 Rajashri R. Naik
Rajashri R. Naik Ashok K. Shakya
Ashok K. Shakya
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 09 January 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1064472
This article is part of the Research Topic Pharmacological and Biochemical Perspectives of Kinase Inhibitors in Cancer and COVID-19 Therapeutics, Volume II View all 7 articles
Protein kinases are enzymes that transfer phosphate to protein, resulting in the modification of the protein. The human genome encodes approximately 538 kinases. Kinases play a role in maintaining a number of cellular processes, including control of the cell cycle, metabolism, survival, and differentiation. Protein kinase dysregulation causes several diseases, and it has been shown that numerous kinases are deregulated in cancer. The oncogenic potential of these kinases is increased by a number of processes, including overexpression, relocation, fusion point mutations, and the disruption of upstream signaling. Understanding of the mechanism or role played by kinases has led to the development of a large number of kinase inhibitors with promising clinical benefits. In this review, we discuss FDA-approved kinase inhibitors and their mechanism, clinical benefits, and side effects, as well as the challenges of overcoming some of their side effects and future prospects for new kinase inhibitor discovery.
Kinases are enzymes that transfer the phosphate group from ATP to a specific substrate during phosphorylation. Depending on the nature of the chemical, the kinases involved may be protein kinases, lipid kinases, carbohydrate kinases, nucleophosphate kinases, or nucleoside diphosphate kinases. The phosphorylatable sites in eukaryotes include serine (ser) threonine (Thr), and tyrosine (tyr) (Adam and Hunter, 2018). Other amino acids that are phosphorylatable include arginine (Arg), Lysine (Lys), and cysteine (Cys) (Ciesla et al., 2011). Phosphorylation alters the substrate’s functionality by controlling the signaling pathway through cellular location, amplification, or interactions with regulatory proteins. In the human genome, protein kinases account for approximately 2% of the genetic material. These kinases are further classified into groups, families, and subfamilies (Manning et al., 2002a; Manning et al., 2002b). More than 500 protein kinases have been identified and their conformational changes and structure have been identified using x-ray crystallography. Protein kinases play a vital role in various cellular functions, such as metabolism, cell cycle regulation, cell survival, and cell differentiation. The target proteins undergo conformational changes as a result of the kinases’ activation, which include the phosphorylation of substrates (amino acids) such as serine, threonine, and tyrosine (Johnson and Lewis, 2001). The process of phosphorylation by kinase on the substrate (target proteins) is a controlled process, and any changes in the regulatory processes leads to disease. There are several mechanisms that lead to kinase dysregulation, such as overexpression, fusion point mutation, and dysregulation of signaling pathway, and these all increase the oncogenic capabilities of the kinases. Deregulation can lead to altered expression of a kinase and change its function, as well as affect its initiation and survival. One example of deregulation is BRAF, a proto-oncogene mutant that encodes serine/threonine protein kinase B-Raf and is found in 40%–50% of melanoma cases (Dankner et al., 2018). V600E is the mutation in BRAF that makes MAPK (mitogen-activated protein kinase) constitutively active, resulting in enhanced cell proliferation of the cell (Śmiech et al., 2020). Epidermal Growth Factor Receptor (EGFR) is another oncogene that belongs to the ErbB family of tyrosine kinases. Mutation in EGFR and overexpression of EGFR protein leads to the dysregulation of signals that favor tumor cells in terms of proliferation, survival, and metastasis (Thomas and Weihua, 2019).
The development of small molecules to target kinases has significantly increased due to the success of imatinib, the first tyrosine kinase inhibitor, which targeted the Bcr-Abl receptor. Since its discovery 20 years ago, large numbers of small-molecule kinase inhibitors have been developed to treat cancer. It was the first tyrosine kinase inhibitor to be approved by the US FDA (in 2001) for treating chronic myelogenous leukemia. In this review, we will discuss the different types of KI, examples of FDA-approved kinase inhibitors, some of the clinical trials that led to their approval, their side effects, and their limitations. Finally, we will emphasize how some of these limitations can be overcome by understanding the mechanism that leads to resistance.
There are many kinase inhibitors (Tables 1–Tables 3), which depending on their mode of target binding and mechanism of action can be divided into seven types. The different types of inhibitor, along with an example and mode of action, are summarized in Table 4.
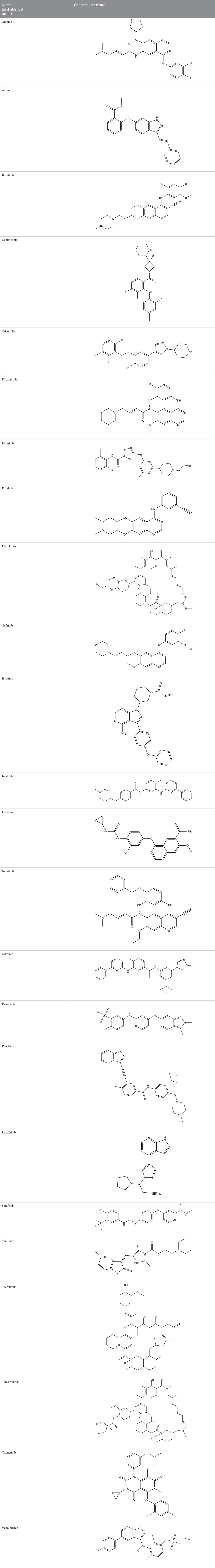
TABLE 1. Chemical structures of tyrosine kinase inhibitors used in chemotherapy.
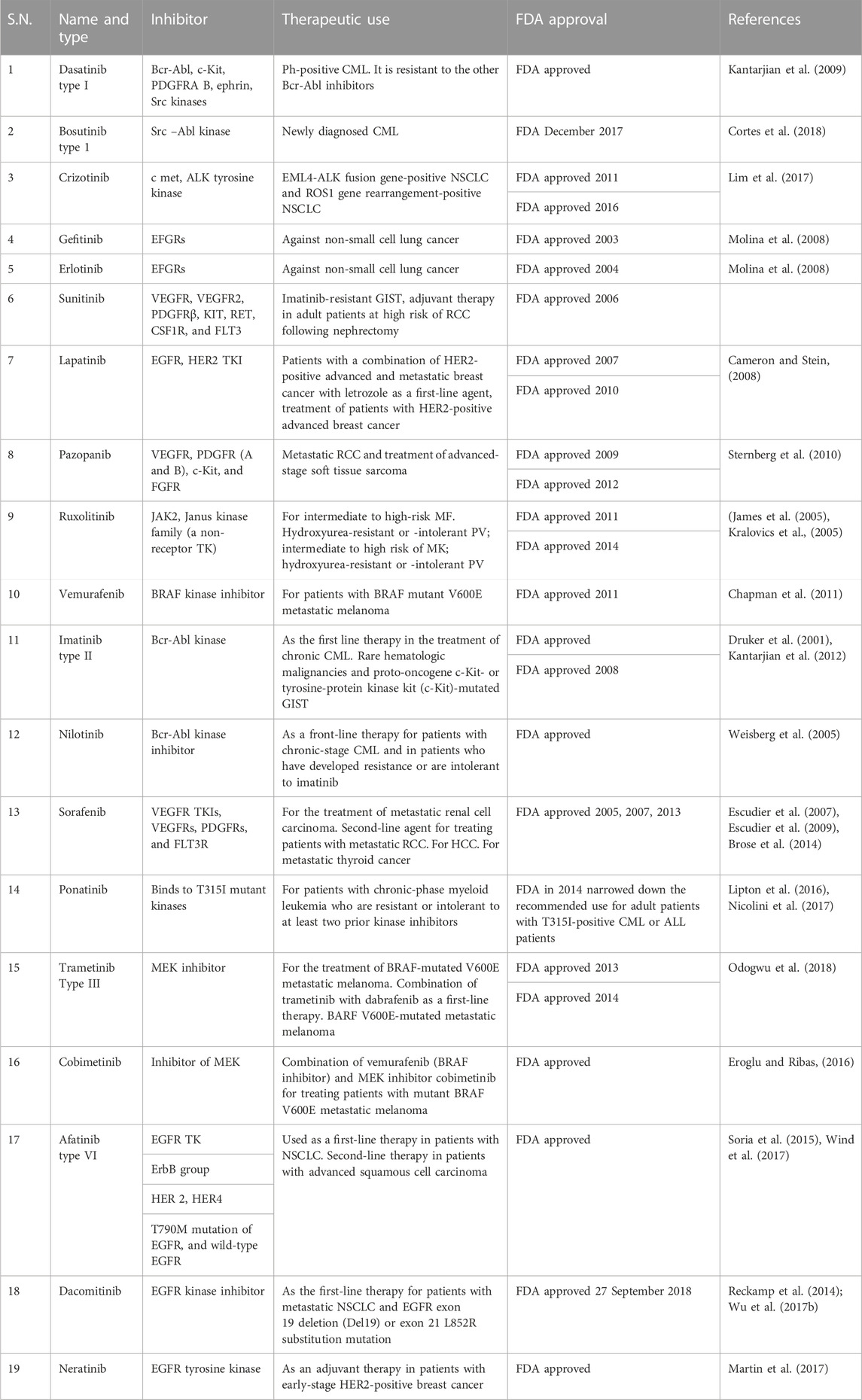
TABLE 2. Therapeutic uses of FDA-approved kinase inhibitors.
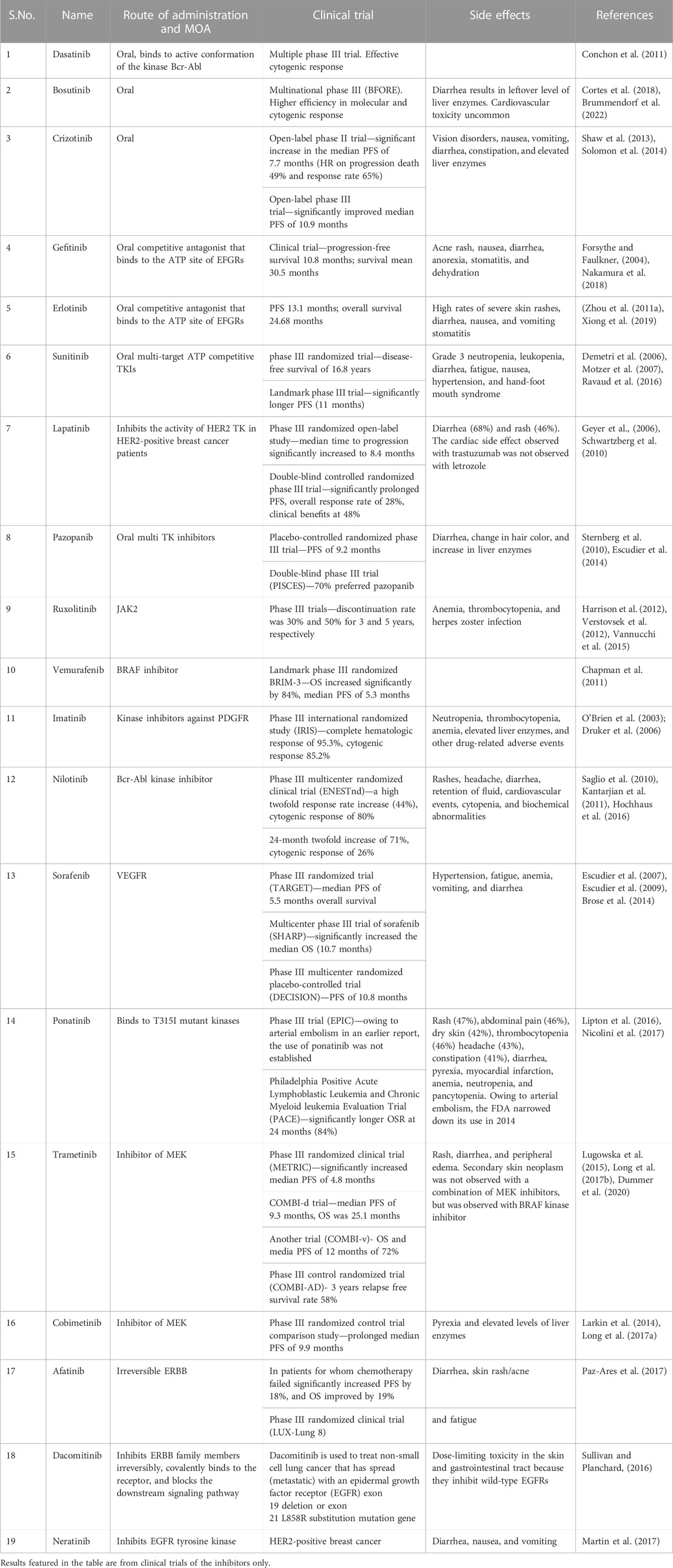
TABLE 3. Mechanism of action, results of clinical trials, and side effects of kinase inhibitors.
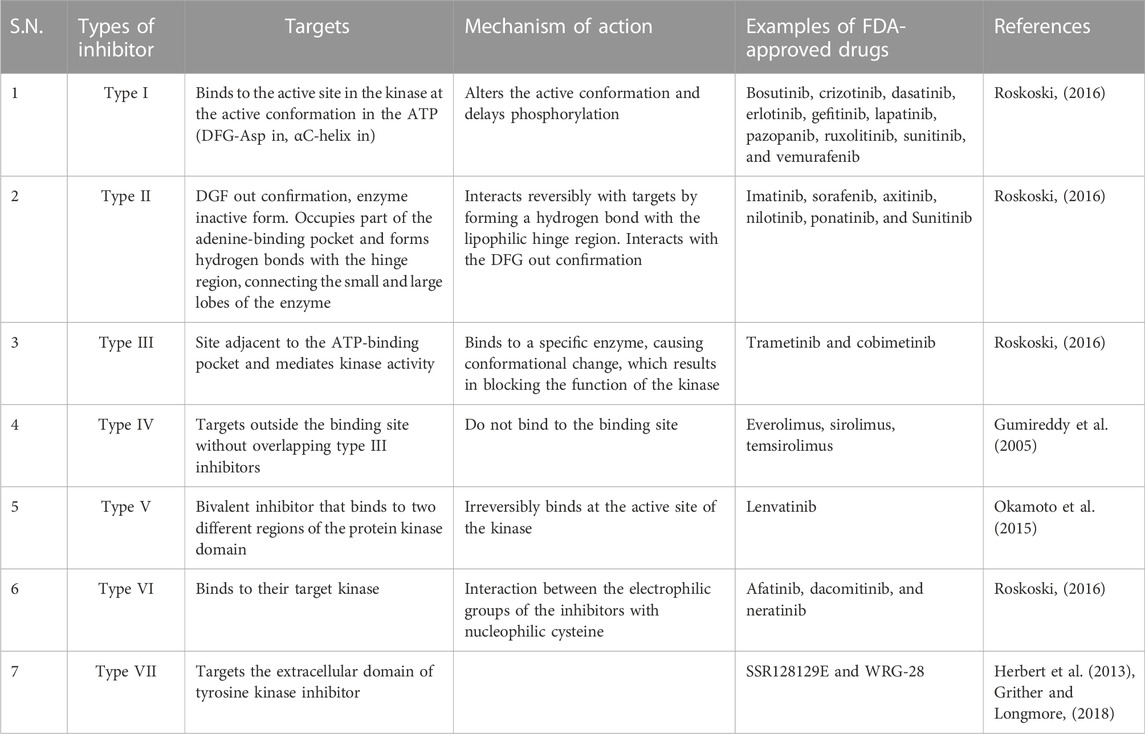
TABLE 4. Targets and mechanism of action of different types of FDA-approved tyrosine kinase inhibitors.
Type I inhibitors bind to the active conformation of the kinase in the ATP (DFG-IN). This binding can alter the active conformation and delay phosphorylation. Currently, there are 10 FDA-approved tyrosine kinase inhibitors: bosutinib, crizotinib, dasatinib, erlotinib, gefitinib, lapatinib, pazopanib, ruxolitinib, sunitinib, and vemurafenib.
Type II inhibitors target the DFG-out conformation, the inactive form of the enzyme (Kufareva and Abagyan, 2008). They interact reversibly with the targets by forming a hydrogen bond with the lipophilic hinge region. The high specificity of type II inhibitors is due to their lipophilic interactions, which contribute to the decreased toxicity of type II inhibitors compared with type I inhibitors (Bhullar et al., 2018). Imatinib, sorafenib, axitinib, nilotinib, ponatinib, and sunitinib are examples of type II kinase inhibitors.
Type III inhibitors bind to the site adjacent to the ATP-binding pocket and mediate kinase activity (Amato et al., 2014). They are non-competitive inhibitors and bind to a specific kinase and exhibit high specificity. Binding to a specific enzyme causes conformational change that blocks the function of kinase. Type III inhibitors are classified into two subtypes: Type IIIA inhibitors bind to the adenine-binding site next to the ATP-binding site, and type IIIB inhibitors bind to other sites, then the site elsewhere. MEK1/2 are well-known type III inhibitors that bind to the cavity adjacent to the binding site of ATP. FDA-approved type III kinase inhibitors include trametinib and cobimetinib (Fasano et al., 2014).
This type of inhibitor binds sites that are far removed from the ATP-binding site without overlapping with type III inhibitors. Everolimus, sirolimus, and temsirolimus are all examples of FDA-approved type IV kinase inhibitors (Gumireddy et al., 2005).
Type V inhibitors are bivalent and bind to the active site of the kinase irreversibly. These types of inhibitors are generally specific and potent.
This type of inhibitor binds to their target kinase through interaction between the electrophilic groups of the inhibitors with a nucleophilic cysteine. FDA-approved drugs that belong to this group include afatinib, dacomitinib, and neratinib, which all target EGFR (Roskoski, 2019). Neratinib is a recently approved kinase inhibitor that inhibits human epidermal growth factor receptor two and prevents the recurrence of early stage HER2-positive breast cancer in patients (Rabindran et al., 2004).
These types of inhibitors are defined as non-classical allosteric inhibitors that target the extracellular domain of tyrosine kinase inhibitors. They do not directly block the binding of the kinase domain/ligand-polypeptide site and are smaller than other inhibitors. Type VII inhibitors include SSR128129E (Herbert et al., 2013), which targets the fibroblast growth factor receptor, and WRG-28, which inhibits discoidin domain receptors (DDRs) (Grither and Longmore, 2018).
Since the discovery of imatinib, several TKIs have been developed to inhibit Bcr-Abl kinases. These second generation TKIs were found to be more effective in treating patients whose treatment with imatinib failed or are resistant to imatinib (Shah et al., 2004). They are more potent at inhibiting Bcr-Abl than imatinib and nilotinib. Along with Bcr-Abl, c-Kit, PDGFRA, and B (the ephrin kinases), dasatinib also inhibits Src-kinase, which is associated with imatinib resistance. Dasatininb binds to the active conformation of the kinase Bcr-Abl. In vitro studies have shown that it can inhibit the activity of 14 out of 15 Bcr-Abl kinases, except for the mutant kinase T315I; therefore, it should be more efficient than imatinib. It may also be more efficient against imatinib-resistant CML (Shah et al., 2004). Dasatinib is approved by the FDA for treating Ph-positive CML, which is resistant to other Bcr-Abl inhibitors developed earlier. Various phase III clinical trials have shown a beneficial effect of dasatinib in CML patients who were resistant or intolerant to imatinib. Dasatinib and imatinib have been compared as first-line treatments (Kantarjian et al., 2009). Dasatinib was shown to have superior cytogenic responses to nilotinib, less efficient transformation to rapid or blast phase transformation, and higher PFS and OS rates (Kantarjian et al., 2009).
Bosutinib is an orally administered third generation tyrosine kinase inhibitor. It is an Src-Abl kinase inhibitor like dasatinib but is more potent in inhibiting Bcr-Abl than imatinib. However, it does not have the potency to inhibit the activity of c-Kit or PDGFR kinase. The side effects of imatinib are caused by its inhibitory activity of c-Kit and PDGFR. A third generation TKI called bosutinib was designed as a result and has the same effectiveness as imatinib but with lesser side effects and a better safety profile than earlier Bcr-Abl inhibitors (Golas et al., 2003; Kantarjian et al., 2012). Bosutinib was efficacious at treating patients who were resistant to imatinib or intolerant to imatinib in phase I and II clinical trials (Kantarjian et al., 2014).
Bosutinib and imatinib were compared for their efficacy in treating newly diagnosed CML patients in a multinational phase III trial of the bosutinib-line in first-line chronic myelogenous leukemia treatment (B-FORE). Over 12 months, bosutinib exhibited higher efficiency with respect to the rate of molecular response (47.2% in the bosutinib group vs 39.9% in the imatinib group, p = 0.02). Similar results were observed in the complete cytogenic response (77.2% in the bosutinib group vs. 66.4% in the imatinib group, p = 0.0078). Based on the encouraging results of this trial, in December 2017, the FDA approved bosutinib for the treatment of newly diagnosed CML (Brümmendorf et al., 2015; Cortes et al., 2018).
Bosutinib has been found to be superior to other tyrosine kinase inhibitors because of its distinct side effects. The commonly encountered side effect with bosutinib is diarrhea, which can be addressed by a supportive measure, and leftover liver enzymes. Additionally, it is noteworthy that cardiovascular toxicity is rare with bosutinib (Brümmendorf et al., 2015; Cortes et al., 2018).
Crizotinib was originally developed to inhibit c-Met. It also inhibits ALK tyrosine kinase activity and has been shown to be effective in NCSLC patients with an ALK fusion gene (Kwon and Meagher, 2012). Crizotinib has a beneficial effect on ROS-1 gene rearrangement, resulting in positive NSCLC due to its sequence similarity to the AKL and c-ros oncogene (ROS1) (Lim et al., 2017). Crizotinib was authorized by the FDA in 2011 for NSCLC with the EML4-ALK fusion gene, and again in 2016 for NSCLC with the ROS1 gene rearrangement.
In an open-label phase II trial, crizotinib was tested against pemetrexed in patients with NSCLC positive for the EML4-ALK fusion gene. A significant increase was observed in the median PFS in the crizotinib group (7.7 months) compared with pemetrexed (3 months). The HR on progression death with crizotinib was 49% and the response rate was 65%, compared with 20% for pemetrexed (Shaw et al., 2013).
Criozotinib was compared with chemotherapy (pemetrex plus cisplatin or carboplatin) in an open-label phase III randomized clinical trial in patients with ALK-positive NSCLC (profile 1,014). Crizotinib significantly improved the median PFS (10.9 months) compared with chemotherapy (7 months), and it had a risk of progression or death of 0.45 (CI 0.35–0.60, p < 0.001) (Solomon et al., 2014). This kinase inhibitor can cause vision problems, nausea, vomiting, diarrhea, constipation, and elevated liver enzyme levels (Shaw et al., 2013).
Gefitinib and erlotinib are competitive antagonists that bind to the ATP site of EGFRs and are first-generation EGFR-TKIs. Both drugs are used against non-small cell lung cancer, which accounts for 80% of lung cancers (Molina et al., 2008). In May 2003 and November 2004, the FDA approved gefitinib and erlotinib, respectively. Many studies are being conducted to examine the reuse of these drugs in cancers that involve the activation of EGFRs.
These TKIs are well-established treatments for NSCLC and are used as a first-line treatment for patients with EGFR-mutated NSCLC (Sebastian et al., 2014). EGFR mutation is common in the Asian population, especially in female non-smokers and non-smokers in general (Tseng et al., 2017; Graham et al., 2018). Anti-apoptotic properties and uncontrolled cell growth are all associated with EGFR mutation. Adenocarcinoma is the most common type of NSCLS and the most common type of EGFR-mutated NSCLS. Gefitinib was initially approved for treating NSCLS but was later withdrawn from the market due to a lack of evidence that it had a beneficial effect on survival rate in unselected patients. However, it was approved once again, as the mutation is key to the response of gefitinib. Gefitinib is significantly more efficacious against NSCLC than chemotherapy; the progression-free survival rate was 10.8 months vs. 5.4 months, respectively, and the survival mean was 30.5 months vs. 23.6 months (Maemondo et al., 2010), respectively. In combination with chemotherapy, gefitinib was superior to gefitinib monotherapy (Nakamura et al., 2018).
Erlotinib was significantly superior to chemotherapy in advanced EGFR-positive NSCLC (PFS 13.1 vs. 4.6 months, respectively) (Zhou et al., 2011a), but its overall survival was lower than chemotherapy (24.68 vs. 26.16 months, respectively); it was superior to chemotherapy compared with chemotherapy with enhanced PS but not OS. A meta-analysis of gefitinib and erlotinib revealed that the efficacy of these two TKIs was comparable, although erlotinib caused more adverse effects.
Although they have not yet received therapy approval, gefitinib and erlotinib are being tested for their effectiveness against different malignancies. Although gefitinib was approved in 2005 for the treatment of pancreatic cancer in combination with chemotherapy, much of the research is still at the preclinical stage and the results are not promising. Gefitinib and erlotinib are being tested as treatments for various cancers, including nasopharyngeal cancer (Chua et al., 2008), gastric cancer (Dragovich et al., 2006), esophageal cancer (Wainberg et al., 2011), cervical cancer (Schilder et al., 2009), renal cell carcinoma (Gordon et al., 2009), and hepatocellular carcinoma (HCC) (Llovet and Hernandez-Gea, 2014). They have yet to receive approval for treatment and the results are not encouraging.
The most common side effects associated with gefitinib include acne rash, nausea, diarrhea, anorexia, stomatitis, and dehydration. These side effects were well tolerated by patients and the drug was withdrawn from patients who could not tolerate it. Overall, gefitinib is one of the more well-tolerated cytotoxic drugs (Forsythe and Faulkner, 2004).
Erlotinib has more severe adverse effects than gefitinib; however, like gefitinib, it is well tolerated. Dose reduction due to intolerance to side effects was more frequent with Erlotinib. According to reports, this drug caused significantly high rates of severe skin rashes, diarrhea, nausea, and vomiting, as well as stomatitis (Zhang et al., 2018).
Sunitinib is a first-generation VEGFR TKI and is a multi-target ATP competitive TKI. In various solid tumors, VEGFR is overexpressed and induces angiogenesis by binding to the vascular endothelium. Sunitinib is known to inhibit various tyrosine kinases, such as VEGFR2, PDGFRβ, KIT, RET, CSF1R, and FLT3 (O’Farrell et al., 2003; Faivre et al., 2007; Polyzos, 2008). In 2006, the FDA approved sunitinib for the treatment of metastatic RCC (mRCC) based on a phase III trial in which sunitinib exhibited superiority over IFN therapy. Sunitinib has also been approved for the treatment of imatinib-resistant gastrointestinal stromal tumors (GISTs). Furthermore, sunitinib was approved by the FDA in 2017 as an adjuvant therapy for adult patients at high risk of CCR after nephrectomy. In addition, in a phase III randomized trial, sunitinib was evaluated in patients who had nephrectomy and loco-regional RCC. Sunitinib patients exhibited a significant increase in disease-free survival compared with the placebo group (HR-0.76; 6.8 vs. 5.6 years, respectively [95% CI 0.59–0.98; p = 0.03]) (Ravaud et al., 2016).
In another landmark phase III trial, sunitinib was tested against interferon-α in patients with advanced metastatic RCC. Sunitinib patients showed significantly longer PFS compared with interferon patients (11 months vs. 5 months, respectively (HR-0.42 [95% CI 0.32–0.54; p < 0·0001]) (Motzer et al., 2007). Common side effects associated with sunitinib include grade 3 neutropenia, leukopenia, diarrhea, fatigue, nausea, hypertension, and hand-foot mouth syndrome (Demetri et al., 2006).
Lapatinib is a next-generation inhibitor that also targets HER2 TK in addition to EGFR. Lapatinib was designed and developed to inhibit the action of HER2 TK in patients with HER2-positive breast cancer (Cameron and Stein, 2008). FDA approval was given to lapatinib in 2007 to treat HER2 patients with metastatic and advanced breast cancer. This approval was followed by a phase III open-label study that compared the use of lapatinib and capecitabine with capecitabine alone in HER2 patients with advanced metastatic breast cancer who had previously been treated with anthracycline, taxanes, and trastuzumab. With the combination therapy, the median time to progression increased significantly to 8.4 months from 4.4 months, without increasing the toxic effect or systematic cardiac effect (Geyer et al., 2006). In 2010, lapatinib was approved for the treatment of patients with HER2-positive advanced breast cancer in combination with letrozole as a first-line agent. This approval was based on the results of a double-blind randomized phase III trial in postmenopausal women with HER2-positive advanced breast cancer. PFS was significantly prolonged in the combination therapy group compared with the letrozole and placebo groups, and the total response rate was 28% vs. 15%, respectively. Additionally, the combination therapy group had a clinical advantage of 48%, compared with 29% for letrozole alone, and experienced diarrhea 68% of the time and a rash 46% of the time. The cardiac side effect observed in trastuzumab was not observed in the letrozole group (Schwartzberg et al., 2010).
Pazopanib is a multi TKI. It is an inhibitor of VEGFR, PDGFR (A and B), c-Kit, and fibroblast growth factor receptor (FGFR) (van Geel et al., 2012). For metastatic RCC, pazopanib was approved by the FDA in 2009, and in 2012 it was approved by the FDA for the treatment of advanced-stage soft tissue sarcoma. A global multicenter placebo-controlled randomized phase III double-blind trial was conducted with patients with locally advanced and metastatic RCC. Pazopanib significantly improved PFS compared with the control group (9.2 months vs. 4.2 months, respectively), with a median duration response of more than 1 year (Sternberg et al., 2010). The FDA approved pazopanib as a first-line therapy for patients with metastatic RCC based on the findings of this study. By contrast, in a placebo-controlled phase III randomized trial, in which adjuvant sunitinib or sorafenib was tested in patients with high-risk non-metastatic RCC (ASSURE), pazopanib did not outperform the placebo (Motzer et al., 2017).
In a randomized controlled double-blind phase III trial that studied the preference of treatments in patients with metastatic RCC (PISCES), more patients preferred pazopanib (approximately 70%) than sunitinib (22%), whereas approximately 8% had no preference (Escudier et al., 2014). This proved that there was a clinical preference for pazopanib, which had the same therapeutical potentials but with a different profile of side effects. The side effects exhibited with pazopanib include diarrhea, change in hair color, and increased enzymes in the liver (Sternberg et al., 2010).
A mutation in the JAK2 kinase domain is found in approximately 95% of patients with polycythemia vera (PV) and 50% of patients with essential thrombocytosis (ET) and myelofibrosis (MF), which causes pathogenesis at the molecular level (James et al., 2005; Kralovics et al., 2005). Ruxolitinib is a non-receptor TKI of JAK2 and Janus kinase. Several studies have tested the efficacy of ruxolitinib in all three myeloproliferative diseases. Ruxolitinib is approved for treating polycythemia vera (PV) patients who have hydroxyurea resistance or intolerance. Additionally, in 2011, ruxolitinib was approved for patients with intermediate to high-risk myelofibrosis (MF) and who were PV resistant or intolerant to hydroxyurea (2014). However, it has been noted that a significant number of patients receiving ruxolitinib lose their hair, experience a subpar response, or develop cytopenia, and this causes them to stop taking the medication after a few months. In fact, in some of the phase III trials, the discontinuation rate was 50% and 70% at 3 and 5 years, respectively. Some of the side effects reported for this drug include anemia, thrombocytopenia, and herpes zoster infection (Harrison et al., 2012; Verstovsek et al., 2012; Vannucchi et al., 2015).
Vemurafenib was the first BRAF kinase inhibitor approved (by the FDA in 2011) for the treatment of metastatic melanoma patients with the BRAF V600E mutation. A landmark phase III randomized control trial compared Vemurafenib (RO5185426) with dacarbazine in patients with BRAF V600E mutant metastatic melanoma who had not previously received any treatment. At 6 months, the OS in the vemurafenib group increased significantly to 84% (95% CI 78–89) compared with 64% in the dacarbazine group (95% CI 56–73, HR-0.37; 95% CI 0.26–0.55; p < 0.001), and the median PFS for vemurafenib was 5.3 months compared with 1.6 months for dacarbazine (Chapman et al., 2011). In the extended study, there was a significant increase in the median OS in the vemurafenib group (95% CI 12.0–15.2) compared with the dacarbazine group (13.6 months vs. 9.6 months, respectively; 95% CI 7.9–12.8; HR-0.70; 95% CI 0.57–0.87; p = 0.0008). The median PFS also increased significantly in the vemurafenib group (6.9 months; 95% CI 6.1–7.0) compared with the dacarbazine group (1.6 months; 95% CI 1.6–2.1; HR-0.38; 95% CI 0.32–0.46, p < 0.0001 (Chapman et al., 2011; McArthur et al., 2014).
Ciba-Geigy discovered imatinib while screening for platelet-derived growth factor receptor (PDGFR) kinase inhibitors. Interestingly, the compound CGP53,716 demonstrated significant activity against Abl-kinase; further modification of this compound to specifically target Bcr-Abl kinase resulted in the development and discovery of imatinib (ST571) (Wong and Witte, 2004). Owing to previous studies demonstrating its efficacy in patients for whom first-line therapy failed, imatinib was tested against standard therapy (interferon and cytarabine) in patients with previously untreated CML (Druker et al., 2001; Kantarjian et al., 2002). The FDA later approved imatinib as a first-line therapy for chronic CML. Additionally, it has since been approved for treating rare hematologic malignancies, as well as proto-oncogene c-Kit or tyrosine-protein kinase Kit-mutated GIST (Imatinib, 2008).
In a landmark phase III international multicenter crossover randomized study of interferon and STI571 (IRIS), imatinib was compared with a combination of interferon and cytarabine in patients with chronic CML. There was a significant increase in complete hematologic response in the imatinib group (95.3%; 95% CI 93.2–96.9) compared with the combination therapy group (interferon and cytarabine) (27.3%; 95% CI 6.0–61.0). Additionally, there was a major cytogenic response in the imatinib group (85.2%; 95% CI 81.9–88.0) compared with the combination group (22.1%; 95% CI 18.7–25.8). Although there was no significant difference in OS, it should be noted that around 89.2% (493 out of 553) patients, either discontinued or shifted to other group (O'Brien et al., 2003). Sixty-month follow-up showed that only 3% of the patients remained in the combination group. Furthermore, the follow-up showed a complete hematologic response of 98%, a cytogenic response of 92%, and an overall survival rate (OSR) of 89% (95% CI 86–92) in the imatinib group (Druker et al., 2006). Follow-up at 120 months showed that 48.3% of patients who were randomly assigned to the imatinib group completed the treatment regime assigned to them, whereas 1.3% completed the treatment regime in the interferon and cytarabine group. The OSR in the imatinib group was 83.3% (95% CI 80.1–86.6) (Hochhaus et al., 2017). Neutropenia, thrombocytopenia, anemia, elevated liver enzymes, and other drug-related adverse events were observed (Druker et al., 2006). In a 10-year follow-up study, no new additional side effects were observed (Hochhaus et al., 2017).
Nilotinib is a second-generation Bcr-Abl kinase inhibitor that was developed to overcome the imatinib mutant. Nilotinib is 20-fold more efficacious than imatinib. An in vitro study of the inhibitory activity of nilotinib suggested that it was able to inhibit most of the 15 Bcr-Abl mutant resistant receptors (Weisberg et al., 2005). It was designed to be more efficacious than imatinib and with lesser side effects than its counterpart. Nilotinib was approved by the FDA as a first-line therapy for patients with chronic CML and for patients who develop resistance or are intolerant to imatinib. In a phase III multicenter randomized clinical trial (ENESTnd), the safety and efficacy of 300 mg BID and 400 mg BID of nilotinib was compared with 400 mg BID of imatinib in patients with advanced-stage CML. The 12-month study revealed that 300 mg BID of nilotinib resulted in a response rate (44%) double that of imatinib 400 mg BID (22%). The cytogenic response was 80% in the 300 mg BID nilotinib group and 65% in the imatinib group; a similar response was observed in a 24-month follow-up, which showed a twofold increase of 71% in the nilotinib group and 44% in the imatinib group. Complete cytogenic response was 26% in the nilotinib group and 10% in the imatinib group. However, although there was a lower rate of death related to CML, there was no improvement in OS in the nilotinib group (Saglio et al., 2010; Kantarjian et al., 2011). The molecular response in the 300 mg BID nilotinib group was significantly higher in the 60-month follow-up study (Hochhaus et al., 2016). Rashes, headaches, diarrhea, fluid retention, cardiovascular events, cytopenia, and biochemical abnormalities were all reported as side effects (Hochhaus et al., 2016).
Sorafenib, like sunitinib, is a first-generation VEGFR TKI. It inhibits VEGFRs, PDGFRs, and Fms-related tyrosine kinase/Flk2/Stk-2-receptor (FLT3R). It was the first TKI to be approved for the treatment of metastatic RCC in 2005, and it is currently used as a second-line agent in the treatment of patients with metastatic RCC. Sorafenib was approved for HCC in 2007, and for metastatic thyroid cancer in 2013.
In a phase III randomized double-blind placebo-controlled trial of advanced RCC named TARGET (Approaches in Renal Cancer Global Evaluation Trial), sorafenib was compared with placebo. The results showed that sorafenib significantly improved median PFS (5.5 months vs. 2.8 months; HR-0.44, 95% CI 0.35 to 0.55; p < 0.01) (Escudier et al., 2007), but the efficacy and the safety evaluation did not show any benefits with respect to OS. However, when the post crossover placebo survival data were censored, there was a significant improvement in OS in the sorafenib group (17.8 vs. 14.3 in the control; HR-0.78, p < 0.029) (Escudier et al., 2009). Additionally, sorafenib was compared with a placebo in a multicenter phase III double-blind placebo trial of sorafenib in patients with advanced HCC (Hepatocellular Carcinoma Assessment Randomized Protocol [SHARP]) and was shown to significantly increase the median OS (7.9 months–10.7 months) (Llovet et al., 2008).
The efficacy of sorafenib was tested in patients with radioactive iodine-refractory locally advanced or metastatic differentiated thyroid cancer in the multicenter phase III randomized placebo-controlled trial Nexavar versus Placebo in Locally Advanced/Metastatic Radio-active Iodine Refractory Differentiated Thyroid Cancer (DECISION). The results showed that sorafenib was associated with a significantly longer PFS than the placebo group (10.8 months vs. 5.8 months; HR-0.59, 95% CI 0.45–0.76; p < 0.0001) (Brose et al., 2014). Side effects that are commonly encountered with this inhibitor include hypertension, fatigue, anemia, vomiting, and diarrhea (Escudier et al., 2007; Escudier et al., 2009; Brose et al., 2014).
Owing to the steric hindrance caused by the bulky isoleucine residue at the 315 position, all previously produced Bcr-Abl kinases find it difficult to bind to this mutant T315I kinase. To overcome this steric hindrance, a computational and structure-based approach was used (Zhou et al., 2011b). Ponatinib was developed specifically to bind to T315I mutant kinases. The results of in vitro studies demonstrated that it was active against all mutant T315I Bcr-Abl kinases (O'Hare et al., 2009).
A significant response to ponatinib was observed in patients who did not respond to therapy, patients with a T315I mutation, and patients who exhibited refractory therapy to TKI with an undetectable mutation in Bcr-Abl (Cortes et al., 2012; Cortes et al., 2013). In 66% of patients with a T315I mutation, ponatinib was able to induce the cytogenic response, proving its clinical efficacy. However, in generalized CML-CP patients it did not show superior efficacy to previous TKIs. Therefore, this suggests that it may be used as first-line therapy in patients with a T315I mutation or otherwise just as a second-line therapy following the use of first- and second-generation TKIs (Cortes et al., 2012). The phase III trial for the evaluation of ponatinib vs imatinib in chronic myeloid leukemia (EPIC), designed to study the safety evaluation and clinical efficacy, was terminated due to previous reports of arterial thrombosis (Lipton et al., 2016). Therefore, the use of ponatinib in patients with newly diagnosed chronic myeloid lymphoma has not yet been established.
Ponatinib was compared with Allo-SCT in the Philadelphia Positive Acute Lymphoblastic Leukemia and Chronic Myeloid Leukemia Evaluation Trial (PACE). Ponatinib exhibited a significantly longer OSR at 24 and 48 months (24 months, 84% vs. 60.5%, p = 0.004; 48 months, 72.7% vs. 55.8%, p = 0.013; HR −0.37, 95% CI 0.16–0.84; p = 0.017) (Nicolini et al., 2017). To overcome the resistance of ponatinib, it is necessary to further understand the mechanism by which resistance occurs (Zabriskie et al., 2014; Wang et al., 2017; BCR-ABL1 compound mutations drive ponatinib, 2014). In 2014, the FDA narrowed the recommended use to ponatinib for adult patients with T315I-positive CML or ALL who had no other TKI options due to the risk of arterial embolism. Ponatinib side effects include rash (47%), abdominal pain (46%), dry skin (42%), thrombocytopenia (46%), and headache (43%). Additionally, constipation (41%), diarrhea, pyrexia, myocardial infarction, anemia, neutropenia, and pancytopenia have been reported. The arterial thrombotic event resulting from the treatment was observed by the onsite investigators in 2.2%, 0.7%, and 1.6% patients (Lipton et al., 2016). The most severe adverse effect observed in approximately 31% of the patients was arterial occlusive events (Cortes et al., 2013).
Trametinib is an inhibitor of MEK and was developed to treat BRAF-mutated metastatic melanoma by targeting MEK. In 2013, the FDA approved trametinib for the treatment of BRAF V600E mutant metastatic melanoma, and in 2014 the FDA approved its use in combination with dabrafenib. This combination is currently being approved as a first-line treatment for BARF V600E mutant metastatic melanoma.
In a phase III randomized clinical trial (METRIC), the efficacy of trametinib vs. dacarbazine was tested in patients with advanced stage or metastatic BRAF V600 E/K mutant-positive melanoma. The results of the clinical trials showed that trametinib significantly increased the median PFS (4.8 months in the trametinib group and 1.5 months in the dacarbazine group). In the trametinib group, the HR for progression of disease or death was 0.45 (95% CI 0.33–0.63; p < 0.001) (Flaherty et al., 2012).
In another study, a double-blind phase III randomized control trial, a comparison was made between dabrafenib monotherapy and trametinib and dabrafenib combination therapy in patients with BRAF mutant melanoma (COMBI-d). The results of the primary analysis at 9 months follow-up showed a median PFS of 9.3 months for combination therapy and 8.8 months for dabrafenib monotherapy (HR-0.75; 95% CI 0.57–0.99, p = 0·0348) (Long et al., 2014). OS was 47% in the combination group vs., 58% in the monotherapy group (HR-0.71; 95% CI 0.55–0.92, p = 0·0107). The median OS was 25.1 months for the combination group vs. 18 months for the dabrafenib group (Long et al., 2014).
The results of a double-blind phase III randomized control trial (COMBI-v) of trametinib in combination with dabrafenib against vemurafenib alone were similar for each treatment in patients with unresectable or metastatic BRAF V600 E/K cutaneous melanoma. The results showed a significant increase in the OS and median PFS at 12 months in the combination group (72% vs. 65% for vemurafenib alone).
A phase III double-blind placebo controlled randomized trial of dabrafenib with trametinib, an MEK inhibitor, was carried out for adjuvant therapy for patients with high chances of BRAF V600 mutant-positive stage III melanoma after surgical resection (COMBI-AD). The result of the 2.8-year follow-up study showed that the estimated 3-year free survival rate in the combination group was 58% compared with 39% in the placebo control group (Long et al., 2015; Long et al., 2017a).
Some of the common side effects associated with trametinib include rash, diarrhea, and peripheral edema. Asymptomatic and reversible reduction in cardiac ejection fraction and toxic ocular effect were not frequent. It may be interesting to note that the secondary skin neoplasm observed in patients with the BRAF KI was not observed in patients who received a combination of MEK inhibitors (Flaherty et al., 2012).
The FDA approved the MEK inhibitor cobimetinib in combination with vemurafenib (a BRAF inhibitor) for the treatment of patients with mutant BRAF V600E metastatic melanoma after a phase III controlled randomized trial comparison study between vemurafenib alone and vemurafenib and cobimetinib in combination. In the study, the combination group showed a prolonged median PFS of 9.9 months compared with 6.2 months in the vemurafenib group (HR for death or disease progression was 0.51; 95% CI 0.39–0.68, p < 0.001) (Long et al., 2017a). The common side effects observed were pyrexia and elevated levels of liver enzymes (Larkin et al., 2014; Long et al., 2017a).
The first mTOR inhibitor approved was rapamycin, also known as sirolimus. It is produced by Streptomyces hygroscopicus (Sehgal et al., 1975) and is an antifungal agent with poor pharmacokinetic properties. However, much of the research has been focused on the development of synthetic analogs of rapamycin that have a good pharmacokinetic profile suitable for therapy (Liu et al., 2009). These synthetic analogs, such as everolimus, temsirolimus, and radaforolimus, differ from sirolimus at the C-40-O position, resulting in superior pharmacokinetic and pharmacodynamic profiles, with suitable therapeutic potentials. These drugs are used in the treatment of solid tumors, such as RCC, breast cancer, pancreatic neuroendocrine tumors, and tuberous sclerosis complex (Awada et al., 2008).
Targeting of mTOR kinase may benefit the management of mRCC, and temsirolimus is one of the available inhibitors. Significant results from phase III trials led, in 2007, to FDA approval of temsirolimus as a single agent in poor rim RCC patients Bergmann et al. (2014). The results showed superior OS with temsirolimus compared with IFN-α or temsirolimus plus IFN-α. Additionally, temsirolimus showed benefits against clear cell carcinoma and papillary RCC.
Lenvatinib is a TKI that inhibits VEGFR and various other FGFRs, such as FGFR1, FGFR2, FGFR3, FGFR4, PDGFRs, RET, and c-Kit (Okamoto et al., 2015; Kannaiyan and Mahadevan, 2018). It was approved for the treatment of radioactive iodine-refractory differentiated thyroid cancer by the FDA in 2015, and in 2016, the FDA approved it as a second-line therapy in combination with everolimus for treating metastatic RCC after anti-angiogenic therapy.
In a phase II randomized open-label multicenter trial, lenvatinib was tested as a second-line therapy for patients with metastatic RCC who had undergone anti-angiogenic therapy; lenvatinib was compared with everolimus for its efficacy, as well as the combination of everolimus and lenvatinib. The combination of lenvatinib and everolimus increased the median PFS to 14.6 months compared with 5.5 months for everolimus alone, whereas a significant improvement in the median PFS (7.4 months) was observed in the lenvatinib monotherapy group compared with the everolimus group (Motzer et al., 2015).
In a phase III randomized double-blind trial, lenvetanib was investigated against the placebo in patients with progressive thyroid cancer refractive to radioactive iodine. Lenvatinib showed a median PFS of 18.3 months compared with 3.6 months with the placebo (HR for progression was 0.21) (Schlumberger et al., 2015). The side effects that were observed in patients included hypertension, fatigue, nausea, vomiting, lack of appetite, diarrhea, and hand-foot syndrome (Motzer et al., 2015).
Afatinib is an orally available second-generation EGFR TKI. The second-generation EGFR TKIs were designed to target other members of ErbB group, including HER2. They not only target the T790M mutation of EGFR, but other EGFR-activating mutations and wild-type EFGR (Wind et al., 2017).
Afatinib is an irreversible inhibitor of the ERBB family that includes HER1 (EGFR), HER2, and HER4. Compared with chemotherapy, afatinib was effective in prolonging PFS (11.1 months vs. 6.9 months, respectively) (Yang et al., 2013). Like erlotinib, afatinib also increases PFS significantly by 18% (19% OS).
In NSCLC patients for whom chemotherapy failed, afatinib, like erolitinib, significantly increased PFS by 18% and OS by 19%, and it was better in terms of disease control rate (51% vs. 40%). Subsequently, the FDA approved the use of afatinib as a first-line therapy in patients with NSCLC.
In the phase III randomized clinical trial LUX-Lung 8, afatinib was compared with erolitinib in patients with advanced squamous cell carcinoma as a second-line therapy. Subsequently, afatinib was approved as a second-line treatment due to the findings of the study (Soria et al., 2015). Diarrhea, rashes or pimples on the skin, and weariness are among the side effects frequently reported with afatinib (Paz-Ares et al., 2017).
Dacomitinib is an oral EGFR KI that irreversibly inhibits ERBB family members; it covalently binds to the receptor and blocks the downstream signaling pathway. It is more effective than the first-generation EGFR TKIs against all wild-type EGFRs and EGFR with Del19 or L858R mutations, with broad activity against multiple ErbB receptors, such as EGFR, HER2, and HER4. On 27 September 2018, the FDA approved dacomitinib (Vizimpro, Pfizer) as a first-line therapy for patients with metastatic NSCLC or EGFR with Del19 or exon 21 L858R mutations, as detected by an FDA-approved test (Reckamp et al., 2014; Wu et al., 2017a). The dose-limiting toxicity exhibited by second-generation EGFR TKIs is mainly exhibited in the skin and gastrointestinal tract because they also inhibit wild-type EGFRs (Sullivan and Planchard, 2016).
Neratinib is a TKI that targets all EGFRs and also inhibits EGFR tyrosine kinase. It is approved by the FDA as an adjuvant therapy for patients with early-stage breast cancer who test positive for HER2 after trastuzumab adjuvant therapy. This approval was based on the findings of a phase III randomized clinical trial that looked at the efficacy and safety of neratinib after adjuvant therapy with neoadjuvant and trastuzumab in patients with HER2-positive early-stage (I-III) breast cancer. Some of the side effects observed included diarrhea, nausea, and vomiting (Martin et al., 2017).
These small molecules interact with different receptors in various proteins. The interaction of these TKIs has been well studied using various molecular modeling software. Figure 1 shows one of the options in which molecular modeling and docking studies are carried out using Schrodinger software (Maestro 12.1, United States). In this review, these molecules have been redocked.
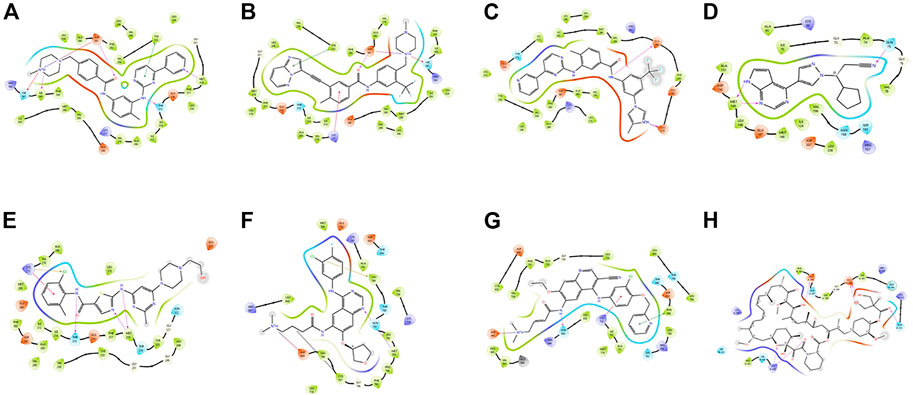
FIGURE 1. Docking poses of different proteins with drugs.(A) Imatinib. (B) Ponatib. (C) Nilotinib docked to human Abl kinase (2HYY). (D) Ruxolitinib dock to c-Jun N-terminal kinase (2P33). (E) Dasatinib docked to activated ABL kinase domain (2GQG). (F) Afatinib with EGFR kinase (4G5J). (G) Neratilib docked to HER2(3RCD). (H) Temsirolimus with human serum albumin (1AO6).
The success of imatinib has led to the development of various KIs; currently (as of 29 October 2021) there are approximately 68 FDA-approved KIs in clinical use (http://www.brimr.org/PKI/PKIs.htm). All of these KIs are delivered orally, with a few exceptions, such as temsirolimus and trilaciclib, and netarsudil, which is available as an ophthalmic solution for eye drops. The development of KIs in cancer has led to a new approach in this regard. It may be noted that they are generally less toxic and have more chemotherapeutic potential than conventional chemotherapy. However, like conventional therapy, KIs have some limitations, such as the development of resistance, and some adverse effects.
Resistance developed during treatment can be addressed through the clonal expansion of cells that do not respond to specific kinase or secondary kinase mutations that render the kinase inhibitor ineffective. Understanding the resistance mechanism has resulted in the design and development of new KIs or combination therapies. For instance, development of the new Bcr-Abl KI ponatinib targeted the T315I mutant gene that was resistance to first- and second-generation Bcr-Abl inhibitors. Understanding the mechanism of resistance of EGFR T790M to the TKI of EGFR has led to the development of osimertinib, which targets the mutant gene of EGFR T790M. Combination therapy is another option for overcoming resistance; for example, understanding the mechanism of the MEK inhibitor that causes reactivation of the MAPK pathway, which in turn activates MEK towards the resistance of the BRAF inhibitors (in melanoma), prompted the use of combination therapy consisting of BRAF and MEK inhibitors, which resulted in better clinical outcomes than single therapy.
One of the main concerns associated with the use of KIs is adverse effects. As mentioned above, owing to the risk of arterial embolism, the FDA had to narrow down the recommended use of ponatinib. This issue can be addressed by choosing more specific KIs. Additionally, understanding the mechanisms of side effects may help to lessen their effect.
The remaining option for overcoming the above concerns is to continuously and thoroughly screen the patient’s condition, plasma concentration, and personalized treatment approach. Even if the stage and the cancer are the same, there may be differences depending on the patient’s status and tumor, hence a personalized treatment regime may be one of the best approaches.
Owing to the success of KIs, there is a rise in the design and development of new small molecule inhibitors. The use of new technologies, such as mass spectrometry-based proteomics, may help facilitate the development of more specific target-oriented inhibitors.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
APC (in part) is supported by the Deanship of University Scientific Research.
The authors would like to express their gratitude to the Deans of the Faculty of Pharmacy and Allied Medical Sciences and the Dean of Scientific Research at Al-Ahliyya Amman University in Amman, Jordan, for providing the required facilities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adam, K., and Hunter, T. (2018). Histidine kinases and the missing phosphoproteome from prokaryotes to eukaryotes. Lab. Invest. 98, 233–247. doi:10.1038/labinvest.2017.118
Amato, R. J., Stepankiw, M., and Ochuwa, N. S. (2014). “Chapter 8 - molecular-targeted therapy for renal cell carcinoma,” in Renal disease in cancer patients. Editors K. W. Finkel, and S. C. Howard (United States: Academic Press), 115–127.
Awada, A., Cardoso, F., Fontaine, C., Dirix, L., De Grève, J., Sotiriou, C., et al. (2008). The oral mTOR inhibitor RAD001 (everolimus) in combination with letrozole in patients with advanced breast cancer: Results of a phase I study with pharmacokinetics. Eur. J. Cancer 44, 84–91. doi:10.1016/j.ejca.2007.10.003
BCR-ABL1 compound mutations drive ponatinib resistance, BCR-ABL1 compound mutations drive ponatinib resistance. Cancer Discov. 4 (2014) Of13, doi:10.1158/2159-8290.CD-RW2014-186
Bergmann, L., Maute, L., and Guschmann, M. (2014). Temsirolimus for advanced renal cell carcinoma. Expert Rev. Anticancer Ther. 14, 9–21. doi:10.1586/14737140.2014.864562
Bhullar, K. S., Lagarón, N. O., McGowan, E. M., Parmar, I., Jha, A., Hubbard, B. P., et al. (2018). Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 17, 48. doi:10.1186/s12943-018-0804-2
Brose, M. S., Nutting, C. M., Jarzab, B., Elisei, R., Siena, S., Bastholt, L., et al. (2014). Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 384, 319–328. doi:10.1016/S0140-6736(14)60421-9
Brümmendorf, T. H., Cortes, J. E., de Souza, C. A., Guilhot, F., Duvillié, L., Pavlov, D., et al. (2015). Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukaemia: Results from the 24-month follow-up of the BELA trial. Br. J. Haematol. 168, 69–81. doi:10.1111/bjh.13108
Brummendorf, T. H., Cortes, J. E., Milojkovic, D., Gambacorti-Passerini, C., Clark, R. E., le Coutre, P., et al. (2022). Bosutinib versus imatinib for newly diagnosed chronic phase chronic myeloid leukemia: Final results from the BFORE trial. Leukemia 36, 1825–1833. doi:10.1038/s41375-022-01589-y
Cameron, D. A., and Stein, S. (2008). Drug insight: Intracellular inhibitors of HER2--clinical development of lapatinib in breast cancer. Nat. Clin. Pract. Oncol. 5, 512–520. doi:10.1038/ncponc1156
Chapman, P. B., Hauschild, A., Robert, C., Haanen, J. B., Ascierto, P., Larkin, J., et al. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516. doi:10.1056/NEJMoa1103782
Chua, D. T., Wei, W. I., Wong, M. P., Sham, J. S., Nicholls, J., and Au, G. K. (2008). Phase II study of gefitinib for the treatment of recurrent and metastatic nasopharyngeal carcinoma. Head. Neck 30, 863–867. doi:10.1002/hed.20792
Ciesla, J., Fraczyk, T., and Rode, W. (2011). Phosphorylation of basic amino acid residues in proteins: Important but easily missed. Acta Biochim. Pol. 58, 137–148. doi:10.18388/abp.2011_2258
Conchon, M., Freitas, C. M., Rego, M. A., and Braga Junior, J. W. (2011). Dasatinib - clinical trials and management of adverse events in imatinib resistant/intolerant chronic myeloid leukemia. Rev. Bras. Hematol. Hemoter. 33, 131–139. doi:10.5581/1516-8484.20110034
Cortes, J. E., Gambacorti-Passerini, C., Deininger, M. W., Mauro, M. J., Chuah, C., Kim, D. W., et al. (2018). Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: Results from the randomized BFORE trial. J. Clin. Oncol. 36, 231–237. doi:10.1200/JCO.2017.74.7162
Cortes, J. E., Kantarjian, H., Shah, N. P., Bixby, D., Mauro, M. J., Flinn, I., et al. (2012). Ponatinib in refractory Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 367, 2075–2088. doi:10.1056/NEJMoa1205127
Cortes, J. E., Kim, D. W., Pinilla-Ibarz, J., le Coutre, P., Paquette, R., Chuah, C., et al. (2013). A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 369, 1783–1796. doi:10.1056/NEJMoa1306494
Dankner, M., Rose, A. A. N., Rajkumar, S., Siegel, P. M., and Watson, I. R. (2018). Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 37, 3183–3199. doi:10.1038/s41388-018-0171-x
Demetri, G. D., van Oosterom, A. T., Garrett, C. R., Blackstein, M. E., Shah, M. H., Verweij, J., et al. (2006). Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 368, 1329–1338. doi:10.1016/S0140-6736(06)69446-4
Dragovich, T., McCoy, S., Fenoglio-Preiser, C. M., Wang, J., Benedetti, J. K., Baker, A. F., et al. (2006). Phase II trial of erlotinib in gastroesophageal junction and gastric adenocarcinomas: Swog 0127. J. Clin. Oncol. 24, 4922–4927. doi:10.1200/JCO.2006.07.1316
Druker, B. J., Guilhot, F., O'Brien, S. G., Gathmann, I., Kantarjian, H., Gattermann, N., et al. (2006). Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 355, 2408–2417. doi:10.1056/NEJMoa062867
Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., et al. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037. doi:10.1056/NEJM200104053441401
Dummer, R., Brase, J. C., Garrett, J., Campbell, C. D., Gasal, E., Squires, M., et al. (2020). Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAF(V600)-mutant, stage III melanoma (COMBI-AD): Exploratory biomarker analyses from a randomised, phase 3 trial. Lancet. Oncol. 21, 358–372. doi:10.1016/S1470-2045(20)30062-0
Eroglu, Z., and Ribas, A. (2016). Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 8, 48–56. doi:10.1177/1758834015616934
Escudier, B., Eisen, T., Stadler, W. M., Szczylik, C., Oudard, S., Siebels, M., et al. (2007). Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 356, 125–134. doi:10.1056/NEJMoa060655
Escudier, B., Eisen, T., Stadler, W. M., Szczylik, C., Oudard, S., Staehler, M., et al. (2009). Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 27, 3312–3318. doi:10.1200/JCO.2008.19.5511
Escudier, B., Porta, C., Bono, P., Powles, T., Eisen, T., Sternberg, C. N., et al. (2014). Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES study. J. Clin. Oncol. 32, 1412–1418. doi:10.1200/JCO.2013.50.8267
Faivre, S., Demetri, G., Sargent, W., and Raymond, E. (2007). Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 6, 734–745. doi:10.1038/nrd2380
Fasano, M., Della Corte, C. M., Califano, R., Capuano, A., Troiani, T., Martinelli, E., et al. (2014). Type III or allosteric kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin. Investig. Drugs 23, 809–821. doi:10.1517/13543784.2014.902934
Flaherty, K. T., Robert, C., Hersey, P., Nathan, P., Garbe, C., Milhem, M., et al. (2012). Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 367, 107–114. doi:10.1056/NEJMoa1203421
Forsythe, B., and Faulkner, K. (2004). Overview of the tolerability of gefitinib (IRESSA) monotherapy : Clinical experience in non-small-cell lung cancer. Drug Saf. 27, 1081–1092. doi:10.2165/00002018-200427140-00002
Geyer, C. E., Forster, J., Lindquist, D., Chan, S., Romieu, C. G., Pienkowski, T., et al. (2006). Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743. doi:10.1056/NEJMoa064320
Golas, J. M., Arndt, K., Etienne, C., Lucas, J., Nardin, D., Gibbons, J., et al. (2003). SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 63, 375–381.
Gordon, M. S., Hussey, M., Nagle, R. B., Lara, P. N., Mack, P. C., Dutcher, J., et al. (2009). Phase II study of erlotinib in patients with locally advanced or metastatic papillary histology renal cell cancer: Swog S0317. J. Clin. Oncol. 27, 5788–5793. doi:10.1200/JCO.2008.18.8821
Graham, R. P., Treece, A. L., Lindeman, N. I., Vasalos, P., Shan, M., Jennings, L. J., et al. (2018). Worldwide frequency of commonly detected EGFR mutations. Arch. Pathol. Lab. Med. 142, 163–167. doi:10.5858/arpa.2016-0579-CP
Grither, W. R., and Longmore, G. D. (2018). Inhibition of tumor-microenvironment interaction and tumor invasion by small-molecule allosteric inhibitor of DDR2 extracellular domain. Proc. Natl. Acad. Sci. U. S. A. 115, E7786–E7794. doi:10.1073/pnas.1805020115
Gumireddy, K., Baker, S. J., Cosenza, S. C., John, P., Kang, A. D., Robell, K. A., et al. (2005). A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc. Natl. Acad. Sci. U. S. A. 102, 1992–1997. doi:10.1073/pnas.0408283102
Harrison, C., Kiladjian, J. J., Al-Ali, H. K., Gisslinger, H., Waltzman, R., Stalbovskaya, V., et al. (2012). JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 366, 787–798. doi:10.1056/NEJMoa1110556
Herbert, C., Schieborr, U., Saxena, K., Juraszek, J., De Smet, F., Alcouffe, C., et al. (2013). Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell 23, 489–501. doi:10.1016/j.ccr.2013.02.018
Hochhaus, A., Larson, R. A., Guilhot, F., Radich, J. P., Branford, S., Hughes, T. P., et al. (2017). Long-Term outcomes of imatinib treatment for chronic myeloid leukemia. N. Engl. J. Med. 376, 917–927. doi:10.1056/NEJMoa1609324
Hochhaus, A., Saglio, G., Hughes, T. P., Larson, R. A., Kim, D. W., Issaragrisil, S., et al. (2016). Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 30, 1044–1054. doi:10.1038/leu.2016.5
Imatinib (2008). Imatinib: New indication. New indications, but not robust evidence Prescrire Int. 17, 91–94.
James, C., Ugo, V., Le Couédic, J. P., Staerk, J., Delhommeau, F., Lacout, C., et al. (2005). A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148. doi:10.1038/nature03546
Johnson, L. N., and Lewis, R. J. (2001). Structural basis for control by phosphorylation. Chem. Rev. 101, 2209–2242. doi:10.1021/cr000225s
Kannaiyan, R., and Mahadevan, D. (2018). A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 18, 1249–1270. doi:10.1080/14737140.2018.1527688
Kantarjian, H., Cortes, J., Kim, D. W., Dorlhiac-Llacer, P., Pasquini, R., DiPersio, J., et al. (2009). Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood 113, 6322–6329. doi:10.1182/blood-2008-11-186817
Kantarjian, H. M., Cortes, J. E., Kim, D. W., Khoury, H. J., Brümmendorf, T. H., Porkka, K., et al. (2014). Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood 123, 1309–1318. doi:10.1182/blood-2013-07-513937
Kantarjian, H. M., Hochhaus, A., Saglio, G., De Souza, C., Flinn, I. W., Stenke, L., et al. (2011). Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet. Oncol. 12, 841–851. doi:10.1016/S1470-2045(11)70201-7
Kantarjian, H. M., Shah, N. P., Cortes, J. E., Baccarani, M., Agarwal, M. B., Undurraga, M. S., et al. (2012). Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood 119, 1123–1129. doi:10.1182/blood-2011-08-376087
Kantarjian, H., Sawyers, C., Hochhaus, A., Guilhot, F., Schiffer, C., Gambacorti-Passerini, C., et al. (2002). Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 346, 645–652. doi:10.1056/NEJMoa011573
Kralovics, R., Passamonti, F., Buser, A. S., Teo, S. S., Tiedt, R., Passweg, J. R., et al. (2005). A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790. doi:10.1056/NEJMoa051113
Kufareva, I., and Abagyan, R. (2008). Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J. Med. Chem. 51, 7921–7932. doi:10.1021/jm8010299
Kwon, J., and Meagher, A. (2012). Crizotinib: A breakthrough for targeted therapies in lung cancer. J. Adv. Pract. Oncol. 3, 267–272. doi:10.6004/jadpro.2012.3.4.8
Larkin, J., Ascierto, P. A., Dréno, B., Atkinson, V., Liszkay, G., Maio, M., et al. (2014). Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 371, 1867–1876. doi:10.1056/NEJMoa1408868
Lim, S. M., Kim, H. R., Lee, J. S., Lee, K. H., Lee, Y. G., Min, Y. J., et al. (2017). Open-label, multicenter, phase II study of ceritinib in patients with non-small-cell lung cancer harboring ROS1 rearrangement. J. Clin. Oncol. 35, 2613–2618. doi:10.1200/JCO.2016.71.3701
Lipton, J. H., Chuah, C., Guerci-Bresler, A., Rosti, G., Simpson, D., Assouline, S., et al. (2016). Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: An international, randomised, open-label, phase 3 trial. Lancet. Oncol. 17, 612–621. doi:10.1016/S1470-2045(16)00080-2
Liu, Q., Thoreen, C., Wang, J., Sabatini, D., and Gray, N. S. (2009). mTOR mediated anti-cancer drug discovery. Drug Discov. Today. Ther. Strateg. 6, 47–55. doi:10.1016/j.ddstr.2009.12.001
Llovet, J. M., and Hernandez-Gea, V. (2014). Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. 20, 2072–2079. doi:10.1158/1078-0432.CCR-13-0547
Llovet, J. M., Ricci, S., Mazzaferro, V., Hilgard, P., Gane, E., Blanc, J. F., et al. (2008). Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390. doi:10.1056/NEJMoa0708857
Long, G. V., Flaherty, K. T., Stroyakovskiy, D., Gogas, H., Levchenko, E., de Braud, F., et al. (2017). Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 28, 1631–1639. doi:10.1093/annonc/mdx176
Long, G. V., Hauschild, A., Santinami, M., Atkinson, V., Mandalà, M., Chiarion-Sileni, V., et al. (2017). Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 377, 1813–1823. doi:10.1056/NEJMoa1708539
Long, G. V., Stroyakovskiy, D., Gogas, H., Levchenko, E., de Braud, F., Larkin, J., et al. (2014). Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 371, 1877–1888. doi:10.1056/NEJMoa1406037
Long, G. V., Stroyakovskiy, D., Gogas, H., Levchenko, E., de Braud, F., Larkin, J., et al. (2015). Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386, 444–451. doi:10.1016/S0140-6736(15)60898-4
Lugowska, I., Koseła-Paterczyk, H., Kozak, K., and Rutkowski, P. (2015). Trametinib: A MEK inhibitor for management of metastatic melanoma. Onco. Targets. Ther. 8, 2251–2259. doi:10.2147/OTT.S72951
Maemondo, M., Inoue, A., Kobayashi, K., Sugawara, S., Oizumi, S., Isobe, H., et al. (2010). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388. doi:10.1056/NEJMoa0909530
Manning, G., Plowman, G. D., Hunter, T., and Sudarsanam, S. (2002). Evolution of protein kinase signaling from yeast to man. Trends biochem. Sci. 27, 514–520. doi:10.1016/s0968-0004(02)02179-5
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. doi:10.1126/science.1075762
Martin, M., Holmes, F. A., Ejlertsen, B., Delaloge, S., Moy, B., Iwata, H., et al. (2017). Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 18, 1688–1700. doi:10.1016/S1470-2045(17)30717-9
McArthur, G. A., Chapman, P. B., Robert, C., Larkin, J., Haanen, J. B., Dummer, R., et al. (2014). Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet. Oncol. 15, 323–332. doi:10.1016/S1470-2045(14)70012-9
Molina, J. R., Yang, P., Cassivi, S. D., Schild, S. E., and Adjei, A. A. (2008). Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 83, 584–594. doi:10.4065/83.5.584
Motzer, R. J., Haas, N. B., Donskov, F., Gross-Goupil, M., Varlamov, S., Kopyltsov, E., et al. (2017). Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma. J. Clin. Oncol. 35, 3916–3923. doi:10.1200/JCO.2017.73.5324
Motzer, R. J., Hutson, T. E., Glen, H., Michaelson, M. D., Molina, A., Eisen, T., et al. (2015). Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet. Oncol. 16, 1473–1482. doi:10.1016/S1470-2045(15)00290-9
Motzer, R. J., Hutson, T. E., Tomczak, P., Michaelson, M. D., Bukowski, R. M., Rixe, O., et al. (2007). Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356, 115–124. doi:10.1056/NEJMoa065044
Nakamura, A., Inoue, A., Morita, S., Hosomi, Y., Kato, T., Fukuhara, T., et al. (2018). Phase III study comparing gefitinib monotherapy (G) to combination therapy with gefitinib, carboplatin, and pemetrexed (GCP) for untreated patients (pts) with advanced non-small cell lung cancer (NSCLC) with EGFR mutations (NEJ009). J. Clin. Oncol. 36, 9005. doi:10.1200/jco.2018.36.15_suppl.9005
Nicolini, F. E., Basak, G. W., Kim, D. W., Olavarria, E., Pinilla-Ibarz, J., Apperley, J. F., et al. (2017). Overall survival with ponatinib versus allogeneic stem cell transplantation in Philadelphia chromosome-positive leukemias with the T315I mutation. Cancer 123, 2875–2880. doi:10.1002/cncr.30558
O'Brien, S. G., Guilhot, F., Larson, R. A., Gathmann, I., Baccarani, M., Cervantes, F., et al. (2003). Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 348, 994–1004. doi:10.1056/NEJMoa022457
O'Farrell, A. M., Abrams, T. J., Yuen, H. A., Ngai, T. J., Louie, S. G., Yee, K. W., et al. (2003). SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 101, 3597–3605. doi:10.1182/blood-2002-07-2307
O'Hare, T., Shakespeare, W. C., Zhu, X., Eide, C. A., Rivera, V. M., Wang, F., et al. (2009). AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16, 401–412. doi:10.1016/j.ccr.2009.09.028
Odogwu, L., Mathieu, L., Blumenthal, G., Larkins, E., Goldberg, K. B., Griffin, N., et al. (2018). FDA approval summary: Dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist 23, 740–745. doi:10.1634/theoncologist.2017-0642
Okamoto, K., Ikemori-Kawada, M., Jestel, A., von Konig, K., Funahashi, Y., Matsushima, T., et al. (2015). Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med. Chem. Lett. 6, 89–94. doi:10.1021/ml500394m
Paz-Ares, L., Tan, E. H., O'Byrne, K., Zhang, L., Hirsh, V., Boyer, M., et al. (2017). Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Overall survival data from the phase IIb LUX-lung 7 trial. Ann. Oncol. 28, 270–277. doi:10.1093/annonc/mdw611
Polyzos, A. (2008). Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma and various other solid tumors. J. Steroid Biochem. Mol. Biol. 108, 261–266. doi:10.1016/j.jsbmb.2007.09.004
Rabindran, S. K., Discafani, C. M., Rosfjord, E. C., Baxter, M., Floyd, M. B., Golas, J., et al. (2004). Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 64, 3958–3965. doi:10.1158/0008-5472.CAN-03-2868
Ravaud, A., Motzer, R. J., Pandha, H. S., George, D. J., Pantuck, A. J., Patel, A., et al. (2016). Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. N. Engl. J. Med. 375, 2246–2254. doi:10.1056/NEJMoa1611406
Reckamp, K. L., Giaccone, G., Camidge, D. R., Gadgeel, S. M., Khuri, F. R., Engelman, J. A., et al. (2014). A phase 2 trial of dacomitinib (PF-00299804), an oral, irreversible pan-HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non-small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer 120, 1145–1154. doi:10.1002/cncr.28561
Roskoski, R. (2016). Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 103, 26–48. doi:10.1016/j.phrs.2015.10.021
Roskoski, R. (2019). Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 139, 395–411. doi:10.1016/j.phrs.2018.11.014
Saglio, G., Kim, D. W., Issaragrisil, S., le Coutre, P., Etienne, G., Lobo, C., et al. (2010). Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 362, 2251–2259. doi:10.1056/NEJMoa0912614
Schilder, R. J., Sill, M. W., Lee, Y. C., and Mannel, R. (2009). A phase II trial of erlotinib in recurrent squamous cell carcinoma of the cervix: A gynecologic oncology group study. Int. J. Gynecol. Cancer 19, 929–933. doi:10.1111/IGC.0b013e3181a83467
Schlumberger, M., Tahara, M., Wirth, L. J., Robinson, B., Brose, M. S., Elisei, R., et al. (2015). Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 372, 621–630. doi:10.1056/NEJMoa1406470
Schwartzberg, L. S., Franco, S. X., Florance, A., O'Rourke, L., Maltzman, J., Johnston, S., et al. (2010). Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist 15, 122–129. doi:10.1634/theoncologist.2009-0240
Sebastian, M., Schmittel, A., and Reck, M. (2014). First-line treatment of EGFR-mutated nonsmall cell lung cancer: Critical review on study methodology. Eur. Respir. Rev. 23, 92–105. doi:10.1183/09059180.00008413
Sehgal, S. N., Baker, H., and Vézina, C. (1975). Rapamycin (AY-22, 989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 28, 727–732. doi:10.7164/antibiotics.28.727
Shah, N. P., Tran, C., Lee, F. Y., Chen, P., Norris, D., and Sawyers, C. L. (2004). Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399–401. doi:10.1126/science.1099480
Shaw, A. T., Kim, D. W., Nakagawa, K., Seto, T., Crinó, L., Ahn, M. J., et al. (2013). Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 368, 2385–2394. doi:10.1056/NEJMoa1214886
Śmiech, M., Leszczyński, P., Kono, H., Wardell, C., and Taniguchi, H. (2020). Emerging BRAF mutations in cancer progression and their possible effects on transcriptional networks. Genes (Basel) 11, 1342. doi:10.3390/genes11111342
Solomon, B. J., Mok, T., Kim, D. W., Wu, Y. L., Nakagawa, K., Mekhail, T., et al. (2014). First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 371, 2167–2177. doi:10.1056/NEJMoa1408440
Soria, J. C., Felip, E., Cobo, M., Lu, S., Syrigos, K., Lee, K. H., et al. (2015). Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): An open-label randomised controlled phase 3 trial. Lancet. Oncol. 16, 897–907. doi:10.1016/S1470-2045(15)00006-6
Sternberg, C. N., Davis, I. D., Mardiak, J., Szczylik, C., Lee, E., Wagstaff, J., et al. (2010). Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 28, 1061–1068. doi:10.1200/JCO.2009.23.9764
Sullivan, I., and Planchard, D. (2016). Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front. Med. 3, 76. doi:10.3389/fmed.2016.00076
Thomas, R., and Weihua, Z. (2019). Rethink of EGFR in cancer with its kinase independent function on board. Front. Oncol. 9, 800. doi:10.3389/fonc.2019.00800
Tseng, C. H., Chiang, C. J., Tseng, J. S., Yang, T. Y., Hsu, K. H., Chen, K. C., et al. (2017). EGFR mutation, smoking, and gender in advanced lung adenocarcinoma. Oncotarget 8, 98384–98393. doi:10.18632/oncotarget.21842
van Geel, R. M., Beijnen, J. H., and Schellens, J. H. (2012). Concise drug review: Pazopanib and axitinib. Oncologist 17, 1081–1089. doi:10.1634/theoncologist.2012-0055
Vannucchi, A. M., Kiladjian, J. J., Griesshammer, M., Masszi, T., Durrant, S., Passamonti, F., et al. (2015). Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 372, 426–435. doi:10.1056/NEJMoa1409002
Verstovsek, S., Mesa, R. A., Gotlib, J., Levy, R. S., Gupta, V., DiPersio, J. F., et al. (2012). A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 366, 799–807. doi:10.1056/NEJMoa1110557
Wainberg, Z. A., Lin, L. S., DiCarlo, B., Dao, K. M., Patel, R., Park, D. J., et al. (2011). Phase II trial of modified FOLFOX6 and erlotinib in patients with metastatic or advanced adenocarcinoma of the oesophagus and gastro-oesophageal junction. Br. J. Cancer 105, 760–765. doi:10.1038/bjc.2011.280
Wang, D., Pan, H., and Wang, Y. (2017). T315L: A novel mutation within BCR-ABL kinase domain confers resistance against ponatinib. Leuk. Lymphoma 58, 1733–1735. doi:10.1080/10428194.2016.1251591
Weisberg, E., Manley, P. W., Breitenstein, W., Brüggen, J., Cowan-Jacob, S. W., Ray, A., et al. (2005). Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7, 129–141. doi:10.1016/j.ccr.2005.01.007
Wind, S., Schnell, D., Ebner, T., Freiwald, M., and Stopfer, P. (2017). Clinical pharmacokinetics and pharmacodynamics of afatinib. Clin. Pharmacokinet. 56, 235–250. doi:10.1007/s40262-016-0440-1
Wong, S., and Witte, O. N. (2004). The BCR-ABL story: Bench to bedside and back. Annu. Rev. Immunol. 22, 247–306. doi:10.1146/annurev.immunol.22.012703.104753
Wu, L., Leng, D., Cun, D., Foged, C., and Yang, M. (2017). Advances in combination therapy of lung cancer: Rationales, delivery technologies and dosage regimens. J. Control. Release 260, 78–91. doi:10.1016/j.jconrel.2017.05.023
Wu, Y.-L., Cheng, Y., Zhou, X., Lee, K. H., Nakagawa, K., Niho, S., et al. (2017). Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet. Oncol. 18, 1454–1466. doi:10.1016/S1470-2045(17)30608-3
Xiong, L., Li, R., Sun, J., Lou, Y., Zhang, W., Bai, H., et al. (2019). Erlotinib as neoadjuvant therapy in stage IIia (N2) EGFR mutation-positive non-small cell lung cancer: A prospective, single-arm, phase II study. Oncologist 24, 157–e64. doi:10.1634/theoncologist.2018-0120
Yang, J. C., Hirsh, V., Schuler, M., Yamamoto, N., O'Byrne, K. J., Mok, T. S., et al. (2013). Symptom control and quality of life in LUX-lung 3: A phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 31, 3342–3350. doi:10.1200/JCO.2012.46.1764
Zabriskie, M. S., Eide, C. A., Tantravahi, S. K., Vellore, N. A., Estrada, J., Nicolini, F. E., et al. (2014). BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 26, 428–442. doi:10.1016/j.ccr.2014.07.006
Zhang, W., Wei, Y., Yu, D., Xu, J., and Peng, J. (2018). Gefitinib provides similar effectiveness and improved safety than erlotinib for advanced non-small cell lung cancer: A meta-analysis. Med. Baltim. 97, e0460. doi:10.1097/MD.0000000000010460
Zhou, C., Wu, Y. L., Chen, G., Feng, J., Liu, X. Q., Wang, C., et al. (2011). Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet. Oncol. 12, 735–742. doi:10.1016/S1470-2045(11)70184-X
Keywords: FDA, cancer kinase inhibitors, BCR-ABL, EGFR, BRAF, MEK, ALK, BTK inhibitor
Citation: Naik RR and Shakya AK (2023) Exploring the chemotherapeutic potential of currently used kinase inhibitors: An update. Front. Pharmacol. 13:1064472. doi: 10.3389/fphar.2022.1064472
Received: 08 October 2022; Accepted: 28 November 2022;
Published: 09 January 2023.
Edited by:
Muthupandian Saravanan, Saveetha Dental College And Hospitals, IndiaReviewed by:
Surya Amarachintha, Georgia Southwestern State University, United StatesCopyright © 2023 Naik and Shakya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashok K. Shakya, YXNob2trdW1hcjI4MTFAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.