 Amy M. Chinn
Amy M. Chinn Cristina Salmerón
Cristina Salmerón Jihyung Lee2
Jihyung Lee2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Pharmacol., 21 March 2022
Sec. Inflammation Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.833832
This article is part of the Research TopicCyclic nucleotide phosphodiesterases (PDEs) in immune regulation and inflammationView all 8 articles
Chronic decreases in the second messenger cyclic AMP (cAMP) occur in numerous settings, but how cells compensate for such decreases is unknown. We have used a unique system—murine dendritic cells (DCs) with a DC-selective depletion of the heterotrimeric GTP binding protein Gαs—to address this issue. These mice spontaneously develop Th2-allergic asthma and their DCs have persistently lower cAMP levels. We found that phosphodiesterase 4B (PDE4B) is the primary phosphodiesterase expressed in DCs and that its expression is preferentially decreased in Gαs-depleted DCs. PDE4B expression is dynamic, falling and rising in a protein kinase A-dependent manner with decreased and increased cAMP concentrations, respectively. Treatment of DCs that drive enhanced Th2 immunity with a PDE4B inhibitor ameliorated DC-induced helper T cell response. We conclude that PDE4B is a homeostatic regulator of cellular cAMP concentrations in DCs and may be a target for treating Th2-allergic asthma and other settings with low cellular cAMP concentrations.
Cellular concentration of the second messenger 3′,5′-cyclic adenosine monophosphate (cyclic AMP, cAMP) can profoundly impact organismal physiology. Maintaining cellular cAMP homeostasis is thus essential (Raker et al., 2016). Decreased and increased cAMP levels in immune cells generally produce pro- and anti-inflammatory effects, respectively (Bariagaber and Whalen 2003; Bodor et al., 2012; Lee et al., 2015; Peters-Golden 2009; Rueda et al., 2016). Extracellular stimuli perturb intracellular concentrations of cAMP and trigger signaling cascades. Such perturbations are often transient: after removal of the stimulus, cAMP concentrations return to basal levels even though cellular microdomains may be sites of cAMP action (Bock et al., 2020; Tsvetanova et al., 2021; Zhang et al., 2020). By contrast, chronic dosing with drugs can persistently alter cAMP concentrations. Antagonists of Gs-coupled G protein-coupled receptors (GPCRs) and agonists of Gi-coupled GPCRs decrease cellular cAMP concentrations and are used to treat disease settings that include hypertension, congestive heart failure, and chronic pain (Law et al., 2000; Pierre et al., 2009; Rehsia and Dhalla 2010). Decreased cellular cAMP as a result of certain mutations in the gene Gnas result in clinical disorders, including pseudohypoparathyroidism, Albright hereditary osteodystrophy, and progressive osseus heteroplasia. Expression of certain cyclic nucleotide phosphodiesterases (PDEs) can increase in response to treatment with cAMP-elevating drugs; however, prior work has not defined how cells compensate for decreased cAMP levels (D’Sa et al., 2002; Lee et al., 2002; Liu et al., 2000). We hypothesized that previously unrecognized mechanisms may mediate compensation for a reduction in cAMP concentration and thereby contribute to cellular homeostasis. Here, we used a primary cell model to test this hypothesis: bone marrow-derived dendritic cells (DCs) from CD11cΔGnas (ΔGnas) mice that have a depletion of Gαs, an essential subunit of heterotrimeric (αβγ) Gs proteins that stimulate adenylyl cyclase to convert ATP to cAMP and activate cellular cAMP signaling (Figure 1A, Supplementary Figure S1A) (Lee et al., 2015; Lee et al., 2020). Our studies identify a novel role for PDE4B as a key contributor to cellular cAMP homeostasis and regulator of functional activity in DCs.
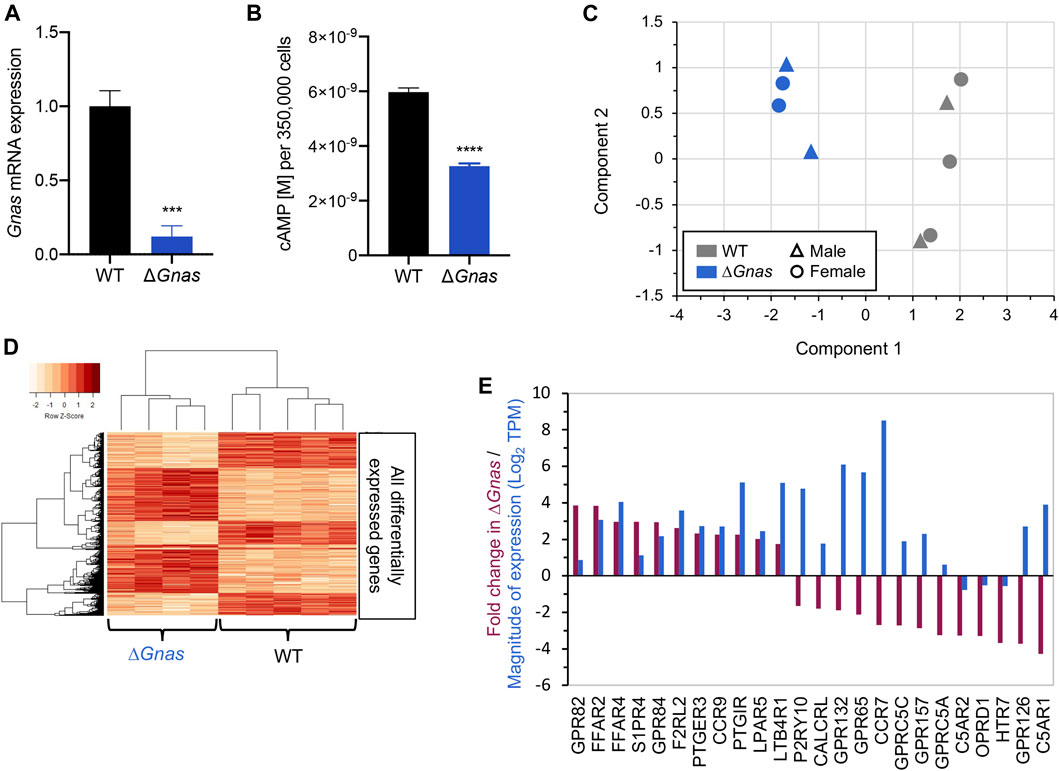
FIGURE 1. ΔGnas DCs have decreased cAMP levels and associated changes in gene expression. (A) Gnas gene expression in wild-type (WT) and ΔGnas DCs; n = 6, p < 0.001. (B) Basal cAMP levels in WT and ΔGnas DCs; n = 5-6, p < 0.0001. (C) WT and ΔGnas DC RNA-Seq samples as visualized in a Multidimensional Scaling (MDS) plot wherein a data set is analyzed to cluster the results into principal components (component 1 and component 2). These components explain the greatest degree of variance across the samples analyzed. The data show that WT and ΔGnas DCs form two separate clusters based on gene expression. WT n = 5, ΔGnas n = 4. (D) Heatmap of differentially expressed genes. Each column lists the gene expression from a single biological replicate. WT n = 5, ΔGnas n = 4. (E) Fold-change (Purple) and level of expression (Blue) of differentially expressed GPCRs in ΔGnas DCs compared to WT cells. WT n = 5, ΔGnas n = 4, FDR< 0.05.
All animal procedures were approved by the University of California San Diego Institutional Animal Care and Use Committee (animal protocol S04173) and in accordance with the National Institutes of Health guide for the care and use of Laboratory animals. C57BL/6J mice and B6. Cg-Tg(TcraTcrb)425Cbn/J (OT-II) mice were purchased from the Jackson Laboratory. CD11cΔGnas (ΔGnas) mice were generated as described previously (Lee et al., 2015). DCs were isolated from both male and female mice for all experiments as RNA-Seq results did not indicate that samples clustered according to gender (Figure 1C).
Mouse femurs and tibiae were cleaned of attached muscle and then briefly dipped in 70% ethanol. Bone marrow was collected by cutting the ends of the bones off with surgical scissors and flushing with 2% Fetal Bovine Serum (FBS, ThermoFisher Scientific, 16000044) in Phosphate Buffered Saline (PBS, ThermoFisher Scientific, 14040133). Cells were spun down at 1,220 rpm for 9 min at 4°C then passed through a 100 μm cell strainer (Corning, 431,752) and rinsed with RP10 media consisting of RPMI 1640 (ThermoFisher Scientific, 22400089) supplemented with 10% FBS (ThermoFisher Scientific, 16000044), 10% penicillin-streptomycin (ThermoFisher Scientific, 15140122), and 50 μM 2-Mercaptoethanol (Sigma Aldrich, M3148-100 ML). Cells were spun down at 1,220 rpm for 9 min at 4°C and then plated in Falcon Petri Dishes (Corning, 351,058) and cultured for 6 days at 5% CO2 in RP10 media with 10 ng/ml recombinant mouse Granulocyte-macrophage colony-stimulating factor (GM-CSF, ThermoFisher Scientific, 14833162). Cells were refed on day 4 of the culture with additional RP10 media and GM-CSF. On day 6, all the media and floating cells were removed and centrifuged at 1,220 rpm for 9 min at 4°C. CD11c+ DCs were then isolated using EasySep Mouse CD11c Positive Selection Kit II (StemCell Technologies, 18,780).
Spleens from OT-II mice were removed and a single cell suspension of splenocytes was generated. Red blood cells were lysed using ACK Lysing Buffer (ThermoFisher Scientific, A1049201) before CD4+ T cells were isolated from this splenocyte population using EasySep Mouse CD4+ T cell Isolation Kit (StemCell Technologies, 19,852).
Isolated DCs were treated with 100 μg/ml ovalbumin (Sigma Aldrich, O1641) for 24 h and then co-cultured with 1 × 105 isolated naïve OT-II CD4+ T cells in a 1:1 ratio for 3 days in RP10 media (see above formulation). CD4+ T cells were then removed from the culture and stimulated with 10 μg/ml plate-bound anti-CD3e antibody (clone 2c11) (BioXcell, BE0001-1) and 1 μg/ml anti-CD28 antibody (ThermoFisher Scientific, 16-0,289-81) for 24 h. Cytokine levels in the supernatant were assayed by ELISA (ThermoFisher Scientific, 88-7044-22 and 88-7371-22) per the manufacturer’s instructions.
To investigate the effects of PDE4 inhibitors on DC-induced T cell differentiation, WT or ΔGnas DCs were isolated and then incubated with 100 μg/ml ovalbumin (Sigma Aldrich, O1641) and either 10 μM A33 (provided by Tetra Therapeutics, now commercially available at Tocris (Tocris, 6313)), 10 μM Ro 20-1724 (Sigma Aldrich, 557,502), or DMSO vehicle control for 24 h. DCs were then washed with RP10 media and co-cultured with OT-II CD4+ T cells. After 3 days, cytokine levels in the supernatant were assayed by ELISA.
DC2.4 cells were a kind gift from Kenneth Rock (cells first described in (Shen et al., 1997)). DC2.4 cells were cultured at 5% CO2 in RPMI 1640 (ThermoFisher Scientific, 21870076) supplemented with 10% FBS (ThermoFisher Scientific, 16000044), 2 mM L-glutamine (ThermoFisher Scientific, 25030081), 10% penicillin-streptomycin (ThermoFisher Scientific, 15140122), 1X NEAA (ThermoFisher Scientific, 11140050), 10 mM HEPES (ThermoFisher Scientific, 15630080), and 55 μM 2-Mercaptoethanol (Sigma Aldrich, M3148-100 ML).
WT and Kin- S49 cells (Insel et al., 1975) were cultured in 10% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/L glucose (ThermoFisher Scientific, 11960044) supplemented with 2 mM L-glutamine (ThermoFisher Scientific, 25030081), 10% heat-inactivated horse serum (Sigma Aldrich, H1138), 1 mM sodium pyruvate (ThermoFisher Scientific, 11360070), and 10 mM HEPES (ThermoFisher Scientific, 15630080). Cells were continuously maintained in logarithmic growth.
DCs were isolated as stated above. For DCs that were treated with drugs, the cells were plated in a 24-well plate and cultured for the specified length of time at 5% CO2 in RPMI 1640 (ThermoFisher Scientific, 22400089) supplemented with 10% FBS (ThermoFisher Scientific, 16000044), 10% penicillin-streptomycin (ThermoFisher Scientific, 15140122), and 50 μM 2-Mercaptoethanol (Sigma Aldrich, M3148-100 ML). In different experiments, DCs were treated with Prostaglandin E2 (PGE2, Sigma Aldrich, P0409), CPT (8-(4-Chlorophenylthio)adenosine 3′,5′-cyclic monophosphate sodium salt; Sigma Aldrich, C3912), 6 MB (N6-Monobutyryladenosine 3′:5′-cyclic monophosphate sodium salt; Sigma Aldrich, M1380), 8 ME (8-(4-Chlorophenylthio)-2′-O-methyladenosine-3′, 5′-cyclic monophosphate, sodium salt; Axxora BLG-C041-05), MDL-12,330 A (Sigma Aldrich, M182), or vehicle control. After treatment, cells were washed 2x with DPBS before being lysed with RLT Buffer from the RNeasy Mini Kit (Qiagen, 74,104).
All RNA was isolated using RNeasy Mini Kit (Qiagen, 74,104) with on-column DNase digestion using RNase-Free DNase (Qiagen, 79,254). After isolation, 1 μg of RNA was converted into cDNA using iScript cDNA Synthesis Kit (Bio-Rad, 1708891). qPCR samples were run in triplicates on a Bio-Rad CFX Connect Real-Time PCR Detection System using PerfeCTa SYBR Green SuperMix (VWR, 101414-146) with 0.5 μM primers and 8 ng cDNA in a 20 μl reaction volume. dCt values were calculated by subtracting the Ct value for the housekeeping gene 18S from the Ct value of the gene being evaluated. A dCt <25 was used as the threshold of detection. Fold-change between two genes was calculated by formula 2-(dCTGene 1 – dCtGene 2). Unless otherwise stated, data were normalized to WT or vehicle control.
Primers were designed with Primer-BLAST using thermodynamic oligo alignment and analyzed with Integrated DNA Technologies (IDT) OligoAnalyzer Tool to minimize self- and heterodimer tendencies for each primer set. Primer sequences are shown in Supplementary Table S3.
Libraries for RNA-Sequencing (RNA-Seq) were prepared at the University of California San Diego Institute for Genomic Medicine Core Facility, using the Illumina Truseq stranded mRNA kit (Illumina, 20022371) per manufacturer’s protocols. Libraries were sequenced using a NovaSeq 6000 sequencing system (Illumina) as per manufacturer’s protocols, with 150 base pair, paired-end reads, at the University of California, San Francisco Center for Advanced Technology core facility.
Following sequencing, FASTQ files were analyzed as follows. First, files were inspected for sequencing quality using FASTQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). All files cleared standard quality assessment, with no quality trimming or other steps needed, apart from removal of adapters. Adapters were trimmed using BBDuk, using standard commands (jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/) and removing sequences corresponding to known Illumina adapters, yielding cleaned-up FASTQ files, which were rechecked via FASTQC to verify adapter removal.
From these files, we quantified gene expression via Kallisto (v0.43.1) with bias detection on (Bray et al., 2016), using the Ensembl v79 reference transcriptome for mice. Transcript expression from Kallisto was converted to gene-level expression (in transcripts per million [TPM] and estimated counts) via the Tximport package (Soneson et al., 2015) in R, using standard commands in the Tximport vignette. Estimated counts were used as input to the edgeR package (Robinson et al., 2010) in R, counts were normalized using the “TMM” method (trimmed mean of M-values), followed by assessment of differential expression (DE), via the inbuilt “exact test” method in edgeR. Low expressed genes (i.e., those not expressed >1 count per million in at least three samples) were filtered out prior to DE analysis. Genes with false discovery rate (FDR) < 0.05 were considered to have statistically significant DE. RNA-Seq data of WT and ΔGnas DCs were deposited in NCBI’s Gene Expression Omnibus under accession number GSE158783.
Single-cell RNA-Seq (scRNA-Seq) analysis of WT CD11c+/ CD45.1+/CD45.2+ DCs from mouse lungs were obtained from GEO, accession number GSE149617 (PMID 32392463). Processed data from 10x scRNA-Seq of these cells were available from GEO, with filtered UMIs (unique molecular identifiers) stored as h5 files. These h5 files were analyzed in R, via Seurat (PMID: 31178118). In brief, files were converted to Seurat objects in R, followed by normalization using relative counts, with a scale factor = 10,000. These normalized counts were used as input for Supplementary Figure S2A which shows expression for PDEs. In addition, the data were rescaled using the SCtransform tool within Seurat, followed by dimension reduction via UMAP (Uniform Manifold Approximation and Projection) using 30 dimensions. The UMAP reduced data were then plotted using the FeaturePlot tool in Seurat to show the spatial variation of PDE expression in the DC population analyzed in Supplementary Figure S2B.
Data from RNA-Seq analysis of human epidermal dendritic cells were obtained from GEO, accession number GSE130804 (Bertram et al., 2019). Processed data in gene-level counts were available from GEO; these data were analyzed via edgeR (Robinson et al., 2010), allowing for expression to be normalized via the TMM method (as above) and expressed in counts per million (CPM). Expression for PDEs was extracted from these normalized data in CPM and plotted in Figure S2C. This dataset had n = 4 donors with four replicates of CD11c+ DC samples sequenced from each donor, yielding n = 16 samples.
Isolated CD11c+ DCs were seeded at 350,000 cells/well in a white bottom, white walled 96-well plate (Millipore Sigma, M0187) and cultured overnight in RP10 media at 37°C, 5% CO2. To assay basal cAMP levels, media was removed and the assay performed on the cells attached to the bottom of the wells so as to only measure intracellular cAMP. Intracellular cAMP levels were then assayed by the HitHunter cAMP Assay for Biologics (DiscoverX, 90-0075LM25) per the manufacturer’s instructions.
Isolated CD11c+ DCs were washed twice with PBS and re-suspended in RIPA buffer (Cell Signaling Technology, 9806S) with 1X protease inhibitor cocktail (Sigma Aldrich, P8340-1 ML) and 1X phosphatase inhibitor cocktail (PhosSTOP, Sigma Aldrich, 4906845001). Samples were sonicated 3X and then centrifuged at max speed for 15 min at 4°C. Supernatants were removed and stored at −70°C. Protein was quantified by Pierce BCA Protein Assay Kit (ThermoFisher Scientific 23,225). Samples were mixed with 4x Laemmli Sample Buffer (Bio-Rad, 1610747) with 2-mercaptoethanol and boiled at 95°C for 10 min. 20 μg of protein was loaded on a 12% Criterion TGX Precast Protein Gel (Bio-Rad, 5671043) and run using Bio-Rad Criterion Vertical Electrophoresis Cell on 85 V for 10 min, and then 15 W, max amp, 200 V for 45 min. Precision Plus Protein Kaleidoscope Prestained Protein Standards (Bio-Rad, 1610375) was used for the ladder. Membranes were activated in methanol and blots were transferred with Bio-Rad Criterion Blotter (100 V for 45 min). Membranes were blocked in 5% milk/1X TBST (Cell Signaling Technology, 9997S) at room temperature and incubated with primary rabbit anti-GNAS antibody (Abcam, ab83735; 1:500 dilution) in 2.5% milk/TBST at 4°C overnight on a shaker. Blots were then washed with 1X TBST, incubated with secondary anti-Rabbit-HRP (Cell Signaling, 70,745; 1:5000 dilution) in 2.5% milk/TBST for 1 h at room temperature on a shaker, washed with TBST, and then visualized using 1 ml of ECL Ultra chemiluminescent reagent (Lumigen, TMA-6) on a Fluorchem HD2 imaging system, using Alphaview 3.4.0.0 software.
Blots were then stripped with Restore Western Blot Stripping Buffer (ThermoFisher Scientific, 21,059), washed, blocked with 5% milk/1X TBST, and re-incubated with goat anti-β-tubulin antibody (Abcam, ab21057; 1 μg/ml) in 2.5% milk/TBST at 4°C overnight on a shaker. Blots were then washed with 1X TBST, incubated with secondary anti-Goat-HRP antibody (Abcam, ab6013; 1:5000 dilution) for 1 h at room temperature on a shaker, washed with TBST, and then visualized. Afterwards, they were stripped (ThermoFisher Scientific, 21,059) per manufacturer’s instructions, washed, blocked with 5% milk/1X TBST, and re-incubated with mouse anti-PDE4B antibody (LSBio, LS-B11018; 1:500 dilution) in 2.5% milk/TBST at 4°C overnight on a shaker. Blots were then washed with 1X TBST, incubated with secondary anti-mouse-HRP antibody (Cell Signaling, 70,765; 1:3,000 dilution) for 1 h at room temp on a shaker, washed with TBST, and visualized. ImageJ was used to analyze densitometry.
PDE activity was measured using Cyclic Nucleotide phosphodiesterase Assay Kit (Enzo Life Sciences, BML-AK800-0,001). Isolated CD11c+ DCs were resuspended in the PDE Assay Buffer and 1X protease inhibitor cocktail (Sigma Aldrich, P8340-1 ML) and sonicated. Samples were then centrifuged at 10,000 rpm for 10 min at 4°C. Lysates were removed and desalted per the manufacturer’s instructions before being stored at −70°C. Protein was quantified by Pierce BCA Protein Assay Kit (ThermoFisher Scientific, 23,225). PDE activity in 2.5 μg protein was assayed per the manufacturer’s instructions.
Data were analyzed with Prism 8.0 software (GraphPad Software, La Jolla, CA) and are presented as mean ± SEM. Statistical analysis was done using two-way ANOVA with Sidak’s multiple comparisons test to correct for multiple comparisons. Unpaired t tests were used to determine statistical significance when comparing two groups. A value of p < 0.05 was considered statistically significant.
ΔGnas mice, generated by crossing Gnasfl/fl mice with CD11c-Cre mice, have reduced basal cAMP levels (Figure 1B) and cAMP accumulation in response to stimulation with Gs-coupled GPCR agonists and forskolin (Lee et al., 2015). The small amount of Gnas present in ΔGnas DCs (Figure 1A) is due to the leakiness of CD11c-Cre (Lee et al., 2015). ΔGnas DCs thus have persistent, low cAMP concentrations and provide a primary cell model to investigate compensation for chronically decreased cAMP levels. RNA-Seq of WT and ΔGnas DCs revealed that WT and ΔGnas DCs cluster separately from one another (Figure 1C). Differentially expressed genes of WT and ΔGnas DCs form distinct groups, and groups of co-regulated genes form a consistent pattern between the two genotypes (Figure 1D).
To assess for compensatory changes, we used RNA-Seq and qPCR to evaluate upstream and downstream components of the cAMP signaling pathway. We found that WT and ΔGnas DCs express a similar number of GPCRs (Supplementary Tables S1,S2), with Gi-linked GPCRs being the largest group of GPCRs (Supplementary Figures S1B,C). Twenty GPCRs had >2-fold increased or decreased expression in ΔGnas DCs (FDR< 0.05) (Figure 1E). Seven of these decreased GPCRs couple to Gi (S1PR4, GPR84, PTGER3, CCR9, CCR7, OPRD1, C5AR1) and are predicted to inhibit adenylyl cyclase activity and might contribute to compensation for decreased cAMP levels by blunting further decrease of cAMP. Expression of Gα proteins (other than Gαs) and adenylyl cyclase isoforms were not significantly different in ΔGnas DCs (Supplementary Figures S1D–H).
cAMP primarily mediates its effects via protein kinase A (PKA) and exchange protein directly activated by cAMP (Epac). The two Epac genes, EPAC1 and EPAC2, were not detectable, implicating PKA as the mediator of cAMP signaling in murine DCs. ΔGnas DCs had >55% reduction in PRKAR2B mRNA which encodes the PKA regulatory subunit RIIβ, suggesting possible feedback between decreased cAMP concentration and its expression (Figure 2A, Supplementary Figure S1I). However, elevation in cAMP levels did not increase PKA RIIβ expression in ΔGnas DCs treated for 24 h with CPT (a non-selective cAMP analog), 6MB (a PKA-selective cAMP analog), and 8ME (an Epac-selective cAMP analog) (Figure 2B). A-kinase anchoring proteins (AKAPs) are scaffolds that tether protein complexes with PKA in subcellular regions (Esseltine and Scott 2013). Expression of the 14 murine AKAPs was unaltered in ΔGnas DCs (Supplementary Figures S1J,K).
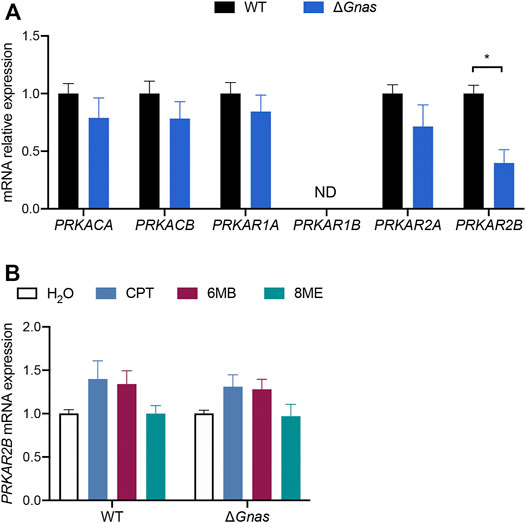
FIGURE 2. ΔGnas DCs have decreased expression of PRKAR2B, whose expression is insensitive to increases in cAMP levels. (A) Protein kinase A (PKA) subunit expression in WT and ΔGnas DCs; n = 5, p < 0.05. (B) PRKAR2B expression in WT and ΔGnas DCs after treatment with the cAMP analogs CPT (non-selective), 6MB (PKA-selective), and 8ME (Epac-selective), (each 50 μM, 24 h); n = 3, data are not statistically significant.
Cyclic AMP is removed from cells via efflux by the multidrug resistance-associated protein 4 (MRP4/ABCC4) transporter and via hydrolysis by PDEs. MRP4 expression was not changed in ΔGnas DCs (Supplementary Figures S1L,M). Assessment of the 21 murine PDEs revealed that PDE4B is the predominant isoform in WT and ΔGnas DCs and that PDE4B is expressed many-fold higher than other PDE isoforms (Figures 3A,B). Single cell RNA-Seq data of mouse lung DCs (Bosteels et al., 2020) (Supplementary Figures S2A,B) and bulk RNA-Seq data of human epidermal DCs (Bertram et al., 2019) (Supplementary Figure S2C) confirm that PDE4B is the highest expressed PDE isoform in both murine tissue-resident and human DCs. We found that ΔGnas DCs have decreased expression of 2 PDEs: PDE4B is reduced >50% and PDE4D is reduced 60% compared to WT cells (Figure 3C). Since PDE4B is predominant in DCs and 20-fold higher expressed than PDE4D, our subsequent studies focused on PDE4B. ΔGnas DCs also had reduced PDE4B protein expression and decreased overall PDE activity, consistent with the prominent expression of PDE4B mRNA in DCs (Figures 3D–F). RNA-Seq data revealed that murine DCs express 4 PDE4B transcripts; the 2 longest transcripts account for >85% of PDE4B expression in both WT and ΔGnas DCs with no differences between genotypes in the prevalence of transcript variants (Supplementary Figures S2D-F).
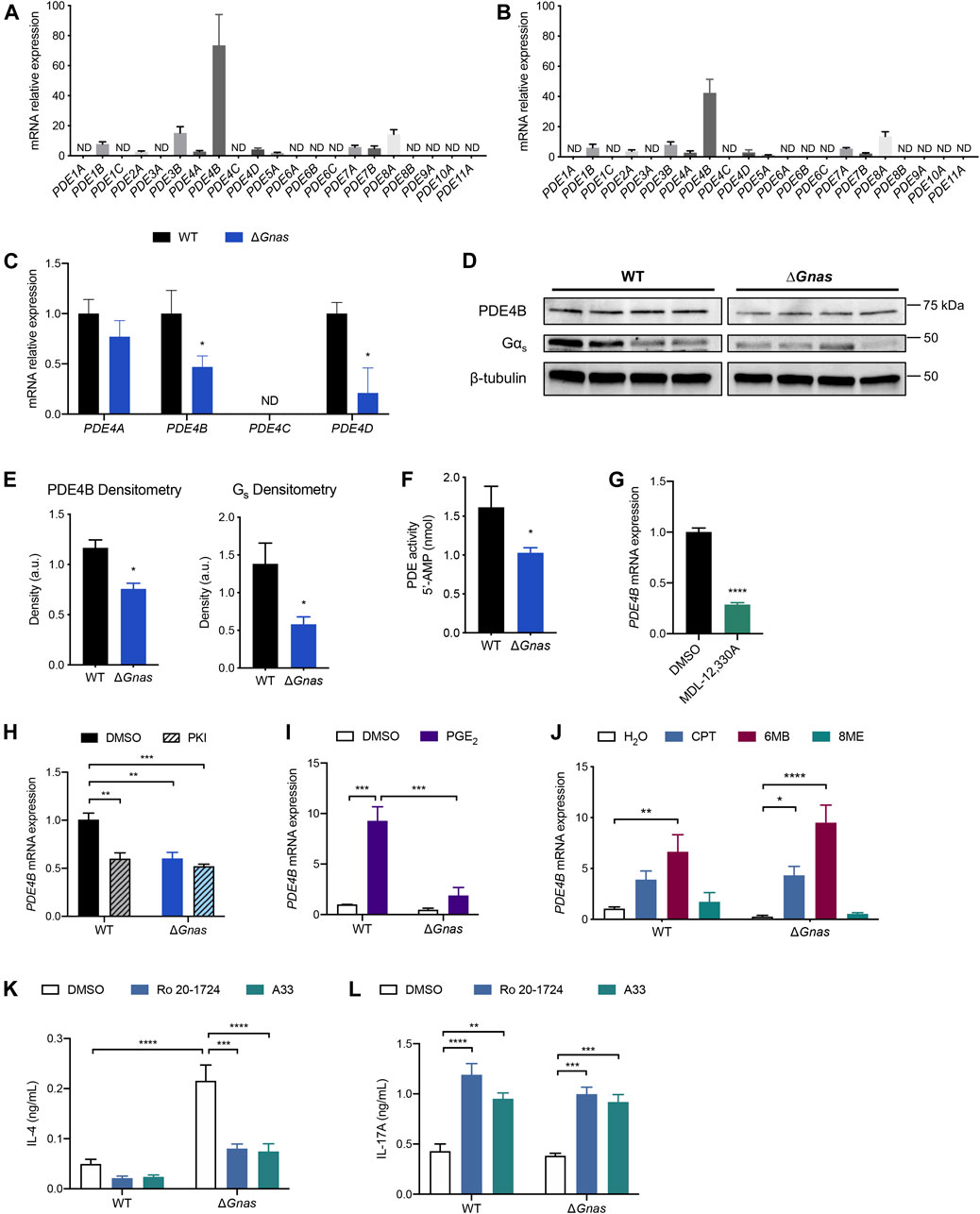
FIGURE 3. PDE4B expression regulates cAMP levels and PDE4B inhibition decreases Th2 differentiation and increases Th17 differentiation in DCs. (A,B) Expression profile of PDE mRNA expression in (A) WT DCs; n = 7 and (B) ΔGnas DCs; n = 6. ND = not detected; data normalized to lowest-expressing detectable gene. (C) PDE4 isoform fold change in ΔGnas DCs compared to WT cells; n = 5–6. (D) Western blots and (E) densitometry of Gαs and PDE4B protein expression in WT and ΔGnas DCs; n = 4. (F) PDE activity in WT and ΔGnas DCs; n = 5–6. (G) PDE4B expression after treatment of WT DCs with the adenylyl cyclase inhibitor MDL-12,330A (10μM, 16 h); n = 3. (H) PDE4B expression in WT and ΔGnas DCs treated with a PKA inhibitor (PKI, 10 μM) for 24 h; n = 4. Data normalized to WT DMSO control. (I,J) Fold change in PDE4B expression after treatment of WT and ΔGnas DCs with (I) PGE2 (10μM, 24 h); n = 2-4, and (J) cAMP analogs CPT (non-selective, 50 μM, 24 h), 6MB (PKA-selective, 50 μM), and 8ME (Epac-selective, 50 μM) for 24 h; n = 3–5. Data normalized to WT vehicle control. (K,L) WT and ΔGnas DCs were treated with DMSO (vehicle control), Ro 20-1724 (pan-PDE4 inhibitor, 10 μM), or A33 (PDE4B-selective inhibitor, 10 μM) for 24 h before culturing with CD4+ OT-II T cells. (K) IL-4 secretion and (L) IL-17 A secretion from co-cultured T cells was quantified; n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Decreased cAMP in WT DCs in response to treatment with the adenylyl cyclase inhibitor MDL-12,330A lowered PDE4B mRNA expression, demonstrating that reduced PDE4B expression in ΔGnas DCs stems from the decrease in cAMP and not from depletion of Gαs (Figure 3G, Supplementary Figure S3A). Treatment of WT DCs for 24 h with PKI, an endogenous PKA inhibitor, reduced PDE4B mRNA, implying that PKA mediates the decrease in PDE4B expression (Figure 3H). To test if reduced PKA activation decreases PDE4B levels in other cell types, we assessed its expression in WT and Kin- S49 T lymphoma cells. Kin- S49 cells, which lack PKA activity but have functional Gαs (Orellana and McKnight 1990), have an 80% decrease in PDE4B expression compared to WT S49 cells, consistent with the findings in ΔGnas DCs (Supplementary Figure S3B).
To determine if PDE4B expression is bidirectionally regulated by cAMP, we treated WT and ΔGnas DCs with PGE2 for 24 h. PGE2 treatment increased PDE4B expression in WT DCs, a response that was blunted in ΔGnas DCs (Figure 3I). To investigate which cAMP effector mediated this increase in expression, we treated WT and ΔGnas DCs for 24 h with the cAMP analogs CPT and 6MB, but not 8ME, increased PDE4B expression in WT and ΔGnas DCs, thus implicating PKA as the mediator of the increase in PDE4B expression (Figure 3J). We obtained similar results using DC2.4 cells (a DC cell line): increased cAMP via PKA increased PDE4B levels (Supplementary Figures S3C,D). Thus, expression of PDE4B is dynamic and regulated by cAMP/PKA, decreasing and increasing in cells with lower and higher cAMP concentrations, respectively.
We next assessed whether PDE4B influences DC function, in particular the ability of DCs to regulate Th2 immunity (Lee et al., 2015). Increased cAMP levels have an anti-inflammatory effect on DCs, so antagonism of PDE4B could potentially reduce DC-induced Th2 inflammation (Galgani et al., 2004; Jin et al., 2010; Lee et al., 2015; Rueda et al., 2016; Chinn and Insel 2020). ΔGnas DCs have reduced PDE4B expression compared to WT cells, but PDE4B is still the predominant PDE in the Gαs-depleted cells (Figure 3B, Figures 3D,E).
Allergic asthma is largely driven by Th2-mediated inflammation (Fajt and Wenzel 2015; Tran et al., 2016). ΔGnas mice develop allergic asthma in response to ovalbumin (OVA) immunization, as shown by increased IgE serum levels, airway hyperresponsiveness, and airway inflammation (Lee et al., 2015). These mice also spontaneously develop asthma at age 6 months without OVA immunization, mimicking the spontaneous development of asthma in humans. Allergic asthma in ΔGnas mice is driven by preferential differentiation of Th2 cells by ΔGnas DCs. By contrast, WT DCs do not induce naïve CD4+ T cells to differentiate into Th2 cells ex vivo (Lee et al., 2015).
We treated WT and ΔGnas DCs with ovalbumin plus either vehicle control, the pan-PDE4 inhibitor Ro 20-1724, or the PDE4B-selective inhibitor A33 (Naganuma et al., 2009). DCs were then co-cultured with naïve CD4+ T cells from OT-II mice to determine if PDE4 inhibition in DCs affects helper T cell differentiation. Treatment of ΔGnas DCs with either Ro 20-1724 or A33 equally decreased IL-4 secretion by co-cultured CD4+ T cells, implying that PDE4B likely accounts for the majority of the PDE4 activity in ΔGnas DCs and that PDE4/PDE4B inhibition in DCs reduces Th2 differentiation (Figure 3K). Decreased DC-induced Th2 differentiation can abrogate allergic lung inflammation in vivo (Lee et al., 2015). Thus, targeting PDE4B may be a novel means to reduce DC-mediated Th2 inflammation–even in DCs with decreased expression from compensation for lower cAMP concentrations. Ro 20-1724 and A33 treatment of WT and ΔGnas DCs also increased T cell-secreted IL-17 A which is indicative of increased Th17 differentiation, consistent with data showing that increased cAMP levels in DCs promote Th17 differentiation (Figure 3L) (Datta et al., 2010; Lee et al., 2020).
Altered concentrations of cAMP can regulate many cellular functions and phenotypes; maintaining cAMP homeostasis is thus a critical feature of cell and tissue physiology (Raker et al., 2016; Chinn and Insel 2020). Moreover, altering cAMP levels is used to treat many diseases, including pulmonary, cardiovascular, renal, gastrointestinal, dermatologic, and neuropsychiatric disorders (Law et al., 2000; Pierre et al., 2009; Rehsia and Dhalla 2010). Prior work has demonstrated that cells can compensate (decrease toward basal levels) drug-induced increases in cAMP by increasing RNA expression of PDE isoforms (D’Sa et al., 2002; Le Jeune et al., 2002; Catherin jin and Conti 2002; Liu et al., 2000; Torphy et al., 1995). The current data identify a mechanism by which cells can compensate for decreased cAMP in a primary cell system, in particular in DCs, and have implications for cellular homeostasis with respect to settings with altered cAMP levels.
The current data are the first of which we are aware that have defined PDE expression and functional responses in DCs. A recent review that discussed roles for PDE4 in multiple immune cell types did not include discussion of DCs (Zuo et al., 2019). The prominent expression of PDE4B in DCs is consistent with its detection in other immune and inflammatory cells (Jiang et al., 1998; Catherin jin and Conti 2002; Maurice et al., 2014).
How does cAMP regulate PDE4B expression? PDE4B has a CRE site in its promoter region, suggesting its expression is mediated by a CREB family member (D’Sa et al., 2002; Wang et al., 2003). Our findings support the idea that PDE4B is responsible for 1) reduced PDE activity in ΔGnas DCs, and 2) compensation to increase cAMP levels in a setting of persistent decrease in cAMP concentration. The decrease in PDE4B expression does not restore “normal” cAMP levels, since cAMP concentration in ΔGnas DCs is less than that of WT DCs, but the decrease in PDE4B may partially blunt physiological effects resulting from decreased cAMP levels. PDE4B thus acts as a homeostatic regulator of cellular cAMP concentrations, rising with increased and falling with decreased cAMP levels. Other cell types may use additional compensatory mechanisms, e.g., elevated cAMP can increase MRP4 mRNA in HeLa and vascular smooth muscle cells via Epac-mediated actions (Bröderdorf et al., 2014).
Many marketed drugs alter cAMP levels. Our findings suggest a targeted strategy directed at components that maintain cAMP homeostasis. PDE4B appears to be a key regulator of both increased and decreased cAMP levels in DCs; other components might be targeted in other cell types. PDE4 is implicated in multiple disease settings, with 3 FDA-approved PDE4 inhibitors marketed for the treatment of atopic dermatitis, chronic obstructive pulmonary disorder, psoriasis, and psoriatic arthritis; several PDE4B-selective inhibitors are in development (Li, Zuo, and Tang 2018; Tibbo and Baillie 2020). These and other findings support the evidence that PDE4B and other PDEs are key contributors to cellular homeostasis, including in cellular microdomains/nanodomains (Bock et al., 2020; Zhang et al., 2020). PDEs, including PDE4B in at least certain cell types, thus appear to be fundamental for modulating cellular function and ultimately organismal health. In this regard, the current study identifies a potential role of PDE4B inhibition as a therapeutic approach to increase cAMP in DCs as a means to blunt Th2-mediated responses.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, GSE158783.
The animal study was reviewed and approved by University of California San Diego Institutional Animal Care and Use Committee.
Conceptualization: AMC and PAI; Methodology: AMC, PAI, JL, ER, and KS; Investigation: AMC, KS, JL, and CS; Formal analysis: AMC, KS; Writing–original draft preparation: AMC, PAI; Supervision: PAI; Funding acquisition: PAI and ER; All authors reviewed the manuscript.
This research was funded by National Institutes of Health (NIH) grants R56AI110505, T32GM007752, RO1HL14199, and UO1AI125860.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank their colleague Nicholas Webster for many useful discussions regarding this topic. A33 compound kindly provided by Tetra Therapeutics, Inc.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.833832/full#supplementary-material
Bariagaber, A. K., and Whalen, M. M. (2003). Decreased Adenylyl Cyclase and CAMP-dependent Protein Kinase Activities Inhibit the Cytotoxic Function of Human Natural Killer Cells. Hum. Immunol. 64 (9), 866–873. doi:10.1016/s0198-8859(03)00154-x
Bertram, K. M., Botting, R. A., Baharlou, H., Rhodes, J. W., Rana, H., Graham, J. D., et al. (2019). Identification of HIV Transmitting CD11c+ Human Epidermal Dendritic Cells. Nat. Commun. 10 (1). doi:10.1038/s41467-019-10697-w
Bock, Andreas., Annibale, Paolo., Konrad, Charlotte., Hannawacker, Annette., Selma, E., Isabella Maiellaro, Anton., et al. (2020). Optical Mapping of CAMP Signaling at the Nanometer Scale. Cell 182 (6), 1519–1530.e17. doi:10.1016/j.cell.2020.07.035
Bodor, Josef., Bopp, Tobias., Vaeth, Martin., Klein, Matthias., Edgar, Serfling., Hünig, Thomas., et al. (2012). Cyclic AMP Underpins Suppression by Regulatory T Cells. Eur. J. Immunol. 42 (6), 1375–1384. doi:10.1002/eji.201141578
Bosteels, Cedric., Neyt, Katrijn., Vanheerswynghels, Manon., van Helden, M. J., Sichien, D., et al. (2020). Inflammatory Type 2 CDCs Acquire Features of CDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 52 (6), 1039–1056.e9. doi:10.1016/j.immuni.2020.04.005
Bray, Nicolas. L., Pimentel, Harold., Melsted, Páll., and Pachter, Lior. (2016). Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 34 (5), 525–527. doi:10.1038/nbt.3519
Bröderdorf, Susanne., Zang, Sebastian., Schaletzki, Yvonne., Grube, Markus., Kroemer, Heyo. K., and Jedlitschky, Gabriele. (2014). CAMP Regulates Expression of the Cyclic Nucleotide Transporter MRP4 (ABCC4) through the EPAC Pathway. Pharmacogenetics and Genomics 24 (10), 522–526. doi:10.1097/FPC.0000000000000084
Catherin Jin, S. L., and Conti, Marco. (2002). Induction of the Cyclic Nucleotide Phosphodiesterase PDE4B Is Essential for LPS-Activated TNF-α Responses. Proc. Natl. Acad. Sci. United States America 99 (11), 7628–7633. doi:10.1073/pnas.122041599
Chinn, Amy. M., and Insel, Paul. A. (2020). Cyclic AMP in Dendritic Cells: A Novel Potential Target for Disease-Modifying Agents in Asthma and Other Allergic Disorders. Br. J. Pharmacol. 177 (15), 3363–3377. doi:10.1111/bph.15095
Datta, Sandip. K., Sabet, Mojgan., Nguyen, Kim. Phung. L., Valdez, Patricia. A., Gonzalez-Navajas, Jose. M., Islam, Shamima., et al. (2010). Mucosal Adjuvant Activity of Cholera Toxin Requires Th17 Cells and Protects against Inhalation Anthrax. Proc. Natl. Acad. Sci. United States America 107 (23), 10638–10643. doi:10.1073/pnas.1002348107,
D’Sa, Carrol., Tolbert, Lara. M., Conti, Marco., and Duman, Ronald. S. (2002). Regulation of CAMP-specific Phosphodiesterases Type 4B and 4D (PDE4) Splice Variants by CAMP Signaling in Primary Cortical Neurons. J. Neurochem. 81 (4), 745–757.
Esseltine, Jessica. L., and Scott, John. D. (2013). AKAP Signaling Complexes: Pointing towards the Next Generation of Therapeutic Targets? Trends Pharmacol. Sci. 34 (12), 648–655. doi:10.1016/j.tips.2013.10.005
Fajt, Merritt. L., and Wenzel, Sally. E. (2015). Asthma Phenotypes and the Use of Biologic Medications in Asthma and Allergic Disease: The Next Steps toward Personalized Care. J. Allergy Clin. Immunol. 135 (2), 299–310. doi:10.1016/J.JACI.2014.12.1871
Galgani, Mario., De Rosa, Veronica., De Simone, Salvatore., Leonardi, Antonio., Ugo, D’Oro., Napolitani, Giorgio., et al. (2004). Cyclic AMP Modulates the Functional Plasticity of Immature Dendritic Cells by Inhibiting Src-like Kinases through Protein Kinase A-Mediated Signaling. J. Biol. Chem. 279 (31), 32507–32514. doi:10.1074/jbc.M403355200
Insel, Paul. A., Bourne, Henry. R., Coffino, Philip., and Tomkins, Gordon. M. (1975). Cyclic AMP-dependent Protein Kinase: Pivotal Role in Regulation of Enzyme Induction and Growth. Science 190 (4217), 896–898. doi:10.1126/science.171770
Jiang, X., Paskind, M., Weltzien, R., and Epstein, P. M. (1998). Expression and Regulation of MRNA for Distinct Isoforms of CAMP-specific PDE-4 in Mitogen-Stimulated and Leukemic Human Lymphocytes. Cel Biochem. Biophys. 28 (2), 135–160. doi:10.1007/BF02737809
Jin, S. L. Catherine., Goya, Sho., Nakae, Susumu., Wang, Dan., Bruss, Matthew., Hou, Chiaoyin., et al. (2010). Phosphodiesterase 4B Is Essential for TH2-Cell Function and Development of Airway Hyperresponsiveness in Allergic Asthma. J. Allergy Clin. Immunol. 126 (6), 1252–1259.e12. doi:10.1016/J.JACI.2010.08.014
Law, Ping-Yee., Wong, Yung. H., and Loh, Horace. H. (2000). Molecular Mechanisms and Regulation of Opioid Receptor Signaling. Annu. Rev. Pharmacol. Toxicol. 40 (1), 389–430. doi:10.1146/annurev.pharmtox.40.1.389
Le Jeune, I. R., Shepherd, M., Van Heeke, G., Houslay, M. D., and Hall, I. P. (2002). Cyclic AMP-dependent Transcriptional Up-Regulation of Phosphodiesterase 4D5 in Human Airway Smooth Muscle Cells. Identification and Characterization of a Novel PDE4D5 Promoter. J. Biol. Chem. 277 (39), 35980–35989. doi:10.1074/jbc.M204832200
Lee, J., Kim, T. H., Murray, F., Li, X., Choi, S. S., Broide, D. H., et al. (2015). Cyclic AMP Concentrations in Dendritic Cells Induce and Regulate Th2 Immunity and Allergic Asthma. Proc. Natl. Acad. Sci. 112 (5), 1529–1534. doi:10.1073/pnas.1417972112
Lee, J., Zhang, J., Chung, Y.-J., Kim, J. H., Min Kook, C., González-Navaja, J. M., et al. (2020). Inhibition of IRF4 in Dendritic Cells by PRR-independent and -Dependent Signals Inhibit Th2 and Promote Th17 Responses. ELife 9, e49416. doi:10.7554/eLife.49416
Lee, Richard., Wolda, Sharon., Moon, Eunyi., Esselstyn, James., Hertel, Carmen., and Adam, Lerner. (2002). PDE7A Is Expressed in Human B-Lymphocytes and Is Up-Regulated by Elevation of Intracellular CAMP. Cell Signal. 14 (3), 277–284. doi:10.1016/S0898-6568(01)00250-9
Li, Heng., Zuo, Jianping., and Tang, Wei. (2018). Phosphodiesterase-4 Inhibitors for the Treatment of Inflammatory Diseases. Front. Pharmacol. 9 (OCT), 1048. doi:10.3389/fphar.2018.01048
Liu, Hanguan., Palmer, Daniel., Jimmo, Sandra. L., Tilley, Douglas. G., Dunkerley, Heather. A., Pang, Stephen. C., et al. 2000. “Expression of Phosphodiesterase 4D (PDE4D) Is Regulated by Both the Cyclic AMP-dependent Protein Kinase and Mitogen-Activated Protein Kinase Signaling Pathways.” doi:10.1074/jbc.M001634200
Maurice, Donald. H., Ke, Hengming., Ahmad, Faiyaz., Wang, Yousheng., Chung, Jay., and Manganiello, V. C. (2014). Advances in Targeting Cyclic Nucleotide Phosphodiesterases. Nat. Rev. Drug Discov. 13 (4), 290. doi:10.1038/NRD4228
Naganuma, Kenji., Omura, Akifumi., Maekawara, Naomi., Saitoh, Masahiro., Ohkawa, Naoto., Kubota, Takashi., et al. (2009). Discovery of Selective PDE4B Inhibitors. Bioorg. Med. Chem. Lett. 19 (12), 3174–3176. doi:10.1016/j.bmcl.2009.04.121
Orellana, S. A., and McKnight, G. S. (1990). The S49 Kin- Cell Line Transcribes and Translates a Functional MRNA Coding for the Catalytic Subunit of CAMP-dependent Protein Kinase. J. Biol. Chem. 265 (6), 3048–3053. doi:10.1016/s0021-9258(19)39731-5
Peters-Golden, Marc. (2009). Putting on the Brakes: Cyclic AMP as a Multipronged Controller of Macrophage Function. Sci. Signaling 2 (75), pe37. doi:10.1126/scisignal.275pe37
Pierre, Sandra., Eschenhagen, Thomas., Geisslinger, Gerd., and Scholich, Klaus. (2009). Capturing Adenylyl Cyclases as Potential Drug Targets. Nat. Rev. Drug Discov. 8 (4), 321–335. doi:10.1038/nrd2827
Raker, K., Verena, K., Becker, C., and Steinbrink, K. (2016). The CAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 7, 123. doi:10.3389/fimmu.2016.00123
Rehsia, Navneet. S., and Dhalla, Naranjan. S. (2010). Mechanisms of the Beneficial Effects of Beta-Adrenoceptor Antagonists in Congestive Heart Failure. Exp. Clin. Cardiol. 15 (4), e86–95. doi:10.1007/s10741-009-9152-z
Robinson, Mark. D., McCarthy, Davis. J., and Smyth, Gordon. K. (2010). EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics (Oxford, England) 26 (1), 139–140. doi:10.1093/bioinformatics/btp616
Rueda, Cesar. M., Jackson, Courtney. M., and Chougnet, Claire. A. (2016). Regulatory T-Cell-Mediated Suppression of Conventional T-Cells and Dendritic Cells by Different CAMP Intracellular Pathways. Front. Immunol. 7, 216. doi:10.3389/fimmu.2016.00216
Shen, Z., Reznikoff, G., Dranoff, G., and Rock, K. L. (1997). Cloned Dendritic Cells Can Present Exogenous Antigens on Both MHC Class I and Class II Molecules. J. Immunol. 158 (6).
Soneson, Charlotte., Love, Michael. I., and Robinson, Mark. D. (2015). Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 4, 1521. doi:10.12688/f1000research.7563.2
Tibbo, Amy. J., and Baillie, George. S. (2020). Phosphodiesterase 4B: Master Regulator of Brain Signaling. Cells 9 (5). doi:10.3390/cells9051254
Torphy, T. J., Zhou, H. L., Foley, J. J., Sarau, H. M., Manning, C. D., and Barnette, M. S. (1995). Salbutamol Up-Regulates PDE4 Activity and Induces a Heterologous Desensitization of U937 Cells to Prostaglandin E2. Implications for the Therapeutic Use of Beta-Adrenoceptor Agonists. J. Biol. Chem. 270 (40), 23598–23604. doi:10.1074/jbc.270.40.23598
Tran, Trung. N., Zeiger, Robert. S., Peters, Stephen. P., Colice, G., Newbold, P., and Goldman, M. (2016). Overlap of Atopic, Eosinophilic, and TH2-High Asthma Phenotypes in a General Population with Current Asthma. Ann. Allergy Asthma Immunol. 116 (1), 37–42. doi:10.1016/J.ANAI.2015.10.027
Tsvetanova, Nikoleta. G., Trester-Zedlitz, Michelle., Newton, Billy. W., Peng, Grace. E., Johnson, Jeffrey. R., Jimenez-Morales, David., et al. (2021). Endosomal CAMP Production Broadly Impacts the Cellular Phosphoproteome. J. Biol. Chem. 297 (1), 100907. doi:10.1016/J.JBC.2021.100907
Wang, Daguang., Deng, Chengjun., Bugaj-Gaweda, Bozena., Kwan, May., Gunwaldsen, Caryn., Leonard, Chris., et al. (2003). Cloning and Characterization of Novel PDE4D Isoforms PDE4D6 and PDE4D7. Cell Signal. 15 (9), 883–891. doi:10.1016/S0898-6568(03)00042-1
Zhang, Jason. Z., Lu, Tsan-Wen., Stolerman, L. M., Taylor, Susan. S., Mehta, Sohum., and Zhang, Jin. (2020). Phase Separation of a PKA Regulatory Subunit Controls CAMP Compartmentation and Oncogenic Signaling. Cell 182 (6), 1531–1544.e15. doi:10.1016/j.cell.2020.07.043,
Keywords: cyclic AMP (cAMP), compensation, phosphodiesterase (PDE), PDE4B, Th2 immunity
Citation: Chinn AM, Salmerón C, Lee J, Sriram K, Raz E and Insel PA (2022) PDE4B Is a Homeostatic Regulator of Cyclic AMP in Dendritic Cells. Front. Pharmacol. 13:833832. doi: 10.3389/fphar.2022.833832
Received: 12 December 2021; Accepted: 01 February 2022;
Published: 21 March 2022.
Edited by:
Robert B. Nelson, MindImmune Therapeutics, Inc., United StatesReviewed by:
Evi Kostenis, University of Bonn, GermanyCopyright © 2022 Chinn, Salmerón, Lee, Sriram, Raz and Insel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul A. Insel, cGluc2VsQGhlYWx0aC51Y3NkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.