 Jianjun Gao1
Jianjun Gao1 Zhaoyan Gu2,3*
Zhaoyan Gu2,3*- 1Department of Nephrology, Chinese PLA Strategic Support Force Characteristic Medical Center, Beijing, China
- 2Department of Endocrinology, Second Medical Center, Chinese PLA General Hospital, Beijing, China
- 3National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital, Beijing, China
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily of ligand-activated transcription factors. Accumulating evidence suggests that PPARs may play an important role in the pathogenesis of kidney disease. All three members of the PPAR subfamily, PPARα, PPARβ/δ, and PPARγ, have been implicated in many renal pathophysiological conditions, including acute kidney injury, diabetic nephropathy, and chronic kidney disease, among others. Emerging data suggest that PPARs may be potential therapeutic targets for renal disease. This article reviews the physiological roles of PPARs in the kidney and discusses the therapeutic utility of PPAR agonists in the treatment of kidney disease.
Introduction
Peroxisome proliferator-activated receptors (PPARs), a group of nuclear hormone receptors, consist of threeisotypes, i.e., PPARα, PPARβ/δ, and PPARγ (Xi et al., 2020). PPARs can regulate gene transcription in either ligand-dependent or -independent manner. The target genes are critical for fatty acid oxidation (FAO) and transportation, glucose metabolism, adipogenesis, cholesterol transportation and biosynthesis, apoptosis, and the inflammatory response (Tyagi et al., 2011; Derosa et al., 2018). Numerous studies employing experimental and clinical models have shown that PPARs play important roles in lipid metabolism and energy homeostasis in the kidney (Tovar-Palacio et al., 2012; Corrales et al., 2018) (Table 1). This review focuses on the roles of PPARs in renal metabolism as well as therapeutic strategies targeting the activation of PPARs in kidney disease.
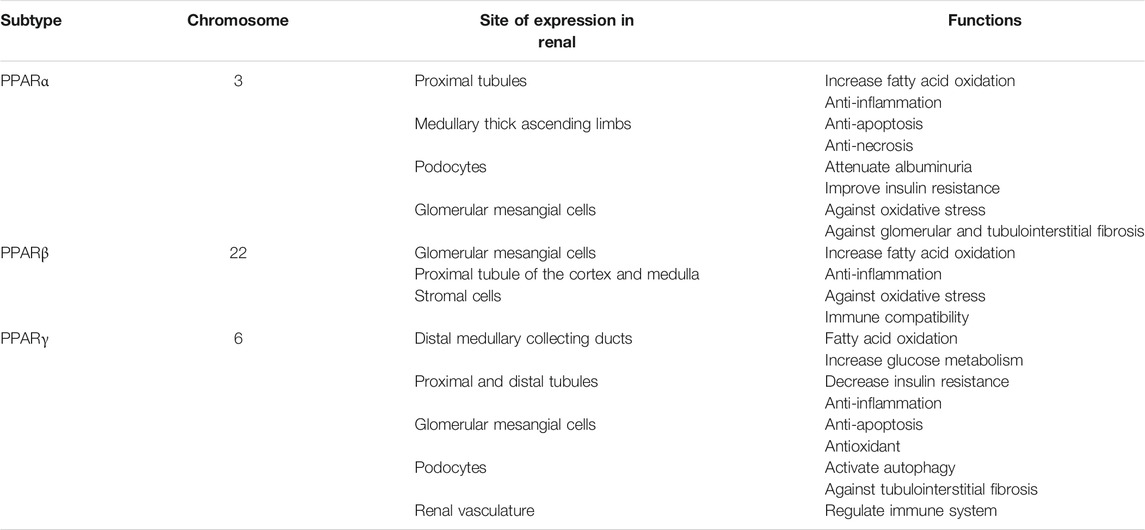
TABLE 1. PPAR subtypes, chromosome location, expression site, and their functions in kidney diseases.
PPAR Family
PPAR family proteins have four main functional segments: the N-terminal ligand-independent transactivation domain (AF1, A/B domain), DNA-binding domain (DBD or C domain), co-factor docking domain (D domain), and C-terminal E/F domain that includes the ligand-binding domain (LBD) and ligand-dependent transactivation domain (AF2 domain; Figure 1) (Miyachi, 2021). The LBD and C-terminal activation functional domain form a large ligand-binding pocket. After the ligand-binding pocket interacts with a ligand, PPARs are translocated to the nucleus, either homodimerize or heterodimerize with another nuclear receptor, the retinoid X receptor (RXR) (Grygiel-Górniak, 2014). The PPRA or PPAR/RXR dimer binds to specific DNA response elements, peroxisome proliferator response elements (PPREs), to activate gene transcription. The conserved DBD is the domain involved in binding to PPREs (Berger and Moller, 2002). PPREs most commonly consist of a direct repeat of hexameric core recognition elements with a 1-bp spacer (DR1, 5′-AGGTCANAGGTCA-3′) located in the promoter regions of PPAR target genes (Dixon et al., 2021). After activation of the PPAR/RXR complex at the PPRE, the PPAR/RXR heterodimer can recruit diverse nuclear receptor co-factors, including coactivators, such as PPARγ coactivator-1α (PGC-1α), or co-repressors, such as the nuclear co-repressor and the silencing mediator for retinoid and thyroid hormone receptors (Dowell et al., 1999; Qi et al., 2000; Petr et al., 2018).
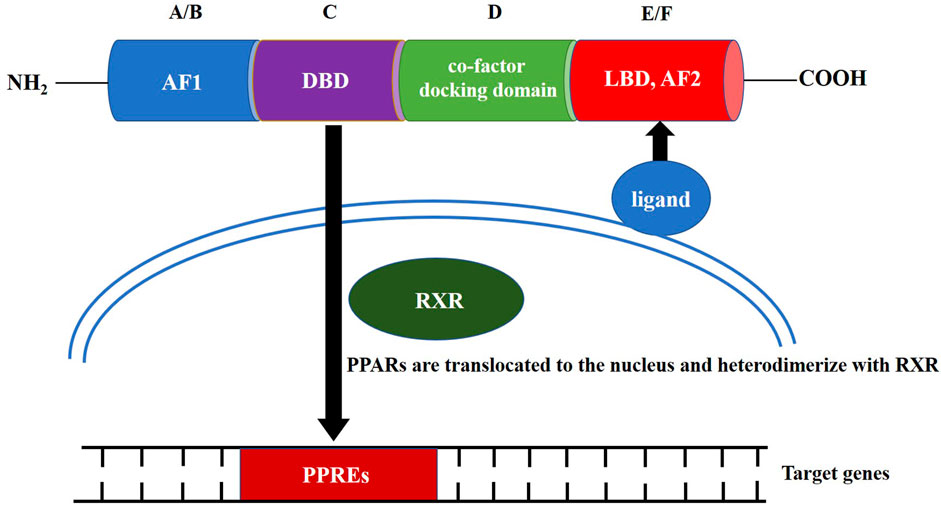
FIGURE 1. Structure and molecular mechanism of action of peroxisome proliferator-activated receptors.
Although PPARs show a high degree of identity at the amino acid level and very similar structures, each isoform has unique tissue distribution, ligand selectivity, and biological functions (Ruan et al., 2008). The selectivity of each of the three PPAR isotypesis dependent on the divergent amino-acid sequences in the LBD. Fatty acids and their metabolites, synthetic pharmaceutical agents, including hypolipidemic fibrates, and the antidiabetic agent thiazolidinedione (TZD) have been shown to bind to and activate PPAR (Ricote et al., 2004; Monsalve et al., 2013). In recent years, many PPAR-null mice models have been available to study their implicaton in kidney physiology (Table 2).
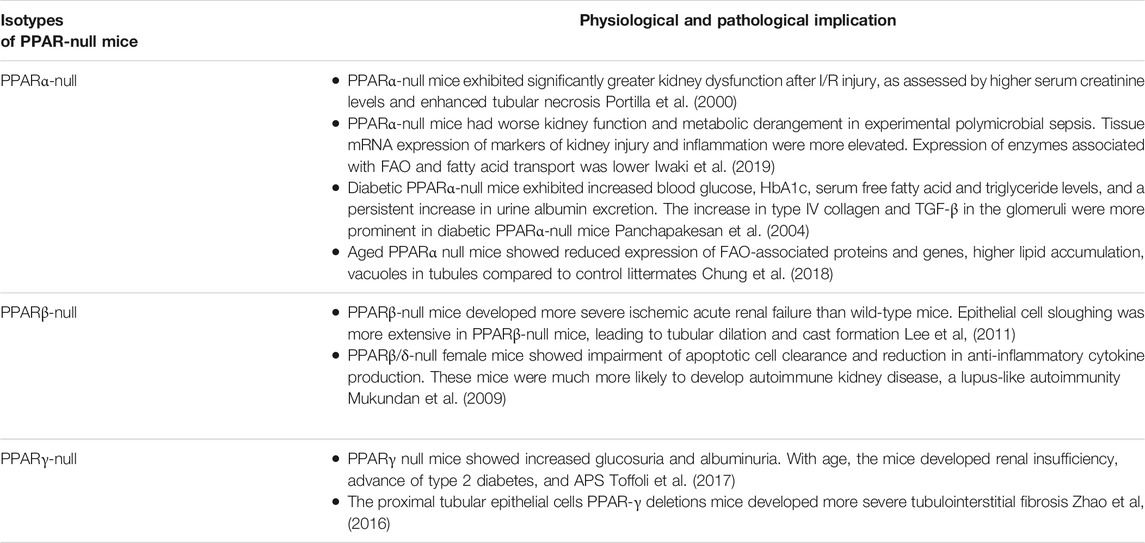
TABLE 2. Physiological and pathological implications in kidney of PPAR-null mice for the different isotypes.
PPARα
PPARα, the first member of the PPAR subfamily identified, is highly expressed in tissues that exhibit high levels of mitochondrial and FAO activity, including those of the liver, kidney, intestinal mucosa, and heart (Dixon et al., 2021). Lower levels of PPARα expression have also been detected in several other tissues. Within the kidney, PPARα is abundant in the proximal tubules and medullary thick ascending limbs, with much lower levels in glomerular mesangial cells (Guan et al., 1997; Kamijo et al., 2002). Given the high levels of expression in proximal tubules and medullary thick ascending limbs, PPARα has been implicated in metabolic regulation of the kidney. Many types of fatty acids and synthetic lipid-lowering fibrates (e.g., fenofibrate, clofibrate) can serve as PPARα agonists and regulate the transcription of several genes involved in FAO and the inflammatory response in the kidney (Di Paola and Cuzzocrea, 2007). It has been reported that activation of PPARα by clofibrate significantly induced the expression of β-oxidation enzymes in the renal cortex, including long-chain acyl-CoA dehydrogenase, medium-chain acyl-CoA dehydrogenase, and acyl-CoA oxidase (Ouali et al., 1998). PGC-1α, a coactivator of PPARα, is one of the main upstream transcriptional regulators of mitochondrial biogenesis and activity (Wu et al., 1999; Arany et al., 2005). In the kidney, PGC-1α is predominantly expressed in proximal tubules and medullary thick ascending limbs. Recent evidence supports the suggestion that PPARα and PGC-1α are critical regulators of the kidney involved in maintaining the balance of energy production and consumption (Portilla, 2003; Chambers and Wingert, 2020).
PPARα and Acute Kidney Injury
PPARα-mediated FAO has been suggested to play an important regulatory role in the pathogenesis of acute kidney injury (AKI). PPARα null mice subjected to ischemia/reperfusion (I/R) injury exhibited significantly enhanced cortical necrosis and poorer kidney function in comparison to wild-type controls (Portilla et al., 2000). Upon cisplatin-induced AKI, the binding of PPARα to its target genes was inhibited and the expression of its coactivator, PGC-1α, was decreased in the mouse kidney and proximal tubule cells in culture, suggesting that FAO is suppressed in cisplatin-induced AKI (Portilla et al., 2002). Mice deficient in PPARα have poorer kidney function with sepsis-induced AKI, which is also related to reduced FAO and increased inflammation (Iwaki et al., 2019). Clinical data indicated that genome-wide expression profiles are characterized by repression of the PPARα signaling pathway with increased incidence of severe sepsis in AKI (Wong et al., 2009). Lipopolysaccharide (LPS)-treated mice exhibited a 40% decrease in renal FAO and inhibition of the expression of key transcription factors required for FAO. LPS also caused reductions in renal PPARα, RXR, and PGC-1α mRNA levels (Feingold et al., 2008). PPARα exhibits a protective role against sepsis-associated AKI by improving reduced FAO and increased inflammation (Iwaki et al., 2019). The increased expression of PPARα in proximal tubular epithelial cells in mice was shown to be sufficient to maintain FAO and protect kidney function and morphology in AKI (Li et al., 2009). PPARα ligands attenuate cisplatin-induced AKI by preventing the inhibition of FAO (Li et al., 2004a), reducing apoptosis and necrosis in proximal tubule cells through a decrease in endonuclease G activity (Li et al., 2004b), and limiting inflammatory processes by blocking NF-κB activity (Li et al., 2005; Baud and Letavernier, 2007). The activation of NF-κB, apoptosis, and oxidative stress induced by fatty acid-bound albumin (FA-BSA) in HK-2 cells was also markedly suppressed by fenofibrate (Zuo et al., 2015). Therefore, PPARα may be considered as a novel therapeutic target for preventing AKI. The mitochondrial matrix protein cyclophilin D (CypD) binds to PPARα and inhibits its nuclear translocation as well as the transcription of PPARα-regulated FAO genes during cisplatin-induced AKI. The genetic or pharmacological inhibition of CypD was reported to preserve PPARα transcriptional activity and prevent FAO impairment (Jang et al., 2020). The upregulation of PGC-1α can improve FAO and renal recovery from I/R injury by regulating NAD biosynthesis (Tran et al., 2016). Therefore, the regulation of PGC-1α was also identified as a new way to improve AKI.
PPARα and Diabetic Nephropathy
Recent studies suggested important roles of FAO dysfunction and insulin resistance in the pathogenesis and progression of diabetic nephropathy (DN). PPARα may be a promising therapeutic target for treating diabetic renal complications. Herman et al. reported heavy lipid deposition and increased amounts of intracellular lipid droplets in kidney biopsies of patients with DN. It is also worth noting that several genes involved in FAO pathways, including PPARα, were downregulated (Herman-Edelstein et al., 2014). In an experimental study, more severe glomerular structural changes as well as albuminuria were noted in diabetic PPARα-knockout mice, with increased type IV collagen and transforming growth factor (TGF)-β expression detected in the glomerular lesions (Park et al., 2006). The PPARα agonist fenofibrate has been shown to reduce fasting blood glucose and insulin resistance, decrease urinary albumin excretion and glomerular mesangial expansion, suppress oxidative stress, and attenuate inflammation in diabetic animals (Park et al., 2006; Zuo et al., 2015; Yaribeygi et al., 2018). In addition, fenofibrate can downregulate the TGF-β signaling pathway, which plays a key role in the progression of DN (Wilmer et al., 2002). Endothelial dysfunction-induced M1 macrophage recruitment has been shown to play a key role in the development of DN. Fenofibrate can prevent DN by reducing M1 macrophage recruitment through regulating endothelial cell function as observed in a mouse model of type 2 diabetes (Feng et al., 2021). The PPARα activator gemfibrozil not only alleviated dyslipidemia but also attenuated albuminuria in normotensive noninsulin-dependent diabetic patients (Smulders et al., 1997). The Action to Control Cardiovascular Risk in Diabetes (ACCORD) study additionally demonstrated that fibrate therapy with intensive glucose control could significantly reduce microalbuminuria and macroalbuminuria in patients with type 2 diabetes (Ginsberg et al., 2010; Ismail-Beigi et al., 2010). It was reported that the downregulation of PGC-1α significantly increased reactive oxygen species in high glucose-stimulated renal mesangial cells, and endogenous PGC-1α expression resulted in protective effects against oxidative stress, glomerulosclerosis, and tubulointerstitial fibrosis in experimental DN (Zhang et al., 2018).
PPARα and Chronic Kidney Disease
PPARα was shown to be downregulated in aggressive mouse models of autosomal dominant polycystic kidney disease (ADPKD) and primary human ADPKD cells, suggesting that decreased PPARα function may underlie the impaired FAO and oxidative phosphorylation in ADPKD (Hajarnis et al., 2017; Lakhia et al., 2018).
Meanwhile, PPARα also regulates age-associated renal fibrosis. PPARα and FAO-associated gene expression was decreased with age, which is directly related to the lipid metabolic disturbances in renal diseases in aged (Tovar-Palacio et al., 2012). PPRAα null mice exhibited higher lipid accumulation in renal tubule compared with littermates, which suggested the importance of PPRAα in the development of age-related renal fibrosis (Chung et al., 2018). Targeting PPARα is also useful for preventing age-associated CKD.
In unilateral ureteral obstruction models, preserving the expression of PPARα led to a reduction in tubulointerstitial fibrosis and inflammation. Further analyses reveal decreased production of TGF-β, IL-1b, IL-6, and TNF-α, reduced macrophage infiltration (Li et al., 2013). PPARα also plays an important role in glomerulonephritis. Saga et al. reported that bezafibrate, a PPARα agonist, attenuated the severity and extent of diseased glomeruli and decreased the number of CD8+ cells in the glomeruli in anti-glomerular basement membrane (GBM) crescentic glomerulonephritis. Moreover, the urinary protein level was diminished after bezafibrate treatment, in parallel with the attenuation of glomerular injury (Saga et al., 2005). Plasma free fatty acid and triglyceride levels were elevated in relation to a decrease in PPARα expression in high-fat diet (HFD) models. In these models, treatment with fenofibrate increased PPARα expression, prevented HFD-induced renal lipotoxicity, reduced oxidative stress and lipid accumulation in the glomeruli, and prevented the development of albuminuria and glomerular fibrosis (Tanaka et al., 2011; Chung et al., 2012). Taken together, these findings suggest that PPARα may be a novel therapeutic target for the treatment of kidney disease.
PPARβ/δ
PPARβ/δ plays a key role in a number of biological processes, including fertility, lipid metabolism, bone formation, mast cell immunity, skin and brain development, and tumorigenesis (Wu et al., 2009). Although PPARβ/δ mRNA has been detected in almost all tissues and cells examined, it is relatively abundant in the kidney, with ubiquitous expression in all nephron segments, including glomerular mesangial cells, medullary interstitial cells, and stromal cells (Guan et al., 1997). However, the role of PPARβ/δ in the kidney has not been investigated in detail.
PPARβ/δ and Acute Kidney Injury
Letavernier et al. reported that PPARβ/δ-knockdown mice exhibited much greater kidney dysfunction and exacerbated injury compared to their wild-type counterparts after I/R injury. PPARβ/δ may protect the kidney against I/R injury by activating the antiapoptotic Akt signaling pathway and increasing the spread of tubular epithelial cells (Letavernier et al., 2005). PPARβ/δ agonist treatment has also been shown to attenuate renal dysfunction, leukocyte infiltration, and the formation of interleukin (IL)-6 and tumor necrosis factor-α (TNF-α) in the diabetic kidney during I/R injury. The expression of suppressor of cytokine signaling-3, which plays important roles in the cytokine-activated signaling pathway, was increased after PPARβ/δ agonist treatment in I/R injury models (Collino et al., 2011).
PPARβ/δ and Diabetic Nephropathy
The expression of PPARβ/δ in the kidney was downregulated in type 1 diabetic Akita and OVE26 mice, which might have been associated with decreased FAO and increased renal triglyceride accumulation (Proctor et al., 2006). The expression of PPARβ/δ was found to be increased in renal medullary interstitial cells under hypertonic conditions, with the overexpression of PPARβ/δ protecting cultured medullary interstitial cells from hypertonicity-induced cell death. These results indicate that PPARβ/δ is an important survival factor for medullary interstitial cells under hypertonic conditions in the renal medulla (Han et al., 2011).
PPARβ/δ and Chronic Kidney Disease
PPARβ/δ attenuates kidney injury through inhibiting inflammatory and immune system. In a mouse model of protein-overload nephropathy, mice receiving the PPARβ agonist GW501516, developed less severe tubulointerstitial lesions, macrophage infiltration, and decreased mRNA expression of monocyte chemotactic protein (MCP-1) and TNFα. In vitro, results of the study showed that GW501516 attenuated MCP-1 expression via direct inhibition of the TGF-β activated kinase (TAK1)-NF-κB pathway, a common signaling pathway of inflammatory (Yang et al., 2011). PPARβ/δ has a pivotal role in maintain self-tolerance. PPARβ/δ-deficient female mice decreased expression of opsonins such as complement component-1qb (C1qb), which resulted impairment of apoptotic cell clearance and reduction in anti-inflammatory cytokine production. These mice were much more likely to develop autoimmune kidney disease, a lupus-like autoimmunity (Mukundan et al., 2009). Treatment of lupus mice with PPARβ/δ agonist reduced incidence of hypertension, endothelial disfunction, renal inflammation, and organ damage of mice, which was associated with decreased plasma anti-double-stranded DNA autoantibodies and anti-inflammatory, antioxidant effects (Romero et al., 2017).
PPARγ
PPARγ is constitutively expressed throughout the kidney, predominantly in the distal medullary collecting ducts, and at low levels in many other nephron segments, such as the proximal tubules and renal vasculature (Yang et al., 2012; Zhou et al., 2013). In accordance with the results of other studies, PPARγ expression has also been reported in cultured glomerular mesangial cells, podocytes, and proximal epithelial cells (Zhang and Guan, 2005). Accumulating evidence has revealed the renoprotective effects of PPARγ activation (Doi et al., 2007; Miyazaki et al., 2007; Corrales et al., 2018).
PPARγ and Acute Kidney Injury
PPARγ agonists were reported to protect the kidney against I/R injury by inhibiting I/R injury-induced diffuse tubular necrosis and acute inflammation (Reel et al., 2013), and reducing nitric oxide plasma levels, ED-1+ cell infiltration, and cleaved caspase-3 expression (Betz et al., 2012). A recent study showed that the PPARγ agonist pioglitazone decreased the expression of NF-κB-related proteins and the mRNA expression of inflammatory cytokines, including TNF-α and monocyte chemotactic protein-1 (MCP-1) in a renal I/R model (Zou et al., 2021). These data suggest that PPARγ agonists may be helpful in reducing renal I/R injury because of their anti-inflammatory, antioxidant, and anti-apoptosis effects. Moreover, PPARγ agonists have been found to increase AMP-activated protein kinase phosphorylation, inhibit p62 and cleaved caspase-3/8 protein expression, reduce cell apoptosis, and activate two autophagy-related proteins, LC3 II and Beclin-1, in the kidneys and proximal tubular cells of rats with an I/R injury (Chen et al., 2018; Xi et al., 2019). Therefore, PPARγ agonists exert renoprotective effects via the activation of autophagy. The PPARγ agonist pioglitazone protects against histological alterations in the kidney and ameliorates decreases in glutathione (GSH) and ascorbic acid levels induced by cisplatin treatment by preventing a decline in antioxidant status (Jesse et al., 2014). Pioglitazone can also decrease the expression of NF-κB p65 target genes (e.g., IL-6, IL-1β, and TNF-α) and inhibit histological injury and inflammatory cell infiltration in rats with cisplatin-induced AKI (Zhang et al., 2016). Medic et al. reported that pioglitazone reduced serum urea and creatinine levels, as well as the urinary level of kidney injury molecule-1, thus mitigating histological injury in response to gentamicin-induced kidney injury in rats (Medic et al., 2019).
Cyclooxygenase-2 (COX-2) is an inducible enzyme, which is constitutively expressed and highly regulated in response to alterations in intravascular volume (Rios et al., 2012). The protective effect of COX-2 on renal vascular function was associated with prostacyclin signaling through PPARβ/δ. PPARβ/δ activation conferred renal vasodilatory effects by regulating COX-2 and offered a potential strategy for treatment of acute renal failure (Kirkby et al., 2018). PPARα or PPARγ agonists (fenofibrate, rosiglitazone) lowered blood pressure through anti-inflammatory effect by reducing COX-2 expression in the kidney, which may be one of the indirect mechanisms of renal protection of PPARs (Bae et al., 2010; Lee et al., 2011). More researches are needed to ascertain the role of PPARs and COX-2 in renal disease.
PPARγ and Diabetic Nephropathy
PPARγ is upregulated in the presence of high glucose in HK-2 cells. Activation of PPARγ reverses G1 phase cell cycle arrest and suppresses high-glucose-induced TGF-β and MCP-1 levels in these cells (Panchapakesan et al., 2004). Therefore, PPARγ has been postulated to be involved in the pathogenesis of DN. Stimulation of PPARγ may protect against the development of DN. PPARγ agonist treatment has been reported to be effective in improving microalbuminuria, intrarenal nitric oxide bioavailability, and protecting renal function in patients with DN (Bakris et al., 2003; Grossman, 20032003; Tang et al., 2010; Pistrosch et al., 2012). In animal experiments, PPARγ agonists were shown to improve the effects of kidney injury by preventing mesangial expansion, glomerulosclerosis, tubulointerstitial inflammation and fibrosis, and tubular dilation and atrophy, with partial improvements observed after the downregulation of renal disintegrin and metalloprotease-17 as well as angiotensin-converting enzyme-2 shedding (Ohga et al., 2007; Bilan et al., 2011; Chodavarapu et al., 2013). The PPARγ agonist rosiglitazone, which induces PGC-1α expression, may ameliorate podocyte impairment, GBM thickening, and kidney fibrosis in DN (Zhang et al., 2018). The PPAR Pro12Ala gene polymorphism was shown to be significantly associated with a decreased risk of developing DN (Tönjes and Stumvoll, 2007). It has been reported that telmisartan, a weak PPARγ agonist, can slow the progression of DN (Matsui et al., 2007). The PPARα/γ dual agonist tesaglitazar not only improved lipid metabolism and increased adiponectin levels but also prevented albuminuria and renal glomerular fibrosis in diabetic mice (Yang et al., 2009). Another study reported a similar result, that a combination of low doses of the PPARα agonist fenofibrate and the PPARγ agonist rosiglitazone attenuated diabetic kidney injury to a greater extent than did either drug alone (Arora et al., 2010).
PPARγ null mice showed increased glucosuria and albuminuria in 3 weeks old. With age the mice developed renal insufficiency, advance of type 2 diabetes, and anti-phospholipid syndrome (APS), an autoimmune disorder associated with glomerular injury and microthrombi. The results reflected PPARγ activities in systemic metabolic hemostasis, and in the immune and inflammatory system (Toffoli et al., 2017).
PPARγ and Chronic Kidney Disease
There is a growing body of evidence showing that the activation of PPARγ plays a protective role in renal interstitial fibrosis disease. The expression of PPARγ is increased in glomeruli in a substantial proportion of patients with chronic kidney disease (CKD), particularly in macrophages, podocytes, and some parietal epithelial cells (Revelo et al., 2005; Paueksakon et al., 2142). PPARγ activation can delay the progression of CKD by inducing klotho restoration (Lin et al., 2017) and inhibiting Wnt signaling-mediated fibrogenesis (Maquigussa et al., 2018). PPARγ in renal tubular epithelial play an important role of maintaining the normal epithelial phenotype and opposing fibrogenesis (Zhao et al., 2016). In an open-label randomized crossover study in nondiabetic obese patients with proteinuric CKD, rosiglitazone treatment was shown to decrease proteinuria (Kincaid-Smith et al., 2008). In a randomized, double-blind, placebo-controlled study, rosiglitazone was also shown to lower the homeostasis model assessment score, an indicator of insulin sensitivity, in patients with CKD (Chan et al., 2011). Rosiglitazone attenuated the progression of hyperuricemic nephrophathy rat model through inhibiting TGF-β and NF-κB signaling, suppressing epithelial-to-mesenchymal transition (EMT), reducing inflammation, and lowered serum uric acid levels (Wang et al., 2020). The PPARγ agonist troglitazone ameliorated both glomerulosclerosis and aortic medial thickening in spontaneously hypertensive rats subjected to 5/6 nephrectomy (Yoshida et al., 2001). Troglitazone also attenuated renal interstitial fibrosis and inflammation in the model of unilateral ureteral obstruction (UUO) through reduction of TGF-β expression (Kawai et al., 2009). Another PPARγ agonists, pioglitazone, reduced renal fibrosis and its progression. It was recently demonstrated that pioglitazone in TGFβ transgenic mice inhibited the renal mRNA expression of all the profibrotic effectors, and TGFβ-STAT3 and TGFβ-EGR1 transcriptional activation pathways (Nemeth et al., 2019). Pioglitazone treatment of male Zucker diabetic fatty (ZDF) rats ameliorated diabetic kidney disease, improved renal blood flow and renal fibrosis, which was associated with lower renal expression of Twist-1, an evolutionarily conserved protein that can accelerate renal EMT and interstitial fibrosis (Wang et al., 2019). Moreover, similar results have been confirmed in vitro, with thiaziolidinediones (TZD) shown to prevent increases in TGF-β and extracellular matrix components in cultured human mesangial cells (Maeda et al., 2005) and inhibit mesangial cell and fibroblast proliferation (Nicholas et al., 2001; Zafiriou et al., 2005). Activation of PPARγ by rosiglitazone attenuated primary renal fibrolasts proliferation by suppressing AKT phosphorylation and skp2 production (Lu et al., 2016). PPARγ agonists were reported to delay the progression of polycystic kidney disease in a rat model by inhibiting cell proliferation and fibrosis (Yoshihara et al., 2011). PPARγ agonists were also found to inhibit renal interstitial macrophage infiltration, downregulate the expression of downstream target genes, and upregulate bone morphogenetic protein-7 expression, eventually blocking renal fibrosis (Lin et al., 2005).
Podocyte damage is the crucial step in the pathogenesis of CKD and progression of end stage renal disease (ESRD). Some studies have focused on the role of PPARγ activation in preventing podocyte injury. It was reported that PPARγ agonist protected podocytes in acute nephric syndrome, which was dependent partially on restoration of podocyte structure (Zuo et al., 2012). Rosiglitazone completely restored the reduced nephrin expression, and prevented MtD and oxidative stress in podocytes exposed to the mineralocorticoid aldosterone (Aldo). It was suggested that rosiglitazone might protect podocytes from injury by improving mitochondrial function (Zhu et al., 2020-31). Pioglitazone decreased puromycin aminonucleoside (PAN)-induced podocyte apoptosis and necrosis, while restoring podocyte differentiation (Kanjanabuch et al., 2007). PPARγ activation in the podocyte seems to be a key protective response after injury. PPARγ agonists might be effective in the treatment of CKD by protecting podocytes.
PPARγ activation plays a potently role in preservation of renal function of kidney allografts. Rosiglitazone has the immunosuppressive, antifibrotic, antiproliferative, anti-inflammatory actions, which are the leading causes of chronic allograft failure. It was reported that rosiglitazone treatment reduced serum creatinine, albuminuria, chronic allograft damage in the rat renal transplantation models. Meanwhile, the deposition of extracellular matrix proteins such as collagen, fibronectin, decorin was lowered (Kiss et al., 2010). Administration of rosiglitazone also reduced proteinuria and decreased interstitial collagen deposition in renal allograft transplantation model. It was suggested that rosiglitazone attenuated the development of chronic renal allograft dysfunction via inhibition of TGF-β and NF-κB pathway activation, the renal EMT, and inflammation (Deng et al., 2019).
Klotho is an anti-aging protein mainly expression in the kidney. A decreased expression of Klotho has been reported in aging and CKD. Therapeutic approaches to stimulate Klotho expression in CKD can exert vasculo-protective effects (Buchanan et al., 2020). The Klotho gene has two upstream non-canonical PPARγ binding sites. Influencing the PPARγ pathway might result in an increased renal tubular Klotho mRNA and protein expression (Zhang et al., 2008). PPARγ activation was attributed to increased renal Klotho expression and reduced oxidative stress, which effectively ameliorated the age-related nephrosclerosis in ApoE-null mice (Shen et al., 2018). Acetylation of PPARγ could prevent Klotho loss, and attenuate renal damage in CKD mouse model consequentially (Lin et al., 2017).
Conclusion
The kidney is a highly metabolic organ and consumes a large amount of energy to maintain fluid and electrolyte homeostasis. All three PPAR isotypes perform complementary physiological functions and may confer therapeutic benefits in kidney disease. The activation of PPAR isotypescan result in distinct biological processes in kidney disease. Agonists of PPARs show considerable promise for the treatment of AKI, DN, glomerulonephritis, and CKD. However, some undesirable severe side effects of PPAR agonists have been reported. For example, they can result in increased serum levels of creatinine and cystatin C, and potentially decrease the estimated glomerular filtration rate and creatinine clearance (Hiukka et al., 2010). Therefore, these agonists should be used with caution in clinical therapy for kidney diseases. Further large-scale, prospective, randomized trials are necessary to evaluate the effects of these agonists on renal outcomes in patients with kidney disease.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This research was funded by research projects from Chinese PLA Strategic Support Force Characteristic Medical Center, grant number No19ZX65.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arany, Z., He, H., Lin, J., Hoyer, K., Handschin, C., Toka, O., et al. (2005). Transcriptional Coactivator PGC-1 Alpha Controls the Energy State and Contractile Function of Cardiac Muscle. Cell Metab 1, 259–271. doi:10.1016/j.cmet.2005.03.002
Arora, M. K., Reddy, K., and Balakumar, P. (2010). The Low Dose Combination of Fenofibrate and Rosiglitazone Halts the Progression of Diabetes-Induced Experimental Nephropathy. Eur. J. Pharmacol. 636, 137–144. doi:10.1016/j.ejphar.2010.03.002
Bae, E. H., Kim, I. J., Ma, S. K., and Kim, S. W. (2010). Rosiglitazone Prevents the Progression of Renal Injury in DOCA-Salt Hypertensive Rats. Hypertens. Res. 33, 255–262. doi:10.1038/hr.2009.217
Bakris, G., Viberti, G., Weston, W. M., Heise, M., Porter, L. E., and Freed, M. I. (2003). Rosiglitazone Reduces Urinary Albumin Excretion in Type II Diabetes. J. Hum. Hypertens. 17, 7–12. doi:10.1038/sj.jhh.1001444
Baud, L., and Letavernier, E. (2007). PPARalpha Contributes to Tubular protection. J. Am. Soc. Nephrol. 18, 3017–3018. doi:10.1681/ASN.2007091036
Berger, J., and Moller, D. E. (2002). The Mechanisms of Action of PPARs. Annu. Rev. Med. 53, 409–435. doi:10.1146/annurev.med.53.082901.104018
Betz, B., Schneider, R., Kress, T., Schick, M. A., Wanner, C., and Sauvant, C. (2012). Rosiglitazone Affects Nitric Oxide Synthases and Improves Renal Outcome in a Rat Model of Severe Ischemia/reperfusion Injury. PPAR Res. 2012, 219319. doi:10.1155/2012/219319
Bilan, V. P., Salah, E. M., Bastacky, S., Jones, H. B., Mayers, R. M., Zinker, B., et al. (2011). Diabetic Nephropathy and Long-Term Treatment Effects of Rosiglitazone and Enalapril in Obese ZSF1 Rats. J. Endocrinol. 210, 293–308. doi:10.1530/JOE-11-0122
Buchanan, S., Combet, E., Stenvinkel, P., and Shiels, P. G. (2020). Klotho, Aging, and the Failing Kidney. Front. Endocrinol. (Lausanne) 11, 560. doi:10.3389/fendo.2020.00560
Chambers, J. M., and Wingert, R. A. (2020). PGC-1α in Disease: Recent Renal Insights into a Versatile Metabolic Regulator. Cells 9, 2234. doi:10.3390/cells9102234
Chan, D. T., Watts, G. F., Irish, A. B., and Dogra, G. K. (2011). Rosiglitazone Does Not Improve Vascular Function in Subjects with Chronic Kidney Disease. Nephrol. Dial. Transpl. 26, 3543–3549. doi:10.1093/ndt/gfr049
Chen, W., Xi, X., Zhang, S., Zou, C., Kuang, R., Ye, Z., et al. (2018). Pioglitazone Protects against Renal Ischemia-Reperfusion Injury via the AMP-Activated Protein Kinase-Regulated Autophagy Pathway. Front. Pharmacol. 9, 851. doi:10.3389/fphar.2018.00851
Chodavarapu, H., Grobe, N., Somineni, H. K., Salem, E. S., Madhu, M., and Elased, K. M. (2013). Rosiglitazone Treatment of Type 2 Diabetic Db/db Mice Attenuates Urinary Albumin and Angiotensin Converting Enzyme 2 Excretion. PloS one 8, e62833. doi:10.1371/journal.pone.0062833
Chung, H. W., Lim, J. H., Kim, M. Y., Shin, S. J., Chung, S., Choi, B. S., et al. (2012). High-fat Diet-Induced Renal Cell Apoptosis and Oxidative Stress in Spontaneously Hypertensive Rat Are Ameliorated by Fenofibrate through the PPARα-FoxO3a-PGC-1α Pathway. Nephrol. Dial. Transpl. 27, 2213–2225. doi:10.1093/ndt/gfr613
Chung, K. W., Lee, E. K., Lee, M. K., Oh, G. T., Yu, B. P., and Chung, H. Y. (2018). Impairment of PPARα and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 29, 1223–1237. doi:10.1681/ASN.2017070802
Collino, M., Benetti, E., Miglio, G., Castiglia, S., Rosa, A. C., Aragno, M., et al. (2011). Peroxisome Proliferator-Activated Receptor β/δ Agonism Protects the Kidney against Ischemia/reperfusion Injury in Diabetic Rats. Free Radic. Biol. Med. 50, 345–353. doi:10.1016/j.freeradbiomed.2010.10.710
Corrales, P., Izquierdo-Lahuerta, A., and Medina-Gomez, G. (2018). Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma. Int. J. Mol. Sci. 19, 63. doi:10.3390/ijms19072063
Deng, J., Xia, Y., Zhou, Q., Wang, X., Xiong, C., Shao, X., et al. (2019). Protective Effect of Rosiglitazone on Chronic Renal Allograft Dysfunction in Rats. Transpl. Immunol. 54, 20–28. doi:10.1016/j.trim.2019.01.002
Derosa, G., Sahebkar, A., and Maffioli, P. (2018). The Role of Various Peroxisome Proliferator-Activated Receptors and Their Ligands in Clinical Practice. J. Cel Physiol 233, 153–161. doi:10.1002/jcp.25804
Di Paola, R., and Cuzzocrea, S. (2007). Peroxisome Proliferator-Activated Receptors Ligands and Ischemia-Reperfusion Injury. Naunyn Schmiedebergs Arch. Pharmacol. 375, 157–175. doi:10.1007/s00210-007-0141-2
Dixon, E. D., Nardo, A. D., Claudel, T., and Trauner, M. (2021). The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes (Basel) 12, 645. doi:10.3390/genes12050645
Doi, S., Masaki, T., Arakawa, T., Takahashi, S., Kawai, T., Nakashima, A., et al. (2007). Protective Effects of Peroxisome Proliferator-Activated Receptor Gamma Ligand on Apoptosis and Hepatocyte Growth Factor Induction in Renal Ischemia-Reperfusion Injury. Transplantation 84, 207–213. doi:10.1097/01.tp.0000269614.21367.3f
Dowell, P., Ishmael, J. E., Avram, D., Peterson, V. J., Nevrivy, D. J., and Leid, M. (1999). Identification of Nuclear Receptor Corepressor as a Peroxisome Proliferator-Activated Receptor Alpha Interacting Protein. J. Biol. Chem. 274, 15901–15907. doi:10.1074/jbc.274.22.15901
Feingold, K. R., Wang, Y., Moser, A., Shigenaga, J. K., and Grunfeld, C. (2008). LPS Decreases Fatty Acid Oxidation and Nuclear Hormone Receptors in the Kidney. J. Lipid Res. 49, 2179–2187. doi:10.1194/jlr.M800233-JLR200
Feng, X., Gao, X., Wang, S., Huang, M., Sun, Z., Dong, H., et al. (2021). PPAR-alpha Agonist Fenofibrate Prevented Diabetic Nephropathy by Inhibiting M1 Macrophages via Improving Endothelial Cell Function in Db/db Mice. Front. Med. 8, 652558. doi:10.3389/fmed.2021.652558
Ginsberg, H. N., Ginsberg, H. N., Elam, M. B., Lovato, L. C., Crouse, J. R., Leiter, L. A., et al. (2010). Effects of Combination Lipid Therapy in Type 2 Diabetes Mellitus. N. Engl. J. Med. 362, 1563–1574. doi:10.1056/NEJMoa1001282
Grossman, E. (20032003). Rosiglitazone reduces blood pressure and urinary albumin excretion in type 2 diabetes: G Bakris et al. J. Hum. Hypertens. 17, 5–6. doi:10.1038/sj.jhh.1001474
Grygiel-Górniak, B. (2014). Peroxisome Proliferator-Activated Receptors and Their Ligands: Nutritional and Clinical Implications-Aa Review. Nutr. J. 13, 17. doi:10.1186/1475-2891-13-17
Guan, Y., Zhang, Y., Davis, L., and Breyer, M. D. (1997). Expression of Peroxisome Proliferator-Activated Receptors in Urinary Tract of Rabbits and Humans. Am. J. Physiol. 273, F1013–F1022. doi:10.1152/ajprenal.1997.273.6.F1013
Hajarnis, S., Lakhia, R., Yheskel, M., Williams, D., Sorourian, M., Liu, X., et al. (2017). microRNA-17 Family Promotes Polycystic Kidney Disease Progression through Modulation of Mitochondrial Metabolism. Nat. Commun. 8, 14395. doi:10.1038/ncomms14395
Han, Q., Zhang, X., Xue, R., Yang, H., Zhou, Y., Kong, X., et al. (2011). AMPK Potentiates Hypertonicity-Induced Apoptosis by Suppressing NFκB/COX-2 in Medullary Interstitial Cells. J. Am. Soc. Nephrol. 22, 1897–1911. doi:10.1681/asn.2010080822
Herman-Edelstein, M., Scherzer, P., Tobar, A., Levi, M., and Gafter, U. (2014). Altered Renal Lipid Metabolism and Renal Lipid Accumulation in Human Diabetic Nephropathy. J. Lipid Res. 55, 561–572. doi:10.1194/jlr.P040501
Hiukka, A., Maranghi, M., Matikainen, N., and Taskinen, M. R. (2010). PPARalpha: an Emerging Therapeutic Target in Diabetic Microvascular Damage. Nat. Rev. Endocrinol. 6, 454–463. doi:10.1038/nrendo.2010.89
Ismail-Beigi, F., Craven, T., Banerji, M. A., Basile, J., Calles, J., Cohen, R. M., et al. (2010). Effect of Intensive Treatment of Hyperglycaemia on Microvascular Outcomes in Type 2 Diabetes: an Analysis of the ACCORD Randomised Trial. Lancet 376, 419–430. doi:10.1016/s0140-6736(10)60576-4
Iwaki, T., Bennion, B. G., Stenson, E. K., Lynn, J. C., Otinga, C., Djukovic, D., et al. (2019). PPARα Contributes to protection against Metabolic and Inflammatory Derangements Associated with Acute Kidney Injury in Experimental Sepsis. Physiol. Rep. 7, e14078. doi:10.14814/phy2.14078
Jang, H. S., Noh, M. R., Jung, E. M., Kim, W. Y., Southekal, S., Guda, C., et al. (2020). Proximal Tubule Cyclophilin D Regulates Fatty Acid Oxidation in Cisplatin-Induced Acute Kidney Injury. Kidney Int. 97, 327–339. doi:10.1016/j.kint.2019.08.019
Jesse, C. R., Bortolatto, C. F., Wilhelm, E. A., Roman, S. S., Prigol, M., and Nogueira, C. W. (2014). The Peroxisome Proliferator-Activated Receptor-γ Agonist Pioglitazone Protects against Cisplatin-Induced Renal Damage in Mice. J. Appl. Toxicol. 34, 25–32. doi:10.1002/jat.2818
Kamijo, Y., Hora, K., Tanaka, N., Usuda, N., Kiyosawa, K., Nakajima, T., et al. (2002). Identification of Functions of Peroxisome Proliferator-Activated Receptor Alpha in Proximal Tubules. J. Am. Soc. Nephrol. 13, 1691–1702. doi:10.1097/01.asn.0000018403.61042.56
Kanjanabuch, T., Ma, L. J., Chen, J., Pozzi, A., Guan, Y., Mundel, P., et al. (2007). PPAR-gamma Agonist Protects Podocytes from Injury. Kidney Int. 71, 1232–1239. doi:10.1038/sj.ki.5002248
Kawai, T., Masaki, T., Doi, S., Arakawa, T., Yokoyama, Y., Doi, T., et al. (2009). PPAR-gamma Agonist Attenuates Renal Interstitial Fibrosis and Inflammation through Reduction of TGF-Beta. Lab. Invest. 89, 47–58. doi:10.1038/labinvest.2008.104
Kincaid-Smith, P., Fairley, K. F., Farish, S., Best, J. D., and Proietto, J. (2008). Reduction of Proteinuria by Rosiglitazone in Non-diabetic Renal Disease. Nephrology (Carlton) 13, 58–62. doi:10.1111/j.1440-1797.2007.00903.x
Kirkby, N. S., Sampaio, W., Etelvino, G., Alves, D. T., Anders, K. L., Temponi, R., et al. (2018). Cyclooxygenase-2 Selectively Controls Renal Blood Flow through a Novel PPARβ/δ-dependent Vasodilator Pathway. Hypertension 71, 297–305. doi:10.1161/HYPERTENSIONAHA.117.09906
Kiss, E., Popovic, Z. V., Bedke, J., Adams, J., Bonrouhi, M., Babelova, A., et al. (2010). Peroxisome Proliferator-Activated Receptor (PPAR)gamma Can Inhibit Chronic Renal Allograft Damage. Am. J. Pathol. 176, 2150–2162. doi:10.2353/ajpath.2010.090370
Lakhia, R., Yheskel, M., Flaten, A., Quittner-Strom, E. B., Holland, W. L., and Patel, V. (2018). PPARα Agonist Fenofibrate Enhances Fatty Acid β-oxidation and Attenuates Polycystic Kidney and Liver Disease in Mice. Am. J. Physiol. Ren. Physiol 314, F122–f31. doi:10.1152/ajprenal.00352.2017
Lee, D. L., Wilson, J. L., Duan, R., Hudson, T., and El-Marakby, A. (2011). Peroxisome Proliferator-Activated Receptor-α Activation Decreases Mean Arterial Pressure, Plasma Interleukin-6, and COX-2 while Increasing Renal CYP4A Expression in an Acute Model of DOCA-Salt Hypertension. PPAR Res. 2011, 502631. doi:10.1155/2011/502631
Letavernier, E., Perez, J., Joye, E., Bellocq, A., Fouqueray, B., Haymann, J. P., et al. (2005). Peroxisome Proliferator-Activated Receptor Beta/delta Exerts a strong protection from Ischemic Acute Renal Failure. J. Am. Soc. Nephrol. 16, 2395–2402. doi:10.1681/ASN.2004090802
Li, S., Basnakian, A., Bhatt, R., Megyesi, J., Gokden, N., Shah, S. V., et al. (2004). PPAR-alpha Ligand Ameliorates Acute Renal Failure by Reducing Cisplatin-Induced Increased Expression of Renal Endonuclease G. Am. J. Physiol. Ren. Physiol 287, F990–F998. doi:10.1152/ajprenal.00206.2004
Li, S., Gokden, N., Okusa, M. D., Bhatt, R., and Portilla, D. (2005). Anti-inflammatory Effect of Fibrate Protects from Cisplatin-Induced ARF. Am. J. Physiol. Ren. Physiol 289, F469–F480. doi:10.1152/ajprenal.00038.2005
Li, S., Mariappan, N., Megyesi, J., Shank, B., Kannan, K., Theus, S., et al. (2013). Proximal Tubule PPARα Attenuates Renal Fibrosis and Inflammation Caused by Unilateral Ureteral Obstruction. Am. J. Physiol. Ren. Physiol 305, F618–F627. doi:10.1152/ajprenal.00309.2013
Li, S., Nagothu, K. K., Desai, V., Lee, T., Branham, W., Moland, C., et al. (2009). Transgenic Expression of Proximal Tubule Peroxisome Proliferator-Activated Receptor-Alpha in Mice Confers protection during Acute Kidney Injury. Kidney Int. 76, 1049–1062. doi:10.1038/ki.2009.330
Li, S., Wu, P., Yarlagadda, P., Vadjunec, N. M., Proia, A. D., Harris, R. A., et al. (2004). PPAR Alpha Ligand Protects during Cisplatin-Induced Acute Renal Failure by Preventing Inhibition of Renal FAO and PDC Activity. Am. J. Physiol. Ren. Physiol 286, F572–F580. doi:10.1152/ajprenal.00190.2003
Lin, Q., Gu, Y., Ma, J., and Lin, S. Y. (2005). Protection of Rosiglitazone against Renal Interstitial Lesion and its Mechanism. Zhonghua Yi Xue Za Zhi 85, 1618–1624.
Lin, W., Zhang, Q., Liu, L., Yin, S., Liu, Z., and Cao, W. (2017). Klotho Restoration via Acetylation of Peroxisome Proliferation-Activated Receptor γ Reduces the Progression of Chronic Kidney Disease. Kidney Int. 92, 669–679. doi:10.1016/j.kint.2017.02.023
Lu, J., Shi, J., Gui, B., Yao, G., Wang, L., Ou, Y., et al. (2016). Activation of PPAR-γ Inhibits PDGF-Induced Proliferation of Mouse Renal Fibroblasts. Eur. J. Pharmacol. 789, 222–228. doi:10.1016/j.ejphar.2016.06.051
Maeda, A., Horikoshi, S., Gohda, T., Tsuge, T., Maeda, K., and Tomino, Y. (2005). Pioglitazone Attenuates TGF-Beta(1)-Induction of Fibronectin Synthesis and its Splicing Variant in Human Mesangial Cells via Activation of Peroxisome Proliferator-Activated Receptor (PPAR)gamma. Cell Biol Int 29, 422–428. doi:10.1016/j.cellbi.2005.01.005
Maquigussa, E., Paterno, J. C., de Oliveira Pokorny, G. H., da Silva Perez, M., Varela, V. A., da Silva Novaes, A., et al. (2018). Klotho and PPAR Gamma Activation Mediate the Renoprotective Effect of Losartan in the 5/6 Nephrectomy Model. Front. Physiol. 9, 1033. doi:10.3389/fphys.2018.01033
Matsui, T., Yamagishi, S., Ueda, S., Nakamura, K., Imaizumi, T., Takeuchi, M., et al. (2007). Telmisartan, an Angiotensin II Type 1 Receptor Blocker, Inhibits Advanced Glycation End-Product (AGE)-induced Monocyte Chemoattractant Protein-1 Expression in Mesangial Cells through Downregulation of Receptor for AGEs via Peroxisome Proliferator-Activated Receptor-Gamma Activation. J. Int. Med. Res. 35, 482–489. doi:10.1177/147323000703500407
Medic, B., Stojanovic, M., Rovcanin, B., Kekic, D., Skodric, S. R., Jovanovic, G. B., et al. (2019). Pioglitazone Attenuates Kidney Injury in an Experimental Model of Gentamicin-Induced Nephrotoxicity in Rats. Scientific Rep. 9, 13689. doi:10.1038/s41598-019-49835-1
Miyachi, H. (2021). Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 22, 9223. doi:10.3390/ijms22179223
Miyazaki, Y., Cersosimo, E., Triplitt, C., and DeFronzo, R. A. (2007). Rosiglitazone Decreases Albuminuria in Type 2 Diabetic Patients. Kidney Int. 72, 1367–1373. doi:10.1038/sj.ki.5002516
Monsalve, F. A., Pyarasani, R. D., Delgado-Lopez, F., and Moore-Carrasco, R. (2013). Peroxisome Proliferator-Activated Receptor Targets for the Treatment of Metabolic Diseases. Mediators Inflamm. 2013, 549627. doi:10.1155/2013/549627
Mukundan, L., Odegaard, J. I., Morel, C. R., Heredia, J. E., Mwangi, J. W., Ricardo-Gonzalez, R. R., et al. (2009). PPAR-delta Senses and Orchestrates Clearance of Apoptotic Cells to Promote Tolerance. Nat. Med. 15, 1266–1272. doi:10.1038/nm.2048
Nemeth, A., Mozes, M. M., Calvier, L., Hansmann, G., and Kokeny, G. (2019). The PPARgamma Agonist Pioglitazone Prevents TGF-Beta Induced Renal Fibrosis by Repressing EGR-1 and STAT3. BMC Nephrol. 20, 245. doi:10.1186/s12882-019-1431-x
Nicholas, S. B., Kawano, Y., Wakino, S., Collins, A. R., and Hsueh, W. A. (2001). Expression and Function of Peroxisome Proliferator-Activated Receptor-Gamma in Mesangial Cells. Hypertension 37, 722–727. doi:10.1161/01.hyp.37.2.722
Ohga, S., Shikata, K., Yozai, K., Okada, S., Ogawa, D., Usui, H., et al. (2007). Thiazolidinedione Ameliorates Renal Injury in Experimental Diabetic Rats through Anti-inflammatory Effects Mediated by Inhibition of NF-kappaB Activation. Am. J. Physiol. Ren. Physiol 292, F1141–F1150. doi:10.1152/ajprenal.00288.2005
Ouali, F., Djouadi, F., Merlet-Bénichou, C., and Bastin, J. (1998). Dietary Lipids Regulate Beta-Oxidation Enzyme Gene Expression in the Developing Rat Kidney. Am. J. Physiol. 275, F777–F784. doi:10.1152/ajprenal.1998.275.5.F777
Panchapakesan, U., Pollock, C. A., and Chen, X. M. (2004). The Effect of High Glucose and PPAR-Gamma Agonists on PPAR-Gamma Expression and Function in HK-2 Cells. Am. J. Physiol. Ren. Physiol 287, F528–F534. doi:10.1152/ajprenal.00445.2003
Park, C. W., Kim, H. W., Ko, S. H., Chung, H. W., Lim, S. W., Yang, C. W., et al. (2006). Accelerated Diabetic Nephropathy in Mice Lacking the Peroxisome Proliferator-Activated Receptor Alpha. Diabetes 55, 885–893. doi:10.2337/diabetes.55.04.06.db05-1329
Paueksakon, P., Revelo, M. P., Ma, L. J., Marcantoni, C., and Fogo, A. B. (2142). Microangiopathic Injury and Augmented PAI-1 in Human Diabetic Nephropathy. Kidney Int. 61, 2142–2148. doi:10.1046/j.1523-1755.2002.00384.x
Petr, M., Stastny, P., Zajac, A., Tufano, J. J., and Maciejewska-Skrendo, A. (2018). The Role of Peroxisome Proliferator-Activated Receptors and Their Transcriptional Coactivators Gene Variations in Human Trainability: A Systematic Review. Int. J. Mol. Sci. 19, 1472. doi:10.3390/ijms19051472
Pistrosch, F., Passauer, J., Herbrig, K., Schwanebeck, U., Gross, P., and Bornstein, S. R. (2012). Effect of Thiazolidinedione Treatment on Proteinuria and Renal Hemodynamic in Type 2 Diabetic Patients with Overt Nephropathy. Horm. Metab. Res. 44, 914–918. doi:10.1055/s-0032-1314836
Portilla, D., Dai, G., McClure, T., Bates, L., Kurten, R., Megyesi, J., et al. (2002). Alterations of PPARalpha and its Coactivator PGC-1 in Cisplatin-Induced Acute Renal Failure. Kidney Int. 62, 1208–1218. doi:10.1111/j.1523-1755.2002.kid553.x
Portilla, D., Dai, G., Peters, J. M., Gonzalez, F. J., Crew, M. D., and Proia, A. D. (2000). Etomoxir-induced PPARalpha-Modulated Enzymes Protect during Acute Renal Failure. Am. J. Physiol. Ren. Physiol 278, F667–F675. doi:10.1152/ajprenal.2000.278.4.F667
Portilla, D. (2003). Energy Metabolism and Cytotoxicity. Semin. Nephrol. 23, 432–438. doi:10.1016/s0270-9295(03)00088-3
Proctor, G., Jiang, T., Iwahashi, M., Wang, Z., Li, J., and Levi, M. (2006). Regulation of Renal Fatty Acid and Cholesterol Metabolism, Inflammation, and Fibrosis in Akita and OVE26 Mice with Type 1 Diabetes. Diabetes 55, 2502–2509. doi:10.2337/db05-0603
Qi, C., Zhu, Y., and Reddy, J. K. (2000). Peroxisome Proliferator-Activated Receptors, Coactivators, and Downstream Targets. Cell Biochem Biophys 32 Spring, 187–204. doi:10.1385/cbb:32:1-3:187
Reel, B., Guzeloglu, M., Bagriyanik, A., Atmaca, S., Aykut, K., Albayrak, G., et al. (2013). The Effects of PPAR-γ Agonist Pioglitazone on Renal Ischemia/reperfusion Injury in Rats. J. Surg. Res. 182, 176–184. doi:10.1016/j.jss.2012.08.020
Revelo, M. P., Federspiel, C., Helderman, H., and Fogo, A. B. (2005). Chronic Allograft Nephropathy: Expression and Localization of PAI-1 and PPAR-Gamma. Nephrol. Dial. Transpl. 20, 2812–2819. doi:10.1093/ndt/gfi172
Ricote, M., Valledor, A. F., and Glass, C. K. (2004). Decoding Transcriptional Programs Regulated by PPARs and LXRs in the Macrophage: Effects on Lipid Homeostasis, Inflammation, and Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 24, 230–239. doi:10.1161/01.ATV.0000103951.67680.B1
Rios, A., Vargas-Robles, H., Gámez-Méndez, A. M., and Escalante, B. (2012). Cyclooxygenase-2 and Kidney Failure. Prostaglandins Other Lipid Mediat 98, 86–90. doi:10.1016/j.prostaglandins.2011.11.004
Romero, M., Toral, M., Robles-Vera, I., Sánchez, M., Jiménez, R., O'Valle, F., et al. (2017). Activation of Peroxisome Proliferator Activator Receptor β/δ Improves Endothelial Dysfunction and Protects Kidney in Murine Lupus. Hypertension 69, 641–650. doi:10.1161/HYPERTENSIONAHA.116.08655
Ruan, X., Zheng, F., and Guan, Y. (2008). PPARs and the Kidney in Metabolic Syndrome. Am. J. Physiol. Ren. Physiol 294, F1032–F1047. doi:10.1152/ajprenal.00152.2007
Saga, D., Sakatsume, M., Ogawa, A., Tsubata, Y., Kaneko, Y., Kuroda, T., et al. (2005). Bezafibrate Suppresses Rat Antiglomerular Basement Membrane Crescentic Glomerulonephritis. Kidney Int. 67, 1821–1829. doi:10.1111/j.1523-1755.2005.00280.x
Shen, D., Li, H., Zhou, R., Liu, M. J., Yu, H., and Wu, D. F. (2018). Pioglitazone Attenuates Aging-Related Disorders in Aged Apolipoprotein E Deficient Mice. Exp. Gerontol. 102, 101–108. doi:10.1016/j.exger.2017.12.002
Smulders, Y. M., van Eeden, A. E., Stehouwer, C. D., Weijers, R. N., Slaats, E. H., and Silberbusch, J. (1997). Can Reduction in Hypertriglyceridaemia Slow Progression of Microalbuminuria in Patients with Non-insulin-dependent Diabetes Mellitus? Eur. J. Clin. Invest. 27, 997–1002. doi:10.1046/j.1365-2362.1997.2330779.x
Tanaka, Y., Kume, S., Araki, S., Isshiki, K., Chin-Kanasaki, M., Sakaguchi, M., et al. (2011). Fenofibrate, a PPARα Agonist, Has Renoprotective Effects in Mice by Enhancing Renal Lipolysis. Kidney Int. 79, 871–882. doi:10.1038/ki.2010.530
Tang, S. C., Leung, J. C., Chan, L. Y., Cheng, A. S., Lan, H. Y., and Lai, K. N. (2010). Renoprotection by Rosiglitazone in Accelerated Type 2 Diabetic Nephropathy: Role of STAT1 Inhibition and Nephrin Restoration. Am. J. Nephrol. 32, 145–155. doi:10.1159/000316056
Toffoli, B., Gilardi, F., Winkler, C., Soderberg, M., Kowalczuk, L., Arsenijevic, Y., et al. (2017). Nephropathy in Pparg-Null Mice Highlights PPARgamma Systemic Activities in Metabolism and in the Immune System. PloS one 12, e0171474. doi:10.1371/journal.pone.0171474
Tönjes, A., and Stumvoll, M. (2007). The Role of the Pro12Ala Polymorphism in Peroxisome Proliferator-Activated Receptor Gamma in Diabetes Risk. Curr. Opin. Clin. Nutr. Metab. Care 10, 410–414. doi:10.1097/MCO.0b013e3281e389d9
Tovar-Palacio, C., Torres, N., Diaz-Villaseñor, A., and Tovar, A. R. (2012). The Role of Nuclear Receptors in the Kidney in Obesity and Metabolic Syndrome. Genes Nutr. 7, 483–498. doi:10.1007/s12263-012-0295-5
Tran, M. T., Zsengeller, Z. K., Berg, A. H., Khankin, E. V., Bhasin, M. K., Kim, W., et al. (2016). PGC1α Drives NAD Biosynthesis Linking Oxidative Metabolism to Renal protection. Nature 531, 528–532. doi:10.1038/nature17184
Tyagi, S., Gupta, P., Saini, A. S., Kaushal, C., and Sharma, S. (2011). The Peroxisome Proliferator-Activated Receptor: A Family of Nuclear Receptors Role in Various Diseases. J. Adv. Pharm. Technol. Res. 2, 236–240. doi:10.4103/2231-4040.90879
Wang, X., Deng, J., Xiong, C., Chen, H., Zhou, Q., Xia, Y., et al. (2020). Treatment with a PPAR-γ Agonist Protects against Hyperuricemic Nephropathy in a Rat Model. Drug Des. Devel Ther. 14, 2221–2233. doi:10.2147/DDDT.S247091
Wang, Z., Liu, Q., Dai, W., Hua, B., Li, H., and Li, W. (2019). Pioglitazone Downregulates Twist-1 Expression in the Kidney and Protects Renal Function of Zucker Diabetic Fatty Rats. Biomed. Pharmacother. 118, 109346. doi:10.1016/j.biopha.2019.109346
Wilmer, W. A., Dixon, C. L., Hebert, C., Lu, L., and Rovin, B. H. (2002). PPAR-alpha Ligands Inhibit H2O2-Mediated Activation of Transforming Growth Factor-Beta1 in Human Mesangial Cells. Antioxid. Redox Signal. 4, 877–884. doi:10.1089/152308602762197416
Wong, H. R., Cvijanovich, N., Allen, G. L., Lin, R., Anas, N., Meyer, K., et al. (2009). Genomic Expression Profiling across the Pediatric Systemic Inflammatory Response Syndrome, Sepsis, and Septic Shock Spectrum. Crit. Care Med. 37, 1558–1566. doi:10.1097/CCM.0b013e31819fcc08
Wu, J., Chen, L., Zhang, D., Huo, M., Zhang, X., Pu, D., et al. (2009). Peroxisome Proliferator-Activated Receptors and Renal Diseases. Front. Biosci. (Landmark Ed. 14, 995–1009. doi:10.2741/3291
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 98, 115–124. doi:10.1016/S0092-8674(00)80611-X
Xi, X., Zou, C., Ye, Z., Huang, Y., Chen, T., and Hu, H. (2019). Pioglitazone Protects Tubular Cells against Hypoxia/reoxygenation Injury through Enhancing Autophagy via AMPK-mTOR Signaling Pathway. Eur. J. Pharmacol. 863, 172695. doi:10.1016/j.ejphar.2019.172695
Xi, Y., Zhang, Y., Zhu, S., Luo, Y., Xu, P., and Huang, Z. (2020). PPAR-mediated Toxicology and Applied Pharmacology. Cells 9, 352. doi:10.3390/cells9020352
Yang, J., Zhang, D., Li, J., Zhang, X., Fan, F., and Guan, Y. (2009). Role of PPARgamma in Renoprotection in Type 2 Diabetes: Molecular Mechanisms and Therapeutic Potential. Clin. Sci. (Lond) 116, 17–26. doi:10.1042/cs20070462
Yang, J., Zhou, Y., and Guan, Y. (2012). PPARγ as a Therapeutic Target in Diabetic Nephropathy and Other Renal Diseases. Curr. Opin. Nephrol. Hypertens. 21, 97–105. doi:10.1097/MNH.0b013e32834de526
Yang, X., Kume, S., Tanaka, Y., Isshiki, K., Araki, S., Chin-Kanasaki, M., et al. (2011). GW501516, a PPARδ Agonist, Ameliorates Tubulointerstitial Inflammation in Proteinuric Kidney Disease via Inhibition of TAK1-Nfκb Pathway in Mice. PloS one 6, e25271. doi:10.1371/journal.pone.0025271
Yaribeygi, H., Mohammadi, M. T., Rezaee, R., and Sahebkar, A. (2018). Fenofibrate Improves Renal Function by Amelioration of NOX-4, IL-18, and P53 Expression in an Experimental Model of Diabetic Nephropathy. J. Cel Biochem 119, 7458–7469. doi:10.1002/jcb.27055
Yoshida, K., Kohzuki, M., Xu, H. L., Wu, X. M., Kamimoto, M., and Sato, T. (2001). Effects of Troglitazone and Temocapril in Spontaneously Hypertensive Rats with Chronic Renal Failure. J. Hypertens. 19, 503–510. doi:10.1097/00004872-200103000-00019
Yoshihara, D., Kurahashi, H., Morita, M., Kugita, M., Hiki, Y., Aukema, H. M., et al. (2011). PPAR-gamma Agonist Ameliorates Kidney and Liver Disease in an Orthologous Rat Model of Human Autosomal Recessive Polycystic Kidney Disease. Am. J. Physiol. Ren. Physiol 300, F465–F474. doi:10.1152/ajprenal.00460.2010
Zafiriou, S., Stanners, S. R., Saad, S., Polhill, T. S., Poronnik, P., and Pollock, C. A. (2005). Pioglitazone Inhibits Cell Growth and Reduces Matrix Production in Human Kidney Fibroblasts. J. Am. Soc. Nephrol. 16, 638–645. doi:10.1681/asn.2004040278
Zhang, H., Li, Y., Fan, Y., Wu, J., Zhao, B., Guan, Y., et al. (2008). Klotho Is a Target Gene of PPAR-Gamma. Kidney Int. 74, 732–739. doi:10.1038/ki.2008.244
Zhang, J., Zhang, Y., Xiao, F., Liu, Y., Wang, J., Gao, H., et al. (2016). The Peroxisome Proliferator-Activated Receptor γ Agonist Pioglitazone Prevents NF-Κb Activation in Cisplatin Nephrotoxicity through the Reduction of P65 Acetylation via the AMPK-SIRT1/p300 Pathway. Biochem. Pharmacol. 101, 100–111. doi:10.1016/j.bcp.2015.11.027
Zhang, L., Liu, J., Zhou, F., Wang, W., and Chen, N. (2018). PGC-1α Ameliorates Kidney Fibrosis in Mice with Diabetic Kidney Disease through an Antioxidative Mechanism. Mol. Med. Rep. 17, 4490–4498. doi:10.3892/mmr.2018.8433
Zhang, Y., and Guan, Y. (2005). PPAR-gamma Agonists and Diabetic Nephropathy. Curr. Diab Rep. 5, 470–475. doi:10.1007/s11892-005-0057-5
Zhao, M., Chen, Y., Ding, G., Xu, Y., Bai, M., Zhang, Y., et al. (2016). Renal Tubular Epithelium-Targeted Peroxisome Proliferator-Activated Receptor-γ Maintains the Epithelial Phenotype and Antagonizes Renal Fibrogenesis. Oncotarget 7, 64690–64701. doi:10.18632/oncotarget.11811
Zhou, T. B., Drummen, G. P., Jiang, Z. P., Long, Y. B., and Qin, Y. H. (2013). Association of Peroxisome Proliferator-Activated Receptors/retinoic Acid Receptors with Renal Diseases. J. Recept Signal. Transduct Res. 33, 349–352. doi:10.3109/10799893.2013.838786
Zhu, C., Huang, S., Yuan, Y., Ding, G., Chen, R., Liu, B., et al. (2020). Mitochondrial Dysfunction Mediates Aldosterone-Induced Podocyte Damage: a Therapeutic Target of PPARγ. Am. J. Pathol. 178, 2020–2031. doi:10.1016/j.ajpath.2011.01.029
Zou, G., Zhou, Z., Xi, X., Huang, R., and Hu, H. (2021). Pioglitazone Ameliorates Renal Ischemia-Reperfusion Injury via Inhibition of NF-Κb Activation and Inflammation in Rats. Front. Physiol. 12, 707344. doi:10.3389/fphys.2021.707344
Zuo, N., Zheng, X., Liu, H., and Ma, X. (2015). Fenofibrate, a PPARα Agonist, Protect Proximal Tubular Cells from Albumin-Bound Fatty Acids Induced Apoptosis via the Activation of NF-kB. Int. J. Clin. Exp. Pathol. 8, 10653–10661. doi:10.1124/jpet.109.151225
Keywords: peroxisome proliferator-activated receptors, kidney disease, acute kidney injury, diabetic nephropathy, chronic kidney disease
Citation: Gao J and Gu Z (2022) The Role of Peroxisome Proliferator-Activated Receptors in Kidney Diseases. Front. Pharmacol. 13:832732. doi: 10.3389/fphar.2022.832732
Received: 14 January 2022; Accepted: 14 February 2022;
Published: 04 March 2022.
Edited by:
David E. Stec, University of Mississippi Medical Center, United StatesReviewed by:
Walter Wahli, University of Lausanne, SwitzerlandGabor Kokeny, Semmelweis University, Hungary
Copyright © 2022 Gao and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaoyan Gu, Z3V6aGFveWFuMzAxQDE2My5jb20=