 Joaquim Trigo Marquês1
Joaquim Trigo Marquês1 Catarina Frazão De Faria1
Catarina Frazão De Faria1 Marina Reis1,2
Marina Reis1,2 Diana Machado3Susana Santos1Maria da Soledade Santos1
Diana Machado3Susana Santos1Maria da Soledade Santos1 Miguel Viveiros3
Miguel Viveiros3 Filomena Martins1*
Filomena Martins1* Rodrigo F. M. De Almeida1*
Rodrigo F. M. De Almeida1*- 1Centro de Química Estrutural, Institute of Molecular Sciences, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade de Lisboa, Lisboa, Portugal
- 2Instituto Superior de Educação e Ciências (ISEC Lisboa), Lisboa, Portugal
- 3Unidade de Microbiologia Medica, Global Health and Tropical Medicine, Instituto de Higiene e Medicina Tropical, Universidade Nova de Lisboa, Lisboa, Portugal
The upsurge of multidrug-resistant tuberculosis has toughened the challenge to put an end to this epidemic by 2030. In 2020 the number of deaths attributed to tuberculosis increased as compared to 2019 and newly identified multidrug-resistant tuberculosis cases have been stably close to 3%. Such a context stimulated the search for new and more efficient antitubercular compounds, which culminated in the QSAR-oriented design and synthesis of a series of isoniazid derivatives active against Mycobacterium tuberculosis. From these, some prospective isonicotinoyl hydrazones and isonicotinoyl hydrazides are studied in this work. To evaluate if the chemical derivatizations are generating compounds with a good performance concerning several in vitro assays, their cytotoxicity against human liver HepG2 cells was determined and their ability to bind human serum albumin was thoroughly investigated. For the two new derivatives presented in this study, we also determined their lipophilicity and activity against both the wild type and an isoniazid-resistant strain of Mycobacterium tuberculosis carrying the most prevalent mutation on the katG gene, S315T. All compounds were less cytotoxic than many drugs in clinical use with IC50 values after a 72 h challenge always higher than 25 µM. Additionally, all isoniazid derivatives studied exhibited stronger binding to human serum albumin than isoniazid itself, with dissociation constants in the order of 10−4–10−5 M as opposed to 10−3 M, respectively. This suggests that their transport and half-life in the blood stream are likely improved when compared to the parent compound. Furthermore, our results are a strong indication that the N′ = C bond of the hydrazone derivatives of INH tested is essential for their enhanced activity against the mutant strain of M. tuberculosis in comparison to both their reduced counterparts and INH.
Introduction
As the 2030 deadline established by WHO to eradicate tuberculosis (TB) approaches, no significant advances concerning the mitigation of the disease have been achieved. Unfortunately, although TB is curable and preventable, the numbers associated to this disease are still quite alarming and, consequently, it remains as one of the top 10 causes of death worldwide and, until 2019, the main cause of mortality from a single infectious agent. The figures recently published in the WHO global TB report 2021 (which refers to the 2020 situation) (World Health Organization, 2021) show a practically immutable situation over the last few years up to 2019, that only changed dramatically in 2020 (7.1 million new case notifications of people newly diagnosed with TB in 2019 as opposed to 5.8 million in 2020) due to the high prevalence of COVID-19, which caused a disruption in the notification of new TB cases. The number of total cases per year, remains clearly above 10 million since 2000 (World Health Organization, 2021). An encouraging fact, on the other hand, was the progressive decrease of total deaths attributed to TB, which, in 2020, were around 1.3 million among HIV-negative people, a reduction of 0.6 million as compared to 2000. However, as compared to the 2019’s situation, there was an increase in the total number of TB-induced deaths. This scenario arose due to the significant decrease in TB treatment as a consequence of the much smaller number of TB cases detected and notified as a result of the COVID-19 pandemic (Ong et al., 2020; World Health Organization, 2021). Additionally, a rather preoccupying situation that might help explain the unsuccess in effectively controlling and reducing these numbers is the proliferation of multidrug-resistant tuberculosis (MDR-TB), a form of TB which is not countered by the two most powerful first-line antituberculars, isoniazid (INH) and rifampicin. In fact, ∼3% of new TB cases had MDR or rifampicin-resistant-TB (World Health Organization, 2021). Recently, two new drugs, Bedaquiline and Delamanid, have been introduced in a combination regimen to treat MDR-TB in adults, though without ensuring a side effect-free therapy (Brigden et al., 2015; Zumla et al., 2015). In 2019, another drug, Pretomanid, was approved by the United States FDA for the treatment of pulmonary treatment-intolerant or nonresponsive MDR-TB or extensively drug-resistant tuberculosis (XDR-TB) in adults (Keam, 2019).
The effectiveness of INH to fight TB is known since the 1950’s when its high activity against Mycobacterium tuberculosis (Mtb) was first described (Bernstein et al., 1952; Vilchèze and Jacobs, 2007). INH is a pro-drug that, when activated by the Mtb catalase-peroxidase enzyme KatG, leads to the inhibition of the biosynthesis of mycolic acids and, consequently, cell lysis (Winder and Collins, 1970; Heym et al., 1993; Rawat et al., 2003; Zhao et al., 2006; Vilchèze and Jacobs, 2007; Wiseman et al., 2010). Mutations in the katG gene represent the most frequent mechanism of resistance to INH (Hazbón et al., 2006). Thus, the overall picture of TB demands the development of new, effective and low toxicity antitubercular compounds and, within this scope, the design of new INH derivatives which can be an effective route to overcome Mtb drug resistance. With that in mind, we have previously described the QSAR-oriented design and synthesis of several new and more active INH derivatives with enhanced lethality against MDR-TB (Martins et al., 2014). Although the most active compound against the most frequent katG mutation, i.e., the katG S315T mutant was a compound with a lengthy C10 alkyl chain, lipophilicity was not found as an important determinant of the antitubercular activity of these derivatives (Martins et al., 2014), neither the steric hindrance due to larger substituents seemed to be a ruling factor as regards the activity of the derivatives against the mutant strain (Teixeira et al., 2015). A recent lipophilicity assessment of a series of these isoniazid derivatives led to the conclusion that zwitterionic hydrazones partition to the outer palisade region of the sodium dodecyl sulfate micelles, whereas neutral hydrazides tend to penetrate further into the micellar core (Santos et al., 2020). INH, on its turn, shows a pH-dependent affinity for the membrane/water interface of a phospholipid bilayer (Pinheiro et al., 2014). Therefore, even though lipophilicity is not a factor influencing the activity of the compounds, it seems to promote an enhancement in their bioavailability inside the cell (Vila-Viçosa et al., 2017; Machado et al., 2018).
The selectivity index (SI) and the ability to bind plasma proteins are important parameters to predict the in vivo potency of a compound (Muller and Milton, 2012). SI translates quantitatively the relationship between the safety (toxicity) of a drug and its efficacy. It can help guiding the development of new drugs and a high SI is always desirable since it indicates a drug with a more favorable safety profile. However, in the case of life-threatening diseases for which no adequate treatment exists, drugs with low SI may also be acceptable (Muller and Milton, 2012).
On the other hand, the study of the interaction of a drug with plasma proteins may provide important insights concerning the pharmacokinetics (absorption, distribution, metabolism, and excretion—ADME processes) of a pharmaceutical agent within the body (Smith et al., 2010; Pellegatti et al., 2011; Wanat, 2020). The free drug hypothesis advocates that it is the free drug concentration, at the site of action, that exerts the biological activity, not the total drug concentration or the bound drug concentration to either plasma or tissue proteins (Smith et al., 2010). According to this hypothesis, envisaging drug candidates exhibiting reduced plasma protein binding is not the most fruitful strategy, since the binding to plasma proteins increases the solubility of the prospective pharmaceutical agent, extends its in vivo half-life by avoiding premature clearance, and slows down or prevents its passive extravasation to the target tissues. All of this has a remarkable impact on the bioavailability of the drug. In fact, the report on plasma protein binding is an FDA requirement in screening potential therapeutic agents.
Human serum albumin (HSA) is the most abundant protein in the plasma (35–50 mg/ml) (Wanat, 2020) and, although it plays a myriad of other functions, excels at being the most important non-specific transporter protein in the blood (Colmenarejo, 2003; Kanakis et al., 2006; Maiti et al., 2006; Pessoa and Tomaz, 2010; Schmidt et al., 2010; Kratz and Elsadek, 2012). HSA is comprised of a single chain containing three structurally similar domains termed I, II and III. In turn, each of these domains is constituted by two helical subdomains (A and B) (Curry et al., 1998; Ascenzi and Fasano, 2010). The crystal structure of HSA revealed the presence of binding sites for aromatic and heterocyclic ligands within subdomains IIA and IIIA, corresponding to Sudlow proposed sites I and II, respectively (Sudlow et al., 1976; Dockal et al., 1999; Petitpas et al., 2001).
Considering all the above, we provide a detailed biophysical study of the interaction of a series of INH derivatives—Figure 1 - with this plasma protein and a correlation study with the corresponding lipophilicity is undertaken. We also determined their cytotoxicity towards HepG2 Cells. From these assays, a SI was calculated considering the minimum inhibitory concentration (MIC) against both the wild type Mtb and the mutant S315T katG Mtb.
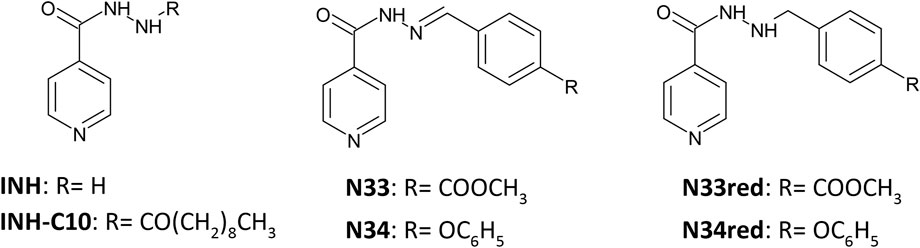
FIGURE 1. Structure of the compounds studied in this work.
Materials and Methods
General
All reagents and solvents were obtained from commercial suppliers with analytical grade and used without further purification. HSA (lyophilized powder, ≥ 97% pure) was obtained from Merck (ref. A9511, Sigma-Aldrich®). The phosphate buffered saline (PBS: 0.01 M Na2HPO4, 0.0018 M KH2PO4, 0.0027 M KCl and 0.137 M NaCl, pH = 7.4) was prepared by dissolving pre-prepared tablets from VWR in MilliQ® water (resistivity >18 MΩ cm). Chromatographic purification was performed on silica-gel 60, 0.004–0.066 mm, 230–400 mesh ASTM (Merck ref. 1.09385, Millipore®). Analytical thin layer chromatographies (TLC) were carried out on precoated silica gel 60 F254 aluminium sheets (Merck ref. 1.05554, Millipore®) and visualized with UV light (254 nm).
1H NMR (400.1 MHz) and 13C NMR (100.6 MHz) were recorded on a Bruker Avance 400 spectrometer; chemical shifts were expressed as δ values and referenced to the residual solvent peak (DMSO-d6, δH = 2.50, δC = 39.5) and coupling constants were reported in units of Hertz (Hz).
The signals of 1H and 13C spectra were unambiguously assigned using 2D NMR techniques (COSY, HMQC, HMBC and NOESY) – see Supplementary Figures S1–S4 for 1H and 13C NMR spectra in Supplementary Material. FTIR spectra were obtained using a Nicolet 6700 (Thermo Electron Corp. Madison. WI) in KBr pellets and only the diagnostic absorption bands were reported in cm−1. HR-ESI-MS spectra were acquired in the positive mode on FT-ICR-MS Solarix XR Bruker Daltonics, 7 Tesla at the FCUL node of the Portuguese Mass Spectrometry Network.
The purity of the compounds (>98.1%) was evaluated by GC-MS analysis using a Shimadzu® GCMS-QP2010 Plus, coupled with an autosampler AOC-20s, an automatic injector AOC-20i and a Teknokroma® Sapiens-5MS capillary column (30 m × 0.25 mm × 0.25 µm; dimethyl/diphenylpolysiloxane 95%:5%), and operating with an EI source. The carrier gas was nitrogen with a flow of 1 ml/min. 1 μl of each sample was directly injected via the autosampler into a split/splitless programmable-temperature mode (10°C/min until 200°C, then held at this temperature for 2 min, raised to 300°C at 10°C/min and maintained for 15 min at this final temperature); the mass spectrometer transfer line was set at 250°C.
Synthesis
INH-C10, N33 and N34 were obtained as fully described in reference (Martins et al., 2014). N33red and N34red were obtained by reduction of the parent Schiff base. Sodium cyanoborohydride (2 equiv.) was added portion wise to a solution of N33 or N34 in hot methanol. The reaction mixture was acidified with methanolic HCl 5M until pH 3; at that point the solution color changed to dark yellow. The reaction was kept under reflux and monitored by TLC until the starting material was consumed. The solvent was then evaporated under reduced pressure, giving an orange-yellow oil. This residue was dissolved in water and basified to pH > 7 by addition of a few drops of NaOH 6 M, to break up the possible cyanoborate adducts. The solution was then washed with brine and extracted several times with dichloromethane, the combined organic extracts were dried with MgSO4 and the solvent evaporated under reduced pressure affording crude yellowish solids. Pure N33red and N34red were obtained after column chromatography with Et2O/MeOH 96.5:3.5 and by recrystallization from CH2Cl2/petroleum ether 2:3, respectively (adapted from (Borch et al., 1971)). Both N33red and N34red are very stable in aqueous solution (PBS with 5% DMSO) for at least 48 h (Supplementary Figure S5).
Methyl 4-((2-isonicotinoylhydrazinyl)methyl)benzoate (N33red) Amorphous white powder η= 62.6% IR (KBr): ν (cm−1) = 3,283 (N-H amine), 3,238 (N-H amide), 3,049 (Ar C-H), 1718 (C=O ester), 1,667 (C=O amide), 1,280 (C-O aryl ester); 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 10.30 (d, 1H, J = 4.8 Hz, C=ONH), 8.69 (AA′ part of a AA′BB′ system, 2H, J = 4.6, 1.7 Hz, H-2/H-6), 7.92 (AA′ part of a AA′BB′ system, 2H, J = 7.9 Hz, H-3’/H-5′), 7.66 (BB″ part of a AA′BB′ system, 2H J = 4.6; 1.8 Hz, H-3/H-5), 7.53 (BB″ part of a AA′BB′ system, 2H, J = 7.9 Hz, H-2′/H-6′), 5.74 (m, 1H, NHCH2), 4.08 (d, 2H, J = 4.1 Hz, CH2), 3.84 (s, 3H,CH3); 13C NMR (100.6 MHz, DMSO-d6): δ (ppm) = 166.15 (OC= O), 163.92 (NHC= O), 150.19 (C-2/C-6), 144.24 (C-1′), 140.11 (C-4), 129.00 (C-3′/C-5′), 128.74 (C-2′/C-6′), 128.31 (C-4′), 121.04 (C-3/C-5), 53.95 (CH2), 52.02 (CH3). HR-ESI-MS: 286.11903 m/z [M+H]+ (calcd. for C15H15N3O3 +H, 286.11862)
N'-(4-phenoxybenzyl)isonicotinohydrazide (N34red) Amorphous white powder η= 50.6%, recrystallization from CH2Cl2: petroleum ether (2:3); IR (KBr): ν (cm−1) = 3,308 (N-H amine), 3,276 (N-H amide), 3,039 (Ar C-H), 1,640 (C=O amide), 1,247 (C-O aryl ether); 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 10.32 (d, 1H, J = 3.6 Hz, C=ONH), 8.70 (AA′ part of a AA’BB’ system, 2H, J = 4.6 Hz, H-2/H-6), 7.69 (BB″ part of a AA’BB’ system, 2H, J = 4.7 Hz, H-3/H-5), 7.38 (m, 4H, H-2’/H-6′, H-3″/H-5″), 7.12 (t, 1H, J = 7.1 Hz, H-4″), 6.97 (m, 4H, H-3’/H-5′, H-2″/H-6″), 5.55 (sb, 1H, J = 4.2 Hz, NHCH2), 3.97 (s, 2H, CH2); 13C NMR (100.6 MHz, DMSO-d6): δ (ppm) = 163.77 (NHC = O), 156.83 (COC), 155.63 (COC), 150.19 (C-2/C-6), 140.17 (C-4), 133.48 (C-1′), 130.34 (C-2′/C-6′), 129.97 (C-3″/C-5″), 123.28 (C-4″), 121.04 (C-3/C-5), 118.46 (C-2″/C-6″), 118.39 (C-3′/C-5′), 53.86 (CH2). HR-ESI-MS: 320.13929 m/z [M+H]+ (calcd. for C19H17N3O2+H, 320.13935).
Lipophilicity Measurements
Lipophilicity was evaluated as log Po/w, the octanol-water partition coefficient obtained by the shake-flask method (Ràfols et al., 2012; Martins et al., 2014), and as log Kp(NaDS/w) the partition coefficient towards sodium dodecyl sulfate (NaDS) anionic micelles obtained by differential spectrophotometry for INH, INH-C10, N33 and N34 in previous works (Ràfols et al., 2012; Martins et al., 2014; Santos et al., 2020). For the new compounds, N33red and N34red, a preliminary assessment of their log Po/w was achieved in this work, on the basis of the same general procedure already reported (Ràfols et al., 2012; Martins et al., 2014). For both compounds, samples were dissolved in water-saturated octanol at pH 7.5 (KH2PO4/NaOH) to guarantee negligible ionization. The partition to octanol-saturated aqueous buffer was evaluated, at room temperature, using a volume ratio, ro/w, of 5/30 and 10/30 for N33red and 2.5/40, 5/40 for N34red, after sonication of the solutions and partition on a mechanical tumbler. Equilibrium concentrations were determined by UV-Vis spectrophotometry between 200 and 500 nm after phase separation.
HepG2 Cell Viability Assay
HepG2 cells were plated on 384-well tissue culture treated polystyrene plates at 2000 cells in 25 μl of HepG2 maintenance medium per well. After an overnight incubation at 37°C, the cells were dosed with test compounds in dimethyl sulfoxide (DMSO) and controls over wide ranges of concentrations (Table 1) and incubated for 72 h at 37°C. DMSO was at 0.5% in the final solution. The aqueous solubility of the compounds limited the range of concentrations tested. Cell viability was measured using the Promega CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT) kit by adding the Dye Solution to each well and incubating for 3 h at 37°C. After incubation, the Solubilization Solution/Stop Mix was added to each well. Plates were incubated at 37°C for 1 h, mixed on a plate shaker for 10 min and then absorbance was read at 570 nm. Cell viability was determined by comparison against control cells in the presence of DMSO only and was measured in three independent replicates for each compound concentration. The IC50 was calculated with GraphPad Prism using a Sigmoidal Dose-response (variable slope) algorithm. Chlorpromazine, a drug with a well-known and characterized hepatotoxicity (Frötschl et al., 2005; Gandhi et al., 2013; Morgan et al., 2019), was used as the positive control.
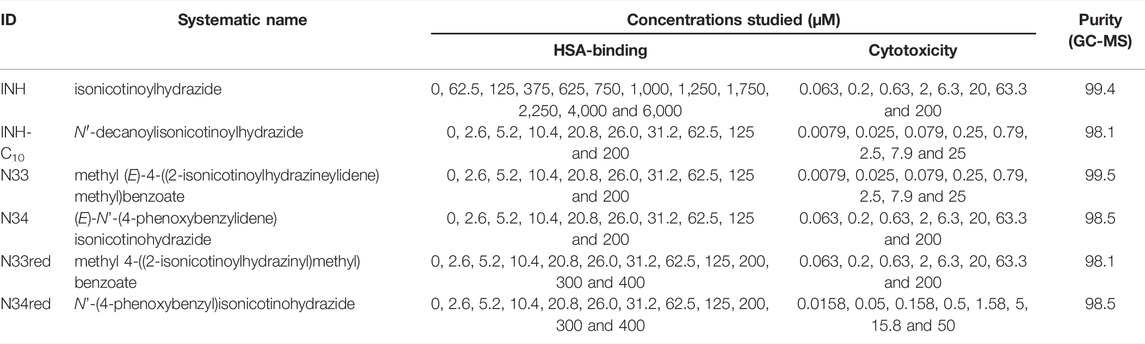
TABLE 1. Abbreviation (ID), systematic name, concentrations in HSA-binding and cytotoxicity studies and purity of the compounds used in this work.
Determination of Minimum Inhibitory Concentrations
The isoniazid derivatives were screened in vitro for its activity against M. tuberculosis by broth macrodilution using the BACTEC MGIT 960 system and the growth monitored, at 37°C, with the Epicenter V5.80A software (Becton Dickinson, Diagnostic Systems, Sparks, MD, United States) as previously described (Springer et al., 2009; Martins et al., 2014). The MGIT tubes were inoculated with 0.8 ml of OADC (oleic acid, albumin, dextrose, catalase; Becton and Dickinson), 0.1 ml of the compound at the pretended concentration and 0.5 ml of the strain suspension. The compounds were tested from 0.03125 to 128 µM. At the time of testing, two-fold serial dilutions were prepared to achieve the desired concentrations. When positive growth was observed in the drug containing tube (GU ≥ 100) before the positivity of the proportional growth control (containing a 1/100 dilution of the suspension of the strain), this indicates that more than 1% of the population was able to grow in the presence of the concentration of the anti-TB drug and, as per the proportion testing method definition recommended by WHO, the strain is considered resistant at the corresponding drug concentration (Canetti et al., 1963). Thus, the MIC was the lowest concentration with GU < 100 when the drug-free 1/100 proportional control tube reached the positivity threshold of 400 GU. An absolute growth control (undiluted) was included in every assay to monitor the normal growth of each strain. The assays were performed in triplicate (biological replicates) and the final value was given as the result of two concordant values (Machado et al., 2016).
The M. tuberculosis H37Rv ATCC27294T reference strain was obtained from the American Type Culture Collection (Virginia, United States). The M. tuberculosis clinical strain, monorresistant to isoniazid due to the presence of the mutation S315T in the katG gene (Machado et al., 2018, Machado et al., 2016), was isolated in 2003 from a Portuguese patient as part of the routine mycobacteriology laboratory services provided by Universidade NOVA de Lisboa (Lisboa, Portugal) to the local hospitals. Informed consent was not required for this study as it involves only anonymized bacterial isolates.
Sample Preparation for Spectroscopic Measurements With Albumin
HSA stock solutions were prepared by gently dissolving the protein in PBS (Matos et al., 2013). The concentration of the HSA stock solutions was 10.4 µM as determined by spectrophotometry using the molar absorption coefficient reported by Pace et al. (ε280 nm = 35,219 M−1 cm−1) (Pace et al., 1995).
INH and INH derivatives stock solutions were prepared in DMSO. From the stock solutions, appropriate dilutions were prepared so that each compound was added to HSA in PBS at different concentrations ensuring that DMSO was at a fixed concentration of 5% (v/v) in the final solution. HSA concentration in the final solution was 5.2 µM. Due to limitations imposed by the solubility of each compound, the range of concentrations assayed varied for each compound as shown in Table 1. Prior to measurements, the samples were incubated at 37.0 ± 0.5°C for 24 h. Other incubation times were tested as well – 48, 72 and 96 h – and since no changes in the data obtained were observed the incubation time was set to 24 h. Three or more independent replicates were prepared and analyzed for each system.
For the competitive binding assays with warfarin the same preparation protocol was carried out, except that the solution also contained warfarin in equimolar proportion to HSA (5.2 µM) equilibrated prior to incubation with the compound. Warfarin stock solution was also prepared with DMSO.
Spectroscopic Measurements and Data Analysis
Fluorescence measurements were performed with a Fluorolog 3.22 spectrofluorimeter (Horiba Jobin Yvon, Villeneuve D'ascq, France) at 24.0 ± 0.5°C in a sample compartment with temperature-control. Quartz Suprasil® cuvettes with 1 × 0.4 cm path length were used to gather the spectroscopic data.
For steady-state fluorescence intensity measurements, the excitation wavelength was set to 295 nm and emission was collected in the range 310–550 nm. The bandwidth was 4 nm in both excitation and emission paths. The fluorescence intensity was corrected for the absorption and emission inner filter effects using the absorption spectra recorded for each sample (Lakowicz, 2006). Electronic absorption spectra were recorded at room temperature with a Jasco V-560 spectrophotometer (Hiroshima, Japan) in the range from 250 to 500 nm.
Time-resolved fluorescence measurements were carried out by the single photon counting technique, using a nanoLED N-280 (Horiba Jobin Yvon, Villeneuve D'ascq, France) for the excitation of the protein. The emission wavelength was set to 350 nm in order to avoid the contribution from tyrosine residues and collect only the photons emitted by the tryptophane residue 214Trp (Starosta et al., 2020). The bandwidth was 11 nm and a timescale of 55ps/channel (1,024 channels) was employed. Ludox® was the scattering agent used to obtain the instrumental response function. The experimental fluorescence intensity decays were analysed with the software TRFA® version 1.4 (Minsk, Belarus). The fluorescence intensity decays, I(t), were analysed by fitting a sum of exponentials, and the decay law was then obtained according to Eq. 1:
where αi and τi are the normalized amplitude and lifetime of component i, respectively.
The changes in quantum yield due to processes affecting the fluorescence lifetime were assessed from the amplitude-weighted mean fluorescence lifetime,
The criteria to ascertain the good quality of the fitting were the reduced χ2 value close to 1 and the random distributions of weighted residuals and residuals autocorrelation.
Regarding both steady-state and time-resolved measurements, an adequate blank was subtracted from each reading and at least three independent experiments were performed for each compound. The results are presented as mean ± standard deviation. Unless otherwise stated, statistical significance versus INH was determined by one-way ANOVA with Tukey’s post-hoc test (*, p < 0.05; **, p < 0.01; ***p < 0.001) (Mishra et al., 2019).
Results and Discussion
Four different and complementary in vitro approaches, deemed to be suitable considering the current stage of development of this project, were employed to assess the potential in vivo application of the INH derivatives under study to treat MDR-TB.
In vitro Evaluation of Hepatotoxicity
In early screening of potential drugs, it is important to assess their in vitro cytotoxicity to be able to justify further investments in their development. In fact, cell viability assays to evaluate drug toxicity are frequently employed during the process of drug development (Riss et al., 2013; Jakštys et al., 2015; Niepel et al., 2019). In general terms, drugs can exert their toxicity through on-target pharmacology, i.e., by an exacerbated action on the primary pharmacological target, or through off-target toxicity, which is a common mode of toxicity for small-molecule drugs (and/or their metabolites) as a result of their pharmacological promiscuity and/or their unintended chemical reactivity with biomolecules (Muller and Milton, 2012).
Many drugs in clinical use exhibit, at some point, some degree of hepatotoxicity (Devarbhavi, 2012). To evaluate the in vitro toxicity of this series of INH derivatives, the viability of HepG2 cells, a human liver cancer cell line, was determined by the MTT tetrazolium reduction assay (Riss et al., 2013), a colorimetric assay to assess cell metabolic activity, and the IC50 value, i.e., the concentration for 50% reduction in cell viability, was obtained for each compound (Supplementary Figure S6). The IC50 value for the positive control, chlorpromazine, a drug mainly used in the treatment of psychotic disorders, was 13.9 µM, which is consistent with a previous assessment of IC50—34.7 µM against the same cell line, but a shorter incubation time (24 h instead of 72 h) (Wang et al., 2002). The hepatoxicity of INH is well described (Girling, 1978; Wu and Cederbaum, 1996; Huang et al., 2003; Bhadauria et al., 2007; Singh et al., 2011) and is much lower than that of chlorpromazine. From studies using HepG2 cells it has been suggested that the main mechanisms by which INH may exert toxic effects is by inducing oxidative stress, mitochondria dysfunction and apoptosis (Bhadauria et al., 2007).
Here, we intended to assess the toxicity of these INH derivatives as a pre-screening approach to evaluate the suitability of these compounds to be considered in the treatment of TB. The results obtained in cell viability assays (Supplementary Figure S6, Table 2 (in µM) and Supplementary Table S1 (in μg ml−1)) suggest that most compounds tested are not toxic within the concentration ranges studied. The parental drug INH as well as compounds N34 and N33red did not significantly affect HepG2 cell viability up to 200 µM. Due to limitations imposed by the aqueous solubility of the compounds, the effects of INH-C10 and N33 were only tested up to 25 µM. At this concentration INH-C10 only marginally affected cell viability, whereas N33 had no significant impact (Supplementary Figure S6). N34red was the only INH derivative for which it was possible to determine an IC50 value, i.e., 48.5 µM. All the IC50 values obtained show that INH and the studied derivatives are less toxic than drugs currently in clinical use. Rifampin, an antibiotic employed for the treatment of mycobacterial infections, has an IC50 value of 25.5 µM (Vahdati-Mashhadian et al., 2013). More importantly, all of these INH derivatives seem to be safer than bedaquiline, which has an IC50 of 17.4 µM (Lupien et al., 2018) and half of them are certainly better, in terms of cytotoxicity, than delamanid, which presents an IC50 of 98.9 µM (Lupien et al., 2018). The IC50 presented for bedaquiline and delamanid were obtained in the same conditions, i.e., against the same cell line, HepG2, and the same incubation time (72 h) (Lupien et al., 2018).
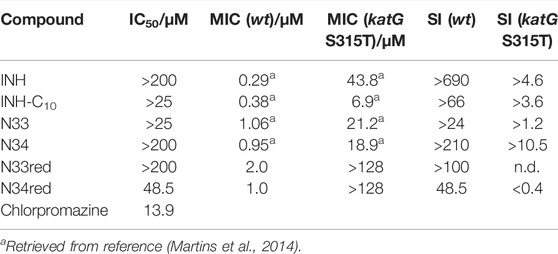
TABLE 2. IC50 values of the compounds studied in this work against HepG2 cells (including the positive control, chlorpromazine), MIC values against wt (MIC (wt)) and katG (S315T) (MIC (katG S315T)) strains of Mtb and selectivity index computed with the activity against wt (SI (wt)) and katG S135T (SI (katG S315T)).
The selectivity index, SI, which depends on the toxicity of the compound, is also an important parameter when considering the pharmaceutical application of new drugs. In a previous work we determined the activity of a series of INH derivatives against the wild type strain of Mtb (H37Rv) and the katG (S315T) mutant strain that lies as the most frequent and important cause of INH resistance worldwide (Martins et al., 2014). The minimum inhibitory concentrations (MIC), defined as the lowest concentration necessary to inhibit 99% of the bacterial population, established in (Martins et al., 2014) using the BACTEC™ MGIT™ 960 system (BACTEC 960) and the Epicenter V5.53A software, together with the IC50 values determined in the present work against HepG2 cells, can be used to estimate the in vitro SI according to Eq. 3:
In Table 2, the SIs considering the activity against both the wild type and mutant strains are presented. The data gathered so far indicates that, by comparison with the other studied compounds, INH presents the best performance against the wt Mtb; however, when considering the mutant strain katG (S315T), the SI of N34 makes it a very promising compound. In addition, two other INH derivatives also present higher activity against this strain when compared with INH, which in the case of INH-C10 corresponds to a 6-fold gain in efficacy. At this point, the solubility limit of this compound in buffer with 0.5% DMSO prevents us to elaborate further on the SI for this compound. Yet, the in vitro efficiency of a drug can greatly differ from that in vivo (Braggio et al., 2010). Drug efficiency is defined as the fraction of the dose administered that effectively reaches the site of action (Braggio et al., 2010). While in vitro this parameter is expected to be near 1, in vivo it greatly depends on the ADME processes, which can be profoundly influenced by the plasma protein binding ability of the drugs.
Albumin Binding Studies and Correlations With Compound Lipophilicity
It is common understanding that the binding to plasma proteins can not only modulate the pharmacokinetics of a drug, but also lower its toxicity (Schmidt et al., 2010; Wanat, 2020). Thus, in addition to the SI, plasma protein binding is also an important aspect to take into consideration in the evaluation of potential drugs. In fact, many of the highly prescribed drugs bind to plasma proteins in a percentage greater than 98% (Smith et al., 2010). This constitutes the rationale behind the evaluation of HSA interactions with INH and its derivatives, since HSA is the most abundant protein in the plasma, and, as already referred, also the most important non-specific drug carrier.
HSA contains 18 tyrosine residues and a single tryptophane residue, 214Trp, located in subdomain IIA. The interaction between INH or its derivatives and HSA can be studied by fluorescence spectroscopy by selective excitation of 214Trp at 295 nm and emission collection at 340 nm or longer wavelengths (Luís et al., 2014; Teale and Weber, 1957). Although 214Trp is in the vicinity of Sudlow’s binding site I, its photophysics also reports changes occurring in binding site II due to its high sensitivity to changes in the environment either as a result of drug binding or of structural alterations of the protein (Luís et al., 2014). The emission spectra of HSA in the presence of increasing concentrations of each compound can be found in Figure 2. The maximum emission intensity of HSA in the absence of any compound was observed at 334 nm in agreement with previous observations (Demoro et al., 2013; Luís et al., 2014), indicating that 214Trp is shielded from the aqueous solvent, since tryptophane has a typical emission maximum close to 350 nm in water (Teale and Weber, 1957). As a general acknowledgment, with the increment in the concentration of the compounds there is a pronounced decrease in the emission intensity of HSA, thus all compounds have the ability to quench the fluorescence of 214Trp. Moreover, in the normalized emission spectra, it can be observed that INH, INH-C10 N33red and N34red did not promote any spectral shifts (Figures 3A,B,E,F), whereas N33 induced a red shift (Figure 3C) and N34 may have caused a small blue shift (Figure 3D). Therefore, while INH, INH-C10, N33red and N34red binding do not seem to induce structural alterations in the protein that change the exposure degree of 214Trp, the same is not verified for N33 and possibly N34. The red shift in the maximum emission upon the binding of N33 points to a structural change that seems to leave 214Trp exposed to a more polar environment, suggesting a greater water accessibility in the vicinity of that amino acid residue. In fact, for the highest concentration of N33, the maximum emission is shifted to ∼350 nm, which is close to the maximum emission of 214Trp in water. On the other hand, the blue shift effect of N34 is much smaller (ca. 5 nm).
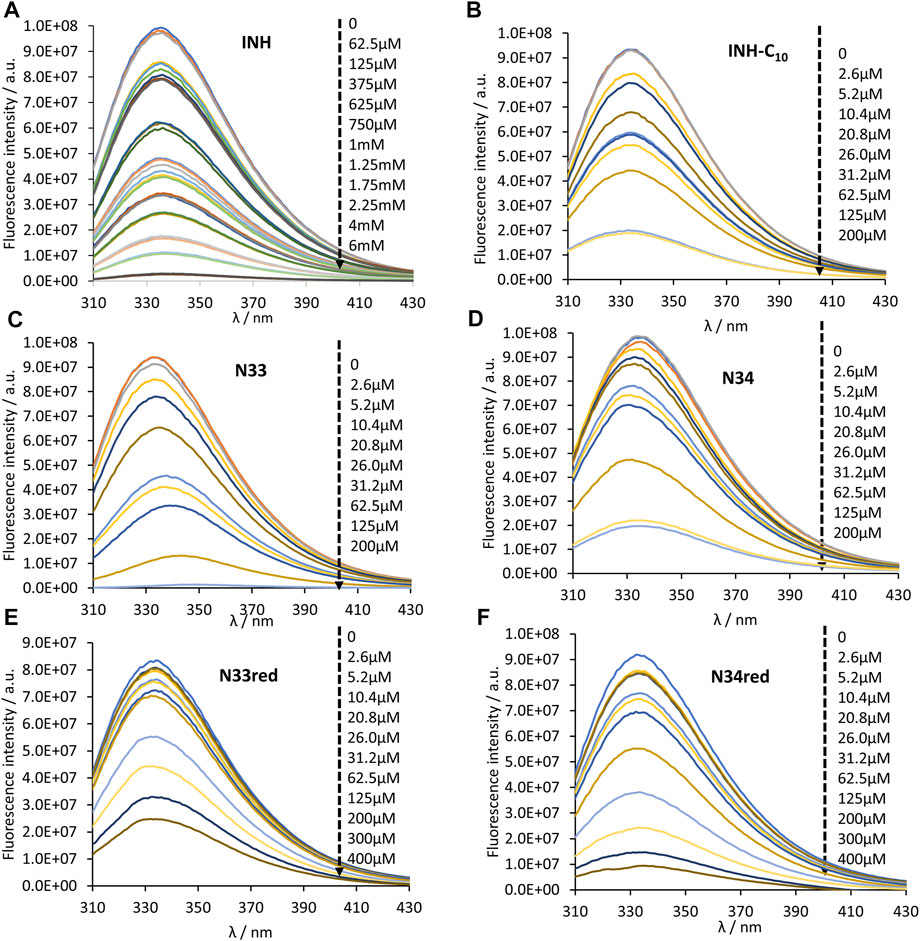
FIGURE 2. Steady-state emission spectra of HSA 214Trp in the absence and presence of (A) INH, (B) INH-C10, (C) N33, (D) N34, (E) N33red and (F) N34red. 214Trp fluorescence quenching (λex = 295 nm) is observed upon compound binding (black dashed arrow indicates the direction of increasing compound concentration). The conditions were as follows: [HSA] = 5.2 µM, kept constant; samples prepared in PBS, pH 7.4; 24 h incubation at (37.0 ± 0.5)°C; measurements at room temperature, (24.0 ± 0.5)°C.
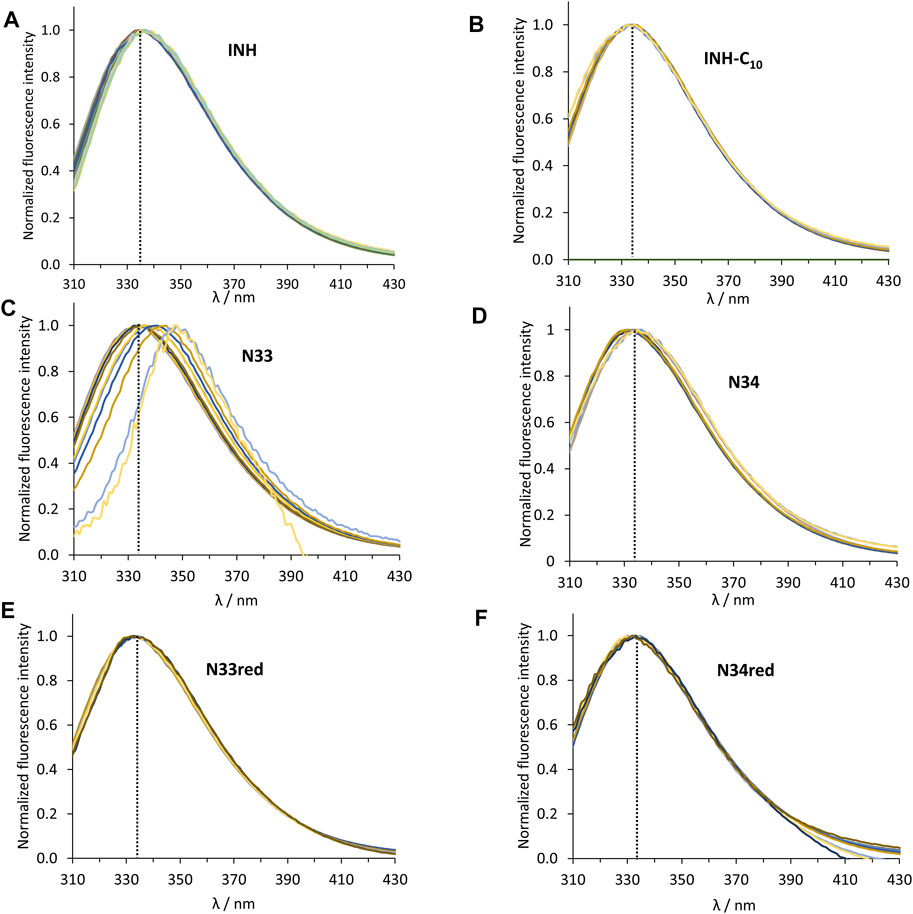
FIGURE 3. Normalized steady-state emission spectra of HSA 214Trp in the absence and presence of (A) INH, (B) INH-C10, (C) N33, (D) N34, (E) N33red and (F) N34red. The dashed black line indicates the maximum emission of HSA in the absence of any compound (λ = 334 nm). The conditions were as follows: [HSA] = 5.2 µM, kept constant; λex = 295 nm; samples prepared in PBS, pH 7.4; 24 h incubation at (37.0 ± 0.5)°C; measurements at room temperature, (24.0 ± 0.5)°C.
For further analysis, the fluorescence intensity values were taken at excitation and emission wavelengths of λex = 295 nm and λem = 340 nm and corrected for the inner filter effects, yielding a corrected fluorescence intensity value, F, for each sample. The variation of such fluorescence intensity (ΔF) between the samples containing only HSA (F0) and HSA + compound (F) can be calculated for each concentration of the compound to estimate the dissociation constant (Kd) for the equilibrium HSA + compound. As detailed in (Luís et al., 2014), plotting ΔF as a function of compound concentration ([C]), Eq. 4:
enables the computation of Kd values and of the maximum variation in fluorescence intensity (ΔFmax) for unlimited compound concentration, by a non-linear fit with Eq. 4. In Figure 4 the graphical representation of ΔF as a function of [C] is shown for INH (Figure 4A) and all INH derivatives (Figure 4B) studied in this work, and Kd values for all the compounds are presented in Table 3.
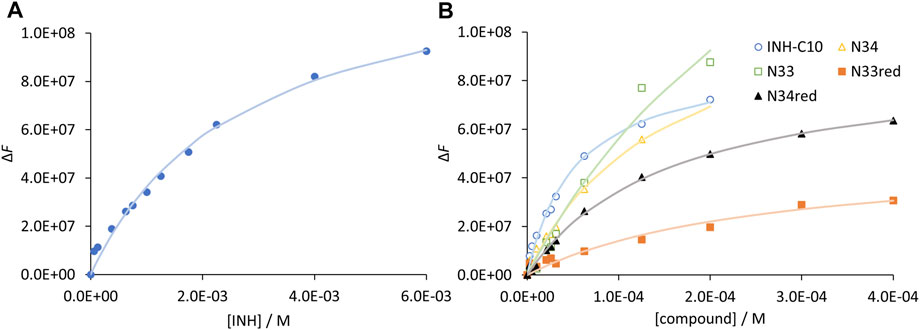
FIGURE 4. Compound binding to HSA as assessed by steady-state fluorescence spectroscopy. Variation of fluorescence intensity for increasing concentration of (A) INH and (B) INH derivatives. The dots correspond to experimental data and the lines correspond to the non-linear fit of Eq. 5. Analysis is based on the fluorescence intensity at λem = 340 nm (λex = 295 nm), under the following conditions: [HSA] = 5.2 µM; samples prepared in PBS, pH 7.4; 24 h incubation at (37.0 ± 0.5) °C; measurements at room temperature, (24.0 ± 0.5)°C. INH was represented separately due to the very different concentration range assayed.
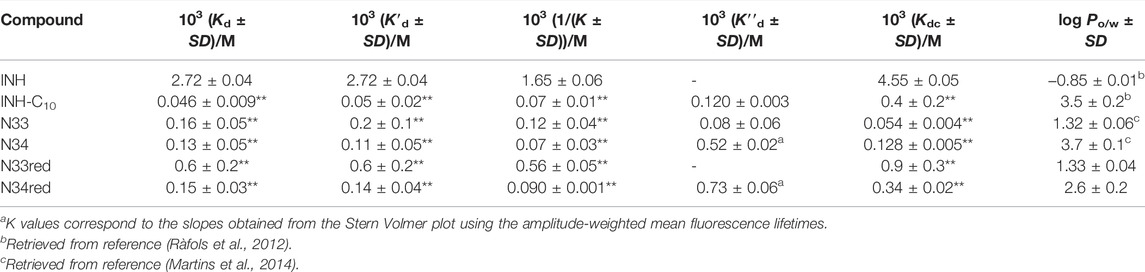
TABLE 3. Dissociation constants of the compounds studied in this work for HSA retrieved from the variation in the steady-state fluorescence intensity (Kd), from the variation in the fluorescence intensity corrected regarding the variation in fluorescence lifetime (K′d) and from the variation in
Regarding the data in Figure 4 the emission intensity at 340 nm was used instead of that at 334 nm to avoid water Raman scattering and improve the signal-to-background ratio, while maintaining very good sensitivity (close to maximum) (Demoro et al., 2013; Matos et al., 2013). The Kd of INH in the range of 10−3M (Table 3), as determined by us, represents a weak interaction as compared to other drugs, which typically display Kd in the range of 10−4–10−6M (Sowell et al., 2001; Chen and Hage, 2006; Kanakis et al., 2006; Kim and Wainer, 2008; Mallik et al., 2008; Joseph et al., 2010; Joseph et al., 2011; Matsuda et al., 2011; Yang et al., 2014). A Kd value of 3.9 ± 0.4 × 10−4 M has been previously reported for INH (Ascenzi et al., 2011). However, it is not mentioned in that study if the fluorescence intensity was corrected for inner filter effects. In fact, if we use the uncorrected fluorescence intensity data to calculate INH Kd, we obtain a value of 6.5 × 10−4 M (Supplementary Figure S7).
Notably, all INH derivatives exhibit a more favourable interaction with HSA as compared to INH (Table 3). INH-C10, which was the compound with the strongest antimicrobial activity against the katG S315T mutant strain of Mtb, is also the one with more affinity for HSA. Compound N34, the one that displayed the most promising SI regarding the activity against the mutant strain, also presents a Kd value that is one order of magnitude lower than that of INH, meaning that binding is one order of magnitude stronger. The Kd determined for the INH derivatives lay within the range of many clinically used drugs, 10−4–10−6, (Smith et al., 2010) which is another encouraging result regarding future efforts in the development of alternative therapies to treat MDR-TB based on the chemical tailoring of INH. The binding of the compounds tested to HSA displays a good correlation with their lipophilicity, as expressed by their octanol-water partition coefficients (Table 3), which is clearly shown in the plot of log Kd vs. log Po/w (Figure 5). N33 deviates from the linear trend and was not considered in the linear regression. Such deviation probably occurs because this compound induces a structural change in the protein that leaves 214Trp exposed to water as suggested by the strong emission red shift (Figure 3C), unlike all other compounds. The correlation found between log Kd and log Po/w follows the typical trend for compound-plasma protein binding, i.e., as compounds are more lipophilic the more significant becomes their binding to plasma proteins (Croom, 2012). As mentioned, the range of Kd of the derivatives is equivalent to other drugs. Higher Kd would result in an increased free fraction of the drug, whereas much lower Kd hinders the release of the compound by HSA. The same rational can be applied to lipophilicity, since a low lipophilicity implies a low partition towards lipid membranes, thus a limited ability to reach the target inside Mtb. On the other hand, a very high lipophilicity corresponds to a very high affinity for lipid membranes, which may result in a compound being trapped in the membrane once it penetrates it. We have previously shown that the lipophilicity of INH-C10 (Santos et al., 2020) is important for the accumulation of this compound in lipid membranes with a concomitant increase in bioavailability inside the cell (Vila-Viçosa et al., 2017).
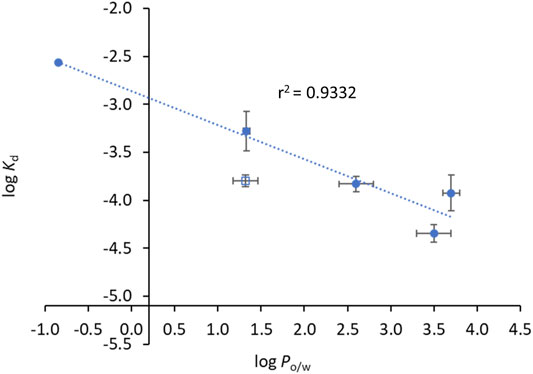
FIGURE 5. Compounds’ binding to HSA is correlated with their lipophilicity. The dots correspond to experimental data and the line corresponds to a linear fit to the data points. log Po/w and log Kd values can be found in Table 3. Data points used for the linear fit included INH, INH-C10, N34, N33red and N34red. The rational as to why N33, open square, is not used for the linear fit is given in the text. The relationship between log Kd and log Po/w is given by log Kd = -(0.35 ± 0.05) log Po/w – (2.86 ± 0.14).
Regarding the fluorescence intensity decays of HSA 214Trp, the best fit was always obtained using three exponential terms. The normalized amplitude and lifetime of each component (α1 = 0.261, α2 = 0.428, α3 = 0.314, τ1 = 0.662 ns, τ2 = 3.443 ns, τ3 = 6.935 ns), that best described the fluorescence intensity decay of HSA alone are in good agreement with the ones previously reported (Luís et al., 2014; Starosta et al., 2020). As depicted in Figure 6 and Supplementary Figure S8 not all compounds have the same efficiency reducing the fluorescence lifetime of 214Trp. The invariance of the amplitude-weighted mean fluorescence lifetime (
where F0 and F are the fluorescence intensities in the absence and presence of a concentration of quencher compound, [C]. When used to analyse time-resolved fluorescence data, F is replaced by
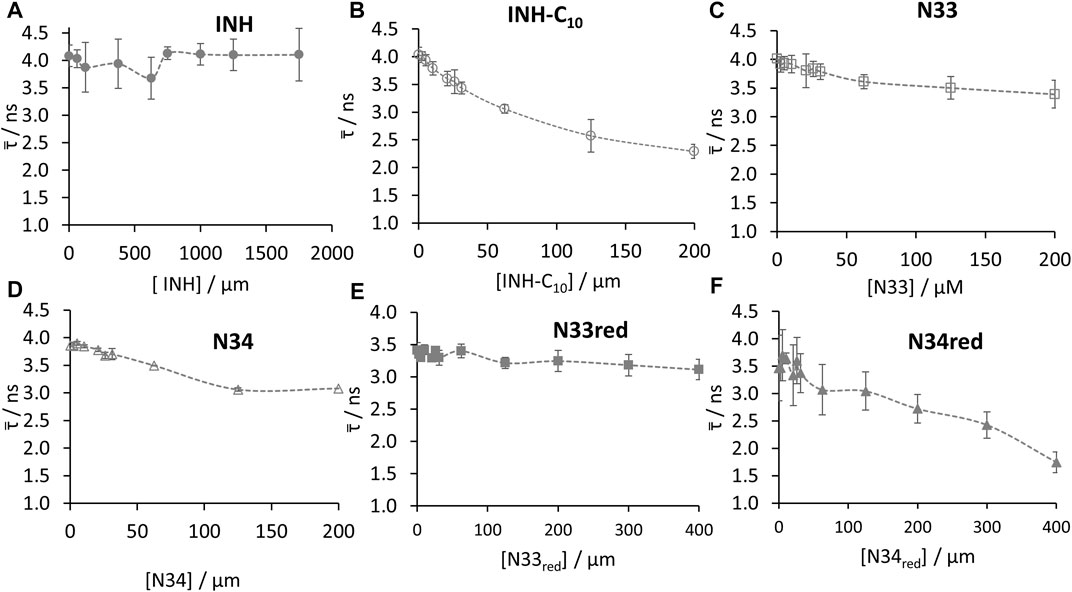
FIGURE 6. Amplitude-weighted mean fluorescence lifetime (
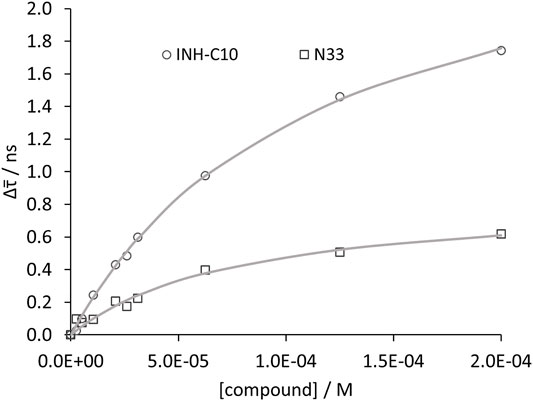
FIGURE 7. Compound binding to HSA as assessed by time resolved fluorescence spectroscopy. Variation of
Whenever the decrease in
The Stern–Volmer plots for HSA static quenching by the compounds, i.e., the quenching obtained from the steady-state fluorescence intensity corrected for the decrease of the fluorescence lifetimes are shown in Figure 8.
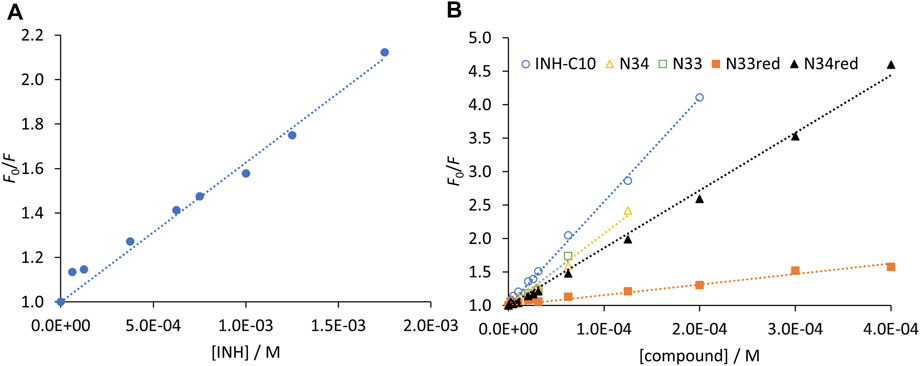
FIGURE 8. Stern-Volmer plots of HSA 214Trp obtained from the steady-state fluorescence intensity measurements after removing the contribution of the fluorescence lifetimes decrease for (A) INH and (B) INH derivatives. The dots correspond to experimental data and the lines correspond to the linear fit to Eq. 6. Analysis based on the fluorescence intensity at λem = 340 nm (λex = 295 nm), under the following conditions: [HSA] = 5.2 µM; samples prepared in PBS, pH 7.4; 24 h incubation at (37.0 ± 0.5)°C; measurements at room temperature, (24.0 ± 0.5)°C. INH was represented separately due to the very different concentration range assayed.
For a static quenching mechanism, K represents the association constant for the formation of non-fluorescent HSA-compound ground-state complex. Therefore, the reverse of K gives an estimate of the dissociation constant. By fitting a straight line to the data in Figure 8 forcing a unitary intercept, i.e., assuming a Stern-Volmer-like behaviour, a reasonable correlation coefficient was obtained in all cases. It is worth noting that the reverse of the Stern-Volmer constants, 1/K (Table 3), yields values mostly identical to the K′d determined by the non-linear fit according to Eq. 4.
The interaction of INH and its derivatives with HSA was further examined regarding their putative binding site. INH is known to bind to the warfarin binding pocket of site I (Wang et al., 2016). Thus, to assess if INH derivatives would bind to the same site as INH (and warfarin), fluorescence spectroscopy measurements were carried out in the presence of the fluorescent site-marker warfarin in the same molar concentration as HSA (5.2 µm). Here, to avoid any contribution from the 214Trp (or tyrosine residues) and ensure that only the fluorescence from warfarin was being collected, the excitation wavelength used was 320 nm (Figure 9).
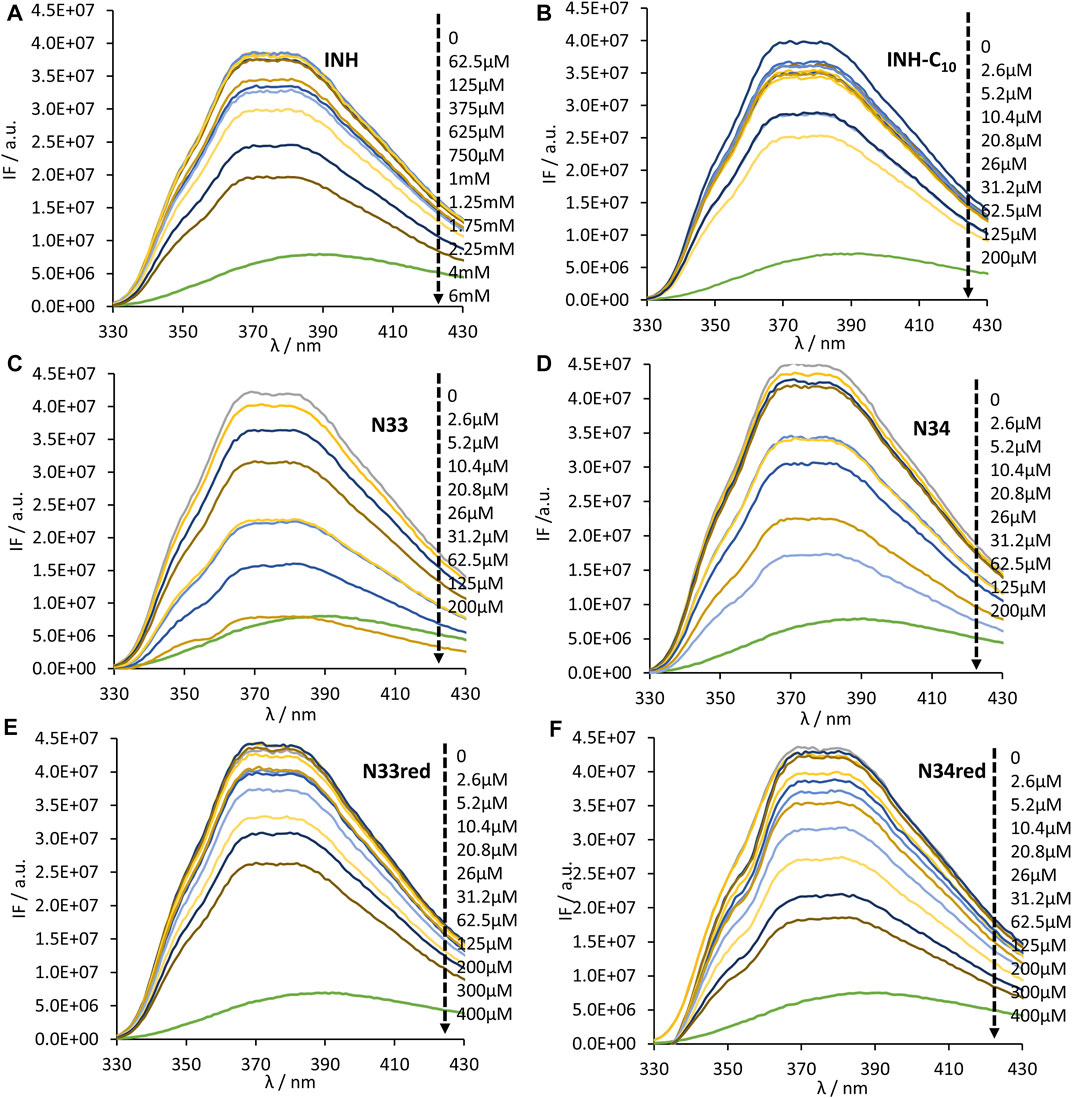
FIGURE 9. Steady-state emission spectra of the complex HSA-bound warfarin in the absence and presence of (A) INH, (B) INH-C10, (C) N33, (D) N34, (E) N33red and (F) N34red. λex = 320 nm to ensure that only warfarin is excited. The green line in all plots is the emission spectrum of warfarin in water. A decrease in fluorescence intensity is observed upon compound binding (black large arrow indicates the direction of increasing compound concentration). The conditions were as follows: [warfarin] = [HSA] = 5.2 µM, kept constant; samples prepared in PBS, pH 7.4; 24 h incubation at (37.0 ± 0.5)°C; measurements at room temperature, (24.0 ± 0.5)°C.
As can be observed, an increase in the concentration of the compounds results in a decrease in the intensity of fluorescence emission by warfarin, which is due to the displacement of warfarin from the HSA-warfarin complex to water, where it has a much lower fluorescence quantum yield (Vasquez et al., 2009). Such decrease in the fluorescence intensity supports the hypothesis that all compounds compete with warfarin for the same binding site, or, in other words, that INH derivatives will bind at the same site as its parental compound, INH. Using the dampening in the fluorescence intensity of warfarin it is possible to retrieve the HSA-compound dissociation constant for the binding site of warfarin (Kdc) - Table 3. With this purpose, the following equation, that is valid for pure competition at a single site, can be used (Cheng and Prusoff, 1973):
where C50, the concentration of compound that displaces half of the initially bound warfarin, is determined from the variation of the fluorescence intensity, as exemplified in Supplementary Figure S9 and Kdw is the dissociation constant of warfarin. The calculated Kdw is 9 × 10−6 M, which is in good agreement with previous determinations (Loun and Hage, 1994; Chen and Hage, 2006; Yoo et al., 2010).
For an evaluation of the contribution of the binding to the warfarin binding site to total binding, the corresponding binding constants (the reverse of the respective dissociation constants) are presented in Supplementary Table S2. Considering these values, for the compounds that do not promote a change in the fluorescence lifetime of 214Trp, INH and N33red, the Kb (binding constant retrieved from the variation in the steady-state fluorescence intensity) is close to Kbc (binding constant for the warfarin binding site), which suggests that the main binding process of these compounds involves HSA Sudlow’s binding site I. For the other compounds, for which a variation in the fluorescence lifetime of HSA upon their binding was observed, both binding processes must be considered—the binding that induces only a decrease in the fluorescence intensity of 214Trp (K′b – binding constant from the variation in the fluorescence intensity corrected regarding the variation in fluorescence lifetime) and the binding that induces a decrease in the fluorescence lifetime of 214Trp (K″b – binding constant retrieved from the variation in
Interestingly, the binding to the warfarin site also shows some correlation with the lipophilicity of the compounds (Figure 10). Plotting log Kdc vs. log Po/w for all compounds except N33 yields a reasonably linear correlation (Figure 10, blue data). Additionally, since INH-C10 is the only compound for which the binding to Sudlow’s site I accounts for only a minor fraction of the binding to HSA (Kbc clearly smaller than both Kb′ and Kb″), we also performed a linear regression of log Kdc vs. log Po/w excluding this compound (Figure 10, open circle), which significantly improved the correlation (r2 = 0.9993).
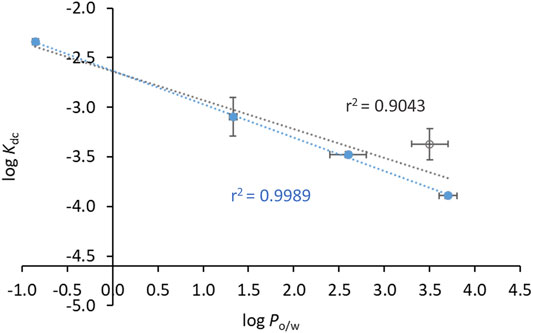
FIGURE 10. Compounds’ affinity to the binding site of warfarin in HSA (Sudlow’s binding site I) is correlated with their lipophilicity. The dots correspond to experimental data and the lines to linear fits. log Po/w and log Kdc values can be found in Table 3. Data in closed circles are for INH, N34, N33red and N34red, and the open circle is for INH-C10. The linear fit either includes all the points (black line) or all points except INH-C10 (blue line). For the latter, the relationship between log Kd and log Po/w is given by log Kdc = -(0.34 ± 0.01) log Po/w - (2.63 ± 0.02).
Nonetheless, the result to be highlighted is that all derivatives studied in this work bind to warfarin binding site and all of them do it with higher affinity than INH. Since HSA is known to influence ADME processes in vivo, the more effective binding of the derivatives to HSA suggests that these compounds might be more efficiently transported through the blood stream, while being more protected than INH against early clearance or involvement in unintended chemical reactivity with biomolecules. In addition, the enhanced permeability and retention-like (EPR) effect, consisting in the accumulation of macromolecules such as albumin in the altered tissue, most commonly described in the context of cancer therapeutics, may be also relevant for drug targeting in tuberculosis, given the inflammatory processes associated with this infectious disease (Fenaroli et al., 2018).
Antimycobacterial Activity
A total of six compounds were tested for their ability to inhibit the growth of actively replicating Mtb strains, the wild type H37Rv reference strain and a clinical strain harbouring the most common mutation on the katG gene, S315T. Compounds N33 and N34 are more active against the mutated strain than INH for the same strain. Compounds N33red and N34red are active against the wild-type strain, whereas their activity is completely abolished in the mutated strain. Moreover, N33 and N34 are still active against the mutated strain despite an increase in their MICs when compared with values in the wild type strain. Interestingly, for the three compounds INH-C10, N33 and N34, the MICs undergo a similar 20-fold increase going from wt to S315T, whereas for INH this increase is of approximately 150-fold. This suggests that the underlying features that render these compounds more active than INH against the mutated strain do share some similarities. All these compounds are more hydrophobic than INH (see Table 3). This can promote a better permeation of Mtb cell wall and plasma membrane, allowing a larger intracellular accumulation of the compounds. A previous study has shown that the hydrophobic nature of INH-C10 promotes a better trafficking across the Mtb membrane, which compensates for its smaller reactivity (the N′-acyl group stabilizes the molecule, resulting in slower spontaneous radical formation) when compared with INH (Vila-Viçosa et al., 2017), resulting in lower MIC values (Martins et al., 2014). The only structural difference between N33red and N34red and the parent hydrazones N33 and N34 is the nature of the bond between the N′ atom of the isonicotinhydrazide moiety and its substituents (Figure 1). The lack of the N′ = C bond in the reduced compounds allows the free rotation around the N′-C single bond in N33red and N34red. This stereo arrangement can hinder membrane permeability, i.e., compounds may still be adsorbed at the membrane but crossing will be much slower, thus diminishing the ability of N33red and N34red to reach and/or interact favourably with the active site of Mtb S315T strain, leading to higher MIC values as compared to the corresponding non-reduced compounds. The reason for the higher activity of N33 and N34 vis-à-vis INH in the mutant is still not totally rationalized, hinting that more than one factor is probably responsible for this behaviour (e.g., a balance between trafficking and reactivity).
Conclusion
In this work the in vitro toxicity and plasma protein binding of a series of isoniazid derivatives were investigated. Even though some of these INH derivatives might present a higher degree of toxicity than INH itself, they have shown to be less aggressive in vitro than some therapeutic agents currently in use, including the two new drugs that have been recently introduced as part of combination regimens to treat MDR-TB in adults, bedaquiline and delamanid. Although this type of comparison might have some limitations due to differences in experimental settings/conditions, the same cell type and incubation time have been employed in both cases, allowing thus a reasonable judgement about the potential safety of these derivatives. This outcome is especially relevant in the case of compounds INH-C10, N33 and N34, which are more active than INH against the most frequent Mtb katG mutation (the S315T mutant). Therefore, the rational used for the design of these INH derivatives, and their experimentally verified ability to overcome limitations caused by the S315T mutation with acceptable selectivity indexes, may be considered as strategies to improve INH activity, especially in cases where acquired resistance is emerging due to selection during treatment of a S315T mutated population.
In addition to their promising safety data, these INH derivatives also proved to have a greater binding affinity towards HSA than INH itself, with dissociation constants that in the case of INH-C10 are two orders of magnitude more favourable. Furthermore, the efficient binding of INH derivatives to HSA is expected to improve their half-life and solubility and lower their toxicity, allowing us to predict that in an in vivo scenario Mtb may be exposed to high and safe doses of these compounds for lengthy periods of time. The albumin binding behaviour of the studied compounds correlates with their lipophilicity, which in the case of INH-C10 is in harmony with prior molecular dynamic simulations predicting a higher partition to a phospholipid bilayer as opposed to INH (Vila-Viçosa et al., 2017). Nevertheless, it should be emphasized that the increased activity of N33 and N34 towards the mutated Mtb does not correlate with their lipophilicity. N33 and N33red have the same log Po/w, and although N34red is less lipophilic than N34, it still presents a higher log Po/w than both N33 and N33red. However, in both cases (i.e., for N33red and N34red) there is a dramatic loss of activity against the mutated Mtb when reducing the hydrazones to hidrazides. This is most likely related with the spatial arrangement of the molecules and their affinity towards the S1315T katG, as discussed above.
All summed up, we hypothesize that these INH derivatives might be able to reach and accumulate near the KatG active site more efficiently than INH, while simultaneously reducing adverse effects. These results may be of particular importance in the context of MDR-TB, for which alternatives to INH are especially urgent in order to overcome the reduced catalytic activity of the mutated KatG. The fact that some of the compounds studied here have lower MIC values against the mutant Mtb than INH and, simultaneously, present both a higher affinity for HSA and a potential to permeate more easily membrane barriers due to their enhanced lipophilicity, makes rather interesting to continue exploring the chemical space around INH based on the structures of the two derivatives INH-C10 and N34, in view of future development of new MDR-TB agents.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
JM: conceptualization, formal analysis, investigation, methodology, validation, visualization, writing—original draft; CF: formal analysis, investigation, methodology, visualization; MR: formal analysis, investigation, methodology, visualization; DM: formal analysis, investigation, methodology, visualization, writing—review and editing; SS: formal analysis, investigation, supervision, visualization, writing—review and editing, MS: formal analysis, investigation, validation, writing—review and editing, MV: funding acquisition, investigation, methodology, resources, supervision, validation, writing—review and editing; FM: conceptualization, formal analysis, funding acquisition, investigation, project administration, resources, validation, writing - review and editing; RA: conceptualization, formal analysis, funding acquisition, investigation, methodology, resources, supervision, validation, writing—original draft.
Funding
Financed by Fundação para a Ciência e a Tecnologia, I.P./MCTES through national funds (PIDDAC, PT2020) under projects PTDC/MED-QUI/29036/2017, PTDC/BIA-MIC-30692/2017, EXPL/BIA-BFS/1034/2021, UIDB/00100/2020, UIDP/00100/2020, LA/P/0056/2020, UID/Multi/04413/2020, CEECIND/03247/2018 and DL57/CEECIND/0256/2017.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.868545/full#supplementary-material
References
Ascenzi, P., Bolli, A., Di Masi, A., Tundo, G. R., Fanali, G., Coletta, M., et al. (2011). Isoniazid and Rifampicin Inhibit Allosterically Heme Binding to Albumin and Peroxynitrite Isomerization by Heme-Albumin. J. Biol. Inorg. Chem. 16, 97–108. doi:10.1007/s00775-010-0706-2
Ascenzi, P., and Fasano, M. (2010). Allostery in a Monomeric Protein: the Case of Human Serum Albumin. Biophys. Chem. 148, 16–22. doi:10.1016/j.bpc.2010.03.001
Bernstein, J., Lott, W. A., Steinberg, B. A., and Yale, H. L. (1952). Chemotherapy of Experimental Tuberculosis. V. Isonicotinic Acid Hydrazide (Nydrazid) and Related Compounds. Am. Rev. Tuberc. 65, 357–364. doi:10.1164/art.1952.65.4.357
Bhadauria, S., Singh, G., Sinha, N., and Srivastava, S. (2007). Isoniazid Induces Oxidative Stress, Mitochondrial Dysfunction and Apoptosis in Hep G2 Cells. Cel Mol Biol (Noisy-le-grand) 53 (1), 102–114. doi:10.1170/T781
Borch, R. F., Bernstein, M. D., and Durst, H. D. (1971). Cyanohydridoborate Anion as a Selective Reducing Agent. J. Am. Chem. Soc. 93, 2897–2904. doi:10.1021/ja00741a013
Braggio, S., Montanari, D., Rossi, T., and Ratti, E. (2010). Drug Efficiency: a New Concept to Guide lead Optimization Programs towards the Selection of Better Clinical Candidates. Expert Opin. Drug Discov. 5, 609–618. doi:10.1517/17460441.2010.490553
Brigden, G., Hewison, C., and Varaine, F. (2015). New Developments in the Treatment of Drug-Resistant Tuberculosis: Clinical Utility of Bedaquiline and Delamanid. Infect. Drug Resist. 8, 367–378. doi:10.2147/IDR.S68351
Canetti, G., Froman, S., Grosset, J., Hauduroy, P., Langerova, M., Mahler, H. T., et al. (1963). Mycobacteria: Laboratory Methods for Testing Drug Sensitivity and Resistance. Bull. World Health Organ. 29, 565–578.
Chen, J., and Hage, D. S. (2006). Quantitative Studies of Allosteric Effects by Biointeraction Chromatography: Analysis of Protein Binding for Low-Solubility Drugs. Anal. Chem. 78, 2672–2683. doi:10.1021/ac052017b
Cheng, Y., and Prusoff, W. H. (1973). Relationship between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 22, 3099–3108. doi:10.1016/0006-2952(73)90196-2
Colmenarejo, G. (2003). In Silico prediction of Drug-Binding Strengths to Human Serum Albumin. Med. Res. Rev. 23, 275–301. doi:10.1002/med.10039
Croom, E. (2012). “Chapter Three - Metabolism of Xenobiotics of Human Environments,” in Progress in Molecular Biology and Translational Science. Editor E. HODGSON (Cambridge, MA, United States: Academic Press).
Curry, S., Mandelkow, H., Brick, P., and Franks, N. (1998). Crystal Structure of Human Serum Albumin Complexed with Fatty Acid Reveals an Asymmetric Distribution of Binding Sites. Nat. Struct. Biol. 5, 827–835. doi:10.1038/1869
Demoro, B., De Almeida, R. F., Marques, F., Matos, C. P., Otero, L., Costa Pessoa, J., et al. (2013). Screening Organometallic Binuclear Thiosemicarbazone Ruthenium Complexes as Potential Anti-tumour Agents: Cytotoxic Activity and Human Serum Albumin Binding Mechanism. Dalton Trans. 42, 7131–7146. doi:10.1039/c3dt00028a
Devarbhavi, H. (2012). An Update on Drug-Induced Liver Injury. J. Clin. Exp. Hepatol. 2, 247–259. doi:10.1016/j.jceh.2012.05.002
Dockal, M., Carter, D. C., and Rüker, F. (1999). The Three Recombinant Domains of Human Serum Albumin. Structural Characterization and Ligand Binding Properties. J. Biol. Chem. 274, 29303–29310. doi:10.1074/jbc.274.41.29303
Fenaroli, F., Repnik, U., Xu, Y., Johann, K., Van Herck, S., Dey, P., et al. (2018). Enhanced Permeability and Retention-like Extravasation of Nanoparticles from the Vasculature into Tuberculosis Granulomas in Zebrafish and Mouse Models. ACS Nano 12, 8646–8661. doi:10.1021/acsnano.8b04433
Frötschl, R., Weickardt, S., Staszewski, S., Kaufmann, G., and Kasper, P. (2005). Effects of Chlorpromazine with and without UV Irradiation on Gene Expression of HepG2 Cells. Mutat. Res. 575, 47–60. doi:10.1016/j.mrfmmm.2005.03.002
Gandhi, A., Guo, T., Shah, P., Moorthy, B., and Ghose, R. (2013). Chlorpromazine-induced Hepatotoxicity during Inflammation Is Mediated by TIRAP-dependent Signaling Pathway in Mice. Toxicol. Appl. Pharmacol. 266, 430–438. doi:10.1016/j.taap.2012.11.030
Girling, D. J. (1978). The Hepatic Toxicity of Antituberculosis Regimens Containing Isoniazid, Rifampicin and Pyrazinamide. Tubercle 59, 13–32. doi:10.1016/0041-3879(77)90022-8
Hazbón, M. H., Brimacombe, M., Bobadilla Del Valle, M., Cavatore, M., Guerrero, M. I., Varma-Basil, M., et al. (2006). Population Genetics Study of Isoniazid Resistance Mutations and Evolution of Multidrug-Resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 50, 2640–2649. doi:10.1128/AAC.00112-06
Heym, B., Zhang, Y., Poulet, S., Young, D., and Cole, S. T. (1993). Characterization of the katG Gene Encoding a Catalase-Peroxidase Required for the Isoniazid Susceptibility of Mycobacterium tuberculosis. J. Bacteriol. 175, 4255–4259. doi:10.1128/jb.175.13.4255-4259.1993
Huang, Y. S., Chern, H. D., Su, W. J., Wu, J. C., Chang, S. C., Chiang, C. H., et al. (2003). Cytochrome P450 2E1 Genotype and the Susceptibility to Antituberculosis Drug-Induced Hepatitis. Hepatology 37, 924–930. doi:10.1053/jhep.2003.50144
Jakštys, B., Ruzgys, P., Tamošiūnas, M., and Šatkauskas, S. (2015). Different Cell Viability Assays Reveal Inconsistent Results after Bleomycin Electrotransfer In Vitro. J. Membr. Biol. 248, 857–863. doi:10.1007/s00232-015-9813-x
Joseph, K. S., Anguizola, J., and Hage, D. S. (2011). Binding of Tolbutamide to Glycated Human Serum Albumin. J. Pharm. Biomed. Anal. 54, 426–432. doi:10.1016/j.jpba.2010.09.003
Joseph, K. S., Anguizola, J., Jackson, A. J., and Hage, D. S. (2010). Chromatographic Analysis of Acetohexamide Binding to Glycated Human Serum Albumin. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 878, 2775–2781. doi:10.1016/j.jchromb.2010.08.021
Kanakis, C. D., Tarantilis, P. A., Polissiou, M. G., Diamantoglou, S., and Tajmir-Riahi, H. A. (2006). Antioxidant Flavonoids Bind Human Serum Albumin. J. Mol. Struct. 798, 69–74. doi:10.1016/j.molstruc.2006.03.051
Kim, H. S., and Wainer, I. W. (2008). Rapid Analysis of the Interactions between Drugs and Human Serum Albumin (HSA) Using High-Performance Affinity Chromatography (HPAC). J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 870, 22–26. doi:10.1016/j.jchromb.2008.05.029
Kratz, F., and Elsadek, B. (2012). Clinical Impact of Serum Proteins on Drug Delivery. J. Control. Release 161, 429–445. doi:10.1016/j.jconrel.2011.11.028
Loun, B., and Hage, D. S. (1994). Chiral Separation Mechanisms in Protein-Based HPLC Columns. 1. Thermodynamic Studies of (R)- and (S)-warfarin Binding to Immobilized Human Serum Albumin. Anal. Chem. 66, 3814–3822. doi:10.1021/ac00093a043
Luís, D. V., Silva, J., Tomaz, A. I., De Almeida, R. F., Larguinho, M., Baptista, P. V., et al. (2014). Insights into the Mechanisms Underlying the Antiproliferative Potential of a Co(II) Coordination Compound Bearing 1,10-Phenanthroline-5,6-Dione: DNA and Protein Interaction Studies. J. Biol. Inorg. Chem. 19, 787–803. doi:10.1007/s00775-014-1110-0
Lupien, A., Vocat, A., Foo, C. S., Blattes, E., Gillon, J. Y., Makarov, V., et al. (2018). Optimized Background Regimen for Treatment of Active Tuberculosis with the Next-Generation Benzothiazinone Macozinone (PBTZ169). Antimicrob. Agents Chemother. 62 (11), e00840–18. doi:10.1128/AAC.00840-18
Machado, D., Girardini, M., Viveiros, M., and Pieroni, M. (2018). Challenging the Drug-Likeness Dogma for New Drug Discovery in Tuberculosis. Front Microbiol. 9, 1367. doi:10.3389/fmicb.2018.01367
Machado, D., Pires, D., Perdigão, J., Couto, I., Portugal, I., Martins, M., et al. (2016). Ion Channel Blockers as Antimicrobial Agents, Efflux Inhibitors, and Enhancers of Macrophage Killing Activity against Drug Resistant Mycobacterium tuberculosis. PloS one 11 (2), e0149326. doi:10.1371/journal.pone.0149326
Maiti, T. K., Ghosh, K. S., Debnath, J., and Dasgupta, S. (2006). Binding of All-Trans Retinoic Acid to Human Serum Albumin: Fluorescence, FT-IR and Circular Dichroism Studies. Int. J. Biol. Macromol 38, 197–202. doi:10.1016/j.ijbiomac.2006.02.015
Mallik, R., Yoo, M. J., Chen, S., and Hage, D. S. (2008). Studies of Verapamil Binding to Human Serum Albumin by High-Performance Affinity Chromatography. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 876, 69–75. doi:10.1016/j.jchromb.2008.10.022
Martins, F., Santos, S., Ventura, C., Elvas-Leitão, R., Santos, L., Vitorino, S., et al. (2014). Design, Synthesis and Biological Evaluation of Novel Isoniazid Derivatives with Potent Antitubercular Activity. Eur. J. Med. Chem. 81, 119–138. doi:10.1016/j.ejmech.2014.04.077
Matos, C. P., Valente, A., Marques, F., Adão, P., Paula Robalo, M., De Almeida, R. F. M., et al. (2013). New Polydentate Ru(III)-Salan Complexes: Synthesis, Characterization, Anti-tumour Activity and Interaction with Human Serum Proteins. Inorg. Chim. Acta 394, 616–626. doi:10.1016/j.ica.2012.09.026
Matsuda, R., Anguizola, J., Joseph, K. S., and Hage, D. S. (2011). High-performance Affinity Chromatography and the Analysis of Drug Interactions with Modified Proteins: Binding of Gliclazide with Glycated Human Serum Albumin. Anal. Bioanal. Chem. 401, 2811–2819. doi:10.1007/s00216-011-5382-8
Mishra, P., Singh, U., Pandey, C. M., Mishra, P., and Pandey, G. (2019). Application of Student's T-Test, Analysis of Variance, and Covariance. Ann. Card. Anaesth. 22 (4), 407–411. doi:10.4103/aca.ACA_94_19
Morgan, K., Martucci, N., Kozlowska, A., Gamal, W., Brzeszczyński, F., Treskes, P., et al. (2019). Chlorpromazine Toxicity Is Associated with Disruption of Cell Membrane Integrity and Initiation of a Pro-inflammatory Response in the HepaRG Hepatic Cell Line. Biomed. Pharmacother. 111, 1408–1416. doi:10.1016/j.biopha.2019.01.020
Muller, P. Y., and Milton, M. N. (2012). The Determination and Interpretation of the Therapeutic index in Drug Development. Nat. Rev. Drug Discov. 11, 751–761. doi:10.1038/nrd3801
Niepel, M., Hafner, M., Mills, C. E., Subramanian, K., Williams, E. H., Chung, M., et al. (2019). A Multi-center Study on the Reproducibility of Drug-Response Assays in Mammalian Cell Lines. Cell Syst 9, 35–48. e5. doi:10.1016/j.cels.2019.06.005
Ong, C. W. M., Migliori, G. B., Raviglione, M., Macgregor-Skinner, G., Sotgiu, G., Alffenaar, J. W., et al. (2020). Epidemic and Pandemic Viral Infections: Impact on Tuberculosis and the Lung: A Consensus by the World Association for Infectious Diseases and Immunological Disorders (WAidid), Global Tuberculosis Network (GTN), and Members of the European Society of Clinical Microbiology and Infectious Diseases Study Group for Mycobacterial Infections (ESGMYC). Eur. Respir. J. 56, 2001727. doi:10.1183/13993003.01727-2020
Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995). How to Measure and Predict the Molar Absorption Coefficient of a Protein. Protein Sci. 4, 2411–2423. doi:10.1002/pro.5560041120
Pellegatti, M., Pagliarusco, S., Solazzo, L., and Colato, D. (2011). Plasma Protein Binding and Blood-free Concentrations: Which Studies Are Needed to Develop a Drug? Expert Opin. Drug Metab. Toxicol. 7, 1009–1020. doi:10.1517/17425255.2011.586336
Pessoa, J. C., and Tomaz, I. (2010). Transport of Therapeutic Vanadium and Ruthenium Complexes by Blood Plasma Components. Curr. Med. Chem. 17, 3701–3738. doi:10.2174/092986710793213742
Petitpas, I., Bhattacharya, A. A., Twine, S., East, M., and Curry, S. (2001). Crystal Structure Analysis of Warfarin Binding to Human Serum Albumin: Anatomy of Drug Site I. J. Biol. Chem. 276, 22804–22809. doi:10.1074/jbc.M100575200
Pinheiro, M., Silva, A. S., Pisco, S., and Reis, S. (2014). Interactions of Isoniazid with Membrane Models: Implications for Drug Mechanism of Action. Chem. Phys. Lipids 183, 184–190. doi:10.1016/j.chemphyslip.2014.07.002
Ràfols, C., Bosch, E., Ruiz, R., Box, K. J., Reis, M., Ventura, C., et al. (2012). Acidity and Hydrophobicity of Several New Potential Antitubercular Drugs: Isoniazid and Benzimidazole Derivatives. J. Chem. Eng. Data 57, 330–338.
Rawat, R., Whitty, A., and Tonge, P. J. (2003). The Isoniazid-NAD Adduct Is a Slow, Tight-Binding Inhibitor of InhA, the Mycobacterium tuberculosis Enoyl Reductase: Adduct Affinity and Drug Resistance. Proc. Natl. Acad. Sci. U S A. 100, 13881–13886. doi:10.1073/pnas.2235848100
Riss, T. L. M., Niles, A. L., Duellman, S., Benink, H. A., Worzella, T. J., and Minor, L. (2013). “Cell Viability Assays,” in Assay Guidance Manual. S. S. Markossian, A. Grossman, and K. Brimacombe. Editors (Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences).
Santos, M. S. C. S., Matos, A. M., Reis, M., and Martins, F. (2020). Lipophilicity Assessment of Some Isoniazid Derivatives Active against Mycobacterium tuberculosis. Colloids Surf. A: Physicochemical Eng. Aspects 599, 124820. doi:10.1016/j.colsurfa.2020.124820
Schmidt, S., Gonzalez, D., and Derendorf, H. (2010). Significance of Protein Binding in Pharmacokinetics and Pharmacodynamics. J. Pharm. Sci. 99, 1107–1122. doi:10.1002/jps.21916
Singh, M., Sasi, P., Rai, G., Gupta, V. H., Amarapurkar, D., and Wangikar, P. P. (2011). Studies on Toxicity of Antitubercular Drugs Namely Isoniazid, Rifampicin, and Pyrazinamide in an In Vitro Model of HepG2 Cell Line. Med. Chem. Res. 20, 1611–1615. doi:10.1007/s00044-010-9405-3
Smith, D. A., Di, L., and Kerns, E. H. (2010). The Effect of Plasma Protein Binding on In Vivo Efficacy: Misconceptions in Drug Discovery. Nat. Rev. Drug Discov. 9, 929–939. doi:10.1038/nrd3287
Sowell, J., Mason, J. C., Strekowski, L., and Patonay, G. (2001). Binding Constant Determination of Drugs toward Subdomain IIIA of Human Serum Albumin by Near-Infrared Dye-Displacement Capillary Electrophoresis. Electrophoresis 22, 2512–2517. doi:10.1002/1522-2683(200107)22:12<2512::AID-ELPS2512>3.0.CO;2-9
Springer, B., Lucke, K., Calligaris-Maibach, R., Ritter, C., and Böttger, E. C. (2009). Quantitative Drug Susceptibility Testing of Mycobacterium tuberculosis by Use of MGIT 960 and EpiCenter Instrumentation. J. Clin. Microbiol. 47 (6), 1773–1780. doi:10.1128/JCM.02501-08
Starosta, R., Santos, F. C., and De Almeida, R. F. M. (2020). Human and Bovine Serum Albumin Time-Resolved Fluorescence: Tryptophan and Tyrosine Contributions, Effect of DMSO and Rotational Diffusion. J. Mol. Struct. 1221, 128805. doi:10.1016/j.molstruc.2020.128805
Sudlow, G., Birkett, D. J., and Wade, D. N. (1976). Further Characterization of Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 12, 1052–1061.
Teale, F. W., and Weber, G. (1957). Ultraviolet Fluorescence of the Aromatic Amino Acids. Biochem. J. 65, 476–482. doi:10.1042/bj0650476
Teixeira, V. H., Ventura, C., Leitão, R., Ràfols, C., Bosch, E., Martins, F., et al. (2015). Molecular Details of INH-C10 Binding to Wt KatG and its S315T Mutant. Mol. Pharm. 12, 898–909. doi:10.1021/mp500736n
Vahdati-Mashhadian, N., Jafari, M. R., Sharghi, N., and Sanati, T. (2013). Protective Effects of Vitamin C and NAC on the Toxicity of Rifampin on Hepg2 Cells. Iran J. Pharm. Res. 12, 141–146.
Vasquez, J. M., Vu, A., Schultz, J. S., and Vullev, V. I. (2009). Fluorescence Enhancement of Warfarin Induced by Interaction with Beta-Cyclodextrin. Biotechnol. Prog. 25, 906–914. doi:10.1002/btpr.188
Vila-Viçosa, D., Victor, B. L., Ramos, J., Machado, D., Viveiros, M., Switala, J., et al. (2017). Insights on the Mechanism of Action of INH-C10 as an Antitubercular Prodrug. Mol. Pharm. 14, 4597–4605. doi:10.1021/acs.molpharmaceut.7b00719
Vilchèze, C., and Jacobs, W. R. (2007). The Mechanism of Isoniazid Killing: Clarity through the Scope of Genetics. Annu. Rev. Microbiol. 61, 35–50. doi:10.1146/annurev.micro.61.111606.122346
Wanat, K. (2020). Biological Barriers, and the Influence of Protein Binding on the Passage of Drugs across Them. Mol. Biol. Rep. 47, 3221–3231. doi:10.1007/s11033-020-05361-2
Wang, K., Shindoh, H., Inoue, T., and Horii, I. (2002). Advantages of In Vitro Cytotoxicity Testing by Using Primary Rat Hepatocytes in Comparison with Established Cell Lines. J. Toxicol. Sci. 27, 229–237. doi:10.2131/jts.27.229
Wang, Y. R., Fang, Q., Hu, T. Y., and Liu, Y. (2016). Spectroscopic and Molecular Docking Study on Specific Binding and Inhibition of Isoniazid to Human Serum Albumin and Catalase. Guang Pu Xue Yu Guang Pu Fen Xi 36, 3789–3795.
Winder, F. G., and Collins, P. B. (1970). Inhibition by Isoniazid of Synthesis of Mycolic Acids in Mycobacterium tuberculosis. J. Gen. Microbiol. 63, 41–48. doi:10.1099/00221287-63-1-41
Wiseman, B., Carpena, X., Feliz, M., Donald, L. J., Pons, M., Fita, I., et al. (2010). Isonicotinic Acid Hydrazide Conversion to Isonicotinyl-NAD by Catalase-Peroxidases. J. Biol. Chem. 285, 26662–26673. doi:10.1074/jbc.M110.139428
World Health Organization (2021). Global Tuberculosis Report 2021. Available at: https://www.who.int/publications/i/item/9789240037021.
Wu, D., and Cederbaum, A. I. (1996). Ethanol Cytotoxicity to a Transfected HepG2 Cell Line Expressing Human Cytochrome P4502E1. J. Biol. Chem. 271, 23914–23919. doi:10.1074/jbc.271.39.23914
Yang, F., Zhang, Y., and Liang, H. (2014). Interactive Association of Drugs Binding to Human Serum Albumin. Int. J. Mol. Sci. 15, 3580–3595. doi:10.3390/ijms15033580
Yoo, M. J., Schiel, J. E., and Hage, D. S. (2010). Evaluation of Affinity Microcolumns Containing Human Serum Albumin for Rapid Analysis of Drug-Protein Binding. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 878, 1707–1713. doi:10.1016/j.jchromb.2010.04.028
Zhao, X., Yu, H., Yu, S., Wang, F., Sacchettini, J. C., and Magliozzo, R. S. (2006). Hydrogen Peroxide-Mediated Isoniazid Activation Catalyzed by Mycobacterium tuberculosis Catalase-Peroxidase (KatG) and its S315T Mutant. Biochemistry 45, 4131–4140. doi:10.1021/bi051967o
Keywords: antimycobacterial activity, Mycobacterium tuberculosis, isoniazid resistance, human serum albumin binding, cytotoxicity, lipophilicity
Citation: Marquês JT, Frazão De Faria C, Reis M, Machado D, Santos S, Santos MdS, Viveiros M, Martins F and De Almeida RFM (2022) In vitro Evaluation of Isoniazid Derivatives as Potential Agents Against Drug-Resistant Tuberculosis. Front. Pharmacol. 13:868545. doi: 10.3389/fphar.2022.868545
Received: 02 February 2022; Accepted: 08 April 2022;
Published: 04 May 2022.
Edited by:
Yurong Lai, Gilead, United StatesReviewed by:
Guilherme F. S. Fernandes, University College London, United KingdomKelvin Wang, Auckland University of Technology, New Zealand
Copyright © 2022 Marquês, Frazão De Faria, Reis, Machado, Santos, Santos, Viveiros, Martins and De Almeida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filomena Martins, filomena.martins@fc.ul.pt; Rodrigo F. M. De Almeida, rfalmeida@fc.ul.pt