
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 11 October 2022
Sec. Respiratory Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1015926
This article is part of the Research Topic Novel Therapeutic Strategies for the Treatment of Respiratory Diseases View all 8 articles
 Hongshu Sui1*†
Hongshu Sui1*† Xinghua Xu1†
Xinghua Xu1† Yanping Su1
Yanping Su1 Zhaoqing Gong1Minhua Yao1
Zhaoqing Gong1Minhua Yao1 Xiaocui Liu1Ting Zhang1Ziyao Jiang1Tianhao Bai2Junzuo Wang3Jingjun Zhang4
Xiaocui Liu1Ting Zhang1Ziyao Jiang1Tianhao Bai2Junzuo Wang3Jingjun Zhang4 Changlong Xu5,6*
Changlong Xu5,6* Mingjiu Luo2*
Mingjiu Luo2*Cystic fibrosis (CF) is a life-threatening autosomal-recessive disease caused by mutations in a single gene encoding cystic fibrosis transmembrane conductance regulator (CFTR). CF effects multiple organs, and lung disease is the primary cause of mortality. The median age at death from CF is in the early forties. CF was one of the first diseases to be considered for gene therapy, and efforts focused on treating CF lung disease began shortly after the CFTR gene was identified in 1989. However, despite the quickly established proof-of-concept for CFTR gene transfer in vitro and in clinical trials in 1990s, to date, 36 CF gene therapy clinical trials involving ∼600 patients with CF have yet to achieve their desired outcomes. The long journey to pursue gene therapy as a cure for CF encountered more difficulties than originally anticipated, but immense progress has been made in the past decade in the developments of next generation airway transduction viral vectors and CF animal models that reproduced human CF disease phenotypes. In this review, we look back at the history for the lessons learned from previous clinical trials and summarize the recent advances in the research for CF gene therapy, including the emerging CRISPR-based gene editing strategies. We also discuss the airway transduction vectors, large animal CF models, the complexity of CF pathogenesis and heterogeneity of CFTR expression in airway epithelium, which are the major challenges to the implementation of a successful CF gene therapy, and highlight the future opportunities and prospects.
Cystic fibrosis (CF) is an autosomal-recessive disease affecting over 80,000 people worldwide (Brown et al., 2017; Scotet et al., 2020a). CF is caused by mutations in a single gene encoding a product named cystic fibrosis transmembrane conductance regulator (CFTR) (Riordan et al., 1989; Rommens et al., 1989). CFTR is as a cyclic adenosine monophosphate (cAMP)-dependent ion channel protein that conducts chloride (Cl−) and bicarbonate (HCO3−) ions across the epithelia, and it plays an important role in epithelial ion transport and fluid homeostasis (Welsh and Smith, 1993; Zabner et al., 1998; Gadsby et al., 2006; Pezzulo et al., 2012; Stoltz et al., 2015; Kunzelmann et al., 2017). Mutations in CFTR can result in reduced or absent expression, or malfunction, leading to CF. Nearly 400 of the ∼2,000 identified CFTR mutations have been confirmed to be disease-causing mutations (http://www.genet.sickkids.on.ca/app). The classification system of the Cystic Fibrosis Foundation (www.cff.org) groups these mutations into five classes by the problems in: 1) protein production (no synthesis), 2) defective protein processing, 3) defective gating, 4) low conductance, and 5) insufficient quantities of protein expression (low synthesis or increased turnover). Worldwide, the distribution and frequency of CFTR variants vary in different countries and ethnic groups, but only a limited number of mutant alleles display a frequency higher than 1% (Ferec and Cutting, 2012). An early study conducted in United States found that CF more frequently occurred in non-Hispanic Caucasian (1/2,500) and Ashkenazi Jews (1/2,300), compared to that in Hispanic Caucasian (1:13,500), African Americans (1:15,100), and Asian Americans (1:35,100) (Palomaki et al., 2004). Figure 1 presents the common CF-related CFTR mutations of >1% frequency in population worldwide. The most common CF-associated mutation is F508del, an example of a Class 2 processing-defect mutation. F508del accounts for approximately two-thirds of CF alleles (66.8%) (Bobadilla et al., 2002). The other common mutations include G542X (2.6%, Class 1), N1303K (1.6%, Class 2), G551D (1.5%, Class 3), W1282X (1.0%, Class 1) (Zvereff et al., 2014; Chamayou et al., 2020; Scotet et al., 2020a; Scotet et al., 2020b; Petrova et al., 2021; Yiallouros et al., 2021).
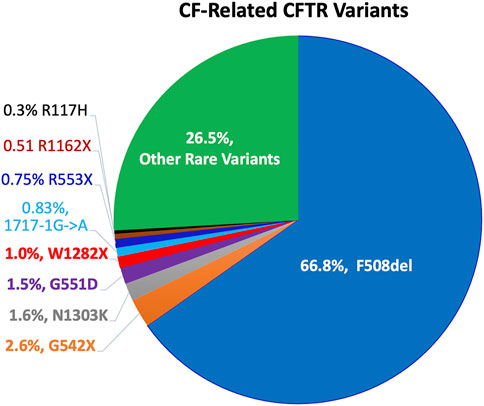
FIGURE 1. Worldwide distribution of disease-causing CFTR variants. More than 2,000 different mutations in CFTR have been identified, and ∼400 mutations are confirmed to be disease-causing. (http://www.genet.sickkids.on.ca/app). Pie chart represents the frequencies of common CF mutations (>0.3%) in population worldwide. Notably, the distribution and frequency of CFTR variants vary in different countries and ethnic groups.
CF affects various organ systems and is associated with dehydration of the epithelial surface, reduction in the pH of secretions, and excessive mucus production and obstruction (Shteinberg et al., 2021). Lung disease is the primary cause of CF morbidity and mortality (Stoltz et al., 2015). Patients with CF suffer progressive damage in the lung from recurrent respiratory infections and exaggerated inflammatory response, eventually, respiratory failure. In the past, therapies for patients with CF aimed to provide symptomatic care. With improved treatments and better healthcare, now the median age of death from CF is in the early forties (Keogh et al., 2018). Recent advances in pharmaceutical rescue of mutant CFTR function using CFTR modulators have made it possible to target the underlying genetic cause of CF and further increase the quality of life for patients. CFTR modulators are small molecule drugs that were identified from high throughput screening for correcting the impaired CFTR trafficking to the plasmid membrane (corrector) or for increasing the CFTR channel conductance (potentiator) (Lopes-Pacheco, 2019). Thus, CFTR modulator therapies are genotype-dependent, effective on patients who are with the CFTR mutants affecting protein trafficking (Class II), gating and conductance (Class III and IV). To date, four CFTR modulator therapies have been approved and ∼90% CF patients with certain CFTR mutants are benefited with therapeutic outcomes (Ramsey et al., 2011; Rowe et al., 2017; Keating et al., 2018; Taylor-Cousar et al., 2018; Heijerman et al., 2019; Middleton et al., 2019). While the potentiator Ivacaftor is eligible for patients with the G551D mutation (Ramsey et al., 2011), Trikafta, the combination of three correctors and potentiator, has been used to treat the patients carrying one copy of the most common F508del mutant (Middleton et al., 2019). However, CFTR modulator therapies require life-long drug administration, their long-term potential side effects remain unclear (Lopes-Pacheco, 2019). Currently, approximately 10% of patients with CF, who have drug-refractory missense mutations, who produce little to no CFTR protein, or who cannot tolerate CFTR modulators, are without an option of CFTR modulator therapy and still rely on symptomatic treatment (Clancy et al., 2019; Middleton et al., 2019). For treatments to the patients left behind, the interest of pharmaceutical industry to explore novel CF modulators to target those rare CFTR mutations is still growing (Fajac and Sermet, 2021; Laselva et al., 2021). The RNA-based techniques such as engineered tRNA (Lueck et al., 2019) or antisense oligonucleotides (ASOs) (Michaels et al., 2020) to mask the nonsense mutation or to mediate splicing modulation are potentially useful as therapeutic approaches. Although the emerging mRNA delivery technique is thought to be feasible to treating any CF patients, independent of underlying mutations, using the mRNA encoding CFTR (Haque et al., 2018; Robinson et al., 2018), this treatment necessitates life-time administration, similarly as do the CFTR modulator therapies, engineered tRNA or ASOs. In contrast, gene therapy, either via gene addition or gene editing, is able to alter the CFTR expression at DNA level, and such alternation is expected to be persistent in whole life of the recipient cells. As the mutation-agnostic, gene therapy is suitable for all patients with CF, regardless of their genotype (Griesenbach et al., 2016). Thus, gene therapy for CF lung disease remains an attractive approach for treating all the population with CF, not only for other currently lacked of the option of CFTR modulator therapy (Choi and Engelhardt, 2021; Mercier et al., 2021). The goal for gene therapy as a cure for CF lung disease has been pursuing for almost 30 years, desirable outcome has not yet met. Researchers in both industry and academic have contributed substantial effort and talents to tackle the challenges encountered during this long journey. New animal models and more efficacious gene transfer agents have been developed to move the field in the path to success. Several recent excellent review articles have summarized and discussed the lessons and achievements from the last 3 decades (Cooney et al., 2018a; Donnelley and Parsons, 2018; Yan et al., 2019; Tang et al., 2020a; Allan et al., 2021; Lee et al., 2021). In this review, after a brief description of the lessons learned from the previous unsuccessful clinical trials of CF gene therapy in an alignment with recent progress in animal models and viral vectors as tools of gene delivery, we will focus on the major challenges to the implementation of the gene therapy for CF lung disease. We will discuss why such difficulties are hindering the advance of CFTR gene addition therapies toward a successful clinical trial and how researchers are tackling these problems now. We also discuss the novel non-viral vector delivery strategies, and the potential and limitation of the CRISPR-based gene editing as a path toward a cure for CF.
CF was first described as a specific disease in 1938 (Andersen, 1938), and the CFTR gene was discovered in 1989, with multiple mutations identified responsible for CF, including the most common F508del (Riordan et al., 1989; Rommens et al., 1989). Shortly after the cloning of CFTR, the goal to develop gene therapy as a cure for CF lung disease was eagerly pursued by both academia and industry (Flotte, 1993; Cooney et al., 2018a). In 1993, a pioneer study intended to compensate for defective CFTR in the nasal airway via gene replacement using an E1-deleted recombinant adenoviral (rAd) vector to deliver a normal copy of CFTR complementary DNA (cDNA) to the nasal epithelium of three patients with CF. Albeit not placebo-controlled, this study demonstrated proof-of-concept for gene therapy; specifically, for the transient correction of Cl− transport after vector inoculation (Zabner et al., 1993). Since then, nine further rAd vector trials to treat CF lung disease were conducted, but it turned out that the first generation of rAd vectors were not suitable for gene therapy because of transient gene expression and the strong immunogenicity (Joseph et al., 2001; Perricone et al., 2001).
Recombinant adeno-associated virus (rAAV) vectors are the most promising vectors for human gene therapy. Several rAAV-based gene therapies have been approved for clinical treatments (Kuzmin et al., 2021). CF lung disease gene therapy using rAAV-2 vector was evaluated in clinics from 1998 to 2007, led by the Targeted Genetics Corporation (Guggino and Cebotaru, 2017, 2020). Phase I single-dose studies demonstrated the safety of the gene transfer agent, tgAAVCF, in human application and its successful delivery to the maxillary sinuses and lungs of patients with CF. Although vector-derived CFTR mRNA expression was beyond the detection limit, the sinus transepithelial potential was assessable on days 7 and 14 after infection, indicating the transient functional restoration (Wagner et al., 1998; Aitken et al., 2001; Flotte et al., 2003). Phase II trials of two doses of vectors with an interval of 30 days found that 25% of the patients treated with tgAAVCF demonstrated an improvement of >10% in forced expiratory volume in 1 s (FEV1) while no improvement was observed in the placebo group (Wagner et al., 2002; Moss et al., 2004; Moss et al., 2007). Despite these promising results, the overall outcomes did not meet the primary efficacy endpoint of an improvement in lung function.
Non-viral vectors are advantageous because they are less immunogenic than viral vectors for repeat dosing (Yin et al., 2014). Studies on non-viral CFTR delivery revealed that the cationic liposome (GL67A)-formulated plasmid-based CFTR gene transfer to the airway epithelia (nose and lung) was safe (Alton et al., 1999) and also feasible for repeat dosing (Hyde et al., 2000). Using the most potent non-viral vector (GL67A complexed with pGM169, a CpG nucleotide-free CTTR expression plasmid) (Hyde et al., 2008; Alton et al., 2013; Alton et al., 2014), the United Kingdom Cystic Fibrosis Gene Therapy Consortium (UK CFGTC) conducted a double-blinded, placebo-controlled, multidose trial to assess its clinical benefit. This is to date the largest CF gene therapy phase IIb trial with 116 patients (62 active, 54 placebo). In 2015, the results of the trials were published: patients with CF who received nebulized vectors at 28-day intervals for 12 months demonstrated an increase in FEV1 of 3.7% (0.07%–7.25%) (Alton et al., 2015). However, such improvement was still insufficient to restore function in CF lungs.
Collectively, the goal of gene therapy as a cure for CF lung disease has been pursued for almost 3 decades. Selected clinical trials of the gene therapy for CF lung disease are listed in Table 1. Although some promising results have been obtained from 36 gene therapy clinical trials involving ∼600 patients with CF (Ginn et al., 2018), desirable outcomes are yet to be demonstrated. Nevertheless, these early attempts established the proof-of-concept for CF gene therapy through gene replacement, and it has taught the field many important lessons, which are discussed in the next section.
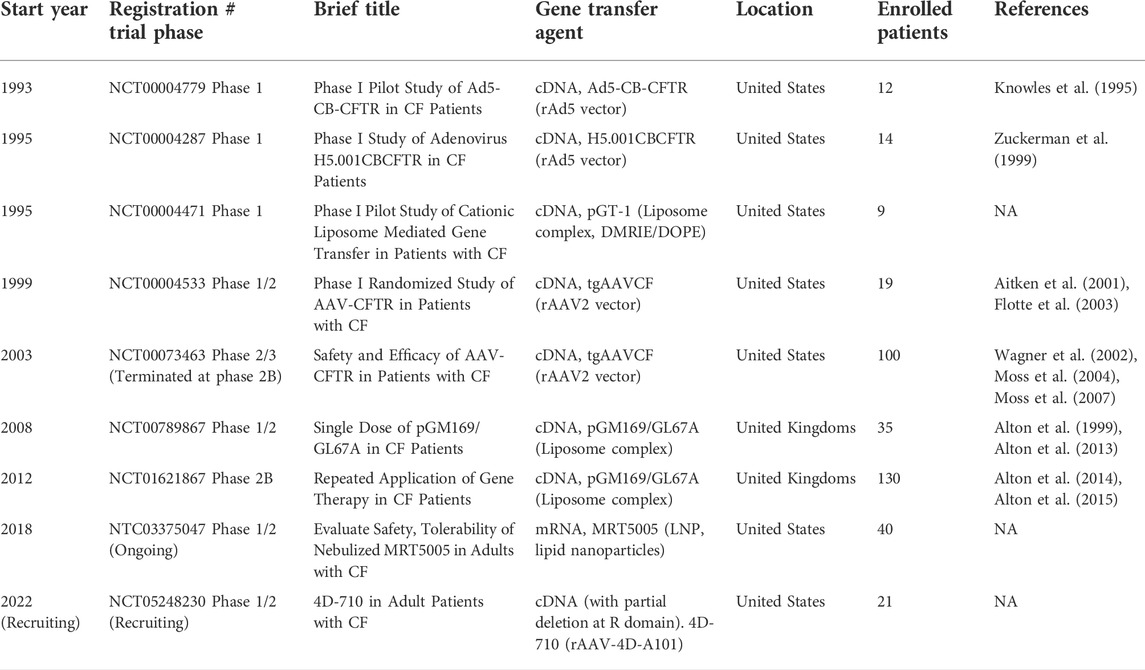
TABLE 1. Selected clinical trials of gene therapy for cystic fibrosis lung disease.
Although the airways are non-invasively accessible, pulmonary gene transfer has proven more difficult than anticipated. The lung airways have evolved a complex array of extracellular and intracellular mechanisms to protect against pathogens and foreign invaders (Ferrari et al., 2003). Airway epithelial cells, the primary recipients of CFTR gene transfer, play an indispensable role as the first-line host defense in the lung and have a critical role in innate antiviral responses to infection, and thus are resistant to vector transduction or transfection (Hewitt and Lloyd, 2021). For the accession of cell receptors on the apical surface, transgene vectors must be able to penetrate the airway mucus layer (Cone, 2009) and overcome the innate defenses, e.g., mucociliary clearance (MCC), which is a self-clearing mechanism of the airways (Bustamante-Marin and Ostrowski, 2017). In airways with CF, these physical barriers are exacerbated because of the increased thickness and viscosity of the mucus and sputum (Kreda et al., 2012; Shteinberg et al., 2021). The CFTR transfer vectors used in previous trials have proven inefficient to transduce or transfect CF airways via apical luminal delivery. Researches into the transduction biology of these vectors in different airway epithelium model systems in vitro and in the lungs of experimental animals in vivo have provided valuable clues to guide the developments of next-generation airway transduction vectors and well-designed delivery approaches that use pharmacological interventions to overcome the extracellular and intracellular barriers to productive transduction.
rAd vectors were the first viral vectors tested in patients with CF (Zabner et al., 1993; Crystal et al., 1994). Although respiratory tract infections are the most common manifestation of adenovirus infection, partial correction of the Cl− transport defect in nasal epithelium with CF using rAd-CFTR was only observed when the nasal epithelium was damaged during delivery (Joseph et al., 2001). This clinical finding was supported by the later discovery that the coxsackievirus and adenovirus receptor (CAR), which mediates the attachment and infection of adenoviruses type-2 and -5, is localized to the basolateral membrane of the human airway epithelium (Walters et al., 1999; Excoffon, 2020). Other disadvantages are that rAd-mediated CFTR expression in post-mitotic airway epithelial cells is transient and that rAd infection promotes strong cellular and humoral immune responses, which cause the destruction of transduced cell, as well as prevent repeated dosing (Harvey et al., 1999a; Harvey et al., 1999b). The anti-adenovirus immune responses might be further enhanced if the host has preexisting Pseudomonas infection (Tosi et al., 2004), which is a hallmark of CF.
In attempts to solve the disadvantages of rAd for human gene therapy, helper-dependent adenovirus vector (HD-Ad) was developed (Brunetti-Pierri and Ng, 2006). The HD-Ad, in which all viral-encoded genes are removed, obviates the T-cell responses to cryptic viral protein expression; thus, it induces less inflammation and prolongs gene expression in the airways (Cao et al., 2005; Kushwah et al., 2008). It was reported that HD-Ad vectors were effective to express CFTR in the lungs of CF knockout mice (Koehler et al., 2003) and efficiently transduced large experimental animals like rabbits (Koehler et al., 2005) and ferrets (Yan et al., 2015a) via aerosol delivery. The large capacity of HD-Ad vectors (up to 37 kb) can be used to deliver both a gene editing endonuclease system and donor DNA for homologous recombination in a single vector (Xia et al., 2018; Zhou et al., 2019). It also enables the application of piggyBac transposase-mediated integration (Cooney et al., 2018b) or programmable nuclease-mediated targeted insertion (Xia et al., 2019; Bandara et al., 2021), which migrate the transgene cassette from the rAd genome to the chromosome, providing a solution to transient expression. Additionally, the application of sodium caprate (Gregory et al., 2003) or lysophosphatidylcholine (LPC) (Koehler et al., 2005) disrupts the tight junctions and allows for rAd vector to access the CAR at the basolateral cell surface to initiate transduction. Notably, the administration of HD-Ad vector formulated with LPC efficiently transduced airway basal cells in lungs of mice and pigs (Cao et al., 2018). Given that the airway basal cells are a multipotent progenitor for airway repair and regeneration (Hong et al., 2004; Hajj et al., 2007; Rock et al., 2009), integration of a CFTR expression cassette into basal cells allows the functional CFTR expression in their differentiated progeny.
When AAV-based CF gene therapy began in 1990s, rAAV2 was the only available serotype vector. Although the airways are not a natural host of AAV2, its application in lung gene transfer was rationalized by its wide tropism, and the results from preclinical studies demonstrated that rAAV2 was able to productively transduce the lungs of rabbits and rhesus macaques (Conrad et al., 1996; Beck et al., 1999; Beck et al., 2002; Fischer et al., 2003). However, later studies on rAAV2 transduction biology in the cell culture model of polarized human airway epithelium (HAE) at an airway-liquid interface (ALI) discovered that rAAV2 poorly transduced human epithelium from the apical membrane. The apical transduction of rAAV2 in HAE-ALI was 100-fold less efficient than that in the polarized cultures derived from the primary airway epithelial cells of rhesus macaques (Liu et al., 2007). Studies also found that, while transgene expression of rAAV2 apical infection was 200-fold lower than that of basolateral infection in HAE-ALI, a substantial amount of apically internalized rAAV2 genomes was detected (Duan et al., 1998). The lack of a correlation between vector entry and transgene expression suggested that the inefficiency was not primarily due to vector binding and endocytosis; rather, it was due to the failure to establish productive transduction. This observation was consistent with the outcome of clinical trials, specifically, the persistence of the tgAAVCF genomes but undetectable level of vector-derived CFTR mRNA in the recipients’ airways (Aitken et al., 2001). Subsequent research on vector intracellular trafficking revealed that impaired vector nuclear transport was the rate-limiting step in rAAV productive transduction in polarized airway epithelium (Duan et al., 2000). This post-entry block is responsive to the inhibition of proteasome activity, and it can be overcome by transient pharmacological intervention during or post transduction (Yan et al., 2002; Yan et al., 2004; Ding et al., 2005; Jennings et al., 2005; Monahan et al., 2010).
Another limitation of rAAV vectors in CFTR gene transfer is their relatively small package capacity (∼4.9 kb) (Dong et al., 1996; Flotte, 2000). The 4.5-kb CFTR coding sequence approaches the size of the AAV genome (4.68 kb); thus, tgAAVCF used the intrinsic promoter activity of the AAV2 inserted terminal repeat sequence (ITR) for CFTR expression (Flotte et al., 1993). Although studies in non-human primates supported this application, subsequent studies found that the cryptic promoter activity within AAV ITR was too weak in human airways and that the incorporation of an 83-bp synthesized sequence of essential transcriptional motifs (tg83 promoter) to tgAAVCF achieved a three-fold increase in the efficacy of CFTR functional expression in HAE-ALI (Zhang et al., 2004). To free up room for a stronger promoter, research for CFTR structure and function attempted to identify shortened CFTR minigene with only minimal reduction in its function (Carroll et al., 1995; Zhang et al., 1998; Sirninger et al., 2004; Cebotaru and Guggino, 2014). The most promising version is the CFTR∆R, whose size was reduced by 156 bp with a partial deletion (amino acid residues: 708–759) at the N-terminal portion of the R-domain. CFTR∆R generated similar Cl− channel function in vitro and rescued the lethal intestinal phenotype in CFTR knockout mice in vivo (Ostedgaard et al., 2002). The use of CFTR∆R released enough room for the incorporation of another 100-bp enhancer element to strengthen the activity of the tg83 promoter. The resultant rAAV genome (AV2. F5tg83CFTR∆R) of 4.89 kb proved effective for the functional expression of CFTR in human CF airway epithelial cultures in vitro and transcription of CFTR mRNA in ferret airways in vivo (Yan et al., 2015b; Tang et al., 2020b).
The toolbox of rAAV-based vectors has largely expanded since 2000 (Asokan et al., 2012). Now, it is known that rAAV1 (a naturally occurring serotype) (Yan et al., 2006; Yan et al., 2013b) and rAAV2.5T (a variant selected from directed evaluation of an AAV capsid library in HAE-ALI) (Excoffon et al., 2009) are much more efficient than rAAV2 in apically transducing HAE-ALI. The post-entry block responsive for proteasome inhibition appears ubiquitous for parvovirus capsids: it was effective for most AAV serotypes and variants (Yan et al., 2002; Yan et al., 2006), as well as human bocavirus 1 (HBoV1) (Yan et al., 2013a). Composed of the human airway tropic AAV2.5T capsid and the most potent rAAV-CFTR construct, the next-generation rAAV-based CFTR transfer vector (AV2/2.5T.F5tg83CFTR∆R) was developed (Tang et al., 2020b). Currently, preclinical studies are being conducted by Spirovant Sciences, Inc. (Philadelphia, PA) to establish the efficacy of this new delivery system formulated with transduction augmenter (proteasome inhibitors) in the lungs of CF ferrets
Lentiviral vectors derived from immunodeficiency viruses can transduce both dividing and non-dividing cells, and transgene expression from the integrated viral genome likely persists for the entire life cycle of recipient cells (Milone and O'Doherty, 2018). Although the insertional mutagenesis is an inherent safety concern, it has not yet been proven in clinical trials using lentiviral vectors (Schlimgen et al., 2016; Biasco et al., 2018). Because the lung airways are not a natural lentivirus host, lentiviral vectors used for airway transduction are necessitated to pseudotype with appropriate envelope proteins (Marquez Loza et al., 2019). As the hurdles to the success of non-viral strategies are poor transfection efficiency and transient expression, the UK CFGTC has since pursued a phase I/IIa lentiviral vector gene therapy trial for CF (Alton et al., 2017) with rSIV.F/HN-hCEF-CFTR (Mitomo et al., 2010; Griesenbach et al., 2012). rSIV.F/HN-hCEF-CFTR harboring the CFTR expression cassette from the clinically tested non-viral vector pGM169 is pseudotyped with F and HN (fusion and hemagglutinin-neuraminidase) proteins from the Sendai virus (SeV), which is a murine parainfluenza that causes mild disease in humans. A direct comparison between GL67A formulated pGM169 and rSIV.F/HN-hCEF-CFTR indicated that the lentiviral strategy was several log orders more efficient than the non-viral strategy in transducing airway epithelial cells (Alton et al., 2017).
Despite the lungs having defense mechanisms against foreign invaders, respiratory viruses can circumvent these barriers to establish infection and replicate progeny (Nichols et al., 2008). Indeed, recombinant vectors from paramyxoviruses, such as human parainfluenza virus (PIV) (Zhang et al., 2005), respiratory syncytial virus (RSV) (Kwilas et al., 2010), and murine SeV (Ferrari et al., 2004), are able to efficiently transduce airway epithelium in vitro and in the lungs of mice in vivo. However, the vectors derived from these negative-sense, single-stranded RNA viruses only achieved transient expression, and their transduction induced strong adaptive immune responses. These disadvantages and the difficulties in vector production limit their path to clinical application.
DNA viruses associated with human respiratory disease include the nonenveloped double-stranded DNA (dsDNA) viruses from the family Adenoviridae and the newly discovered HBoV1, a single-stranded DNA virus from the family Parvoviridae (Allander et al., 2005). HBoV1 causes acute respiratory tract infection in young children (Kapoor et al., 2010; Peltola et al., 2013). The seroprevalence of HBoV1 capsid-specific immunoglobulin G is 13% and 59% in children and adults, respectively. However, seroconversion does not appear to prevent repeat infection by HBoV1 (Meriluoto et al., 2012). The full-length genome of HBoV1 has been cloned. While HEK293 cells are not permissive to HBoV1 infection, the transfection of the cloned HBoV1 duplex genome to these cells can produce infectious virions of HBoV1 (Huang et al., 2012). In vitro, HBoV1 efficiently infects polarized human HAE and produces progeny (Deng et al., 2013). As a relative of AAV, but an autonomous parvovirus, HBoV1 is easy to manipulate as a recombinant vector by the co-transfection of a rHBoV1 transfer plasmid and a helper plasmid, which provides viral components in trans, without the need for an adenovirus helper. rHBoV1 is able to efficiently transduce polarized HAE-ALI from the apical surface, but replication-competent rHBoV1 genomes would be expected in rHBoV1 viral stocks (Yan et al., 2013a). To eliminate this safety concern, a chimeric parvoviral vector, rAAV/HBoV1, was developed through parvovirus cross genera pseudopackaging. Composed of the clinically proven rAAV2 genome and the airway tropic HBoV1 capsid, rAAV2/HBoV1 maintains rHBoV1’s ability to efficiently transduce HAE-ALI from the apical membrane. Furthermore, the size of the HBoV1 genome is 5.5 kb; thus, the HBoV1 capsid can comfortably accommodate an oversized rAAV genome of up to 5.9 kb. With this 20% increased package capacity, rAAV2/HBoV1 vectors can use a strong promoter (and incorporate necessary transcriptional regulation elements) to express the full-length CFTR (Yan et al., 2013a). rAAV2/HBoV1 has demonstrated transduction in ferret lungs in vivo, enabling preclinical studies in CF ferret models (Yan et al., 2013a; Yan et al., 2017).
CF animal models are essential in preclinical studies to validate the therapeutic strategy, to determine gene transfer agent dosing, administration routes, and application schedules in clinical trials. Soon after the discovery of CFTR, many CF mouse models were developed. However, because mice with CFTR mutations did not develop “spontaneous” chronic bacterial infection and/or inflammation in the lungs, CF mice are not a suitable animal model for testing gene therapies of CF lung disease (Wilke et al., 2011). To date, this problem has been solved by the developments of large-animal CF models (Yan et al., 2015a). CFTR knock-out (KO) ferrets (Sun et al., 2008) and pigs (Rogers et al., 2008) recapitulate many features of the lung disease phenotype observed in humans with CF. Lentivirus-, rAd- and rAAV-mediated CFTR lung gene transfer in CFTR-KO pigs have demonstrated the proof-of-concept for gene therapy in the hostile environment of CF lungs (Cooney et al., 2016; Steines et al., 2016; Cooney et al., 2018b). However, CF is a progressive disease, the lung disease phenotypes do not present at newborn and young age, in both ferret and pig models. For ferret, it usually takes a year or more to develop an infected CF lung. Both pig and ferret CFTR-KO models have severe gastrointestinal defects before and after birth, this makes them particularly difficult to rear beside of the low survival rate of the newborns. Thus, the utility of the first-generation CF ferrets and pigs as preclinical models for gene therapy has been limited (Yan et al., 2015a). The newly developed CFTR modulator-responsive CF models, e.g. CFTRG551D ferret (Sun et al., 2019), are even more suitable for preclinical studies with greatly increased survival rates and reduced difficulty in caring for sick animals. Administration of Ivacaftor (VX-770) during the gestation period protects the pancreas and intestine of the CF kits. After birth, CFTRG551D ferrets grow normally when Ivacaftor is given. Withdrawal of the drug reinitiates disease in the pancreas, gut, and lung at any age. Furthermore, these models express malfunction CFTR proteins, thus, immune responses to the transgene product, CFTR, are not expected to occur (Yan et al., 2019).
Previous rAAV CF lung gene therapy clinical trials have highlighted the importance of the compatibility of delivery systems and animal models in preclinical studies. Before entering clinical trials, tgAAVCF was tested in rabbits and rhesus macaques with a single dose and repeat dosing. Vector-specific CFTR mRNA expression and the viral genome were detected in the macaques up to 180 days after infection (Conrad et al., 1996; Beck et al., 1999). These studies supported the utilities of AAV2 capsid for transduction and the AAV2 ITR promoter for transgene expression. However, the outcomes of CFTR expression from clinical trials were undesirable. Although it could be argued that the preclinical studies were not conducted in CF lungs, side-by-side direct comparison of the rAAV2 transductions in polarized human and monkey airway epithelial cultures revealed a large cross-species tropism difference of this AAV serotype capsid: The apical transduction of rAAV2 in the ALI cultures differentiated from primary airway cells of rhesus macaques was 100-fold more efficient than that in polarized HAE-ALI cultures (Liu et al., 2007). Similarly, a new AAV capsid variant (H22) identified from directed evolution of the AAV capsid library in the pig airway in vivo did not effectively transduce polarized HAE-ALI in vitro. Thus, despite the fact that AAVH22 vector, AAVH22/F5tg83CFTRΔR, effectively expressed CFTR in lungs of CF pigs to correct the defects of Cl−transport, pH of the airway surface fluid, and bacterial killing (Steines et al., 2016), its potential for application in humans is limited. Of note, rAAV2.5T and AAV/HBoV1 vectors are able to transduce ferret lungs in vivo (Yan et al., 2017; Tang et al., 2020b). This cross-species compatibility in vector tropisms enabled a more predictive preclinical path to evaluate the efficacy of gene therapy and to optimize vector delivery in the CF lungs of the CFTRG551D ferret.
Early studies in humans carrying splice variants in CFTR (c.1393-1G>A [1525-1G>A], an exon 10 in-frame skipping) suggested that as little as 8% of the normal CFTR transcript was sufficient to preserve normal lung function (Chu et al., 1992; Chu et al., 1993). In vitro reconstitution experiments showed that polarized HAE cultures derived from mixtures of non-CF and ∆F508/∆F508 primary human airway epithelial cells at a 2:8 ratio (WT:CF) restored ∼70% of CFTR-mediated Cl−transport in 100% wild-type cultures (Farmen et al., 2005). Although these lines of evidence suggest that modest endogenous CFTR expression may suffice to improve CF lung disease, complementation of the defect in lung function by ectopic CFTR expression in CF airways is complicated because CFTR expression is highly regulated at different levels in the airways and in different epithelial cell types (Jiang and Engelhardt, 1998; Davis and Wypych, 2021). Additionally, the majority of cell types on the airway surfaces that are accessible to CFTR gene transfer vectors are terminally differentiated with a defined lifespan; thus, gene therapy necessitates repeat dosing to achieve unintermitted CFTR expression in the lungs with CF.
The respiratory system is divided into the upper airways and lower airways. The conducting airways are defined as those sections of the respiratory tract which do not directly participate in gas exchange, and the distal airways include terminal and respiratory bronchioles and alveoli. The airway epithelium begins as a ciliated pseudostratified columnar epithelium in the trachea and slowly transition to that of a non-ciliated simple cuboidal epithelium in the terminal bronchioles. Human airways are lined by a variety of cell types, primarily including ciliated, mucous, and secretory cells on the surface layer, as well as the basal cells that are secured to the basement membrane (Figure 2). Each of these cell types plays a functionally distinct role and expresses different levels of CFTR (Davis and Wypych, 2021). Ciliated columnar cells are the dominant cell type throughout the surface epithelium of the large and small airways and thereby play a central role in the first line of lung defense and a pivotal role in airway homeostasis through MCC. Their amount increases with airway branching, from 47% ± 2% in the trachea to 73% ± 1% in the small airway epithelium (Raman et al., 2009). Goblet cells are the primary secretory cells in proximal cartilaginous airways, which also contain numerous submucosal glands (SMG) (Widdicombe and Wine, 2015). SMG consist of acini and tubules connected to the surface epithelium via collecting and ciliated ducts and house diverse cell types: serous, secretory, goblet, myoepithelial and ciliated cells, as well as basal cells. The terminal bronchioles contain basal cells, secretory club cells, ciliated cells, and neuroendocrine cells, whereas the respiratory bronchioles contain primarily club cells. Alveoli represent the most distal portion of the respiratory tract, where the major cell types are type I and type II pneumocytes and alveolar macrophages, which are the most abundant innate immune cells in the distal lung parenchyma (Whitsett et al., 2019).
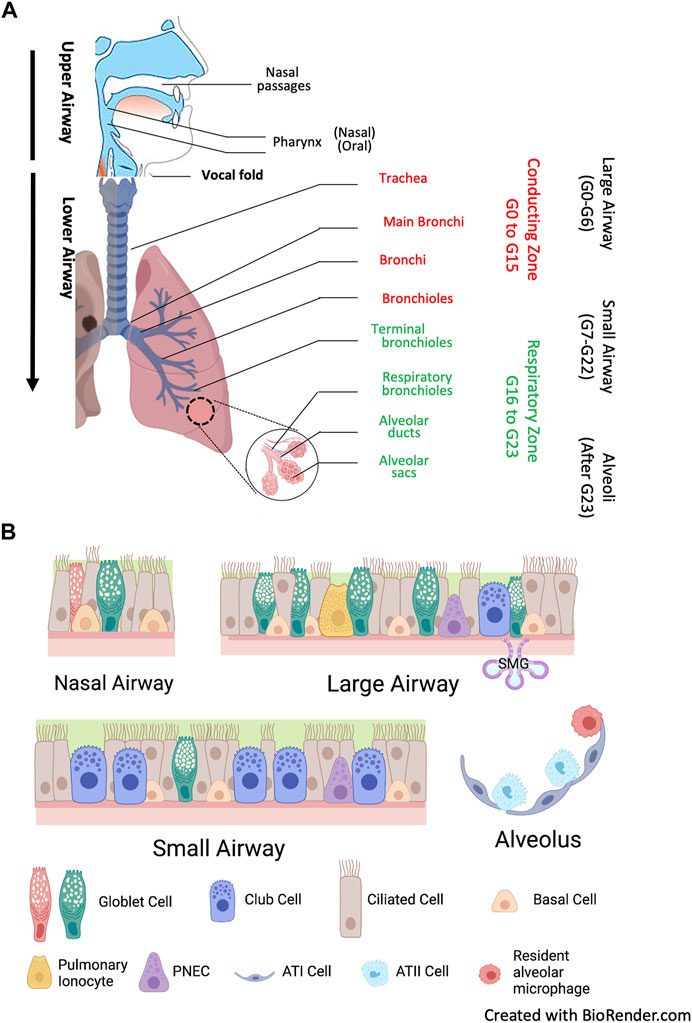
FIGURE 2. Human airways and major airway epithelial cell types. (A) Anatomy. The human airways are divided into the upper airways, lower airways and the lung parenchyma. The upper airways include the nose and nasal passages, paranasal sinuses, the pharynx, and the portion of the larynx above the vocal folds (cords). The lower airways include conducting zone from generation (G) G0 to G16, and respiratory zone from G17 and beyond. The trachea and proximal bronchi (generation G0 to G6) are considered as large airways. The small airways are distal bronchioles whose diameter is smaller than 2mm, starting from G7 the terminal bronchioles in conducting zone to the respiratory bronchioles in respiratory zone. After G23, the airway epithelium merges with the alveolar epithelium with pneumocyte type I and type II cells (alveolar type I and II cells, ATI and ATII cells) in lung parenchyma. (B) Airway epithelium is a continuous cellular layer with epithelial cell populations and functions vary along the respiratory tree. It is pseudostratified in the nasal and large airways, becoming columnar and cuboidal in the small airways. Submucosal glands (SMG) are majorly found in large airways. Major surface epithelial cell types are columnar ciliated cells, goblet cells, club cells and basal cells. Their composition is similar in large and small airways, but with gradually less ciliated cells, and the secretory cells shift from goblet cells to the dome-shaped club cells. Airway basal cells have the capacity to differentiate into the major cell types of the airways, and a subset of club cells can differentiate to ciliated cells and goblet cells. Pulmonary ionocytes express high level CFTR, but the mechanism by which they contributed to cystic fibrosis are still largely unknown. Ionocytes may be absent in the small airways. Pulmonary neuroendocrine cells (PNEC) serve as key communicators between the immune and nervous system.
CFTR protein and mRNA expression are abundant in type II pneumocytes (alveolar type II cells, ATII) in the lung parenchyma, club cells in the respiratory bronchioles, and serous cells of SMG (Engelhardt et al., 1992; McCray et al., 1992; Engelhardt et al., 1994). However, CFTR is only expressed in a small subset of cells in the surface airway epithelium. Recent research using single-cell RNA sequencing (scRNA-seq) has uncovered more cellular heterogeneity within the airways and the correlations with CFTR expression (Montoro et al., 2018; Plasschaert et al., 2018). The newly identified pulmonary ionocytes express CFTR at the highest level per cell; thus, they represent a relatively large proportion of the total CFTR transcripts in the airways. However, ionocytes comprise <1% of epithelial cells along the conducting airways, even though they are abundant in the submucosal gland ducts and the surface airway surrounding these glands (Yu et al., 2022). Ionocytes are involved in pH regulation, ion transport, and hydration of the airways, however, it remains unclear whether ionocyte depletion in human airways leads to chronic inflammation and spontaneous bacterial infection, which are the hallmarks of CF. In mice, FOXI1 gene is necessary for the expression of ionocyte markers in the trachea. Knockout of FOXI1 in mouse caused a loss of CFTR expression and disrupts airway fluid and mucus physiology (Montoro et al., 2018). However, there are differences in airway epithelial cell biology between mice and humans (Pan et al., 2019). Patients with homozygous missense mutations in FOXI1 present with early-onset sensorineural deafness and distal renal tubular acidosis, but not associate with lung disease (Enerback et al., 2018). As ionocytes are not the sole CFTR-expressing airway epithelial cell type, targeting ionocytes in the treatment of the lungs with CF in humans may not be crucially necessary (Barbry et al., 2021).
Analyses from scRNA-seq combining cell number and level of CFTR expression suggested that a rank order for total CFTR expression in human superficial airway epithelium was secretory cells, basal cells, ionocytes, and ciliated cells, respectively (Okuda et al., 2021). Several scRNA-seq studies have drawn a consistent picture with secretory cells as major sites for CFTR expression in human surface airways (Deprez et al., 2020; Goldfarbmuren et al., 2020; Habermann et al., 2020; Miller et al., 2020). Due to the rarity of ionocytes, it is possible that the proportion of CFTR signals transcribed from other cell types that express lower levels of CFTR but are abundant in the surface airways may better correlate with CFTR protein function required for airway surface fluid homeostasis. Indeed, a recent study suggested that secretory cells dominate CFTR expression and function in human airway epithelia, and that CFTR-mediated hydration is associated with secretory cells (Okuda et al., 2021). Thus, while CF therapy may need to restore CFTR function in multiple cell types, including ionocytes, a major target likely is the airway secretory cells. However, secretory cells in the conducting airways (goblet cells) also differ in their biology to those in the bronchioles (club cells) (Dean and Snelgrove, 2018); thus, CFTR may play a different role in these two types of secretory cell. Additionally, it is difficult for gene transfer agents to access the secretory cells underneath the surface airways, such as in areas surrounding the SMG.
Ciliated cells are the first contact accessible to the gene transfer agents delivered from the airway lumen. They had long been thought to be a target for CF gene therapy (Kreda et al., 2005; Zhang et al., 2009). However, more recent studies by scRNA-seq discovered that little to no CFTR mRNA transcripts are detected in this cell type. In light of this, it remains unclear whether ectopic expression of CFTR in ciliated cells are able to compensate the CF lung function if CFTR expression is not sufficiently reinstalled in secretory cells and inonocytes. It is also unknown whether the vector infection and the exogenous CFTR expression in this cell type will cause ciliopathies that impact its functions in MCC and normal innate immunity. Comprehensive preclinical studies of CFTR delivery in animal models of CF and an understanding of CFTR function at the cellular level to control airway clearance and innate immunity will help to resolve these unanswered questions.
CF is a multi-organ disease, and pulmonary disease is the main cause of morbidity and mortality. While CFTR is commonly known an ion channel protein that conducts chloride (Cl−) and bicarbonate (HCO3−) transport on the apical membrane of the epithelial cells, the pathophysiology of CF is more challenging than a mere dysregulation of epithelial ion transport. CFTR also interacts with other ion channels, such as epithelial sodium channel (ENaC) (Shei et al., 2018), to regulate the airway surface fluid (ASL) movement, pH homeostasis and mucus viscosity (Wu et al., 2018). Therefore, this interaction plays a major role in the MCC and innate immunity to defend the lung infections. The loss of functional CFTR in CF results in dysregulation of their functions. In the lung airways, the CFTR gene defect results in the loss of a functional CFTR expression (either absence or malfunction of CFTR protein). The abnormal ion conductance leads to dehydration of ASL, impaired MCC, airway obstruction by viscous sputum and abnormally thick and sticky mucus promotes chronic infection and inflammation. The consequence of this cascade is inflammation and progressive chronic endobronchial bacterial infection, the hallmarks of CF lungs, resulting in permanent lung damages (Ratjen, 2009; Huang et al., 2021). Additionally, CFTR is also expressed by immune cells, including alveolar macrophages and neutrophils, which help maintaining immunological and physiological homeostasis in the lungs and are the front line of cellular defense against pathogens that were not eliminated by the mechanical defenses of the airways (Yoshimura et al., 1991). Impaired immune cellular defense has been reported in CF (Leveque et al., 2017). While the current gene therapy development efforts target life-threatening lung disease via CFTR replacement (gene addition) or mutation repair (gene editing), it is worth mentioning that Cl− secretion in airway epithelial cells is not limited to CFTR. CFTR knockout mice do not develop the typical lung disease phenotype seen in humans with CF, which is probably at least partially attributable to up-regulation of the alternative chloride channels (Guilbault et al., 2007). CFTR has also been proposed to inhibit ENaC activity through direct physical interactions, and dehydration of CF ASL is driven by ENaC (Stutts et al., 1995). Thus, stimulation of alternative chloride channels (Xu et al., 2005; Liu et al., 2015; Quesada and Dutzler, 2020) or inhibition of sodium absorption (Blacona et al., 2022) may be able to compensate for the lack of CFTR-mediate Cl− secretion in CF airways or to maximize the efficacy of gene therapies aiming at CFTR. Notably, it was reported that proteasome modulation agent doxorubicin facilitated long-term functional inhibition of ENaC currents. Doxorubicin has been used as an augmenter to enhance rAAV-mediated CFTR transfer in human airway epithelium and it was found that the inhibition of ENaC activity was predominantly attributed to a doxorubicin-dependent decrease in γ-ENaC subunit mRNA expression and an increase in γ-ENaC promoter methylation, independent of CFTR vector administration (Zhang et al., 2004).
Ciliated cells, goblet cells, and club secretory cells on surface airway epithelia are the primary cell types accessible to the CFTR transfer vectors applied luminally. These cell types are terminally differentiated with a defined lifespan, except for a subset of club cells in small airways which may have the ability to differentiate into other cell types (Rawlins et al., 2009; Dean and Snelgrove, 2018). Even if basal cells, the stem cell progenitors underneath the surface layer, could be exposed to the vectors by transient disruption of the tight junctions, the episomal rAAV genomes in the transduced cells will be diluted out on serial passage during the cell proliferation. Because of the relatively slow turnover rate of epithelia (Rawlins and Hogan, 2008), treatment of CF patients can be periodic as durable, functional complementation in the transduced cells likely persists for a certain period or through their lifespan. Nevertheless, using rAAV for the treatments to CF lungs requires multiple repeated dosing to achieve sustained CFTR expression for the life of the individual. This notion is likely also true for the integrating lentiviral vectors, unless the basal cells are transduced.
Pre-existing humoral immunity to AAV capsids is a potential barrier to rAAV vector-mediated gene therapy, especially following re-administration of the same rAAV vector (Boutin et al., 2010; Masat et al., 2013; Mingozzi and High, 2013). Besides the elicitation of neutralizing antibodies (NAbs), vector immunogenicity also represents the major cause of the destruction of transduced cells, which is mediated by the induced cytotoxic T lymphocytes (CTL) response after transduction (Vandamme et al., 2017; Ronzitti et al., 2020; Verdera et al., 2020). While little is known of the effects of T cells on the duration of rAAV transduction administrated through airways, the involvement of CD8+ T cells in the destruction of rAAV-transduced hepatocytes was reported in the rAAV2 hemophilia B clinical trial (Mingozzi et al., 2007). Transgene protein product-specific CTL was also observed in rAAV trials for Duchenne’s Muscular Dystrophy (Mendell et al., 2010) and α1-antitrypsin (Calcedo et al., 2017). Lentiviral vectors may face similar or the same immune response problems as rAAV vectors.
Pharmaceutical immunosuppression is likely able to dampen the immune response to repeat administration of rAAV, through global effects, T-Cell or/and B-Cell specific effects, or other mechanisms (Jiang et al., 2006; Arruda et al., 2009; Mingozzi et al., 2013; Chu and Ng, 2021). However, the approaches that had been tested in gene therapy trials via local (intramuscular injection) or systemic (intravenous injection) rAAV delivery necessitate patients to be maintained with daily administration of a regime of immunosuppressants for a relative long period (Corti et al., 2014; Corti et al., 2015). Such treatment might result in chronic immune suppression that is intolerable in CF patients, as the CF lungs are prone to bacterial infections. A recent study of repeat dosing with rAAV1 in the lungs of rhesus macaques demonstrated that a brief course of treatment with methylprednisolone succinate prior to infection boosted the efficiency of transduction and increased the longevity of transgene expression (Yanda et al., 2022). Methylprednisolone is a mild immunosuppressant, it has been used in patients with CF for several indications, including bronchiolitis, bronchial hyperreactions, aspergillosis, and mild-to-moderate obstructive pulmonary disease (Lai et al., 2000; Thomson et al., 2006). However, as the CF condition is not duplicated in non-human primates, the application of methylprednisolone for rAAV repeated dosing requires studies using CF animal models (pigs or ferrets) to establish the minimal effective dose and to clarify the safety and feasibility.
Theoretically, genome manipulation is able to precisely correct (edit) any mutations of a target gene via homologous recombination (HR) without altering its endogenous patterns of expression. Although the efficiency of HR in mammalian cells is extremely low, it was found that induction of double strand DNA break (DSB) could raise the incidence of HR by multiple orders of magnitude. Thus, a key step to genome manipulation or gene editing is the generation of a sequence-specific DSB at the target gene. Thus, gene editing requires sequence-specific endonucleases to create DSB at desired site and relies on the followed repair of the DSB by two cellular DNA damage repair mechanisms: the non-homologous end joining (NHEJ) and the homology-directed repair (HDR) (Ceccaldi et al., 2016). However, the gene therapy field long lacked such a tool of “molecular scissors” until the development of engineered programable nucleases in 2010. Zinc finger nucleases (ZFN) (Urnov et al., 2010) and transcription activator-like effector nucleases (TALEN) (Zhang et al., 2011) are the first two programable nucleases developed to manipulate a gene of interest with practically high specificity and efficiency. ZFNs are fusion proteins built with an array of zinc finger domains, that each recognizes a 3 bp DNA motif, attached to the endonuclease domain of the bacterial FokI restriction enzyme. TALENs are also proteins fused to the catalytic domain of the FokI nuclease. They use transcription activator-like effectors (TALEs) rather than zinc fingers to recognize specific DNA sequences. ZFN- and TALEN -mediated HDR to correct the mutations at endogenous CFTR loci in CF patient-derived iPSCs (Crane et al., 2015; Fleischer et al., 2020) and TALEN-mediated site-specific integration of a CFTR minigene at the AAVS1 safe harbor (Xia et al., 2019) are the examples of using these tools in the research for CF gene therapy.
The later application of the bacterial “adaptive immune system”, namely the clustered regularly interspaced short palindromic repeats (CRISPR) system, in mammalian genetic engineering has exhibited a facile, highly versatile and efficient gene editing tool that the field of gene therapy has long desired (Cox et al., 2015; Araldi et al., 2020). Distinct from the ZFNs and TALENs that require custom protein design for each target, CRISPR-associated proteins (Cas) are enzymes that associate with CRISPR RNAs to bind to and alter DNA or RNA target sequences. The CRISPR system for gene editing is comprised of a Cas and a single guide RNA (sgRNA) that site-specifically guides the complex to the gene of interest. Acting as a programmable endonuclease (with Cas) (Jinek et al., 2012) or nickase (with nCas) (Ran et al., 2013), the complex is able to generate a DSB or create a single-stranded break in the targeted gene in a site-specific manner. When catalytically inactive Cas (dCas) is used, the complex binds to the target without creating strand breaks (Liu et al., 2016). CRISPR-based gene therapy has shown promise in sickle cell disease by correcting the mutation in hematopoietic stem cells ex vivo (Frangoul et al., 2021) and in vivo in Leber congenital amaurosis (LCA) by deleting delete the dominantly inherited intronic IVS26 loss-of-function mutation (Maeder et al., 2019) and in transthyretin amyloidosis by disrupting the disease-causing allele (Gillmore et al., 2021). It presents new opportunities to advance CF gene therapy from conventional gene addition to precise gene correction of any CFTR mutation at the endogenous locus, by which, theoretically, the complexities of CFTR regulation at the cellular level in the lung could be solved (Yan et al., 2019; Vu and McCray, 2020).
The CRISPR/Cas-nuclease approach relies on the generation of a DSB at the desired site of a target gene. The DSB activates the cellular DNA damage response (DDR) by the recruitment of repair factors to the site of DNA damage, and thus, gene correction is enabled by the cellular DSB repair machinery, either through homology-directed repair (HDR) or non-homologous end joining (NHEJ) (Lieber, 2010). With the provided template for gene correction, the HDR response enables homologous recombination (HR) between the template and the target gene at the cleaved site to introduce desired correction, insertion, or deletion (Paquet et al., 2016). NHEJ directly ligates the ends of the breaks, however, it can generate unpredictable insertions and deletions of various lengths (indels) (Betermier et al., 2014). HDR and NHEJ are competing processes. Under most conditions, NHEJ is more efficient than HDR. HDR is highly cell cycle-dependent; it occurs only in the G2 and S phases of the replication cycle (Heyer et al., 2010). Thus, most mitotically quiescent cells and well-differentiated cells do not support HDR. Although CRISPR/Cas-nuclease generates DSBs in a site-specific manner, unintended off-target cleavage is a safety concern (Fu et al., 2013). Moreover, unpredictable rearrangement and translocation, as well as a high ratio of undesired indel byproducts, are potential results of the repair (Jeggo, 1998; Kosicki et al., 2018).
Base editing (BE) (Gaudelli et al., 2017) and prime editing (PE) (Anzalone et al., 2019) are the next generation of CRISPR-based gene editing without the need for DSB. BE utilizes a dCas9 or nCas9 fused with a deaminase domain to edit specific loci, allowing for the introduction of point mutations through direct enzymatic C>T base conversion (cytidine base editor) or A>G base conversion (adenine base editor). However, the application of BE is limited by the presence of a compatible protospacer adjacent motif (PAM) at the mutation to be edited and by the availability of A>G and C>T transitions and C>G transversions (Rees and Liu, 2018). PE enables single-base substitution for all 12 potential transition and transversion reactions. Moreover, it facilitates alterations in short insertions or deletions without the need for a donor DNA template. PE consists of a Cas9 nickase fused with a modified reverse transcriptase and a multifunctional prime editing guide RNA (pegRNA), which directs the fusion complex to the target site for flexible PAM recognition and also encodes an RNA template for editing as a contiguous extension of the gRNA. These CRISPR-based gene editing tools are outlined in Figure 3.
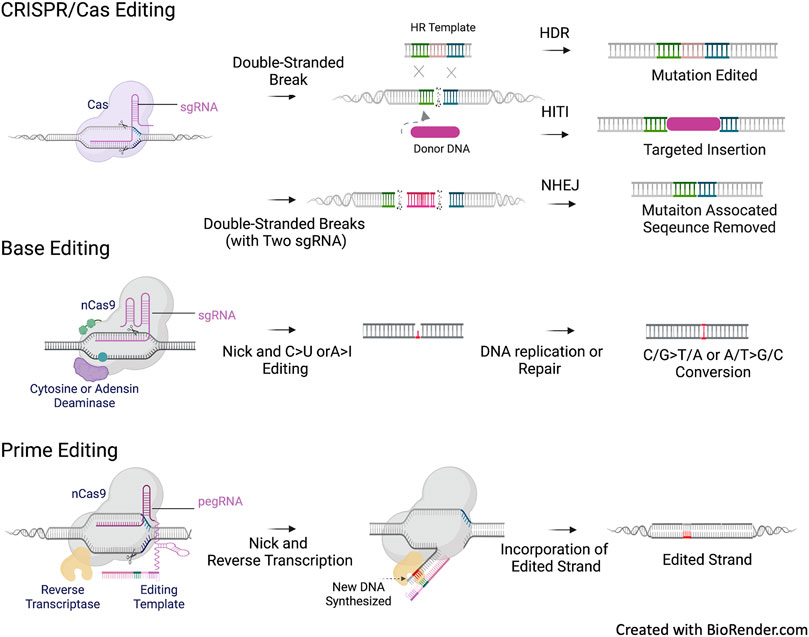
FIGURE 3. CFTR mutation types and CREIPSR-based Gene Editing. CRIPSR/Cas editing approaches rely on the generation of double stranded DNA break (DSB) and the following repair via non-homologous end joining (NHEJ) or homologous recombination (HR). NHEJ can be used for the allele specific repair of splicing mutation in the intronic sequence, such as the 3272-26A>G (c.3140–26A>G) and 3,849 + 10kbC>T (c.3718–2477C>T) CFTR mutations. Homology-independent targeted insertion (HITI) is an approach based on NHEJ. Theoretically, HITI can universally address all CFTR variants independent of genotypes, the donor DNA can be a splicing acceptor associated CFTR cDNA sequence to be inserted into the intronic sequence of CFTR to reconstitute a mutation free mini gene, or a CFTR expression cassette to be inserted into the safe harbor of the chromosome, such as AAVS1. Homology-directed repair (HDR) requires a template for HR to correct point mutation or small deletion/deletion. Base editing and prime editing do not require the generation of DSB and DNA template for correction. Base editing is only used to correct single base point mutation, and primer editing can be used to correct point mutation as well as short deletion/insertion. Same as HDR, the correction options of base editing and prime editing are mutation dependent.
CRISPR-based gene editing approaches have been explored to correct certain CFTR mutations in cultured airway basal cells, iPS cells, and advanced cellular models, such as organoid cultures derived from patients with CF (Ensinck et al., 2021; Lee et al., 2021). HDR-based CFTR corrections have demonstrated the capacities to correct missense or nonsense mutations in exons, to ablate splice mutations, and to insert “super exons” from the DNA donor template provided along with the CRISPR components (Schwank et al., 2013; Firth et al., 2015). HDR has succeeded in repairing F508del CFTR in primary human airway basal cell cultures (Suzuki et al., 2020; Vaidyanathan et al., 2020) and G551D CFTR in proliferating ferret airway basal cells, to which the components of the CRISPR system were delivered by either transfection or rAAV infection. However, the efficiency of HDR is low because the error-prone NHEJ dominates the cellular DNA repair machinery. Next-generation sequencing analyses of the targeted amplicon from rAAV-infected ferret airway basal cells (without selection or enrichment) revealed a high fidelity with HDR of only 0.55% corrected G551 alleles associated with additional modifications, but the 13.21% correction rate of the G551D mutation with HDR (including imperfect HDR events) was lower than the 22.57% rate of undesired mutagenesis caused by NHEJ (Yan et al., 2022). Because NHEJ is efficient in both dividing and non-dividing cells, NHEJ-based gene editing, which only requires the expression of a Cas protein with pairs of gRNAs, appears practicable, particularly in the repair of aberrant CFTR mRNA transcripts caused by intronic splicing mutations. The efficacy of this gene-correction strategy has been verified in intestinal organoids and airway epithelial cells derived from patients with CF, who carry the mutations of c.1679 + 1.6kbA>G (1811 + 1.6kbA>G), or c.3140–26A>G (3272-26A>G) or c.3717 + 12191C>T (3,849 + 10kbC>T), with an allele-specific repair efficiency of ∼40% and complete functional recovery of the CFTR channel (Sanz et al., 2017; Maule et al., 2019). A more complicated NHEJ editing strategy is homology-independent targeted insertion (HITI) (Suzuki et al., 2016). HITI requires a donor DNA and targeted integration with correct orientation. It has been reported that targeted insertion of a splicing acceptor-associated CFTR minigene of exons 9–27 into CFTR intron eight of F508del/F508del airway basal cells was able to reconstitute the functional CFTR expression in the edited cells, although the efficiency was low. Other approaches include targeted insertion of a CFTR minigene expression cassette into a genome safe-harbor or the CFTR locus (Xia et al., 2019; Zhou et al., 2019).
BE is an attractive approach for gene editing based CF gene therapy because many disease-causing CFTR variants can be corrected using single base conversion (Geurts et al., 2020). Adenine base editor has been used to correct premature stop mutations in R553X and W1282X, as well as the 3,849 + 10 kb C>T splicing mutation in human airway epithelial cells at 38%–82% efficiency with minimal bystander edits and indels (Krishnamurthy et al., 2021). The repair of the CFTR-R785X mutation has been compared between BE and PE approaches in patient-derived intestinal organoids (Geurts et al., 2021). Adenine base editor demonstrated a correction efficiency of 9.1%, whereas the best-performing PE achieved a correction efficiency of 5.7%. Although both approaches outperformed the conventional HDR, which demonstrated a correction efficiency of 1.22%, BE appears to be superior to prime editing in terms of both safety and efficiency when the mutation is targetable. Nevertheless, PE can theoretically correct mutations that cannot be corrected with BE. PE has demonstrated its capacity to repair the most common CFTR mutation (F508del) in patient-derived intestinal organoids, although the efficiency was lower than that of HDR (Geurts et al., 2021).
The applications of gene editing in humans are undergoing with several clinical trials: one is an ex vivo mutation repair strategy, using autologous CRISPR-Cas9 modified hematopoietic stem cells to treat hematopoietic stem cells (NCT04208529); another two are in vivo strategies for LCA, using rAAV to express spCas9 and two gRNAs in eyes (NCT03872479), and for transthyretin amyloidosis [(hereditary transthyretin amyloidosis with polyneuropathy (ATTRv-PN) and transthyretin amyloidosis-related cardiomyopathy (ATTR-CM)), using lipid nanoparticle (LNP) encapsulating Cas9 mRNA and sgRNA (NCT04601051). While these trials are promising, the implementation of CRISPR-based editing to many inherited diseases with more complex etiologies, like CF, will require more challenging approaches.
CRISPR/Cas9 HDR approaches can be used to target CFTR gene correction to airway stem cells. Currently, the CFTR gene editing approaches have been explored in cultured primary airway basal cells, iPS cells, and organoid cultures derived from patients with CF. Theoretically, gene editing in airway basal cells in vitro can be used for autologous cell therapy to regenerate airways with CFTR functional cells. It requires autologous corrected cells to generate a sufficient cell quantity through ex vivo expansion and to have the capacity to engraft, proliferate, and persist long-term in recipient airways without complications (Berical et al., 2019; King et al., 2020). However, unlike the current cell-based therapies such as the hematopoietic cell transplantation, an practical strategy to deliver autologous gene-edited airway basal cells for CF and any lung disease has not yet developed. As well, there is a lack of method to control the mucus and infection to allow engraftment of cells. An engraftment strategy would involve minimal airway injure, but the prospect of deliberately injuring the lungs of CF recipients to ablate the endogenous cells is daunting. (Berical et al., 2019).
Notably, for in vivo application, HDR is inefficient in the repair of DSBs in quiescent and differentiated cell types of the airway epithelium. The HITI approaches (Suzuki et al., 2016; Xia et al., 2019; Suzuki et al., 2020) appear compatible in editing basal cells and post-mitotic airway epithelia, however, a lifelong cure is only achievable if the airway basal cells are efficiently targeted for permanent correction. Editing of CFTR in vector-accessible surface airway epithelial cell types may produce durable CFTR activity for a certain period, but similar to the gene addition approaches, repeat dosing is required. Because in vivo lung gene editing encounters the similar dilemma in delivery as do the gene addition approaches and also the efficiency of current HITI efficiency to reconstitute a function expression of CFTR in vitro is still sufficiently low, studies have not yet progressed toward CFTR correction in vivo due to the lack of robust delivery methods to efficiently target basal cells. The current available viral vector systems have limited utility to deliver all the components of the CRISPR-based gene editing system with in a single vector, especially for the base- and prime editors that are large size cas9-based fusion proteins, thus, engineered DNA-free virus-like particles that can be used for in vivo editing may offer additional therapeutic options (Banskota et al., 2022). Although it is more challenging to edit basal progenitor cells in vivo than proliferating cells in vitro, the development of a robust delivery system enabling efficient basal cell targeting and correction will translate current proof-of-concept studies into real therapies for patients with CF.
Restoration of lung function in patients with CF relies on rebuilding normal homeostatic mechanisms that require the interaction between transgene-expressed CFTR and other ion channels to regulate effective airway clearance and innate immunity (Shah et al., 2016). Effective gene therapies for CF lung disease have been pursued since the discovery of CFTR in 1989. Researchers in both academia and industry have contributed significant resources and talent to tackling the challenges encountered in the long journey toward a cure for CF. Currently, gene editing is still in its infancy. rAAV based CFTR-replacement strategies for CF remain the primary hope for clinical success. Although now the field of CF gene therapy is equipped with the next generation rAAV based transduction vectors highly tropic to human airway, the greatest challenge is to deliver a vector to as many of the epithelial cells that line the surface airway as possible, and permanent correction requires the airway basal progenitor cells to be targeted. There is also an urgent need of safe and effective strategies to reduce the vector immunogenicity for repeated dosing. The safety is particularly important for the application of immunomodulation in CF lungs, which are prone to bacterial infection. While the discussion of this review is mostly focused on the applications of viral vector strategies, it is worth to note here that development of the polymeric nanoparticles as new delivery tool for gene therapy. A recent study showed that intratracheal delivery of nanoparticles-formulated thymulin-expressing plasmids was capable to penetrate the airway mucus barrier, which therapeutically reversed the key pathology of experimental allergic asthma in mice (da Silva et al., 2020). The nanoparticles-encapsulating mRNA delivery techniques are getting matured now, its application expand in the field of gene therapy rapidly. Currently, there is an ongoing CF clinical trial in phase1/2 to test the delivery of a drug called MRT5005, which uses lipid nanoparticles to deliver an mRNA encoding the full-length CFTR, to the CF lungs via nebulizer (NTC03375047; https://www.cff.org/Trials/Pipeline/details/10157/MRT5005). Thus, LPN-mediated Cas9 mRNA delivery for in vivo lung gene editing could become possible. The availability of large-animal CF models enables preclinical studies to validate new therapies, and also facilitates the basic biomedicine research to identify the pathophysiologically relevant target cell types for effective CF gene therapy and to test the new delivery tools in CF airways. CF is a complex disease, and the success of gene therapy for CF will hopefully also define a path for the treatment of other complex and devastating diseases.
HS: Writing original draft, editing and finalizing, supervision, funding acquisition; XX: Literature collecting and reviewing, writing original draft; YS, JW,TB, MY, XL, TZ, and ZJ: Literature collecting, discussion, and reviewing; JZ, CX and ML: discussion, editing, final draft proof reading and funding acquisition.
This work was supported by the National Natural Science Foundation of China grants #32072738 (ML) and #81670004 (HS); Doctoral Startup Fund of Shandong First Medical University #001003053 (XX); Academic Promotion Programme of Shandong First Medical University #2019QL013 (JZ); the Scientific and Technological Innovation Major Base of Guangxi #2018-15-Z04 (CX); the State Project for Essential Drug Research and Development #2019ZX09301132 (CX), and Guangxi Key Research and Development Project # AB20117001 (CX).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aitken, M. L., Moss, R. B., Waltz, D. A., Dovey, M. E., Tonelli, M. R., McNamara, S. C., et al. (2001). A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum. Gene Ther. 12 (15), 1907–1916. doi:10.1089/104303401753153956
Allan, K. M., Farrow, N., Donnelley, M., Jaffe, A., and Waters, S. A. (2021). Treatment of cystic fibrosis: From gene- to cell-based therapies. Front. Pharmacol. 12, 639475. doi:10.3389/fphar.2021.639475
Allander, T., Tammi, M. T., Eriksson, M., Bjerkner, A., Tiveljung-Lindell, A., and Andersson, B. (2005). Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. U. S. A. 102 (36), 12891–12896. doi:10.1073/pnas.0504666102
Alton, E. W., Stern, M., Farley, R., Jaffe, A., Chadwick, S. L., Phillips, J., et al. (1999). Cationic lipid-mediated CFTR gene transfer to the lungs and nose of patients with cystic fibrosis: a double-blind placebo-controlled trial. Lancet 353 (9157), 947–954. doi:10.1016/s0140-6736(98)06532-5
Alton, E. W., Baker, A., Baker, E., Boyd, A. C., Cheng, S. H., Coles, R. L., et al. (2013). The safety profile of a cationic lipid-mediated cystic fibrosis gene transfer agent following repeated monthly aerosol administration to sheep. Biomaterials 34 (38), 10267–10277. doi:10.1016/j.biomaterials.2013.09.023
Alton, E. W., Boyd, A. C., Cheng, S. H., Davies, J. C., Davies, L. A., Dayan, A., et al. (2014). Toxicology study assessing efficacy and safety of repeated administration of lipid/DNA complexes to mouse lung. Gene Ther. 21 (1), 89–95. doi:10.1038/gt.2013.61
Alton, E., Armstrong, D. K., Ashby, D., Bayfield, K. J., Bilton, D., Bloomfield, E. V., et al. (2015). Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet. Respir. Med. 3 (9), 684–691. doi:10.1016/S2213-2600(15)00245-3
Alton, E. W., Beekman, J. M., Boyd, A. C., Brand, J., Carlon, M. S., Connolly, M. M., et al. (2017). Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 72 (2), 137–147. doi:10.1136/thoraxjnl-2016-208406
Andersen, D. (1938). Cystic fibrosis of the pancreas and its relation to celiac disease. Am. J. Dis. Child. 56 (2), 344–399. doi:10.1001/archpedi.1938.01980140114013
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576 (7785), 149–157. doi:10.1038/s41586-019-1711-4
Araldi, R. P., Khalil, C., Grignet, P. H., Teixeira, M. R., de Melo, T. C., Modolo, D. G., et al. (2020). Medical applications of clustered regularly interspaced short palindromic repeats (CRISPR/Cas) tool: A comprehensive overview. Gene 745, 144636. doi:10.1016/j.gene.2020.144636
Arruda, V. R., Favaro, P., and Finn, J. D. (2009). Strategies to modulate immune responses: a new frontier for gene therapy. Mol. Ther. 17 (9), 1492–1503. doi:10.1038/mt.2009.150
Asokan, A., Schaffer, D. V., and Samulski, R. J. (2012). The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther. 20 (4), 699–708. doi:10.1038/mt.2011.287
Bandara, R. A., Chen, Z. R., and Hu, J. (2021). Potential of helper-dependent Adenoviral vectors in CRISPR-cas9-mediated lung gene therapy. Cell Biosci. 11 (1), 145. doi:10.1186/s13578-021-00662-w
Banskota, S., Raguram, A., Suh, S., Du, S. W., Davis, J. R., Choi, E. H., et al. (2022). Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 185 (2), 250–265.e16. doi:10.1016/j.cell.2021.12.021
Barbry, P., Marcet, B., and Caballero, I. (2021). Where is the cystic fibrosis transmembrane conductance regulator? Am. J. Respir. Crit. Care Med. 203 (10), 1214–1216. doi:10.1164/rccm.202012-4434ED
Beck, S. E., Jones, L. A., Chesnut, K., Walsh, S. M., Reynolds, T. C., Carter, B. J., et al. (1999). Repeated delivery of adeno-associated virus vectors to the rabbit airway. J. Virol. 73 (11), 9446–9455. doi:10.1128/JVI.73.11.9446-9455.1999
Beck, S. E., Laube, B. L., Barberena, C. I., Fischer, A. C., Adams, R. J., Chesnut, K., et al. (2002). Deposition and expression of aerosolized rAAV vectors in the lungs of Rhesus macaques. Mol. Ther. 6 (4), 546–554. doi:10.1006/mthe.2002.0698
Berical, A., Lee, R. E., Randell, S. H., and Hawkins, F. (2019). Challenges facing airway epithelial cell-based therapy for cystic fibrosis. Front. Pharmacol. 10, 74. doi:10.3389/fphar.2019.00074
Betermier, M., Bertrand, P., and Lopez, B. S. (2014). Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 10 (1), e1004086. doi:10.1371/journal.pgen.1004086
Biasco, L., Rothe, M., Buning, H., and Schambach, A. (2018). Analyzing the genotoxicity of retroviral vectors in hematopoietic cell gene therapy. Mol. Ther. Methods Clin. Dev. 8, 21–30. doi:10.1016/j.omtm.2017.10.002
Blacona, G., Raso, R., Castellani, S., Pierandrei, S., Del Porto, P., Ferraguti, G., et al. (2022). Downregulation of epithelial sodium channel (ENaC) activity in cystic fibrosis cells by epigenetic targeting. Cell. Mol. Life Sci. 79 (5), 257. doi:10.1007/s00018-022-04190-9
Bobadilla, J. L., Macek, M., Fine, J. P., and Farrell, P. M. (2002). Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum. Mutat. 19 (6), 575–606. doi:10.1002/humu.10041
Boutin, S., Monteilhet, V., Veron, P., Leborgne, C., Benveniste, O., Montus, M. F., et al. (2010). Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 21 (6), 704–712. doi:10.1089/hum.2009.182
Brown, S. D., White, R., and Tobin, P. (2017). Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment. JAAPA 30 (5), 23–27. doi:10.1097/01.JAA.0000515540.36581.92
Brunetti-Pierri, N., and Ng, P. (2006). Progress towards the clinical application of helper-dependent adenoviral vectors for liver and lung gene therapy. Curr. Opin. Mol. Ther. 8 (5), 446–454.
Bustamante-Marin, X. M., and Ostrowski, L. E. (2017). Cilia and mucociliary clearance. Cold Spring Harb. Perspect. Biol. 9 (4), a028241. doi:10.1101/cshperspect.a028241
Calcedo, R., Somanathan, S., Qin, Q., Betts, M. R., Rech, A. J., Vonderheide, R. H., et al. (2017). Class I-restricted T-cell responses to a polymorphic peptide in a gene therapy clinical trial for alpha-1-antitrypsin deficiency. Proc. Natl. Acad. Sci. U. S. A. 114 (7), 1655–1659. doi:10.1073/pnas.1617726114
Cao, H. B., Wang, A., Martin, B., Koehler, D. R., Zeitlin, P. L., Tanawell, A. K., et al. (2005). Down-regulation of IL-8 expression in human airway epithelial cells through helper-dependent adenoviral-mediated RNA interference. Cell Res. 15 (2), 111–119. doi:10.1038/sj.cr.7290275
Cao, H., Ouyang, H., Grasemann, H., Bartlett, C., Du, K., Duan, R., et al. (2018). Transducing airway basal cells with a helper-dependent adenoviral vector for lung gene therapy. Hum. Gene Ther. 29 (6), 643–652. doi:10.1089/hum.2017.201
Carroll, T. P., Morales, M. M., Fulmer, S. B., Allen, S. S., Flotte, T. R., Cutting, G. R., et al. (1995). Alternate translation initiation codons can create functional forms of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 270 (20), 11941–11946. doi:10.1074/jbc.270.20.11941
Cebotaru, L., and Guggino, W. B. (2014). Complement yourself: Transcomplementation rescues partially folded mutant proteins. Biophys. Rev. 6 (1), 169–180. doi:10.1007/s12551-014-0137-3
Ceccaldi, R., Rondinelli, B., and D'Andrea, A. D. (2016). Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26 (1), 52–64. doi:10.1016/j.tcb.2015.07.009
Chamayou, S., Sicali, M., Lombardo, D., Maglia, E., Liprino, A., Cardea, C., et al. (2020). The true panel of cystic fibrosis mutations in the Sicilian population. BMC Med. Genet. 21 (1), 89. doi:10.1186/s12881-020-0958-9
Choi, S. H., and Engelhardt, J. F. (2021). Gene therapy for cystic fibrosis: Lessons learned and paths forward. Mol. Ther. 29 (2), 428–430. doi:10.1016/j.ymthe.2021.01.010
Chu, W. S., and Ng, J. (2021). Immunomodulation in administration of rAAV: Preclinical and clinical adjuvant pharmacotherapies. Front. Immunol. 12, 658038. doi:10.3389/fimmu.2021.658038
Chu, C. S., Trapnell, B. C., Curristin, S. M., Cutting, G. R., and Crystal, R. G. (1992). Extensive posttranscriptional deletion of the coding sequences for part of nucleotide-binding fold 1 in respiratory epithelial mRNA transcripts of the cystic fibrosis transmembrane conductance regulator gene is not associated with the clinical manifestations of cystic fibrosis. J. Clin. Invest. 90 (3), 785–790. doi:10.1172/JCI115952
Chu, C. S., Trapnell, B. C., Curristin, S., Cutting, G. R., and Crystal, R. G. (1993). Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat. Genet. 3 (2), 151–156. doi:10.1038/ng0293-151
Clancy, J. P., Cotton, C. U., Donaldson, S. H., Solomon, G. M., VanDevanter, D. R., Boyle, M. P., et al. (2019). CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 18 (1), 22–34. doi:10.1016/j.jcf.2018.05.004
Cone, R. A. (2009). Barrier properties of mucus. Adv. Drug Deliv. Rev. 61 (2), 75–85. doi:10.1016/j.addr.2008.09.008
Conrad, C. K., Allen, S. S., Afione, S. A., Reynolds, T. C., Beck, S. E., Fee-Maki, M., et al. (1996). Safety of single-dose administration of an adeno-associated virus (AAV)-CFTR vector in the primate lung. Gene Ther. 3 (8), 658–668.
Cooney, A. L., Abou Alaiwa, M. H., Shah, V. S., Bouzek, D. C., Stroik, M. R., Powers, L. S., et al. (2016). Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight 1, 88730. doi:10.1172/jci.insight.88730
Cooney, A. L., McCray, P. B., and Sinn, P. L. (2018a). Cystic fibrosis gene therapy: Looking back, looking forward. Genes (Basel) 9 (11), 538. doi:10.3390/genes9110538
Cooney, A. L., Singh, B. K., Loza, L. M., Thornell, I. M., Hippee, C. E., Powers, L. S., et al. (2018b). Widespread airway distribution and short-term phenotypic correction of cystic fibrosis pigs following aerosol delivery of piggyBac/adenovirus. Nucleic Acids Res. 46 (18), 9591–9600. doi:10.1093/nar/gky773
Corti, M., Elder, M., Falk, D., Lawson, L., Smith, B., Nayak, S., et al. (2014). B-cell depletion is protective against anti-AAV capsid immune response: A human subject case study. Mol. Ther. Methods Clin. Dev. 1, 14033. doi:10.1038/mtm.2014.33
Corti, M., Cleaver, B., Clement, N., Conlon, T. J., Faris, K. J., Wang, G., et al. (2015). Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in pompe disease: Preclinical to clinical planning. Hum. Gene Ther. Clin. Dev. 26 (3), 185–193. doi:10.1089/humc.2015.068
Cox, D. B., Platt, R. J., and Zhang, F. (2015). Therapeutic genome editing: prospects and challenges. Nat. Med. 21 (2), 121–131. doi:10.1038/nm.3793
Crane, A. M., Kramer, P., Bui, J. H., Chung, W. J., Li, X. S., Gonzalez-Garay, M. L., et al. (2015). Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 4 (4), 569–577. doi:10.1016/j.stemcr.2015.02.005
Crystal, R. G., McElvaney, N. G., Rosenfeld, M. A., Chu, C. S., Mastrangeli, A., Hay, J. G., et al. (1994). Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat. Genet. 8 (1), 42–51. doi:10.1038/ng0994-42
da Silva, A. L., de Oliveira, G. P., Kim, N., Cruz, F. F., Kitoko, J. Z., Blanco, N. G., et al. (2020). Nanoparticle-based thymulin gene therapy therapeutically reverses key pathology of experimental allergic asthma. Sci. Adv. 6, eaay7973. doi:10.1126/sciadv.aay7973
Davis, J. D., and Wypych, T. P. (2021). Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 14 (5), 978–990. doi:10.1038/s41385-020-00370-7
Dean, C. H., and Snelgrove, R. J. (2018). New rules for club development: New insights into human small airway epithelial club cell ontogeny and function. Am. J. Respir. Crit. Care Med. 198 (11), 1355–1356. doi:10.1164/rccm.201805-0925ED
Deng, X., Yan, Z., Luo, Y., Xu, J., Cheng, F., Li, Y., et al. (2013). In vitro modeling of human bocavirus 1 infection of polarized primary human airway epithelia. J. Virol. 87 (7), 4097–4102. doi:10.1128/JVI.03132-12
Deprez, M., Zaragosi, L. E., Truchi, M., Becavin, C., Ruiz Garcia, S., Arguel, M. J., et al. (2020). A single-cell atlas of the human healthy airways. Am. J. Respir. Crit. Care Med. 202 (12), 1636–1645. doi:10.1164/rccm.201911-2199OC
Ding, W., Zhang, L., Yan, Z., and Engelhardt, J. F. (2005). Intracellular trafficking of adeno-associated viral vectors. Gene Ther. 12 (11), 873–880. doi:10.1038/sj.gt.3302527
Dong, J. Y., Fan, P. D., and Frizzell, R. A. (1996). Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 7 (17), 2101–2112. doi:10.1089/hum.1996.7.17-2101
Donnelley, M., and Parsons, D. W. (2018). Gene therapy for cystic fibrosis lung disease: Overcoming the barriers to translation to the clinic. Front. Pharmacol. 9, 1381. doi:10.3389/fphar.2018.01381
Duan, D., Yue, Y., Yan, Z., McCray, P. B., and Engelhardt, J. F. (1998). Polarity influences the efficiency of recombinant adenoassociated virus infection in differentiated airway epithelia. Hum. Gene Ther. 9 (18), 2761–2776. doi:10.1089/hum.1998.9.18-2761
Duan, D., Yue, Y., Yan, Z., Yang, J., and Engelhardt, J. F. (2000). Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Invest. 105 (11), 1573–1587. doi:10.1172/JCI8317
Enerback, S., Nilsson, D., Edwards, N., Heglind, M., Alkanderi, S., Ashton, E., et al. (2018). Acidosis and deafness in patients with recessive mutations in FOXI1. J. Am. Soc. Nephrol. 29 (3), 1041–1048. doi:10.1681/ASN.2017080840
Engelhardt, J. F., Yankaskas, J. R., Ernst, S. A., Yang, Y., Marino, C. R., Boucher, R. C., et al. (1992). Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet. 2 (3), 240–248. doi:10.1038/ng1192-240
Engelhardt, J. F., Zepeda, M., Cohn, J. A., Yankaskas, J. R., and Wilson, J. M. (1994). Expression of the cystic fibrosis gene in adult human lung. J. Clin. Invest. 93 (2), 737–749. doi:10.1172/JCI117028
Ensinck, M., Mottais, A., Detry, C., Leal, T., and Carlon, M. S. (2021). On the corner of models and cure: Gene editing in cystic fibrosis. Front. Pharmacol. 12, 662110. doi:10.3389/fphar.2021.662110
Excoffon, K. J., Koerber, J. T., Dickey, D. D., Murtha, M., Keshavjee, S., Kaspar, B. K., et al. (2009). Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc. Natl. Acad. Sci. U. S. A. 106 (10), 3865–3870. doi:10.1073/pnas.0813365106
Excoffon, K. (2020). The coxsackievirus and adenovirus receptor: virological and biological beauty. FEBS Lett. 594 (12), 1828–1837. doi:10.1002/1873-3468.13794
Fajac, I., and Sermet, I. (2021). Therapeutic approaches for patients with cystic fibrosis not eligible for current CFTR modulators. Cells 10, 2793. doi:10.3390/cells10102793
Farmen, S. L., Karp, P. H., Ng, P., Palmer, D. J., Koehler, D. R., Hu, J., et al. (2005). Gene transfer of CFTR to airway epithelia: Low levels of expression are sufficient to correct Cl- transport and overexpression can generate basolateral CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 289 (6), L1123–L1130. doi:10.1152/ajplung.00049.2005
Ferec, C., and Cutting, G. R. (2012). Assessing the disease-liability of mutations in CFTR. Cold Spring Harb. Perspect. Med. 2, a009480. doi:10.1101/cshperspect.a009480
Ferrari, S., Griesenbach, U., Geddes, D. M., and Alton, E. (2003). Immunological hurdles to lung gene therapy. Clin. Exp. Immunol. 132 (1), 1–8. doi:10.1046/j.1365-2249.2003.02124.x
Ferrari, S., Griesenbach, U., Shiraki-Iida, T., Shu, T., Hironaka, T., Hou, X., et al. (2004). A defective nontransmissible recombinant Sendai virus mediates efficient gene transfer to airway epithelium in vivo. Gene Ther. 11 (22), 1659–1664. doi:10.1038/sj.gt.3302334
Firth, A. L., Menon, T., Parker, G. S., Qualls, S. J., Lewis, B. M., Ke, E., et al. (2015). Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 12 (9), 1385–1390. doi:10.1016/j.celrep.2015.07.062
Fischer, A. C., Beck, S. E., Smith, C. I., Laube, B. L., Askin, F. B., Guggino, S. E., et al. (2003). Successful transgene expression with serial doses of aerosolized rAAV2 vectors in rhesus macaques. Mol. Ther. 8 (6), 918–926. doi:10.1016/j.ymthe.2003.08.015
Fleischer, A., Vallejo-Diez, S., Martin-Fernandez, J. M., Sanchez-Gilabert, A., Castresana, M., Del Pozo, A., et al. (2020). iPSC-Derived Intestinal Organoids from Cystic Fibrosis Patients Acquire CFTR Activity upon TALEN-Mediated Repair of the p.F508del Mutation. Mol. Ther. Methods Clin. Dev. 17, 858–870. doi:10.1016/j.omtm.2020.04.005
Flotte, T. R., Afione, S. A., Solow, R., Drumm, M. L., Markakis, D., Guggino, W. B., et al. (1993). Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J. Biol. Chem. 268 (5), 3781–3790. doi:10.1016/s0021-9258(18)53762-5
Flotte, T. R., Zeitlin, P. L., Reynolds, T. C., Heald, A. E., Pedersen, P., Beck, S., et al. (2003). Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients: a two-part clinical study. Hum. Gene Ther. 14 (11), 1079–1088. doi:10.1089/104303403322124792
Flotte, T. R. (1993). Prospects for virus-based gene therapy for cystic fibrosis. J. Bioenerg. Biomembr. 25 (1), 37–42. doi:10.1007/BF00768066
Flotte, T. R. (2000). Size does matter: overcoming the adeno-associated virus packaging limit. Respir. Res. 1 (1), 16–18. doi:10.1186/rr6
Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y. S., Domm, J., Eustace, B. K., et al. (2021). CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med. 384 (3), 252–260. doi:10.1056/NEJMoa2031054
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31 (9), 822–826. doi:10.1038/nbt.2623
Gadsby, D. C., Vergani, P., and Csanady, L. (2006). The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440 (7083), 477–483. doi:10.1038/nature04712
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2017). Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551 (7681), 464–471. doi:10.1038/nature24644
Geurts, M. H., de Poel, E., Amatngalim, G. D., Oka, R., Meijers, F. M., Kruisselbrink, E., et al. (2020). CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell 26 (4), 503–510. doi:10.1016/j.stem.2020.01.019
Geurts, M. H., de Poel, E., Pleguezuelos-Manzano, C., Oka, R., Carrillo, L., Andersson-Rolf, A., et al. (2021). Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci. Alliance 4, e202000940. doi:10.26508/lsa.202000940
Gillmore, J. D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M. L., et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385 (6), 493–502. doi:10.1056/NEJMoa2107454
Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M., and Abedi, M. R. (2018). Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 20 (5), e3015. doi:10.1002/jgm.3015
Goldfarbmuren, K. C., Jackson, N. D., Sajuthi, S. P., Dyjack, N., Li, K. S., Rios, C. L., et al. (2020). Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 11 (1), 2485. doi:10.1038/s41467-020-16239-z
Gregory, L. G., Harbottle, R. P., Lawrence, L., Knapton, H. J., Themis, M., and Coutelle, C. (2003). Enhancement of adenovirus-mediated gene transfer to the airways by DEAE dextran and sodium caprate in vivo. Mol. Ther. 7 (1), 19–26. doi:10.1016/s1525-0016(02)00021-7
Griesenbach, U., Inoue, M., Meng, C., Farley, R., Chan, M., Newman, N. K., et al. (2012). Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am. J. Respir. Crit. Care Med. 186 (9), 846–856. doi:10.1164/rccm.201206-1056OC
Griesenbach, U., Davies, J. C., and Alton, E. (2016). Cystic fibrosis gene therapy: a mutation-independent treatment. Curr. Opin. Pulm. Med. 22 (6), 602–609. doi:10.1097/MCP.0000000000000327
Guggino, W. B., and Cebotaru, L. (2017). Adeno-associated virus (AAV) gene therapy for cystic fibrosis: current barriers and recent developments. Expert Opin. Biol. Ther. 17 (10), 1265–1273. doi:10.1080/14712598.2017.1347630
Guggino, W. B., and Cebotaru, L. (2020). Gene therapy for cystic fibrosis paved the way for the use of adeno-associated virus in gene therapy. Hum. Gene Ther. 31 (9-10), 538–541. doi:10.1089/hum.2020.046
Guilbault, C., Saeed, Z., Downey, G. P., and Radzioch, D. (2007). Cystic fibrosis mouse models. Am. J. Respir. Cell Mol. Biol. 36 (1), 1–7. doi:10.1165/rcmb.2006-0184TR
Habermann, A. C., Gutierrez, A. J., Bui, L. T., Yahn, S. L., Winters, N. I., Calvi, C. L., et al. (2020). Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 6, eaba1972. doi:10.1126/sciadv.aba1972
Hajj, R., Baranek, T., Le Naour, R., Lesimple, P., Puchelle, E., and Coraux, C. (2007). Basal cells of the human adult airway surface epithelium retain transit-amplifying cell properties. Stem Cells 25 (1), 139–148. doi:10.1634/stemcells.2006-0288
Haque, A., Dewerth, A., Antony, J. S., Riethmuller, J., Schweizer, G. R., Weinmann, P., et al. (2018). Chemically modified hCFTR mRNAs recuperate lung function in a mouse model of cystic fibrosis. Sci. Rep. 8 (1), 16776. doi:10.1038/s41598-018-34960-0
Harvey, B. G., Hackett, N. R., El-Sawy, T., Rosengart, T. K., Hirschowitz, E. A., Lieberman, M. D., et al. (1999a). Variability of human systemic humoral immune responses to adenovirus gene transfer vectors administered to different organs. J. Virol. 73 (8), 6729–6742. doi:10.1128/JVI.73.8.6729-6742.1999
Harvey, B. G., Leopold, P. L., Hackett, N. R., Grasso, T. M., Williams, P. M., Tucker, A. L., et al. (1999b). Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J. Clin. Invest. 104 (9), 1245–1255. doi:10.1172/JCI7935
Heijerman, H. G. M., McKone, E. F., Downey, D. G., Van Braeckel, E., Rowe, S. M., Tullis, E., et al. (2019). Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 394 (10212), 1940–1948. doi:10.1016/S0140-6736(19)32597-8
Hewitt, R. J., and Lloyd, C. M. (2021). Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 21 (6), 347–362. doi:10.1038/s41577-020-00477-9
Heyer, W. D., Ehmsen, K. T., and Liu, J. (2010). Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139. doi:10.1146/annurev-genet-051710-150955
Hong, K. U., Reynolds, S. D., Watkins, S., Fuchs, E., and Stripp, B. R. (2004). In vivo differentiation potential of tracheal basal cells: evidence for multipotent and unipotent subpopulations. Am. J. Physiol. Lung Cell. Mol. Physiol. 286 (4), L643–L649. doi:10.1152/ajplung.00155.2003
Huang, Q., Deng, X., Yan, Z., Cheng, F., Luo, Y., Shen, W., et al. (2012). Establishment of a reverse genetics system for studying human bocavirus in human airway epithelia. PLoS Pathog. 8 (8), e1002899. doi:10.1371/journal.ppat.1002899
Huang, E. N., Quach, H., Lee, J. A., Dierolf, J., Moraes, T. J., and Wong, A. P. (2021). A developmental role of the cystic fibrosis transmembrane conductance regulator in cystic fibrosis lung disease pathogenesis. Front. Cell Dev. Biol. 9, 742891. doi:10.3389/fcell.2021.742891
Hyde, S. C., Southern, K. W., Gileadi, U., Fitzjohn, E. M., Mofford, K. A., Waddell, B. E., et al. (2000). Repeat administration of DNA/liposomes to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 7 (13), 1156–1165. doi:10.1038/sj.gt.3301212
Hyde, S. C., Pringle, I. A., Abdullah, S., Lawton, A. E., Davies, L. A., Varathalingam, A., et al. (2008). CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat. Biotechnol. 26 (5), 549–551. doi:10.1038/nbt1399
Jeggo, P. A. (1998). DNA breakage and repair. Adv. Genet. 38, 185–218. doi:10.1016/s0065-2660(08)60144-3
Jennings, K., Miyamae, T., Traister, R., Marinov, A., Katakura, S., Sowders, D., et al. (2005). Proteasome inhibition enhances AAV-mediated transgene expression in human synoviocytes in vitro and in vivo. Mol. Ther. 11 (4), 600–607. doi:10.1016/j.ymthe.2004.10.020
Jiang, Q., and Engelhardt, J. F. (1998). Cellular heterogeneity of CFTR expression and function in the lung: implications for gene therapy of cystic fibrosis. Eur. J. Hum. Genet. 6 (1), 12–31. doi:10.1038/sj.ejhg.5200158
Jiang, H., Couto, L. B., Patarroyo-White, S., Liu, T., Nagy, D., Vargas, J. A., et al. (2006). Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood 108 (10), 3321–3328. doi:10.1182/blood-2006-04-017913
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337 (6096), 816–821. doi:10.1126/science.1225829
Joseph, P. M., O'Sullivan, B. P., Lapey, A., Dorkin, H., Oren, J., Balfour, R., et al. (2001). Aerosol and lobar administration of a recombinant adenovirus to individuals with cystic fibrosis. I. Methods, safety, and clinical implications. Hum. Gene Ther. 12 (11), 1369–1382. doi:10.1089/104303401750298535
Kapoor, A., Simmonds, P., Slikas, E., Li, L., Bodhidatta, L., Sethabutr, O., et al. (2010). Human bocaviruses are highly diverse, dispersed, recombination prone, and prevalent in enteric infections. J. Infect. Dis. 201 (11), 1633–1643. doi:10.1086/652416
Keating, D., Marigowda, G., Burr, L., Daines, C., Mall, M. A., McKone, E. F., et al. (2018). VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 379 (17), 1612–1620. doi:10.1056/NEJMoa1807120
Keogh, R. H., Szczesniak, R., Taylor-Robinson, D., and Bilton, D. (2018). Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J. Cyst. Fibros. 17 (2), 218–227. doi:10.1016/j.jcf.2017.11.019
King, N. E., Suzuki, S., Barilla, C., Hawkins, F. J., Randell, S. H., Reynolds, S. D., et al. (2020). Correction of airway stem cells: Genome editing approaches for the treatment of cystic fibrosis. Hum. Gene Ther. 31 (17-18), 956–972. doi:10.1089/hum.2020.160
Knowles, M. R., Hohneker, K. W., Zhou, Z., Olsen, J. C., Noah, T. L., Hu, P. C., et al. (1995). A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N. Engl. J. Med. 333 (13), 823–831. doi:10.1056/NEJM199509283331302
Koehler, D. R., Sajjan, U., Chow, Y. H., Martin, B., Kent, G., Tanswell, A. K., et al. (2003). Protection of Cftr knockout mice from acute lung infection by a helper-dependent adenoviral vector expressing Cftr in airway epithelia. Proc. Natl. Acad. Sci. U. S. A. 100 (26), 15364–15369. doi:10.1073/pnas.2436478100
Koehler, D. R., Frndova, H., Leung, K., Louca, E., Palmer, D., Ng, P., et al. (2005). Aerosol delivery of an enhanced helper-dependent adenovirus formulation to rabbit lung using an intratracheal catheter. J. Gene Med. 7 (11), 1409–1420. doi:10.1002/jgm.797
Kosicki, M., Tomberg, K., and Bradley, A. (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36 (8), 765–771. doi:10.1038/nbt.4192
Kreda, S. M., Mall, M., Mengos, A., Rochelle, L., Yankaskas, J., Riordan, J. R., et al. (2005). Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell 16 (5), 2154–2167. doi:10.1091/mbc.e04-11-1010
Kreda, S. M., Davis, C. W., and Rose, M. C. (2012). CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb. Perspect. Med. 2, a009589. doi:10.1101/cshperspect.a009589
Krishnamurthy, S., Traore, S., Cooney, A. L., Brommel, C. M., Kulhankova, K., Sinn, P. L., et al. (2021). Functional correction of CFTR mutations in human airway epithelial cells using adenine base editors. Nucleic Acids Res. 49 (18), 10558–10572. doi:10.1093/nar/gkab788
Kunzelmann, K., Schreiber, R., and Hadorn, H. B. (2017). Bicarbonate in cystic fibrosis. J. Cyst. Fibros. 16 (6), 653–662. doi:10.1016/j.jcf.2017.06.005
Kushwah, R., Cao, H., and Hu, J. (2008). Characterization of pulmonary T cell response to helper-dependent adenoviral vectors following intranasal delivery. J. Immunol. 180 (6), 4098–4108. doi:10.4049/jimmunol.180.6.4098
Kuzmin, D. A., Shutova, M. V., Johnston, N. R., Smith, O. P., Fedorin, V. V., Kukushkin, Y. S., et al. (2021). The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 20 (3), 173–174. doi:10.1038/d41573-021-00017-7
Kwilas, A. R., Yednak, M. A., Zhang, L., Liesman, R., Collins, P. L., Pickles, R. J., et al. (2010). Respiratory syncytial virus engineered to express the cystic fibrosis transmembrane conductance regulator corrects the bioelectric phenotype of human cystic fibrosis airway epithelium in vitro. J. Virol. 84 (15), 7770–7781. doi:10.1128/JVI.00346-10
Lai, H. C., FitzSimmons, S. C., Allen, D. B., Kosorok, M. R., Rosenstein, B. J., Campbell, P. W., et al. (2000). Risk of persistent growth impairment after alternate-day prednisone treatment in children with cystic fibrosis. N. Engl. J. Med. 342 (12), 851–859. doi:10.1056/NEJM200003233421204
Laselva, O., Ardelean, M. C., and Bear, C. E. (2021). Phenotyping rare CFTR mutations reveal functional expression defects restored by TRIKAFTA(TM). J. Pers. Med. 11, 301. doi:10.3390/jpm11040301
Lee, J. A., Cho, A., Huang, E. N., Xu, Y., Quach, H., Hu, J., et al. (2021). Gene therapy for cystic fibrosis: new tools for precision medicine. J. Transl. Med. 19 (1), 452. doi:10.1186/s12967-021-03099-4
Leveque, M., Le Trionnaire, S., Del Porto, P., and Martin-Chouly, C. (2017). The impact of impaired macrophage functions in cystic fibrosis disease progression. J. Cyst. Fibros. 16 (4), 443–453. doi:10.1016/j.jcf.2016.10.011
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. doi:10.1146/annurev.biochem.052308.093131
Liu, X., Luo, M., Trygg, C., Yan, Z., Lei-Butters, D. C., Smith, C. I., et al. (2007). Biological differences in rAAV transduction of airway epithelia in humans and in old world non-human primates. Mol. Ther. 15 (12), 2114–2123. doi:10.1038/sj.mt.6300277
Liu, X., Li, T., Riederer, B., Lenzen, H., Ludolph, L., Yeruva, S., et al. (2015). Loss of Slc26a9 anion transporter alters intestinal electrolyte and HCO3(-) transport and reduces survival in CFTR-deficient mice. Pflugers Arch. 467 (6), 1261–1275. doi:10.1007/s00424-014-1543-x
Liu, X. S., Wu, H., Ji, X., Stelzer, Y., Wu, X., Czauderna, S., et al. (2016). Editing DNA methylation in the mammalian genome. Cell 167 (1), 233–247. doi:10.1016/j.cell.2016.08.056
Lopes-Pacheco, M. (2019). CFTR modulators: The changing face of cystic fibrosis in the era of precision medicine. Front. Pharmacol. 10, 1662. doi:10.3389/fphar.2019.01662
Lueck, J. D., Yoon, J. S., Perales-Puchalt, A., Mackey, A. L., Infield, D. T., Behlke, M. A., et al. (2019). Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 10 (1), 822. doi:10.1038/s41467-019-08329-4
Maeder, M. L., Stefanidakis, M., Wilson, C. J., Baral, R., Barrera, L. A., Bounoutas, G. S., et al. (2019). Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25 (2), 229–233. doi:10.1038/s41591-018-0327-9
Marquez Loza, L. I., Yuen, E. C., and McCray, P. B. (2019). Lentiviral vectors for the treatment and prevention of cystic fibrosis lung disease. Genes 10, E218. doi:10.3390/genes10030218
Masat, E., Pavani, G., and Mingozzi, F. (2013). Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discov. Med. 15 (85), 379–389.
Maule, G., Casini, A., Montagna, C., Ramalho, A. S., De Boeck, K., Debyser, Z., et al. (2019). Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat. Commun. 10 (1), 3556. doi:10.1038/s41467-019-11454-9
McCray, P. B., Wohlford-Lenane, C. L., and Snyder, J. M. (1992). Localization of cystic fibrosis transmembrane conductance regulator mRNA in human fetal lung tissue by in situ hybridization. J. Clin. Invest. 90 (2), 619–625. doi:10.1172/JCI115901
Mendell, J. R., Campbell, K., Rodino-Klapac, L., Sahenk, Z., Shilling, C., Lewis, S., et al. (2010). Dystrophin immunity in Duchenne's muscular dystrophy. N. Engl. J. Med. 363 (15), 1429–1437. doi:10.1056/NEJMoa1000228
Mercier, J., Ruffin, M., Corvol, H., and Guillot, L. (2021). Gene therapy: A possible alternative to CFTR modulators? Front. Pharmacol. 12, 648203. doi:10.3389/fphar.2021.648203
Meriluoto, M., Hedman, L., Tanner, L., Simell, V., Makinen, M., Simell, S., et al. (2012). Association of human bocavirus 1 infection with respiratory disease in childhood follow-up study, Finland. Emerg. Infect. Dis. 18 (2), 264–271. doi:10.3201/eid1802.111293
Michaels, W. E., Bridges, R. J., and Hastings, M. L. (2020). Antisense oligonucleotide-mediated correction of CFTR splicing improves chloride secretion in cystic fibrosis patient-derived bronchial epithelial cells. Nucleic Acids Res. 48 (13), 7454–7467. doi:10.1093/nar/gkaa490
Middleton, P. G., Mall, M. A., Drevinek, P., Lands, L. C., McKone, E. F., Polineni, D., et al. (2019). Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 381 (19), 1809–1819. doi:10.1056/NEJMoa1908639
Miller, A. J., Yu, Q., Czerwinski, M., Tsai, Y. H., Conway, R. F., Wu, A., et al. (2020). In vitro and in vivo development of the human airway at single-cell resolution. Dev. Cell 53 (1), 818–128.e6. doi:10.1016/j.devcel.2020.09.012
Milone, M. C., and O'Doherty, U. (2018). Clinical use of lentiviral vectors. Leukemia 32 (7), 1529–1541. doi:10.1038/s41375-018-0106-0
Mingozzi, F., and High, K. A. (2013). Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122 (1), 23–36. doi:10.1182/blood-2013-01-306647
Mingozzi, F., Maus, M. V., Hui, D. J., Sabatino, D. E., Murphy, S. L., Rasko, J. E., et al. (2007). CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13 (4), 419–422. doi:10.1038/nm1549
Mingozzi, F., Chen, Y., Edmonson, S. C., Zhou, S., Thurlings, R. M., Tak, P. P., et al. (2013). Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther. 20 (4), 417–424. doi:10.1038/gt.2012.55
Mitomo, K., Griesenbach, U., Inoue, M., Somerton, L., Meng, C., Akiba, E., et al. (2010). Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol. Ther. 18 (6), 1173–1182. doi:10.1038/mt.2010.13
Monahan, P. E., Lothrop, C. D., Sun, J., Hirsch, M. L., Kafri, T., Kantor, B., et al. (2010). Proteasome inhibitors enhance gene delivery by AAV virus vectors expressing large genomes in hemophilia mouse and dog models: a strategy for broad clinical application. Mol. Ther. 18 (11), 1907–1916. doi:10.1038/mt.2010.170
Montoro, D. T., Haber, A. L., Biton, M., Vinarsky, V., Lin, B., Birket, S. E., et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560 (7718), 319–324. doi:10.1038/s41586-018-0393-7
Moss, R. B., Rodman, D., Spencer, L. T., Aitken, M. L., Zeitlin, P. L., Waltz, D., et al. (2004). Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest 125 (2), 509–521. doi:10.1378/chest.125.2.509
Moss, R. B., Milla, C., Colombo, J., Accurso, F., Zeitlin, P. L., Clancy, J. P., et al. (2007). Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 18 (8), 726–732. doi:10.1089/hum.2007.022
Nichols, W. G., Peck Campbell, A. J., and Boeckh, M. (2008). Respiratory viruses other than influenza virus: impact and therapeutic advances. Clin. Microbiol. Rev. 21 (2), 274–290. doi:10.1128/CMR.00045-07,
Okuda, K., Dang, H., Kobayashi, Y., Carraro, G., Nakano, S., Chen, G., et al. (2021). Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med. 203 (10), 1275–1289. doi:10.1164/rccm.202008-3198OC
Ostedgaard, L. S., Zabner, J., Vermeer, D. W., Rokhlina, T., Karp, P. H., Stecenko, A. A., et al. (2002). CFTR with a partially deleted R domain corrects the cystic fibrosis chloride transport defect in human airway epithelia in vitro and in mouse nasal mucosa in vivo. Proc. Natl. Acad. Sci. U. S. A. 99 (5), 3093–3098. doi:10.1073/pnas.261714599
Palomaki, G. E., FitzSimmons, S. C., and Haddow, J. E. (2004). Clinical sensitivity of prenatal screening for cystic fibrosis via CFTR carrier testing in a United States panethnic population. Genet. Med. 6 (5), 405–414. doi:10.1097/01.gim.0000139505.06194.39
Pan, H., Deutsch, G. H., and Wert, S. E.Ontology SubcommitteeNHLBI Molecular Atlas of Lung Development Program Consortium (2019). Comprehensive anatomic ontologies for lung development: A comparison of alveolar formation and maturation within mouse and human lung. J. Biomed. Semant. 10 (1), 18. doi:10.1186/s13326-019-0209-1
Paquet, D., Kwart, D., Chen, A., Sproul, A., Jacob, S., Teo, S., et al. (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533 (7601), 125–129. doi:10.1038/nature17664
Peltola, V., Soderlund-Venermo, M., and Jartti, T. (2013). Human bocavirus infections. Pediatr. Infect. Dis. J. 32 (2), 178–179. doi:10.1097/INF.0b013e31827fef67
Perricone, M. A., Morris, J. E., Pavelka, K., Plog, M. S., O'Sullivan, B. P., Joseph, P. M., et al. (2001). Aerosol and lobar administration of a recombinant adenovirus to individuals with cystic fibrosis. II. Transfection efficiency in airway epithelium. Hum. Gene Ther. 12 (11), 1383–1394. doi:10.1089/104303401750298544
Petrova, N., Balinova, N., Marakhonov, A., Vasilyeva, T., Kashirskaya, N., Galkina, V., et al. (2021). Ethnic differences in the frequency of CFTR gene mutations in populations of the European and north caucasian part of the Russian federation. Front. Genet. 12, 678374. doi:10.3389/fgene.2021.678374
Pezzulo, A. A., Tang, X. X., Hoegger, M. J., Abou Alaiwa, M. H., Ramachandran, S., Moninger, T. O., et al. (2012). Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487 (7405), 109–113. doi:10.1038/nature11130
Plasschaert, L. W., Zilionis, R., Choo-Wing, R., Savova, V., Knehr, J., Roma, G., et al. (2018). A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560 (7718), 377–381. doi:10.1038/s41586-018-0394-6
Quesada, R., and Dutzler, R. (2020). Alternative chloride transport pathways as pharmacological targets for the treatment of cystic fibrosis. J. Cyst. Fibros. 19 Suppl 1, S37–S41. doi:10.1016/j.jcf.2019.10.020
Raman, T., O'Connor, T. P., Hackett, N. R., Wang, W., Harvey, B. G., Attiyeh, M. A., et al. (2009). Quality control in microarray assessment of gene expression in human airway epithelium. BMC Genomics 10, 493. doi:10.1186/1471-2164-10-493
Ramsey, B. W., Davies, J., McElvaney, N. G., Tullis, E., Bell, S. C., Drevinek, P., et al. (2011). A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365 (18), 1663–1672. doi:10.1056/NEJMoa1105185
Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154 (6), 1380–1389. doi:10.1016/j.cell.2013.08.021
Ratjen, F. A. (2009). Cystic fibrosis: pathogenesis and future treatment strategies. Respir. Care 54 (5), 595–605. doi:10.4187/aarc0427
Rawlins, E. L., and Hogan, B. L. (2008). Ciliated epithelial cell lifespan in the mouse trachea and lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 295 (1), L231–L234. doi:10.1152/ajplung.90209.2008
Rawlins, E. L., Okubo, T., Xue, Y., Brass, D. M., Auten, R. L., Hasegawa, H., et al. (2009). The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4 (6), 525–534. doi:10.1016/j.stem.2009.04.002
Rees, H. A., and Liu, D. R. (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19 (12), 770–788. doi:10.1038/s41576-018-0059-1
Riordan, J. R., Rommens, J. M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., et al. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245 (4922), 1066–1073. doi:10.1126/science.2475911
Robinson, E., MacDonald, K. D., Slaughter, K., McKinney, M., Patel, S., Sun, C., et al. (2018). Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol. Ther. 26 (8), 2034–2046. doi:10.1016/j.ymthe.2018.05.014
Rock, J. R., Onaitis, M. W., Rawlins, E. L., Lu, Y., Clark, C. P., Xue, Y., et al. (2009). Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. U. S. A. 106 (31), 12771–12775. doi:10.1073/pnas.0906850106
Rogers, C. S., Hao, Y., Rokhlina, T., Samuel, M., Stoltz, D. A., Li, Y., et al. (2008). Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J. Clin. Invest. 118 (4), 1571–1577. doi:10.1172/JCI34773
Rommens, J. M., Iannuzzi, M. C., Kerem, B., Drumm, M. L., Melmer, G., Dean, M., et al. (1989). Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245 (4922), 1059–1065. doi:10.1126/science.2772657
Ronzitti, G., Gross, D. A., and Mingozzi, F. (2020). Human immune responses to adeno-associated virus (AAV) vectors. Front. Immunol. 11, 670. doi:10.3389/fimmu.2020.00670
Rowe, S. M., Daines, C., Ringshausen, F. C., Kerem, E., Wilson, J., Tullis, E., et al. (2017). Tezacaftor-Ivacaftor in residual-function heterozygotes with cystic fibrosis. N. Engl. J. Med. 377 (21), 2024–2035. doi:10.1056/NEJMoa1709847
Sanz, D. J., Hollywood, J. A., Scallan, M. F., and Harrison, P. T. (2017). Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS One 12 (9), e0184009. doi:10.1371/journal.pone.0184009
Schlimgen, R., Howard, J., Wooley, D., Thompson, M., Baden, L. R., Yang, O. O., et al. (2016). Risks associated with lentiviral vector exposures and prevention strategies. J. Occup. Environ. Med. 58 (12), 1159–1166. doi:10.1097/JOM.0000000000000879
Schwank, G., Koo, B. K., Sasselli, V., Dekkers, J. F., Heo, I., Demircan, T., et al. (2013). Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13 (6), 653–658. doi:10.1016/j.stem.2013.11.002
Scotet, V., Gutierrez, H., and Farrell, P. M. (2020a). Newborn screening for CF across the globe-where is it worthwhile? Int. J. Neonatal Screen. 6 (1), 18. doi:10.3390/ijns6010018
Scotet, V., L'Hostis, C., and Ferec, C. (2020b). The changing epidemiology of cystic fibrosis: Incidence, survival and impact of the CFTR gene discovery. Genes 11, E589. doi:10.3390/genes11060589
Shah, V. S., Ernst, S., Tang, X. X., Karp, P. H., Parker, C. P., Ostedgaard, L. S., et al. (2016). Relationships among CFTR expression, HCO3- secretion, and host defense may inform gene- and cell-based cystic fibrosis therapies. Proc. Natl. Acad. Sci. U. S. A. 113 (19), 5382–5387. doi:10.1073/pnas.1604905113
Shei, R. J., Peabody, J. E., Kaza, N., and Rowe, S. M. (2018). The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr. Opin. Pharmacol. 43, 152–165. doi:10.1016/j.coph.2018.09.007
Shteinberg, M., Haq, I. J., Polineni, D., and Davies, J. C. (2021). Cystic fibrosis. Lancet 397 (10290), 2195–2211. doi:10.1016/S0140-6736(20)32542-3
Sirninger, J., Muller, C., Braag, S., Tang, Q., Yue, H., Detrisac, C., et al. (2004). Functional characterization of a recombinant adeno-associated virus 5-pseudotyped cystic fibrosis transmembrane conductance regulator vector. Hum. Gene Ther. 15 (9), 832–841. doi:10.1089/hum.2004.15.832
Steines, B., Dickey, D. D., Bergen, J., Excoffon, K. J., Weinstein, J. R., Li, X., et al. (2016). CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight 1 (14), e88728. doi:10.1172/jci.insight.88728
Stoltz, D. A., Meyerholz, D. K., and Welsh, M. J. (2015). Origins of cystic fibrosis lung disease. N. Engl. J. Med. 372 (16), 1574–1575. doi:10.1056/NEJMc1502191
Stutts, M. J., Canessa, C. M., Olsen, J. C., Hamrick, M., Cohn, J. A., Rossier, B. C., et al. (1995). CFTR as a cAMP-dependent regulator of sodium channels. Science 269 (5225), 847–850. doi:10.1126/science.7543698
Sun, X., Yan, Z., Yi, Y., Li, Z., Lei, D., Rogers, C. S., et al. (2008). Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J. Clin. Invest. 118 (4), 1578–1583. doi:10.1172/JCI34599
Sun, X., Yi, Y., Yan, Z., Rosen, B. H., Liang, B., Winter, M. C., et al. (2019). In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med. 11, eaau7531. doi:10.1126/scitranslmed.aau7531
Suzuki, K., Tsunekawa, Y., Hernandez-Benitez, R., Wu, J., Zhu, J., Kim, E. J., et al. (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540 (7631), 144–149. doi:10.1038/nature20565
Suzuki, S., Crane, A. M., Anirudhan, V., Barilla, C., Matthias, N., Randell, S. H., et al. (2020). Highly efficient gene editing of cystic fibrosis patient-derived airway basal cells results in functional CFTR correction. Mol. Ther. 28 (7), 1684–1695. doi:10.1016/j.ymthe.2020.04.021
Tang, Y., Yan, Z., and Engelhardt, J. F. (2020a). Viral vectors, animal models, and cellular targets for gene therapy of cystic fibrosis lung disease. Hum. Gene Ther. 31 (9-10), 524–537. doi:10.1089/hum.2020.013
Tang, Y., Yan, Z., Lin, S., Huntemann, E. D., Feng, Z., Park, S. Y., et al. (2020b). Repeat dosing of AAV2.5T to ferret lungs elicits an antibody response that diminishes transduction in an age-dependent manner. Mol. Ther. Methods Clin. Dev. 19, 186–200. doi:10.1016/j.omtm.2020.09.008
Taylor-Cousar, J. L., Jain, M., Barto, T. L., Haddad, T., Atkinson, J., Tian, S., et al. (2018). Lumacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease homozygous for F508del-CFTR. J. Cyst. Fibros. 17 (2), 228–235. doi:10.1016/j.jcf.2017.09.012
Thomson, J. M., Wesley, A., Byrnes, C. A., and Nixon, G. M. (2006). Pulse intravenous methylprednisolone for resistant allergic bronchopulmonary aspergillosis in cystic fibrosis. Pediatr. Pulmonol. 41 (2), 164–170. doi:10.1002/ppul.20333
Tosi, M. F., van Heeckeren, A., Ferkol, T. W., Askew, D., Harding, C. V., Kaplan, J. M., et al. (2004). Effect of Pseudomonas-induced chronic lung inflammation on specific cytotoxic T-cell responses to adenoviral vectors in mice. Gene Ther. 11 (19), 1427–1433. doi:10.1038/sj.gt.3302290
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., and Gregory, P. D. (2010). Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11 (9), 636–646. doi:10.1038/nrg2842
Vaidyanathan, S., Salahudeen, A. A., Sellers, Z. M., Bravo, D. T., Choi, S. S., Batish, A., et al. (2020). High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell 26 (2), 161–171.e4. doi:10.1016/j.stem.2019.11.002
Vandamme, C., Adjali, O., and Mingozzi, F. (2017). Unraveling the complex story of immune responses to AAV vectors trial after trial. Hum. Gene Ther. 28 (11), 1061–1074. doi:10.1089/hum.2017.150
Verdera, H. C., Kuranda, K., and Mingozzi, F. (2020). AAV vector immunogenicity in humans: A long journey to successful gene transfer. Mol. Ther. 28 (3), 723–746. doi:10.1016/j.ymthe.2019.12.010
Vu, A., and McCray, P. B. (2020). New directions in pulmonary gene therapy. Hum. Gene Ther. 31 (17-18), 921–939. doi:10.1089/hum.2020.166
Wagner, J. A., Reynolds, T., Moran, M. L., Moss, R. B., Wine, J. J., Flotte, T. R., et al. (1998). Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet 351 (9117), 1702–1703. doi:10.1016/S0140-6736(05)77740-0
Wagner, J. A., Nepomuceno, I. B., Messner, A. H., Moran, M. L., Batson, E. P., Dimiceli, S., et al. (2002). A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum. Gene Ther. 13 (11), 1349–1359. doi:10.1089/104303402760128577
Walters, R. W., Grunst, T., Bergelson, J. M., Finberg, R. W., Welsh, M. J., and Zabner, J. (1999). Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J. Biol. Chem. 274 (15), 10219–10226. doi:10.1074/jbc.274.15.10219
Welsh, M. J., and Smith, A. E. (1993). Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73 (7), 1251–1254. doi:10.1016/0092-8674(93)90353-r
Whitsett, J. A., Kalin, T. V., Xu, Y., and Kalinichenko, V. V. (2019). Building and regenerating the lung cell by cell. Physiol. Rev. 99 (1), 513–554. doi:10.1152/physrev.00001.2018
Widdicombe, J. H., and Wine, J. J. (2015). Airway gland structure and function. Physiol. Rev. 95 (4), 1241–1319. doi:10.1152/physrev.00039.2014
Wilke, M., Buijs-Offerman, R. M., Aarbiou, J., Colledge, W. H., Sheppard, D. N., Touqui, L., et al. (2011). Mouse models of cystic fibrosis: phenotypic analysis and research applications. J. Cyst. Fibros. 10 Suppl 2, S152–S171. doi:10.1016/S1569-1993(11)60020-9
Wu, D., Boucher, R. C., Button, B., Elston, T., and Lin, C. L. (2018). An integrated mathematical epithelial cell model for airway surface liquid regulation by mechanical forces. J. Theor. Biol. 438, 34–45. doi:10.1016/j.jtbi.2017.11.010
Xia, E., Duan, R., Shi, F., Seigel, K. E., Grasemann, H., and Hu, J. (2018). Overcoming the undesirable CRISPR-cas9 expression in gene correction. Mol. Ther. Nucleic Acids 13, 699–709. doi:10.1016/j.omtn.2018.10.015
Xia, E., Zhang, Y., Cao, H., Li, J., Duan, R., and Hu, J. (2019). TALEN-mediated gene targeting for cystic fibrosis-gene therapy. Genes 10, E39. doi:10.3390/genes10010039
Xu, J., Henriksnas, J., Barone, S., Witte, D., Shull, G. E., Forte, J. G., et al. (2005). SLC26A9 is expressed in gastric surface epithelial cells, mediates Cl-/HCO3- exchange, and is inhibited by NH4+. Am. J. Physiol. Cell Physiol. 289 (2), C493–C505. doi:10.1152/ajpcell.00030.2005
Yan, Z., Zak, R., Luxton, G. W., Ritchie, T. C., Bantel-Schaal, U., and Engelhardt, J. F. (2002). Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J. Virol. 76 (5), 2043–2053. doi:10.1128/jvi.76.5.2043-2053.2002
Yan, Z., Zak, R., Zhang, Y., Ding, W., Godwin, S., Munson, K., et al. (2004). Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surfaces of human airway epithelia. J. Virol. 78 (6), 2863–2874. doi:10.1128/jvi.78.6.2863-2874.2004
Yan, Z., Lei-Butters, D. C., Liu, X., Zhang, Y., Zhang, L., Luo, M., et al. (2006). Unique biologic properties of recombinant AAV1 transduction in polarized human airway epithelia. J. Biol. Chem. 281 (40), 29684–29692. doi:10.1074/jbc.M604099200
Yan, Z., Keiser, N. W., Song, Y., Deng, X., Cheng, F., Qiu, J., et al. (2013a). A novel chimeric adenoassociated virus 2/human bocavirus 1 parvovirus vector efficiently transduces human airway epithelia. Mol. Ther. 21 (12), 2181–2194. doi:10.1038/mt.2013.92
Yan, Z., Lei-Butters, D. C., Keiser, N. W., and Engelhardt, J. F. (2013b). Distinct transduction difference between adeno-associated virus type 1 and type 6 vectors in human polarized airway epithelia. Gene Ther. 20 (3), 328–337. doi:10.1038/gt.2012.46
Yan, Z., Stewart, Z. A., Sinn, P. L., Olsen, J. C., Hu, J., McCray, P. B., et al. (2015a). Ferret and pig models of cystic fibrosis: prospects and promise for gene therapy. Hum. Gene Ther. Clin. Dev. 26 (1), 38–49. doi:10.1089/humc.2014.154
Yan, Z., Sun, X., Feng, Z., Li, G., Fisher, J. T., Stewart, Z. A., et al. (2015b). Optimization of recombinant adeno-associated virus-mediated expression for large transgenes, using a synthetic promoter and tandem array enhancers. Hum. Gene Ther. 26 (6), 334–346. doi:10.1089/hum.2015.001
Yan, Z., Feng, Z., Sun, X., Zhang, Y., Zou, W., Wang, Z., et al. (2017). Human bocavirus type-1 capsid facilitates the transduction of ferret airways by adeno-associated virus genomes. Hum. Gene Ther. 28 (8), 612–625. doi:10.1089/hum.2017.060
Yan, Z., McCray, P. B., and Engelhardt, J. F. (2019). Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 28 (R1), R88–R94. doi:10.1093/hmg/ddz139
Yan, Z., Vorhies, K., Feng, Z., Park, S. Y., Choi, S. H., Zhang, Y., et al. (2022). Recombinant adeno-associated virus-mediated editing of the G551D cystic fibrosis transmembrane conductance regulator mutation in ferret airway basal cells. Hum. Gene Ther. doi:10.1089/hum.2022.036
Yanda, M. K., Tomar, V., Cebotaru, C. V., Guggino, W. B., and Cebotaru, L. (2022). Short-term steroid treatment of rhesus macaque increases transduction. Hum. Gene Ther. 33 (3-4), 131–147. doi:10.1089/hum.2021.239
Yiallouros, P. K., Matthaiou, A., Anagnostopoulou, P., Kouis, P., Libik, M., Adamidi, T., et al. (2021). Demographic characteristics, clinical and laboratory features, and the distribution of pathogenic variants in the CFTR gene in the Cypriot cystic fibrosis (CF) population demonstrate the utility of a national CF patient registry. Orphanet J. Rare Dis. 16 (1), 409. doi:10.1186/s13023-021-02049-z
Yin, H., Kanasty, R. L., Eltoukhy, A. A., Vegas, A. J., Dorkin, J. R., and Anderson, D. G. (2014). Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 15 (8), 541–555. doi:10.1038/nrg3763
Yoshimura, K., Nakamura, H., Trapnell, B. C., Chu, C. S., Dalemans, W., Pavirani, A., et al. (1991). Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 19 (19), 5417–5423. doi:10.1093/nar/19.19.5417
Yu, W., Moninger, T. O., Thurman, A. L., Xie, Y., Jain, A., Zarei, K., et al. (2022). Cellular and molecular architecture of submucosal glands in wild-type and cystic fibrosis pigs. Proc. Natl. Acad. Sci. U. S. A. 119, e2119759119. doi:10.1073/pnas.2119759119
Zabner, J., Couture, L. A., Gregory, R. J., Graham, S. M., Smith, A. E., and Welsh, M. J. (1993). Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 75 (2), 207–216. doi:10.1016/0092-8674(93)80063-k
Zabner, J., Smith, J. J., Karp, P. H., Widdicombe, J. H., and Welsh, M. J. (1998). Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol. Cell 2 (3), 397–403. doi:10.1016/s1097-2765(00)80284-1
Zhang, L., Wang, D., Fischer, H., Fan, P. D., Widdicombe, J. H., Kan, Y. W., et al. (1998). Efficient expression of CFTR function with adeno-associated virus vectors that carry shortened CFTR genes. Proc. Natl. Acad. Sci. U. S. A. 95 (17), 10158–10163. doi:10.1073/pnas.95.17.10158
Zhang, L. N., Karp, P., Gerard, C. J., Pastor, E., Laux, D., Munson, K., et al. (2004). Dual therapeutic utility of proteasome modulating agents for pharmaco-gene therapy of the cystic fibrosis airway. Mol. Ther. 10 (6), 990–1002. doi:10.1016/j.ymthe.2004.08.009
Zhang, L., Bukreyev, A., Thompson, C. I., Watson, B., Peeples, M. E., Collins, P. L., et al. (2005). Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J. Virol. 79 (2), 1113–1124. doi:10.1128/JVI.79.2.1113-1124.2005
Zhang, L., Button, B., Gabriel, S. E., Burkett, S., Yan, Y., Skiadopoulos, M. H., et al. (2009). CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 7 (7), e1000155. doi:10.1371/journal.pbio.1000155
Zhang, F., Cong, L., Lodato, S., Kosuri, S., Church, G. M., and Arlotta, P. (2011). Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 29 (2), 149–153. doi:10.1038/nbt.1775
Zhou, Z. P., Yang, L. L., Cao, H., Chen, Z. R., Zhang, Y., Wen, X. Y., et al. (2019). In vitro validation of a CRISPR-mediated CFTR correction strategy for preclinical translation in pigs. Hum. Gene Ther. 30 (9), 1101–1116. doi:10.1089/hum.2019.074
Zuckerman, J. B., Robinson, C. B., McCoy, K. S., Shell, R., Sferra, T. J., Chirmule, N., et al. (1999). A phase I study of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator gene to a lung segment of individuals with cystic fibrosis. Hum. Gene Ther. 10 (18), 2973–2985. doi:10.1089/10430349950016384
Keywords: cystic fibrosis, gene therapy, viral vectors, animal models, airway delivery
Citation: Sui H, Xu X, Su Y, Gong Z, Yao M, Liu X, Zhang T, Jiang Z, Bai T, Wang J, Zhang J, Xu C and Luo M (2022) Gene therapy for cystic fibrosis: Challenges and prospects. Front. Pharmacol. 13:1015926. doi: 10.3389/fphar.2022.1015926
Received: 10 August 2022; Accepted: 29 September 2022;
Published: 11 October 2022.
Edited by:
Elisabetta Pace, National Research Council (CNR), ItalyReviewed by:
Aja Aravamudhan, University of Minnesota Twin Cities, United StatesCopyright © 2022 Sui, Xu, Su, Gong, Yao, Liu, Zhang, Jiang, Bai, Wang, Zhang, Xu and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongshu Sui, aHNzdWlAc2RmbXUuZWR1LmNu; Changlong Xu, eHVjaGFuZ2xvbmcyMDExQGhvdG1haWwuY29t; Mingjiu Luo, bHVvbWpAc2RhdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.