
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 21 April 2022
Sec. Translational Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.873082
This article is part of the Research Topic Kappa Opioid Receptors Revealed: Disentangling the Pharmacology to Open Up New Therapeutic Strategies View all 7 articles
 Chiara Sturaro1
Chiara Sturaro1 Davide Malfacini2*Michela Argentieri1
Davide Malfacini2*Michela Argentieri1 Francine M. Djeujo2Erika Marzola3Valentina Albanese3Chiara Ruzza1,4Remo Guerrini3,4
Francine M. Djeujo2Erika Marzola3Valentina Albanese3Chiara Ruzza1,4Remo Guerrini3,4 Girolamo Calo’2
Girolamo Calo’2 Paola Molinari5
Paola Molinari5The present study investigated the in vitro pharmacology of the human kappa opioid receptor using multiple assays, including calcium mobilization in cells expressing chimeric G proteins, the dynamic mass redistribution (DMR) label-free assay, and a bioluminescence resonance energy transfer (BRET) assay that allows measurement of receptor interaction with G protein and β-arrestin 2. In all assays, dynorphin A, U-69,593, and [D-Pro10]dyn(1-11)-NH2 behaved as full agonists with the following rank order of potency [D-Pro10]dyn(1-11)-NH2 > dynorphin A ≥ U-69,593. [Dmt1,Tic2]dyn(1-11)-NH2 behaved as a moderate potency pure antagonist in the kappa-β-arrestin 2 interaction assay and as low efficacy partial agonist in the other assays. Norbinaltorphimine acted as a highly potent and pure antagonist in all assays except kappa-G protein interaction, where it displayed efficacy as an inverse agonist. The pharmacological actions of novel kappa ligands, namely the dynorphin A tetrameric derivative PWT2-Dyn A and the palmitoylated derivative Dyn A-palmitic, were also investigated. PWT2-Dyn A and Dyn A-palmitic mimicked dynorphin A effects in all assays showing similar maximal effects but 3–10 fold lower potency. In conclusion, in the present study, multiple in vitro assays for the kappa receptor have been set up and pharmacologically validated. In addition, PWT2-Dyn A and Dyn A-palmitic were characterized as potent full agonists; these compounds are worthy of further investigation in vivo for those conditions in which the activation of the kappa opioid receptor elicits beneficial effects e.g. pain and pruritus.
The kappa opioid receptor was cloned in 1993 (Meng et al., 1993), while its pharmacological identification spans more than two-decades before. Portoghese and others proposed more than a single opioid receptor type existed (Portoghese, 1965). The earliest direct demonstrations of multiple opioid receptor binding sites were obtained with radiolabelled naloxone and etorphine molecules (Pert and Snyder, 1973; Simon et al., 1973; Terenius, 1973). The first definitive evidence that these receptors did not form a homogeneous population was presented in 1976 (Martin et al., 1976), and kappa opioid receptors as specific subtype confirmed with structure-activity relationship studies on the dynorphin(1-13) scaffold (Chavkin and Goldstein, 1981). Subsequent medicinal chemistry and molecular biology studies identified major ligand binding and receptor domains (Hjorth et al., 1995; Hjorth et al., 1996; Thirstrup et al., 1996; Cheng et al., 1998; Coward et al., 1998; Jones et al., 1998; Bonner et al., 2000; Celver et al., 2002; Owens and Akil, 2002; Zhang et al., 2002; Pascal and Milligan, 2005; Yan et al., 2005): discoveries recently confirmed via X-ray crystallography (Wu et al., 2012; Che et al., 2020).
Kappa opioid receptor is a G protein-coupled receptor (GPCR), whose activation mediates guanosine diphosphate (GDP) to guanosine triphosphate (GTP) exchange in the alpha subunit of pertussis toxin-sensitive heterotrimeric G proteins. These G proteins dissociate and activate downstream cascades, including depression of cyclic adenosine monophosphate (cAMP) formation, opening of potassium and closing of voltage-gated calcium channels. Activated kappa opioid receptors interact with G protein-coupled receptor kinases (GRK: GRK2,3,5,6, (Chen et al., 2016; Chiu et al., 2017)) leading to C-terminal phosphorylation; this allows β-arrestin interaction.
Kappa opioid receptor agonists were identified to differentially trigger G protein or β-arrestin coupling (Mores et al., 2019; Faouzi et al., 2020); this capacity, i.e. ligand-dependent activation of some but not other specific transducer of a given GPCR, is called biased agonism (Kenakin, 2017). Very intriguingly, efforts have been made to model biased agonists activity and opioid receptors domain binding: G protein bias involves transmembrane domain V and extracellular loop II, while β-arrestin bias transmembrane domains II and III (Uprety et al., 2021). Moreover, several ligands were studied for their coupling preferences towards the different Gi/o (Olsen et al., 2020). The reason for this focus on the different transductional fingerprints of the kappa opioid receptor lies in the possibility to discriminate its in vivo effects i.e. therapeutic vs side-effects. The kappa opioid receptor is a key player in pain and mood modulation. Kappa opioid receptor agonists display a potent antinociceptive effect; however, their clinical use has been strongly limited because dysphoria, hallucinations, and dissociation accompany therapeutic effects. For the kappa opioid receptor, G protein engagement over β-arrestins interaction has been proposed as the preferred profile (Bruchas and Chavkin, 2010). Importantly, therapeutic advantage by kappa opioid receptor activation might be obtained through the development of 1) G protein-biased, 2) peripherally restricted, and 3) mixed opioid agonists (Paton et al., 2020). Intriguingly, nalfurafine is marketed (in Japan) as an antipruritic drug. It is a selective kappa opioid agonist (Inui, 2015), reportedly biased towards G protein (Mores et al., 2019), particularly at human compared to rodent receptors (Schattauer et al., 2017), and it is currently the sole marketed selective kappa opioid receptor agonist. Captivating insights come from novel omics approaches are modifying the scenario of GPCR biased agonism by studying several (virtually all) possible transduction mechanism at a time. Such studies linked the mammalian Target of Rapamycin pathway to the aversive behavior of kappa opioid receptor agonists (Liu et al., 2019).
Blocking the kappa opioid receptor is recommended to treat depressive, anxiety, and substance use disorders (Lutz and Kieffer, 2013; Carlezon and Krystal, 2016). GPCRs residence time (reciprocal of the dissociation rate constant) has been described as good predictor of in vivo efficacy (Strasser et al., 2017; van der Velden et al., 2020). However, as stated by Page and co-workers the peculiarly slow pharmacodynamic of kappa opioid receptor antagonists complicate their early phase clinical investigation; therefore, the development of shorter-acting antagonists is needed and might provide insight into whether these drugs are efficacious as predicted in preclinical studies (Carroll and Carlezon, 2013; Page et al., 2019).
In this study we deployed multiple assays to study the pharmacology of standard kappa opioid receptor ligands with differing pharmacological properties then to characterize and compare two novel dynorphin A (Dyn A) derivatives: PWT2-Dyn A and Dyn A-palmitic. PWT2-Dyn A is a tetrabranched derivative obtained by jointing four molecules of [Cys18]Dyn A to a PWT2 core. Dyn A-palmitic was obtained by reaction of [Cys18]Dyn A with a palmitoyl functionalized maleimide moiety. Specifically, we report data using calcium mobilization, dynamic mass redistribution (DMR), and bioluminescence resonance energy transfer (BRET) receptor-transducer interaction assays.
Peptides Dyn A, [D-Pro10]Dyn(1-11)-NH2 ([D-Pro10]) and [Dmt1,Tic2]Dyn(1-11)-NH2 ([Dmt1,Tic2]) were synthesized in line with solid phase peptide synthesis methodology previously reported (Gairin et al., 1986; Guerrini et al., 1998). The homo tetrameric PWT2-Dyn A was obtained grafting four molecules of [Cys18]Dyn A with the PWT2 core in a classical thiol-Michael reaction using experimental conditions previously optimized for the synthesis of nociceptin/orphanin FQ tetra branched derivatives (Guerrini et al., 2014). Similarly, the Dyn A analogue, Dyn A-palmitic was synthesized reacting in solution [Cys18]Dyn A with a palmitoyl functionalized maleimide moiety (Pacifico et al., 2020).
The non-peptide molecules U-69,593, Nor-Binaltorphimine (Nor-BNI), and GDP were from Tocris bioscience (Bristol, United Kingdom). All tissues culture media and supplements were from Invitrogen (Paisley, United Kingdom). Reagents used were from Sigma Chemical Co. (Poole, United Kingdom) and were of the highest purity available.
Concentrated solutions of ligands were made in ultrapure water (1 mM, peptides; 10 mM, GDP) or dimethyl sulfoxide (10 mM) and kept at - 20°C until use.
Chinese Hamster Ovary (CHO) cells stably co-expressing the human kappa or mu opioid receptors and the Gαqi5 protein or the human delta and the GαqG66Di5 protein were used in this assay. Cells were generated as described by Camarda and co-workers (Camarda and Calo, 2013). They were cultured in DMEM/F-12 (1:1) medium supplemented with 10% FBS, 2 mM l-glutamine, 200 mg/ml G418, 100 IU/ml penicillin, 100 IU/ml streptomycin, and 1 μg/ml fungizone. In the assays, cells were maintained at 37°C in a humidified atmosphere with 5% CO2. After reaching confluence, cells were detached by trypsinization, and 50,000 cells/well were seeded into 96-well black, clear-bottom plates 24 h before the test. At the assay time, cells were pre-incubated for 30 min at 37°C protected from light with a loading solution consisting of HBSS supplemented with 2.5 mM probenecid, 3 μM Fluo-4 AM, and 0.01% pluronic acid. The loading solution was subsequently discarded, and 100 μL/well of assay buffer consisting of HBSS with 20 mM HEPES, 2.5 mM probenecid, and 500 µM Brilliant Black (Sigma-Aldrich, St. Louis, United States) was dispensed and incubated for an additional 10 min under the same conditions. Serial dilutions of ligands were prepared in HBSS buffer with 20 mM HEPES and 0.02% of bovine serum albumin (BSA) to minimize ligands’ stickiness to plasticware. The automated microplate reader FlexStation II (Molecular Device, Union City, CA 94587, United States) was employed to detect changes in fluorescence intensity. Experiments were carried out at 37°C. Automated additions were carried out in a volume of 50 μL/well. In antagonism experiments, ligands tested as antagonists were administered 24 min before adding the agonist. Three cycles of mixing were performed immediately after injection to ensure proper drug diffusion into the well. The effects of all compounds were expressed as the maximum change in percentage over the baseline fluorescence measured in samples treated with vehicle.
DMR experiments were conducted as previously described (Malfacini et al., 2018). Chinese Hamster Ovary (CHO) cells stably expressing the human kappa receptor were kindly provided by L Toll (Torrey Pines Institute for Molecular Studies, Port St. Lucie, United States). Cells were cultured in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12) supplemented with 10% (v/v) Fetal Calf Serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 15 mM HEPES. The medium was supplemented with 400 μg/ml G418 to maintain expression. Cells were seeded at a density of 45,000 cells/well in 120 μL into fibronectin-coated EnspireTM-LC 96-wells plates and cultured for 20 h to form a confluent monolayer. On the day of the experiment, cells were washed twice and maintained with assay buffer: HBSS with 20 mM HEPES, 0.01% BSA fraction V; for 90 min before the experiment. DMR was monitored in real-time with a temporal resolution of 44 s throughout the assay. Experiments were performed at 37°C, using an EnSight Multimode Plate Reader (PerkinElmer). A 5 min baseline was first established, followed by adding compounds manually in a volume of 40 μL and recording compound-triggered DMR signal for 60 min. In antagonism experiments, ligands tested as antagonists were added 30 min before agonist administration. Maximum picometers (pm) modification (peak) were used to determine agonist response after baseline normalization.
In vitro pharmacological effects of ligands at kappa opioid receptors were assessed by evaluating receptor interaction with G-protein and β-arrestin 2 with a BRET assay previously set up and validated for delta and mu (Molinari et al., 2010) and NOP (Malfacini et al., 2015) receptors.
SH-SY5Y human neuroblastoma cells permanently co-expressing the different pairs of fusion proteins were prepared using the pantropic retroviral expression system by Clontech as described previously (Molinari et al., 2010). Specifically, human kappa-RLuc fusion protein was made by linking the C-terminal of the receptor sequence without its stop codon, to the N-terminal of RLuc through a 13-mer linker peptide (RTEEQKLISEEDL) and cloned into the retroviral expression vector pQIXN (Clontech). The construction of the plasmids encoding the bovine Gβ1 and the human β-Arrestin 2 N-terminal-tagged with RGFP (Prolume, Pinetop, United States) were previously detailed (Molinari et al., 2010).
SH-SY5Y cells stably co-expressing the fusoproteins kappa-RLuc and Gβ1-RGFP or kappa-RLuc and β-arrestin 2-RGFP were grown in DMEM/F12 (1:1) medium supplemented with 10% FBS, 2 mM l-Glutamine, 100 μg/ml hygromycin B, 400 μg/ml G418, 100 units/ml penicillin G, 100 μg/ml streptomycin and 1 μg/ml Fungizone at 37°C in a humidified atmosphere with of 5% CO2. Enriched plasma membrane samples from kappa-RLuc/Gβ1-RGFP expressing cells for receptor-G protein interaction assay were prepared by differential centrifugation and stored at −80°C until use as previously described. Total protein concentration in membrane preparations was determined by colorimetric method with Quantum Protein-BCA kit (EuroClone, Pero (MI), IT) using the multiplate reader Victor Nivo (PerkinElmer, Walthman, MA, United States). BRET assays were performed in white opaque 96 wells microplates (PerkinElmer, Walthman, MA, United States) at room temperature. In kappa opioid receptor-G protein interaction experiments, cell membranes were thawed and resuspended in PBS supplemented with 0.02% BSA, and an amount of 5 μg of total protein was dispensed in each 96 well. In kappa opioid receptor/β-arrestin 2 recruitment experiments 100,000 cells/well were seeded 24 h prior to the BRET test. On the day of the experiment, the complete medium was discarded, and cells were washed with PBS supplemented with 0.5 mM MgCl2, 0.9 mM CaCl2. Cells were subsequently incubated with 2 μM Prolume Purple Coelenterazine (NanoLight Technology, White Mountain, AZ; United States) for 10 min before bioluminescence reading. Agonists were added and incubated for 5 min (or 30 min where indicated) before microplate reading. In antagonism experiments, antagonists were dispensed into wells 15 min before coelenterazine incubation, Dyn A was subsequently added, and luminescence was acquired. Counts per second (CPS) were detected by Victor luminometer, emissions were selected using 405(10) and 510(30) bandpass filters for Rluc and RGFP, respectively. Acquired data were computed as BRET ratios calculated as follow:
Agonists effects were expressed as a fraction of the maximal effect induced by Dynorphin A following blank (vehicle) subtraction.
RLuc interference experiments were performed on SH-SY5Y cell membranes prepared and quantified as described above. Because several ligands were described to directly interact with RLuc or generate unspecific luminescence artifacts (Auld et al., 2008), the amplitude of ligand-RLuc light alteration was quantified. Cells used expressed the kappa-RLuc and the β-Arrestin 2-RGFP fusoproteins. Following the membrane extraction routine, cell membranes did lose the soluble protein β-Arrestin 2-RGFP but not the kappa-RLuc allowing for precise quantification of direct RLuc light emission alteration. High concentrations of ligands were tested with membranes in the presence of CNTZ, and results were expressed in relation to vehicle/buffer set at 100% (Supplementary Figure S1). 15% alteration of vehicle/buffer RLuc-CNTZ emission was considered as the threshold for excluding a given compound’s concentration from the concentration-response curves in following pharmacological experiments.
The pharmacological terminology is consistent with the International Union of Basic and Clinical Pharmacology (IUPHAR) recommendations (Neubig et al., 2003). Concentration-response curves to agonists were fitted to the four-parameter logistic nonlinear regression model as follows:
Curve fitting was performed using PRISM 6.0 (GraphPad Software Inc., San Diego, CA).
In antagonism experiments, concentration-response curves to Dyn A in the absence and in the presence of increasing concentrations of antagonists (Schild analysis) were performed and antagonist potency derived as pA2 intercept to the y-axis of the linear regression in the Schild plot. If unsurmountable antagonist behavior was detected, the pKB value was derived as follows: a double-reciprocal plot of equi-effective concentrations of agonist (A) in the absence (1/A) and presence (1/A’) of the antagonist (B) was constructed and pKB was derived from the equation pKB = log {(slope-1)/[B]} (Kenakin, 2009).
To quantify the differences in agonist efficacies for G protein and arrestin interactions, bias factors were calculated by choosing the endogenous kappa opioid ligand Dyn A as standard unbiased ligand. For this analysis, the Emax and EC50 of the agonist were derived using a 3-parameter logistic model with slope values equal to unity. Although several agonist curves displayed slope values different from 1, refitting the curves with the parameter fixed to unity did not produce a statistically significant reduction of the goodness of fit. Under such conditions, the relative ratio (Emax/EC50)lig/(Emax/EC50)Dyn A is equivalent to the relative (τ/K)lig/(τ/K)Dyn A ratio as defined by the operational model (Black and Leff, 1983; Griffin et al., 2007). Taking ratios of these values between G protein and arrestin can cancel the common K and yield the ratio of ligand intrinsic efficacy across the two transduction proteins. Thus, the following formula was used for calculating agonist bias factors in log10 units:
Bias factors were considered significantly different from the reference ligand when a ligand’s CL95% did not include zero. The pharmacological terminology and computations related to biased agonism were consistent with IUPHAR recommendations (Kolb et al., 2022).
Data are expressed as mean ± sem of n experiments and were analyzed statistically using one-way analysis of variance followed by Dunnett’s test for multiple comparisons. Potency values and bias factors are expressed as mean (CL95%). p values <0.05 were considered statistically significant.
The endogenous peptide Dyn A, the synthetic peptide [D-Pro10], and the non-peptide arylacetamide U-69,593 were chosen as standard kappa opioid agonists and tested on CHO cells stably co-expressing the kappa opioid receptor together with signaling altered chimeric G protein (Gαqi5) enabling calcium mobilization assaying. In this system, all compounds evoked high maximal effects (Emax, approx. 300% fluorescence intensity units (FIU) over the baseline) and displayed high potency ranging from 9.08 for [D-Pro10] to 8.09 for U-69,593 (Figure 1Ai). Conversely, peptide [Dmt1,Tic2] and non-peptide Nor-BNI antagonists did not show high stimulation of calcium flux, with [Dmt1,Tic2] increasing the calcium levels to 50% over the baseline only at micromolar concentrations and Nor-BNI being completely inactive (Figure 1Aii). Same antagonists were investigated for their capacity to block kappa opioid receptor activation in Schild protocol experiments where the effects of increasing concentrations of Dyn A were challenged with different concentrations of the two molecules. Nor-BNI produced a dextral shift in the Dyn A concentration-response curve with a progressive depression of maximal effects (Figure 1Bi); for this reason, the estimation of Nor-BNI potency as shown in the Schild plot (Figure 1Bii) was further corroborated by the approach shown in Figure 1Biii. Both computations returned very high potency values (pA2 10.1 and pKB 10.9, respectively). [Dmt1,Tic2] produced a rightward shift in Dyn A concentration-response curve without modifying the agonist maximal effects (Figure 1Ci); a pA2 value of 6.6 was derived from these experiments (Figure 1Cii).
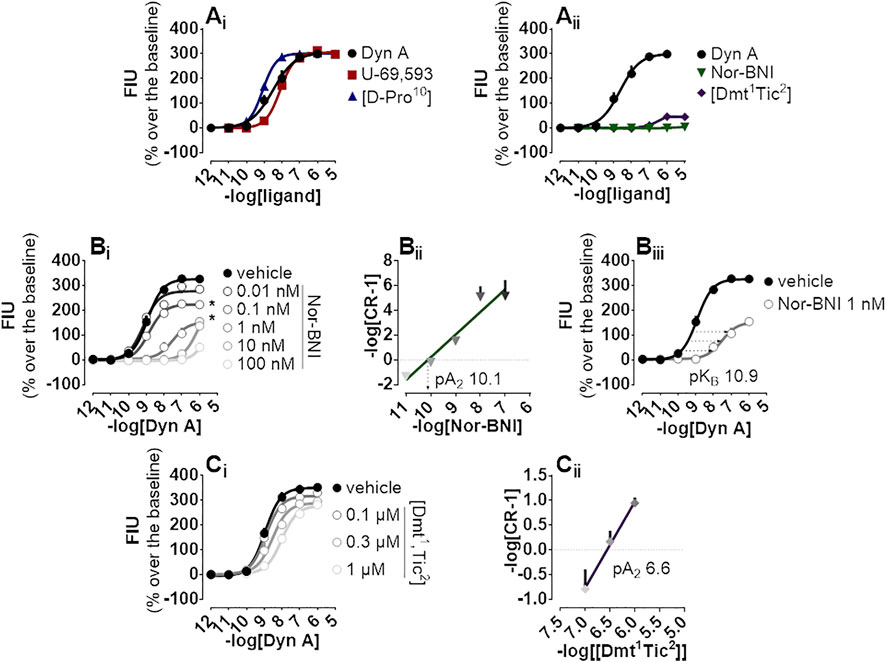
FIGURE 1. Calcium mobilization experiments carried out in CHO cells stably expressing the human kappa opioid receptor and the Gαqi5 chimeric G protein (Ai) Dyn A, U-69,593, and [D-Pro10], and (Aii) Nor-BNI and [Dmt1,Tic2] concentration-response curves (Bi) Concentration-response curves to Dyn A in the absence and in the presence of increasing (10 p.m.—100 nM) concentrations of Nor-BNI, corresponding (Bii) Schild analysis and (Biii) computation of antagonist potency for non-competitive antagonists (Ci) Concentration-response curves to Dyn A in the presence of increasing concentrations (0.1—1 µM) of [Dmt1,Tic2] with corresponding (Ci) Schild analysis. Data are mean +sem of three to six independent experiments performed in duplicate and Emax were analyzed statistically using one-way analysis of variance followed by Dunnett’s test for multiple comparisons. p values <0.05 were considered statistically significant and labelled with *.
DMR is a noninvasive, unbiased, label-free approach that was employed to assess the pharmacological profile of human kappa opioid receptors expressed in CHO cells. Dyn A (Figure 2Ai), U-69,593 (Figure 2Aii), and [D-Pro10] (Figure 2Aiii) elicited a concentration-dependent positive DMR signal, with Emax spanning from 234 to 251 pm (pm) for Dyn A and [D-Pro10], respectively. Potencies for Dyn A, U-69,593, and [D-Pro10] were 9.15, 8.60, and 9.84 (Figure 2Aiv). Nor-BNI (Figure 2Bi) did not modify DMR baseline while [Dmt1,Tic2] (Figure 2Bii) slightly stimulated the DMR response only at micromolar concentrations (Figure 2Biii).
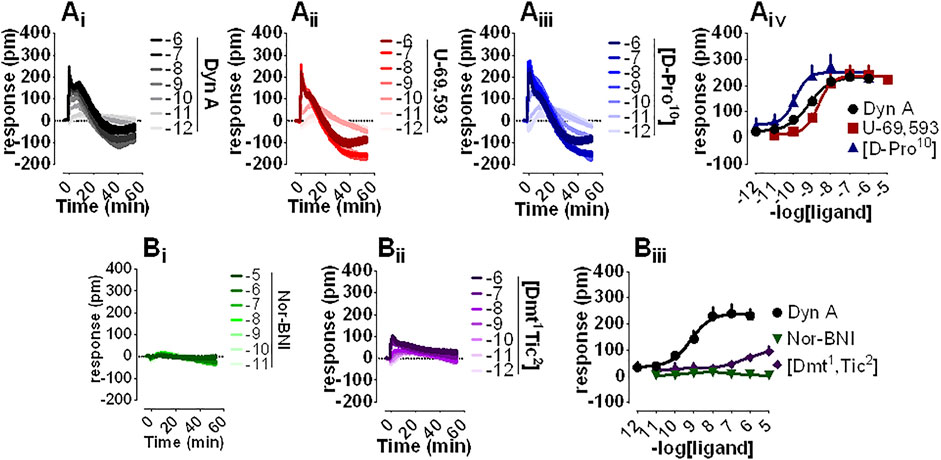
FIGURE 2. DMR experiments in CHO cells stably expressing the human kappa opioid receptor (Ai-iii) Averaged kinetics of increasing concentrations of Dyn A, U-69,593, and [D-Pro10], and (Aiv) corresponding concentration-response curves (Bi-ii) Averaged effects of Nor-BNI and [Dmt1,Tic2], and (Biii) corresponding concentration-response curves. Data represented are mean +sem of four to seven independent experiments performed in duplicate and Emax were analyzed statistically using one-way analysis of variance followed by Dunnett’s test for multiple comparisons.
Nor-BNI and [Dmt1,Tic2] antagonist properties were evaluated by Schild analysis. Comparison of representative Dyn A concentration-effect curve alone (Figure 3Ai) to that of Dyn A in the presence of single concentrations of Nor-BNI (1—100 nM, Figure 3Aii-iv) are shown. Nor-BNI concentration-dependently shifted the sigmoidal curves of Dyn A to the right without significantly affecting Emax (Figure 3Bi). pA2 from Schild plot extrapolation was 10.7 (Figure 3Bii). Dyn A alone (Figure 3Ci) and following preincubation with [Dmt1,Tic2] at different single concentrations (0.1—1 μM, Figure 3Cii-iv) are shown. DMR response to Dyn A was slightly depressed by all concentrations of antagonist used (Figure 3Di), likely because the weak stimulatory effect of [Dmt1,Tic2] (as in Figure 2Bii, Biii) is subtracted from the baseline by the instrument software. In Schild analysis of [Dmt1,Tic2] against Dyn Aa pA2 of 7.7 was calculated.
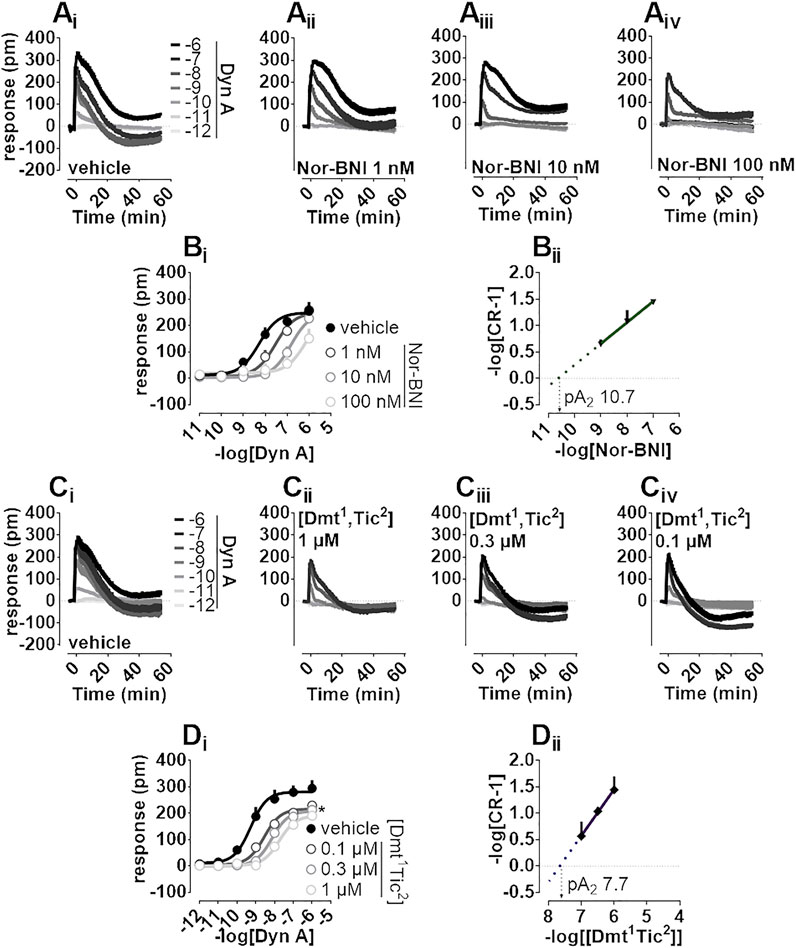
FIGURE 3. DMR experiments in CHO cells stably expressing the human kappa opioid receptor (Ai-iv) Averaged kinetic traces of increasing (1 p.m.—1 µM) concentrations of Dyn A in the absence and in the presence of different (1–100 nM) concentrations of Nor-BNI, corresponding (Bi) concentration-response curves and (Bii) Schild analysis (Ci-iv) Averaged kinetic traces of increasing Dyn A concentrations in the absence and in the presence of different (0.1–1 µM) concentrations of [Dmt1,Tic2] with corresponding (Di) concentration-response curves and (Dii) Schild analysis. Data represented are mean +sem of five to seven independent experiments performed in duplicate and Emax were analyzed statistically using one-way analysis of variance followed by Dunnett’s test for multiple comparisons. p values <0.05 were considered statistically significant and labelled with *.
A BRET approach enabling the measurement of kappa opioid receptor to G protein (Gβ1) or to β-Arrestin 2 interaction was employed to test the activity of kappa opioid receptor ligands. In the kappa-G protein interaction assay, Dyn A, U-69,593, and [D-Pro10] displayed similar maximal effects and potency (pEC50) of 8.21, 8.52, and 8.36, respectively (Figure 4Ai). The interaction of the β-Arrestin 2 to the kappa opioid receptor was also promoted by Dyn A, U-69,593, and [D-Pro10] with similar high maximal effects, and pEC50 values of 7.74, 6.72, and 8.07, respectively (Figure 4Aii). The computation of bias factors (towards G protein) for U-69,593 and [D-Pro10] returned values of 1.22 and 0.29, respectively (Supplementary Figure S2).
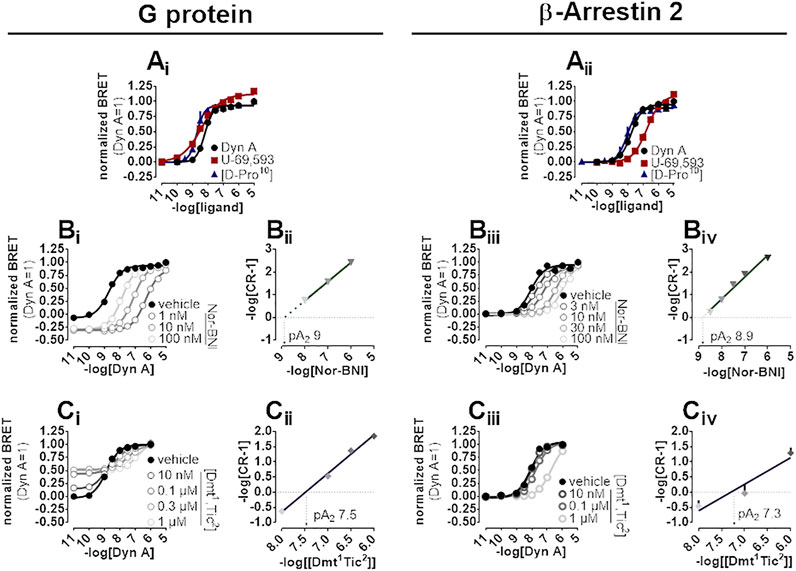
FIGURE 4. BRET experiments in SH-SY5Y cells stably expressing the human kappa opioid-RLuc tagged receptor together with either the Gβ1-RGFP or the β-Arrestin 2-RGFP fusoprotein. Concentration-response curves to Dyn A, U-69,593, and [D-Pro10] on the stimulation of kappa-G protein (Ai) and kappa-β-Arrestin 2 (Aii) interaction. Concentration-response curves to Dyn A in the absence and in the presence of increasing concentrations of Nor-BNI on the stimulation of kappa-G protein (Bi) and kappa-β-Arrestin 2 (Biii) interactions with corresponding Schild plots (Bii and Biv, respectively). Concentration-response curves to Dyn A in the absence and in the presence of increasing concentrations of [Dmt1,Tic2] on the stimulation of kappa-G protein (Ci) and kappa-β-Arrestin 2 (Ciii) interaction, with corresponding Schild analysis (Cii and Civ, respectively). Data represented are mean +sem of three to five independent experiments performed in duplicate.
In kappa-G protein interaction experiments, Nor-BNI (1–100 nM) shifted the concentration-effect curves to Dyn A to the right without altering maximal effects (Figure 4Bi). A pA2 of nine was extrapolated from the Schild plot (Figure 4Bii). Because Nor-BNI appeared to depress the baseline of kappa-G protein interaction, and, since this assay is performed on cell membranes, GDP can be applied to investigate receptor constitutive activity (see (Vezzi et al., 2013)) for similar experiments at delta and mu opioid receptors), Nor-BNI activity was compared to that of GDP at 5 and 30 min. Experiments (Supplementary Figure S3, Supplementary Table S1) showed how Nor-BNI depression of baseline, detected also with GDP, was visible only with prolonged incubation time. Nor-BNI, in addition, generated a robust shift to the right of Dyn A response in kappa-β-Arrestin 2 interaction, with no modification of maximal effects (Figure 4Biii). pA2 value obtained from the Schild plot was of 8.9 (Figure 4Biv). In kappa-G protein interaction experiments [Dmt1,Tic2] concentration-dependently increased baseline (Supplementary Figure S3) and elicited a dextral displacement of the concentration-response curve to Dyn A without altering maximal effects (Figure 4Ci). Schild analysis of these results yielded a pA2 of 7.5 (Figure 4Cii). In kappa-β-Arrestin 2 interaction experiments, [Dmt1,Tic2] did not show any residual agonist activity but produced a dextral displacement of the Dyn A concentration-response curve without alteration of the agonist maximal effect (Figure 4Ciii); a pA2 value of 7.3 was derived from these experiments (Figure 4Civ).
PWT2-Dyn A is a tetrabranched derivative of Dyn A (Figure 5Ai) whose high-performance liquid chromatography and mass spectrometric analysis are shown in Supplementary Figure S4. PWT2-Dyn A was capable of evoking concentration-dependent increase of calcium mobilization in CHO cells stably expressing kappa opioid receptors and chimeric G proteins. The effects of PWT2-Dyn A were assayed up to 100 nM, and maximal effects were constrained to be the same as Dyn A (299% over the baseline) with a potency (pEC50) derived for PWT2-Dyn A of 7.51 (Dyn A 8.53) (Figure 5Bi). CHO cells stably expressing either the mu opioid receptor and the Gαqi5 chimeric G protein or the delta opioid receptor and the GαqG66Di5 chimeric G protein were employed to study the selectivity of action of PWT2-Dyn A over the mu and the delta opioid receptors. Dermorphin displayed high maximal effects (294% over the baseline) and moderate potency (pEC50 7.57) in cells expressing the mu opioid receptor. Dyn A produced a stimulation (154% over the baseline) only at the highest concentration tested. PWT2-Dyn A was inactive (Figure 5Bii). DPDPE was tested as a standard delta opioid agonist, its maximal effects (202% over the baseline) and potency (pEC50 7.74) were found high, Dyn A also displayed high maximal effects (171% over the baseline) albeit with lower (pEC50 7.00) potency and PWT2-Dyn A exerted a slight (77% over the baseline) stimulatory effect only at the highest concentration tested (Figure 5Biii).
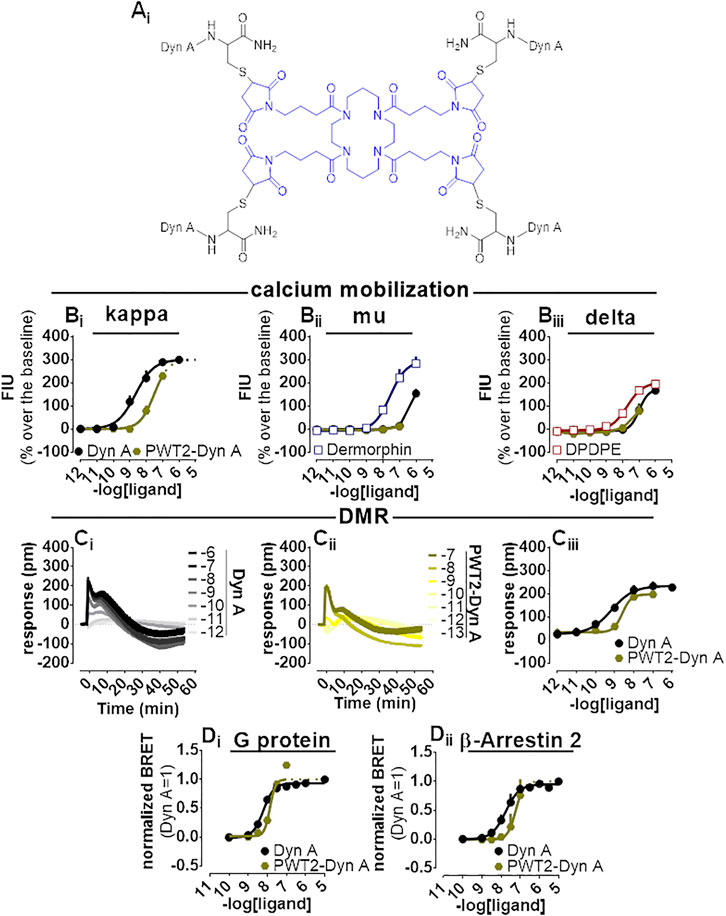
FIGURE 5. Pharmacological characterization of PWT2-Dyn A. Molecular structure (Ai). Calcium mobilization experiments in CHO stably expressing the human kappa (B), mu (Bii)i and delta (Bii) opioid receptor and chimeric G proteins, concentration-response curves to Dyn A and PWT2-Dyn A. Averaged DMR traces obtained in CHO cells stably expressing the human kappa opioid for increasing concentration of Dyn A (Ci) and PWT2-Dyn A (Cii), with corresponding concentration-response curves (Ciii). Concentration response curves to Dyn A and PWT2-Dyn A performed with a BRET assay on SH-SY5Y cells stably expressing the human kappa opioid-RLuc tagged receptor together with either the Gβ1-RGFP (Di) or the β-Arrestin 2-RGFP fusoprotein (Dii). Data are mean +sem of three to six independent experiments performed in duplicate.
When Dyn A and PWT2-Dyn A activities were compared in the DMR assay with CHO cells stably expressing the kappa opioid receptor (Figure 5Ci-iii), PWT2-Dyn A mimicked the stimulatory effect exerted by the natural peptide Dyn A with similar maximal effects and 4-fold lower potency (pEC50 8.51).
In the BRET assay, only concentrations of PWT2-Dyn A up to 10 nM could be tested because of unclear alteration of the RLuc emitted light (Supplementary Figure S1). Nevertheless, 10 nM PWT2-Dyn A reached the maximal effects of Dyn A in the kappa-G protein (Figure 5Di) and kappa-β-Arrestin 2 (Figure 5Dii) interaction assays, with derived potency values 3-fold lower than Dyn A in both assays.
A palmitoylated derivative of Dyn A, Dyn A-palmitic (Figure 6Ai), was examined. Dyn A-palmitic high-performance liquid chromatography and mass spectrometric analysis are shown in Supplementary Figure S5. In calcium mobilization studies performed with kappa receptor-expressing cells, Dyn A-palmitic displayed high maximal effects similar to that of Dyn A with only 2-fold loss of potency (Figure 6Bi). The selectivity of Dyn A-palmitic over the mu and the delta opioid receptors was assayed as for PWT2-Dyn A. Dyn A-palmitic, mimicked the stimulatory effects of dermorphin, with an approximately 6-fold lower potency (Figure 6Bii). At delta opioid receptor-expressing cells Dyn A-palmitic was 10-fold less potent than DPDPE (Figure 6Biii).
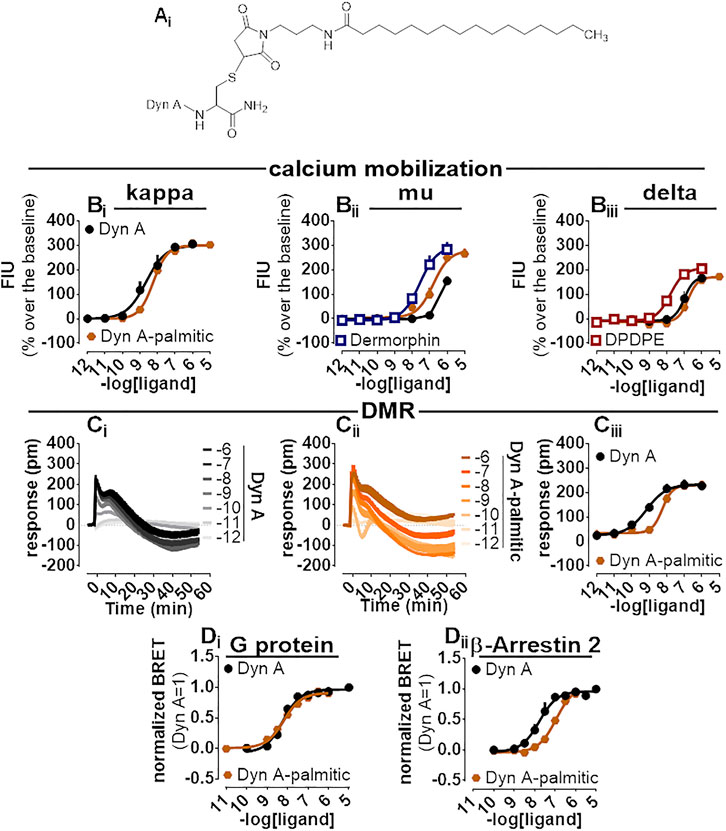
FIGURE 6. Pharmacological characterization of Dyn A-palmitic. Molecular structure (Ai). Calcium mobilization experiments in CHO cells stably expressing the human kappa (Bi), mu (Bii), and delta (Biii) opioid receptors and chimeric G proteins. Concentration-response curves to Dyn A and Dyn A-palmitic. Averaged DMR traces obtained in CHO cells stably expressing the human kappa opioid for increasing concentration of Dyn A (Ci) and Dyn A-palmitic (Cii), with corresponding concentration-response curves (Ciii). Concentration response curves to Dyn A and Dyn A-palmitic performed with a BRET assay on SH-SY5Y cells stably expressing the human kappa opioid-RLuc tagged receptor together with either the Gβ1-RGFP (Di) or the β-Arrestin 2-RGFP fusoprotein (Dii). Data are mean +sem of three to six independent experiments performed in duplicate.
Dyn A-palmitic was tested on the DMR assay together with Dyn A (Figure 6Ci, Cii), both compounds elicited high maximal effects in CHO cells expressing the kappa opioid receptor, with Dyn A-palmitic showing 6-fold lower potency than Dyn A (Figure 6Ciii).
In the BRET assay for kappa-G protein interaction, Dyn A and Dyn A-palmitic concentration-response curves were superimposable (potency ∼6 nM) (Figure 6Di). In kappa-β Arrestin-2 interaction experiments and despite mimicking the stimulatory effects of Dyn A with similar maximal effects, Dyn A-palmitic showed 7-fold lower potency than the natural peptide (Figure 6Dii). A bias factor (towards G protein) of 0.74 was derived from these experiments (Supplementary Figure S2).
All pharmacological parameters with corresponding CL95% or sem are summarized in Table 1. Results obtained in terms of biased agonism are plotted as bias plot and bias factors bar diagrams in Supplementary Figure S2.
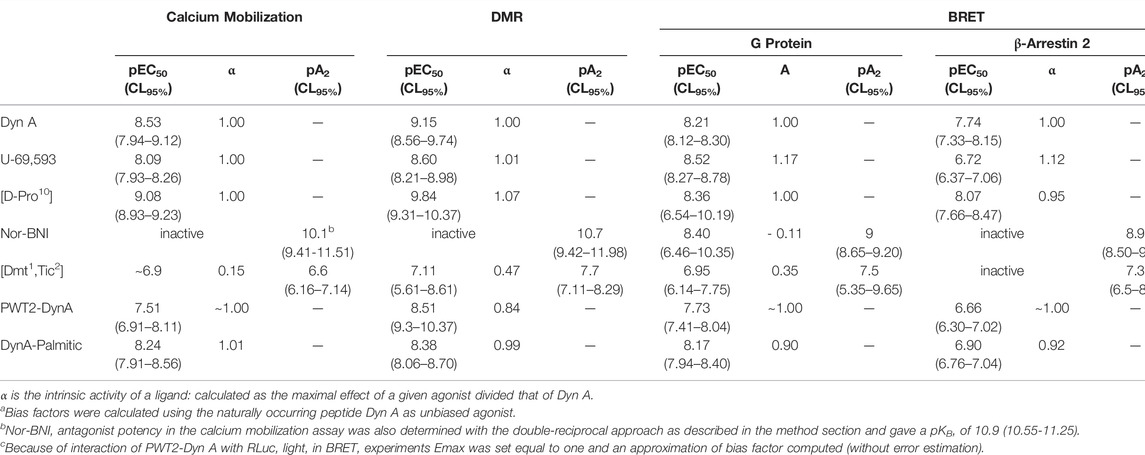
TABLE 1. Pharmacological parameters and relative CL95% for Dyn A, U-69,593, [D-Pro10], Nor-BNI, [Dmt1, Tic2], PWT2-DynA, and DynA-Palmitic in the indicated assays.
The kappa opioid receptor controls important biological functions, and its pharmacological modulation has therapeutic potential. For instance, selective kappa receptor antagonists promote robust anxiolytic and antidepressant-like effects and can also be investigated as innovative drugs to treat substance use disorders (Lutz and Kieffer, 2013; Carlezon and Krystal, 2016). Whilst, kappa receptor agonists display analgesic/antinociceptive and antipruritic (e.g. the Japanese marketed nalfurafine (Inui, 2015)) properties, clinical development is limited by important side effects such as dysphoria, sedation, hallucinations, and diuresis. To increase the therapeutic index of kappa receptor agonists, different strategies have been proposed, including G protein-biased agonists, peripherally restricted molecules, and mixed opioid receptor agonists (Paton et al., 2020).
The aim of this study was twofold, establishing a panel of assays to finely study kappa opioid receptor pharmacology and characterize novel ligands: PWT2-Dyn A and Dyn A-palmitic. We, therefore, deployed an array of assays to investigate the pharmacological profile of the human kappa opioid receptor in recombinant cells. The kappa opioid receptor natively couples to Gi/o heterotrimeric G proteins (Paton et al., 2020); therefore, we could measure calcium mobilization (a prototypical Gq signal) only adopting chimeric Gq/i proteins as described before by us (Camarda and Calo, 2013) and others (Zhang et al., 2012). DMR, an integrated, unbiased approach to assess real-time activation of intracellular signaling, was also employed as previously described for different GPCRs (Schroder et al., 2011), including opioid receptors (Morse et al., 2011; Grundmann et al., 2018). Finally, as previously reported for other opioid receptors (Molinari et al., 2010; Vezzi et al., 2013; Malfacini et al., 2015), two BRET approaches for measuring kappa-G protein and kappa-β-Arrestin 2 interaction were used to further investigate the pharmacology of kappa ligands, and eventually estimate their bias factor. Collectively, the pharmacological fingerprint of the kappa opioid receptor, obtained with potent and selective agonists and antagonists, is in line to that reported in the literature (Alexander et al., 2021). Novel ligands, PWT2-Dyn A and Dyn A-palmitic, showed similar pharmacology to that of the parental natural peptide Dyn A although with slight decrease of potencies. In addition, Dyn A-palmitic showed a significant bias towards G protein.
Collectively, in our assays, Dyn A, U-69,593, and [D-Pro10] behaved as full agonists of the human kappa opioid receptor with the following rank order of potency [D-Pro10] > Dyn A ≥ U-69,593, which is in line with several studies employing different assays and preparations (cells and isolated tissues), and multiple isoforms of the kappa opioid receptor (human, mouse, and guinea pig) (Gairin et al., 1984; Lahti et al., 1985; Gairin et al., 1988b; Meng et al., 1993; Yasuda et al., 1993; Soderstrom et al., 1997). Dyn A elicited robust activation of the kappa opioid receptor with potency ranging from 1 to 6 nM, except for the kappa-β-arrestin 2 interaction assay with a slightly lower potency of 18 nM. Similar results were obtained in calcium mobilization studies (Camarda and Calo, 2013). For stimulation of [35S]GTPγS binding, potency values obtained were approximately one order of magnitude higher (Zhu et al., 1997). U-69,593 potencies ranked from 3 to 8 nM with 191 nM in the kappa-β-arrestin 2 interaction assay, demonstrating a robust bias towards G protein activation. Literature data showed similar potency values in receptor binding assay of 4 nM, and forskolin stimulated inhibition of cAMP of 17 nM (Katsumata et al., 1996). However, G protein-biased behavior of U-69,593 is not corroborated by literature findings. In CHO cells expressing the human kappa receptor, U-69,593 displayed similar potency and efficacy in [35S]GTPγS stimulation and β-arrestin 2 recruitment experiments; however, the effects of U-69,593 were not compared to those of Dyn A (Ho et al., 2018). In the study by White and co-workers (White et al., 2014), U-69,593 has been classified as an unbiased kappa agonist, but Salvinorin A rather than Dyn A was used as a reference ligand. Finally, U-69,593 was also reported to be biased for internalization versus [35S]GTPγS stimulation (DiMattio et al., 2015). These inconsistencies underline the need of adopting more stringent rules in the design and reporting of biased agonism studies; these rules have been recently made available by IUPHAR (Kolb et al., 2022).
[D-Pro10] is a relatively poorly studied, highly potent Dyn A derivative. Developed in 1984 (Arimitsu et al., 1977), non-amidated [D-Pro10]Dyn(1-11) displayed a 7-fold higher affinity to the kappa opioid receptor (detected with [3H]Bremazocine binding) than Dyn A and showed 62- and 233-fold selectivity over the mu and delta opioid receptors, respectively. This peptide has been used as a probe to visualize kappa-opioid receptor sites in autoradiographic studies (Jomary et al., 1988). Intracerebroventricular administration of [D-Pro10]Dyn(1-11) in mice did not show any activity against thermal stimulus (tail-flick), but produced a dose-dependent antinociceptive effect in the acetic acid-induced writhing model of visceral pain, in line with a kappa-driven agonist activity (Gairin et al., 1988a). Our results confirm the high potency and full agonist activity of this peptide as a kappa agonist. In addition, the present findings suggest that the [D-Pro10] chemical modification does not affect the unbiased agonist features of the naturally occurring peptide Dyn A.
As far as antagonism is concerned, Dyn A concentration-response curve was challenged by increasing concentrations of Nor-BNI and [Dmt1,Tic2] in all assays adopted. Nor-BNI displayed potency values ranging 0.01–1.26 nM. [Dmt1,Tic2] potency, was lower and ranged from 20 to 251 nM. The pharmacological activity of Nor-BNI is in line with the literature, its affinity reported is approximately 1 nM (Simonin et al., 1995) and potency 0.1 nM (Zhu et al., 1997) (Birch et al., 1987). Regarding the type of antagonism, because the calcium mobilization assay was the only one showing an unsurmountable type of antagonism for Nor-BNI, we still classify Nor-BNI as a competitive antagonist. Unsurmountable/noncompetitive-like behaviors can be measured when antagonist-receptor complexes are reversible per se but dissociate too slowly that only a fraction of the receptors are accessible to subsequent stimulation by the agonist. In the calcium mobilization assay, the short time in which agonist effects are measured does not allow equilibration to be reached, especially when the antagonist slowly dissociates from the receptor (Charlton and Vauquelin, 2010). In fact, Nor-BNI is a very long-lasting antagonist in vivo, displaying an irreversible-like behavior (Jones and Holtzman, 1992) [Dmt1,Tic2] was developed in 1998 (Guerrini et al., 1998) and showed affinity values of 3.3—4.3 nM, with equal affinity at the mu opioid receptor and 10-fold higher affinity at the delta opioid receptor. Bioassay experiments on isolated tissues gave antagonist potency values of 90 and 8 nM for this peptide in guinea pig ileum and rabbit jejunum, respectively. The present results demonstrated that [Dmt1,Tic2] is actually a low efficacy (α 0.15–0.3) partial agonist at the human kappa receptor; of note the peptide behaved as a pure antagonist only in the kappa-β-arrestin 2 interaction assay. As expected partial agonist potency of [Dmt1,Tic2] was close to antagonistic potency values measured in the same assay. Compounds like [Dmt1,Tic2] characterized by mixed opioid receptor activity and different degrees of efficacy are currently considered of great interest (Paton et al., 2020); clearly further studies are needed to investigate the beneficial versus unwanted effects elicited in vivo by this opioid receptor ligand.
In kappa-G protein interaction experiments, Nor-BNI not only antagonized the effects of Dyn A but produced a concentration-dependent reduction in the baseline, thus behaving as an inverse agonist. Interestingly, in line with the slow kinetics (see before) of this molecule, this effect was evident only with prolonged incubation time. In addition, kappa opioid receptor constitutive activity is confirmed by experiments with GDP that, being capable of inhibiting the activated G protein, can elicit a condition of virtual zero receptor activation (Vezzi et al., 2013). Since the maximal inhibitory effect of Nor-BNI is similar to that of GDP, Nor-BNI should be classified as a full inverse agonist. As expected, the pEC50 of Nor-BNI studied as inverse agonist was close to the pA2 obtained investigating the molecule as antagonist. The present results corroborate previous findings obtained in [35S]GTPγS binding studies performed with HEK cells expressing the kappa receptor both in term of inverse agonist behavior of Nor-BNI and amount of kappa receptor constitutive activity (10–15% of basal values) (Wang et al., 2007). The liability of the kappa receptor to display constitutive activity seems to be similar to that of the mu receptor and much lower to that of the delta opioid receptor (Wang et al., 2007; Vezzi et al., 2013). Indeed, constitutive activity at GPCRs was first described in cells expressing delta receptors (Costa and Herz, 1989). As far as the possible physiological role of kappa receptor constitutive activity is concerned, little information is available in the literature; however, there is evidence suggesting that constitutively active kappa receptors are expressed in the ventral tegmental area (Polter et al., 2017) and in the medial prefrontal cortex (Sirohi and Walker, 2015) where may regulate response to stress in relation to substance use disorders and impulsivity, respectively.
Finally, we assessed how PWT2-Dyn A and Dyn A-palmitic behave in our pharmacological assays. PWT stands for peptide welding technology, and it is a relatively novel approach to generate with high yield and purity tetrabranched peptide molecules (Guerrini et al., 2014). PWT derivatives of nociceptin/orphanin FQ and related peptides, opioid peptides, tachykinins, and neuropeptide S were generated and characterized pharmacologically in previous studies (reviewed in (Calo et al., 2018)). In vitro these PWT derivatives mimicked the effects of the native peptides with no major changes of affinity, pharmacological activity, potency, or selectivity of action. However, in vivo studies demonstrated that PWT peptides are generally characterized by increased potency and, more importantly, long-lasting effects (Calo et al., 2018). Accordingly, in this study, PWT2-Dyn A showed pharmacology very similar to Dyn A, and similar selectivity for kappa over mu and delta opioid receptors. In addition, although caution should be exercised in evaluating PWT2-Dyn A data in the BRET assay (i.e. alteration of RLuc light), PWT2-Dyn A seemed to maintain the unbiased features of Dyn A. Clearly, further studies are needed to evaluate the in vivo actions of PWT2-Dyn A, assessing the usefulness of this kappa receptor ligand as a novel research tool.
We synthesized and characterized the palmitoylated derivative of Dyn A (Dyn A-palmitic). In the frame of a previous structure-activity relationship study on nociceptin/orphanin FQ, C-terminus palmitoylation has been identified as the chemical modification leading to the highest increase in G protein bias at the NOP receptor (Pacifico et al., 2020). In the same study, C-terminal palmitoylation was also applied at other opioid peptides including dermorphin, deltorphin A, and Leu-enkephalin, which were tested at mu and delta receptors. Palmitoylated peptides consistently displayed high potency in receptor-G protein interaction experiments while their effects in receptor-β-arrestin 2 interaction experiments were variable: they behaved as G protein biased agonists at NOP and delta receptors and as unbiased agonists at the mu receptor (Pacifico et al., 2020). Results obtained at the kappa opioid receptor with Dyn A-palmitic are therefore only partially in line with the findings mentioned above. In fact, C-terminal palmitoylation of Dyn A generated a kappa selective full agonist, but its potency was slightly lower than that of the native peptide. On the other hand, Dyn A-palmitic displayed a statistically significant bias factor towards G protein at kappa receptors, similar to that reported for palmitoylated peptides at NOP and delta (but not mu) receptors. Thus, the present results corroborated previous findings suggesting that C-terminal palmitoylation is a valuable strategy for altering the signaling properties of opioid receptor ligands, although this is not always associated with increased potency. The mechanism by which C-terminal modifications of peptide agonists for opioid receptors promote biased agonism toward G protein is at present unknown. The discussion of this specific issue and some speculative considerations have been reported in (Pacifico et al., 2020). Dyn A-palmitic could be added to the available research tools for investigating functional selectivity and its potential in drug discovery in the opioid receptor field (Dogra and Yadav, 2015; Gomes et al., 2020; Che et al., 2021; French and van Rijn, 2022).
The overall results obtained in the different assays with standard kappa receptor ligands are in line with literature findings both in terms of rank order of potency of agonists and potency of selective and competitive antagonists. In addition, the use of multiple assays and standard ligands allows detailed pharmacological investigation of activity and classification of ligands as full and partial agonists, neutral antagonists, and inverse agonists. We generated and studied PWT2-Dyn A and Dyn A-palmitic, which demonstrated selective kappa receptor full agonist behavior. Moreover, BRET receptor-transducer interaction studies suggested that PWT2-Dyn A acts as an unbiased agonist while Dyn A-palmitic as an agonist biased toward G protein. Further studies should assess the features of these novel compounds in vivo in animal models of pain and pruritus, conditions in which the selective activation of kappa receptors elicits beneficial effects.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Conceptualization: GC’, PM Formal analysis: CS, DM, CR, RG Funding acquisition: CR, RG, GC’, PM Investigation: CS, DM, MA, FD, EM, VA, CR, PM Resources: EM, VA, RG, PM Supervision: CR, GC’, PM Writing—original draft: DM Writing—review and editing: DM, CR, RG, GC’, PM.
This study was funded by grants from the University of Padova (Padua, IT) (DOR 2020 grant to GC’), from the University of Ferrara (Ferrara, IT) (FAR and FIR grants to CR, and FAR 2020 grant to RG), and supported by Istituto Superiore di Sanità (Rome, IT) (intramural funds to PM). DM position was funded by the Department of Pharmaceutical and Pharmacological Sciences, University of Padua (ARD-B).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank D. G. Lambert (University of Leicester, Leicester, United Kingdom) for helpful discussion and proofreading the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.873082/full#supplementary-material
Alexander, S. P., Christopoulos, A., Davenport, A. P., Kelly, E., Mathie, A., Peters, J. A., et al. (2021). The Concise Guide to Pharmacology 2021/22: G Protein-Coupled Receptors. Br. J. Pharmacol. 178 (Suppl. 1), S27–S156. doi:10.1111/bph.15538
Arimitsu, T., Di Chiro, G., Brooks, R. A., and Smith, P. B. (1977). White-gray Matter Differentiation in Computed Tomography. J. Comput. Assist. Tomogr 1 (4), 437–442. doi:10.1097/00004728-197710000-00009
Auld, D. S., Southall, N. T., Jadhav, A., Johnson, R. L., Diller, D. J., Simeonov, A., et al. (2008). Characterization of Chemical Libraries for Luciferase Inhibitory Activity. J. Med. Chem. 51 (8), 2372–2386. doi:10.1021/jm701302v
Birch, P. J., Hayes, A. G., Sheehan, M. J., and Tyers, M. B. (1987). Norbinaltorphimine: Antagonist Profile at Kappa Opioid Receptors. Eur. J. Pharmacol. 144 (3), 405–408. doi:10.1016/0014-2999(87)90397-9
Black, J. W., and Leff, P. (1983). Operational Models of Pharmacological Agonism. Proc. R. Soc. Lond. B Biol. Sci. 220 (1219), 141–162. doi:10.1098/rspb.1983.0093
Bonner, G., Meng, F., and Akil, H. (2000). Selectivity of Mu-Opioid Receptor Determined by Interfacial Residues Near Third Extracellular Loop. Eur. J. Pharmacol. 403 (1-2), 37–44. doi:10.1016/s0014-2999(00)00578-1
Bruchas, M. R., and Chavkin, C. (2010). Kinase Cascades and Ligand-Directed Signaling at the Kappa Opioid Receptor. Psychopharmacology (Berl) 210 (2), 137–147. doi:10.1007/s00213-010-1806-y
Calo', G., Rizzi, A., Ruzza, C., Ferrari, F., Pacifico, S., Gavioli, E. C., et al. (2018). Peptide Welding Technology - A Simple Strategy for Generating Innovative Ligands for G Protein Coupled Receptors. Peptides 99, 195–204. doi:10.1016/j.peptides.2017.10.004
Camarda, V., and Calo', G. (2013). Chimeric G Proteins in Fluorimetric Calcium Assays: Experience with Opioid Receptors. Methods Mol. Biol. 937, 293–306. doi:10.1007/978-1-62703-086-1_18
Carlezon, W. A., and Krystal, A. D. (2016). Kappa-Opioid Antagonists for Psychiatric Disorders: From Bench to Clinical Trials. Depress. Anxiety 33 (10), 895–906. doi:10.1002/da.22500
Carroll, F. I., and Carlezon, W. A. (2013). Development of κ Opioid Receptor Antagonists. J. Med. Chem. 56 (6), 2178–2195. doi:10.1021/jm301783x
Celver, J., Vishnivetskiy, S. A., Chavkin, C., and Gurevich, V. V. (2002). Conservation of the Phosphate-Sensitive Elements in the Arrestin Family of Proteins. J. Biol. Chem. 277 (11), 9043–9048. doi:10.1074/jbc.M107400200
Charlton, S. J., and Vauquelin, G. (2010). Elusive Equilibrium: the challenge of Interpreting Receptor Pharmacology Using Calcium Assays. Br. J. Pharmacol. 161 (6), 1250–1265. doi:10.1111/j.1476-5381.2010.00863.x
Chavkin, C., and Goldstein, A. (1981). Specific Receptor for the Opioid Peptide Dynorphin: Structure-Aactivity Relationships. Proc. Natl. Acad. Sci. U S A. 78 (10), 6543–6547. doi:10.1073/pnas.78.10.6543
Che, T., Dwivedi-Agnihotri, H., Shukla, A. K., and Roth, B. L. (2021). Biased Ligands at Opioid Receptors: Current Status and Future Directions. Sci. Signal. 14 (677). doi:10.1126/scisignal.aav0320
Che, T., English, J., Krumm, B. E., Kim, K., Pardon, E., Olsen, R. H. J., et al. (2020). Nanobody-enabled Monitoring of Kappa Opioid Receptor States. Nat. Commun. 11 (1), 1145. doi:10.1038/s41467-020-14889-7
Chen, C., Chiu, Y. T., Wu, W., Huang, P., Mann, A., Schulz, S., et al. (2016). Determination of Sites of U50,488H-Promoted Phosphorylation of the Mouse κ Opioid Receptor (KOPR): Disconnect between KOPR Phosphorylation and Internalization. Biochem. J. 473 (4), 497–508. doi:10.1042/BJ20141471
Cheng, Z. J., Yu, Q. M., Wu, Y. L., Ma, L., and Pei, G. (1998). Selective Interference of Beta-Arrestin 1 with Kappa and delta but Not Mu Opioid Receptor/G Protein Coupling. J. Biol. Chem. 273 (38), 24328–24333. doi:10.1074/jbc.273.38.24328
Chiu, Y. T., Chen, C., Yu, D., Schulz, S., and Liu-Chen, L. Y. (2017). Agonist-Dependent and -Independent κ Opioid Receptor Phosphorylation: Distinct Phosphorylation Patterns and Different Cellular Outcomes. Mol. Pharmacol. 92 (5), 588–600. doi:10.1124/mol.117.108555
Costa, T., and Herz, A. (1989). Antagonists with Negative Intrinsic Activity at delta Opioid Receptors Coupled to GTP-Binding Proteins. Proc. Natl. Acad. Sci. U S A. 86 (19), 7321–7325. doi:10.1073/pnas.86.19.7321
Coward, P., Wada, H. G., Falk, M. S., Chan, S. D., Meng, F., Akil, H., et al. (1998). Controlling Signaling with a Specifically Designed Gi-Coupled Receptor. Proc. Natl. Acad. Sci. U S A. 95 (1), 352–357. doi:10.1073/pnas.95.1.352
DiMattio, K. M., Ehlert, F. J., and Liu-Chen, L. Y. (2015). Intrinsic Relative Activities of κ Opioid Agonists in Activating Gα Proteins and Internalizing Receptor: Differences between Human and Mouse Receptors. Eur. J. Pharmacol. 761, 235–244. doi:10.1016/j.ejphar.2015.05.054
Dogra, S., and Yadav, P. N. (2015). Biased Agonism at Kappa Opioid Receptors: Implication in Pain and Mood Disorders. Eur. J. Pharmacol. 763 (Pt B), 184–190. doi:10.1016/j.ejphar.2015.07.018
Faouzi, A., Varga, B. R., and Majumdar, S. (2020). Biased Opioid Ligands. Molecules 25 (18). doi:10.3390/molecules25184257
French, A. R., and van Rijn, R. M. (2022). An Updated Assessment of the Translational Promise of G-Protein-Biased Kappa Opioid Receptor Agonists to Treat Pain and Other Indications without Debilitating Adverse Effects. Pharmacol. Res. 177, 106091. doi:10.1016/j.phrs.2022.106091
Gairin, J. E., Gouarderes, C., Mazarguil, H., Alvinerie, P., and Cros, J. (1984). [D-Pro10]Dynorphin-(1-11) Is a Highly Potent and Selective Ligand for Kappa Opioid Receptors. Eur. J. Pharmacol. 106 (2), 457–458. doi:10.1016/0014-2999(84)90741-6
Gairin, J. E., Gout, R., Meunier, J. C., and Cros, J. (1988a). [D-Pro10]-dynorphin(1-11) Is a Kappa-Selective Opioid Analgesic in Mice. J. Pharmacol. Exp. Ther. 245 (3), 995–1001.
Gairin, J. E., Jomary, C., Pradayrol, L., Cros, J., and Meunier, J. C. (1986). 125I-DPDYN, monoiodo[D-Pro10]dynorphin(1-11): a Highly Radioactive and Selective Probe for the Study of Kappa Opioid Receptors. Biochem. Biophys. Res. Commun. 134 (3), 1142–1150. doi:10.1016/0006-291x(86)90370-0
Gairin, J. E., Mazarguil, H., Alvinerie, P., Botanch, C., Cros, J., and Meunier, J. C. (1988b). N,N-diallyl-tyrosyl Substitution Confers Antagonist Properties on the Kappa-Selective Opioid Peptide [D-Pro10]dynorphin A(1-11). Br. J. Pharmacol. 95 (4), 1023–1030. doi:10.1111/j.1476-5381.1988.tb11735.x
Gomes, I., Sierra, S., Lueptow, L., Gupta, A., Gouty, S., Margolis, E. B., et al. (2020). Biased Signaling by Endogenous Opioid Peptides. Proc. Natl. Acad. Sci. U S A. 117 (21), 11820–11828. doi:10.1073/pnas.2000712117
Griffin, M. T., Figueroa, K. W., Liller, S., and Ehlert, F. J. (2007). Estimation of Agonist Activity at G Protein-Coupled Receptors: Analysis of M2 Muscarinic Receptor Signaling through Gi/o,Gs, and G15. J. Pharmacol. Exp. Ther. 321 (3), 1193–1207. doi:10.1124/jpet.107.120857
Grundmann, M., Merten, N., Malfacini, D., Inoue, A., Preis, P., Simon, K., et al. (2018). Lack of Beta-Arrestin Signaling in the Absence of Active G Proteins. Nat. Commun. 9 (1), 341. doi:10.1038/s41467-017-02661-3
Guerrini, R., Capasso, A., Marastoni, M., Bryant, S. D., Cooper, P. S., Lazarus, L. H., et al. (1998). Rational Design of Dynorphin A Analogues with delta-receptor Selectivity and Antagonism for delta- and Kappa-Receptors. Bioorg. Med. Chem. 6 (1), 57–62. doi:10.1016/s0968-0896(97)10008-6
Guerrini, R., Marzola, E., Trapella, C., Pela', M., Molinari, S., Cerlesi, M. C., et al. (2014). A Novel and Facile Synthesis of Tetra Branched Derivatives of Nociceptin/orphanin FQ. Bioorg. Med. Chem. 22 (14), 3703–3712. doi:10.1016/j.bmc.2014.05.005
Hjorth, S. A., Thirstrup, K., Grandy, D. K., and Schwartz, T. W. (1995). Analysis of Selective Binding Epitopes for the Kappa-Opioid Receptor Antagonist Nor-Binaltorphimine. Mol. Pharmacol. 47 (6), 1089–1094.
Hjorth, S. A., Thirstrup, K., and Schwartz, T. W. (1996). Radioligand-dependent Discrepancy in Agonist Affinities Enhanced by Mutations in the Kappa-Opioid Receptor. Mol. Pharmacol. 50 (4), 977–984.
Ho, J. H., Stahl, E. L., Schmid, C. L., Scarry, S. M., Aubé, J., and Bohn, L. M. (2018). G Protein Signaling-Biased Agonism at the κ-opioid Receptor Is Maintained in Striatal Neurons. Sci. Signal. 11 (542). doi:10.1126/scisignal.aar4309
Inui, S. (2015). Nalfurafine Hydrochloride to Treat Pruritus: a Review. Clin. Cosmet. Investig. Dermatol. 8, 249–255. doi:10.2147/CCID.S55942
Jomary, C., Gairin, J. E., Cros, J., and Meunier, J. C. (1988). Autoradiographic Localization of Supraspinal Kappa-Opioid Receptors with [125I-Tyr1, D-Pro10]dynorphin A-(1-11). Proc. Natl. Acad. Sci. U S A. 85 (2), 627–631. doi:10.1073/pnas.85.2.627
Jones, D. N., and Holtzman, S. G. (1992). Long Term Kappa-Opioid Receptor Blockade Following Nor-Binaltorphimine. Eur. J. Pharmacol. 215 (2-3), 345–348. doi:10.1016/0014-2999(92)90055-9
Jones, R. M., Hjorth, S. A., Schwartz, T. W., and Portoghese, P. S. (1998). Mutational Evidence for a Common Kappa Antagonist Binding Pocket in the Wild-type Kappa and Mutant mu[K303E] Opioid Receptors. J. Med. Chem. 41 (25), 4911–4914. doi:10.1021/jm9805182
Katsumata, S., Ienaga, Y., Minami, M., Nakagawa, T., Kanematsu, K., and Satoh, M. (1996). Pharmacological Characterization of KT-90 Using Cloned Mu-, delta- and Kappa-Opioid Receptors. Eur. J. Pharmacol. 312 (3), 349–355. doi:10.1016/0014-2999(96)00469-4
Kenakin, T. (2017). Theoretical Aspects of GPCR-Ligand Complex Pharmacology. Chem. Rev. 117 (1), 4–20. doi:10.1021/acs.chemrev.5b00561
Kenakin, T. P. (2009). “Orthosteric Drug Antagonism,” in A Pharmacology Primer. Editor T. P. Kenakin. Third Edition (New York: Academic Press), 101–127. doi:10.1016/b978-0-12-374585-9.00006-2
Kolb, P., Kenakin, T., Alexander, S. P. H., Bermudez, M., Bohn, L. M., Breinholt, C. S., et al. (2022). Community Guidelines for GPCR Ligand Bias: IUPHAR Review XXXII. Br. J Pharmacol. 1–24. doi:10.1111/bph.15811
Lahti, R. A., Mickelson, M. M., McCall, J. M., and Von Voigtlander, P. F. (1985). [3H]U-69593 a Highly Selective Ligand for the Opioid Kappa Receptor. Eur. J. Pharmacol. 109 (2), 281–284. doi:10.1016/0014-2999(85)90431-5
Liu, J. J., Chiu, Y. T., DiMattio, K. M., Chen, C., Huang, P., Gentile, T. A., et al. (2019). Phosphoproteomic Approach for Agonist-specific Signaling in Mouse Brains: mTOR Pathway Is Involved in κ Opioid Aversion. Neuropsychopharmacology 44 (5), 939–949. doi:10.1038/s41386-018-0155-0
Lutz, P. E., and Kieffer, B. L. (2013). Opioid Receptors: Distinct Roles in Mood Disorders. Trends Neurosci. 36 (3), 195–206. doi:10.1016/j.tins.2012.11.002
Malfacini, D., Ambrosio, C., Gro', M. C., Sbraccia, M., Trapella, C., Guerrini, R., et al. (2015). Pharmacological Profile of Nociceptin/Orphanin FQ Receptors Interacting with G-Proteins and β-Arrestins 2. PLoS One 10 (8), e0132865. doi:10.1371/journal.pone.0132865
Malfacini, D., Simon, K., Trapella, C., Guerrini, R., Zaveri, N. T., Kostenis, E., et al. (2018). NOP Receptor Pharmacological Profile - A Dynamic Mass Redistribution Study. PLoS One 13 (8), e0203021. doi:10.1371/journal.pone.0203021
Martin, W. R., Eades, C. G., Thompson, J. A., Huppler, R. E., and Gilbert, P. E. (1976). The Effects of Morphine- and Nalorphine- like Drugs in the Nondependent and Morphine-dependent Chronic Spinal Dog. J. Pharmacol. Exp. Ther. 197 (3), 517–532.
Meng, F., Xie, G. X., Thompson, R. C., Mansour, A., Goldstein, A., Watson, S. J., et al. (1993). Cloning and Pharmacological Characterization of a Rat Kappa Opioid Receptor. Proc. Natl. Acad. Sci. U S A. 90 (21), 9954–9958. doi:10.1073/pnas.90.21.9954
Molinari, P., Vezzi, V., Sbraccia, M., Grò, C., Riitano, D., Ambrosio, C., et al. (2010). Morphine-like Opiates Selectively Antagonize Receptor-Arrestin Interactions. J. Biol. Chem. 285 (17), 12522–12535. doi:10.1074/jbc.M109.059410
Mores, K. L., Cummins, B. R., Cassell, R. J., and van Rijn, R. M. (2019). A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists. Front. Pharmacol. 10, 407. doi:10.3389/fphar.2019.00407
Morse, M., Tran, E., Sun, H., Levenson, R., and Fang, Y. (2011). Ligand-directed Functional Selectivity at the Mu Opioid Receptor Revealed by Label-free Integrative Pharmacology On-Target. PLoS One 6 (10), e25643. doi:10.1371/journal.pone.0025643
Neubig, R. R., Spedding, M., Kenakin, T., and Christopoulos, A. (2003). International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology. Pharmacol. Rev. 55 (4), 597–606. doi:10.1124/pr.55.4.4
Olsen, R. H. J., DiBerto, J. F., English, J. G., Glaudin, A. M., Krumm, B. E., Slocum, S. T., et al. (2020). TRUPATH, an Open-Source Biosensor Platform for Interrogating the GPCR Transducerome. Nat. Chem. Biol. 16 (8), 841–849. doi:10.1038/s41589-020-0535-8
Owens, C. E., and Akil, H. (2002). Determinants of Ligand Selectivity at the Kappa-Receptor Based on the Structure of the Orphanin FQ Receptor. J. Pharmacol. Exp. Ther. 300 (3), 992–999. doi:10.1124/jpet.300.3.992
Pacifico, S., Ferrari, F., Albanese, V., Marzola, E., Neto, J. A., Ruzza, C., et al. (2020). Biased Agonism at Nociceptin/Orphanin FQ Receptors: A Structure Activity Study on N/OFQ(1-13)-NH2. J. Med. Chem. 63 (19), 10782–10795. doi:10.1021/acs.jmedchem.9b02057
Page, S., Mavrikaki, M. M., Lintz, T., Puttick, D., Roberts, E., Rosen, H., et al. (2019). Behavioral Pharmacology of Novel Kappa Opioid Receptor Antagonists in Rats. Int. J. Neuropsychopharmacol. 22 (11), 735–745. doi:10.1093/ijnp/pyz054
Pascal, G., and Milligan, G. (2005). Functional Complementation and the Analysis of Opioid Receptor Homodimerization. Mol. Pharmacol. 68 (3), 905–915. doi:10.1124/mol.105.013847
Paton, K. F., Atigari, D. V., Kaska, S., Prisinzano, T., and Kivell, B. M. (2020). Strategies for Developing κ Opioid Receptor Agonists for the Treatment of Pain with Fewer Side Effects. J. Pharmacol. Exp. Ther. 375 (2), 332–348. doi:10.1124/jpet.120.000134
Pert, C. B., and Snyder, S. H. (1973). Opiate Receptor: Demonstration in Nervous Tissue. Science 179 (4077), 1011–1014. doi:10.1126/science.179.4077.1011
Polter, A. M., Barcomb, K., Chen, R. W., Dingess, P. M., Graziane, N. M., Brown, T. E., et al. (2017). Constitutive Activation of Kappa Opioid Receptors at Ventral Tegmental Area Inhibitory Synapses Following Acute Stress. Elife 6. doi:10.7554/eLife.23785
Portoghese, P. S. (1965). A New Concept on the Mode of Interaction of Narcotic Analgesics with Receptors. J. Med. Chem. 8 (5), 609–616. doi:10.1021/jm00329a013
Schattauer, S. S., Kuhar, J. R., Song, A., and Chavkin, C. (2017). Nalfurafine Is a G-Protein Biased Agonist Having Significantly Greater Bias at the Human Than Rodent Form of the Kappa Opioid Receptor. Cell Signal 32, 59–65. doi:10.1016/j.cellsig.2017.01.016
Schröder, R., Schmidt, J., Blättermann, S., Peters, L., Janssen, N., Grundmann, M., et al. (2011). Applying Label-free Dynamic Mass Redistribution Technology to Frame Signaling of G Protein-Coupled Receptors Noninvasively in Living Cells. Nat. Protoc. 6 (11), 1748–1760. doi:10.1038/nprot.2011.386
Simon, E. J., Hiller, J. M., and Edelman, I. (1973). Stereospecific Binding of the Potent Narcotic Analgesic (3H) Etorphine to Rat-Brain Homogenate. Proc. Natl. Acad. Sci. U S A. 70 (7), 1947–1949. doi:10.1073/pnas.70.7.1947
Simonin, F., Gavériaux-Ruff, C., Befort, K., Matthes, H., Lannes, B., Micheletti, G., et al. (1995). Kappa-Opioid Receptor in Humans: cDNA and Genomic Cloning, Chromosomal Assignment, Functional Expression, Pharmacology, and Expression Pattern in the central Nervous System. Proc. Natl. Acad. Sci. U S A. 92 (15), 7006–7010. doi:10.1073/pnas.92.15.7006
Sirohi, S., and Walker, B. M. (2015). Maturational Alterations in Constitutive Activity of Medial Prefrontal Cortex Kappa-Opioid Receptors in Wistar Rats. J. Neurochem. 135 (4), 659–665. doi:10.1111/jnc.13279
Soderstrom, K., Choi, H., Berman, F. W., Aldrich, J. V., and Murray, T. F. (1997). N-alkylated Derivatives of [D-Pro10]dynorphin A-(1-11) Are High Affinity Partial Agonists at the Cloned Rat Kappa-Opioid Receptor. Eur. J. Pharmacol. 338 (2), 191–197. doi:10.1016/s0014-2999(97)81948-6
Strasser, A., Wittmann, H. J., and Seifert, R. (2017). Binding Kinetics and Pathways of Ligands to GPCRs. Trends Pharmacol. Sci. 38 (8), 717–732. doi:10.1016/j.tips.2017.05.005
Terenius, L. (1973). Characteristics of the "receptor" for Narcotic Analgesics in Synaptic Plasma Membrane Fraction from Rat Brain. Acta Pharmacol. Toxicol. (Copenh) 33 (5), 377–384. doi:10.1111/j.1600-0773.1973.tb01539.x
Thirstrup, K., Elling, C. E., Hjorth, S. A., and Schwartz, T. W. (1996). Construction of a High Affinity Zinc Switch in the Kappa-Opioid Receptor. J. Biol. Chem. 271 (14), 7875–7878. doi:10.1074/jbc.271.14.7875
Uprety, R., Che, T., Zaidi, S. A., Grinnell, S. G., Varga, B. R., Faouzi, A., et al. (2021). Controlling Opioid Receptor Functional Selectivity by Targeting Distinct Subpockets of the Orthosteric Site. Elife 10. doi:10.7554/eLife.56519
van der Velden, W. J. C., Heitman, L. H., and Rosenkilde, M. M. (2020). Perspective: Implications of Ligand-Receptor Binding Kinetics for Therapeutic Targeting of G Protein-Coupled Receptors. ACS Pharmacol. Transl Sci. 3 (2), 179–189. doi:10.1021/acsptsci.0c00012
Vezzi, V., Onaran, H. O., Molinari, P., Guerrini, R., Balboni, G., Calò, G., et al. (2013). Ligands Raise the Constraint that Limits Constitutive Activation in G Protein-Coupled Opioid Receptors. J. Biol. Chem. 288 (33), 23964–23978. doi:10.1074/jbc.M113.474452
Wang, D., Sun, X., and Sadee, W. (2007). Different Effects of Opioid Antagonists on Mu-, delta-, and Kappa-Opioid Receptors with and without Agonist Pretreatment. J. Pharmacol. Exp. Ther. 321 (2), 544–552. doi:10.1124/jpet.106.118810
White, K. L., Scopton, A. P., Rives, M. L., Bikbulatov, R. V., Polepally, P. R., Brown, P. J., et al. (2014). Identification of Novel Functionally Selective κ-opioid Receptor Scaffolds. Mol. Pharmacol. 85 (1), 83–90. doi:10.1124/mol.113.089649
Wu, H., Wacker, D., Mileni, M., Katritch, V., Han, G. W., Vardy, E., et al. (2012). Structure of the Human κ-opioid Receptor in Complex with JDTic. Nature 485 (7398), 327–332. doi:10.1038/nature10939
Yan, F., Mosier, P. D., Westkaemper, R. B., Stewart, J., Zjawiony, J. K., Vortherms, T. A., et al. (2005). Identification of the Molecular Mechanisms by Which the Diterpenoid Salvinorin A Binds to Kappa-Opioid Receptors. Biochemistry 44 (24), 8643–8651. doi:10.1021/bi050490d
Yasuda, K., Raynor, K., Kong, H., Breder, C. D., Takeda, J., Reisine, T., et al. (1993). Cloning and Functional Comparison of Kappa and delta Opioid Receptors from Mouse Brain. Proc. Natl. Acad. Sci. U S A. 90 (14), 6736–6740. doi:10.1073/pnas.90.14.6736
Zhang, F., Li, J., Li, J.-G., and Liu-Chen, L.-Y. (2002). (−)U50,488H [(trans)-3,4-Dichloro-N-Methyl-N-[2-(1-Pyrrolidinyl)-Cyclohexyl]benzeneacetamide] Induces Internalization and Down-Regulation of the Human, but Not the Rat, κ-Opioid Receptor: Structural Basis for the Differential Regulation. J. Pharmacol. Exp. Ther. 302 (3), 1184–1192. doi:10.1124/jpet.302.3.1184
Zhang, Y., Wang, Z., Cox, D. P., and Civelli, O. (2012). Study on the Activation of the Opioid Receptors by a Set of Morphine Derivatives in a Well-Defined Assay System. Neurochem. Res. 37 (2), 410–416. doi:10.1007/s11064-011-0627-7
Keywords: kappa opioid receptor, G protein-coupled receptor, label-free, BRET, calcium mobilization, biased agonism, PWT2-dyn A, dyn A-palmitic
Citation: Sturaro C, Malfacini D, Argentieri M, Djeujo FM, Marzola E, Albanese V, Ruzza C, Guerrini R, Calo’ G and Molinari P (2022) Pharmacology of Kappa Opioid Receptors: Novel Assays and Ligands. Front. Pharmacol. 13:873082. doi: 10.3389/fphar.2022.873082
Received: 10 February 2022; Accepted: 24 March 2022;
Published: 21 April 2022.
Edited by:
Wendy Margaret Walwyn, University of California, Los Angeles, United StatesReviewed by:
Susruta Majumdar, St. Louis College of Pharmacy, United StatesCopyright © 2022 Sturaro, Malfacini, Argentieri, Djeujo, Marzola, Albanese, Ruzza, Guerrini, Calo’ and Molinari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Malfacini, ZGF2aWRlLm1hbGZhY2luaUB1bmlwZC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.