Corrigendum: Propofol Protects Myocardium From Ischemia/Reperfusion Injury by Inhibiting Ferroptosis Through the AKT/p53 Signaling Pathway
 Shengqiang Li1
Shengqiang Li1 Zhen Lei1
Zhen Lei1 Xiaomei Yang1
Xiaomei Yang1 Meng Zhao1
Meng Zhao1 Yonghao Hou1
Yonghao Hou1 Di Wang2
Di Wang2 Shuhai Tang1
Shuhai Tang1 Jingxin Li3*
Jingxin Li3* Jingui Yu1*
Jingui Yu1*- 1Department of Anesthesiology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, China
- 2Department of Anesthesiology, The Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, China
- 3Department of Physiology, School of Basic Medical Science, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China
The molecular mechanism underlying the protective role of propofol against myocardial ischemia/reperfusion (I/R) injury remains poorly understood. Previous studies have shown that ferroptosis is an imperative pathological process in myocardial I/R injury. We hypothesized that propofol prevents myocardial I/R injury by inhibiting ferroptosis via the AKT/p53 signaling pathway. The ferroptosis-inducing agent erastin (E) and AKT inhibitor MK2206 (MK) were used to investigate the role of propofol in myocardial I/R injury. H9C2 cells treated without any reagents, erastin for 24 h, propofol for 1 h before adding erastin were assigned as the control (C), E, and E + P group, respectively. Cell viability, reactive oxygen species (ROS), and the expression of antioxidant enzymes, including ferritin heavy chain 1 (FTH1), cysteine/glutamate transporter (XCT), and glutathione peroxidase 4 (GPX4) in H9C2 cells. Rat hearts from the I/R + P or I/R groups were treated with or without propofol for 20 min before stopping perfusion for 30 min and reperfusion for 60 min. Rat hearts from the I/R + P + MK or I/R + MK groups were treated with or without propofol for 20 min, with a 10-min treatment of MK2206 before stopping perfusion. Myocardial histopathology, mitochondrial structure, iron levels, and antioxidant enzymes expression were assessed. Our results demonstrated that erastin increased H9C2 cell mortality and reduced the expression of antioxidant enzymes. I/R, which reduced the expression of antioxidant enzymes and increased iron or p53 (p < 0.05), boosted myocardium pathological and mitochondrion damage. Propofol inhibited these changes; however, the effects of propofol on I/R injury were antagonized by MK (p < 0.05). In addition, AKT siRNA inhibited the propofol-induced expression of antioxidant enzymes (p < 0.05). Our findings confirm that propofol protects myocardium from I/R injury by inhibiting ferroptosis via the AKT/p53 signal pathway.
Introduction
Propofol (2,6-diisopropyl phenol), a popular intravenous anesthetic, is widely used in the induction and maintenance of general anesthesia (Bovill, 2006). Emerging data have shown that propofol exerts profound protective effects on myocardial ischemia/reperfusion (I/R) injury (Suleiman et al., 2015) by scavenging ROS (Zhao et al., 2019), reducing calcium overload (Zhou et al., 1997), and inhibiting polymorphonuclear neutrophils (PMN) adhesion (Szekely et al., 2000). Nevertheless, the specific underlying mechanism remains unclear.
Ferroptosis is a newly recognized form of cell death (Chen et al., 2020) that is distinct from apoptosis, necrosis, and autophagy (Macías-Rodríguez et al., 2020), characterized by cell membrane rupture and vesiculation, mitochondrial atrophy, ridge reduction, and nuclear-lacking chromatin aggregation (Yagoda et al., 2007; Yang and Stockwell, 2008). Under the action of divalent iron (Dixon et al., 2012) or iron-containing enzymes (Yang et al., 2016), unsaturated fatty acids of the cell membrane undergo lipid peroxidation to induce ferroptosis. The expression level of glutathione peroxidase 4 (GPX4) (Zhang et al., 2021), as the core enzyme of the antioxidant system, decreases during ferroptosis.
Ferroptosis is associated with a range of diseases, such as myocardial I/R injury, cancer, degenerative disease, and acute kidney injury (Han et al., 2020). As a critical role in the pathological process of myocardial injury (Stockwell et al., 2017), ferroptosis is closely related to p53 (Kruse and Gu, 2009). When cells are hypoxic, the stability and activity of p53 are enhanced. Subsequently, p53 enters the nucleus to regulate the expression of its downstream targets and promote the development of ferroptosis (Jiang et al., 2015). The activity of p53 is negatively regulated by AKT (also known as protein kinase B, PKB), a serine/threonine-specific protein kinase that is critical for regulating myocardial I/R injury (Yin et al., 2013). Phosphorylation of AKT promotes the combination of murine double minute 2 (MDM2) and p53, which leads to the degradation of p53 to inhibit ferroptosis (Freedman et al., 1999). A previous study has shown that propofol protects hepatic I/R injury by activating AKT phosphorylation (Wei et al., 2021) and p53 expression. Nevertheless, the effect of propofol on myocardial I/R injury remains unclear. Thus, we hypothesized that propofol may protect the myocardium by inhibiting myocardial ferroptosis through the AKT/p53 signaling pathway.
To test the above hypothesis, a series of in vitro and in vivo experiments were performed to investigate the effect and underlying mechanism of propofol on ferroptosis. This study demonstrates that propofol inhibits ferroptosis of both H9C2 cells and rat myocardium via the AKT/p53 signaling pathway, and it protects the myocardium from I/R injury.
Materials and Methods
Cell Culture
H9C2 cells were obtained from KeyGEN BioTECH (KG444, Nanjing, China), and cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, 0030034DJ, GIBCO, New York, USA) supplemented with 10% fetal bovine serum (FBS, 16140071, GIBCO, New York, USA) and 1% penicillin/streptomycin (15140122, GIBCO, New York, USA). The cells were incubated in a tri-gas incubator with 5% CO2 at 37°C.
Cell Viability Assay
Cells were inoculated at 3,000–4,000/well in 96-well plates. After treatment, 10% cell counting kit-8 (CCK-8, HY-K0301, MCE, New Jersey, United States) reagent was added and incubated for 1–3 h. The absorbance was measured using a Microplate Reader (Thermo Fisher Scientific, Massachusetts, United States) at 450 nm.
Experimental Grouping and Corresponding Treatment
H9C2 cells were treated with no reagents (C), erastin (5 μM, GC16630, California, GLPBIO, United States) for 24 h (E), and propofol (50 μM, D126608, Sigma, Missouri, United States) for 1 h before erastin (E + P). Each experiment was repeated three times (Figure 1A).
FIGURE 1
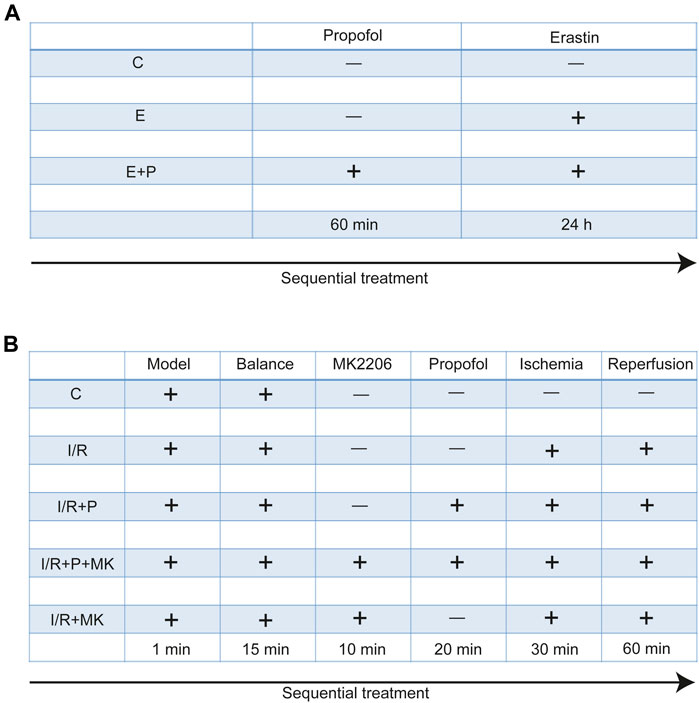
FIGURE 1. Experimental grouping and corresponding treatment. Experimental grouping and corresponding treatment on H9C2 cells (A) and rat hearts (B).
Trypan Blue Assay
H9C2 cells were inoculated in 12-well plates and treated as described above. Next, 0.4% trypan blue solution (T8070, Solarbio, Beijing, China) was added to the cells for 3 min. Cell viability was calculated by a cell counter (Countstar® BioTech, ALIT Life Science, Shanghai, China).
Assessment of ROS Production
H9C2 cells were inoculated into 12-well plates overnight and treated as described above, then 10 µM dihydroethidium solution (DHE, S0063, Beyotime, Shanghai, China) was added to each well and the plates were incubated in the dark for 30 min at 37°C. The cells were observed under a fluorescence microscope (Nikon, Tokyo, Japan) and analyzed with Image J software (National Institutes of Health, Maryland, United States).
Malondialdehyde, Iron Levels, and Superoxide Dismutase Activity Measurement
H9C2 cells from each group were collected and used to generate a 10% homogenate, which was placed at −20°C and repeatedly thawed on ice 3 times. The supernatant was centrifuged at 2,500 r/min for 10 min at 4°C. The MDA levels and SOD activity were determined according to the kit instruction (A003-1-2 and A001-3-2, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The iron levels in H9C2 cells were detected using a kit (DIFE-250, BioAssay Systems, California, USA) and in the myocardium were determined according to the kit instructions (A039-2-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The protein concentration was measured with a BCA kit (P0012S, Beyotime, Shanghai, China).
Detection of Antioxidant Expressions in AKT siRNA-Transfecting H9C2 Cells
After incubation in 12-well plates for 24 h, the H9C2 cell medium was replaced with a serum-free medium (Opti-MEM I, Thermo Fisher Scientific, Massachusetts, United States). The siRNA (Gene Pharma, Shanghai, China) was diluted in serum-free medium and mixed with diluted Lipofectamine 2000 (Lipo 2000, Thermo Fisher Scientific, Massachusetts, USA) for 20 min at room temperature. Then, the mixture was added into 12-well plates and incubated for 6–8 h, replacing the medium with a complete medium once. After being incubated for 24 h, the cells were treated for follow-up experiments as the design above. Proteins were extracted from H9C2 cells and handled as western blot analysis. The siRNA sequence of AKT was as follows: F: 5′-CCAAGCACCGUGUGACCAUTT-3′, R: 5′-AUGGUCACACGGUGCUUGGTT-3′.
Animals
Adult male rats (Wistar, weight 180–240 g), purchased from the Experimental Animal Center of Shandong University, were housed in a constant temperature (25 ± 2°C) room, fed with standard water and laboratory diet.
Langendorff Heart Preparation
Rats were acclimatized to the laboratory environment for 3 days, anesthetized by 10% chloral hydrate, and sequentially injected with heparin (250 U/kg, H8060, Solarbio, Beijing, China). The hearts were rapidly resected and dipped into 40 ml ice-cold KH solution (pH 7.4, containing in mM: NaCl 115, MgSO4 1.2, KCl 4.7, KH2PO4 1.2, CaCl2 1.8, glucose 11, and NaHCO3 25). Finally, the hearts were cannulated using a retrofitted Langendorff apparatus and perfused with KH at a velocity of 10 ml/min, which was gassed with 5% CO2 and 95% O2 at 37°C. Myocardial ischemia was caused by stopping perfusion for 30 min, and reperfusion resulted from recanalization for another 60 min.
Animal Grouping and Treatment
All rats were randomly divided into five groups. After stabilizing for 15 min, rat hearts were treated with KH for 120 min (C), with (I/R + P) or without (I/R) propofol for 20 min before stopping perfusion for 30 min and reperfusion for 60 min, with (I/R + P + MK) or without propofol (I/R + MK) after MK2206 (15 nM, GC16304, New York, GLPBIO, USA) for 10 min before stopping perfusion (N = 6, Figure 1B).
MDA, Iron Levels, and Glutathione/Oxidized Glutathione (GSH/GSSG) Ratio Determination
After recanalization, the rat heart left ventricle anterior wall was cut. MDA, iron levels (A039-2-1, A039-2-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China), and the GSH/GSSG ratio (S0053, Beyotime, Shanghai, China) were assessed using their respective assay kits. The concentration of protein was detected by a BCA kit and the absorbance was determined with a microplate reader.
Myocardial Fiber Assessment
Hearts were fixed overnight in 4% paraformaldehyde, dehydrated with alcohol, and made transparent with xylene. They were then fixed in paraffin and finally cut into 3–4-µm-thick slices, which were placed in xylene and alcohol and then stained with hematoxylin-eosin (HE) (C0105S, Beyotime, Shanghai, China). Finally, the sections were sealed with neutral resin and imaged under a microscope (Nikon, Tokyo, Japan).
Mitochondrial Morphology Assessment
After reperfusion as described above, the left ventricle anterior wall was cut into a size of 2 mm × 5 mm × 10 mm and fixed overnight in electron microscope fixative (G1102, Servicebio, Wuhan, China). The sections were then embedded in phosphate buffer and baked in the oven. Sequentially, they were cut to a size 50–60 nm and stained with 2% uranyl acetate lead. Images were acquired under a transmission electron microscope (HITACHI, Tokyo, Japan).
Western Blot Analysis
Proteins were extracted from H9C2 cells and myocardium lysates of the left ventricle anterior wall. Protein was separated by 12% SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membranes, blocked with 5% non-fatty milk for 60 min, and incubated with primary antibodies overnight at 4°C. Then, the membranes were incubated with secondary antibodies for 60 min and detected with enhanced chemiluminescence (ECL) detection system. Images were analyzed by ImageJ software.
Immunofluorescence Assessment
After dewaxing and rehydrating according to a previous description, paraffin sections were repaired in citrate solution (P0083, Beyotime, Shanghai, China) for 30 min, blocked with 5% bovine serum albumin (BSA) for 60 min, and incubated with primary antibodies overnight at 4°C. Subsequently, they were incubated with secondary antibodies for 60 min and DAPI (C1002, Beyotime, Shanghai, China) for 15 min, followed by visualization using an immunofluorescence microscope and analyzed with ImageJ software.
Antibodies
The following antibodies were used: anti-AKT (ab179463, Abcam, Cambridgeshire, UK), anti-AKT (phospho Ser473, 4060, CST, Boston, United States), anti-P53 (2,524, CST, Boston, United States), anti-P53 (phospho S392, ab33889, Abcam, Cambridgeshire, UK), anti-Ferritin (ab75973, Abcam, Cambridgeshire, UK), anti-GPX4 (ab125066, Abcam, Cambridgeshire, UK), anti-XCT (ab175186, Abcam, Cambridgeshire), anti-α-tubulin (ab7291,Abcam, Cambridgeshire, UK). HRP-conjugated secondary antibodies (ZB-2301 or ZB-2305, ZSGB-BIO, Beijing, China) and fluorochrome conjugated secondary antibody (ab150064, Abcam, Cambridgeshire).
Statistical Analysis
Data are expressed as mean ± standard deviation (SD) and were analyzed by one-way ANOVA with Tukey’s post hoc test or the t-test. GraphPad Prism version 9.0 (San Diego, California, United States) was used for analysis, and a p value < 0.05 was considered to indicate a significant difference.
Results
Effect of Propofol on Erastin-Induced H9C2 Cells Ferroptosis
First, we examined the effect of erastin on H9C2 cells and found a linear inhibition of cell viability with increasing erastin concentrations using the CCK-8 assay. We found that 2.5 µM erastin markedly decreased cell viability; hence it was selected for follow-up experiments (Figure 2A). The cell viability was lower in Group E than in Group C, as revealed by the optical microscope and the trypan blue assay, which was increased by propofol pretreatment (Figures 2B,D,E). Similarly, the ROS, MDA, and iron levels were higher in Group E than in Group C as revealed by DHE staining, MDA, and iron test. All of these parameters were also reduced by propofol pretreatment (Figures 2C,F–H). The change of SOD activity was similar to the trypan blue results (Figure 2I). In addition, western bolt analysis showed a significant decrease in the expression of anti-ferroptosis enzymes (FTH1 and GPX4) in Group E compared with Group C, which was again reduced by propofol pretreatment (Figures 2J–L). Taken together, these results demonstrated that erastin induced ferroptosis in H9C2 cells and propofol pretreatment alleviates this form of death, suggesting an important protective role of propofol in the H9C2 cells ferroptosis.
FIGURE 2
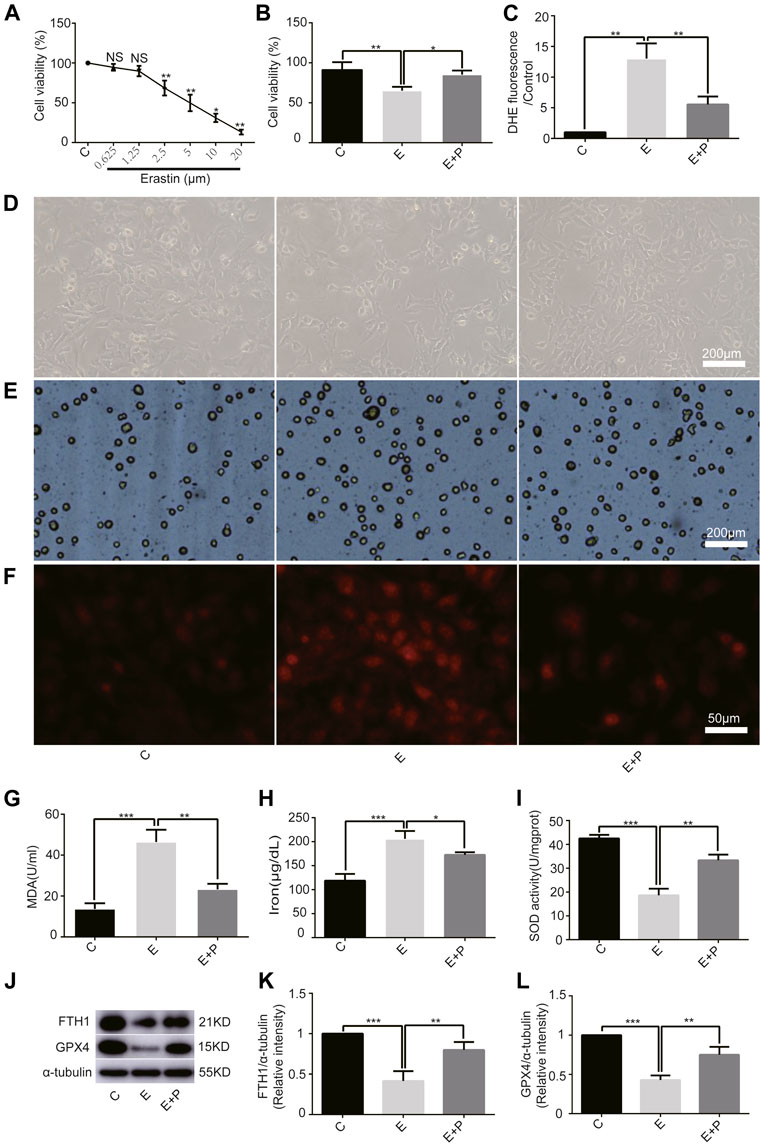
FIGURE 2. Effect of propofol on erastin-induced H9C2 cell ferroptosis. (A). Cell viability of H9C2 cells treated with erastin (0–20 μM). (B,D,E): Effect of propofol on erastin-induced cell death. Dead cells were stained with trypan blue. Scale bar: 200 μm. (C,F): Effect of propofol on reactive oxygen species (ROS) production. Quantification of ROS is expressed as DHE. Scale bar: 50 μm. Effect of propofol on MDA (G), iron (H), and SOD (I) production. (J–L): Influence of propofol on antioxidant enzymes (FTH1 and GPX4). N = 3. Data are expressed as the mean ± SD. Significance was calculated using one-way ANOVA with Tukey’s post hoc test or the t-test. p-values < 0.05 were considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001.
Effect of Propofol on AKT Knockdown in H9C2 Cells
Given the central role of AKT in myocardial protection, we investigated whether AKT was involved in the anti-ferroptosis effect of propofol. We first knocked out the AKT gene of H9C2 cells with AKT siRNA. Western bolt analysis showed that AKT siRNA significantly reduced AKT and P-AKT expressions levels (Figures 3A–C). In addition, it showed that AKT siRNA significantly reduced the expression of anti-ferroptosis enzymes (FTH1, XCT, and GPX4) in H9C2 cells, which was further reduced by erastin, while propofol slightly inhibited the effect of erastin (Figures 3D–G). Similarly, the cell viability of H9C2 cells in the CCK8 experiment was similar to the above enzymes (Figure 3H). These results indicated that propofol might inhibit ferroptosis in H9C2 cells through the AKT signaling pathway.
FIGURE 3
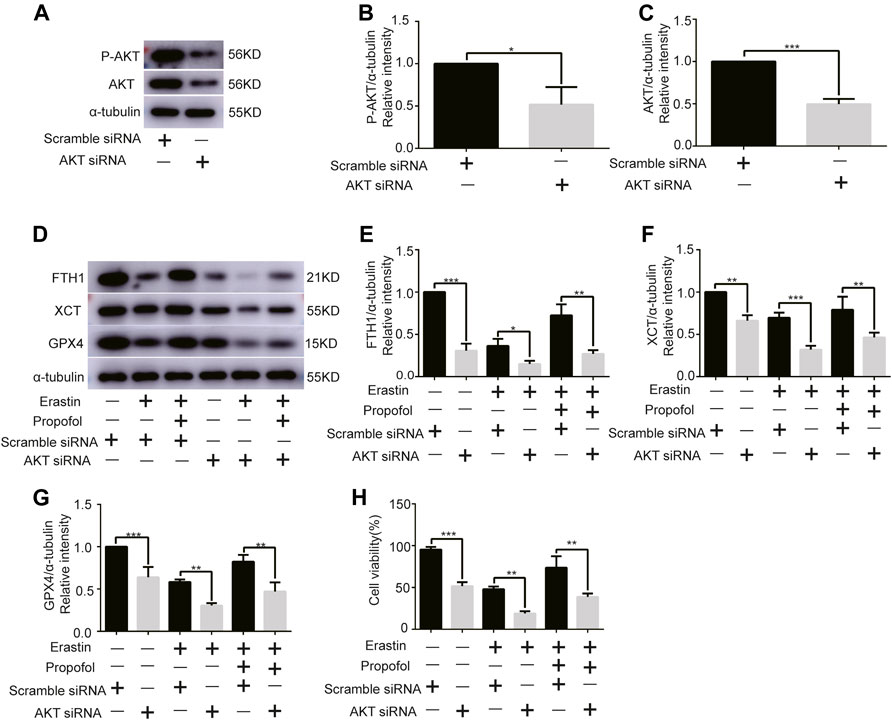
FIGURE 3. Effect of AKT knockdown on H9C2 cell ferroptosis with propofol pre-treatment. (A–C). Expression of AKT and p-AKT in H9C2 cells transfected with AKT and scramble siRNA. (D–G). Levels of FTH1, XCT, and GPX4 in H9C2 cells transfected with AKT and scramble siRNA with propofol pre-treatment. (H). Change in the viability of H9C2 cells transfected with AKT and scramble siRNA with propofol pre-treatment. N = 3. Data are expressed as the mean ± SD. Significance was calculated using one-way ANOVA with Tukey’s post hoc test or the t-test. p-values < 0.05 were considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001.
Influence of I/R-Induced Myocardial Ferroptosis
To understand the effect of propofol on myocardial tissue ferroptosis, we established an isolated myocardial I/R model with the Langendorff system. Similarly, MDA and iron levels were higher in Group I/R than in Group C, as revealed by the MDA and iron test; they were also reduced by propofol pretreatment (Figures 4A,B). In addition, western bolt analysis revealed a significant decrease in the expression of anti-ferroptosis enzymes (FTH1, XCT, and GPX4) in the I/R compared with Group C, which was also reduced by propofol pretreatment (Figures 4C–F). Taken together, these results demonstrated that I/R induced myocardial tissue ferroptosis and propofol pretreatment alleviates this phenomenon.
FIGURE 4
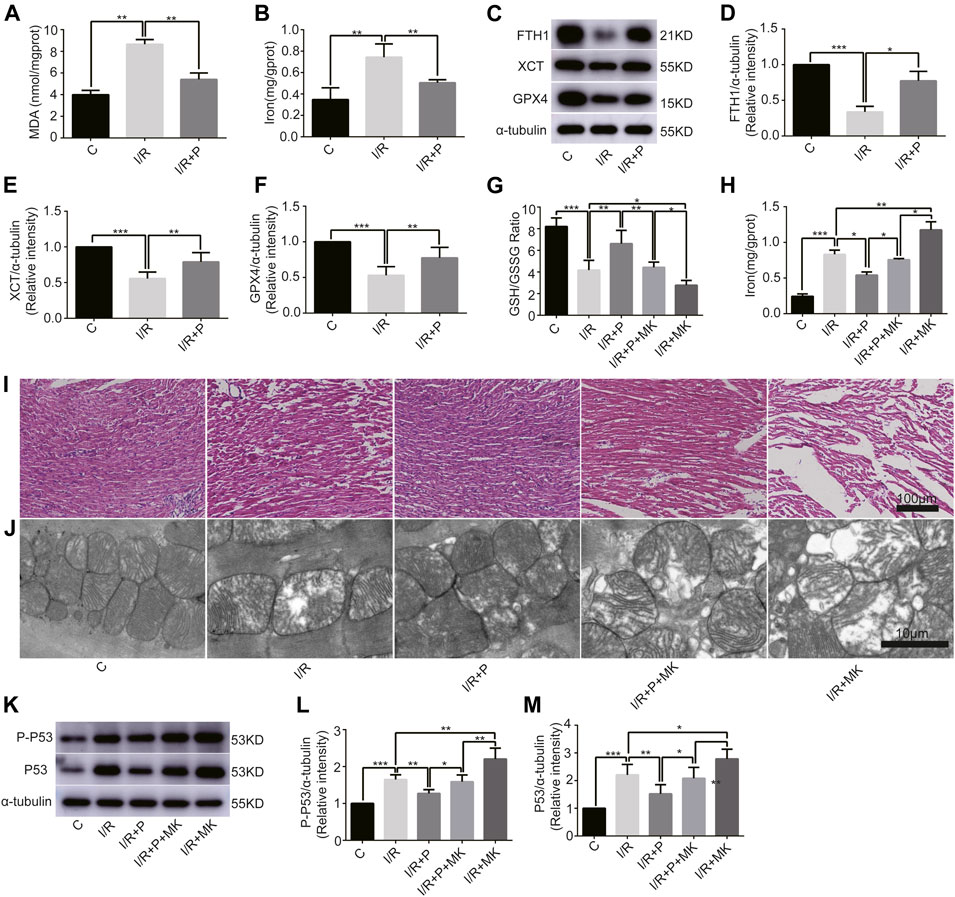
FIGURE 4. Influence of AKT inhibitor on myocardial ferroptosis with propofol pretreatment. (A–F): Effect of propofol on I/R-induced myocardial MDA, iron, and antioxidant enzymes expressions. (G–H): GSH/GSSG ratio and iron changes in the myocardium. (I) Structural changes in myocardial fibers based on HE staining. Scale bar: 100 μm. (J) Mitochondrial changes under electron microscopy. Scale bar: 10 μm. (K–M): Expression of p53 and p-p53 in myocardium based on the western blot assay. N = 6. Data are expressed as the mean ± SD. Significance was calculated using one-way ANOVA with Tukey’s post hoc test or the t-test. p-values < 0.05 were considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001.
Influence of AKT Inhibitor on Myocardium Ferroptosis With Propofol Pre-Treatment
Next, we investigated whether AKT was involved in the anti-ferroptosis effect of propofol on the myocardium. We pretreated myocardial tissue with AKT inhibitor (MK2206). I/R showed a lower GSH/GSSG ratio than C, which was reversed with propofol. However, MK2206 inhibited all the effects of propofol (Figure 4G). MK2206 significantly increased myocardium iron levels in Group I/R + MK compared with Group I/R and inhibited the effect of propofol (Figure 4H). Similarly, HE staining showed an abundance of severely wavy and injured myofibers in Group I/R compared with Group C, which was prevented by propofol (Figure 4I). TEM revealed that enlarging or distorting myocardial mitochondrial ridges in Group I/R compared with Group C, which was significantly diminished in Group I/R + P. MK2206 attenuated all these effects (Figure 4J). Changes in p53 and p-p53 protein were common with increased myocardium iron (Figure 4K–M). In addition, western blot showed that the changes in anti-ferroptosis enzymes (FTH1, XCT, and GPX4) were similar to those in the GSH/GSSG ratio (Figures 5A–D). These results suggest that propofol pretreatment inhibits ferroptosis in myocardial tissue through the AKT/p53 signaling pathway.
FIGURE 5
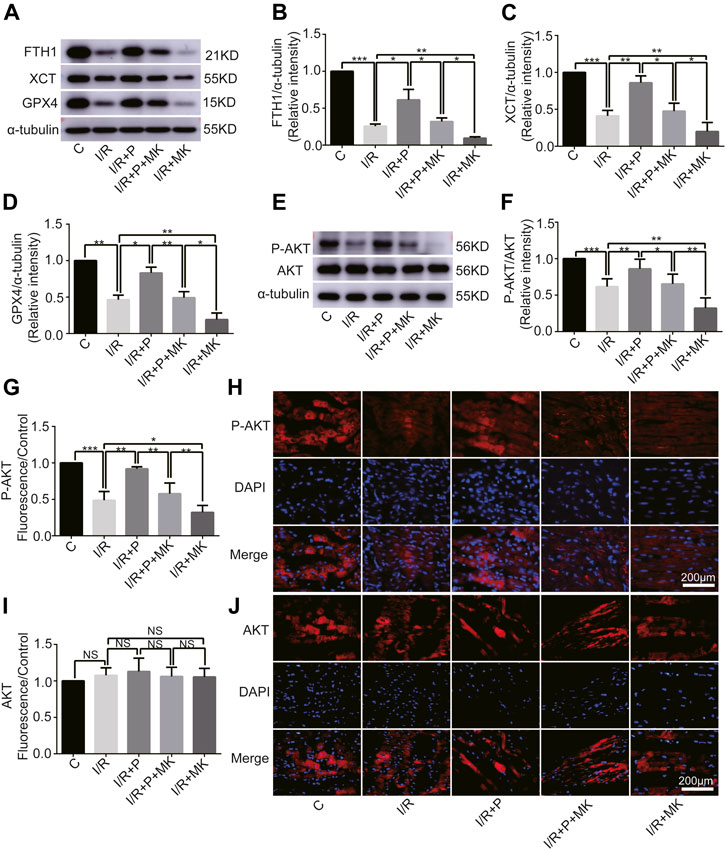
FIGURE 5. Changes in protein levels in response to propofol. (A–D): Influence of propofol on I/R-induced antioxidant enzyme expression. (E–F). Expression of AKT and p-AKT in myocardium based on western blot analysis. (G–J). Expression of AKT and P-AKT by immunofluorescence. N = 6. Data are expressed as the mean ± SD. Scale bar: 20 µm. Significance was calculated using one-way ANOVA with Tukey’s post hoc test or the t-test. p-values < 0.05 were considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001.
Change of AKT Activity by Propofol
Western blot analysis showed that AKT phosphorylation, which was inhibited by I/R or MK2206, was activated by propofol (Figures 5E,F). Immunofluorescence levels showing P-AKT phosphorylation were identical to the western blot results shown above (Figures 5G,H). There was no difference in immunofluorescence levels of AKT among the groups (Figures 5I,J).
Discussion
The major finding of this study was that propofol pretreatment inhibited ferroptosis in H9C2 cells and rat myocardium by reducing the expression of ROS, iron, and lipid peroxidation as well as increasing the expression of anti-ferroptosis enzymes. Therefore, propofol protects the myocardium from I/R injury through the AKT/P53 signaling pathway. To the best of our knowledge, this is the first study demonstrating that propofol inhibits myocardial ferroptosis and the underlying molecular mechanism.
Investigating the death processes and underlying mechanisms of cardiomyocytes may provide insights into novel therapeutic approaches for heart diseases, especially I/R injury (Whelan et al., 2010). Ferroptosis, which is distinct from conventional cell death, including apoptosis, necroptosis, and autophagy-dependent cell death, has recently been identified as a key pathological process connecting oxidative stress, inflammation, and cardiovascular diseases (Yu et al., 2021). Ferroptosis, an iron-dependent type of cell death that is mainly caused by oxidation-reduction imbalance, differs from conventional cell death processes characterized by typical cell shrinkage, mitochondrial fragmentation, and nuclear condensation. With the increasing understanding of the molecular mechanisms of ferroptosis, ferroptosis has become a potential novel diagnostic and therapeutic target of cardiomyopathy. In this study, our results demonstrated that ferroptosis played an essential role in myocardial injury induced by erastin or I/R following previous findings (Fang et al., 2019; Li et al., 2020). Briefly, erastin or I/R triggered iron accumulation and then damaged cardiomyocytes due to oxidative stress (Olivieri, 1994).
A number of studies have reported that propofol protects the myocardium from I/R injury (Kobayashi et al., 2008; Li et al., 2019), but the mechanism is not clear. Our study showed that p53, which promoted fatal lipid ROS accumulation and resulted in cardiomyocyte’s ferroptosis, was upregulated in I/R-induced myocardial injury, which is consistent with the previous study (Tang et al., 2021). Notably, we found that propofol, which significantly inhibited p53 expression, inhibited myocardial ferroptosis caused by erastin or I/R.
Our results also demonstrated that erastin or I/R induced antioxidant enzyme damage and ROS accumulation, resulting in cell ferroptosis. Recent studies have shown that GPX4 improves iron absorption by directly decomposing peroxides, catalyzing the transformation of reduced glutathione (GSH) to oxidized GSH (GSSG) (Imai et al., 2017), and inhibiting the expression of ferritin heavy chain 1 (FTH1) (Sun et al., 2016). In the present study, we found that propofol improved the antioxidant capacity of cardiomyocytes by improving the levels of FTH1, XCT, or GPX4 and reducing iron levels. Another study has shown that p53 inhibits cysteine absorption by downregulating XCT expression (Magri et al., 2021), resulting in the inhibition of cystine-dependent glutathione oxidase activity and increased cell lipid ROS, leading to ferroptosis (Jiang et al., 2015). Our results showing propofol downregulation of p53 and reduction of cardiomyocyte dysfunction are consistent with the previous study (Lai et al., 2011).
A recent study showed that AKT plays a pivotal role in p53 degradation (Chibaya et al., 2021), which is inconsistent with the present findings. The p53 was degraded by binding with MDM2 (Wang et al., 2020), and this process was accelerated by AKT phosphorylation at ser166 or 188 (Feng et al., 2004). As shown in our study, propofol protected the myocardium from I/R-induced injury by AKT phosphorylation (Shravah et al., 2014). The expression of p53 increased after MK2206 pretreatment, indicating that signaling pathways other than AKT were involved in p53 degradation. When DNA is damaged, ataxia telangiectasia-mutated (ATM) activation induces p53 activation, resulting in cell aging or apoptosis (Jiang and Chen, 2020). Calcium-dependent kinase 5 (CDK5) and CDK9 activate p53 by phosphorylating ser33, which enhances the binding abilities of cyclic adenosine monophosphate (AMP) response element-binding protein (CREB) binding protein (CBP), thus raising p53 transcription activity (Zhang et al., 2002) (Figure 6).
FIGURE 6
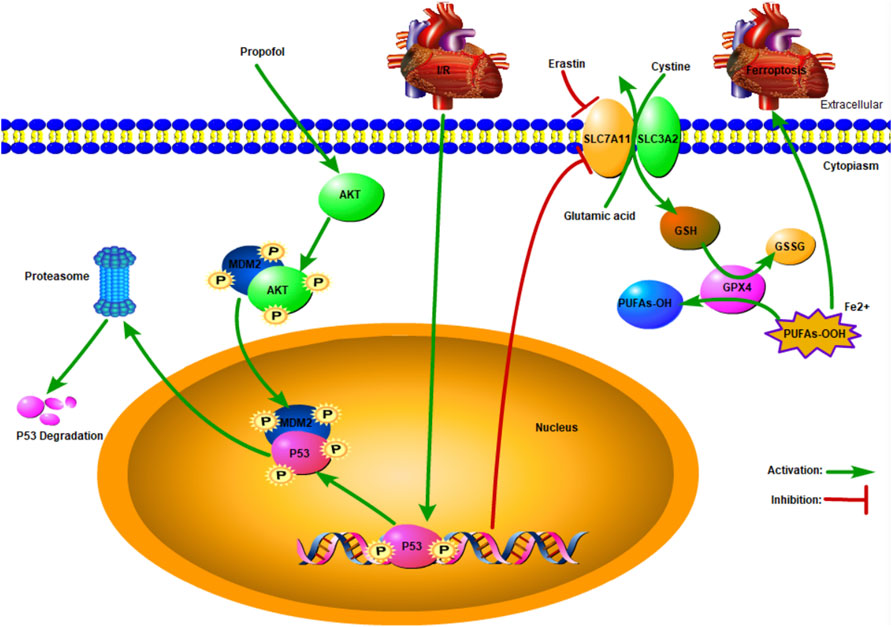
FIGURE 6. Schematic: Propofol protects the myocardium from ischemia/reperfusion injury by influencing an AKT/p53 signaling pathway-mediated ferroptosis mechanism.
Our study has some limitations. First, the Langendorff model rather than the rat I/R model was used in this study. However, the rat Langendorff model is easy to prepare, and the experimental results are reproducible (Finegan et al., 2000), which is consistent with best practices for reduction fine displacement (3R) (Flecknell, 2002). Second, the protective effects of AKT, which is the key survival signaling component in the heart, are associated with numerous factors. Recent studies have shown that propofol plays a protective role in the heart by activating the AKT/eNOS (Yan et al., 2021), ser473/thr308 (Lou et al., 2015), or PI3K/AKT (Wu et al., 2012) signal pathways. In our study, we only explored one biological process of AKT; other regulatory effects require further investigation. Third, the animal experiments were complicated and difficult, with a low success rate and inconsistent results (De Villiers and Riley, 2020). Fourth, propofol was used at a single concentration (50 µM) according to a previous study (Yu et al., 2018), in which propofol (50 µM) pretreatment was able to protect myocardium from I/R injury in the rat Langendorff model. The single concentration of propofol used in this study could limit our understanding of its role in I/R injury. Finally, we measured only the expression levels of FTH1, XCT, and GPX4 but not those of other antioxidant enzymes.
In conclusion, propofol pretreatment inhibits myocardial ferroptosis through the AKT/p53 signaling pathway, reducing ROS, iron, and lipid peroxidation and increasing antioxidant enzyme expression. These data may explain why propofol reduces perioperative complications associated with myocardial I/R injury, including arrhythmias, decreasing systolic and diastolic function, and myocardial stunning.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by The Experimental Animals Medical Ethics Committee in Shandong University (ECSBMSSDU 2019-2-048).
Author Contributions
Study conception/design: SL and JY. Experimental execution: SL, ZL, and XY. Data analysis: SL, MZ, and YH. Drafting of paper: SL, DW, and ST. Manuscript editing/revision: SL, JL, and JY. Manuscript reading/final approval: all authors.
Funding
This study was supported by the National Natural Science Foundation of China (No. 26010105131548), Shandong Province Natural Science Foundation (ZR2017BH041), and Key Research and Development Program of Shandong Province (No. 2018GSF118079).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the colleagues at Shandong University and the reviewers for their insightful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.841410/full#supplementary-material
References
Bovill, J. G. (2006). Intravenous Anesthesia for the Patient with Left Ventricular Dysfunction. Semin. Cardiothorac. Vasc. Anesth. 10, 43–48. doi:10.1177/108925320601000108
Chen, Y., Hua, Y., Li, X., Arslan, I. M., Zhang, W., and Meng, G. (2020). Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front. Pharmacol. 11, 42. doi:10.3389/fphar.2020.00042
Chibaya, L., Karim, B., Zhang, H., and Jones, S. N. (2021). Mdm2 Phosphorylation by Akt Regulates the P53 Response to Oxidative Stress to Promote Cell Proliferation and Tumorigenesis. Proc. Natl. Acad. Sci. USA 118 (2021), e2003193118. doi:10.1073/pnas.2003193118
De Villiers, C., and Riley, P. R. (2020). Mouse Models of Myocardial Infarction: Comparing Permanent Ligation and Ischaemia-Reperfusion. Dis. Model. Mech. 13, dmm046565. doi:10.1242/dmm.046565
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An Iron-dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a Target for protection against Cardiomyopathy. Proc. Natl. Acad. Sci. U S A. 116, 2672–2680. doi:10.1073/pnas.1821022116
Feng, J., Tamaskovic, R., Yang, Z., Brazil, D. P., Merlo, A., Hess, D., et al. (2004). Stabilization of Mdm2 via Decreased Ubiquitination Is Mediated by Protein Kinase B/Akt-dependent Phosphorylation. J. Biol. Chem. 279, 35510–35517. doi:10.1074/jbc.M404936200
Finegan, B. A., Gandhi, M., Clanachan, A. S., and cardiology, c. (2000). Phentolamine Prevents the Adverse Effects of Adenosine on Glycolysis and Mechanical Function in Isolated Working Rat Hearts Subjected to Antecedent Ischemia. J. Mol. Cell Cardiol 32, 1075–1086. doi:10.1006/jmcc.2000.1144
Freedman, D. A., Wu, L., Levine, A. J., and Cmls, m. l. s. (1999). Functions of the MDM2 Oncoprotein. Cell Mol Life Sci 55, 96–107. doi:10.1007/s000180050273
Han, C., Liu, Y., Dai, R., Ismail, N., Su, W., and Li, B. (2020). Ferroptosis and its Potential Role in Human Diseases. Front. Pharmacol. 11, 239. doi:10.3389/fphar.2020.00239
Imai, H., Matsuoka, M., Kumagai, T., Sakamoto, T., and Koumura, T. (2017). Lipid Peroxidation-dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 403, 143–170. doi:10.1007/82_2016_508
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 520, 57–62. doi:10.1038/nature14344
Jiang, S., and Chen, J. (2020). WRN Inhibits Oxidative Stress-Induced Apoptosis of Human Lensepithelial Cells through ATM/p53 Signaling Pathway and its Expression Is Downregulated by DNA Methylation. Mol. Med. 26, 68. doi:10.1186/s10020-020-00187-x
Kobayashi, I., Kokita, N., and Namiki, A. (2008). Propofol Attenuates Ischaemia-Reperfusion Injury in the Rat Heart In Vivo. Eur. J. Anaesthesiol 25, 144–151. doi:10.1017/S0265021507001342
Kruse, J. P., and Gu, W. (2009). Modes of P53 Regulation. Cell 137, 609–622. doi:10.1016/j.cell.2009.04.050
Lai, H. C., Yeh, Y. C., Wang, L. C., Ting, C. T., Lee, W. L., Lee, H. W., et al. (2011). Propofol Ameliorates Doxorubicin-Induced Oxidative Stress and Cellular Apoptosis in Rat Cardiomyocytes. Toxicol. Appl. Pharmacol. 257, 437–448. doi:10.1016/j.taap.2011.10.001
Li, W., Li, W., Leng, Y., Xiong, Y., and Xia, Z. (2020). Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury through Endoplasmic Reticulum Stress. DNA Cell Biol 39, 210–225. doi:10.1089/dna.2019.5097
Li, Y. m., Sun, J. g., Hu, L. h., Ma, X. c., Zhou, G., and Physiology, X. z. H. J. J. o. C. (2019). Propofol‐mediated Cardioprotection Dependent of microRNA‐451/HMGB1 against Myocardial Ischemia‐reperfusion Injury. J. Cell Physiol 234, 23289–23301. doi:10.1002/jcp.28897
Lou, P. H., Lucchinetti, E., Zhang, L., Affolter, A., Gandhi, M., Zhakupova, A., et al. (2015). Propofol (Diprivan®) and Intralipid® Exacerbate Insulin Resistance in Type-2 Diabetic Hearts by Impairing GLUT4 Trafficking. Anesth. Analg 120, 329–340. doi:10.1213/ane.0000000000000558
Macías-Rodríguez, R. U., Inzaugarat, M. E., Ruiz-Margáin, A., Nelson, L. J., and Cubero, F. J. J. I. J. o. M. S. (2020). Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-apoptotic Cell Death? Int. J. Mol. Sci. 21, 1651.
Magri, J., Gasparetto, A., Conti, L., Calautti, E., Cossu, C., Ruiu, R., et al. (2021). Tumor-Associated Antigen xCT and Mutant-P53 as Molecular Targets for New Combinatorial Antitumor Strategies. Cells 10, 108. doi:10.3390/cells10010108
Shravah, J., Wang, B., Pavlovic, M., Kumar, U., Chen, D. D., Luo, H., et al. (2014). Propofol Mediates Signal Transducer and Activator of Transcription 3 Activation and Crosstalk with Phosphoinositide 3-kinase/AKT. JAKSTAT 3, e29554. doi:10.4161/jkst.29554
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. doi:10.1016/j.cell.2017.09.021
Suleiman, M. S., Underwood, M., Imura, H., and Caputo, M. (2015). Cardioprotection during Adult and Pediatric Open Heart Surgery. Biomed. Res. Int. 2015, 712721. doi:10.1155/2015/712721
Sun, X., Ou, Z., Chen, R., Niu, X., Chen, D., Kang, R., et al. (2016). Activation of the P62-Keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology 63, 173–184. doi:10.1002/hep.28251
Szekely, A., Heindl, B., Zahler, S., Conzen, P. F., and Becker, B. F. (2000). Nonuniform Behavior of Intravenous Anesthetics on Postischemic Adhesion of Neutrophils in the guinea Pig Heart. Anesth. Analg 90, 1293–1300. doi:10.1097/00000539-200006000-00007
Tang, L.-J., Zhou, Y.-J., Xiong, X.-M., Li, N.-S., Zhang, J.-J., Luo, X.-J., et al. (2021). Ubiquitin-specific Protease 7 Promotes Ferroptosis via Activation of the p53/TfR1 Pathway in the Rat Hearts after Ischemia/reperfusion. Free Radic. Biol. Med. 162, 339–352. doi:10.1016/j.freeradbiomed.2020.10.307
Wang, W., Qin, J. J., Rajaei, M., Li, X., Yu, X., Hunt, C., et al. (2020). Targeting MDM2 for Novel Molecular Therapy: Beyond Oncology. Med. Res. Rev. 40, 856–880. doi:10.1002/med.21637
Wei, L., Chen, W., Hu, T., Tang, Y., Pan, B., Jin, M., et al. (2021). Effect and Mechanism of Propofol in Hepatic Ischemia/reperfusion Injury of Rat. J. Cell Physiol 25, 4185. doi:10.26355/eurrev_202106_26115
Whelan, R. S., Kaplinskiy, V., and Kitsis, R. N. (2010). Cell Death in the Pathogenesis of Heart Disease: Mechanisms and Significance. Annu. Rev. Physiol. 72, 19–44. doi:10.1146/annurev.physiol.010908.163111
Wu, K. L., Chen, C. H., and Shih, C. D. (2012). Nontranscriptional Activation of PI3K/Akt Signaling Mediates Hypotensive Effect Following Activation of Estrogen Receptor β in the Rostral Ventrolateral Medulla of Rats. J. Biomed. Sci. 19, 76. doi:10.1186/1423-0127-19-76
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent Oxidative Cell Death Involving Voltage-dependent Anion Channels. Nature 447, 864–868. doi:10.1038/nature05859
Yan, J.-H., Tang, Y., and Guo, K. (2021). Evaluation of Propofol in Inhibiting Proliferation of Cardiac Fibroblasts in Angiotensin II-Induced Mouse. Crit. Rev. Eukaryot. Gene Expr. 31, 71–78. doi:10.1615/CritRevEukaryotGeneExpr.2021037483
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. U S A. 113, E4966–E4975. doi:10.1073/pnas.1603244113
Yang, W. S., and Stockwell, B. R. (2008). Synthetic Lethal Screening Identifies Compounds Activating Iron-dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 15, 234–245. doi:10.1016/j.chembiol.2008.02.010
Yin, Y., Guan, Y., Duan, J., Wei, G., Zhu, Y., Quan, W., et al. (2013). Cardioprotective Effect of Danshensu against Myocardial Ischemia/reperfusion Injury and Inhibits Apoptosis of H9c2 Cardiomyocytes via Akt and ERK1/2 Phosphorylation. Eur. J. Pharmacol. 699, 219–226. doi:10.1016/j.ejphar.2012.11.005
Yu, X., Sun, X., Zhao, M., Hou, Y., Li, J., Yu, J., et al. (2018). Propofol Attenuates Myocardial Ischemia Reperfusion Injury Partly through Inhibition of Resident Cardiac Mast Cell Activation. Int. Immunopharmacol 54, 267–274. doi:10.1016/j.intimp.2017.11.015
Yu, Y., Yan, Y., Niu, F., Wang, Y., Chen, X., Su, G., et al. (2021). Ferroptosis: a Cell Death Connecting Oxidative Stress, Inflammation and Cardiovascular Diseases. Cell Death Discov 7, 193. doi:10.1038/s41420-021-00579-w
Zhang, J., Krishnamurthy, P. K., and Johnson, G. V. (2002). Cdk5 Phosphorylates P53 and Regulates its Activity. J. Neurochem. 81, 307–313. doi:10.1046/j.1471-4159.2002.00824.x
Zhang, Y., Swanda, R. V., Nie, L., Liu, X., Wang, C., Lee, H., et al. (2021). mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 12, 1589. doi:10.1038/s41467-021-21841-w
Zhao, L., Zhuang, J., Wang, Y., Zhou, D., Zhao, D., Zhu, S., et al. (2019). Propofol Ameliorates H9c2 Cells Apoptosis Induced by Oxygen Glucose Deprivation and Reperfusion Injury via Inhibiting High Levels of Mitochondrial Fusion and Fission. Front. Pharmacol. 10, 61. doi:10.3389/fphar.2019.00061
Keywords: Akt, ferroptosis, heart, ischemia/reperfusion injury, propofol, p53
Citation: Li S, Lei Z, Yang X, Zhao M, Hou Y, Wang D, Tang S, Li J and Yu J (2022) Propofol Protects Myocardium From Ischemia/Reperfusion Injury by Inhibiting Ferroptosis Through the AKT/p53 Signaling Pathway. Front. Pharmacol. 13:841410. doi: 10.3389/fphar.2022.841410
Received: 22 December 2021; Accepted: 21 February 2022;
Published: 16 March 2022.
Edited by:
Xianwei Wang, Xinxiang Medical University, ChinaReviewed by:
Ruo-Peng Liang, First Affiliated Hospital of Zhengzhou University, ChinaHuan He, Nanchang University, China
Copyright © 2022 Li, Lei, Yang, Zhao, Hou, Wang, Tang, Li and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingxin Li, bGppbmd4aW5Ac2R1LmVkdS5jbg==; Jingui Yu, eXVqaW5ndWkxMTA5QDEyNi5jb20=