 Wentao Shi
Wentao Shi Manali Singha2
Manali Singha2 Limeng Pu
Limeng Pu Michal Brylinski
Michal Brylinski
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 11 March 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.837715
This article is part of the Research TopicChemoinformatics Approaches to Structure- and Ligand-Based Drug Design, Volume IIView all 20 articles
Computational modeling is an essential component of modern drug discovery. One of its most important applications is to select promising drug candidates for pharmacologically relevant target proteins. Because of continuing advances in structural biology, putative binding sites for small organic molecules are being discovered in numerous proteins linked to various diseases. These valuable data offer new opportunities to build efficient computational models predicting binding molecules for target sites through the application of data mining and machine learning. In particular, deep neural networks are powerful techniques capable of learning from complex data in order to make informed drug binding predictions. In this communication, we describe Pocket2Drug, a deep graph neural network model to predict binding molecules for a given a ligand binding site. This approach first learns the conditional probability distribution of small molecules from a large dataset of pocket structures with supervised training, followed by the sampling of drug candidates from the trained model. Comprehensive benchmarking simulations show that using Pocket2Drug significantly improves the chances of finding molecules binding to target pockets compared to traditional drug selection procedures. Specifically, known binders are generated for as many as 80.5% of targets present in the testing set consisting of dissimilar data from that used to train the deep graph neural network model. Overall, Pocket2Drug is a promising computational approach to inform the discovery of novel biopharmaceuticals.
Recent developments in genomics revealed novel disease-related molecular targets, many of which are yet to be characterized with respect to the possibility of modulating their functions with pharmaceutical agents. Another challenge in pharmacotherapy arises from resistance effects to existing drugs complicating the treatment of particularly infectious diseases (Trebosc et al., 2019) and cancer (Shou et al., 2004). Therefore, many drug development projects are focused on the discovery of small molecule therapeutics with new mode of action (Gerry and Schreiber, 2018). Generating novel small molecules is a difficult endeavor due to the high complexity of biological systems and the enormous size of chemical space of organic compounds. Traditional experimental techniques can be used to identify drug-like molecules performing specific biochemical tasks by binding to macromolecular targets with a high specificity in order to modulate their cellular functions. Nonetheless, even advanced high-throughput screening methods have notable limitations due to the long time and high costs of screening a large number of drug candidates.
To make the drug discovery process more efficient, modern approaches incorporate miscellaneous computational components. Virtual screening (VS) is perhaps the most widely used strategy to help identify potentially bioactive molecules from large collections of commercially available as well as virtual compounds (Segler et al., 2018). Despite its utility, this technology has certain drawbacks such as high false-positive rates, the requirement of predefined ligand libraries for structure-based VS, oversimplified scoring functions, and protein structure frameworks absent in ligand-based VS (Wu et al., 2019). More recently, machine learning (ML) methods addressing many of these issues have become available for drug discovery. New ML techniques include a quantitative structure-activity relationship model to predict the target affinity, toxicity, and side effects (Mouchlis et al., 2021) and an approach to model polypharmacy side effects with graph convolutional networks (GCN) (Zitnik et al., 2018).
Deep learning (DL) is a family of modern machine leaning models utilizing deep neural networks (DNNs). DL models have been demonstrated to be powerful feature extractors for ligand binding site classifiers (Jiménez et al., 2017; Pu et al., 2019; Shi et al., 2020) and metric learning models for binding sites in proteins (Simonovsky and Meyers, 2020). Recurrent neural networks (RNNs) are iterative DL models that generate sequences through multiple iterations. In each iteration, the RNN model generates an output of time
In order to achieve this goal, we developed Pocket2Drug, a new deep generative model with the encoder-decoder architecture. Inspired by the framework of image captioning models taking images as the input to generate corresponding captions (Vinyals et al., 2015; Xu et al., 2015), the basic idea is to provide RNN with the prior information on ligand binding pockets to improve the chances of finding bioactive molecules. A typical image captioning model consists of two parts, an encoder/feature extractor and a decoder. A convolutional neural network (CNN) is often used as the encoder extracting fixed-size latent feature vectors from the input images containing the prior information that can subsequently be decoded by an RNN to generate image captions. Formally, image captioning models learn the probability of sequences conditioned on prior information, i.e.,
Pocket2Drug has a similar encoder-decoder architecture consisting of an encoder to extract features and a decoder to generate molecules. Nonetheless, Pocket2Drug differs from typical image captioning models in that it employs a graph representation of drug binding sites instead of images. Consequently, a GNN is employed as the encoder to extract the prior information from input pockets followed by an RNN decoder to generate molecule strings, which are the equivalents of image captions. In comprehensive benchmarking simulations against ligand-bound, ligand-free, and low-homology datasets of binding sites, we show that Pocket2Drug employing the encoder-decoder DNN effectively predicts binding drugs for input pocket structures.
Datasets used in this study were compiled from a non-redundant library of 51,677 pockets with bound ligands constructed for binding site prediction with eFindSite (Brylinski and Feinstein, 2013). The redundancy in the original library was already removed by excluding proteins with the template modeling (TM)-score, measuring the structure similarity (Zhang and Skolnick, 2004), of ≥0.4 and the 3D Tanimoto coefficient (TC), measuring the ligand similarity (Kawabata, 2011), of ≥0.7. We further filtered the dataset based on the synthetic accessibility (SA) score (Ertl and Schuffenhauer, 2009) removing low- and high-complexity compounds whose SA scores are ≤1 and ≥6, respectively. This procedure resulted in a high-quality dataset of 48,365 pockets binding small organic compounds, which were randomly split into training (90%) and testing (10%) subsets. The training subset of 43,529 pockets is referred to as the Pocket2Drug-train dataset while the remaining 4,836 (testing) pockets are called the Pocket2Drug-holo dataset.
Next, 433 pockets having a protein sequence identity of ≤0.5 with pockets in the training subset were selected from the Pocket2Drug-holo dataset creating the Pocket2Drug-lowhomol dataset to evaluate the ability to generalize to unseen data. Finally, the basic local alignment search tool (BLAST) (Altschul et al., 1990) was used with a sequence identity threshold of 95% to identify the apo structures of Pocket2Drug-holo proteins in the Protein Data Bank (PDB) (Berman et al., 2002). Ligand-free structures were then aligned on the corresponding holo-proteins with TM-align (Zhang and Skolnick, 2005) and those producing significant alignments with a TM-score of ≥0.5 (Xu and Zhang, 2010) were retained. This procedure resulted in 828 ligand-free pockets referred to as the Pocket2Drug-apo dataset.
Binding pockets are represented as graphs, in which nodes are non-hydrogen atoms and edges connect pairs of atoms spatially located within 4.5 Å from one another (Shi et al., 2021). Node features include the hydrophobicity (Mahn et al., 2009), the charge, the binding probability (Jian et al., 2016), the solvent accessible surface area (Ali et al., 2014), and the sequence entropy (Liao et al., 2005), whereas the edge attribute is the bond multiplicity for covalently bonded atoms and 0 for atoms interacting non-covalently. Pockets are centered at the origin with principal axes aligned to Cartesian axes. The coordinates of individual atoms are also used as node features in order to provide the additional 3D information on binding pockets. This graph representation of ligand binding sites was used to accurately classify pockets in protein structures with GraphSite (Shi et al., 2021).
Pocket2Drug is implemented in PyTorch v1.7.1 (Paszke et al., 2019) and employs a DNN with the encoder-decoder architecture. The model learns the probability distribution of molecules conditioned on ligand binding pockets,
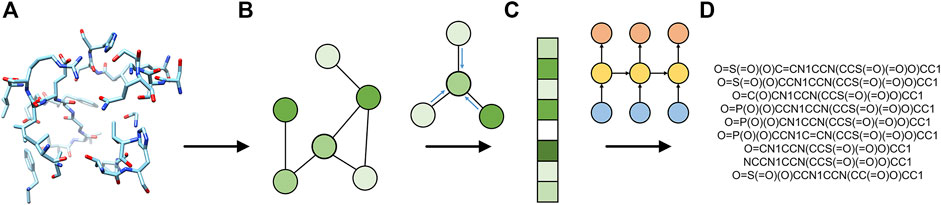
FIGURE 1. Flowchart of Pocket2Drug. The input ligand-binding pocket (A) is first represented as a graph (B) and then used by the encoder graph neural network to generate a fixed-size graph embedding (C). The decoder recurrent neural network generates molecule strings (D) from the graph embedding.
The GNN encoder extracts latent features from the input pocket graphs. We use the embedding network implemented in the GraphSite classifier as the feature extractor with the last fully connected layer removed and the remaining parts of the classifier employed as the feature extractor. The message passing function utilizes weighted neighbor node features, in which weights are generated by a two-layer, fully connected neural network taking edge features as the input. Updated node features in
are first computed as a weighted sum of the first-order neighbors. The features of
As a decoder, we use the gated recurrent unit (GRU), which is a variation of the vanilla RNN (Cho et al., 2014). The decoder network models a conditional probability of the output sequence based on the prior information on a ligand binding pocket:
where
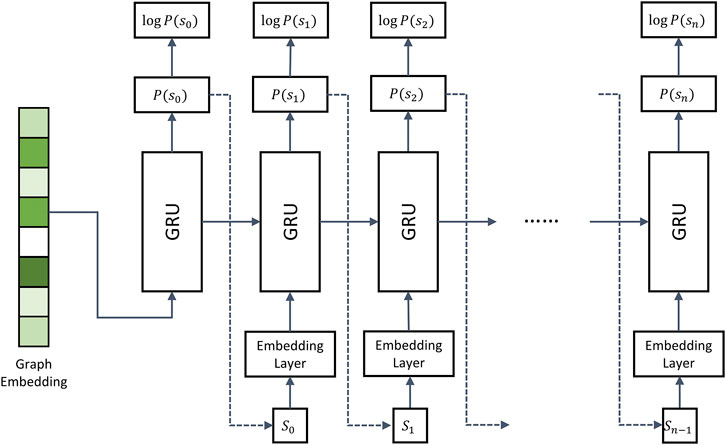
FIGURE 2. Architecture of the recurrent neural network decoder. The decoder employs multiple gated recurrent units (GRUs). During model training, the molecule strings of binding drugs are used as the input. Dashed arrows represent the inference stage, in which the token sampled from
Dashed arrows in Figure 2 represent the inference stage. Here, the first iteration is the same as during training, i.e., the encoder generates graph embeddings used as the input in the first iteration. However, in the subsequent iterations, the RNN model takes the token
Molecules can be represented by strings encoded by different tokenization schemes. Although SMILES is a widely used molecular string system, it was not designed for ML applications. Because of a strict syntax of SMILES, a significant portion of molecules generated by machine learning models are invalid. In addition, parentheses and ring indicators may be separated by long distances in SMILES strings causing problems for RNNs that have difficulty learning long-term dependencies (Öztürk et al., 2020). This issue can be addressed by improving either the RNN model or the tokenization scheme. For instance, RNN variants implementing “shortcuts” were developed to model long-term dependencies (Hochreiter and Schmidhuber, 1997). A long short-term memory (LSTM) model can also be used instead of a vanilla RNN in de novo drug design applications to learn the distribution of a drug dataset (Ertl et al., 2017). Another workaround is to improve the tokenization scheme to make the string representation of molecules more suitable for ML applications. An example is DeepSMILES developed to enhance DL-based models taking SMILES as the input (O’Boyle and Dalke, 2018).
Pocket2Drug employs SELF-referencing Embedding Strings (SELFIES), another molecule tokenization scheme designed for machine learning applications (Krenn et al., 2020). The SELFIES method was selected because of several important properties. Not only any molecule can be represented by a SELFIES string, but also all virtual molecules generated by an ML model are valid. Importantly, the information on rings and branches in SELFIES is localized by storing the branch size and ring size together with their identifiers. This tokenization scheme makes it easier for RNNs to learn from the “past” information compared to, e.g., SMILES that require RNNs to infer ring/branch indicators based on non-localized information.
Pocket2Drug was trained on the Pocket2Drug-train dataset and validated against Pocket2Drug-holo, -apo, and -lowhomol datasets. We first analyze the size of molecules generated for the Pocket2Drug-holo dataset. Figure 3 shows that there is a notable correlation between the size of pockets and the size of binding molecules, referred to as label ligands, across experimental complex structures (blue bars). Encouragingly, the size of ligands constructed by Pocket2Drug is also correlated with the pocket size, although these molecules tend to be somewhat smaller than the corresponding label ligands (green bars). This result can be attributed to the fact that capturing longer dependencies in molecular strings is more difficult for the RNN trained to minimize the sum of cross-entropy loss function. In other words, the model makes fewer mistakes by generating smaller molecules.
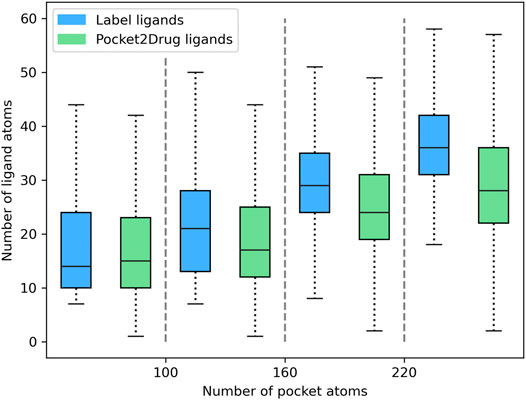
FIGURE 3. Relationship between the ligand size and the size of binding pockets. The size of ligands and pockets is quantified by the number of non-hydrogen atoms. Binding pockets are assigned to four size groups: <100, 100–160, 161–220, and >220 atoms. For each pocket group, quartiles and the interquartile range are calculated for the size of label ligands (blue bars) and those molecules generated by Pocket2Drug (green bars).
Next, the quality of molecules generated for the Pocket2Drug-holo dataset is evaluated using two complementing protocols, one based on the chemical similarity of binding molecules (Baldi and Nasr, 2010) and another utilizing the structure alignments of protein pockets (Yeturu and Chandra, 2011). Pocket2Drug is compared to two baselines. The first method randomly selects drug candidates from the ZINC database, a curated collection of commercially available chemical compounds prepared specifically for virtual screening (Irwin and Shoichet, 2005). The second baseline method selects drug candidates from the output of a vanilla RNN (Segler et al., 2018) representing a typical DL-based approach for de novo drug design.
The performance of Pocket2Drug, ZINC, and vanilla RNN are evaluated with the TC between the generated molecules and label ligands. For each pocket in the Pocket2Drug-holo dataset, TC values are calculated for a specified number of molecules sampled from the model output and the highest TC is selected as the final score. Table 1 reports the percentage of Pocket2Drug-holo pockets with the corresponding score greater than or equal to a TC threshold ranging from 0.7 to 1.0. Encouragingly, using Pocket2Drug significantly improves chances to find binding molecules compared to ZINC and vanilla RNN. For a sample size of 20,480 (10 batches of 2,048 molecules each to maximize the GPU utilization), Pocket2Drug generates at least one molecule which a TC of ≥0.7 to the label ligand for as many as 95.9% pockets. Note that two molecules sharing chemical similarity with a TC of ≥0.7 tend to have a similar bioactivity (Kumar, 2011; Ben Lo, 2016). For the majority of pockets (52.5%), Pocket2Drug selects the label ligand itself (a TC of 1.0). This performance is significantly higher than that of ZINC/vanilla RNN that selects ligands with a TC of ≥0.7 for 58.9%/57.1% of pockets and label ligands for merely 0.4%/0.1% of pockets. Increasing the sample size to 81,920 slightly improves the performance because four times more molecules are used to select that with the highest TC value. A significantly improved performance of Pocket2Drug over vanilla RNN can be attributed to the effective utilization of the prior information on ligand binding pockets learned by the ML model.
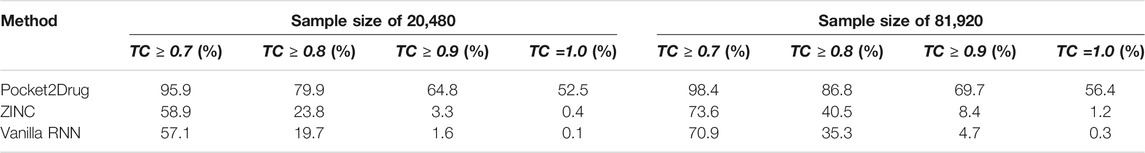
TABLE 1. Hit rates for the Pocket2Drug-holo dataset.
Next, the performance of Pocket2Drug is assessed against the Pocket2Drug-apo dataset. The mean root-mean-square deviation (RMSD) (Kabsch, 1976) of ligand-free structures against ligand-bound conformations is 1.2 Å ± 0.9. This low RMSD is expected because, with a few exceptions, the structures of apo- and holo-proteins tend to be highly similar (Brylinski and Skolnick, 2008). Table 2 reports hit rates for molecules generated by Pocket2Drug using ligand-free and the corresponding ligand-bound pockets in the Pocket2Drug-holo dataset. Encouragingly, the performance of Pocket2Drug is independent on the ligand binding state of target proteins, therefore, the model does not require input proteins to be co-crystallized with ligands in order to successfully generate binding molecules.
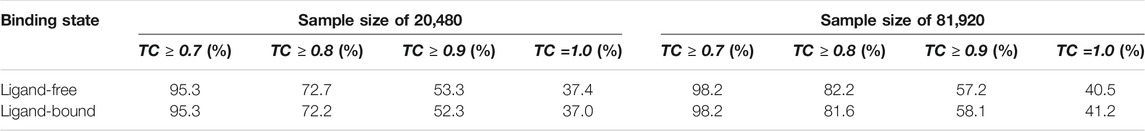
TABLE 2. Hit rates for the Pocket2Drug-apo dataset. For each ligand-free structure, the corresponding ligand-bound pocket is selected from the Pocket2Drug-holo dataset for the apples-to-apples comparison.
We also evaluate the ability of Pocket2Drug to generalize to unseen data by measuring its performance against the Pocket2Drug-lowhomol dataset. As reported in Table 3, label ligands (a TC of 1.0) are generated by Pocket2Drug in 77.1%/80.5% of the cases when the sample size is 20,480/81,920. This performance represents a notable improvement over ZINC and vanilla RNN selecting a very few label ligands. Pocket2Drug also achieves the highest performance for other TC thresholds ranging from 0.7 to 0.9. These results show that Pocket2Drug not only performs exceptionally well against Pocket2Drug-holo and -apo datasets, but also against the Pocket2Drug-lowhomol dataset comprising proteins with a low sequence homology to the training subset demonstrating that it generalizes well to unseen data.
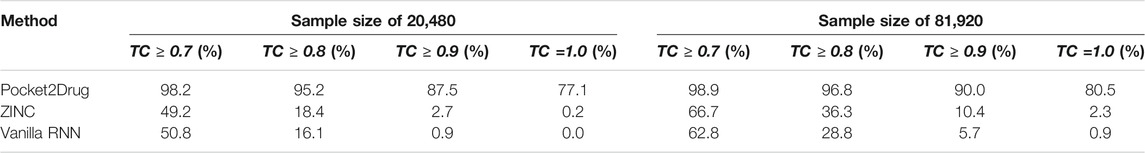
TABLE 3. Hit rates for the Pocket2Drug-lowhomol dataset.
Two representative examples of pockets in the Pocket2Drug-lowhomol dataset are discussed in detail, a nucleotide binding site in the human mitogen and stress activated protein kinase 1 (MSK1) and a sugar binding site in d-allose binding protein (ALBP) from E. coli. MSK1 is involved in the regulation of mitogen activated kinases and it is required by the tumor-promoter-induced neoplastic cell transformation (Malakhova et al., 2010). The complex structure of MSK1 and the phospho-amino-phosphonic acid-adenylate ester (AMP-PNP) (Malakhova et al., 2010) was chosen as the target. AMP-PNP is a competitive ATPase inhibitor blocking the ATP-dependent oxidative phosphorylation (Lardy et al., 1975). Figure 4A shows the distribution of TC similarities between the label ligand, AMP-PNP, and molecules generated by Pocket2Drug and two baseline methods. Although most virtual molecules have relatively low TC similarities to AMP-PNP, more molecules with high TC vales are sampled from the Pocket2Drug model compared to ZINC and vanilla RNN. According to the Fisher-Pitman permutation test (Neuhäuser and Manly, 2004), the difference between Pocket2Drug and vanilla RNN is statistically significant with a p-value close to 0 and that between Pocket2Drug and ZINC is insignificant with a p-value of 0.1.
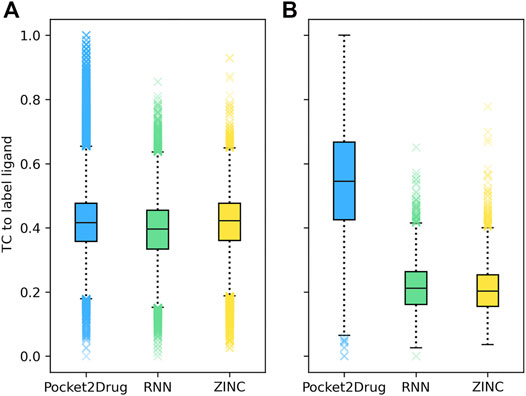
FIGURE 4. Chemical similarity of molecules generated by Pocket2Drug to label ligands. Label ligands are molecules bound to target pockets in experimental complex structures, (A) AMP-PNP binding to MSK1 and (B) ß-d-allose binding to ALBP. Chemical similarity is measured with the Tanimoto coefficient (TC).
To better understand the biological relevance of molecules generated by Pocket2Drug, five representative compounds with TC similarities against AMP-PNP ranging from 1.0 to 0.8 are presented in Figure 5. Figure 5A shows AMP-PNP, which is a nonhydrolyzable ATP analogue forming hydrogen bonds with MSK1 pocket residues through several moieties, NH2 in adenine, 3′ OH in pentose sugar, OH in ß-phosphate, NH linking ß- and γ-phosphates and OH in γ-phosphate in the complex crystal structure (Malakhova et al., 2010). Interestingly, several molecules generated by Pocket2Drug have common substructures with either substitutions in the adenine moiety (Figures 5E,F) and the terminal phosphate group (Figure 5B) or sharing the PNP subunit (Figures 5C,D). These virtual molecules contain groups forming important hydrogen bonds with MSK1 pocket residues. To further evaluate the possibility of binding, all molecules were docked into the AMP-PNP pocket of MSK1 with fkcombu (Kawabata and Nakamura, 2014). The docking scores of the generated molecules are 12.5, 18.1, 21.8, 17.6, and 13.0 (Figures 5B–F, respectively). These results indicate that molecules generated by Pocket2Drug dock favorably to the target pocket with the compound shown in Figures 5B,G having the best docking score due to the substitution in ß-phosphate group.

FIGURE 5. Examples of molecules generated by Pocket2Drug for a binding site in MSK1. (A) The label ligand, AMP-PNP. (B–F) Molecules constructed by Pocket2Drug with maximum common substructures to the label ligand highlighted in cyan. (G) Molecule shown in B (ice blue) docked to the binding site in MSK1 (orange).
The improvement of Pocket2Drug over baseline methods is even more perceptible for ALBP where the distribution of TC similarities to the label ligand is shifted toward much higher values for molecules sampled from the Pocket2Drug model (Figure 4B). Differences between Pocket2Drug and both baseline methods are statistically significant with p-values close to 0. ALBP is a member of the ATP-binding cassette (ABC) transporter family facilitating the import and export of various molecules across the cell membrane (Fath and Kolter, 1993). ALBP binds ß-d-allose, shown in Figure 6A, with a Kd of 0.33 μm (Chaudhuri et al., 1999). In the crystal complex structure, ß-d-allose forms multiple interactions with the pocket residues of ALBP through the ring oxygen and five hydroxyl moieties (Chaudhuri et al., 1999). Selected compounds generated by Pocket2Drug are presented in Figures 6B–F. In addition to a substituted cyclohexane (Figure 6B), several substituted allose molecules (Figures 6C–F) sharing a high chemical similarity with the label ligand, ß-d-allose (Figure 6A), were constructed. Most of these molecules dock well to ALBP pocket with docking scores of 4.1, 3.7, 20.9, 3.5, and 9.8 for compounds shown in Figures 6B–F, respectively. Interestingly, a substituted cyclohexane in the molecule shown in Figure 6B adopts the chair conformation similarly to ß-d-allose bound to ALBP in the experimental complex structure. A compound shown in Figures 6E,G has the best docking score, whereas that shown in Figure 6D has less favorable docking score than those ligands having a comparable size to ß-d-allose because of the large substitution at 5′ position that does not fit in the binding pocket of ALBP. Docking results suggest that molecules generated by Pocket2Drug are capable of forming favorable interactions with the target pocket.
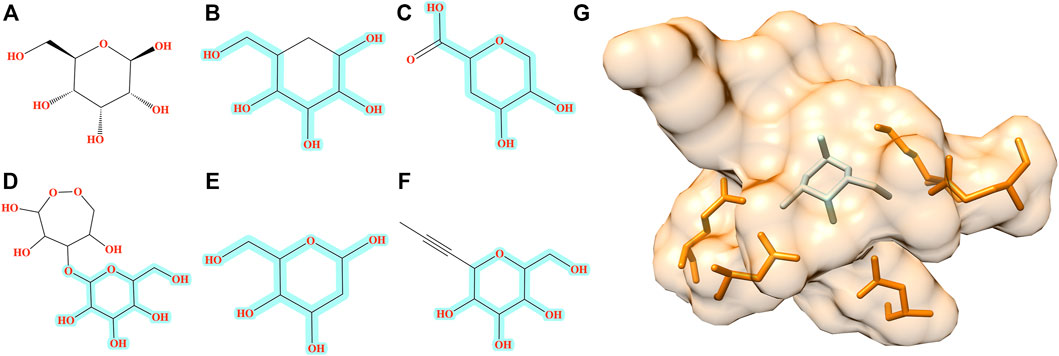
FIGURE 6. Examples of molecules generated by Pocket2Drug for a binding site in ALBP. (A) The label ligand, ß-d-allose. (B–F) Molecules constructed by Pocket2Drug with maximum common substructures to the label ligand highlighted in cyan. (G) Molecule shown in E (ice blue) docked to the binding site in ALBP (orange).
In addition to the assessment by ligand chemical similarity described above, the performance of Pocket2Drug is also evaluated with pocket structure alignments. This approach is based on an assumption that a molecule generated for the target pocket is a hit if a similar molecule binds to a site that is structurally similar to the target pocket (Govindaraj and Brylinski, 2018; Gaieb et al., 2019). A flowchart of the evaluation procedure is shown in Figure 7. For a target pocket in the testing set (Figure 7A), molecules generated by Pocket2Drug are ranked according to their frequencies and 100 of the most frequent molecules are selected. For each drug candidate (Figure 7B), chemically similar ligands with a TC of ≥0.7 are identified in the PubChem BioAssay dataset comprising 73,021 active interactions involving 919 unique proteins and 17,367 unique compounds (Wang et al., 2012). Next, the experimental complex structures of these ligands bound to similar proteins with a sequence identity of ≥70% to PubChem BioAssay targets are retrieved from the PDB. The extracted binding sites (Figure 7C) are finally structurally aligned to the initial target pocket with PocketAlign, an accurate method to superpose ligand binding sites in a sequence order-independent manner (Yeturu and Chandra, 2011). Essentially, this procedure validates molecules generated for target pockets by finding similar interactions that have already been determined experimentally through binding assays and protein crystallography.
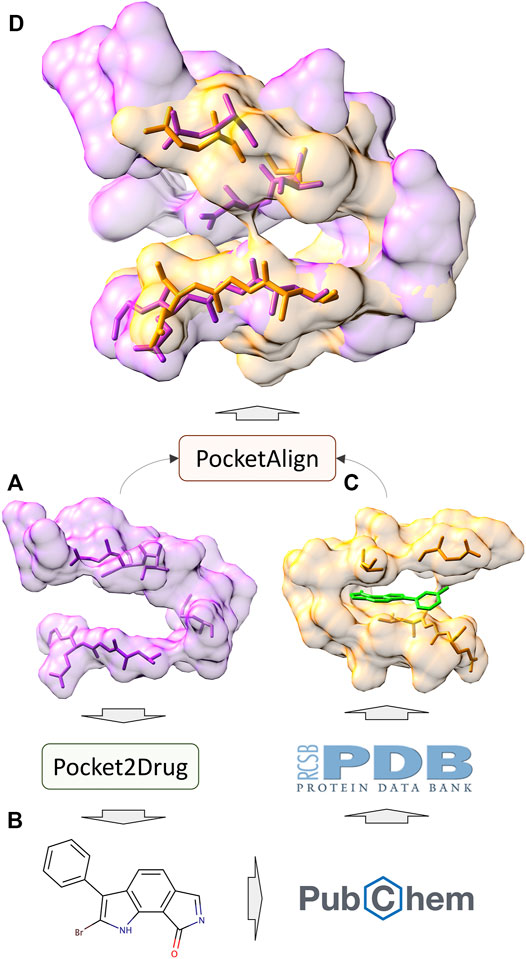
FIGURE 7. Flowchart of the evaluation by pocket structure alignments. For a target pocket (A), a molecule is generated by Pocket2Drug (B). This compound is then scanned through the PubChem BioAssay for similar molecules for which experimental complex structures are available in the Protein Data Bank. The extracted binding site (C) corresponding to the know interaction in PubChem BioAssay is structurally aligned to the target pocket by PocketAlign. A high-quality alignment (D) indicates that the generated molecule is likely to bind to the target pocket.
Similar to the evaluation protocol by ligand chemical similarity, Pocket2Drug is compared to ZINC and vanilla RNN. For each target pocket, 100 molecules from the ZINC database and 100 molecules generated by vanilla RNN are selected so that their molecular weight distributions match those calculated for compounds selected by Pocket2Drug. In terms of statistics, the number of pocket pairs used as input for structure alignments is 17,620 for Pocket2Drug, 6,307 for ZINC, and 6,694 for vanilla RNN. The number of valid pocket alignments constructed by PocketAlign (Yeturu and Chandra, 2011) are 16,987 (Pocket2Drug), 741 (ZINC), and 4,902 (vanilla RNN). A valid pocket alignment has the RMSD of ≤2 Å; higher RMSD values indicate that two pockets are structurally dissimilar. According to this criterion, as many as 96.4% of validation pairs of pockets identified using output molecules generated by Pocket2Drug produce valid structure alignments, while these percentages are notably lower for ZINC (11.7%) and vanilla RNN (73.2%). The distribution of the RMSD scores of pocket alignments for all tested methods is presented in Figure 8. Not only using molecules selected by Pocket2Drug results in the highest percentage of valid structure alignments, but also RMSD values for these superpositions are generally much lower compared to ZINC and vanilla RNN. The mean RMSD scores for pocket2Drug, ZINC, and vanilla RNN are 1.1 Å, 1.6 Å, and 1.6 Å, respectively.
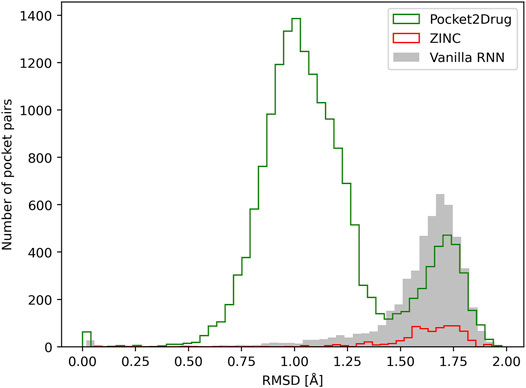
FIGURE 8. Assessment of the quality of pocket alignments constructed with PocketAlign. Alignment quality is evaluated by the root-mean-square deviation (RMSD) calculated over non-hydrogen atoms of binding residues. Target pockets are aligned to binding sites identified in the Protein Data Bank for molecules generated by Pocket2Drug (green) and two baselines, ZINC (red) and vanilla RNN (gray).
Structure alignment results demonstrate that for a large number of molecules generated by Pocket2Drug for target pockets, there are experimentally determined interactions between chemically similar ligands binding to structurally similar pockets. Two representative cases are selected to exemplify the evaluation by pocket structure alignments. The first target pocket is a nucleotide binding site in MSK1 used in the previous section to illustrate the results of the evaluation by ligand chemical similarity. Among molecules generated by Pocket2Drug, a compound ranked 12 with the frequency of 21 (Figure 9A) is chemically similar to midostaurin (PubChem-CID: 9829523, Figure 9B), a protein kinase C (PKC) inhibitor (Eder et al., 2004) used to treat systemic mastocytosis, acute myeloid leukemia, and mast cell leukemia (National Cancer Institute Dictionary, 2021). According to the bioassay data (PubChem-BAID: 208295368), midostaurin inhibits PKC-α isoform with the half-maximal inhibitory concentration (IC50) of 22 nm (Millward et al., 2006). Midostaurin has been co-crystalized with the human dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A, 25% sequence identity with PKC-α) with the equilibrium dissociation constant (Kd) of 100 nm (PDB-ID: 4nct) (Alexeeva et al., 2015). Figure 9C shows the structure alignment constructed by PocketAlign between AMP-PNP binding pocket in MSK1 and midostaurin binding pocket in DYRK1A. Despite a low global sequence identity between these proteins of only 26%, their binding pockets are structurally highly similar with the RMSD of 0.90 Å. The compound generated by Pocket2Drug docks to the AMP-PNP binding pocket in MSK1 with a score of 58.5 (Figure 9D).
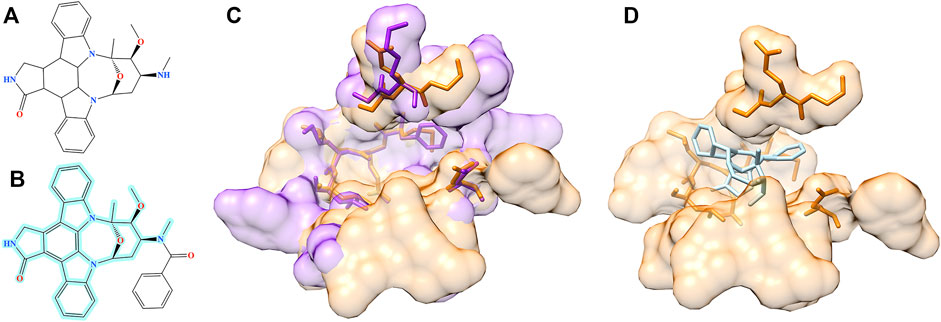
FIGURE 9. Example of the evaluation by pocket alignment for a binding site in MSK1. (A) A molecule generated by Pocket2Drug at rank 12. (B) A similar compound, midostaurin, with the maximum common substructure to Pocket2Drug molecule highlighted in cyan. (C) A structure alignment between the target binding site in MSK1 (orange) and midostaurin binding pocket in DYRK1A (purple). (D) The molecule generated by Pocket2Drug (ice blue) docked to the target site in MSK1 (orange) with fkcombu.
The second example is the human angiopoietin-1 receptor (Tie-2), an enzyme involved in vessel remodeling, branching, stability, and maturation (Yu, 2005). Using the binding site of Tie-2 as the input, Pocket2Drug generated a molecule shown in Figure 10A at rank 9 with a frequency of 5. This compound is chemically similar to doramapimod (PubChem-CID: 156422, Figure 10B), an inhibitor of ephrin type-A receptor 2 (EphA2) with a TC of 0.73. According to the bioassay data (PubChem-BAID: 40394839), doramapimod binds to EphA2 with a Kd of 0.37 nm and has been tested for its anti-proliferative activity in the SF-268 cell line. It inhibits the viability of EphA2 growth dependent glioblastoma cells with a half-maximal effective concentration (EC50) of 5 μm (Heinzlmeir et al., 2017). Despite a low global sequence identity of 37%, the structure alignment of binding sites in Tie-2 (PDB-ID: 2oo8) and EphA2 (PDB-ID: 5nkd) yields an RMSD of 0.95 Å (Figure 10C). Docking simulations with fkcombu confirmed that the molecule generated by Pocket2Drug fits well into the binding site of Tie-2 with a score of 24.3 (Figure 10D).
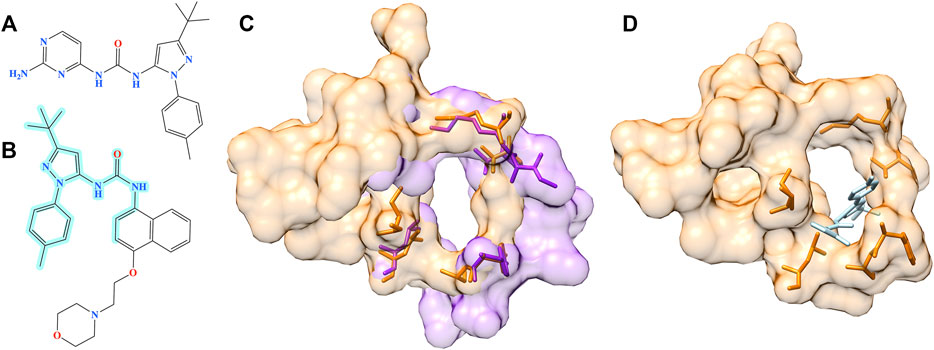
FIGURE 10. Example of the evaluation by pocket alignment for a binding site in Tie-2. (A) A molecule generated by Pocket2Drug at rank 9. (B) A similar compound, doramapimod, with the maximum common substructure to Pocket2Drug molecule highlighted in cyan. (C) A structure alignment between the target binding site in Tie-2 (orange) and doramapimod binding pocket in EphA2 (purple). (D) The molecule generated by Pocket2Drug (ice blue) docked to the target site in Tie-2 (orange) with fkcombu.
In this communication, we describe Pocket2Drug, a novel deep learning model employing an encoder-decoder architecture to predict binding molecules for a ligand binding site. Pocket2Drug was trained in an end-to-end supervised manner against a large collection of ligand-pocket pairs. The analysis of molecules generated by Pocket2Drug using two evaluation protocols based on ligand chemical similarity and pocket structure alignments revealed that this algorithm significantly improves the chances of finding binding ligands compared to traditional techniques. Pocket2Drug not only yields a high accuracy against ligand-free structures, but it also generalizes well to unseen data, viz. those pockets extracted from proteins that are different from training instances. These findings are particularly important in drug discovery against novel protein structures, where it can help significantly reduce the search space of drug candidates. In contrast to traditional virtual screening typically employing a library of 200,000 to over 1,000,000 molecules (Hughes et al., 2011), Pocket2Drug generates molecules that have high chances to bind to target pockets within a smaller sample of 81,920 compounds. Therefore, it can potentially decrease the number of molecules to be subjected to structure-based virtual screening from millions to tens of thousands.
Pocket2Drug can be improved by incorporating reinforcement learning imposing additional restraints on the synthetic accessibility, solubility, and toxicity of generated molecules, depending on a specific application. Additional improvements can also be achieved by applying a framework similar to the conditional recurrent neural network (cRNN), utilizing the RNN with the prior information (Xu et al., 2021), to the heterogeneous input data. In contrast to cRNN, in which the pre-computed information is used as the prior condition for RNN, Pocket2Drug is an end-to-end DNN, therefore the encoder is updated during training. Another difference is the data representation; cRNN uses a voxel representation as the prior information, whereas Pocket2Drug employs a computationally more efficient graph representation. Nonetheless, the heterogeneous pocket data can be combined by concatenating embedding vectors generated by different feature extractors in order to provide the prior information on ligand binding sites.
An attention mechanism was shown to significantly improve the performance of image captioning because it helps the model capture more semantically meaningful parts of images (Xu et al., 2015). We expect that the same methodology can be implemented in Pocket2Drug since pocket residues contribute differently to the formation of molecular interactions with binding ligands. These are examples of future research directions that will be explored to further improve the performance of Pocket2Drug in the discovery of novel biopharmaceuticals.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/shiwentao00/Pocket2Drug, https://osf.io/qacwj/.
Conceptualization: WS; Methods: WS and LP; Dataset: MB; Evaluation and case studies: WS, MS, and GS; Supervision: MB; Funding requisition: JR and MB; Manuscript draft: WS, LP, MS, and GS; Final manuscript: MB.
This work has been supported in part by the National Institute of General Medical Sciences of the National Institutes of Health award R35GM119524, the US National Science Foundation award CCF1619303, the Louisiana Board of Regents contract LEQSF(2016-19)-RD-B03 and by the Center for Computation and Technology, Louisiana State University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alexeeva, M., Åberg, E., Engh, R. A., and Rothweiler, U. (2015). The Structure of a Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A-Pkc412 Complex Reveals Disulfide-Bridge Formation with the Anomalous Catalytic Loop HRD(HCD) Cysteine. Acta Crystallogr. D Biol. Crystallogr. 71 (Pt 5), 1207–1215. doi:10.1107/S1399004715005106
Ali, S. A., Hassan, M. I., Islam, A., and Ahmad, F. (2014). A Review of Methods Available to Estimate Solvent-Accessible Surface Areas of Soluble Proteins in the Folded and Unfolded States. Curr. Protein Pept. Sci. 15 (5), 456–476. doi:10.2174/1389203715666140327114232
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215 (3), 403–410. doi:10.1016/S0022-2836(05)80360-2
Baldi, P., and Nasr, R. (2010). When Is Chemical Similarity Significant? the Statistical Distribution of Chemical Similarity Scores and its Extreme Values. J. Chem. Inf. Model. 50 (7), 1205–1222. doi:10.1021/ci100010v
Ben Lo, J. Z. T. (2016). “Chemical Similarity Networks for Drug Discovery,” in Special Topics in Drug Discovery (Intech), 53–72. doi:10.5772/65106
Berman, H. M., Battistuz, T., Bhat, T. N., Bluhm, W. F., Bourne, P. E., Burkhardt, K., et al. (2002). The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 58 (Pt 6 No 1), 899–907. doi:10.1107/s0907444902003451
Brylinski, M., and Feinstein, W. P. (2013). eFindSite: Improved Prediction of Ligand Binding Sites in Protein Models Using Meta-Threading, Machine Learning and Auxiliary Ligands. J. Comput. Aided Mol. Des. 27 (6), 551–567. doi:10.1007/s10822-013-9663-5
Brylinski, M., and Skolnick, J. (2008). What Is the Relationship between the Global Structures of Apo and Holo Proteins. Proteins 70 (2), 363–377. doi:10.1002/prot.21510
Chaudhuri, B. N., Ko, J., Park, C., Jones, T. A., and Mowbray, S. L. (1999). Structure of D-Allose Binding Protein from Escherichia coli Bound to D-Allose at 1.8 A Resolution. J. Mol. Biol. 286 (5), 1519–1531. doi:10.1006/jmbi.1999.2571
Cho, K., van Merrienboer, B., Gulcehre, C., Bahdanau, D., Bougares, F., Schwenk, H., et al. (2014). “Learning Phrase Representations Using RNN Encoder-Decoder for Statistical Machine Translation,” in Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, October 25–29, 2014.
Eder, J. P., Garcia-Carbonero, R., Clark, J. W., Supko, J. G., Puchalski, T. A., Ryan, D. P., et al. (2004). A Phase I Trial of Daily Oral 4'- N -Benzoyl-Staurosporine in Combination with Protracted Continuous Infusion 5-fluorouracil in Patients with Advanced Solid Malignancies. Invest. New Drugs 22 (2), 139–150. doi:10.1023/B:DRUG.0000011790.31292.ef
Ertl, P., and Schuffenhauer, A. (2009). Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform 1 (1), 8. doi:10.1186/1758-2946-1-8
Ertl, P., Lewis, R., Martin, E., and Polyakov, V. (2017). In Silico Generation of Novel, Drug-like Chemical Matter Using the LSTM Neural Network. arXiv preprint arXiv:1712.07449.
Fath, M. J., and Kolter, R. (1993). ABC Transporters: Bacterial Exporters. Microbiol. Rev. 57 (4), 995–1017. doi:10.1128/mr.57.4.995-1017.1993
Gaieb, Z., Parks, C., and Amaro, R. (2019). Evaluation of Binding Site Comparison Algorithms and Proteometric Machine Learning Models in the Detection of Protein Pockets Capable of Binding the Same Ligand. ChemRxiv preprint ChemRxiv:9178136.
Gerry, C. J., and Schreiber, S. L. (2018). Chemical Probes and Drug Leads from Advances in Synthetic Planning and Methodology. Nat. Rev. Drug Discov. 17 (5), 333–352. doi:10.1038/nrd.2018.53
Govindaraj, R. G., and Brylinski, M. (2018). Comparative Assessment of Strategies to Identify Similar Ligand-Binding Pockets in Proteins. BMC Bioinformatics 19 (1), 91. doi:10.1186/s12859-018-2109-2
Graves, A. (2013). Generating Sequences with Recurrent Neural Networks. arXiv preprint arXiv:1308.0850.
Gupta, A., Müller, A. T., Huisman, B. J. H., Fuchs, J. A., Schneider, P., and Schneider, G. (2018). Generative Recurrent Networks for De Novo Drug Design. Mol. Inform. 37 (1-2), 1700111. doi:10.1002/minf.201700111
Heinzlmeir, S., Lohse, J., Treiber, T., Kudlinzki, D., Linhard, V., Gande, S. L., et al. (2017). Chemoproteomics-Aided Medicinal Chemistry for the Discovery of EPHA2 Inhibitors. ChemMedChem 12 (12), 999–1011. doi:10.1002/cmdc.201700217
Hochreiter, S., and Schmidhuber, J. (1997). Long Short-Term Memory. Neural Comput. 9 (8), 1735–1780. doi:10.1162/neco.1997.9.8.1735
Hughes, J. P., Rees, S., Kalindjian, S. B., and Philpott, K. L. (2011). Principles of Early Drug Discovery. Br. J. Pharmacol. 162 (6), 1239–1249. doi:10.1111/j.1476-5381.2010.01127.x
Irwin, J. J., and Shoichet, B. K. (2005). ZINC--a Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 45 (1), 177–182. doi:10.1021/ci049714+
Jian, J. W., Elumalai, P., Pitti, T., Wu, C. Y., Tsai, K. C., Chang, J. Y., et al. (2016). Predicting Ligand Binding Sites on Protein Surfaces by 3-dimensional Probability Density Distributions of Interacting Atoms. PloS one 11 (8), e0160315. doi:10.1371/journal.pone.0160315
Jiménez, J., Doerr, S., Martínez-Rosell, G., Rose, A. S., and De Fabritiis, G. (2017). DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 33 (19), 3036–3042. doi:10.1093/bioinformatics/btx350
Kabsch, W. (1976). A Solution for the Best Rotation to Relate Two Sets of Vectors. Acta Cryst. Sect A. 32 (5), 922–923. doi:10.1107/s0567739476001873
Kawabata, T. (2011). Build-up Algorithm for Atomic Correspondence between Chemical Structures. J. Chem. Inf. Model. 51 (8), 1775–1787. doi:10.1021/ci2001023
Kawabata, T., and Nakamura, H. (2014). 3D Flexible Alignment Using 2D Maximum Common Substructure: Dependence of Prediction Accuracy on Target-Reference Chemical Similarity. J. Chem. Inf. Model. 54 (7), 1850–1863. doi:10.1021/ci500006d
Krenn, M., Häse, F., Nigam, A., Friederich, P., and Aspuru-Guzik, A. (2020). Self-Referencing Embedded Strings (SELFIES): A 100% Robust Molecular String Representation. Machine Learn. Sci. Techn. 1 (4), 045024. doi:10.1088/2632-2153/aba947
Kumar, A. (2011). Chemical Similarity Methods : A Tutorial Review. The Chem. educator 16, 46–50. doi:10.1333/s00897112344a
Lardy, H. A., Schuster, S. M., and Ebel, R. E. (1975). Exploring Sites on Mitochondrial ATPase for Catalysis, Regulation, and Inhibition. J. Supramol Struct. 3 (3), 214–221. doi:10.1002/jss.400030303
Liao, H., Yeh, W., Chiang, D., Jernigan, R. L., and Lustig, B. (2005). Protein Sequence Entropy Is Closely Related to Packing Density and Hydrophobicity. Protein Eng. Des. Sel 18 (2), 59–64. doi:10.1093/protein/gzi009
Mahn, A., Lienqueo, M. E., and Salgado, J. C. (2009). Methods of Calculating Protein Hydrophobicity and Their Application in Developing Correlations to Predict Hydrophobic Interaction Chromatography Retention. J. Chromatogr. A. 1216 (10), 1838–1844. doi:10.1016/j.chroma.2008.11.089
Malakhova, M., D'Angelo, I., Kim, H. G., Kurinov, I., Bode, A. M., and Dong, Z. (2010). The crystal Structure of the Active Form of the C-Terminal Kinase Domain of Mitogen- and Stress-Activated Protein Kinase 1. J. Mol. Biol. 399 (1), 41–52. doi:10.1016/j.jmb.2010.03.064
Millward, M. J., House, C., Bowtell, D., Webster, L., Olver, I. N., Gore, M., et al. (2006). The Multikinase Inhibitor Midostaurin (PKC412A) Lacks Activity in Metastatic Melanoma: a Phase IIA Clinical and Biologic Study. Br. J. Cancer 95 (7), 829–834. doi:10.1038/sj.bjc.6603331
Mouchlis, V. D., Afantitis, A., Serra, A., Fratello, M., Papadiamantis, A. G., Aidinis, V., et al. (2021). Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 22 (4), 1676. doi:10.3390/ijms22041676
National Cancer Institute Dictionary (2021). Available from: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/n-benzoyl-staurosporine (Accessed December 4, 2021).
Neuhäuser, M., and Manly, B. F. (2004). The Fisher-Pitman Permutation Test when Testing for Differences in Mean and Variance. Psychol. Rep. 94 (1), 189–194. doi:10.2466/pr0.94.1.189-194
O'Boyle, N., and Dalke, A. (2018). DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures. ChemRxiv. doi:10.26434/chemrxiv.7097960.v1
Öztürk, H., Özgür, A., Schwaller, P., Laino, T., and Ozkirimli, E. (2020). Exploring Chemical Space Using Natural Language Processing Methodologies for Drug Discovery. Drug Discov. Today 25 (4), 689–705. doi:10.1016/j.drudis.2020.01.020
Paszke, A., Gross, S., Massa, F., Lerer, A., Bradbury, J., Chanan, G., et al. (2019). “PyTorch: An Imperative Style, High-Performance Deep Learning Library,” in Proceedings of the Thirty-third Conference on Neural Information Processing Systems (NeurIPS), Vancouver, BC, December 8–14, 2019.
Pu, L., Govindaraj, R. G., Lemoine, J. M., Wu, H. C., and Brylinski, M. (2019). DeepDrug3D: Classification of Ligand-Binding Pockets in Proteins with a Convolutional Neural Network. Plos Comput. Biol. 15 (2), e1006718. doi:10.1371/journal.pcbi.1006718
Segler, M. H. S., Kogej, T., Tyrchan, C., and Waller, M. P. (2018). Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 4 (1), 120–131. doi:10.1021/acscentsci.7b00512
Shi, W., Lemoine, J. M., Shawky, A. A., Singha, M., Pu, L., Yang, S., et al. (2020). BionoiNet: Ligand-Binding Site Classification with Off-The-Shelf Deep Neural Network. Bioinformatics 36 (10), 3077–3083. doi:10.1093/bioinformatics/btaa094
Shi, W., Singha, M., Pu, L., Ramanujam, J., and Brylinski, M. (2021). Graphsite: Ligand-Binding Site Classification Using Deep Graph Neural Network. bioRxiv, 2021.12.06.471420.
Shou, J., Massarweh, S., Osborne, C. K., Wakeling, A. E., Ali, S., Weiss, H., et al. (2004). Mechanisms of Tamoxifen Resistance: Increased Estrogen Receptor-HER2/neu Cross-Talk in ER/HER2-positive Breast Cancer. J. Natl. Cancer Inst. 96 (12), 926–935. doi:10.1093/jnci/djh166
Simonovsky, M., and Meyers, J. (2020). DeeplyTough: Learning Structural Comparison of Protein Binding Sites. J. Chem. Inf. Model. 60 (4), 2356–2366. doi:10.1021/acs.jcim.9b00554
Trebosc, V., Gartenmann, S., Tötzl, M., Lucchini, V., Schellhorn, B., Pieren, M., et al. (2019). Dissecting Colistin Resistance Mechanisms in Extensively Drug-Resistant Acinetobacter Baumannii Clinical Isolates. mBio 10 (4), e01083. doi:10.1128/mBio.01083-19
Vinyals, O., Toshev, A., Bengio, S., and Erhan, D. (2015). “Show and Tell: A Neural Image Caption Generator,” in Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, June 8–10, 2015, 3156–3164. doi:10.1109/cvpr.2015.7298935
Vinyals, O., Bengio, S., and Kudlur, M. (2016). “Order Matters: Sequence to Sequence for Sets,” in Proceedings of the International Conference on Learning Representations, San Juan, Puerto Rico, May 2–4, 2016, 3156–3164. doi:10.1109/cvpr.2015.7298935 arXiv preprint arXiv:1511.06391.
Wang, Y., Xiao, J., Suzek, T. O., Zhang, J., Wang, J., Zhou, Z., et al. (2012). PubChem's BioAssay Database. Nucleic Acids Res. 40 (Database issue), D400–D412. doi:10.1093/nar/gkr1132
Weininger, D. (1988). SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 28 (1), 31–36. doi:10.1021/ci00057a005
Wu, K. J., Lei, P. M., Liu, H., Wu, C., Leung, C. H., and Ma, D. L. (2019). Mimicking Strategy for Protein-Protein Interaction Inhibitor Discovery by Virtual Screening. Molecules 24 (24), 4428. doi:10.3390/molecules24244428
Xu, J., and Zhang, Y. (2010). How Significant Is a Protein Structure Similarity with TM-Score = 0.5? Bioinformatics 26 (7), 889–895. doi:10.1093/bioinformatics/btq066
Xu, K., Ba, J., Kiros, R., Cho, K., Courville, A., Salakhutdinov, R., et al. (2015). “Show, Attend and Tell: Neural Image Caption Generation with Visual Attention,” in International conference on machine learning (Lille, France: PMLR).
Xu, K., Li, C., Tian, Y., Sonobe, T., Kawarabayashi, K., and Jegelkaet, S. (2018). “Representation Learning on Graphs with Jumping Knowledge Networks,” in International Conference on Machine Learning (Stockholm, Sweden: PMLR).
Xu, M., Ran, T., and Chen, H. (2021). De Novo molecule Design through the Molecular Generative Model Conditioned by 3D Information of Protein Binding Sites. J. Chem. Inf. Model. 61 (7), 3240–3254. doi:10.1021/acs.jcim.0c01494
Yasonik, J. (2020). Multiobjective De Novo Drug Design with Recurrent Neural Networks and Nondominated Sorting. J. Cheminform 12 (1), 14–19. doi:10.1186/s13321-020-00419-6
Yeturu, K., and Chandra, N. (2011). PocketAlign a Novel Algorithm for Aligning Binding Sites in Protein Structures. J. Chem. Inf. Model. 51 (7), 1725–1736. doi:10.1021/ci200132z
Yu, Q. (2005). The Dynamic Roles of Angiopoietins in Tumor Angiogenesis. Future Oncol. 1 (4), 475–484. doi:10.2217/14796694.1.4.475
Zhang, Y., and Skolnick, J. (2004). Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins 57 (4), 702–710. doi:10.1002/prot.20264
Zhang, Y., and Skolnick, J. (2005). TM-align: a Protein Structure Alignment Algorithm Based on the TM-Score. Nucleic Acids Res. 33 (7), 2302–2309. doi:10.1093/nar/gki524
Keywords: ligand binding sites, drug discovery and development, in silico drug design, deep learning, graph neural network, recurrent neural network, generative model, machine learning
Citation: Shi W, Singha M, Srivastava G, Pu L, Ramanujam J and Brylinski M (2022) Pocket2Drug: An Encoder-Decoder Deep Neural Network for the Target-Based Drug Design. Front. Pharmacol. 13:837715. doi: 10.3389/fphar.2022.837715
Received: 17 December 2021; Accepted: 10 February 2022;
Published: 11 March 2022.
Edited by:
Adriano D. Andricopulo, University of Sao Paulo, BrazilCopyright © 2022 Shi, Singha, Srivastava, Pu, Ramanujam and Brylinski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michal Brylinski, bWljaGFsQGJyeWxpbnNraS5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.