
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 17 May 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.855626
This article is part of the Research TopicMulti-targeted Natural Products as Cancer Therapeutics: Challenges and Opportunities, Volume IIView all 17 articles
 Xun Zhang1,2
Xun Zhang1,2 Zhong Yao1,2Zhiyi Xue1,2Shuai Wang3Xuemeng Liu1,2Yaotian Hu1,2Yan Zhang1,2
Zhong Yao1,2Zhiyi Xue1,2Shuai Wang3Xuemeng Liu1,2Yaotian Hu1,2Yan Zhang1,2 Jian Wang1,2,4
Jian Wang1,2,4 Xingang Li1,2*
Xingang Li1,2* Anjing Chen1,2*
Anjing Chen1,2*Resibufogenin (RB) is a major active ingredient in the traditional Chinese medicine Chansu and has garnered considerable attention for its efficacy in the treatment of cancer. However, the anticancer effects and underlying mechanisms of RB on glioblastoma (GBM) remain unknown. Here, we found that RB induced G2/M phase arrest and inhibited invasion in a primary GBM cell line, P3#GBM, and two GBM cell lines, U251 and A172. Subsequently, we demonstrated that RB-induced G2/M phase arrest occurred through downregulation of CDC25C and upregulation of p21, which was caused by activation of the MAPK/ERK pathway, and that RB inhibited GBM invasion by elevating intercellular Ca2+ to suppress the Src/FAK/Paxillin focal adhesion pathway. Intriguingly, we confirmed that upon RB binding to ATP1A1, Na+-K+-ATPase was activated as a receptor and then triggered the intracellular MAPK/ERK pathway and Ca2+-mediated Src/FAK/Paxillin focal adhesion pathway, which led to G2/M phase arrest and inhibited the invasion of GBM cells. Taken together, our findings reveal the antitumor mechanism of RB by targeting the ATP1A1 signaling cascade and two key signaling pathways and highlight the potential of RB as a new class of promising anticancer agents.
Glioblastoma multiforme (GBM, WHO IV) accounts for nearly 51.4% of all kinds of brain cancer and remains the leading cause of cancer-related death due to intracranial malignant disease (Preusser et al., 2011). Currently, the standard of therapy for newly diagnosed patients is maximum safe resection followed by adjuvant ionizing radiotherapy and temozolomide chemotherapy (Stupp et al., 2005). However, median survival has remained stagnant over the last decade, remaining at 14.6 months (Hegi et al., 2005; Linz 2010; Stupp et al., 2014). Developing novel anticancer strategies in addition to chemotherapy, radiation and immunotherapy will be critical for patients with glioma. Therefore, low-toxicity, effective drugs to treat glioma and prolong the survival of patients are urgently needed.
Natural ingredient extracts have historically been an important resource for potential cancer therapies. Chansu, a dried secretion of Bufo gargarizans Cantor or B. melanostictus Schneider, is a widely used traditional Chinese medicine that exhibits a variety of pharmacological activities, including anti-inflammatory, detoxifying, analgesic, stimulatory, and anticancer properties (Sun et al., 2016; Apryani et al., 2020; Song et al., 2021). Chansu was reported to significantly inhibit the malignant progression of glioma (Lan et al., 2018), lung cancer (Yu et al., 2014) and breast cancer (Dong et al., 2011). Resibufogenin (RB), alternatively known as bufogenin and recibufogenin, is a major active ingredient in cinobufacini. RB has been commonly used for the treatment of a variety of tumors in recent years. For instance, RB was demonstrated to inhibit TAK1-mediated NF-κB activity through protein kinase C-dependent repression of GSK-3 in pancreatic cancer (Liu et al., 2018). Moreover, RB inhibited the malignant progression of ovarian clear cell carcinoma by suppressing the PI3K/AKT and actin cytoskeleton signaling pathways. Nevertheless, the effect and the molecular mechanisms of RB on glioma are currently unclear.
The dysregulated cell cycle of glioma cells is the main cause of their malignant proliferation and is a classic therapeutic target for oncology drug development (Thomas et al., 2014). The cell G2/M phase transition is mediated by the activation of the CDK1-Cyclin B complex (Chang and Ferrell 2013). Activation of CDK1 is mediated by dephosphorylation of CDK1 at Tyr15 by CDC25C (Tsuchiya, Murai and Yamashita 2015). Additionally, the elevated expression of p21 can downregulate the expression of CDK1-Cyclin B complex to induce G2/M phase arrest (Chang et al., 2000). It has been demonstrated that signaling mediated by mitogen-activated protein kinases (MAPKs) plays an important role in regulating the cell cycle and responding to DNA damage (Schaeffer and Weber 1999). MAPKs are classified into three subfamilies based on their sequence similarity and the nature of their upstream activators: JNK/SAPK, ERK1/2 and p38. Extracellular signal-regulated kinase (ERK) is a major transmitter of extracellular signals that connect cellular membrane receptor stimulation to alterations in cellular function (Robinson and Cobb 1997; Roux and Blenis 2004; Ramos 2008). The transient activation of ERK is critical in cell proliferation, whereas continuous ERK activation triggers cell G2/M phase arrest by promoting CDC25C ubiquitination and proteasomal degradation (Eymin et al., 2006; Kar et al., 2006; Guo et al., 2011; Wang et al., 2016) and by upregulating p21 expression (Karkhanis and Park 2015). Additionally, the newly developed clinical drug Ibrance, the first CDK4/6 inhibitor approved by the FDA in 2015, targets the cell cycle and may be effective and safe against certain types of breast cancer (Thomas et al., 2014).
In addition to the uncontrollable proliferation of glioma, invasion is another distinctive feature of its malignant phenotype. The focal adhesion complex composed of integrins serves as the primary site of cell adhesion to the extracellular matrix and interacts with the actin cytoskeleton to regulate cell motility. The dynamic regulation of focal adhesion and the associated actin cytoskeleton reorganization are critical factors for cell invasion (Carragher and Frame 2004). Focal adhesion kinase (FAK) was discovered to associate with Src via a Src homology domain-mediated interaction after it was initially identified as a tyrosine phosphorylated protein in cells transformed by the Src oncogene (Kanner et al., 1990; Schaller et al., 1992; Cobb et al., 1994). The FAK-Src complex interacts with and phosphorylates a number of adaptor proteins, including P130cas (Hsia et al., 2005) and Paxillin (Schlaepfer, Mitra and Ilic 2004), to promote tumor invasion. Furthermore, the ever-present second messenger Ca2+ has been identified as a vital regulator of cancer cell invasion by regulating the dynamic cycle of focal adhesion complex assembly and disassembly (Chang et al., 2017; Wu et al., 2021). Exploring RB’s target proteins in glioma cells will help us better understand RB’s anticancer mechanisms and generate new treatment options for glioma. Chansu has been reported to inhibit Na+-K+-ATPase (Bick et al., 2002; Wang, Sun and Heinbockel 2014). Na+-K+-ATPase, which is found in the plasma membrane, is a classic ion transporter enzyme that exchanges Na+ for K+ in cells. ATP1A1 is one of 4 isoforms of the Na+-K+-ATPase alpha subunit identified in humans (Clausen, et al., 2017). When Na+-K+-ATPase is only partially inhibited, there is no significant disruption of the homeostasis of intracellular Na+ and K+. Depending on the cell after activation by the appropriate ligand, Na+-K+-ATPase stimulates the proliferation of healthy cells (Aydemir-Koksoy, Abramowitz and Allen 2001; Chen et al., 2019) or, in contrast, inhibits the proliferation of tumor cells (Prassas et al., 2011). Moreover, ATP1A1 can activate the MAPK/ERK pathway (Ono et al., 2016), and Na+-K+-ATPase can alter intracellular Ca2+ concentrations (Bick et al., 2002). Therefore, exploring the mechanisms of RB in glioma will identify new clinical agents for the targeted treatment of glioma.
Taken together, our study shows that when RB binds to ATP1A1, Na+-K+-ATPase is activated as a receptor, accompanied by activation of the intracellular MAPK/ERK pathway and inhibition of the Ca2+-regulated Src/FAK/Paxillin focal adhesion pathway, after which GBM cells undergo G2/M phase arrest and inhibition of invasion.
All animal procedures were approved by the Ethics Committee of Qilu Hospital of Shandong University (permit number: DWLL-2-21-042). This study was conducted in full adherence to relevant regulations and guidelines.
The human GBM cell lines U251 and A172 were purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Normal human astrocyte (NHA) cells were kindly provided by Prof. Rolf Bjerkvig at the Department of Biomedicine, University of Bergen, Norway. Complete medium was used to maintain the GBM cell lines and NHA cells: Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific; Waltham, MA, United States) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), streptomycin (100 g/ml), and penicillin (100 U/mL). Prof. Rolf Bjerkvig of the Department of Biomedicine at the University of Bergen, Norway kindly provided the patient-derived GBM stem-like cell (GSC) line P3#GBM. P3#GBM was obtained from Haukeland University Hospital (Bergen, Norway) following approval of the local ethics committee (approval number 2009/117) (Golebiewska et al., 2020). In addition, P3#GBM primary cells have been cultured in Prof. Rolf Bjerkvig’s laboratory since 2009, and several associated articles have been published (Wang et al., 2009; Han et al., 2020). P3#GBM cells were cultured in serum-free Neurobasal™ medium (Gibco/Thermo Fisher Scientific) supplemented with 2% B-27 Neuro Mix (Thermo Fisher Scientific), 20 ng/ml epidermal growth factor (EGF; Thermo Fisher Scientific), 10 ng/ml basic fibroblast growth factor (bFGF; PeproTech; Rocky Hill, NJ, United States), and streptomycin (100 g/ml), and the cells were incubated at 37°C in a humidified atmosphere composed of 95% air and 5% CO2.
RB (purity ≥99.69%, CAS# 465-39-4), purchased from MedChemExpress (MCE; Monmouth, NJ, United States), was dissolved in dimethyl sulfoxide (DMSO; Sigma–Aldrich; St. Louis, MO, United States) and stored at −20°C as a stock solution (10 and 50 mM). Ruthenium red (RR; purity ≥95.0%, CAS# 11,103-72-3) purchased from MCE was dissolved in DMSO and stored at −20°C as a 10 mM stock solution. U0126 (CAS# 109,511-58-2) purchased from Beyotime (Shanghai, China) was dissolved in DMSO as a 10 mM stock solution and stored at −20°C. The stock solutions were diluted directly in complete medium, and the final DMSO concentration was <0.1%.
The following antibodies were used: MMP-2 (#87809S, CST; Boston, MA, United States), Vimentin (#5741S, CST), N-cadherin (#13116S, CST), Snail (#3879S, CST), phospho-ERK (p-ERK,Thr202/Tyr204, #4370S, CST), ERK (#4695S, CST), phosphor-JNK (p-JNK,Thr183/Tyr185, #4668S, CST), JNK (#9552S, CST), phospho-p38 (p-p38, Thr180/Tyr182, #4511S, CST), p38 (#8690S, CST), GAPDH (#5174S, CST), p21 (#2947S, CST), CDC25C (#4688S, CST), phospho-CDK1 (p-CDK1, Tyr15, #9111S, CST), CDK1 (#28439S, CST), Cyclin B1 (#12231S, CST), Src (#2109S, CST), phosphor-Src (p-Src, Ser17, #12432SS, CST), ATP1A1 (#23565S, CST) Cyclin B2 (#ab185622, Abcam; Cambridge, MA, United States), FAK (#ab40794, Abcam), phospho-FAK (p-FAK, Y397, #ab81298, Abcam), Paxillin (#ab32084, Abcam), and phospho-Paxillin (p-Paxillin, Y118, #ab109547, Abcam). Secondary antibodies against goat anti-rabbit/mouse were purchased from Zhongshan Golden Bridge Biotechnology (Zhongshan Golden Bridge; Beijing, China). The luminance was determined using a chemiluminescence imager (Bio–Rad ChemiDoc XRS+; Hercules, CA, United States) in accordance with the manufacturer’s protocol.
The Cell Counting Kit-8 assay (CCK-8; Dojindo, Kumamoto, Japan) was performed to determine cell viability. To obtain an accurate IC50 of each cell, we experimented with different concentration gradients. For P3#GBM cells, we used a concentration gradient of RB at 0, 0.5, 1, 1.5, 2, 2.5, 3 and 3.5 μM for 48 h. For the GBM cell lines U251 and A172, we used RB concentrations of 0, 1.5, 3, 4.5, 6, 7.5 and 9 μM for 48 h. For NHA cells, we used a concentration gradient of RB at 0, 5, 15, 20, 25, 30, 35, 40 and 45 μM for 48 h. All final DMSO concentrations were <0.1%. P3#GBM, U251 and A172 cells (4 × 103 cells/well) were inoculated in 96-well plates and incubated at 37°C. After 24 h, the original culture medium was replaced with 100 μL of culture medium containing various doses of RB or vehicle control (diluted DMSO). After 24, 48 and 72 h of treatment, P3#GBM, U251 and A172 cells were incubated in 10 μL of CCK-8 reagent with 90 μL of serum-free DMEM for 1 h. An EnSight multimode plate scanner was used to detect the absorbance at 450 nm (PerkinElmer, Hopkinton, MA, United States).
U251 and A172 cells (800 cells/well) were inoculated in 6-well plates. After cell attachment, U251 and A172 cells were treated with DMSO or RB (2 or 4 μM), and then fresh medium was replaced once every 3 days for the duration of the expression. After incubation for 14 days, 4% paraformaldehyde was used to fix U251 and A172 colonies for 5 min, and then 0.5% crystal violet was used to stain U251 and A172 colonies for 15 min and washed three times with PBS. A bright-field microscope was used to count colonies with more than 50 cells (Leica; Solms, Germany).
5-ethynyl-2′-deoxyuridine (EdU) binding assays were used to detect the binding of EdU, a thymidine analog, to proliferating cells via a catalytic reaction between EdU and Apollo fluorescent dye (RiboBio; Guangzhou, China). The nuclei of the cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). The number of EdU-positive cells in each well was determined using a fluorescence microscope (Leica, Solms, Germany).
Western blotting was performed as previously described (Yao et al., 2020). P3#GBM, U251 and A172 cells were treated with RB for 48 h and then lysed in RIPA lysis buffer (Beyotime) with protease and phosphatase inhibitors (Beyotime) for 30 min and sonicated to enhance lysis. The luminescence intensity was detected with a chemiluminescence imager (Bio–Rad ChemiDoc XRS+; Hercules, CA, United States).
P3#GBM, U251 and A172 (4 × 105) were inoculated in 6-well plates. After treating cells with (diluted DMSO) or RB (2 and 4 μM) for 48 h, the cells were digested, centrifuged, and incubated overnight at 4°C in cold 70% ethanol. Samples were centrifuged, stained with propidium iodide (BD Biosciences; San Jose, CA, United States) for 10-15 min protected from light and subjected to cell cycle analysis by flow cytometry (Accuri C6, BD Biosciences). ModFit software (Becton Dickinson; San Diego, CA, United States) was used to identify P3#GBM, U251 and A172 cell cycle distributions.
To determine how RB affected GBM cell invasion, we carried out three invasion assays: transwell invasion assays, 3D spheroid invasion assays and GBM-brain organoid coculture invasion assays.
The transwell invasion assay was performed using 24-well Matrigel invasion chambers (Costar, Cambridge, MA, United States). Briefly, Matrigel was mixed with serum-free cell culture medium at 4°C in a 1:8 dilution ratio. One hundred microliters were applied uniformly to the surface of the polycarbonate membrane in the upper chamber and left at 37°C for 1 h to polymerize into a gel. The cells were suspended in inserts (2 × 104 cells/200 μL). DMEM containing 30% fetal bovine serum was added to the lower chamber (600 μL). U251 and A172 cells were treated with vehicle control (diluted DMSO) or RB (2 or 4 μM) for 48 h. Fixation, staining, digital imaging and counting of cells under invading membranes.
In the 3D spheroid invasion assay, P3#GBM, U251 or A172 cells (3,000 cells/well) were inoculated in a U-shaped 96-well plate (Trevigen; Gaithersburg, MD, United States) to develop tumor spheroids for 3 days, and then tumor spheroids were embedded in invasive matrix (Trevigen; Gaithersburg, MD, United States). Medium containing vector control (diluted DMSO) or RB (2 or 4 μM) was replaced with fresh complete medium, and 0-h spheroids were pictured as a reference point to measure the invasive area. The relative invasion area of tumor spheroids was measured on 6th day.
Our previous work described the protocol for culturing 18-days rat fetal brain organoids for the GBM brain organoid coculture invasion ex vivo system (Bjerkvig, Laerum and Mella 1986). After 3 weeks in culture, the differentiation of cells in normal brain organoids was complete, and brain organoids could be exposed to tumor organoids. U251-GFP and A172-GFP cells (n = 3,000), established using GFP lentiviruses, were plated in 96-well plates for 3 days to generate tumor spheroids and then cocultured with mature brain organoids for 24 h. GBM organoids were treated with diluted DMSO or 4 μM RB for 0, 48 and 96 h. Confocal microscopy captured coculture images of tumor cell invasion (Leica DMi8; Solms, Germany). ImageJ (National Institutes of Health; Bethesda, United States) software was used to analyze the GBM cell-related invasive area.
Na+-K+-ATPase activities were determined by a Na+-K+-ATPase kit (A070-2-2; Nanjing Jiancheng Bioengineering Institute; Nanjing, China). The sample and mixed reagents were added according to the manufacturer’s instructions and reacted precisely at 37°C for 10 min. Then, R4 was added and centrifuged for 10 min at 3,500 rpm, and the supernatant was collected. Then, the color developer was added to the supernatant at room temperature for 2 min, and R6 was added for 5 min at room temperature. An EnSight multimode plate scanner was used to detect the absorbance at 636 nm and 1 cm optical diameter (PerkinElmer, Hopkinton, MA, United States).
A total of 2–4 ×105 GBM cells were inoculated in each 6-well plate, and the cells were treated with diluted DMSO or 4 μM RB for 48 h. After 48 h of treatment, the cells were washed three times with PBS and incubated with 10 μM 2′,7′-dichlorofluorescein diacetate (DCFH-DA, Sigma–Aldrich) for 30 min. After incubation, DCFH-DA was replaced with PBS. The fluorescence intensity of DCFH-DA was detected by flow cytometry (BD C6, San Jose, CA, United States). FlowJo software was used to analyze flow cytometry data (Treestar; Ashland, OR, United States), and at least 1 × 105 cells were analyzed.
RNA sequencing (LC-Bio; Shanghai, China) was performed on RB-treated P3#GBM cells (4 μM RB) and U251 cells (4 μM RB) and their parent cell lines. Full data accompanying this experiment are available in the Sequence Read Archive (SRA). The accession number is PRJNA796785.
Fluo-4/AM (MCE) was added to the cells after they had been treated with diluted DMSO or RB (2 or 4 μM) for 48 h. The solution was kept in the dark at room temperature for 30 min. Heated Hank’s balanced salt solution was used to wash U251 and A172 cells. Intracellular Ca2+ levels in GBM cells were detected by measuring the fluorescence intensity of the calcium indicator Fluo-4/AM with fluorescence microscopy and flow cytometry. FlowJo software was used to analyze flow cytometry data.
Short interfering RNA sequences (siRNAs) silencing ATP1A1 (GenePharma Gene; Shanghai, China) were transfected into cells with Lipofectamine 3,000 reagent (Thermo Fisher Scientific). The silencing efficiency in U251 and A172 cells was assessed by immunoblotting 48 h after transfection. The siRNA sequence was as follows: siATP1A1#3 5′-GGAGGCUUCUUUACUUACUTT-3′.
We constructed P3#GBM cells infected with lentivirus expressing fluorescein-GFP for animal studies. A total of 4 × 105 fluorescein-GFP P3#GBM cells in a 10 μl cell suspension were stereotaxically inoculated into 4-week-old BALB/c nude male mouse brains (1 mm posterior to bregma, 2 mm right of midline suture, depth 1.5 mm) (Han et al., 2019; Johannessen et al., 2019). Male mice were chosen because of the uneven tumor formation in the animal model due to individual differences in sex hormones in female mice. On the 7th day, after determining tumor size, the mice were randomly divided into two groups: the NC (n = 10) and RB groups (n = 10). Mice were administered daily intraperitoneal injections of NC (DMSO) or RB (10 mg/kg/day). Bioluminescence imaging was used to evaluate the size of the tumor every week (IVIS Spectrum, Hopkinton, MA, United States). During imaging, mice were intraperitoneally administered D-fluorescein and potassium salt D (150 mg/kg; Yeasen Biotech; Ltd., Shanghai, China) under isoflurane gas anesthesia. The survival of each mouse was recorded after death. Kaplan–Meier analysis was performed on the median survival of the tumor-bearing mice, and log-rank analysis was used to measure the statistical significance of the differences. Finally, the tumors were cut up and frozen in liquid nitrogen or placed in formalin for immunohistochemistry.
Tumors were separated from the sacrificed mice, fixed in 4% paraformaldehyde and embedded in paraffin. The paraffin-embedded samples were sectioned (4 μm) and secured on glass slides. The sections were heated in a microwave oven at pH 7.2 in 10 mM citric acid buffer for epitope retrieval. The slides were then incubated with primary antibodies (rabbit anti-Ki-67 1:200 dilution; rabbit anti-ATP1A1 1:100 dilution) overnight at 4°C, followed by incubation with HRP-conjugated secondary antibodies for 1 h at room temperature. Antibodies were detected using the substrate diaminobenzidine (Beyotime), and slides were counterstained with hematoxylin (Beyotime).
Two pathologists independently evaluated each immunostained section under light microscopy. Immunoreactive scores were calculated by multiplying the intensity of staining (from 0 to 4; 0 = negative, 1 = weakly positive; light yellow, 2 = moderately positive; yellowish-brown, 3 = strongly positive; brown) by the percentages of stained-positive cells (from 0 to 100).
At least three biological replicates of each experiment were performed. Data are provided as the mean ± SEM. Statistical analysis was performed using GraphPad Prism 8. The following p values were considered significant: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
The chemical structure of RB is shown in Figure 1A. The half-maximal inhibitory concentration (IC50) values were 2.29, 3.05 and 6.21 µM in the primary GBM cell line P3#GBM and in two GBM cell lines, U251 and A172. However, the IC50 value was 32.66 µM in NHA cells. NHA cells showed proliferation inhibition only at higher concentrations compared to GBM cells, suggesting that RB may be more sensitive to inhibiting GBM cell proliferation at certain concentrations. To determine the cytotoxic effects and inhibitory activity of RB in GBM cells, we treated P3#GBM, U251 and A172 cells with RB at 2 or 4 µM concentrations for 24, 48 and 72 h in vitro. CCK-8 assays showed that RB significantly inhibited the proliferation of GBM cells in a concentration-dependent manner (Figure 1B and Supplementary Figure S1A).
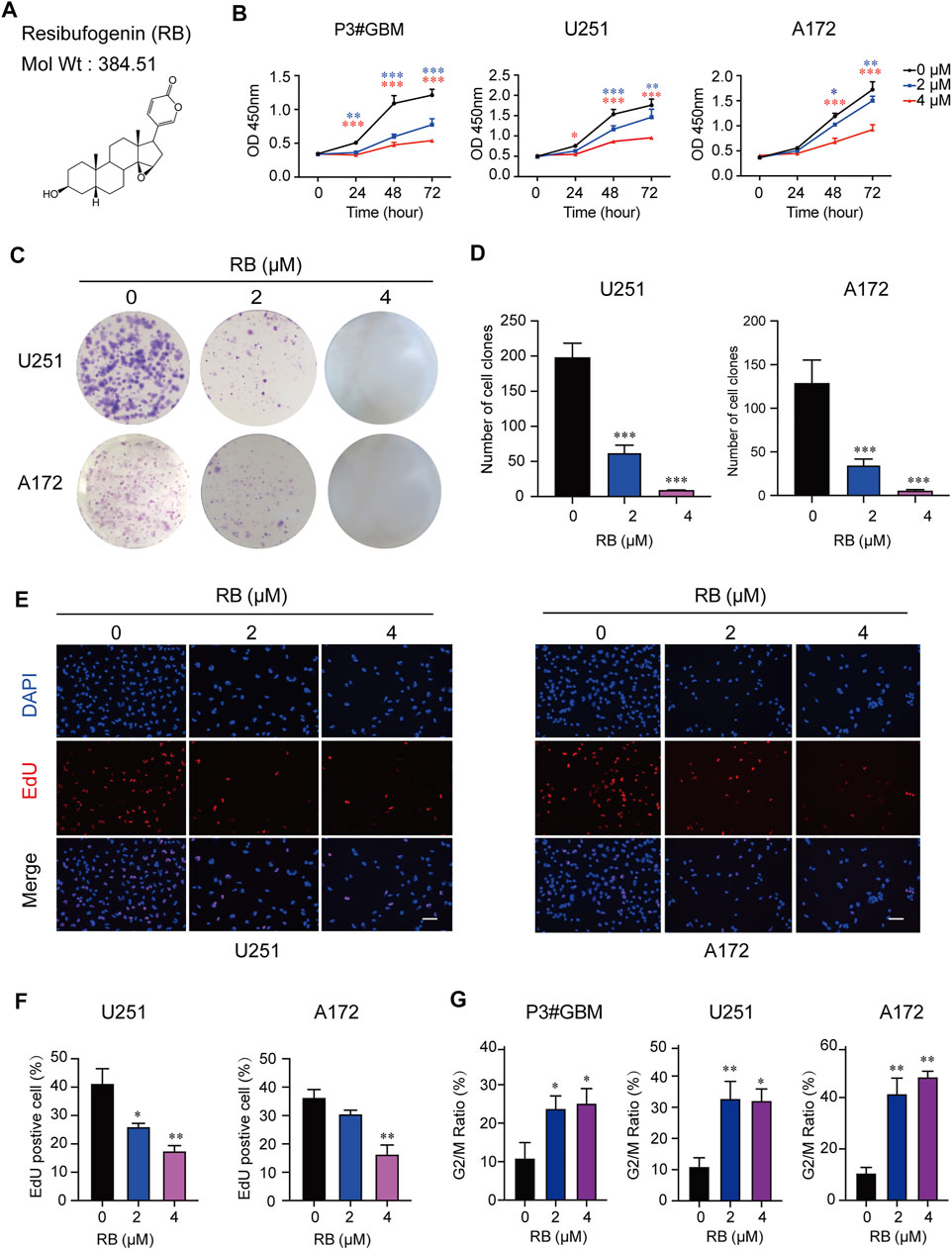
FIGURE 1. RB inhibits GBM cell proliferation and induces GBM cell cycle arrest in the G2/M phase. (A) Chemical structure of RB drawn through ChemDraw 20.0. The molecular weight of RB is 384.51 g/mol. The topological polar surface area of RB is 59.06. P3#GBM, U251 and A172 GBM cells were treated with 2 μM or 4 μM RB under normal culture conditions. (B) Using the CCK-8 assay, proliferation curves of P3#GBM, U251 and A172 GBM cells treated with vehicle, DMSO (0), 2 μM or 4 μM RB for 0, 24, 48 or 72 h. (C) Representative images of the cell clone formation assay for U251 and A172 cells given DMSO (0), 2 μM or 4 μM RB. (D) Graphic representation of the number of colonies shown in (C). (E) EdU assays conducted on U251 and A172 cells treated with vehicle, DMSO (0) and 2 or 4 μM RB for 48 h. EdU was detected by Apollo 567 staining (red), nuclei were highlighted by DAPI staining (blue), and images were merged. Scale bar, 50 μm. (F) Graphic representation of the percentages of EdU-positive U251 and A172 cells shown in (E). (G) Graphic representation of the percentages of G2/M phase cells calculated from each group (Supplementary Figure S1B). Three independent experiments were performed with data shown as the mean ± SEM by two-way ANOVA (B) or one-way ANOVA (D,F,G). *p < 0.05; **p < 0.01 and ***p < 0.001.
To determine whether RB inhibited the proliferation of GBM cells, we performed a colony formation assay. The results showed that RB at 2 μM significantly reduced the number of U251 and A172 cell colonies, and no colonies appeared under 4 μM RB treatment (Figures 1C,D). These results were also confirmed in EdU assays, where the percentages of EdU-positive cells were significantly reduced in the U251 and A172 cells treated with 2 and 4 μM RB (Figures 1E,F), indicating that RB significantly inhibited the proliferation of human GBM cells.
To further determine whether proliferation inhibition induced by RB was associated with cell cycle arrest, we examined the effect of RB on the cell cycle of P3#GBM, U251 and A172 cells. Flow cytometry data indicated that RB significantly inhibited cell cycle progression in P3#GBM, U251 and A172 cells. A significant increase in the G2/M phase fraction was coupled with a decrease in the G0/G1 phase fraction (Figure 1G and Supplementary Figure S1B). Taken together, these results revealed that RB inhibited cell proliferation and induced G2/M-phase cell cycle arrest in GBM cells.
To investigate whether RB affects cell invasion, we performed transwell assays and 3D spheroid-based invasion assays. The results showed that in both transwell (Figures 2A,B) and 3D spheroid-based invasion assays (Figures 2C,D), treatment with RB significantly reduced GBM cell invasion by ∼2–4-fold compared with that in the control cells. Next, to further demonstrate the inhibitory effect of RB on GBM cell invasion, we conducted coculture experiments with GBM spheroid and brain organoid coculture assays. GBM cells were cultured for 3 days to develop tumor spheroids, which were then cocultured with mature rat brain organoids and treated with diluted DMSO or RB (4 μM). As shown in Figures 2E,G, after 48 and 96 h of treatment, the invasive area by GBM spheroids was decreased significantly in the 4 μM RB-treated U251 spheroids compared to the control U251 spheroids. We found the same results in 4 μM RB-treated A172 spheroids, where RB also reduced the area invaded by tumor cells after 48 and 96 h (Figures 2F,G).
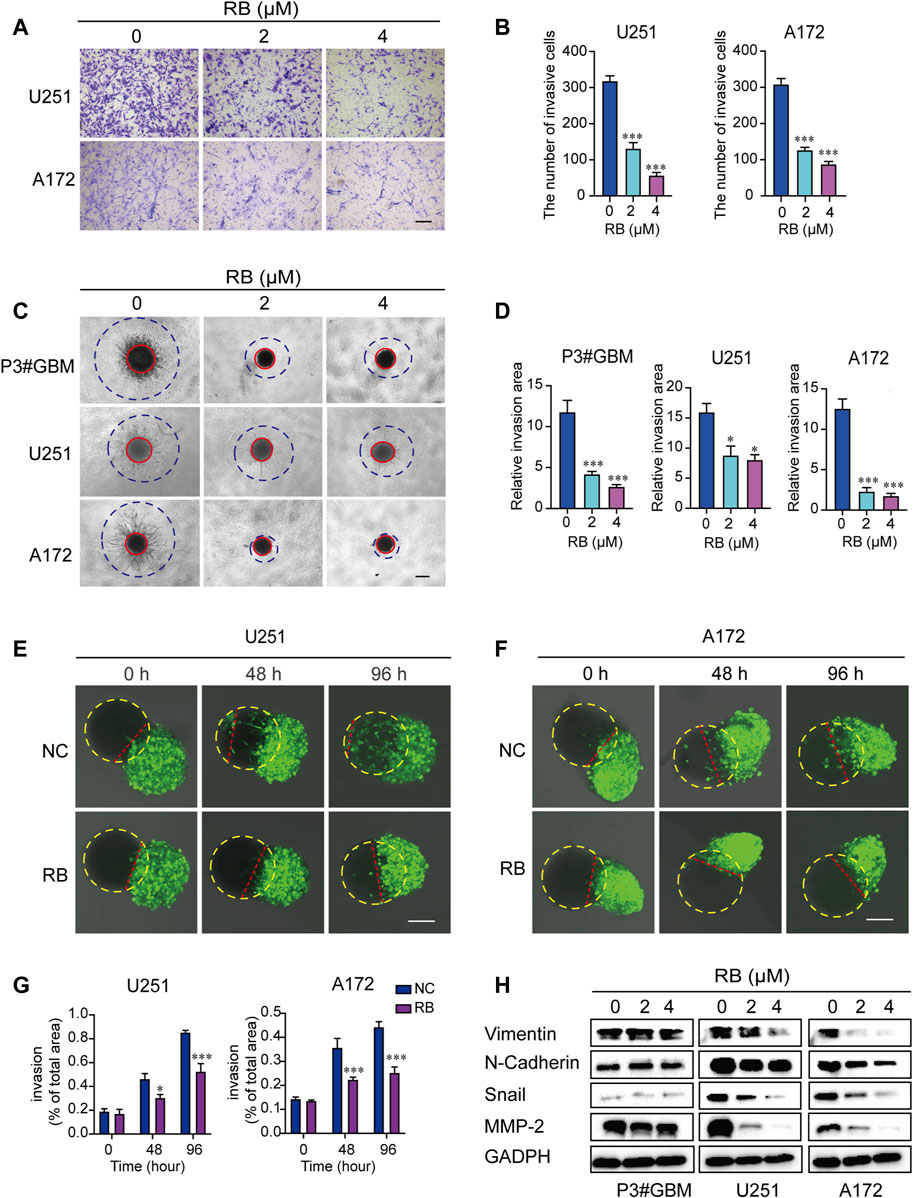
FIGURE 2. RB inhibits the invasion of GBM cells. (A) Representative images of transwell invasion assays. Scale bar, 100 μm. (B) The numbers of invasive GBM cells shown in (A). (C) Representative images of the 3D spheroid invasion assay performed on P3#GBM, U251 and A172 cells treated with vehicle control (diluted DMSO) or RB (2 or 4 μM) and evaluated on the 6th day. Scale bar, 200 μm. (D) Graphic representation of the relative invasive area shown in (C). (E) Representative images of coculture invasion assays for U251 cells treated with vehicle, DMSO (0) and 4 μM RB and evaluated at 0, 48, and 96 h. The yellow dotted line represents brain organoids, and the red dotted line represents the U251 cell invasion range. Scale bar, 100 μm. (F) Representative images of coculture invasion assays for A172 cells treated with vehicle, DMSO (0) and 4 μM RB and evaluated at 0, 48, and 96 h. The yellow dotted line represents brain organoids, and the red dotted line represents the A172 cell invasion range. Scale bar, 100 μm. (G) Quantification of the invasive area after 0, 48, and 96 h of 4 μM RB treatment shown in (E,F). (H) Western blot analysis of MMP2 and EMT-related proteins after 4 μM RB treatment for 48 h. Three independent experiments were performed with data shown as the mean ± SEM by two-way ANOVA (G) or one-way ANOVA (B,D). *p < 0.05; **p < 0.01, and ***p < 0.001.
In addition, we examined the effect of RB on the protein expression levels of several mesenchymal markers in GBM cells. We found that RB inhibited GBM cell invasion by decreasing the expression of Vimentin, N-cadherin, Snail and MMP-2 (Figure 2H). These data suggested that RB suppressed the invasion of GBM cells.
As shown in Figure 1G and Supplementary Figure S1B, flow cytometry analysis indicated that G2/M phase arrest occurred in GBM cells following 2 or 4 μM RB treatment. To elucidate the mechanisms of RB-induced G2/M arrest in GBM cells, we performed whole transcriptome sequencing in vehicle control (diluted DMSO) and 4 μM RB-treated P3#GBM and U251 cells. The differentially expressed mRNAs were chosen using the cutoffs of p value <0.05 and log2 fold change >1. Figure 3A shows that 913 and 812 mRNAs had upregulated expression in P3#GBM and U251 cells, respectively, and that the number of intersecting genes with upregulated expression between the two GBM cell lines was 509. To shed light on the biological functions of the intersecting genes, we performed KEGG analysis of the intersecting genes with upregulated expression and found that the MAPK pathway was enriched as the top pathway (Figure 3B). In light of the fact that MAPK families have been shown to play a vital role in cycle transition (Zhang and Liu 2002; Wang et al., 2007), we predicted that the activation of the MAPK pathway might be a key determinant of RB-induced G2/M phase arrest. Then, we examined which kinds of MAPKs mediate RB-induced G2/M phase arrest in GBM cells. As shown in Figure 3C, the total amount of ERK1/2, JNK and p38 MAP kinases did not change significantly. The significant activation of ERK1/2 was observed in P3#GBM, U251 and A172 cells, but not JNK and p38. Only activation of ERK was consistent with the sequencing result that MAPK signaling pathway was significantly upregulated.
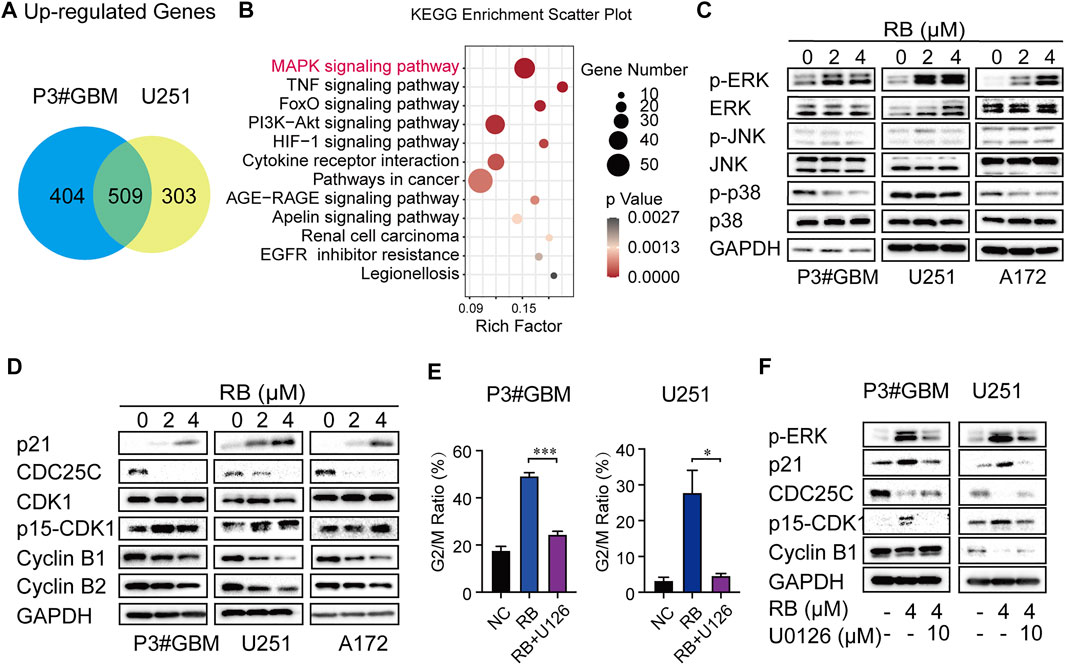
FIGURE 3. Activation of the ERK/MAPK pathway involves RB-induced G2/M arrest in GBM cells. (A) Intersections of genes with upregulated expression in P3#GBM and U251 cells treated with 4 μM RB compared with control cells. (B) KEGG pathway enrichment analysis of intersecting genes with upregulated expression. (C) Western blot analysis of total and phosphorylated protein levels of ERK, JNK and p38 after 2 or 4 μM RB treatment for 48 h in P3#GBM, U251 and A172 cells compared with control cells. (D) G2/M phase-related protein expression after RB treatment for 48 h in P3#GBM, U251 and A172 cells analyzed by western blot. (E) P3#GBM and U251 cells were pretreated with U0126 (10 μM; ERK inhibitor) for 6 h, and then, 4 μM RB was incubated for another 48 h. Graphic representation of percentages of G2/M phase cells calculated from each group were shown in Supplementary Figure S1C. (F) Western blot analysis of the protein expression of p-ERK, p21, CDC25C, p15-CDK1 and cyclin B1 in P3#GBM and U251 cells pretreated for 6 h with U0126 (10 μM) and then treated with 4 μM RB. Data from three independent experiments were displayed as the mean ± SEM by one-way ANOVA (E). *p < 0.05; **p < 0.01, and ***p < 0.001.
Activation of the MAPK/ERK pathway has been reported to induce cancer cells to arrest in G2/M phase by regulating the protein expression levels of p21 and CDC25C (Zhang and Liu 2002; Wang et al., 2007). The levels of several G2/M-related proteins in GBM cells treated with RB were then determined using western blotting. Cyclin B1, Cyclin B2, and CDC25C levels were decreased, whereas p21 and Tyr15-phospho-CDK1 levels were increased and total CDK1 remained unchanged (Figure 3D), indicating that G2/M phase arrest induced by RB was associated with altered levels of key checkpoint-related proteins.
We hypothesized that the activation of the MAPK/ERK pathway contributed to RB-mediated G2/M phase arrest. To ascertain the function of ERK in RB-induced G2/M phase arrest, we pretreated P3#GBM and U251 cells for 6 h with the specific inhibitor of ERK, U0126 (10 μM). As shown by flow cytometry, the ratios of cells in the G2/M phase were markedly increased in cultures treated with 4 μM RB compared to the DMSO control, but RB-mediated G2/M phase arrest was significantly rescued by U0126 in P3#GBM and U251 cells (Figure 3E and Supplementary Figure S1C). Next, we examined whether an inhibitor of ERK modulates the expression of key checkpoint proteins in RB-induced G2/M phase arrest. As shown in Figure 3F, the protein expression level of p-ERK was significantly attenuated by treatment with the ERK inhibitor U0126, indicating that the MAPK/ERK pathway was engaged in RB-induced GBM cell G2/M phase arrest. U0126 restored RB-induced inhibition of Cyclin B1, Cyclin B2 and CDC25C expression and blocked Tyr15-phospho-CDK1 and p21 expression in P3#GBM and U251 cells. These results suggested that RB induced GBM cells to arrest in G2/M phase via the MAPK/ERK/CDC25C/p21/CDK1/Cyclin B pathway.
Figure 4A reveals that a total of 2018 and 1938 mRNAs had downregulated expression in P3#GBM and U251 cells, respectively, and that the number of intersecting genes with downregulated expression between the two GBM cell lines was 904. To probe the mechanism of RB-mediated inhibition of GBM cell invasion, we performed KEGG pathway enrichment of intersecting genes with downregulated expression and found that focal adhesion was enriched as the top pathway (Figure 4B).
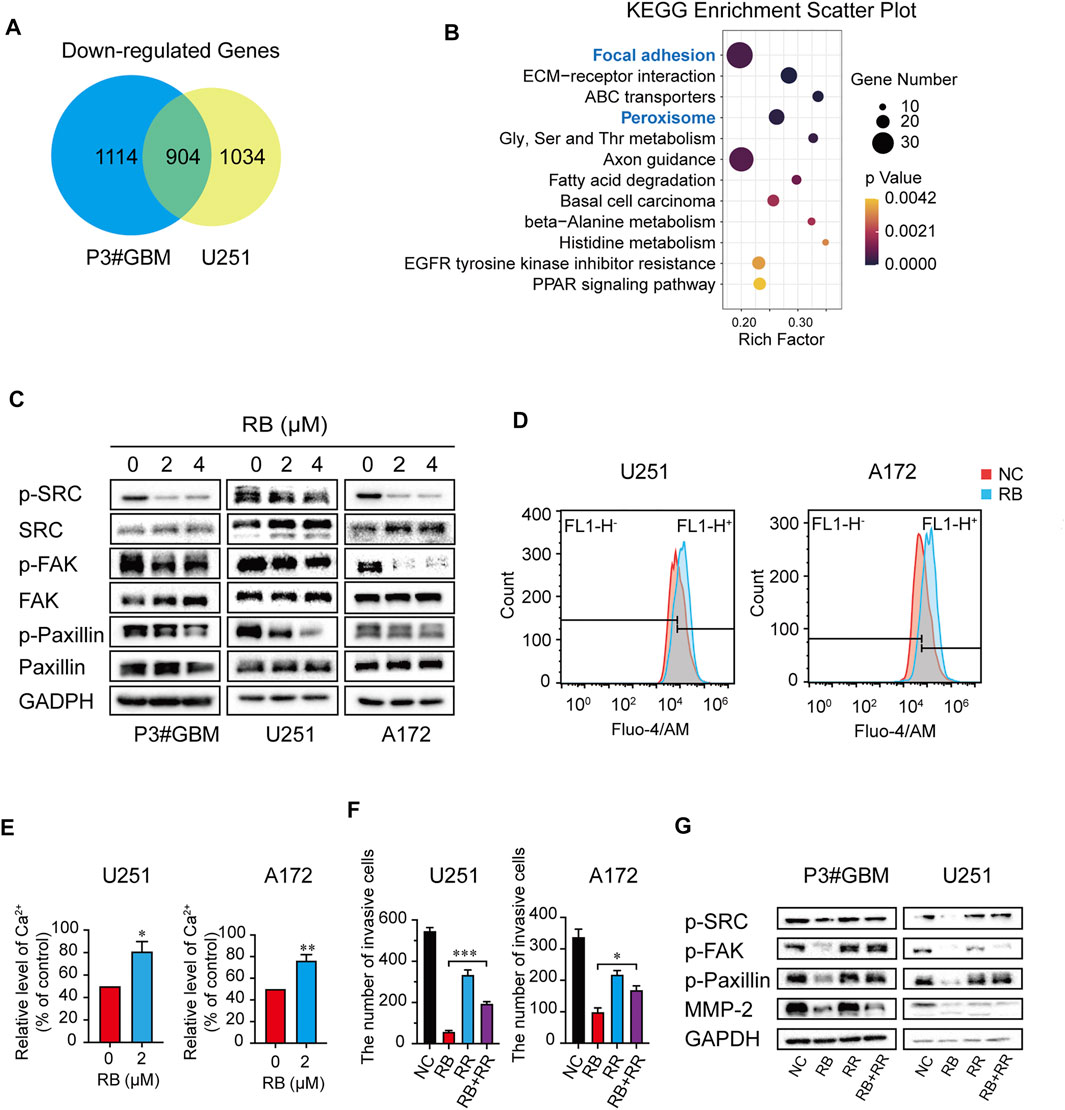
FIGURE 4. RB inhibits GBM cell invasion partially by elevating intracellular Ca2+ levels to suppress the Src/FAK/Paxillin focal adhesion pathway. (A) Intersections of genes with downregulated expression in P3#GBM and U251 cells treated with 4 μM RB compared with control cells. (B) KEGG pathway enrichment analysis of intersecting genes with downregulated expression. (C) P3#GBM, U251 and A172 glioma cells were treated with RB for 48 h. Western blot analysis was used to determine focal adhesion-associated protein expression levels. (D) Intracellular Ca2+ was measured by Fluo-4/AM with flow cytometry. (E) Graphic representation of the mean fluorescence intensity of Fluo-4/AM shown in (D). (F) The numbers of invasive GBM cells were shown in Supplementary Figure S2G. (G) Western blot analysis showed that blocking Ca2+ with RR treatment for 48 h rescued RB-induced inhibition of invasion in U251 and A172 cells. Three independent experiments were performed with data shown as the mean ± SEM by unpaired Student’s t test (E) or one-way ANOVA (F). *p < 0.05; **p < 0.01 and ***p < 0.001.
Src, FAK, and Paxillin are the main elements of focal adhesion, a high-intensity attachment points of cells to the extracellular matrix formed by the leading edge of the invading or attached cells (Stallcup 2017; Schiffer et al., 2018). We hypothesized that RB inhibits GBM cell invasion partly by suppressing the Src/FAK/Paxillin focal adhesion pathway in GBM cells. Our results showed that RB treatment decreased the phospho-Src, FAK, and Paxillin levels, although it had no effect on focal adhesion-related total Src, FAK, and Paxillin levels (Figure 4C), suggesting that RB inhibited GBM cell invasion by suppressing the Src/FAK/Paxillin focal adhesion pathway.
A previous study reported that maintained Ca2+ influx can destroy the components of FAK’s focal connections in the presence of Src deficiency, markedly inhibiting cell invasion and motility (Jeong et al., 2020). We further investigated whether RB altered intracellular Ca2+ levels to suppress the Src/FAK/Paxillin focal adhesion pathway, thereby inhibiting the invasion of GBM cells.
To determine whether Ca2+ in cells was a key target for RB treatment, we first assessed intracellular Ca2+ levels using flow cytometry and fluorescence microscopy. As shown in Figures 4D,E and Supplementary Figures S2A,B, intracellular Ca2+ levels were significantly elevated in the 4 μM RB-treated cells compared with the control cells. To investigate whether Ca2+ elevation was a crucial factor in the RB-inhibited focal adhesion pathway, we first treated RB-treated U251 and A172 cells with RR (3 μM), an L-type calcium current blocker, and decreased Ca2+ levels were observed after treatment with the combination of 4 μM RB and 3 μM RR compared to RB treatment alone (Supplementary Figures S2C,D). These same results were also confirmed by flow cytometry, where 3 μM RR blocked the Ca2+ elevation induced by RB (Supplementary Figures S2E,F). Furthermore, transwell assays revealed that the combination of 4 μM RB and 3 μM RR significantly rescued invasion in U251 and A172 cells compared to RB treatment alone (Figure 4F and Supplementary Figures S2G), suggesting that elevated Ca2+ inhibited invasion in treated cells. Additionally, western blot revealed that the activity of focal adhesion-related proteins inhibited by RB could be rescued by reducing intracellular Ca2+ by RR (Figure 4G). Taken together, our results demonstrated that RB inhibited GBM cell invasion partly by elevating intracellular Ca2+ levels to suppress the Src/FAK/Paxillin focal adhesion pathway.
We found that RB-mediated G2/M phase arrest inhibited GBM cell proliferation by activating the MAPK/ERK pathway, which altered p21/CDC25C/CDK1/Cyclin B protein expression, and RB inhibited GBM invasion by increasing intercellular Ca2+ to suppress the Src/FAK/Paxillin focal adhesion pathway. Thus, exploring RB target proteins in GBM cells would provide insight into the mechanisms underlying the inhibition of GBM malignant progression. The traditional Chinese medicine Chansu is known to be a specific inhibitor of Na+-K+-ATPase. Moreover, previous research showed that RB could alter intracellular K+ and Na+ concentrations by affecting the function of Na+-K+-ATPase (Hao et al., 2011). To predict the most likely Na+-K+-ATPase-related target proteins of RB, we used BioSolveIT SeeSAR software, which indicated that RB may target ATP1A1 in GBM cells (Figure 5A).
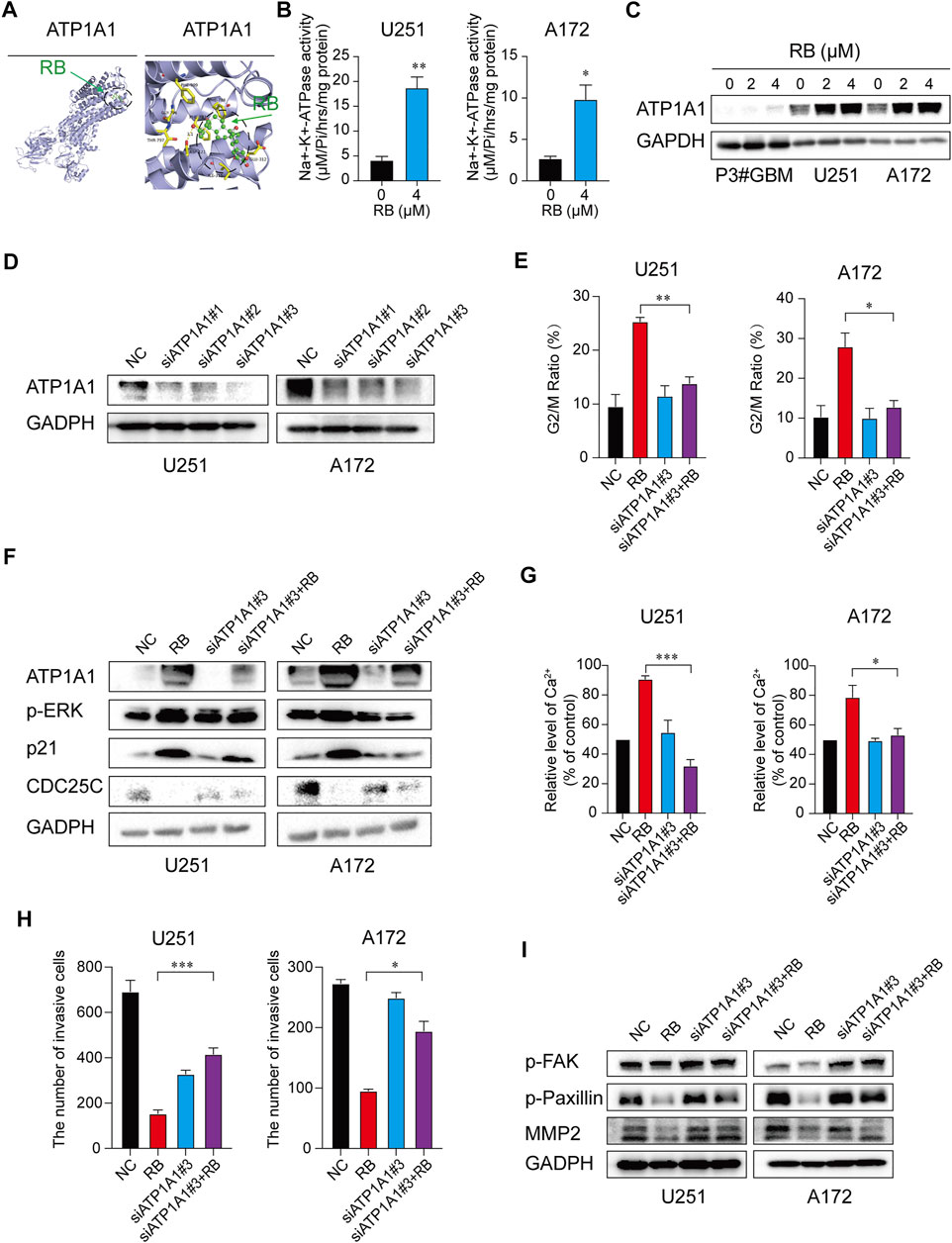
FIGURE 5. RB inhibits malignant progression of GBM by targeting ATP1A1 and increasing Na+-K+-ATPase activity in GBM cells. (A) The most likely predicted target of RB. The binding energy of RB with the ATP1A1 protein was −8.7 kcal/mol, proving a good binding interaction. RB interacts with the ATP1A1 protein mainly through the formation of hydrogen bonds as well as hydrophobic interactions. The hydrogen bond length with ASP-121 was 3.1 Å; hydrophobic interactions were formed with Ile-315, Glu-312, Phe-785, Phe-783, Phe-909, Gly-796, and Thr-797. (B) Graphic representation of Na+-K+-ATPase activity. (C) Representative western blot analysis of total ATP1A1 protein expression. (D) Western blotting analysis validated the efficiency of siATP1A1s in GBM cells. (E) GBM cells were transfected with siATP1A1#3 for 48 h and incubated with 4 μM RB for 48 h. Graphic representation of the percentages of G2/M phase cells calculated from the NC, RB, siATP1A1#3 and siATP1A1#3 + RB cells (Supplementary Figure S3D). (F) Western blot analysis of ATP1A1, p21, CDC25C and G2/M-related proteins from NC, RB, siATP1A1#3 and siATP1A1#3 + RB in U251 and A172 cells. (G) Graphic representation of intracellular Ca2+ levels from the NC, RB, siATP1A1#3 and siATP1A1#3 + RB groups in U251 and A172 cells (Supplementary Figure S3E). (H) The numbers of invasive cells as shown in Supplementary Figure S3F. (I) Western blot analysis of the protein expression levels of p-FAK, p-Paxillin and MMP2 in NC, RB, siATP1A1#3 and siATP1A1#3 + RB in U251 and A172 cells. Three independent experiments were performed with data shown as the mean ± SEM by unpaired Student’s t test (B) or one-way ANOVA (E,G,H). *, p < 0.05; **, p < 0.01 and ***, p < 0.001.
First, we tested whether RB regulated Na+-K+-ATPase enzyme activity in GBM cells and found that treatment with 4 μM RB significantly enhanced Na+-K+-ATPase activity by ∼4-fold in U251 cells and by ∼6-fold in A172 cells (Figure 5B), indicating that RB significantly increased Na+-K+-ATPase activity in GBM cells. ROS and Na+-K+-ATPase develop a feed-forward cycle (Yan et al., 2013; Sodhi et al., 2015; Yan et al., 2017). Activation of Na+-K+-ATPase could block the Na+-K+-ATPase/ROS amplification loop as well as increase ATP1A1 expression (Yan et al., 2017). Whole transcriptome sequencing analysis suggested that peroxisomes were inactivated in GBM cells following treatment with RB (Figure 4B and Supplementary Figure S3A). Thus, we detected the levels of intracellular ROS in P3#GBM, U251 and A172 cells treated with RB. As expected, the levels of intracellular ROS were significantly decreased following treatment with RB (Supplementary Figures S3B,C). Furthermore, we tested ATP1A1 expression and found that RB significantly increased the expression of ATP1A1 (Figure 5C). These results showed that activation of Na+-K+-ATPase with RB upregulated ATP1A1 expression by blocking the Na+-K+-ATPase/ROS amplification loop. ATP1A1 may be the target by which RB inhibits the malignant progression of GBM.
To further clarify the target of RB in GBM cells, we knocked down ATP1A1 using siRNA in U251 and A172 cells. Our results showed that siATP1A1#3 efficiently knocked down ATP1A1 at the protein level in U251 and A172 cells (Figure 5D). In the cells transfected with ATP1A1 siRNAs, RB treatment markedly alleviated G2/M phase arrest induced by RB alone (Figure 5E and Supplementary Figure S3D). Next, after silencing ATP1A1, RB treatment markedly blocked the protein levels of p21, CDC25C, p-ERK and CDK1 compared to RB treatment alone in U251 and A172 cells (Figure 5F).
Furthermore, in cells transfected with ATP1A1 siRNAs, RB treatment substantially alleviated intracellular Ca2+ levels (Figure 5G and Supplementary Figure S3E) and restored the invasiveness of GBM cells (Figure 5H and Supplementary Figure S3F). Furthermore, in U251 and A172 cells transfected with ATP1A1 siRNAs, treatment with RB substantially restored the protein expression of MMP2 and the phosphorylation of FAK and Paxillin compared to RB treatment alone (Figure 5I). These results showed that RB inhibited the malignant progression of GBM by targeting the ATP1A1 signaling cascade and increasing Na+-K+-ATPase activity in GBM cells.
The efficacy of RB was evaluated using an orthotopic tumor modeling approach from P3#GBM-luciferase-expressing cells inoculated in BALB/c nude mice bregma. As shown in Figure 6A, after the 7th day of implantation, mice were selected randomly into two groups (n = 10/group): the NC (DMSO) and RB groups (10 mg/kg/day), and tumor growth was evaluated using luciferase bioluminescence. The bioluminescence values of the two groups of mice showed a significant difference by 21 days (Figure 6B). On the 21st day after implantation, bioluminescence values were nearly 42% lower in the RB-treated group than in the DMSO-treated group (Figure 6C). Kaplan–Meier analysis was performed on the median survival of the tumor-bearing mice, and log-rank analysis was used to measure the statistical significance of the differences. As shown in Figure 6D, RB prolonged the median survival of the tumor-bearing mice from 26 to 29 days (p = 0.0082). We also investigated the expression of Ki-67 and ATP1A1 by immunohistochemical (IHC) staining. The expression of Ki-67 was markedly lower under RB treatment, suggesting that RB suppresses the proliferation of GBM in vivo (Figures 6E,F). The expression of ATP1A1 was increased after RB treatment, suggesting that RB inhibited the growth of GBM by targeting Na+-K+-ATPase.
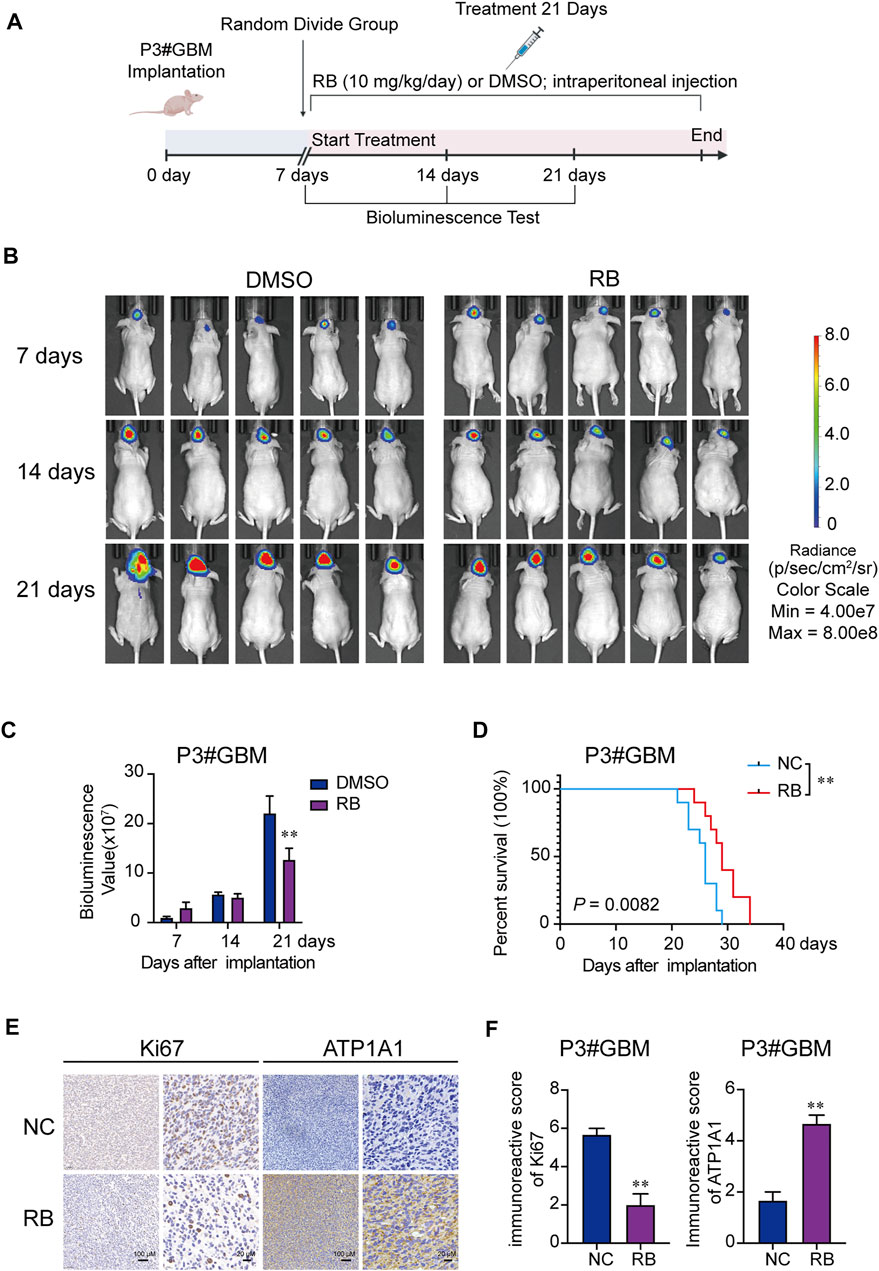
FIGURE 6. RB exerts antitumor efficacy in vivo. (A) Scheme of tumor inoculation and systemic injection. (B) P3#GBM-luciferase cells were orthotopically inoculated into BALB/c nude mice. PerkinElmer IVIS Spectrum monitored tumor growth by detecting bioluminescence. (C) Bioluminescence values on the 7th, 14th, and 21st days after implantation. (D) Kaplan–Meier analysis was performed on the median survival of the tumor-bearing mice, and log-rank analysis was used to measure the statistical significance of the differences. (E) Images of IHC staining for ATP1A1 and Ki-67 in sections from brains of orthotopic P3#GBM tumor-bearing nude mice treated with DMSO or RB. Scale bars, 100 and 20 μm. (F) Immunoreactive scores of Ki-67 and ATP1A1 in sections from the brains of orthotopic P3#GBM tumor-bearing nude mice treated with DMSO or RB in (E). Three independent experiments were performed with data shown as the mean ± SEM by two-way ANOVA (C) or unpaired Student’s t test (F). Survival differences between groups were assessed by the log-rank test (D). *p < 0.05; **p < 0.01 and ***p < 0.001.
In recent years, the use of RB in cancer treatment has attracted increasing attention worldwide. Previous studies have shown that RB has anticancer properties. Intriguingly, we provided the first systematic analysis of the anticancer property of RB on glioblastoma in vitro and in vivo. This study demonstrated that RB binds to ATP1A1 and increases Na+-K+-ATPase activity, which activates the MAPK/ERK pathway, which regulates the expression of CDC25C and p21, followed by a reduction in the activity of the CDK1-Cyclin B complex, ultimately leading to G2/M phase arrest. Furthermore, the increase in Na+-K+-ATPase elevates intracellular Ca2+ accumulation to inhibit invasion by suppressing the Src/FAK/Paxillin focal adhesion pathway in GBM cells (Figure 7). This study not only contributes to research on RB in glioma but is also the first report to elucidate the target of RB in the inhibition of the malignant progression of GBM.
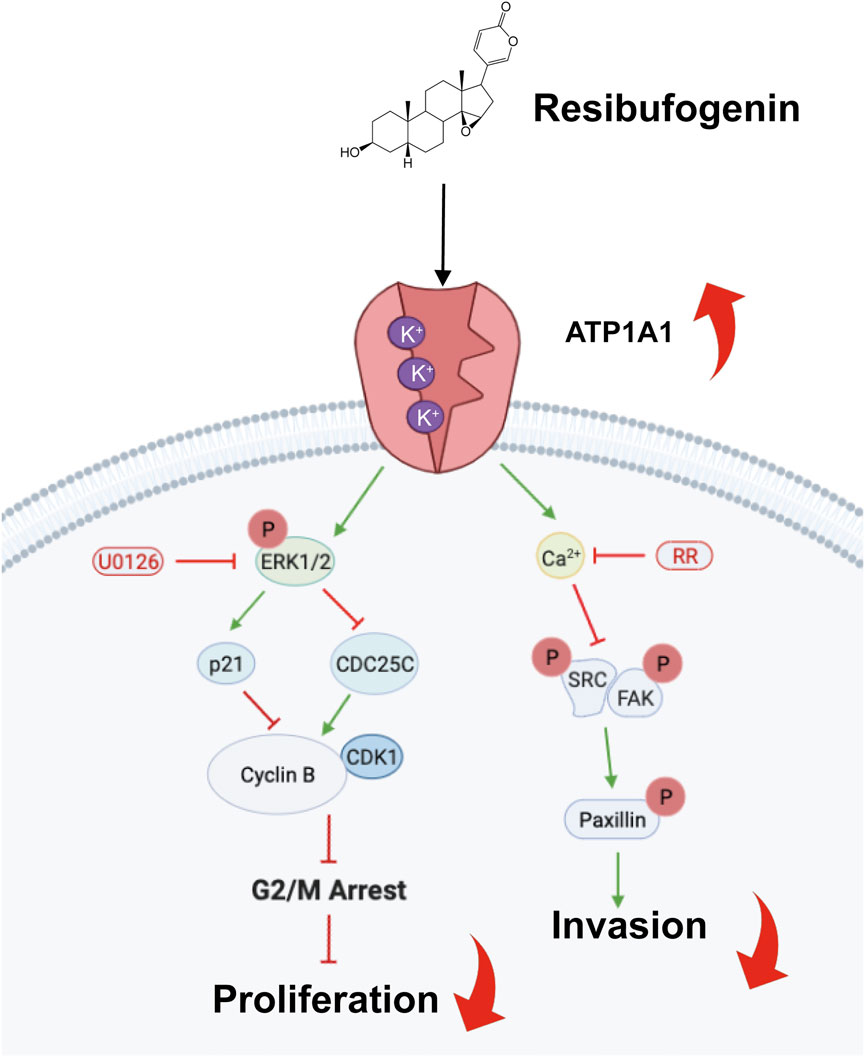
FIGURE 7. Scheme showing the central role of RB targeting ATP1A1 and inducing G2/M phase arrest and inhibiting cell invasion.
Chansu, known as a cardiac glycoside, has long been used in the treatment of disease. Digoxin, ouabain, and digitoxin are common cardiac drugs used to treat heart disease by blocking Na+-K+-ATPase and raising intracellular Ca2+ concentrations (Ahmed et al., 2008; Gjesdal, Feyzi and Olsson 2008). Additionally, these drugs were shown to be beneficial for patients with breast cancer (Stenkvist et al., 1979) and have been related to a reduced risk of lymphoma, leukemia, and urothelial and kidney cancers (Haux et al., 2001). Therefore, we further explored the antitumor potential of RB in glioma and identified a potential strategy for GBM therapy.
We found that at lower concentrations of 2 and 4 μM RB, Na+- K+-ATPase activity was activated. This result is in accordance with reports that the cardiotonic steroids ouabain, digoxin, and bufadienolides all have a reversed U-shaped dosage curve with suppression of Na+-K+-ATPase activity at higher doses and increased Na+-K+-ATPase activity at low doses (Bagrov, Shapiro and Fedorova 2009; Oselkin, Tian and Bergold 2010). We elucidated that 2 and 4 μM RB exhibited antitumor effects by activating Na+-K+-ATPase in cancer cells. In the future, we will explore the effects of wide-range concentrations of RB on glioma cell proliferation and invasion.
Inhibition of Na+- K+-ATPase generally leads to an increase in intracellular Na+ and a reduction in intracellular K+ and then induces an increase in intracellular Ca2+, which activates various pathways with a combination of genomic and nongenomic effects (Bagrov et al., 2009). However, in RB-treated glioma cells, we found that Ca2+ was significantly elevated after activation of Na+-K+-ATPase by RB, which inhibited the Src/FAK/Paxillin focal adhesion pathway. We hypothesize the existence of two possible mechanisms. A previous study hypothesized that Chansu induces the accumulation of intracellular Ca2+ possibly by acting at sites beyond Na+-K+-ATPase, either directly or indirectly through alteration in Ca2+ concentration (Bick et al., 2002). Another hypothesis is that activation of Na+-K+ ATPase by RB may stimulate the Na+- Ca2+ exchanger reverse mode, causing Ca2+ elevation. Na+-K+-ATPase is a special cell membrane protein that can breakdown ATP to obtain energy and use this energy for the active transport of Na+ and K+. The inactivation of Na+-K+-ATPase increases Na+ and attenuates Ca2+ extrusion through the Na+-Ca2+ exchanger, causing Ca2+ accumulation (Li et al., 2014). The mechanisms by which RB leads to the accumulation of Ca2+ in tumor cells deserve further study.
We observed a decrease in p-p38 in P3#GBM and A172 cells but not in U251 cells after treatment with RB. We thought that heterogeneity between GBM cells resulted in different phosphorylation levels of p38 after RB treatment. Therefore, the downregulation of p-p38 is not the key pathway of RB-induced GBM cell cycle arrest. We considered that the underlying reasons for the downregulation of p-p38 could be that 1) the inactivation of the FAK pathway induced by RB inhibiting the phosphorylation level of p38 (Alam et al., 2019) or 2) the decrease in intracellular ROS by RB-mediated activation of Na+-K+-ATPase leading to a downregulation in the phosphorylation of p38 (Maradagi et al., 2022).
This is the first preclinical study investigating the anticancer property of RB on GBM, and we investigated putative pathways induced by this compound. However, a limitation of this study is that the effects of RB in GBM were studied alone and not compared with those of temozolomide or the combination with temozolomide. Furthermore, Src/FAK/Paxillin focal adhesion pathways were detected via mRNA sequencing and Western blot assays. We need to further confirm that RB functions in GBM via Src/FAK/Paxillin focal adhesion pathways through well-designed experiments involving overexpression and knockdown of key components of these signaling pathways.
In summary, although our research on the functions of Chansu is far limited, the value of this class of compounds has raised increasing attention. These molecules play an important role not only in anesthesia and heart diseases but also in malignant tumors. Further investigation of these compounds will result in new and effective therapeutic strategies that are critical for optimizing health and treating disease. Further, our research demonstrates that RB inhibits GBM growth in tumor-bearing mice and prolongs animal survival. To expand the scope of the clinical application of RB, researchers must conduct large-scale, multicenter collaborative clinical trials in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Materials and Methods 2.11.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Qilu Hospital affiliated to Shandong University.
Conception and design: XZ, XL, and AC. Acquisition of data: XZ, ZY, ZX, SW, XL, and YZ. Analysis and interpretation of data: XZ and ZY. Writing and review of the manuscript: XZ, ZY, ZX, SW, YH, XL, and JW. Revision of the manuscript and study supervision: AC and XL.
This work was supported by the National Natural Science Foundation of China (81972351), the Department of Science and Technology of Shandong Province (ZR2019ZD33), the Special Foundation for Taishan Scholars (tsqn201909173, ts20110814 and tshw201502056), the China Postdoctoral Science Foundation (2020T130371 and 2018M642666), the Clinical Research Center of Shandong University (2020SDUCRCB002), the Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL-2022003A) and the Shandong Research Institute of Industrial Technology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.855626/full#supplementary-material
ANOVA: Analysis of variance; RB, Resibufogenin; RR: Ruthenium red; GBM: Glioblastoma; ROS: Reactive oxygen species; ATP1A1: Na+-K+-ATPase α1; WHO: World Health Organization; TAK1: Transforming growth factor beta-activated kinase 1; GSK-3: Glycogen synthase kinase-3; Tyr: Tyrosine; Ile: Isoleucine; Glu: Glutamic acid; Phe: Phenylalanine; Gly: Glycine; Thr: Threonine; Src: Nonreceptor tyrosine kinase; Ca2+: Calcium; Na+: Sodium; K+: Potassium; GSCs: Glioblastoma stem-like cells; MMP-2: Matrix metallopeptidase 2; JNK: C-Jun n-terminal kinase; DCFH-DA: 2′,7′-Dichlorodihydrofluorescein; KEGG: Kyoto Encyclopedia of Genes and Genomes; siRNA: Small interfering RNA.
Ahmed, A., Pitt, B., Rahimtoola, S. H., Waagstein, F., White, M., Love, T. E., et al. (2008). Effects of Digoxin at Low Serum Concentrations on Mortality and Hospitalization in Heart Failure: a Propensity-Matched Study of the DIG Trial. Int. J. Cardiol. 123, 138–146. doi:10.1016/j.ijcard.2006.12.001
Alam, S., Liu, Q., Liu, S., Liu, Y., Zhang, Y., Yang, X., et al. (2019). Up-regulated Cathepsin C Induces Macrophage M1 Polarization through FAK-Triggered P38 MAPK/NF-κB Pathway. Exp. Cel Res 382, 111472. doi:10.1016/j.yexcr.2019.06.017
Apryani, E., Ali, U., Wang, Z. Y., Wu, H. Y., Mao, X. F., Ahmad, K. A., et al. (2020). The Spinal Microglial IL-10/β-endorphin Pathway Accounts for Cinobufagin-Induced Mechanical Antiallodynia in Bone Cancer Pain Following Activation of α7-nicotinic Acetylcholine Receptors. J. Neuroinflammation 17, 75. doi:10.1186/s12974-019-1616-z
Aydemir-Koksoy, A., Abramowitz, J., and Allen, J. C. (2001). Ouabain-induced Signaling and Vascular Smooth Muscle Cell Proliferation. J. Biol. Chem. 276, 46605–46611. doi:10.1074/jbc.M106178200
Bagrov, A. Y., Shapiro, J. I., and Fedorova, O. V. (2009). Endogenous Cardiotonic Steroids: Physiology, Pharmacology, and Novel Therapeutic Targets. Pharmacol. Rev. 61, 9–38. doi:10.1124/pr.108.000711
Bick, R. J., Poindexter, B. J., Sweney, R. R., and Dasgupta, A. (2002). Effects of Chan Su, a Traditional Chinese Medicine, on the Calcium Transients of Isolated Cardiomyocytes: Cardiotoxicity Due to More Than Na, K-ATPase Blocking. Life Sci. 72, 699–709. doi:10.1016/s0024-3205(02)02302-0
Bjerkvig, R., Laerum, O. D., and Mella, O. (1986). Glioma Cell Interactions with Fetal Rat Brain Aggregates In Vitro and with Brain Tissue In Vivo. Cancer Res. 46, 4071–4079.
Carragher, N. O., and Frame, M. C. (2004). Focal Adhesion and Actin Dynamics: a Place where Kinases and Proteases Meet to Promote Invasion. Trends Cel Biol 14, 241–249. doi:10.1016/j.tcb.2004.03.011
Chang, B. D., Broude, E. V., Fang, J., Kalinichenko, T. V., Abdryashitov, R., Poole, J. C., et al. (2000). p21Waf1/Cip1/Sdi1-induced Growth Arrest Is Associated with Depletion of Mitosis-Control Proteins and Leads to Abnormal Mitosis and Endoreduplication in Recovering Cells. Oncogene 19, 2165–2170. doi:10.1038/sj.onc.1203573
Chang, J. B., and Ferrell, J. E. (2013). Mitotic Trigger Waves and the Spatial Coordination of the Xenopus Cell Cycle. Nature 500, 603–607. doi:10.1038/nature12321
Chang, S. J., Chen, Y. C., Yang, C. H., Huang, S. C., Huang, H. K., Li, C. C., et al. (2017). Revealing the Three Dimensional Architecture of Focal Adhesion Components to Explain Ca2+-Mediated Turnover of Focal Adhesions. Biochim. Biophys. Acta Gen. Subj. 1861, 624–635. doi:10.1016/j.bbagen.2017.01.002
Chen, L., Jiang, P., Li, J., Xie, Z., Xu, Y., Qu, W., et al. (2019). Periplocin Promotes Wound Healing through the Activation of Src/ERK and PI3K/Akt Pathways Mediated by Na/K-ATPase. Phytomedicine 57, 72–83. doi:10.1016/j.phymed.2018.12.015
Clausen, M. V., Hilbers, F., and Poulsen, H. (2017). The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 8, 371.)
Cobb, B. S., Schaller, M. D., Leu, T. H., and Parsons, J. T. (1994). Stable Association of Pp60src and Pp59fyn with the Focal Adhesion-Associated Protein Tyrosine Kinase, pp125FAK. Mol. Cel Biol 14, 147–155. doi:10.1128/mcb.14.1.147
Dong, Y., Yin, S., Li, J., Jiang, C., Ye, M., and Hu, H. (2011). Bufadienolide Compounds Sensitize Human Breast Cancer Cells to TRAIL-Induced Apoptosis via Inhibition of STAT3/Mcl-1 Pathway. Apoptosis 16, 394–403. doi:10.1007/s10495-011-0573-5
Eymin, B., Claverie, P., Salon, C., Brambilla, C., Brambilla, E., and Gazzeri, S. (2006). p14ARF Triggers G2 Arrest through ERK-Mediated Cdc25C Phosphorylation, Ubiquitination and Proteasomal Degradation. Cell Cycle 5, 759–765. doi:10.4161/cc.5.7.2625
Gjesdal, K., Feyzi, J., and Olsson, S. B. (2008). Digitalis: a Dangerous Drug in Atrial Fibrillation? an Analysis of the SPORTIF III and V Data. Heart 94, 191–196. doi:10.1136/hrt.2006.108399
Golebiewska, A., Hau, A. C., Oudin, A., Stieber, D., Yabo, Y. A., Baus, V., et al. (2020). Patient-derived Organoids and Orthotopic Xenografts of Primary and Recurrent Gliomas Represent Relevant Patient Avatars for Precision Oncology. Acta Neuropathol. 140, 919–949. doi:10.1007/s00401-020-02226-7
Guo, Y., Zhang, X., Meng, J., and Wang, Z. Y. (2011). An Anticancer Agent Icaritin Induces Sustained Activation of the Extracellular Signal-Regulated Kinase (ERK) Pathway and Inhibits Growth of Breast Cancer Cells. Eur. J. Pharmacol. 658, 114–122. doi:10.1016/j.ejphar.2011.02.005
Han, D., Yu, T., Dong, N., Wang, B., Sun, F., and Jiang, D. (2019). Napabucasin, a Novel STAT3 Inhibitor Suppresses Proliferation, Invasion and Stemness of Glioblastoma Cells. J. Exp. Clin. Cancer Res. 38, 289. doi:10.1186/s13046-019-1289-6
Han, M., Wang, S., Fritah, S., Wang, X., Zhou, W., Yang, N., et al. (2020). Interfering with Long Non-coding RNA MIR22HG Processing Inhibits Glioblastoma Progression through Suppression of Wnt/β-Catenin Signalling. Brain 143, 512–530. doi:10.1093/brain/awz406
Hao, S., Bao, Y. M., Zhao, R. G., Wang, H. S., Bi, J., An, L. J., et al. (2011). Effects of Resibufogenin on Voltage-Gated Sodium Channels in Cultured Rat Hippocampal Neurons. Neurosci. Lett. 501, 112–116. doi:10.1016/j.neulet.2011.06.059
Haux, J., Klepp, O., Spigset, O., and Tretli, S. (2001). Digitoxin Medication and Cancer; Case Control and Internal Dose-Response Studies. BMC cancer 1, 11. doi:10.1186/1471-2407-1-11
Hegi, M. E., Diserens, A. C., Gorlia, T., Hamou, M. F., de Tribolet, N., Weller, M., et al. (2005). MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 352, 997–1003. doi:10.1056/NEJMoa043331
Hsia, D. A., Lim, S. T., Bernard-Trifilo, J. A., Mitra, S. K., Tanaka, S., den Hertog, J., et al. (2005). Integrin Alpha4beta1 Promotes Focal Adhesion Kinase-independent Cell Motility via Alpha4 Cytoplasmic Domain-specific Activation of C-Src. Mol. Cel Biol 25, 9700–9712. doi:10.1128/MCB.25.21.9700-9712.2005
Jeong, K. Y., Park, M., Sim, J. J., and Kim, H. M. (2020). Combination Antitumor Effect of Sorafenib via Calcium-dependent Deactivation of Focal Adhesion Kinase Targeting Colorectal Cancer Cells. Molecules 25, e5299. doi:10.3390/molecules25225299
Johannessen, T. C., Hasan-Olive, M. M., Zhu, H., Denisova, O., Grudic, A., Latif, M. A., et al. (2019). Thioridazine Inhibits Autophagy and Sensitizes Glioblastoma Cells to Temozolomide. Int. J. Cancer 144, 1735–1745. doi:10.1002/ijc.31912
Kanner, S. B., Reynolds, A. B., Vines, R. R., and Parsons, J. T. (1990). Monoclonal Antibodies to Individual Tyrosine-Phosphorylated Protein Substrates of Oncogene-Encoded Tyrosine Kinases. Proc. Natl. Acad. Sci. U S A. 87, 3328–3332. doi:10.1073/pnas.87.9.3328
Kar, S., Wang, M., Ham, S. W., and Carr, B. I. (2006). H32, a Non-quinone Sulfone Analog of Vitamin K3, Inhibits Human Hepatoma Cell Growth by Inhibiting Cdc25 and Activating ERK. Cancer Biol. Ther. 5, 1340–1347. doi:10.4161/cbt.5.10.3223
Karkhanis, M., and Park, J. I. (2015). Sp1 Regulates Raf/MEK/ERK-induced p21(CIP1) Transcription in TP53-Mutated Cancer Cells. Cell Signal 27, 479–486. doi:10.1016/j.cellsig.2015.01.005
Lan, Y. L., Wang, X., Lou, J. C., Xing, J. S., Yu, Z. L., Wang, H., et al. (2018). Bufalin Inhibits Glioblastoma Growth by Promoting Proteasomal Degradation of the Na+/K+-ATPase α1 Subunit. Biomed. Pharmacother. 103, 204–215. doi:10.1016/j.biopha.2018.04.030
Li, Q., Pogwizd, S. M., Prabhu, S. D., and Zhou, L. (2014). Inhibiting Na+/K+ ATPase Can Impair Mitochondrial Energetics and Induce Abnormal Ca2+ Cycling and Automaticity in guinea Pig Cardiomyocytes. PLoS One 9, e93928. doi:10.1371/journal.pone.0093928
Linz, U. (2010). Commentary on Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-year Analysis of the EORTC-NCIC Trial (Lancet Oncol. 2009;10:459-466). Cancer 116, 1844–1846. doi:10.1002/cncr.24950
Liu, L., Liu, Y., Liu, X., Zhang, N., Mao, G., Zeng, Q., et al. (2018). Resibufogenin Suppresses Transforming Growth Factor-β-Activated Kinase 1-mediated Nuclear Factor-Κb Activity through Protein Kinase C-dependent Inhibition of Glycogen Synthase Kinase 3. Cancer Sci. 109, 3611–3622. doi:10.1111/cas.13788
Maradagi, T., Kumar, R., and Ponesakki, G. (2022). Hyperglycaemia-induced Human Hepatocellular Carcinoma (HepG2) Cell Proliferation through ROS-Mediated P38 Activation Is Effectively Inhibited by a Xanthophyll Carotenoid, Lutein. Diabetic Med. : a J. Br. Diabetic Assoc. 39, e14713. doi:10.1111/dme.14713
Ono, Y., Tsuruma, K., Takata, M., Shimazawa, M., and Hara, H. (2016). Glycoprotein Nonmetastatic Melanoma Protein B Extracellular Fragment Shows Neuroprotective Effects and Activates the PI3K/Akt and MEK/ERK Pathways via the Na+/K+-ATPase. Sci. Rep. 6, 23241. doi:10.1038/srep23241
Oselkin, M., Tian, D., and Bergold, P. J. (2010). Low-dose Cardiotonic Steroids Increase Sodium-Potassium ATPase Activity that Protects Hippocampal Slice Cultures from Experimental Ischemia. Neurosci. Lett. 473, 67–71. doi:10.1016/j.neulet.2009.10.021
Prassas, I., Karagiannis, G. S., Batruch, I., Dimitromanolakis, A., Datti, A., and Diamandis, E. P. (2011). Digitoxin-induced Cytotoxicity in Cancer Cells Is Mediated through Distinct Kinase and Interferon Signaling Networks. Mol. Cancer Ther. 10, 2083–2093. doi:10.1158/1535-7163.MCT-11-0421
Preusser, M., de Ribaupierre, S., Wöhrer, A., Erridge, S. C., Hegi, M., Weller, M., et al. (2011). Current Concepts and Management of Glioblastoma. Ann. Neurol. 70, 9–21. doi:10.1002/ana.22425
Ramos, J. W. (2008). The Regulation of Extracellular Signal-Regulated Kinase (ERK) in Mammalian Cells. Int. J. Biochem. Cel Biol 40, 2707–2719. doi:10.1016/j.biocel.2008.04.009
Robinson, M. J., and Cobb, M. H. (1997). Mitogen-activated Protein Kinase Pathways. Curr. Opin. Cel Biol 9, 180–186. doi:10.1016/s0955-0674(97)80061-0
Roux, P. P., and Blenis, J. (2004). ERK and P38 MAPK-Activated Protein Kinases: a Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 68, 320–344. doi:10.1128/MMBR.68.2.320-344.2004
Schaeffer, H. J., and Weber, M. J. (1999). Mitogen-activated Protein Kinases: Specific Messages from Ubiquitous Messengers. Mol. Cel Biol 19, 2435–2444. doi:10.1128/mcb.19.4.2435
Schaller, M. D., Borgman, C. A., Cobb, B. S., Vines, R. R., Reynolds, A. B., and Parsons, J. T. (1992). pp125FAK a Structurally Distinctive Protein-Tyrosine Kinase Associated with Focal Adhesions. Proc. Natl. Acad. Sci. U S A. 89, 5192–5196. doi:10.1073/pnas.89.11.5192
Schiffer, D., Mellai, M., Boldorini, R., Bisogno, I., Grifoni, S., Corona, C., et al. (2018). The Significance of Chondroitin Sulfate Proteoglycan 4 (CSPG4) in Human Gliomas. Int. J. Mol. Sci. 19, e2724. doi:10.3390/ijms19092724
Schlaepfer, D. D., Mitra, S. K., and Ilic, D. (2004). Control of Motile and Invasive Cell Phenotypes by Focal Adhesion Kinase. Biochim. Biophys. Acta 1692, 77–102. doi:10.1016/j.bbamcr.2004.04.008
Sodhi, K., Maxwell, K., Yan, Y., Liu, J., Chaudhry, M. A., Getty, M., et al. (2015). pNaKtide Inhibits Na/K-ATPase Reactive Oxygen Species Amplification and Attenuates Adipogenesis. Sci. Adv. 1, e1500781. doi:10.1126/sciadv.1500781
Song, J., Zou, D., Zhao, X., Chen, Y., Lv, F., Wang, S., et al. (2021). Bufalin Inhibits Human Diffuse Large B-Cell Lymphoma Tumorigenesis by Inducing Cell Death through the Ca2+/NFATC1/cMYC Pathway. Carcinogenesis 42, 303–314. doi:10.1093/carcin/bgaa108
Stallcup, W. B. (2017). NG2 Proteoglycan Enhances Brain Tumor Progression by Promoting Beta-1 Integrin Activation in Both Cis and Trans Orientations. Cancers (Basel) 9, e31. doi:10.3390/cancers9040031
Stenkvist, B., Bengtsson, E., Eriksson, O., Holmquist, J., Nordin, B., and Westman-Naeser, S. (1979). Cardiac Glycosides and Breast Cancer. Lancet 1, 563. doi:10.1016/s0140-6736(79)90996-6
Stupp, R., Brada, M., van den Bent, M. J., Tonn, J. C., and Pentheroudakis, G. (2014). High-grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 25 (Suppl. 3), iii93–101. iii93-ii101. doi:10.1093/annonc/mdu050
Stupp, R., Mason, W. P., van den Bent, M. J., Weller, M., Fisher, B., Taphoorn, M. J., et al. (2005). Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 352, 987–996. doi:10.1056/NEJMoa043330
Sun, P., Feng, L. X., Zhang, D. M., Liu, M., Liu, W., Mi, T., et al. (2016). Bufalin Derivative BF211 Inhibits Proteasome Activity in Human Lung Cancer Cells In Vitro by Inhibiting β1 Subunit Expression and Disrupting Proteasome Assembly. Acta Pharmacol. Sin 37, 908–918. doi:10.1038/aps.2016.30
Thomas, A. A., Brennan, C. W., DeAngelis, L. M., and Omuro, A. M. (2014). Emerging Therapies for Glioblastoma. JAMA Neurol. 71, 1437–1444. doi:10.1001/jamaneurol.2014.1701
Tsuchiya, Y., Murai, S., and Yamashita, S. (2015). Dual Inhibition of Cdc2 Protein Kinase Activation during Apoptosis in Xenopus Egg Extracts. FEBS J. 282, 1256–1270. doi:10.1111/febs.13217
Wang, J., Miletic, H., Sakariassen, P. Ø., Huszthy, P. C., Jacobsen, H., Brekkå, N., et al. (2009). A Reproducible Brain Tumour Model Established from Human Glioblastoma Biopsies. BMC cancer 9, 465. doi:10.1186/1471-2407-9-465
Wang, L. H., Jiang, X. R., Chen, G. L., Guo, W., Zhang, J. Y., Cui, L. J., et al. (2016). Anti-tumor Activity of SL4 against Breast Cancer Cells: Induction of G2/M Arrest through Modulation of the MAPK-dependent P21 Signaling Pathway. Sci. Rep. 6, 36486. doi:10.1038/srep36486
Wang, R., He, G., Nelman-Gonzalez, M., Ashorn, C. L., Gallick, G. E., Stukenberg, P. T., et al. (2007). Regulation of Cdc25C by ERK-MAP Kinases during the G2/M Transition. Cell 128, 1119–1132. doi:10.1016/j.cell.2006.11.053
Wang, Z. J., Sun, L., and Heinbockel, T. (2014). Resibufogenin and Cinobufagin Activate central Neurons through an Ouabain-like Action. PloS one 9, e113272. doi:10.1371/journal.pone.0113272
Wu, S., Chen, M., Huang, J., Zhang, F., Lv, Z., Jia, Y., et al. (2021). ORAI2 Promotes Gastric Cancer Tumorigenicity and Metastasis through PI3K/Akt Signaling and MAPK-dependent Focal Adhesion Disassembly. Cancer Res. 81, 986–1000. doi:10.1158/0008-5472.CAN-20-0049
Yan, X., Xun, M., Dou, X., Wu, L., Zhang, F., and Zheng, J. (2017). Activation of Na+-K+-ATPase with DRm217 Attenuates Oxidative Stress-Induced Myocardial Cell Injury via Closing Na+-K+-ATPase/Src/Ros Amplifier. Apoptosis 22, 531–543. doi:10.1007/s10495-016-1342-2
Yan, Y., Shapiro, A. P., Haller, S., Katragadda, V., Liu, L., Tian, J., et al. (2013). Involvement of Reactive Oxygen Species in a Feed-Forward Mechanism of Na/K-ATPase-Mediated Signaling Transduction. J. Biol. Chem. 288, 34249–34258. doi:10.1074/jbc.M113.461020
Yao, Z., Zhang, X., Zhao, F., Wang, S., Chen, A., Huang, B., et al. (2020). Ursodeoxycholic Acid Inhibits Glioblastoma Progression via Endoplasmic Reticulum Stress Related Apoptosis and Synergizes With the Proteasome Inhibitor Bortezomib. ACS Chem. Neurosci. 11, 1337–1346. doi:10.1021/acschemneuro.0c00095
Yu, Z., Guo, W., Ma, X., Zhang, B., Dong, P., Huang, L., et al. (2014). Gamabufotalin, a Bufadienolide Compound from Toad Venom, Suppresses COX-2 Expression through Targeting IKKβ/NF-Κb Signaling Pathway in Lung Cancer Cells. Mol. Cancer 13, 203. doi:10.1186/1476-4598-13-203
Keywords: resibufogenin, GBM, proliferation, invasion, ATP1A1
Citation: Zhang X, Yao Z, Xue Z, Wang S, Liu X, Hu Y, Zhang Y, Wang J, Li X and Chen A (2022) Resibufogenin Targets the ATP1A1 Signaling Cascade to Induce G2/M Phase Arrest and Inhibit Invasion in Glioma. Front. Pharmacol. 13:855626. doi: 10.3389/fphar.2022.855626
Received: 15 January 2022; Accepted: 08 April 2022;
Published: 17 May 2022.
Edited by:
Wei-Dong Zhang, Second Military Medical University, ChinaReviewed by:
Luis Eduardo M. Quintas, Federal University of Rio de Janeiro, BrazilCopyright © 2022 Zhang, Yao, Xue, Wang, Liu, Hu, Zhang, Wang, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingang Li, bGl4Z0BzZHUuZWR1LmNu; Anjing Chen, Y2hlbmFqQHNkdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.