 Hui Jiang
Hui Jiang Meiqi Li
Meiqi Li Guohua Wang
Guohua Wang Qianqian Luo
Qianqian Luo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 12 April 2022
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.869794
This article is part of the Research Topic Nutraceuticals: New Perspectives and Approaches in Human Health and Disease View all 12 articles
Background and Aims: Hepatic iron overload always leads to oxidative stress, which has been found to be involved in the progression of liver disease. However, whether iron disorder is involved in acute liver disease and the further molecular mechanisms remain unclear.
Methods: A mice model of acute liver injury (ALI) was established via intraperitoneal injection of thioacetamide (TAA) (250 mg/kg/day) for 3 consecutive days. Ferrostatin-1 (Fer-1) was administered intraperitoneally (2.5 μM/kg/day) starting 3 days before TAA treatment. Deferoxamine (DFO) was intraperitoneally injected (200 mg/kg/day) with TAA treatment for 3 days. We further observed the effect of Fer-1 on TAA model with high-iron diet feeding. ALI was confirmed using histological examination and liver function activity. Moreover, expressions of iron metabolism and ferroptosis proteins were measured by Western blot analysis.
Results: The study revealed that the iron accumulation and ferroptosis contributed to TAA-induced ALI pathogenesis. TAA induced prominent inflammation and vacuolar degeneration in the liver as well as liver dysfunction. In addition, protein expression of the cystine/glutamate antiporter SLC7A11 (xCT) and glutathione peroxidase 4 (GPX4) was significantly decreased in the liver, while transferrin receptor 1 (TfR1), ferroportin (Fpn) and light chain of ferritin (Ft-L) expression levels were increased after TAA exposure. As the same efficiency as DFO, pre-administration of Fer-1 significantly decreased TAA-induced alterations in the plasma ALT, AST and LDH levels compared with the TAA group. Moreover, both Fer-1 and DFO suppressed TfR1, Fpn and Ft-L protein expression and decreased iron accumulation, but did not affect xCT or GPX4 expression in the liver. Both Fer-1and DFO prevented hepatic ferroptosis by reducing the iron content in the liver. Furthermore, Fer-1 also reduced iron and reversed liver dysfunction under iron overload conditions.
Conclusion: These findings indicate a role of TAA-induced iron accumulation and ferroptosis in the pathogenesis of ALI model. The effect of Fer-1 was consistent with that of DFO, which prevented hepatic ferroptosis by reducing the iron content in the liver. Thus, Fer-1 might be a useful reagent to reverse liver dysfunction and decreasing the iron content of the liver may be a potential therapeutic strategy for ALI.
The liver is one of the vital organs in the body and plays a vital role in the detoxification of foreign substances, secretion of bile for digestion, metabolic functions of various nutrients, and regulation of iron metabolism. The liver is not only the major iron storage site but also where the iron regulation hormone hepcidin is synthesized (Nicolas et al., 2001; Sikorska et al., 2016). Systemic and cellular iron homeostasis is sustained through several main iron-related proteins, including iron importer, exporter and storage proteins. Iron is taken up into cells via transferrin receptor 1 (TfR1) and sequestered by ferritin light chain (Ft-L) (Hentze et al., 2010) or exported out of cells via ferroportin (Fpn). Iron homeostasis is controlled through regulation of duodenal iron absorption, macrophage iron release and hepatocyte iron storage, which is mainly carried out by the iron-related proteins above (Hentze et al., 2010; Ganz 2011).
Acute liver injury (ALI) often develops rapidly and may involve drug-induced liver failure or cholestasis. Animals that develop ALI show enhanced generation of hepatic ROS and enhanced lipid peroxidation with the formation of lipid peroxides. ALI may be caused by viruses, drugs or toxins. Among the various toxins, a thiono-sulfur-containing compound, thioacetamide (TAA), has been used extensively in the development of animal models of ALI (Chu et al., 2001; Bruck et al., 2007) because it induces lipid peroxidation, oxidative stress, and inflammation, ultimately causing functional hepatocyte death and liver dysfunction (Muller et al., 1991; Grek and Arasi 2016; Thawley 2017).
Ferroptosis, which is a recently identified novel form of regulated cell death, proceeds differently from apoptosis, necrosis, and autophagic cell death (Dixon et al., 2012). It is mediated by the iron-dependent oxidative degeneration of lipids and leads to mitochondrial shrinkage, with increased mitochondrial membrane density and outer mitochondrial membrane rupture (Xie et al., 2016). It has been reported that excess cellular iron is the main driver of the Fenton reaction and the production of reactive oxygen species (ROS) (He et al., 2020). Moreover, ferroptosis was demonstrated to be induced by phospholipid peroxidation associated with free iron-mediated Fenton reactions (Hadian and Stockwell 2020). Thus, we hypothesized that TAA-induced ALI is mediated by excess iron, causing substantial oxidative stress/lipid peroxidation and further inducing ferroptosis in hepatocytes, which finally results in liver injury and dysfunction. In this study, iron and ferroptosis were identified as the cornerstones for detecting the mechanism of ALI. TAA was administered intraperitoneally to induce the ALI model in mice. We found that iron content and mobilization were significantly enhanced, as well as liver dysfunction, while protein expression of the cystine/glutamate antiporter SLC7A11 (xCT) and glutathione peroxidase 4 (GPX4) in the liver was decreased in the TAA-induced ALI model. However, both deferoxamine (DFO) and ferrostatin-1 (Fer-1) suppressed the liver dysfunction induced by TAA by reducing iron accumulation in the liver but did not affect xCT or GPX4 expression. Our data suggest that decreasing the iron content in the liver may be a potential therapeutic strategy for ALI.
All animals were provided by the animal experimental centre of Nantong University. Mice were maintained in stainless steel cages with a relative humidity of 55–60% at 21 ± 2 C with 12-h rotation periods of light and dark. All animal handling procedures were performed according to approved guidelines. All chemicals and reagents used in this study were purchased from Sigma (St. Louis, MO, United States) unless otherwise stated. The ferroptosis inhibitor ferrostatin-1 (Fer-1) was obtained from Selleck Chemicals (S7243, TX, United States).
Male Institute of Cancer Research mice (ICR, 8 weeks old) were randomly divided into different groups, including control group, TAA group, Fer-1 pre-treatment and TAA group, or TAA with DFO group (n = 6-9 mice per group). According to previous studies (França et al., 2019; Han et al., 2019; Liu et al., 2021), TAA was injected (i.p., 250 mg/kg/day) for 3 consecutive days to induce acute liver injury. Fer-1 was administered (i.p., 2.5 μM/kg/day) for 3 days before TAA treatment (Wang et al., 2017), and DFO (200 mg/kg/day, i. p) was injected with TAA for 3 days (Mansour 2000; Wu et al., 2011). TAA, DFO and Fer-1 were all dissolved in normal saline. The control group was injected with equal volumes of normal saline solution, using the same injection schedule. To compare the effects of normal-iron diet (NID) and high-iron diet (HID) on TAA-induced ALI model, we next fed the iron content of diets according to previous protocol (Wang et al., 2017). Briefly, male ICR mice were fed with either a standard AIN-76A diet (50 mg Fe/kg; Research Diets, Inc., New Brunswick, United States) or high-iron AIN-76A diet (8.3 g Fe/kg; Research Diets, Inc.) for 14 days. TAA was administered (i.p., 250 mg/kg/day) for 3 consecutive days after HID intervention. The mice were finally anaesthetized with 1% pentobarbital sodium (40 mg/kg body weight, i. p.) and received myocardial perfusion using phosphate-buffered saline (PBS), after which the liver tissues were collected for measurements.
Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin, and glutathione (GSH) were measured following the protocol of an ALT kit (Cat# C009-1-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China), AST kit (Cat# C010-1-1, Nanjing Jiancheng Bioengineering Institute), albumin kit (Cat# A028-1-1, Nanjing Jiancheng Bioengineering Institute), and GSH assay kit (Cat# A006-1-1, Nanjing Jiancheng Bioengineering Institute), respectively. The optical densities were measured with a Synergy 2 multi-mode microplate reader (Agilent Technologies, Inc.). Each sample was measured three times, and the average absorbance values were calculated for every sample. The concentrations in the samples were determined using the standard curves.
After paraformaldehyde fixation, the hepatic tissue was embedded in paraffin and sectioned with at 5-μm thickness. Liver sections were then mounted onto slides and taken for haematoxylin-eosin (H&E) staining. Briefly, slides were deparaffinized in xylene (5 min, twice) and hydrated by passing through decreasing concentration of alcohol baths (100, 90, 80, 70%) and water, followed by haematoxylin staining for 2 min. The sections were then washed in running tap water until sections “blue” for 5 min. The sections were then rinsed with distilled water, rinsed with 0.1% hydrochloric acid in 50% ethanol, rinsed with tap water for 15 min, stained with eosin for 1 min, and rinsed again with tap water. Next, the slides were dehydrated with 95 and 100% ethanol successively followed by xylene (5 min, twice) for clearing. Finally, sections were mounted with neutral balata and covered with a cover-slip.
Paraformaldehyde-fixed paraffin-embedded tissues were sectioned into 20 μm sections and stored at room temperature. Slides were blocked for nonspecific binding using a solution block, and endogenous peroxidase activity was quenched. 3,3′-Diaminobenzidine (DAB)-enhanced Perls’ staining was performed to observe iron accumulation in paraffin-embedded liver sections following the manufacturer’s instructions. Briefly, sections of liver tissue were washed with PBS and incubated in freshly prepared Perls’ solution (1% potassium ferricyanide in 0.1-M hydrochloric acid buffer) for 1 h, followed by a 15 min incubation in DAB. Slides were immersed for 1 hour and then stained with DAB. All slides were counterstained with haematoxylin and visualized under a DM4000B microscope (Leica, Germany) at a final magnification of ×200. The data were collected from three fields of view per mice, and analyzed semi-quantitatively with ImageJ software. As described previously (Moos and Møllgård 1993), the quantitative analysis of Perls’ staining was used to reflect iron deposition levels. Integrated Density of different treatment groups was finally normalized to control group. The analysis was done by an investigator masked to experimental group.
Proteins were collected and homogenized in RIPA lysis buffer (Beyotime, PRC) and sonicated with a sonifier. The BCA (Pierce, Rockford) detection method was employed to detect protein content. A sample containing 30 μg protein was loaded and run in each well of SDS–PAGE gels. The membranes were incubated with primary antibodies (1:1,000) against TfR1 (Cat. 13–6800, Thermo Fisher Scientific, MA, United States), Fpn1 (Cat. NBP1-21502, Novus, Centennial, CO, United States), ferritin-L (Cat. 10727-1-AP, Proteintech, Chicago, United States), xCT (Cat. 26864-1-AP, Proteintech) and GPx4 (Cat. ab125066, Abcam, Cambridge, United Kingdom) at 4°C overnight. Blots were then incubated with goat anti-rabbit or anti-mouse IRDye 800 CW secondary antibody at a 1:10,000 dilution (Li-Cor, Lincoln Co., Ltd., United States) at room temperature for 1 h. GAPDH (MAB374, Merck, State of New Jersey, United States) monoclonal antibody (1:10,000) was used as a loading control. The band densities of the specific blots were scanned via Odyssey CLx infrared imaging system (LI-COR Biosciences) and analyzed with ImageJ software. Results were shown as the optical density ratio normalized to GAPDH.
Iron in the liver was detected using the tissue iron measurement method as follows. Liver tissue (0.1 g) was obtained, and then, 1 ml of tissue digestive liquid (3 M hydrochloric acid and 0.61 M trichloroacetic acid) was added to the liver for digestion at 65 C for 60 h to ensure complete digestion of the liver tissue. After digestion, the volume of the tissue digestion liquid was fixed to 1.5 ml, and the digestion mixture was centrifuged at 10,000 g for 10 min. The supernatant was removed and collected for detection. Iron developing color working solution was freshly prepared for each experiment, and consisted of 100 mg disodium-4,7-diphenyl-1,10-phenanthroline disulfonate (Cat. 146617, sigma, State of New Jersey, United States), 60 ml ddH2O, 1.429 ml 70% thioglycollic acid (S8750, sigma, State of New Jersey, United States), supplement with ddH2O to 100 ml. A 96-well plate was further used for detection, and 200 ml iron developing colour working solution was added to each well. Then, 10 ml ddH2O, 10 ml tissue digestion solution or 10 ml iron standard solution (500 μg/dl) was added to different wells. Finally, 10 ml of sample was added to the sample well, fully mixed, and incubated at room temperature for 10 min. Absorbance was measured at 535 nm. The method of iron quantification was according to the formula, iron content (Ug/g Tissue wet weight) = OD/tissue weight × (1.5–0.25 × tissue weight) × (1/iron standard 500 OD × 4.77).
Statistical analysis was performed with GraphPad Prism 8.0 software. All the data in this study are shown as the mean ± SEM. Data of two groups were analyzed for statistical significance with Student’s t-test (non-directional). Variations between the means in multiple groups were analysed via one-way analysis of variance, and then, Tukey’s post-hoc test was performed for multiple comparisons. A probability value of p < 0.05 was viewed as statistically significant.
TAA induced liver dysfunction, suppressed anti-ferroptosis-related protein expression and enhanced iron-related protein expression in the liver.
Injection of TAA induced prominent inflammation and vacuolar degeneration in the liver within 3 days compared with the control group (Figure 1A). TAA induced obvious liver dysfunction, which was reflected by increased plasma ALT and AST levels in the TAA model (Figures 1B,C, p < 0.001). In addition, TAA significantly reduced anti-ferroptosis-related protein (xCT) expression but not GPX4 expression compared to the control (Figures 1D–F). In addition, TAA induced a significant increase in iron-related proteins, including TfR1, Fpn and Ft-L expression, compared to the control (Figures 1G–J, p < 0.05 or p < 0.01).
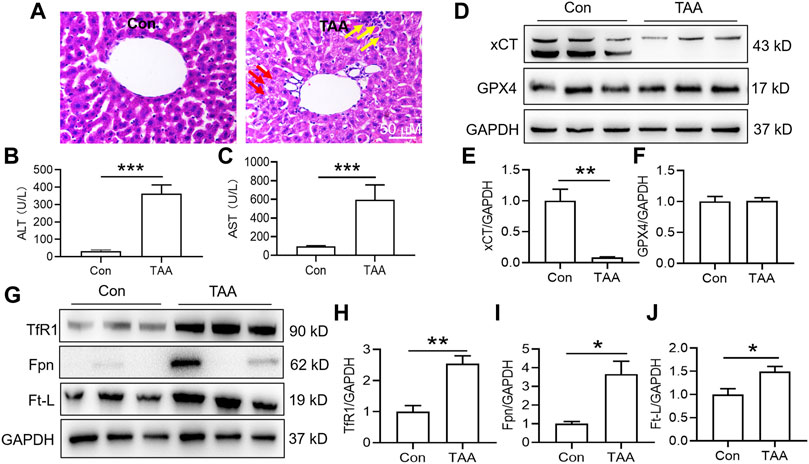
FIGURE 1. TAA induced liver dysfunction, suppressed anti-ferroptosis-related protein expression and enhanced iron-related protein expression in the liver. (A) Liver injury was assessed by H&E staining and histological examination in mice with or without TAA administration for 3 days (bar = 50 μm). TAA induced prominent inflammation (yellow arrow) and vacuolar degeneration (red arrow) in the liver within 3 days compared with the control group. (B) Plasma ALT and (C) AST levels in control and TAA-injected mice. (D) Western blotting analysis of xCT and GPX4 expression in the livers of mice with or without TAA treatment. (E–F) Relative protein expression levels of xCT and GPX4. (G) Western blotting analysis of TfR1, Fpn and Ft-L expression in the livers of mice with or without TAA treatment. (H–J) Relative protein expression levels of TfR1, Fpn and Ft-L. All data are presented as the mean ± SEM (n = 6-9 mice per group); *p < 0.05, **p < 0.01 and ***p < 0.001 versus the indicated group.
Both Fer-1 and DFO suppressed TAA-induced liver dysfunction but did not affect anti-ferroptosis-related protein expression in the liver.
In comparison with the control, injection of TAA induced a profound increase in plasma levels of ALT and AST concomitant with a marked increase in the plasma LDH level. Pre-administration of Fer-1 for 3 days significantly decreased TAA-induced alterations in the plasma ALT, AST and LDH levels compared with the TAA group (Figures 2A–C, p < 0.001). TAA significantly reduced xCT but not GPX4 expression in the livers of mice. However, pre-administration of Fer-1 did not affect xCT or GPX4 expression in the liver compared to TAA-treated mice (Figures 2D–F).
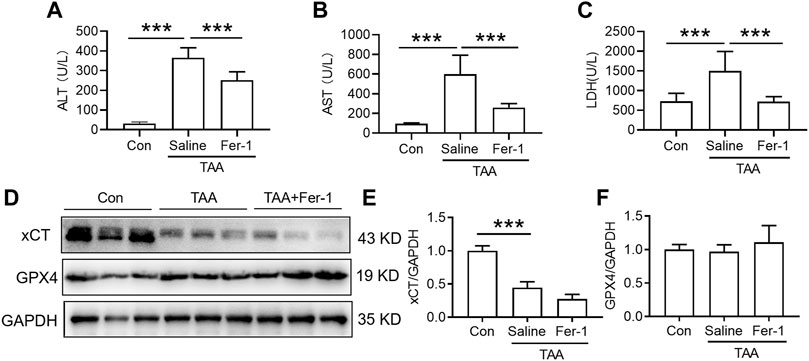
FIGURE 2. Effects of Fer-1 on liver function and anti-ferroptosis-related protein expression in TAA-induced ALI mice. (A) Plasma ALT, (B) AST and (C) LDH levels in the control and TAA model mice with or without Fer-1 pre-treatment. (D) Western blotting analysis of xCT and GPX4 expression in the liver of control and ALI model mice with or without Fer-1 pre-treatment. (E–F) Relative protein expression levels of xCT and GPX4 expression in (D). All data are presented as the mean ± SEM (n = 6-9 mice per group); ***p < 0.001 versus the indicated group.
Administration of DFO for 3 days with TAA treatment significantly attenuated the TAA-induced increase in plasma ALT, AST and LDH (Figures 3A–C, p < 0.001). In addition, administration of DFO did not alter xCT or GPX4 expression induced by TAA in the liver compared to TAA-treated mice (Figures 3D–F).
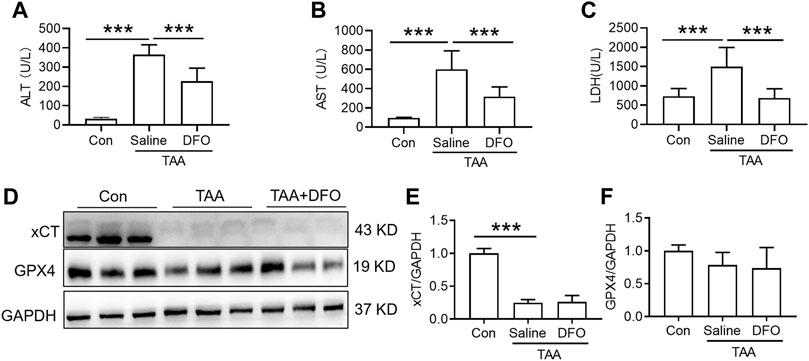
FIGURE 3. Effects of DFO on liver function and anti-ferroptosis-related protein expression in TAA-induced ALI mice. (A) Plasma ALT, (B) AST and (C) LDH levels in the control and TAA model mice with or without DFO treatment. (D) Western blotting analysis of xCT and GPX4 expression in the livers of the control and ALI model mice with or without DFO treatment. (E–F) Relative protein expression of xCT and GPX4 in (D). All data are presented as the mean ± SEM (n = 6-9 mice per group); ***p < 0.001 versus the indicated group.
TAA induced a significant increase in iron uptake, iron export and iron storage in the liver, which were reflected by increased TfR1, Fpn and Ft-L expression, respectively (Figures 4A–D). In addition, iron staining of the liver showed that TAA induced significant iron accumulation (Figures 4E,F, p < 0.001), especially positive centrolobular deposition of iron and liver nonheme iron content (Figure 4G, p < 0.05). However, Fer-1 pre-treatment reduced the TAA-induced increase in Fpn and Ft-L but did not affect TfR1 expression. In addition, compared to TAA alone, Fer-1 pre-treatment decreased the TAA-induced iron levels and increased iron accumulation in the liver (Figure 4).
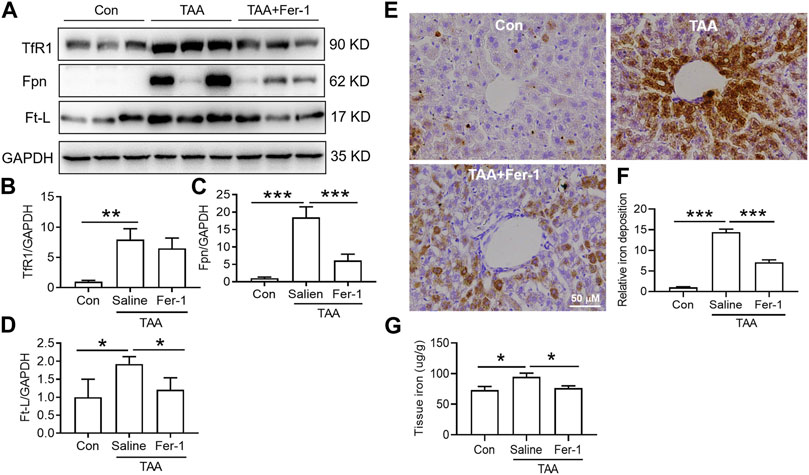
FIGURE 4. Effects of Fer-1 on iron-related protein expression and iron content in TAA-induced ALI mice. (A) Western blotting analysis of TfR1, Fpn and Ft-L expression in the livers of mice after different treatments. (B–D) Relative protein expression of TfR1, Fpn and Ft-L in (A) (E) Fer-1 suppressed the increase in positive centrolobular iron deposition induced by TAA. (F) Semiquantitative iron levels in the mouse livers described in (E) (G) Iron content detection in the livers of mice with different treatments. All data are presented as the mean ± SEM (n = 6-9 mice per group); *p < 0.05, **p < 0.01, and ***p < 0.001 versus the indicated group.
TAA similarly upregulated TfR1, Fpn and Ft-L expression in the liver (Figures 5A–D, p < 0.01 or p < 0.05), and also induced significant iron accumulation (Figures 5E,F, p < 0.001) and increased liver iron content (Figure 5G, p < 0.05). However, DFO pre-treatment before TAA administration significantly decreased Fpn and Ft-L but did not affect TfR1 expression (Figures 5A–D). Additionally, DFO decreased the TAA-induced increase in the iron level and iron accumulation in the liver (Figures 5E–G).
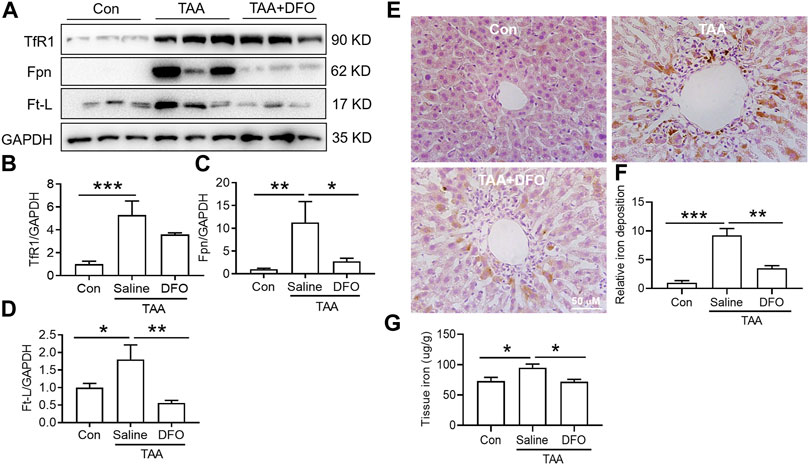
FIGURE 5. Effects of DFO on iron-related protein expression and iron content in TAA-induced ALI mice. (A) Western blotting analysis of TfR1, Fpn and Ft-L expression in the livers of mice with different treatments. (B–D) Relative protein exprssion of TfR1, Fpn and Ft-L in (A) (E) DFO suppressed the increase in positive centrolobular iron deposition induced by TAA. (F) Semiquantitative iron levels of the mice liver in (E) (G) Iron content detection in the livers of mice with different treatments. All data are presented as the mean ± SEM (n = 6-9 mice per group); *p < 0.05, **p < 0.01, and ***p < 0.001 versus the indicated group.
Fer-1 reversed TAA-induced liver dysfunction by reducing iron but not activating anti-ferroptosis-related protein expression in the liver under iron overload conditions.
Compared with NID, HID did not increase the levels of hepatic damage biomarkers (ALT, AST and LDH), and this diet was not inducing the same acute liver damage as TAA (Figures 6A–C). In addition, HID downregulated the protein expression of TfR1 (Figures 6D,E, p < 0.05), but did not influence the expression of Fpn compared to NID (Figures 6D,F). Unlike TfR1 level, Ft-L expression was significantly enhanced by HID in the liver (Figure 6G, p < 0.01). HID-fed mice had developed a significant iron increase than NID mice (Figures 6H–J, p < 0.01).
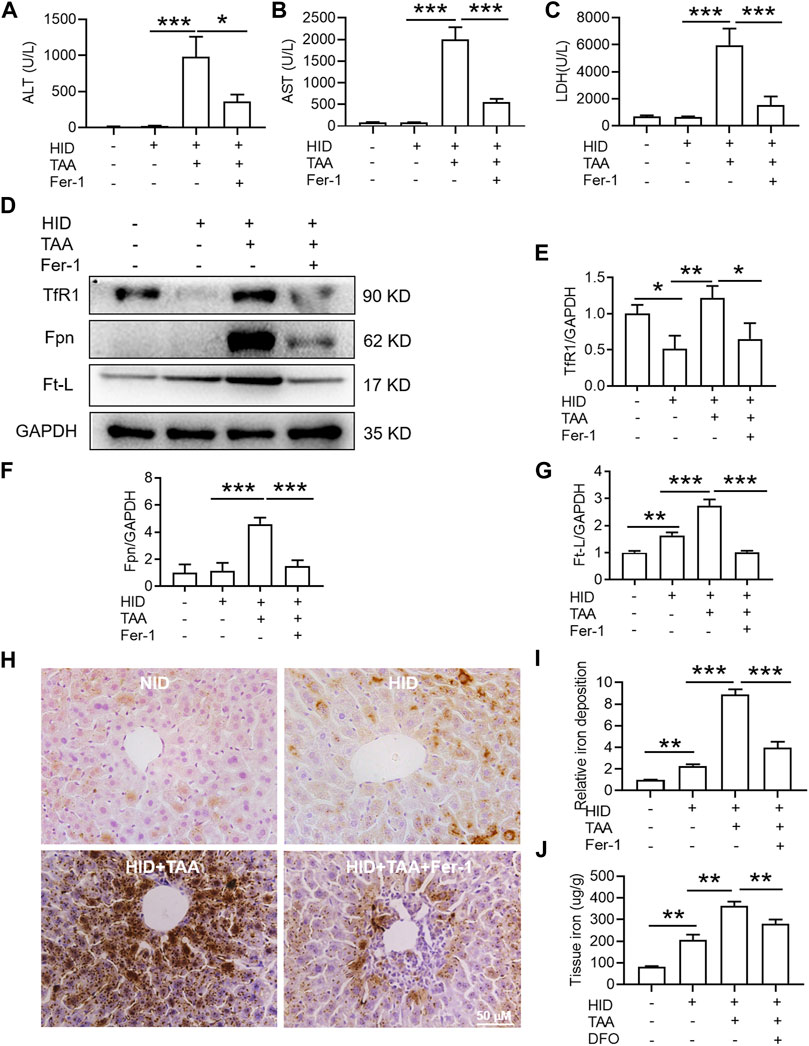
FIGURE 6. Effects of Fer-1 on liver function, hepatic iron content and anti-ferroptosis-related protein expression in the liver under iron overload conditions. (A) Plasma ALT (B) AST and (C) LDH levels in the control and TAA model mice with or without Fer-1 pre-treatment under high iron diet (HID) conditions. (D) Western blotting analysis of TfR1, Fpn and Ft-L expression in the livers of mice with different treatments under HID conditions. (E–G) Relative protein expression of TfR1, Fpn and Ft-L in (D) (H) Fer-1 inhibited the increase in positive centrolobular iron deposition induced by TAA under HID conditions in the liver. (I) Semiquantitative analysis of iron deposition levels in (H) (J) Iron content detection in the livers of mice with different treatments. All data are presented as the mean ± SEM (n = 6-9 mice per group); *p < 0.05, **p < 0.01, and ***p < 0.001 versus the indicated group.
Consistent with the effects of TAA under NID conditions, TAA induced profound liver dysfunction, which was shown by an increase in plasma ALT, AST and LDH levels under iron overload conditions (Figures 6A–C). Pre-administration of Fer-1 for 3 days before TAA injection significantly attenuated the TAA-induced increase in plasma ALT, AST and LDH compared with the TAA treatment group under high iron conditions (Figures 6A–C). Similar to TAA treatment under NID conditions, TAA induced a significant enhancement in TfR1, Fpn and Ft-L expression in the liver (Figures 6D–G). Moreover, compared to the HID group, iron staining results suggested that TAA induced significant iron accumulation under high iron conditions (Figures 6H,I) and increased liver iron detection (Figure 6J). However, pre-treatment of Fer-1 reversed the increases in the expression levels of the above three proteins and iron accumulation in the liver under HID conditions (Figures 6D–J).
TAA is a potent toxicant that causes oxidative stress and further induces liver damage, which is highly similar to human acute liver damage (Zargar et al., 2019). The TAA-induced liver injury model has been widely studied for its biochemical and histological effects in animals and is usually used to establish liver injury models (Rahman and Hodgson 2003). However, administration of TAA to mice causes either chronic/acute liver failure or cirrhosis depending on the dose and duration of TAA exposure (Shapiro et al., 2006). In previous studies, high-dosage TAA is known to induce lipid peroxidation and oxidative stress, and the pathological effects are mainly limited to ALI rather than causing direct damage to other organs (Muller et al., 1991; Mladenovic et al., 2012). Thus, according to previous studies (França et al., 2019; Han et al., 2019; Liu et al., 2021), we administered 250 mg/kg/day TAA to mice via intraperitoneal injection for 3 days to establish the ALI model in our present study.
Although TAA-induced ALI was primarily provoked by oxidative stress (Zhan et al., 2019), whether it is partially an iron-dependent process is still uncertain. To investigate whether iron disorder is involved in liver dysfunction, we evaluated liver function and liver iron content in TAA-induced ALI mice. Because the liver is the major extraerythrocyte storage organ for iron and most of the iron within the liver is stored in Ft-L, which is often used to indicate iron levels (Ishizaka et al., 2005; Galaris and Pantopoulos 2008). TAA not only significantly induced iron mobilization, which was reflected as increased transferrin receptor 1 (TfR1), but also increased iron storage, which was reflected as increased ferritin-L (Ft-L) expression in the liver. These results were consistent with those of other studies suggesting that TAA induces iron accumulation in the liver of ALI mice (Ackerman et al., 2015). However, we also noticed that TAA induced the increase of ferroportin (Fpn) expression in the liver, which usually facilitate iron efflux to lower tissue iron. Actually, iron is tightly controlled at cell and systemic levels to prevent both deficiency and overload under physiological conditions. Iron regulatory proteins post-transcriptionally control genes encoding proteins that modulate iron transport, use and storage and are themselves regulated by iron (Gao et al., 2019). Thus, the elevated Fpn may represent an endogenous effort to maintain iron homoeostasis (Anderson and Frazer 2017). It is also understandable that both Fer-1 and DFO decreased iron content, while reducing Fpn in the liver after TAA exposure. Intracellular iron can be stored in the protein shell of ferritin (Ft) as a crystalline core of ferric (Fe3+) ions, and the iron must first be released from the core to catalyze oxidative reactions (Skaper 2019). Thus, Ft is able to restrict the availability of iron to participate in redox active iron species in the cytosol. However, more ferritin subunits are synthesized using translational and transcriptional mechanisms in response to increased cell iron (Badu-Boateng and Naftalin 2019). Increased intracellular ferritin may occur in response to oxidative stress provoked by accumulated iron, which could be result of various stressors such as UV light, increased temperature, and inflammation (Vile and Tyrrell 1993; Tisma et al., 2009). Consistent with previous findings, we found that TAA significantly induced iron increase in the liver and also promoted Ft-L expression.
Although ferroptosis was reported to be involved in the pathogenesis of many injury models (Skouta et al., 2014; Liu et al., 2020), it is still unclear whether ferroptosis is involved in the TAA-induced ALI model. In addition, iron accumulation is considered to be the main cause of ferroptosis initiation (Li et al., 2020). This prompted us to wonder whether TAA induces iron accumulation and ultimately initiates ferroptosis in the liver. The synthetic antioxidant Fer-1 was reported to act as a ferroptosis inhibitor via a reductive mechanism to prevent damage to membrane lipids, thereby inhibiting ferroptosis (Dixon et al., 2012). The iron chelator DFO is widely used to reduce iron in tissue after injection or oral administration (Buss et al., 2003). In the present study, we introduced Fer-1 and DFO to investigate whether ferroptosis occurred because these agents can decrease iron content to achieve an anti-ferroptotic effect in the TAA-induced ALI model. We found that both Fer-1 and DFO rescued liver dysfunction and inhibited iron content and accumulation in the liver. However, it was previously reported that Fer-1 can attenuate oxidative, iron-dependent cancer cell death by blocking cystine import and glutathione production (Skouta et al., 2014). Our data demonstrated that Fer-1 and DFO did not play anti-ferroptotic roles by affecting cystine import or glutathione production in a TAA-induced ALI model. This result may suggest that Fer-1 plays an anti-ferroptotic role similar to DFO in the liver in the ALI model and that the mechanism involves chelation of hepatic iron. Moreover, a recent study demonstrated that Fer-1, in the presence of ferrous iron, produces the most relevant anti-ferroptotic effect by forming a complex with iron (Miotto et al., 2020).
Ferroptosis is a multi-step regulated cell death that is characterized by excessive iron accumulation and lipid peroxidation (Yu et al., 2020). Lipid peroxide accumulation is mainly through the xCT and GPX4-dependent mechanisms (Kim et al., 2021). The process of ferroptosis is also dependent on intracellular iron because the accumulation of iron acts as a catalyst for converting peroxides into free radicals (Kim et al., 2021). Oxidative degradation of lipids occurs when there is depletion of the antioxidant glutathione and a loss of activity of the lipid repair enzyme GPX4. Lipid peroxidation then leads to cell membrane denaturation (Cao and Dixon 2016). The results were also consistent with other studies, which confirmed that ferroptosis can be induced by a loss of activity of xCT and suppression of GPX4, followed by accumulation of lipid reactive oxygen species (ROS) (Ma et al., 2008; Yang and Stockwell 2016; Latunde-Dada 2017). However, both anti-ferroptotic proteins xCT and GPX4 were not modulated by Fer-1 investigated. It was concluded that the outcome of TAA-induced acute liver injury may depend on the level of hepatic iron concentration and iron overload may exacerbate the injury. However, it’s not entirely clear that whether Fer-1 or DFO block iron mobilisation into the liver instead. Thus, more study is necessary to investigate their effects on the expression of molecules that play key roles in iron transport, use and storage.
In addition, the study revealed HID-fed mice had developed a significant iron increase. In addition, HID did not increase the protein expression of TfR1 and Fpn in the same magnitude of TAA. Indeed, HID even downregulated TfR1 compared to NID. Similar with previous research (Weiss et al., 2018), the decreased TfR1 after HID may also represent endogenous feedback to maintain the iron balance. Unlike TfR1 level, Ft-L expression was significantly enhanced by HDI in the liver, which directly indicated increased iron levels. We further found that HID did not increase the levels of hepatic damage biomarkers (ALT, AST and LDH), and this diet was not inducing the same acute liver damage as TAA. However, previous study has revealed that an increase in hepatic iron concentration might exacerbate TAA-induced live injury (Ackerman et al., 2015). We confirmed that TAA aggravated the damage to the liver with iron overload. We further revealed that Fer-1 still had an anti-ferroptotic effect on TAA-induced liver ferroptosis by reducing the iron content in high iron-fed ALI mice. All these results provide new evidence of the anti-ferroptotic effect of Fer-1 in the TAA-induced ALI model, the mechanism of which is mediated by the ability of Fer-1 to reduce iron content and iron accumulation in the liver (Figure 7).
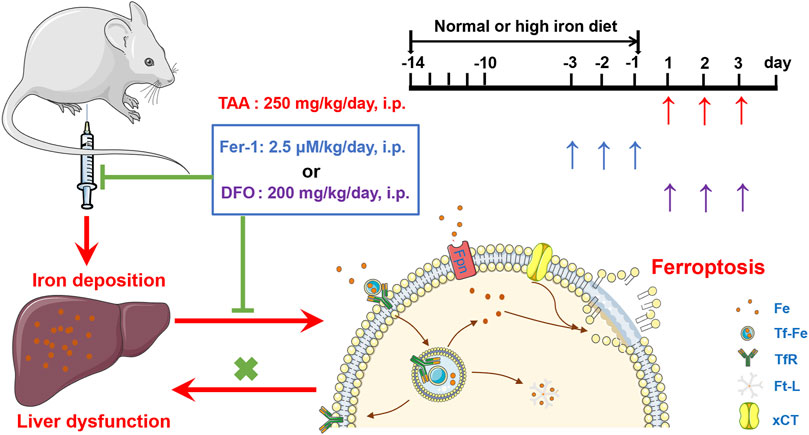
FIGURE 7. Proposed mechanisms of the ferroptosis inhibitor Fer-1 against the TAA-induced ALI mice model.
Taken together, these findings indicate a role of TAA-induced iron deposition and ferroptosis in the pathogenesis of ALI model. Ferroptosis inhibitor Fer-1 plays a role in rescuing liver injury and dysfunction by reducing liver iron levels and inhibiting TAA-induced ferroptosis in the liver. Similar to DFO, Fer-1 might be a useful reagent to reverse liver dysfunction under iron overload conditions.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Research Ethics Committee of the Institute of Nantong University.
QL and GW conceived, organized, and supervised the work; HJ and WY, performed the experiments; XZ and ML contributed to the analysis of data; QL and GW prepared, wrote and revised the manuscript.
This work was funded by National Natural Science Foundation of China (81873924, 82171190); Open Cooperation Program from Key Laboratory of Extreme Environmental Medicine, Ministry of Education (KL2019GY011); High-level Innovation and Entrepreneurship Talents Introduction Program of Jiangsu Province of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ackerman, Z., Pappo, O., Link, G., Glazer, M., and Grozovski, M. (2015). Liver Toxicity of Thioacetamide Is Increased by Hepatocellular Iron Overload. Biol. Trace Elem. Res. 163 (1-2), 169–176. doi:10.1007/s12011-014-0110-9
Anderson, G. J., and Frazer, D. M. (2017). Current Understanding of Iron Homeostasis. Am. J. Clin. Nutr. 106 (Suppl. 6), 1559s–1566s. doi:10.3945/ajcn.117.155804
Badu-Boateng, C., and Naftalin, R. J. (2019). Ascorbate and Ferritin Interactions: Consequences for Iron Release In Vitro and In Vivo and Implications for Inflammation. Free Radic. Biol. Med. 133, 75–87. doi:10.1016/j.freeradbiomed.2018.09.041
Bruck, R., Ashkenazi, M., Weiss, S., Goldiner, I., Shapiro, H., Aeed, H., et al. (2007). Prevention of Liver Cirrhosis in Rats by Curcumin. Liver Int. 27 (3), 373–383. doi:10.1111/j.1478-3231.2007.01453.x
Buss, J. L., Torti, F. M., and Torti, S. V. (2003). The Role of Iron Chelation in Cancer Therapy. Curr. Med. Chem. 10 (12), 1021–1034. doi:10.2174/0929867033457638
Cao, J. Y., and Dixon, S. J. (2016). Mechanisms of Ferroptosis. Cell Mol Life Sci 73 (11-12), 2195–2209. doi:10.1007/s00018-016-2194-1
Chu, C. J., Wang, S. S., Lee, F. Y., Chang, F. Y., Lin, H. C., Hou, M. C., et al. (2001). Detrimental Effects of Nitric Oxide Inhibition on Hepatic Encephalopathy in Rats with Thioacetamide-Induced Fulminant Hepatic Failure. Eur. J. Clin. Invest. 31 (2), 156–163. doi:10.1046/j.1365-2362.2001.00775.x
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an Iron-dependent Form of Nonapoptotic Cell Death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
França, M. E. R., Ramos, R. K. L. G., Oliveira, W. H., Duarte-Silva, E., Araújo, S. M. R., Lós, D. B., et al. (2019). Tadalafil Restores Long-Term Memory and Synaptic Plasticity in Mice with Hepatic Encephalopathy. Toxicol. Appl. Pharmacol. 379, 114673. doi:10.1016/j.taap.2019.114673
Galaris, D., and Pantopoulos, K. (2008). Oxidative Stress and Iron Homeostasis: Mechanistic and Health Aspects. Crit. Rev. Clin. Lab. Sci. 45 (1), 1–23. doi:10.1080/10408360701713104
Ganz, T. (2011). Hepcidin and Iron Regulation, 10 Years Later. Blood 117 (17), 4425–4433. doi:10.1182/blood-2011-01-258467
Gao, G., Li, J., Zhang, Y., and Chang, Y. Z. (2019). Cellular Iron Metabolism and Regulation. Adv. Exp. Med. Biol. 1173, 21–32. doi:10.1007/978-981-13-9589-5_2
Grek, A., and Arasi, L. (2016). Acute Liver Failure. AACN Adv. Crit. Care 27 (4), 420–429. doi:10.4037/aacnacc2016324
Hadian, K., and Stockwell, B. R. (2020). SnapShot: Ferroptosis. Cell 181 (5), 1188–e1. doi:10.1016/j.cell.2020.04.039
Han, J. H., Kim, O. H., Lee, S. C., Kim, K. H., Park, J. H., Lee, J. I., et al. (2019). A Novel Hepatic Anti-fibrotic Strategy Utilizing the Secretome Released from Etanercept-Synthesizing Adipose-Derived Stem Cells. Int. J. Mol. Sci. 20 (24), 6302. doi:10.3390/ijms20246302
He, Y. J., Liu, X. Y., Xing, L., Wan, X., Chang, X., and Jiang, H. L. (2020). Fenton Reaction-independent Ferroptosis Therapy via Glutathione and Iron Redox Couple Sequentially Triggered Lipid Peroxide Generator. Biomaterials 241, 119911. doi:10.1016/j.biomaterials.2020.119911
Hentze, M. W., Muckenthaler, M. U., Galy, B., and Camaschella, C. (2010). Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 142 (1), 24–38. doi:10.1016/j.cell.2010.06.028
Ishizaka, N., Saito, K., Noiri, E., Sata, M., Ikeda, H., Ohno, A., et al. (2005). Administration of ANG II Induces Iron Deposition and Upregulation of TGF-Beta1 mRNA in the Rat Liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288 (4), R1063–R1070. doi:10.1152/ajpregu.00281.2004
Kim, S., Kang, S. W., Joo, J., Han, S. H., Shin, H., Nam, B. Y., et al. (2021). Characterization of Ferroptosis in Kidney Tubular Cell Death under Diabetic Conditions. Cell Death Dis 12 (2), 160. doi:10.1038/s41419-021-03452-x
Latunde-Dada, G. O. (2017). Ferroptosis: Role of Lipid Peroxidation, Iron and Ferritinophagy. Biochim. Biophys. Acta Gen. Subj 1861 (8), 1893–1900. doi:10.1016/j.bbagen.2017.05.019
Li, J., Cao, F., Yin, H. L., Huang, Z. J., Lin, Z. T., Mao, N., et al. (2020). Ferroptosis: Past, Present and Future. Cel Death Dis 11 (2), 88. doi:10.1038/s41419-020-2298-2
Liu, P., Feng, Y., Li, H., Chen, X., Wang, G., Xu, S., et al. (2020). Ferrostatin-1 Alleviates Lipopolysaccharide-Induced Acute Lung Injury via Inhibiting Ferroptosis. Cell Mol Biol Lett 25, 10. doi:10.1186/s11658-020-00205-0
Liu, R., Cui, J., Sun, Y., Xu, W., Wang, Z., Wu, M., et al. (2021). Autophagy Deficiency Promotes M1 Macrophage Polarization to Exacerbate Acute Liver Injury via ATG5 Repression during Aging. Cell Death Discov 7 (1), 397. doi:10.1038/s41420-021-00797-2
Ma, J., Chen, J. Y., Idowu, M., and Nyokong, T. (2008). Generation of Singlet Oxygen via the Composites of Water-Soluble Thiol-Capped CdTe Quantum Dots-Sulfonated Aluminum Phthalocyanines. J. Phys. Chem. B 112 (15), 4465–4469. doi:10.1021/jp711537j
Mansour, M. A. (2000). Protective Effects of Thymoquinone and Desferrioxamine against Hepatotoxicity of Carbon Tetrachloride in Mice. Life Sci. 66 (26), 2583–2591. doi:10.1016/s0024-3205(00)00592-0
Miotto, G., Rossetto, M., Di Paolo, M. L., Orian, L., Venerando, R., Roveri, A., et al. (2020). Insight into the Mechanism of Ferroptosis Inhibition by Ferrostatin-1. Redox Biol. 28, 101328. doi:10.1016/j.redox.2019.101328
Mladenovic, D., Krstic, D., Colovic, M., Radosavljevic, T., Rasic-Markovic, A., Hrncic, D., et al. (2012). Different Sensitivity of Various Brain Structures to Thioacetamide-Induced Lipid Peroxidation. Mc 8 (1), 52–58. doi:10.2174/157340612799278603
Moos, T., and Møllgård, K. (1993). A Sensitive post-DAB Enhancement Technique for Demonstration of Iron in the central Nervous System. Histochemistry 99 (6), 471–475. doi:10.1007/bf00274100
Müller, D., Sommer, M., Kretzschmar, M., Zimmermann, T., Buko, V. U., Lukivskaya, O., et al. (1991). Lipid Peroxidation in Thioacetamide-Induced Macronodular Rat Liver Cirrhosis. Arch. Toxicol. 65 (3), 199–203. doi:10.1007/BF02307309
Nicolas, G., Bennoun, M., Devaux, I., Beaumont, C., Grandchamp, B., Kahn, A., et al. (2001). Lack of Hepcidin Gene Expression and Severe Tissue Iron Overload in Upstream Stimulatory Factor 2 (USF2) Knockout Mice. Proc. Natl. Acad. Sci. U S A. 98 (15), 8780–8785. doi:10.1073/pnas.151179498
Rahman, T. M., and Hodgson, H. J. (2003). The Effects of Early and Late Administration of Inhibitors of Inducible Nitric Oxide Synthase in a Thioacetamide-Induced Model of Acute Hepatic Failure in the Rat. J. Hepatol. 38 (5), 583–590. doi:10.1016/s0168-8278(03)00050-3
Shapiro, H., Ashkenazi, M., Weizman, N., Shahmurov, M., Aeed, H., and Bruck, R. (2006). Curcumin Ameliorates Acute Thioacetamide-Induced Hepatotoxicity. J. Gastroenterol. Hepatol. 21 (2), 358–366. doi:10.1111/j.1440-1746.2005.03984.x
Sikorska, K., Bernat, A., and Wroblewska, A. (2016). Molecular Pathogenesis and Clinical Consequences of Iron Overload in Liver Cirrhosis. Hepatobiliary Pancreat. Dis. Int. 15 (5), 461–479. doi:10.1016/s1499-3872(16)60135-2
Skaper, S. D. (2019). Oligodendrocyte Precursor Cells as a Therapeutic Target for Demyelinating Diseases. Prog. Brain Res. 245, 119–144. doi:10.1016/bs.pbr.2019.03.013
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J. Am. Chem. Soc. 136 (12), 4551–4556. doi:10.1021/ja411006a
Thawley, V. (2017). Acute Liver Injury and Failure. Vet. Clin. North. Am. Small Anim. Pract. 47 (3), 617–630. doi:10.1016/j.cvsm.2016.11.010
Tisma, V. S., Basta-Juzbasic, A., Jaganjac, M., Brcic, L., Dobric, I., Lipozencic, J., et al. (2009). Oxidative Stress and Ferritin Expression in the Skin of Patients with Rosacea. J. Am. Acad. Dermatol. 60 (2), 270–276. doi:10.1016/j.jaad.2008.10.014
Vile, G. F., and Tyrrell, R. M. (1993). Oxidative Stress Resulting from Ultraviolet A Irradiation of Human Skin Fibroblasts Leads to a Heme Oxygenase-dependent Increase in Ferritin. J. Biol. Chem. 268 (20), 14678–14681. doi:10.1016/s0021-9258(18)82386-9
Wang, H., An, P., Xie, E., Wu, Q., Fang, X., Gao, H., et al. (2017). Characterization of Ferroptosis in Murine Models of Hemochromatosis. Hepatology 66 (2), 449–465. doi:10.1002/hep.29117
Weiss, A., Spektor, L., Cohen, L. A., Magid Gold, I., Zhang, D. L., Truman-Rosentsvit, M., et al. (2018). Orchestrated Regulation of Iron Trafficking Proteins in the Kidney during Iron Overload Facilitates Systemic Iron Retention. PLoS One 13 (10), e0204471. doi:10.1371/journal.pone.0204471
Wu, H., Wu, T., Xu, X., Wang, J., and Wang, J. (2011). Iron Toxicity in Mice with Collagenase-Induced Intracerebral Hemorrhage. J. Cereb. Blood Flow Metab. 31 (5), 1243–1250. doi:10.1038/jcbfm.2010.209
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: Process and Function. Cell Death Differ 23 (3), 369–379. doi:10.1038/cdd.2015.158
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: Death by Lipid Peroxidation. Trends Cel Biol 26 (3), 165–176. doi:10.1016/j.tcb.2015.10.014
Yu, Y., Jiang, L., Wang, H., Shen, Z., Cheng, Q., Zhang, P., et al. (2020). Hepatic Transferrin Plays a Role in Systemic Iron Homeostasis and Liver Ferroptosis. Blood 136 (6), 726–739. doi:10.1182/blood.2019002907
Zargar, S., Alonazi, M., Rizwana, H., and Wani, T. A. (20192019). Resveratrol Reverses Thioacetamide-Induced Renal Assault with Respect to Oxidative Stress, Renal Function, DNA Damage, and Cytokine Release in Wistar Rats. Oxid Med. Cel Longev 2019, 1702959. doi:10.1155/2019/1702959
Zhan, F., Zhao, G., Li, X., Yang, S., Yang, W., Zhou, S., et al. (2019). Inositol-requiring Enzyme 1 Alpha Endoribonuclease Specific Inhibitor STF-083010 Protects the Liver from Thioacetamide-Induced Oxidative Stress, Inflammation and Injury by Triggering Hepatocyte Autophagy. Int. Immunopharmacol 73, 261–269. doi:10.1016/j.intimp.2019.04.051
ALI acute liver injury
ALT alanine aminotransferase
AST aspartate aminotransferase
DFO deferoxamine
Fer-1 ferrostatin-1
Fpn ferroportin
Ft-L ferritin-L
GPX4 glutathione peroxidase 4
GSH aseglutathione
TAA thioacetamide
TfR1 transferrin receptor 1
xCT cystine/glutamate antiporter SLC7A11
Keywords: thioacetamide, ferroportin, transferrin receptor 1, ferroptosis inhibitor, acute liver injury, deferoxamine, iron
Citation: Jiang H, Zhang X, Yang W, Li M, Wang G and Luo Q (2022) Ferrostatin-1 Ameliorates Liver Dysfunction via Reducing Iron in Thioacetamide-induced Acute Liver Injury in Mice. Front. Pharmacol. 13:869794. doi: 10.3389/fphar.2022.869794
Received: 05 February 2022; Accepted: 29 March 2022;
Published: 12 April 2022.
Edited by:
Antimo Cutone, University of Molise, ItalyReviewed by:
Gladys Oluyemisi Latunde-Dada, King’s College London, United KingdomCopyright © 2022 Jiang, Zhang, Yang, Li, Wang and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guohua Wang, d2doQG50dS5lZHUuY24=; Qianqian Luo, cWlhbnFpYW5sdW9AbnR1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.