 Shimeng Jiao1,2,3
Shimeng Jiao1,2,3 Ting Cao
Ting Cao Hualin Cai
Hualin Cai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 12 October 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1005702
This article is part of the Research Topic The Pros and Cons of Psychotropic Drug-Induced Changes in Periphery and Central Nervous System: Elucidating Structural and Molecular Mechanisms View all 8 articles
Treatment-resistant schizophrenia (TRS) often results in severe disability and functional impairment. Currently, the diagnosis of TRS is largely exclusionary and emphasizes the improvement of symptoms that may not be detected early and treated according to TRS guideline. As the gold standard, clozapine is the most prescribed selection for TRS. Therefore, how to predict TRS in advance is critical for forming subsequent treatment strategy especially clozapine is used during the early stage of TRS. Although mounting studies have identified certain clinical factors and neuroimaging characteristics associated with treatment response in schizophrenia, the predictors for TRS remain to be explored. Biomarkers, particularly for peripheral biomarkers, show great potential in predicting TRS in view of their predictive validity, noninvasiveness, ease of testing and low cost that would enable their widespread use. Recent evidence supports that the pathogenesis of TRS may be involved in abnormal neurotransmitter systems, inflammation and stress. Due to the heterogeneity of TRS and the lack of consensus in diagnostic criteria, it is difficult to compare extensive results among different studies. Based on the reported neurobiological mechanisms that may be associated with TRS, this paper narratively reviews the updates of peripheral biomarkers of TRS, from genetic and other related perspectives. Although current evidence regarding biomarkers in TRS remains fragmentary, when taken together, it can help to better understand the neurobiological interface of clinical phenotypes and psychiatric symptoms, which will enable individualized prediction and therapy for TRS in the long run.
Schizophrenia is a severe, lifelong condition characterized by poor behavioral, emotional, and cognitive function that affects about 1% of the world’s population (Marder and Cannon, 2019). With the development of antipsychotic drugs in the 1950s, patients with schizophrenia were given the opportunity to be treated and rehabilitated (Nucifora et al., 2019). Unfortunately, not all patients respond to antipsychotic drugs, and around a third of patients were considered treatment-resistant after failing to respond to two different antipsychotic drugs (Faden and Citrome, 2019). Compared with schizophrenia in remission, the treatment cost for TRS is 3-11 fold higher, accounting for 60%–80% of the entire cost of schizophrenia management, which places a heavy financial burden on families and society (Kennedy et al., 2014). Patients who are resistant to treatment have much severer symptoms, disabilities, higher risk of suicide, lower quality of life as well as worse prognosis than patients who are responsive to treatment (Correll and Howes, 2021).
The definition of TRS has been continuously updated since 1966 (Itil et al., 1966), and the clinical focus on TRS switches from the persistence of positive symptoms to a stronger emphasis on comprehensive assessment of psychosocial functioning. The available standards for understanding and defining TRS have been illustrated in several reviews (Vita et al., 2019; Correll and Howes, 2021; Howes et al., 2022). However, the existent standards vary with studies, especially in the recommendations of optimum drug therapy and the methods for assessment of treatment response, which limited their research and clinical transformation, as well as restricted the formation of consistent clinical management (Howes et al., 2022). In order to address this issue, the Treatment Response and Resistance in Psychosis (TRRIP) Working Group has updated the consensus standards and guidelines based on the assessment of previously defined approaches (Howes et al., 2017). The term “treatment-resistant schizophrenia (TRS)” has been proposed to this category of schizophrenia patients who match the following characteristics: 1) at least two different antipsychotic drugs were used, 2) prescribed dose was the target dose for acute phase of schizophrenia or a total daily dose equivalent to 600 mg chlorpromazine, 3) each antipsychotic treatment algorism lasted at least 6 weeks and required at least 12 weeks of treatment time and 4) during the prospective observational period, the symptom improvement was less than 20% and the severity still reached the treatment-resistant threshold (moderate severity or above according to the standardized rating scale).
The only antipsychotic drug that has been proven to be effective in TRS is clozapine (Rajagopal et al., 2018). However, it is only effective in 40%–70% of patients with TRS (Samanaite et al., 2018). These patients who are resistant to clozapine were called ultra–treatment resistant schizophrenia (Howes et al., 2017). Patients who have not reacted to previous antipsychotic medications can benefit from clozapine, but it carries a risk of major side effects and demands frequent blood testing (Samanaite et al., 2018). For these reasons, clozapine treatment may be difficult for both patients and doctors. Therefore, there is a significant delay in starting clozapine treatment (mean delay 2–5 years) in regular clinical practice (Yoshimura et al., 2017). However, clozapine should be used for TRS patients especially during the early stage according to research (Horsdal et al., 2017; Egerton et al., 2021). Biomarkers of TRS can forecast the appearance of TRS and offer proof for clinicians to recommend clozapine. Additionally, if tests could be created to assist doctors in anticipating whether or not a patient will respond to clozapine, this would significantly shorten the time before clozapine is used and allow for selective use of clozapine in the subset of patients where it is most likely to be effective.
Currently, several demographic and clinical factors have been identified to predict worse response to treatment for schizophrenia, such as family history of psychosis, earlier age of onset, poor social functioning prior to onset, longer untreated time, more severe negative symptoms, and extrapyramidal symptoms shown early in treatment (Nucifora et al., 2019; Takeuchi et al., 2019; Correll and Howes, 2021; Cai et al., 2022). As for the neurobiological mechanisms underlying TRS, several hypotheses are proposed including dopamine hypersensitivity, high dopaminergic and normal dopaminergic subtypes, glutamate/GABAergic disorders, serotonin disorders, inflammation and stress (Potkin et al., 2020). However, the majority of available studies focused on how the preset treatment strategy improves psychiatric symptoms rather than on exploring the underlying neurobiological alterations, the biological mechanisms of the above mentioned risk factors (Bumb et al., 2015). As evidenced by the variability of antipsychotic treatment responsiveness, it has come to a consensus that TRS actually represents a distinct biological category with its own pathophysiology (Iasevoli et al., 2016; Nucifora et al., 2019). On one hand, the three elements, including a standard diagnosis of schizophrenia, antipsychotic drug use and duration of treatment, need to be present at the same time according to the most recent consensus standards defining TRS. However, several factors, such as poor patient compliance or a protracted patient response time, can influence this procedure. In addition, patients who developed TRS only after the initial response to antipsychotic medications fell into this category, making it impossible to diagnose TRS at the beginning of the first antipsychotic treatment (Howes et al., 2017; Howes et al., 2022). On the other hand, an accurate prediction of poor response to the treatment of schizophrenia based on clinical factors requires profound experiences and skills of the psychiatrists, and will also be affected by the subjective attitude of patients (Bumb et al., 2015). In this context, there is an increasing clinical need to predict the development of TRS with novel biomarkers.
The neuroimaging technology of TRS is relatively mature. It potentially serves as a tool for distinguishing TRS from non-TRS depending on brain structural and functional abnormalities. Most studies reported cerebral structure abnormalities include atrophied brain, enlarged ventricular, reduced cortical thickness, compromised integrity of gray matter and white matter (Vita et al., 2019). TRS showed a wide range of brain activity patterns and connectivity abnormalities, particularly in the frontotemporal and subcortical networks (Molent et al., 2019). The state-of-the-art neuroimaging technologies such as magnetic resonance spectroscopy (MRS or MRI), Computed Tomography (CT), functional Magnetic Resonance Imaging (fMRI), Positron Emission Tomography (PET), and Single Photon Emission Computed Tomography (SPECT) usually require professionals and are often associated with a high cost (Samanaite et al., 2018; Vita et al., 2019; Cao et al., 2020), despite the fact that they are noninvasive.
A biomarker is a biological, genetic, epigenetic, or chemical trait that can be used to diagnose, monitor, and predict disease (Kraguljac et al., 2021). Compared with neuroimaging biomarkers from central nervous system, peripheral biomarkers are more widely available, and cost-effective. Although the molecular causes and pathways of TRS are still unclear, accumulating studies are focusing on the exploration of validated, selective, and specific peripheral biomarkers for TRS in the hope of future routine use (Nedic Erjavec et al., 2018). This paper mainly reviews the scientific development of peripheral genetic, inflammation and stress biomarkers related to TRS, based on the neurobiological hypotheses currently implicated in TRS. The goal of this review is to provide new insights in monitoring and diagnosing TRS, which will allow for earlier intervention and noticing of a change in the disease’s course and severity (Nucifora et al., 2019). When taken together the data of peripheral TRS biomarkers, it will help us better understand the underlying neurological mechanisms of TRS and guide the development of future treatments (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014).
The literature search was performed at the PubMed, Embase, BIOSIS and PsychInfo databases using the following keywords “schizophrenia,” “resistant,” “refractory,” “response,” “treatment,” “clozapine,” “biomarkers” and “peripheral biomarkers” as well as different combinations of these keywords. References are papers published before May 2022 including Clinical Trial, Meta-analysis, Randomized Controlled Trial and Review. The search results were deduplicated and screened. According to the inclusion criteria and exclusion criteria, two authors S.J. and T.C. independently screened the retrieved literature, and discussed together when the results were inconsistent. Selection criteria are as follows: 1) It is an original English paper. 2) The results are from clinical studies. 3) Therapeutic resistant schizophrenia was included only when clearly defined in the study according to criteria customized at the time. Exclusion criteria were: 1) The inclusion criteria for TRS were not clearly defined. 2) Studies on animals. In total, 64 studies were selected for this review (see Figure 1 for details).
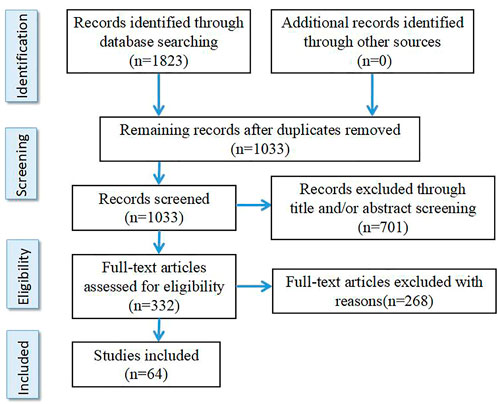
FIGURE 1. Flowchart of study selection.
High heritability for schizophrenia emphasizes the important role of genetic variation in the etiology of this devastating disease (Legge et al., 2021). TRS may be even more heritable, with more significantly genetic underpinnings (Reynolds et al., 2006; Gillespie et al., 2017). Due to the variations in the effectiveness of antipsychotic drugs on controlling symptoms and inducing adverse side effects, antipsychotic pharmacogenetics issues have received considerable attention in recent years. The researchers exerted their energy primarily on identifying candidate genes that have been associated with the pharmacological actions and mechanisms of these antipsychotic drug receptors (Reynolds, 2004; Reynolds et al., 2006). A recent study utilized two large and orthogonally created databases to collate drug targets into 167 gene sets targeted by pharmacologically similar antipsychotic drugs, and then examined the enrichment of schizophrenia risk loci in these sets. Interestingly, the findings support the notion that there are specific genetic overlaps between schizophrenia pathogenesis and antipsychotic mechanism of action (Ruderfer et al., 2016). To identify genes with a role in both the risks of developing schizophrenia and treatment resistance, they tested gene sets that were previously enriched for the singleton disruptive mutations in schizophrenia, gene sets implicated in antipsychotic efficacy by PharmGKB and genes with known roles in ADME (absorption, distribution, metabolism, excretion) (Dahl, 2002; Purcell et al., 2014). The results indicated that the genes related to the targets of antipsychotic drugs are the only overlapped set showing enrichment for both disease risk and treatment resistance (Ruderfer et al., 2016).
Pharmacogenetic biomarkers from peripheral blood leucocytes can predict TRS through genetic variation by analyzing the correlation between the risk genes, namely single nucleotide polymorphisms (SNPs) and the efficacy of antipsychotics (Pouget et al., 2014). Genetic studies have focused on the association between TRS and multiple candidate genes, involving multiple neurotransmitter systems that are consistent with the etiological hypothesis of TRS. It can be demonstrated by the pharmacodynamics of new antipsychotics affecting not only dopaminergic receptors but also serotonergic and glutamate receptors (Ji et al., 2008b; Kohlrausch et al., 2008; Bilic et al., 2014; Zink et al., 2014).
While altered dopaminergic function is the main feature of schizophrenia, patients with TRS might have distinct patterns in dopaminergic abnormality (Kogure et al., 2021). Specifically, as compared with patients with treatment-responsive schizophrenia, decreased dopamine synthetic capacity in striatum, decreased dopaminergic synaptic density and dopamine transporter protein expression in caudate nucleus have been reported in TRS patients (Demjaha et al., 2012; Sagud et al., 2018). It is well acknowledged that dopamine hypersensitivity induced by the compensatory upregulation of dopamine D2 receptors (DRD2) caused by the over-blockade of antipsychotics perhaps play a role in the pathogenesis of TRS (Potkin et al., 2020). Available evidence indicates that dopamine dysregulation associated with TRS may result from expression and/or functional changes in dopamine synthase, receptor, transporter, and catabolic enzyme (Purves-Tyson et al., 2017).
Several studies have suggested that the COMT gene is known to be one of the key factors in the regulation of dopamine level in the PFC, which is associated with drug response. Besides, COMT is believed to be the carriers of the Met allele closely related to treatment resistance (Kirenskaya et al., 2018; Hajj et al., 2019). Met/Met of COMT was proved as predictors of TRS (Escamilla et al., 2018). It has been demonstrated that the occurrence of TRS tended to be higher in patients with the COMT L/L genotype than in the other patients. These findings suggested that the COMT genotype with low-activity is more common in TRS (Inada et al., 2003). Another study has identified the associations between COMT rs4680 and rs4818 polymorphisms and TRS, which were gender dependent. In female TRS patients, the COMT rs4680 AA genotype carriers were more frequent than G allele carriers, as the same with COMT rs4818 CC genotype. However, there was no such association in males (Sagud et al., 2018).
The DRD2 density has been found to be lower in A1 allele carriers of Taq1A and Del allele noncarriers of—141C Ins/Del for DRD2 gene polymorphisms. Evidence suggested that haplotype polymorphisms of Taq1A and -141c Ins/Del DRD2 genes contributed to DRD2 antagonist resistance, especially linked to anxiety and depressive symptoms in schizophrenia (Kondo et al., 2003). For A-241G of DRD2, the G allele frequency was higher in the TRS patients than in the responsive schizophrenic group (Escamilla et al., 2018). Among the haplotypes observed in the DRD3 gene, the T/A/G/A/C haplotype was related to TRS (Kohlrausch et al., 2008). Besides, the Ser/Gly genotype of DRD3 may predict TRS (Escamilla et al., 2018). Data also showed that the sensitivity to treatment for schizophrenia was affected by DRD1 rs4532 polymorphism with the presence of G-allele increasing the risk of TRS (Ota et al., 2012).
The pathogenesis of schizophrenia is related to the abnormality of the serotonergic system (Herken et al., 2003). Components of the serotonin system are being investigated as a risk factor for schizophrenia and are believed to play an important role in the clinical efficacy of antipsychotics (Lieberman et al., 1998). This is supported by the fact that some atypical antipsychotic drugs such as risperidone and clozapine modulate serotonin for antipsychotic effects (Roth et al., 2004). Altered serotonin responses in the brains of TRS patients have been reported (Anttila et al., 2007). Serotonin (5-HT) receptor and serotonin transporter (5-HTT) are the key components of the serotonergic neurotransmitter system (Wang et al., 2007). In recent years, the role of their gene polymorphisms in TRS has been studied. For example, clozapine exerts antipsychotic effects by binding to several serotonin receptor subtypes that can reduce dopaminergic neurotransmission (Ji et al., 2008b; Kohlrausch et al., 2010). It is easy to think of studying the relationship between serotonin receptor subtypes and TRS. However, in a study analyzed the mutations in three serotonin receptors (HTR2A, HTR3A, and HTR4), even though patients with the T/T genotype of the HTR3A polymorphism received significantly higher daily antipsychotic doses, no significant association was observed between TRS and each allele, genotype, and haplotype (Ji et al., 2008b).
The 5-hydroxytryptamine receptor 2A (HTR2A) gene polymorphism was implicated to play a role in the pathogenesis of schizophrenia. Although the findings were mixed, HTR2A polymorphisms could predict TRS in general. Consistent with the previous result, evidence showed that T102C polymorphism in the HTR2A gene did not accelerate susceptibility to schizophrenia. However, the frequency of hospitalization was higher in the patients with T/C and T/T genotypes compared with the patients with C/C genotype, suggesting that it may represent a poorer prognosis and help distinguish TRS (Herken et al., 2003). Another study hypothesized that HTR2A T102C polymorphism, tryptophan hydroxylase 1 (TPH1) gene polymorphism and the G-protein beta-3 subunit (GNB3) C825T polymorphism alter serotonergic neurotransmission and are related to treatment response in schizophrenia. The HTR2A T102C C/C genotype or the TPH1 C/A genotype, and the GNB3 C825T C/T genotype were found in TRS patients when compared with responders. In addition, 5-HT2A T102C polymorphism and the GNB3 C825T C/T polymorphism were associated with genders (Anttila et al., 2007). Besides, the deletion allele at the 3 bp-insertion/deletion polymorphism site (-100_-102delAAG) encoding HTR3B was significantly more frequent in the TRS group than the insertion allele by a single-marker comparison (Ji et al., 2008a).
The 5-HTT, encoded by the SLC6A4 gene, is responsible for reabsorption of serotonin into the presynaptic neuron and is a major regulator of serotonin function (Lesch et al., 1996). In addition, 5-HTTLPR is a 44 bp insertion/deletion polymorphism in the promoter region of SLC6A4, in which the short and long HTTRLP variants differentially modulate transcriptional activity of the 5HTT gene promoter (Sookoian et al., 2008). Another 5-HTT polymorphism is a variable number of tandem repeat elements known as HTTVNTR in the intron2. Compared with all other serotonin transporter-promoter region (SERT-PR) genotypes, “SS” SERT-PR carriers were less likely to develop TRS, while “LaS” SERT-PR carriers were almost three times more likely to develop TRS than those with the SS genotype (short‘S’ and long“L” allele) (Bilic et al., 2014). However, some inconsistent results were obtained. It was revealed that polymorphisms of rs6295 which encoded 5-hydroxytryptamine 1A receptor (5-HT1A) and 5-HTTLPR can influence some clinical symptoms of schizophrenia but no association was found in the progression of TRS (Terzić et al., 2015). Following risperidone treatment, the 5-HTTRLP patients without the L allele showed worse improvement on the Brief Psychiatric Rating Scale (BPRS) than patients with the L allele but no such relationship was found for the HTTVNTR. In haplotype analysis, the frequency of L-12 (the size of HTTVNTR fragment was 300bp) haplotype showed significant differences between responders and non-responders (Wang et al., 2007).
The combined role of genes in the serotonin and dopamine systems has been reported in several studies (Kohlrausch et al., 2008; Zink et al., 2014). TRS is attributed to an interaction between the dopamine transporter (DAT) and a variable number tandem repeat (VNTR) polymorphism inside intron 2 (SERT-in2). Patients with SERT-in2ll and DAT 9/10, 9/9, 9/9, and 6/6 genotypes were more likely to develop TRS, as were those with DAT 10/10 or 10/12 genotypes paired with SERT-in2ls or ss genotypes (Bilic et al., 2014).
The dopaminergic system of schizophrenia has been studied for a long time, which can indeed explain some pathogenesis and therapeutic targets of schizophrenia. However, it is accepted that dopamine dysfunction cannot explain all aspects of schizophrenia (Bartolomeis et al., 2019). A large number of studies have shown that the pathophysiology of schizophrenia involves not just dysregulation of dopaminergic, but aberrant γ-aminobutyric acid (GABA), glutamatergic neurotransmission and their interactions (Lin et al., 2012). Several studies of pharmacological models and clinical trials have elucidated the role of excitatory glutamatergic signaling in the pathophysiology of schizophrenia from the angles of neurodevelopment risks, pathologic precipitations, and genetic susceptibility (Lin et al., 2012; Zink et al., 2014; Uno and Coyle, 2019). There is considerable evidence indicating that treatment resistance is associated with elevated glutamate levels without altered dopaminergic function (Demjaha et al., 2014; Zink et al., 2014). In support, a series of cross-sectional studies have shown that poorer antipsychotic responses are associated with higher levels of glutamate metabolites in the anterior cingulate cortex (ACC) compared to patients who exhibit good responses or healthy volunteers (Leung et al., 2019; Tarumi et al., 2020; Egerton et al., 2021). Higher levels of glutamate metabolites also predicted the adverse effects after re-initiation of antipsychotics (Egerton et al., 2021). Inhibitory GABAergic system has also been reported to participate in TRS (Tan et al., 2019; Bojesen et al., 2020). The GABAergic system dysfunction is associated with cognitive dysfunction and patients with TRS often have severe cognitive impairment, suggesting that GABAergic dysfunction may be more closely related to TRS patients (Miyazawa et al., 2022).
N-methyl-D-aspartic acid (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are the key components of glutamatergic neurotransmission (Uno and Coyle, 2019). Multiple evidence suggested that the N-methyl D-aspartate (NMDA) receptor mediated function is reduced in schizophrenia (Lin et al., 2012). For example, the NMDA receptor antagonists, PCP and ketamine, induce psychiatric and behavioral changes related to schizophrenia, even TRS in animals models (Moghaddam and Javitt, 2012; Rajagopal et al., 2022). An interesting study aimed to examine whether polymorphisms in the 366C/G and 2664C/T genes of glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B) were associated with TRS in Chinese patients. Although no significant association was found, the results suggested that patients with GRIN2B 2664C/C genotype require a higher dose of clozapine (Chiu et al., 2003).
There is also some indication that the glutamate metabolism receptor 3 (GRM3) gene, which controls the signaling of NMDA receptors, is relevant to antipsychotic response. Two GRM3 markers (rs1989796 and rs1476455) were linked to the refractory global symptoms. Specifically, participants with an rs1476455_CC genotype or rs1989796_CC genotype had significantly higher BPRS scores than A-carriers or T-carriers respectively, as indicated in a cohort of ninety-five TRS patients (Bishop et al., 2011). Another study revealed that GRM3 rs1468412 was linked to the changes in cognition and symptom response after antipsychotic medication, potentially assisting in the identification of patients with poor symptom improvement (Bishop et al., 2015). Furthermore, data suggests that synergistic association of PI4KA and GRM3 genetic polymorphisms may influence antipsychotic reactions (Kaur et al., 2014).
An association study for five SNPs including Glutamate decarboxylase 1 (GAD1), Gamma-Aminobutyric Acid Type B Receptor Subunit 1 (GABBR1) and Gamma-Aminobutyric Acid Type B Receptor Subunit 2 (GABBR2) genes was performed among 357 patients with TRS, 682 non-TRS patients and 508 healthy controls. The findings demonstrated that rs3749034 on GAD1 and rs10985765 on GABBR2 could be related to TRS. However, no significant differences in allelic and/or genetic distributions for any of the five SNPs were revealed. Thus, further studies in other ethnic groups and with larger samples are needed (Miyazawa et al., 2022). Interestingly, the evidence confirmed that the interaction between dopaminergic signaling and GABAergic gene expression affects the clinical phenotype of schizophrenia which can help identify TRS patients. Specifically, the percentage of subjects with Met allele of rs4680 on the COMT gene and C/C homozygote of rs3470934 on the GAD1 gene was significantly higher in the TRS group than non-TRS group and HC group (Kogure et al., 2021).
Muscarinic activity might contribute to the clinical signs and symptoms of schizophrenia. Type I muscarinic receptor (CHRM1) is the prefrontal cortex’s main cholinoceptive target and linked to cognitive performance (Marino et al., 1998). C267A is one of the genes that codes for it. The heterozygote group (267C/A) performed better than the homozygote group (267C/C) in terms of accurate replies and persistent errors of the Wisconsin card sorting test. However, the genotype distribution of schizophrenia patients in this study did not differ from that of healthy controls, proving that the CHRM1 C267A polymorphism did not significantly increase the susceptibility to schizophrenia (Liao et al., 2003).
Neurobiological studies have shown that the endocannabinoid system plays a significant role in both the propensity for schizophrenia and the efficacy of the response to antipsychotic medicines (Fernandez-Espejo et al., 2009). Endogenous cannabinoids bind to and activate two distinct receptors: the mostly central cannabinoid receptor type 1 (CB1) and the primarily peripherally expressed cannabinoid receptor type 2 (CB2) (Ishiguro et al., 2010). The majority of studies concentrated on the CB1 gene polymorphisms (Ferretjans et al., 2012). Only one investigation supported the association between cannabinoid receptors and drug responsiveness. The 1359 G/A (rs1049353) G allele of the CB1 gene was much more frequent in non-responsive individuals than in responsive patients (Hamdani et al., 2008). There was no correlation between the reaction to antipsychotics and three additional genetic variants (rs806368, rs806379, and rs806380) coding the CB1 (Hamdani et al., 2008).
The H1, H2, H3, and H4 histamine receptors are activated by the central nervous system (CNS) histaminergic system, which controls sleep-wake, attention, and metabolic balance (Raddatz et al., 2012). The evidence found the efficacy of risperidone was affected by the histamine receptor H4(HRH4) polymorphism rs4483927. Patients with the TT genotype of rs4483927 had poor Positive and Negative Syndrome Scale (PANSS) scores. However, because 113 Chinese Han patients with schizophrenia who had never before encountered antipsychotic drug resistance followed an 8-week course of risperidone monotherapy in this study, no concrete proof of histamine receptor variation for TRS was offered (Wei et al., 2013).
The cell signaling and neural development is modulated by AKT1 gene which is a member of the serine-threonine protein kinase. This disorder is associated with developmental, structural, and functional abnormalities of the hippocampus (Balu et al., 2012). AKT1 genes were upregulated in the TRS group in comparison to the healthy group. However, the other genes including neurotransmission, inflammation, neurodevelopment and protein degradation were not found positive results (Moretti et al., 2018).
The dopaminergic, serotonergic, and glutamate/GABA systems have been the focus of recent advances in pharmacogenetics to identify specific risk genes for TRS. There is only weak evidence that genetic markers for other systems such muscarinic, histamine, cannabinoids, etc. are involved in TRS. However, certain findings supported the discovery of genes associated with risk for schizophrenia and the pathophysiology of schizophrenia (Scarr et al., 2013; Brown et al., 2014). These regions now serve as a starting point for investigation into genetic TRS markers and pharmaceutical targets.
The genetic risks of TRS and the underlying mechanisms are more likely to involve the superposition or interaction of multiple genetic loci (Ruderfer et al., 2016; Samanaite et al., 2018). Genome-wide association studies (GWAS) becomes an important tool for understanding the biological underpinnings of schizophrenia. The combinations of the risk functional SNPs observed in this study may be useful as new biomarkers in clinical settings. Pharmacogenomic studies use genome-wide data instead of a candidate gene approach to understand the mechanisms of antipsychotic response and are being applied to identify polymorphisms in TRS (Ackenheil and Weber, 2004; McClay et al., 2011). Polygenic risk score (PRS)analysis captures the genetic load of alleles associated with traits across multiple loci representing an approximation of the genetic risk burden (Euesden et al., 2015), and thus can aggregate the effects of multiple SNPs on the genome to create the sum of alleles associated phenotype through GWAS (Nucifora et al., 2019). There is some evidence that a greater genetic burden equates to a greater likelihood of developing TRS (Nucifora et al., 2019; Pisanu and Squassina, 2019). However, most studies only used PRS method to investigate risk variants in schizophrenia, and no GWAS studies specifically conducted for TRS. It is speculated that the development of a PRS specifically for TRS may show higher predictive accuracy.
In spite of SNPs, rare mutations are also shown to increase the risk of schizophrenia (O'Tuathaigh et al., 2017; Purcell et al., 2014). Genes encoding calcium channels, and proteins involved in glutamatergic neurotransmission and synaptic plasticity have been independently implicated (Fromer et al., 2014; Purcell et al., 2014). The role of rare disruptive mutations was extensively investigated in TRS. Compared with the non-clozapine treated patients (treatment responsive), the 347 gene targets of antipsychotic drugs were enriched for singleton disruptive mutations in TRS patients (Cao et al., 2020). Another study reported a large exome sequencing study of de novo mutations in schizophrenia, comprising 482 nonsynonymous mutations, in which 64 were de novo loss-of-function (LoF) occurred preferentially in schizophrenia cases with premorbid cognitive impairment (Fromer et al., 2014). Interestingly, they also found overlap between genes affected by rare variants in schizophrenia and those within GWAS loci, suggesting that common and rare variants are complementary rather than contradictory (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014).
Copy number variants (CNVs) are essentially deletions and duplications in the human genome that fall between large (chromosomal aberrations >1 Mbp) and small (insertions or deletions 1–50 bp) aberrations (Girirajan et al., 2011). Recent studies of schizophrenia (SCZ) have reported an increased burden of de novo CNVs (International Schizophrenia Consortium, 2008). The relationship between CNV and TRS was supported by two studies. A high-resolution genome-wide CNV analysis was carried out on a predominantly Japanese population with 1699 schizophrenia patients and 824 controls using array comparative genomic hybridization. TRS was seen in individuals with a 22q11.21 deletion XXX/XXY, and two clinically significant CNVs (Kushima et al., 2017). This result supported patients with clinically significant CNVs were more likely to experience TRS. Another study confirmed a connection between the overall amounts of genome-wide duplications and TRS in 612 schizophrenia patients. The results suggested an increase in rare replication burden across the genome of TRS patients (Martin and Mowry, 2016).
Although the two studies did not include TRS as a separate subgroup, they provided detailed evidence for CNV involvement in the pathogenesis of schizophrenia. The first study measured the amount of fragment of the human satellite III (f-SatIII) in leukocytes from 401 healthy individuals and 840 schizophrenia patients (Ershova et al., 2019). The patients generally differ from the healthy group significantly because their repeat content values were lower. Another study reported that in contrast to 3181 controls, 4 out of 3391 schizophrenia patients had the serine/threonine kinase gene unc-51-like kinase 4 (ULK4) deletions spanning exons 21 to 34. The big splice variant of ULK4’s deletions that remove exons 33 and 34 were also more prevalent in schizophrenia patients (Lang et al., 2014).
We also found some evidence that involves epigenetics including DNA methylation and microRNAs (miRNAs) (You et al., 2020; Hannon et al., 2021). One concrete example was the ribonuclease DICER1, which plays a key role in the production of miRNAs. In comparison to the healthy group, DICER1 genes were increased in the TRS group (Moretti et al., 2018). The top three miRNAs with the highest fold change values were screened out from peripheral blood mononuclear cells, hsa-mir-218-5p and hsa-mir-1262 of them were biomarkers for an earlier and more accurate diagnosis of TRS (You et al., 2020). It is interesting to note that, compared to non-TRS instances, TRS cases appear to have much greater proportions of granulocytes and lower proportions of CD8+ T-cells based on cellular composition variables derived from DNA methylation data. TRS was linked to considerably higher smoking score determined from DNA methylation than non-TRS (Hannon et al., 2021).
To sum up, the development of a PRS specifically for TRS through GWAS, the mechanism-driven approach to identify rare mutations and the epigenetics will provide more information for TRS in addition to exploring candidate genes SNPs (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Pisanu and Squassina, 2019; Pardiñas et al., 2022). Much of the evidence for the types of these genetic variants focuses on schizophrenia now, and exploring the association of TRS with these variants as a separate subgroup would provide a more thorough genetic model for TRS.
Immune reactions and inflammation have long been implicated in the pathophysiology of schizophrenia, according to the available epidemiological, clinical, and postmortem evidence (Bechter et al., 2010). Prenatal and early life are vulnerable periods for precipitating factors, like infection, autoimmune or psychological stress to result in inflammation at later stage of mental illness (Möller et al., 2015). For example, abuse in childhood may have long-term consequences, leading to psychotic onset during adolescence or adults (Leboyer et al., 2016). Systemic immune-inflammatory cells can pass through the blood-brain barrier, altering central inflammatory processes, particularly in astrocytes and microglia (Leonard and Maes, 2012). Moreover, increased peripheral and cerebral inflammatory processes were found to play a role in TRS (Roomruangwong et al., 2020). Inflammatory indicators are linked to cognitive deficits and negative symptoms in schizophrenia, both of which are notoriously difficult to treat (Garcia-Rizo et al., 2012). Previous research has similarly associated higher inflammatory status with a worse clinical outcome in TRS patients (Momtazmanesh et al., 2019). More than 60% of TRS patients had monocyte activation and proteome changes in blood, as well as moderate cerebrospinal fluid pathology, microglia activation, or dysconnectivity in neuroimaging (Bechter, 2013).
Biomarkers of psychotic condition can be traced in peripheral immune system with different phenotypes stratified by blood cells (Miller et al., 2013). There are few studies focusing on the relationship between therapeutic responsiveness and inflammatory complete blood count (CBC) markers in patients with schizophrenia. Schizophrenia patients treated with clozapine had increased relative numbers of NK cells, naive B cells, CXCR5+ memory T cells and classical monocytes, while had decreased numbers of dendritic cells (DC), HLA-DR+ regulatory T-cells (Tregs), and CD4+memory T cells. More severe negative and cognitive symptoms were linked to the above mentioned decreased cells (Fernandez-Egea et al., 2016). In clinical settings, the neutrophil to lymphocyte ratio (NLR), monocyte to lymphocyte ratio (MLR), and platelet to lymphocyte ratio (PLR) derived from CBC data can be used as proxies for systemic inflammation. In a six-year retrospective study, although baseline NLR was similar, only the responsive subgroup showed decrement in NLR after treatment, suggesting that persistent inflammation is associated with treatment resistance in schizophrenia. Meanwhile, PLR was higher on admission in the responsive group and significantly decreased after treatment, while no change was observed in the TRS patients due to lower PRL at admission (Labonté et al., 2022). These findings suggested that CBC could be a feasible biomarker and used to track TRS patients for targeted therapies (Fernandez-Egea et al., 2016; Labonté et al., 2022).
The immune response is largely regulated by interleukin-6 (IL-6), which plays a vital role in the immunological response during the body’s anti-infection process. A great number of studies on this cytokine have consistently found that TRS patients have considerably greater serum IL-6 levels than normal controls or treatment-responsive patients (Maes, 2000; Roomruangwong et al., 2020). Interestingly, elevated IL-6 levels were found to be inversely related to the endogenous anti-cytokine CC16, which may constitute a hallmark of schizophrenia (Lin et al., 1998). Dickkopf-related protein (DDK1) and IL-6 levels in the non-responders to treatment group were significantly higher than in the partial responsive and control groups. Further meta-logistic regression analysis revealed that IL-6 in combination with CCL11 (eotaxin) was the greatest predictor of non-responders when compared to partial-responders (Al-Dujaili et al., 2021). In addition, IL-6 may result in downregulation of Wnt signaling, thus leading to a disruption of the blood-brain barrier (BBB) and neuroinflammation (Al-Dujaili et al., 2021). TRS appeared to be linked to other elevated cytokine levels of IL-12/IL-23p40, IL-17A, and beta 2 macroglobulin (B2M) in blood. These cytokines have a role in the establishment and differentiation of Th17 immune pathways, suggesting that the activation of the IL-17 pathway may present at onset of the disease and increase as the disease progresses to drug resistance (Leboyer et al., 2021). Besides, several other studies patients with TRS had higher levels of IL-2, IL-8, IL-10, soluble tumor necrosis factor receptor 1 (sTNF-R1), sTNF-R2, monocyte chemotactic protein 1 (MCP-1) and IL-1R antagonist (IL-1RA) than the control group, indicating immunological inflammation is one of the important components of TRS (Lin et al., 1998; Maes, 2000; Zhang et al., 2004; Miller et al., 2013; Noto et al., 2015; Al-Dujaili et al., 2021; Leboyer et al., 2021; Labonté et al., 2022).
Furthermore, evidence shows that individuals with high levels of C-reactive protein (CRP), a biomarker of chronic or acute inflammation, were more likely to develop schizophrenia (Horsdal et al., 2017). When compared to healthy controls, TRS patients had considerably higher levels of high-sensitivity C-reactive protein (hsCRP)(Challa et al., 2021). However, they discovered no link between CRP levels and TRS at the time of the first schizophrenia diagnosis (Horsdal et al., 2017). Finally, patients with severe mental illnesses had shorter telomeres and more inflammation. It revealed that telomere attrition was also caused by a number of biological injuries, including inflammatory processes (Squassina et al., 2020).
In short, a growing body of evidence supported the association of inflammatory markers with disease risk, severity, and treatment resistance of schizophrenia. Finally, due to the role of the immune system in the physiopathology of schizophrenia and the relative availability of testing methods, immune markers are considered as potential peripheral biomarkers for TRS patients.
Oxidative stress has been repeatedly identified as one of the most important biological underpinnings of the pathophysiology of schizophrenia (Cui et al., 2020), which is evident by malfunction of antioxidant system and the change of activity of different antioxidant enzymes (Pinheiro et al., 2017). The levels of antioxidant, catalase, and glutathione peroxide are reduced in schizophrenia. It has been found that antioxidative status is negatively associated with specific cognitive abnormalities, whereas glutathione levels are positively correlated with executive function (Martínez-Cengotitabengoa et al., 2012). Because of its abundance of polyunsaturated fatty acids, high consumption of oxygen, and low quantities of antioxidative enzymes, the brain is particularly susceptible to oxidative stress (Möller et al., 2015). Reduced antioxidant levels in the brain, combined with increased production of reactive oxygen species, put neurons in greater danger of injury, especially in TRS (Pinheiro et al., 2017).
In TRS patients, lipid peroxidation and neuronal damage were shown to be higher than in non-refractory individuals (Medina-Hernández et al., 2007). This study found elevated serum levels of lipid peroxidation byproducts malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) and neuron-specific enzymes in TRS patients compared with healthy controls indicated greater systemic oxidative stress and greater neuronal metabolic changes in TRS patients (Keller et al., 1999; Medina-Hernández et al., 2007). Moreover, telomere length shortening in peripheral leukocytes is a marker of poor treatment response in patients with chronic schizophrenia, which may be caused by oxidative stress-induced cellular dysfunction leading to the exacerbation of TRS (Yu et al., 2008).
From another perspective, the composition of fatty acids is associated with neurocognition and mood regulation in the brain (Assies et al., 2014). Changes in enzyme-induced and non-enzyme-induced antioxidant systems generated increased lipid peroxidation, leading to reduced levels of fatty acids in schizophrenia (Bitanihirwe and Woo, 2011). For example, oxidative stress reduced the content of polyunsaturated fatty acids (especially omega-3 polyunsaturated fatty acids) and increased lipid peroxidation byproducts (Assies et al., 2014). Messamore and McNamara collected more than 130 blood samples from psychosis patients with treatment-resistant mood disorders. The majority of these patients had extremely low levels of eicosapenaenoic acid (EPA) and docosahexaenoic acid (DHA), indicating long-chain omega-3 fatty acid deficiency in these patients (Messamore and McNamara, 2016). Our group studied the contents of fatty acids in the erythrocyte membrane of schizophrenia. Patients were grouped into different episodes, including the first episode, 2–3 episodes, 4–6 episodes, and more than 6 episodes. Overall, there were reductions in both saturated and unsaturated fatty acids at baseline compared with healthy controls. Follow-up assessment after 1 month showed an increase in membrane fatty acids in patients treated with atypical antipsychotics only within 3 episodes. In addition, changes in C22:5n3 and omega 3 index, as well as gender and frequency of episodes, were all significant risk variables for diminished treatment response. Using a targeted metabolomics approach, the study revealed abnormalities in fatty acid metabolism that lead to a reduced response to treatment in patients with multiple schizophrenia (Li et al., 2022).
Carbonyl stress is a metabolic abnormality characterized by increased reactive carbonyl compounds (RCOs) and their attendant protein modifications, which leads to the accumulation of advanced glycation end products (AGEs) such pentosidine (Ohnuma et al., 2018). Oxidative stress is a central mediator of AGEs formation which converts glucose and lipids to reactive carbonyl compounds (RCOs), and RCOs are transformed to AGEs and advanced lipid peroxidation end products (Ohnuma et al., 2018). Glyoxalase breaks down the RCO into lactic acid and glutathione for detoxification and the rate-limiting enzymes in this metabolic pathway are glyoxalase 1 and 2 (GLO1 and GLO2)(Kouidrat et al., 2015; Rodrigues et al., 2020). Besides, Pyridoxamine, one of the three forms of vitamin B6, can detoxify RCO, including AGEs (Li et al., 2022). There is increasing evidence that carbonyl stress is closely associated with schizophrenia accompanied by exhibiting high plasma pentosidine and low serum vitamin B6 levels (Miyashita et al., 2014).
In the first study to observe the clinical features of schizophrenia with increased carbonyl stress, patients with carbonyl stress were found to be less educated, have significantly higher rates of hospitalization, stay longer in hospital, and take higher doses of antipsychotic medications than patients without carbonyl stress (Miyashita et al., 2014). The severe clinical symptoms identified in these carbonyl stress patients are very comparable to the definition of TRS (Kane et al., 1988). Serum pyridoxal, which is measurably stable, is converted from pyridoxamine (Son et al., 2020). Another study from the same research group also mentioned that high pentosidine and low pyridoxal levels in the peripheral blood could be state markers of TRS (Katsuta et al., 2014). Pyridoxal levels have been shown to decline with clinical progression and are linked to less symptom alleviation, whereas pentosidine levels are higher, possibly due to higher antipsychotic doses (Katsuta et al., 2014).
Metabolites, intermediates, enzymes and their activity in many metabolic pathways are altered due to high levels of reactive oxygen species (ROS)(Nedic Erjavec et al., 2018). However, there are only a few studies on the role of this process in the TRS. As summarized above, enzymatic (superoxide dismutase (SOD) and catalase) and non-enzymatic antioxidants (e.g., glutathione (GSH)) may participate in this pathological process (Do et al., 2009). The relationship among neuronal and systemic oxidative stress, lipid peroxidation and treatment-resistant psychiatric disorders is largely unknown. Hopefully, the focus on carbonyl stress in the field of schizophrenia has broadened the scope of therapeutic resistance markers and neurobiology for schizophrenia.
Besides, some evidence suggests that therapeutic approaches for alleviating carbonyl stress, such as vitamin B6 supplementation, could offer novel therapeutic benefits (Miyashita et al., 2014). In a 24-week open trial, ten Japanese schizophrenic patients with high plasma pentosidine were recruited and given a high dose of pyridoxamine. According to the findings, this medication is a viable treatment for TRS caused by elevated carbonyl stress (Itokawa et al., 2018). Several other clinical studies have also looked into the effectiveness of vitamin B6 supplementation in schizophrenia, however the results were mixed (Miyashita et al., 2014). In the future, prospective and longitudinal designs are needed for carbonyl stress to clarify the exact clinical relationship between TRS and two proposed biomarkers, as well as to validate these new findings, such as pyridoxamine supplementation, which may have new therapeutic benefits for this subset of patients.
It is likely that clozapine impacts a complex network of neurotransmitter pathways given its affinity for various targets (Miller, 2009). Clozapine is known to exert its action on adrenergic, histamine, and muscarinic receptors in addition to the three primary neurotransmitter systems indicated above (Nair et al., 2020).
Rudi Hwang et al. concentrated on the reaction to clozapine and the dopamine receptor gene polymorphisms. Their findings were unique in that they were based on diverse populations consisting of 232 subjects with 183 Caucasians and 49 African Americans. Three dopamine receptors tested in the African American population showed promising findings. The A/C genotype of SNP rs265976 in DRD1 was more prevalent in non-responders (Hwang et al., 2007). The impact of 12 SNPs across the whole DRD2 gene on clozapine response was examined in this exploratory investigation. Being a responder was linked to having the allele 2 (B2) of TaqIB, allele A of rs1125394, and not having the TaqIA1 (T) allele (Hwang et al., 2005). Additionally, it was found that the DRD4 120-bp 1-copy allele and intron I (G)n 142-bp/140-bp genotype were related to non-responder status (Hwang et al., 2012). Two dopamine receptor gene polymorphisms were found in Caucasian individuals. The DRD3 rs2134655 A allele was linked to improved responsiveness. The T allele of rs1394016 and the GG genotype of rs2399504 both showed the same tendency (Hwang et al., 2010). Besides, exon III 4R allele carriers responded better than non-4R allele carriers. All DRD5 tests were negative (Hwang et al., 2012).
There was additional evidence that the reaction to clozapine is related to dopamine receptors. The 2,2 DRD1 homozygotes have responded to clozapine with a more BPRS symptom improvement compared to 1,2 DRD2 subjects (Potkin et al., 2003). A recent genome-wide association study found an SNP, rs2514218, 47 kb upstream of the DRD2 gene, was linked to risk for schizophrenia. The rs2514218 risk allele A was linked to a larger decline in BPRS scores in the Caucasian group with clozapine (Huang et al., 2016). Compared to the D2 receptor, the DRD3 receptor exhibits a higher affinity for the clozapine. In comparison to the responders, the non-responders had a much greater genotype of DRD3 rs9680 AA gene (M et al., 2020). A Ser9Gly point mutation that changes the amino acid serine (Ser-9) to glycine (Gly-9) in the N-terminal extracellular region of DRD3. Ser-9/Ser-9 genotypes were prevalent in non-responders, and Ser-9/Gly-9 genotypes were prevalent in respondents (Scharfetter et al., 1999).
Two studies discovered a link between the COMT Val158Met polymorphism and clozapine responses. In the first study, clinical responses to clozapine were superior in those Val/Val patients compared to the val/met or met/met patients who had one or two met alleles in the PANSS Negative Subscale (Bosia et al., 2015). The second investigation did not discover an independent relationship between the COMT Val158Met polymorphism and clozapine responsiveness. However, participants with the COMT Val/Met or Met/Met genotype as well as the DRD4 120-bp alleles (120/240 and 120/120) demonstrated a significantly improved clinical response to clozapine (Rajagopal et al., 2018).
The serotonergic system appears to be the primary mechanism through which clozapine exerts its effects, and changes in serotonergic synaptic levels may have an impact on the antipsychotic response (Kohlrausch et al., 2010). The link between the serotonin and dopamine systems is mediated by the 5-HT1A-R -1019 C/G polymorphism. There was a higher improvement in the PANSS Negative Subscale in 5-HT1A-R G/G patients with clozapine treatment (Bosia et al., 2015). The association between 5-HT2A polymorphisms and clozapine responsiveness was verified by three trials, and the outcomes were consistent (Arranz et al., 1996; Arranz et al., 1998; Masellis, 1998). The frequency of the allele Tyr452 described in the coding region of the 5-HT2A receptor gene and homozygosity for the allele G-1438 identified in the promoter region were both higher in non-responders than in responders (Arranz et al., 1998). The 5-HT3A receptor, which is the only receptor-coupled ion channel among the serotonin receptors, has emerged as a key target in schizophrenia patients (Souza et al., 2010; Rajkumar et al., 2012). It is well documented that clozapine treatment responsiveness has been linked to alleles of three HTR3A variations, including rs2276302, rs1062613, and rs1150226. The G-T-T haplotype, which spans rs2276302-rs1062613-rs1150226, was correlated to a poorer response (Souza et al., 2010). Consistent with this, this study also discovered that minor alleles of both rs1062613 and rs2276302 were closely linked with clozapine responses. What is innovative about this study is that HTR3A pharmacogenetics combined with clinical predictors can more strongly explain differences in clinical response to clozapine (Rajkumar et al., 2012). Additionally, because HTTLPR regulates the transcriptional activity of the 5-HTT gene, genotypes with low expression such S'/S′ or S'/L′ would be more likely to have poor responses to clozapine (Kohlrausch et al., 2010).
Clozapine affects multiple glutamate/GABA signal transduction receptors, transporters, and enzymes, drawing attention to the interaction between SNP gene variations and clozapine sensitivity in these pathways (Daskalakis and George, 2009; Gammon et al., 2021). However, there are no ideal positive results on the relationship between the variation of genes in this system and clozapine response. Using standard genotyping procedures, the relationship between clozapine response and ten glutamate system gene variants of GRM2, SLC1A2, SLC6A9, and GAD1 (encoding metabotropic glutamate receptor 2 (mGluR2), glutamate transporter 1 (GLT1), glycine transporter 1 (GlyT1), and GAD1, respectively) was investigated. Surprisingly, no significant association was observed except that CC homozygous of rs16831558, which is located in the GlyT1 gene (SLC6A9), showed allele-dose-dependent improvement in positive symptoms as compared with T allele carriers (Taylor et al., 2017). In another study, whereas the data showed that an allele carrier of the GRIN2B intron variant rs1072388 responded to clozapine slightly better than G homozygous, there was no significant correlation between the seven GRIN2B variants and treatment response to clozapine (Taylor et al., 2016). Another study that included 45 patients taking clozapine was unable to confirm the presence of any of the four single-nucleotide variations in the GRIN1, GRIN2A, or GRIN2B subunits of the NMDA-R.
It has been suggested that variations in cytochrome P450 (CYP) enzyme activity have a major impact on the pharmacokinetics of clozapine. Several cytochrome P450 (CYP) enzymes, including CYP1A2, CYP2C19, CYP2D6, and CYP3A4, have been shown to involve clozapine metabolism. The low CYP3A4 express patients needed a much lower dose (50%) of clozapine to achieve the ideal blood level than other expressers (Tóth et al., 2017). When compared to the control group and those who responded to clozapine, the distributions of alleles and genotypes revealed a higher frequency of CYP2C19*17 in the patients who had partial response to clozapine (Rodrigues-Silva et al., 2020). The genotypes of CYP1A2 and CYP2D6 expression in the patients were not found positive results (Tóth et al., 2017).
Because clozapine affects the Hypothalamic–pituitary–adrenal (HPA) axis, the FK506 binding protein 5 (FKBP5) modulating HPA axis is of particular importance. When compared to C-carriers, FKBP5 rs1360780 TT-homozygotes had a 2.11 times higher risk of nonresponse. Neurotrophic tyrosine kinase receptor 2 (NTRK2) is BDNF’s high-affinity receptor. When compared to C-carriers, TT-homozygous for rs1778929 in the NTRK2 gene showed a 1.7-fold increased risk of nonresponse. In contrast, rs10465180 CC-homozygous individuals had a 2.15-fold increased risk of nonresponse compared to T-carriers (Mitjans et al., 2015). Adenosine triphosphate-binding cassette class B (ABCB1) is necessary for the passage of clozapine through a number of bodily barriers. The non-responder subgroup had a considerably higher frequency of the TT genotype allele than the responder subgroup (M et al., 2020). A genome-wide investigation identified risk loci that have an impact on each of the five main psychiatric disorders. The intronic rs2535629 polymorphism in Inter-alpha-trypsin inhibitor heavy chain H3 (ITIH3) is the region’s strongest association discovery (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). This variation has also been linked to schizophrenia in Japanese patients. Patients with homozygous for the minor A allele of rs2535629 had the highest improvement in their negative BPRS scores after receiving clozapine medication (Brandl et al., 2016).
Brain-derived neurotrophic factor (BDNF) is crucial for neurogenesis and maturation of neural developmental pathways (Yamamori et al., 2013). A study found that compared to non-responders, responders had higher serum levels of BDNF (Krivoy et al., 2018). The study discovered that the start of clozapine treatment in patients with TRS was highly associated with a brief increase in WBC, neutrophil, and platelet count. The study showed that clozapine had early hematological effects, but it was not determined if these changes predict how a patient will respond to treatment (Blackman et al., 2021). When compared to non-TRS patients, those who did not respond to clozapine had higher levels of IL-12/IL-23p40, IL-17A, IL-6, IL-10, IFN, and B2M (Leboyer et al., 2021). The relationship between lipid dysregulation and the therapeutic response to clozapine in TRS was investigated. A rise in triglyceride levels was highly indicative of both clinical and PANSS score improvements (Lally et al., 2013). It was found that lower CLZ: NDMC (N-desmethylclozapine, the main metabolite of clozapine) ratios were related to higher cognitive function. This result indicated the CLZ:NDMC ratio may be helpful for maximizing clozapine’s potential cognitive advantage (Costa-Dookhan et al., 2020).
As we searched for neurotransmitter data, we discovered that histamine and muscarinic receptor pathways had been implicated increasingly (Mancama et al., 2003). However, the majority of clozapine and these receptor pathways evidence was being conducted on animals examining the mechanism of clozapine in these systems (Digby et al., 2012). The acetylcholine (ACh) innervation of five different receptor subtypes, CHRM1-CHRM5, causes the muscarinic system’s actions (Vilaró et al., 1994). Muscarinic pathways are crucial for memory, learning, and attention (Hagan et al., 1987). NDMC has been demonstrated to be an M1 muscarinic receptor partial agonist while clozapine was an M1 antagonist in vitro and in vivo, it is probable that NDMC agonism leads to the drug’s particularly advantageous effects (Davies et al., 2005; Li et al., 2005). Through M1 receptor activation, NDMC promoted the release of ACh in brain areas important for cognition, including prefrontal cortex and hippocampus (Carruthers et al., 2015). NDMC ameliorated novel object recognition impairment induced by phencyclidine, and these findings provided more evidence that M1 agonism was a possible target for the treatment of cognitive impairment in schizophrenia (Miyauchi et al., 2017). Additionally, we discovered some evidence that, at clinically meaningful concentrations, clozapine and its active metabolite NDMC interact with the four human histamine receptors (Mancama et al., 2002; Humbert-Claude et al., 2012). The literature’s findings, like those for muscarine, mostly explored the mechanism of this receptor in animal studies. There are few clinical studies on peripheral markers of these two major receptor pathways to predict or reflect the therapeutic effect of TRS.
The above-mentioned discoveries on peripheral biomarkers of TRS provide new information on the neurobiological process of TRS. These neurobiological processes are interrelated, which result in abnormalities of neurological functioning, leading to the pathogenesis of TRS, as shown in Figure 2.
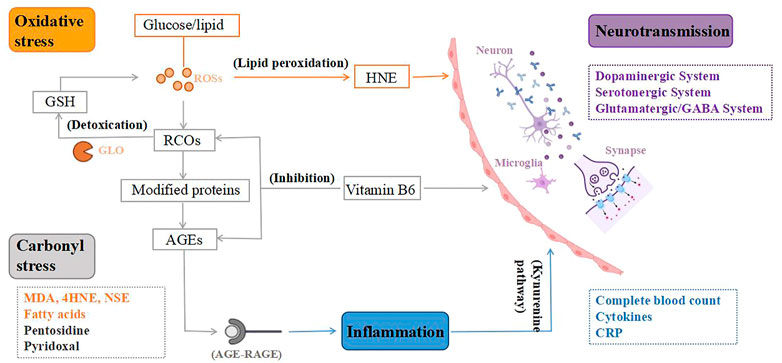
FIGURE 2. From peripheral biomarkers to neurobiology of treatment-resistant schizophrenia (TRS). To begin with, the lipid peroxidation product 4-hydroxynonenal (HNE) created by oxidative stress can alter the blood-brain barrier, and peroxidation also causes aberrant fatty acid levels in the synaptic membrane, leading to an altered neuronal microenvironment. Second, oxidative stress is a key mediator of carbonyl stress, as glucose and lipids are transformed to reactive carbonyl compounds (RCOs), and excess RCOs are converted to advanced glycation end products (AGEs). Glyoxalase converts RCOs to glutathione (GSH), which is an antioxidant. Vitamin B6 detoxifies RCOs and AGEs, and its deficiency can impact the metabolism of many neurotransmitters, resulting in various neurotransmission deficits in brain function. Third, AGEs interact with the AGE receptor (RAGE), causing inflammation to rise. Increased neurotoxic metabolites as a result of inflammatory damage mediated by the Kynurenine pathway. Microglia are also activated, releasing inflammatory cytokines that cause dysregulation of neuronal circuits and neurodegeneration. Therefore, TRS, a neurological disorder, is caused by the combination of mentioned mechanisms. ROSs, reactive oxygen species; GLO, glyoxalase.
First, in schizophrenia, dysregulation of glutamate activity in neural circuits may be caused by an initial inflammatory injury mediated by the kynurenine pathway (Pedraz-Petrozzi et al., 2020), which is accompanied by a decrease in 5-HT synthesis but an increase in neurotoxic metabolites derived from tryptophan, such as 3-hydroxy-kynurenine (3HK), 3-hydroxy-anthranilic acid (3-OHAA), anthranilic acid and quinolinic acid (QA). Ultimately, this leads to neuronal circuit dysregulation and neurodegeneration (Möller et al., 2015). Second, in addition to factors involved in oxidative stress that are thought to cause monoamine changes, oxidative stress itself causes neurochemical and neuroanatomical changes (Nedic Erjavec et al., 2018). Furthermore, the increased peroxidation in TRS may indicate an aberrant fatty acid level in the synaptic membrane, resulting in neuronal dysfunction and associated abnormality in the microenvironment (Medina-Hernández et al., 2007). There’s also evidence that changes in the physicochemical milieu of lipids and fatty acids affect the affinity of membrane receptors for neurotransmitters including dopamine, GABAergic, and glutamate, which could be linked to adverse reactions to antipsychotic therapy in TRS patients (Fenton et al., 2000). The lipid peroxidation product 4-hydroxynonenal induced by oxidative stress can compromise the integrity of the blood-brain barrier and increase its permeability (Möller et al., 2015), resulting in elevated inflammatory response and systemic stress response (Nedic Erjavec et al., 2018). Third, in carbonyl stress, AGEs interact with the AGE receptor (RAGE), causing increased oxidative stress and inflammatory mediators (Kouidrat et al., 2015). Microglia get engaged when the brain is under psychological stress, releasing inflammatory cytokines that cause aberrant neurogenesis and neuronal degeneration (Katsuta et al., 2014). Because pyridoxal is a key coenzyme in the production of several neurotransmitters, including dopamine, serotonin, and GABA, VB6 deficiency can impact the metabolism of many neurotransmitters, resulting in various neurotransmission deficits in brain function (Itokawa et al., 2018; Toriumi et al., 2021).
A series of peripheral biomarkers with high availability and feasibility have been demonstrated for TRS and were summarized in Table 1–3. First of all, molecular and genetic methods, such as pharmacogenomics, provide a means of predicting the molecular basis of treatment response. Furthermore, detection of peripheral immune system and stress biomarkers can be used to study the heterogeneity of psychotropic medication reactions in a simple and cost-effective manner. However, up to date, most biomarkers are still premature and no marker has been validated, for selectively and specificity in replication, or fully used in clinical practice on a consistent basis. A combination/panel of markers could be a more effective strategy for predicting and responding to treatment outcomes.

TABLE 1. Genetic biomarkers of treatment resistant schizophrenia.
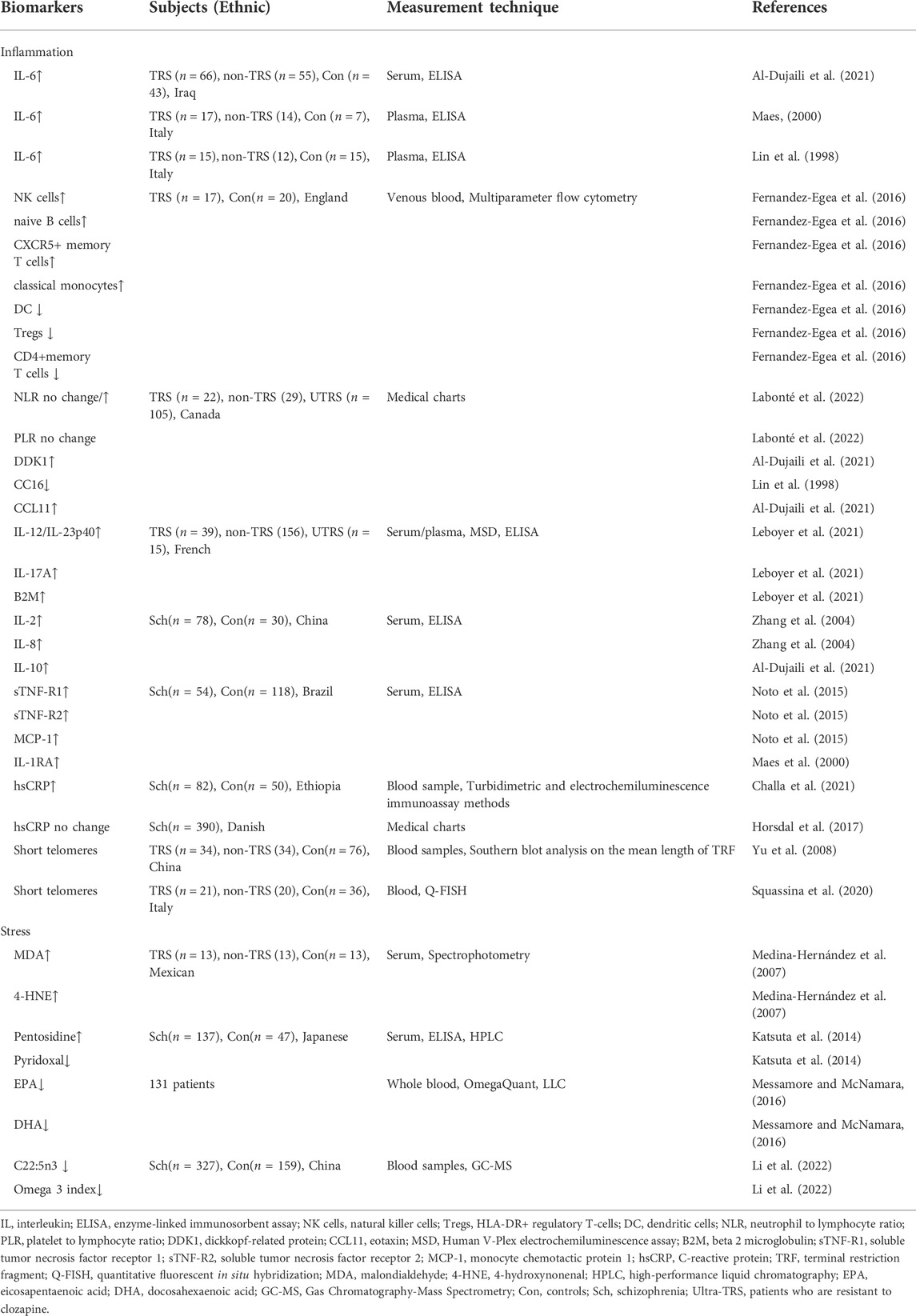
TABLE 2. Inflammation and stress biomarkers of treatment resistant schizophrenia.

TABLE 3. Genetic biomarkers of treatment resistant schizophrenia with clozapine treatment.
TRS may be a subtype of schizophrenia with distinct neurological foundations, based on the mounting results of antipsychotic responsiveness and biomarkers. However, compared with a portion of patients who fail to respond to initial treatment, many patients develop TRS as the disease progressed, which may be caused by the aggravation of neurobiological impairment caused by recurrence or long-term untreated psychosis (Takeuchi et al., 2019; Li et al., 2022). These two features cannot be disentangled according to the content of this review, the available data favors the combined effects of the two aspects.
First, the biggest limitation in terms of the evidence was the difference in the definition of the TRS and the clozapine resistant patients, although they were defined according to the standards. The different study results may result from the treatment characteristics of the sample, such as treatment duration and single-drug or multi-drug treatment. Second, many cases need to be included in studies of genetic markers, especially rare CNVS, and thousands of carefully validated cases and matched controls need to be screened for unusual mutations. However, the number of large-scale prospective studies of genetic markers is relatively small. Third, on the one hand, some first-episode patients were resistant to treatment, while others responded to first-episode treatment and then did not respond to atypical antipsychotics. On the other hand, even response to clozapine varies after diagnosis of TRS. Patients were not stratified in these studies, and these differences resulted in a lack of replicated findings. Fourth, the use of specific prognostic biomarkers regarding the response to treatment in TRS should accordingly reflect the neurobiology of the specific phenotype. The identification of biomarkers reflecting treatment response may be impeded by the lack of established, accurate neurobiology of TRS. Finally, the CNS can immediately reflect pathological and biochemical abnormalities in patients with TRS because it is fundamentally a mental condition. However, due to the low cost and simple accessibility of peripheral biomarkers, as well as the high cost of neuroimaging technology and the impracticality of brain biopsy, researchers are very interested in peripheral biomarkers. Even while we recognize the importance of peripheral biomarkers, we may acknowledge that they are unable to correctly reflect the structural and functional alterations of the CNS. It is unclear if changes in peripheral biomarkers of TRS correspond to alterations in the CNS. The difficulty of biomarker identification lies in the development of a set of high-quality biomarker methods to ensure the measurement of real, accurate, sensitive and long-term stability.
When examining pertinent peripheral biomarkers of TRS, future studies should consider adhering to the TRS diagnostic consensus and recommendations. High sample sizes and reliable peripheral biomarker measuring and analysis techniques should be used to produce more precise and reproducible peripheral biomarkers. Stratification of schizophrenia patients in research is also critical. The subtypes of TRS may 1 day be distinguishable based on peripheral biomarkers and neurobiological mechanisms in addition to clinical factors (illness duration, number of episodes, predominance of negative symptoms, etc.). During the literature survey, we found that there is almost no animal model developed for TRS, which is an interesting topic that future researchers should strive to address. In a recent study, Rajagopal et al. recently used sub-chronic treatment of rodents with phencyclidine plus physiological stress to develop an antipsychotic-resistant model of schizophrenia in mice, which is worth digging up underlying mechanisms and clinical similarities (Rajagopal et al., 2022). Animal models for TRS, particularly those with cognitive impairment and negative symptoms, could provide novel insight into the pathophysiology, therapeutic strategy, and new pharmacological development for resistant individuals.
SJ and HC designed the structure of the manuscript, SJ and TC searched and reviewed the literature, SJ wrote the manuscript, TC assisted with the improvement of the manuscript, HC provided overall guidance and feedback on revisions of the manuscript. All authors contributed to and have approved the final manuscript.
This work was supported in part by the grants from Hunan Provincial Natural Science Foundation of China (2021JJ30922), Hunan Provincial Health Commission Research Project (202113010595), Changsha Municipal Natural Science Foundation (kq2007045), Talent Project established by Chinese Pharmaceutical Association Hospital Pharmacy Department (No. CPA-Z05-ZC-2021-003), and New Clinical Medical Technology Project of the Second Xiangya Hospital of Central South University ([2021]94).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ackenheil, M., and Weber, K. (2004). Differing response to antipsychotic therapy in schizophrenia: Pharmacogenomic aspects. Dialogues Clin. Neurosci. 6, 71–77. doi:10.31887/DCNS.2004.6.1/mackenheil
Al-Dujaili, A. H., Mousa, R. F., Al-Hakeim, H. K., and Maes, M. (2021). High mobility group protein 1 and dickkopf-related protein 1 in schizophrenia and treatment-resistant schizophrenia: Associations with interleukin-6, symptom domains, and neurocognitive impairments. Schizophr. Bull. 47, 530–541. doi:10.1093/schbul/sbaa136
Anttila, S., Kampman, O., Illi, A., Rontu, R., Lehtimäki, T., and Leinonen, E. (2007). Association between 5-ht2a, TPH1 and GNB3 genotypes and response to typical neuroleptics: A serotonergic approach. BMC psychiatry 7, 22. doi:10.1186/1471-244X-7-22
Arranz, M. J., Collier, D. A., Munro, J., Sham, P., Kirov, G., Sodhi, M., et al. (1996). Analysis of a structural polymorphism in the 5-HT2A receptor and clinical response to clozapine. Neurosci. Lett. 217, 177–178. doi:10.1016/0304-3940(96)13094-9
Arranz, M. J., Munro, J., Owen, M. J., Spurlock, G., Sham, P. C., Zhao, J., et al. (1998). Evidence for association between polymorphisms in the promoter and coding regions of the 5-HT2A receptor gene and response to clozapine. Mol. Psychiatry 3, 61–66. doi:10.1038/sj.mp.4000348
Assies, J., Mocking, R. J. T., Lok, A., Ruhé, H. G., Pouwer, F., and Schene, A. H. (2014). Effects of oxidative stress on fatty acid- and one-carbon-metabolism in psychiatric and cardiovascular disease comorbidity. Acta Psychiatr. Scand. 130, 163–180. doi:10.1111/acps.12265
International Schizophrenia Consortium (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241. doi:10.1038/nature07239
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi:10.1038/nature13595
Cross-Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 381, 1371–1379. doi:10.1016/S0140-6736(12)62129-1
Balu, D. T., Carlson, G. C., Talbot, K., Kazi, H., Hill-Smith, T. E., Easton, R. M., et al. (2012). Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus 22, 230–240. doi:10.1002/hipo.20887
Bartolomeis, A., Avagliano, C., Vellucci, L., D'Ambrosio, L., Manchia, M., D'Urso, G., et al. (2019). Translating preclinical findings in clinically relevant new antipsychotic targets: Focus on the glutamatergic postsynaptic density. Implications for treatment resistant schizophrenia. Neurosci. Biobehav. Rev. 107, 795–827. doi:10.1016/j.neubiorev.2019.08.019
Bechter, K., Reiber, H., Herzog, S., Fuchs, D., Tumani, H., and Maxeiner, H. G. (2010). Cerebrospinal fluid analysis in affective and schizophrenic spectrum disorders: Identification of subgroups with immune responses and blood-CSF barrier dysfunction. J. Psychiatr. Res. 44, 321–330. doi:10.1016/j.jpsychires.2009.08.008
Bechter, K. (2013). Schizophrenia--a mild encephalitis? [Schizophrenie--eine milde enzephalitis?]. Fortschr. Neurol. Psychiatr. 81, 250–259. doi:10.1055/s-0033-1335253
Bilic, P., Jukic, V., Vilibic, M., Savic, A., and Bozina, N. (2014). Treatment-resistant schizophrenia and DAT and SERT polymorphisms. Gene 543, 125–132. doi:10.1016/j.gene.2014.03.050
Bishop, J. R., Del Miller, D., Ellingrod, V. L., and Holman, T. (2011). Association between type-three metabotropic glutamate receptor gene (GRM3) variants and symptom presentation in treatment refractory schizophrenia. Hum. Psychopharmacol. 26, 28–34. doi:10.1002/hup.1163
Bishop, J. R., Reilly, J. L., Harris, M. S. H., Patel, S. R., Kittles, R., Badner, J. A., et al. (2015). Pharmacogenetic associations of the type-3 metabotropic glutamate receptor (GRM3) gene with working memory and clinical symptom response to antipsychotics in first-episode schizophrenia. Psychopharmacology 232, 145–154. doi:10.1007/s00213-014-3649-4
Bitanihirwe, B. K. Y., and Woo, T.-U. W. (2011). Oxidative stress in schizophrenia: An integrated approach. Neurosci. Biobehav. Rev. 35, 878–893. doi:10.1016/j.neubiorev.2010.10.008
Blackman, G., Lisshammar, J. E. L., Zafar, R., Pollak, T. A., Pritchard, M., Cullen, A. E., et al. (2021). Clozapine response in schizophrenia and hematological changes. J. Clin. Psychopharmacol. 41, 19–24. doi:10.1097/JCP.0000000000001329
Bojesen, K. B., Ebdrup, B. H., Jessen, K., Sigvard, A., Tangmose, K., Edden, R. A. E., et al. (2020). Treatment response after 6 and 26 weeks is related to baseline glutamate and GABA levels in antipsychotic-naïve patients with psychosis. Psychol. Med. 50, 2182–2193. doi:10.1017/S0033291719002277
Bosia, M., Lorenzi, C., Pirovano, A., Guglielmino, C., Cocchi, F., Spangaro, M., et al. (2015). COMT Val158Met and 5-ht1a-R -1019 C/G polymorphisms: Effects on the negative symptom response to clozapine. Pharmacogenomics 16, 35–44. doi:10.2217/pgs.14.150
Brandl, E. J., Lett, T. A., Chowdhury, N. I., Tiwari, A. K., Bakanidze, G., Meltzer, H. Y., et al. (2016). The role of the ITIH3 rs2535629 variant in antipsychotic response. Schizophr. Res. 176, 131–135. doi:10.1016/j.schres.2016.06.032
Brown, J. A., Horváth, S., Garbett, K. A., Schmidt, M. J., Everheart, M., Gellért, L., et al. (2014). The role of cannabinoid 1 receptor expressing interneurons in behavior. Neurobiol. Dis. 63, 210–221. doi:10.1016/j.nbd.2013.11.001
Bumb, J. M., Enning, F., and Leweke, F. M. (2015). Drug repurposing and emerging adjunctive treatments for schizophrenia. Expert Opin. Pharmacother. 16, 1049–1067. doi:10.1517/14656566.2015.1032248
Cai, H., Zeng, C., Zhang, X., Liu, Y., Wu, R., Guo, W., et al. (2022). Diminished treatment response in relapsed versus first-episode schizophrenia as revealed by a panel of blood-based biomarkers: A combined cross-sectional and longitudinal study. Psychiatry Res. 316, 114762. doi:10.1016/j.psychres.2022.114762
Cao, T., Li, N., and Cai, H. (2020). Candidate metabolic biomarkers for schizophrenia in CNS and periphery: Do any possible associations exist? Schizophr. Res. 226, 95–110. doi:10.1016/j.schres.2019.03.009
Carruthers, S. P., Gurvich, C. T., and Rossell, S. L. (2015). The muscarinic system, cognition and schizophrenia. Neurosci. Biobehav. Rev. 55, 393–402. doi:10.1016/j.neubiorev.2015.05.011
Challa, F., Seifu, D., Sileshi, M., Getahun, T., Geto, Z., Kassa, D., et al. (2021). Serum level of high sensitive C-reactive protein and IL - 6 markers in patients with treatment-resistant schizophrenia in Ethiopia: A comparative study. BMC psychiatry 21, 428. doi:10.1186/s12888-021-03443-4
Chiu, H.-J., Wang, Y.-C., Liou, Y.-J., Lai, I.-C., and Chen, J.-Y. (2003). Association analysis of the genetic variants of the N-methyl D-aspartate receptor subunit 2b (NR2b) and treatment-refractory schizophrenia in the Chinese. Neuropsychobiology 47, 178–181. doi:10.1159/000071211
Correll, C. U., and Howes, O. D. (2021). Treatment-resistant schizophrenia: Definition, predictors, and therapy options. J. Clin. Psychiatry 82, MY20096AH1C. doi:10.4088/JCP.MY20096AH1C
Costa-Dookhan, K. A., Agarwal, S. M., Chintoh, A., Tran, V. N., Stogios, N., Ebdrup, B. H., et al. (2020). The clozapine to norclozapine ratio: A narrative review of the clinical utility to minimize metabolic risk and enhance clozapine efficacy. Expert Opin. Drug Saf. 19, 43–57. doi:10.1080/14740338.2020.1698545
Cui, G., Qing, Y., Hu, X., Wang, P., Sun, L., Yang, X., et al. (2020). Serum metabolomic profiling based on fourier transform-ion cyclotron resonance-mass Spectrometry: Do the dysfunctions of metabolic pathways reveal a universal risk of oxidative stress in schizophrenia? Antioxid. Redox Signal. 33, 679–688. doi:10.1089/ars.2020.8141
Dahl, M.-L. (2002). Cytochrome p450 phenotyping/genotyping in patients receiving antipsychotics: Useful aid to prescribing? Clin. Pharmacokinet. 41, 453–470. doi:10.2165/00003088-200241070-00001
Daskalakis, Z. J., and George, T. P. (2009). Clozapine, GABA(B), and the treatment of resistant schizophrenia. Clin. Pharmacol. Ther. 86, 442–446. doi:10.1038/clpt.2009.115
Davies, M. A., Compton-Toth, B. A., Hufeisen, S. J., Meltzer, H. Y., and Roth, B. L. (2005). The highly efficacious actions of N-desmethylclozapine at muscarinic receptors are unique and not a common property of either typical or atypical antipsychotic drugs: Is M1 agonism a pre-requisite for mimicking clozapine's actions? Psychopharmacology 178, 451–460. doi:10.1007/s00213-004-2017-1
Demjaha, A., Egerton, A., Murray, R. M., Kapur, S., Howes, O. D., Stone, J. M., et al. (2014). Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol. Psychiatry 75, e11–e13. doi:10.1016/j.biopsych.2013.06.011
Demjaha, A., Murray, R. M., McGuire, P. K., Kapur, S., and Howes, O. D. (2012). Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. Am. J. Psychiatry 169, 1203–1210. doi:10.1176/appi.ajp.2012.12010144
Digby, G. J., Noetzel, M. J., Bubser, M., Utley, T. J., Walker, A. G., Byun, N. E., et al. (2012). Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J. Neurosci. 32, 8532–8544. doi:10.1523/JNEUROSCI.0337-12.2012
Do, K. Q., Cabungcal, J. H., Frank, A., Steullet, P., and Cuenod, M. (2009). Redox dysregulation, neurodevelopment, and schizophrenia. Curr. Opin. Neurobiol. 19, 220–230. doi:10.1016/j.conb.2009.05.001
Egerton, A., Murphy, A., Donocik, J., Anton, A., Barker, G. J., Collier, T., et al. (2021). Dopamine and glutamate in antipsychotic-responsive compared with antipsychotic-nonresponsive psychosis: A multicenter Positron emission Tomography and magnetic resonance spectroscopy study (strata). Schizophr. Bull. 47, 505–516. doi:10.1093/schbul/sbaa128
Ershova, E. S., Agafonova, O. N., Zakharova, N. V., Bravve, L. V., Jestkova, E. M., Golimbet, V. E., et al. (2019). Copy number variation of satellite III (1q12) in patients with schizophrenia. Front. Genet. 10, 1132. doi:10.3389/fgene.2019.01132
Escamilla, R., Camarena, B., Saracco-Alvarez, R., Fresán, A., Hernández, S., and Aguilar-García, A. (2018). Association study between COMT, DRD2, and DRD3 gene variants and antipsychotic treatment response in Mexican patients with schizophrenia. Neuropsychiatr. Dis. Treat. 14, 2981–2987. doi:10.2147/NDT.S176455
Euesden, J., Lewis, C. M., and O'Reilly, P. F. (2015). PRSice: Polygenic risk score software. Bioinforma. Oxf. Engl. 31, 1466–1468. doi:10.1093/bioinformatics/btu848
Faden, J., and Citrome, L. (2019). Resistance is not futile: Treatment-refractory schizophrenia - overview, evaluation and treatment. Expert Opin. Pharmacother. 20, 11–24. doi:10.1080/14656566.2018.1543409
Fenton, W. S., Hibbeln, J., and Knable, M. (2000). Essential fatty acids, lipid membrane abnormalities, and the diagnosis and treatment of schizophrenia. Biol. Psychiatry 47, 8–21. doi:10.1016/s0006-3223(99)00092-x
Fernandez-Egea, E., Vértes, P. E., Flint, S. M., Turner, L., Mustafa, S., Hatton, A., et al. (2016). Peripheral immune cell populations associated with cognitive deficits and negative symptoms of treatment-resistant schizophrenia. PloS one 11, e0155631. doi:10.1371/journal.pone.0155631
Fernandez-Espejo, E., Viveros, M.-P., Núñez, L., Ellenbroek, B. A., and Rodriguez de Fonseca, F. (2009). Role of cannabis and endocannabinoids in the Genesis of schizophrenia. Psychopharmacology 206, 531–549. doi:10.1007/s00213-009-1612-6
Ferretjans, R., Moreira, F. A., Teixeira, A. L., and Salgado, J. V. (2012). The endocannabinoid system and its role in schizophrenia: A systematic review of the literature. Braz. J. Psychiatry. 34 (2), S163–S177. doi:10.1016/j.rbp.2012.07.003
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi:10.1038/nature12929
Gammon, D., Cheng, C., Volkovinskaia, A., Baker, G. B., and Dursun, S. M. (2021). Clozapine: Why is it so uniquely effective in the treatment of a range of neuropsychiatric disorders? Biomolecules 11, 1030. doi:10.3390/biom11071030
Garcia-Rizo, C., Fernandez-Egea, E., Oliveira, C., Justicia, A., Bernardo, M., and Kirkpatrick, B. (2012). Inflammatory markers in antipsychotic-naïve patients with nonaffective psychosis and deficit vs. nondeficit features. Psychiatry Res. 198, 212–215. doi:10.1016/j.psychres.2011.08.014
Gillespie, A. L., Samanaite, R., Mill, J., Egerton, A., and MacCabe, J. H. (2017). Is treatment-resistant schizophrenia categorically distinct from treatment-responsive schizophrenia? A systematic review. BMC psychiatry 17, 12. doi:10.1186/s12888-016-1177-y
Girirajan, S., Campbell, C. D., and Eichler, E. E. (2011). Human copy number variation and complex genetic disease. Annu. Rev. Genet. 45, 203–226. doi:10.1146/annurev-genet-102209-163544
Hagan, J. J., Jansen, J. H., and Broekkamp, C. L. (1987). Blockade of spatial learning by the M1 muscarinic antagonist pirenzepine. Psychopharmacology 93, 470–476. doi:10.1007/BF00207237
Hajj, A., Obeid, S., Sahyoun, S., Haddad, C., Azar, J., Rabbaa Khabbaz, L., et al. (2019). Clinical and genetic factors associated with resistance to treatment in patients with schizophrenia: A case-control study. Int. J. Mol. Sci. 20, E4753. doi:10.3390/ijms20194753
Hamdani, N., Tabeze, J.-P., Ramoz, N., Ades, J., Hamon, M., Sarfati, Y., et al. (2008). The CNR1 gene as a pharmacogenetic factor for antipsychotics rather than a susceptibility gene for schizophrenia. Eur. Neuropsychopharmacol. 18, 34–40. doi:10.1016/j.euroneuro.2007.05.005
Hannon, E., Dempster, E. L., Mansell, G., Burrage, J., Bass, N., Bohlken, M. M., et al. (2021). DNA methylation meta-analysis reveals cellular alterations in psychosis and markers of treatment-resistant schizophrenia. eLife 10, e58430. doi:10.7554/eLife.58430
Herken, H., Erdal, M. E., Erdal, N., and Aynacioglu, S. (2003). T102C polymorphisms at the 5-ht2a receptor gene in Turkish schizophrenia patients: A possible association with prognosis. Neuropsychobiology 47, 27–30. doi:10.1159/000068872
Horsdal, H. T., Wimberley, T., Benros, M. E., and Gasse, C. (2017). C-reactive protein levels and treatment resistance in schizophrenia-A Danish population-based cohort study. Hum. Psychopharmacol. Clin. Exp. 32, e2632. doi:10.1002/hup.2632
Howes, O. D., McCutcheon, R., Agid, O., Bartolomeis, A., Beveren, N. J. M., Birnbaum, M. L., et al. (2017). Treatment-resistant schizophrenia: Treatment response and resistance in psychosis (TRRIP) working group consensus guidelines on diagnosis and terminology. Am. J. Psychiatry 174, 216–229. doi:10.1176/appi.ajp.2016.16050503
Howes, O. D., Thase, M. E., and Pillinger, T. (2022). Treatment resistance in psychiatry: State of the art and new directions. Mol. Psychiatry 27, 58–72. doi:10.1038/s41380-021-01200-3
Huang, E., Maciukiewicz, M., Zai, C. C., Tiwari, A. K., Li, J., Potkin, S. G., et al. (2016). Preliminary evidence for association of genome-wide significant DRD2 schizophrenia risk variant with clozapine response. Pharmacogenomics 17, 103–109. doi:10.2217/pgs.15.155
Humbert-Claude, M., Davenas, E., Gbahou, F., Vincent, L., and Arrang, J.-M. (2012). Involvement of histamine receptors in the atypical antipsychotic profile of clozapine: A reassessment in vitro and in vivo. Psychopharmacology 220, 225–241. doi:10.1007/s00213-011-2471-5
Hwang, R., Shinkai, T., Luca, V., Müller, D. J., Ni, X., Macciardi, F., et al. (2005). Association study of 12 polymorphisms spanning the dopamine D(2) receptor gene and clozapine treatment response in two treatment refractory/intolerant populations. Psychopharmacology 181, 179–187. doi:10.1007/s00213-005-2223-5
Hwang, R., Shinkai, T., Luca, V., Ni, X., Potkin, S. G., Lieberman, J. A., et al. (2007). Association study of four dopamine D1 receptor gene polymorphisms and clozapine treatment response. J. Psychopharmacol. 21, 718–727. doi:10.1177/0269881106072341
Hwang, R., Tiwari, A. K., Zai, C. C., Felsky, D., Remington, E., Wallace, T., et al. (2012). Dopamine D4 and D5 receptor gene variant effects on clozapine response in schizophrenia: Replication and exploration. Prog. Neuropsychopharmacol. Biol. Psychiatry 37, 62–75. doi:10.1016/j.pnpbp.2011.11.018
Hwang, R., Zai, C., Tiwari, A., Müller, D. J., Arranz, M. J., Morris, A. G., et al. (2010). Effect of dopamine D3 receptor gene polymorphisms and clozapine treatment response: Exploratory analysis of nine polymorphisms and meta-analysis of the Ser9Gly variant. Pharmacogenomics J. 10, 200–218. doi:10.1038/tpj.2009.65
Iasevoli, F., Giordano, S., Balletta, R., Latte, G., Formato, M. V., Prinzivalli, E., et al. (2016). Treatment resistant schizophrenia is associated with the worst community functioning among severely-ill highly-disabling psychiatric conditions and is the most relevant predictor of poorer achievements in functional milestones. Prog. Neuropsychopharmacol. Biol. Psychiatry 65, 34–48. doi:10.1016/j.pnpbp.2015.08.010
Inada, T., Nakamura, A., and Iijima, Y. (2003). Relationship between catechol-O-methyltransferase polymorphism and treatment-resistant schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 1, 35–39. doi:10.1002/ajmg.b.20023
Ishiguro, H., Horiuchi, Y., Ishikawa, M., Koga, M., Imai, K., Suzuki, Y., et al. (2010). Brain cannabinoid CB2 receptor in schizophrenia. Biol. Psychiatry 67, 974–982. doi:10.1016/j.biopsych.2009.09.024
Itil, T. M., Keskiner, A., and Fink, M. (1966). Therapeutic studies in “therapy resistant” schizophrenic patients. Compr. Psychiatry 7, 488–493. doi:10.1016/s0010-440x(66)80028-7
Itokawa, M., Miyashita, M., Arai, M., Dan, T., Takahashi, K., Tokunaga, T., et al. (2018). Pyridoxamine: A novel treatment for schizophrenia with enhanced carbonyl stress. Psychiatry Clin. Neurosci. 72, 35–44. doi:10.1111/pcn.12613
Ji, X., Takahashi, N., Branko, A., Ishihara, R., Nagai, T., Mouri, A., et al. (2008a). An association between serotonin receptor 3B gene (HTR3B) and treatment-resistant schizophrenia (TRS) in a Japanese population. Nagoya J. Med. Sci. 70, 11–17. doi:10.18999/NAGJMS.70.1-2.11
Ji, X., Takahashi, N., Saito, S., Ishihara, R., Maeno, N., Inada, T., et al. (2008b). Relationship between three serotonin receptor subtypes (HTR3A, HTR2A and HTR4) and treatment-resistant schizophrenia in the Japanese population. Neurosci. Lett. 435, 95–98. doi:10.1016/j.neulet.2008.01.083
Kane, J., Honigfeld, G., Singer, J., and Meltzer, H. (1988). Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch. Gen. Psychiatry 45, 789–796. doi:10.1001/archpsyc.1988.01800330013001
Katsuta, N., Ohnuma, T., Maeshima, H., Takebayashi, Y., Higa, M., Takeda, M., et al. (2014). Significance of measurements of peripheral carbonyl stress markers in a cross-sectional and longitudinal study in patients with acute-stage schizophrenia. Schizophr. Bull. 40, 1366–1373. doi:10.1093/schbul/sbt234
Kaur, H., Jajodia, A., Grover, S., Baghel, R., Jain, S., and Kukreti, R. (2014). Synergistic association of PI4KA and GRM3 genetic polymorphisms with poor antipsychotic response in south Indian schizophrenia patients with low severity of illness. Am. J. Med. Genet. B Neuropsychiatr. Genet. 165B, 635–646. doi:10.1002/ajmg.b.32268
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (1999). 4-hydroxynonenal increases neuronal susceptibility to oxidative stress. J. Neurosci. Res. 58, 823–830. doi:10.1002/(sici)1097-4547(19991215)58:6<823::aid-jnr9>3.0.co;2-t
Kennedy, J. L., Altar, C. A., Taylor, D. L., Degtiar, I., and Hornberger, J. C. (2014). The social and economic burden of treatment-resistant schizophrenia: A systematic literature review. Int. Clin. Psychopharmacol. 29, 63–76. doi:10.1097/YIC.0b013e32836508e6
Kirenskaya, A. V., Storozheva, Z. I., Gruden, M. A., and Sewell, R. D. E. (2018). COMT and GAD1 gene polymorphisms are associated with impaired antisaccade task performance in schizophrenic patients. Eur. Arch. Psychiatry Clin. Neurosci. 268, 571–584. doi:10.1007/s00406-018-0881-7
Kogure, M., Kanahara, N., Miyazawa, A., Oishi, K., Nakata, Y., Oda, Y., et al. (2021). Interacting roles of COMT and GAD1 genes in patients with treatment-resistant schizophrenia: A genetic association study of schizophrenia patients and healthy controls. J. Mol. Neurosci. 71, 2575–2582. doi:10.1007/s12031-021-01866-y
Kohlrausch, F. B., Gama, C. S., Lobato, M. I., Belmonte-de-Abreu, P., Callegari-Jacques, S. M., Gesteira, A., et al. (2008). Naturalistic pharmacogenetic study of treatment resistance to typical neuroleptics in European-Brazilian schizophrenics. Pharmacogenet. Genomics 18, 599–609. doi:10.1097/FPC.0b013e328301a763
Kohlrausch, F. B., Salatino-Oliveira, A., Gama, C. S., Lobato, M. I., Belmonte-de-Abreu, P., and Hutz, M. H. (2010). Influence of serotonin transporter gene polymorphisms on clozapine response in Brazilian schizophrenics. J. Psychiatr. Res. 44, 1158–1162. doi:10.1016/j.jpsychires.2010.04.003
Kondo, T., Mihara, K., Suzuki, A., Yasui-Furukori, N., and Kaneko, S. (2003). Combination of dopamine D2 receptor gene polymorphisms as a possible predictor of treatment-resistance to dopamine antagonists in schizophrenic patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 921–926. doi:10.1016/S0278-5846(03)00151-9
Kouidrat, Y., Amad, A., Arai, M., Miyashita, M., Lalau, J.-D., Loas, G., et al. (2015). Advanced glycation end products and schizophrenia: A systematic review. J. Psychiatr. Res. 66-67, 112–117. doi:10.1016/j.jpsychires.2015.04.023
Kraguljac, N. V., McDonald, W. M., Widge, A. S., Rodriguez, C. I., Tohen, M., and Nemeroff, C. B. (2021). Neuroimaging biomarkers in schizophrenia. Am. J. Psychiatry 178, 509–521. doi:10.1176/appi.ajp.2020.20030340
Krivoy, A., Hochman, E., Sendt, K.-V., Hollander, S., Vilner, Y., Selakovic, M., et al. (2018). Association between serum levels of glutamate and neurotrophic factors and response to clozapine treatment. Schizophr. Res. 192, 226–231. doi:10.1016/j.schres.2017.05.040
Kushima, I., Aleksic, B., Nakatochi, M., Shimamura, T., Shiino, T., Yoshimi, A., et al. (2017). High-resolution copy number variation analysis of schizophrenia in Japan. Mol. Psychiatry 22, 430–440. doi:10.1038/mp.2016.88
Labonté, C., Zhand, N., Park, A., and Harvey, P. D. (2022). Complete blood count inflammatory markers in treatment-resistant schizophrenia: Evidence of association between treatment responsiveness and levels of inflammation. Psychiatry Res. 308, 114382. doi:10.1016/j.psychres.2021.114382
Lally, J., Gallagher, A., Bainbridge, E., Avalos, G., Ahmed, M., and McDonald, C. (2013). Increases in triglyceride levels are associated with clinical response to clozapine treatment. J. Psychopharmacol. 27, 401–403. doi:10.1177/0269881112472568
Lang, B., Pu, J., Hunter, I., Liu, M., Martin-Granados, C., Reilly, T. J., et al. (2014). Recurrent deletions of ULK4 in schizophrenia: A gene crucial for neuritogenesis and neuronal motility. J. Cell Sci. 127, 630–640. doi:10.1242/jcs.137604
Leboyer, M., Godin, O., Terro, E., Boukouaci, W., Lu, C.-L., Andre, M., et al. (2021). Immune signatures of treatment-resistant schizophrenia: A FondaMental academic centers of expertise for schizophrenia (FACE-SZ) study. Schizophr. Bull. Open 2, sgab012. doi:10.1093/schizbullopen/sgab012
Leboyer, M., Oliveira, J., Tamouza, R., and Groc, L. (2016). Is it time for immunopsychiatry in psychotic disorders? Psychopharmacology 233, 1651–1660. doi:10.1007/s00213-016-4266-1
Legge, S. E., Santoro, M. L., Periyasamy, S., Okewole, A., Arsalan, A., and Kowalec, K. (2021). Genetic architecture of schizophrenia: A review of major advancements. Psychol. Med. 51, 2168–2177. doi:10.1017/S0033291720005334
Leonard, B., and Maes, M. (2012). Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci. Biobehav. Rev. 36, 764–785. doi:10.1016/j.neubiorev.2011.12.005
Lesch, K. P., Bengel, D., Heils, A., Sabol, S. Z., Greenberg, B. D., Petri, S., et al. (1996). Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Sci. (New York, N.Y.) 274, 1527–1531. doi:10.1126/science.274.5292.1527
Leung, C. C.-Y., Gadelrab, R., Ntephe, C. U., McGuire, P. K., and Demjaha, A. (2019). Clinical course, neurobiology and therapeutic approaches to treatment resistant schizophrenia. Toward an integrated view. Front. Psychiatry 10, 601. doi:10.3389/fpsyt.2019.00601
Li, N., Yang, P., Tang, M., Liu, Y., Guo, W., Lang, B., et al. (2022). Reduced erythrocyte membrane polyunsaturated fatty acid levels indicate diminished treatment response in patients with multi- versus first-episode schizophrenia. Schizophrenia 8, 7. doi:10.1038/s41537-022-00214-2
Li, Z., Huang, M., Ichikawa, J., Dai, J., and Meltzer, H. Y. (2005). N-desmethylclozapine, a major metabolite of clozapine, increases cortical acetylcholine and dopamine release in vivo via stimulation of M1 muscarinic receptors. Neuropsychopharmacology 30, 1986–1995. doi:10.1038/sj.npp.1300768
Liao, D.-L., Hong, C.-J., Chen, H.-M., Chen, Y.-E., Lee, S.-M., Chang, C.-Y., et al. (2003). Association of muscarinic m1 receptor genetic polymorphisms with psychiatric symptoms and cognitive function in schizophrenic patients. Neuropsychobiology 48, 72–76. doi:10.1159/000072880
Lieberman, J. A., Mailman, R. B., Duncan, G., Sikich, L., Chakos, M., Nichols, D. E., et al. (1998). Serotonergic basis of antipsychotic drug effects in schizophrenia. Biol. Psychiatry 44, 1099–1117. doi:10.1016/S0006-3223(98)00187-5
Lin, A., Kenis, G., Bignotti, S., Tura, G. J., Jong, R., Bosmans, E., et al. (1998). The inflammatory response system in treatment-resistant schizophrenia: Increased serum interleukin-6. Schizophr. Res. 32, 9–15. doi:10.1016/s0920-9964(98)00034-6
Lin, C.-H., Lane, H.-Y., and Tsai, G. E. (2012). Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol. Biochem. Behav. 100, 665–677. doi:10.1016/j.pbb.2011.03.023
M, N., Patil, A. N., Pattanaik, S., Kaur, A., Banerjee, D., and Grover, S. (2020). ABCB1 and DRD3 polymorphism as a response predicting biomarker and tool for pharmacogenetically guided clozapine dosing in Asian Indian treatment resistant schizophrenia patients. Asian J. Psychiatr. 48, 101918. doi:10.1016/j.ajp.2019.101918
Maes, M., Bocchio Chiavetto, L., Bignotti, S., Battisa Tura, G., Pioli, R., Boin, F., et al. (2000). Effects of atypical antipsychotics on the inflammatory response system in schizophrenic patients resistant to treatment with typical neuroleptics. Eur. Neuropsychopharmacol. 10, 119–124. doi:10.1016/s0924-977x(99)00062-0
Maes, M., Bocchio Chiavetto, L., Bignotti, S., Battisa Tura, G., Pioli, R., BoinF., , et al. (2000). Effects of atypical antipsychotics on the inflammatory response system in schizophrenic patients resistant to treatment with typical neuroleptics. Eur. Neuropsychopharmacol. 10, 119–124. doi:10.1016/S0924-977X(99)00062-0
Mancama, D., Arranz, M. J., and Kerwin, R. W. (2003). Genetic perspectives of histamine and muscarinic receptors in schizophrenia and clozapine response. Drug Dev. Res. 60, 119–126. doi:10.1002/ddr.10291
Mancama, D., Arranz, M., Munro, J., Osborne, S., Makoff, A., Collier, D., et al. (2002). Investigation of promoter variants of the histamine 1 and 2 receptors in schizophrenia and clozapine response. Neurosci. Lett. 333, 207–211. doi:10.1016/s0304-3940(02)00178-7
Marder, S. R., and Cannon, T. D. (2019). Schizophrenia. N. Engl. J. Med. 381, 1753–1761. doi:10.1056/NEJMra1808803
Marino, M. J., Rouse, S. T., Levey, A. I., Potter, L. T., and Conn, P. J. (1998). Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc. Natl. Acad. Sci. U. S. A. 95, 11465–11470. doi:10.1073/pnas.95.19.11465
Martin, A. K., and Mowry, B. (2016). Increased rare duplication burden genomewide in patients with treatment-resistant schizophrenia. Psychol. Med. 46, 469–476. doi:10.1017/S0033291715001701
Martínez-Cengotitabengoa, M., Mac-Dowell, K. S., Leza, J. C., Micó, J. A., Fernandez, M., Echevarría, E., et al. (2012). Cognitive impairment is related to oxidative stress and chemokine levels in first psychotic episodes. Schizophr. Res. 137, 66–72. doi:10.1016/j.schres.2012.03.004
Masellis, M., Basile, V., Meltzer, H. Y., Lieberman, J. A., Sevy, S., Macciardi, F. M., et al. (1998). Serotonin subtype 2 receptor genes and clinical response to clozapine in schizophrenia patients. Neuropsychopharmacology 19, 123–132. doi:10.1016/S0893-133X(98)00007-4
McClay, J. L., Adkins, D. E., Aberg, K., Stroup, S., Perkins, D. O., Vladimirov, V. I., et al. (2011). Genome-wide pharmacogenomic analysis of response to treatment with antipsychotics. Mol. Psychiatry 16, 76–85. doi:10.1038/mp.2009.89
Medina-Hernández, V., Ramos-Loyo, J., Luquin, S., Sánchez, L. F. C., García-Estrada, J., and Navarro-Ruiz, A. (2007). Increased lipid peroxidation and neuron specific enolase in treatment refractory schizophrenics. J. Psychiatr. Res. 41, 652–658. doi:10.1016/j.jpsychires.2006.02.010
Messamore, E., and McNamara, R. K. (2016). Detection and treatment of omega-3 fatty acid deficiency in psychiatric practice: Rationale and implementation. Lipids Health Dis. 15, 25. doi:10.1186/s12944-016-0196-5
Miller, B. J., Gassama, B., Sebastian, D., Buckley, P., and Mellor, A. (2013). Meta-analysis of lymphocytes in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 73, 993–999. doi:10.1016/j.biopsych.2012.09.007
Miller, R. (2009). Mechanisms of action of antipsychotic drugs of different classes, refractoriness to therapeutic effects of classical neuroleptics, and individual variation in sensitivity to their actions: Part I. Curr. Neuropharmacol. 7, 302–314. doi:10.2174/157015909790031229
Mitjans, M., Catalán, R., Vázquez, M., González-Rodríguez, A., Penadés, R., Pons, A., et al. (2015). Hypothalamic-pituitary-adrenal system, neurotrophic factors and clozapine response: Association with FKBP5 and NTRK2 genes. Pharmacogenet. Genomics 25, 274–277. doi:10.1097/FPC.0000000000000132
Miyashita, M., Arai, M., Kobori, A., Ichikawa, T., Toriumi, K., Niizato, K., et al. (2014). Clinical features of schizophrenia with enhanced carbonyl stress. Schizophr. Bull. 40, 1040–1046. doi:10.1093/schbul/sbt129
Miyauchi, M., Neugebauer, N. M., Sato, T., Ardehali, H., and Meltzer, H. Y. (2017). Muscarinic receptor signaling contributes to atypical antipsychotic drug reversal of the phencyclidine-induced deficit in novel object recognition in rats. J. Psychopharmacol. 31, 1588–1604. doi:10.1177/0269881117731278
Miyazawa, A., Kanahara, N., Kogure, M., Otsuka, I., Okazaki, S., Watanabe, Y., et al. (2022). A preliminary genetic association study of GAD1 and GABAB receptor genes in patients with treatment-resistant schizophrenia. Mol. Biol. Rep. 49, 2015–2024. doi:10.1007/s11033-021-07019-z
Moghaddam, B., and Javitt, D. (2012). From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37, 4–15. doi:10.1038/npp.2011.181
Molent, C., Olivo, D., Wolf, R. C., Balestrieri, M., and Sambataro, F. (2019). Functional neuroimaging in treatment resistant schizophrenia: A systematic review. Neurosci. Biobehav. Rev. 104, 178–190. doi:10.1016/j.neubiorev.2019.07.001
Möller, M., Swanepoel, T., and Harvey, B. H. (2015). Neurodevelopmental animal models reveal the convergent role of neurotransmitter systems, inflammation, and oxidative stress as biomarkers of schizophrenia: Implications for novel drug development. ACS Chem. Neurosci. 6, 987–1016. doi:10.1021/cn5003368
Momtazmanesh, S., Zare-Shahabadi, A., and Rezaei, N. (2019). Cytokine alterations in schizophrenia: An updated review. Front. Psychiatry 10, 892. doi:10.3389/fpsyt.2019.00892
Moretti, P. N., Ota, V. K., Gouvea, E. S., Pedrini, M., Santoro, M. L., Talarico, F., et al. (2018). Accessing gene expression in treatment-resistant schizophrenia. Mol. Neurobiol. 55, 7000–7008. doi:10.1007/s12035-018-0876-4
Nair, P. C., McKinnon, R. A., Miners, J. O., and Bastiampillai, T. (2020). Binding of clozapine to the GABAB receptor: Clinical and structural insights. Mol. Psychiatry 25, 1910–1919. doi:10.1038/s41380-020-0709-5
Nedic Erjavec, G., Konjevod, M., Nikolac Perkovic, M., Svob Strac, D., Tudor, L., Barbas, C., et al. (2018). Short overview on metabolomic approach and redox changes in psychiatric disorders. Redox Biol. 14, 178–186. doi:10.1016/j.redox.2017.09.002
Noto, C., Maes, M., Ota, V. K., Teixeira, A. L., Bressan, R. A., Gadelha, A., et al. (2015). High predictive value of immune-inflammatory biomarkers for schizophrenia diagnosis and association with treatment resistance. World J. Biol. Psychiatry 16, 422–429. doi:10.3109/15622975.2015.1062552
Nucifora, F. C., Woznica, E., Lee, B. J., Cascella, N., and Sawa, A. (2019). Treatment resistant schizophrenia: Clinical, biological, and therapeutic perspectives. Neurobiol. Dis. 131, 104257. doi:10.1016/j.nbd.2018.08.016
O'Tuathaigh, C. M. P., Moran, P. M., Zhen, X. C., and Waddington, J. L. (2017). Translating advances in the molecular basis of schizophrenia into novel cognitive treatment strategies. Br. J. Pharmacol. 174, 3173–3190. doi:10.1111/bph.13938
Ohnuma, T., Nishimon, S., Takeda, M., Sannohe, T., Katsuta, N., and Arai, H. (2018). Carbonyl stress and microinflammation-related molecules as potential biomarkers in schizophrenia. Front. Psychiatry 9, 82. doi:10.3389/fpsyt.2018.00082
Ota, V. K., Spíndola, L. N., Gadelha, A., dos Santos Filho, A. F., Santoro, M. L., Christofolini, D. M., et al. (2012). DRD1 rs4532 polymorphism: A potential pharmacogenomic marker for treatment response to antipsychotic drugs. Schizophr. Res. 142, 206–208. doi:10.1016/j.schres.2012.08.003
Pardiñas, A. F., Smart, S. E., Willcocks, I. R., Holmans, P. A., Dennison, C. A., Lynham, A. J., et al. (2022). Interaction testing and polygenic risk scoring to estimate the association of common genetic variants with treatment resistance in schizophrenia. JAMA psychiatry 79, 260–269. doi:10.1001/jamapsychiatry.2021.3799
Pedraz-Petrozzi, B., Elyamany, O., Rummel, C., and Mulert, C. (2020). Effects of inflammation on the kynurenine pathway in schizophrenia - a systematic review. J. Neuroinflammation 17, 56. doi:10.1186/s12974-020-1721-z
Pinheiro, D. S., Da Santos, R. S., Brito, R. B., Aline, H. D. S., Ghedini, P. C., and Reis, A. A. S. (2017). GSTM1/GSTT1 double-null genotype increases risk of treatment-resistant schizophrenia: A genetic association study in Brazilian patients. PloS one 12, e0183812. doi:10.1371/journal.pone.0183812
Pisanu, C., and Squassina, A. (2019). Treatment-resistant schizophrenia: Insights from genetic studies and machine learning approaches. Front. Pharmacol. 10, 617. doi:10.3389/fphar.2019.00617
Potkin, S. G., Basile, V. S., Jin, Y., Masellis, M., Badri, F., Keator, D., et al. (2003). D1 receptor alleles predict PET metabolic correlates of clinical response to clozapine. Mol. Psychiatry 8, 109–113. doi:10.1038/sj.mp.4001191
Potkin, S. G., Kane, J. M., Correll, C. U., Lindenmayer, J.-P., Agid, O., Marder, S. R., et al. (2020). The neurobiology of treatment-resistant schizophrenia: Paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr. 6, 1. doi:10.1038/s41537-019-0090-z
Pouget, J. G., Shams, T. A., Tiwari, A. K., and Müller, D. J. (2014). Pharmacogenetics and outcome with antipsychotic drugs. Dialogues Clin. Neurosci. 16, 555–566. doi:10.31887/DCNS.2014.16.4/jpouget
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi:10.1038/nature12975
Purves-Tyson, T. D., Owens, S. J., Rothmond, D. A., Halliday, G. M., Double, K. L., Stevens, J., et al. (2017). Putative presynaptic dopamine dysregulation in schizophrenia is supported by molecular evidence from post-mortem human midbrain. Transl. Psychiatry 7, e1003. doi:10.1038/tp.2016.257
Raddatz, R., Hudkins, R. L., Mathiasen, J. R., Gruner, J. A., Flood, D. G., Aimone, L. D., et al. (2012). CEP-26401 (irdabisant), a potent and selective histamine H₃ receptor antagonist/inverse agonist with cognition-enhancing and wake-promoting activities. J. Pharmacol. Exp. Ther. 340, 124–133. doi:10.1124/jpet.111.186585
Rajagopal, L., Ryan, C., Elzokaky, A., Burstein, E. S., and Meltzer, H. Y. (2022). Pimavanserin augments the efficacy of atypical antipsychotic drugs in a mouse model of treatment-refractory negative symptoms of schizophrenia. Behav. Brain Res. 422, 113710. doi:10.1016/j.bbr.2021.113710
Rajagopal, V. M., Rajkumar, A. P., Jacob, K. S., and Jacob, M. (2018). Gene-gene interaction between DRD4 and COMT modulates clinical response to clozapine in treatment-resistant schizophrenia. Pharmacogenet. Genomics 28, 31–35. doi:10.1097/FPC.0000000000000314
Rajkumar, A. P., Poonkuzhali, B., Kuruvilla, A., Srivastava, A., Jacob, M., and Jacob, K. S. (2012). Outcome definitions and clinical predictors influence pharmacogenetic associations between HTR3A gene polymorphisms and response to clozapine in patients with schizophrenia. Psychopharmacology 224, 441–449. doi:10.1007/s00213-012-2773-2
Reynolds, G. P. (2004). Receptor mechanisms in the treatment of schizophrenia. J. Psychopharmacol. 18, 340–345. doi:10.1177/026988110401800303
Reynolds, G. P., Templeman, L. A., and Godlewska, B. R. (2006). Pharmacogenetics of schizophrenia. Expert Opin. Pharmacother. 7, 1429–1440. doi:10.1517/14656566.7.11.1429
Rodrigues, T., Borges, P., Mar, L., Marques, D., Albano, M., Eickhoff, H., et al. (2020). GLP-1 improves adipose tissue glyoxalase activity and capillarization improving insulin sensitivity in type 2 diabetes. Pharmacol. Res. 161, 105198. doi:10.1016/j.phrs.2020.105198
Rodrigues-Silva, C., Semedo, A. T., Da Neri, H. F. S., Vianello, R. P., Galaviz-Hernández, C., Sosa-Macías, M., et al. (2020). The CYP2C19*2 and CYP2C19*17 polymorphisms influence responses to clozapine for the treatment of schizophrenia. Neuropsychiatr. Dis. Treat. 16, 427–432. doi:10.2147/NDT.S228103
Roomruangwong, C., Noto, C., Kanchanatawan, B., Anderson, G., Kubera, M., Carvalho, A. F., et al. (2020). The role of aberrations in the immune-inflammatory response system (IRS) and the compensatory immune-regulatory reflex system (CIRS) in different phenotypes of schizophrenia: The IRS-CIRS theory of schizophrenia. Mol. Neurobiol. 57, 778–797. doi:10.1007/s12035-019-01737-z
Roth, B. L., Sheffler, D. J., and Kroeze, W. K. (2004). Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 3, 353–359. doi:10.1038/nrd1346
Ruderfer, D. M., Charney, A. W., Readhead, B., Kidd, B. A., Kähler, A. K., Kenny, P. J., et al. (2016). Polygenic overlap between schizophrenia risk and antipsychotic response: A genomic medicine approach. Lancet. Psychiatry 3, 350–357. doi:10.1016/S2215-0366(15)00553-2
Sagud, M., Tudor, L., Uzun, S., Perkovic, M. N., Zivkovic, M., Konjevod, M., et al. (2018). Haplotypic and genotypic association of catechol-O-methyltransferase rs4680 and rs4818 polymorphisms and treatment resistance in schizophrenia. Front. Pharmacol. 9, 705. doi:10.3389/fphar.2018.00705
Samanaite, R., Gillespie, A., Sendt, K.-V., McQueen, G., MacCabe, J. H., and Egerton, A. (2018). Biological predictors of clozapine response: A systematic review. Front. Psychiatry 9, 327. doi:10.3389/fpsyt.2018.00327
Scarr, E., Um, J. Y., Cowie, T. F., and Dean, B. (2013). Cholinergic muscarinic M4 receptor gene polymorphisms: A potential risk factor and pharmacogenomic marker for schizophrenia. Schizophr. Res. 146, 279–284. doi:10.1016/j.schres.2013.01.023
Scharfetter, J., Chaudhry, H. R., Hornik, K., Fuchs, K., Sieghart, W., Kasper, S., et al. (1999). Dopamine D3 receptor gene polymorphism and response to clozapine in schizophrenic Pakastani patients. Eur. Neuropsychopharmacol. 10, 17–20. doi:10.1016/S0924-977X(99)00044-9
Son, S., Arai, M., Miyata, J., Toriumi, K., Mizuta, H., Hayashi, T., et al. (2020). Enhanced carbonyl stress and disrupted white matter integrity in schizophrenia. Schizophr. Res. 223, 242–248. doi:10.1016/j.schres.2020.08.007
Sookoian, S., Gianotti, T. F., Gemma, C., Burgueño, A., and Pirola, C. J. (2008). Contribution of the functional 5-HTTLPR variant of the SLC6A4 gene to obesity risk in male adults. Obes. (Silver Spring, Md.) 16, 488–491. doi:10.1038/oby.2007.64
Souza, R. P., Luca, V. de, Meltzer, H. Y., Lieberman, J. A., and Kennedy, J. L. (2010). Influence of serotonin 3A and 3B receptor genes on clozapine treatment response in schizophrenia. Pharmacogenet. Genomics 20, 274–276. doi:10.1097/FPC.0b013e328337ce3e
Squassina, A., Manchia, M., Pisanu, C., Ardau, R., Arzedi, C., Bocchetta, A., et al. (2020). Telomere attrition and inflammatory load in severe psychiatric disorders and in response to psychotropic medications. Neuropsychopharmacology 45, 2229–2238. doi:10.1038/s41386-020-00844-z
Takeuchi, H., Siu, C., Remington, G., Fervaha, G., Zipursky, R. B., Foussias, G., et al. (2019). Does relapse contribute to treatment resistance? Antipsychotic response in first- vs. second-episode schizophrenia. Neuropsychopharmacology 44, 1036–1042. doi:10.1038/s41386-018-0278-3
Tan, T., Wang, W., Williams, J., Ma, K., Cao, Q., and Yan, Z. (2019). Stress exposure in dopamine D4 receptor knockout mice induces schizophrenia-like behaviors via disruption of GABAergic transmission. Schizophr. Bull. 45, 1012–1023. doi:10.1093/schbul/sby163
Tarumi, R., Tsugawa, S., Noda, Y., Plitman, E., Honda, S., Matsushita, K., et al. (2020). Levels of glutamatergic neurometabolites in patients with severe treatment-resistant schizophrenia: A proton magnetic resonance spectroscopy study. Neuropsychopharmacology 45, 632–640. doi:10.1038/s41386-019-0589-z
Taylor, D. L., Tiwari, A. K., Lieberman, J. A., Potkin, S. G., Meltzer, H. Y., Knight, J., et al. (2016). Genetic association analysis of N-methyl-D-aspartate receptor subunit gene GRIN2B and clinical response to clozapine. Hum. Psychopharmacol. 31, 121–134. doi:10.1002/hup.2519
Taylor, D. L., Tiwari, A. K., Lieberman, J. A., Potkin, S. G., Meltzer, H. Y., Knight, J., et al. (2017). Pharmacogenetic analysis of functional glutamate system gene variants and clinical response to clozapine. Mol. Neuropsychiatry 2, 185–197. doi:10.1159/000449224
Terzić, T., Kastelic, M., Dolžan, V., and Plesničar, B. K. (2015). Influence of 5-HT1A and 5-HTTLPR genetic variants on the schizophrenia symptoms and occurrence of treatment-resistant schizophrenia. Neuropsychiatr. Dis. Treat. 11, 453–459. doi:10.2147/NDT.S76494
Toriumi, K., Miyashita, M., Suzuki, K., Yamasaki, N., Yasumura, M., Horiuchi, Y., et al. (2021). Vitamin B6 deficiency hyperactivates the noradrenergic system, leading to social deficits and cognitive impairment. Transl. Psychiatry 11, 262. doi:10.1038/s41398-021-01381-z
Tóth, K., Csukly, G., Sirok, D., Belic, A., Kiss, Á., Háfra, E., et al. (2017). Potential role of patients' CYP3A-status in clozapine pharmacokinetics. Int. J. Neuropsychopharmacol. 20, 529–537. doi:10.1093/ijnp/pyx019
Uno, Y., and Coyle, J. T. (2019). Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 73, 204–215. doi:10.1111/pcn.12823
Vilaró, M., Palacios, J., and Mengod, G. (1994). Multiplicity of muscarinic autoreceptor subtypes? Comparison of the distribution of cholinergic cells and cells containing mRNA for five subtypes of muscarinic receptors in the rat brain. Brain Res. Mol. Brain Res. 21, 30–46. doi:10.1016/0169-328X(94)90375-1
Vita, A., Minelli, A., Barlati, S., Deste, G., Giacopuzzi, E., Valsecchi, P., et al. (2019). Treatment-resistant schizophrenia: Genetic and neuroimaging correlates. Front. Pharmacol. 10, 402. doi:10.3389/fphar.2019.00402
Wang, L., Yu, L., He, G., Zhang, J., Zhang, A.-P., Du, J., et al. (2007). Response of risperidone treatment may be associated with polymorphisms of HTT gene in Chinese schizophrenia patients. Neurosci. Lett. 414, 1–4. doi:10.1016/j.neulet.2006.09.014
Wei, Z., Wang, L., Yu, T., Wang, Y., Sun, L., Wang, T., et al. (2013). Histamine H4 receptor polymorphism: A potential predictor of risperidone efficacy. J. Clin. Psychopharmacol. 33, 221–225. doi:10.1097/JCP.0b013e318283963b
Yamamori, H., Hashimoto, R., Ishima, T., Kishi, F., Yasuda, Y., Ohi, K., et al. (2013). Plasma levels of mature brain-derived neurotrophic factor (BDNF) and matrix metalloproteinase-9 (MMP-9) in treatment-resistant schizophrenia treated with clozapine. Neurosci. Lett. 556, 37–41. doi:10.1016/j.neulet.2013.09.059
Yoshimura, B., Yada, Y., So, R., Takaki, M., and Yamada, N. (2017). The critical treatment window of clozapine in treatment-resistant schizophrenia: Secondary analysis of an observational study. Psychiatry Res. 250, 65–70. doi:10.1016/j.psychres.2017.01.064
You, X., Zhang, Y., Long, Q., Liu, Z., Ma, X., Lu, Z., et al. (2020). Investigating aberrantly expressed microRNAs in peripheral blood mononuclear cells from patients with treatment-resistant schizophrenia using miRNA sequencing and integrated bioinformatics. Mol. Med. Rep. 22, 4340–4350. doi:10.3892/mmr.2020.11513
Yu, W.-Y., Chang, H.-W., Lin, C.-H., and Cho, C.-L. (2008). Short telomeres in patients with chronic schizophrenia who show a poor response to treatment. J. Psychiatry Neurosci. 33, 244–247. doi:10.1016/j.schres.2017.03.007
Zhang, X. Y., Zhou, D. F., Cao, L. Y., Zhang, P. Y., Wu, G. Y., and Shen, Y. C. (2004). Changes in serum interleukin-2, -6, and -8 levels before and during treatment with risperidone and haloperidol: Relationship to outcome in schizophrenia. J. Clin. Psychiatry 65, 940–947. doi:10.4088/jcp.v65n0710
Keywords: treatment resistant schizophrenia (TRS), gene, inflammation, stress, neurobiology, peripheral biomarkers
Citation: Jiao S, Cao T and Cai H (2022) Peripheral biomarkers of treatment-resistant schizophrenia: Genetic, inflammation and stress perspectives. Front. Pharmacol. 13:1005702. doi: 10.3389/fphar.2022.1005702
Received: 28 July 2022; Accepted: 26 September 2022;
Published: 12 October 2022.
Edited by:
Tomiki Sumiyoshi, National Center of Neurology and Psychiatry, JapanReviewed by:
Frederick Charles Nucifora, Johns Hopkins Medicine, United StatesCopyright © 2022 Jiao, Cao and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hualin Cai, aHVhbGluY2FpQGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.