 Zhipeng Zhao1†
Zhipeng Zhao1† Rongsheng Tong
Rongsheng Tong Lei Zhong
Lei Zhong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 13 December 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1073713
This article is part of the Research Topic Epigenetic Regulation and Therapy Resistance in Cancer View all 5 articles
Epigenetic alterations are implicated in tumour immune evasion and immune checkpoint blockade (ICB) resistance. SET domain bifurcated histone methyltransferase 1 (SETDB1) is a histone lysine methyltransferase that catalyses histone H3K9 di- and tri-methylation on euchromatin, and growing evidence indicates that SETDB1 amplification and abnormal activation are significantly correlated with the unfavourable prognosis of multiple malignant tumours and contribute to tumourigenesis and progression, immune evasion and ICB resistance. The main underlying mechanism is H3K9me3 deposition by SETDB1 on tumour-suppressive genes, retrotransposons, and immune genes. SETDB1 targeting is a promising approach to cancer therapy, particularly immunotherapy, because of its regulatory effects on endogenous retroviruses. However, SETDB1-targeted therapy remains challenging due to potential side effects and the lack of antagonists with high selectivity and potency. Here, we review the role of SETDB1 in tumourigenesis and immune regulation and present the current challenges and future perspectives of SETDB1 targeted therapy.
Immune checkpoint blockade (ICB) therapy eradicates tumour cells via releasing the brake on the adaptive immune response and has revolutionized clinical treatments for multiple malignancies (Deng and Zhang, 2018; Bagchi et al., 2021). To date, 18 immune checkpoint inhibitors (ICIs) have been approved as cancer therapeutics, including 12 programmed cell death protein 1 (PD-1) monoclonal antibodies (pembrolizumab, nivolumab, cemiplimab, toripalimab, sintilimab, camrelizumab, tislelizumab, zimberelimab, penpulimab, dostarlimab, serplulimab and prolgolimab), five programmed cell death ligand 1 (PD-L1) monoclonal antibodies (atezolizumab, durvalumab, avelumab, envafolimab and sugemalimab), and one monoclonal antibody that blocks cytotoxic T-lymphocyte associated protein 4 (ipilimumab) (Dhillon, 2021; Dhillon and Duggan, 2022; Lee, 2022; Markham, 2022; Yi et al., 2022). Despite considerable advancements, the response rate to ICIs is currently limited to 10%–25% in most tumour types (Schoenfeld and Hellmann, 2020), and those with deficient immunogenic epitopes (low mutational burden) (Verdegaal et al., 2016; Tran et al., 2017), impoverished tumour-infiltrating lymphocytes (Galluzzi et al., 2018; Fanale et al., 2022), or profuse immunosuppressive factors (such as PD-L1, CD73, and indoleamine 2,3-dioxygenase 1) (Young et al., 2016; Chen and Mellman, 2017; Song et al., 2021) are less likely to respond (Galluzzi et al., 2018). Furthermore, initial ICI responders may develop acquired resistance (Schoenfeld and Hellmann, 2020). These limitations have motivated efforts to explore novel immunosuppressive mechanisms and immunotherapy approaches to complement current ICB treatments.
Epigenetic dysregulation is one of the most important hallmarks of tumourigenesis (Hanahan and Weinberg, 2011). Since accumulating evidence has identified epigenetic alterations as drivers of immune escape, epigenetic therapies could enhance response and overcome ICB resistance (Gomez et al., 2020; Topper et al., 2020). Unlike gene mutations, epigenetic modifications are heritable phenotypic alterations that do not change the nucleotide sequence and mainly include histone modification, DNA methylation and changes in chromatin structure and microRNA levels (Cheng et al., 2019; Cao and Yan, 2020). These modifications mediate cancer immunoediting, characterised by decreased antigenicity and immunogenicity, disruption of key interactions for immune response, establishment of T cell exhaustion, and creation of an immunosuppressive tumour microenvironment (TME) (Gomez et al., 2020; Gangoso et al., 2021). The reversibility and targetability of epigenetic alterations make them attractive interventions for promoting tumour cell response to immunotherapy. SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) was recently identified as a critical epigenetic regulator that contributes to the immunosuppressive TME and ICB resistance (Griffin et al., 2021; Zhang et al., 2021). Here, we review the effects of SETDB1 on tumourigenesis and progression, particularly its role in regulating the tumour immune response, and present the current challenges and future perspectives of antitumour therapy targeting SETDB1.
SETDB1, also known as ERG-associated protein with SET domain (ESET), belongs to the histone lysine methyltransferase family. It maps to chromosome 1q21.3 and comprises 1291 amino acids with a molecular weight of 143.1 kDa (Markouli et al., 2021b). SETDB1 is highly evolutionarily conserved, and mouse SETDB1 shows 92% similarity with human SETDB1 at the amino acid level (Yang et al., 2002). SETDB1 consists of an N-terminus containing two nuclear export signal (NES) domains, two nuclear localisation signal (NLS) domains, two Tudor domains and a methyl-CpG-binding domain (MBD). The C-terminus contains the pre-SET, bifurcated SET and post-SET domains (Supplementary Figure 1A) (Batham et al., 2019; Markouli et al., 2021a). The NES and NLS domains in the N-terminus regulate SETDB1 localisation (Cho et al., 2013). The Tudor domains may anchor SETDB1 to arginine and lysine residues on histone or nonhistone substrates and are involved in forming transcriptional repression-associated multiprotein complexes, such as the Kruppel-associated box (KRAB) zinc finger protein-KRAB-associated protein-1 (KRAB-ZFP-KAP1) complex (Schultz et al., 2002). The MBD CpG domain contains two arginine residues that bind to methylated DNA (Ho et al., 2008), thereby regulating the interaction with DNA (cytosine-5) methyltransferase 3, heterochromatin formation and gene silencing by coordinating CpG methylation with H3K9 trimethylation (Li et al., 2006). The C-terminus domains of SETDB1 are responsible for its methyltransferase catalytic activity (Yang et al., 2002). The bifurcated SET domain is the main region of catalytic activity and is separated by a 347-amino acid insert. Although the exact function of this insertion remains unelucidated, SETDB1 ubiquitination at lysine 867 within the SET insertion is essential for the acquisition of complete enzymatic activity of SETDB1 in mammals (Ishimoto et al., 2016). Additionally, SETDB1 recruits S-adenosine methionine (SAM) and human homolog of murine activating transcription factor a (ATFa)-associated modulator (hAM) as cofactors during the catalytic process (Supplementary Figure S1B) (Wang et al., 2003).
SETDB1 is an epigenetic modulator that transfers methyl groups to histones and represses target gene transcription. Histones are proteins that provide structural support, around which DNA wraps to generate nucleosomes and subsequently tightly packed chromatin (Kouzarides, 2007). They can be chemically modified by some enzymes to modulate gene transcription, and methylation of lysine residues is one of the most common modifications of histones, which can affect chromatin structure and the interaction of transcription factors with nucleosomes (Hyun et al., 2017). The nucleosome is a histone octamer that consists of two duplicates of each core histones H2A, H2B, H3, and H4 (Hyun et al., 2017). SETDB1 is a histone H3 lysine 9 (H3K9) methyltransferase responsible for the chemical modifications of H3K9 di- and tri-methylation on euchromatin (Yang et al., 2002; Wang et al., 2003). This post-translational modification recruits heterochromatin protein 1 (HP1) to euchromatic promoters and establishes a heterochromatin-like silenced state at euchromatin with the coordination of KAP1 (Schultz et al., 2002; Ayyanathan et al., 2003).
Growing evidence has revealed the close correlation of SETDB1 overexpression and abnormal activity in a variety of malignancies with an unfavourable prognosis of cancer patients (Figure 1) (Wong et al., 2016; Orouji et al., 2019; Yu et al., 2020; Strepkos et al., 2021). Additionally, SETDB1 amplification plays pivotal roles in tumourigenesis and progression, such as promoting cell proliferation, migration, invasion, epithelial-mesenchymal transition (EMT), metastasis, resistance, and immune evasion (Figure 1). Mechanically, elevated SETDB1 facilitates H3K9 methylation at the promoter regions of multiple tumour-suppressive genes, leading to gene silencing and tumourigenesis (Cao et al., 2020; Ogawa et al., 2020; Strepkos et al., 2021). Meanwhile, various other mechanisms are also involved. For example, activated AKT kinase is beneficial for cancer cell proliferation and survival, while SETDB1 could activate AKT by mediating methylation of AKT K64 residue and promote the development of non-small-cell lung carcinoma (NSCLC) (Wang et al., 2019). Leonard I. Zon group identified SETDB1 as a cooperator of BRAF (V600E) mutation to accelerate melanoma formation (Ceol et al., 2011). In this study, SETDB1 was found to form a complex with other H3K9 methyltransferases, such as SUV39H1, and they acted together to alter gene transcription in a manner that promotes onset and invasiveness of melanoma. Moreover, SETDB1 is also considered an oncogene in hepatocellular carcinoma (HCC) cells because it boosts their proliferation and migration via interaction with T-cell lymphoma invasion and metastasis-inducing protein 1 (Tiam1) (Zhang et al., 2018). SETDB1 upregulation was reported to be closely related to the progression and poor prognosis of patients with HCC and was found in all metastatic foci, demonstrating the important role of SETDB1 in HCC aggressiveness. miR-29 was identified as a negative modulator of SETDB1 in this process (Wong et al., 2016).
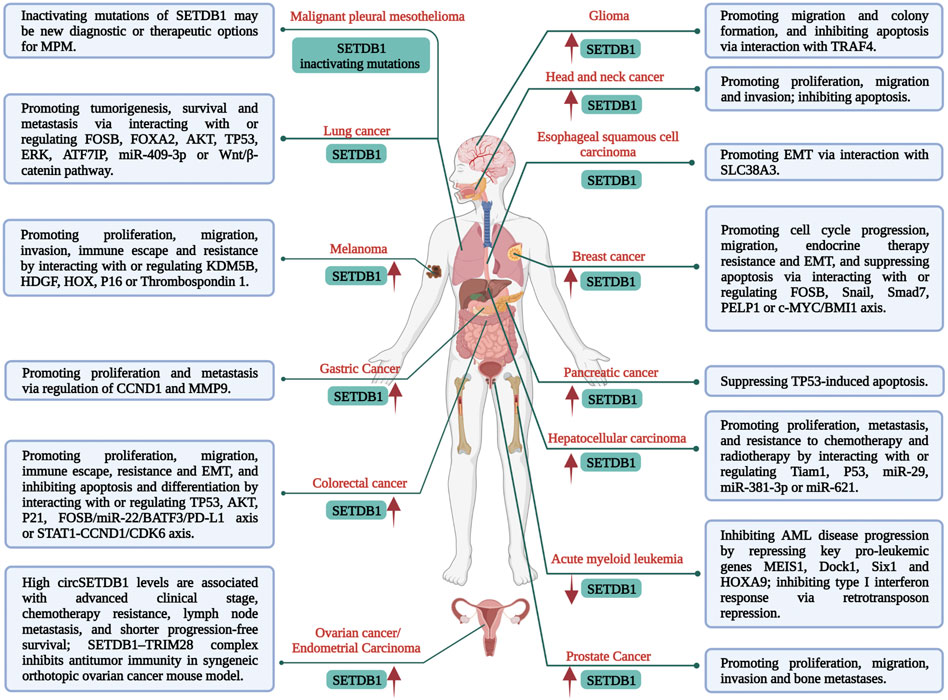
FIGURE 1. Multiple functions and mechanisms of SETDB1 regulation in malignancies.
The initial step in metastasis is EMT, which leads to a motile phenotype in cancer cells. SETDB1 was reported to participate in regulating EMT in colorectal cancer (CRC) cells by binding to the P21 promoter and affecting its activity (Cao et al., 2020), in esophageal squamous cell carcinoma via interaction with SLC38A3 (Liu et al., 2020), and in HCC by interacting with Tiam1 (Zhang et al., 2018). Treatment resistance in cancer may be intrinsic or acquired, which immensely restricts the efficacy of anti-cancer therapeutics. SETDB1 has also been implicated in tumour resistance. Guler et al. (2017) found elevated levels of SETDB1 in multiple drug-tolerant cancer cells, and increased H3K9me3 was enriched at long interspersed nuclear element-1 (LINE-1) genomic loci. Thus, derepression of LINE-1 elements by SETDB1 or G9a loss could restore treatment sensitivity. SETDB1 overexpression was also found to mediate the resistance of breast cancer cells to tamoxifen. This is attributed to the SETDB1 regulation on the target genes of estrogen receptor (ER) and AKT, while ER coregulator PELP1 interacts directly with SETDB1 and is necessary for SETDB1-mediated regulatory effects and tamoxifen resistance (Liu et al., 2022). Notably, although less reported, SETDB1 can also play a tumour-suppressive role, such as in acute myeloid leukaemia (AML) (Ropa et al., 2020) and some metastatic lung cancer cells (Wu et al., 2014). Thus, the role of SETDB1 in cancer may depend on its type and stage.
The regulation of immune responses mediated by SETDB1 is complex and involves multiple mechanisms. The most prominent is the limitation of endogenous retrotransposon expression, which enables tumour cells to evade innate immune sensing (Zhang et al., 2021; Pan et al., 2022). Retrotransposons are transposable elements (TEs) that include LINEs and endogenous retroviruses (ERVs). They amplify via reverse transcription of RNA into DNA using an RNA transposition intermediate (Wu et al., 2021). Retrotransposons only exist in eukaryotes, and over 40% of the human genome is derived from retrotransposons. Due to their viral origin, the nucleic acids that they produce are recognised as ‘non-self’ pathogen-specific molecules by the innate sensors and trigger an immune response (Tie and Rowe, 2017; Wu et al., 2021). Specifically, double-stranded RNA (dsRNA) derived from endogenous retroviral elements can be induced by low-dose 5-aza-2-deoxycytidine (a DNA methylation inhibitor), and then activate interferon through MDA5/MAVS/IRF7 sensors, thereby exhibiting anti-tumour effects (Roulois et al., 2015). Additionally, accumulation of cytoplasmic DNA resulting from retroelements or DNA damage was also reported as a pathogen-associated molecular pattern. It could be recognized by cGAS/STING signalling axis, which then activated type I interferon and promoted responses to innate immunity (Sun et al., 2013; Härtlova et al., 2015).
Cuellar et al. first reported the function of SETDB1-mediated ERV suppression in cancer cells in 2017 when they discovered that SETDB1 silencing in AML cells elevated the expression of IFN-β and IFN-stimulated genes. IFN induction by SETDB1 disruption was attributed to decreased H3K9me3 at repetitive loci, upregulation of some ERV subfamilies, LINEs and satellite repeats, and subsequent production of dsRNA that elicited a cytosolic, nucleic acid-sensing cascade and IFN-mediated cellular apoptosis (Cuellar et al., 2017). Meanwhile, Hu et al. found that activating transcription factor 7-interacting protein (Atf7ip) and its binding partner SETDB1 were chromatin modifiers that mediated tumour immune escape, and deletion of either Atf7ip or SETDB1 stimulated antigen expression and presentation, promoted T-cell activation and infiltration, resulting in augmented antitumour immune responses. In this process, SETDB1 deficiency plays an essential role in decreasing H3K9me3 deposition on the chromosomes encoding ERV-derived antigens and up-regulating tumour immunogenicity (Hu et al., 2021). Recently, two studies published in Nature systematically revealed the effects of and mechanisms behind SETDB1 regulation of tumour-intrinsic immunogenicity (Griffin et al., 2021; Zhang et al., 2021). They identified SETDB1 (or SETDB1 recruited by KDM5B) as a chromatin regulator that depresses tumour-intrinsic immunogenicity and mediates immune tolerance in both mice and human tumours, and SETDB1 depletion potently boosted the efficacy of ICB in mouse models. Mechanistically, SETDB1 epigenetically silences broad domains, mainly at segmental duplications-evolving loci that are enriched with TEs and immune genes. SETDB1 knockout recovers the potential of multiple TEs in encoding retroviral antigens and generating major histocompatibility complex class I (MHCI) peptides and derepresses immunostimulatory genes, thereby triggering T cell responses (Griffin et al., 2021). Moreover, SETDB1 was also found to repress the expression of radiation-induced ERVs, and SETDB1 deletion potently enhanced ERV expression and increased type I interferons, promoting antitumour immunity and sensitizing cancer radiotherapy (Pan et al., 2022).
Besides the ERV-related mechanism, SETDB1 also mediates immune response by regulating cytokine expression and some signalling axes (Hachiya et al., 2016; Lin et al., 2021). In macrophages, SETDB1 effectively inhibits Toll-like receptor 4 (TLR4)-mediated interleukin 6 (IL-6) expression by increasing H3K9 methylation levels on the IL-6 promoter and restraining its transcription. SETDB1 loss reduces H3K9me3 and enhances TLR4-mediated nuclear factor-κB (NF-κB) recruitment to the IL-6 promoter, which increases IL-6 expression in vitro and in vivo (Hachiya et al., 2016). T helper 17 (Th17) cells are implicated in multiple autoimmune diseases and cancers. OX40 inhibits Th17 cell function in a RelB (an NF-κB family member)-dependent manner, which recruits SETDB1 and G9a to the IL-17 locus to induce suppressive chromatin modifications, depressing IL-17 expression (Xiao et al., 2016). Moreover, Lin et al. (2021) reported that ablation of tripartite motif-containing 28 (TRIM28), a component of the human silencing hub (HUSH) complex, substantially boosted PD-L1 expression in ovarian cancer cells. As the catalytic subunit of HUSH, SETDB1 loss phenocopied TRIM28 silencing and elevated PD-L1 expression. Mechanistic studies revealed that SETDB1-TRIM28 suppression accelerated the formation of micronuclei in the cytoplasm and activated the cGAS-STING cascade-mediated innate immune response, thereby promoting PD-L1 expression and CD8+ T cell infiltration. Inconsistently, high levels of SETDB1 expression were positively related to PD-L1 expression in CRC. SETDB1 silencing markedly suppressed PD-L1 expression and boosted the cytotoxicity of T cells to CRC cells through the FOSB/miR-22/BATF3 cascade (Tian et al., 2022). The opposing expression of PD-L1 upon SETDB1 loss in these two studies may be attributed to the distinct tumour types and regulatory mechanisms.
Noteworthily, SETDB1 is also essential for the development of B cells and T cells (Pasquarella et al., 2016; Takikita et al., 2016). SETDB1 deficiency has been reported to impair B-cell development through unfolded protein response (UPR)-mediated pro-B cell apoptosis (Pasquarella et al., 2016). Mechanistically, SETDB1 functions as a repressor of specific ERV families and exogenous retroviruses in mouse B lymphocytes, such as the retrotransposon murine leukaemia virus (MLV), while SETDB1 depletion derepresses the expression of MLV proteins, leading to UPR-mediated apoptosis (Collins et al., 2015; Pasquarella et al., 2016). Likewise, impaired T-cell development was also observed in thymocyte-specific SETDB1 knockout mice, particularly CD8-lineage T cells, which exhibited increased apoptosis (Takikita et al., 2016). This is partially caused by SETDB1 deficiency-mediated derepression of FcγRIIb expression, and FcγRIIb can suppress ERK activation and induce cell apoptosis (Takikita et al., 2016). Martin et al. (2015) also reported the role of SETDB1 repression of FcγRIIb in thymocyte development. They revealed that SETDB1 deletion activated FcγRIIb expression, which then led to increased ZAP70 phosphorylation and apoptosis of CD4/CD8 double-positive T cells. Additionally, Jarid2 directly interacts with SETDB1 and recruits it to the Zbtb16 locus that encodes promyelocytic leukemia zinc finger (PLZF) protein, a signature transcription factor of immature invariant natural killer T (iNKT) cells, and its deficiency in thymocytes will affect the proper development of iNKT cells through decreasing H3K9me3 and upregulating PLZF levels (Pereira et al., 2014). These findings reveal the essential role of SETDB1 in immune cell development, which cannot be ignored in SETDB1 targeted therapy.
Given the important role of SETDB1 in tumourigenesis, progression, and tumour immune escape, developing SETDB1 antagonists is a promising strategy for cancer chemotherapy and immunotherapy. However, there are not so many studies concerning SETDB1 antagonists at present, particularly the specific SETDB1 inhibitors. This is due to the lack of SET domain crystal structure, which is challenged by its bifurcated characteristic. Several reported SETDB1 selective inhibitors do not directly target the SET domain and are still in the stage of biochemical activity testing (Table 1). Park et al. (2017) screened peptide-competitive small molecule SETDB1 inhibitors using an approach of pharmacophore model-based virtual screening combined with biological evaluation. Two compounds VH01 and VH06 were identified as the final hits that obviously reduced H3K9me3 levels and had neuronal effects. In the surface plasmon resonance assay, VH01 and VH06 bound to SETDB1 with the KD values of 3.26 ± 1.71 μM and 0.232 ± 0.146 μM, respectively. The tandem Tudor domain (TTD) of SETDB1 can recognize methylated lysine. Specifically intercepting the interaction of SETDB1-TTD with the endogenous binders is an attractive approach for the development of small molecule SETDB1 antagonists (Mader et al., 2019; Guo et al., 2021). (R, R)-59 was a selective small-molecule SETDB1-TTD inhibitor screened based on this concept, which exhibited a KD value of 0.088 ± 0.045 μM in the isothermal titration calorimetry (ITC) detection (Guo et al., 2021). In addition, 5-allyloxy-2-(pyrrolidin-1-yl) quinoline (APQ) was also identified as a negative regulator of SETDB1 activity. It effectively decreased H3K9me3 levels and ameliorated Huntington’s disease symptoms in vitro and in vivo (Hwang et al., 2021).
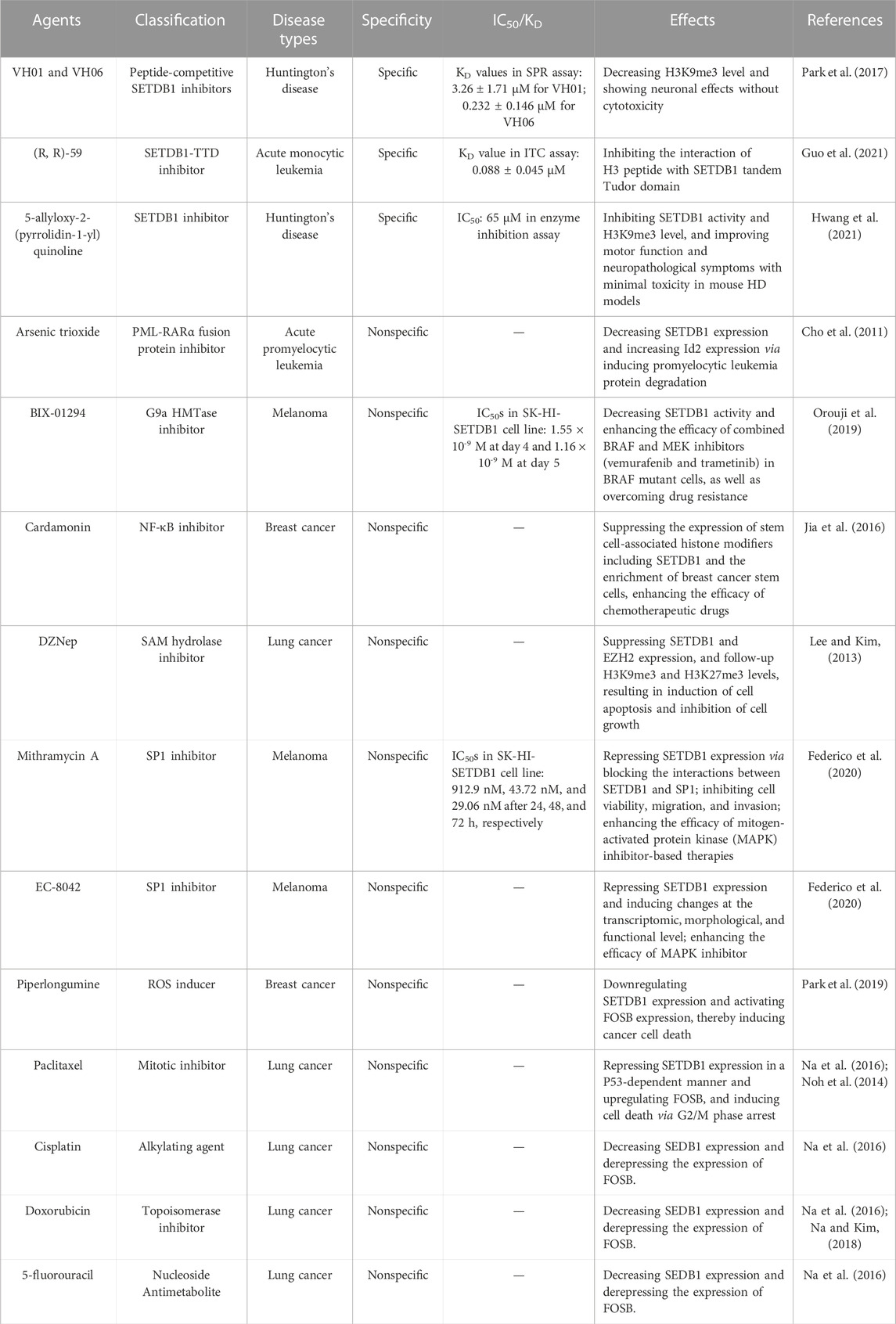
TABLE 1. List of the reported small molecule SETDB1 antagonists.
Recently, most available SETDB1 inhibitors are non-specific (Table 1), and they affect SETDB1 function by inhibiting SETDB1 expression or histone methyltransferase (HMTase) activity. Arsenic trioxide (As2O3) showed inhibitory effect on SETDB1 signalling through inducing the degradation of promyelocytic leukemia protein (Cho et al., 2011). BIX-01294 (CAS 935693-62-2), a G9a HMTase inhibitor, also had the ability to block SETDB1 activity. The combination of BIX-01294 with BRAF and MEK inhibitors exhibited a high level of synergistic effects in killing melanoma cells and even resistant cells (Orouji et al., 2019). Cardamonin could suppress the expression of multiple stem cell-associated histone modifying genes including SETDB1, and prevent the enrichment of breast cancer stem-like cells when combined with chemotherapeutic drugs (Jia et al., 2016). DZNep, a SAM hydrolase inhibitor, efficaciously depressed H3K9me3 and H3K27me3 expression through downregulation of SETDB1 and EZH2 expression. The inhibition of DZNep against multiple HMTases contributed to its anti-proliferative and pro-apoptotic activity on lung cancer cells (Lee and Kim, 2013). Mithramycin A and mithramycin analog EC-8042 were reported to inhibit SETDB1 expression and cause changes at transcriptomic and functional levels. They could sensitize melanoma cells to mitogen-activated protein kinase inhibitor (Ryu et al., 2006; Federico et al., 2020). Piperlongumine (PL), a natural alkaloid compound, was also reported to be potent in downregulating SETDB1 expression in breast cancer cells. Reduced SETDB1 induced caspase 9-dependent PARP cleavage and FOSB expression, which mediated the anti-breast cancer effects of PL (Park et al., 2019). Additionally, several chemotherapeutic agents, including paclitaxel (PTX), cisplatin, doxorubicin, and 5-fluorouracil, were identified as potential antagonists against SETDB1 expression at both transcription and protein levels (Noh et al., 2014; Na et al., 2016; Na and Kim, 2018). Of these, PTX repressed SETDB1 expression in a p53-dependent manner, and PTX-induced p53 expression might involve in heterochromatic suppression on SETDB1 promoter (Noh et al., 2014).
Overall, SETDB1 amplification is closely correlated with poor prognosis of multiple malignancies and contributes to tumourigenesis, tumour progression and immune evasion. This is mainly attributed to H3K9me3 deposition by SETDB1 on tumour-suppressive genes, retrotransposons, and immune clusters. SETDB1 depletion in mouse tumours derepresses immunostimulatory genes and TE-encoded retroviral antigens and MHCI peptides, which will increase CD8+ T cells and p15E-specific CD8+ T cells expressing canonical cytotoxicity genes, triggering cytotoxic T cell responses and sensitizing tumours to ICB (Griffin et al., 2021). Therefore, SETDB1 targeting is an attractive strategy for cancer therapy, particularly in combination with immunotherapy and radiotherapy, because of its regulatory effects on ERVs. Nevertheless, SETDB1-targeted therapy remains challenging.
First, various SETDB1-associated regulatory mechanisms remain unelucidated. As already mentioned, SETDB1 is not positively associated with tumourigenesis and progression of all malignancies, and it may act as a tumour suppressor in certain tumour types or at certain stages. Meanwhile, SETDB1 is essential for immune cell development and mammalian genome stability, and activated ERVs caused by SETDB1 deletion in somatic cells may provoke carcinogenic mutations through ERV insertions. These challenges highlight the need to elucidate the differences and specific mechanisms of SETDB1 interaction with relevant target proteins, ERVs and immune genes across different tumour types and noncancerous cells. Second, SETDB1-targeted therapy is hampered by the lack of SETDB1 inhibitors with high selectivity and activity. The recently reported potent SETDB1 antagonists are mostly non-specific, and since most are cytotoxic chemotherapy drugs, off-target effects and side effects are inevitable. A potential approach to develop selective SETDB1 inhibitors is the specific blocking of SETDB1-TTD interaction. Alternatively, SETDB1 activity could be depressed by the development of selective antagonists against SAM (a methyl donor for SETDB1-mediated methylation). Moreover, the elucidation of SETDB1 regulation-related molecular mechanisms and specific targets, as well as the resolution of SET domain crystal structure will benefit the future development of therapeutics targeting SETDB1.
In summary, SETDB1 is considered an oncogene in multiple malignant tumours. Although challenged by its functions in normal cells and potential side effects, increasing evidence nevertheless strongly supports SETDB1 as a candidate target for cancer therapy, particularly immunotherapy. A deeper understanding of the molecular mechanisms of SETDB1 that regulate ERVs and immune genes in both tumour and normal cells and the availability of more selective and effective SETDB1 inhibitors may lead to SETDB1 targeting as a promising antitumour epigenetic therapy.
Conception and design: LZ, ZZ, and LF; acquisition of data: ZZ, LF, XP, and TM; analysis and interpretation of data: LZ and ZZ; writing and review of the manuscript: LZ, ZZ, and LF; study supervision and funding acquisition: LZ and RT.
This work was supported by the Research Funds of Science & Technology Department of Sichuan Province (2021YJ0134, 2019YFS0514), the Research Project of Chengdu Science and Technology Bureau (2022-YF05-01611-SN), the National Key Research and Development Program of China (2020YFC2005500), and the Open Project of Radiation Oncology Key Laboratory of Sichuan Province (2020FSZLX04).
We sincerely thank all of our colleagues for their generous supports.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.1073713/full#supplementary-material
Ayyanathan, K., Lechner, M. S., Bell, P., Maul, G. G., Schultz, D. C., Yamada, Y., et al. (2003). Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: A mammalian cell culture model of gene variegation. Genes Dev. 17 (15), 1855–1869. doi:10.1101/gad.1102803
Bagchi, S., Yuan, R., and Engleman, E. G. (2021). Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu. Rev. Pathol. 16, 223–249. doi:10.1146/annurev-pathol-042020-042741
Batham, J., Lim, P. S., and Rao, S. (2019). SETDB-1: A potential epigenetic regulator in breast cancer metastasis. Cancers (Basel) 11 (8), 1143. doi:10.3390/cancers11081143
Cao, J., and Yan, Q. (2020). Cancer epigenetics, tumor immunity, and immunotherapy. Trends Cancer 6 (7), 580–592. doi:10.1016/j.trecan.2020.02.003
Cao, N., Yu, Y., Zhu, H., Chen, M., Chen, P., Zhuo, M., et al. (2020). SETDB1 promotes the progression of colorectal cancer via epigenetically silencing p21 expression. Cell Death Dis. 11 (5), 351. doi:10.1038/s41419-020-2561-6
Ceol, C. J., Houvras, Y., Jane-Valbuena, J., Bilodeau, S., Orlando, D. A., Battisti, V., et al. (2011). The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471 (7339), 513–517. doi:10.1038/nature09806
Chen, D. S., and Mellman, I. (2017). Elements of cancer immunity and the cancer-immune set point. Nature 541 (7637), 321–330. doi:10.1038/nature21349
Cheng, Y., He, C., Wang, M., Ma, X., Mo, F., Yang, S., et al. (2019). Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 4, 62. doi:10.1038/s41392-019-0095-0
Cho, S., Park, J. S., and Kang, Y.-K. (2011). Dual functions of histone-lysine N-methyltransferase Setdb1 protein at promyelocytic leukemia-nuclear body (PML-NB): Maintaining PML-NB structure and regulating the expression of its associated genes. J. Biol. Chem. 286 (47), 41115–41124. doi:10.1074/jbc.M111.248534
Cho, S., Park, J. S., and Kang, Y.-K. (2013). Regulated nuclear entry of over-expressed Setdb1. Genes 18 (8), 694–703. doi:10.1111/gtc.12068
Collins, P. L., Kyle, K. E., Egawa, T., Shinkai, Y., and Oltz, E. M. (2015). The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 112 (27), 8367–8372. doi:10.1073/pnas.1422187112
Cuellar, T. L., Herzner, A.-M., Zhang, X., Goyal, Y., Watanabe, C., Friedman, B. A., et al. (2017). Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 216 (11), 3535–3549. doi:10.1083/jcb.201612160
Deng, H., and Zhang, Z. (2018). The application of nanotechnology in immune checkpoint blockade for cancer treatment. J. Control. Release 290, 28–45. doi:10.1016/j.jconrel.2018.09.026
Dhillon, S., and Duggan, S. (2022). Sugemalimab: First approval. Drugs 82 (5), 593–599. doi:10.1007/s40265-022-01693-4
Dhillon, S. (2021). Penpulimab: First approval. Drugs 81 (18), 2159–2166. doi:10.1007/s40265-021-01640-9
Fanale, D., Dimino, A., Pedone, E., Brando, C., Corsini, L. R., Filorizzo, C., et al. (2022). Prognostic and predictive role of tumor-infiltrating lymphocytes (TILs) in ovarian cancer. Cancers (Basel) 14 (18), 4344. doi:10.3390/cancers14184344
Federico, A., Steinfass, T., Larribère, L., Novak, D., Morís, F., Núñez, L.-E., et al. (2020). Mithramycin A and mithralog EC-8042 inhibit SETDB1 expression and its oncogenic activity in malignant melanoma. Mol. Ther. Oncolytics 18, 83–99. doi:10.1016/j.omto.2020.06.001
Galluzzi, L., Chan, T. A., Kroemer, G., Wolchok, J. D., and López-Soto, A. (2018). The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 10 (459), eaat7807. doi:10.1126/scitranslmed.aat7807
Gangoso, E., Southgate, B., Bradley, L., Rus, S., Galvez-Cancino, F., McGivern, N., et al. (2021). Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 184 (9), 2454–2470.e26. doi:10.1016/j.cell.2021.03.023
Gomez, S., Tabernacki, T., Kobyra, J., Roberts, P., and Chiappinelli, K. B. (2020). Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. 65, 99–113. doi:10.1016/j.semcancer.2019.12.019
Griffin, G. K., Wu, J., Iracheta-Vellve, A., Patti, J. C., Hsu, J., Davis, T., et al. (2021). Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 595 (7866), 309–314. doi:10.1038/s41586-021-03520-4
Guler, G. D., Tindell, C. A., Pitti, R., Wilson, C., Nichols, K., KaiWai Cheung, T., et al. (2017). Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer Cell 32 (2), 221–237. doi:10.1016/j.ccell.2017.07.002
Guo, Y., Mao, X., Xiong, L., Xia, A., You, J., Lin, G., et al. (2021). Structure-guided discovery of a potent and selective cell-active inhibitor of SETDB1 tudor domain. Angew. Chem. Int. Ed. Engl. 60 (16), 8760–8765. doi:10.1002/anie.202017200
Hachiya, R., Shiihashi, T., Shirakawa, I., Iwasaki, Y., Matsumura, Y., Oishi, Y., et al. (2016). The H3K9 methyltransferase Setdb1 regulates TLR4-mediated inflammatory responses in macrophages. Sci. Rep. 6, 28845. doi:10.1038/srep28845
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Härtlova, A., Erttmann, S. F., Raffi, F. A., Schmalz, A. M., Resch, U., Anugula, S., et al. (2015). DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42 (2), 332–343. doi:10.1016/j.immuni.2015.01.012
Ho, K. L., McNae, I. W., Schmiedeberg, L., Klose, R. J., Bird, A. P., and Walkinshaw, M. D. (2008). MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol. Cell 29 (4), 525–531. doi:10.1016/j.molcel.2007.12.028
Hu, H., Khodadadi-Jamayran, A., Dolgalev, I., Cho, H., Badri, S., Chiriboga, L. A., et al. (2021). Targeting the atf7ip-setdb1 complex augments antitumor immunity by boosting tumor immunogenicity. Cancer Immunol. Res. 9 (11), 1298–1315. doi:10.1158/2326-6066.CIR-21-0543
Hwang, Y. J., Hyeon, S. J., Kim, Y., Lim, S., Lee, M. Y., Kim, J., et al. (2021). Modulation of SETDB1 activity by APQ ameliorates heterochromatin condensation, motor function, and neuropathology in a Huntington's disease mouse model. J. Enzyme Inhib. Med. Chem. 36 (1), 856–868. doi:10.1080/14756366.2021.1900160
Hyun, K., Jeon, J., Park, K., and Kim, J. (2017). Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49 (4), e324. doi:10.1038/emm.2017.11
Ishimoto, K., Kawamata, N., Uchihara, Y., Okubo, M., Fujimoto, R., Gotoh, E., et al. (2016). Ubiquitination of lysine 867 of the human SETDB1 protein upregulates its histone H3 lysine 9 (H3K9) methyltransferase activity. PLoS One 11 (10), e0165766. doi:10.1371/journal.pone.0165766
Jia, D., Tan, Y., Liu, H., Ooi, S., Li, L., Wright, K., et al. (2016). Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget 7 (1), 771–785. doi:10.18632/oncotarget.5819
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128 (4), 693–705. doi:10.1016/j.cell.2007.02.005
Lee, A. (2022). Serplulimab: First approval. Drugs 82 (10), 1137–1141. doi:10.1007/s40265-022-01740-0
Lee, J.-K., and Kim, K.-C. (2013). DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem. Biophys. Res. Commun. 438 (4), 647–652. doi:10.1016/j.bbrc.2013.07.128
Li, H., Rauch, T., Chen, Z.-X., Szabó, P. E., Riggs, A. D., and Pfeifer, G. P. (2006). The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 281 (28), 19489–19500. doi:10.1074/jbc.M513249200
Lin, J., Guo, D., Liu, H., Zhou, W., Wang, C., Müller, I., et al. (2021). The SETDB1-TRIM28 complex suppresses antitumor immunity. Cancer Immunol. Res. 9 (12), 1413–1424. doi:10.1158/2326-6066.CIR-21-0754
Liu, R., Hong, R., Wang, Y., Gong, Y., Yeerken, D., Yang, D., et al. (2020). Defect of SLC38A3 promotes epithelial-mesenchymal transition and predicts poor prognosis in esophageal squamous cell carcinoma. Chin. J. Cancer Res. 32 (5), 547–563. doi:10.21147/j.issn.1000-9604.2020.05.01
Liu, Z., Liu, J., Ebrahimi, B., Pratap, U. P., He, Y., Altwegg, K. A., et al. (2022). SETDB1 interactions with PELP1 contributes to breast cancer endocrine therapy resistance. Breast Cancer Res. 24 (1), 26. doi:10.1186/s13058-022-01520-4
Mader, P., Mendoza-Sanchez, R., Iqbal, A., Dong, A., Dobrovetsky, E., Corless, V. B., et al. (2019). Identification and characterization of the first fragment hits for SETDB1 Tudor domain. Bioorg. Med. Chem. 27 (17), 3866–3878. doi:10.1016/j.bmc.2019.07.020
Markham, A. (2022). Envafolimab: First approval. Drugs 82 (2), 235–240. doi:10.1007/s40265-022-01671-w
Markouli, M., Strepkos, D., Chlamydas, S., and Piperi, C. (2021a). Histone lysine methyltransferase SETDB1 as a novel target for central nervous system diseases. Prog. Neurobiol. 200, 101968. doi:10.1016/j.pneurobio.2020.101968
Markouli, M., Strepkos, D., and Piperi, C. (2021b). Structure, activity and function of the SETDB1 protein methyltransferase. Life (Basel) 11 (8), 817. doi:10.3390/life11080817
Martin, F. J., Xu, Y., Lohmann, F., Ciccone, D. N., Nicholson, T. B., Loureiro, J. J., et al. (2015). KMT1E-mediated chromatin modifications at the FcγRIIb promoter regulate thymocyte development. Genes Immun. 16 (2), 162–169. doi:10.1038/gene.2014.70
Na, H.-H., and Kim, K.-C. (2018). SETDB1-mediated FosB regulation via ERK2 is associated with an increase in cell invasiveness during anticancer drug treatment of A549 human lung cancer cells. Biochem. Biophys. Res. Commun. 495 (1), 512–518. doi:10.1016/j.bbrc.2017.10.176
Na, H.-H., Noh, H.-J., Cheong, H.-M., Kang, Y., and Kim, K.-C. (2016). SETDB1 mediated FosB expression increases the cell proliferation rate during anticancer drug therapy. BMB Rep. 49 (4), 238–243. doi:10.5483/bmbrep.2016.49.4.031
Noh, H.-J., Kim, K.-A., and Kim, K.-C. (2014). p53 down-regulates SETDB1 gene expression during paclitaxel induced-cell death. Biochem. Biophys. Res. Commun. 446 (1), 43–48. doi:10.1016/j.bbrc.2014.02.053
Ogawa, S., Fukuda, A., Matsumoto, Y., Hanyu, Y., Sono, M., Fukunaga, Y., et al. (2020). SETDB1 inhibits p53-mediated apoptosis and is required for formation of pancreatic ductal adenocarcinomas in mice. Gastroenterology 159 (2), 682–696. e613. doi:10.1053/j.gastro.2020.04.047
Orouji, E., Federico, A., Larribère, L., Novak, D., Lipka, D. B., Assenov, Y., et al. (2019). Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int. J. Cancer 145 (12), 3462–3477. doi:10.1002/ijc.32432
Pan, D., Bao, X., Hu, M., Jiao, M., Li, F., and Li, C.-Y. (2022). SETDB1 restrains endogenous retrovirus expression and antitumor immunity during radiotherapy. Cancer Res. 82 (15), 2748–2760. doi:10.1158/0008-5472.CAN-21-3523
Park, I., Hwang, Y. J., Kim, T., Viswanath, A. N. I., Londhe, A. M., Jung, S. Y., et al. (2017). In silico probing and biological evaluation of SETDB1/ESET-targeted novel compounds that reduce tri-methylated histone H3K9 (H3K9me3) level. J. Comput. Aided. Mol. Des. 31 (10), 877–889. doi:10.1007/s10822-017-0052-3
Park, J.-A., Na, H.-H., Jin, H.-O., and Kim, K.-C. (2019). Increased expression of FosB through reactive oxygen species accumulation functions as pro-apoptotic protein in piperlongumine treated MCF7 breast cancer cells. Mol. Cells 42 (12), 884–892. doi:10.14348/molcells.2019.0088
Pasquarella, A., Ebert, A., Pereira de Almeida, G., Hinterberger, M., Kazerani, M., Nuber, A., et al. (2016). Retrotransposon derepression leads to activation of the unfolded protein response and apoptosis in pro-B cells. Development 143 (10), 1788–1799. doi:10.1242/dev.130203
Pereira, R. M., Martinez, G. J., Engel, I., Cruz-Guilloty, F., Barboza, B. A., Tsagaratou, A., et al. (2014). Jarid2 is induced by TCR signalling and controls iNKT cell maturation. Nat. Commun. 5, 4540. doi:10.1038/ncomms5540
Ropa, J., Saha, N., Hu, H., Peterson, L. F., Talpaz, M., and Muntean, A. G. (2020). SETDB1 mediated histone H3 lysine 9 methylation suppresses MLL-fusion target expression and leukemic transformation. Haematologica 105 (9), 2273–2285. doi:10.3324/haematol.2019.223883
Roulois, D., Loo Yau, H., Singhania, R., Wang, Y., Danesh, A., Shen, S. Y., et al. (2015). DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162 (5), 961–973. doi:10.1016/j.cell.2015.07.056
Ryu, H., Lee, J., Hagerty, S. W., Soh, B. Y., McAlpin, S. E., Cormier, K. A., et al. (2006). ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc. Natl. Acad. Sci. U. S. A. 103 (50), 19176–19181. doi:10.1073/pnas.0606373103
Schoenfeld, A. J., and Hellmann, M. D. (2020). Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37 (4), 443–455. doi:10.1016/j.ccell.2020.03.017
Schultz, D. C., Ayyanathan, K., Negorev, D., Maul, G. G., and Rauscher, F. J. (2002). SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16 (8), 919–932. doi:10.1101/gad.973302
Song, X., Si, Q., Qi, R., Liu, W., Li, M., Guo, M., et al. (2021). Indoleamine 2, 3-dioxygenase 1: A promising therapeutic target in malignant tumor. Front. Immunol. 12, 800630. doi:10.3389/fimmu.2021.800630
Strepkos, D., Markouli, M., Klonou, A., Papavassiliou, A. G., and Piperi, C. (2021). Histone methyltransferase SETDB1: A common denominator of tumorigenesis with therapeutic potential. Cancer Res. 81 (3), 525–534. doi:10.1158/0008-5472.CAN-20-2906
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339 (6121), 786–791. doi:10.1126/science.1232458
Takikita, S., Muro, R., Takai, T., Otsubo, T., Kawamura, Y. I., Dohi, T., et al. (2016). A histone methyltransferase ESET is critical for T cell development. J. Immunol. 197 (6), 2269–2279. doi:10.4049/jimmunol.1502486
Tian, J., Wang, W., Zhu, J., Zhuang, Y., Qi, C., Cai, Z., et al. (2022). Histone methyltransferase SETDB1 promotes immune evasion in colorectal cancer via FOSB-mediated downregulation of MicroRNA-22 through BATF3/PD-L1 pathway. J. Immunol. Res. 2022, 4012920. doi:10.1155/2022/4012920
Tie, C. H., and Rowe, H. M. (2017). Epigenetic control of retrotransposons in adult tissues: Implications for immune regulation. Curr. Opin. Virol. 25, 28–33. doi:10.1016/j.coviro.2017.06.007
Topper, M. J., Vaz, M., Marrone, K. A., Brahmer, J. R., and Baylin, S. B. (2020). The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 17 (2), 75–90. doi:10.1038/s41571-019-0266-5
Tran, E., Robbins, P. F., and Rosenberg, S. A. (2017). 'Final common pathway' of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 18 (3), 255–262. doi:10.1038/ni.3682
Verdegaal, E. M. E., de Miranda, N. F. C. C., Visser, M., Harryvan, T., van Buuren, M. M., Andersen, R. S., et al. (2016). Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536 (7614), 91–95. doi:10.1038/nature18945
Wang, G., Long, J., Gao, Y., Zhang, W., Han, F., Xu, C., et al. (2019). SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 21 (2), 214–225. doi:10.1038/s41556-018-0266-1
Wang, H., An, W., Cao, R., Xia, L., Erdjument-Bromage, H., Chatton, B., et al. (2003). mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell 12 (2), 475–487. doi:10.1016/j.molcel.2003.08.007
Wong, C.-M., Wei, L., Law, C.-T., Ho, D. W.-H., Tsang, F. H.-C., Au, S. L.-K., et al. (2016). Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology 63 (2), 474–487. doi:10.1002/hep.28304
Wu, P.-C., Lu, J.-W., Yang, J.-Y., Lin, I. H., Ou, D.-L., Lin, Y.-H., et al. (2014). H3K9 histone methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3 pathway to suppress lung cancer metastasis. Cancer Res. 74 (24), 7333–7343. doi:10.1158/0008-5472.CAN-13-3572
Wu, Y., Wu, X., and Li, S. (2021). Retrotransposons: Jump to cancer? Trends Cancer 7 (7), 577–579. doi:10.1016/j.trecan.2021.04.004
Xiao, X., Shi, X., Fan, Y., Wu, C., Zhang, X., Minze, L., et al. (2016). The costimulatory receptor OX40 inhibits interleukin-17 expression through activation of repressive chromatin remodeling pathways. Immunity 44 (6), 1271–1283. doi:10.1016/j.immuni.2016.05.013
Yang, L., Xia, L., Wu, D. Y., Wang, H., Chansky, H. A., Schubach, W. H., et al. (2002). Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 21 (1), 148–152. doi:10.1038/sj.onc.1204998
Yi, M., Zheng, X., Niu, M., Zhu, S., Ge, H., and Wu, K. (2022). Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 21 (1), 28. doi:10.1186/s12943-021-01489-2
Young, A., Ngiow, S. F., Barkauskas, D. S., Sult, E., Hay, C., Blake, S. J., et al. (2016). Co-Inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 30 (3), 391–403. doi:10.1016/j.ccell.2016.06.025
Yu, L., Ye, F., Li, Y.-Y., Zhan, Y.-Z., Liu, Y., Yan, H.-M., et al. (2020). Histone methyltransferase SETDB1 promotes colorectal cancer proliferation through the STAT1-CCND1/CDK6 axis. Carcinogenesis 41 (5), 678–688. doi:10.1093/carcin/bgz131
Zhang, S.-M., Cai, W. L., Liu, X., Thakral, D., Luo, J., Chan, L. H., et al. (2021). KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 598 (7882), 682–687. doi:10.1038/s41586-021-03994-2
Keywords: SETDB1, immune escape, immune checkpoint blockade resistance, endogenous retrovirus, epigenetic therapy
Citation: Zhao Z, Feng L, Peng X, Ma T, Tong R and Zhong L (2022) Role of histone methyltransferase SETDB1 in regulation of tumourigenesis and immune response. Front. Pharmacol. 13:1073713. doi: 10.3389/fphar.2022.1073713
Received: 18 October 2022; Accepted: 29 November 2022;
Published: 13 December 2022.
Edited by:
Ting Wang, Sichuan Cancer Hospital, ChinaReviewed by:
Xiaonan Zhang, Uppsala University, SwedenCopyright © 2022 Zhao, Feng, Peng, Ma, Tong and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhong, emhsNzIzNTMwMUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.