
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 25 October 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1020556
This article is part of the Research Topic New Insights into the Histone Lysine Specific Demethylase (LSD1) Inhibitors for Cancer Treatment View all 4 articles
 Yu Li1
Yu Li1 Yuanyuan Zhao1Xiaona Li1Liuqun Zhai1Hua Zheng1Ying Yan1Qiang Fu2Jinlian Ma3Haier Fu1*Zhenqiang Zhang3*
Yuanyuan Zhao1Xiaona Li1Liuqun Zhai1Hua Zheng1Ying Yan1Qiang Fu2Jinlian Ma3Haier Fu1*Zhenqiang Zhang3* Zhonghua Li3*
Zhonghua Li3*Alzheimer’s disease (AD) is a common chronic neurodegenerative disease characterized by cognitive learning and memory impairments, however, current treatments only provide symptomatic relief. Lysine-specific demethylase 1 (LSD1), regulating the homeostasis of histone methylation, plays an important role in the pathogenesis of many neurodegenerative disorders. LSD1 functions in regulating gene expression via transcriptional repression or activation, and is involved in initiation and progression of AD. Pharmacological inhibition of LSD1 has shown promising therapeutic benefits for AD treatment. In this review, we attempt to elaborate on the role of LSD1 in some aspects of AD including neuroinflammation, autophagy, neurotransmitters, ferroptosis, tau protein, as well as LSD1 inhibitors under clinical assessments for AD treatment.
Alzheimer’s disease (AD) is a chronic and complex neurodegenerative disease, most often associated with cognitive function disorders including memory decline, learning deficits, psychiatric disorders, and spatial disorientation, etc., AD is most popular in people over the age of 65, and the risk of AD increases with age. With the aggravation of the aging problem worldwide, AD population is estimated to reach more than 131 million by 2050 barring the development of effective treatment to prevent AD (Cummings et al., 2016). Pathologically, AD is a complex and multifactorial disease, with a number of factors accounting for the prognosis of AD, such as chronic inflammation, synaptic dysfunction, amyloid plaques accumulation, neurofibrillary tangles, and neuronal death (Long and Holtzman, 2019; Ju and Tam, 2022). Over a long period of time, much attention has been paid to AD treatments based on the classical amyloid cascade and tau aggregation hypothesis. However, despite considerable research efforts to tackle these issues, there has been a paucity of effective treatment options for AD patients, and over 200 AD investigational programs have failed over the past decades (Yiannopoulou et al., 2019; Majernikova et al., 2021). Therefore, there are urgent needs for effective treatments for this condition.
In recent years, epigenetic mechanisms have emerged as an important player in central nervous system (CNS) disorders (Coppede, 2021). Epigenetic regulations shape the phenotype without altering the genotype, and mainly include DNA methylation, histone modifications, chromatin remodeling, nucleosome positioning and non-coding RNAs (Stoccoro and Copped, 2018). Histone methylation represents one of the best characterized post-transcriptional modifications on histone tail residues, and is associated with neural functions related to cognitive abilities (Bannister and Kouzarides, 2011; Collins et al., 2019; Lopez-Tobon et., 2020). Increasing studies suggest that histone methylations play essential roles in neuronal differentiation and plasticity, as well as in memory and learning process, while impaired regulation of histone methylation are tightly associated with neurodegenerative disorders (Fischer et al., 2007; Herre and Korb, 2019). Thus, targeting histone methylation has been proposed to be a novel therapeutic strategy for AD (Lin et al., 2019; Zheng et al., 2019).
Lysine methyltransferases and demethylases are responsible for catalyzing the process of N-methylation and N-demethylation of histone lysine, respectively (Kaniskan et al., 2018). Unlike histone acetylation, phosphorylation and ubiquitination, which were identified as a dynamic process, histone methylation has historically been considered as an irreversible mark until the discovery of lysine specific demethylase 1 (LSD1) in 2004 (Shi et al., 2004). Through maintaining a proper situation of histone methylation, LSD1 makes a broad scope of regulatory effects on histone or nonhistone proteins (Huang et al., 2007; Wang et al., 2009; Ciccone et al., 2009; Kontaki and Taliandis, 2010). Several lines of research have revealed that, by forming distinct protein complexes, LSD1 contributes to various biological processes including cell proliferation, stem cell biology, autophagy, metabolism and neural development (Wang et al., 2007; Stefano et al., 2016; Maes et al., 2015). The aberrant function of LSD1 has been found to be able to stimulate many negative biological processes such as tumor initiation and progression, and is closely connected with various malignant tumors, especially lung cancer and leukemia (Wang et al., 2009; Magerl et al., 2010; Lokken and Zeleznik-Le, 2012). Either genetic depletion of LSD1 or pharmacological inhibition has been shown to suppress the tumor growth, and therefore LSD1 is becoming a promising therapeutic target for cancer treatment (Schmitt et al., 2013).
In CNS, LSD1 plays a crucial regulatory role in the maintenance of pluripotency and in specification of neuronal commitment of multipotent cells, which is proposed to be an important hallmark of the situation and progression of many neurodegenerative disorders such as AD (Maiques-Diaz et al., 2018). There are currently no findings about genetic disorders affecting brain development or function caused by LSD1 mutations (Maes et al., 2015). However, by forming distinct protein complexes probably affected by mutation or epigenetic modification, modulation of LSD1 activity is proposed to be able to improve the symptoms of neurodegenerative disease. For instance, the repressor complex of LSD1-REST-CoREST-HDAC1/2 functions primarily in controlling developmental program and modulating neuronal morphology in CNS diseases (Toffolo et al., 2014; Saez et al., 2015). Christopher et al. (2017) reported that inducible depletion of LSD1 in mice led to paralysis along with neurodegeneration of hippocampus and cortex, causing learning and memory deficits. Mechanistically, they found that LSD1 depletion could induce transcriptional changes in neurodegenerative pathways as well as re-activation of stem cell genes in the degenerating hippocampus. Furthermore, the authors detected that LSD1 protein mislocalized in the cytoplasm associated with pathological protein aggregates in brains of AD patients. Although it remains unclear whether the functional impairment of LSD1 is directly connected with the neurodegeneration, the sustained amount of LSD1 in the brain is required to maintain the neuronal function (Christopher et al., 2017; Swahari and West, 2019). Noteworthy, several lines of studies have shown that specific pharmacological inhibition of LSD1 displayed significant neuroprotective effects in CNS disorders, underlying the therapeutic potential of LSD1 intervention in such diseases related to epigenetic dysregulation (Matsuda et al., 2019; Maes et al., 2020; Baba et al., 2021).
Here, this review focuses on the recent hallmark findings on the functional role of LSD1 in neurodegenerative disorders, particularly AD, along with its therapeutic potential in the treatment alternatives of AD (Figure 1).
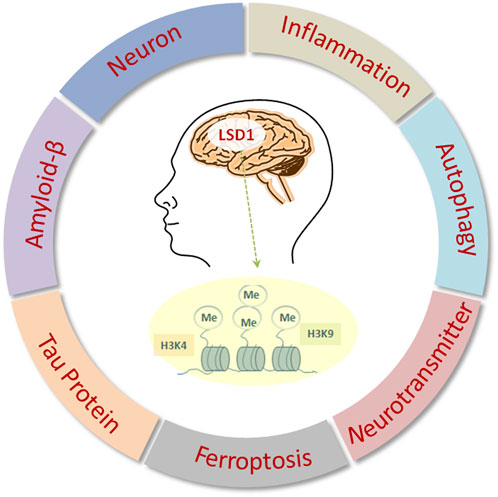
FIGURE 1. Biological functions of LSD1 in AD.
LSD1 belongs to the FAD-dependent amine oxidase superfamily. The structure of LSD1 is highly conserved, and is divided into three major protein domains: an N-terminal SWIRM (Swi3p/Rsc8p/Morira) structural domain, a central protruding Tower domain, and a C-terminal amine oxidase like (AOL) domain (Figure 2A) (Chen et al., 2006).
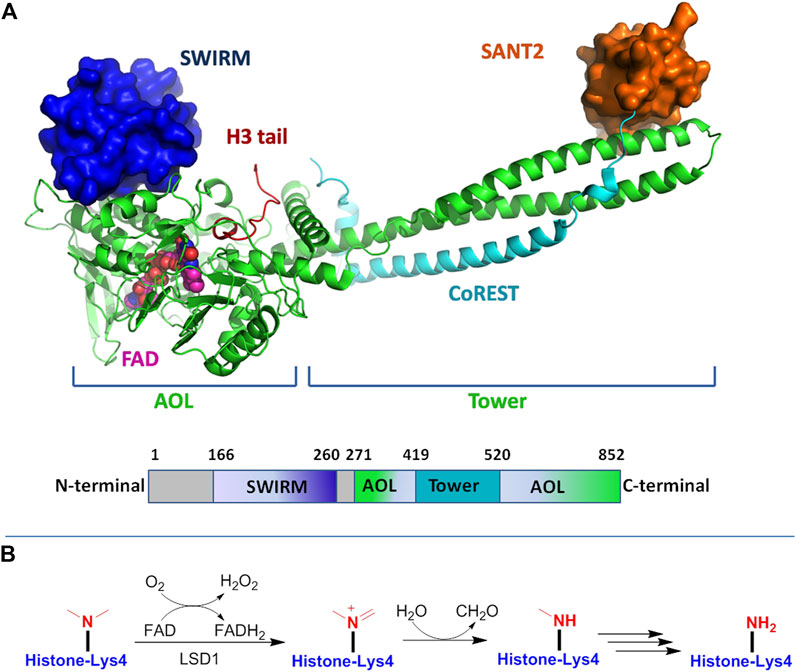
FIGURE 2. (A) Structure of LSD1 protein: up for the 3D model and down for the schematic illustration of LSD1 domains. Figures were created using PyMol (http://pymol.sourceforge.net); (B) FAD-dependent demethylation mechanism of mono-methylated lysine residue by LSD1.
The SWIRM domain interacts tightly with AOL domain to form a core structure, which covalently binds to FAD responsible for the enzymatic catalysis. The Tower domain protrudes from the AOL region, offering a binding platform to interaction with the LSD1 partners and CoREST. With FAD as coenzyme, LSD1 specifically catalyzes the demethylation process of mono- and di-methylated histone H3 lysine 4 (Shi et al., 2004). As shown in Figure 2B, in this catalytic reaction, the methyl lysine residue is oxidized by FAD to producing an imine intermediate, which is subsequently subjected to hydrolysis reaction to form a demethylated lysine and a formaldehyde molecule, while the reduced FAD is recovered by reoxidation. Due to the reaction requiring a hydride transfer or sing electron for the imine formation, LSD1 only enables the demethylation of mono-/di-methylated lysine, but not tri-methylated ones (Forneris et al., 2005; Chen et al., 2006; Forneris et al., 2008). LSD1 can also remove mono- and dimethylated K9 of histone 3 by forming complexes with androgen receptor or other nuclear receptors (Metzger et al., 2005; Garcia-Bassets et al., 2007). On the other hand, the demethylation activity of LSD1 may be negatively impacted by other histone modifications such as H3K9 deacetylation or H3S10 phosphorylation (Perillo et al., 2008; Binda et al., 2010). In addition to histone substrates, LSD1 could also demethylate nonhistone proteins such as p53, E2F transcription factor, DNA methyltransferases (DNMTs) and further modulate their activity (Ooi et al., 2007; Ciccone et al., 2009).
Moreover, histone modification is usually associated with the activation or repression of gene transcription. LSD1 plays a repressive role in gene transcription through the interaction with CoREST, nucleosome remodeling and other deacetylase complex, and LSD1 is also capable of promoting transcriptional activation by binding to androgen receptor or estrogen receptor, thereby regulating gene expression (Figure 3) (Metzger et al., 2005). Besides, LSD1 is recruited by zinc finger transcription factors into repressive complexes to interact with RCOR and HDAC1/2 to demethylate H3K4me1/2 marks associated with the active transcription state (Barski et al., 2007; Upadhyay et al., 2014). By regulating the expression of target genes, LSD1 is closely related to tumorigenesis, pluripotent stem cells, and neurodegenerative diseases (Maes et al., 2015; Stefano et al., 2016).

FIGURE 3. Dual functions of LSD1 as transcriptional repressor or activator by binding distinct cofactors.
Neuroinflammation is a defensive response of the immune system of the brain, protecting the CNS against infectious insults, injury and disease (Spencer et al., 2012; Zhang and Jiang, 2015). Current studies show that a significant portion of the occurrence and progression of AD is attributed to neuroinflammation (Culibrk and Hahn, 2020). Neuroinflammation is not a resultant role driven by emerging senile plaques and neurofibrillar tangles, but promotes the pathogenesis of AD as to the plaques and tangles themselves (Zhang et al., 2013). An analysis of clinical manifestation including mild cognitive impairments prior to dementia stage further supports the involvement of neuroinflammation in AD pathogenesis (Heneka et al., 2015). Neuroinflammation is a chronic response, during which microglia might have been initiated to cause a rapid switch to a damaging phenotype (Colton et al., 2006). Thus, neuroinflammation is usually found to occur at the early stage of AD progress prior to the presence of cognitive deterioration (Fu et al., 2019). In AD, microglia are able to bind to soluble Aβ oligomers and Aβ fibrils via cell surface receptors such as SCARA1, CD36, CD14 as well as Toll-like receptors, and these receptor engagement could induce the production of proinflammatory cytokines to partly contribute the inflammatory response (Liu et al., 2005; Stewart et al., 2010). Besides, accumulation of Aβ and neuronal debris could also activate inflammatory factors to stimulate chronic inflammation (Streit et al., 2009; Krabbe et al., 2013).
Emerging evidences have showed that histone modifications are closely associated with chromatin remodeling and gene transcription, and further play critical roles in regulating inflammation associated diseases (Sun et al., 2019; Surace and Hedrich, 2019). Kim et al. (2018) identified PKCα-LSD1-NF-κB axis as a new epigenetic pathway to activate and amplify the inflammation reaction, and pharmacological inhibition of LSD1 significantly reduced the excessive systemic inflammatory response. Zhuo et al. (2019) reported that inhibition of LSD1 could ameliorate Ox-LDL-stimulated NLRP3 activation and inflammation by promoting autophagy via PI3K/Akt/mTOR signaling pathway. In addition, LSD1 inactivation was also found to reduce inflammatory cell recruitment to tissues through mediating NF-κB signaling cascade and suppressing the production of cytokines (Wang et al., 2009). Lorenzo et al. (2021) found that the chromatin remodeling factor HMG20A mainly expressed in hypothalamic astrocytes, could affect the CNS development by inhibiting LSD1-CoREST complex, and inactivation of HMG20A may reduce neuroinflammation.
The current findings support neuroinflammation as a causal role in the pathogenesis of AD, however the identification of inflammatory biomarkers has not yet been established as a valuable indication for diagnosis of AD, and it is supposed to be the focus of future studies (Heneka et al., 2015).
Autophagy is a natural and evolutionarily conserved catabolic process through a lysosome dependent regulated mechanism, which degrades the damaged and dysfunctional components and protein aggregates (Klionsky, 2008; Vijayakumar and Cho, 2019). Autophagy plays an important cytoprotective role in responding to the environmental change, and has a tight connection with cell survival and cellular homeostasis (Yu et al., 2018). The autophagy deficiency is closely associated with multiple kinds of human diseases such as caner and CNS diseases, and the regulation of autophagy has emerged as a potential strategy for treating these diseases (Djajadikerta et al., 2020).
In AD, autophagy is a key modulator of Aβ production and clearance. The aberrant metabolism of Aβ peptides and tau proteins is found to be affected by autophagy, which can cause the accumulation and aggregation of toxic proteins to aggravate AD pathogenesis (Nilsson et al., 2013; Nilsson and Sarido, 2014). Autophagy is usually considered as a cytoplasmic event, increasing findings indicate that epigenetic regulation is also linked to the autophagy process. In particular, histone methylations have crucial roles in regulating autophagy at multiple levels, and is involved in the transcriptional control of multiple downstream effectors (Fullgrabe et al., 2014). Multiple lines of studies suggest that LSD1 is a negative regulator of genes required for autophagic process, and that LSD1 depletion or inhibition is able to induce autophagy in a range of human cell lines (Byun et al., 2017; Chao et al., 2017). Periz et al. (2015) found that LSD1 with ubiquitination factor E4B (UBE4B) exerted synergistic effect to increase the clearance of misfolded proteins through the control of the p53-dependent transcription program. Moreover, Ambrosio et al. (2017) reported that LSD1 inhibition could elevate the expression of SENE2 and consequent inhibition of mTORC1 to trigger the induction of autophagy.
Neurotransmitters are a class of signaling molecules secreted by neurons to impact cells across synapses, and are essential for signal transduction within the whole nervous system (Erum et al., 2019; Stolero et al., 2021; Lourenco et al., 2021). Neurotransmitter is released from presynaptic neuron and bound to postsynaptic receptors such as G-protein coupled receptors or ligand-gated ion channels, subsequently priming the intracellular signaling pathway (Predescu et al., 2019; Boczek et al., 2021). Neurotransmitters including glutamate, GABA, acetylcholine, glycine and norepinephrine, play key roles in neuronal communications to maintain the balance between excitatory and inhibitory synaptic transmission, connecting with many human diseases and mental disorders (Erum et al., 2019; Kandimalla and Reddy, 2017; Reddy, 2017). For instance, in the CNS, glutamate with its receptors regulates the majority of excitatory neurotransmission and synaptic plasticity (Riedel, 2003). The dysfunction of the normal signaling pathway mediated by glutamate is closely involved in a variety of neuropathological disorders (Bleich et al., 2003).
LSD1 with its paralog LSD2 belongs to the FAD-dependent amine oxidase enzyme family comprising monoamine oxidases, which enable the metabolization of norepinephrine, related neurotransmitters as well as polyamine oxidases that metabolize spermidine and other alkylamines (Hou et al., 2010; Edmondson et al., 2004). LSD1 structure contains an amine oxidase domain which recognizes a class of enzymes that may catalyze the FAD-dependent oxidation of amine substrates ranging from amino acids to aromatic neurotransmitters (Forneris et al., 2009). Compared to WT mice, Lim et al. (2017) found that the LSD1 knock-in mice has higher probability of presynaptic neurotransmitter release, without differences in amplitude between the genotypes. Moreover, Longaretti et al. (2020a) discovered that glutamatergic transmission could regulate the ratio of LSD1 and neuroLSD1 associated with a prototypic negative feedback mechanism, in which the activation of N-methyl-D-aspartate receptor induces downregulation of neurolLSD1 to further limit postsynaptic glutamate responses. In addition, they also found that the splicing mechanism in chronic social defeat stress, transiently affected the amount of LSD1/neuroLSD1 to undergo desensitization (Longaretti et al., 2020b).
Ferroptosis is a newly identified mode of cell death dependent on iron and has distinct morphological and biochemical features different from cell apoptosis, necrosis, autophagy, etc., (Dixon et al., 2012; Xie et al., 2016; Weiland et al., 2019). Ferroptosis is initiated by the imbalance of redox homeostasis due to aberrant increase of iron-dependent ROS production, leading to the accumulation of iron ions, increase of lipid peroxidation products and depletion of glutathione (Dixon et al., 2012; Kraft et al., 2020; Chen et al., 2021). The accumulation of lipid peroxides is proposed to be one of the hallmark events in the initiation and progression of ferroptosis (Cheng and Li, 2007). Emerging studies have shown that ferroptosis is involved with the pathophysiological mechanism of cognitive dysfunction such as vascular dementia and senile dementia (Yan and Zhang, 2019; Derry et al., 2020). In brain, ferroptosis induced by depletion of neuronal glutathione peroxidease 4 (GPX4), could lead to cognitive impairment, while ferroptosis inhibition alleviates cognitive disorder (Hambright et al., 2017). A number of evidences have demonstrated that ferroptosis is closely associated with AD pathogenesis and development. In the AD brain, the accumulation of pathological neuronal iron largely contributes to the oxidative damage through the overproduction of free radicals (Smith et al., 1997). In addition, ferroptosis also mediates the cytotoxicity of Aβ peptides, and the slow accumulation of Aβ peptides can cause the prolonged ferroptosis in neurons to induce additional toxicity (Huang et al., 2020). Although increasing findings have revealed the essential role of ferroptosis in the pathogenesis of AD, the exact mechanism of action of ferroptosis in AD remains unclear (Chen et al., 2021).
Feng et al. found that LSD1 could promote both ferroptosis and oxidative stress through the activation of TLR4/NOX4 pathway, and LSD1 inactivation could alleviate ferroptosis (Feng et al., 2022). In addition, Li et al. (2015) reported LSD1 inhibition could elevate the expression of antioxidant gene SLC7A11, an important ferroptosis regulator, which is crucial for GSH synthesis/release as well as confer potent GSH-dependent neuroprotective effects. The transcriptional factor Nrf2 is a crucial regulator factor of ferroptosis process, and the activation of NRF2-LSD1 complex can regulate downstream antioxidant gene expression (Lin et al., 2021). Overall, there is currently little research on molecular mechanism of LSD1-mediated ferroptosis, especially in AD field, and it is worthy of further investigation.
Hyperphosphorlation and aggregation of tau protein in brain are a well-known pathological hallmark of AD, which is associated with synaptic loss, neuroinflammation, and neuronal death. The aberrant tau phosphorylation disrupts its binding to microtubules to promote microtubule accumulation, leading to its aggregation into neurofibrillary tangles mostly known as a primary marker of AD (Cho and Johnson, 2003). Although a great progress has been made in the understanding of AD pathogenesis, the precise mechanism of how tau protein contributes to neurodegeneration remains poorly understood (Engstrom et al., 2020).
LSD1 is mainly localized in the nucleus, while pharmacological intervention could cause a translocation of LSD1 from the nucleus to the cytoplasm (Yabuta and Shidoji, 2019). In tau pathology, Christopher et al. (2017) found LSD1 is localized to tau aggregates and sequestered in the cytoplasm, and proposed that the reduction of LSD1 in nucleus impairs the survival of hippocampal and cortical. Engstrom et al. (2020) discovered that pathological tau protein excluded LSD1 from the nucleus in neurons to cause neuronal cell death, since nuclear LSD1 is required for neuronal survival. In addition, they also demonstrated that the reduction of LSD1 could accelerate the tauopathy phenotype, while the extra supply of LSD1 slowed down the process of neuronal cell death, indicating LSD1 as a critical mediator of pathological tau-induced neurodegeneration (Engstrom et al., 2020). Thus LSD1 could be a potential therapeutic strategy for AD treatment.
LSD1 plays an essential regulatory role in neural stem cell proliferation and neuronal development, and is continuously required for neuronal progenitor cell maintenance and terminal differentiation (Sun et al., 2010; Zibetti et al., 2010; Zhang et al., 2014). LSD1 is recruited by orphan nuclear receptor TLX, essential for neuronal stem cell, to the promoters of its target genes for transcriptional repression, thereby promoting neural stem cell growth (Yokoyama et al., 2008; Sun et al., 2010). In addition, Zhang et al. (2014) reported that LSD1 is essential for neuronal progenitor cell maintenance during cortical development, through modulating H3K4 methylation of the LSD1-binding site downstream of atrophin 1. Genetic or pharmacological intervention of LSD1 could cause the elevated methylation of this binding site as well as reduction of atrophin 1, thereby leading to neuronal progenitor cell differentiation (Zhang et al., 2014). Moreover, LSD1 degradation mediated by JADE-2, an E3 ubiquitin ligase, has a unique role in regulating neurogenesis and neural differentiation (Han et al., 2014). Christopher et al. (2017) reported that inducible depletion of LSD1 of adult mice mainly occurs in neurons, suggesting that neurons are possibly more vulnerable to LSD1 expression. They also found that LSD1 inhibition caused by pathological tau aggregates in aging neurons of AD could lead to neuronal cell death and dementia. Laurent et al. (2015) identified LSD1+8a, an isoform of LSD1 in neuronal cells, regulates H3K9me2 methylation in collaboration with supervillin as a cofactor, playing a unique role in neurodegeneration. Another isoform of LSD1, LSDn enables the regulation of H4K20 methylation relating to gene transcription for neuronal function, while depletion of LSDn could cause severe cognitive impairments (Wang et al., 2015).
As aforementioned, many genetic evidences reveal that LSD1 has an important protective role in neurodegeneration. In contrast with genetic depletion of LSD1, pharmacological inhibition of LSD1 with specific LSD1 inhibitors produces completely different outcomes, showing a promising treatment potential in CNS diseases. Such that huge discrepancy mainly lies in the fact genetic intervention and a small molecule inhibitor can perturb a protein’s activity in different ways to further result in different or even opposite results about the protein’s biological function (Knight and Shokat, 2007). In most cases, intervention with small molecules do not affect the expression of the protein of interest but catalytic activity of the protein, while genetic intervention such as gene knockout could lead to complete loss of the protein to further cause protein complex dissociation to cause the release of cofactors to activate other signal pathways (Knight et al., 2007). In terms of LSD1, a number of findings have shown that LSD1 depletion can cause embryonic lethality, and non-specific LSD1 inhibitors may lead to severe hematological toxicity due to the dissociation of LSD1-contained complex (Foster et al., 2010; Sprussel et al., 2012). For example, LSD1 in complex with grow factor-independent 1B (GFI1B), is reported to be responsible for hematopoietic cell development and differentiation, and LSD1 knockdown causes derepression of GFI1B-mediated genes to lead to hematopoietic stem cell expansion and abnormal hematological differentiation (Saleque et al., 2007; Sprussel et al., 2012; Ishikawa et al., 2017).
On the other hand, tranylcypromine (TCP)-derived irreversible LSD1 inhibitors have been reported to exert hematological toxicities, probably due to the formation of TCP-FAD adduct to disrupt the interaction between LSD1 and cofactors (Maiques-Diaz et al., 2018; Matsuda et al., 2019). To address this issue, the researchers of Takeda Pharmaceutical Company have reported that their TCP-like LSD1 inhibitors such as TAK-448 and TAK-418 could normalize the aberrant epigenetic control of gene expression and efficiently improve cognitive deficits in animal models of CNS disorders, without causing hematological toxicity in vivo (Matsuda et al., 2019; Baba et al., 2021; Baba et al., 2022). Further investigation showed that both LSD1 inhibitors mechanistically formed unique compact formly FAD adduct (Figure 4), which posed the minimal impacts on LSD1-GFI1B complex. ORY-2001 of Oryzon Genomics is a brain penetrant LSD1 inhibitor with dual LSD1/MAO-B inhibition, and it is able to inactivate LSD1 in brain to improve memory deficit, behavior abnormalities and social obstacles of SMAP8 mice model (Maes et al., 2020). Overall, the alternation of the interactions of LSD1 with its cofactors is a highly interesting issue to address in terms of CNS field, and the role of LSD1 in different tissues and cell context also needs to be further elucidated (Maiques-Diaz and Somerrvaille, 2016).
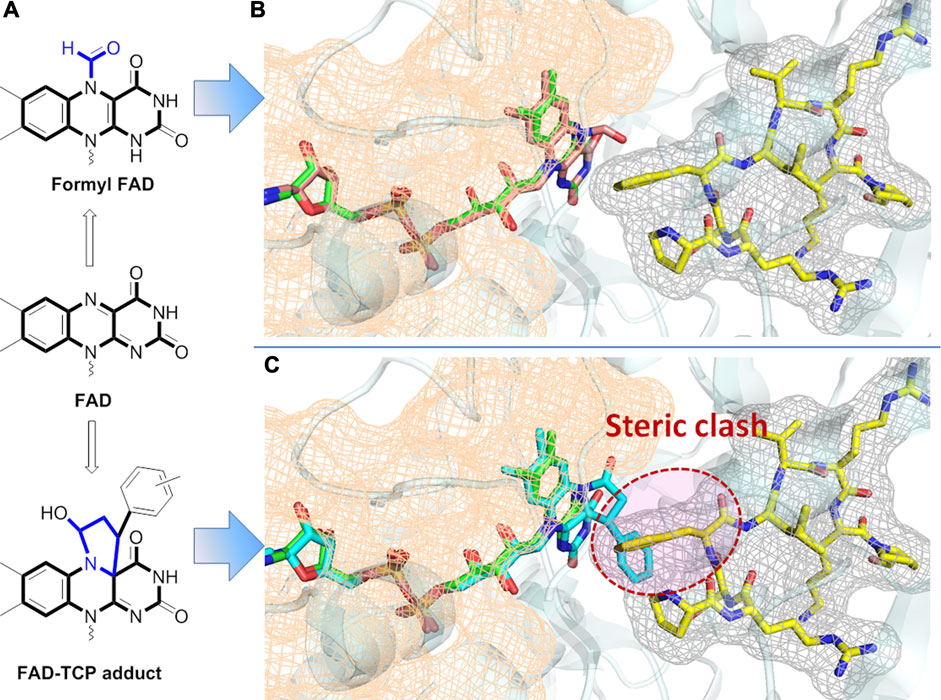
FIGURE 4. (A) Chemical structures of adducts of FAD with TCP in two different ways; (B) Superimposed structure of formyl FAD in LSD1 with GFI1B peptide (PDB: 7E0G); (C) Superimposed structure of FAD-TCP in LSD1 with GFI1B peptide (PDB:2Z5U). FAD is presented as stick model in green, formyl FAD in brown (B) and FAD-TCP adduct in cyan (C).
To date, many findings have shed light on the importance of LSD1 in various human diseases such as cancers and neurodegeneration, and targeting LSD1 has already become a powerful and promising therapeutic approach for such diseases. A lot of efforts have been made towards the development of potent and specific pharmacological inhibitors of LSD1 over the past decade.
TCP is the first identified LSD1 inhibitor with weak inhibitory activity and poor specificity, which abrogates the catalytic activity by forming covalent adduct with the coenzyme FAD (Schmidt and McCafferty, 2007; Binada et al., 2010). Despite its insufficient activity, TCP has opened a path for the development of mechanism-based irreversible LSD1 inhibitors, making TCP an attractive chemical starting point for designing such inhibitors (Yang et al., 2015). Up to now, a large number of TCP-derived LSD1inhibitors with improved affinity and specificity, have been well developed, and many of which have laid a solid foundation for in-depth biological studies on the LSD1 inhibition in various diseases (Mould et al., 2015). Currently, TCP-derived LSD1 inhibitors such as ORY-1001, IMG7289, GSK2879552 and INCB059872 (Figure 5A) alone or in combination with other agents such as all-trans retinoic acid (ATRA), cytarabine or azacitidine, etc., are undergoing clinical assessments mainly for treatment of leukemias and solid tumors (Lee et al., 2016; Somervaille et al., 2016; Zheng et al., 2016). Apart from irreversible LSD1 inhibitors, the development of reversible inhibitors has also greatly stimulated interest in this field, albeit with more challenging (Mould et al., 2015). Compared with irreversible inhibitors, the development of reversible inhibitors lags far behind, and there are currently only two compounds SP-2577 and CC-90011 in clinical trials for cancer treatment (Figure 5B).
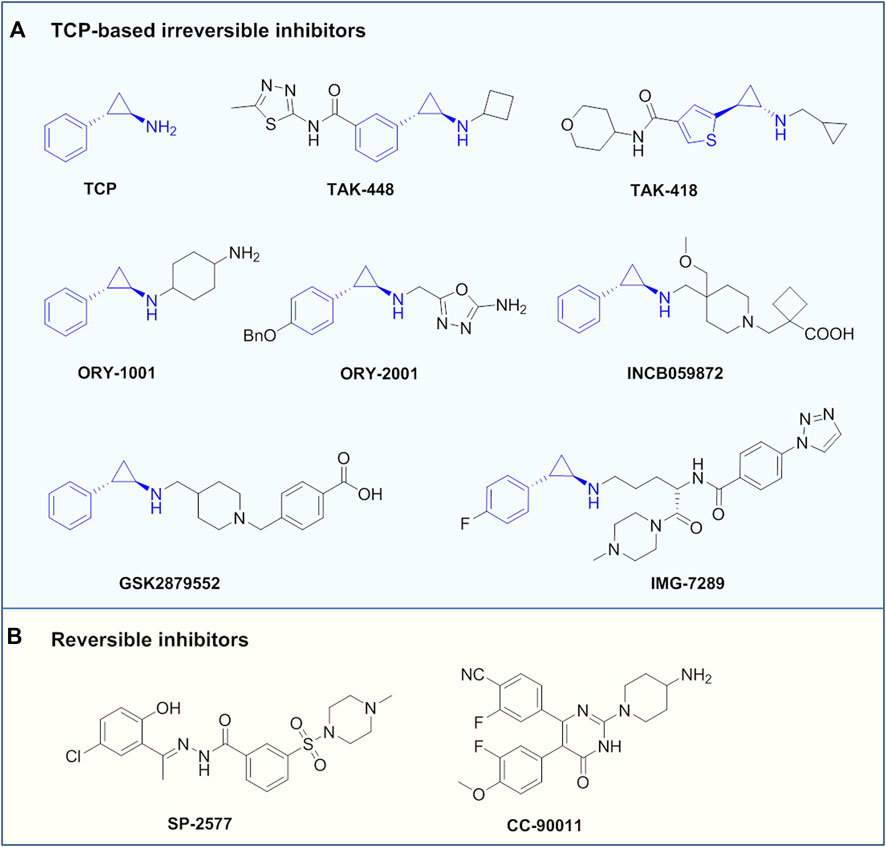
FIGURE 5. Representative LSD1 inhibitors under clinical assessment.
In addition, TAK-418 and TAK-448 (Figure 5A) are highly enzyme-specific LSD1 inhibitors without impact on LSD1 complex, and both compounds are reported to show therapeutic potentials for cancer, AD, autism symptom disorder and Kabuki syndrome et al. (Baba et al., 2021; Yin et al., 2021; Baba et al., 2022). ORY-2001 (also known as Vafidemstat, Figure 5A) is being evaluated in clinical assessments for treating CNS diseases such as AD and borderline personality disorder. ORY-2001 is a highly potent and orally active dual LSD1/MAO-B inhibitor developed by Oryzon Genomics SA, of note, it is so far the only LSD1 inhibitor currently under clinical assessment for treating AD. Preclinical investigation of ORY-2001 showed that it could mitigate cognitive impairment and reduce neuroinflammation, as well as normalize genes related to cognitive function, neuroplasticity and memory (Maes et al., 2020). Of note is that the in vivo pharmacological activity of ORY-2001 was mainly due to its inhibition of LSD1, while MAO-B inhibition had a minor contribution (Maes et al., 2020). In animal models, ORY-2001 could restore memory deficit and reduce aggression, as well as improve social behavior. The first-in-human clinical assessment demonstrated that ORY-2001 was safe and well tolerated, and possessed good PK/PD profile with rapid oral absorption, long half-life of 22 h, moderate systemic accumulation (mean AUC ration <2) after 5 days of administration. In particular, ORY-2001 did not cause apparent hematological toxicity, and transient reversible thrombocytopenia was observed in the treatment at 4 mg (Fang et al., 2019). ORY-2001 is currently in phaseⅡclinical trial (NCT03867253) to test the safety and preliminary efficacy in mild to moderate AD patients, and part of the clinical results demonstrated that ORY-2001 significantly reduced neuroinflammation after 6 and 12 months of treatment (Cummings et al., 2022). A summary of LSD1 inhibitors in clinical trails is provided in Table 1.
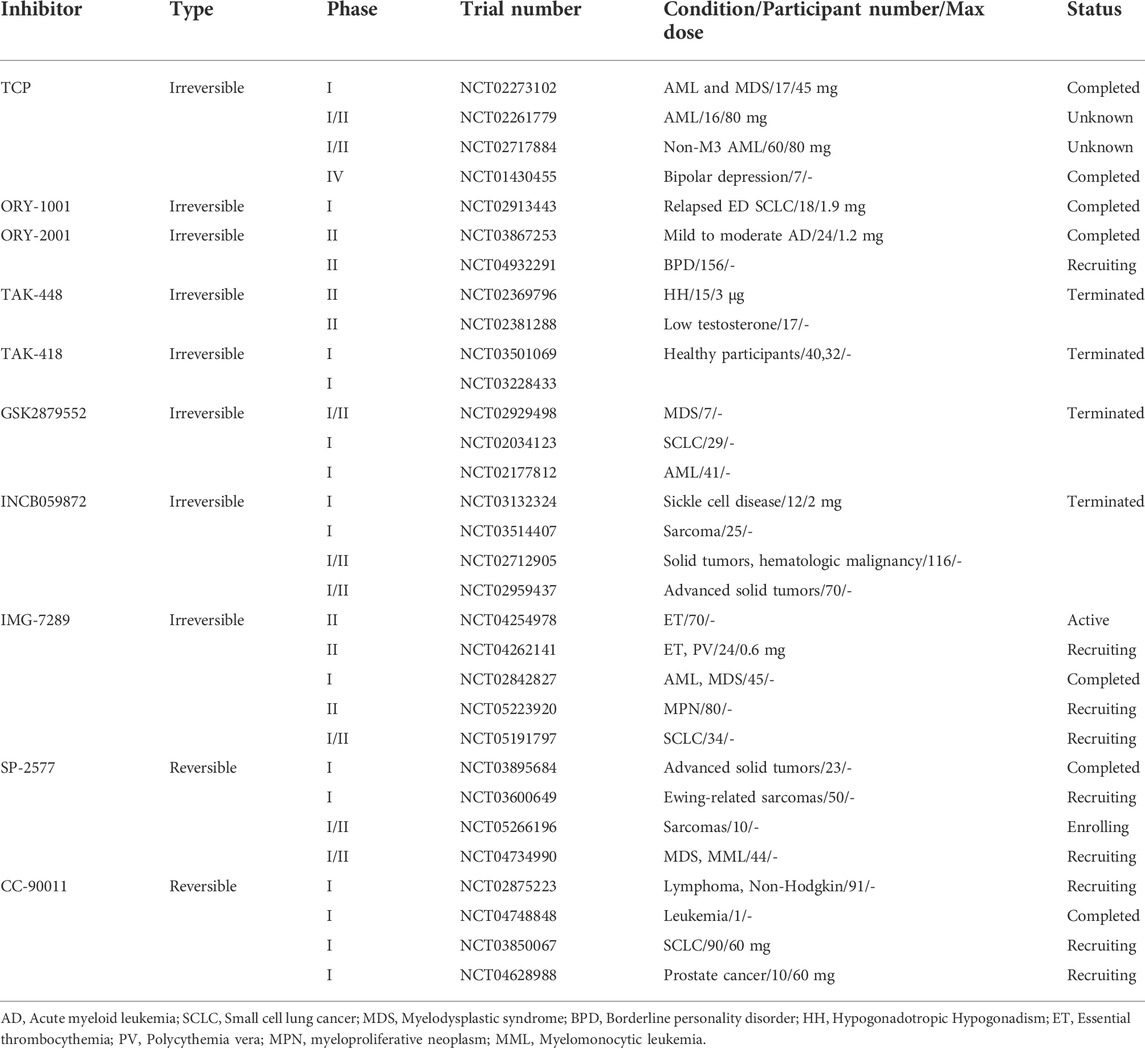
TABLE 1. LSD1 inhibitors in clinical trials (ClinicalTrails.gov in May 2022).
AD represents a complex and inexorable neurodegenerative disease. Although considerable efforts have been made to treat AD, poor progress has been achieved regarding to new therapeutics over the past decade (Long and Holtzman, 2019; Yiannopoulou and Papagergiou, 2020). LSD1 is an important epigenetic regulator and has a versatile role in mammalian biology and diseases. LSD1-mediated histone demethylation is closely associated with the pathogenesis of neurodegenerative diseases. In AD, LSD1 plays an important role in neural stem cell proliferation, neuronal differentiation and neuroprotection, and management of LSD1 provides an additional opportunity for developing therapeutic approaches in AD. It is important to note that genetic depletion of LSD1 may cause the pathogenesis of neurodegenerative disease, while enzymatic activity-specific inhibition of LSD1 demonstrates potential therapeutics for neurodegenerative diseases. These opposite results could be attributed to enzyme activity and complex function affected in different ways (Knight et al., 2007). Specific inhibitors of LSD1 form unique adduct with cofactor FAD to reduce its catalytic activity without affecting enzyme expression, while LSD1 depletion by genetic methods disrupt the interaction of LSD1 with cofactors to further activate other signal pathways.
To date, a fair number of LSD1 inhibitors have been developed, and some of them are currently undergoing clinical evaluation for cancers and CNS diseases. Despite remarkable advances of irreversible TCP-based inhibitors, there is only ORY-2001 in clinical trial for AD therapy. Mechanistically, the chemical process of LSD1 inhibition by TCP involves a single electron reduction mechanism via a homolytic cleavage of cyclopropyl ring to generate different FAD adducts directly impacting LSD1 complex (Yang et al., 2007; Fang et al., 2019). However, the exact mechanism of ring cleavage of TCP to form FAD adducts needs to be further studied, which is important to design potent and specific inhibitors without disrupting the interaction of LSD1 with cofactors like GFI1B, thereby reducing blood toxicity (Maiques-Diaz and Somervaille, 2016; Matsuda et al., 2019). Besides, the mechanism of action of TCP-like LSD1 inhibitors should be further elucidated in order to develop novel chemical entities possessing increased affinity and enhanced specificity. In addition, the high-resolution crystal structure of LSD1 protein with ORY-2001 is believed to provide valuable guideline for developing enzyme activity-specific LSD1 inhibitors for AD treatment.
To sum up, we propose the following general directions to consider for AD research: A) Investigations of reversible LSD1 inhibitors in AD treatment will be highly interesting to provide new pharmacological phenotypes for AD treatment; B) The way of TCP to form FAD adduct should be further elucidated to develop new LSD1 inhibitors with high affinity and cell-context specificity; C) Dual/multiple-targeted LSD1 inhibitors is of interest to combat complicated disease like AD, since this strategy has been largely developed for cancer treatment; D) Genetic depletion of LSD1 yields completely different results in AD compared with that of pharmacological intervention, and thus the research model should be carefully evaluated.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by Joint Construction Project of Medical Science and Technology Research Plan of Henan Province, China (LHGJ20210614); Scientific and Technological Project of Henan Province (212102310314); Joint Research Fund of Science and Technology R&D Plan of Henan Province (222301420068); Program for Science&Technology Innovation Talents in Universities of Henan Province (No. 23HASTIT044); Henan Engineering Research Center for Prevention and Treatment of Major Chronic Diseases with Chinese Medicine.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ambrosio, S., Sacca, C. D., Amente, S., Paladino, S., Lania, L., and Majello, B. (2017). Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 36 (48), 6701–6711. doi:10.1038/onc.2017.267
Baba, R., Matsuda, S., Arakawa, Y., Yamada, R., Suzuki, N., Ando, T., et al. (2021). LSD1 enzyme inhibitor TAK-418 unlocks aberrant epigenetic machinery and improves autism symptoms in neurodevelopmental disorder models. Sci. Adv. 7 (11), eaba1187. doi:10.1126/sciadv.aba1187
Baba, R., Matsuda, S., Maeda, R., Murakami, K., Yamamoto, Y., Nakatani, A., et al. (2022). Investigating the therapeutic potential of LSD1 enzyme activity-specific inhibition by TAK-418 for social and memory deficits in rodent disease models. ACS Chem. Neurosci. 13 (3), 313–321. doi:10.1021/acschemneuro.1c00713
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21 (3), 381–395. doi:10.1038/cr.2011.22
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129 (4), 823–837. doi:10.1016/j.cell.2007.05.009
Binda, C., Valente, S., Romanenghi, M., Pilotto, S., Cirilli, R., Karytinos, A., et al. (2010). Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 132 (19), 6827–6833. doi:10.1021/ja101557k
Bleich, S., Romer, K., Wiltfang, J., and Kornhuber, J. (2003). Glutamate and the glutamate receptor system: A target for drug action. Int. J. Geriatr. Psychiatry 18 (1), S33–S40. doi:10.1002/gps.933
Boczek, T., Mackiewicz, J., Sobolczyk, M., Wawrzyniak, J., Lisek, M., Ferenc, B., et al. (2021). The role of G protein-coupled receptors (GPCRs) and calcium signaling in schizophrenia. Focus on GPCRs activated by neurotransmitters and chemokines. Cells 10 (5), 1228. doi:10.3390/cells10051228
Byun, S., Kim, Y. C., Zhang, Y., Kong, B., Guo, G., Sadoshima, J., et al. (2017). A postprandial FGF19-SHP-LSD1 regulatory Axis mediates epigenetic repression of hepatic autophagy. EMBO J. 36 (12), 1755–1769. doi:10.15252/embj.201695500
Chao, A., Lin, C. Y., Chao, A. N., Tsai, C. L., Chen, M. Y., Lee, L. Y., et al. (2017). Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget 8 (43), 74434–74450. doi:10.18632/oncotarget.20158
Chen, K., Jiang, X., Wu, M., Cao, X., Bao, W., and Zhu, L. Q. (2021a). Ferroptosis, a potential therapeutic target in Alzheimer's disease. Front. Cell Dev. Biol. 9, 704298. doi:10.3389/fcell.2021.704298
Chen, X., Comish, P. B., Tang, D., and Kang, R. (2021b). Characteristics and biomarkers of ferroptosis. Front. Cell Dev. Biol. 9, 637162. doi:10.3389/fcell.2021.637162
Chen, Y., Yang, Y., Wang, F., Wan, K., Yamane, K., Zhang, Y., et al. (2006). Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc. Natl. Acad. Sci. U. S. A. 103 (38), 13956–13961. doi:10.1073/pnas.0606381103
Cheng, Z., and Li, Y. (2007). What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: An update. Chem. Rev. 107 (3), 748–766. doi:10.1021/cr040077w
Cho, J. H., and Johnson, G. V. (2003). Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J. Biol. Chem. 278 (1), 187–193. doi:10.1074/jbc.M206236200
Christopher, M. A., Myrick, D. A., Barwick, B. G., Engstrom, A. K., Porter-Stransky, K. A., Boss, J. M., et al. (2017). LSD1 protects against hippocampal and cortical neurodegeneration. Nat. Commun. 8 (1), 805. doi:10.1038/s41467-017-00922-9
Ciccone, D. N., Su, H., Hevi, S., Gay, F., Lei, H., Bajko, J., et al. (2009). KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461 (7262), 415–418. doi:10.1038/nature08315
Collins, B. E., Greer, C. B., Coleman, B. C., and Sweatt, J. D. (2019). Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin 12 (1), 7. doi:10.1186/s13072-018-0251-8
Colton, C. A., Mott, R. T., Sharpe, H., Xu, Q., Van Nostrand, W. E., and Vitek, M. P. (2006). Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J. Neuroinflammation 3, 27. doi:10.1186/1742-2094-3-27
Coppede, F. (2021). Epigenetic regulation in Alzheimer's disease: Is it a potential therapeutic target? Expert Opin. Ther. Targets 25 (4), 283–298. doi:10.1080/14728222.2021.1916469
Culibrk, R. A., and Hahn, M. S. (2020). The role of chronic inflammatory bone and Joint disorders in the pathogenesis and progression of Alzheimer's disease. Front. Aging Neurosci. 12, 583884. doi:10.3389/fnagi.2020.583884
Cummings, J., Aisen, P. S., DuBois, B., Frolich, L., Jack, C. R., Jones, R. W., et al. (2016). Drug development in Alzheimer's disease: The path to 2025. Alzheimers Res. Ther. 8, 39. doi:10.1186/s13195-016-0207-9
Cummings, J., Lee, G., Nahed, P., Kambar, M., Zhong, K., Fonseca, J., et al. (2022). Alzheimer's disease drug development pipeline: 2022. Alzheimers Dement. 8 (1), e12295. doi:10.1002/trc2.12295
Derry, P. J., Hegde, M. L., Jackson, G. R., Kayed, R., Tour, J. M., Tsai, A. L., et al. (2020). Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer's disease from a ferroptosis perspective. Prog. Neurobiol. 184, 101716. doi:10.1016/j.pneurobio.2019.101716
Di Stefano, B., Collombet, S., Jakobsen, J. S., Wierer, M., Sardina, J. L., Lackner, A., et al. (2016a). C/EBP alpha creates elite cells for iPSC reprogramming by upregulating Klf4 and increasing the levels of Lsd1 and Brd4. Nat. Cell Biol. 18 (4), 371–381. doi:10.1038/ncb3326
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Djajadikerta, A., Keshri, S., Pavel, M., Prestil, R., Ryan, L., and Rubinsztein, D. C. (2020). Autophagy induction as a therapeutic strategy for neurodegenerative diseases. J. Mol. Biol. 432 (8), 2799–2821. doi:10.1016/j.jmb.2019.12.035
Edmondson, D. E., Mattevi, A., Binda, C., Li, M., and Hubálek, F. (2004). Structure and mechanism of monoamine oxidase. Curr. Med. Chem. 11, 1983–1993. doi:10.2174/0929867043364784
Engstrom, A. K., Walker, A. C., Moudgal, R. A., Myrick, D. A., Kyle, S. M., Bai, Y., et al. (2020). The inhibition of LSD1 via sequestration contributes to tau-mediated neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 117 (46), 29133–29143. doi:10.1073/pnas.2013552117
Fang, Y., Liao, G., and Yu, B. (2019). LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 12 (1), 129. doi:10.1186/s13045-019-0811-9
Feng, R., Xiong, Y., Lei, Y., Huang, Q., Liu, H., Zhao, X., et al. (2022). Lysine-specific demethylase 1 aggravated oxidative stress and ferroptosis induced by renal ischemia and reperfusion injury through activation of TLR4/NOX4 pathway in mice. J. Cell. Mol. Med. 26, 4254–4267. doi:10.1111/jcmm.17444
Fischer, A., Sananbenesi, F., Wang, X., Dobbin, M., and Tsai, L. H. (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447 (7141), 178–182. doi:10.1038/nature05772
Forneris, F., Battaglioli, E., Mattevi, A., and Binda, C. (2009). New roles of flavoproteins in molecular cell biology: Histone demethylase LSD1 and chromatin. FEBS J. 276 (16), 4304–4312. doi:10.1111/j.1742-4658.2009.07142.x
Forneris, F., Binda, C., Battaglioli, E., and Mattevi, A. (2008). LSD1: Oxidative chemistry for multifaceted functions in chromatin regulation. Trends biochem. Sci. 33 (4), 181–189. doi:10.1016/j.tibs.2008.01.003
Forneris, F., Binda, C., Vanoni, M. A., Mattevi, A., and Battaglioli, E. (2005). Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 579 (10), 2203–2207. doi:10.1016/j.febslet.2005.03.015
Foster, C. T., Dovey, O. M., Lezina, L., Luo, J. L., Gant, T. W., Barlev, N., et al. (2010). Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol. Cell. Biol. 30 (20), 4851–4863. doi:10.1128/MCB.00521-10
Fu, W. Y., Wang, X., and Ip, N. Y. (2019). Targeting neuroinflammation as a therapeutic strategy for Alzheimer's disease: Mechanisms, drug candidates, and new opportunities. ACS Chem. Neurosci. 10 (2), 872–879. doi:10.1021/acschemneuro.8b00402
Fullgrabe, J., Klionsky, D. J., and Joseph, B. (2014). The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 15 (1), 65–74. doi:10.1038/nrm3716
Garcia-Bassets, I., Kwon, Y. S., Telese, F., Prefontaine, G. G., Hutt, K. R., Cheng, C. S., et al. (2007). Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 128 (3), 505–518. doi:10.1016/j.cell.2006.12.038
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi:10.1016/j.redox.2017.01.021
Han, X., Gui, B., Xiong, C., Zhao, L., Liang, J., Sun, L., et al. (2014). Destabilizing LSD1 by jade-2 promotes neurogenesis: An antibraking system in neural development. Mol. Cell 55 (3), 482–494. doi:10.1016/j.molcel.2014.06.006
Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet. Neurol. 14 (4), 388–405. doi:10.1016/s1474-4422(15)70016-5
Herre, M., and Korb, E. (2019). The chromatin landscape of neuronal plasticity. Curr. Opin. Neurobiol. 59, 79–86. doi:10.1016/j.conb.2019.04.006
Hou, H., and Yu, H. (2010). Structural insights into histone lysine demethylation. Curr. Opin. Struct. Biol. 20 (6), 739–748. doi:10.1016/j.sbi.2010.09.006
Huang, J., Sengupta, R., Espejo, A. B., Lee, M. G., Dorsey, J. A., Richter, M., et al. (2007). p53 is regulated by the lysine demethylase LSD1. Nature 449 (7158), 105–108. doi:10.1038/nature06092
Huang, L., McClatchy, D. B., Maher, P., Liang, Z., Diedrich, J. K., Soriano-Castell, D., et al. (2020). Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis. 11 (10), 828. doi:10.1038/s41419-020-03020-9
Ishikawa, Y., Gamo, K., Yabuki, M., Takagi, S., Toyoshima, K., Nakayama, K., et al. (2017). A novel LSD1 inhibitor T-3775440 disrupts GFI1B-containing complex leading to transdifferentiation and impaired growth of AML cells. Mol. Cancer Ther. 16 (2), 273–284. doi:10.1158/1535-7163.MCT-16-0471
Jingjing, W., Zhikai, W., Xingyi, Z., Peixuan, L., Yiwu, F., Xia, W., et al. (2021). Lysine-specific demethylase 1 (LSD1) serves as an potential epigenetic determinant to regulate inflammatory responses in mastitis. Int. Immunopharmacol. 91, 107324. doi:10.1016/j.intimp.2020.107324
Ju, Y., and Tam, K. Y. (2022). Pathological mechanisms and therapeutic strategies for Alzheimer's disease. Neural Regen. Res. 17 (3), 543–549. doi:10.4103/1673-5374.320970
Kandimalla, R., and Reddy, P. H. (2017). Therapeutics of neurotransmitters in Alzheimer's disease. J. Alzheimers Dis. 57 (4), 1049–1069. doi:10.3233/JAD-161118
Kaniskan, H. U., Martini, M. L., and Jin, J. (2018). Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 118 (3), 989–1068. doi:10.1021/acs.chemrev.6b00801
Kim, D., Nam, H. J., Lee, W., Yim, H. Y., Ahn, J. Y., Park, S. W., et al. (2018). PKCα-LSD1-NF-κB-Signaling cascade is crucial for epigenetic control of the inflammatory response. Mol. Cell 69 (3), 398–411. e396. doi:10.1016/j.molcel.2018.01.002
Klionsky, D. J. (2008). Autophagy revisited: A conversation with christian de Duve. Autophagy 4 (6), 740–743. doi:10.4161/auto.6398
Knight, Z. A., and Shokat, K. M. (2007). Chemical genetics: Where genetics and Pharmacology meet. Cell 128 (3), 425–430. doi:10.1016/j.cell.2007.01.021
Kontaki, H., and Talianidis, I. (2010). Lysine methylation regulates E2F1-induced cell death. Mol. Cell 39 (1), 152–160. doi:10.1016/j.molcel.2010.06.006
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with beta-amyloid deposition in mice with alzheimer-like pathology. PLoS One 8 (4), e60921. doi:10.1371/journal.pone.0060921
Kraft, V. A. N., Bezjian, C. T., Pfeiffer, S., Ringelstetter, L., Muller, C., Zandkarimi, F., et al. (2020). GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 6 (1), 41–53. doi:10.1021/acscentsci.9b01063
Laurent, B., Ruitu, L., Murn, J., Hempel, K., Ferrao, R., Xiang, Y., et al. (2015). A specific LSD1/kdm1a isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell 57 (6), 957–970. doi:10.1016/j.molcel.2015.01.010
Lee, S. H., Liu, X. M., Diamond, M., Dostalik, V., Favata, M., He, C., et al. (2016). TPL2 is an oncogenic driver in keratocanthoma and squamous cell carcinoma. Cancer Res. 76, 6712–6722. doi:10.1158/0008-5472.CAN-15-3274
Li, A., He, Y., Sun, S., Cai, C., and Li, H. (2015). Lysine-specific demethylase 1 inhibitors protect cochlear spiral ganglion neurons against cisplatin-induced damage. Neuroreport 26 (9), 539–547. doi:10.1097/WNR.0000000000000386
Lim, C. S., Nam, H. J., Lee, J., Kim, D., Choi, J. E., Kang, S. J., et al. (2017). PKCα-mediated phosphorylation of LSD1 is required for presynaptic plasticity and hippocampal learning and memory. Sci. Rep. 7 (1), 4912. doi:10.1038/s41598-017-05239-7
Lin, C. Y., Chang, C. B., Wu, R. C., Chao, A., Lee, Y. S., Tsai, C. N., et al. (2021). Glucose activates lysine-specific demethylase 1 through the KEAP1/p62 pathway. Antioxidants (Basel) 10 (12), 1898. doi:10.3390/antiox10121898
Lin, L., Liu, A., Li, H., Feng, J., and Yan, Z. (2019). Inhibition of histone methyltransferases EHMT1/2 reverses amyloid-beta-induced loss of AMPAR currents in human stem cell-derived cortical neurons. J. Alzheimers Dis. 70 (4), 1175–1185. doi:10.3233/JAD-190190
Liu, Y., Walter, S., Stagi, M., Cherny, D., Letiembre, M., Schulz-Schaeffer, W., et al. (2005). LPS receptor (CD14): A receptor for phagocytosis of Alzheimer's amyloid peptide. Brain 128, 1778–1789. doi:10.1093/brain/awh531
Lokken, A. A., and Zeleznik-Le, N. J. (2012). Breaking the LSD1/kdm1a addiction: Therapeutic targeting of the epigenetic modifier in AML. Cancer Cell 21 (4), 451–453. doi:10.1016/j.ccr.2012.03.027
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179 (2), 312–339. doi:10.1016/j.cell.2019.09.001
Longaretti, A., Forastieri, C., Gabaglio, M., Rubino, T., Battaglioli, E., and Rusconi, F. (2020a). Termination of acute stress response by the endocannabinoid system is regulated through lysine-specific demethylase 1-mediated transcriptional repression of 2-AG hydrolases ABHD6 and MAGL. J. Neurochem. 155 (1), 98–110. doi:10.1111/jnc.15000
Longaretti, A., Forastieri, C., Toffolo, E., Caffino, L., Locarno, A., Miseviciute, I., et al. (2020b). LSD1 is an environmental stress-sensitive negative modulator of the glutamatergic synapse. Neurobiol. Stress 13, 100280. doi:10.1016/j.ynstr.2020.100280
Lopez-Tobon, A., Trattaro, S., and Testa, G. (2020). The sociability spectrum: Evidence from reciprocal genetic copy number variations. Mol. Autism 11 (1), 50. doi:10.1186/s13229-020-00347-0
Lorenzo, P. I., Martin Vazquez, E., Lopez-Noriega, L., Fuente-Martin, E., Mellado-Gil, J. M., Franco, J. M., et al. (2021). The metabesity factor HMG20A potentiates astrocyte survival and reactive astrogliosis preserving neuronal integrity. Theranostics 11 (14), 6983–7004. doi:10.7150/thno.57237
Lourenco, M. V., Ribeiro, F. C., Santos, L. E., Beckman, D., Melo, H. M., Sudo, F. K., et al. (2021). Cerebrospinal fluid neurotransmitters, cytokines, and chemokines in Alzheimer's and lewy body diseases. J. Alzheimers Dis. 82 (3), 1067–1074. doi:10.3233/JAD-210147
Maes, T., Mascaro, C., Ortega, A., Lunardi, S., Ciceri, F., Somervaille, T. C., et al. (2015). KDM1 histone lysine demethylases as targets for treatments of oncological and neurodegenerative disease. Epigenomics 7 (4), 609–626. doi:10.2217/epi.15.9
Maes, T., Mascaro, C., Rotllant, D., Lufino, M. M. P., Estiarte, A., Guibourt, N., et al. (2020). Modulation of KDM1A with Vafidemstat rescues memory deficit and behavioral alterations. PLoS One 15 (5), e0233468. doi:10.1371/journal.pone.0233468
Magerl, C., Ellinger, J., Braunschweig, T., Kremmer, E., Koch, L. K., Holler, T., et al. (2010). H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Hum. Pathol. 41 (2), 181–189. doi:10.1016/j.humpath.2009.08.007
Maiques-Diaz, A., and Somervaille, T. C. (2016). LSD1: Biologic roles and therapeutic targeting. Epigenomics 8 (8), 1103–1116. doi:10.2217/epi-2016-0009
Maiques-Diaz, A., Spencer, G. J., Lynch, J. T., Ciceri, F., Williams, E. L., Amaral, F. M. R., et al. (2018). Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 22 (13), 3641–3659. doi:10.1016/j.celrep.2018.03.012
Majernikova, N., den Dunnen, W. F. A., and Dolga, A. M. (2021). The potential of ferroptosis-targeting therapies for Alzheimer's disease: From mechanism to transcriptomic analysis. Front. Aging Neurosci. 13, 745046. doi:10.3389/fnagi.2021.745046
Matsuda, S., Baba, R., Oki, H., Morimoto, S., Toyofuku, M., Igaki, S., et al. (2019). T-448, a specific inhibitor of LSD1 enzyme activity, improves learning function without causing thrombocytopenia in mice. Neuropsychopharmacology 44 (8), 1505–1512. doi:10.1038/s41386-018-0300-9
Metzger, E., Wissmann, M., Yin, N., Muller, J. M., Schneider, R., Peters, A. H., et al. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437 (7057), 436–439. doi:10.1038/nature04020
Mould, D. P., McGonagle, A. E., Wiseman, D. H., Williams, E. L., and Jordan, A. M. (2015). Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: Clinical significance and progress to date. Med. Res. Rev. 35 (3), 586–618. doi:10.1002/med.21334
Nilsson, P., Loganathan, K., Sekiguchi, M., Matsuba, Y., Hui, K., Tsubuki, S., et al. (2013). Aβ secretion and plaque formation depend on autophagy. Cell Rep. 5 (1), 61–69. doi:10.1016/j.celrep.2013.08.042
Nilsson, P., and Saido, T. C. (2014). Dual roles for autophagy: Degradation and secretion of Alzheimer's disease Aβ peptide. Bioessays 36 (6), 570–578. doi:10.1002/bies.201400002
Ooi, S. K., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z., et al. (2007). DNMT3L Connects Unmethylated Lysine 4 of Histone H3 to de novo Methylation of DNA. Nature 448 (7154), 714–717. doi:10.1038/nature05987
Perillo, B., Ombra, M. N., Bertoni, A., Cuozzo, C., Sacchetti, S., Sasso, A., et al. (2008). DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319 (5860), 202–206. doi:10.1126/science.1147674
Periz, G., Lu, J., Zhang, T., Kankel, M. W., Jablonski, A. M., Kalb, R., et al. (2015). Regulation of protein quality control by UBE4B and LSD1 through p53-mediated transcription. PLoS Biol. 13 (4), e1002114. doi:10.1371/journal.pbio.1002114
Predescu, D. V., Cretoiu, S. M., Cretoiu, D., Pavelescu, L. A., Suciu, N., Radu, B. M., et al. (2019). G protein-coupled receptors (GPCRs)-Mediated calcium signaling in ovarian cancer: Focus on GPCRs activated by neurotransmitters and inflammation-associated molecules. Int. J. Mol. Sci. 20 (22), E5568. doi:10.3390/ijms20225568
Reddy, P. H. (2017). A critical assessment of research on neurotransmitters in Alzheimer's disease. J. Alzheimers Dis. 57 (4), 969–974. doi:10.3233/JAD-170256
Riedel, G., Platt, B., and Micheau, J. (2003). Glutamate receptor function in learning and memory. Behav. Brain Res. 140 (1-2), 1–47. doi:10.1016/s0166-4328(02)00272-3
Saez, J. E., Gomez, A. V., Barrios, A. P., Parada, G. E., Galdames, L., Gonzalez, M., et al. (2015). Decreased expression of CoREST1 and CoREST2 together with LSD1 and HDAC1/2 during neuronal differentiation. PLoS One 10 (6), e0131760. doi:10.1371/journal.pone.0131760
Saleque, S., Kim, J., Rooke, H. M., and Orkin, S. H. (2007). Epigenetic regulation of hematopoietic differentiation by gfi-1 and gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 27 (4), 562–572. doi:10.1016/j.molcel.2007.06.039
Schmidt, D. M., and McCafferty, D. G. (2007). Trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 46 (14), 4408–4416. doi:10.1021/bi0618621
Schmitt, M. L., Hauser, A. T., Carlino, L., Pippel, M., Schulz-Fincke, J., Metzger, E., et al. (2013). Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity. J. Med. Chem. 56 (18), 7334–7342. doi:10.1021/jm400792m
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119 (7), 941–953. doi:10.1016/j.cell.2004.12.012
Smith, M. A., Harris, P. L., Sayre, L. M., and Perry, G. (1997). Iron accumulation in alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U. S. A. 94 (18), 9866–9868. doi:10.1073/pnas.94.18.9866
Somervaille, T., Salamero, O., Montesinos, P., Willekens, C., Simon, J. A. P., Pigneux, A., et al. (2016). Safety, phamacokinetics (PK), pharmacodynamics (PD) and preliminary activity in acute leukemia of ory-1001, a first-in-class inhibitor of lysine-specific histone demethylase 1A (LSD1/KDM1A): Initial results from a first-in-human phase 1 study. Blood 128 (22), 4060. doi:10.1182/blood.V128.22.4060.4060
Spencer, J. P., Vafeiadou, K., Williams, R. J., and Vauzour, D. (2012). Neuroinflammation: Modulation by flavonoids and mechanisms of action. Mol. Asp. Med. 33 (1), 83–97. doi:10.1016/j.mam.2011.10.016
Sprussel, A., Schulte, J. H., Weber, S., Necke, M., Handschke, K., Thor, T., et al. (2012). Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 26 (9), 2039–2051. doi:10.1038/leu.2012.157
Stewart, C. R., Stuart, L. M., Wilkinson, K., van Gils, J. M., Deng, J., Halle, A., et al. (2010). CD36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11 (2), 155–161. doi:10.1038/ni.1836
Stoccoro, A., and Coppede, F. (2018). Role of epigenetics in Alzheimer's disease pathogenesis. Neurodegener. Dis. Manag. 8 (3), 181–193. doi:10.2217/nmt-2018-0004
Stolero, N., and Frenkel, D. (2021). The dialog between neurons and microglia in Alzheimer's disease: The neurotransmitters view. J. Neurochem. 158 (6), 1412–1424. doi:10.1111/jnc.15262
Streit, W. J., Braak, H., Xue, Q. S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 118 (4), 475–485. doi:10.1007/s00401-009-0556-6
Sun, G., Alzayady, K., Stewart, R., Ye, P., Yang, S., Li, W., et al. (2010). Histone demethylase LSD1 regulates neural stem cell proliferation. Mol. Cell. Biol. 30 (8), 1997–2005. doi:10.1128/MCB.01116-09
Sun, P., Zhang, S. J., Maksim, S., Yao, Y. F., Liu, H. M., and Du, J. (2019). Epigenetic modification in macrophages: A promising target for tumor and inflammation-associated disease therapy. Curr. Top. Med. Chem. 19 (15), 1350–1362. doi:10.2174/1568026619666190619143706
Surace, A. E. A., and Hedrich, C. M. (2019). The role of epigenetics in autoimmune/inflammatory disease. Front. Immunol. 10, 1525. doi:10.3389/fimmu.2019.01525
Swahari, V., and West, A. E. (2019). Histone demethylases in neuronal differentiation, plasticity, and disease. Curr. Opin. Neurobiol. 59, 9–15. doi:10.1016/j.conb.2019.02.009
Toffolo, E., Rusconi, F., Paganini, L., Tortorici, M., Pilotto, S., Heise, C., et al. (2014). Phosphorylation of neuronal lysine-specific demethylase 1 LSD1/kdm1a impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. J. Neurochem. 128 (5), 603–616. doi:10.1111/jnc.12457
Upadhyay, G., Chowdhury, A. H., Vaidyanathan, B., Kim, D., and Saleque, S. (2014). Antagonistic actions of rcor proteins regulate LSD1 activity and cellular differentiation. Proc. Natl. Acad. Sci. U. S. A. 111 (22), 8071–8076. doi:10.1073/pnas.1404292111
Van Erum, J., Van Dam, D., and De Deyn, P. P. (2019). Alzheimer's disease: Neurotransmitters of the sleep-wake cycle. Neurosci. Biobehav. Rev. 105, 72–80. doi:10.1016/j.neubiorev.2019.07.019
Vijayakumar, K., and Cho, G. W. (2019). Autophagy: An evolutionarily conserved process in the maintenance of stem cells and aging. Cell biochem. Funct. 37 (6), 452–458. doi:10.1002/cbf.3427
Wang, G. G., Allis, C. D., and Chi, P. (2007). Chromatin remodeling and cancer, Part I: Covalent histone modifications. Trends Mol. Med. 13 (9), 363–372. doi:10.1016/j.molmed.2007.07.003
Wang, J., Hevi, S., Kurash, J. K., Lei, H., Gay, F., Bajko, J., et al. (2009a). The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 41 (1), 125–129. doi:10.1038/ng.268
Wang, J., Telese, F., Tan, Y., Li, W., Jin, C., He, X., et al. (2015). LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 18 (9), 1256–1264. doi:10.1038/nn.4069
Wang, Y., Zhang, H., Chen, Y. P., Sun, Y. M., Yang, F., Yu, W. H., et al. (2009b). LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138 (4), 660–672. doi:10.1016/j.cell.2009.05.050
Weiland, A., Wang, Y., Wu, W., Lan, X., Han, X., Li, Q., et al. (2019). Ferroptosis and its role in diverse brain diseases. Mol. Neurobiol. 56 (7), 4880–4893. doi:10.1007/s12035-018-1403-3
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: Process and function. Cell Death Differ. 23 (3), 369–379. doi:10.1038/cdd.2015.158
Yabuta, S., and Shidoji, Y. (2019). Cytoplasmic translocation of nuclear LSD1 (KDM1A) in human hepatoma cells is induced by its inhibitors. Hepat. Oncol. 6 (2), HEP13. doi:10.2217/hep-2018-0008
Yan, N., and Zhang, J. J. (2019). The emerging roles of ferroptosis in vascular cognitive impairment. Front. Neurosci. 13, 811. doi:10.3389/fnins.2019.00811
Yang, J. H., Lou, Q. X., Chen, Y. X., and Tang, K. K. (2015). Brønsted acid–catalyzed friedel–crafts reaction of indoles to α-ketimino esters. Synth. Commun. 45 (16), 1887–1892. doi:10.1080/00397911.2015.1039033
Yang, M., Culhane, J. C., Szewczuk, L. M., Jalili, P., Ball, H. L., Machius, M., et al. (2007). Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 46 (27), 8058–8065. doi:10.1021/bi700664y
Yiannopoulou, K. G., Anastasiou, A. I., Zachariou, V., and Pelidou, S. H. (2019). Reasons for failed trials of disease-modifying treatments for alzheimer disease and their contribution in recent research. Biomedicines 7 (4), E97. doi:10.3390/biomedicines7040097
Yiannopoulou, K. G., and Papageorgiou, S. G. (2020). Current and future treatments in alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 12, 1179573520907397–12. doi:10.1177/1179573520907397
Yin, W., Arkilo, D., Khudyakov, P., Hazel, J., Gupta, S., Quinton, M. S., et al. (2021). Safety, pharmacokinetics and pharmacodynamics of TAK-418, a novel inhibitor of the epigenetic modulator lysine-specific demethylase 1A. Br. J. Clin. Pharmacol. 87 (12), 4756–4768. doi:10.1111/bcp.14912
Yokoyama, A., Takezawa, S., Schule, R., Kitagawa, H., and Kato, S. (2008). Transrepressive function of TLX requires the histone demethylase LSD1. Mol. Cell. Biol. 28 (12), 3995–4003. doi:10.1128/MCB.02030-07
Yu, L., Chen, Y., and Tooze, S. A. (2018). Autophagy pathway: Cellular and molecular mechanisms. Autophagy 14 (2), 207–215. doi:10.1080/15548627.2017.1378838
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153 (3), 707–720. doi:10.1016/j.cell.2013.03.030
Zhang, F., and Jiang, L. (2015). Neuroinflammation in Alzheimer's disease. Neuropsychiatr. Dis. Treat. 11, 243–256. doi:10.2147/NDT.S75546
Zhang, F., Xu, D., Yuan, L., Sun, Y., and Xu, Z. (2014). Epigenetic regulation of atrophin1 by lysine-specific demethylase 1 is required for cortical progenitor maintenance. Nat. Commun. 5, 5815. doi:10.1038/ncomms6815
Zheng, Y. C., Yu, B., Chen, Z. S., Liu, Y., and Liu, H. M. (2016). TCPs: Privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapy. Epigenomics 8 (5), 651–666. doi:10.2217/epi-2015-0002
Zheng, Y., Liu, A., Wang, Z. J., Cao, Q., Wang, W., Lin, L., et al. (2019). Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer's disease. Brain 142 (3), 787–807. doi:10.1093/brain/awy354
Zhuo, X., Wu, Y., Yang, Y., Gao, L., Qiao, X., and Chen, T. (2019). Knockdown of LSD1 meliorates ox-LDL-stimulated NLRP3 activation and inflammation by promoting autophagy via SESN2-mesiated PI3K/Akt/mTOR signaling pathway. Life Sci. 233, 116696. doi:10.1016/j.lfs.2019.116696
Zibetti, C., Adamo, A., Binda, C., Forneris, F., Toffolo, E., Verpelli, C., et al. (2010). Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J. Neurosci. 30 (7), 2521–2532. doi:10.1523/JNEUROSCI.5500-09.2010
Keywords: alzheimer’s disease, epigenetics, demethylase, LSD1, inhibitors
Citation: Li Y, Zhao Y, Li X, Zhai L, Zheng H, Yan Y, Fu Q, Ma J, Fu H, Zhang Z and Li Z (2022) Biological and therapeutic role of LSD1 in Alzheimer’s diseases. Front. Pharmacol. 13:1020556. doi: 10.3389/fphar.2022.1020556
Received: 16 August 2022; Accepted: 13 October 2022;
Published: 25 October 2022.
Edited by:
Chao Han, China Pharmaceutical University, ChinaCopyright © 2022 Li, Zhao, Li, Zhai, Zheng, Yan, Fu, Ma, Fu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haier Fu, ZnVoYWllcjAzMzBAMTYzLmNvbQ==; Zhenqiang Zhang, enpxa3ljQGhhY3RjbS5lZHUuY24=; Zhonghua Li, bGl6aEBoYWN0Y20uZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.