 Hongyu Qin1†
Hongyu Qin1† Lei Zhang
Lei Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 09 January 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1054616
This article is part of the Research TopicMedicinal chemistry of the PARP-1 inhibitor anti-tumor agents and other natural product derivativesView all 4 articles
Introduction: Development of Poly (ADP-ribose) polymerase (PARP) inhibitors has been extensively studied in cancer treatment. Olaparib, the first approved PARP inhibitor, showed potency in the inhibition of both BRCA (breast cancer associated)-mutated and BRCA-unmutated cancers.
Methods: Aiming to the discovery of olaparib analogs for the treatment of cancer, structural modifications were performed based on the scaffold of olaparib. In the first series, reduction of carbonyl group to CH2 led to decrease of PARP1 inhibitory activity. Preserving the original carbonyl group, molecules with potent PARP1 inhibitory activities were derived by introduction of hydrazide and aromatic nitrogen mustard groups. The synthesized compounds were evaluated in the in the PARP1 enzyme inhibitory screening, cancer cell based antiproliferative assay, cell cycle arrest and apoptosis studies.
Results: It is remarkable that, molecule C2 with chlorambucil substitution, exhibited potent PARP1 inhibitory activity and a broad-spectrum of anticancer potency in the in vitro antiproliferative assay. Compared with olaparib and chlorambucil, molecule C2 also showed significant potency in inhibition of a variety of BRCA-unmutated cell lines. Further analysis revealed the effects of C2 in induction of G2/M phase cell cycle arrest and promotion of apoptosis.
Discussion: Collectively, the olaparib-chlorambucil hybrid molecule (C2) could be utilized as a lead compound for further drug design.
Poly (ADP-ribose) polymerases (PARPs) are a group of enzymes with the biological function of transferring ADP-ribose to target proteins (Krishnakumar and Kraus, 2010). PARPs play crucial roles in a variety of cellular processes, including regulation of transcription, replication, recombination, apoptosis, and DNA damage response (Pines et al., 2012; Hu et al., 2014). Within the PARP family, PARP1 serves a critical function in DNA repair and has thus received considerable interest as a major target of inhibitor development (Chaudhuri and Nussenzweig, 2017). For more than a decade, inhibition of PARP1 has been a promising therapeutic intervention strategy for cancer (Coyne et al., 2015).
PARP inhibitors (PARPis) have been widely recognized for their significance in inhibition of homologous recombination repair (HR)-deficient tumors (Wang et al., 2016; Rose et al., 2020). In synthetically lethal interactions, tumor cells with essential HR gene mutations in the breast cancer-associated 1 and 2 (BRCA1 and BRCA2) genes can be targeted by PARPis (Bryant et al., 2005). PARPis are the first approved antitumor agents targeting the DNA damage response in BRCA1/2-mutated breast and ovarian cancers (Dockery et al., 2017). Several PARP inhibitors, including olaparib (Menear et al., 2008), rucaparib (Thomas et al., 2007), niraparib (Jones et al., 2015), and talazoparib (Shen et al., 2013), are approved for the treatment of ovarian, breast, and pancreatic cancers with BRCA mutations (Figure 1).
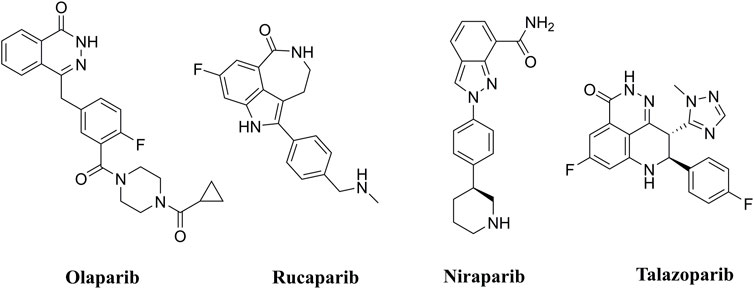
FIGURE 1. FDA approved PARP inhibitors utilized for the treatment of cancer.
Olaparib, the first approved small molecule PARPi, is utilized for the treatment of germline BRCA-mutated ovarian and breast cancers. However, there are also some drawbacks that limit the wide application of olaparib, such as its low solubility in both aqueous and lipophilic solutions (Gala et al., 2020). Thus, olaparib was characterized with relatively poor pharmacokinetics profiles and bioavailability (Jackson and Chester, 2015). Therefore, in the search for novel olaparib analogs, we have tested molecules derived from structural modification of olaparib. First, the carbonyl group connected to the piperazine ring was reduced to CH2, and a series of olaparib derivatives were synthesized to improve solubility (Figure 2). In further structural derivatization, hydrazide and aromatic nitrogen mustard groups were introduced without reduction of the carbonyl group. The synthesized molecules were evaluated using PARP1 enzyme inhibitory screening and cancer cell-based antiproliferative assays and apoptosis studies.
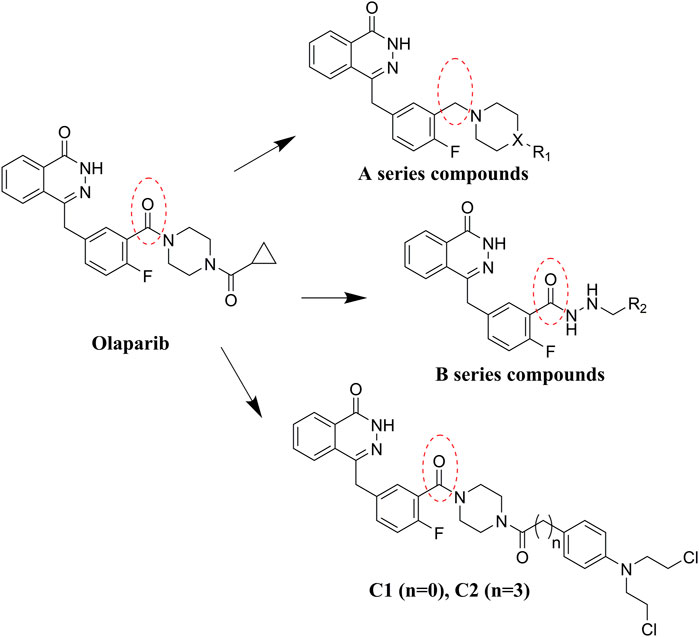
FIGURE 2. Design of olaparib derivatives in the SAR study.
Commercially available compound 2 was used as the starting material for the synthesis of the key intermediates in the synthesis (Scheme 1). Intermediates 2a and 2e were obtained by esterification and condensation of starting material 2, respectively. In the synthesis of A series target compounds, compound 2a was treated by LiAlH4 reduction and subsequent bromination of the hydroxyl group to generate intermediate 2c. The other parts (1b–12b) of the target molecules were synthesized by the coupling of 1-Boc-piperazine with substituted benzoyl chlorides and deprotection of the Boc group. Target compounds A1–A12 were derived by condensation of 2c with 1b–12b, and compounds A13–A23 were synthesized by reacting 2c with various substituted piperazines and piperidines. Target compounds B1–B6 were synthesized by hydrazinolysis of intermediate 2a, followed by condensation reaction with various aliphatic aldehydes and reduction of Schiff base. Preparation of target compounds C1 and C2 were accomplished by removal of the Boc group from the structure of intermediate 2e and subsequent condensation with substituted nitrogen mustards.
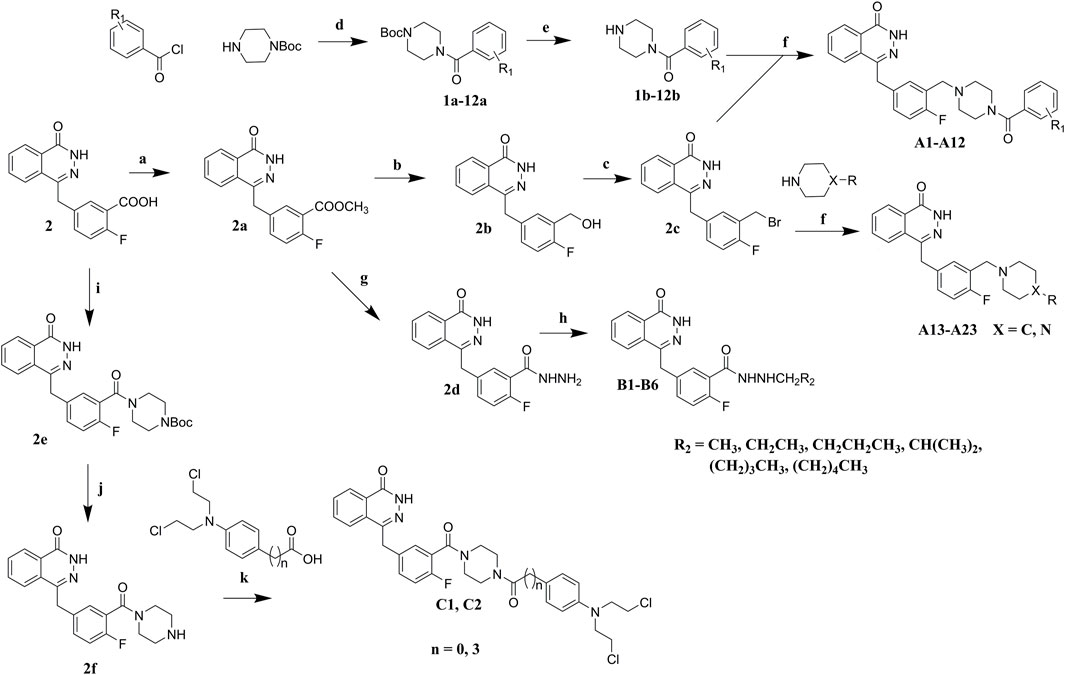
SCHEME 1. Synthesis of target compounds. Reagents and conditions: (a) H2SO4, MeOH, reflux; (b) LiAlH4, THF, 0°C, RT; (c) PBr3, DMF, 0°C; (d) Et3N, DCM, 0°C, RT; (e) TFA, DCM, RT; (f) K2CO3, DCM, RT; (g) NH2NH2.H2O, MeOH, 80°C; (h) R2CHO, NaBH4, MeOH; (i) Et3N, TBTU, Boc-piperazine, DCM, 0°C, RT; (j) TFA, DCM, RT; (k) Et3N, TBTU, DCM, 0°C, RT.
In development of olaparib analogs with improved solubility, the carbonyl group in the structure of olaparib was reduced to CH2. A series of olaparib derivatives were synthesized with improved water solubility and liposolubility. To evaluate the PARP1 inhibitory activity of the synthesized compounds, the enzyme-based test was performed. As shown in Table 1, all the derived A series compounds exhibited decreased potency compared with olaparib. In this series, cycloalkane formyl substitutions in piperazine showed relatively high inhibitory activity; these included molecules A21, A22, and A23. Among A1–A3, fluorine substitution in the meta-position of the phenyl ring resulted in improved PARP1 inhibitory activity. The CF3 substitution in the benzene ring (A4–A6) led to decreased activity compared with the fluorine substitution. A methyl group in the ortho-position of the phenyl ring (A9) increased inhibitory activity. Compounds with a methoxy group in the phenyl ring (A11 and A13) did not show favorable activity, while the 1,3-dioxole substitution (A20) increased inhibitory potency. Replacement of the carbonyl group with a methylene group between piperazine and the phenyl ring (A15 and A18) decreased PARP1 inhibitory potency. The substituted piperidine R groups (A14 and A16–A19) exhibited reduced inhibitory potency compared with the substituted piperazine groups.

TABLE 1. Structure and PARP1 inhibitory activities of A series PARP inhibitors.
The binding of inhibitors to the active site of receptors may be key factors that influence inhibitory enzyme potency. Therefore, the crystal structure of olaparib in the active site of PARP1 (PDB entry: 7AAD) was analyzed to identify the structural elements that affect PARP1 inhibitory activity. As shown in Figure 3, the carbonyl group of olaparib could form H-bond interaction with the amino group of Tyr896. The generated H-bond interaction makes an important contribution to the binding of olaparib to the active site of PARP1, with a distance of 2.811 Å between atoms (between the oxygen atom of the carbonyl group and the nitrogen atom of Tyr896). Compared with olaparib, the decreased activity of A series compounds was deduced to be due to the loss of H-bond interaction resulting from the reduction of the carbonyl group.
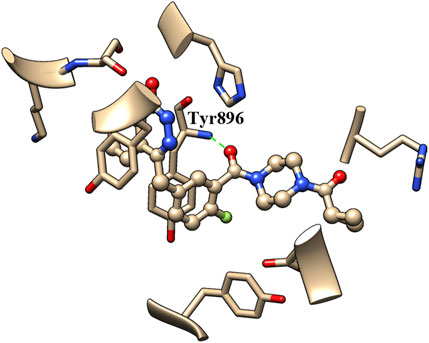
FIGURE 3. Binding pattern analysis of olaparib in the active site of PARP1.
Considering the importance of the carbonyl group in the structure of olaparib for PARP1 binding, the CO group was retained for further olaparib analog design. Hydrazides with various alkane substitutions were then introduced. The corresponding B series molecules were synthesized, and alkanes of different lengths were utilized to regulate the binding and solubility of target compounds. Nitrogen mustards are known alkylating agents that exert their anticancer activities depending on DNA damage. The aromatic nitrogen mustard groups have been widely used in the design of hybrid anticancer molecules (Chen et al., 2018). It is significant that nitrogen mustards exert their function by damaging DNA, while PARP1 inhibitors prevent DNA repair. Therefore, development of bifunctional alkylating-PARP1 inhibitor molecules may lead to therapeutic agents with synergistic anticancer activities. In this study, to verify the proposed theory, aromatic nitrogen mustard pharmacophores were introduced into the structure of olaparib.
The PARP1 inhibitory activities of B series compounds and nitrogen mustard containing C1 and C2 were determined using the enzymatic assay (Table 2). Among the B series molecules, compound B3, with an alkyl chain length of four carbon atoms, exhibited the highest inhibitory activity, with an IC50 value of 2.8 nM, compared with olaparib (IC50 value of 1.0 nM). Increase or decrease of the alkyl chain length resulted in reduction of PARP1 inhibitory potency. It is also important to note that the compound with branched alkane (B4) had reduced activity compared with compounds with straight alkanes. Overall, the B series compounds exhibited potent PARP1 inhibitory activity compared with olaparib.
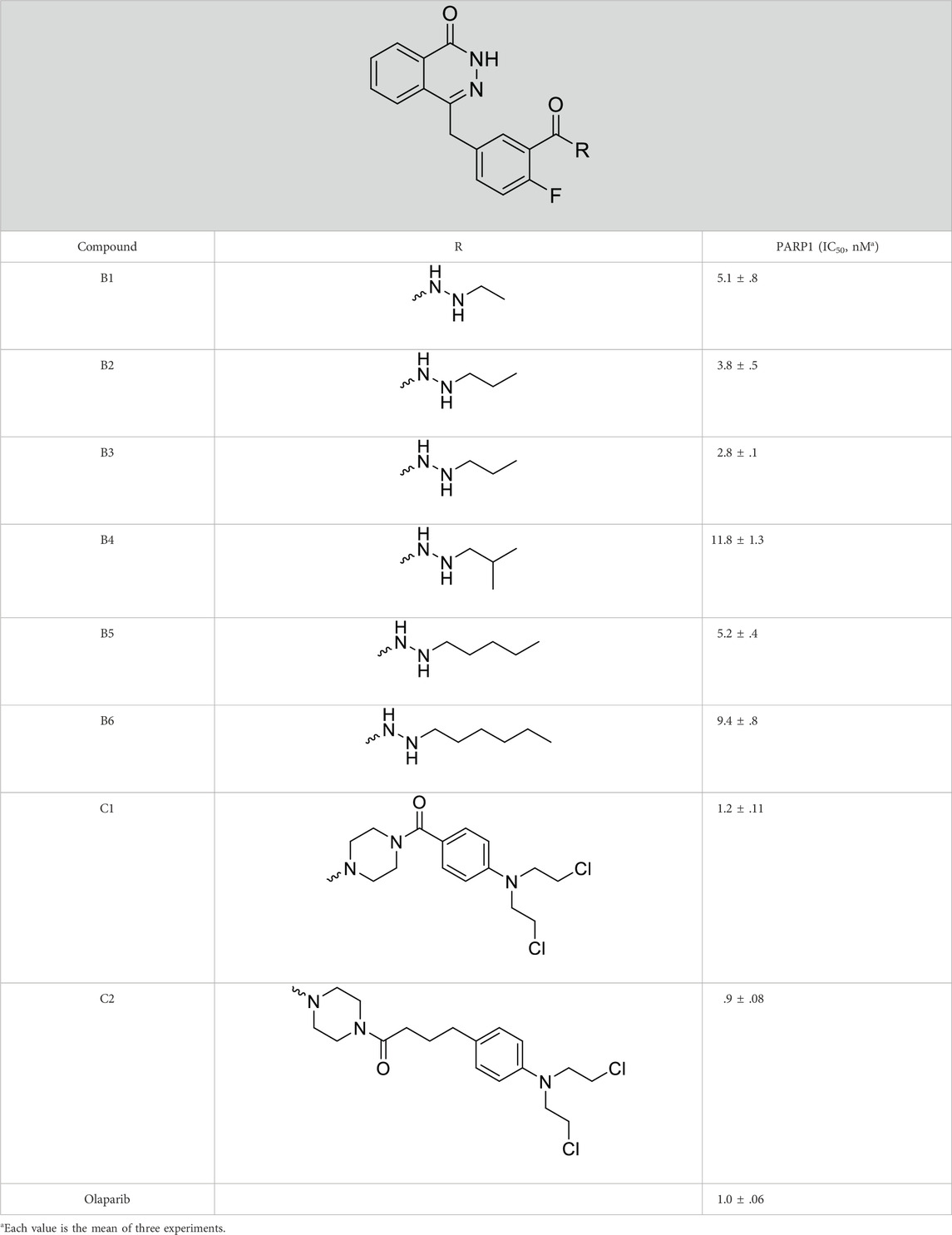
TABLE 2. Structure and PARP1 inhibitory activities of B and C series PARP inhibitors.
C series compounds are hybrid molecules with aromatic nitrogen mustard substituted in the piperazine of olaparib. The compound with a long linker (C2) exhibited better enzyme inhibitory activity than the compound with a short linker (C1). Compared with olaparib (IC50 value of 1.0 nM), molecule C2 had an IC50 value of .9 nM in inhibition of PARP1 activity. These data suggest that introduction of an aromatic nitrogen mustard group improves PARP1 inhibitory activity and that the olaparib–chlorambucil hybrid molecule C2 could be utilized for further evaluation as a potent PARP1 inhibitor.
It has been reported that olaparib has potential efficacy in tumors lacking DNA repair defects (Gelmon et al., 2011; Poveda et al., 2019). The existence of non-DNA repair functions of PARP1 provides targetable opportunities for anticancer drug development (Hanahan and Weinberg, 2011; Weaver and Yang, 2013). Therefore, both BRCA-mutated cell lines (MDA-MB-436, Capan-1, and HCC 1937) and cell lines without BRCA mutations (K562, Raji, 8226, U87, B16, SKOV3, MCF-7, SW620, A549, MDA-MB-231, CH151, U251, and Hela) were utilized for the antiproliferative assay. Olaparib and the alkylating agent chlorambucil were used as the positive control. Only molecules with high PARP1 inhibitory activities (B3, C1, and C2) were selected for the cancer cell-based assay. As shown in Table 3, compound B3 exhibited very limited antiproliferative activity against the tested cell lines compared with olaparib. These data suggest that the performed hydrazide substitutions could not improve the potency of olaparib in both enzymatic and antiproliferative tests. In inhibition of the BRCA-mutated cell lines (MDA-MB-436, Capan-1, and HCC 1937), molecule C2 showed activity similar to olaparib. Compared with the positive control, molecule C2 exhibited high potency in inhibition of several BRCA-unmutated cell lines, such as K562, Raji, U87, MCF-7, A549, CH151, U251, and Hela cells. It was deduced that the chlorambucil part of C2 contributed to the antiproliferative activity against BRCA-unmutated cells. It was interesting that molecule C1 selectively inhibited the growth of A549 cells without obvious inhibitory activity against other BRCA-unmutated cells. These data suggest that molecule C1 could be used as a lead compound for the design of anticancer drugs for the treatment of lung cancer. In summary, molecule C2 exhibited high anticancer activity in vitro in the antiproliferative test, and the olaparib–nitrogen mustard hybridization strategy demonstrated good potential as anticancer therapy.
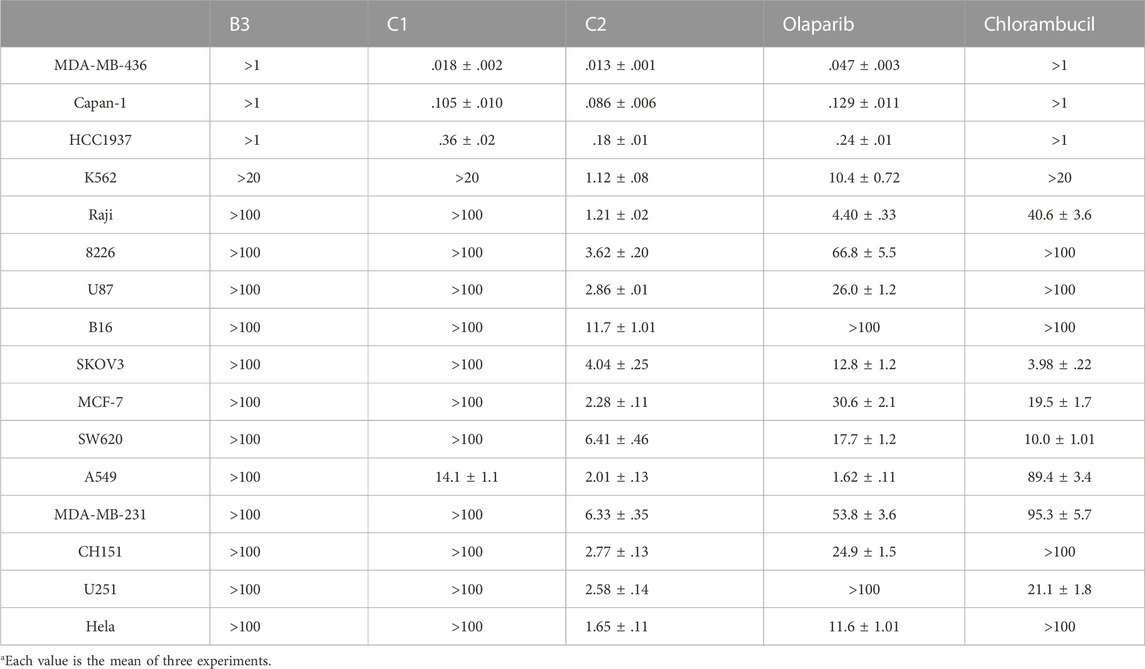
TABLE 3. In vitro antiproliferative activities of representative compounds (IC50, μMa).
A characteristic change in tumor cells is dysregulation of the cell cycle due to genetic mutations, resulting in uncontrolled cell proliferation. Cell cycle arrest is usually induced by chemotherapeutic drugs in cancer treatment. Therefore, in this study, the effect of C2 on cell cycle progression was evaluated in K562 cells at doses of 1.25–5.0 µM. As shown in Figure 4, C2 and chlorambucil increased cell number at the G2/M phase in a dose-dependent manner. The proportion of cells in the G2/M phase increased from 0% to 45.9% with C2 treatment (corresponding to concentration increase from 1.25 to 5.0 µM); with chlorambucil treatment, cells at the G2/M phase increased from 0% to 61.60% when concentration was increased from 2.5 to 10 µM. However, at the tested concentrations of 2.5–10 µM, olaparib did not exhibit any effects on regulation of cell cycle in K562 cells. The BRCA1-mutated HCC1937 human breast cancer cell line was also utilized to evaluate the cell cycle arrest patterns induced by molecule C2, olaparib, and chlorambucil. Figure 5 reveals that all the tested compounds increased cell proportion in the G2/M phase. These results suggest that induction of G2/M phase arrest plays an important role in the in vitro anticancer effects of molecule C2.
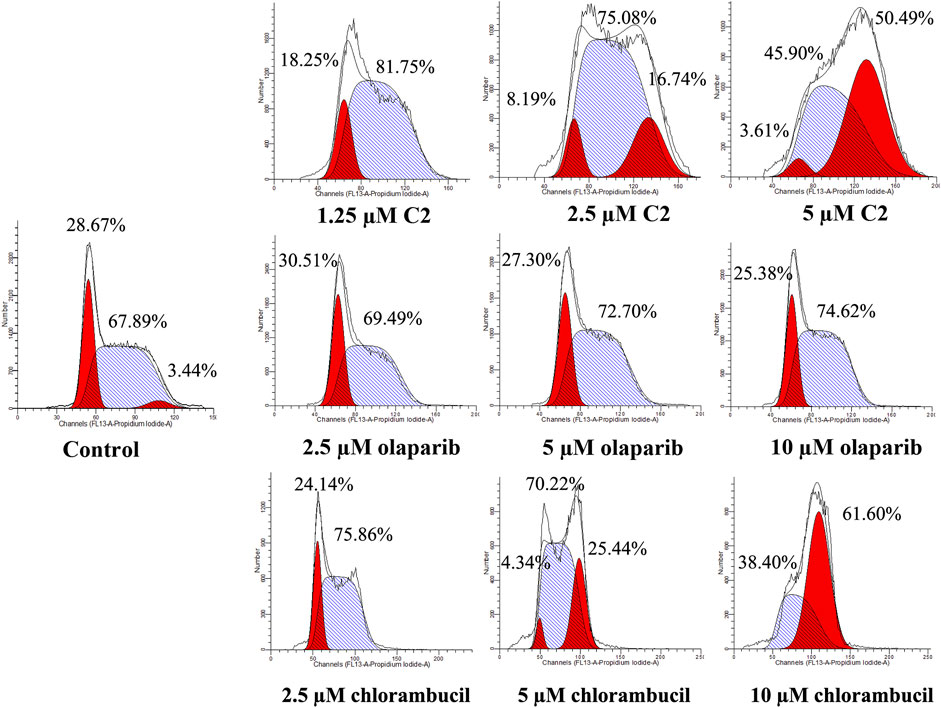
FIGURE 4. Cell cycle analysis of K562 cells treated with molecule C2, olaparib, or chlorambucil. Cells were treated with molecule C2 (1.25, 2.5, or 5.0 µM), olaparib, or chlorambucil (2.5, 5.0, or 10 µM) at different concentrations for 24 h. The results were evaluated using flow cytometry.
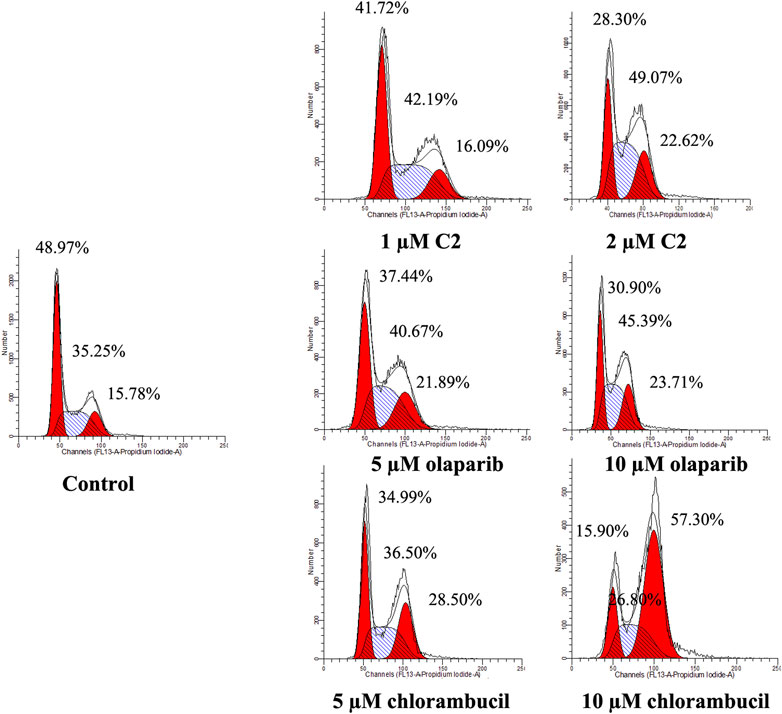
FIGURE 5. Cell cycle analysis of HCC1937 cells treated with molecule C2, olaparib, or chlorambucil. Cells were treated with molecule C2 (1 or 2 µM), olaparib, or chlorambucil (5 or 10 µM) at different concentrations for 24 h. The results were evaluated using flow cytometry.
Apoptosis plays an important role in the inhibition of cancer. PARP inhibitors have been used to induce apoptosis in different cancer cell lines. Therefore, the effect of molecule C2 in the induction of cancer cell apoptosis was evaluated. K562 cells, which were sensitive to the test compounds, were selected for the apoptosis study and analyzed by annexin V staining. The results demonstrated that an increased number of apoptotic K562 cells were detected after treatment with different doses of C2, olaparib, or chlorambucil (Figure 6). It is noteworthy that molecule C2 induced K562 cell apoptosis at higher rates with lower doses than olaparib and chlorambucil. The percentage of apoptotic cells was significantly increased from .90% in the control to 10.96% and 12.69% after treatment with 1 μM and 2 µM of C2, respectively, compared with olaparib (apoptotic rates of 1.53% and 1.91% at doses of 5 μM and 10 µM, respectively) and chlorambucil (apoptotic rates of 2.49% and 2.96% at doses of 5 μM and 10 µM, respectively). These data suggest that induction of apoptosis plays an important role in the anticancer effects of molecule C2.
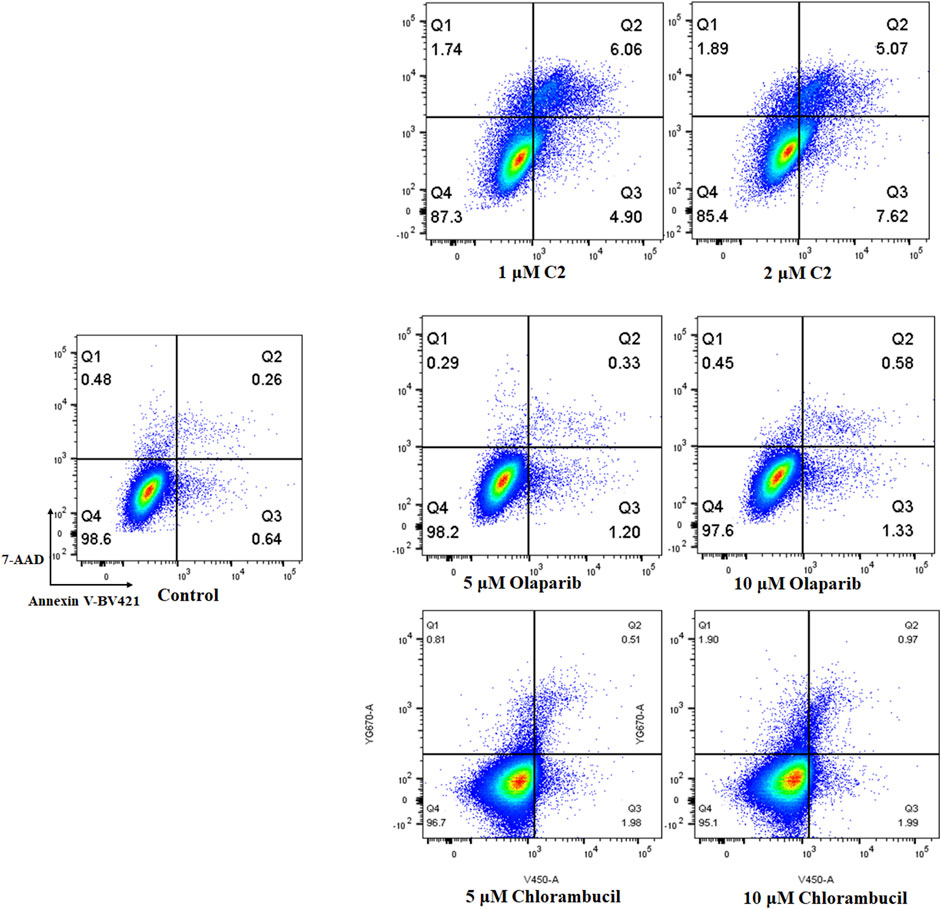
FIGURE 6. Proapoptotic effects of molecule C2. K562 cells were treated with molecule C2 (1 or 2 µM), olaparib (5 or 10 µM), or chlorambucil (5 or 10 µM) for 24 h. The cells were then stained with Annexin V–BV421/7-AAD, and the apoptotic status of the cells was assessed using flow cytometry.
Inhibition of PARPs has emerged as a research focus in the development of targeted cancer treatments. Four PARP inhibitors are currently approved for clinical use: olaparib, rucaparib, niraparib, and talazoparib. As the first approved PARP inhibitor, olaparib exhibited potency in the inhibition of both BRCA-mutated and BRCA-unmutated cancers. In development of novel PARP inhibitors as anticancer drugs, modifications have been made to the structure of olaparib. In the current study, 31 compounds were designed, synthesized, and tested for activity. Among the derived molecules, an olaparib–chlorambucil hybrid molecule (C2) exhibited high potency in the inhibition of PARP1 activity and showed high antiproliferative activity against a group of cancer lines. Notably, in the inhibition of multiple BRCA-unmutated cancer cells, molecule C2 showed improved anticancer activity in vitro compared with olaparib and chlorambucil. Analysis of the cell cycle and apoptosis showed that G2/M phase cell cycle arrest and apoptosis are involved in the anticancer effects of C2. Our study led to the discovery of a potent PARP1 inhibitor–chlorambucil hybrid compound with strong anticancer effects. Our results also revealed the potential of PARP inhibitor–chemotherapeutic drug hybrids in the treatment of cancer.
All commercially available materials, reagents, and solvents were used without further purification. All reactions were monitored using TLC with .25-mm silica gel plates (60GF-254). UV light and ferric chloride were used to visualize the spots. 1H NMR and 13C NMR spectra were recorded on a Bruker DRX spectrometer at 500 MHz, using TMS as an internal standard. High-resolution mass spectra were produced at Weifang Medical University.
Tert-butyl 4-(4-fluorobenzoyl)piperazine-1-carboxylate (1a). To a solution of 1-Boc-piperazine (1.86 g, 10 mmol) in DCM, the following was added dropwise: 4-fluorobenzoyl chloride (1.90 g, 12 mmol), followed by Et3N (3.03 g, 30 mmol) at 0°C; the mixture was stirred for 4 h. The solvent was then evaporated, and the residue was taken up in EtOAc (50 ml). The EtOAc solution was washed with saturated citric acid (3 × 50 ml), NaHCO3 (3 × 50 ml), and saturated brine solution (3 × 50 ml), dried over MgSO4, and evaporated under a vacuum. The crude product was used directly in the next step.
(4-Fluorophenyl)(piperazin-1-yl)methanone (1b). To a solution of 1a in DCM with TFA at RT, and the mixture was stirred for 2 h. The mixture was then concentrated in vacuo. The crude oil was used for the following step without purification.
Methyl 2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoate (2a). Three drops of concentrated sulfuric acid were added to the methanol solution of dissolved compound 2 (2.98 g, 10 mmol). The mixture was heated at reflux for 5 h in an oil bath. TLC analysis indicated the complete disappearance of the starting material. Upon cooling to ambient temperature, the solvent was evaporated under vacuum. The residue was extracted with EtOAc (50 ml) and saturated NaHCO3 (3 × 50 ml), dried over MgSO4, and evaporated under vacuum. The desired compound 2a (2.97 g, 95% yield) was derived as white powder. HRMS m/z: 313.09586 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.62 (s, 1H), 8.27 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.93–7.83 (m, 3H), 7.63 (s, 1H), 7.30 (t, J = 8.0 Hz, 1H), 4.39 (s, 2H), and 3.84 (s, 3H).
(5-Fluoro-3-(hydroxymethyl)benzyl)phthalazin-1(2H)-one (2b). A solution of LiAlH4 (.12 g, 3.20 mmol) in THF was stirred at 0°C; after 10 min, compound 2a (.50 g, 1.60 mmol) was added. The reaction solution was stirred at room temperature for 30 min. Then ice water was poured into the reaction mixture, and the solution was adjusted to pH 5-6 using 1 M HCl. The target compound 2b (.36 g, 80% yield) was obtained by filtering. HRMS m/z: 283.08902 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26 (q, J = 4.0 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.89–7.79 (m, 2H), 7.40–7.38 (m, 1H), 7.28–7.24 (m, 1H), 7.08–7.04 (m, 1H), 5.23–5.20 (m, 1H), 4.48 (d, J = 8.0 Hz, 2H), and 4.29 (s, 2H).
4-(3-(Bromomethyl)-4-fluorobenzyl)phthalazin-1(2H)-one (2c). The starting material, 2b (.20 g, .71 mmol), was dissolved in DMF, and the solution was cooled to 0°C. PBr3 (.29 g, 1.07 mmol) was added to the solution at 0°C, and then, the mixture was stirred for 30 min. The reaction was quenched by addition of H2O and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and evaporated under vacuum. The target compound 2c (.18 g, 74% yield) was derived by crystallization in ether. HRMS m/z: 347.01743 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.95–7.90 (m, 1H), 7.85 (dd, J = 8.0, 1.2 Hz, 1H), 7.81 (dd, J = 6.0, 1.2 Hz, 1H), 7.45 (dd, J = 8.0, 2.0 Hz, 1H), 7.36 (dd, J = 2.8, 2.4 Hz, 1H), 7.34–7.33 (m, 1H), 7.19–7.14 (m, 1H), 4.64 (s, 2H), and 4.29 (s, 2H).
4-(4-Fluoro-3-((4-(4-fluorobenzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A1). To a solution of 2c (.15 g, .43 mmol) and 1b (.10 g, .48 mmol) in DCM, K2CO3 (.18 g, 1.29 mmol) was added at RT, and the mixture was stirred overnight. The solvent was evaporated, and the residue was dissolved in EtOAc (20 ml). The EtOAc solution was then washed with saturated brine (3 × 20 ml), dried over MgSO4, and evaporated under vacuum. The desired compound was derived by crystallization in EtOAc as white powder. HRMS (AP-ESI) m/z calcd for C27H24F2N4O2 [M + H]+ 475.19473, found 475.19174. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.88–7.77 (m, 2H), 7.46–7.42 (m, 2H), 7.33–7.24 (m, 4H), 7.11–7.07 (m, 1H), 4.30 (s, 2H), 3.50–3.16 (m, 6H), and 2.33 (s, 2H). 13C-NMR (400 MHz, DMSO) δ 168.451, 164.208, 161.756, 161.164, 159.870, 145.629, 134.439, 133.838, 132.655 (d, J = 74.2 Hz), 130.042 (d, J = 8.5 Hz), 129.655, 128.364, 126.486, 126.088, 124.516, 115.954, 65.385, 54.677, 52.773, 40.398 (d, J = 21.0 Hz), 39.980, and 37.257 ppm.
4-(4-Fluoro-3-((4-(2-fluorobenzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A2). HRMS (AP-ESI) m/z calcd for C27H24F2N4O2 [M + H]+ 475.19473, found 475.19235 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25–8.24 (m, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.86–7.77 (m, 2H), 7.51–7.49 (m, 2H), 7.38–7.24 (m, 4H), 7.11–7.07 (m, 1H), 4.30 (s, 2H), 3.58 (s, 2H), 3.50 (s, 2H), 3.15 (s, 2H), 2.37 (s, 2H), and 2.28–2.26 (m, 2H). 13C-NMR (400 MHz, DMSO) δ 164.273, 161.164, 159.862, 159.168, 156.729, 145.622, 134.472, 133.829, 132.064, 131.909, 131.871 (d, J = 11.2 Hz), 129.278, 128.365, 126.483, 126.099, 125.423, 124.569, 116.353, 115.835, 54.669, 52.903, 52.337, 46.926, 41.682, and 37.247 ppm.
4-(4-Fluoro-3-((4-(3-fluorobenzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A3). HRMS (AP-ESI) m/z calcd for C27H24F2N4O2 [M + H]+ 475.19473, found 475.19229 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.58 (s, 1H), 8.26–8.23 (m, 1H), 7.92–7.90 (m, 1H), 7.87–7.76 (m, 2H), 7.53–7.47 (m, 1H), 7.33–7.19 (m, 5H), 7.11–7.07 (m, 1H), 4.29 (s, 2H), 3.54 (s, 2H), 3.50 (s, 2H), 3.24 (s, 2H), 2.36 (s, 2H), and 2.23 (s, 2H). 13C-NMR (400 MHz, DMSO) δ 167.851 (d, J = 2.1 Hz), 163.500, 161.164 (d, J = 10.3 Hz), 161.062, 159.871, 145.612, 138.656 (d, J = 6.8 Hz), 134.458 (d, J = 3.1 Hz), 133.816, 132.016, 131.885, 131.122 (d, J = 8.1 Hz), 129.733, 128.370, 126.077, 124.370 (d, J = 15.1 Hz), 123.402, 116.945, 115.604, 114.501, 54.677, 52.846, 52.306, 47.441, 41.906, and 37.263 ppm.
4-(4-Fluoro-3-((4-(4-(trifluoromethyl)benzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A4). HRMS (AP-ESI) m/z calcd for C28H24F4N4O2 [M + H]+ 525.19154, found 525.18939 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (dd, J1 = 6.8 Hz, J2 = 1.2 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.87–7.77 (m, 4H), 7.59 (d, J = 8.0 Hz, 2H), 7.33–7.24 (m, 2H), 7.12–7.07 (m, 1H), 4.30 (s, 2H), 3.58 (s, 2H), 3.51 (s, 2H), 3.22 (s, 2H), 2.39 (s, 2H), and 2.30 (s, 2H). 13C-NMR (400 MHz, DMSO) δ 167.966, 161.170, 159.869, 158.751, 145.620, 140.434, 134.465 (d, J = 3.1 Hz), 133.839, 132.044, 131.899, 130.248, 129.681, 128.187 (d, J = 17.9 Hz), 126.479, 125.977, 124.348 (d, J = 15.1 Hz), 123.043, 115.828, 115.607, 54.669, 52.857, 52.277, 47.434, 41.890, and 37.252 ppm.
4-(4-Fluoro-3-((4-(2-(trifluoromethyl)benzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A5). HRMS (AP-ESI) m/z calcd for C28H24F4N4O2 [M + H]+ 525.19154, found 525.18878 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.24 (dd, J1 = 8.0 Hz, J2 = 2.8 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.86–7.73 (m, 4H), 7.66 (d, J = 8.0 Hz, 1H), 7.44–7.42 (m, 1H), 7.33–7.31 (m, 1H), 7.25–7.23 (m, 1H), 7.11–7.06 (m, 1H), 4.29 (s, 2H), 3.57 (s, 2H), 3.49 (s, 2H), 3.05–2.97 (m, 2H), and 2.41–2.19 (m, 4H). 13C-NMR (400 MHz, DMSO) δ 166.462, 161.155, 159.869, 159.735, 145.596, 135.171, 134.462, 133.329, 132.006, 131.967, 129.923, 129.651 (d, J = 8.6 Hz), 128.363, 127.009, 126.060, 125.536, 124.359, 122.836, 115.808, 54.685, 52.360, 52.161, 46.989, 41.464, and 37.250 ppm.
4-(4-Fluoro-3-((4-(3-(trifluoromethyl)benzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A6). HRMS (AP-ESI) m/z calcd for C28H24F4N4O2 [M + H]+ 525.19154, found 525.18993 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.24 (q, J = 4.0 Hz, 1H), 7.91 (J = dd, J1 = 8.0 Hz, J2 = 2.8 Hz, 1H), 7.86–7.76 (m, 3H), 7.72–7.68 (m, 2H), 7.34–7.23 (m, 2H), 7.09 (m, 1H), 4.30 (s, 2H), 3.57 (s, 2H), 3.50 (s, 2H), 3.24 (s, 2H), 2.39 (s, 2H), and 2.30 (s, 2H). 13C-NMR (400 MHz, DMSO) δ 168.096, 161.647, 159.928, 159.190, 145.215, 136.444, 135.119, 134.753, 134.024, 132.867, 131.938, 131.562, 130.244, 129.486 (d, J = 12.6 Hz), 128.378, 127.076, 126.246, 125.693, 124.393, 122.984, 116.198, 52.306, 50.519, and 37.383 ppm.
4-(3-((4-(2-Chlorobenzoyl)piperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A7). HRMS (AP-ESI) m/z calcd for C27H24ClFN4O2 [M + H]+ 491.16518, found 491.16260 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.58 (s, 1H), 8.25 (q, J = 4.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.86–7.76 (m, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.47–7.40 (m, 2H), 7.40–7.31 (m, 2H), 7.27–7.23 (m, 1H), 7.11–7.06 (m, 1H), 4.29 (s, 2H), 3.58 (s, 2H), 3.49 (s, 2H), 3.06 (s, 2H), 2.38 (s, 2H), and 2.30 (s, 2H). 13C-NMR (400 MHz, DMSO) δ 165.874, 161.156, 159.863, 158.733, 145.617, 136.162, 134.472, 133.839, 131.912, 130.963, 129.877, 129.559, 128.361, 128.062, 126.490, 126.096, 124.339, 115.609 (d, J = 22.3 Hz), 54.688, 52.817, 52.264, 46.615, 41.425, and 37.246 ppm.
4-(3-((4-(3-Bromobenzoyl)piperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A8). HRMS (AP-ESI) m/z calcd for C27H24BrFN4O2 [M + H]+ 535.11467, found 535.11224 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.60 (s, 1H), 8.25 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.87–7.81 (m, 2H), 7.79–7.66 (m, 1H), 7.56 (s, 1H), 7.44–7.25 (m, 5H), 4.30 (s, 3H), 3.50 (s, 4H), 3.24 (s, 2H), and 2.31 (s, 3H). 13C-NMR (400 MHz, DMSO) δ 167.670, 159.889, 134.458, 133.887, 131.929, 131.189, 130.035, 129.546, 128.372, 126.491 (d, J = 14.5 Hz), 126.347, 126.129, 122.195, 52.811, 40.600, 40.392, 39.975, 39.766, and 37.285 ppm.
4-(4-Fluoro-3-((4-(2-methylbenzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A9). HRMS (AP-ESI) m/z calcd for C28H27FN4O2 [M + H]+ 471.21980, found 471.21762 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.58 (s, 1H), 8.24 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.91–7.90 (m, 1H), 7.85–7.75 (m, 2H), 7.32–7.21 (m, 5H), 7.12–7.06 (m, 2H), 4.29 (s, 2H), 3.58 (s, 2H), 3.49 (s, 2H), 3.04 (t, J = 4.8 Hz, 2H), 2.37 (s, 2H), 2.22 (s, 2H), and 2.18 (s, 3H). 13C-NMR (400 MHz, DMSO) δ 168.873, 161.152, 159.862, 158.730, 145.620, 136.764, 134.428, 131.993, 131.899, 130.622, 129.065, 128.365, 126.068, 124.370, 115.602, 54.709, 53.056, 52.491, 46.628, 41.219, 37.255, and 19.052 ppm.
4-(3-((4-(4-Ethylbenzoyl)piperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A10). HRMS (AP-ESI) m/z calcd for C29H29FN4O2 [M + H]+ 485.23545, found 485.23273 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.86–7.76 (m, 2H), 7.32–7.25 (m, 6H), 7.11–7.07 (m, 1H), 4.30 (s, 2H), 3.50 (s, 4H), 2.64 (q, J = 8.0 Hz, 2H), 2.23 (s, 4H), and 1.20 (t, J = 7.6 Hz, 3H). 13C-NMR (400 MHz, DMSO) δ 169.453, 161.164, 159.869, 158.754, 145.827, 134.435, 133.839, 133.622 (d, J = 21.8 Hz), 131.982, 131.914, 129.650, 129.559, 128.183 (d, J = 18.7 Hz), 127.587, 126.105, 124.525, 115.835, 54.711, 40.604, 40.396, 40.187, 37.258, 28.450, and 15.863 ppm.
4-(4-Fluoro-3-((4-(2-methoxybenzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A11). HRMS (AP-ESI) m/z calcd for C28H27FN4O3 [M + H]+ 487.21472, found 487.21201 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (q, J = 4.0 Hz, 1H), 7.91 (d, J = 4.0 Hz, 1H), 7.86–7.78 (m, 2H), 7.41–7.39 (m, 1H), 7.37–7.31 (m, 1H), 7.25–7.24 (m, 1H), 7.15–7.06 (m, 3H), 7.01–6.97 (m, 1H), 4.30 (s, 2H), 3.76 (s, 3H), 3.54 (s, 2H), 3.49 (s, 2H), 3.05 (s, 2H), and 2.36–2.33 (s, 4H). 13C-NMR (400 MHz, DMSO) δ 166.762, 161.153, 159.869, 158.733, 155.311, 145.623, 134.415, 133.812, 131.938 (d, J = 9.2 Hz), 131.892, 130.776, 129.557, 128.114, 126.035 (d, J = 4.7 Hz), 124.379 (d, J = 15.0 Hz), 121.075, 115.806 (d, J = 22.3 Hz), 111.770, 55.833, 54.738, 52.919, 52.425, 46.617, 41.365, and 37.260 ppm.
4-(3-((4-Benzoylpiperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A12). HRMS (AP-ESI) m/z calcd for C27H25FN4O2 [M + H]+ 457.20415, found 457.20154 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (q, J = 4.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.86–7.76 (m, 2H), 7.46–7.43 (m, 3H), 7.37–7.23 (m, 4H), 7.19 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 4.30 (s, 2H), 3.54 (s, 2H), 3.50 (s, 2H), 3.25 (s, 2H), and 2.30 (s, 4H). 13C-NMR (400 MHz, DMSO) δ 169.328, 161.162, 159.868, 158.743, 145.628, 136.319, 134.435 (d, J = 3.1 Hz), 133.824, 132.016, 131.907, 129.963, 129.562, 128.370, 127.355, 126.096, 124.519 (d, J = 15.1 Hz), 124.369, 115.832, 115.611, 54.697, 52.919, 52.431, 47.499, 41.919, and 37.260 ppm.
4-(4-Fluoro-3-((4-(4-methoxybenzoyl)piperazin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A13). HRMS (AP-ESI) m/z calcd for C28H27FN4O3 [M + H]+ 487.21472, found 487.21152 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.92–7.91 (m, 1H), 7.87–7.77 (m, 2H), 7.35–7.31 (m, 3H), 7.28–7.24 (m, 1H), 7.11–7.07 (m, 1H), 7.00–6.96 (m, 2H), 4.30 (s, 2H), 3.80 (s, 3H), 3.50 (s, 2H), 3.41 (s, 4H), and 2.32 (s, 4H). 13C-NMR (400 MHz, DMSO) δ 169.306, 161.162, 160.637, 159.891, 158.743, 145.652, 134.412, 133.829, 132.008, 131.910, 129.712, 129.482, 128.174 (d, J = 18.2 Hz), 126.075, 124.365 (d, J = 15.0 Hz), 115.820, 114.081, 55.695, 54.700, 52.704, 40.573, 40.156, 39.947, and 37.262 ppm.
1-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzyl)piperidine-4-carboxamide (A14). HRMS (AP-ESI) m/z calcd for C22H23FN4O2 [M + H]+ 395.18850, found 395.18613 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.93 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.87–7.79 (m, 2H), 7.34 (q, J = 4.0 Hz, 1H), 7.24–7.21 (m, 2H), 7.18–7.04 (m, 1H), 6.71 (s, 1H), 4.30 (s, 2H), 3.42 (s, 2H), 2.75–2.72 (m, 2H), 2.09–1.97 (m, 1H), 1.90–1.85 (m, 2H), 1.62–1.59 (m, 2H), and 1.54–1.45 (m, 2H). 13C-NMR (400 MHz, DMSO) δ 176.952, 159.853, 145.706, 134.390, 133.812, 131.941, 131.880, 129.559, 129.416, 128.372, 126.488, 126.100, 125.228, 115.728, 115.499, 55.150, 52.968, 42.037, 40.395, 39.769, 37.335, and 28.899 ppm.
4-(3-((4-(Benzo[d][1,3]dioxol-5-ylmethyl)piperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A15). HRMS (AP-ESI) m/z calcd for C28H27FN4O3 [M + H]+ 487.21472, found 487.21194 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.58 (s, 1H), 8.26–8.24 (m, 1H), 7.91–7.90 (m, 1H), 7.89–7.77 (m, 2H), 7.35–7.31 (m, 1H), 7.29–7.21 (m, 1H), 7.09–7.05 (m, 1H), 6.85–6.81 (m, 2H), 6.73–6.71 (m, 1H), 5.98 (s, 2H), 4.29 (s, 2H), and 2.27 (s, 8H). 13C-NMR (400 MHz, DMSO) δ 161.092, 159.870, 158.674, 148, 147.638, 146.566, 134.372, 133.777, 132.493, 131.688, 129.567, 129.344, 128.391, 126.483, 124.938, 122.349, 115.735, 109.470, 108.262, 101.218, 62.153, 54.698, 52.795, and 37.308 ppm.
4-(3-([1,4′-Bipiperidin]-1′-ylmethyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A16). HRMS (AP-ESI) m/z calcd for C26H31FN4O [M + H]+ 435.25616, found 435.25369 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26–8.24 (m, 1H), 7.91–7.79 (m, 3H), 7.28–7.21 (m, 2H), 7.09–7.04 (m, 1H), 4.29 (s, 2H), 3.40 (s, 2H), 2.73–2.70 (m, 2H), 2.39 (s, 4H), 2.08 (m, 1H), 1.83 (t, J = 12.0 Hz, 2H), 1.56 (d, J = 12.0 Hz, 2H), 1.46–1.45 (m, 4H), and 1.37–1.31 (m, 4H). 13C-NMR (400 MHz, DMSO) δ 159.856, 145.680, 134.406, 133.803, 131.919, 131.660, 129.571, 129.271, 128.383, 126.464, 126.133, 125.171, 115.709, 115.476, 62.293, 54.798, 53.044, 50.133, 40.608, 40.400 (d, J = 21.0 Hz), 39.983, 39.357, 37.308, 27.843, 26.556, and 25.074 ppm.
Methyl1-(2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzyl)piperidine-4-carboxylate (A17). HRMS (AP-ESI) m/z calcd for C23H24FN3O3 [M + H]+ 410.18907, found 410.18555 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26–8.24 (m, 1H), 7.93–7.91 (m, 1H), 7.87–7.79 (m, 2H), 7.33–7.30 (m, 1H), 7.24–7.21 (m, 1H), 7.09–7.05 (m, 1H), 4.30 (s, 2H), 3.59 (s, 3H), 3.43 (s, 2H), 2.68–2.65 (m, 2H), 2.26 (s, 1H), 1.95 (dd, J1 = 9.6 Hz, J2 = 8.8 Hz, 2H), 1.75–1.72 (m, 2H), and 1.52–1.47 (m, 2H). 13C-NMR (400 MHz, DMSO) δ 175.251, 159.856, 145.689, 134.432, 133.815, 131.938, 131.814, 129.570, 129.447, 128.374, 126.483, 126.111, 115.523, 54.998, 52.403, 51.858, 40.606 (d, J = 21.0 Hz), 40.188, 39.771, 39.354, 37.294, and 28.353 ppm.
4-(3-((4-Benzylpiperidin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A18). HRMS (AP-ESI) m/z calcd for C28H28FN3O [M + H]+ 442.22964, found 442.22733 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26–8.24 (m, 1H), 7.90–7.89 (m, 1H), 7.88–7.77 (m, 2H), 7.30–7.14 (m, 7H), 7.08–7.03 (m, 1H), 4.29 (s, 2H), 3.39 (s, 2H), 2.66–2.63 (m, 2H), 2.45 (s, 2H), 1.82–1.79 (m, 2H), and 1.44–1.41 (m, 2H). 13C-NMR (400 MHz, DMSO) δ 159.882, 145.666, 140.753, 133.786, 131.897, 129.561, 129.442, 128.580, 128.397, 126.480, 126.208, 126.124, 115.732, 115.526, 65.386, 54.943, 53.305, 42.738, 37.326, and 32.086 ppm.
4-(4-Fluoro-3-((4-(hydroxymethyl)piperidin-1-yl)methyl)benzyl)phthalazin-1(2H)-one (A19). HRMS (AP-ESI) m/z calcd for C22H24FN3O2 [M + H]+ 382.19326, found 382.19049 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26–8.24 (m, 1H), 7.93–7.91 (m, 1H), 7.87–7.79 (m, 2H), 7.32–7.30 (m, 1H), 7.22–7.21 (m, 1H), 7.09–7.04 (m, 1H), 4.39 (t, J = 4.0 Hz, 1H), 4.29 (s, 2H), 3.34 (s, 2H), 3.21 (d, J = 9.6 Hz, 2H), 2.71 (d, J = 12.0 Hz, 2H), 1.85 (dd, J1 = 9.6 Hz, J2 = 1.2 Hz, 2H), 1.57–1.54 (m, 2H), 1.27 (s, 1H), and 1.05–1.01 (m, 2H). 13C-NMR (400 MHz, DMSO) δ 161.106, 159.865, 158.692, 145.724, 134.365 (d, J = 3.1 Hz), 133.804, 131.937, 131.772, 131.727, 129.244 (d, J = 8.3 Hz), 128.370, 126.482, 125.315 (d, J = 14.7 Hz), 115.477, 66.394, 55.220, 53.298, 40.587, 39.335, 38.679, 37.315, and 29.129 ppm.
4-(3-((4-(Benzo[d][1,3]dioxole-5-carbonyl)piperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A20). HRMS (AP-ESI) m/z calcd for C28H25FN4O4 [M + H]+ 501.19398, found 501.19138 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.25 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1H), 7.93–7.90 (m, 1H), 7.87–7.79 (m, 2H), 7.33–7.31 (m, 1H), 7.25 (s, 1H), 7.09–7.07 (m, 1H), 6.97–6.93 (m, 2H), 6.88–6.86 (m, 1H), 6.07 (s, 2H), 4.30 (s, 2H), 3.50 (s, 2H), 3.40 (m, 4H), and 2.32 (s, 4H). 13C-NMR (400 MHz, DMSO) δ 168.816, 161.157, 159.868, 158.737, 148.656, 147.567, 145.635, 134.432, 133.844, 132.001, 131.922, 129.715, 128.363, 126.101, 124.403 (d, J = 14.8 Hz), 121.828, 115.829, 115.609, 108.559, 108.185, 101.891, 54.703, 52.653, and 37.255 ppm.
4-(3-((4-(Cyclopropanecarbonyl)piperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A21). HRMS (AP-ESI) m/z calcd for C24H25FN4O4 [M + H]+ 421.20415, found 421.20166 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.27–8.24 (m, 1H), 7.94–7.92 (m, 1H), 7.88–7.79 (m, 2H), 7.36–7.33 (m, 1H), 7.28–7.23 (m, 1H), 7.12–7.09 (m, 1H), 4.30 (s, 2H), 3.60 (s, 2H), 3.50 (s, 2H), 3.41–3.36 (m, 2H), 2.34 (s, 2H), 2.25 (s, 2H), 1.94–1.91 (m, 1H), and .71–.67 (m, 4H). 13C-NMR (400 MHz, DMSO) δ 171.328, 161.164, 159.866, 158.744, 145.659, 134.466 (d, J = 3.1 Hz), 133.851, 132.010, 131.953 (d, J = 5.7 Hz), 129.561, 128.372, 126.097, 124.398 (d, J = 15.1 Hz), 115.821, 115.599, 54.708, 53.203, 52.490, 45.229, 41.942, 40.607, 37.271, 10.600, and 7.444 ppm.
4-(3-((4-(4,4-Difluorocyclohexane-1-carbonyl)piperazin-1-yl)methyl)-4-fluorobenzyl) phthalazin-1(2H)-one (A22). HRMS (AP-ESI) m/z calcd for C27H29F3N4O2 [M + H]+ 499.23226, found 499.22986 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.27–8.25 (m, 1H), 7.93–7.91 (m, 1H), 7.88–7.80 (m, 2H), 7.34–7.32 (m, 1H), 7.28–7.24 (m, 1H), 7.12–7.07 (m, 1H), 4.30 (s, 2H), 3.50 (s, 2H), 3.45 (s, 2H), 3.38 (s, 2H), 2.75 (s, 1H), 2.33 (s, 2H), 2.24 (s, 2H), 2.03–2.02 (m, 2H), 2.00–1.81 (s, 2H), 1.71–1.67 (m, 2H), and 1.60–1.53 (m, 2H). 13C-NMR (400 MHz, DMSO) δ 173.714, 161.158, 159.867, 158.739, 145.644, 134.424, 133.849, 132.000, 131.932, 129.613 (d, J = 13.2 Hz), 129.562, 128.372, 126.102, 124.398 (d, J = 15.2 Hz), 115.814 (d, J = 22.3 Hz), 54.705, 53.350, 52.602, 45.142, 41.304, 40.600, 40.391, 40.183, 37.270, 29.581, 26.046, and 25.615 ppm.
4-(3-((4-(Cyclohexanecarbonyl)piperazin-1-yl)methyl)-4-fluorobenzyl)phthalazin-1(2H)-one (A23). HRMS (AP-ESI) m/z calcd for C27H31FN4O2 [M + H]+ 463.25110, found 463.24875 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.27–8.25 (m, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.87 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 2H), 7.83 (dd, J1 = 4.0 Hz, J2 = 1.2 Hz, 2H), 7.33–7.32 (m, 1H), 7.28–7.24 (m, 1H), 7.12–7.07 (m, 1H), 4.30 (s, 2H), 3.48 (s, 2H), 3.40 (s, 2H), 3.36 (s, 2H), 2.51 (s, 2H), 2.30 (s, 2H), 2.23 (s, 2H), 1.69 (s, 2H), 1.64 (s, 1H), 1.59 (d, J = 8.0 Hz, 2H), and 1.34 (s, 4H). 13C-NMR (400 MHz, DMSO) δ 172.500, 161.165, 159.868, 158.744, 145.650, 134.444, 133.857, 131.954, 129.726, 128.369, 126.495, 126.097, 124.384 (d, J = 15.1 Hz), 115.826, 115.604, 54.675, 53.327, 52.505, 45.178, 41.430, 37.270, 32.907, 32.676, 32.437, 26.017, and 25.919 (d, J = 9.8 Hz) ppm.
2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzohydrazide (2d). Compound 2a (.50 g, 1.60 mmol) and hydrazine hydrate (.32 g, 8.00 mmol, 80%) were mixed in methanol. The mixture was then heated up to 80°C and stirred for 24 h. After cooling to room temperature, the desired compound 2d (.42 g, 84%) was derived by crystallization as white powder. HRMS m/z: 313.10779 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.60 (s, 1H), 9.48 (s, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.91–7.80 (m, 2H), 7.51–7.44 (m, 2H), 7.20–7.17 (m, 1H), 4.50 (s, 2H), and 4.32 (s, 2H).
N′-ethyl-2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzohydrazide (B1). To a stirred solution of 2d (0.31 g, 1.00 mmol) in MeOH, acetaldehyde (0.07 g, 1.50 mmol) was added and stirred at room temperature for 1 h. Then NaBH4 (0.08 g, 2.00 mmol) was added to the solution. After 4 h of stirring, the solvent was evaporated under vacuum, and water was added. The desired compound, B1 (0.31 g, 91%), was derived by filtering. HRMS (AP-ESI) m/z calcd for C18H17FN4O2 [M + H]+ 341.14155, found 341.13947 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.60 (s, 1H), 9.73 (d, J = 4.0 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.91–7.81 (m, 2H), 7.52 (d, J = 8.0 Hz, 1H), 7.47–7.44 (m, 1H), 7.23–7.18 (m, 1H), 5.10–5.08 (m, 1H), 4.33 (s, 2H), 2.83–2.76 (s, 2H), and 1.01 (t, J = 8.0 Hz, 3H). 13C-NMR (400 MHz, DMSO) δ 163.109, 159.860, 159.510, 157.046, 145.394, 134.816, 134.783, 133.976, 132.010, 130.398, 128.355, 127, 126.521, 123.487, 116.483, 45.727, 36.889, and 13.451 ppm.
2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)-N′-propylbenzohydrazide (B2). HRMS (AP-ESI) m/z calcd for C19H19FN4O2 [M + H]+ 355.15720, found 355.15543 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.60 (s, 1H), 9.73 (d, J = 4.0 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.90 (t, J = 8.0 Hz, 1H), 7.84–7.81 (m, 2H), 7.51 (d, J = 8.0 Hz, 1H), 7.45 (s, 1H), 7.22–7.17 (m, 1H), 5.11 (s, 1H), 4.32 (s, 2H), 2.73 (q, J1 = 8.0 Hz, 2H), 1.48–1.39 (m, 2H), and .88 (t, J = 8.0 Hz, 3H). 13C-NMR (400 MHz, DMSO) δ 163.073, 159.864, 159.515, 157.051, 145.389, 134.808 (d, J = 3.4 Hz), 134.775, 133.969, 132.927(d, J = 8.3 Hz), 130.386 (d, J = 2.9 Hz), 128.354, 126.519, 123.487 (d, J = 15.7 Hz), 116, 53.287, 36.893, 21.174, and 12.048 ppm.
N′-butyl-2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzohydrazide (B3). HRMS (AP-ESI) m/z calcd for C20H21FN4O2 [M + H]+ 369.17285, found 369.17093 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.60 (s, 1H), 9.73 (s, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.91–7.87 (m, 1H), 7.84–7.81 (m, 1H), 7.51–7.44 (m, 2H), 7.22–7.17 (m, 1H), 5.08 (s, 1H), 4.33 (s, 2H), 2.76–2.73 (m, 2H), 1.44–1.28 (m, 4H), and .88 (t, J = 8.0 Hz, 3H). 13C-NMR (400 MHz, DMSO) δ 163.074, 159.870, 159.523, 157.057, 145.384, 134.808 (d, J = 3.3 Hz), 134.775, 133.960, 132.854 (d, J = 8.3 Hz), 131.995, 130.378 (d, J = 2.8 Hz), 128.351, 126.518, 123.470 (d, J = 15.7 Hz), 116.704, 51.088, 36.901, 30.084, 20.203, and 14.353 ppm.
2-Fluoro-N′-isobutyl-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzohydrazide (B4). HRMS (AP-ESI) m/z calcd for C20H21FN4O2 [M + H]+ 369.17285, found 369.17078 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.60 (s, 1H), 9.74 (s, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.89 (t, J = 8.0 Hz, 1H), 7.83 (t, J = 8.0 Hz, 1H), 7.51–7.45 (m, 2H), 7.22–7.17 (m, 1H), 5.10 (s, 1H), 4.33 (s, 2H), 2.60–2.57 (m, 2H), 1.77–1.67 (m, 1H), and .90 (d, J = 8.0 Hz, 6H). 13C-NMR (400 MHz, DMSO) δ 163.067, 159.859, 159.509, 157.046, 145.392, 134.803, 134.770 (d, J = 3.4 Hz), 133.982, 132.846 (d, J = 8.3 Hz), 130.356 (d, J = 2.8 Hz), 128.352, 126.521, 126.009, 123.487 (d, J = 15.7 Hz), 116.707 (d, J = 22.5 Hz), 59.376, 36.885, 26.871, and 21.043 ppm.
2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)-N′-pentylbenzohydrazide (B5). HRMS (AP-ESI) m/z calcd for C21H23FN4O2 [M + H]+ 383.18850, found 383.18677 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 9.72 (d, J = 8.0 Hz, 1H), 8.27–8.25 (m, 1H), 7.98–7.96 (m, 1H), 7.91–7.81 (m, 2H), 7.52–7.43 (m, 2H), 7.20 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 1H), 5.10–5.01 (m, 1H), 4.33 (s, 2H), 2.75 (dd, J1 = 6.8 Hz, J2 = 6.4 Hz, 2H), 1.44–1.41 (m, 2H), 1.30–1.29 (m, 4H), and .88 (d, J = 8.0 Hz, 3H). 13C-NMR (400 MHz, DMSO) δ 163.052, 159.851, 159.496, 157.032, 145.391, 134.814, 134.781, 133.987, 132.027, 130.380, 128.354, 126.523, 126.017, 123.337, 116.489, 51.361, 36.886, 29.234, 27.575, 22.497, and 14.382 ppm.
2-Fluoro-N′-hexyl-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzohydrazide (B6). HRMS (AP-ESI) m/z calcd for C22H25FN4O2 [M + H]+ 397.20415, found 397.20239 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 9.72 (d, J = 8.0 Hz, 1H), 8.27–8.25 (m, 1H), 7.98–7.96 (m, 1H), 7.91–7.81 (m, 2H), 7.51–7.43 (m, 2H), 7.20 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 1H), 5.10–5.05 (m, 1H), 4.33 (s, 2H), 2.77–2.72 (m, 2H), 1.44–1.34 (m, 2H), 1.32–1.22 (m, 6H), and .86 (d, J = 8.0 Hz, 3H). 13C-NMR (400 MHz, DMSO) δ 163.036, 159.849, 159.506, 157.041, 145.381, 134.807, 134.775 (d, J = 3.2 Hz), 133.975, 132.846 (d, J = 8.2 Hz), 130.364, 128.357, 126.521, 126.005, 123.326 (d, J = 15.6 Hz), 116.482, 51.413, 36.893, 31.672, 27.883, 26.714, 22.529, and 14.392 ppm.
Tert-butyl 4-(2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazine-1-carboxylate (2e). To the solution of compound 2 (.50 g, 1.68 mmol) in DCM, Et3N (.51 g, 5.04 mmol) and TBTU (.65 g, 2.02 mmol) were added at 0°C. The solution was kept at 0°C for 20 min, and then 1-Boc-piperazine (.38 g, 2.02 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvent was then evaporated, and the compound was dissolved in EtOAc (30 ml). The EtOAc solution was washed with 1 M citric acid (3 × 30 ml), NaHCO3 (3 × 30 ml), and brine (3 × 30 ml). The organic phase layer was dried over Mg2SO4 and concentrated in vacuo. The desired compound 2e (.64 g, 78%) was derived by crystallization in EtOAc as white powder. HRMS m/z: 489.18884 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.60 (s, 1H), 8.27 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.91–7.88 (m, 1H), 7.84 (t, J = 8.0 Hz, 1H), 7.46–7.43 (m, 1H), 7.36 (d, J = 4.0 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 4.33 (s, 2H), 3.59 (s, 2H), 3.38 (s, 2H), 3.23 (s, 2H), 3.15 (s, 2H), and 1.41 (s, 9H).
4-(4-Fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (2f). The solution of 2e in DCM with TFA was stirred for 2 h. Then the solution was concentrated in vacuo to get 2f, which was used for the next step reaction without purification.
4-(3-(4-(4-(Bis(2-chloroethyl)amino)benzoyl)piperazine-1-carbonyl)-4-fluorobenzyl) phthalazin-1(2H)-one (C1). To the solution of 4-[bis(2-chlorethyl)amino]benzoic acid (.27 g, 1.02 mmol) in DCM, Et3N (.31 g, 3.06 mmol) and TBTU (.39 g, 1.22 mmol) were added at 0°C. The solution was kept at 0°C for 20 min, and then 2f (.47 g, 1.22 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvent was then evaporated, with the residue taken up in EtOAc (30 ml) and then washed with 1 M citric acid solution (3 × 30 ml), saturated NaHCO3 solution (3 × 30 ml), and saturated brine solution (3 × 30 ml). The organic phase layer was dried over Mg2SO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography (PE:EA, 1: 4 v/v) to obtain the pure product C1 (0.39 g, 63%) as white powder. HRMS (AP-ESI) m/z calcd for C31H30Cl2FN5O3 [M + H]+ 610.17897, found 610.17621 [M + H]+. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.88 (t, J = 8.0 Hz, 1H), 7.81 (t, J = 8.0 Hz, 1H), 7.44 (s, 1H), 7.36 (d, J = 4.0 Hz, 1H), 7.31 (d, J = 8.0 Hz, 2H), 7.24 (t, J = 8.0 Hz, 1H), 6.78 (d, J = 8.0 Hz, 2H), 4.33 (s, 2H), 3.76 (s, 8H), 3.65 (s, 2H), 3.57 (s, 2H), 3.42 (s, 2H), and 3.22 (s, 2H). 13C-NMR (400 MHz, DMSO) δ 170.047, 164.508, 159.855, 148.180, 145.315, 135.295, 133.952, 132.016, 130.018, 129.550, 128.366, 126.544, 125.946, 123.928, 123.180, 111.422, 52.312, 41.420, 40.609, 40.400, 40.192, 39.983, 39.775, 39.358, and 36.901 ppm.
4-(3-(4-(4-(4-(Bis(2-chloroethyl)amino)phenyl)butanoyl)piperazine-1-carbonyl)-4-fluorobenzyl)phthalazin-1(2H)-one (C2). HRMS (AP-ESI) m/z calcd for C34H36Cl2FN5O3 [M-H]- 650.20992, found 650.21179 [M-H]-. 1H-NMR (400 MHz, DMSO) δ 12.59 (s, 1H), 8.26 (dd, J = 8.0, 1.2 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.90 (t, J = 7.8, 1.5 Hz, Hz, 1H), 7.87–7.80 (m, 1H), 7.44 (s, 1H), 7.36 (t, J = 3.0 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 7.03 (d, J = 4.0 Hz, 2H), 6.67 (q, J = 4.0 Hz, 2H), 4.33 (s, 2H), 3.69 (d, J = 4.0 Hz, 8H), 3.62–3.51 (m, 4H), 3.37 (s, 2H), 3.15 (s, 2H), 2.47 (d, J = 4.0 Hz, 2H), 2.34 (t, J = 8.0 Hz, 1H), 2.27 (t, J = 6.9 Hz, 1H), and 1.73 (d, J = 8.0 Hz, 2H). 13C-NMR (400 MHz, DMSO) δ 171.126, 164.441, 159.859, 155.612, 145.302, 144.866, 135.278, 133.954, 132.180 (d, J = 15.8 Hz), 130.415, 129.769, 129.553, 129.395 (d, J = 15.9 Hz), 128.374, 126.549, 124.133, 116.508, 116.295, 112.351, 52.673, 47.099, 45.361, 44.946, 42.092, 41.609, 40.611, 40.194, 36.908, 34.042, 32.154, and 27.240 ppm.
The proliferation of cancer cells was tested by CCK-8 assay. Briefly, cells were seeded in 96-well plates at approximately 5 × 103 cells per well. The cells were treated with test compounds after 24 h of incubation. CCK-8 reagent (10 ml) was added to each well after 72 h of incubation, and the cells were then incubated at 37°C for 4 h. The light absorbance at 450 nm was measured using an Opsys microplate reader (Dynex Technologies, Chantilly, VA, United States). Results are illustrated as percentage viability normalized to DMSO-treated control cells.
K562 and HCC1937 cells were incubated with different doses of molecule C2, olaparib, or chlorambucil for 24 h. After treatment, cells were collected and fixed with 70% pre-chilled ethanol in PBS and stored at −20°C overnight. The cells were then washed twice with PBS, incubated with 100 μg/ml RNase I (Solarbio, China) at 37°C for 1 h, and stained with propidium iodide (PI, 10 μg/ml, Solarbio, China) for 30 min, in the dark, at room temperature. Finally, the DNA content was measured using flow cytometry (FACSAria III, Becton Dickinson, United States). The data were analyzed and fitted using ModFit software.
K562 cells were treated with various concentrations of molecule C2, olaparib, or chlorambucil for 24 h. Cells were then harvested and washed twice with PBS and then resuspended with binding buffer (Becton Dickinson, United States). Cells were incubated with Annexin V-BV421 (Becton Dickinson, United States) and 7-AAD (Becton Dickinson, United States) for double labeling for 30 min, in the dark, at room temperature, followed by analysis using flow cytometry (FACSAria III, Becton Dickinson, United States). The data were analyzed using FlowJo v10 software.
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding authors.
LeZ designed the project. LiZ performed enzymatic screening; HQ synthesized the molecules; JF performed the in vitro antitumor experiments. JZ and LeZ analyzed the data and wrote the manuscript.
This work was supported by the Science and Technology support plan for Youth Innovation in Universities of Shandong Province (Grant No. 2019KJM001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035), 913–917. doi:10.1038/nature03443
Chaudhuri, A. R., and Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18 (10), 610–621. doi:10.1038/nrm.2017.53
Chen, Y. M., Jia, Y. P., Song, W. G., and Zhang, L. (2018). Therapeutic potential of nitrogen mustard based hybrid molecules. Front. Pharmacol. 9, 1453. ARTN 1453. doi:10.3389/fphar.2018.01453
Coyne, G. O., Chen, A., and Kummar, S. (2015). Delivering on the promise: Poly ADP ribose polymerase inhibition as targeted anticancer therapy. Curr. Opin. Oncol. 27 (6), 475–481. doi:10.1097/Cco.0000000000000238
Dockery, L. E., Gunderson, C. C., and Moore, K. N. (2017). Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. Onco Targets Ther. 10, 3029–3037. doi:10.2147/OTT.S114714
Gala, U. H., Miller, D. A., and Williams, R. O. (2020). Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochimica Biophysica Acta-Reviews Cancer 1873 (1), 188319. ARTN 188319. doi:10.1016/j.bbcan.2019.188319
Gelmon, K. A., Tischkowitz, M., Mackay, H., Swenerton, K., Robidoux, A., Tonkin, K., et al. (2011). Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 12 (9), 852–861. doi:10.1016/S1470-2045(11)70214-5
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Hu, Y. D., Petit, S. A., Ficarro, S. B., Toomire, K. J., Xie, A. Y., Lim, E., et al. (2014). PARP1-Driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 4 (12), 1430–1447. doi:10.1158/2159-8290.Cd-13-0891
Jackson, S. E., and Chester, J. D. (2015). Personalised cancer medicine. Int. J. Cancer 137 (2), 262–266. doi:10.1002/ijc.28940
Jones, P., Wilcoxen, K., Rowley, M., and Toniatti, C. (2015). Niraparib: A poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J. Med. Chem. 58 (8), 3302–3314. doi:10.1021/jm5018237
Krishnakumar, R., and Kraus, W. L. (2010). The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39 (1), 8–24. doi:10.1016/j.molcel.2010.06.017
Menear, K. A., Adcock, C., Boulter, R., Cockcroft, X. L., Copsey, L., Cranston, A., et al. (2008). 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 51 (20), 6581–6591. doi:10.1021/jm8001263
Pines, A., Vrouwe, M. G., Marteijn, J. A., Typas, D., Luijsterburg, M. S., Cansoy, M., et al. (2012). PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 199 (2), 235–249. doi:10.1083/jcb.201112132
Poveda, A. M., Davidson, R., Blakeley, C., and Milner, A. (2019). Olaparib maintenance monotherapy in platinum-sensitive, relapsed ovarian cancer without germline BRCA mutations: OPINION phase IIIb study design. Future Oncol. 15 (32), 3651–3663. doi:10.2217/fon-2019-0343
Rose, M., Burgess, J. T., O'Byrne, K., Richard, D. J., and Bolderson, E. (2020). PARP inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front. Cell Dev. Biol. 8, 564601. doi:10.3389/fcell.2020.564601
Shen, Y. Q., Rehman, F. L., Feng, Y., Boshuizen, J., Bajrami, I., Elliott, R., et al. (2013). BMN673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 19 (18), 5003–5015. doi:10.1158/1078-0432.Ccr-13-1391
Thomas, H. D., Calabrese, C. R., Batey, M. A., Canan, S., Hostomsky, Z., Kyle, S., et al. (2007). Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther. 6 (3), 945–956. doi:10.1158/1535-7163.MCT-06-0552
Wang, Y. Q., Wang, P. Y., Wang, Y. T., Yang, G. F., Zhang, A., and Miao, Z. H. (2016). An update on poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors: Opportunities and challenges in cancer therapy. J. Med. Chem. 59 (21), 9575–9598. doi:10.1021/acs.jmedchem.6b00055
Keywords: PARP, olaparib, chlorambucil, cancer, structural modification
Citation: Qin H, Zhang J, Zhao Y, Zhang L, Feng J and Zhang L (2023) Discovery of a potent olaparib–chlorambucil hybrid inhibitor of PARP1 for the treatment of cancer. Front. Pharmacol. 13:1054616. doi: 10.3389/fphar.2022.1054616
Received: 27 September 2022; Accepted: 12 December 2022;
Published: 09 January 2023.
Edited by:
Olivier Feron, Université catholique de Louvain, BelgiumReviewed by:
Alexander Tikhomirov, Russian Academy of Medical Sciences, RussiaCopyright © 2023 Qin, Zhang, Zhao, Zhang, Feng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhong Feng, ZmVuZ2ppbmhvbmc1MjBAMTYzLmNvbQ==; Lei Zhang, bGVpemhhbmdjaGVtaWNhbEBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.