (5), 614–627. doi:10.1021/mp060032e
Bromley, S. K., Thomas, S. Y., and Luster, A. D. (2005). Chemokine Receptor CCR7 Guides T Cell Exit from Peripheral Tissues and Entry into Afferent Lymphatics. Nat. Immunol. 6 (9), 895–901. doi:10.1038/ni1240
Chanana, A. D., and Joel, D. D. (1985). The Ovine Bronchopulmonary Lymph Drainage Pathways: a Model to Study Pulmonary Immune Responses. Exp. Lung Res. 9 (3-4), 211–220. doi:10.3109/01902148509057524
Choi, H. S., Ashitate, Y., Lee, J. H., Kim, S. H., Matsui, A., Insin, N., et al. (2010). Rapid Translocation of Nanoparticles from the Lung Airspaces to the Body. Nat. Biotechnol. 28 (12), 1300–1303. doi:10.1038/nbt.1696
Enkhbaatar, P., Murakami, K., Shimoda, K., Mizutani, A., Traber, L., Phillips, G. B., et al. (2003). The Inducible Nitric Oxide Synthase Inhibitor BBS-2 Prevents Acute Lung Injury in Sheep after Burn and Smoke Inhalation Injury. Am. J. Respir. Crit. Care Med. 167 (7), 1021–1026. doi:10.1164/rccm.200209-1031PP
Fanciullino, R., Ciccolini, J., and Milano, G. (2021). COVID-19 Vaccine Race: Watch Your Step for Cancer Patients. Br. J. Cancer 124 (5), 860–861. doi:10.1038/s41416-020-01219-3
Glukhova, O. E. (2020). Liposome Drug Delivery System across Endothelial Plasma Membrane: Role of Distance between Endothelial Cells and Blood Flow Rate. Molecules 25 (8), 1875. doi:10.3390/molecules25081875
Han, S., Hu, L., Quach, T., Simpson, J. S., Edwards, G. A., Trevaskis, N. L., et al. (2016). Lymphatic Transport and Lymphocyte Targeting of a Triglyceride Mimetic Prodrug Is Enhanced in a Large Animal Model: Studies in Greyhound Dogs. Mol. Pharm. 13 (10), 3351–3361. doi:10.1021/acs.molpharmaceut.6b00195
Haque, S., Feeney, O., Meeusen, E., Boyd, B. J., McIntosh, M. P., Pouton, C. W., et al. (2019). Local Inflammation Alters the Lung Disposition of a Drug Loaded Pegylated Liposome after Pulmonary Dosing to Rats. J. Control Release 307, 32–43. doi:10.1016/j.jconrel.2019.05.043
Haque, S., Whittaker, M., McIntosh, M. P., Pouton, C. W., Phipps, S., and Kaminskas, L. M. (2018). A Comparison of the Lung Clearance Kinetics of Solid Lipid Nanoparticles and Liposomes by Following the 3H-Labelled Structural Lipids after Pulmonary Delivery in Rats. Eur. J. Pharm. Biopharm. 125, 1–12. doi:10.1016/j.ejpb.2018.01.001
Haque, S., Whittaker, M. R., McIntosh, M. P., Pouton, C. W., and Kaminskas, L. M. (2016). Disposition and Safety of Inhaled Biodegradable Nanomedicines: Opportunities and Challenges. Nanomedicine 12 (6), 1703–1724. doi:10.1016/j.nano.2016.03.002
Hardy, C. L., Lemasurier, J. S., Mohamud, R., Yao, J., Xiang, S. D., Rolland, J. M., et al. (2013). Differential Uptake of Nanoparticles and Microparticles by Pulmonary APC Subsets Induces Discrete Immunological Imprints. J. Immunol. 191 (10), 5278–5290. doi:10.4049/jimmunol.1203131
Kaminskas, L. M., Kota, J., McLeod, V. M., Kelly, B. D., Karellas, P., and Porter, C. J. (2009). PEGylation of Polylysine Dendrimers Improves Absorption and Lymphatic Targeting Following SC Administration in Rats. J. Control Release 140 (2), 108–116. doi:10.1016/j.jconrel.2009.08.005
Kaminskas, L. M., Landersdorfer, C. B, Bischof, R. J, Leong, N, Ibrahim, J, Davies, A. N, et al. (2020). Aerosol Pirfenidone Pharmacokinetics after Inhaled Delivery in Sheep: a Viable Approach to Treating Idiopathic Pulmonary Fibrosis. Pharm. Res. 37 (1), 1–11. doi:10.1007/s11095-019-2732-2
Ke, X., Howard, G. P., Tang, H., Cheng, B., Saung, M. T., Santos, J. L., et al. (2019). Physical and Chemical Profiles of Nanoparticles for Lymphatic Targeting. Adv. Drug Deliv. Rev. 151-152, 72–93. doi:10.1016/j.addr.2019.09.005
Kirby, A. C., Coles, M. C., and Kaye, P. M. (2009). Alveolar Macrophages Transport Pathogens to Lung Draining Lymph Nodes. J. Immunol. 183 (3), 1983–1989. doi:10.4049/jimmunol.0901089
Li, C., Chen, Y., Zhao, Y., Lung, D. C., Ye, Z., Song, W., et al. (2021). Intravenous Injection of COVID-19 mRNA Vaccine Can Induce Acute Myopericarditis in Mouse Model. Clin. Infect. Dis. official Publ. Infect. Dis. Soc. Am., ciab707. doi:10.1093/cid/ciab707
Liu, Q., Guan, J., Qin, L., Zhang, X., and Mao, S. (2020). Physicochemical Properties Affecting the Fate of Nanoparticles in Pulmonary Drug Delivery. Drug Discov. Today 25 (1), 150–159. doi:10.1016/j.drudis.2019.09.023
Marasini, N., and Kaminskas, L. M. (2019). Subunit-based Mucosal Vaccine Delivery Systems for Pulmonary Delivery - Are They Feasible? Drug Dev. Ind. Pharm. 45 (6), 882–894. doi:10.1080/03639045.2019.1583758
Marasini, N., Haque, S., and Kaminskas, L. M. (2017). Polymer-drug Conjugates as Inhalable Drug Delivery Systems: A Review. Curr. Opin. Colloid & Interface Sci. 31, 18–29. doi:10.1016/j.cocis.2017.06.003
Partanen, T. A., Arola, J., Saaristo, A., Jussila, L., Ora, A., Miettinen, M., et al. (2000). VEGF-C and VEGF-D Expression in Neuroendocrine Cells and Their Receptor, VEGFR-3, in Fenestrated Blood Vessels in Human Tissues. FASEB J. 14 (13), 2087–2096. doi:10.1096/fj.99-1049com
Pereira, E. R., Jones, D., Jung, K., and Padera, T. P. (2015). The Lymph Node Microenvironment and its Role in the Progression of Metastatic Cancer. Semin. Cell Dev. Biol. 38, 98–105. Elsevier. doi:10.1016/j.semcdb.2015.01.008
Ryan, G. M., Bischof, R. J., Enkhbaatar, P., McLeod, V. M., Chan, L. J., Jones, S. A., et al. (2016). A Comparison of the Pharmacokinetics and Pulmonary Lymphatic Exposure of a Generation 4 PEGylated Dendrimer Following Intravenous and Aerosol Administration to Rats and Sheep. Pharm. Res. 33 (2), 510–525. doi:10.1007/s11095-015-1806-z
Schraufnagel, D. E. (2010). Lung Lymphatic Anatomy and Correlates. Pathophysiology 17 (4), 337–343. doi:10.1016/j.pathophys.2009.10.008
Siafaka, P. I., Okur, N. Ü., Karantas, I. D., Okur, M. E., and Gündoğdu, E. A. (2021). Current Update on Nanoplatforms as Therapeutic and Diagnostic Tools: A Review for the Materials Used as Nanotheranostics and Imaging Modalities. Asian J. Pharm. Sci. 16 (1), 24–46. doi:10.1016/j.ajps.2020.03.003
Thomson, J. (1999). Sample Preparation and Counting of Biological Samples. Packard Instrument Application Note AN9002-CSR. Waltham, MA: PerkinElmer Inc.
Trevaskis, N. L., Kaminskas, L. M., and Porter, C. J. (2015). From Sewer to Saviour - Targeting the Lymphatic System to Promote Drug Exposure and Activity. Nat. Rev. Drug Discov. 14 (11), 781–803. doi:10.1038/nrd4608
Videira, M., Almeida, A. J., and Fabra, A. (2012). Preclinical Evaluation of a Pulmonary Delivered Paclitaxel-Loaded Lipid Nanocarrier Antitumor Effect. Nanomedicine 8 (7), 1208–1215. doi:10.1016/j.nano.2011.12.007
Videira, M. A., Botelho, M. F., Santos, A. C., Gouveia, L. F., de Lima, J. J., and Almeida, A. J. (2002). Lymphatic Uptake of Pulmonary Delivered Radiolabelled Solid Lipid Nanoparticles. J. Drug Target 10 (8), 607–613. doi:10.1080/1061186021000054933
Vu, M. N., Rajasekhar, P., Poole, D. P., Khor, S. Y., Truong, N. P., Nowell, C. J., et al. (2019). Rapid Assessment of Nanoparticle Extravasation in a Microfluidic Tumor Model. ACS Appl. Nano Mat. 2 (4), 1844–1856. doi:10.1021/acsanm.8b02056
Weber, E., Sozio, F., Borghini, A., Sestini, P., and Renzoni, E. (2018). Pulmonary Lymphatic Vessel Morphology: a Review. Ann. Anat. 218, 110–117. doi:10.1016/j.aanat.2018.02.011
Zhang, Y., Huo, M., Zhou, J., and Xie, S. (2010). PKSolver: An Add-In Program for Pharmacokinetic and Pharmacodynamic Data Analysis in Microsoft Excel. Comput. Methods Programs Biomed. 99 (3), 306–314. doi:10.1016/j.cmpb.2010.01.007
Keywords: lung lymph, pharmacokinetics, sheep, inhalation, mediastinal lymph, Nanoparticles, liposomes
Citation: Ibrahim JP, Haque S, Bischof RJ, Whittaker AK, Whittaker MR and Kaminskas LM (2022) Liposomes are Poorly Absorbed via Lung Lymph After Inhaled Administration in Sheep. Front. Pharmacol. 13:880448. doi: 10.3389/fphar.2022.880448
Received: 21 February 2022; Accepted: 22 April 2022;
Published: 02 June 2022.
Edited by:
Leonid Kagan, Rutgers, The State University of New Jersey, United StatesReviewed by:
Medha D. Joshi, Midwestern University, United StatesCopyright © 2022 Ibrahim, Haque, Bischof, Whittaker, Whittaker and Kaminskas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M Kaminskas, bC5rYW1pbnNrYXNAdXEuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.