 Victoria A. Macht
Victoria A. Macht Ryan P. Vetreno
Ryan P. Vetreno Fulton T. Crews
Fulton T. Crews
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 02 March 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.849997
This article is part of the Research Topic Adolescent Brain and Alcohol View all 9 articles
Alcohol (ethanol) use and misuse is a costly societal issue that can affect an individual across the lifespan. Alcohol use and misuse typically initiates during adolescence and generally continues into adulthood. Not only is alcohol the most widely abused drug by adolescents, but it is also one of the most widely abused drugs in the world. In fact, high rates of maternal drinking make developmental ethanol exposure the most preventable cause of neurological deficits in the Western world. Preclinical studies have determined that one of the most consistent effects of ethanol is its disruption of hippocampal neurogenesis. However, the severity, persistence, and reversibility of ethanol’s effects on hippocampal neurogenesis are dependent on developmental stage of exposure and age at assessment. Complicating the neurodevelopmental effects of ethanol is the concurrent development and maturation of neuromodulatory systems which regulate neurogenesis, particularly the cholinergic system. Cholinergic signaling in the hippocampus directly regulates hippocampal neurogenesis through muscarinic and nicotinic receptor actions and indirectly regulates neurogenesis by providing anti-inflammatory regulatory control over the hippocampal environmental milieu. Therefore, this review aims to evaluate how shifting maturational patterns of the cholinergic system and its regulation of neuroimmune signaling impact ethanol’s effects on adult neurogenesis. For example, perinatal ethanol exposure decreases basal forebrain cholinergic neuron populations, resulting in long-term developmental disruptions to the hippocampus that persist into adulthood. Exaggerated neuroimmune responses and disruptions in adult hippocampal neurogenesis are evident after environmental, developmental, and pharmacological challenges, suggesting that perinatal ethanol exposure induces neurogenic deficits in adulthood that can be unmasked under conditions that strain neural and immune function. Similarly, adolescent ethanol exposure persistently decreases basal forebrain cholinergic neuron populations, increases hippocampal neuroimmune gene expression, and decreases hippocampal neurogenesis in adulthood. The effects of neither perinatal nor adolescent ethanol are mitigated by abstinence whereas adult ethanol exposure-induced reductions in hippocampal neurogenesis are restored following abstinence, suggesting that ethanol-induced alterations in neurogenesis and reversibility are dependent upon the developmental period. Thus, the focus of this review is an examination of how ethanol exposure across critical developmental periods disrupts maturation of cholinergic and neuroinflammatory systems to differentially affect hippocampal neurogenesis in adulthood.
The birth, maturation, and functional integration of new neurons, termed neurogenesis, is a critical developmental process originally thought to be isolated to fetal and early neonatal development wherein bursts of new neurons aggregate to form the various regions of the central nervous system (Altman and Das, 1965). In humans, neurogenesis associated with the developmental formation of the brain occurs across gestation, with cortical structures forming prior to the hippocampus and the hippocampal dentate gyrus not demonstrating mature cytoarchitecture until 34 weeks, making it one of the last structures to mature in humans (Arnold and Trojanowski, 1996) and rodents (for review see (Snyder, 2019)). This rapid maturation of the human nervous system during the human third trimester of pregnancy has been colloquially termed the “brain growth spurt” and corresponds to the first 10 days of postnatal life in rodents, where rodent hippocampal development similarly peaks (West et al., 1986). The developmental view that neurogenesis terminates near birth in humans and in the early neonatal period in rodents theoretically left mammals with a finite number of neurons for the duration of their lifespan and led to the erroneous conclusion that any subsequent loss of neurons through drug use, stress, traumatic brain injury, or insult was permanent. However, this dogma has been challenged over the last several decades with emerging evidence indicating that select mammalian brain regions continue to generate and functionally integrate new neurons to varying degrees throughout the lifespan (for review see (Gross, 2000; Ming and Song, 2005)). One region of continuous neurogenesis is the subventricular zone of the lateral ventricles; the other, which is the focus of the current review, is the subgranular zone of the hippocampal dentate gyrus (Altman and Das, 1965). It is important to note that while this review will focus on findings surrounding markers of adult neurogenesis from rodent studies, the prevalence of adult neurogenesis in humans remains an ongoing scientific debate. For example, while doublecortin is a conventional marker of neurogenesis in rodents, recent single-nucleus RNA-seq data question its validity as a comparable marker of adult neurogenesis in humans (Franjic et al., 2022). These findings highlight the center of this debate, which predominantly consists of technical issues in identification and labeling of adult newborn neurons in humans. See Kempermann et al. (2018) for more insight regarding this critical discussion.
In contrast, functional integration of adult hippocampal newborn neurons in rodents has been critically linked to brain health, plasticity, and cognitive function, broadly, but also more specifically to discrete roles in spatial processing and pattern separation (Clelland et al., 2009) as well as cognitive flexibility (Anacker and Hen, 2017) and reversal learning (Garthe and Kempermann, 2013). The unique role of adult hippocampal neurogenesis in cellular and behavioral plasticity is supported by evidence that loss of neurogenesis, which occurs with age as well as with exposure to stress, drugs, or disease, is tightly coupled to loss of cognitive function in these domains (Burghardt et al., 2012; Winner and Winkler, 2015; Anacker and Hen, 2017). Similarly, restoration of hippocampal neurogenesis through lifestyle or therapeutic interventions can recover cognitive functioning (Abdipranoto et al., 2008). As such, restoration of neurogenesis has emerged as a central factor to be considered in various cognitive therapeutic interventions (Abdipranoto et al., 2008).
Disentangling the mediators of hippocampal neurogenesis has led to a pharmacopeia of drug manipulations, highlighting the complex interweaving of multiple systems including trophic support, proinflammatory factors, and various neuromodulatory systems (Macht et al., 2020a). Of the neuromodulatory systems which influence neurogenesis, cholinergic regulation of hippocampal neurogenesis is unique in that cholinergic receptors not only directly regulate proliferation of neuroprogenitor cells (Kotani et al., 2006, 2008), but the cholinergic system also indirectly modulates the hippocampal environmental milieu through anti-inflammatory feedback actions (Conejero-Goldberg et al., 2008; Rosas-Ballina and Tracey, 2009; Gnatek et al., 2012; Li L. et al., 2019) and mediation of glial-derived trophic support (Blondel et al., 2000; Wu et al., 2004; Takarada et al., 2012; Pöyhönen et al., 2019). As such, co-disruption of the basal forebrain cholinergic system and neuroimmune signaling pathways can have cascading repercussions on neural health and cognitive function, leading to a predisposition toward neurological disorders that manifest across development and aging.
In this review, we will focus on a common disruptor of both the basal forebrain cholinergic system and hippocampal neurogenesis: ethyl alcohol (ethanol). Ethanol exposure exerts adverse consequences on the brain throughout the lifespan, with deficits evident from exposure during perinatal development through adulthood. The spectrum of ethanol’s adverse effects produces enormous individual, interpersonal, and societal costs. For example, the robust teratogenic effects of ethanol exposure in utero coupled with high rates of ethanol intake in pregnant women (30.3% drink alcohol at some point during pregnancy; 8.3% binge drink at some point during pregnancy) (Ethen et al., 2009) makes fetal alcohol spectrum disorder (FASD) the most preventable neurological disorder in the Western world (Clarke and Gibbard, 2003). Of note, these estimates on drinking during pregnancy can vary slightly across studies depending on inclusion criteria, type of assessment, and whether estimates are assessing changes over time within a population or comparisons between populations. Similarly, ethanol is one of the first and most prevalent drugs to be used and misused by youth (Patrick and Schulenberg, 2014). Furthermore, adolescents are also more likely than any other age group to engage in binge (4–5+ drinks in 2 h) and high-intensity (10+ drinks/session) drinking patterns, which result in high blood ethanol concentrations (BECs) (Chung et al., 2018; Patrick and Terry-McElrath, 2019). The prevalence and adverse effects of ethanol intake continue into adulthood where ethanol use and misuse is associated with even more fatalities than the ongoing opioid epidemic (Esser et al., 2020; Spencer et al., 2020; Mattson, 2021). However, the long-term effects of ethanol exposure on adult hippocampal neurogenesis, cholinergic function, and subsequently on adult cognitive function are highly dependent on the developmental window of exposure. In this review, we will examine the interaction of ethanol exposure across distinct perinatal, adolescent, and adult developmental windows on the maturing cholinergic neurotransmitter and neuroimmune signaling systems in relation to alterations in adult hippocampal neurogenesis (see Figure 1).

FIGURE 1. Neurodevelopmental consequences of alcohol exposure on the adult hippocampus. This review aims to compare the long-term effects of perinatal, adolescent, and adult ethanol exposure on adult hippocampal neurogenesis. The developmental impact of ethanol on the basal forebrain cholinergic system and cholinergic signaling within the adult hippocampus will be further discussed in the context of modulation of neuroinflammatory signaling as well as mediation of hippocampal neuroprogenitors and neurogenesis Section 1. The first section will discuss how perinatal ethanol exposure produces maturational changes throughout adolescence and into adulthood, resulting in long-term disruptions in adult hippocampal neurogenesis. Rodent perinatal ethanol exposure models the teratogenic effects of ethanol in utero in the human, which often results in a diagnosis of fetal alcohol spectrum disorders (FASD). Of note, the human third trimester, which is the brain growth spurt, corresponds neurodevelopmentally with the first 10 postnatal days (P) in the rat. Therefore, rodent models of human prenatal development must encompass both the prenatal period as well as early neonatal development, collectively termed perinatal exposure. It is important to note that due to methodological considerations, the vast majority of rodent models of FASD use either a prenatal or a postnatal design due to confounds in maternal behavior with ethanol-exposed dams Section 2. The second section will discuss the impact of adolescent ethanol exposure on adult hippocampal neurogenesis. Adolescent development is defined in rodents and humans by a collective set of behavioral, cognitive, and physiological characteristics which do not have concrete endpoints. As such, while there is some variation in the cut-off range for this period, traditionally this has been defined from human ages 10–19 years and rodent ages P28-P59, although some human researchers consider adolescent development to continue until through 24 years Section 3. The third section will discuss the impact of adult ethanol exposure on adult hippocampal neurogenesis. In humans, young adulthood (years 20–24) corresponds with rodent P60-P89. Adulthood, which is typically considered at least 25 years of age, corresponds with approximately P90 in the rat. A central theme of this review is that the long-term effects of ethanol exposure depend on the developmental events occurring during ethanol exposure. Therefore, while the age of ethanol exposure will vary by section, all sections will focus on the long-term effects of ethanol with endpoints in adulthood.
Each section of this review will discuss the adult impact of ethanol exposure on these systems during discrete neurodevelopmental windows: perinatal, adolescence, and adulthood. Incubation effects, persistence, and reversibility will be discussed, with sex differences highlighted when applicable. The goal of this review is to highlight the molecular underpinnings of ethanol’s impact on hippocampal neurogenesis across the continuum of the lifespan.
Teratogenic disruptions during perinatal (i.e., surrounding birth, which includes in utero and neonatal) development induce long-lasting neurodevelopmental consequences on neurological structure and function due to alterations in development of neurocircuitry and other neurobiological systems, permanently shifting maturational trajectories in both the brain and body. One of the most ubiquitously known teratogens is ethanol, and the consequences of ethanol exposure during perinatal development are devastating. These consequences were first documented in 1968 by Paul Lemoine (Lemoine, 1968) in France and later in 1973 by Jones and Smith (Jones and Smith, 1973) in the United States. Since then, clinical and preclinical studies have identified a range of physical, physiological, and behavioral alterations induced by ethanol during perinatal development that are collectively termed FASD; these include physical alterations (e.g., facial dysmorphology, microcephaly, low birth weight) as well as cognitive-behavioral deficits (e.g., hyperactivity, attentional deficits, psychosocial deficits, impaired executive function, learning difficulties, and impaired memory, etc.) (Clarke and Gibbard, 2003). Somewhat surprisingly, despite the global identification and dissemination of information about alcohol’s teratogenic effects to physicians, scientists, and the public, alcohol use and misuse during pregnancy has remained relatively unchanged through the decades (Bhuvaneswar et al., 2007). In fact, alcohol is not only the most commonly used drug by females of reproductive age (24.4% prevalence), but Kanny et al. (2013) found that approximately 12.5% of women continue to drink at binge levels (4+ drinks in 2 h) during pregnancy, resulting in high BECs (0.08% or greater) in relatively short periods of time (Kanny et al., 2013). Investigations into this continuance of alcohol use by women both during pregnancy and while breastfeeding despite alcohol’s known teratogenicity have revealed a complex interplay of factors influencing this ongoing prevalence in behavior, including but not limited to societal pressure, internal and external stress, alcohol dependence, poor understanding of scientific data on substance use during pregnancy, and even inaccurate advice from medical practitioners (Latuskie et al., 2019; Hernandez et al., 2021; Popova et al., 2021). These findings highlight the importance of greater dissemination scientific data on alcohol’s teratogenicity to lay populations and medical practitioners. Alcohol use and binge drinking cause devastating effects to fetal development, resulting in early miscarriage, stillbirth, or upon survival, physical, neurocognitive, and behavioral deficits that are progressively defined along the FASD spectrum, with the most severe deficits resulting in a diagnosis of fetal alcohol syndrome (FAS) (Henderson et al., 2007). Despite these regrettable facts, alcohol use during pregnancy remains the most preventable source of neurological deficits in the Western world (Abel and Sokol, 1987) with estimates of up to 4.8% of school-age children in the United States exhibiting some characteristics of FASD (May et al., 2014).
This section will focus on findings from rodent models of FASD. More specifically, this section will focus on the long-term impact of perinatal ethanol exposure on adult hippocampal neurogenesis, cholinergic function, neuroimmune signaling, and the relationship between these alterations and cognitive-behavioral deficits.
The vast majority of newborn neurons are generated in the mammalian brain during perinatal development (Bayer, 1989). This rapid rate of neurogenesis in humans results from an astounding rate of cell division, with approximately 250,000 nerve cells formed every minute and over 80 billion neurons formed in a newborn human, 70 million in a newborn mouse, and 200 million in a newborn rat brain (Bandeira et al., 2009; von Bartheld et al., 2016). Cortical neurons populate their respective regions first, whereas dentate granule neurons of the hippocampus develop later, beginning in utero in humans during the third trimester and in the rodent at birth (Bayer, 1980a; 1980b). Concurrently, the hippocampus also begins to express nicotinic cholinergic receptors that regulate hippocampal neurogenesis and regional neurodevelopment (Naeff et al., 1992; Zhang et al., 1998; Adams et al., 2002). Thus, the human third trimester and the first 10 days of rodent postnatal development are equivalent, and constitute the developmental timeframe known as the brain’s growth spurt, which is characterized by rapid hippocampal maturation and the beginnings of hippocampal cholinergic innervation (Dobbing and Sands, 1979; Matthews et al., 1974; Nadler et al., 1974) (see Figure 2). In fact, unilateral hippocampal ablation during this early postnatal developmental window reduces cholinergic immunoreactivity in the basal forebrain by 40% in adulthood, highlighting the importance of the reciprocal feedback between the hippocampus and basal forebrain during perinatal neurodevelopment (Plaschke et al., 1997). This reciprocal development between the hippocampus and basal forebrain-to-hippocampus cholinergic projections is dependent upon the production and release of nerve growth factor (NGF), which is selectively produced and released by cells targeted by these cholinergic projections (Higgins et al., 1989). NGF binds to tropomyosin receptor kinase A (TrkA) receptors on cholinergic terminals, providing critical signaling feedback that regulates gene expression necessary for cellular differentiation, influencing somal size, and neurite outgrowth and arborization (as reviewed by (Niewiadomska et al., 2011)), thereby providing continual reciprocal maintenance between cholinergic projection neurons with their target regions during development (Li et al., 1995).
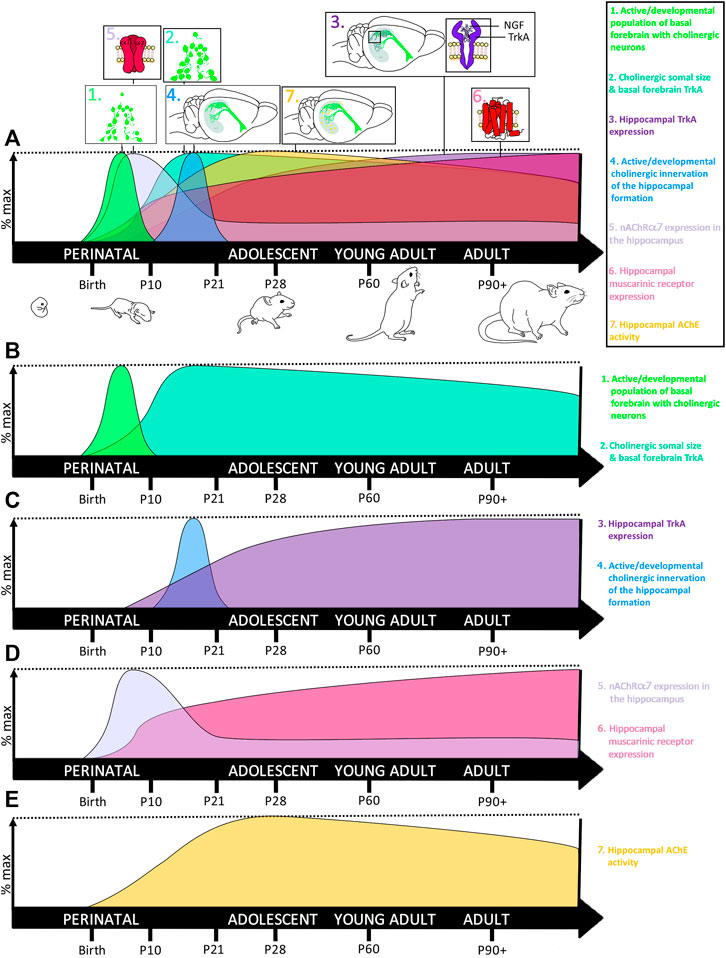
FIGURE 2. Development of the basal forebrain-hippocampal cholinergic system. (A) The cholinergic system undergoes several waves of rapid neurodevelopment during early neonatal and early preadolescent developmental windows, with changes to the hippocampus often lagging behind the basal forebrain. (B) 1. Newly differentiated cholinergic neurons aggregate to form the basal forebrain in early neonatal development, with the most rapid increase in ChAT+IR evidenced around the rat postnatal day 7 (Li et al., 1995), during the brain’s growth spurt. 2. Somal size of these newborn basal forebrain cholinergic neurons continues to increase until weaning, with somal size decreasing slightly into adulthood. (C) 3. Somal size is tightly coupled to local increases in NGF binding to TrkA receptors, signaling both cellular differentiation and survival TrkA receptors peak in the medial septum at approximately 21 days and then remain high throughout adulthood. In contrast, TrkA receptors in the hippocampus remain low during neonatal development, begin increasing across adolescence, where they do not reach maximal levels until adulthood (Li et al., 1995). 4. Rising increases in hippocampal TrkA coincide with onset of basal forebrain innervation of the hippocampal formation, which occurs most robustly between P10-21 and peaks around P17 (Matthews et al., 1974; Nadler et al., 1974). (D) 5. Interestingly, nicotinic alpha-7 receptor (nAChR-α7) expression in the hippocampus rises rapidly during early neonatal development, after which hippocampal nAChR-α7 declines, stabilizing to adult levels by preadolescence (Ben-Barak and Dudai, 1979; Court et al., 1997). These reductions in hippocampal nAChR-α7 are thought to parallel developmental periods of synaptic pruning. 6. In contrast, muscarinic receptor expression in the hippocampus goes through a brief acceleration around P7, and then slowly increases throughout adulthood (Ben-Barak and Dudai, 1979; Court et al., 1997). (E) 7. Cholinergic activity within the hippocampus becomes more tightly regulated during adolescence where its extracellular enzymatic degradation by acetylcholinesterase (AChE) peaks during early adolescence, around P30. Thus collectively, the basal forebrain cholinergic system reaches peak maturity during the neonatal period of the brain’s growth spurt, but the cholinergic innervation and regulation of the hippocampus matures during pre- and early adolescence. This early critical window of cholinergic system neurodevelopment makes it sensitive to developmental insults, including ethanol.
The correlation of human third trimester to the first 10 postnatal days of rodent neurodevelopment (Dobbing and Sands, 1979) has engendered technical complications for the development of FASD rodent models with variations in patterns of ethanol administration (e.g., acute, repeated), developmental period of ethanol exposure (e.g., prenatal, neonatal, or perinatal), and route of ethanol administration (e.g., vapor, intragastric intubation, intraperitoneal injection) (for review see (Patten et al., 2014)). In addition, models of FASD must contend with the complications associated with untangling neurological and behavioral outcomes driven by ethanol itself versus alterations in maternal behavior in ethanol-exposed dams, variation in maternal-pup dynamics in ethanol-exposed pups in neonatal split litter models, and other considerations that arise with cross-fostering and artificial rearing. As such, the vast majority of rodent models of FASD use either prenatal (human first and second trimester equivalent) or neonatal (colloquially termed “third-trimester” models) with few studies utilizing models that encompass the entire human three trimesters of pregnancy due to the aforementioned complications. Thus, for the purpose of this review, rodent models of FASD will be referred to generally as perinatal exposure paradigms unless otherwise stated.
The myriad of developmental and pharmacological variations in rodent FASD models complicates translation of findings to the human literature, particularly when results vary by experimental design. For example, the characteristic facial dysmorphology evidenced in children with FASD (i.e., microcephaly, smooth philtrum, small eyes, short nose with a low nasal bridge, and thin upper vermilion) has been specifically linked to ethanol-induced cell death during early embryonic exposure (mouse embryonic days 7–9) (Sulik, 1984) are not evident in rodent neonatal exposure studies that model the human third trimester. These findings highlight alcohol’s unique pathogenic constellation of teratogenic effects that are often dependent on the developmental window of alcohol exposure. However, some consistencies have emerged in the field that are strengthened by their reliability across models despite these technical nuances. For example, reductions in hippocampal volume and cell number as well as deficits on hippocampal-dependent cognitive-behavioral tasks are consistent across various preclinical models of FASD (West et al., 1986; Tran and Kelly, 2003; Gil-Mohapel et al., 2014) and also corroborate findings from humans studies on FASD (Willoughby et al., 2008). This suggests that the hippocampus and related cognitive behavioral outcomes are especially sensitive to the teratogenic effects of ethanol.
Some of the molecular deficits induced by perinatal ethanol persist long after the cessation of exposure whereas other deficits only emerge later under system challenges, suggesting that perinatal ethanol exposure can induce latent neurological deficits which manifest over time. For example, while hippocampal cell death and neuronal loss is a common immediate finding in perinatal ethanol exposure models (for review see (Gil-Mohapel et al., 2010)), findings regarding the lasting impact of perinatal ethanol exposure on hippocampal neurogenesis in adolescence and adulthood have yielded mixed results. The vast majority of long-term studies on the effects of perinatal ethanol on adult hippocampal neurogenesis have been performed using the thymidine analog 5-bromo-2′-deoxyuridine (BrdU), which is incorporated into dividing cells during the S-phase of mitosis to permanently label newly synthesized DNA, allowing assessments of either proliferation or survival of those cells depending on timing of euthanasia (for a review of findings, see Table 1). Although a large percentage of these BrdU+ cells (∼90%) become neurons, a population of these cells also become glia (Nixon and Crews, 2002). As such, BrdU studies in the absence of secondary neuronal markers, such as neuronal nuclei (NeuN) or doublecortin (DCX), the microtubule-associated protein marker of immature neurons, cannot definitively differentiate between neurogenesis and gliogenesis (Nixon and Crews, 2002). The lack of secondary confirmation that changes in BrdU+ cells in adulthood after perinatal ethanol exposure reflect persistent changes in adult hippocampal neurogenesis rather than gliogenesis remains a limitation of the field that needs to be addressed in future studies.
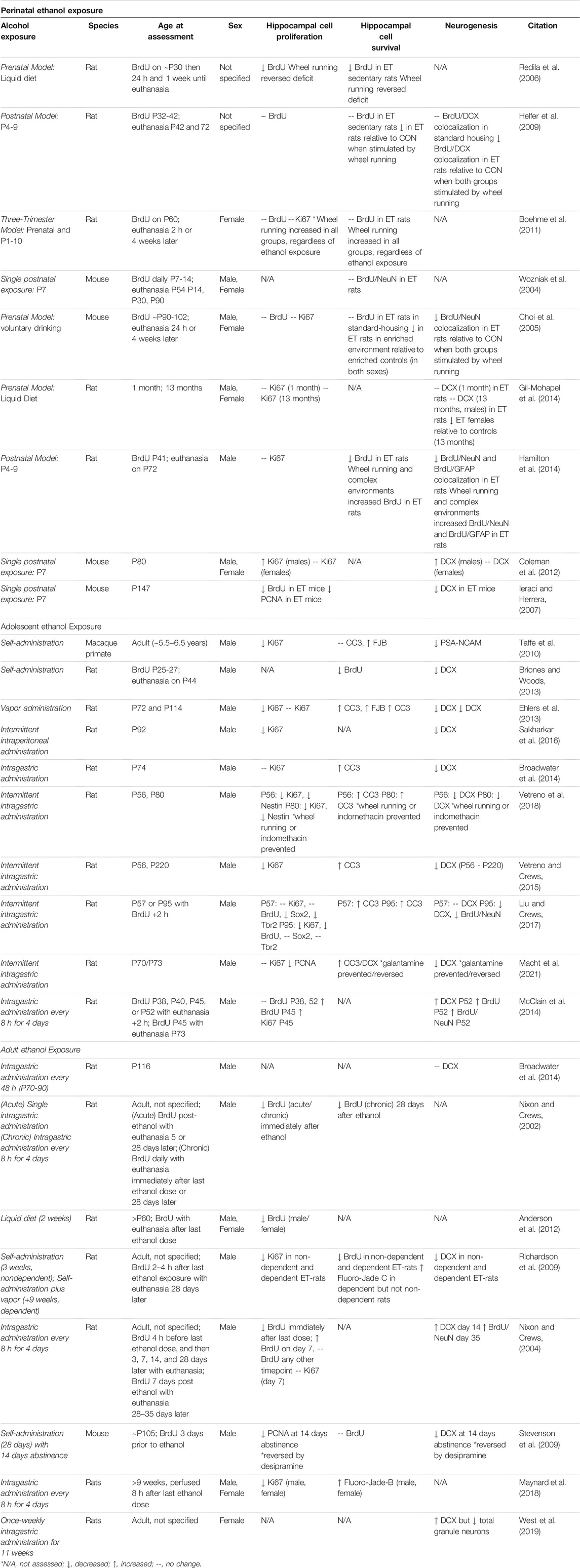
TABLE 1. Impact of developmental ethanol exposure on adult hippocampal neurogenesis.
Human studies of FASD indicate prior alcohol exposure results in attenuated age-related increases in hippocampal volume across adolescent development (Willoughby et al., 2008). However, few preclinical studies have reported that perinatal ethanol exposure itself causes persistent reductions of BrdU+ cell proliferation or survival (Redila et al., 2006; Ieraci and Herrera, 2007; Hamilton et al., 2014). For example, rat models of FASD using prenatal ethanol exposure, a three-trimester liquid diet exposure, or a third-trimester neonatal binge exposure have reported no changes in BrdU+ cell proliferation or survival when examined during either adolescence or adulthood under standard housing conditions (Choi et al., 2005; Helfer et al., 2009; Boehme et al., 2011; Gil-Mohapel et al., 2014). Some of these studies did examine co-localization of BrdU+ cells with NeuN and/or examined DCX. In contrast, many of the FASD rodent studies that did not find impaired cell proliferation and/or survival under normal housing conditions reported impairments in cell survival specifically within the hippocampal neurogenic niche when unmasked under positive regulators, including environmental enrichment and exercise, or negative regulators, such as aging (Gil-Mohapel et al., 2014). Several of these studies supporting an unmasking of adult neurogenic deficits after perinatal ethanol exposure similarly report that this deficit was specifically related to reduced cell survival of newborn neurons using co-localization of BrdU with DCX and/or NeuN (Choi et al., 2005; Helfer et al., 2009; Boehme et al., 2011; Gil-Mohapel et al., 2014). This suggests that perinatal ethanol exposure sensitizes the adult neurogenic niche to insults, where deficits become more robust under conditions that challenge neurogenesis either acutely or across the lifespan. Moreover, these studies indicate that unmasking of the reductions in hippocampal neurogenesis after perinatal ethanol exposure is associated with impaired progenitor survival rather than reductions in the proliferating neuroprogenitor pool, suggesting that perinatal ethanol induces subtle long-term disruptions to the hippocampal neurogenic milieu to persistently impact cell death cascades in newborn neurons (see Table 1).
Future studies will need to elaborate on these findings, particularly in relation to perinatal ethanol-driven effects on adult hippocampal neurogenesis versus gliogenesis as well as potential sensitivity differences in males versus females. Examination and reporting of both sexes is particularly critical, as some studies have found differences solely in one sex but not the other (see Table 1). For example, females seem to be particularly sensitive to age-related unmasking of neurogenic deficits after prenatal ethanol liquid diet exposure, as 13-month-old females but not males exhibited emergent deficits in DCX immunoreactivity that were not evident in early adolescence (Gil-Mohapel et al., 2014). In addition, some variation in outcomes may be due to developmental differences in ethanol’s impact on other concurrently developing neural systems, such as the cholinergic system. For example, differences in perinatal ethanol-induced disruption of hippocampal neurogenesis may shift across adolescence into adulthood when hippocampal innervation becomes refined, or become further exasperated with age as cholinergic systems exhibit age-related neurodegeneration (Schliebs and Arendt, 2011). As such, divergence in outcomes could be partially attributable to differences in prenatal versus postnatal ethanol exposure on brain development, suggesting that more studies encompassing the entirety of the spectrum of human development are critical to reconciling divergent findings due to variations in prenatal versus postnatal exposure on adult hippocampal neurogenesis. Only one study to date has examined the molecular consequences of perinatal ethanol exposure on adult neurogenesis using a three-trimester model (Boehme et al., 2011) – this study examined BrdU in adulthood selectively in females and found that a three-trimester model of FASD did not impact BrdU+ immunoreactivity in adult females (P60), and wheel running increased BrdU+ immunoreactivity across all ages. As it is unclear whether this finding is specific for this age, sex, or glial versus neurogenesis, more studies, and in particular studies that use a three-trimester model, are necessary to clarify these discrepancies.
The cholinergic system rapidly develops during the early neonatal period in rodents (see Figure 2), and is a key regulator of both hippocampal neuroimmune signaling and neurogenesis (Kaneko et al., 2006; Field et al., 2012; Terrando et al., 2014), suggesting that disruptions in maturation of the cholinergic system may have cascading consequences on adult hippocampal neurogenesis in models of FASD. Perinatal ethanol exposure disrupts basal forebrain cholinergic system development, as a single ethanol exposure on postnatal day (P)7 was reported to decrease basal forebrain cholinergic neurons in adulthood (P70) by 34–42% in both male and female rats, relative to age-matched controls (Smiley et al., 2021).
Perinatal ethanol exposure also induces large-scale induction of apoptotic cascades within 24 h after acute ethanol, with variations in regional sensitivity to induction of cell death cascades corresponding with variations in developmental timing of various brain regions (Ikonomidou et al., 2000). For example, peak ethanol-induced induction of the apoptotic marker cleaved caspase-3 in the basal forebrain is most prominent on P7, corresponding with development of the basal forebrain system (Ikonomidou et al., 2000; Olney et al., 2002; Young et al., 2005; Farber et al., 2010). The persistent loss of basal forebrain cholinergic neurons after neonatal ethanol exposure suggests that 1) ethanol-induced decreases in adult basal forebrain cholinergic neurons may result from caspase-3-mediated apoptotic cell death during critical neonatal developmental windows, and 2) reductions in adult populations of basal forebrain cholinergic neurons after neonatal ethanol exposure do not recover despite long periods of abstinence. These findings have been reproduced across a variety of mammalian species, from mice to primates, suggesting a high level of congruency in this literature. Moreover, neonatal ethanol-induced loss of basal forebrain cholinergic neurons is accompanied by diminished evoked acetylcholine efflux in the adult hippocampus as assessed using in vivo microdialysis, indicating that perinatal ethanol exposure persistently disrupts basal forebrain-hippocampal cholinergic neurocircuitry (Perkins et al., 2015). These findings further highlight that discrepancies in the effects of perinatal ethanol exposure on adult hippocampal neurogenesis may vary across FASD models – particularly prenatal versus postnatal exposure models – as prenatal exposure models miss critical neonatal developmental periods of cholinergic systems, which could subsequently shift the ability of acetylcholine to modulate neuroprogenitors and the hippocampal environmental milieu to regulate adult hippocampal neurogenesis.
Loss of basal forebrain cholinergic neurons and subsequent diminution of acetylcholine efflux in the hippocampus have critical implications for cognitive and behavioral deficits in FASD and preclinical models of FASD. For example, deprivation of choline, which is an essential nutrient and critical component of acetylcholine synthesis, exacerbates perinatal ethanol-induced deficits in motor development in early life (Idrus et al., 2017). Conversely, choline supplementation is one of the most researched preclinical interventions in models of FASD and is currently under investigation in clinical trials (Wozniak et al., 2013; Nguyen et al., 2016, Jacobson et al., 2018a; Jacobson et al., 2018b, Wozniak et al., 2020). Clinical evaluation of the efficacy of dietary choline supplementation is supported by a bevy of preclinical research indicating that dietary choline supplementation mitigates cognitive deficits in preclinical models of FASD, particularly on tasks requiring cognitive flexibility. In third-trimester models of FASD, choline supplementation mitigates the long-term effects of neonatal ethanol across both sexes on deficits in discrimination learning (Thomas et al., 2000), spatial working memory (Thomas et al., 2010), spatial reversal learning (Thomas et al., 2004), and trace fear conditioning (Wagner and Hunt, 2006), with findings suggesting that early rodent postnatal choline supplementation is critical (Ryan et al., 2008). A caveat to these findings is that choline is known to exhibit multiple mechanisms of action beyond increasing acetylcholine synthesis, including operating as a methyl donor to regulate epigenetic changes and influencing lipid metabolism due to its role as a precursor for phosphatidylcholine (Bekdash et al., 2013; da Silva et al., 2015; Balaraman et al., 2017). Furthermore, phosphatidylcholine is a primary structural component in both cell and myelin membranes (Sakurai and Kawamura, 1984), making it a critical contributing player to neuronal development. However, choline’s role in acetylcholine synthesis is likely important for its beneficial effects on brain development as cholinesterase inhibitors have revealed convergent beneficial effects in models of FASD. For example, the cholinesterase inhibitor galantamine, which is FDA-approved for the treatment of Alzheimer’s disease (Lilienfeld, 2002; Hampel et al., 2018; Haake et al., 2020), recovered the perinatal ethanol-induced diminution of acetylcholine efflux in the adolescent hippocampus (Perkins et al., 2015). Increasing cholinergic output in the adult hippocampus via choline supplementation or cholinesterase inhibition may also mitigate perinatal ethanol-induced deficits in hippocampal neurogenesis, as the cholinesterase inhibitors galantamine and donepezil both increase cholinergic neurotransmission and hippocampal neurogenesis in adult rats (Kotani et al., 2006, Kotani et al., 2008). This suggests that broadly increasing acetylcholine neurotransmission may produce downstream beneficial effects on hippocampal neurogenesis (Kotani et al., 2006, Kotani et al., 2008; Kita et al., 2014; Madrid et al., 2021), further highlighting 1) the critical role of acetylcholine in neurogenesis, 2) the likely contribution of acetylcholine reductions to loss of neurogenesis in these model, and 3) the therapeutic potential of compounds which target the cholinergic system in FASD.
One of the mechanisms by which perinatal ethanol-induced disruption of basal forebrain-hippocampal cholinergic neurocircuitry may affect adult hippocampal neurogenesis is through long-term alterations in cholinergic receptor activation in adulthood. Both muscarinic and nicotinic receptors directly affect neuroprogenitor pools and indirectly regulate the hippocampal environmental milieu. Muscarinic M1 receptors are abundant in the adult hippocampus, where they colocalize with newborn cells in the dentate gyrus to play a key role in hippocampal cell proliferation (Levey et al., 1995; Mohapel et al., 2005). In fact, M1 receptor activation is sufficient to rescue cell proliferation deficits in a model of basal forebrain cholinergic denervation (Van Kampen and Eckman, 2010). However, there are conflicting findings regarding the impact of perinatal ethanol on adult muscarinic receptor expression, often reflecting a divergence in adult outcomes due to differing developmental periods of ethanol exposure across FASD rodent models. For example, ethanol exposure across prenatal development reduced adult (i.e., P90) hippocampal muscarinic receptor density in male and female rats (Black et al., 1995), whereas ethanol exposure from P4 to P10 using intragastric intubation which resulted in cyclical, high blood ethanol concentrations increased hippocampal muscarinic receptor density and decreased their dissociation constant in adulthood (P90) across both sexes (Kelly et al., 1989). Interestingly, muscarinic receptor dynamics are not affected by dosing regimens that result in consistent BECs. Non-cyclical, stable level BECs were achieved by spreading 12 feedings of 2.5% (v/v) ethanol consistently across a 24-h period, suggesting that muscarinic receptors are impacted not only by developmental window, but also by circulating BECs. These results highlight that both developmental timing and dosing regimen of ethanol exposure can create complex alterations in muscarinic receptor expression and affinity dynamics (Kelly et al., 1989).
Interestingly, the sole study reporting that ethanol exposure across gestation decreases adult cell proliferation (Redila et al., 2006) parallels the finding that ethanol exposure across gestation similarly decreases hippocampal muscarinic receptor density in adulthood (Black et al., 1995). Likewise, FASD models that use the third-trimester neonatal binge paradigm (i.e., P4-P10) typically do not report long-term deficits in hippocampal cell proliferation, with one study reporting increased cell proliferation in adult males after a single ethanol dose on P7 (Coleman et al., 2012). These discrepancies highlight that a full three-trimester model, which more accurately reflects human prenatal development by encompassing both the prenatal and postnatal periods, is critical to reconcile the prenatal versus postnatal literature discrepancies regarding hippocampal muscarinic receptor dynamics, and therefore is an important future direction for the field.
In contrast to muscarinic receptors, nicotinic receptors have received little attention in relation to the lasting effects of perinatal ethanol exposure, with a single study reporting that hippocampal α7 nicotinic receptor density in adulthood is unaffected by perinatal (i.e., P2-P10) ethanol exposure (Perkins et al., 2015). However, reductions of basal forebrain cholinergic tone in the hippocampus may have cascading consequences on nicotinic receptor activation that are similarly important for adult neurogenesis. In fact, choline is also a selective agonist at the α7 nicotinic receptor (Alkondon et al., 1997), and loss of cholinergic activation of the α7 nicotinic receptor could impair important anti-inflammatory feedback onto microglia (Li L. et al., 2019), disrupting the inflammatory balance in the hippocampal environmental milieu. This suggests that a more thorough assessment of perinatal ethanol’s impact on nicotinic receptor dynamics and the long-term mediation of nicotinic signaling the adult neurogenic niche remains an important future direction for the field.
A growing body of evidence has implicated the innate immune system not only in pathogen defense, but also in neurodevelopment (for review see (Macht, 2016)). While microglia are classically associated with the brain’s immune response, it is becoming increasingly appreciated that astrocytes and neurons can also contribute to proinflammatory signaling cascades. However, the acute effect of ethanol on innate immune induction in preclinical models of FASD may depend on the timing of microglial regional increases in population across the CNS. For example, while neurons and astrocytes exhibit common neuroepithelial origins, ontogeny of neurons precedes that of astrocytes, with the vast majority of astrocytes not produced until first postnatal month in the rodent (Qian et al., 2000; Sauvageot and Stiles, 2002). Microglia, in contrast, are derived from developing macrophages in the yolk sac during early embryogenesis (peaking on approximately embryonic day [E]7) and then migrate to the developing nervous system whereupon they continue to proliferate, populating developing brain regions in a caudal-rostral manner (Alliot et al., 1999; Ginhoux et al., 2010). Interestingly, a single 50-min exposure to ethanol in a vapor chamber on gestational day 12.5 increased microglia undergoing the S phase of the cell cycle, which includes critical periods of DNA synthesis (Salem et al., 2021). Future studies would need to examine if these changes in the microglial cell cycle underlie congruent findings from several studies that ethanol also acutely increases proinflammatory gene induction in the developing prenatal and neonatal brain, likely reflecting varying levels of neuroimmune induction within both neurons and glia, including microglia.
Specifically, prenatal ethanol exposure increased CCL6, IL-21, IL-10ra, and TNFα expression in the developing brain of male and female rats, relative to age-matched controls, on E17 (Terasaki and Schwarz, 2016), with female rats exposed to ethanol also exhibiting increases in CCL2, CCL5, CCL9, CXCL10, and IL-5 in whole brain homogenates. Postnatal ethanol exposure on P5 also acutely increases hippocampal proinflammatory gene expression of IL-1β and CCL4 in male and female rats (Ruggiero et al., 2018), with multiple exposures to ethanol (P4-P9) resulting in even more dramatic induction of proinflammatory signaling cascades, including IL-1β, TNFα, CD11b, and CCL4 in both sexes (Boschen et al., 2016). Similarly, a single exposure to ethanol on P4 in mice results in significant increases in CCL2 and monocyte chemotactic protein-induced protein (MCPIP) in the brain and in cultured microglial cell lines over the course of several hours (Zhang et al., 2018). Increased CCL2 signaling after ethanol is particularly important as it regulates acute induction of apoptotic cascades after developmental (P4) ethanol exposure (Zhang et al., 2018), suggesting that ethanol induction of neuroimmune cascades early in development may directly contribute to neuronal loss, including loss of developing cholinergic neurons. Indeed, persistent increases in CCL2 signaling (Pascual et al., 2017) by perinatal ethanol may increase sensitivity of adult newborn hippocampal neurons to cleaved caspase-3-induced apoptosis, reducing their successful integration into hippocampal neurocircuitry.
The effects of perinatal ethanol on innate immune activation are further exacerbated in adulthood. This is in part due to the fact that perinatal ethanol has cascading repercussions on immune regulation of the developing cholinergic system to impact later neuroimmune signaling dynamics in adulthood. For example, some studies suggest perinatal ethanol sensitizes later innate immune gene responses in adulthood, in part due to diminished capacity for cholinergic anti-inflammatory feedback that leads to an exaggerated proinflammatory neuroimmune response. Prenatal ethanol exposure (2 g/kg ethanol, twice daily, E10-E16) dramatically exacerbates the adult response to modest innate immune challenges (25 μg/kg lipopolysaccharide [LPS]), evidenced by exaggerated hippocampal gene expression of IL-1β and IL-6 in male rats but not female rats, relative to controls (Terasaki and Schwarz, 2016). Similar long-term alterations in the peripheral immune system in adulthood (approximately 4 months old rats) have been evidenced after gestational exposure to ethanol vapor across days 8–19, with adult males but not adult females exhibiting increased levels of circulating monocytes (Bake et al., 2021). Conversely, adult males exhibited decreased basal levels of peripherally circulating cytokines whereas adult females exhibited elevated peripheral levels of circulating cytokines. The authors speculate that low circulating peripheral cytokines in adult males under basal conditions could reflect inappropriate immune responsivity to infection, possibly contributing to higher rates of persistent systemic infections in males with FASD. These findings that prenatal ethanol exposure increases sensitivity to neuroimmune signaling induction in adult males, but not females, may have critical implications, as human studies report that males are more likely to receive an early diagnosis of FASD, reflective of increased phenotypic severity (Thanh et al., 2014; DiPietro and Voegtline, 2017; Osborne et al., 2018).
In addition, persistent increases in CCL2 signaling (Pascual et al., 2017) by perinatal ethanol may increase sensitivity of adult newborn hippocampal neurons to cleaved caspase-3-induced apoptotic cascades, reducing their successful integration into hippocampal circuitry. These persistent proinflammatory effects induced by ethanol appear to be mediated by TLR4 signaling cascades, as TLR4-deficient mice do not exhibit long-term upregulation of several cytokines and chemokines, including IL-1β and CCL2, in models of perinatal ethanol exposure (Pascual et al., 2017). Thus, not only does ethanol’s acute induction of CCL2 and TLR4 signaling pathways during perinatal development contribute to acute increases in proinflammatory gene expression, but these effects also persist into adulthood. Adult exposure to LPS induces a sensitized CCL2 and TLR4 signaling response in the hippocampus in rodent models of FASD, contributing to increased apoptosis in the adult neurogenic niche and greater sensitivity to proinflammatory-induced loss of adult neurogenesis.
Collectively, these findings indicate that perinatal ethanol-induced acute induction of proinflammatory signaling cascades may be an early underlying factor in developmental cholinergic deficits by contributing to their programmed cell death during rodent neonatal or human third-trimester development. In adulthood, these developmental deficits in cholinergic function may unmask greater impairments under conditions of neuronal stress, potentially through alterations in muscarinic and nicotinic receptor activation and consequential augmented disruption of the proinflammatory signaling in the hippocampal environmental milieu, reflecting a vicious cycle that may drive both impairments in neurogenesis and greater cognitive deficits in high-complexity tasks (see Figure 3).
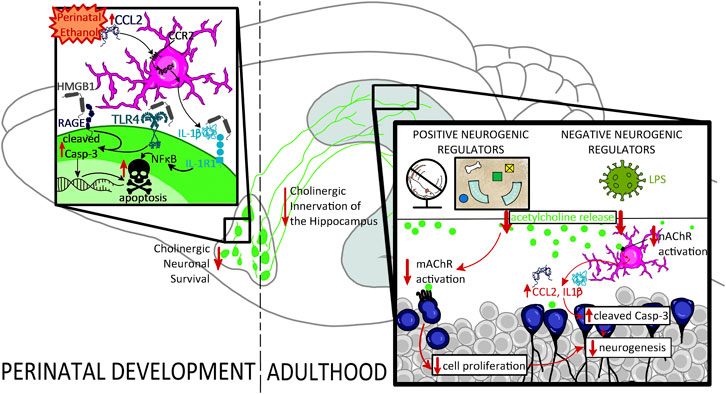
FIGURE 3. Proposed mechanism underlying the impact of perinatal ethanol on adult hippocampal neurogenesis. Perinatal ethanol acutely increases CCL2 proinflammatory cascades, resulting in increases in IL-1β and HMGB1 gene expression as well as activation of TLR4, RAGE, and IL-1R1 receptor signaling, leading to increased activation of cleaved caspase-3 (Casp-3) pathways, and resulting in cellular apoptosis. Activation of cell death pathways results in basal forebrain cholinergic cell death during critical developmental periods. This persistent ethanol-induced loss of basal forebrain cholinergic neurons persists into adulthood and results in long-lasting neurodevelopmental repercussions. A long-term consequence of these perinatal ethanol effects is a hypofunctioning cholinergic network evidenced by decreased capacity for stimulated acetylcholine release in the hippocampus in adulthood. Acetylcholine release is stimulated by positive neurogenic regulatory such as voluntary exercise and environmental enrichment, as well as negative neurogenic regulatory factors such as LPS. Loss of hippocampal cholinergic signaling can unmask deficits in adult neurogenesis, decreasing either cell proliferation, or, more frequently, increasing cell death through hypersensitivity to proinflammatory gene induction and activation of Casp-3 apoptotic pathways.
Children with FASD have several complications due to large-scale cell death induced by prenatal alcohol exposure coupled with broad shifts in epigenetic regulation of gene expression. Epigenetics is the modulation of gene expression in the absence of alterations to DNA, making it a master director of brain development by altering gene accessibility to transcriptional machinery across time (Podobinska et al., 2017). For example, the most frequently investigated epigenetic modification is changes in patterns of methylation across the genome. Increases in DNA methylation on CpG dinucleotides at gene promoter regions restrict transcriptional machinery’s access to specific genes, thereby reducing transcription and translation into protein. In contrast, removal of these methyl groups at CpG-rich promoter islands increases gene access to transcription machinery, resulting in increased gene transcription. Changes in methylation at various gene loci are a critical part of normal neurodevelopment and can be further modified by environmental exposures, including maternal behavior (Weaver et al., 2004) and alcohol exposure (Kobor and Weinberg, 2011), with increased sensitivity across different periods over the lifespan. Epigenetics, including DNA methylation, therefore has the capacity to alter gene expression with tight temporal regulation during critical developmental periods. These neurological and epigenetic changes during critical brain developmental windows shift the brain’s developmental trajectory in children with FASD, resulting in a variety of social, emotional, cognitive, and behavioral deficits that emerge with age (Kodituwakku, 2009). This suggests several considerations in the context of FASD: 1) ethanol-induced alterations during perinatal development persistently impact development by changing gene transcription, 2) some consequences of perinatal ethanol will emerge over time in conjunction with developmental changes in epigenetic regulation of gene expression, and 3) pharmacological interventions which aim to reverse perinatal ethanol-induced developmental effects will be most effective if they also reverse perinatal ethanol-induced epigenetic changes.
The emergence of ethanol’s cascading consequences on neurodevelopment over time complicates interventions aimed at reversal of neurological and behavioral deficits in models of FASD. In fact, the majority of current pharmacological intervention strategies approved for children with FASD target other neuropsychiatric comorbidities, such as the use of stimulants for the highly co-morbid diagnosis of attention deficit hyperactivity disorder in FASD children, with few treatment strategies aimed to prevent or reverse FASD pathophysiology itself (Martinez and Egea, 2007; Wozniak et al., 2019). These challenges in specifically treating FASD are highlighted in preclinical findings surrounding the cholinergic system. For example, using a third-trimester model (P4-P9), Milbocker and Klintsova (Milbocker and Klintsova, 2021) reported that ethanol reduced cholinergic neuron populations (identified by choline acetyltransferase (ChAT)) in the young adult basal forebrain (P72). This deficit was not recovered with either voluntary exercise, complex environmental housing, or a combination of the two (P30-72). Interestingly, choline supplementation has yielded some success with preclinical studies, suggesting that prenatal choline supplements given in conjunction with ethanol exposure across gestation can mitigate loss of brain weight in models of FASD (Thomas et al., 2009) as well as prevent adult working memory deficits in the Morris water maze (Thomas et al., 2010). Choline supplementation also attenuates deficits in cholinergic muscarinic receptor binding (Monk et al., 2012) and causes broad changes in brain gene regulation including cholinergic content as well as receptor expression and function (Otero et al., 2012). However, while one mechanism of choline supplementation is through enhanced acetylcholine synthesis, choline supplementation also reverses perinatal ethanol’s increases in DNA methylation in the hippocampus (Otero et al., 2012). The ability of choline supplementation to reverse alternations in hippocampal DNA methylation after perinatal ethanol is an exciting finding, but these interactions are complex as choline supplementation exhibited opposing effects in the absence of ethanol, highlighting that the role of choline supplementation in DNA methylation must be taken in context with expression of methyltransferase activity and other epigenetic regulators. To further complicate the mechanism of choline supplementation as a therapeutic in models of FASD, choline also affects lipid metabolism and enhances liver oxidation of fatty acids. This suggests that choline supplementation may affect hippocampal gene transcription indirectly through broader impacts on the body in general. These results highlight the multifaceted mechanisms by which choline supplementation may help mitigate developmental consequences of ethanol, as choline is a precursor for the synthesis of acetylcholine, a methyl donor, influencing epigenetic regulation of gene transcription (Zeisel, 2006), and a regulator of lipid metabolism. Collectively, this suggests that ethanol and choline supplementation effect gene transcription broadly, and hippocampal gene transcription specifically.
The positive preclinical effects of choline supplementation on FASD behavioral and neurological outcomes have given it a spotlight in clinical trials, several of which are ongoing (Wozniak et al., 2020). However, clinical trials have further highlighted that choline supplementation efficacy relies heavily on a preventative and/or early intervention strategy and has not yielded clinical success when implemented later in life (e.g., in school-age children) (Nguyen et al., 2016), suggesting the effective therapeutic window for choline supplementation is narrow. The narrow therapeutic window in FASD remains an ongoing clinical challenge (Idrus and Thomas, 2011), particularly for intervention strategies aimed at mitigating deficits in females with FASD, as they are often diagnosed later in life than males (DiPietro and Voegtline, 2017; Osborne et al., 2018). Thus, sex differences in FASD presentation and biological alterations are important to consider in the search for novel treatments for FASD in older populations.
Adolescence is a period of rapid brain maturation with extensive myelination, synaptic pruning, and neurogenesis, highlighting that remodeling of molecular circuitry is a critical component of this developmental period that parallels rapid behavioral and cognitive growth (Arain et al., 2013). Adolescence is also a period when alcohol experimentation and use is typically initiated across both sexes (Petit et al., 2013). As adolescents are less sensitive than adults to the soporific effects of alcohol, they tend to achieve higher BECs than adults in a single drinking session. This means that adolescents are far more likely than adults to consume alcohol in a binge drinking pattern (Kuntsche and Gmel, 2013), which is defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) as consuming 4+/5+ drinks in a 2-h period for women and men, respectively (Alcohol Policy Information System, 2020; Drinking Levels Defined, 2021). In fact, the vast majority of alcohol consumption in adolescents is characterized by intake during weekend binge drinking sessions (Chung et al., 2018), where periods of high levels of alcohol intake are intermittently dispersed with short periods of abstinence. The percentage of individuals engaged in binge drinking sessions escalates across adolescence and into college age, with approximately 4% of eighth grade, 9% of 10th grade, 14% of 12th grade, and 44% of college students reporting recent binge drinking episodes (Wechsler et al., 1995; O’Malley et al., 1998; Johnston et al., 2019). Moreover, adolescent-specific neuromaturation persists in humans through age 25 (Crews et al., 2019), well beyond the legal drinking age, suggesting that these persistent neurodevelopmental disruptions which are continuing to be elucidated in clinical and preclinical studies potentially impact a broad spectrum of the legal drinking population.
The long-term consequences of alcohol drinking in adolescence have primarily been examined through two collaborative consortiums: 1) National Consortium on Alcohol and Neurodevelopment in Adolescence (NCANDA), which evaluates the progressive disruption of neurodevelopment and behavioral alterations with alcohol exposure across adolescence in humans, and 2) the Neurobiology of Adolescent Drinking in Adulthood (NADIA) consortium, which uses rodent models of adolescent intermittent binge ethanol exposure to examine mechanisms underlying ethanol’s long-term molecular and cognitive-behavioral changes in adulthood (Crews et al., 2019). As adolescents who binge drink often continue drinking in adulthood, discerning the discrete effects of adolescent versus adult alcohol exposure can be complex in human studies. Thus, the NADIA consortium has modeled patterns of human adolescent binge drinking in rodents using a paradigm of adolescent intermittent ethanol (AIE) exposure in rats. In this model, ethanol exposure occurs specifically across periadolescent development (P25-P55) via an intermittent dosing regimen that results in the high BECs (>150 mg/dl) that are achieved in human adolescents (Lamminpää et al., 1993; Kraus et al., 2005; White et al., 2006; Donovan, 2009; Patrick et al., 2013; Orio et al., 2018), followed by a period of abstinence to determine the long-lasting, persistent effects of adolescent ethanol exposure on the brain and behavior in adulthood (for a review of the model, see (Crews et al., 2019)). Findings from the NADIA consortium and other rodent models of adolescent ethanol exposure have indicated that the effects of ethanol on the brain and behavior during adolescence are distinct from perinatal or even adult exposure, instead being molded from the landscape of adolescent development where neurocircuitry is being remodeled and refined versus formed. The result is a plethora of cognitive-behavioral and neurobiological changes that persist into adulthood despite abstinence (as reviewed by (Crews et al., 2019)). This section will highlight some of these persistent effects, particularly in relation to adult hippocampal neurogenesis, the cholinergic system, and neuroinflammation.
Neurogenesis during adolescent development is on average four-fold greater than during early adulthood (He and Crews, 2007; Kozareva et al., 2019), and this developmental finding is consistent across mammalian species (Snyder, 2019). High levels of adolescent neurogenesis are thought to reflect a heightened need for neuroplasticity during this period, which corresponds with adolescent-related behavioral changes (Kozareva et al., 2019). Generally, adolescence is characterized by increased exploration, risk-taking behaviors, inhibition of juvenile behavioral patterns, and acquisition of new behaviors essential for transition from parental care to independence as an adult (Spear L. P., 2000). As neurogenesis is an index of neuroplasticity and facilitates pattern separation, spatial learning and memory, and cognitive flexibility, it follows that heightened neurogenesis during adolescence contributes to adaptive behavioral development during this period. However, the increased need for hippocampal neuroplasticity during normal adolescent neurodevelopment also confers a period of neurogenic vulnerability with ensuing consequences on behaviors that rely upon this mechanism of cellular plasticity.
Newborn neurons during adolescence are particularly sensitive to ethanol, showing dose-dependent reductions in hippocampal neurogenesis after acute ethanol exposure (Crews et al., 2006). The magnitude of this deficit in hippocampal neurogenesis after acute adolescent ethanol exposure is somewhat extraordinary, as identical amounts of ethanol acutely reduce neurogenesis by 80% in adolescence versus 30% in adulthood when normalized to respective developmental control levels of neurogenesis (Crews et al., 2006). More concerning are the long-term consequences of adolescent binge ethanol exposure. Our laboratory has found that not only does AIE exposure reduce adult expression of DCX, a neuroprogenitor cytoskeleton protein, in late adolescence (24 h post-AIE), but this reduction in DCX expression persists well into adulthood (P220) in both the dorsal and ventral dentate gyrus (Nixon and Crews, 2002; Crews et al., 2006). This finding reproduces across species (mice, rats, and nonhuman primates) as well as across various routes of administration (intragastric intubation, intraperitoneal injection, vapor) (see (Macht et al., 2020a) for review), and is unique to this developmental window. For example, as previously discussed, perinatal ethanol exposure only mildly reduces adult hippocampal neurogenesis, often requiring a challenge to unmask deficits. Similarly, chronic but not intermittent ethanol exposure in adulthood reduces neurogenesis in a transient manner which recovers following periods of abstinence (Nixon and Crews, 2002; Broadwater et al., 2014). This is not true following intermittent alcohol exposure across adolescence, where reductions in hippocampal neurogenesis are persistent, lasting well into adulthood, perhaps for the duration of the organism’s life, despite abstinence (Nixon and Crews, 2002; Crews et al., 2006).
The enduring loss of hippocampal newborn neurons after AIE most likely involves both subtle disruptions in the neuroprogenitor cell proliferating pool as well as more robust findings involving decreased survival of neuroprogenitors as they differentiate into neurons and integrate into hippocampal circuitry (for summary see Table 1). Cell proliferation after AIE has most frequently been evaluated using staining of the nuclear protein Ki67, which has high fidelity with BrdU expression patterns for proliferation (Kee et al., 2002). Ki67 is expressed during mitosis across mammalian species and exhibits a very short half-life, making it a conservative but accurate estimate of actively dividing cells in the subgranular zone of the neurogenic niche. Some (Nixon and Crews, 2002; Taffe et al., 2010; Vetreno and Crews, 2015; Liu and Crews, 2017) but not all (Ehlers et al., 2013; Broadwater et al., 2014; Macht et al., 2021) studies have found that adolescent binge ethanol exposure decreases Ki67 immunoreactivity in adulthood. Conversely, adolescent binge ethanol exposure has consistently been found to induce activation of the apoptotic executioner caspase cleaved caspase-3 (Ehlers et al., 2013; Broadwater et al., 2014; Vetreno and Crews, 2015; Liu and Crews, 2017; Macht et al., 2021), and/or to increase hippocampal necrotic cell death, as marked by Fluoro-Jade B (Taffe et al., 2010; Ehlers et al., 2013), across species and across variations in route of administration. Moreover, AIE increases expression of cleaved caspase-3 specifically within DCX-labeled neurons, suggesting that activation of cell death machinery directly contributes to loss of newborn hippocampal neurons (Macht et al., 2021). Collectively, these findings suggest that adolescent binge ethanol exposure disrupts the neurogenic niche in ways that interfere with the successful maturation of adult newborn neurons, potentially through mechanisms involving either 1) a failure to assimilate signals driving successful integration into existing hippocampal circuitry or 2) hypersensitivity to the stimulation of cell death executioner pathways.
One mechanism that activates cell death pathways in adult newborn neurons is induction of proinflammatory signaling cascades, resulting in cleavage of caspase-3 and consequential catalyzation and cleavage of critical cell proteins, chromatin condensation, DNA fragmentation, and ultimately cellular apoptosis (Porter and Jänicke, 1999; Macht et al., 2021). Chronic induction of proinflammatory signaling cascades within the hippocampus are a persistent molecular consequence of adolescent ethanol exposure (Crews et al., 2016), and AIE-induced upregulation of proinflammatory gene expression is evident across multiple signaling steps, including increasing the nuclear histone-binding protein high mobility group box protein 1 (HMGB1) (Swartzwelder et al., 2019; Macht et al., 2021), CCL2 (Macht et al., 2021), Toll-like receptor 4 (TLR4), the canonical neuroimmune gene transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells p65 (pNFκB p65) (Li Q. et al., 2019), and cyclooxygenase-2 (COX-2) (Pascual et al., 2007; Macht et al., 2021).
HMGB1 is a central player in these molecular cascades within the hippocampus, with multiple studies demonstrating AIE increases hippocampal granule cell expression of HMGB1 immunoreactivity in the adult dentate gyrus (Vetreno et al., 2018; Swartzwelder et al., 2019; Macht et al., 2021). Upon stimulation, HMGB1 is translocated from the nucleus to the cytoplasm whereupon it is actively and passively secreted into the extracellular space. Once in the extracellular space, HMGB1 functions are multifaceted as HMGB1 has the potential to act as a solo player wherein it binds to and activates a variety of innate immune receptors, including TLR4 and the receptor for advanced glycation end products (RAGE) (Miyata et al., 1996; Yanai et al., 2009), or it can act in concert with other extracellular factors, forming complexes with other immune molecules including IL-1β to subsequently potentiate their responses (as reviewed by (Bianchi, 2009)). Thus, induction of HMGB1→TLR4/RAGE→pNFκB p65 signaling cascades may be a critical mediator in AIE-induced cell death of neuroprogenitors, as expression of hippocampal pNFκB p65 and cleaved caspase-3 are highly correlated (He and Crews, 2007). Furthermore, reversal of hippocampal proinflammatory cascades through the cholinesterase inhibitor galantamine prevents and reverses both AIE induction of HMGB1, COX-2, and CCL2 as well as reduces AIE induction of cleaved caspase-3 in DCX-labeled immature neurons in male rats (Macht et al., 2021), further highlighting that disruption of cholinergic signaling contributes to AIE induction of neuronal inflammatory proinflammatory cascades, cell death, and adult hippocampal neurogenesis.
Hippocampal microgliogenesis and partial microglial activation are also evident in adulthood after AIE, evidenced by increases in the number of hippocampal Iba+ cells as well as shifts in these Iba1+ cells towards a less ramified morphological state and increases in microglial (Iba-1+immunoreactive) expression of pNF-κB p65 (McClain et al., 2011; Liu and Crews, 2017). This phenotypic shift of hippocampal microglia by adolescent binge ethanol exposure may contribute to the observed increases in expression of several proinflammatory cytokines in the adult hippocampus after adolescent binge ethanol exposure, including IL-1β, TNFα, and IL-6 (Gómez et al., 2018). Consequences of increased extracellular IL-1β are magnified by concurrent increases in HMGB1, as IL-1β and HMGB1 form complexes to increase affinity at TLR4 (Coleman et al., 2018), potentiating proinflammatory signaling. This suggests that neurons and microglia work in concert to potentiate proinflammatory responses and activate apoptotic pathways in newborn hippocampal neurons in adulthood.
Adolescent binge ethanol exposure causes a loss of basal forebrain cholinergic neurons immediately following the conclusion of ethanol treatment (i.e., P55) that persists well into adulthood (i.e., P220 [165 days post-ethanol]), paralleling AIE-induced lasting reductions in hippocampal neurogenesis and proinflammatory induction. The observed 20–30% reduction in basal forebrain cholinergic neuron number following adolescent binge ethanol exposure is consistent across both mouse and rat studies (Coleman et al., 2011; Vetreno and Crews, 2018; Macht et al., 2020a; Vetreno et al., 2020; Crews et al., 2021) in both sexes (Vetreno and Crews, 2018). However, unlike in models of FASD, the reduction in cholinergic neuronal markers after adolescent binge ethanol exposure is preventable and reversible with exercise (Vetreno and Crews, 2018) as well as with repeated treatment with the cholinesterase inhibitor galantamine (Crews et al., 2021) and the non-steroidal anti-inflammatory compound indomethacin (Vetreno and Crews, 2018). Restoration of basal forebrain cholinergic neurons after AIE also restores hippocampal neurogenesis, highlighting the reciprocal dynamic between these systems (Macht et al., 2021). These findings suggest not only that the persistent loss of cholinergic anti-inflammatory feedback after AIE is a central mechanism in the long-term induction of hippocampal proinflammatory signaling cascades, but also that proinflammatory signaling plays a role in AIE-induced cholinergic pathology more locally within the basal forebrain, evidenced by AIE increased phosphorylation of NF-κB p65 within cholinergic neurons (Vetreno et al., 2020).
One could assume that loss of expression of ChAT immunoreactivity after AIE – as with findings in fetal alcohol models – suggests cell death of this neuronal population. However, this conclusion from the perinatal literature is fueled by 1) concurrent induction of caspase activity of these neurons during critical neurodevelopmental windows during the brain’s growth spurt, and 2) failure to restore ChAT immunoreactivity with pharmacological or environmental interventions (Ikonomidou et al., 2000; Olney et al., 2002; Young et al., 2005; Farber et al., 2010; Smiley et al., 2021). Findings from AIE do not parallel the fetal alcohol field in this regard. For example, there is no loss of total neuronal number in the basal forebrain after AIE, and expression of cholinergic cells can be recovered with exercise or pharmacological interventions (Vetreno and Crews, 2018; Vetreno et al., 2020; Crews et al., 2021). While neurogenesis could restore cell loss due to cell death, this process is highly confounded by development, and shortly after P10 in rodents, is solely restricted to the subventricular zone and the dentate gyrus of the hippocampus. This suggests that AIE-induced loss and restoration of cholinergic neurons in the basal forebrain is not due to either cell death or spontaneous adult neurogenesis in this region. Rather, the ability to restore ethanol-induced loss of expression of basal forebrain cholinergic neurons in adulthood after adolescent but not perinatal ethanol exposure suggests different mechanisms underlying these long-term changes in cholinergic cell expression. The groundbreaking discovery that cholinergic neurons can be recovered after adolescent but not perinatal ethanol exposure reveals a unique mechanism of neuroplasticity that is centered on epigenetics (Vetreno et al., 2020).
Significant epigenetic remodeling occurs across adolescence in response to hormonal changes that occur with sexual maturation as well as with environmental maturational events, reflecting a critical window of cellular plasticity related to neuronal circuit maturation during this developmental period (Spear L. P., 2000; Lister et al., 2013; Mychasiuk and Metz, 2016). These modifications are sensitive to ethanol-induced increases in neuroimmune gene expression, and epigenetic modifications induced by ethanol are evident across a variety of brain regions including the amygdala, hippocampus, prefrontal cortex, and basal forebrain (Coleman et al., 2011; Montesinos et al., 2016). Expression of the cholinergic neuronal phenotype requires epigenetic modifications that allow continued expression of several genes necessary for the execution of the synthesis, release, and reuptake of acetylcholine as well as the maintenance of cholinergic projections at target regions. Four of these critical proteins are choline acetyltransferase (ChAT), nerve growth factor (NGF), the high-affinity NGF receptor tropomyosin receptor kinase A (TrkA), and the low-affinity NGF receptor p75NTR, which also can function as a death receptor. Recent findings indicate that adolescent binge ethanol exposure suppresses the cholinergic neuronal phenotype via modulation of epigenetic machinery regulating cholinergic genes in both the hippocampus and basal forebrain (as reviewed by (Crews et al., 2019)). In particular, AIE increases histone 3 lysine 9 dimethylation (H3K9me2) by 2.5-fold at the CpG island of the ChAT gene promoter, restricting ChAT gene access to transcriptional machinery, thereby silencing ChAT protein expression as evidenced via immunohistochemistry in the basal forebrain (Vetreno et al., 2020). Similarly, AIE increases H3K9me2 occupation 1.7-fold at the CpG island of the TrkA promoter, suggesting reduced gene transcription of this receptor which is critical to NGF-mediated signaling of trophic support. Furthermore, increased H3K9me2 occupation at both of these cholinergic gene promoter regions is associated with increased phosphorylation of NFκB p65 and HMGB1 in basal forebrain cholinergic neurons, suggesting AIE induction of proinflammatory transcription factors may mediate epigenetic suppression of the cholinergic phenotype (Vetreno et al., 2020).
Both HMGB1 and phosphorylation of NFκB p65/p50 have been implicated in pathways that modify epigenetic machinery, although their roles are complex, depending in part on co-activation of other factors. Thus, HMGB1 and NFκB p65/p50 can have distinct functions under normal physiological versus pathological conditions. For example, under normal physiological developmental conditions, nuclear HMGB1 promotes the formation of transcription complexes by overcoming limitations with tightly bent DNA (Crothers, 1993). In contrast, NFκB in combination with HMGB1 can facilitate the formation of repressome complexes wherein HMGB1 binds to G9a, resulting in histone deacetylation and increases in H3K9 and H3K27 methylation that close chromatin suppressing gene transcription (Abhimanyu et al., 2021). This pathway has been best elucidated in studies on peripheral leukocytes, where this pathway functions as a suppressor of proinflammatory response after LPS, reflecting endotoxin tolerance (Gazzar et al., 2009). However, in the brain, regulation of NFκB and HMGB1 complexes are beginning to be elucidated, with evidence mounting that this pathway is particularly important to the suppression of the cholinergic phenotype after AIE (Crews et al., 2021). Of note, prior AIE studies have solely focused on upregulation of NFκB p65; future studies should also investigate p50 due to their somewhat divergent impact on HMGB1-mediated epigenetic modulation of gene transcription.
Ethanol-induced epigenetic silencing shines light on potential mechanisms for recovery with interventions that reverse this epigenetic silencing (Vetreno et al., 2020), and in fact, both voluntary exercise and galantamine administration reverse epigenetic silencing of basal forebrain cholinergic neurons (Vetreno et al., 2020; Crews et al., 2021). These findings highlight a novel mechanism underlying the perseverance of effects of adolescent binge ethanol on the brain (see Figure 4).
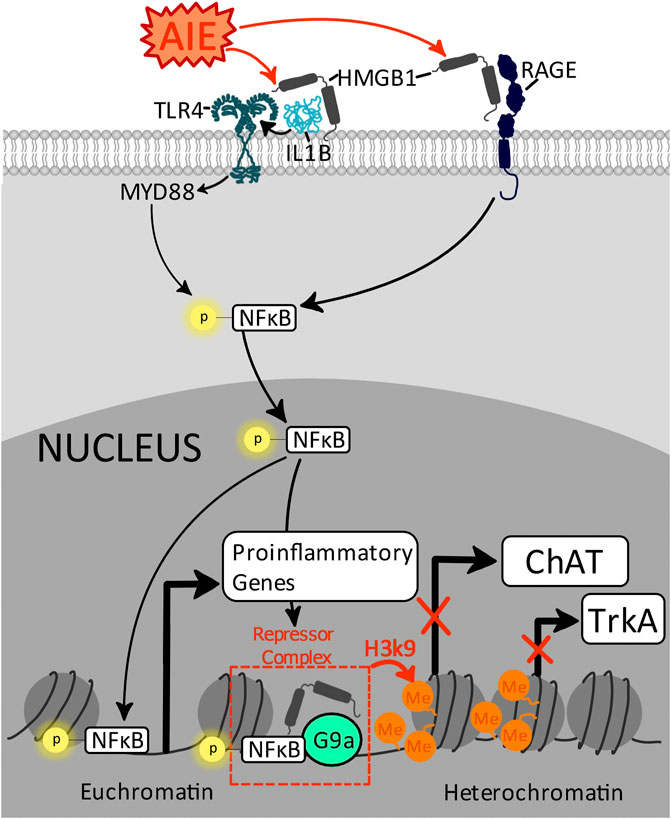
FIGURE 4. Proposed mechanism by which AIE increases proinflammatory gene transcription and suppresses cholinergic gene transcription through distinct epigenetic modifications. AIE increases extracellular HMGB1 and proinflammatory cytokines IL-1β, which activate TLR4 and RAGE receptors, activating intracellular signaling cascades resulting in the phosphorylation and nuclear translocation of NFĸB p65/50. Within the nucleus, NFĸB is a master regulator of gene transcription with specific effects depending on the types of complexes formed by NFĸB p65/50. For example, NFĸB p65 increases gene transcription of a variety of proinflammatory genes, including TLR4, IL-1β, TNFα, CCL2, and COX-2. However, in the presence of HMGB1, NFĸB p50 forms a repressome complex with G9a (Abhimanyu et al., 2021), driving H3K9 methylation at the ChAT and TrkA promotors, reducing cholinergic gene transcription and suppressing the cholinergic neuronal phenotype (Vetreno et al., 2020; Crews et al., 2021).
Despite the loss of basal forebrain cholinergic neuron phenotype, behaviorally-evoked acetylcholine in the hippocampus during reversal learning in adulthood is slightly, but non-significantly, blunted by prior adolescent binge ethanol exposure (Fernandez and Savage, 2017). This finding needs to be explored further as effects could be distinct across various conditions which evoke acetylcholine release (e.g., cognitive task versus stress versus innate immune challenge; simple task versus complex task), and it is unknown whether these findings extend to females. Regardless, hippocampal cholinergic receptors are adversely impacted by adolescent ethanol exposure (Crews et al., 2016), indicating that even in the absence of overt AIE-induced changes in evoked acetylcholine release in the hippocampus, there may be critical underlying changes in cholinergic signaling dynamics which may be amplified by alterations in cholinergic receptor expression.
Adolescent brain maturation shows developmental reductions in nicotinic cholinergic receptor expression (Coleman et al., 2011). Interestingly, one study found that adolescent binge ethanol exposure accelerates the maturational decline in gene expression for both muscarinic and nicotinic cholinergic receptor subtypes, resulting in even greater reductions in both cholinergic neurons and receptors in adulthood (Coleman et al., 2011). As activation of nicotinic α7 receptors has been identified as an interface in cholinergic anti-inflammatory feedback on microglia (Pavlov et al., 2009; Gnatek et al., 2012; Terrando et al., 2014; Li L. et al., 2019, 7), AIE-induced loss of hippocampal nicotinic α7 receptors could adversely impact microglial ability to regulate innate immune gene expression within the neurogenic niche in adulthood, disrupting the hippocampal microenvironment and facilitating activation of apoptotic cascades. This suggests that AIE-induced loss of cholinergic anti-inflammatory feedback via decreased nAChRα7 activation on immunocompetent glia is at the juncture of both persistent inductions in neuroinflammatory signaling cascades, induction of apoptotic executioner caspases, and reductions in survival of adult newborn neurons.
AIE persistent induction of neuroimmune genes as well as reduction in hippocampal neurogenesis corresponds with impairments in a variety of learning and memory tasks, including discrimination learning (Pascual et al., 2007) and reversal learning in males (Crews et al., 2016; Vetreno et al., 2018; Crews et al., 2019). In fact, not only do these neurogenic deficits persist into middle age despite abstinence (Vetreno et al., 2014; Crews et al., 2016), but AIE similarly impairs performance in cognitive flexibility-related tasks in male and female rats long into middle age (Macht et al., 2020b) suggesting that neither AIE-induced deficits in neurogenesis nor AIE-related cognitive flexibility deficits recover with abstinence alone. However, several intervention strategies have emerged that can prevent and/or restore both adult neurogenesis and behavioral flexibility deficits in adulthood in rats exposed to AIE. For example, pre-treatment with the non-steroidal anti-inflammatory drug indomethacin blocks AIE induction of hippocampal inflammatory cascades, restores expression of basal forebrain cholinergic neurons, restores hippocampal neurogenesis (as evidenced by increases in DCX+ immunoreactivity), and restores behavioral deficits (Vetreno and Crews, 2018; Vetreno et al., 2018). The cholinesterase inhibitor galantamine similarly blocks and reverses AIE-induced neuroimmune gene induction as well as blocks and reverses adolescent ethanol-induced loss of neurogenesis (Macht et al., 2021). These findings suggest that AIE-induced reductions in the cholinergic system are a central mechanistic player in both hippocampal neuroinflammatory increases as well as the loss of adult hippocampal neurogenesis and persistence of behavioral flexibility deficits (see Figure 5). Adolescent development therefore presents a unique period of vulnerability to the detrimental effects of binge ethanol on adult behavioral and cellular plasticity deficits in the neurogenic niche.
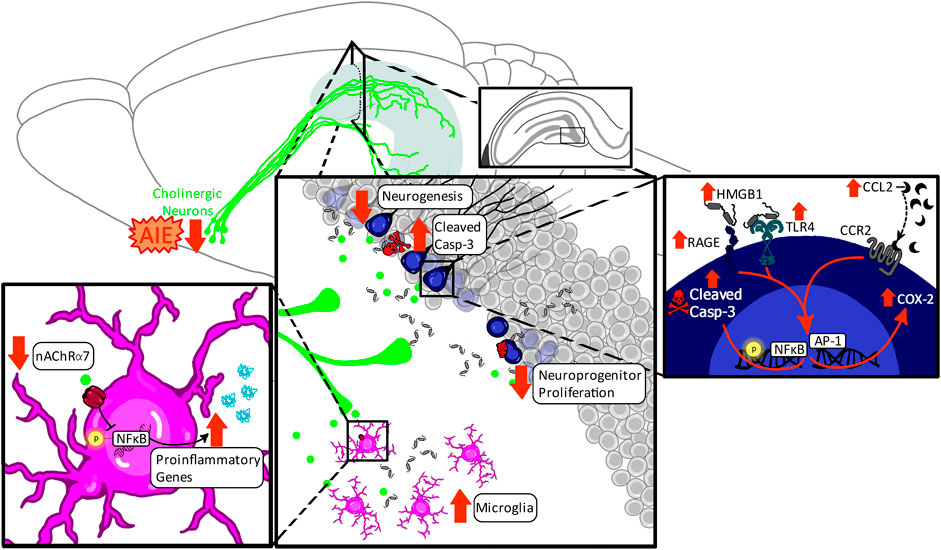
FIGURE 5. Proposed mechanism underlying the persistent loss of adult hippocampal neurogenesis after AIE. AIE persistently decreases phenotypic expression of cholinergic neurons in the basal forebrain and decreases nAChRα7 expression in the hippocampus, consistent with reduced forebrain-hippocampal cholinergic inhibitory feedback of inflammatory responses. This results in increases in microglia number and proinflammatory microglial phenotypes, and increases hippocampal proinflammatory gene expression in the environmental milieu, including IL-1β, TNFα, and CCL2. Shifts in the hippocampal environmental milieu towards a proinflammatory state has several negative consequences on adult neurogenesis. Increases in extracellular HMGB1 in combination with IL-1β and CCL2 increase neuronal proinflammatory signaling through TLR4/RAGE and CCR2 receptors, respectively. Activation of these receptors results in increases in phosphorylation of NFĸB p65 and translocation to the nucleus where it further potentiates proinflammatory gene expression and induces activation of caspase-3 to initiate cell-death cascades. Increases of caspase-3 in newborn neurons suggest these neurons undergo apoptosis during maturation, resulting in decreased adult hippocampal neurogenesis.
During the adolescent developmental period, changes are governed by alterations in epigenetic regulation of cholinergic and neuroimmune genes, driving an environmental imbalance in the hippocampal environmental milieu which is unfavorable to the successful adult birth, maturation, and integration of newborn hippocampal neurons. This suggests that in the absence of intervention, both adolescent ethanol-induced loss of neurogenesis (Vetreno and Crews, 2015), basal forebrain cholinergic neuron expression, and cognitive-behavioral deficits (Macht et al., 2020b) persist long into adulthood, suggesting a possible permanence of these neurodevelopmental alterations. This highlights the necessity for active interventions following adolescent ethanol exposure to restore both hippocampal neurogenesis and cognitive function. Surprisingly, as the loss of basal forebrain cholinergic neurons is mediated by epigenetic mechanisms, restoration of cholinergic function after adolescent ethanol has the potential to be restored by reversing epigenetic suppression of the cholinergic phenotype. Collectively, these results suggest enhanced sensitivity of neuroprogenitors to ethanol during adolescence. The persistence of these effects suggest that this period confers a unique developmental susceptibility to ethanol with potential long-lasting consequences on hippocampal neurocircuitry and behavior. However, the reversibility of these factors through voluntary exercise gives hope for interventions aimed at restoring these deficits in adulthood.
Alcohol use disorder (AUD) is highly prevalent in Western society, with estimates of up to 29% of adults suffering from AUD at some point in their lifetime (Grant et al., 2015). This disorder is characterized by patterns of high levels of alcohol intake despite adverse social, occupational, economic, and health consequences. While AUD has historically been more prevalent in adult males, prevalence of AUD in adult females has been rapidly rising over the last decades (Roberta et al., 2017). The issue of AUD has become increasingly prevalent in the face of the COVID-19 pandemic as studies estimate that ∼24% of individuals reported increases in alcohol intake irrespective of prior diagnosis of AUD, and 17% of previously abstinent individuals with AUD relapsed during pandemic lockdowns (Kim et al., 2020). These increases in adult binge drinking escalate over time, with 1.19-fold greater odds of individuals binge drinking for every week at home (Weerakoon et al., 2021). Adult binge alcohol consumption is concerning as it results in a host of neurological changes and damage, including increases in neuroinflammatory factors as well as decreases in hippocampal neurogenesis and some subtle changes in the central cholinergic system. Therefore, understanding the long-term neurobiological consequences to binge drinking in adulthood is critical to intervention strategies, and of escalating importance in today’s society.
Although neurogenesis was originally thought to be isolated to the neonate, Altman and Das (Altman and Das, 1965) revolutionized this view with their 1965 seminal findings. It is now estimated that up to 6% of granular cell neurons are replaced every month in adulthood in rodents as a result of hippocampal neurogenesis (Cameron and Mckay, 2001). Although the prevalence and function of adult neurogenesis in humans is an ongoing scientific debate centered on technical discrepancies (for review see (Kempermann et al., 2018)), rodents have routinely demonstrated quantifiable neurogenesis in adulthood with emerging work centered instead on elucidating the functional roles of neurogenesis in relation to hippocampal circuitry and behavior (van Praag et al., 2002; Clelland et al., 2009; Garthe and Kempermann, 2013). Collectively, results seem to indicate that these new neurons play critical roles in hippocampal physiology and neuronal plasticity, and as with other developmental periods, adult newborn neurons are sensitive to the effects of ethanol.
Preclinical studies indicate that adult chronic binge ethanol exposure decreases hippocampal neuroprogenitor cell proliferation and neurogenesis (Nixon and Crews, 2002) even in models of moderate alcohol blood ethanol concentrations (Anderson et al., 2012), although the severity of effects increases with prolonged dependence (Richardson et al., 2009). However, these results become more complex when comparing these models of adult AUD to adolescent intermittent exposure paradigms as there are important distinctions in the models used, particularly in relation to ethanol dependence and withdrawal symptoms. For example, 4-day binge and liquid diet paradigms which model human AUD result in physical dependence to ethanol in conjunction with pronounced withdrawal symptoms (Morris et al., 2010). Models of AIE do not result in dependence or severe withdrawal symptoms. In fact, when ethanol is administered intermittently across adulthood (P70-90), there are no reductions in hippocampal neurogenesis, unlike in adolescent intermittent ethanol models (Broadwater et al., 2014). This suggests that intermittent ethanol exposure in adulthood is not sufficiently severe to induce long-term neurogenic deficits, and instead models of AUD, which result in physical dependence and/or severe withdrawal systems, are necessary for inducing deficits in adult hippocampal neurogenesis after adult ethanol exposure (Table 1).
Adult ethanol-induced suppression of hippocampal cell proliferation is further complicated by withdrawal periods, as there is some evidence for a brief burst in cell proliferation 1 week into abstinence (Nixon and Crews, 2004). However, this effect appears to be insufficient to recover neurogenesis as 2 weeks of abstinence is insufficient to recover ethanol-induced loss of DCX immunoreactivity after 1 month of ethanol self-administration (Stevenson et al., 2009). Moreover, there is some evidence that loss of neuroprogenitor proliferation after binge ethanol is more dramatic in females than males (Maynard et al., 2018), and that this decrease in female progenitors is similarly due to greater sensitivity to ethanol’s impairment in the availability of trophic support. Unlike perinatal or adolescent binge alcohol exposure, adult binge alcohol exposure does not increase cellular apoptosis in the adult dentate gyrus (Obernier et al., 2002), suggesting a divergence of mechanisms between developmental and adult alcohol impairments in hippocampal neurogenesis. However, adult binge alcohol does induce necrotic cell death in the hippocampus with females again being particularly sensitive to this effect, resulting in greater loss in hippocampal cell number after binge alcohol specifically in females (Maynard et al., 2018). This suggests that although adult binge ethanol exposure reduces neurogenesis in males and females, this effect could be particularly devastating in females due to an inability to repopulate the granular cell layer over time. In support of this, some recent findings suggest that recovery of neurogenesis after recurrent binge adult alcohol exposure by short-term voluntary wheel running (3 days) is insufficient to recover granular layer cell loss in females (West et al., 2019). Therefore, alcohol-induced granular cell layer loss in females may require more prolonged or intensive therapeutic intervention strategies (for summary, see Figure 6).
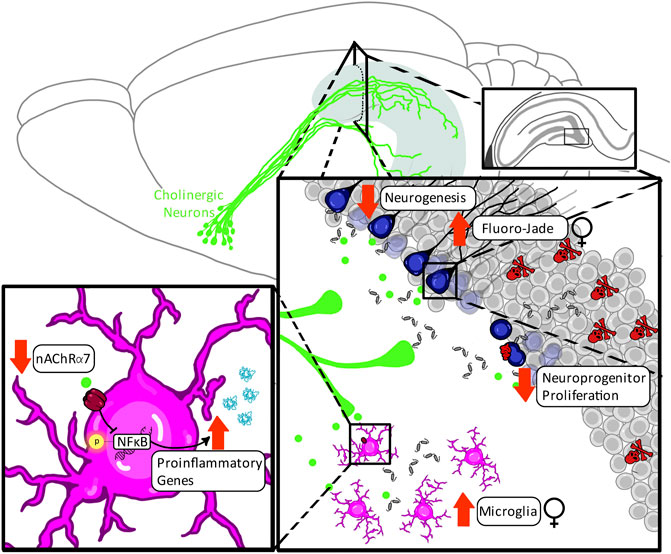
FIGURE 6. Proposed model of chronic adult ethanol impact on hippocampal neuroinflammation and neurogenesis. The adult neurogenic niche is sensitive to neuroinflammatory insults with females being particularly sensitive to these effects. Heightened sensitivity to inflammatory factors may be mediated by reductions in nicotinic α7 receptor expression in the adult hippocampus (Robles and Sabriá, 2008), resulting in poor cholinergic regulation of neuroinflammation that is independent from overarching loss of cholinergic cells. This ethanol-induced disruption in the hippocampal environmental milieu increases necrotic cell death in the granule cell layer of the hippocampus with females being particularly sensitive to ethanol-induced granule cell loss in adulthood (West et al., 2019). However, the impact of chronic ethanol in adulthood on the neurogenic niche is complex. Non-dependent or dependent, but short-term binge ethanol models in adulthood result in either no changes in adult neurogenesis or a transient burst-like increase in neurogenesis that is associated acutely with the withdrawal period (Nixon and Crews, 2004; West et al., 2019). In contrast, long-term dependence models reveal reductions in hippocampal neurogenesis, which are primarily driven by ethanol-induced reductions in the neuroprogenitor pool (Richardson et al., 2009; Stevenson et al., 2009).
The basal forebrain cholinergic system is profoundly affected by ethanol during early development, with both perinatal and AIE exposure reducing basal forebrain cholinergic neuron expression in addition to decreasing cholinergic regulation of the hippocampus. However, adult alcohol exposure does not produce as rapid or robust an effect on the basal forebrain cholinergic system. Just as intermittent ethanol exposure across adolescence but not adulthood produces deficits in adult hippocampal neurogenesis (Vetreno et al., 2018), adolescent but not adult intermittent ethanol exposure reduces basal forebrain ChAT immunoreactivity (Vetreno et al., 2014). Even more dramatically, 20 days of repeated adult binge ethanol exposure in adulthood is still insufficient to reduce basal forebrain cholinergic neuron numbers (Vetreno et al., 2014). Only studies that have employed a forced sole-source ethanol drinking paradigm of 12+ weeks have shown ethanol-reduced basal forebrain cholinergic neuron expression in adults (Arendt et al., 1988b), indicating that these cells are far less susceptible to ethanol-induced damage in adulthood. However, chronic adult ethanol consumption does impact axonal projections, with evidence of decreased cholinergic innervation of the hippocampus (Cadete-Leite et al., 1995). This suggests that even in the absence of reductions in cholinergic cell numbers, chronic adult ethanol exposure can reduce cholinergic projections resulting in impaired function at target regions, including the hippocampus. In support of this, in in vivo microdialysis studies in adults after long-term 3-month ethanol exposure in drinking water, acetylcholine efflux in the hippocampus was significantly reduced by 57%, relative to controls (Casamenti et al., 1993). This effect was reversible with abstinence, suggesting a greater degree of plasticity in cholinergic systems in adulthood, and increased recoverability following ethanol exposure.
Chronic ethanol exposure in adulthood can also disrupt cholinergic receptors, but this finding is complex and largely depends on subtype. For example, 10 weeks of ethanol consumption decreases hippocampal nicotinic receptor expression independently from altering binding affinity (Robles and Sabriá, 2008). In contrast, rodent models using a 28-week liquid diet of ethanol have not reported alterations in number of muscarinic receptor subtypes in the hippocampus (Rothberg et al., 1993, Rothberg et al., 1996). Despite evidence suggesting that there is no impact of ethanol on binding affinity or number of muscarinic receptor subtype, models of chronic ethanol exposure in adulthood do impair acetylcholine-mediated evoked electrophysiological post-synaptic potential responses in pyramidal cells of the CA1 region of the hippocampus, suggesting impaired cholinergic function in this region (Rothberg and Hunter, 1991). Cholinergic activation of pyramidal cells is thought to be mediated through muscarinic receptors, as these effects can be blocked by the muscarinic atropine but not nicotinic agonists (Cole and Nicoll, 1984; Valentino and Dingledine, 2021). This reduced hippocampal cellular responsivity to acetylcholine is postulated to be due to impaired intracellular transduction mechanisms following cholinergic muscarinic receptor activation, suggesting that chronic ethanol may alter muscarinic receptor function in the absence of overarching loss of receptor number. As proliferation of neuroprogenitor pools is dependent on cholinergic activation of muscarinic receptors and subsequent mobilization of intracellular signaling cascades (Ma et al., 2000), these findings suggest that alterations in responsivity to acetylcholine in the hippocampus could underlie some of the deficits in neuroprogenitor proliferation evidenced after chronic ethanol exposure in adulthood.
The loss of hippocampal nicotinic receptor expression could underlie sensitivity to neuroinflammatory induction after chronic ethanol in adulthood, as microglial nicotinic α7 receptors play critical negative feedback role for proinflammatory cascades. Thus, loss of activation at nicotinic α7 receptors is suggestive of a loss of anti-inflammatory feedback and chronic induction of proinflammatory signaling cascades in the brain of individuals with AUD. In support of this concept, an increase in neuroinflammation in post-mortem human brains with AUD is one of the most prevalent clinical findings (Coleman and Crews, 2018), and preclinical models further suggest that neuroinflammation is tightly coupled to continuation of drinking behaviors in nonhuman primates (Beattie et al., 2018). Preclinical models have replicated these results: 6 months of ethanol treatment followed by 2 months of withdrawal in male rats resulted in long-term induction on the activated microglial marker CD11b and the cytokine IL-15 (Cruz et al., 2017). More recent evidence suggests that females may be even more sensitive to this effect as a 4-day binge is sufficient to increase microglial number and frequency of activated phenotypes, indicated by increases in microglia expressing major histocompatibility complex II (MHC II) in the hippocampus of female but not male rats (Barton et al., 2017). These findings support a growing body of clinical and preclinical findings indicating that females may be more sensitive to ethanol-induced damage in adulthood (Alfonso-Loeches et al., 2013).
The effect of ethanol on hippocampal neuroinflammation can be magnified under conditions which challenge the immune system, such as a LPS or polyI:C innate immune challenge. Ten days of intragastric administration of ethanol in mice potentiates proinflammatory responses in brain to a lipopolysaccharide-TLR4 innate immune challenge which included CCL2, COX-2, gp91phox NADPH oxidase subunit, TNFα, and IL-1β as well as greater morphological changes in microglia (Qin et al., 2008). Similarly, a polyI:C-TLR3 challenge after 10 days of ethanol administration also results in an exaggerated increase in the microglial marker ionized calcium binding adaptor molecule 1 (Iba1) in the dentate gyrus of the hippocampus (Qin and Crews, 2012). These neuroinflammatory inductions after ethanol were also associated with reductions in the newborn neuronal marker DCX and greater induction of necrotic and apoptotic cell death in the hippocampus, further highlighting that induction of neuroinflammation, sensitivity to activation of cell death cascades, and loss of neurogenesis are tightly coupled. In fact, in studies of rat hippocampal brain slice cultures, blocking IL-1β with either an antagonist or neutralizing antibody blocked adult ethanol inhibition of hippocampal neurogenesis (Zou and Crews, 2012). Neuroimmune signaling involves feed-forward processes that increase expression of proinflammatory agonists and receptors, particularly TLR receptors. Inhibition of proinflammatory signaling at multiple components of the amplification process are effective. These findings highlight that neuroinflammation is a particularly crucial mediator of neurogenic deficits in adulthood.
Therapeutics under investigation for AUD often exhibit anti-inflammatory components. Given the critical role of the cholinergic system in regulating anti-inflammatory feedback, it is unsurprising that many of these restorative interventions center around the cholinergic systems. Rodent and human studies suggest successful therapeutic targets at both cholinergic muscarinic M4 receptors (Walker et al., 2020) and nicotinic α7 and α7β3 receptors through varenicline (Witkiewitz et al., 2019), and since the late 1980s, rat models of AUD have even been treated with cholinergic-rich brain transplants (Arendt et al., 1988a). While the majority of therapies for AUD focus on reducing relapse behaviors and overall ethanol intake, the ability to reverse molecular cascades involved in cognitive deficits after long-term ethanol use is another important consideration. The mechanisms underlying the efficacy of cholinergic-based therapeutics continue to be investigated for use in alcohol-related disorders, but an emerging theme is their regulation of ethanol-induced neuroinflammation. Hippocampal neuroinflammation is a critical long-term consequence of adult ethanol intake, partially through its mediation of caspase-activated apoptotic cascades and reductive consequences on hippocampal neurogenesis (Liu et al., 2021). In fact, targeting cholinergic systems in adulthood after ethanol use may be effective not through direct increases in activation of cholinergic circuits, but rather indirectly through their mediating factors on both neuroinflammation and neurogenesis. To emphasize this point, targeting ethanol’s induction of IL-1β is sufficient for restoration of hippocampal neurogenesis in adulthood in rodents (Zou and Crews, 2012). This suggests that ethanol induction of neuroinflammatory markers and hypersensitivity of inflammatory systems in adulthood is a central mediator to the loss of hippocampal neurogenesis.
These mechanisms linking neuroinflammation and neurogenesis are particularly important when examining sex differences as human females are more likely than men to experience organ damage following long-term alcohol use, including greater brain damage in general and more specifically loss of hippocampal volume (Agartz et al., 1999; Hommer et al., 2001; Mann et al., 2005; Sharrett-Field et al., 2013), and are more likely to need active interventions beyond abstinence than males. For example, preclinical models indicate that abstinence is insufficient for granule cell layer recovery in females in the absence of an intervention such as voluntary exercise (Maynard and Leasure, 2013). Voluntary exercise after adult ethanol exposure restores the proinflammatory-trophic balance in the hippocampal environmental milieu – a balance which exhibits greater ethanol-induced disruptions in females than males (Maynard et al., 2018). These alarming emerging findings suggest that examining the mechanisms for reversing ethanol-related brain damage in females is becoming an increasingly critical area of investigation as females close the gender gap in AUDs.
The molecular mechanisms of ethanol-related neuronal damage and related intervention strategies circulate around the cholinergic system, neuroinflammation, and the mechanistic mediators of these systems on hippocampal neurogenesis regardless of developmental window, suggestive of certain central commonalities within the effects of ethanol on these systems. However, across the neurodevelopmental landscape, while the mechanisms may overarchingly be consistent, various systems display differing sensitivities in the magnitude and reversibility of ethanol-related disruptions. For example, in models of FASD, ethanol produces seemingly irreversible deficits in the cholinergic system, and one of the key factors in treatment for FASD focuses on preventing rather than reversing this neuronal damage. If unmitigated, the irrecoverable loss of cholinergic neurons is a hindrance to the functional recovery of ethanol-related damage, and later life consequences of this disruption include greater neuroinflammatory responses and decreases in neurogenesis when neural systems are challenged. As females tend to be diagnosed with FASD later than males, this suggests that intervention strategies may be particularly difficult for this population as females have a greater likelihood of missing early intervention therapeutic windows.
In contrast, adolescence is marked by epigenetic-mediated suppression of basal forebrain cholinergic phenotypes, which is exciting from a therapeutic standpoint as reversal of epigenetic machinery in cholinergic neurons similarly restores their phenotypic expression, which consequently also restores the hippocampal neuroinflammatory-trophic balance as well as ethanol-mediated deficits in hippocampal neurogenesis. This balance between persistence and reversibility after adolescent ethanol exposure highlights a shift in the mechanisms driving ethanol-induced loss of hippocampal neurogenesis and neurocognitive impairments from the cholinergic system as the central mediator to neuroimmune dysregulation playing a greater role.
This shift becomes more apparent in adulthood, where hippocampal neuroimmune induction in both preclinical models and in human postmortem tissue from individuals diagnosed with AUD persists long after ethanol exposure and seem to drive long-term cellular consequences, including hippocampal damage and deficits in neurogenesis. While abstinence reverses some of these deficits in males, females exhibit greater sensitivity to ethanol-related brain damage, with emerging studies finding that females require more aggressive intervention strategies than males.
The shifts in vulnerability of cholinergic versus neuroimmune systems over development suggests that efficacy of interventions aimed at restoring ethanol-related damage to hippocampal neurogenesis and cognitive function must take a developmental approach. However, a limitation of preclinical models of FASD, adolescent binge drinking, and AUD is that often alcohol exposure in humans is not isolated to a single developmental period. In fact, prior exposure to ethanol either in utero or in adolescence increases drinking behaviors at later developmental windows (Moore and Riley, 2015; Crews et al., 2019). Thus, an individual with AUD is more likely to have engaged in binge drinking during adolescence. Similarly, an individual who drinks in adolescence is likely to continue drinking into adulthood, and an individual with FASD is significantly more likely to misuse alcohol as adolescents and develop AUD as an adult (Baer et al., 2003; Alati et al., 2008). This double- or even triple hit of ethanol-mediated neuronal disruption over time adds layers of complication to the impact of ethanol on neural systems in clinical populations, and remains an important future direction in preclinical research. For example, the persistent decrease in cholinergic systems after perinatal ethanol exposure may create a vulnerability for even more dramatic neuroimmune induction and loss of neurogenesis following subsequent adolescent binge drinking. Sex differences in vulnerability to ethanol’s disruption of developing neural and immune systems may similarly compound over the lifespan, shifting the severity, perseverance, and/or reversibility of ethanol’s molecular and behavioral consequences, suggesting that the focus of intervention strategies may need to shift over time across sexes but also even within individuals depending on their age and history of exposure.
All authors contributed to the intellectual and conceptual design and writing of this review manuscript. All authors have read and approved the final manuscript.
This review was funded by grants T32 AA007573, K01 AA025713, the Bowles Center for Alcohol Studies, and U01 AA020023.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Jennie Vaughn for her help in copyediting this manuscript.
Abdipranoto, A., Wu, S., Stayte, S., and Vissel, B. (2008). The Role of Neurogenesis in Neurodegenerative Diseases and its Implications for Therapeutic Development. CNS Neurol. Disord. Drug Targets 7, 187–210. doi:10.2174/187152708784083858
Abel, E. L., and Sokol, R. J. (1987). Incidence of Fetal Alcohol Syndrome and Economic Impact of FAS-Related Anomalies. Drug Alcohol Depend. 19, 51–70. doi:10.1016/0376-8716(87)90087-1
Abhimanyu, C. O., Ontiveros, R. S., Nishiguchi, T., Ladki, M., Hilton, I. B., Schlesinger, L. S., et al. (2021). Reversing Post-Infectious Epigenetic-Mediated Immune Suppression. Front. Immunol. 12, 2093. doi:10.3389/fimmu.2021.688132
Adams, C. E., Broide, R. S., Chen, Y., Winzer-Serhan, U. H., Henderson, T. A., Leslie, F. M., et al. (2002). Development of the Alpha7 Nicotinic Cholinergic Receptor in Rat Hippocampal Formation. Brain Res. Dev. Brain Res. 139, 175–187. doi:10.1016/s0165-3806(02)00547-3
Agartz, I., Momenan, R., Rawlings, R. R., Kerich, M. J., and Hommer, D. W. (1999). Hippocampal Volume in Patients with Alcohol Dependence. Arch. Gen. Psychiatry 56, 356–363. doi:10.1001/archpsyc.56.4.356
Alati, R., Clavarino, A., Najman, J. M., O'Callaghan, M., Bor, W., Mamun, A. A., et al. (2008). The Developmental Origin of Adolescent Alcohol Use: Findings from the Mater University Study of Pregnancy and its Outcomes. Drug Alcohol Depend. 98, 136–143. doi:10.1016/j.drugalcdep.2008.05.011
Alcohol Policy Information System (2020). Report to Congress on the Prevention and Reduction of Underage Drinking. Available at: https://alcoholpolicy.niaaa.nih.gov/resource/report-to-congress-on-the-prevention-and-reduction-of-underage-drinking/17 (Accessed October 20, 2021).
Alfonso-Loeches, S., Pascual, M., and Guerri, C. (2013). Gender Differences in Alcohol-Induced Neurotoxicity and Brain Damage. Toxicology 311, 27–34. doi:10.1016/j.tox.2013.03.001
Alkondon, M., Pereira, E. F., Cortes, W. S., Maelicke, A., and Albuquerque, E. X. (1997). Choline Is a Selective Agonist of Alpha7 Nicotinic Acetylcholine Receptors in the Rat Brain Neurons. Eur. J. Neurosci. 9, 2734–2742. doi:10.1111/j.1460-9568.1997.tb01702.x
Alliot, F., Godin, I., and Pessac, B. (1999). Microglia Derive from Progenitors, Originating from the Yolk Sac, and Which Proliferate in the Brain. Brain Res. Dev. Brain Res. 117, 145–152. doi:10.1016/S0165-3806(99)00113-3
Altman, J., and Das, G. D. (1965). Autoradiographic and Histological Evidence of Postnatal Hippocampal Neurogenesis in Rats. J. Comp. Neurol. 124, 319–335. doi:10.1002/cne.901240303
Anacker, C., and Hen, R. (2017). Adult Hippocampal Neurogenesis and Cognitive Flexibility - Linking Memory and Mood. Nat. Rev. Neurosci. 18, 335–346. doi:10.1038/nrn.2017.45
Anderson, M. L., Nokia, M. S., Govindaraju, K. P., and Shors, T. J. (2012). Moderate Drinking? Alcohol Consumption Significantly Decreases Neurogenesis in the Adult hippocampus. Neuroscience 224, 202–209. doi:10.1016/j.neuroscience.2012.08.018
Arain, M., Haque, M., Johal, L., Mathur, P., Nel, W., Rais, A., et al. (2013). Maturation of the Adolescent Brain. Neuropsychiatr. Dis. Treat. 9, 449–461. doi:10.2147/NDT.S39776
Arendt, T., Allen, Y., Sinden, J., Schugens, M. M., Marchbanks, R. M., Lantos, P. L., et al. (1988a). Cholinergic-rich Brain Transplants Reverse Alcohol-Induced Memory Deficits. Nature 332, 448–450. doi:10.1038/332448a0
Arendt, T., Henning, D., Gray, J. A., and Marchbanks, R. (1988b). Loss of Neurons in the Rat Basal Forebrain Cholinergic Projection System after Prolonged Intake of Ethanol. Brain Res. Bull. 21, 563–569. doi:10.1016/0361-9230(88)90193-1
Arnold, S. E., and Trojanowski, J. Q. (1996). Human Fetal Hippocampal Development: I. Cytoarchitecture, Myeloarchitecture, and Neuronal Morphologic Features. J. Comp. Neurol. 367, 274–292. doi:10.1002/(SICI)1096-9861(19960401)367:2<274::AID-CNE9>3.0.CO;2-2
Baer, J. S., Sampson, P. D., Barr, H. M., Connor, P. D., and Streissguth, A. P. (2003). A 21-Year Longitudinal Analysis of the Effects of Prenatal Alcohol Exposure on Young Adult Drinking. Arch. Gen. Psychiatry 60, 377–385. doi:10.1001/archpsyc.60.4.377
Bake, S., Pinson, M. R., Pandey, S., Chambers, J. P., Mota, R., Fairchild, A. E., et al. (2021). Prenatal Alcohol-Induced Sex Differences in Immune, Metabolic and Neurobehavioral Outcomes in Adult Rats. Brain Behav. Immun. 98, 86–100. doi:10.1016/j.bbi.2021.08.207
Balaraman, S., Idrus, N. M., Miranda, R. C., and Thomas, J. D. (2017). Postnatal Choline Supplementation Selectively Attenuates Hippocampal microRNA Alterations Associated with Developmental Alcohol Exposure. Alcohol 60, 159–167. doi:10.1016/j.alcohol.2016.12.006
Bandeira, F., Lent, R., and Herculano-Houzel, S. (2009). Changing Numbers of Neuronal and Non-neuronal Cells Underlie Postnatal Brain Growth in the Rat. Proc. Natl. Acad. Sci. U S A. 106, 14108–14113. doi:10.1073/pnas.0804650106
Barton, E. A., Baker, C., and Leasure, J. L. (2017). Investigation of Sex Differences in the Microglial Response to Binge Ethanol and Exercise. Brain Sci. 7, 139. doi:10.3390/brainsci7100139
Bayer, S. A. (1980b). Development of the Hippocampal Region in the Rat. II. Morphogenesis during Embryonic and Early Postnatal Life. J. Comp. Neurol. 190, 115–134. doi:10.1002/cne.901900108
Bayer, S. A. (1980a). Development of the Hippocampal Region in the Rat. I. Neurogenesis Examined with 3H-Thymidine Autoradiography. J. Comp. Neurol. 190, 87–114. doi:10.1002/cne.901900107
Beattie, M. C., Reguyal, C. S., Porcu, P., Daunais, J. B., Grant, K. A., and Morrow, A. L. (2018). Neuroactive Steroid (3α,5α)3-Hydroxypregnan-20-One (3α,5α-THP) and Pro-inflammatory Cytokine MCP-1 Levels in Hippocampus CA1 Are Correlated with Voluntary Ethanol Consumption in Cynomolgus Monkey. Alcohol. Clin. Exp. Res. 42, 12–20. doi:10.1111/acer.13545
Bekdash, R. A., Zhang, C., and Sarkar, D. K. (2013). Gestational Choline Supplementation Normalized Fetal Alcohol-Induced Alterations in Histone Modifications, DNA Methylation, and Proopiomelanocortin (POMC) Gene Expression in β-endorphin-producing POMC Neurons of the Hypothalamus. Alcohol. Clin. Exp. Res. 37, 1133–1142. doi:10.1111/acer.12082
Ben-Barak, J., and Dudai, Y. (1979). Cholinergic Binding Sites in Rat Hippocampal Formation: Properties and Ontogenesis. Brain Res. 166, 245–257. doi:10.1016/0006-8993(79)90211-7
Bhuvaneswar, C. G., Chang, G., Epstein, L. A., and Stern, T. A. (2007). Alcohol Use during Pregnancy: Prevalence and Impact. Prim. Care Companion J. Clin. Psychiatry 9, 455–460. doi:10.4088/pcc.v09n0608
Black, A. C., Goolsby, L. W., Cohen, G. A., and Young, H. E. (1995). Effects of Prenatal Ethanol Exposure on the Hippocampal Neurochemistry of Albino Rats at 90 Days of Postnatal Age. Am. J. Obstet. Gynecol. 173, 514–519. doi:10.1016/0002-9378(95)90275-9
Blondel, O., Collin, C., McCarran, W. J., Zhu, S., Zamostiano, R., Gozes, I., et al. (2000). A Glia-Derived Signal Regulating Neuronal Differentiation. J. Neurosci. 20, 8012–8020. doi:10.1523/JNEUROSCI.20-21-08012.2000
Boehme, F., Gil-Mohapel, J., Cox, A., Patten, A., Giles, E., Brocardo, P. S., et al. (2011). Voluntary Exercise Induces Adult Hippocampal Neurogenesis and BDNF Expression in a Rodent Model of Fetal Alcohol Spectrum Disorders. Eur. J. Neurosci. 33, 1799–1811. doi:10.1111/j.1460-9568.2011.07676.x
Boschen, K. E., Ruggiero, M. J., and Klintsova, A. Y. (2016). Neonatal Binge Alcohol Exposure Increases Microglial Activation in the Developing Rat hippocampus. Neuroscience 324, 355–366. doi:10.1016/j.neuroscience.2016.03.033
Briones, T. L., and Woods, J. (2013). Chronic Binge-like Alcohol Consumption in Adolescence Causes Depression-like Symptoms Possibly Mediated by the Effects of BDNF on Neurogenesis. Neuroscience 254, 324–334. doi:10.1016/j.neuroscience.2013.09.031
Broadwater, M. A., Liu, W., Crews, F. T., and Spear, L. P. (2014). Persistent Loss of Hippocampal Neurogenesis and Increased Cell Death Following Adolescent, but Not Adult, Chronic Ethanol Exposure. Dev. Neurosci. 36, 297–305. doi:10.1159/000362874
Burghardt, N. S., Park, E. H., Hen, R., and Fenton, A. A. (2012). Adult-born Hippocampal Neurons Promote Cognitive Flexibility in Mice. Hippocampus 22, 1795–1808. doi:10.1002/hipo.22013
Cadete-Leite, A., Andrade, J. P., Sousa, N., Ma, W., and Ribeiro-Da-Silva, A. (1995). Effects of Chronic Alcohol Consumption on the Cholinergic Innervation of the Rat Hippocampal Formation as Revealed by Choline Acetyltransferase Immunocytochemistry. Neuroscience 64, 357–374. doi:10.1016/0306-4522(94)00330-8
Cameron, H. A., and Mckay, R. D. (2001). Adult Neurogenesis Produces a Large Pool of New Granule Cells in the Dentate Gyrus. J. Comp. Neurol. 435, 406–417. doi:10.1002/cne.1040
Casamenti, F., Scali, C., Vannucchi, M. G., Bartolini, L., and Pepeu, G. (1993). Long-term Ethanol Consumption by Rats: Effect on Acetylcholine Release In Vivo, Choline Acetyltransferase Activity, and Behavior. Neuroscience 56, 465–471. doi:10.1016/0306-4522(93)90346-H
Choi, I. Y., Allan, A. M., and Cunningham, L. A. (2005). Moderate Fetal Alcohol Exposure Impairs the Neurogenic Response to an Enriched Environment in Adult Mice. Alcohol. Clin. Exp. Res. 29, 2053–2062. doi:10.1097/01.alc.0000187037.02670.59
Chung, T., Creswell, K. G., Bachrach, R., Clark, D. B., and Martin, C. S. (2018). Adolescent Binge Drinking. Alcohol. Res. 39, 5–15.
Clarke, M. E., and Gibbard, W. B. (2003). Overview of Fetal Alcohol Spectrum Disorders for Mental Health Professionals. Can Child. Adolesc. Psychiatr. Rev. 12, 57–63.
Clelland, C. D., Choi, M., Romberg, C., Clemenson, G. D., Fragniere, A., Tyers, P., et al. (2009). A Functional Role for Adult Hippocampal Neurogenesis in Spatial Pattern Separation. Science 325, 210–213. doi:10.1126/science.1173215
Cole, A. E., and Nicoll, R. A. (1984). Characterization of a Slow Cholinergic post-synaptic Potential Recorded In Vitro from Rat Hippocampal Pyramidal Cells. J. Physiol. 352, 173–188. doi:10.1113/jphysiol.1984.sp015285
Coleman, L. G., and Crews, F. T. (2018). Innate Immune Signaling and Alcohol Use Disorders. Handb. Exp. Pharmacol. 248, 369–396. doi:10.1007/16410.1007/164_2018_92
Coleman, L. G., He, J., Lee, J., Styner, M., and Crews, F. T. (2011). Adolescent Binge Drinking Alters Adult Brain Neurotransmitter Gene Expression, Behavior, Brain Regional Volumes, and Neurochemistry in Mice. Alcohol. Clin. Exp. Res. 35, 671–688. doi:10.1111/j.1530-0277.2010.01385.x
Coleman, L. G., Oguz, I., Lee, J., Styner, M., and Crews, F. T. (2012). Postnatal Day 7 Ethanol Treatment Causes Persistent Reductions in Adult Mouse Brain Volume and Cortical Neurons with Sex Specific Effects on Neurogenesis. Alcohol 46, 603–612. doi:10.1016/j.alcohol.2012.01.003
Coleman, L. G., Zou, J., Qin, L., and Crews, F. T. (2018). HMGB1/IL-1β Complexes Regulate Neuroimmune Responses in Alcoholism. Brain Behav. Immun. 72, 61–77. doi:10.1016/j.bbi.2017.10.027
Conejero-Goldberg, C., Davies, P., and Ulloa, L. (2008). Alpha7 Nicotinic Acetylcholine Receptor: A Link between Inflammation and Neurodegeneration. Neurosci. Biobehav. Rev. 32, 693–706. doi:10.1016/j.neubiorev.2007.10.007
Court, J. A., Lloyd, S., Johnson, M., Griffiths, M., Birdsall, N. J., Piggott, M. A., et al. (1997). Nicotinic and Muscarinic Cholinergic Receptor Binding in the Human Hippocampal Formation during Development and Aging. Brain Res. Dev. Brain Res. 101, 93–105. doi:10.1016/S0165-3806(97)00052-7
Crews, F. T., Mdzinarishvili, A., Kim, D., He, J., and Nixon, K. (2006). Neurogenesis in Adolescent Brain Is Potently Inhibited by Ethanol. Neuroscience 137, 437–445. doi:10.1016/j.neuroscience.2005.08.090
Crews, F. T., Robinson, D. L., Chandler, L. J., Ehlers, C. L., Mulholland, P. J., Pandey, S. C., et al. (2019). Mechanisms of Persistent Neurobiological Changes Following Adolescent Alcohol Exposure: NADIA Consortium Findings. Alcohol. Clin. Exp. Res. 43, 1806–1822. doi:10.1111/acer.14154
Crews, F. T., Vetreno, R. P., Broadwater, M. A., and Robinson, D. L. (2016). Adolescent Alcohol Exposure Persistently Impacts Adult Neurobiology and Behavior. Pharmacol. Rev. 68, 1074–1109. doi:10.1124/pr.115.012138
Crews, F. T., Fisher, R., Deason, C., and Vetreno, R. P. (2021). Loss of Basal Forebrain Cholinergic Neurons Following Adolescent Binge Ethanol Exposure: Recovery with the Cholinesterase Inhibitor Galantamine. Front. Behav. Neurosci. 15, 32. doi:10.3389/fnbeh.2021.652494
Crothers, D. M. (1993). Architectural Elements in Nucleoprotein Complexes. Curr. Biol. 3, 675–676. doi:10.1016/0960-9822(93)90065-V
Cruz, C., Meireles, M., and Silva, S. M. (2017). Chronic Ethanol Intake Induces Partial Microglial Activation that Is Not Reversed by Long-Term Ethanol Withdrawal in the Rat Hippocampal Formation. NeuroToxicology 60, 107–115. doi:10.1016/j.neuro.2017.04.005
da Silva, R. P., Lewis, E. D., Leonard, K. A., Goruk, S., Curtis, J. M., Vine, D. F., et al. (2015). Choline Deficiency Impairs Intestinal Lipid Metabolism in the Lactating Rat. J. Nutr. Biochem. 26, 1077–1083. doi:10.1016/j.jnutbio.2015.04.015
DiPietro, J. A., and Voegtline, K. M. (2017). The Gestational Foundation of Sex Differences in Development and Vulnerability. Neuroscience 342, 4–20. doi:10.1016/j.neuroscience.2015.07.068
Dobbing, J., and Sands, J. (1979). Comparative Aspects of the Brain Growth Spurt. Early Hum. Dev. 3, 79–83. doi:10.1016/0378-3782(79)90022-7
Donovan, J. E. (2009). Estimated Blood Alcohol Concentrations for Child and Adolescent Drinking and Their Implications for Screening Instruments. Pediatrics 123, e975–81. doi:10.1542/peds.2008-0027
Drinking Levels Defined (2021). National Institute on Alcohol Abuse and Alcoholism (NIAAA). Available at: https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking (Accessed October 20, 2021).
Ehlers, C. L., Liu, W., Wills, D. N., and Crews, F. T. (2013). Periadolescent Ethanol Vapor Exposure Persistently Reduces Measures of Hippocampal Neurogenesis that Are Associated with Behavioral Outcomes in Adulthood. Neuroscience 244, 1–15. doi:10.1016/j.neuroscience.2013.03.058
El Gazzar, M., Yoza, B. K., Chen, X., Garcia, B. A., Young, N. L., and McCall, C. E. (2009). Chromatin-Specific Remodeling by HMGB1 and Linker Histone H1 Silences Proinflammatory Genes during Endotoxin Tolerance. Mol. Cel. Biol. 29, 1959–1971. doi:10.1128/MCB.01862-08
Esser, M. B., Sherk, A., Liu, Y., Naimi, T. S., Stockwell, T., Stahre, M., et al. (2020). Deaths and Years of Potential Life Lost from Excessive Alcohol Use - United States, 2011-2015. MMWR Morb Mortal Wkly Rep. 69, 981–987. doi:10.15585/mmwr.mm6930a1
Ethen, M. K., Ramadhani, T. A., Scheuerle, A. E., Canfield, M. A., Wyszynski, D. F., Druschel, C. M., et al. (2009). Alcohol Consumption by Women before and during Pregnancy. Matern. Child. Health J. 13, 274–285. doi:10.1007/s10995-008-0328-2
Farber, N. B., Creeley, C. E., and Olney, J. W. (2010). Alcohol-induced Neuroapoptosis in the Fetal Macaque Brain. Neurobiol. Dis. 40, 200–206. doi:10.1016/j.nbd.2010.05.025
Fernandez, G. M., and Savage, L. M. (2017). Adolescent Binge Ethanol Exposure Alters Specific Forebrain Cholinergic Cell Populations and Leads to Selective Functional Deficits in the Prefrontal Cortex. Neuroscience 361, 129–143. doi:10.1016/j.neuroscience.2017.08.013
Field, R. H., Gossen, A., and Cunningham, C. (2012). Prior Pathology in the Basal Forebrain Cholinergic System Predisposes to Inflammation-Induced Working Memory Deficits: Reconciling Inflammatory and Cholinergic Hypotheses of Delirium. J. Neurosci. 32, 6288–6294. doi:10.1523/JNEUROSCI.4673-11.2012
Franjic, D., Skarica, M., Ma, S., Arellano, J. I., Tebbenkamp, A. T. N., Choi, J., et al. (2022). Transcriptomic Taxonomy and Neurogenic Trajectories of Adult Human, Macaque, and Pig Hippocampal and Entorhinal Cells. Neuron 110, 452–e14. doi:10.1016/j.neuron.2021.10.036
Garthe, A., and Kempermann, G. (2013). An Old Test for New Neurons: Refining the Morris Water Maze to Study the Functional Relevance of Adult Hippocampal Neurogenesis. Front. Neurosci. 7, 63. doi:10.3389/fnins.2013.00063
Gil-Mohapel, J., Boehme, F., Kainer, L., and Christie, B. R. (2010). Hippocampal Cell Loss and Neurogenesis after Fetal Alcohol Exposure: Insights from Different Rodent Models. Brain Res. Rev. 64, 283–303. doi:10.1016/j.brainresrev.2010.04.011
Gil-Mohapel, J., Titterness, A. K., Patten, A. R., Taylor, S., Ratzlaff, A., Ratzlaff, T., et al. (2014). Prenatal Ethanol Exposure Differentially Affects Hippocampal Neurogenesis in the Adolescent and Aged Brain. Neuroscience 273, 174–188. doi:10.1016/j.neuroscience.2014.05.012
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate Mapping Analysis Reveals that Adult Microglia Derive from Primitive Macrophages. Science 330, 841–845. doi:10.1126/science.1194637
Gnatek, Y., Zimmerman, G., Goll, Y., Najami, N., Soreq, H., and Friedman, A. (2012). Acetylcholinesterase Loosens the Brain's Cholinergic Anti-inflammatory Response and Promotes Epileptogenesis. Front. Mol. Neurosci. 5, 66. doi:10.3389/fnmol.2012.00066
Gómez, G. I., Falcon, R. V., Maturana, C. J., Labra, V. C., Salgado, N., Rojas, C. A., et al. (2018). Heavy Alcohol Exposure Activates Astroglial Hemichannels and Pannexons in the Hippocampus of Adolescent Rats: Effects on Neuroinflammation and Astrocyte Arborization. Front. Cel. Neurosci. 12, 472. doi:10.3389/fncel.2018.00472
Grant, B. F., Goldstein, R. B., Saha, T. D., Chou, S. P., Jung, J., Zhang, H., et al. (2015). Epidemiology of DSM-5 Alcohol Use Disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry 72, 757–766. doi:10.1001/jamapsychiatry.2015.0584
Gross, C. G. (2000). Neurogenesis in the Adult Brain: Death of a Dogma. Nat. Rev. Neurosci. 1, 67–73. doi:10.1038/35036235
Haake, A., Nguyen, K., Friedman, L., Chakkamparambil, B., and Grossberg, G. T. (2020). An Update on the Utility and Safety of Cholinesterase Inhibitors for the Treatment of Alzheimer's Disease. Expert Opin. Drug Saf. 19, 147–157. doi:10.1080/14740338.2020.1721456
Hamilton, G. F., Jablonski, S. A., Schiffino, F. L., St Cyr, S. A., Klintsova, A. Y., and Klintsova, A. Y. (2014). Exercise and Environment as an Intervention for Neonatal Alcohol Effects on Hippocampal Adult Neurogenesis and Learning. Neuroscience 265, 274–290. doi:10.1016/j.neuroscience.2014.01.061
Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., et al. (2018). The Cholinergic System in the Pathophysiology and Treatment of Alzheimer's Disease. Brain 141, 1917–1933. doi:10.1093/brain/awy132
He, J., and Crews, F. T. (2007). Neurogenesis Decreases during Brain Maturation from Adolescence to Adulthood. Pharmacol. Biochem. Behav. 86, 327–333. doi:10.1016/j.pbb.2006.11.003
Helfer, J. L., Goodlett, C. R., Greenough, W. T., and Klintsova, A. Y. (2009). The Effects of Exercise on Adolescent Hippocampal Neurogenesis in a Rat Model of Binge Alcohol Exposure during the Brain Growth Spurt. Brain Res. 1294, 1–11. doi:10.1016/j.brainres.2009.07.090
Henderson, J., Kesmodel, U., and Gray, R. (2007). Systematic Review of the Fetal Effects of Prenatal Binge-Drinking. J. Epidemiol. Community Health 61, 1069–1073. doi:10.1136/jech.2006.054213
Hernandez, M., von Sternberg, K., Castro, Y., and Velasquez, M. M. (2021). Reasons and Obstacles for Changing Risky Drinking Behavior Among Latinas at Risk of an Alcohol-Exposed Pregnancy. J. Ethn. Substance Abuse 0, 1–15. doi:10.1080/15332640.2021.1952127
Higgins, G. A., Koh, S., Chen, K. S., and Gage, F. H. (1989). NGF Induction of NGF Receptor Gene Expression and Cholinergic Neuronal Hypertrophy within the Basal Forebrain of the Adult Rat. Neuron 3, 247–256. doi:10.1016/0896-6273(89)90038-X
Hommer, D., Momenan, R., Kaiser, E., and Rawlings, R. (2001). Evidence for a Gender-Related Effect of Alcoholism on Brain Volumes. Am. J. Psychiatry 158, 198–204. doi:10.1176/appi.ajp.158.2.198
Idrus, N. M., Breit, K. R., and Thomas, J. D. (2017). Dietary Choline Levels Modify the Effects of Prenatal Alcohol Exposure in Rats. Neurotoxicol. Teratol. 59, 43–52. doi:10.1016/j.ntt.2016.11.007
Idrus, N. M., and Thomas, J. D. (2011). Fetal Alcohol Spectrum Disorders: Experimental Treatments and Strategies for Intervention. Alcohol. Res. Health 34, 76–85.
Ieraci, A., and Herrera, D. G. (2007). Single Alcohol Exposure in Early Life Damages Hippocampal Stem/progenitor Cells and Reduces Adult Neurogenesis. Neurobiol. Dis. 26, 597–605. doi:10.1016/j.nbd.2007.02.011
Ikonomidou, C., Bittigau, P., Ishimaru, M. J., Wozniak, D. F., Koch, C., Genz, K., et al. (2000). Ethanol-Induced Apoptotic Neurodegeneration and Fetal Alcohol Syndrome. Science 287, 1056–1060. doi:10.1126/science.287.5455.1056
Jacobson, S. W., Carter, R. C., Molteno, C. D., Meintjes, E. M., Senekal, M. S., Lindinger, N. M., et al. (2018a). Feasibility and Acceptability of Maternal Choline Supplementation in Heavy Drinking Pregnant Women: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Alcohol. Clin. Exp. Res. 42, 1315–1326. doi:10.1111/acer.13768
Jacobson, S. W., Carter, R. C., Molteno, C. D., Stanton, M. E., Herbert, J. S., Lindinger, N. M., et al. (2018b). Efficacy of Maternal Choline Supplementation during Pregnancy in Mitigating Adverse Effects of Prenatal Alcohol Exposure on Growth and Cognitive Function: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Alcohol. Clin. Exp. Res. 42, 1327–1341. doi:10.1111/acer.13769
Johnston, L. D., Miech, R. A., O’Malley, P. M., Bachman, J. G., Schulenberg, J. E., and Patrick, M. E. (2019). Monitoring the Future National Survey Results on Drug Use, 1975-2018: Overview, Key Findings on Adolescent Drug Use. Institute for Social Research Available at: https://eric.ed.gov/?id=ED594190 (Accessed October 22, 2021).
Jones, K. L., and Smith, D. W. (1973). Recognition of the Fetal Alcohol Syndrome in Early Infancy. Lancet 302, 999–1001. doi:10.1016/S0140-6736(73)91092-1
Kaneko, N., Okano, H., and Sawamoto, K. (2006). Role of the Cholinergic System in Regulating Survival of Newborn Neurons in the Adult Mouse Dentate Gyrus and Olfactory Bulb. Genes Cells 11, 1145–1159. doi:10.1111/j.1365-2443.2006.01010.x
Kanny, D., Liu, Y., Brewer, R. D., Eke, P. I., Cox, S. N., Cheal, N. E., et al. (2013). Vital Signs: Binge Drinking Among Women and High School Girls — United States, 2011. MMWR Morb. Mortal. Wkly. Rep. 62, 9–13.
Kee, N., Sivalingam, S., Boonstra, R., and Wojtowicz, J. M. (2002). The Utility of Ki-67 and BrdU as Proliferative Markers of Adult Neurogenesis. J. Neurosci. Methods 115, 97–105. doi:10.1016/s0165-0270(02)00007-9
Kelly, J., Black, C., and West, R. (1989). Changes in the Muscarinic Cholinergic Receptors in the Hippocampus of Rats Exposed to Ethyl Alcohol during the Brain Growth Spurt. J. Pharmacol. Exp. Ther. 249, 798–804.
Kempermann, G., Gage, F. H., Aigner, L., Song, H., Curtis, M. A., Thuret, S., et al. (2018). Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 23, 25–30. doi:10.1016/j.stem.2018.04.004
Kim, J. U., Majid, A., Judge, R., Crook, P., Nathwani, R., Selvapatt, N., et al. (2020). Effect of COVID-19 Lockdown on Alcohol Consumption in Patients with Pre-existing Alcohol Use Disorder. Lancet Gastroenterol. Hepatol. 5, 886–887. doi:10.1016/S2468-1253(20)30251-X
Kita, Y., Ago, Y., Higashino, K., Asada, K., Takano, E., Takuma, K., et al. (2014). Galantamine Promotes Adult Hippocampal Neurogenesis via M₁ Muscarinic and α7 Nicotinic Receptors in Mice. Int. J. Neuropsychopharmacol. 17, 1957–1968. doi:10.1017/S1461145714000613
Kobor, M. S., and Weinberg, J. (2011). Focus on: Epigenetics and Fetal Alcohol Spectrum Disorders. Alcohol. Res. Health 34, 29–37.
Kodituwakku, P. W. (2009). Neurocognitive Profile in Children with Fetal Alcohol Spectrum Disorders. Dev. Disabil. Res. Rev. 15, 218–224. doi:10.1002/ddrr.73
Kotani, S., Yamauchi, T., Teramoto, T., and Ogura, H. (2008). Donepezil, an Acetylcholinesterase Inhibitor, Enhances Adult Hippocampal Neurogenesis. Chem. Biol. Interact. 175, 227–230. doi:10.1016/j.cbi.2008.04.004
Kotani, S., Yamauchi, T., Teramoto, T., and Ogura, H. (2006). Pharmacological Evidence of Cholinergic Involvement in Adult Hippocampal Neurogenesis in Rats. Neuroscience 142, 505–514. doi:10.1016/j.neuroscience.2006.06.035
Kozareva, D. A., Cryan, J. F., and Nolan, Y. M. (2019). Born This Way: Hippocampal Neurogenesis across the Lifespan. Aging Cell 18, e13007. doi:10.1111/acel.13007
Kraus, C. L., Salazar, N. C., Mitchell, J. R., Florin, W. D., Guenther, B., Brady, D., et al. (2005). Inconsistencies between Actual and Estimated Blood Alcohol Concentrations in a Field Study of College Students: Do Students Really Know How Much They Drink? Alcohol. Clin. Exp. Res. 29, 1672–1676. doi:10.1097/01.alc.0000179205.24180.4a
Kuntsche, E., and Gmel, G. (2013). Alcohol Consumption in Late Adolescence and Early Adulthood - where Is the Problem? Swiss Med. Wkly. 143, w13826. doi:10.4414/smw.2013.13826
Lamminpää, A., Vilska, J., Korri, U. M., and Riihimäki, V. (1993). Alcohol Intoxication in Hospitalized Young Teenagers. Acta Paediatr. 82, 783–788. doi:10.1111/j.1651-2227.1993.tb12558.x
Latuskie, K. A., Andrews, N. C. Z., Motz, M., Leibson, T., Austin, Z., Ito, S., et al. (2019). Reasons for Substance Use Continuation and Discontinuation during Pregnancy: A Qualitative Study. Women Birth 32, e57–e64. doi:10.1016/j.wombi.2018.04.001
Lemoine, P. (1968). Les Enfants de Parents Alcooliques. Anomalies Observees. A Propos de 127 Cas. Ouest Med. 21, 476–482.
Levey, A. I., Edmunds, S. M., Koliatsos, V., Wiley, R. G., and Heilman, C. J. (1995). Expression of M1-M4 Muscarinic Acetylcholine Receptor Proteins in Rat hippocampus and Regulation by Cholinergic Innervation. J. Neurosci. 15, 4077–4092. doi:10.1523/jneurosci.15-05-04077.1995
Li, L., Liu, Z., Jiang, Y. Y., Shen, W. X., Peng, Y. P., and Qiu, Y. H. (2019a). Acetylcholine Suppresses Microglial Inflammatory Response via α7nAChR to Protect Hippocampal Neurons. J. Integr. Neurosci. 18, 51–56. doi:10.31083/j.jin.2019.01.114
Li, Q., Liu, D., Pan, F., Ho, C. S. H., and Ho, R. C. M. (2019b). Ethanol Exposure Induces Microglia Activation and Neuroinflammation through TLR4 Activation and SENP6 Modulation in the Adolescent Rat Hippocampus. Neural Plast. 2019, 1648736. doi:10.1155/2019/1648736
Li, Y., Holtzman, D. M., Kromer, L. F., Kaplan, D. R., Chua-Couzens, J., Clary, D. O., et al. (1995). Regulation of TrkA and ChAT Expression in Developing Rat Basal Forebrain: Evidence that Both Exogenous and Endogenous NGF Regulate Differentiation of Cholinergic Neurons. J. Neurosci. 15, 2888–2905. doi:10.1523/JNEUROSCI.15-04-02888.1995
Lilienfeld, S. (2002). Galantamine--a Novel Cholinergic Drug with a Unique Dual Mode of Action for the Treatment of Patients with Alzheimer's Disease. CNS Drug Rev. 8, 159–176. doi:10.1111/j.1527-3458.2002.tb00221.x
Lister, R., Mukamel, E. A., Nery, J. R., Urich, M., Puddifoot, C. A., Johnson, N. D., et al. (2013). Global Epigenomic Reconfiguration during Mammalian Brain Development. Science 341, 1237905. doi:10.1126/science.1237905
Liu, W., Vetreno, R. P., and Crews, F. T. (2021). Hippocampal TNF-Death Receptors, Caspase Cell Death Cascades, and IL-8 in Alcohol Use Disorder. Mol. Psychiatry 26, 2254–2262. doi:10.1038/s41380-020-0698-4
Liu, W., and Crews, F. T. (2017). Persistent Decreases in Adult Subventricular and Hippocampal Neurogenesis Following Adolescent Intermittent Ethanol Exposure. Front. Behav. Neurosci. 11. doi:10.3389/fnbeh.2017.00151
Ma, W., Maric, D., Li, B. S., Hu, Q., Andreadis, J. D., Grant, G. M., et al. (2000). Acetylcholine Stimulates Cortical Precursor Cell Proliferation In Vitro via Muscarinic Receptor Activation and MAP Kinase Phosphorylation. Eur. J. Neurosci. 12, 1227–1240. doi:10.1046/j.1460-9568.2000.00010.x
Macht, V., Crews, F. T., and Vetreno, R. P. (2020a). Neuroimmune and Epigenetic Mechanisms Underlying Persistent Loss of Hippocampal Neurogenesis Following Adolescent Intermittent Ethanol Exposure. Curr. Opin. Pharmacol. 50, 9–16. doi:10.1016/j.coph.2019.10.007
Macht, V., Elchert, N., and Crews, F. (2020b). Adolescent Alcohol Exposure Produces Protracted Cognitive-Behavioral Impairments in Adult Male and Female Rats. Brain Sci. 10, 785. doi:10.3390/brainsci10110785
Macht, V., Vetreno, R., Elchert, N., and Crews, F. (2021). Galantamine Prevents and Reverses Neuroimmune Induction and Loss of Adult Hippocampal Neurogenesis Following Adolescent Alcohol Exposure. J. Neuroinflammation 18, 212. doi:10.1186/s12974-021-02243-7
Macht, V. A. (2016). Neuro-immune Interactions across Development: A Look at Glutamate in the Prefrontal Cortex. Neurosci. Biobehav. Rev. 71, 267–280. doi:10.1016/j.neubiorev.2016.08.039
Madrid, L. I., Jimenez-Martin, J., Coulson, E. J., and Jhaveri, D. J. (2021). Cholinergic Regulation of Adult Hippocampal Neurogenesis and Hippocampus-dependent Functions. Int. J. Biochem. Cel Biol. 134, 105969. doi:10.1016/j.biocel.2021.105969
Mann, K., Ackermann, K., Croissant, B., Mundle, G., Nakovics, H., and Diehl, A. (2005). Neuroimaging of Gender Differences in Alcohol Dependence: Are Women More Vulnerable? Alcohol. Clin. Exp. Res. 29, 896–901. doi:10.1097/01.ALC.0000164376.69978.6B
Martínez, S. E., and Egea, G. (2007). Novel Molecular Targets for the Prevention of Fetal Alcohol Syndrome. Recent Pat CNS Drug Discov. 2, 23–35. doi:10.2174/157488907779561763
Matthews, D. A., Nadler, J. V., Lynch, G. S., and Cotman, C. W. (1974). Development of Cholinergic Innervation in the Hippocampal Formation of the Rat. I. Histochemical Demonstration of Acetylcholinesterase Activity. Dev. Biol. 36, 130–141. doi:10.1016/0012-1606(74)90196-1
Mattson, C. L., Tanz, L. J., Quinn, K., Kariisa, M., Patel, P., and Davis, N. L. (2021). Trends and Geographic Patterns in Drug and Synthetic Opioid Overdose Deaths - United States, 2013-2019. MMWR Morb. Mortal. Wkly. Rep. 70, 202–207. doi:10.15585/mmwr.mm7006a4
May, P. A., Baete, A., Russo, J., Elliott, A. J., Blankenship, J., Kalberg, W. O., et al. (2014). Prevalence and Characteristics of Fetal Alcohol Spectrum Disorders. Pediatrics 134, 855–866. doi:10.1542/peds.2013-3319
Maynard, M. E., Barton, E. A., Robinson, C. R., Wooden, J. I., and Leasure, J. L. (2018). Sex Differences in Hippocampal Damage, Cognitive Impairment, and Trophic Factor Expression in an Animal Model of an Alcohol Use Disorder. Brain Struct. Funct. 223, 195–210. doi:10.1007/s00429-017-1482-3
Maynard, M. E., and Leasure, J. L. (2013). Exercise Enhances Hippocampal Recovery Following Binge Ethanol Exposure. PLOS ONE 8, e76644. doi:10.1371/journal.pone.0076644
McClain, J. A., Morris, S. A., Deeny, M. A., Marshall, S. A., Hayes, D. M., Kiser, Z. M., et al. (2011). Adolescent Binge Alcohol Exposure Induces Long-Lasting Partial Activation of Microglia. Brain Behav. Immun. 25 Suppl 1, S120–S128. doi:10.1016/j.bbi.2011.01.006
McClain, J. A., Morris, S. A., Marshall, S. A., and Nixon, K. (2014). Ectopic Hippocampal Neurogenesis in Adolescent Male Rats Following Alcohol Dependence. Addict. Biol. 19, 687–699. doi:10.1111/adb.12075
Milbocker, K. A., and Klintsova, A. Y. (2021). Examination of Cortically Projecting Cholinergic Neurons Following Exercise and Environmental Intervention in a Rodent Model of Fetal Alcohol Spectrum Disorders. Birth Defects Res. 113, 299–313. doi:10.1002/bdr2.1839
Ming, G. L., and Song, H. (2005). Adult Neurogenesis in the Mammalian Central Nervous System. Annu. Rev. Neurosci. 28, 223–250. doi:10.1146/annurev.neuro.28.051804.101459
Miyata, T., Hori, O., Zhang, J., Yan, S. D., Ferran, L., Iida, Y., et al. (1996). The Receptor for Advanced Glycation End Products (RAGE) Is a central Mediator of the Interaction of AGE-Beta2microglobulin with Human Mononuclear Phagocytes via an Oxidant-Sensitive Pathway. Implications for the Pathogenesis of Dialysis-Related Amyloidosis. J. Clin. Invest. 98, 1088–1094. doi:10.1172/JCI118889
Mohapel, P., Leanza, G., Kokaia, M., and Lindvall, O. (2005). Forebrain Acetylcholine Regulates Adult Hippocampal Neurogenesis and Learning. Neurobiol. Aging 26, 939–946. doi:10.1016/j.neurobiolaging.2004.07.015
Monk, B. R., Leslie, F. M., and Thomas, J. D. (2012). The Effects of Perinatal Choline Supplementation on Hippocampal Cholinergic Development in Rats Exposed to Alcohol during the Brain Growth Spurt. Hippocampus 22, 1750–1757. doi:10.1002/hipo.22009
Montesinos, J., Pascual, M., Rodríguez-Arias, M., Miñarro, J., and Guerri, C. (2016). Involvement of TLR4 in the Long-Term Epigenetic Changes, Rewarding and Anxiety Effects Induced by Intermittent Ethanol Treatment in Adolescence. Brain Behav. Immun. 53, 159–171. doi:10.1016/j.bbi.2015.12.006
Moore, E. M., and Riley, E. P. (2015). What Happens when Children with Fetal Alcohol Spectrum Disorders Become Adults? Curr. Dev. Disord. Rep. 2, 219–227. doi:10.1007/s40474-015-0053-7
Morris, S. A., Kelso, M. L., Liput, D. J., Marshall, S. A., and Nixon, K. (2010). Similar Withdrawal Severity in Adolescents and Adults in a Rat Model of Alcohol Dependence. Alcohol 44, 89–98. doi:10.1016/j.alcohol.2009.10.017
Mychasiuk, R., and Metz, G. A. (2016). Epigenetic and Gene Expression Changes in the Adolescent Brain: What Have We Learned from Animal Models? Neurosci. Biobehav. Rev. 70, 189–197. doi:10.1016/j.neubiorev.2016.07.013
Nadler, J. V., Matthews, D. A., Cotman, C. W., and Lynch, G. S. (1974). Development of Cholinergic Innervation in the Hippocampal Formation of the Rat. II. Quantitative Changes in Choline Acetyltransferase and Acetylcholinesterase Activities. Dev. Biol. 36, 142–154. doi:10.1016/0012-1606(74)90197-3
Naeff, B., Schlumpf, M., and Lichtensteiger, W. (1992). Pre- and Postnatal Development of High-Affinity [3H]nicotine Binding Sites in Rat Brain Regions: an Autoradiographic Study. Brain Res. Dev. Brain Res. 68, 163–174. doi:10.1016/0165-3806(92)90058-5
Nguyen, T. T., Risbud, R. D., Mattson, S. N., Chambers, C. D., and Thomas, J. D. (2016). Randomized, Double-Blind, Placebo-Controlled Clinical Trial of Choline Supplementation in School-Aged Children with Fetal Alcohol Spectrum Disorders. Am. J. Clin. Nutr. 104, 1683–1692. doi:10.3945/ajcn.116.142075
Niewiadomska, G., Mietelska-Porowska, A., and Mazurkiewicz, M. (2011). The Cholinergic System, Nerve Growth Factor and the Cytoskeleton. Behav. Brain Res. 221, 515–526. doi:10.1016/j.bbr.2010.02.024
Nixon, K., and Crews, F. T. (2002). Binge Ethanol Exposure Decreases Neurogenesis in Adult Rat hippocampus. J. Neurochem. 83, 1087–1093. doi:10.1046/j.1471-4159.2002.01214.x
Nixon, K., and Crews, F. T. (2004). Temporally Specific Burst in Cell Proliferation Increases Hippocampal Neurogenesis in Protracted Abstinence from Alcohol. J. Neurosci. 24, 9714–9722. doi:10.1523/JNEUROSCI.3063-04.2004
Obernier, J. A., Bouldin, T. W., and Crews, F. T. (2002). Binge Ethanol Exposure in Adult Rats Causes Necrotic Cell Death. Alcohol. Clin. Exp. Res. 26, 547–557. doi:10.1111/j.1530-0277.2002.tb02573.x
Olney, J. W., Tenkova, T., Dikranian, K., Muglia, L. J., Jermakowicz, W. J., D'Sa, C., et al. (2002). Ethanol-Induced Caspase-3 Activation in the In Vivo Developing Mouse Brain. Neurobiol. Dis. 9, 205–219. doi:10.1006/nbdi.2001.0475
O’Malley, P. M., Johnston, L. D., and Bachman, J. G. (1998). Alcohol Use Among Adolescents. Alcohol Health Res. World 22, 85–93.
Orio, L., Antón, M., Rodríguez-Rojo, I. C., Correas, Á., García-Bueno, B., Corral, M., et al. (2018). Young Alcohol Binge Drinkers Have Elevated Blood Endotoxin, Peripheral Inflammation and Low Cortisol Levels: Neuropsychological Correlations in Women. Addict. Biol. 23, 1130–1144. doi:10.1111/adb.12543
Osborne, B. F., Turano, A., and Schwarz, J. M. (2018). Sex Differences in the Neuroimmune System. Curr. Opin. Behav. Sci. 23, 118–123. doi:10.1016/j.cobeha.2018.05.007
Otero, N. K., Thomas, J. D., Saski, C. A., Xia, X., and Kelly, S. J. (2012). Choline Supplementation and DNA Methylation in the Hippocampus and Prefrontal Cortex of Rats Exposed to Alcohol during Development. Alcohol. Clin. Exp. Res. 36, 1701–1709. doi:10.1111/j.1530-0277.2012.01784.x
Pascual, M., Blanco, A. M., Cauli, O., Miñarro, J., and Guerri, C. (2007). Intermittent Ethanol Exposure Induces Inflammatory Brain Damage and Causes Long-Term Behavioural Alterations in Adolescent Rats. Eur. J. Neurosci. 25, 541–550. doi:10.1111/j.1460-9568.2006.05298.x
Pascual, M., Montesinos, J., Montagud-Romero, S., Forteza, J., Rodríguez-Arias, M., Miñarro, J., et al. (2017). TLR4 Response Mediates Ethanol-Induced Neurodevelopment Alterations in a Model of Fetal Alcohol Spectrum Disorders. J. Neuroinflammation 14, 145. doi:10.1186/s12974-017-0918-2
Patrick, M. E., Schulenberg, J. E., Martz, M. E., Maggs, J. L., O'Malley, P. M., and Johnston, L. D. (2013). Extreme Binge Drinking Among 12th-Grade Students in the United States: Prevalence and Predictors. JAMA Pediatr. 167, 1019–1025. doi:10.1001/jamapediatrics.2013.2392
Patrick, M. E., and Schulenberg, J. E. (2014). Prevalence and Predictors of Adolescent Alcohol Use and Binge Drinking in the United States. Alcohol. Res. 35, 193–200.
Patrick, M. E., and Terry-McElrath, Y. M. (2019). Prevalence of High-Intensity Drinking from Adolescence through Young Adulthood: National Data from 2016-2017. Subst. Abuse 13, 117822181882297. doi:10.1177/1178221818822976
Patten, A. R., Fontaine, C. J., and Christie, B. R. (2014). A Comparison of the Different Animal Models of Fetal Alcohol Spectrum Disorders and Their Use in Studying Complex Behaviors. Front. Pediatr. 2, 93. doi:10.3389/fped.2014.00093
Pavlov, V. A., Parrish, W. R., Rosas-Ballina, M., Ochani, M., Puerta, M., Ochani, K., et al. (2009). Brain Acetylcholinesterase Activity Controls Systemic Cytokine Levels through the Cholinergic Anti-inflammatory Pathway. Brain Behav. Immun. 23, 41–45. doi:10.1016/j.bbi.2008.06.011
Perkins, A. E., Fadel, J. R., and Kelly, S. J. (2015). The Effects of Postnatal Alcohol Exposure and Galantamine on the Context Pre-exposure Facilitation Effect and Acetylcholine Efflux Using In Vivo Microdialysis. Alcohol 49, 193–205. doi:10.1016/j.alcohol.2015.01.010
Petit, G., Kornreich, C., Verbanck, P., Cimochowska, A., and Campanella, S. (2013). Why Is Adolescence a Key Period of Alcohol Initiation and Who Is Prone to Develop Long-Term Problem Use? A Review of Current Available Data. Socioaffect Neurosci. Psychol. 3, 21890. doi:10.3402/snp.v3i0.21890
Plaschke, M., Naumann, T., Kasper, E., Bender, R., and Frotscher, M. (1997). Development of Cholinergic and GABAergic Neurons in the Rat Medial Septum: Effect of Target Removal in Early Postnatal Development. J. Comp. Neurol. 379, 467–481. doi:10.1002/(sici)1096-9861(19970324)379:4<467::aid-cne1>3.0.co;2-0
Podobinska, M., Szablowska-Gadomska, I., Augustyniak, J., Sandvig, I., Sandvig, A., and Buzanska, L. (2017). Epigenetic Modulation of Stem Cells in Neurodevelopment: The Role of Methylation and Acetylation. Front. Cel. Neurosci. 11, 23. doi:10.3389/fncel.2017.00023
Popova, S., Dozet, D., Akhand Laboni, S., Brower, K., and Temple, V. (2021). Why Do Women Consume Alcohol during Pregnancy or while Breastfeeding? Drug Alcohol Rev., 1–19. doi:10.1111/dar.13425
Porter, A. G., and Jänicke, R. U. (1999). Emerging Roles of Caspase-3 in Apoptosis. Cell Death Differ. 6, 99–104. doi:10.1038/sj.cdd.4400476
Pöyhönen, S., Er, S., Domanskyi, A., and Airavaara, M. (2019). Effects of Neurotrophic Factors in Glial Cells in the Central Nervous System: Expression and Properties in Neurodegeneration and Injury. Front. Physiol. 10, 486. doi:10.3389/fphys.2019.00486
Qian, X., Shen, Q., Goderie, S. K., He, W., Capela, A., Davis, A. A., et al. (2000). Timing of CNS Cell Generation: A Programmed Sequence of Neuron and Glial Cell Production from Isolated Murine Cortical Stem Cells. Neuron 28, 69–80. doi:10.1016/S0896-6273(00)00086-6
Qin, L., and Crews, F. T. (2012). Chronic Ethanol Increases Systemic TLR3 Agonist-Induced Neuroinflammation and Neurodegeneration. J. Neuroinflammation 9, 130. doi:10.1186/1742-2094-9-130
Qin, L., He, J., Hanes, R. N., Pluzarev, O., Hong, J. S., and Crews, F. T. (2008). Increased Systemic and Brain Cytokine Production and Neuroinflammation by Endotoxin Following Ethanol Treatment. J. Neuroinflammation 5, 10. doi:10.1186/1742-2094-5-10
Redila, V. A., Olson, A. K., Swann, S. E., Mohades, G., Webber, A. J., Weinberg, J., et al. (2006). Hippocampal Cell Proliferation Is Reduced Following Prenatal Ethanol Exposure but Can Be Rescued with Voluntary Exercise. Hippocampus 16, 305–311. doi:10.1002/hipo.20164
Richardson, H. N., Chan, S. H., Crawford, E. F., Lee, Y. K., Funk, C. K., Koob, G. F., et al. (2009). Permanent Impairment of Birth and Survival of Cortical and Hippocampal Proliferating Cells Following Excessive Drinking during Alcohol Dependence. Neurobiol. Dis. 36, 1–10. doi:10.1016/j.nbd.2009.05.021
Roberta, A., Claudia, P., Gian, L. G., and Flavia, F. (2017). Sex Differences in Alcohol Use Disorder. Curr. Med. Chem. 24, 2661–2670. doi:10.2174/0929867323666161202092908
Robles, N., and Sabriá, J. (2008). Effects of Moderate Chronic Ethanol Consumption on Hippocampal Nicotinic Receptors and Associative Learning. Neurobiol. Learn. Mem. 89, 497–503. doi:10.1016/j.nlm.2008.01.006
Rosas-Ballina, M., and Tracey, K. J. (2009). Cholinergic Control of Inflammation. J. Intern. Med. 265, 663–679. doi:10.1111/j.1365-2796.2009.02098.x
Rothberg, B. S., and Hunter, B. E. (1991). Chronic Ethanol Treatment Differentially Affects Muscarinic Receptor Responses in Rat hippocampus. Neurosci. Lett. 132, 243–246. doi:10.1016/0304-3940(91)90311-G
Rothberg, B. S., Hunter, B. E., Walker, D. W., Anderson, J. F., and Anderson, K. J. (1996). Long-Term Effects of Chronic Ethanol on Muscarinic Receptor Binding in Rat Brain. Alcohol. Clin. Exp. Res. 20, 1613–1617. doi:10.1111/j.1530-0277.1996.tb01706.x
Rothberg, B. S., Yasuda, R. P., Satkus, S. A., Wolfe, B. B., and Hunter, B. E. (1993). Effects of Chronic Ethanol on Cholinergic Actions in Rat hippocampus: Electrophysiological Studies and Quantification of M1-M5 Muscarinic Receptor Subtypes. Brain Res. 631, 227–234. doi:10.1016/0006-8993(93)91539-5
Ruggiero, M. J., Boschen, K. E., Roth, T. L., and Klintsova, A. Y. (2018). Sex Differences in Early Postnatal Microglial Colonization of the Developing Rat Hippocampus Following a Single-Day Alcohol Exposure. J. Neuroimmune Pharmacol. 13, 189–203. doi:10.1007/s11481-017-9774-1
Ryan, S. H., Williams, J. K., and Thomas, J. D. (2008). Choline Supplementation Attenuates Learning Deficits Associated with Neonatal Alcohol Exposure in the Rat: Effects of Varying the Timing of Choline Administration. Brain Res. 1237, 91–100. doi:10.1016/j.brainres.2008.08.048
Sakharkar, A. J., Vetreno, R. P., Zhang, H., Kokare, D. M., Crews, F. T., and Pandey, S. C. (2016). A Role for Histone Acetylation Mechanisms in Adolescent Alcohol Exposure-Induced Deficits in Hippocampal Brain-Derived Neurotrophic Factor Expression and Neurogenesis Markers in Adulthood. Brain Struct. Funct. 221, 4691–4703. doi:10.1007/s00429-016-1196-y
Sakurai, I., and Kawamura, Y. (1984). Growth Mechanism of Myelin Figures of Phosphatidylcholine. Biochim. Biophys. Acta 777, 347–351. doi:10.1016/0005-2736(84)90439-5
Salem, N. A., Mahnke, A. H., Konganti, K., Hillhouse, A. E., and Miranda, R. C. (2021). Cell-type and Fetal-sex-specific Targets of Prenatal Alcohol Exposure in Developing Mouse Cerebral Cortex. iScience 24, 102439. doi:10.1016/j.isci.2021.102439
Sauvageot, C. M., and Stiles, C. D. (2002). Molecular Mechanisms Controlling Cortical Gliogenesis. Curr. Opin. Neurobiol. 12, 244–249. doi:10.1016/S0959-4388(02)00322-7
Schliebs, R., and Arendt, T. (2011). The Cholinergic System in Aging and Neuronal Degeneration. Behav. Brain Res. 221, 555–563. doi:10.1016/j.bbr.2010.11.058
Sharrett-Field, L., Butler, T. R., Reynolds, A. R., Berry, J. N., and Prendergast, M. A. (2013). Sex Differences in Neuroadaptation to Alcohol and Withdrawal Neurotoxicity. Pflugers Arch. 465, 643–654. doi:10.1007/s00424-013-1266-4
Smiley, J. F., Bleiwas, C., Canals-Baker, S., Williams, S. Z., Sears, R., Teixeira, C. M., et al. (2021). Neonatal Ethanol Causes Profound Reduction of Cholinergic Cell Number in the Basal Forebrain of Adult Animals. Alcohol 97, 1–11. doi:10.1016/j.alcohol.2021.08.005
Snyder, J. S. (2019). Recalibrating the Relevance of Adult Neurogenesis. Trends Neurosci. 42, 164–178. doi:10.1016/j.tins.2018.12.001
Spear, L. P. (2000b). The Adolescent Brain and Age-Related Behavioral Manifestations. Neurosci. Biobehav. Rev. 24, 417–463. doi:10.1016/S0149-7634(00)00014-2
Spear, L. P. (2000a). Neurobehavioral Changes in Adolescence. Curr. Dir. Psychol. Sci. 9, 111–114. doi:10.1111/1467-8721.00072
Spencer, M. R., Curtin, S., and Hedegaard, H. (2020). Rates of Alcohol-Induced Deaths Among Adults Aged 25 and over in Urban and Rural Areas : United States, 2000–2018. Available at: https://stacks.cdc.gov/view/cdc/95119 (Accessed October 20, 2021).
Stevenson, J. R., Schroeder, J. P., Nixon, K., Besheer, J., Crews, F. T., and Hodge, C. W. (2009). Abstinence Following Alcohol Drinking Produces Depression-like Behavior and Reduced Hippocampal Neurogenesis in Mice. Neuropsychopharmacology 34, 1209–1222. doi:10.1038/npp.2008.90
Sulik, K. K. (1984). Critical Periods for Alcohol Teratogenesis in Mice, with Special Reference to the Gastrulation Stage of Embryogenesis. Ciba Found. Symp. 105, 124–141. doi:10.1002/9780470720868.ch8
Swartzwelder, H. S., Healey, K. L., Liu, W., Dubester, K., Miller, K. M., and Crews, F. T. (2019). Changes in Neuroimmune and Neuronal Death Markers after Adolescent Alcohol Exposure in Rats Are Reversed by Donepezil. Sci. Rep. 9, 12110. doi:10.1038/s41598-019-47039-1
Taffe, M. A., Kotzebue, R. W., Crean, R. D., Crawford, E. F., Edwards, S., and Mandyam, C. D. (2010). Long-lasting Reduction in Hippocampal Neurogenesis by Alcohol Consumption in Adolescent Nonhuman Primates. Proc. Natl. Acad. Sci. U S A. 107, 11104–11109. doi:10.1073/pnas.0912810107
Takarada, T., Nakamichi, N., Kawagoe, H., Ogura, M., Fukumori, R., Nakazato, R., et al. (2012). Possible Neuroprotective Property of Nicotinic Acetylcholine Receptors in Association with Predominant Upregulation of Glial Cell Line-Derived Neurotrophic Factor in Astrocytes. J. Neurosci. Res. 90, 2074–2085. doi:10.1002/jnr.23101
Terasaki, L. S., and Schwarz, J. M. (2016). Effects of Moderate Prenatal Alcohol Exposure during Early Gestation in Rats on Inflammation across the Maternal-Fetal-Immune Interface and Later-Life Immune Function in the Offspring. J. Neuroimmune Pharmacol. 11, 680–692. doi:10.1007/s11481-016-9691-8
Terrando, N., Yang, T., Ryu, J. K., Newton, P. T., Monaco, C., Feldmann, M., et al. (2014). Stimulation of the α7 Nicotinic Acetylcholine Receptor Protects against Neuroinflammation after Tibia Fracture and Endotoxemia in Mice. Mol. Med. 20, 667–675. doi:10.2119/molmed.2014.00143
Thanh, N. X., Jonsson, E., Salmon, A., and Sebastianski, M. (2014). Incidence and Prevalence of Fetal Alcohol Spectrum Disorder by Sex and Age Group in Alberta, Canada. J. Popul. Ther. Clin. Pharmacol. 21, e395–404. Available at: https://www.jptcp.com (Accessed October 1, 2021).
Thomas, J. D., Abou, E. J., and Dominguez, H. D. (2009). Prenatal Choline Supplementation Mitigates the Adverse Effects of Prenatal Alcohol Exposure on Development in Rats. Neurotoxicol. Teratol. 31, 303–311. doi:10.1016/j.ntt.2009.07.002
Thomas, J. D., Garrison, M., and O'Neill, T. M. (2004). Perinatal Choline Supplementation Attenuates Behavioral Alterations Associated with Neonatal Alcohol Exposure in Rats. Neurotoxicol. Teratol. 26, 35–45. doi:10.1016/j.ntt.2003.10.002
Thomas, J. D., Idrus, N. M., Monk, B. R., and Dominguez, H. D. (2010). Prenatal Choline Supplementation Mitigates Behavioral Alterations Associated with Prenatal Alcohol Exposure in Rats. Birth Defects Res. A. Clin. Mol. Teratol 88, 827–837. doi:10.1002/bdra.20713
Thomas, J. D., La Fiette, M. H., Riley, E. P., and Riley, E. P. (2000). Neonatal Choline Supplementation Ameliorates the Effects of Prenatal Alcohol Exposure on a Discrimination Learning Task in Rats. Neurotoxicol. Teratol. 22, 703–711. doi:10.1016/S0892-0362(00)00097-0
Tran, T. D., and Kelly, S. J. (2003). Critical Periods for Ethanol-Induced Cell Loss in the Hippocampal Formation. Neurotoxicol. Teratol. 25, 519–528. doi:10.1016/S0892-0362(03)00074-6
Valentino, R. J., and Dingledine, R. J. (2021). Presynaptic Inhibitory Effects of Acetylcholine in the Hippocampus: A 40-Year Evolution of a Serendipitous Finding. J. Neurosci. 41, 4550–4555. doi:10.1523/JNEUROSCI.3229-20.2021
Van Kampen, J. M., and Eckman, C. B. (2010). Agonist-induced Restoration of Hippocampal Neurogenesis and Cognitive Improvement in a Model of Cholinergic Denervation. Neuropharmacology 58, 921–929. doi:10.1016/j.neuropharm.2009.12.005
van Praag, H., Schinder, A. F., Christie, B. R., Toni, N., Palmer, T. D., and Gage, F. H. (2002). Functional Neurogenesis in the Adult hippocampus. Nature 415, 1030–1034. doi:10.1038/4151030a
Vetreno, R. P., Bohnsack, J. P., Kusumo, H., Liu, W., Pandey, S. C., and Crews, F. T. (2020). Neuroimmune and Epigenetic Involvement in Adolescent Binge Ethanol-Induced Loss of Basal Forebrain Cholinergic Neurons: Restoration with Voluntary Exercise. Addict. Biol. 25, e12731. doi:10.1111/adb.12731
Vetreno, R. P., Broadwater, M., Liu, W., Spear, L. P., and Crews, F. T. (2014). Adolescent, but Not Adult, Binge Ethanol Exposure Leads to Persistent Global Reductions of Choline Acetyltransferase Expressing Neurons in Brain. PLOS ONE 9, e113421. doi:10.1371/journal.pone.0113421
Vetreno, R. P., and Crews, F. T. (2018). Adolescent Binge Ethanol-Induced Loss of Basal Forebrain Cholinergic Neurons and Neuroimmune Activation Are Prevented by Exercise and Indomethacin. PLoS ONE 13, e0204500. doi:10.1371/journal.pone.0204500
Vetreno, R. P., and Crews, F. T. (2015). Binge Ethanol Exposure during Adolescence Leads to a Persistent Loss of Neurogenesis in the Dorsal and Ventral hippocampus that Is Associated with Impaired Adult Cognitive Functioning. Front. Neurosci. 9. doi:10.3389/fnins.2015.00035
Vetreno, R. P., Lawrimore, C. J., Rowsey, P. J., and Crews, F. T. (2018). Persistent Adult Neuroimmune Activation and Loss of Hippocampal Neurogenesis Following Adolescent Ethanol Exposure: Blockade by Exercise and the Anti-inflammatory Drug Indomethacin. Front. Neurosci. 12. doi:10.3389/fnins.2018.00200
von Bartheld, C. S., Bahney, J., and Herculano-Houzel, S. (2016). The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 524, 3865–3895. doi:10.1002/cne.24040
Wagner, A. F., and Hunt, P. S. (2006). Impaired Trace Fear Conditioning Following Neonatal Ethanol: Reversal by Choline. Behav. Neurosci. 120, 482–487. doi:10.1037/0735-7044.120.2.482
Walker, L. C., Berizzi, A. E., Chen, N. A., Rueda, P., Perreau, V. M., Huckstep, K., et al. (2020). Acetylcholine Muscarinic M4 Receptors as a Therapeutic Target for Alcohol Use Disorder: Converging Evidence from Humans and Rodents. Biol. Psychiatry 88, 898–909. doi:10.1016/j.biopsych.2020.02.019
Weaver, I. C., Cervoni, N., Champagne, F. A., D'Alessio, A. C., Sharma, S., Seckl, J. R., et al. (2004). Epigenetic Programming by Maternal Behavior. Nat. Neurosci. 7, 847–854. doi:10.1038/nn1276
Wechsler, H., Dowdall, G. W., Davenport, A., and Castillo, S. (1995). Correlates of College Student Binge Drinking. Am. J. Public Health 85, 921–926. doi:10.2105/AJPH.85.7.921
Weerakoon, S. M., Jetelina, K. K., and Knell, G. (2021). Longer Time Spent at home during COVID-19 Pandemic Is Associated with Binge Drinking Among US Adults. Am. J. Drug Alcohol. Abuse 47, 98–106. doi:10.1080/00952990.2020.1832508
West, J. R., Hamre, K. M., and Cassell, M. D. (1986). Effects of Ethanol Exposure during the Third Trimester Equivalent on Neuron Number in Rat Hippocampus and Dentate Gyrus. Alcohol. Clin. Exp. Res. 10, 190–197. doi:10.1111/j.1530-0277.1986.tb05070.x
West, R. K., Wooden, J. I., Barton, E. A., and Leasure, J. L. (2019). Recurrent Binge Ethanol Is Associated with Significant Loss of Dentate Gyrus Granule Neurons in Female Rats Despite Concomitant Increase in Neurogenesis. Neuropharmacology 148, 272–283. doi:10.1016/j.neuropharm.2019.01.016
White, A. M., Kraus, C. L., and Swartzwelder, H. (2006). Many College Freshmen Drink at Levels Far beyond the Binge Threshold. Alcohol. Clin. Exp. Res. 30, 1006–1010. doi:10.1111/j.1530-0277.2006.00122.x
Willoughby, K. A., Sheard, E. D., Nash, K., and Rovet, J. (2008). Effects of Prenatal Alcohol Exposure on Hippocampal Volume, Verbal Learning, and Verbal and Spatial Recall in Late Childhood. J. Int. Neuropsychol. Soc. 14, 1022–1033. doi:10.1017/S1355617708081368
Winner, B., and Winkler, J. (2015). Adult Neurogenesis in Neurodegenerative Diseases: Figure 1. Cold Spring Harb Perspect. Biol. 7, a021287. doi:10.1101/cshperspect.a021287
Witkiewitz, K., Litten, R. Z., and Leggio, L. (2019). Advances in the Science and Treatment of Alcohol Use Disorder. Sci. Adv. 5. doi:10.1126/sciadv.aax4043
Wozniak, D. F., Hartman, R. E., Boyle, M. P., Vogt, S. K., Brooks, A. R., Tenkova, T., et al. (2004). Apoptotic Neurodegeneration Induced by Ethanol in Neonatal Mice Is Associated with Profound Learning/memory Deficits in Juveniles Followed by Progressive Functional Recovery in Adults. Neurobiol. Dis. 17, 403–414. doi:10.1016/j.nbd.2004.08.006
Wozniak, J. R., Fink, B. A., Fuglestad, A. J., Eckerle, J. K., Boys, C. J., Sandness, K. E., et al. (2020). Four-year Follow-Up of a Randomized Controlled Trial of Choline for Neurodevelopment in Fetal Alcohol Spectrum Disorder. J. Neurodev. Disord. 12, 9. doi:10.1186/s11689-020-09312-7
Wozniak, J. R., Fuglestad, A. J., Eckerle, J. K., Kroupina, M. G., Miller, N. C., Boys, C. J., et al. (2013). Choline Supplementation in Children with Fetal Alcohol Spectrum Disorders Has High Feasibility and Tolerability. Nutr. Res. 33, 897–904. doi:10.1016/j.nutres.2013.08.005
Wozniak, J. R., Riley, E. P., and Charness, M. E. (2019). Clinical Presentation, Diagnosis, and Management of Fetal Alcohol Spectrum Disorder. Lancet Neurol. 18, 760–770. doi:10.1016/S1474-4422(19)30150-4
Wu, H., Friedman, W. J., and Dreyfus, C. F. (2004). Differential Regulation of Neurotrophin Expression in Basal Forebrain Astrocytes by Neuronal Signals. J. Neurosci. Res. 76, 76–85. doi:10.1002/jnr.20060
Yanai, H., Ban, T., Wang, Z., Choi, M. K., Kawamura, T., Negishi, H., et al. (2009). HMGB Proteins Function as Universal Sentinels for Nucleic-Acid-Mediated Innate Immune Responses. Nature 462, 99–103. doi:10.1038/nature08512
Young, C., Roth, K. A., Klocke, B. J., West, T., Holtzman, D. M., Labruyere, J., et al. (2005). Role of Caspase-3 in Ethanol-Induced Developmental Neurodegeneration. Neurobiol. Dis. 20, 608–614. doi:10.1016/j.nbd.2005.04.014
Zeisel, S. H. (2006). The Fetal Origins of Memory: The Role of Dietary Choline in Optimal Brain Development. J. Pediatr. 149, S131–S136. doi:10.1016/j.jpeds.2006.06.065
Zhang, K., Wang, H., Xu, M., Frank, J. A., and Luo, J. (2018). Role of MCP-1 and CCR2 in Ethanol-Induced Neuroinflammation and Neurodegeneration in the Developing Brain. J. Neuroinflammation 15, 197. doi:10.1186/s12974-018-1241-2
Zhang, X., Liu, C., Miao, H., Gong, Z. h., and Nordberg, A. (1998). Postnatal Changes of Nicotinic Acetylcholine Receptor α2, α3, α4, α7 and β2 Subunits Genes Expression in Rat Brain. Int. J. Dev. Neurosci. 16, 507–518. doi:10.1016/S0736-5748(98)00044-6
Keywords: ethanol, hippocampus, acetylcholine, fetal alcohol, adolescence, choline, cytokines, doublecortin
Citation: Macht VA, Vetreno RP and Crews FT (2022) Cholinergic and Neuroimmune Signaling Interact to Impact Adult Hippocampal Neurogenesis and Alcohol Pathology Across Development. Front. Pharmacol. 13:849997. doi: 10.3389/fphar.2022.849997
Received: 07 January 2022; Accepted: 14 February 2022;
Published: 02 March 2022.
Edited by:
Youssef Sari, University of Toledo, United StatesReviewed by:
R. Dayne Mayfield, University of Texas at Austin, United StatesCopyright © 2022 Macht, Vetreno and Crews. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fulton T. Crews, ZnVsdG9uX2NyZXdzQG1lZC51bmMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.