 Motonobu Nakamura
Motonobu Nakamura Nobuhiko Satoh
Nobuhiko Satoh Shoko Horita
Shoko Horita
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 10 October 2022
Sec. Renal Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1015204
This article is part of the Research Topic Receptor Biology and Cell Signaling in Diabetes: Volume II View all 6 articles
Energy is continuously expended in the body, and gluconeogenesis maintains glucose homeostasis during starvation. Gluconeogenesis occurs in the liver and kidneys. The proximal tubule is the primary location for renal gluconeogenesis, accounting for up to 25% and 60% of endogenous glucose production during fasting and after a meal, respectively. The mechanistic target of rapamycin (mTOR), which exists downstream of the insulin pathway, plays an important role in regulating proximal tubular gluconeogenesis. mTOR is an atypical serine/threonine kinase present in two complexes. mTORC1 phosphorylates substrates that enhance anabolic processes such as mRNA translation and lipid synthesis and catabolic processes such as autophagy. mTORC2 regulates cytoskeletal dynamics and controls ion transport and proliferation via phosphorylation of SGK1. Therefore, mTOR signaling defects have been implicated in various pathological conditions, including cancer, cardiovascular disease, and diabetes. However, concrete elucidations of the associated mechanisms are still unclear. This review provides an overview of mTOR and describes the relationship between mTOR and renal.
Gluconeogenesis occurs in the liver and renal proximal tubules (PTs), and the PTs produce a considerable amount of total glucose during fasting. Renal gluconeogenesis accounts for up to 60% and 25% of endogenous glucose production during fasting and after a meal, respectively (Alsahli and Gerich, 2017). Studies have demonstrated that knockout of insulin receptors in the PTs causes hyperglycemia, and insulin suppresses renal gluconeogenesis (Cersosimo et al., 1994; Cersosimo et al., 2000). Insulin and the mechanistic target of rapamycin (mTOR) regulate glucose transporter (GLUT4) translocation via a positive feedback loop between Akt2 and mTORC2. Moreover, several recent studies have reported that insulin regulates renal tubule functions (Nakamura, M. et al., 2020; Hoorn et al., 2020; Sohara et al., 2011; Chávez-Canales et al., 2013)
As such, insulin is an important regulator of PT gluconeogenesis (Humphrey et al., 2013), and the insulin/mTOR pathway has recently been shown to regulate PT gluconeogenesis (Nakamura et al., 2020a).
mTOR complex 1 (mTORC1) consists of mTOR, Raptor, GβL, and DEP domain-containing mTOR-interacting protein (DEPTOR) and is inhibited by rapamycin (Foster and Fingar, 2010). mTORC1 is a key growth regulator that senses and integrates a variety of nutritional and environmental factors, including growth factors, energy levels, cellular stress, and amino acids. In response to these signals, mTORC1 promotes cell proliferation by phosphorylating substrates that enhance anabolic processes, such as mRNA translation and lipid synthesis, or by limiting catabolic processes, such as autophagy. The second complex–mTORC2–is composed of mTOR, Rictor, GβL, SAPK interacting protein 1 (Sin1), PRR5/Protor-1, and DEPTOR. mTORC2 promotes cell survival by activating protein kinase B (Akt), regulating cytoskeletal dynamics via activation of PKCα, and regulating ion transport and proliferation via phosphorylation of SGK1. mTOR signaling defects are associated with many pathological conditions, including cancer, cardiovascular disease, and diabetes (Szwed et al., 2021). However, concrete elucidations of the associated mechanisms remain unclear. This review aimed to describe the current understanding of mTOR with respect to insulin and summarize the relationship between mTOR and the renal tubules in glucose metabolism.
Figure 1 depicts a brief overview of the mTOR cascade. Ras homolog enriched in brain (Rheb), a small GTP-binding protein that has been shown to mediate cell growth and control cell size in mammalian cells, is a potent stimulator required for mTORC1 kinase activity in its GTP-bound state and is negatively regulated by the heterodimer tuberous sclerosis complex (TSC1/2) of the GTPase-activating proteins (GAP-TSC) (Fukuda and Shiozaki, 2018; Zhu and Wang, 2020). Much of the upstream stimulation is transmitted through Akt and TSC1/2, which regulate the nucleotide loading state of Rheb (Tee et al., 2003). In contrast, amino acids signal independently from the PI3K/Akt pathway to mTORC1. Particularly, amino acids facilitate the translocation of mTORC1 to the lysosomal surface, where mTORC1 is activated by binding to Rheb. This process is regulated by the concerted action of multiple complexes, mainly v-ATPase, Ragulator, Rag GTPase, and GATOR1/2 (Saxton and Sabatini, 2017; Wolfson and Sabatini, 2017). The enhancement of anabolism by mTORC1 occurs when insulin is present, this cascade is strongly dependent on feeding. Feeding actives mTORC1, whereas during fasting inhibits to conserve limited resources (Saxton and Sabatini, 2017). The process of mTORC1 activation is outlined ahead.
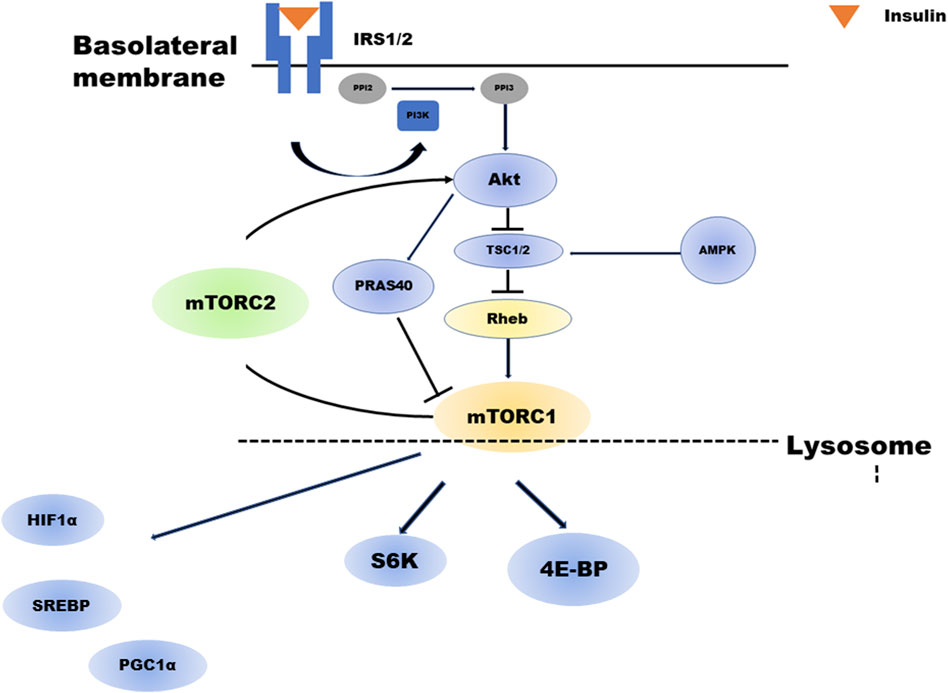
FIGURE 1. Overview of the mTOR cascade. Insulin activates both mTORC1 and mTORC2. Akt regulates mTORC1. TSC2 phosphorylated by insulin-activated Akt will have its activity suppressed. As a result, the activity of GTP-bound Rheb increases, and mTORC1 is activated. In addition, Akt phosphorylates PRAS40, which in turn activates mTORC1. mTORC1 regulates several negative feedback loops that control IRS1/2 and mTORC2 signaling: direct inhibition of IRS1/2 and indirect inhibition of mTORC2 via S6K1. abrAbbreviationsmTORC; mechanistic target of rapamycin complex, Akt; Protein kinase B, TSC; tuberous sclerosis complex, PRAS40; proline-rich Akt substrate of 40 kDa, IRS; insulin receptor substrate, PIP2; Phosphatidylinositol 4,5-bisphosphate, PIP3; Phosphatidylinositol 3,4,5-trisphosphate.
mTORC1 activation is initiated by the low-molecular weight GTP-binding protein Rheb. Rheb activity is regulated by GAPs or TSC1-TSC2 complexes, inhibitory regulators for Rheb. TSC1 and TSC2 are essential for the stability of the TSC2 protein, which has a GAP domain and forms a complex with TSC1. Phosphorylation of TSC2 by insulin-activated Akt suppresses its activity. Consequently, Rheb activity increases (GTP-bound Rheb increases), and mTORC1 is activated. In addition, Akt phosphorylates PRAS40, a component of mTORC1, in turn activating mTORC1.
TSC2 is also phosphorylated by extracellular signal-regulated kinase 1/2 and GSK3β, and its activity is suppressed, resulting in mTORC1 activation. In contrast, depletion of intracellular ATP activates AMP-dependent kinase (AMPK). It is reported that even if Rheb activity increases in the presence of cell growth factors such as insulin and IGF-1, mTORC1 cannot be activated without the amino acid. Rheb is expressed on late endosomal membranes in the cell but cannot recruit mTORC1 from the cytoplasm; thus, mTORC1 cannot be activated without the amino acid. Recently, it has been demonstrated that another GTP-binding protein, Rag, plays a role in this recruitment in an amino acid-dependent manner (Sancak et al., 2010; Zhu and Wang, 2020).
Several studies have shown that mTORC2 is regulated by factors such as phosphatidylinositol-3,4,5-triphosphate (PIP3)-mediated growth factor (Liu et al., 2015), Sin1, phosphorylation (Liu et al., 2013), GβL-ubiquitination (Wang et al., 2017), and reactive oxygen species (Bernard et al., 2020). However, regarding gluconeogenesis in PTs, the molecular mechanism of mTORC2 activation remains largely unknown.
Nutrients and growth factors also influence mTORC2 activation, but how they regulate mTORC2 is more complex and unknown compared to mTORC1. In mammals, increased growth factor/PI3K signaling enhances mTORC2 activity. PIP3 activates mTORC2 at the membrane, and mTORC2 may be regulated in different membrane compartments. The endosomal pool of mTORC2 mediates the PI3K signaling pathway (Ebner et al., 2017), and insulin/PI3K increases mTORC2 activation and binding to ribosomes (Zinzalla et al., 2011). The localization of compartments is pivotal in regulating this endosomal pool (Jia and Bonifacino, 2019). In addition, mTORC2 responds to intracellular nutrient or metabolite status. Activation of mTORC2 is enhanced by removing amino acids (e.g., glutamine) and glucose starvation in the presence of serum (Moloughney et al., 2016). Activation of mTORC2 during glutamine or glucose starvation is essential to maintain flux through the hexosamine biosynthetic pathway. During glucose depletion, AMPK directly activates mTORC2 via phosphorylation of mTOR and rictor in the presence of PI3K signaling (Kazyken et al., 2019).
As shown in Figure 1, mTORC1 regulates several negative feedback loops that control IRS1/2 and mTORC2 signaling (i.e., directly inhibiting IRS1/2 and indirectly inhibiting mTORC2 via S6K1). In β-cell, the mTORC1-S6K1-mediated negative loop constitutes an intracellular regulatory mechanism that suppresses excessive insulin and mTORC2 signaling under physiological conditions and controls cell growth and homeostasis. Meanwhile, chronic mTORC1-S6K1 hyperactivation induced by obesity and overnutrition may attenuate insulin and mTORC2 signaling, resulting in decreased β-cell function and type 2 diabetes (T2D) development. These pathways are not limited to pancreatic beta cells and may also be involved in proximal tubular metabolism (Ardestani et al., 2018).
The key factors in the insulin/mTORC1 pathway are the TSC1/TSC2 complex and Rheb. Recently, the mechanism by which mTORC1 is activated by Rheb and inhibited by proline-rich Akt substrate of 40 kDa (PRAS40) was demonstrated. Rheb is an activator of mTORC1, and GTP-bound Rheb (activated form) binds directly to the kinase catalytic domain of mTORC1 to enhance mTORC1 kinase activity (Long et al., 2005). Further, Rheb increased the binding of mTORC1 to its substrate, 4EBP1 (Sato et al., 2009), and insulin-activated Akt directly phosphorylated TSC2. This phosphorylation directly decreases the GAP activity of TSC2 or changes its subcellular localization, resulting in decreased TSC1/TSC2 activity and consequently increased GTP-bound Rheb (Li et al., 2004). Furthermore, activated Akt activates the mTORC1 pathway by inhibiting PRAS40, an inhibitor that directly binds mTORC1 (Haar et al., 2007; Sancak et al., 2007). PRAS40 has a TOR signaling (TOS) motif found in substrates of mTORC1, and its inhibition is thought to be due to competition for mTORC1 substrates. Only a few proteins are known to be direct substrates of mTORC1, such as 4EBP1, S6K1, STAT3, and Hypoxia Inducible Factor (HIF)1α (Szwed et al., 2021).
In the absence of insulin stimulation, GLUT4 is deposited in intracellular vesicles, but stimulation occurs when insulin binds to its receptor on the plasma membrane, and GLUT4 translocates to the plasma membrane, taking up glucose from the blood into the cell. Intracellular signaling triggered in response to insulin is complex, and Akt, activated by insulin, is phosphorylated by several substrates. This is essential for regulating insulin-dependent processes (Lizcano and Alessi, 2002). In addition, Akt phosphorylates and inhibits AS160, a Rab-GTPase-activating protein. It promotes the translocation of GLUT4 to the plasma membrane by activating the Rab small GTPase and regulating cytoskeletal reorganization (Sano et al., 2003). Furthermore, Akt phosphorylates forkhead box protein O (FOXO), a transcription factor necessary for the expression of gluconeogenic and lipogenic enzymes. For example, FOXO1 activates gluconeogenic genes in the liver (Puigserver et al., 2003), negatively regulates SREBP1c (Kamei et al., 2008), and inhibits lipogenesis (Nakae et al., 2003). Akt activated by insulin phosphorylates serine 256 of FOXO1, inhibiting the expression of gluconeogenic enzymes. Moreover, Akt modulates mTORC1 signaling by phosphorylating and inhibiting TSC1/2, a negative regulator of mTORC1 (Shimobayashi and Hall, 2014). Subsequently, mTORC2 regulates glucose homeostasis via Akt. Akt promotes glucose uptake by increasing GLUT4 translocation to adipocyte membranes (Kohn et al., 1996). Additionally, Akt phosphorylates and deactivates GSK-3 and reduces the phosphorylation rate of glycogen synthase. As a result, glycogen synthase activity increases, leading to increased glycogen accumulation in the muscle and liver (Manning and Cantley, 2007). Akt also regulates glucose homeostasis by phosphorylating and inhibiting FOXO1, a transcription factor that regulates gluconeogenesis (Accili and Arden, 2004).
mTORC1 plays an important role in the development and progression of β-cell damage in T2D. It is possible that mTORC1 functions as a “double-edged sword” by its regulation of β-cell volume and function in response to metabolic stresses (Ardestani et al., 2018). In fact, hyperlipidemia occurs in 75% of patients treated with mTOR inhibitors (Busaidy et al., 2012; Pallet and Legendre, 2013). Surprisingly, 32% of participants treated with everolimus develop hyperglycemia and IR, resulting in new onset of diabetes (Kaplan et al., 2014). Meanwhile, increased mTOR activity is associated with IR (Mordier and Iynedjian, 2007; Hsu et al., 2011), and short-term treatment with caloric restriction and rapamycin leads to increased insulin sensitivity and glucose uptake, suggests the contradictory roles of mTOR and mTOR inhibitors (Ye et al., 2012).
Thus, the chronic effects of rapamycin on insulin sensitivity and glucose metabolism in vivo remain elusive. Chronic rapamycin treatment promoted IR, severe glucose intolerance, and increased gluconeogenesis in a rat model. It was also linked to increased expression of the gluconeogenic enzymes or genes phosphoenolpyruvate carboxykinase (PEPCK) and G6Pase in the liver, increased expression of the transcriptional coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), and enhanced nuclear mobilization of FOXO1, CRTC2, and CREB. These changes were observed despite normal activation of the IRS/PI3k/Akt pathway in the liver of rapamycin-treated rats (Houde et al., 2010). These results demonstrate that chronic inhibition of the mTORC1/S6K1 pathway by rapamycin causes impaired glucose tolerance by inducing transcriptional activation of gluconeogenic genes, primarily through coordinated activation of PGC-1α, CRTC2, CREB, and FOXO1.
Recently, it was suggested that Rheb, which plays a vital role in the insulin/mTOR pathway, is associated with a mechanism by which IR improves in patients undergoing duodenal-jejunal bypass (DJB). Individuals who received DJB demonstrated improved hepatic IR and better insulin signaling, resulting in better control of glucose tolerance. Furthermore, transfection of Wistar rats treated with DJB with a miR-200a inhibitor increased Rheb protein levels and enhanced the feedback effect on insulin receptor substrate-dependent insulin signaling. Meanwhile, transfection with a miR-200a mimic caused the opposite effect. Furthermore, downregulation of miR-200a decreased insulin sensitivity, whereas upregulation of miR-200a in diabetic rats improved diabetes. These results indicate that improvement of hepatic IR is closely associated with Rheb-targeted miRNA-200a. These results could explain the crosstalk between miRNAs and insulin signaling pathways in bariatric surgery (Guo et al., 2016).
The kidneys remove approximately 35% of the secreted insulin. Most insulin is filtered through the glomerulus and completely reabsorbed from the PT’s brush border membrane (BBM). Upon release, insulin is trafficked through endocytic vesicles to the vacuole, where it is disassembled (Kolman et al., 2009). Endocytosis occurs after insulin binds to the Megalin-cubilin complex, which binds to the insulin receptor (IR) present in the BBM of the PT. Megalin is a transmembrane complex protein that salvages most of the serum proteins, including insulin, and is abundantly expressed in the PT segment S1 (Orlando et al., 1998; Bryniarski et al., 2018). Insulin increases its uptake and degradation by inducing an increase in megalin content (Hosojima et al., 2009). Less than 1% of the filtered insulin is transcytosed to the basolateral membrane (Nielsen, 1993), and only 1% is excreted in the urine eventually. Insulin reaches its highest concentration in the PT, where it effects gluconeogenesis suppression (Gatica et al., 2013) and sodium reabsorption (Nakamura et al., 2015a; Nakamura et al., 2020b). Additionally, insulin acts on other tubular sites where IR is abundantly expressed.
PT cells can generate large amounts of energy using β-oxidation of fatty acids and are capable of producing glucose through gluconeogenesis. PTs do not exhibit glycolytic capacity except in the S3 segment; these cells depend on oxidative phosphorylation for metabolism and are able to produce glucose by gluconeogenesis. In acute kidney injury, PT metabolism shifts from fatty acid β-oxidation and gluconeogenesis to glycolysis, whereas in chronic kidney disease, fatty acid oxidation is lost (Faivre et al., 2021; Scholz et al., 2021). Glucose transport in PT is mediated by GLUT and SGLT. GLUT is abundantly expressed in the kidney and is a cell surface glucose transporter. GLUT1 expression is located in the PT S3, TAL, and collecting ducts, and GLUT2 expression is located in PT S1. In cultured mouse PT, insulin increased GLUT1 mRNA, membrane protein content, and glucose uptake (Kang et al., 2010). In HEK cells, GLUT1 traffic to the apical membrane has also been demonstrated under PI3K/AKT signaling with increased glucose uptake (Zambrano et al., 2019). Note that mTOR has recently been implicated in regulating GLUT1 in cancer cells and has been considered a potential therapeutic target, although the association between mTOR and glucose transporters in the PT remains unclear.
Regarding GLUT2, plasma and luminal glucose concentrations have been shown to increase the expression of GLUT2 and translocate it from the basolateral to the BBM side in animal models (Marks et al., 2003). GLUT2 studies in humans have shown that GLUT1 and GLUT2 expressions are remarkably decreased in patients with T2DM compared to those in controls, although there are many unknowns regarding its localization (Norton et al., 2017; Solini et al., 2017).
For GLUT4 trafficking, insulin-dependent GLUT4 trafficking through Ins/Akt signaling occurs in the PTs (Mendes et al., 2010; Coffey et al., 2015), as well as in adipocytes (Sano et al., 2003) and myocytes (Bruss et al., 2005). Although it is clear that the Insulin/mTOR pathway regulates GLUT4 in several cell experiments, whether mTOR pathway also regulates GLUT4 in the PTs remains unclear.
SGLT transports glucose against its concentration gradient using an electrochemical sodium gradient. SGLT1 is found along all PT segments, abundantly expressed in S3 and the outer medulla than in the cortex (Balen et al., 2008). In contrast, SGLT2 is abundantly expressed in the renal cortex, especially in the S1 and S2 segments (Tojo et al., 2015). Insulin regulates SGLT1 directly (Ghezzi and Wright, 2012) and indirectly (Saad et al., 2005). However, there are conflicting reports on SGLT2 regulation. In cultured human renal cells, SGLT2 activity (Ghezzi and Wright, 2012) and protein levels (Nakamura et al., 2015b) were increased independent of insulin and glucose. In contrast, Na+ glucose transport increased by 250% by insulin in HEK cells, while hSGLT1 was relatively insensitive to insulin (Ghezzi and Wright, 2012). Thus, there is still no established theory on the insulin-mediated regulatory mechanism of SGLT, and the relationship between the Insulin/mTOR pathway and SGLT in the PTs remains unclear.
The kidneys, unlike the liver, cannot store large amounts of glycogen and produce glucose through gluconeogenesis. Renal gluconeogenesis is negatively regulated by insulin (Tiwari et al., 2013), while catecholamines, cortisol, thyroid hormone, growth hormone, and parathyroid hormone are positive regulators (Stumvoll et al., 1997; Meyer et al., 2003).
The role of mTORC1 in insulin signaling is multifactorial (Laplante and Sabatini, 2012); mTORC1 induces PPARγ in adipose progenitor cells, promoting their differentiation into adipocytes, in turn leading to fat accumulation (Shan et al., 2016; Soliman 2011). S6 kinase activated by mTORC1 forms a feedback loop by phosphorylating insulin receptor substrate (IRS)1, which mediates insulin receptor signaling, thereby suppressing IRS1 function. This mechanism reportedly plays a vital role in IR. Akt activated by mTORC2 phosphorylates the transcription factors FOXO1 and PPAR to suppress their function. FOXO1 induces PEPCK in the liver. Therefore, mTORC2 may inhibit gluconeogenesis in the liver (Dentin et al., 2007).
Gluconeogenesis occurs in the PTs and liver. mTORC1/2 in PTs may have a different role than the liver. In the liver, insulin regulates gluconeogenesis through the IRS2/PI3K/Akt/FOXO1 pathway (Cheng et al., 2010). Insulin has been reported to suppress gluconeogenesis in PT and the liver (Tiwari et al., 2013). Gluconeogenesis, regulated by insulin, plays an essential role in IR and achieving therapeutic objectives in T2D (Hatting et al., 2018). Indeed, renal gluconeogenesis is enhanced in animal models. Elsewhere, high postprandial insulin levels have been shown to decrease transcription of PT gluconeogenic enzymes in mice (Sasaki et al., 2017) and rabbits (Usarek et al., 2014), and the expression of gluconeogenic genes is reduced by glucose counterregulatory effects in cases of IR and deficiency. Furthermore, primary cultured cells obtained from the kidneys of participants (Pandey et al., 2017) and HK2 cell (Sasaki et al., 2017) exposed to insulin undergo gluconeogenesis reduction.
We recently reported that insulin directly inhibits gluconeogenesis in PTs and that the inhibitory effect of insulin on gluconeogenesis is mediated by the IRS1/Akt2/mTORC1/2 pathway (Nakamura et al., 2020a). Additionally, increased gluconeogenesis in PTs due to mTORC1 and mTORC2 inhibition could contribute to hyperglycemia. It is generally accepted that mTORC1 negatively regulates insulin signaling via S6K phosphorylation in nutrient-saturated states (Um et al., 2004). Prolonged exposure to rapamycin, an mTORC1 antagonist, induces insulin intolerance and hyperglycemia, which has been attributed to mTORC2 inhibition rather than mTORC1 in the liver (Lamming et al., 2012). Moreover, a recent study using a mouse model reported that mTOR was differentially involved in tubular autophagy activity via the insulin/mTOR pathway in the presence of nephropathy, depending on the type of diabetes. This study showed that insulin suppressed autophagy, whereas glucose loading increased it. In T1D, autophagic activity was enhanced and more protective than expected because of hyperglycemia and low insulin; in contrast, in T2D, hyperinsulinemia suppressed autophagy induction (Sakai et al., 2019).
Regarding inflammation, our current understanding of the regulatory mechanisms of mTORC2 is limited. However, mTORC1 and mTORC2 promote T-cell reprogramming, B-cell proliferation, and antibody responses. In macrophages, inhibition of mTOR activity or glycolysis decreases the production of proinflammatory cytokines and the ability to eliminate bacteria. Further, in response to microbial stimuli (e.g., β-glucan), classically-activated macrophages increase aerobic glycolysis via mTORC1, which regulates HIF1. Therefore, it plays an important role in immune cell regulation but is independent of insulin signaling. mTORC1 is potently activated by amino acids, growth factors, T-cell receptors, and interleukin 2.
Regulatory T-cells are strongly regulated by mTORC1, and mTORC1 suppression is known to have immunosuppressive effects (Zeng and Chi, 2017). Based on these mechanisms, therapeutic applications of mTOR inhibitors on inflammation have been investigated. In fact, sirolimus completely suppressed autoimmunity in lupus-prone mice (Warner et al., 1994) and suppressed disease activity in a retrospective study of systemic lupus erythematosus (SLE) participants (Caza et al., 2014). Furthermore, in a recent clinical trial in active symptomatic SLE participants, 12 months of sirolimus treatment was associated with improvement in disease activity, which was suggested to be associated with modification of inflammatory T-cell lineage specification (Lai et al., 2018). In addition to these findings, it has recently been shown that mTORC1 activation in glomeruli is higher in participants with lupus nephritis than in participants with DMN (Mao et al., 2022), that ATG5 and ATG7 and the HRES-1/Rab4A are associated with the molecular pathogenesis of lupus, and genetically driven mTOR activation is associated with fulminant lupus nephritis (Caza et al., 2022).
In this review, we describe the recent findings regarding the relationship between mTOR and renal tubular gluconeogenesis in glucose metabolism. The roles of mTOR in insulin signaling are diverse. The insulin/mTORC1 feedback loop mechanism has been reported to play an essential role in IR. Insulin signals through PI3K; it regulates signaling to mTORC1 through Akt phosphorylation. In addition, Akt, activated by mTORC2, phosphorylates FOXO1 and PPARs, suppressing their function. Therefore, this regulatory mechanism is thought to suppress gluconeogenesis. This mechanism is also active in PTs wherein mTORC1 and mTORC2 are essential for regulating glyconeogenesis and closely involved in regulating acid–base equilibrium and Na transport via v-ATPase, NHE3, and NBCe1. Furthermore, a recent study reported that mTOR was differentially involved in tubular autophagy activity via the insulin/mTOR pathway in the presence of nephropathy, depending on the type of diabetes. (Sakai et al., 2019).
The regulatory mechanisms of mTORC2 remain unclear. In particular, the mechanism of mTORC2 activation during glutamine withdrawal remains to be elucidated but may involve sestrin2. Sestrin2 upregulates the catalytic activity of mTORC2, causing Akt Ser473 phosphorylation. Sestrin2 promotes Akt phosphorylation through Sestrin/GATOR2 and GATOR2/mTORC2 interactions (Kowalsky et al., 2020; Szwed et al., 2021). Thus, activation of mTORC2 during glutamine starvation may be associated with sestrin2-mediated repression of mTORC1. Sestrin3 is also associated with rictor and enhances mTORC2 activation in response to insulin and nutrient stimulation (Tao et al., 2015; Szwed et al., 2021).
The relevance of Rheb, a critical factor in the insulin/mTORC1 pathway in the PTs, remains unclear. mTORC1 enhances the expression of the rate-limiting enzyme of gluconeogenesis via PGC-1α; whether this pathway is also useful in humans and its physiological role in the PTs is also unknown. Inhibition by PRAS40 is thought to be because of competition for substrates of mTORC1, and proteins known to be direct substrates of mTORC1 include 4EBP1, S6K, STAT3, and HIF1α (Szwed et al., 2021). Given that Rheb is involved in substrate association, thus, insulin-induced mTORC1 activation may phosphorylate different substrates compared to amino acid stimulation, which may be associated with PT gluconeogenesis.
Long-term administration of rapamycin to rats promoted IR, severe glucose intolerance, and increased gluconeogenesis (Houde et al., 2010). These changes were observed in the liver of rapamycin-treated rats despite normal activation of the IRS/PI3K/Akt pathway. These results indicated that mTORC1/S6K could regulate gluconeogenesis by modulating several important transcription factors. The mechanism by which DJB improves IR may be related to Rheb, which plays a vital role in the insulin/mTOR pathway. Briefly, DJB may improve insulin resistance through Rheb suppression upstream of mTORC1/S6K (Guo et al., 2016). Although its involvement in PT gluconeogenesis is currently unknown, Rheb inhibition represents a potential new therapeutic target for diabetes and IR.
The effect of insulin on gluconeogenesis suppression differs in the liver and kidney. In the liver, SREBP1c directly suppresses IRS2 transcription and inhibits hepatic insulin signaling (Ide et al., 2004; Tsunekawa et al., 2011). Unlike the liver, the renal cortex is incapable of insulin-induced changes in the expression of IRS2 and SREBP1c (Nakamura N. et al., 2015). Even IR did not change SREBP1c expression in the renal cortex. This indicates that the regulation of IRS2 expression in the renal cortex is quite different from that in the liver. Therefore, the mechanism of gluconeogenesis regulation by the Insulin/mTOR pathway may also differ in the liver and PT.
A recent study showed that mTORC1 might be activated by sensing S-adenosyl methionine (SAM). According to this report, increased catabolism of SAM via the action of glycine N-methyltransferase could extend the lifespan of Drosophila (Obata and Miura, 2015). However, in a cross-sectional study of individuals with metabolic syndrome, plasma SAM levels were reported to be associated with increased fasting insulin levels, IR, and tumor necrosis factor-α (Lind et al., 2018). These results indicate conflicting conclusions, namely insulin mediates mTORC1 through SAM and that SAM concentrations are associated with IR. Although there is no unified perspective, a recent review suggested that selective suppression of mTORC1 may be a potential therapeutic target for aging, metabolic disorders, or aging-related diseases (Kitada et al., 2020). Despite many inconsistencies and unresolved aspects regarding insulin’s regulatory mechanisms on the mTOR signaling pathway, it is clear that mTOR regulation plays a very important role in regulating tubular gluconeogenesis and sodium transport, as earlier described in this review.
Renal gluconeogenesis plays an essential role in normal physiological function, and its impairment has adverse pathological implications. Overall, this review elucidates that mTOR plays an important role in regulating renal gluconeogenesis. Signaling through the insulin/Akt pathway plays an important role in the regulatory mechanism of mTORC1 and mTORC2 in tubular gluconeogenesis; feeding enhances insulin secretion and suppresses proximal tubular gluconeogenesis. By contrast, in the presence of pathologies, such as IR or T2D, this signaling could be attenuated by downregulation of IRS2 expression (Nakamura et al., 2015a; Nakamura et al., 2020b). Furthermore, it suggests that renal gluconeogenesis is enhanced during IR. Such impairment may further contribute to hyperglycemia in T2D (Figure 2). However, further research is required in this area to explain the relevant mechanisms.
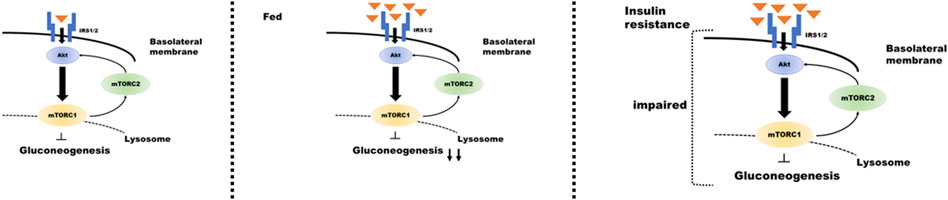
FIGURE 2. Regulation of insulin signaling in the proximal tubule during gluconeogenesis under different conditions. Proximal tubular gluconeogenesis responds differently depending on the state of insulin secretion and the presence of insulin resistance. When insulin is secreted after consuming food (center panel of the figure), insulin/mTOR signaling is enhanced, and gluconeogenesis is inhibited. The condition of insulin resistance (right panel) results in impaired insulin/mTOR signaling and enhanced gluconeogenesis despite high blood glucose levels.
MN, NS, SH, and MN conceived the idea and opinion topic. MN and NS wrote the manuscript. NS, SH, and MN reviewed the manuscript and contributed to its intellectual and scientific content with their domain-specific expertise.
The study was supported by the Japan Society for the Promotion of Science KAKENHI (grant numbers: 19K08671 and 19K17698).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
GAP, GTPase-activating proteins; Sin 1, SAPK interacting protein one; SAM, S-adenosyl Methionine; PT, Proximal tubules; mTOR, Mechanistic target of rapamycin; AMPK, AMP-activated protein kinase; Rheb, Ras homolog enriched in brain; IR, nsulin resistance.
Accili, D., and Arden, K. C. (2004). FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117, 421–426. doi:10.1016/s0092-8674(04)00452-0
Alsahli, M., and Gerich, J. E. (2017). Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 133, 1–9. doi:10.1016/j.diabres.2017.07.033
Ardestani, A., Lupse, B., Kido, Y., Leibowitz, G., and Maedler, K. (2018). mTORC1 signaling: a double-edged sword in diabetic β cells. Cell Metab. 27, 314–331. doi:10.1016/j.cmet.2017.11.004
Balen, D., Ljubojevic, M., Breljak, D., Brzica, H., Zlender, V., Koepsell, H., et al. (2008). Revised immunolocalization of the Na -D-glucose cotransporter SGLT1 in rat organs with an improved antibody 295, C475–C489.
Bernard, M., Yang, B., Migneault, F., Turgeon, J., Dieudé, M., Olivier, M., et al. (2020). Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 16, 2004–2016. doi:10.1080/15548627.2020.1713640
Bruss, M. D., Arias, E. B., Lienhard, G. E., and Cartee, G. D. (2005). Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes 54, 41–50. doi:10.2337/diabetes.54.1.41
Bryniarski, M. A., Yee, B. M., Jaffri, I., Chaves, L. D., Yu, J. A., Guan, X., et al. (2018). Increased megalin expression in early type 2 diabetes: Role of insulin-signaling pathways. Am. J. Physiol. Ren. Physiol. 315, F1191–F1207. doi:10.1152/ajprenal.00210.2018
Busaidy, N. L., Farooki, A., Dowlati, A., Perentesis, J. P., Dancey, J. E., Doyle, L. A., et al. (2012). Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 30, 2919–2928. doi:10.1200/JCO.2011.39.7356
Caza, T. N., Fernandez, D. R., Talaber, G., Oaks, Z., Haas, M., Madaio, M. P., et al. (2014). HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann. Rheum. Dis. 73, 1888–1897. doi:10.1136/annrheumdis-2013-203794
Caza, T., Wijewardena, C., Al-Rabadi, L., and Perl, A. (2022). Cell type-specific mechanistic target of rapamycin-dependent distortion of autophagy pathways in lupus nephritis. Transl. Res. 245, 55–81. doi:10.1016/j.trsl.2022.03.004
Cersosimo, E., Garlick, P., and Ferretti, J. (2000). Regulation of splanchnic and renal substrate supply by insulin in humans. Metabolism. 49, 676–683. doi:10.1016/s0026-0495(00)80048-7
Cersosimo, E., Judd, R. L., and Miles, J. M. (1994). Insulin regulation of renal glucose metabolism in conscious dogs. J. Clin. Invest. 93, 2584–2589. doi:10.1172/JCI117270
Chávez-Canales, M., Arroyo, J. P., Ko, B., Vázquez, N., Bautista, R., Castañeda-Bueno, M., et al. (2013). Insulin increases the functional activity of the renal NaCl cotransporter. J. Hypertens. 31, 303–311. doi:10.1097/HJH.0b013e32835bbb83
Cheng, Z., Tseng, Y., and White, M. F. (2010). Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 21, 589–598. doi:10.1016/j.tem.2010.06.005
Coffey, S., Costacou, T., Orchard, T., and Erkan, E. (2015). Akt links insulin signaling to albumin endocytosis in proximal tubule epithelial cells. Plos One 10, e0140417. doi:10.1371/journal.pone.0140417
Dentin, R., Liu, Y., Koo, S., Hedrick, S., Vargas, T., Heredia, J., et al. (2007). Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature 449, 366–369. doi:10.1038/nature06128
Ebner, M., Sinkovics, B., Szczygieł, M., Ribeiro, D. W., and Yudushkin, I. (2017). Localization of mTORC2 activity inside cells. J. Cell Biol. 216, 343–353. doi:10.1083/jcb.201610060
Faivre, A., Verissimo, T., Auwerx, H., Legouis, D., and de Seigneux, S. (2021). Tubular cell glucose metabolism shift during acute and chronic injuries. Front. Med. (Lausanne) 8, 742072. doi:10.3389/fmed.2021.742072
Foster, K. G., and Fingar, D. C. (2010). Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 285, 14071–14077. doi:10.1074/jbc.R109.094003
Fukuda, T., and Shiozaki, K. (2018). The Rag GTPase-Ragulator complex attenuates TOR complex 1 signaling in fission yeast. Autophagy 14, 1105–1106. doi:10.1080/15548627.2018.1444313
Gatica, R., Bertinat, R., Silva, P., Carpio, D., Ramírez, M. J., Slebe, J. C., et al. (2013). Altered expression and localization of insulin receptor in proximal tubule cells from human and rat diabetic kidney. J. Cell. Biochem. 114, 639–649. doi:10.1002/jcb.24406
Ghezzi, C., and Wright, E. M. (2012). Regulation of the human Na -dependent glucose cotransporter hSGLT2 303, C348–C354.
Guo, W., Han, H., Wang, Y., Zhang, X., Liu, S., Zhang, G., et al. (2016). miR-200a regulates Rheb-mediated amelioration of insulin resistance after duodenal–jejunal bypass. Int. J. Obes. 40, 1222–1232. doi:10.1038/ijo.2016.60
Haar, E. V., Lee, S., Bandhakavi, S., Griffin, T. J., and Kim, D. (2007). Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323. doi:10.1038/ncb1547
Hatting, M., Tavares, C. D., Sharabi, K., Rines, A. K., and Puigserver, P. (2018). Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 1411, 21–35. doi:10.1111/nyas.13435
Hoorn, E. J., Gritter, M., Cuevas, C. A., and Fenton, R. A. (2020). Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol. Rev. 100, 321–356. doi:10.1152/physrev.00044.2018
Hosojima, M., Sato, H., Yamamoto, K., Kaseda, R., Soma, T., Kobayashi, A., et al. (2009). Regulation of megalin expression in cultured proximal tubule cells by angiotensin II type 1A receptor-and insulin-mediated signaling cross talk. Endocrinology 150, 871–878. doi:10.1210/en.2008-0886
Houde, V. P., Brûlé, S., Festuccia, W. T., Blanchard, P., Bellmann, K., Deshaies, Y., et al. (2010). Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59, 1338–1348. doi:10.2337/db09-1324
Hsu, P. P., Kang, S. A., Rameseder, J., Zhang, Y., Ottina, K. A., Lim, D., et al. (2011). The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322. doi:10.1126/science.1199498
Humphrey, S. J., Yang, G., Yang, P., Fazakerley, D. J., Stöckli, J., Yang, J. Y., et al. (2013). Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020. doi:10.1016/j.cmet.2013.04.010
Ide, T., Shimano, H., Yahagi, N., Matsuzaka, T., Nakakuki, M., Yamamoto, T., et al. (2004). SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat. Cell Biol. 6, 351–357. doi:10.1038/ncb1111
Jia, R., and Bonifacino, J. S. (2019). Lysosome positioning influences mTORC2 and AKT signaling. Mol. Cell 75, 26–38. e3. doi:10.1016/j.molcel.2019.05.009
Kamei, Y., Miura, S., Suganami, T., Akaike, F., Kanai, S., Sugita, S., et al. (2008). Regulation of SREBP1c gene expression in skeletal muscle: Role of retinoid X receptor/liver X receptor and forkhead-O1 transcription factor. Endocrinology 149, 2293–2305. doi:10.1210/en.2007-1461
Kang, Y. S., Song, H. K., Lee, M. H., Ko, G. J., Han, J. Y., Han, S. Y., et al. (2010). Visfatin is upregulated in type-2 diabetic rats and targets renal cells. Kidney Int. 78, 170–181. doi:10.1038/ki.2010.98
Kaplan, B., Qazi, Y., and Wellen, J. R. (2014). Strategies for the management of adverse events associated with mTOR inhibitors. Transpl. Rev. 28, 126–133. doi:10.1016/j.trre.2014.03.002
Kazyken, D., Magnuson, B., Bodur, C., Acosta-Jaquez, H. A., Zhang, D., Tong, X., et al. (2019). AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal 12, eaav3249. doi:10.1126/scisignal.aav3249
Kitada, M., Xu, J., Ogura, Y., Monno, I., and Koya, D. (2020). Mechanism of activation of mechanistic target of rapamycin complex 1 by methionine. Front. Cell Dev. Biol. 8, 715. doi:10.3389/fcell.2020.00715
Kohn, A. D., Summers, S. A., Birnbaum, M. J., and Roth, R. A. (1996). Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 271, 31372–31378. doi:10.1074/jbc.271.49.31372
Kolman, P., Pica, A., Carvou, N., Boyde, A., Cockcroft, S., Loesch, A., et al. (2009). Insulin uptake across the luminal membrane of the rat proximal tubule in vivo and in vitro. Am. J. Physiol. Ren. Physiol. 296, F1227–F1237. doi:10.1152/ajprenal.90351.2008
Kowalsky, A. H., Namkoong, S., Mettetal, E., Park, H., Kazyken, D., Fingar, D. C., et al. (2020). The GATOR2–mTORC2 axis mediates Sestrin2-induced AKT Ser/Thr kinase activation. J. Biol. Chem. 295, 1769–1780. doi:10.1074/jbc.RA119.010857
Lai, Z., Kelly, R., Winans, T., Marchena, I., Shadakshari, A., Yu, J., et al. (2018). Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial 391, 1186–1196.
Lamming, D. W., Ye, L., Katajisto, P., Goncalves, M. D., Saitoh, M., Stevens, D. M., et al. (2012). Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643. doi:10.1126/science.1215135
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. 149, 273–293.
Li, Y., Corradetti, M. N., Inoki, K., and Guan, K. (2004). TSC2: Filling the GAP in the mTOR signaling pathway. Trends biochem. Sci. 29, 32–38. doi:10.1016/j.tibs.2003.11.007
Lind, M. V., Lauritzen, L., Vestergaard, H., Hansen, T., Pedersen, O., Kristensen, M., et al. (2018). One-carbon metabolism markers are associated with cardiometabolic risk factors. Nutr. Metab. Cardiovasc Dis. 28, 402–410. doi:10.1016/j.numecd.2018.01.005
Liu, P., Gan, W., Chin, Y. R., Ogura, K., Guo, J., Zhang, J., et al. (2015). PtdIns(3, 4, 5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 35 (45), 1194–1209. doi:10.1158/2159-8290.CD-15-0460
Liu, P., Gan, W., Inuzuka, H., Lazorchak, A. S., Gao, D., Arojo, O., et al. (2013). Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 15, 1340–1350. doi:10.1038/ncb2860
Lizcano, J. M., and Alessi, D. R. (2002). The insulin signalling pathway. Curr. Biol. 12, R236–R238. doi:10.1016/s0960-9822(02)00777-7
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005). Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 15, 702–713. doi:10.1152/physrev.2001.81.2.807
Manning, B. D., and Cantley, L. C. (2007). AKT/PKB signaling: Navigating downstream. Cell 129, 1261–1274. doi:10.1016/j.cell.2007.06.009
Mao, Z., Tan, Y., Tao, J., Li, L., Wang, H., Yu, F., et al. (2022). Renal mTORC1 activation is associated with disease activity and prognosis in lupus nephritis. Rheumatol. Oxf. 61 (9), 3830–3840. doi:10.1093/rheumatology/keac037
Marks, J., Carvou, N. J., Debnam, E. S., Srai, S. K., and Unwin, R. J. (2003). Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J. Physiol. Lond. ) 553, 137–145. doi:10.1113/jphysiol.2003.046268
Mendes, A. I., Matos, P., Moniz, S., and Jordan, P. (2010). Protein kinase WNK1 promotes cell surface expression of glucose transporter GLUT1 by regulating a Tre-2/USP6-BUB2-Cdc16 domain family member 4 (TBC1D4)-Rab8A complex. J. Biol. Chem. 285, 39117–39126. doi:10.1074/jbc.M110.159418
Meyer, C., Stumvoll, M., Welle, S., Woerle, H. J., Haymond, M., and Gerich, J. (2003). Relative importance of liver, kidney, and substrates in epinephrine-induced increased gluconeogenesis in humans. Am. J. Physiol. Endocrinol. Metab. 285, E819–E826. doi:10.1152/ajpendo.00145.2003
Moloughney, J. G., Kim, P. K., Vega-Cotto, N. M., Wu, C., Zhang, S., Adlam, M., et al. (2016). mTORC2 responds to glutamine catabolite levels to modulate the hexosamine biosynthesis enzyme GFAT1. Mol. Cell 63, 811–826. doi:10.1016/j.molcel.2016.07.015
Mordier, S., and Iynedjian, P. B. (2007). Activation of mammalian target of rapamycin complex 1 and insulin resistance induced by palmitate in hepatocytes. Biochem. Biophys. Res. Commun. 362, 206–211. doi:10.1016/j.bbrc.2007.08.004
Nakae, J., Kitamura, T., Kitamura, Y., Biggs, W. H., Arden, K. C., and Accili, D. (2003). The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 4, 119–129. doi:10.1016/s1534-5807(02)00401-x
Nakamura, M., Satoh, N., Suzuki, M., Kume, H., Homma, Y., Seki, G., et al. (2015a). Stimulatory effect of insulin on renal proximal tubule sodium transport is preserved in type 2 diabetes with nephropathy. Biochem. Biophys. Res. Commun. 461, 154–158. doi:10.1016/j.bbrc.2015.04.005
Nakamura, M., Satoh, N., Tsukada, H., Mizuno, T., Fujii, W., Suzuki, A., et al. (2020a). Stimulatory effect of insulin on H -ATPase in the proximal tubule via the Akt/mTORC2 pathway 107, 376–389.
Nakamura, M., Tsukada, H., Seki, G., Satoh, N., Mizuno, T., Fujii, W., et al. (2020b). Insulin promotes sodium transport but suppresses gluconeogenesis via distinct cellular pathways in human and rat renal proximal tubules. Kidney Int. 97, 316–326. doi:10.1016/j.kint.2019.08.021
Nakamura, M., Yamazaki, O., Shirai, A., Horita, S., Satoh, N., Suzuki, M., et al. (2015b). Preserved Na/HCO3 cotransporter sensitivity to insulin may promote hypertension in metabolic syndrome. Kidney Int. 87, 535–542. doi:10.1038/ki.2014.351
Nakamura, N., Matsui, T., Ishibashi, Y., and Yamagishi, S. (2015c). Insulin stimulates SGLT2-mediated tubular glucose absorption via oxidative stress generation. Diabetol. Metab. Syndr. 7, 1–6. doi:10.1186/s13098-015-0044-1
Nielsen, S. (1993). Sorting and recycling efficiency of apical insulin binding sites during endocytosis in proximal tubule cells. Am. J. Physiol. 264, C810–C822. doi:10.1152/ajpcell.1993.264.4.C810
Norton, L., Shannon, C. E., Fourcaudot, M., Hu, C., Wang, N., Ren, W., et al. (2017). Sodium-glucose co-transporter (SGLT) and glucose transporter (GLUT) expression in the kidney of type 2 diabetic subjects. Diabetes Obes. Metab. 19, 1322–1326. doi:10.1111/dom.13003
Obata, F., and Miura, M. (2015). Enhancing S-adenosyl-methionine catabolism extends Drosophila lifespan. Nat. Commun. 6, 1–9. doi:10.1038/ncomms9332
Orlando, R. A., Rader, K., Authier, F., Yamazaki, H., Posner, B. I., Bergeron, J. J., et al. (1998). Megalin is an endocytic receptor for insulin. J. Am. Soc. Nephrol. 9, 1759–1766. doi:10.1681/ASN.V9101759
Pallet, N., and Legendre, C. (2013). Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf. 12, 177–186. doi:10.1517/14740338.2013.752814
Pandey, G., Shankar, K., Makhija, E., Gaikwad, A., Ecelbarger, C., Mandhani, A., et al. (2017). Reduced insulin receptor expression enhances proximal tubule gluconeogenesis. J. Cell. Biochem. 118, 276–285. doi:10.1002/jcb.25632
Puigserver, P., Rhee, J., Donovan, J., Walkey, C. J., Yoon, J. C., Oriente, F., et al. (2003). Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 423, 550–555. doi:10.1038/nature01667
Saad, S., Stevens, V. A., Wassef, L., Poronnik, P., Kelly, D. J., Gilbert, R. E., et al. (2005). High glucose transactivates the EGF receptor and up-regulates serum glucocorticoid kinase in the proximal tubule. Kidney Int. 68, 985–997. doi:10.1111/j.1523-1755.2005.00492.x
Sakai, S., Yamamoto, T., Takabatake, Y., Takahashi, A., Namba-Hamano, T., Minami, S., et al. (2019). Proximal tubule autophagy differs in type 1 and 2 diabetes. J. Am. Soc. Nephrol. 30, 929–945. doi:10.1681/ASN.2018100983
Sancak, Y., Bar-Peled, L., Zoncu, R., Markhard, A. L., Nada, S., and Sabatini, D. M. (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. doi:10.1016/j.cell.2010.02.024
Sancak, Y., Thoreen, C. C., Peterson, T. R., Lindquist, R. A., Kang, S. A., Spooner, E., et al. (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915. doi:10.1016/j.molcel.2007.03.003
Sano, H., Kane, S., Sano, E., Mı̂inea, C. P., Asara, J. M., Lane, W. S., et al. (2003). Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 278, 14599–14602. doi:10.1074/jbc.C300063200
Sasaki, M., Sasako, T., Kubota, N., Sakurai, Y., Takamoto, I., Kubota, T., et al. (2017). Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubules of the kidney. Diabetes 66, 2339–2350. doi:10.2337/db16-1602
Sato, T., Nakashima, A., Guo, L., and Tamanoi, F. (2009). Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem. 284, 12783–12791. doi:10.1074/jbc.M809207200
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi:10.1016/j.cell.2017.02.004
Scholz, H., Boivin, F. J., Schmidt-Ott, K. M., Bachmann, S., Eckardt, K., Scholl, U. I., et al. (2021). Kidney physiology and susceptibility to acute kidney injury: Implications for renoprotection 17, 335–349.
Shan, T., Zhang, P., Jiang, Q., Xiong, Y., Wang, Y., and Kuang, S. (2016). Adipocyte-specific deletion of mTOR inhibits adipose tissue development and causes insulin resistance in mice. 59, 1995–2004.
Shimobayashi, M., and Hall, M. N. (2014). Making new contacts: The mTOR network in metabolism and signalling crosstalk 15, 155–162.
Sohara, E., Rai, T., Yang, S., Ohta, A., Naito, S., Chiga, M., et al. (2011). Acute insulin stimulation induces phosphorylation of the Na-Cl cotransporter in cultured distal mpkDCT cells and mouse kidney. Plos One 6, e24277. doi:10.1371/journal.pone.0024277
Solini, A., Rossi, C., Mazzanti, C. M., Proietti, A., Koepsell, H., and Ferrannini, E. (2017). Sodium-glucose co-transporter (SGLT) 2 and SGLT1 renal expression in patients with type 2 diabetes. Diabetes Obes. Metab. 19, 1289–1294. doi:10.1111/dom.12970
Stumvoll, M., Meyer, C., Mitrakou, A., Nadkarni, V., and Gerich, J. E. (1997). Renal glucose production and utilization: New aspects in humans. Diabetologia 40, 749–757. doi:10.1007/s001250050745
Szwed, A., Kim, E., and Jacinto, E. (2021). Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 101, 1371–1426. doi:10.1152/physrev.00026.2020
Tao, R., Xiong, X., Liangpunsakul, S., and Dong, X. C. (2015). Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes 64, 1211–1223. doi:10.2337/db14-0539
Tee, A. R., Manning, B. D., Roux, P. P., Cantley, L. C., and Blenis, J. (2003). Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268. doi:10.1016/j.cub.2022.01.027
Tiwari, S., Singh, R. S., Li, L., Tsukerman, S., Godbole, M., Pandey, G., et al. (2013). Deletion of the insulin receptor in the proximal tubule promotes hyperglycemia. J. Am. Soc. Nephrol. 24, 1209–1214. doi:10.1681/ASN.2012060628
Tojo, A., Hatakeyama, S., Kinugasa, S., and Nangaku, M. (2015). Angiotensin receptor blocker telmisartan suppresses renal gluconeogenesis during starvation. Diabetes Metab. Syndr. Obes. 8, 103. doi:10.2147/DMSO.S78771
Tsunekawa, S., Demozay, D., Briaud, I., McCuaig, J., Accili, D., Stein, R., et al. (2011). FoxO feedback control of basal IRS-2 expression in pancreatic β-cells is distinct from that in hepatocytes. Diabetes 60, 2883–2891. doi:10.2337/db11-0340
Um, S. H., Frigerio, F., Watanabe, M., Picard, F., Joaquin, M., Sticker, M., et al. (2004). Absence of S6K1 protects against age-and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205. doi:10.1038/nature02866
Usarek, M., Jagielski, A. K., Krempa, P., Dylewska, A., Kiersztan, A., Drozak, J., et al. (2014). Proinsulin C-peptide potentiates the inhibitory action of insulin on glucose synthesis in primary cultured rabbit kidney-cortex tubules: Metabolic studies. Metab. Stud. 92, 1–8. doi:10.1139/bcb-2013-0074
Wang, B., Jie, Z., Joo, D., Ordureau, A., Liu, P., Gan, W., et al. (2017). TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 545, 365–369. doi:10.1038/nature22344
Warner, L. M., Adams, L. M., and Sehgal, S. N. (1994). Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheum. 37, 289–297. doi:10.1002/art.1780370219
Wolfson, R. L., and Sabatini, D. M. (2017). The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. 26, 301–309. doi:10.1016/j.cmet.2017.07.001
Ye, L., Varamini, B., Lamming, D. W., Sabatini, D. M., and Baur, J. A. (2012). Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2. Front. Genet. 3, 177. doi:10.3389/fgene.2012.00177
Zambrano, A., Molt, M., Uribe, E., and Salas, M. (2019). Glut 1 in cancer cells and the inhibitory action of resveratrol as a potential therapeutic strategy. Int. J. Mol. Sci. 20, 3374. doi:10.3390/ijms20133374
Zeng, H., and Chi, H. (2017). mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr. Opin. Immunol. 46, 103–111. doi:10.1016/j.coi.2017.04.005
Zhu, M., and Wang, X. (2020). Regulation of mTORC1 by small GTPases in response to nutrients. J. Nutr. 150, 1004–1011. doi:10.1093/jn/nxz301
Keywords: mTOR, insulin resistance, proximal tubules, gluconeogenesis, diabetes
Citation: Nakamura M, Satoh N, Horita S and Nangaku M (2022) Insulin-induced mTOR signaling and gluconeogenesis in renal proximal tubules: A mini-review of current evidence and therapeutic potential. Front. Pharmacol. 13:1015204. doi: 10.3389/fphar.2022.1015204
Received: 09 August 2022; Accepted: 27 September 2022;
Published: 10 October 2022.
Edited by:
Keizo Kanasaki, Shimane University, JapanReviewed by:
Takahiko Nakagawa, Shiga University of Medical Science, JapanCopyright © 2022 Nakamura, Satoh, Horita and Nangaku. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Motonobu Nakamura, bmFrYW11cmEtc3RtQHVtaW4uYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.