 Nick Evans*
Nick Evans* Ruslan Grygorash
Ruslan Grygorash Paul WilliamsAndrew KyleTerrence KantnerRavindra PathakXiaoBo ShengFabio SimoesHiteshri MakwanaRicardo ResendeElena de JuanAlan JenkinsDavid MorrisAurelie MicheletFrances JewittFelicity Rudge
Paul WilliamsAndrew KyleTerrence KantnerRavindra PathakXiaoBo ShengFabio SimoesHiteshri MakwanaRicardo ResendeElena de JuanAlan JenkinsDavid MorrisAurelie MicheletFrances JewittFelicity Rudge Nicolas CamperAnaïs ManinWilliam McDowell
Nicolas CamperAnaïs ManinWilliam McDowell Martin PabstAntony GodwinMark FrigerioMatthew Bird
Martin PabstAntony GodwinMark FrigerioMatthew Bird- Abzena Ltd., Babraham Research Campus, Babraham, United Kingdom
Antibody-drug conjugates (ADCs) have begun to fulfil their promise as targeted cancer therapeutics with ten clinical approvals to date. As the field matures, much attention has focused upon the key factors required to produce safe and efficacious ADCs. Recently the role that linker-payload reagent design has on the properties of ADCs has been highlighted as an important consideration for developers. We have investigated the effect of incorporating hydrophilic macrocycles into reagent structures on the in vitro and in vivo behavior of ADCs. Bis-sulfone based disulfide rebridging reagents bearing Val-Cit-PABC-MMAE linker-payloads were synthesized with a panel of cyclodextrins and crown ethers integrated into their structures via a glutamic acid branching point. Brentuximab was selected as a model antibody and ten ADCs with a drug-to-antibody ratio (DAR) of 4 were prepared for biological evaluation. In vitro, the ADCs prepared showed broadly similar potency (range: 16–34 pM) and were comparable to Adcetris® (16 pM). In vivo, the cyclodextrin containing ADCs showed greater efficacy than Adcetris® and the most efficacious variant (incorporating a 3′-amino-α-cyclodextrin component) matched a 24-unit poly(ethylene glycol) (PEG) containing comparator. The ADCs bearing crown ethers also displayed enhanced in vivo efficacy compared to Adcetris®, the most active variant (containing a 1-aza-42-crown-14 macrocycle) was superior to an analogous ADC with a larger 24-unit PEG chain. In summary, we have demonstrated that hydrophilic macrocycles can be effectively incorporated into ADC reagent design and offer the potential for enhanced alternatives to established drug-linker architectures.
Introduction
The concept of antibody-drug conjugates (ADCs) as targeted cancer therapeutics combines the unique targeting properties of a monoclonal antibody to deliver a high potency of a cytotoxin attached via an appropriate linker. As the number of ADCs entering the clinic and achieving regulatory approval continues to grow, there remains a concerted research effort to explore alternative drug-linker designs with the aim of enhancing ADC efficacy and safety. A wide array of structural options for the linker component connecting the antibody to the cytotoxic drug exist. Recent attention has focused on the use of site-specific conjugating linkers to improve ADC homogeneity and a variety of both cleavable and non-cleavable linkers have been employed to maximize toxin delivery to tumor cells (Frigerio and Kyle, 2017; Zhou, 2017; Dal Corso et al., 2019).
Recent research has focused on the inclusion of polymeric portions within ADC linker structure to overcome some of the inherent issues surrounding ADC design such as hydrophobicity, aggregation, instability, insufficient drug-loading and reduced circulatory half-life. Typically, the polymers have been amphiphilic in nature, being water-soluble, synthetic and substantially non-antigenic with particular focus on polyalkylene oxides such as poly(ethylene glycol) (PEG). Since the first PEGylated protein was approved in 1990, PEG has become one of the most widely used polymers in biopharmaceutical applications with numerous products entering clinical trials and receiving marketing approval (Veronese and Mero, 2008; Turecek et al., 2016). PEG features within two of the currently approved ADCs, sacituzumab govitecan (Trodelvy®) and loncastuximab tesirine (Zynlonta®), and its utility within ADC reagents has been widely investigated, mainly to improve reagent solubility and to reduce aggregation of the final ADC. The correlation between ADC hydrophobicity and accelerated plasma clearance, particularly for conjugates with high drug-loading, has highlighted the value of incorporating a hydrophilic polymer moiety to mitigate the hydrophobic effects of drug-linkers (Hamblett et al., 2004; Lyon et al., 2015). Recent studies have shown that reagent design, and particularly the positioning of the PEG within the ADC architecture, is an essential consideration when developing the optimal ADC. Some groups describe the use of linear PEG spacers which separates the drug moiety from the antibody (Tiberghien et al., 2016; Bryden et al., 2018; Yang et al., 2019). Others have highlighted an improved reagent format whereby the PEG exists as a pendant molecule which is attached to the reagent via a branching linker (Zhao et al., 2008; Lyon et al., 2015; Burke et al., 2017; Shao et al., 2018; Shao et al., 2020). Our approach has been to use glutamic acid as a branching point to suitably position a PEG chain and a cleavable linker-payload and these reagent structures have been shown to produce efficacious DAR 4, 6 and 8 ADCs (Pabst et al., 2017). ADCs with branched formats have shown improvements in pharmacokinetic (PK) behavior and enhanced in vivo efficacy when compared to ADCs where PEG exists as a spacer segment between the antibody and drug molecules (Lyon et al., 2015; Burke et al., 2017; Pabst et al., 2017). A common feature with these pendant structures is the polymer tethered to the linker has a single point of attachment leaving a free end group in solution. The unattached end of the polymer chain in PEG is typically a methoxy group. It has been suggested that when PEG is arranged in a pendant configuration, it allows for more effective masking of the hydrophobic drug-linker portion of the ADC (Lyon et al., 2015).
Alternative polymers to PEG have also been integrated into ADC reagent design. Submonomer solid phase synthesis has allowed the production of monodisperse polysarcosine (PSAR) oligomers containing 6–24 repeat units (Viricel et al., 2019). Trastuzumab-glucuronide-MMAE based ADCs containing PSAR in branched (n = 6, 12, 18 and 24 units) and spacer (n = 12 units) formats were compared in vivo alongside structurally analogous ADCs with either no polymer or PEG (n = 12 units) in the branched format. ADCs containing the branched reagent structure had improved PK and efficacy compared to the ADCs with no polymer and where the spacer configuration was employed. The branched PSAR(12u) ADC was also found to be more efficacious than a PEG(12u) containing variant. Polyacetal polymers, as part of the Fleximer® technology platform, have also found utility in the development of ADCs with significantly higher drug-to-antibody ratios (DAR ≥20) compared to more common conjugation approaches, which typically produce DARs between 2 to 4 (Yurkovestskiy et al., 2015). Other polymeric reagent architectures have been reported (Zhang et al., 2017; Marcinkowska et al., 2019; Shi et al., 2020), however PEG, with its long-established history and application in the enhancement of biopharmaceuticals, remains at the forefront in the design of more efficacious ADCs. In contrast to the active research interest in the use of polymers in ADC reagents, there are no reports of ADC reagents incorporating pendant macrocyclic moieties where both ends of the polymer chain are tethered to the reagent linker and the pharmacological impact of introducing such a structural constraint.
Macrocyclic molecules, comprising twelve or more atoms in a ring structure, are long established in medicine as drug molecules and drug delivery vehicles. Cyclodextrins, macrocycles constructed of cyclic arrangements of glucopyranose units, have been extensively investigated in drug delivery applications (Challa et al., 2005; Loftsson et al., 2005; Jansook et al., 2018; Kim et al., 2020). Three naturally occurring cyclodextrins (α-, β- and γ-), containing 6, 7 or 8 glucopyranose repeats connected in cycles via α-1,4-glycosidic linkages respectively, are known. As drug carriers, cyclodextrins have been applied to a wide range of biologics including genes, peptides, proteins and oligonucleotides (Irie and Uekama, 1999; Redenti et al., 2001; Ortiz Mellet et al., 2011; Haley et al., 2020). Small molecules, including cytotoxic agents, however, have been the predominant research focus, with over 35 cyclodextrin containing pharmaceutical products gaining market approval (Loftsson and Brewster, 2010; Kurkov and Loftsson, 2013; Gidwani and Vyas, 2015). Crown ethers, cycles consisting of oxyethylene repeat units, are macrocyclic analogues of PEG which have been shown to have a wide variety of activities, ranging from ion complexation and micelle formation (Gokel et al., 1988) to antimicrobial activity (Febles et al., 2016). Substantial research demonstrating the drug delivery potential of crown ethers has also been reported (Jansen et al., 2002; Muzzalupo et al., 2007; Chehardoli and Bahmani, 2019).
With cyclodextrins and crown ethers established in (bio)pharmaceutical applications, the aim of this study was to determine whether these macrocycles may be used as hydrophilic substitutes for the polymeric element present within ADC reagent structures to generate an efficacious drug molecule. In the present study, we demonstrate the effect that integrating such hydrophilic macrocycles into drug-linker reagent structures has on the in vitro and in vivo properties of a series of homogeneous DAR 4 ADCs. The ADCs investigated incorporate the anti-CD30 antibody brentuximab, with the prodrug Val-Cit-PABC-MMAE and either cyclodextrin or crown ether macrocycles linked via a glutamic acid branching point. Conjugation to the antibody was achieved using an established bis-sulfone based disulfide rebridging linker, previously shown to produce stable and homogeneous DAR 4 ADCs (Badescu et al., 2014; Bryant et al., 2015). Analogous ADCs with linear PEGs were prepared as comparators and the biological performance of the conjugates produced were benchmarked against the clinically approved ADC, brentuximab vedotin (Adcetris®).
Materials and Methods
ADC Reagent Synthesis
Reagents 1a-f, 2a-b and 3a-b were prepared using published synthetic methods (Godwin, 2017; Godwin et al., 2017). Preparative silica gel and C18 reverse phase chromatography purifications were conducted on a Reveleris® X2 Flash Chromatography System.
ADC Production
Brentuximab-MMAE ADCs were prepared as previously described (Godwin, 2017; Godwin et al., 2017). Briefly, brentuximab (in 20 mM sodium phosphate, pH 7.5, 150 mM NaCl, 20 mM EDTA) was reduced at 5 mg/ml antibody concentration with TCEP (1.5 equivalents per disulfide) for 1 h at 40°C. Reduced antibody solutions were cooled to 22°C and diluted to 4.21 mg/ml with 20 mM sodium phosphate, pH 7.5, 150 mM NaCl, 20 mM EDTA. Conjugation reagents (1a-f, 2a-b and 3a-b) in MeCN were added (5% v/v MeCN, 1.4 equivalents per disulfide) to the reduced antibody. Conjugation reactions were incubated for16-20 h and subsequently quenched by the addition of 50 mM N-acetyl-l-cysteine (3 mM final concentration). The crude reactions were then purified by hydrophobic interaction chromatography (HIC) using ToyoPearl Phenyl-650S columns, as described previously (Bird et al., 2020). Fractions containing DAR 4 ADC were pooled and buffer exchanged into PBS. Antibody concentration was determined by Bradford assay (Expedeon). DAR 4 was determined by HIC-HPLC, a more quantitative characterisation than SDS-PAGE or UV-vis (see Bird et al., 2020)
In vitro Cytotoxicity Assay
In vitro potency was evaluated in Karpas-229 cells using a CellTiter-Glo® luminescence assay (Promega). Karpas-299 cells were cultured in RPMI-1640 medium (Life Technologies®) supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/ml streptomycin. Karpas-299 cells were counted using disposable Neubauer counting chambers and cell density was adjusted to 0.25 × 104 cells per well (50 μL/well). Cells were incubated for 24 h at 37°C with 5% CO2. The cells were treated by addition of ADCs 4a-f, 5a-b and 6a-b, Adcetris® (Takeda) or MMAE (Concortis) dilution series (50 µL/well) and were then incubated at 37°C/5% CO2 for a further 96 h. Cell viability assays were carried out using CellTiter-Glo® Luminescent reagent as per the manufacturer’s instructions. Briefly, at the end of the 96 h incubation period, an equal volume of CellTiter-Glo® reagent is added to each well and incubated at room temperature. The CellTiter-Glo® reagent produces a luminescent signal which is proportional to the number of viable cells within the well. Luminescence was recorded using a Molecular Devices SpectramaxM3 plate reader and data were processed with GraphPad Prism using a four-parameter non-linear regression.
In vivo Xenograft Studies
In vivo experiments were approved by the Animal Care and Use Committee of Oncodesign (OnComEt). Two Karpas-299 (T-anaplastic large cell lymphoma, ALCL) xenograft studies were performed in healthy female CB17-SCID mice (CBySmn.CB17-Prkdcscid/J, Charles River Laboratories) testing cyclodextrin ADCs (xenograft study 1) and crown ether ADCs (xenograft study 2) and relevant controls. The animals were maintained in SPF health status according to the FELASA guidelines in housing rooms under controlled environmental conditions. The mice were γ-irradiated (1.44 Gy, 60Co) 24–72 h prior to tumor cell injection. Tumors were induced by subcutaneous injection of 107 Karpas-299 cells in 200 µL of RPMI 1640 into the right flank. Tumors were measured in two-dimensions (width (w) and length (L)) twice per week with calipers, and the volume was estimated using the formula:
Treatment tolerability was assessed by bi-weekly body weight measurement and daily observation for clinical signs of treatment-related side effects. Mice were euthanized when a humane endpoint was reached (1,600 mm3 tumor volume) or after a maximum of 71 days post-tumor induction.
Xenograft Study 1 (Cyclodextrin ADCs)
SCID mice with an average body weight of 17.5 g were used for cell inoculation (Day 0). Fourteen days after tumor implantation (Day 14), animals were randomized into groups of eight mice (200 mm3 mean tumor volume) and treatment was initiated. The animals from the vehicle group received a single intravenous (i.v.) injection of PBS. The treated groups were dosed with a single i.v. injection of ADCs 4d, 4b, 4f, 6b and Adcetris® at 0.5 mg/kg or 1 mg/kg.
Xenograft Study 2 (Crown Ether ADCs)
SCID mice with an average body weight of 18.1 g were used for cell inoculation (Day 0). Twelve days after tumor implantation (Day 12), animals were randomized into groups of eight mice (211 mm3 mean tumor volume) and treatment was initiated. The animals from the vehicle group received a single i.v. injection of PBS. The treated groups were dosed with a single i.v. injection of ADCs 5a, 5b, 6a and 6b at 0.4 mg/kg or 0.8 mg/kg, or Adcetris® at 1 mg/kg.
Results
Linker-Payload and ADC Synthesis
Ten reagents with generic structures consisting of a bis-sulfone conjugating linker and a glutamic acid branching point, with the prodrug Val-Cit-PABC-MMAE and either a macrocycle or linear PEG chain appended were designed and synthesized (Figure 1). Six of the reagents (1a-f) contained α-, β- or γ-cyclodextrins and were linked to the reagent via 3′- or 6′-amino modifications on the macrocycles. Two 1-aza-crown ethers were prepared via macrocyclization of ethylene glycol derivatives to prepare 1-aza-24-crown-8 and 1-aza-42-crown-14. These 1-aza-crown ethers were successfully coupled to the branched template structure to yield reagents 2a-b. As comparators, two structurally analogous reagents containing discrete PEGs containing 8 and 24 repeat units were also prepared. The 24-unit reagent (3b), included in this study as a reference, is a known reagent architecture and was previously conjugated to brentuximab variants to produce highly efficacious DAR 4 and DAR 6 ADCs (Pabst et al., 2017). Reagent 3a, containing an 8-unit PEG side-chain, was synthesized to serve as a direct comparator to 24-crown-8 reagent 2a.
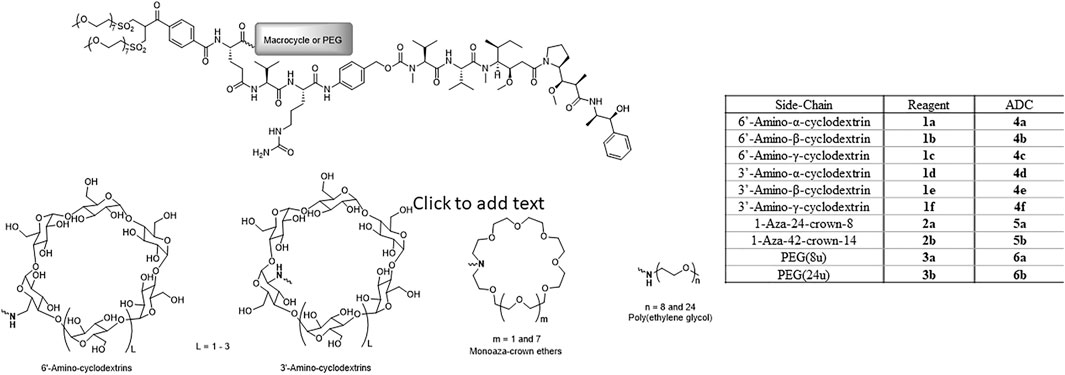
FIGURE 1. Structures of disulfide re-bridging reagents comprising glutamic acid-based branching points with linker-payload (Val-Cit-PAB-MMAE) and macrocycle (cyclodextrin or crown ether) or PEG appended.
Ten brentuximab ADCs with DAR 4 were prepared via an established disulfide re-bridging conjugation method (Bird et al., 2020) using cyclodextrin reagents 1a-f, crown ether reagents 2a-b and PEGylated reagents 3a-b. Each crude conjugation reaction was purified by preparative HIC to isolate the DAR 4 ADC for biological evaluation.
In vitro Cytotoxicity Assay
Prior to in vivo evaluation, the in vitro activity of each ADC was confirmed within a cell viability assay using CD30+ Karpas-299 cells. IC50 values obtained for each ADC are given in Table 1 (representative cell-viability assay curves are given in Supplementary Figure S1). All of the ADCs displayed sub-nanomolar potency (10–11 M range) similar to Adcetris® (16 pM). In all cases, the ADCs displayed greater potency than the payload, MMAE.
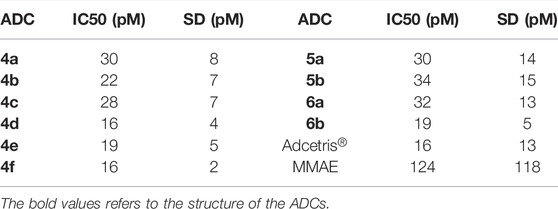
TABLE 1. In vitro cell viability assay results for ADCs 4a-f, 5a-b, 6a-b, Adcertis® and MMAE in Karpas-299 cells. Average IC50 (mean) and standard deviations (SD) were determined from n ≥ 2 experiments.
In vivo Xenograft Study 1
Following the similarities in potencies obtained from the in vitro cell viability assays, in vivo xenograft studies were performed to differentiate between the activities of these conjugates further. Within this first study, cyclodextrin-containing conjugates 4b, 4d, 4f dosed at 0.5 or 1 mg/kg were compared with Adcetris® dosed at 1 mg/kg, or conjugate 6b dosed at 0.5 or 1 mg/kg. Animals were dosed on day 14 following tumor implantation when the tumor volume was approximately 200 mm3.
Figure 2 shows in vivo xenograft mean (+/- SEM) and relative tumor volumes measured over time within mice dosed with either ADC 4b, 4d, 4f and Adcetris® at 0.5 or 1 mg/kg. At 0.5 mg/kg dose, the conjugates reduce tumor growth relative to the vehicle control, but do not prevent tumor growth, as shown by the increasing tumor volumes throughout the duration of the study. ADCs 4d and 4f appear to slow tumor growth to a greater extent than ADC 4b and the positive control comparator, Adcetris®, as shown more clearly by the waterfall plots. At 1 mg/kg dose, the effect of these ADCs upon tumor growth is much more striking, with conjugate 4d showing a complete reduction of tumor volume down to 0 mm3 following dosing on day 14. This complete reduction of tumor growth was maintained throughout the duration of the study until Day 71. The inhibitory effects of conjugates 4b and 4f upon tumor growth were less pronounced than ADC 4d, but nonetheless show a significant reduction in tumor growth throughout the duration of the study, displaying lower mean tumor volume values than the clinical comparator Adcetris® by the end of the study at day 71. The relative tumor volume (%) plots also show that 0 mm3 tumor volume (i.e. complete tumor regression) was achieved within 6 out of 8 animals for conjugates 4b and 4f, whereas only 2 out 8 animals measured 0 mm3 for the Adcetris® treated group. In comparison, for conjugate 4d, complete tumor regression was achieved within 8 out of 8 animals, (0 mm3 final tumor volume), within the 1 mg/kg group and even 4 out of 8 animals achieved this for the 0.5 mg/kg group. Similar data were achieved with conjugate 6b, which showed 1 less animal surviving to the end of the study compared with 4d. These data show that conjugates 4d and 6b have comparable in vivo efficacies. All of these ADCs show greater in vivo efficacy over the clinical comparator Adcetris® within this tumor model. In addition to the tumor volume analyses, body weight measurements taken from each group show that all ADCs were well tolerated in comparison to the vehicle treated group (Supplementary Figure S2).
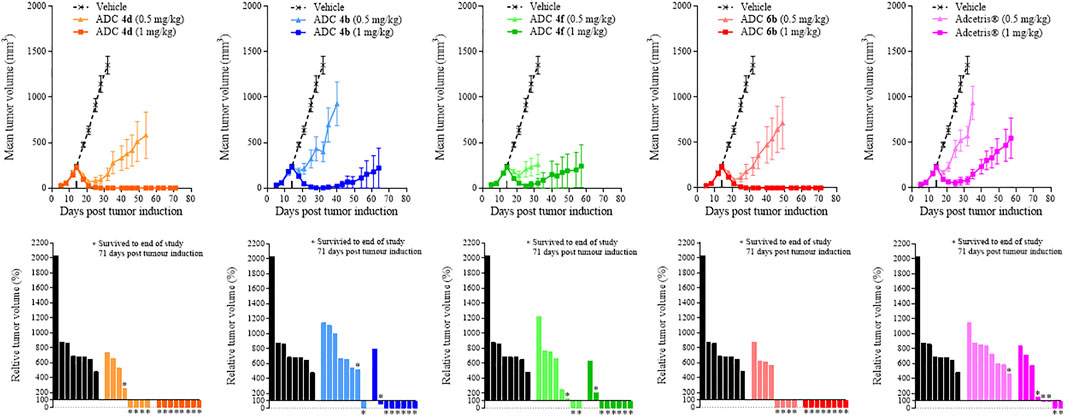
FIGURE 2. Tumor growth curves and relative tumor volume (%) plots taken on the final day of the study (Day 71) for xenograft study 1. Mean tumor volume was plotted until the first death in the group was recorded. Relative tumor volumes were calculated as percentage change on the final day of the study (Day 71) relative to the tumor volume on the day of treatment administration (Day 14).
In vivo Xenograft Study 2
Similarly to the cyclodextrin-containing conjugates, in vivo studies were also required to differentiate between the activities of the crown ether-containing conjugates, 5a and 5b, and their linear PEG-containing comparators 6a, 6b and Adcetris®. In contrast to Study 1, to differentiate the efficacies of these conjugates further, the doses of conjugates 5a, 5b, 6a and 6b were reduced to 0.4 and 0.8 mg/kg (Adcetris® dose was maintained at 1 mg/kg). Animals were dosed on day 12 following tumor implantation when the tumor volume was approximately 200 mm3. Figure 3 shows data obtained with the same in vivo xenograft tumor model as in Study 1, where tumor volumes were measured over time within mice dosed with ADCs 5a, 5b, 6a or 6b at 0.4 or 0.8 mg/kg. At 0.4 mg/kg, these ADCs had little effect upon tumor growth, following a similar growth rate to the vehicle control after Day 20 of the study. As with the previous study, animals were dosed with Adcetris® at 1 mg/kg and the tumor volumes measured over 71 days as a comparator. Adcetris® was given at Day 14 and showed an initial reduction in tumor volume, which was lost shortly after Day 20, resulting in a steady increase in tumor volume thereafter. Within this group, one animal out of the cohort of 8 achieved a complete reduction in tumor volume, where no tumor could be detected by the end of the study (also indicated by the relative tumor volume (%) plot within Figure 3). By comparison, the cohorts treated with ADCs 5a, 5b, 6a and 6b at 0.8 mg/kg dose each showed reductions in mean tumor volume for extended periods. Notably, the efficacy of conjugate 5b was outstanding, achieving a mean tumor volume of 0 mm3, (i.e. complete tumor regression), for the duration of the study up to Day 71. The relative tumor volume (%) plots also show that conjugates 5a and 5b have the greatest efficacy, with 7 out of 8 and 8 out of 8 animals surviving tumor free respectively at 0.8 mg/kg dose, whereas for conjugates 6a and 6b, 5 out of 8 and 6 out of 8 animals survived tumor free at the end of the study respectively. As observed within Study 1 previously, the body weights of each animal were measured throughout and showed that the ADCs were well tolerated, as determined by comparison of the body weights of the treatment groups with the vehicle group, and no treatment related deaths were recorded across all groups (Supplementary Figure S3).
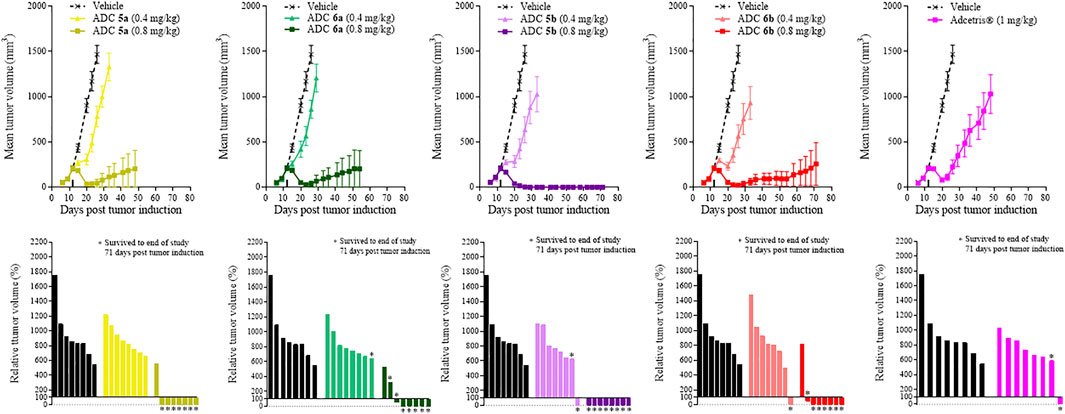
FIGURE 3. Tumor growth curves and relative tumor volume (%) plots taken on the final day of the study (Day 71) for xenograft study 2. Mean tumor volumes was plotted until the first death in the group was recorded. Relative tumor volumes were calculated as percentage change on the final day of the study (Day 71) relative to the tumor volumes on the day of treatment administration (Day 12).
Discussion
Cluster of differentiation 30 (CD30) is a type I transmembrane protein of the tumor necrosis factor receptor (TNFR) superfamily expressed on the medullary of the thymus gland and on a subset of activated T and B cells (Romagnani et al., 1998; van der Weyden et al., 2017). CD30 expression is not normally detected in healthy tissue outside of the immune system, however high levels of CD30 expression are found in Hodgkin lymphoma (HL) and ALCL, making it an attractive target for antibody-based immunotherapy. Numerous naked and conjugated CD30-targeting mAbs have entered clinical development but despite promising results from animal models, early candidates were unsuccessful (Younes, 2011; Schirrmann et al., 2014). Clinical success was eventually achieved with brentuximab vedotin (SGN-35), an ADC comprising a chimeric anti-CD30 IgG1 antibody conjugated to the potent cytotoxin, MMAE, via an enzymatically cleavable MC-Val-Cit-PABC linker. Conjugation of the linker-payload to the antibody is achieved over two steps: 1) partial reduction of the interchain disulfide bonds of the antibody followed by 2) thiol-maleimide conjugation to the unpaired cysteines liberated during reduction. The nature of the conjugation process results in a heterogeneous mixture of differently loaded species ranging from DAR 0 to DAR 8, with an average drug loading of approximately 4. Phase II clinical trials involving brentuximab vedotin as a single agent in patients with relapsed or refractory HL and ALCL resulted in objective response rates of 75 and 86% respectively (Pro et al., 2012; Younes et al., 2012). Based on these impressive results the FDA granted accelerated approval of brentuximab vedotin in 2011 and the ADC has since been marketed as Adcetris®.
Ongoing efforts to improve ADC efficacy have included focussing upon the design of linker-payload reagent architectures. In our previous work we investigated the inclusion of PEG in ADC reagent design, where structures containing branching architectures were revealed to enhance ADC efficacy over structures with no PEG component and where the PEG is included as a spacer within the ADC backbone (Pabst et al., 2017). Other studies have corroborated these results including research from Lyon and coworkers (Lyon et al., 2015; Burke et al., 2017). In the present work, we designed and synthesized eight new reagents using hydrophilic macrocycles in place of the linear PEG component. Each reagent contained identical bis-sulfone disulfide rebridging conjugating groups, glutamic acid branching points connecting Val-Cit-PABC-MMAE as linker-payload. The reagents designed varied only in the nature of the hydrophilic side-chain moiety appended to the branching point. Six new reagents containing cyclodextrins, two reagents for each of α-, β- and γ-cyclodextrin variants with mono-amino substituents in the 3′ or 6′ positions (Figure 1, reagents 1a-f) were produced. Two further reagents bearing 1-aza-crown ethers were synthesized, the smaller macrocycle incorporated into the reagent template was a 24-crown-8 based ring and the larger macrocycle was a 42-crown-14 derivative (Figure 1, reagents 2a-b). Two reagents with 8- and 24-unit linear PEG chains (Figure 1, reagents 3a-b) were also synthesized so comparator ADCs could be prepared. Each of the ten disulfide rebridging reagents described were conjugated to brentuximab via reaction at pre-reduced interchain disulfide bonds to produce ten DAR 4 ADCs for biological evaluation. ADCs 4a-f were prepared using the cyclodextrin based reagents, ADCs 5a-b were synthesized using the crown ether reagents and the linear PEG reagents were used to prepare ADCs 6a-b.
Table 1; Supplementary Figure S1 shows in vitro cell-viability data for the conjugates produced against Karpas-299 cells. Within each figure both Adcetris® and conjugate 6b were used as internal controls as comparators for the test ADCs. From Supplementary Figures S1A–S1C, it can be determined that the cell killing potency of each test ADC is higher than the free payload, MMAE, with each conjugate displaying similar IC50 values in the same range as conjugate 6b and Adcetris®. These data suggest that although the in vitro assay is a reliable test for determining the overall activity of an ADC, as has been demonstrated in previous studies it may not be the best predictor of in vivo efficacy (Hamblett et al., 2004; Pabst et al., 2017).
Figure 2 shows that all of the cyclodextrin-containing ADCs display improved in vivo efficacy over the clinical comparator Adcetris® within this Karpas-299 xenograft tumor model. These data were in contrast to the in vitro data obtained, which showed that all of the cyclodextrin conjugates were of similar or slightly poorer potency to Adcetris®. Figure 2 also shows that 3′-amino-α-cyclodextrin containing conjugate 4d in particular had outstanding efficacy in this model, equivalent to that of the PEG containing conjugate 6b. These data are very encouraging and supportive of future development of cyclodextrin-containing ADCs.
From Supplementary Figure S1C, it can be seen from the in vitro data that all of the PEG-containing ADCs; 5a, 5b, 6a and 6b have comparable IC50s to that of Adcetris®. Once again, this is not reflected within the in vivo efficacy results in Figure 3. It is notable that conjugates 5b and 5a are particularly efficacious in vivo, more so than Adcetris® and even conjugate 6b, which is again in contrast to the in vitro IC50 data. Conjugate 5b appears to be the best of these ADCs, which as with the cyclodextrin-containing ADCs, is very encouraging for further development of crown-ether containing ADCs in the future.
Conclusion
In conclusion, we tested whether hydrophilic macrocycles (cyclodextrin or crown ethers) may be used as hydrophilic substitutes for the polymeric element present in known ADC structures. Within current ADC structures, it is hypothesized that the linear polymer structure, imparts greater efficacy to ADCs in vivo by forming a ‘shield’ around the hydrophobic linker-payload structure, whereby the hydrophobicity of the conjugate is ‘masked’, resulting in a slower clearance of the ADC from the subject (Lyon et al., 2015). It may be considered therefore that in restricting the size and mobility of the polymeric element of the ADC, one may expect decreased shielding of the linker-payload and therefore poorer retention within the subject, resulting in reduced efficacy. However, we have shown that this is not the case for the cyclodextrin- or crown ether- containing ADCs tested in this work.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
NE, MB, RG and PW co-wrote the manuscript. NC and MF edited the manuscript. AK, RP, PW and DM synthesized and characterized reagents for antibody conjugation. TK, XS, FS, HM, RR, ED and AJ prepared ADCs for biological evaluation. AMi, FJ and FR performed in vitro cytotoxicity assays on the ADCs. PW, RG and AK designed and provided managerial oversight to the reagent synthesis work. MB, NC and WM designed and provided managerial oversight to the ADC production work. AMa designed and provided managerial oversight to all biological studies. AG, MP and MF designed and provided managerial oversight to all studies.
Funding
Abzena Ltd. provided all funding for this research.
Conflict of Interest
NE, AK and AG are named inventors on patents relating to the inclusion of macrocycles in ADCs. All authors are or were employees of the Abzena group.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Dr. Rob Holgate and Dr. Tim Jones for the provision of brentuximab. We would also like to thank Dr. Karpas for the use of the Karpas-299 cell line and Oncodesign for performing Karpas-299 xenograft studies.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.764540/full#supplementary-material
References
Badescu, G., Bryant, P., Bird, M., Henseleit, K., Swierkosz, J., Parekh, V., et al. (2014). Bridging Disulfides for Stable and Defined Antibody Drug Conjugates. Bioconjug. Chem. 25, 1124–1136. doi:10.1021/bc500148x
Bird, M., Nunes, J., and Frigerio, M. (2020). Bridged Cysteine Conjugations. Methods Mol. Biol. 2078, 113–129. doi:10.1007/978-1-4939-9929-3_8
Bryant, P., Pabst, M., Badescu, G., Bird, M., McDowell, W., Jamieson, E., et al. (2015). In Vitro and In Vivo Evaluation of Cysteine Rebridged Trastuzumab-MMAE Antibody Drug Conjugates with Defined Drug-To-Antibody Ratios. Mol. Pharm. 12, 1872–1879. doi:10.1021/acs.molpharmaceut.5b00116
Bryden, F., Martin, C., Letast, S., Lles, E., Viéitez-Villemin, I., Rousseau, A., et al. (2018). Impact of Cathepsin B-Sensitive Triggers and Hydrophilic Linkers on In Vitro Efficacy of Novel Site-specific Antibody-Drug Conjugates. Org. Biomol. Chem. 16, 1882–1889. doi:10.1039/c7ob02780j
Burke, P. J., Hamilton, J. Z., Jeffrey, S. C., Hunter, J. H., Doronina, S. O., Okeley, N. M., et al. (2017). Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 16, 116–123. doi:10.1158/1535-7163.MCT-16-0343
Challa, R., Ahuja, A., Ali, J., and Khar, R. K. (2005). Cyclodextrins in Drug Delivery: an Updated Review. AAPS PharmSciTech 6, E329–E357. doi:10.1208/pt060243
Chehardoli, G., and Bahmani, A. (2019). The Role of crown Ethers in Drug Delivery. Supramolecular Chem. 31, 221–238. doi:10.1080/10610278.2019.1568432
Dal Corso, A., Pignataro, L., Belvisi, L., and Gennari, C. (2019). Innovative Linker Strategies for Tumor-Targeted Drug Conjugates. Chemistry 25, 14740–14757. doi:10.1002/chem.201903127
Febles, M., Montalvão, S., Crespín, G. D., Norte, M., Padrón, J. M., Tammela, P., et al. (2016). Synthesis and Biological Evaluation of crown Ether Acyl Derivatives. Bioorg. Med. Chem. Lett. 26, 5591–5593. doi:10.1016/j.bmcl.2016.09.066
Frigerio, M., and Kyle, A. F. (2017). The Chemical Design and Synthesis of Linkers Used in Antibody Drug Conjugates. Curr. Top. Med. Chem. 17, 3393–3424. doi:10.2174/1568026618666180118155847
Gidwani, B., and Vyas, A. (2015). A Comprehensive Review on Cyclodextrin-Based Carriers for Delivery of Chemotherapeutic Cytotoxic Anticancer Drugs. Biomed. Res. Int. 2015, 198268. doi:10.1155/2015/198268
Godwin, A. (2017). Conjugates and Conjugating Reagents. Polytherics LimitedWorld Intellectual Property Organization WO2017/199046 A1.
Godwin, A., Kyle, A., and Evans, N. (2017). Conjugates and Conjugating Reagents Comprising A Linker that Includes at Least Two (-CH2-CH2-O-) Units in A Ring. Polytherics LimitedWorld Intellectual Property Organization WO2017/178828 A1.
Gokel, G. W., Arnold, K. A., Delgado, M., Echeverria, L., Gatto, V. J., Gustowski, D. A., et al. (1988). Lariat Ethers: from Cation Complexation to Supramolecular Assemblies. Pure Appl. Chem. 60, 461–465. doi:10.1351/pac198860040461
Haley, R. M., Gottardi, R., Langer, R., and Mitchell, M. J. (2020). Cyclodextrins in Drug Delivery: Applications in Gene and Combination Therapy. Drug Deliv. Transl. Res. 10, 661–677. doi:10.1007/s13346-020-00724-5
Hamblett, K. J., Senter, P. D., Chace, D. F., Sun, M. M., Lenox, J., Cerveny, C. G., et al. (2004). Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 10, 7063–7070. doi:10.1158/1078-0432.CCR-04-0789
Irie, T., and Uekama, K. (1999). Cyclodextrins in Peptide and Protein Delivery. Adv. Drug Deliv. Rev. 36, 101–123. doi:10.1016/S0169-409X(98)00057-X
Jansen, B. A. J., Wielaard, P., den Dulk, H., Brouwer, J., and Reedijk, J. (2002). Oxa-aza Crown Ethers as Ligands for Mixed-Ligand Cisplatin Derivatives and Dinuclear Platinum Anticancer Drugs. Eur. J. Inorg. Chem. 2002, 2375–2379. doi:10.1002/1099-0682(200209)2002:9<2375:aid-ejic2375>3.0.co;2-b
Jansook, P., Ogawa, N., and Loftsson, T. (2018). Cyclodextrins: Structure, Physicochemical Properties and Pharmaceutical Applications. Int. J. Pharm. 535, 272–284. doi:10.1016/j.ijpharm.2017.11.018
Kim, D.-H., Lee, S.-E., Pyo, Y.-C., Tran, P., and Park, J.-S. (2020). Solubility Enhancement and Application of Cyclodextrins in Local Drug Delivery. J. Pharm. Investig. 50, 17–27. doi:10.1007/s40005-019-00434-2
Kurkov, S. V., and Loftsson, T. (2013). Cyclodextrins. Int. J. Pharmaceutics 453, 167–180. doi:10.1016/j.ijpharm.2012.06.055
Loftsson, T., and Brewster, M. E. (2010). Pharmaceutical Applications of Cyclodextrins: Basic Science and Product Development. J. Pharm. Pharmacol. 62, 1607–1621. doi:10.1111/j.2042-7158.2010.01030.x
Loftsson, T., Jarho, P., Másson, M., and Järvinen, T. (2005). Cyclodextrins in Drug Delivery. Expert Opin. Drug Deliv. 2, 335–351. doi:10.1517/17425247.2.1.335
Lyon, R. P., Bovee, T. D., Doronina, S. O., Burke, P. J., Hunter, J. H., Neff-LaFord, H. D., et al. (2015). Reducing Hydrophobicity of Homogeneous Antibody-Drug Conjugates Improves Pharmacokinetics and Therapeutic index. Nat. Biotechnol. 33, 733–735. doi:10.1038/nbt.3212
Marcinkowska, M., Stanczyk, M., Janaszewska, A., Sobierajska, E., Chworos, A., and Klajnert-Maculewicz, B. (2019). Multicomponent Conjugates of Anticancer Drugs and Monoclonal Antibody with PAMAM Dendrimers to Increase Efficacy of HER-2 Positive Breast Cancer Therapy. Pharm. Res. 36, 154. doi:10.1007/s11095-019-2683-7
Muzzalupo, R., Nicoletta, F. P., Trombino, S., Cassano, R., Iemma, F., and Picci, N. (2007). A New crown Ether as Vesicular Carrier for 5-fluoruracil: Synthesis, Characterization and Drug Delivery Evaluation. Colloids Surf. B Biointerfaces 58, 197–202. doi:10.1016/j.colsurfb.2007.03.010
Ortiz Mellet, C., García Fernández, J. M., and Benito, J. M. (2011). Cyclodextrin-based Gene Delivery Systems. Chem. Soc. Rev. 40, 1586–1608. doi:10.1039/C0CS00019A
Pabst, M., McDowell, W., Manin, A., Kyle, A., Camper, N., De Juan, E., et al. (2017). Modulation of Drug-Linker Design to Enhance In Vivo Potency of Homogeneous Antibody-Drug Conjugates. J. Control Release 253, 160–164. doi:10.1016/j.jconrel.2017.02.027
Pro, B., Advani, R., Brice, P., Bartlett, N. L., Rosenblatt, J. D., Illidge, T., et al. (2012). Brentuximab Vedotin (SGN-35) in Patients with Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma: Results of a Phase II Study. J. Clin. Oncol. 30, 2190–2196. doi:10.1200/JCO.2011.38.0402
Redenti, E., Pietra, C., Gerloczy, A., and Szente, L. (2001). Cyclodextrins in Oligonucleotide Delivery. Adv. Drug Deliv. Rev. 53, 235–244. doi:10.1016/S0169-409X(01)00230-7
Romagnani, P., Annunziato, F., Manetti, R., Mavilia, C., Lasagni, L., Manuelli, C., et al. (1998). High CD30 Ligand Expression by Epithelial Cells and Hassal's Corpuscles in the Medulla of Human Thymus. Blood 91, 3323–3332. doi:10.1182/BLOOD.V91.9.3323.3323_3323_3332
Schirrmann, T., Steinwand, M., Wezler, X., Ten Haaf, A., Tur, M. K., and Barth, S. (2014). CD30 as a Therapeutic Target for Lymphoma. BioDrugs 28, 181–209. doi:10.1007/s40259-013-0068-8
Shao, S., Tsai, M. H., Lu, J., Yu, T., Jin, J., Xiao, D., et al. (2018). Site-specific and Hydrophilic ADCs through Disulfide-Bridged Linker and Branched PEG. Bioorg. Med. Chem. Lett. 28, 1363–1370. doi:10.1016/j.bmcl.2018.03.005
Shao, T., Chen, T., Chen, Y., Liu, X., Chen, Y. L., Wang, Q., et al. (2020). Construction of Paclitaxel-Based Antibody-Drug Conjugates with a PEGylated Linker to Achieve superior Therapeutic index. Signal. Transduct Target. Ther. 5, 132. doi:10.1038/s41392-020-00247-y
Shi, X., Zhang, X. N., Chen, J., Cheng, Q., Pei, H., Louie, S. G., et al. (2020). A Poly-ADP-Ribose Polymer-Based Antibody-Drug Conjugate. Chem. Sci. 11, 9303–9308. doi:10.1039/D0SC01795G
Tiberghien, A. C., Levy, J. N., Masterson, L. A., Patel, N. V., Adams, L. R., Corbett, S., et al. (2016). Design and Synthesis of Tesirine, a Clinical Antibody-Drug Conjugate Pyrrolobenzodiazepine Dimer Payload. ACS Med. Chem. Lett. 7, 983–987. doi:10.1021/acsmedchemlett.6b00062
Turecek, P. L., Bossard, M. J., Schoetens, F., and Ivens, I. A. (2016). PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 105, 460–475. doi:10.1016/j.xphs.2015.11.015
van der Weyden, C. A., Pileri, S. A., Feldman, A. L., Whisstock, J., and Prince, H. M. (2017). Understanding CD30 Biology and Therapeutic Targeting: a Historical Perspective Providing Insight into Future Directions. Blood Cancer J. 7, e603. doi:10.1038/bcj.2017.85
Veronese, F. M., and Mero, A. (2008). The Impact of PEGylation on Biological Therapies. BioDrugs 22, 315–329. doi:10.2165/00063030-200822050-00004
Viricel, W., Fournet, G., Beaumel, S., Perrial, E., Papot, S., Dumontet, C., et al. (2019). Monodisperse Polysarcosine-Based Highly-Loaded Antibody-Drug Conjugates. Chem. Sci. 10, 4048–4053. doi:10.1039/c9sc00285e
Yang, K., Chen, B., Gianolio, D. A., Stefano, J. E., Busch, M., Manning, C., et al. (2019). Convergent Synthesis of Hydrophilic Monomethyl Dolastatin 10 Based Drug Linkers for Antibody-Drug Conjugation. Org. Biomol. Chem. 17, 8115–8124. doi:10.1039/c9ob01639b
Younes, A. (2011). CD30-targeted Antibody Therapy. Curr. Opin. Oncol. 23, 587–593. doi:10.1097/CCO.0b013e32834bb8a7
Younes, A., Gopal, A. K., Smith, S. E., Ansell, S. M., Rosenblatt, J. D., Savage, K. J., et al. (2012). Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients with Relapsed or Refractory Hodgkin's Lymphoma. J. Clin. Oncol. 30, 2183–2189. doi:10.1200/JCO.2011.38.0410
Yurkovetskiy, A. V., Yin, M., Bodyak, N., Stevenson, C. A., Thomas, J. D., Hammond, C. E., et al. (2015). A Polymer-Based Antibody-Vinca Drug Conjugate Platform: Characterization and Preclinical Efficacy. Cancer Res. 75, 3365–3372. doi:10.1158/0008-5472.CAN-15-0129
Zhang, L., Fang, Y., Kopeček, J., and Yang, J. (2017). A New Construct of Antibody-Drug Conjugates for Treatment of B-Cell Non-hodgkin's Lymphomas. Eur. J. Pharm. Sci. 103, 36–46. doi:10.1016/j.ejps.2017.02.034
Zhao, H., Rubio, M. B., and Reddy, P. (2008). Enzon Pharmaceuticals, Inc. Targeted Polymeric Prodrugs Containing Multifunctional LinkersWorld Intellectual Property Organization WO2008/034124 A2.
Keywords: antibody drug conjugate (ADC), crown ether, cyclodextrin, in vivo, xenograft
Citation: Evans N, Grygorash R, Williams P, Kyle A, Kantner T, Pathak R, Sheng X, Simoes F, Makwana H, Resende R, de Juan E, Jenkins A, Morris D, Michelet A, Jewitt F, Rudge F, Camper N, Manin A, McDowell W, Pabst M, Godwin A, Frigerio M and Bird M (2022) Incorporation of Hydrophilic Macrocycles Into Drug-Linker Reagents Produces Antibody-Drug Conjugates With Enhanced in vivo Performance. Front. Pharmacol. 13:764540. doi: 10.3389/fphar.2022.764540
Received: 25 August 2021; Accepted: 28 February 2022;
Published: 17 June 2022.
Edited by:
Lufang Zhou, University of Alabama at Birmingham, United StatesReviewed by:
Abdallah El-Sayed Allam, Tanta University, EgyptMeghdad Abdollahpour-Alitappeh, Larestan University of Medical Sciences, Iran
Floris Van Delft, Synaffix, Netherlands
Copyright © 2022 Evans, Grygorash, Williams, Kyle, Kantner, Pathak, Sheng, Simoes, Makwana, Resende, de Juan, Jenkins, Morris, Michelet, Jewitt, Rudge, Camper, Manin, McDowell, Pabst, Godwin, Frigerio and Bird. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nick Evans, bmljay5ldmFuc0BhYnplbmEuY29t