 Xi Li
Xi Li Xiangyi Ren2†
Xiangyi Ren2† Minfeng Huo
Minfeng Huo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 26 October 2022
Sec. ELSI in Science and Genetics
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1025740
Fabry disease is a monogenic disease characterized by a deficiency or loss of the α-galactosidase A (GLA). The resulting impairment in lysosomal GLA enzymatic activity leads to the pathogenic accumulation of enzymatic substrate and, consequently, the progressive appearance of clinical symptoms in target organs, including the heart, kidney, and brain. However, the mechanisms involved in Fabry disease-mediated organ damage are largely ambiguous and poorly understood, which hinders the development of therapeutic strategies for the treatment of this disorder. Although currently available clinical approaches have shown some efficiency in the treatment of Fabry disease, they all exhibit limitations that need to be overcome. In this review, we first introduce current mechanistic knowledge of Fabry disease and discuss potential therapeutic strategies for its treatment. We then systemically summarize and discuss advances in research on therapeutic approaches, including enzyme replacement therapy (ERT), gene therapy, and chaperone therapy, as well as strategies targeting subcellular compartments, such as lysosomes, the endoplasmic reticulum, and the nucleus. Finally, the future development of potential therapeutic strategies is discussed based on the results of mechanistic studies and the limitations associated with these therapeutic approaches.
Lysosomes are organelles that perform a vital function in the catabolism and recycling of cytosolic compounds (Luzio et al., 2007; Boustany, 2013). Deficiencies in proteins with physiological functionalities can lead to lysosomal storage disorders (LSDs) (Schultz et al., 2011; Platt et al., 2018), with over 70 lysosomal enzyme deficiencies causative of LSDs having been identified to date (Martina et al., 2020). Fabry disease is associated with the reduced activity of lysosomal galactosidase A (GLA), an enzyme involved in the catabolism of globotriaosylceramide (Gb3). GLA deficiency in patients with Fabry disease results in abnormal glycosphingolipid metabolism and the progressive accumulation of Gb3 (Aerts et al., 2008; Kang et al., 2019). With disease progression, patients display multiple clinical phenotypes, such as cardiovascular disease, neuropathic pain, and even increased risk of early death (Frustaci et al., 2014; Aguiar et al., 2018; Uceyler et al., 2019; Weissmann et al., 2021). However, the cellular pathogenic cascade underlying how GLA deficiency results in progressive clinical symptoms remains unclear; moreover, the associated pathogenic cascade has already been initiated before the onset of the organ symptoms of Fabry disease, making de novo diagnosis difficult (Jabbarzadeh-Tabrizi et al., 2020; Spinelli et al., 2020). Thus, to achieve adequate therapeutic outcomes and develop novel therapeutic strategies for the treatment of this disorder, a deeper understanding of the associated pathogenic mechanisms is needed.
Several approaches for the treatment of Fabry disease have been exploited, both preclinically and clinically, including enzyme replacement therapy (ERT), gene therapy, and chaperone therapy (Solomon and Muro, 2017; Castelli et al., 2021; Fernández-Pereira et al., 2021). Despite the success of these therapeutic strategies, each approach has its limitations. For instance, the clinical benefit of ERT is limited by the short half-life of the recombinant enzyme, the production of anti-drug antibodies, and the inability of the enzyme to cross the blood-brain barrier (Lenders and Brand, 2018; Abasolo et al., 2021). The recent rapid advances in nanotechnology and nanoscience have provided novel opportunities for overcoming the limitations of current therapeutics (Ghosh et al., 2010; Gregory et al., 2020). The use of biomaterials and nanotechnology enables the development of therapeutic drugs with reasonable pharmacokinetic characteristics, including sufficient blood retention, good bioavailability, and effective immune-evading abilities (Del Grosso et al., 2019; Li et al., 2021). For instance, preclinical and clinical studies have demonstrated that a polyethylene glycol-modified strategy can improve the therapeutic outcome of ERT (Veronese and Pasut, 2005; Schiffmann et al., 2019). Importantly, the development of targeted approaches in nanoscience allows the spatiotemporal control of drug delivery, potentially resulting in high therapeutic efficacy concomitant with a decreased risk of adverse effects.
In this review, we first analyze and discuss recent mechanistic research into Fabry disease, focusing on lysosomal function, autophagy, and lipid metabolism, as well as their connectivity. Subsequently, we systemically summarize the progress in the development of therapeutic modalities for treating Fabry disease, including ERT, gene therapy, and chaperone therapy (Figure 1), as well as the strategies used for the delivery of therapeutic drugs to target sites via the active or passive targeting of nanotherapeutics. In particular, the advances and remaining challenges associated with each therapeutic approach are also addressed. Finally, we describe the promising prospective therapeutic strategies for the treatment of Fabry disease. Overall, we hope that this review will contribute to increasing our understanding of the mechanisms underlying the development of Fabry disease and provide insights regarding current and future therapeutic strategies for the treatment of this disorder.
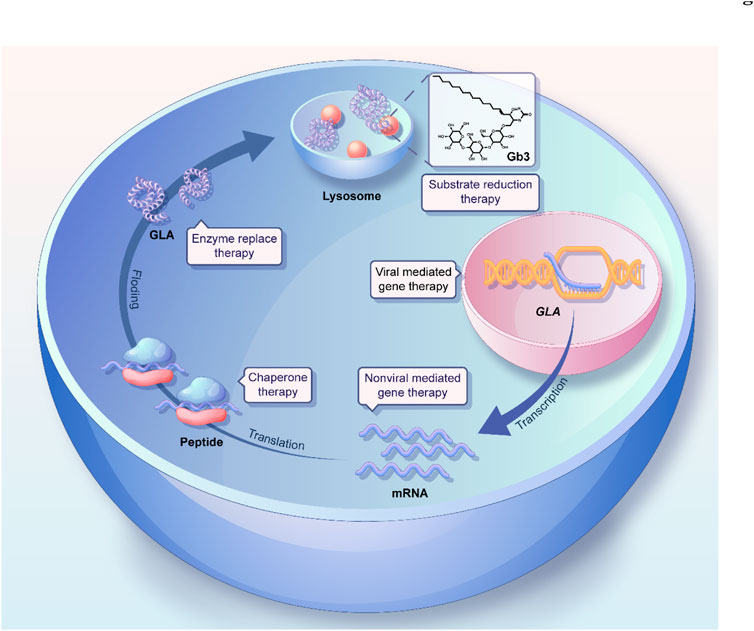
FIGURE 1. Schematic illustration of the GLA synthetic process and the various therapeutic strategies for Fabry disease.
Lysosomes are tightly associated with the degradation and recycling of cellular components, including lipids, proteins, and nucleic acids. The inability to digest Gb3 in lysosomes may lead to the activation of several signaling pathways aimed at reducing the levels of accumulated Gb3, eventually resulting in multiple cellular pathologies (Biancini et al., 2012; Hsu et al., 2019). Autophagy plays a critical role in degrading lysosomal substrates (Festa et al., 2018). Several studies have demonstrated that autophagy is upregulated in response to Gb3 accumulation. Gb3 accumulation can activate autophagy pathway to degrade Gb3, alleviating intracellular stresses simultaneously. Although activating autophagy has beneficial effects on lysosomal stress responses in the short term, long-term activation of this pathway can also result in dysregulated autophagy, which accelerates disease progression and worsens pathogenetic changes. Impaired autophagic activity has been observed in peripheral blood mononuclear cells from patients with Fabry disease as well as in several in vitro models of Fabry disease (Chévrier et al., 2010; Liebau et al., 2013; Ivanova et al., 2019). For instance, Song et al. constructed a model of Fabry disease in human embryonic stem cells (hESCs) via the CRISPR/Cas9-mediated knockout of the GLA gene (Song et al., 2019). The authors found that, compared with their wild-type counterparts, cardiomyocytes differentiated from these hESCs exhibited significantly increased cell size and Gb3 deposition, as well as impaired autophagic flux. These findings indicated that dysregulated autophagy resulting from GLA gene deficiency and consequent Gb3 accumulation plays a role in myocardial hypertrophy. In turn, impaired autophagy can further trigger secondary lysosomal deposition via the accumulation of damaged mitochondria and reduced protein degradation, which may further result in a pathogenic cascade and cellular damage. Xing et al. (Xing et al., 2022) demonstrated that impaired autophagic flux can induce myocardial ischemic/reperfusion injury through the accumulation of damaged mitochondria. The restoration of myocardial autophagy improved cardiomyocyte survival in vitro and heart function in mice in vivo. Combined, these observations suggest that targeting autophagy may represent a potential therapeutic strategy for treating Fabry disease. However, relatively few preclinical or clinical trials have explored the effect of targeting autophagy for the in vivo treatment of this disorder. Moreover, the mechanistic basis of the role of autophagy in Fabry disease has not been fully elucidated, and merits further in vivo investigation.
The mechanism underlying the Gb3 accumulation-induced intracellular pathogenic cascades remains poorly understood and further studies exploring the link between autophagic disorder and upstream lysosomal functions are necessary. Vacuolar (H+)-adenosine triphosphatases (V-ATPases), which use ATP hydrolysis to pump H+ into the lysosome, play a central role in maintaining the acidic environment of this organelle (pH = 5.2–6.1) and normal lysosomal function. Lysosomal substrate accumulation in cells leads to increased expression of V-ATPase. AMP-activated protein kinase (AMPK) is also activated in response to depleted ATP levels, and AMPK phosphorylation was observed to be increased in Fabry cardiomyocytes in an in vitro model of the disease (Chou et al., 2017). Chou et al. constructed in vitro Fabry disease models based on peripheral blood mononuclear cells derived from patients with the disorder (Chou et al., 2017). They found that, compared with controls, Fabry model cells exhibited increased AMPK phosphorylation and elevated glucose transporter 4 (GLUT 4) protein levels, implying that glucose uptake was increased. The authors further investigated energy utilization in Fabry model cells by seahorse metabolic flux assay. Compared with control cells, total energy metabolism, including glycolysis and fatty acid oxidation, was significantly decreased in model cells. Meanwhile, energy metabolism was shifted toward glycolysis from fatty acid β-oxidation. These findings illustrate that GLA gene deficiency can induce AMPK activation and enhance glycolysis; however, further in vivo investigation is required and aberrant fatty acid metabolism in this disorder needs much deeper exploration. Subsequently, activation of AMPK and V-ATPase can lead to the inhibition of mammalian target of rapamycin complex 1 (mTORC1), which is involved in a variety of signal transduction pathways, including the induction of autophagic activation and lysosomal biogenesis. Interestingly, mTOR activity has been reported to be decreased in Fabry podocytes (Liebau et al., 2013). Combined, the results of these studies suggest that autophagic dysfunction likely results from the triggering of the activation of the AMPK-mTOR signaling pathway due to intracellular Gb3 accumulation. These observations further suggest that the targeting of processes upstream of lysosomes to modulate autophagy may represent an attractive therapeutic strategy for Fabry disease.
An increase in the number of lipid droplets is often seen in Fabry tissues and in vitro models of Fabry disease. Recently, Kim et al. generated GLA-mutant kidney organoids via CRISPR-Cas9 technology. Transmission electron microscopy and oil red O staining analysis showed greater lipid accumulation in Fabry disease model organoids compared with that in the controls (Kim et al., 2021), suggesting that lipid metabolism may have been inhibited in the model organoids. However, the mechanism involved in lipid droplet formation in Fabry disease remains elusive, even in preclinical studies. Disordered fatty acid utilization may underlie the observed lipid droplet formation. First, lipid accumulation may indicate that fatty acid oxidation could be blocked. As discussed above, energy demand is mismatched with the decreasing fatty acid β-oxidation. Defects in carnitine palmitoyl transferase 1 (CPT1), a key enzyme in the transportation of fatty acids into mitochondria for β-oxidation, may lead to lipid formation and dysregulated fatty acid utilization. It has been suggested that increased malonyl-CoA synthesis by acetyl CoA carboxylase can block β-oxidation. Abnormal levels of substrate for fatty acid metabolism are also observed in other Fabry cells (Schumann et al., 2021). LC–MS/MS analysis of acylcarnitines isolated from Fabry disease model cells indicated that the levels of short-chain acylcarnitines and free carnitine were significantly elevated in Fabry cells, whereas those of medium- and long-chain acylcarnitines were decreased. The results of recent studies have confirmed that acylcarnitine accumulation leads to mitochondrial dysfunction and impaired fatty acid oxidation (McCoin et al., 2015; Nguyen et al., 2017). Taken together, these reports suggest that fatty acid metabolism is altered in Fabry disease. However, whether CPT1 has a role in Fabry disease is unknown. Additionally, the link between lipid metabolism and Fabry disease phenotypes requires further elucidation in vivo. Secondly, lipid metabolism is strongly associated with autophagy. Under metabolic stress, lipid droplets can be exploited for energy production through autophagy (Rambold et al., 2015; Benador et al., 2019). Blocking autophagy impedes the flux of stored fatty acids from lipid droplets to mitochondria, thus reducing mitochondrial β-oxidation and energy supplementation. Another potential explanation for lipid droplets may be related to mitochondrial biogenesis. Peroxisome proliferator-activated receptor (PPAR) and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α) are the main regulators of mitochondrial biogenesis via gene transcription. When cellular autophagy is inhibited, the levels of the PPARα repressors histone deacetylase 3 (HDAC3) and nuclear receptor co-repressor 1 (NCoR1) continuously increase as they cannot be degraded in the lysosome (Iershov et al., 2019). Evidence from a preclinical study has indicated that the protein expression of PGC1α is upregulated in Fabry cells (Schumann et al., 2021).
Triacylglycerol (TAG), the major component of lipid droplets, can be reused by mitochondria or removed from cells by high-density lipoprotein (HDL). Interestingly, cholesterol metabolism has been reported to fluctuate in various Fabry disease models as well as in patients with Fabry disease (Shirai et al., 2000; Elliott et al., 2006; Kalliokoski et al., 2006). Some studies have also demonstrated that cholesterol levels are 2∼3-fold higher in Fabry cells than in wild-type cells (Rappaport et al., 2016). Patients with Fabry disease may exhibit compensatory mechanisms for cholesterol dyshomeostasis, such as promoting cholesterol efflux and limiting cholesterol synthesis and uptake. It has been demonstrated that Fabry disease is associated with cholesterol transport, which may contribute to alleviating intracellular stress (Glaros et al., 2005). Gb3 is a constituent of lipoproteins. Once it has been deposited in the lysosome, it must be transported out of the lysosome via intracellular lipid-exporting pathways. Several studies have sought to determine whether there is a potential link between Gb3 and cholesterol homeostasis in vitro, with the results showing that the levels of low-density lipoprotein (LDL) are upregulated in glycosphingolipid-treated fibroblasts (Puri et al., 2003; Schueler et al., 2016; van der Veen et al., 2020). Additionally, increasing the levels of LDL can enhance lipid accumulation and facilitate cholesterol influx via cholesterol transport from the circulation (Cedó et al., 2020). Moreover, LDL receptor (LDLR) levels were reported to be increased in cardiomyocytes differentiated from induced pluripotent stem cells of patients with Fabry disease, which contributes to Gb3 storage in vascular cells (Birket et al., 2019). In a retrospective study, total cholesterol and HDL levels were measured before ERT and 10 years after ERT (Stepien and Hendriksz, 2017). Patients who underwent ERT displayed higher HDL levels than those who did not, and ERT was found to (Scharnetzki et al., 2020) exert a cardioprotective effect. These studies indicate that targeting cholesterol metabolism may be a feasible therapeutic option for Fabry disease. In one in vitro Fabry disease model, the authors showed that a peptide mimicking the function of apolipoprotein A1 could mediate cellular Gb3 and cholesterol efflux (Schueler et al., 2016). However, a cause-effect link between Fabry disease and cholesterol transport remains elusive, and further studies are needed to allow the development of a therapeutic strategy targeting cholesterol metabolism in Fabry disease.
Inflammation is a series of adaptive biological responses triggered by several internal or external stimuluses. Controlling inflammatory responses can help restoring host homeostasis and adaptations, while dysregulated inflammatory responses could induce tissue injury and even tissue fibrosis (Medzhitov, 2008; Rozenfeld and Feriozzi, 2017). Progression of Fabry cardiomyopathy is associated with increased inflammatory responses, as demonstrated by elevated inflammatory biomarkers (Yogasundaram et al., 2018). Recently, Song and co-workers reported that pro-inflammation pathway are activated within the cells with Fabry pathology. While the inflammatory responses can be reversed using gene therapy, further confirming the correlation between inflammatory activation and Fabry pathology (Song et al., 2021). Several molecular pathways have been reported to be attributed to the inflammatory activation of Fabry disease, including the nuclear factor kappa B (NF-κB) pathway, oxidative stress, and transforming growth factor-β (TGF-β) pathway. It has also been found that inflammatory activation is closely related to autophagy (defective autophagic protein could enhance inflammasome activation) as well as sphingolipids homeostasis (Levine et al., 2011). Aflaki and co-workers demonstrated that inflammatory response in Gaucher disease is activated through disrupted autophagy (Aflaki et al., 2016). Another study shows that autophagy impairment in Fabry mice exacerbates the pathologies features of renal interstitial fibrosis. Sphingolipids elevate the inflammation responses in many diseases, such as obesity, myocardial infarction, and Parkinson’s disease (Chaurasia et al., 2016; Belarbi et al., 2020; Hadas et al., 2020). For example. Elevated sphingolipids levels (ceramide) were significantly upregulated after myocardial infarction, which triggered severe inflammatory responses and apoptosis of cardiomyocytes (Hadas et al., 2020). Nevertheless, detailed mechanism and consequences about inflammation responses in Fabry disease have not been fully understood. Therapeutic strategy targeting inflammatory to treat Fabry disease is highly demanding and appealing.
ERT involves the exogenous supplementation of the GLA enzyme and has been successfully administrated in the treatment of Fabry disease (Pisani et al., 2017). Fabrazyme (α-GLA, i. v. 1 mg·kg−1 every 2 weeks) and replagal (β- GLA, i. v. 0.2 mg·kg−1 every 2 weeks) have both been approved by the United States Food and Drug Administration for the treatment of Fabry disease, and both have shown the potential to significantly reduce Gb3 deposition and improve the prognosis of patients. However, the clinical application of this approach is partly limited by poor biodistribution, a short half-life, and immunogenic responses after intravenous administration (Pisani et al., 2017). For instance, the liver and spleen play a critical role in the rapid clearance of exogenous Gb3, but the biodistribution of heart, kidney, and brain is insufficient to achieve treatment (Desnick and Schuchman, 2012). Additionally, enzymes are easily degraded in the body, while the cost of using ERT is extremely high (∼$200,000 per year). The effect of ERT is also influenced by the treatment starting age. Initiating ERT at an early age (<25 years) can effectively reduce the deposition of Gb3 and attenuate the progressive clinical manifestations of Farey disease (Arends et al., 2017). ERTs did not show a therapeutic benefit in end-stage organ disease or following the production of antibodies in patients (Biegstraaten et al., 2015; van der Veen et al., 2019). This may be explained by the inability of enzymatic therapy to reverse the clinical manifestations. An in vitro study demonstrated that treating Fabry podocytes with α-GLA could effectively clear high levels of Gb3 but not restore altered cellular signaling (Braun et al., 2019). Accordingly, addressing the above challenges is crucial for maximizing the therapeutic benefits while concomitantly reducing the harmful effects.
Substantial research effort has focused on the development of drug delivery and targeting systems that allow for improved biodistribution and reduced drug clearance (Meka et al., 2016; Gupta et al., 2017). Passive targeting strategies have been proposed focusing on maximizing the effects of ERT (Scaletti et al., 2018). For example, chemical modifications to poly-(ethylene glycol) (PEG) can increase the circulation half-life and reduce the immunogenicity of enzymes in the body (Kizhner et al., 2015). Pegunigalsidase alfa, a PEGylated form of recombinant GLA with a long half-life, has been developed for Fabry disease therapy. In vivo pharmacokinetic results demonstrated that the half-life of pegunigalsidase alfa was significantly increased (approximately 10-fold; t1/2 = 581 min) compared with that for the commercially available enzyme. Importantly, Fabry male mice treated with PEGylated GLA display reduced anti-drug antibody production. Recently, extracellular vehicles (EVs) derived from GLA-overexpressing cells have been developed as a therapeutic vehicle for the treatment of GLA defects (Seras-Franzoso et al., 2021). The EVs not only effectively restored lysosomal function to a greater extent than recombinant GLA, but were also well-tolerated and showed wide organ distribution after in vivo administration (Figure 2).
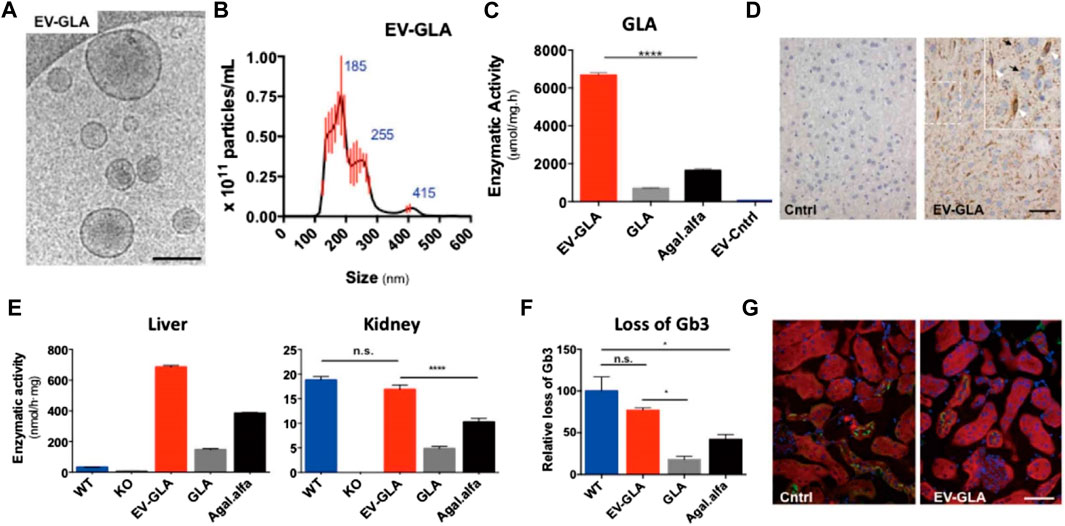
FIGURE 2. CryoTEM imaging (A) and size distribution (B) of EV-GLA. Scale bar: 200 nm. (C) α-GLA activity test for EV-GLA. (D) Immunohistochemistry for the detection of GLA enzyme in liver tissues of GLA knockout (KO) mice after the administration of EV-GLA. Scale bar: 50 μm. (E) Measurement of α-GLA activity in the liver and kidney of various groups. (F) Measurement of Gb3 loss in various groups. (G) Gb3 immunofluorescence (green signal) in the kidneys of KO mice after the administration of a single dose of EV-GLA. Scale bar: 40 μm. Reproduced with permission (Seras-Franzoso et al., 2021). Copyright 2021, The Authors.
Although nanoparticle passive transport has clear advantages for drug delivery and targeting when compared with free drugs, it is still insufficient. Active targeting has been efficiently exploited to increase the bioavailability of nanoparticles and improve their therapeutic effects. In Fabry disease, active targeting strategies are therapeutically attractive options for achieving high drug bioavailability with reduced loss of enzyme activity (Cabrera et al., 2016; Giannotti et al., 2016; Tomsen-Melero et al., 2021). Arginine-glycine-aspartic acid (RGD) peptides are cyclic peptides that display a high affinity for vascular endothelial cells. Recently, RGD-coated hybrid liposomes conjugated with miristalkonium chloride (MKC) were developed for the loading of GLA for Fabry disease-targeting therapy (Tomsen-Melero et al., 2021). In this design, RGD acts as a targeting moiety by recognizing αvβ3 on endothelial cells. Simultaneously, low concentrations of MKC, a cationic surfactant, enhanced loading efficiency and colloidal stability. Although these nanocarriers improved the bioavailability of therapeutic molecules, the crossing of biological membranes still faces challenges.
Once a therapeutic agent has been delivered to a cell, there are barriers that need to be overcome, including biological barriers of steric hindrance intracellularly to exert its therapeutic effects. Many promising methods have already been developed to address these challenges. Lysosomal-targeting drugs that are released via the lysosomal microenvironment have become one of the primary methods for managing Fabry disease. The lysosomal microenvironment is highly acidic, which enables the release of specific molecules when the nanocarrier is internalized by the lysosome. Cationic materials have been widely applied in the construction of lysosome-targeting nanocarriers. Based on these characteristics, pH-responsive or lysosome-targeting nanomaterials have been developed for the delivery of GLA to the lysosome (Giannotti et al., 2011). Negatively charged GLA was encapsulated into cationic trimethyl chitosan to prepare a pH-responsive nanoparticle. In HMEC-1 cells, the self-assembled nanoparticles exhibited excellent cellular uptake efficiency and pH-triggered release of the GLA enzyme in the lysosome. Besides pH-responsive drug release, lysosomal enzyme-responsive systems represent another favorable approach for targeting delivery GLA. Intercellular adhesion molecule-1 (ICAM-1) is overexpressed in endothelial cells and can specially deliver GLA to ICAM-1-expressing cells. Accordingly, an ICAM-1-targeted nanocarrier (anti-ICAM/α-GLA) was constructed via decorating it with an anti-ICAM antibody (Hsu et al., 2011). In a mouse model of Fabry disease, the administration of the anti-ICAM antibody-coated nanocarrier markedly increased the treatment efficacy of GLA. Numerous strategies using a variety of biomaterials have been employed for lysosomal drug delivery for the treatment of a variety of diseases.
Despite the successes of nanoplatforms in achieving GLA delivery, some critical limitations remain to be overcome. First, the design of GLA-based nanomedicine systems is complicated and should be simplified. Secondly, GLA delivery nanoplatforms for Fabry disease therapy have mainly been established using in vitro models. For a better assessment of their therapeutic and clinical translation potential, these nanoplatforms should undergo systemic evaluation in GLA-defective mice, and even in preclinical animal models (e.g., dog, pig, and monkey). Third, the treatment of central nervous system (CNS) manifestations in Fabry disease is hampered by the inability of the enzymes used for ERT to cross the blood-brain barrier; accordingly, strategies that allow these enzymes to access the brain parenchyma to exert their therapeutic effects merit further exploration. Transferrin was recently reported to be able to cross the blood-brain barrier and improve ERT effects through targeting the transferrin receptor that is highly expressed in brain endothelial cells (Ullman et al., 2020). Transferrin may be of great interest in the development of drugs for managing the main manifestations of Fabry disease. Another important issue relates to the biosafety and biocompatibility of nanomedical systems, although advanced nanoscience and materials chemistry can reduce the potential immunogenicity to a certain extent. The long-term biosafety of nanocarriers and materials toxicity of using in synthetic process also need to be considered.
Gene therapy represents an alternative approach for the treatment of Fabry disease through the application of either viral or nonviral vectors (Yasuda et al., 2020). For viral vector-mediated gene therapy, vectors have been developed that allow the highly-efficient expression of target genes in cells. The advantages of using viral vectors, such as adenoviruses, adeno-associated viruses (AVV), and lentiviruses, include that they can directly correct gene defects and their durable efficacy. Khan et al. demonstrated the effectiveness of lentivirus-mediated therapy in five patients with Fabry disease (Khan et al., 2021). α-GLA activity was significantly increased and Gb3 deposition was reduced in these patients after therapy with hematopoietic stem/progenitor cells expressing α-GLA (Figure 3). The immunogenicity of this system was further analyzed using an enzyme-linked immunosorbent assay (ELISA). Among the five patients who underwent gene therapy, only one displayed increased anti-α-GLA antibody production. The results of this study demonstrated the feasibility of utilizing lentivirus-mediated gene therapy for the treatment of Fabry disease. Despite these promising results, the application of lentivirus-based gene therapy in the CNS is hampered by the presence of the blood-brain barrier. In contrast, several adenoviruses and adeno-associated vectors (AAVs) have been demonstrated to efficiently cross the blood-brain barrier. AAVs have been used to correct GLA gene deficiency in the CNS. A recent study explored the therapeutic benefit of AAV9 expressing human α-GLA in Fabry disease model mice (Biferi et al., 2021). After a single intravenous injection, α-GLA activity was significantly increased in several tissues, including the CNS, compared with the control group. A major drawback of this viral vector relates to immunogenicity, random insertion in the host genome, mutagenicity, and organ mistargeting. This highlights the need to take into consideration the safety of viral vectors when designing treatments, thereby facilitating their clinical translation.
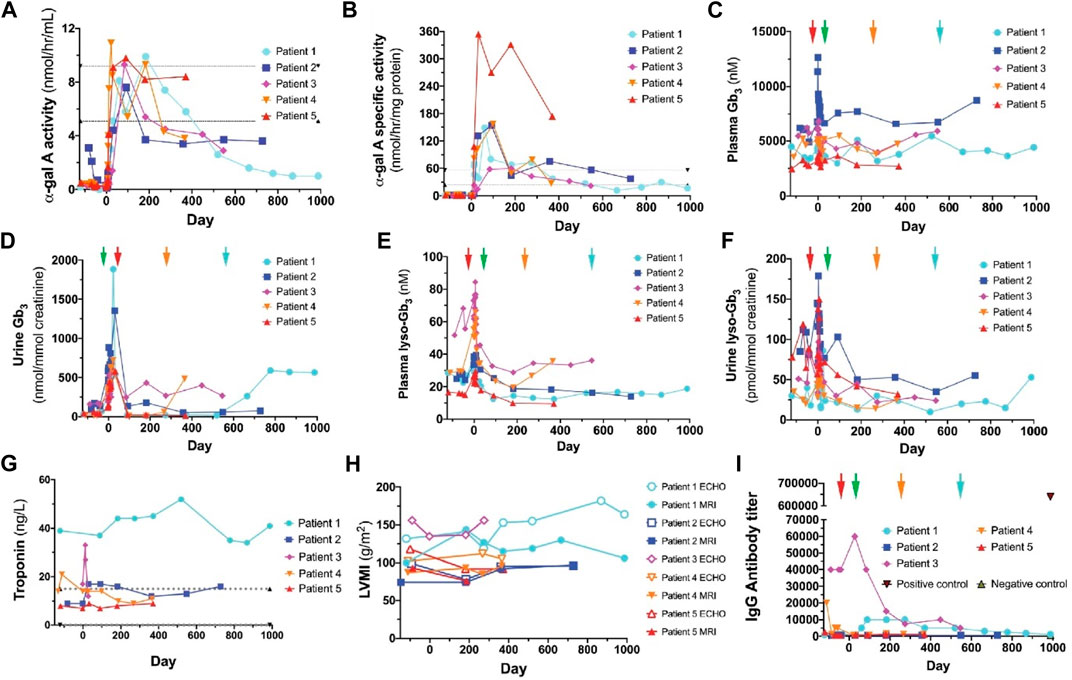
FIGURE 3. α-GLA activity in plasma (A) and leukocytes (B) of patients with Fabry disease following gene therapy. The plasma (C) and urine (D) levels of Gb3 in patients were altered after the initiation of gene therapy. The plasma (E) and urine (F) levels of lyso-Gb3 were altered after the initiation of gene therapy. Detection of troponin levels (G) and left ventricular mass index (LVMI) (H) in patients. Patient three did not attend all the measurements. (I) Anti-α-GLA antibody levels were assessed in five patients following viral vector-mediated therapy (Khan et al., 2021). Copyright 2021, The Authors.
Over recent years, attention has increasingly focused on the use of nanotechnology for gene therapy. Some excellent nanocarriers have achieved prominent therapeutic outcomes concomitant with reduced immunogenicity. To avoid antibody-mediated inactivation, PEGylated AAVs have been engineered through the covalent coating of PEG on the surface of the vectors (Lee et al., 2005). Although the circulation time was improved, transduction efficiency was decreased as binding to cells in target tissues was reduced due to the presence of PEG. This observation stresses the need to strike a balance between the circulation time and transduction efficiency. Recently, tannic acid, as a targeting ligand, was used for the modification of AVV9 to achieve myocardial targeting (Shin et al., 2018). Another approach to providing sufficient therapeutic benefits in gene therapy is the accessibility of the cell nucleus for the correction of gene defects. Once delivered to target cells via a nanocarrier, genes must be transported into the nucleus, and targeting therapeutic genes to the nucleus is important for maximizing therapeutic indexes and reducing drug doses. However, viral vectors or nanoparticles often fail to reach the nucleus, which limits their further applicability. To overcome these limitations, nucleus-targeting strategies have been developed, such as the use of nucleus-targeting ligands, including TAT peptide, R8NLS, and Cr10 (Yang et al., 2012; Li et al., 2015; Tu et al., 2020).
Nonviral treatment modalities can be divided into several categories, including siRNA, mRNA, and naked plasmid DNA. Recent studies have reported the therapeutic potential of mRNA in Fabry disease (Ruiz de Garibay et al., 2012; Zhu et al., 2019). Compared with viral vectors, non-viral vectors display substantially less toxicity and immunogenicity. However, the efficacy of systemically administered free mRNA is restricted by its short half-life and instability. Moreover, the low transfection efficacy of non-viral treatment modalities limits their wide clinical application. Successful in vivo therapy requires the construction of an RNA delivery system with high stability and reasonable delivery efficiency. This goal can be achieved by designing nanocarriers with high cargo-loading abilities, tissue or organelle specificity, and long circulation times. Numerous attempts have been made to develop a nanocarrier aimed at improving protein expression and RNA stability without systemic adverse effects, such as lipid nanoparticles (LNPs), designed as carriers for the delivery of mRNA for nonviral gene therapy (Rizvi et al., 2021; Tenchov et al., 2021). Encapsulation with LNPs limits RNase-mediated enzymatic degradation of mRNA during the delivery process, leading to increased cellular uptake of mRNA and the production of functional protein. Numerous in vitro and in vivo studies have confirmed that LNPs have great potential for mediating Fabry disease progression (DeRosa et al., 2019; Zhu et al., 2019). For instance, Aritz et al. used solid LNPs to encapsulate the pR-M10-α-GLA plasmid for expressing α-GLA in HepG2 cells (Ruiz de Garibay et al., 2012). LNP encapsulation significantly improved the transfection efficacy and the enzymatic activity of α-GLA. Evidence from a study using Fabry disease model mice also confirmed that solid LNPs can act as carriers for nonviral therapy (Figure 4). (Rodríguez-Castejón et al., 2021) Fabry mice were treated with a non-viral vector (plasmid) based on solid LNPs, and then α-GLA enzymatic activity was evaluated in plasma, liver, spleen, and brain. Mice treated with the GLA-expressing plasmid showed higher α-GLA activity than those treated with the naked plasmid. The biosafety of the LNPs was verified via the measurement of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities. These results support that developing nanocarriers for the delivery of nonviral vectors has the potential for markedly improving the therapeutic efficacy of endogenously delivered mRNA.
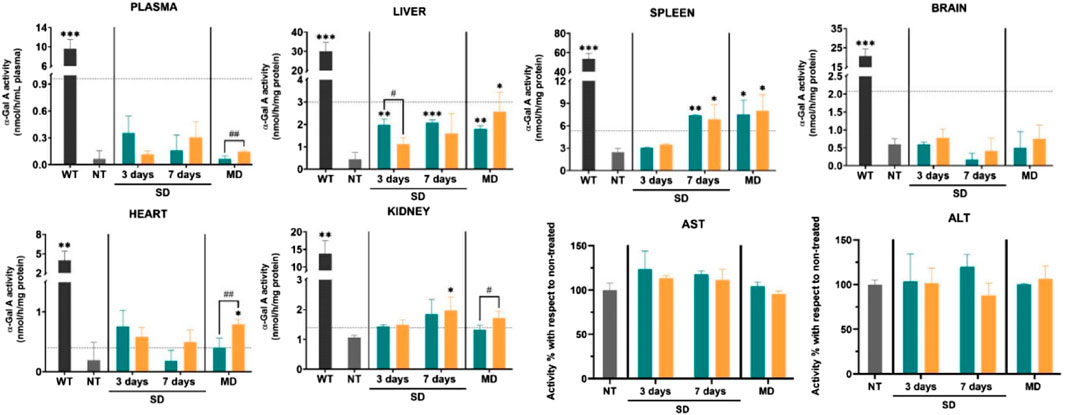
FIGURE 4. α-GLA A enzymatic activity in the plasma and tissues (liver, spleen, brain, heart, kidney) of different groups and aspartate aminotransferase (AST)/alanine aminotransferase (ALT) activity in GLA knockout (KO) mice after various treatments. NT: non-treated; SD: single dose; MD: multiple doses; Green: plasmid DNA; orange: SLP-based vector. Reproduced with permission (Rodríguez-Castejón et al., 2021). Copyright 2021, The Authors.
A key challenge facing the use of LNPs for mRNA delivery lies in that LNPs are mainly confined to hepatocytes, and a lack of cell or organ specificity limits their application (Loughrey and Dahlman, 2022). mRNA taken up by hepatocytes is robustly cleared from the liver, highlighting the importance of improving mRNA delivery to non-liver tissues for systemic mRNA therapy. For Fabry disease, kidney, heart, and CNS are suffering severe pathologies. Therefore, organ targeting and selectiveness are of critically importance for effective mRNA therapy (Knott et al., 2019). To satisfy such demand, LNP-based systems that specifically transport mRNA to specific tissues or organs are urgently demanded. Various LNPs with structural features and biological properties have been developed for organ-targeted mRNA delivery (Li et al., 2019). Notably, LNPs can be modified with various targeting ligands, including active targeting ligands and stimuli-responsive groups. For example, LNPs with RGD modifications can precisely bind vascular endothelial cells, thus improving therapeutic efficacy and reducing off-targeted effects. Moreover, specific organ-targeting LNPs have been developed to facilitate precise mRNA delivery to different organs for treatment (Dilliard et al., 2021; Qiu et al., 2022). These observations demonstrate that targeting specific organs may represent a potential strategy for improving LNP-based mRNA delivery for the treatment of Fabry disease.
Another clinically available approach for the treatment of Fabry disease is chaperone therapy, which involves the stabilization of the conformation of GLA protein in the endoplasmic reticulum to improve its catalytic ability (Riccio et al., 2020). Chaperone therapy can be divided into three types based on the associated mechanism, namely, exogenous competitive chaperones, exogenous non-competitive chaperones, and endogenous molecular chaperones (Suzuki, 2021). Compared with ERT, chaperone therapy has the advantages of excellent bioavailability, oral administration, and the ability to cross the blood-brain barrier (Pereira et al., 2018). Migalastat, an α-galactosidase analog, represents a typical exogenous competitive chaperone for Fabry disease therapy. The efficacy of migalastat has been demonstrated in preclinical studies and patients with Fabry disease. Jonas et al. explored the therapeutic effect of migalastat in patients with Fabry disease and reported that, after 1 year of oral administration of migalastat, the patients showed increased α-GLA levels and reduced serum creatinine concentrations (Müntze et al., 2019). Meanwhile, the myocardial mass index was significantly improved, further demonstrating the efficacy of migalastat. It is generally believed that restoring α-GLA enzymatic activity to over 10% may effectively maintain normal physiological function and ameliorate clinical manifestations in Fabry patients (Parenti et al., 2015). Despite its successful application in the clinic, the benefits of chaperone therapy vary among patients owing to the fluctuating levels of residual enzymatic activity. Migalastat is considered to be an appropriate therapeutic option for the treatment of Fabry patients with amenable mutations.
Heat shock proteins (HSPs) also show promise as small-molecule therapeutic chaperones. HSPs comprise a family of endogenous chaperone proteins that play multiple roles in response to cellular stress, including facilitating protein folding. HSPs can be classified into several groups according to their molecular weight, which ranges from 12 to over 100 kDa. Importantly, HSPs are important mediators of various diseases, including cancer, myocardial infarction, and ischemic stroke (Kumar et al., 2016; Behdarvandy et al., 2020; Wu et al., 2020). Additionally, HSP-based strategies have been developed as therapeutic interventions for the treatment of lysosomal storage diseases. Recombinant human HSP 70 was recently reported as having the capacity to alleviate sphingolipid storage in various lysosomal storage diseases by binding lysosomal bis(monoacylglycero)phosphate to sphingolipid-degrading enzymes and thereby enhancing sphingolipid catabolism (Kirkegaard et al., 2016). In primary fibroblasts from patients with Fabry disease, co-incubation with HSP 70 for 24 h can effectively improve the function of lysosomal-related enzymes, thus effectively alleviating sphingolipid accumulation. Meanwhile, in Fabry disease model mice, glycosphingolipid levels in the kidney, heart, and dorsal root ganglion were significantly reduced following the administration of 5 mg·kg−1 recombinant HSP 70 (Figure 5). These studies provide new insight into HSP-mediated chaperone therapy in Fabry disease. The Chaperone therapy/ERT combination may provide therapeutic advantages over chaperone therapy alone or ERT alone. An enzyme active-site stabilizer (D-galactose-configured a-cyclosulfamidate) has been reported to improve the therapeutic effect of Fabrazyme (Artola et al., 2019). In an in vitro cell model, fibroblasts from Fabry patients co-treated with Fabrazyme and a-cyclosulfamidate displayed significantly enhanced enzyme activity and decreased Gb3/LysoGb3 accumulation. These improved therapeutic effects were ascribed to competitive inhibition of α-GLA and the stabilization of its conformation. These results suggest that combination therapies may provide greater therapeutic benefits than monotherapies in the treatment of Fabry disease.
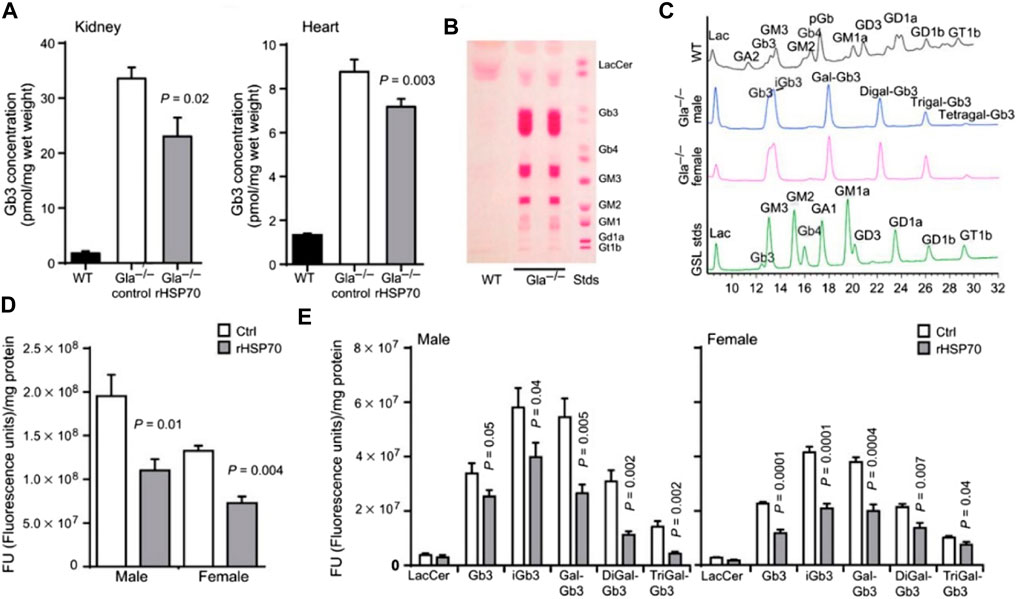
FIGURE 5. (A) Quantification of Gb3 concentrations in the heart and kidneys after various treatments. (B) Thin-layer chromatographic (B) and high-pressure liquid chromatographic (C) analysis of glycosphingolipid storage in wild-type (WT) and GLA knockout (KO) mice. Quantification of glycosphingolipid contents in dorsal root ganglia of KO mice with various treatments. Ctrl: control, PBS; rHSP70: rHSP70 intraperitoneal injection, 5 mg·kg−1. Reproduced with permission (Kirkegaard et al., 2016). Copyright 2016, American Association for the Advancement of Science.
Artificial chaperones that mimic the functions of natural chaperones have been developed for biomedical applications, such as the treatment of tumors and Alzheimer’s disease (Hashimoto et al., 2018; Li et al., 2020; Ma et al., 2020). Several nanomaterials have been designed to achieve chaperone functions. For example, Yang et al. constructed a self-assembling micelle to mimic HSP 70 function by combining poly (-amino ester: hydrophobic microdomains)-block-poly (ε-caprolactone) and poly (ethylene oxide: hydrophilic chain segment)-block-poly (ε-caprolactone). The hydrophobic microdomains could selectively capture amyloid-beta (Aβ) and prevent its aggregation in a mouse model of Alzheimer’s disease (Yang et al., 2019). The concept of artificial chaperones has also been used in facilitating the refolding of lysosomal glycoside hydrolases (Nakamoto et al., 2016). In another report, Kawasaki et al. constructed an artificial nanogel chaperone using polysaccharide and iron oxide nanoparticles to assist β-GLA delivery and folding (Kawasaki et al., 2016). These results suggest that artificial chaperones may provide a feasible alternative to current options in the treatment of Fabry disease.
The endoplasmic reticulum (ER) is where most protein folding occurs, and targeting the delivery of drugs to the ER has been applied in biomedicine (Shi et al., 2021). Shiga toxin comprises a toxic A subunit and a nontoxic B subunit that can specifically bind to Gb3. Therefore, Shiga toxin conjugated medications can be used for ER delivery through Gb3 targeting (El Alaoui et al., 2007). Targeting the ER via Shiga toxin B represents a novel avenue for the treatment of Fabry disease. Apart from Shiga toxin, other compounds such as sulfonyl-containing ligands and sulfunol-peptides can be complexed with nanocarriers to target the ER (Ting et al., 2014; Fang et al., 2019).
Substrate reduction therapy (SRT) refers to the application of molecular medications to reduce the storage of undigested macromolecules by inhibiting the synthesis of the corresponding substrates (Marshall et al., 2019). The advantages of SRT lie on the high bioavailability and its capability in traversing blood-brain barrier. Nevertheless, high residual enzyme activity is typically necessary for effective SRT. Recently, the development of medications for reducing Gb3 accumulation is receiving increasingly attentions in lysosomal storage disease (e.g., Gaucher, Fabry, and GM1-gangliosidosis disease) (Fischetto et al., 2020; Kok et al., 2021). For instance, Lucearstat and Venglustat have been clinically available for Fabry disease to reduce Gb3 accumulations. Lucearstat (N-butyl deoxy galactono-jirimycin) is a molecular inhibitor to competitively inhibit glucosylceramide synthase that catalyzes ceramide into glucosylceramide. The tolerability, biosafety, and efficacy of this medication in treatments against Fabry disease have been investigated in a clinical trial experiment (NCT02930655). Patients with Lucearstat treatment were observed with decreased plasma Gb3 concentration and relieved organ pathologies. Biosafety of Lucearstat has also been validated. These trials show that Lucearstat holds great potentials for clinical treatment of Fabry disease. In addition, phase three clinical trials are focusing the therapeutic effect and management of Fabry neuropathic pain (NCT03425539). Unfortunately, phase three clinical result show that Fabry patient subjected with 6-month Lucearstat treatment fail to alleviate the neuropathic pain of Fabry patients. For Venglustat, another promising SRT drug operated by inhibiting glucosylceramide synthase and reducing Gb3 production. Phase two clinical trials of Venglustat have validated its potential in treatment against Fabry disease (NCT02228460). Currently, trials on long-term effectiveness and safety are on the way (NCT02489344).
The mechanism underlying Gb3-induced organ dysfunction in Fabry disease remains largely unknown. Further studies on the fundamental mechanism involved in the pathophysiology of Fabry disease is expected to lead to improved strategies for its management. Autophagy provides new insights of how Gb3 accumulation and autophagic disorder benefit disease treatments and drug development. In this regard, the use of autophagic inhibitors may represent a promising avenue for the treatment of Fabry disease via reducing secondary lysosomal accumulation. Additional research efforts are required to reveal the therapeutic benefits of autophagic inhibitors. Previous evidence has revealed that activation of the AMPK-mTOR pathway has an important role in the modulation of autophagy. Thus, targeting processes upstream of the lysosomal signal for managing Fabry disease has shown great potential in preclinical studies. Interestingly, some studies have established a connection between Fabry disease and lipid hemostasis, which may provide new insights into disease progression and innovative therapies. Nevertheless, the therapeutic benefits of modulation of lipid hemostasis and inflammatory response have not been demonstrated, even in preclinical studies. Fundamental efforts are needed to further clarify the role of cholesterol metabolism and inflammatory activation in Fabry disease.
With the progress of nanotechnology and nanoscience, the limitation of these modalities can be partly solved. Designing a lysosome-based subcellular-targeted nanoparticle may be a feasible strategy for disease intervention. This targeting strategy has resulted in excellent therapeutic outcomes in lysosomal storage disease by targeting nanoparticles to the lysosome and subsequently triggering high pH-dependent drug release. Several lysosomal targeting delivery systems have been developed in experimental studies. Nevertheless, issues associated with systemic application limit their clinical translation potential. Many medications are still premature for intracellular localization. Therefore, tissue targeting design should be considered prior to the lysosomes targeting design. Accordingly, combining subcellular targeting with active targeting may be an effective strategy for treating Fabry disease. Another constraint to achieving targeted and responsive therapy relates to the complexity of synthetic processing, which can influence the biosafety of nanocarriers. Thus, it is necessary to design simpler nanoplatforms to achieve sufficient therapeutic effects without long-term systemic toxicity.
XL and XR wrote the manuscript, YZ and LD checked different sections of the manuscript. MH and QL edited the manuscript. All authors read and approved the final manuscript.
This work was supported by grants from National Natural Science Foundation of China (Nos. 81873500, 82170360, and 22005327) and the Fundamental Research Funds for the Central Universities (No. 22120220271).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abasolo, I., Seras-Franzoso, J., Moltó-Abad, M., Díaz-Riascos, V., Corchero, J. L., Pintos-Morell, G., et al. (2021). Nanotechnology-based approaches for treating lysosomal storage disorders, a focus on Fabry disease. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 13, e1684. doi:10.1002/wnan.1684
Aerts, J., Groener, J., Kuiper, S., Donker-Koopman, W., Strijland, A., Ottenhoff, R., et al. (2008). Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. U. S. A. 105, 2812–2817. doi:10.1073/pnas.0712309105
Aflaki, E., Moaven, N., Borger, D. K., Lopez, G., Westbroek, W., Chae, J. J., et al. (2016). Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell. 15, 77–88. doi:10.1111/acel.12409
Aguiar, P., Azevedo, O., Pinto, R., Marino, J., Cardoso, C., Sousa, N., et al. (2018). Biomarkers of myocardial fibrosis: Revealing the natural history of fibrogenesis in fabry disease cardiomyopathy. J. Am. Heart Assoc. 7, e007124. doi:10.1161/JAHA.117.007124
Arends, M., Wijburg, F. A., Wanner, C., Vaz, F. M., van Kuilenburg, A. B. P., Hughes, D. A., et al. (2017). Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol. Genet. Metab. 121, 157–161. doi:10.1016/j.ymgme.2017.05.001
Artola, M., Hedberg, C., Rowland, R. J., Raich, L., Kytidou, K., Wu, L., et al. (2019). α-d-Gal-cyclophellitol cyclosulfamidate is a Michaelis complex analog that stabilizes therapeutic lysosomal α-galactosidase A in Fabry disease. Chem. Sci. 10, 9233–9243. doi:10.1039/c9sc03342d
Behdarvandy, M., Karimian, M., Atlasi, M. A., and Azami Tameh, A. (2020). Heat shock protein 27 as a neuroprotective biomarker and a suitable target for stem cell therapy and pharmacotherapy in ischemic stroke. Cell. Biol. Int. 44, 356–367. doi:10.1002/cbin.11237
Belarbi, K., Cuvelier, E., and Bonte, M. A. (2020). Ceramide metabolism and Parkinson’s disease—therapeutic targets. Mol. Neurodegener. 15, 1–16. doi:10.3390/biom11070945
Benador, I., Veliova, M., Liesa, M., and Shirihai, O. (2019). Mitochondria bound to lipid droplets: Where mitochondrial dynamics regulate lipid storage and utilization. Cell. Metab. 29, 827–835. doi:10.1016/j.cmet.2019.02.011
Biancini, G. B., Vanzin, C. S., Rodrigues, D. B., Deon, M., Ribas, G. S., Barschak, A. G., et al. (2012). Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 1822, 226–232. doi:10.1016/j.bbadis.2011.11.001
Biegstraaten, M., Arngrímsson, R., Barbey, F., Boks, L., Cecchi, F., Deegan, P. B., et al. (2015). Recommendations for initiation and cessation of enzyme replacement therapy in patients with fabry disease: The European fabry working group consensus document. Orphanet J. Rare Dis. 10, 36. doi:10.1186/s13023-015-0253-6
Biferi, M. G., Cohen-Tannoudji, M., García-Silva, A., Souto-Rodríguez, O., Viéitez-González, I., San-Millán-Tejado, B., et al. (2021). Systemic treatment of fabry disease using a novel AAV9 vector expressing α-galactosidase A. Mol. Ther. 20, 1. doi:10.1016/j.omtm.2020.10.016
Birket, M. J., Raibaud, S., Lettieri, M., Adamson, A. D., Letang, V., Cervello, P., et al. (2019). A human stem cell model of fabry disease implicates LIMP-2 accumulation in cardiomyocyte pathology. Stem Cell. Rep. 13, 380–393. doi:10.1016/j.stemcr.2019.07.004
Boustany, R. M. (2013). Lysosomal storage diseases—The horizon expands. Nat. Rev. Neurol. 9, 583–598. doi:10.1038/nrneurol.2013.163
Braun, F., Blomberg, L., Brodesser, S., Liebau, M., Schermer, B., Benzing, T., et al. (2019). Enzyme replacement therapy clears Gb3 deposits from a podocyte cell culture model of fabry disease but fails to restore altered cellular signaling. Cell.. Physiol. biochem. 52, 1139–1150. doi:10.33594/000000077
Cabrera, I., Abasolo, I., Corchero, J. L., Elizondo, E., Gil, P. R., Moreno, E., et al. (2016). α-Galactosidase-A loaded-nanoliposomes with enhanced enzymatic activity and intracellular penetration. Adv. Healthc. Mat. 5, 829–840. doi:10.1002/adhm.201500746
Castelli, V., Stamerra, C., Angelo, M., Cimini, A., and Ferri, C. (2021). Current and experimental therapeutics for Fabry disease. Clin. Genet. 100, 239. doi:10.1111/cge.13999
Cedó, L., Metso, J., Santos, D., García-León, A., Plana, N., Sabate-Soler, S., et al. (2020). LDL receptor regulates the reverse transport of macrophage-derived unesterified cholesterol via concerted action of the HDL-LDL Axis: Insight from mouse models. Circ. Res. 127, 778–792. doi:10.1161/CIRCRESAHA.119.316424
Chaurasia, B., Kaddai, V. A., Lancaster, G. I., Henstridge, D. C., Sriram, S., Galam, D. L. A., et al. (2016). Adipocyte ceramides regulate subcutaneous adipose browning, inflammation, and metabolism. Cell. Metab. 24, 820–834. doi:10.1016/j.cmet.2016.10.002
Chévrier, M., Brakch, N., Céline, L., Genty, D., Ramdani, Y., Moll, S., et al. (2010). Autophagosome maturation is impaired in Fabry disease. Autophagy 6, 589–599. doi:10.4161/auto.6.5.11943
Chou, S. J., Yu, W. C., Chang, Y. L., Chen, W. Y., Chang, W. C., Chien, Y., et al. (2017). Energy utilization of induced pluripotent stem cell-derived cardiomyocyte in Fabry disease. Int. J. Cardiol. 232, 255–263. doi:10.1016/j.ijcard.2017.01.009
Del Grosso, A., Galliani, M., Angella, L., Santi, M., Tonazzini, I., Parlanti, G., et al. (2019). Brain-targeted enzyme-loaded nanoparticles: A breach through the blood-brain barrier for enzyme replacement therapy in krabbe disease. Sci. Adv. 5, eaax7462. doi:10.1126/sciadv.aax7462
DeRosa, F., Smith, L., Shen, Y., Huang, Y., Pan, J., Xie, H., et al. (2019). Improved efficacy in a fabry disease model using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol. Ther. 27, 878–889. doi:10.1016/j.ymthe.2019.03.001
Desnick, R. J., and Schuchman, E. H. (2012). Enzyme replacement therapy for lysosomal diseases: Lessons from 20 Years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 13, 307–335. doi:10.1146/annurev-genom-090711-163739
Dilliard, S. A., Cheng, Q., and Siegwart, D. J. (2021). On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 118, 118. doi:10.1073/pnas.2109256118
El Alaoui, A., Schmidt, F., Amessou, M., Sarr, M., Decaudin, D., Florent, J. C., et al. (2007). Shiga toxin-mediated retrograde delivery of a topoisomerase I inhibitor prodrug. Angew. Chem. Int. Ed. Engl. 46, 6469–6472. doi:10.1002/anie.200701270
Elliott, P., Kindler, H., Shah, J., Sachdev, B., Rimoldi, O., Thaman, R., et al. (2006). Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with alpha galactosidase A. Heart 92, 357–360. doi:10.1136/hrt.2004.054015
Fang, L., Trigiante, G., Crespo-Otero, R., Hawes, C. S., Philpott, M. P., Jones, C. R., et al. (2019). Endoplasmic reticulum targeting fluorescent probes to image mobile Zn2. Chem. Sci. 10, 10881–10887. doi:10.1039/c9sc04300d
Fernández-Pereira, C., San Millán-Tejado, B., Gallardo-Gómez, M., Pérez-Márquez, T., Alves-Villar, M., Melcón-Crespo, C., et al. (2021). Therapeutic approaches in lysosomal storage diseases. Biomolecules 11, 1775. doi:10.3390/biom11121775
Festa, B. P., Chen, Z., Berquez, M., Debaix, H., Tokonami, N., Prange, J. A., et al. (2018). Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 9, 161. doi:10.1038/s41467-017-02536-7
Fischetto, R., Palladino, V., Mancardi, M. M., Giacomini, T., Palladino, S., Gaeta, A., et al. (2020). Substrate reduction therapy with miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: A multicenter experience. Mol. Genet. Genomic Med. 8, e1371. doi:10.1002/mgg3.1371
Frustaci, A., Russo, M., Francone, M., and Chimenti, C. (2014). Microvascular angina as prehypertrophic presentation of Fabry disease cardiomyopathy. Circulation 130, 1530–1531. doi:10.1161/CIRCULATIONAHA.114.012178
Ghosh, P., Yang, X., Arvizo, R., Zhu, Z. J., Agasti, S. S., Mo, Z., et al. (2010). Intracellular delivery of a membrane-impermeable enzyme in active form using functionalized gold nanoparticles. J. Am. Chem. Soc. 132, 2642–2645. doi:10.1021/ja907887z
Giannotti, M., Abasolo, I., Oliva, M., Andrade, F., García-Aranda, N., Melgarejo, M., et al. (2016). Highly versatile polyelectrolyte complexes for improving the enzyme replacement therapy of lysosomal storage disorders. ACS Appl. Mat. Interfaces 8, 25741–25752. doi:10.1021/acsami.6b08356
Giannotti, M., Esteban, O., Oliva, M., García-Parajo, M., and Sanz, F. (2011). pH-responsive polysaccharide-based polyelectrolyte complexes as nanocarriers for lysosomal delivery of therapeutic proteins. Biomacromolecules 12, 2524–2533. doi:10.1021/bm2003384
Glaros, E. N., Kim, W. S., Quinn, C. M., Wong, J., Gelissen, I., Jessup, W., et al. (2005). Glycosphingolipid accumulation inhibits cholesterol efflux via the ABCA1/apolipoprotein A-I pathway: 1-phenyl-2-decanoylamino-3-morpholino-1-propanol is a novel cholesterol efflux accelerator. J. Biol. Chem. 280, 24515–24523. doi:10.1074/jbc.M413862200
Gregory, J. V., Kadiyala, P., Doherty, R., Cadena, M., Habeel, S., Ruoslahti, E., et al. (2020). Systemic brain tumor delivery of synthetic protein nanoparticles for glioblastoma therapy. Nat. Commun. 11, 5687. doi:10.1038/s41467-020-19225-7
Gupta, M., Pandey, H., and Sivakumar, S. (2017). Intracellular delivery of β-galactosidase enzyme using arginase-responsive dextran sulfate/poly-l-arginine capsule for lysosomal storage disorder. ACS Omega 2, 9002–9012. doi:10.1021/acsomega.7b01230
Hadas, Y., Vincek, A. S., Youssef, E., Zak, M. M., Chepurko, E., Sultana, N., et al. (2020). Altering sphingolipid metabolism attenuates cell death and inflammatory response after myocardial infarction. Circulation 141, 916–930. doi:10.1161/CIRCULATIONAHA.119.041882
Hashimoto, Y., Mukai, S. a., Sasaki, Y., and Akiyoshi, K. J. A. H. M. (2018). Nanogel tectonics for tissue engineering: Protein delivery systems with nanogel chaperones. Adv. Healthc. Mat. 7, 1800729. doi:10.1002/adhm.201800729
Hsu, J., Serrano, D., Bhowmick, T., Kumar, K., Shen, Y., Kuo, Y. C., et al. (2011). Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J. Control. Release 149, 323–331. doi:10.1016/j.jconrel.2010.10.031
Hsu, M. J., Chang, F. P., Lu, Y. H., Hung, S. C., Wang, Y. C., Yang, A. H., et al. (2019). Identification of lysosomal and extralysosomal globotriaosylceramide (Gb3) accumulations before the occurrence of typical pathological changes in the endomyocardial biopsies of Fabry disease patients. Genet. Med. 21, 224–232. doi:10.1038/s41436-018-0010-z
Iershov, A., Nemazanyy, I., Alkhoury, C., Girard, M., Barth, E., Cagnard, N., et al. (2019). The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARα. Nat. Commun. 10, 1566. doi:10.1038/s41467-019-09598-9
Ivanova, M. M., Changsila, E., Iaonou, C., and Goker-Alpan, O. (2019). Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. Plos One 14, e0210617. doi:10.1371/journal.pone.0210617
Jabbarzadeh-Tabrizi, S., Boutin, M., Day, T., Taroua, M., Schiffmann, R., Auray-Blais, C., et al. (2020). Assessing the role of glycosphingolipids in the phenotype severity of Fabry disease mouse model. J. Lipid Res. 61, 1410–1423. doi:10.1194/jlr.RA120000909
Kalliokoski, R., Kalliokoski, K., Penttinen, M., Kantola, I., Leino, A., Viikari, J., et al. (2006). Structural and functional changes in peripheral vasculature of Fabry patients. J. Inherit. Metab. Dis. 29, 660–666. doi:10.1007/s10545-006-0340-x
Kang, J., Kaissarian, N., Desch, K., Kelly, R., Shu, L., Bodary, P., et al. (2019). α-galactosidase A deficiency promotes von Willebrand factor secretion in models of Fabry disease. Kidney Int. 95, 149–159. doi:10.1016/j.kint.2018.08.033
Kawasaki, R., Sasaki, Y., Katagiri, K., Mukai, S. A., Sawada, S., and Akiyoshi, K. (2016). Magnetically guided protein transduction by hybrid nanogel chaperones with iron oxide nanoparticles. Angew. Chem. Int. Ed. Engl. 55, 11377–11381. doi:10.1002/anie.201602577
Khan, A., Barber, D., Huang, J., Rupar, C., Rip, J., Auray-Blais, C., et al. (2021). Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 12, 1178. doi:10.1038/s41467-021-21371-5
Kim, J. W., Kim, H. W., Nam, S. A., Lee, J. Y., Cho, H. J., Kim, T. M., et al. (2021). Human kidney organoids reveal the role of glutathione in Fabry disease. Exp. Mol. Med. 53, 1580–1591. doi:10.1038/s12276-021-00683-y
Kirkegaard, T., Gray, J., Priestman, D. A., Wallom, K. L., Atkins, J., Olsen, O. D., et al. (2016). Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 8, 355ra118. doi:10.1126/scitranslmed.aad9823
Kizhner, T., Azulay, Y., Hainrichson, M., Tekoah, Y., Arvatz, G., Shulman, A., et al. (2015). Characterization of a chemically modified plant cell culture expressed human α-Galactosidase-A enzyme for treatment of Fabry disease. Mol. Genet. Metab. 114, 259–267. doi:10.1016/j.ymgme.2014.08.002
Knott, K. D., Augusto, J. B., Nordin, S., Kozor, R., Camaioni, C., Xue, H., et al. (2019). Quantitative myocardial perfusion in fabry disease. Circ. Cardiovasc. Imaging 12, e008872. doi:10.1161/CIRCIMAGING.119.008872
Kok, K., Zwiers, K. C., Boot, R. G., Overkleeft, H. S., Aerts, J. M. F. G., and Artola, M. (2021). Fabry disease: Molecular basis, pathophysiology, diagnostics and potential therapeutic directions. Biomolecules 11, 271. doi:10.3390/biom11020271
Kumar, S., Stokes, J., Singh, U. P., Scissum Gunn, K., Acharya, A., Manne, U., et al. (2016). Targeting Hsp70: A possible therapy for cancer. Cancer Lett. 374, 156–166. doi:10.1016/j.canlet.2016.01.056
Lee, G., Maheshri, N., Kaspar, B., and Schaffer, D. (2005). PEG conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol. Bioeng. 92, 24–34. doi:10.1002/bit.20562
Lenders, M., and Brand, E. (2018). Effects of enzyme replacement therapy and antidrug antibodies in patients with fabry disease. J. Am. Soc. Nephrol. 29, 2265–2278. doi:10.1681/asn.2018030329
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335. doi:10.1038/nature09782
Li, B., Zhang, X., and Dong, Y. (2019). Nanoscale platforms for messenger RNA delivery. WIREs Nanomed. Nanobiotechnol. 11, e1530. doi:10.1002/wnan.1530
Li, L., Sun, W., Zhong, J., Yang, Q., Zhu, X., Zhou, Z., et al. (2015). Multistage nanovehicle delivery system based on stepwise size reduction and charge reversal for programmed nuclear targeting of systemically administered anticancer drugs. Adv. Funct. Mat. 25, 4101–4113. doi:10.1002/adfm.201501248
Li, X., Cai, X., Zhang, Z., Ding, Y., Ma, R., Huang, F., et al. (2020). Mimetic heat shock protein mediated immune process to enhance cancer immunotherapy. Nano Lett. 20, 4454–4463. doi:10.1021/acs.nanolett.0c01230
Li, X., Zhang, Y., Ren, X., Wang, Y., Chen, D., Li, Q., et al. (2021). Ischemic microenvironment‐responsive therapeutics for cardiovascular diseases. Adv. Mat. 33, e2105348. doi:10.1002/adma.202105348
Liebau, M. C., Braun, F., Höpker, K., Weitbrecht, C., Bartels, V., Müller, R. U., et al. (2013). Dysregulated autophagy contributes to podocyte damage in Fabry's disease. Plos one 8, e63506. doi:10.1371/journal.pone.0063506
Loughrey, D., and Dahlman, J. E. (2022). Non-liver mRNA delivery. Acc. Chem. Res. 55, 13–23. doi:10.1021/acs.accounts.1c00601
Luzio, J., Pryor, P., and Bright, N. (2007). Lysosomes: Fusion and function. Nat. Rev. Mol. Cell. Biol. 8, 622–632. doi:10.1038/nrm2217
Ma, F. H., Li, C., Liu, Y., and Shi, L. (2020). Mimicking molecular chaperones to regulate protein folding. Adv. Mat. 32, e1805945. doi:10.1002/adma.201805945
Martina, J., Raben, N., and Puertollano, R. (2020). SnapShot: Lysosomal storage diseases. Cell. 180, 602. doi:10.1016/j.cell.2020.01.017
Marshall, J., Nietupski, J. B., Park, H., et al. (2019). Substrate reduction therapy for sandhoff disease through inhibition of glucosylceramide synthase activity. Mol. Thera. 27, 1495–1506. doi:10.1016/j.ymthe.2019.05.018
McCoin, C. S., Knotts, T. A., and Adams, S. H. (2015). Acylcarnitines—Old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 11, 617–625. doi:10.1038/nrendo.2015.129
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. doi:10.1038/nature07201
Meka, A., Abbaraju, P., Song, H., Xu, C., Zhang, J., Zhang, H., et al. (2016). A vesicle supra-assembly approach to synthesize amine-functionalized hollow dendritic mesoporous silica nanospheres for protein delivery. Small 12, 5169–5177. doi:10.1002/smll.201602052
Müntze, J., Gensler, D., Maniuc, O., Liu, D., Cairns, T., Oder, D., et al. (2019). Oral chaperone therapy migalastat for treating fabry disease: Enzymatic response and serum biomarker changes after 1 Year. Clin. Pharmacol. Ther. 105, 1224–1233. doi:10.1002/cpt.1321
Nakamoto, M., Nonaka, T., Shea, K. J., Miura, Y., and Hoshino, Y. (2016). Design of synthetic polymer nanoparticles that facilitate resolubilization and refolding of aggregated positively charged lysozyme. J. Am. Chem. Soc. 138, 4282–4285. doi:10.1021/jacs.5b12600
Nguyen, T. B., Louie, S. M., Daniele, J. R., Tran, Q., Dillin, A., Zoncu, R., et al. (2017). DGAT1-Dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev. Cell. 42, 9–21. doi:10.1016/j.devcel.2017.06.003
Parenti, G., Andria, G., and Valenzano, K. J. (2015). Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 23, 1138–1148. doi:10.1038/mt.2015.62
Pereira, D. M., Valentão, P., and Andrade, P. B. (2018). Tuning protein folding in lysosomal storage diseases: The chemistry behind pharmacological chaperones. Chem. Sci. 9, 1740–1752. doi:10.1039/c7sc04712f
Pisani, A., Bruzzese, D., Sabbatini, M., Spinelli, L., Imbriaco, M., and Riccio, E. (2017). Switch to agalsidase alfa after shortage of agalsidase beta in fabry disease: A systematic review and meta-analysis of the literature. Genet. Med. 19, 275–282. doi:10.1038/gim.2016.117
Platt, F. M., d'Azzo, A., Davidson, B. L., Neufeld, E. F., and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Prim. 4, 27. doi:10.1038/s41572-018-0025-4
Puri, V., Jefferson, J. R., Singh, R. D., Wheatley, C. L., Marks, D. L., and Pagano, R. E. (2003). Sphingolipid storage induces accumulation of intracellular cholesterol by stimulating SREBP-1 cleavage. J. Biol. Chem. 278, 20961–20970. doi:10.1074/jbc.M300304200
Qiu, M., Tang, Y., Chen, J., Muriph, R., Ye, Z., Huang, C., et al. (2022). Lung-selective mRNA delivery of synthetic lipid nanoparticles for the treatment of pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. U. S. A. 119, e2116271119. doi:10.1073/pnas.2116271119
Rambold, A., Cohen, S., and Lippincott-Schwartz, J. (2015). Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell. 32, 678–692. doi:10.1016/j.devcel.2015.01.029
Rappaport, J., Manthe, R., Solomon, M., Garnacho, C., and Muro, S. (2016). A comparative study on the alterations of endocytic pathways in multiple lysosomal storage disorders. Mol. Pharm. 13, 357–368. doi:10.1021/acs.molpharmaceut.5b00542
Riccio, E., Zanfardino, M., Ferreri, L., Santoro, C., Cocozza, S., Capuano, I., et al. (2020). Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: Real-life data. Eur. J. Hum. Genet. 28, 1662–1668. doi:10.1038/s41431-020-0677-x
Rizvi, F., Everton, E., Smith, A., Liu, H., Osota, E., Beattie, M., et al. (2021). Murine liver repair via transient activation of regenerative pathways in hepatocytes using lipid nanoparticle-complexed nucleoside-modified mRNA. Nat. Commun. 12, 613. doi:10.1038/s41467-021-20903-3
Rodríguez-Castejón, J., Alarcia-Lacalle, A., Gómez-Aguado, I., Vicente-Pascual, M., Solinís Aspiazu, M., Del Pozo-Rodríguez, A., et al. (2021). World journal of nephrology. Pharmaceutics 10, 13. doi:10.3390/nano10020364
Rozenfeld, P., and Feriozzi, S. (2017). Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 122, 19–27. doi:10.1016/j.ymgme.2017.09.004
Ruiz de Garibay, A., Delgado, D., Del Pozo-Rodríguez, A., Solinís, M., and Gascón, A. R. (2012). Multicomponent nanoparticles as nonviral vectors for the treatment of Fabry disease by gene therapy. Drug Des. Dev. Ther. 6, 303. doi:10.2147/DDDT.S36131
Scaletti, F., Hardie, J., Lee, Y., Luther, D., Ray, M., and Rotello, V. (2018). Protein delivery into cells using inorganic nanoparticle-protein supramolecular assemblies. Chem. Soc. Rev. 47, 3421–3432. doi:10.1039/c8cs00008e
Scharnetzki, D., Stappers, F., Lenders, M., and Brand, E. (2020). Detailed epitope mapping of neutralizing anti-drug antibodies against recombinant α-galactosidase A in patients with Fabry disease. Mol. Genet. Metab. 131, 229–234. doi:10.1016/j.ymgme.2020.08.005
Schiffmann, R., Goker-Alpan, O., Holida, M., Giraldo, P., Barisoni, L., Colvin, R. B., et al. (2019). Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year phase 1/2 clinical trial. J. Inherit. Metab. Dis. 42, 534. doi:10.1002/jimd.12080
Schueler, U., Kaneski, C., Remaley, A., Demosky, S., Dwyer, N., Blanchette-Mackie, J., et al. (2016). A short synthetic peptide mimetic of apolipoprotein A1 mediates cholesterol and globotriaosylceramide efflux from fabry fibroblasts. JIMD Rep. 29, 69–75. doi:10.1007/8904_2015_507
Schultz, M., Tecedor, L., Chang, M., and Davidson, B. (2011). Clarifying lysosomal storage diseases. Trends Neurosci. 34, 401–410. doi:10.1016/j.tins.2011.05.006
Schumann, A., Schaller, K., Belche, V., Cybulla, M., Grünert, S. C., Moers, N., et al. (2021). Defective lysosomal storage in Fabry disease modifies mitochondrial structure, metabolism and turnover in renal epithelial cells. J. Inherit. Metab. Dis. 44, 1039–1050. doi:10.1002/jimd.12373
Seras-Franzoso, J., Díaz-Riascos, Z., Corchero, J., González, P., Garcia-Aranda, N., Mandana, M., et al. (2021). Extracellular vesicles from recombinant cell factories improve the activity and efficacy of enzymes defective in lysosomal storage disorders. J. Extracell. Vesicles 10, e12058. doi:10.1002/jev2.12058
Shi, Y., Wang, S., Wu, J., Jin, X., and You, J. (2021). Pharmaceutical strategies for endoplasmic reticulum-targeting and their prospects of application. J. Control. Release 329, 337–352. doi:10.1016/j.jconrel.2020.11.054
Shin, M., Lee, H. A., Lee, M., Shin, Y., Song, J., Kang, S., et al. (2018). Targeting protein and peptide therapeutics to the heart via tannic acid modification. Nat. Biomed. Eng. 2, 304–317. doi:10.1038/s41551-018-0227-9
Shirai, T., Ohtake, T., Kimura, M., Iwata, M., Fujigaki, Y., Takayanagi, S., et al. (2000). Atypical Fabry's disease presenting with cholesterol crystal embolization. Intern. Med. 39, 646–649. doi:10.2169/internalmedicine.39.646
Solomon, M., and Muro, S. (2017). Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives. Adv. Drug Deliv. Rev. 118, 109–134. doi:10.1016/j.addr.2017.05.004
Song, H. Y., Yang, Y. P., Chien, Y., Lai, W. Y., Lin, Y. Y., Chou, S. J., et al. (2021). Reversal of the inflammatory responses in fabry patient iPSC-derived cardiovascular endothelial cells by CRISPR/Cas9-Corrected mutation. Int. J. Mol. Sci. 22, 2381. doi:10.3390/ijms22052381
Song, H. Y., Chien, C. S., Yarmishyn, A. A., Chou, S. J., Yang, Y. P., Wang, M. L., et al. (2019). Generation of GLA-knockout human embryonic stem cell lines to model autophagic dysfunction and exosome secretion in fabry disease-associated hypertrophic cardiomyopathy. Cells 8, 327. doi:10.3390/cells8040327
Spinelli, L., Giugliano, G., Imbriaco, M., Esposito, G., Nappi, C., Riccio, E., et al. (2020). Left ventricular radial strain impairment precedes hypertrophy in Anderson-Fabry disease. Int. J. Cardiovasc. Imaging 36, 1465–1476. doi:10.1007/s10554-020-01847-z
Stepien, K. M., and Hendriksz, C. J. (2017). Lipid profile in adult patients with Fabry disease - ten-year follow up. Mol. Genet. Metab. Rep. 13, 3–6. doi:10.1016/j.ymgmr.2017.06.010
Suzuki, Y. (2021). Chaperone therapy for molecular pathology in lysosomal diseases. Brain Dev. 43, 45–54. doi:10.1016/j.braindev.2020.06.015
Tenchov, R., Bird, R., Curtze, A., and Zhou, Q. (2021). Lipid Nanoparticles─From liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS Nano 15, 16982–17015. doi:10.1021/acsnano.1c04996
Ting, C. H., Huang, H. N., Huang, T. C., Wu, C. J., and Chen, J. Y. (2014). The mechanisms by which pardaxin, a natural cationic antimicrobial peptide, targets the endoplasmic reticulum and induces c-FOS. Biomaterials 35, 3627–3640. doi:10.1016/j.biomaterials.2014.01.032
Tomsen-Melero, J., Passemard, S., García-Aranda, N., Díaz-Riascos, Z. V., González-Rioja, R., Nedergaard Pedersen, J., et al. (2021). Impact of chemical composition on the nanostructure and biological activity of α-galactosidase-loaded nanovesicles for fabry disease treatment. ACS Appl. Mat. Interfaces 13, 7825–7838. doi:10.1021/acsami.0c16871
Tu, Z., Donskyi, I., Qiao, H., Zhu, Z., Unger, W., Hackenberger, C., et al. (2020). Graphene oxide‐cyclic R10 peptide nuclear translocation nanoplatforms for the surmounting of multiple‐drug resistance. Adv. Funct. Mat. 30, 2000933. doi:10.1002/adfm.202000933
Uceyler, N., Urlaub, D., Mayer, C., Uehlein, S., Held, M., and Sommer, C. (2019). Tumor necrosis factor-α links heat and inflammation with Fabry pain. Mol. Genet. Metab. 127, 200–206. doi:10.1016/j.ymgme.2019.05.009
Ullman, J., Arguello, A., Getz, J., Bhalla, A., Mahon, C., Wang, J., et al. (2020). Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci. Transl. Med. 12, eaay1163. doi:10.1126/scitranslmed.aay1163
van der Veen, S., Hollak, C., van Kuilenburg, A., and Langeveld, M. (2020). Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 43, 908–921. doi:10.1002/jimd.12228
van der Veen, S., van Kuilenburg, A., Hollak, C., Kaijen, P., Voorberg, J., and Langeveld, M. (2019). Antibodies against recombinant alpha-galactosidase A in Fabry disease: Subclass analysis and impact on response to treatment. Mol. Genet. Metab. 126, 162–168. doi:10.1016/j.ymgme.2018.11.008
Veronese, F. M., and Pasut, G. (2005). PEGylation, successful approach to drug delivery. Drug Discov. Today 10, 1451–1458. doi:10.1016/S1359-6446(05)03575-0
Weissmann, C., Albanese, A., Contreras, N., Gobetto, M., Castellanos, L., and Uchitel, O. (2021). Ion channels and pain in Fabry disease. Mol. Pain 17, 174480692110331. doi:10.1177/17448069211033172
Wu, J., Chen, S., Liu, Y., Liu, Z., Wang, D., and Cheng, Y. (2020). Therapeutic perspectives of heat shock proteins and their protein-protein interactions in myocardial infarction. Pharmacol. Res. 160, 105162. doi:10.1016/j.phrs.2020.105162
Xing, Y., Sui, Z., Liu, Y., Wang, M. M., Wei, X., Lu, Q., et al. (2022). Blunting TRPML1 channels protects myocardial ischemia/reperfusion injury by restoring impaired cardiomyocyte autophagy. Basic Res. Cardiol. 117, 20. doi:10.1007/s00395-022-00930-x
Yang, H., Li, X., Zhu, L., Wu, X., Zhang, S., Huang, F., et al. (2019). Heat shock protein inspired nanochaperones restore amyloid‐β homeostasis for preventative therapy of alzheimer's disease. Adv. Sci. 6, 1901844. doi:10.1002/advs.201901844
Yang, J., Lei, Q., Han, K., Gong, Y., Chen, S., Cheng, H., et al. (2012). Reduction-sensitive polypeptides incorporated with nuclear localization signal sequences for enhanced gene delivery. J. Mat. Chem. 22, 13591. doi:10.1039/c2jm32223d
Yasuda, M., Huston, M. W., Pagant, S., Gan, L., Martin, S., Sproul, S., et al. (2020). AAV2/6 gene therapy in a murine model of fabry disease results in supraphysiological enzyme activity and effective substrate reduction. Mol. Ther. Methods Clin. Dev. 18, 607–619. doi:10.1016/j.omtm.2020.07.002
Yogasundaram, H., Nikhanj, A., Putko, B. N., Boutin, M., Jain-Ghai, S., Khan, A., et al. (2018). Elevated inflammatory plasma biomarkers in patients with fabry disease: A critical link to heart failure with preserved ejection fraction. J. Am. Heart Assoc. 7, e009098. doi:10.1161/JAHA.118.009098
Keywords: fabry disease, lysosomal storage disorder, mechanistic research, fabry therapeutic strategies, alpha-galactosidase A deficiency
Citation: Li X, Ren X, Zhang Y, Ding L, Huo M and Li Q (2022) Fabry disease: Mechanism and therapeutics strategies. Front. Pharmacol. 13:1025740. doi: 10.3389/fphar.2022.1025740
Received: 23 August 2022; Accepted: 10 October 2022;
Published: 26 October 2022.
Edited by:
Guido Iaccarino, University of Naples Federico II, ItalyReviewed by:
Fabian Braun, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2022 Li, Ren, Zhang, Ding, Huo and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Li, aHhsaXFpYW5Ad2Noc2N1LmNu; Minfeng Huo, bWZodW9AdG9uZ2ppLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.