
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 13 January 2022
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.805269
 Fangling Zhang1
Fangling Zhang1 Xiaolin Xiao2
Xiaolin Xiao2 Yong Li1Hefei Wu1Xinyu Deng1
Yong Li1Hefei Wu1Xinyu Deng1 Yinxiao Jiang1Wenwen Zhang1Jian Wang1
Yinxiao Jiang1Wenwen Zhang1Jian Wang1 Xiao Ma1*
Xiao Ma1* Yanling Zhao3*
Yanling Zhao3*GPBAR1, a transmembrane G protein-coupled receptor for bile acids, is widely expressed in multiple tissues in humans and rodents. In recent years, GPBAR1 has been thought to play an important role in bile homeostasis, metabolism and inflammation. This review specifically focuses on the function of GPBAR1 in cholestatic liver disease and summarizes the various pathways through which GPBAR1 acts in cholestatic models. GPBAR1 mainly regulates cholestasis in a holistic system of liver-gallbladder-gut formation. In the state of cholestasis, the activation of GPBAR1 could regulate liver inflammation, induce cholangiocyte regeneration to maintain the integrity of the biliary tree, control the hydrophobicity of the bile acid pool and promote the secretion of bile HCO3−. All these functions of GPBAR1 might be clear ways to protect against cholestatic diseases and liver injury. However, the characteristic of GPBAR1-mediated proliferation increases the risk of proliferation of cholangiocarcinoma in malignant transformed cholangiocytes. This dichotomous function of GPBAR1 limits its use in cholestasis. During disease treatment, simultaneous activation of GPBAR1 and FXR receptors often results in improved outcomes, and this strategy may become a crucial direction in the development of bile acid-activated receptors in the future.
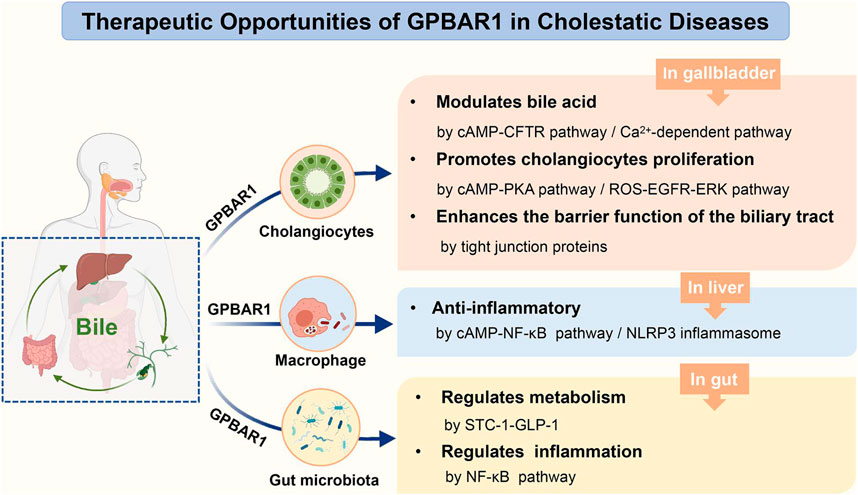
GRAPHICAL ABSTRACT. The role of GPBAR1 in cholestatic diseases.
Cholestasis is defined as the abnormal formation and excretion of bile caused by various internal and external factors in the liver, followed by modification of the bile composition, which may be intrahepatic cholestasis or extrahepatic cholestasis. Viruses, alcohol, stones, drugs, autoimmune and genetic metabolic diseases are common causes of cholestasis. Early clinical manifestations of cholestasis are increases in alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT) levels; then, as the disease develops, symptoms such as jaundice and itching can occur. Severe cholestatic liver disease can lead to hyperbilirubinemia, liver failure, and even death (Chen et al., 2018). The incidence and prevalence of cholestasis have increased globally over the past decades. Cholestasis remains an important public health problem that needs to be effectively addressed. Currently, the most common medical treatment for patients with cholestasis is ursodeoxycholic acid (UDCA), which slows the progression of primary biliary cirrhosis (PBC). However, there are patients who do not respond well to UDCA, and UDCA treatment does not improve survival in primary sclerosing cholangitis (PSC) patients (Ghonem et al., 2015). Due to such limited treatments, alternative therapies are needed. With the development of studies on the aetiology and mechanism of cholestatic liver disease, novel therapies using bile acid (BA)-activated receptors to treat cholestasis have attracted increasing attention.
As the largest superfamily of receptors, G protein-coupled receptors (GPCRs) are involved in almost all aspects of human physiology and myriad disease signalling processes and play essential roles in different cell signalling pathways. Since GPCRs account for approximately 30% of current drug therapeutic targets, these receptors have become one of the most important therapeutic targets for many diseases (Komatsu, 2015). GPBAR1 (also called TGR5, BG37, M-BAR, hGPCR19, and AXOR 109) is a GPCR that was initially discovered in 2002 (Maruyama et al., 2002; Zhong, 2010) GPBAR1 is a transmembrane GPCR that responds to BAs and is widely expressed in various cell types, including cholangiocytes, gallbladder smooth muscle cells and intestinal cells, nerve cells and brown adipose cells (Malhi and Camilleri, 2017; van Nierop et al., 2017). The role of GPBAR1 in metabolic diseases has always been a hot topic of research. As a metabolic regulator, GPBAR1 is involved in the regulation of energy homeostasis and glucose metabolism and has been indicated to have great potential value in the treatment of obesity and type 2 diabetes (Guo et al., 2016a). Furthermore, deoxycholic acid (DCA) and lithocholic acid (LCA), which are natural ligands of GPBAR1, have been shown to have anti-inflammatory effects (Yoneno et al., 2013). Activating GPBAR1 signalling can inhibit the production of proinflammatory cytokines in gastroenteropathy and reduce inflammation in both monocytes and macrophages (Perino and Schoonjans, 2015). To date, the role of GPBAR1 in various physiological and pathological processes has been verified. As various related studies have increased, the role of GPBAR1 in liver disease has also attracted specific attention.
In the liver, GPBAR1 is highly expressed in different nonparenchymal cells, including sinusoidal endothelial cells, Kupffer cells, activated hepatic stellate cells and cholangiocytes, and mediates liver microcirculation, the hepatic inflammatory response and the regulation of biliary function (Keitel and Häussinger, 2018). Cholangiocytes are the main cells that express GPBAR1, which has established the important role of GPBAR1 in biliary secretion, bile duct proliferation and apoptosis as well as other kinds of biliary diseases. In a mouse model of common bile duct ligation (BDL) and BA nourishment, GPBAR1-knockout (KO) mice exhibited more serious liver damage than wild-type (WT) mice, and prolonged cholestasis and an exacerbated inflammatory response were also observed (Péan et al., 2013). This finding provides direct evidence of the potential role of GPBAR1 in the process of cholestatic liver disease, suggesting that GPBAR1 could be an effective target in the treatment of cholestasis.
There is no doubt that the therapeutic efficacy of targeting GPBAR1 in cholestasis has great potential and should be explored. GPBAR1 could protect the liver from BA-induced apoptosis and protect cholangiocytes from BA-induced toxicity. Furthermore, in the pathological state of cholestasis, BA-activated GPBAR1 may mainly affect enterohepatic circulation organs, including the liver, gallbladder and intestine. Since GPBAR1 is widely expressed in these enterohepatic circulation tissues, it tends to regulate cholestasis through a variety of different pathways. In this review, we summarize our understanding of the role of GPBAR1 in the pathophysiology of cholestatic diseases and expound on the different pathways that mediate the effects of GPBAR1 on cholestatic liver disease.
As the main component of bile, BAs are synthesized in the liver and return to the liver via the enterohepatic cycle. During ileal reabsorption, some of the BAs escape into the colon, where they are converted by the intestinal microbiota into secondary BAs. The secondary BA is more hydrophobic and passively interacts with the colon epithelium to form the so-called BA pool with other BAs (Merlen et al., 2020a). Normally, BAs are almost completely confined to enterohepatic circulation, with only trace escapes in the cycle general. The amphiphilic structure of the BA molecule determines whether it is protective or toxic (Portincasa et al., 2020). Hydrophobic BA tends to cause toxic effects, and the stronger the hydrophobicity of the BA pool, the more harmful the impact on liver tissue (Merlen et al., 2020a). Thus, under the pathological condition of cholestasis, regulation is important to prevent excessive hydrophobic BAs from affecting liver repair.
Maruyama et al. showed that compared with that of WT mice, the size of the total BA pool in GPBAR1-deficient mice was significantly decreased by 21–25%, suggesting that GPBAR1 contributes to BA homeostasis (Maruyama et al., 2006). GPBAR1 is hardly expressed in hepatocytes, so GPBAR1 may not be directly associated with BA synthesis and tubule bile secretion. Consistent with this finding, there was no significant difference in the mRNA expression of a key enzyme in the conversion of cholesterol to BA (CYP7a1, CYP8b1, CYP27a1) and BA transporters between GPBAR1-KO mice and WT mice before and after partial hepatectomy (PH) (Péan et al., 2013). However, it is expected that high GPBAR1 expression in cholangiocytes may affect the ductal components of bile secretion (Merlen et al., 2020a). Accordingly, Keitel et al. demonstrated that GPBAR1 mediates chloride secretion in biliary epithelial cells by activating cystic fibrosis transmembrane conductance regulator (CFTR) (Keitel et al., 2009). Péan et al. observed the effect of GPBAR1 on protecting hepatocytes and maintaining remnant liver function after PH. The livers of GPBAR1-KO mice accumulated excessive hydrophobic BA pools and exhibited excessive liver inflammation, suggesting that GPBAR1 could control bile hydrophobicity, as well as cytokine secretion under BA overload after PH. Furthermore, Péan et al. also suggested that GPBAR1 regulates ion exchange to provide further protection against BA overload (Péan et al., 2013). GPBAR1 may regulate ion exchange in bile through cAMP-mediated mechanisms at posttranslational steps. This ion exchange process involves the concept of a HCO3− umbrella. When cholangiocytes are exposed to high concentrations of hydrophobic BAs, the self-protection mechanism of the cell not only forms micelles but also regulates the pH of the apical membrane by secreting HCO3− from the bile to form a HCO3− umbrella (Beuers et al., 2010). This alkaline environment prevents the protonation of glycine-conjugated BAs and their uncontrolled penetration into the apical membrane, which can cause liver damage.
The stability of the HCO3− umbrella is associated with GPBAR1-mediated Cl−/HCO3− exchange and HCO3− secretion (Beuers et al., 2010). Hohenester et al. explored the factors that maintain the integrity of human cholangiocytes in millimolar bile salt monomers in vitro. The results indicated that bile maintained an alkaline pH in the apical membrane of cholangiocytes through the secretion of HCO3−. Anion exchanger 2 (AE2) is a Cl−/HCO3− exchanger that plays a key role in human biliary HCO3− secretion (Hohenester et al., 2012). Thus, GPBAR1 participates in the secretion of HCO3− through CFTR and AE2 in a cAMP-dependent manner (Keitel et al., 2009; Hohenester et al., 2012). Moreover, a biliary glycocalyx may function to protect bile duct cells from bile salt toxicity; this glycocalyx is present in the outer lobes of the apical membrane of cholangiocytes, which enhances the ability of the bile HCO3− umbrella to inhibit the entry of hydrophobic BAs into the cells (Hohenester et al., 2012). The effect of the glycocalyx is similar to that of AE2 and appears crucial for the stability of the GPBAR1-mediated HCO3− umbrella. The concept of the biliary HCO3− umbrella is also applicable to the pathogenesis of various human fibrotic cholangiopathies. In humans, GPBAR1 has been confirmed to be a susceptibility gene in patients with PSC (Karlsen et al., 2010). When GPBAR1 gene is defective, it may affect the formation of the HCO3− umbrella and exacerbate liver injury. The CFTR-dependent chlorine transport pathway mentioned previously for Cl−/HCO3− exchange is considered to be the most typical pathway for HCO3− secretion. In addition, there is a Ca2+-dependent HCO3− secretion pathway (Nathanson et al., 1996). cAMP acts through an autocrine circuit that involves ATP release, P2Y receptor activation, and cytoplasmic Ca2+ increase. ATP release into the lumen stimulates P2Y nucleotide receptors in the apical region of cholangiocytes, which in turn causes an increase in Ca2+ (Figure 1). This series of processes leads to cAMP-induced HCO3− secretion (Minagawa et al., 2007). Furthermore, loss of inositol 1,4,5-trisphosphate receptor (InsP3R) expression can be observed in patients with cholestasis, such as PBC and PSC patients (Shibao et al., 2003). Although it is not clear whether the loss of InsP3R is the result or the cause of cholestasis, studies have shown that secretion mediated by Ca2+ signalling depends on the expression of InsP3R, and the loss of InsP3R reduces the secretion of HCO3− (Minagawa et al., 2007). Since the specific regulatory role of GPBAR1 in Ca2+ signalling is not yet clear, the relationship between GPBAR1 and InsP3R cannot be thoroughly described. If Ca2+-dependent HCO3− secretion is determined to play a necessary role in cholestasis, then InsP3R deficiency may affect the efficacy of GPBAR1 to some extent.
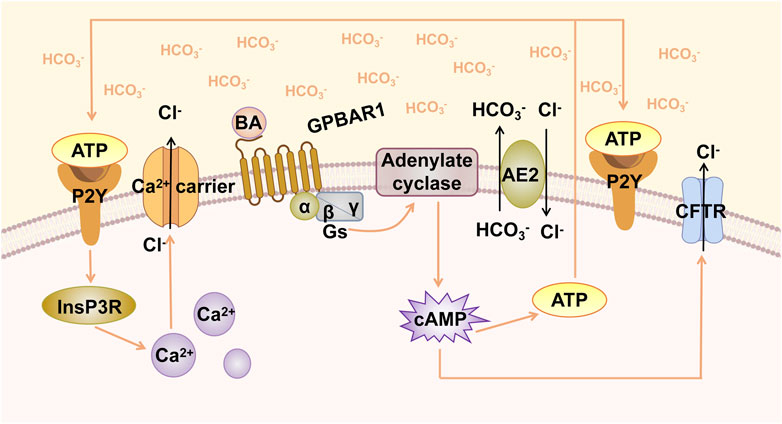
FIGURE 1. GPBAR1-induced biliary HCO3− umbrella. GPBAR1 is activated by BA, leading to the activation of stimulatory G-protein (Gs) and adenylate cyclase, resulting in increased intracellular cAMP levels. cAMP activates CFTR, triggering the secretion of Cl−. AE2 mediates the exchange of Cl−/HCO3− on the apical membrane and promotes the formation of HCO3− umbrella. Increased cAMP levels induce the release of ATP, and ATP binds to the P2Y receptor and activates Ca2+-dependent chloride channels.
There have been few studies on the role of GPBAR1 in the kidney. In the context of BA overload, inadequate elimination of BA in urine exacerbates liver injury after BDL. GPBAR1 may at least promote the excretion of BA in urine by controlling the multidrug resistance-associated protein 2 (MRP2) and MRP4 genes (Péan et al., 2013). In another study, researchers demonstrated that GPBAR1 can regulate the gene and protein expression of aquaporin-2 (AQP2) and intracellular transport through the cAMP-protein kinase A (PKA) pathway and affect the transmembrane transport of water in the kidney. Moreover, glycogen synthase kinase 3β (Gsk3β) may be the downstream target of the GPBAR1 signalling pathway (Li S. et al., 2018). GPBAR1 may have a similar water-regulating role in the biliary epithelium. In line with this hypothesis, GPBAR1 can regulate the distribution of BAs through the selective reabsorption of hydrophobic BAs by the biliary epithelium, which is a process known as cholehepatic shunting (Holter et al., 2020). Specifically, GPBAR1 can promote the insertion of apical sodium-dependent BA transporter (ASBT) into the parietal membrane through cAMP and enhance BA uptake by biliary epithelial cells (Keitel et al., 2009). Cholehepatic shunting is thought to limit the hydrophobicity of the BA pool through the reabsorption of secondary BA by biliary epithelial cells.
In addition, early reports also pointed out that BAs can cause the release of nitric oxide (NO) through the GPBAR1 receptor (Merlen et al., 2020a). In sinusoidal vascular endothelial cells exposed to high concentrations of BA, GPBAR1 can activate and regulate the production of NO through cAMP-dependent endothelial nitric oxide synthase (eNOS); this mechanism that can scavenge BA-induced ROS is also of importance in the protection of the liver parenchyma (Thomas et al., 2008).
Cholangiocytes are a group of epithelial cells and highly specialized cells which line the biliary epithelium; these cells were once considered to be a type of dormant cell but are now considered to be active and hormone-responsive cells (Jones et al., 2015; Banales et al., 2019). During the process of bile duct disease, cholangiocytes respond to exogenous and endogenous damage and directly participate in the progression of bile duct disease; thus, cholangiocytes are considered to be the targets of chronic liver disease in the context of bile duct disease (Guicciardi et al., 2020). Proliferation is a cholangiocyte response that maintains the integrity of the biliary tree during liver injury. Cholangiocyte proliferation could be observed in acute obstructive cholestasis and the early stages of chronic cholestatic liver diseases in humans (Alvaro et al., 2000; Alpini et al., 2002; Lazaridis et al., 2004). Promoting the proliferation of cholangiocytes can benefit the treatment of cholangiopathies, especially PSC and PBC.
GPBAR1 is mainly localized in the apical membrane of gallbladder epithelial cells and primary cilia (Keitel et al., 2009; Keitel and Häussinger, 2012). Several studies have shown that GPBAR1 can promote the proliferation of cholangiocytes, indicating that GPBAR1 can help reduce liver damage under cholestatic conditions in certain circumstances. The intracellular cAMP signalling pathway is responsible for the transmission of BA signals to mediate the cellular functional response. When GPBAR1 is activated, it leads to an increase in the level of intracellular cAMP, followed by an associated cell-specific response (Kawamata et al., 2003; Keitel and Häussinger, 2011). GPBAR1 is linked to cAMP and expressed in cholangiocytes, and cholangiocyte proliferation is closely associated with intracellular cAMP levels (Sand et al., 1992; Lesage et al., 1996; Alpini et al., 1997b). Therefore, GPBAR1 seems to be able to induce cell proliferation through the cAMP pathway (Figure 2). In early experiments, Alpini et al. showed that the effects of both up and downregulation of BAs on cholangiocyte proliferation and secretion are associated with the synthesis of cAMP in cholangiocytes (Alpini et al., 1999). In another study, the researchers mentioned that micromolar concentrations of BA could be used as signals for secretory and proliferative events in large but not small cholangiocytes, while small cholangiocytes may exhibit the phenotype of large cholangiocytes when they proliferate (Alpini et al., 1997a). The responses of large and small cholangiocytes are different in all kinds of bile duct injury experiments, but their responses to BAs are similar (Sato et al., 2018). These studies showed that cAMP is a key messenger for cholangiocyte proliferation. PKA and exchange proteins activated by cAMP (EPACs) may be two important downstream effectors of cAMP-induced cholangiocyte proliferation (Banales et al., 2009). PKA is one of the most well-studied serine/threonine protein kinases and has been reported to be involved in cAMP-mediated proliferation in multiple cell types (Hogarth et al., 2004; Masyuk et al., 2017; Meroni et al., 2019; Smith et al., 2019). The increase in cAMP may activate PKA expression, which in turn promotes cell proliferation. Additionally, two new PKA-independent cAMP effectors, EPAC1 and EPAC2, have also been described to have a variety of cAMP-mediated biological effects including cell proliferation. It should be noted that the activation of EPAC has a cell type-specific effect on proliferation, inducing growth inhibition in vascular smooth muscle cells and growth promotion in other cell types, including endothelial cells (Smith et al., 2019). Therefore, GPBAR1 may induce cholangiocyte proliferation through the cAMP/PKA pathway, while EPAC may participate in regulating proliferation through a signalling pathway parallel to or independent of PKA. Regarding the conditions for cell proliferation, early studies also emphasized the effect of apical BA transporter (ABAT) on the effects of BAs on cholangiocytes. Researchers suggested that BAs can change the function of cholangiocytes only in the presence of sodium in vitro (Alpini et al., 1999). When this sodium-dependent transporter takes up BAs from bile, it may initiate signals to modulate cholangiocyte proliferation and secretion. Recent research on sodium-dependent transporters in cholestasis models has focused on ASBT, and pharmacological ASBT inhibition could attenuate cholestatic liver and bile duct injury by reducing biliary BA concentrations in mice (Baghdasaryan et al., 2016). Whether the existence of such sodium-dependent transporters is conducive to the proliferation of cholangiocytes thus contributes to the treatment of cholestasis seems to be no longer the focus of modern research.
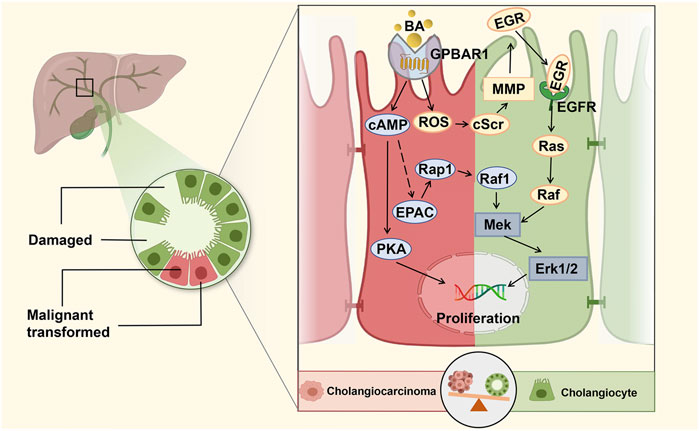
FIGURE 2. GPBAR1 promotes the proliferation of cholangiocyte. GPBAR1 promotes the proliferation of normal cholangiocytes and cholangiocarcinoma cells through a similar mechanism. GPBAR1 mainly depends on the cAMP pathway to play a proliferative role, and PKA and EPAC are two important downstream effectors of cAMP-induced proliferation. In addition, GPBAR1 can also induce proliferation through an increase in ROS and the activation of EGFR.
In a previous study, researchers found that ERK activation in mouse cholangiocytes was a downstream signal of BA-induced cholangiocyte proliferation (Francis et al., 2004). Similarly, in a high-quality study, Reich et al. demonstrated that cholangiocyte proliferation was significantly decreased in GPBAR1-KO mice compared with WT mice under the pathological condition of cholestasis. GPBAR1 induced cholangiocyte proliferation via increased reactive oxygen species (ROS) and the activation of epidermal growth factor receptor (EGFR) and ERK1/2. The specific proliferation pathway can be summarized as the GPBAR1-ROS-Src-MMP-EGFR-ERK-dependent signalling pathway, which indicates that GPBAR1 can trigger cell proliferation independent of adenylate cyclase activation (Reich et al., 2016). Enhancing the barrier function of the biliary tract is an important regulatory mechanism to protect liver cells from the toxic damage induced by BA in cholestatic liver disease. Merlen et al. focused on the relationship between GPBAR1 and tight junction proteins (TJPs), and the experimental results showed that liver injury caused by cholestasis in GPBAR1-KO mice was more serious than that in WT mice. The protective effect of the GPBAR1 agonist on cholestasis-induced liver injury was significantly weakened in JAM-A-KO mice, and GPBAR1 affected the expression and phosphorylation of the main TJP JAM-A to regulate biliary epithelial barrier function. PKCζ is a major kinase involved in JAM-A phosphorylation, which is at least partially activated in the course of JAM-A phosphorylation (Merlen et al., 2020b).
It is worth noting that different localizations of GPBAR1 in cholangiocytes may produce different biological effects. Masyuk et al. showed that GPBAR1 agonists caused opposing changes in cAMP and ERK levels in ciliated and nonciliated H69 cells. GPBAR1 agonists activated the proliferation of nonciliated cells but not ciliated cholangiocytes. This difference may be associated with the coupling of GPBAR1 with the Gαs protein in nonciliated cells and the Gαi protein in ciliated cells (Masyuk et al., 2013). In general, the subcellular localization of GPBAR1 directly affects the functional response of cholangiocytes to BA signals.
Owing to the nearly ubiquitous expression of GPBAR1 in human cells, GPBAR1 shows dichotomous functions in different diseases. As previously mentioned, GPBAR1 activation promotes cholangiocyte proliferation and effectively protects cholangiocytes from BA-induced toxicity in the context of cholestasis. However, GPBAR1 activation can change from beneficial to detrimental in the case of malignant transformed cholangiocytes and promote the risk of cholangiocarcinoma (CCA) proliferation. CCA is a malignant epithelial cell tumour originating from varying locations within the biliary tree and is characterized by the differentiation of cholangiocytes (Razumilava and Gores, 2014). The aetiology of CCA is diverse; here, we focus on the association between cholestasis and CCA. It was reported that CCA may be the consequence of long-term cholestasis and inflammation in the liver (Erice et al., 2018). Most risk factors for CCA can cause cholestasis or chronic inflammation (Labib et al., 2019). Once patients with cholestasis develop CCA at a later stage, the activation of the BA receptor GPBAR1 may exacerbate the situation.
Studies have shown that bile salts facilitate the development of CCA by inducing biliary proliferation, promoting liver inflammation, downregulating FXR and upregulating GPBAR1 (Deutschmann et al., 2018). The expression of GPBAR1 in CCA has been indicated to be increased, and GPBAR1 activation in human CCA cells promotes cell proliferation via a mechanism similar to that in mouse cholangiocytes. GPBAR1 was activated in a CCA cell line, induced the phosphorylation of EGFR and ERK1/2, and further promoted cell proliferation (Reich et al., 2016). At present, there are no clinical data showing that the expression of GPBAR1 is associated with the pathology of CCA patients. In view of this, Li et al. measured the expression of GPBAR1 in 20 pairs of extrahepatic cholangiocarcinoma (ECC) specimens and paratumoural tissues and demonstrated that GPBAR1 was highly expressed in CCA tissues. In addition, GPBAR1 promoted the proliferation, migration and apoptosis resistance of CCA cells. Furthermore, it was observed that GPBAR1 could bind to mortalin and regulate its expression in the CCA cell line; mortalin may be a downstream component of GPBAR1 that promotes CCA cell proliferation, and the interaction between GPBAR1 and mortalin may at least partially promote the occurrence of CCA (Li et al., 2020). Therefore, in the context of CCA, inhibiting GPBAR1 expression at least partially inhibits the proliferation and migration of cancer cells, indicating that GPBAR1 may be a potential therapeutic target for CCA treatment. It is important to note that this effect is the exact opposite of what GPBAR1 needs to do in cholestasis, and the late stage of cholestasis may be the key point for the functional transformation of GPBAR1.
The cause of cholestasis has been widely studied, but the mechanism of liver injury caused by BA is still unclear. BA at the submillimolar level is highly toxic and can directly damage hepatocytes, but even under pathological conditions, toxic BA rarely reaches this submillimolar level; a certain concentration of glycyldeoxycholic acid (GCDCA) can cause apoptosis in rat hepatocytes, but serum GCDCA cannot reach this concentration even in the case of complete biliary obstruction (Li M. et al., 2017). These results suggest that BA-induced liver injury may occur through alternative mechanisms. Recent studies have shown that excess BA is not directly cytotoxic to hepatocytes. During cholestasis, BA, as an inflammatory factor, activates the signalling pathway in hepatocytes and increases the expression of proinflammatory mediators, indicating that BA-mediated proinflammatory effects are the key to causing cholestatic liver injury (Allen et al., 2011; Cai et al., 2017).
The expression of chemokines by hepatocytes is the early initiation event of inflammatory pathogenesis during cholestasis. These specific cytokines can significantly enhance neutrophil chemotaxis and play a crucial role in triggering inflammation (Li M. et al., 2017). In early studies, hydrophilic BAs could not activate GPBAR1, nor could they regulate macrophage activity. Only hydrophobic BAs such as TLC, TC, GCDC, and TCDC have effects on macrophage activation. These BAs are known ligands of GPBAR1, suggesting that BAs may mediate lipopolysaccharide (LPS)-induced inflammation by activating GPBAR1 (Kawamata et al., 2003). In the GPBAR1-KO mouse model, additional direct evidence could be obtained regarding whether GPBAR1 mediates liver inflammation. In the LPS-induced inflammation model, GPBAR1-KO mice showed more severe liver injury and inflammation than WT mice (Wang et al., 2011). Activation of GPBAR1 inhibited the expression of cytokines in mouse macrophages and rat Kupffer cells induced by LPS (Keitel et al., 2008; Wang et al., 2011). GPBAR1 plays the same role in human macrophages (Haselow et al., 2013). GPBAR1 gene KO increased proinflammatory mediators and neutrophil transport in BDL-induced cholestatic mice (Rao et al., 2020). These related studies have shown the anti-inflammatory and hepatoprotective effects mediated by GPBAR1. Considering that GPBAR1 is hardly expressed in hepatocytes but is enriched in macrophages and Kupffer cells, GPBAR1-dependent anti-inflammatory effects during cholestasis may occur downstream of BA-mediated hepatocyte inflammation (Merlen et al., 2020a).
Regarding the relevant mechanism by which GPBAR1 mediates anti-inflammatory in cholestasis, in the macrophages of GPBAR1-KO mice, the RNA levels of various proinflammatory genes, such as inducible nitric oxide synthase (iNOS), interferon-inducible protein and IL-1, which are targeted by NF-κB, were higher than those of WT mouse macrophages, suggesting that the anti-inflammatory effect of GPBAR1 is mediated by inhibiting NF-κB (Calmus and Poupon, 2014) (Figure 3). Wang et al. indicated that GPBAR1 inhibited the NF-κB pathway by mediating the interaction between Iκbα and β-arrestin2 and that GPBAR1 was a negative regulator of NF-κB-mediated liver inflammation (Wang et al., 2011). However, in another study, the role of β-arrestin2 was not confirmed. Pols et al. used β-arrestin2-KO cells to show that the β-arrestin2 molecule cannot be coupled with the anti-inflammatory effect induced by GPBAR1. In macrophages treated with GPBAR1 agonists, the inhibition of cytokine production depends on activation of the cAMP-NF-κB signalling pathway (Pols et al., 2011). cAMP inhibits NF-κB activity, thus effectively inhibiting liver inflammation. Keitel et al. demonstrated that GPBAR1 was located in the plasma membrane of Kupffer cells. BA-induced activation increases the production of cAMP and inhibits the expression of proinflammatory cytokines including IL-1α, IL-1β, IL-6 and TNF-α, induced by LPS (Keitel et al., 2008). The activation of GPBAR1 in macrophages reduces the production of proinflammatory cytokines and maintains the expression of anti-inflammatory cytokines, thereby promoting the development of an anti-inflammatory macrophage phenotype (Li M. et al., 2017). The key anti-inflammatory effects of cAMP are specifically mediated by cAMP-dependent PKA (Wall et al., 2009). In primary human macrophages, inhibition of PKA with a specific inhibitor resulted in an almost complete reduction in TLC-mediated inhibition of LPS-induced cytokine expression. GPBAR1 is at least partially involved in regulating the BA-mediated response to LPS-induced inflammation. This series of processes involves BAs activating GPBAR1, increasing the production of cAMP and the subsequent activation of PKA (Haselow et al., 2013). On the other hand, the PKA-dependent enhancement of LPS-induced anti-inflammatory cytokine (IL-10) expression depends on the presence of CREB (Wall et al., 2009). In LPS-induced inflammation, rescuing inflammatory cytokine expression through the inhibition of PKA is associated with the suppression of BA-induced CREB activation. CREB activation has been shown to inhibit the transcriptional activity of NF-κB by competing with NF-κB for the transcriptional coactivator CBP (Haselow et al., 2013). The upregulation of IL-10 by cAMP may be an important mechanism by which cAMP inhibits the activation of phagocytes. However, cAMP can also inhibit the production of TNF-α and IL-12 by IL-10-deficient dendritic cells (DCs), so there may be another important cAMP-mediated mechanism that inhibits the expression of LPS-induced inflammatory factors (Koga et al., 2009). The transcription factor c-Fos accumulates massively under the stimulation of cAMP and LPS; when the expression of c-Fos is enhanced, c-Fos physically interacts with the p65 protein and inhibits NF-κB-mediated gene expression (Koga et al., 2009; Haselow et al., 2013). Hence, BAs can inhibit the expression of LPS-induced proinflammatory factors through two different cAMP-driven mechanisms. Furthermore, after treatment with BA, the LPS-induced nuclear translocation of NF-κB was delayed. The inhibitory effect of this nuclear translocation is also PKA-dependent, showing a mutual connection with the first two mechanisms (Haselow et al., 2013). In addition, Ichikawa et al. indicated that specific GPBAR1 agonists can induce DCs to exhibit an IL-12 hypoproducing phenotype, resulting in DC differentiation into cells with an anti-inflammatory phenotype. GPBAR1 is downregulated rapidly during the differentiation of monocytes into DCs, and 8-Br-cAMP, which is downstream of the signal, responds to this downregulation, which stimulates and promotes the differentiation of DCs into the IL-12 hypoproducing phenotype (Ichikawa et al., 2012). This process is cAMP-dependent.
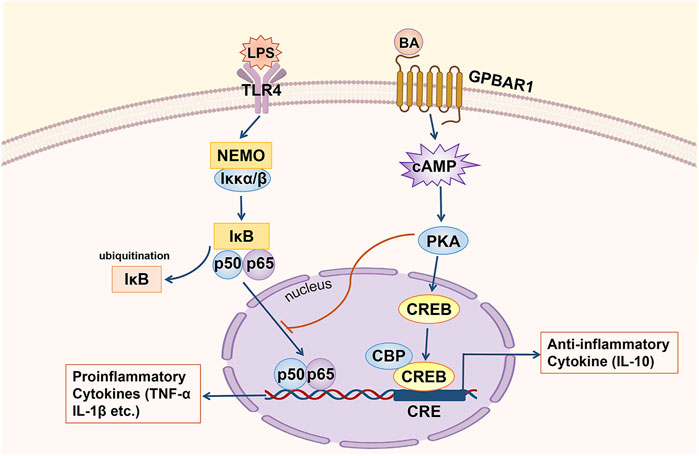
FIGURE 3. GPBAR1 relieves hepatic inflammation. GPBAR1 through the cAMP pathway inhibits the transcription of proinflammatory cytokines mediated by NF-κB (IL-1α, IL-1β and TNF-α, etc.) and maintains the expression of an anti-inflammatory cytokine (IL-10).
Inflammasomes assist host defence against pathogens and play key roles in inflammation (Davis et al., 2011). Previous studies have extensively investigated the NLRP3 inflammasome. The association between GPBAR1 and NLRP3 has also been demonstrated, but most studies have mainly confirmed the effect of GPBAR1 on the NLRP3 inflammasome in the context of neuroinflammation, colitis, pancreatitis and nonalcoholic steatohepatitis (NASH) (Li B. et al., 2018; Chen et al., 2019; Shi et al., 2020; Liang et al., 2021). Few studies have explored the combined effects of GPBAR1 and inflammasomes in cholestasis. Researchers have shown that BA can inhibit the activation of NLRP3 through the GPBAR1-cAMP-PKA axis. GPBAR1-induced PKA activation causes NLRP3 ubiquitination and phosphorylation, while NLRP3 phosphorylation plays a crucial role in the inhibition of NLRP3 inflammasome activation (Guo et al., 2016b). However, scholars have shown that the activation of GPBAR1 activates the NLRP3 inflammasome, causing inflammation in the liver during cholestasis. This effect is contrary to the results of previously mentioned studies, reflecting the controversial role of GPBAR1 activation in inflammation (Gong et al., 2016).
β-catenin signalling has been shown to be necessary to control innate and adaptive immunity during inflammation (Ke et al., 2013). Rao et al. investigated the relationship between GPBAR1 and β-catenin in cholestatic liver injury. The results showed that by interacting with Gsk3β, GPBAR1 activated the β-catenin signalling pathway to control local liver inflammation in immune-mediated cholestatic liver injury. GPBAR1-mediated β-catenin signalling also activated PI3K/AKT and inhibited the TLR4/NF-κB pathway, thus attenuating the inflammatory response, which suggests a new mechanism by which GPBAR1 regulates cholestatic liver disease in a model of cholestatic liver injury induced by BDL (Rao et al., 2020).
After the body ingests food, microbiota converts polysaccharides to short-chain fatty acids and generates amino acids and metabolites, which can be used by the host to harvest energy. Metabolites send signals through their cognate receptors to regulate the metabolism of the host; thus, the gut microbiota is considered a metabolic organ (Wahlström et al., 2016). There is a close relationship between the liver and the gut, which is known as the gut-liver axis. Seventy percent of the blood supply of the liver flows directly from the veins of the intestine (Li Y. et al., 2017). BAs, which are synthesized from cholesterol in the liver and further metabolized by the gut microbiota, are a class of important metabolites that are produced microbially and some of the most abundant metabolites in the intestine (Sorrentino et al., 2020). Evidence has shown that there is an active interaction between the gut microbiota and BAs that plays an important role in the pathogenesis of cholestatic liver diseases such as PBC and PSC (de Aguiar Vallim et al., 2013).
The diversity and richness of the gut microbiota in patients with PSC and PBC were significantly lower than those in healthy individuals (Rossen et al., 2015; Lv et al., 2016). Another study showed that the microbial diversity in PBC patients was significantly decreased, and this effect was reversed after UDCA treatment (Tang et al., 2018). Although the results of current studies on the gut microbiota in PBC and PSC patients are not consistent in terms of changes in species or genera, it is undeniable that the composition of the gut microbiota of patients with cholestasis has changed. On the other hand, studies have shown that the lack of intestinal microbiota can exacerbate hepatobiliary disease in PSC mouse models, indicating the potential protective effect of commensal microbiota against biliary tract injury (Tabibian et al., 2016). Since GPBAR1 is also distributed in the intestine and GPBAR1 regulates cholestasis, is there a close relationship between GPBAR1 and the intestinal microbiota? Does the intestinal microbiota act as another channel through which GPBAR1 regulates cholestasis?
The previous section summarized the role of GPBAR1 in regulating BA homeostasis, cell proliferation and inflammation. In addition, GPBAR1 is also involved in the regulation of metabolism in the body, which seems to connect GPBAR1 and the gut microbiota. Due to the wide diversity of GPCRs and associated ligands, these receptors are almost certainly the basis of the observed ability of the enteroendocrine axis to respond to a wide range of ingested food and luminal components (Gribble and Reimann, 2019). Katsuma et al. showed that BAs promote the secretion of glucagon-like peptide-1 (GLP-1) through GPBAR1 in the enteroendocrine cell line STC-1 (Katsuma et al., 2005). GLP-1 is a pleiotropic hormone with countless metabolic functions (Müller et al., 2019). In terms of intestinal metabolism, studies have shown that diet-induced intestinal microbial dysbiosis in mice is associated with an impaired GLP-1 metabolic response (Grasset et al., 2017). GLP-1 has shown beneficial effects in the treatment of type 2 diabetes and obesity (Gribble and Reimann, 2019). An important study demonstrated that FXR-mediated modification of the intestinal microbiota led to GPBAR1-induced remodelling of the LCA-enriched BA pool and improved glucose tolerance through intestinal GLP-1 secretion. The GPBAR1-GLP-1 axis plays a key role in mediating intestinal BA receptor signal transduction and regulating liver metabolism and homeostasis (Pathak et al., 2018).
Studies have shown that accumulation of BAs in the liver does not contribute to liver damage in the absence of microbiome in vivo, suggesting that gut microbiome is critical to the occurrence of cholestasis (Isaacs-Ten et al., 2020). In fact, there is increasing evidence supporting a close relationship between cholestatic liver injury and gut microbiota. However, the role of gut microbiota in the pathogenesis of cholestasis remains unclear. After BDL in mice, the abundance of an anti-inflammatory gut microbiota (F. prausnitzzi) was significantly reduced, which may contribute to increased intestinal permeability and loss of intestinal barrier function (Cabrera-Rubio et al., 2019). Isaacs-Ten et al. demonstrated that macrophages contribute to promoting intestinal permeability during cholestasis and change the composition of intestinal microorganisms through activation of inflammasome, aggravating cholestatic liver injury (Isaacs-Ten et al., 2020). And in humans, PSC and inflammatory bowel disease (IBD) co-exist frequently, showing significant correlation. All these suggest that the regulation of intestinal inflammation may be another important way to alleviate cholestatic liver injury.
The anti-inflammatory effect of GPBAR1 mediated in macrophages has been discussed above. Considering the high expression of GPBAR1 in the intestine, GPBAR1 seems to be able to improve cholestasis by regulating intestinal inflammation. In the gastrointestinal tract, the anti-inflammatory effect regulated by macrophages was mainly demonstrated by the activation of GPBAR1, and the activation of GPBAR1 significantly inhibited the activation of NF-κB induced by LPS in WT mice. However, it is still important to note that the role of GPBAR1 in gastrointestinal carcinogenesis has not been determined. There are two types of tumor-associated macrophages: M1 macrophages and M2 macrophages. M1 phenotype produces proinflammatory cytokines (IL-1β, IL-6, IL-12 etc.), while M2 has immunosuppressive effect. Macrophages with mixed phenotype of M1/M2 were induced by GPBAR1 activated by bile acid in macrophages. According to the polarization of macrophages, activation of GPBAR1 in colorectal cancer promotes proinflammatory response in M1 macrophages and anti-inflammatory response in M2 macrophages. The activation of GPBAR1 seems to control the balance of proinflammatory and anti-inflammatory factors in the intestine, and the ratio of IL10:IL12 is an important indicator of intestinal mucosal inflammation (Jia et al., 2018).
Cholesterol in the liver synthesizes two main primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA). And intestinal microbiota converts primary BAs to secondary BAs such as deoxycholic acid (DCA) and lithocholic acid (LCA). Different kinds of BAs have different abilities to activate GPBAR1. These BAs that can activate GPBAR1 may be called the most effective natural agonist of GPBAR1. DCA and LCA are physiological ligands of GPBAR1 (Maruyama et al., 2002). Studies have indicated that the order of activation of GPBAR1 by BA is LCA>DCA>CDCA>CA in vitro. In addition, in Chinese hamster ovary cells transfected with human GPBAR1, the order of activation of GPBAR1 by BA is TLCA>LCA>GLCA>TDCA>DCA>GDCA>TCDCA>CDCA>GCDCA>TCA>CA> GCA (Jia et al., 2018). UDCA is a safe and available BA and the mainstream drug for cholestatic liver disease. Studies have shown that UDCA can effectively activate GPBAR1 and play a beneficial role in a NASH mouse model. The EC50 of UDCA is 14 μM, which is 4–7 times higher than the TLCA EC50 (Carino et al., 2019). TUDCA is a conjugate derivative of UDCA that is commonly used in the treatment of gallstones and chronic cholestatic liver disease (Elia et al., 2016). It has also been shown to activate GPBAR1 and induce an increase in the level of cAMP in microglia, thereby exerting an anti-inflammatory effect (Yanguas-Casás et al., 2017).
Semisynthetic BA derivatives can be divided into two kinds of body modifications and side chain modifications (Figure 4; Table 1). INT-777 (6R-ethyl-23(S)-methylcholic acid, S-EMCA, MW 450.65) is a specific GPBAR1 agonist that is widely used in current research and was first described in 2009 (Pellicciari et al., 2009). Studies have shown that GPBAR1 activation by INT-777 can reduce neuroinflammation, improve cognitive impairment, improve glucose tolerance, and increase insulin synthesis (Kumar et al., 2016; Jin et al., 2021). In addition, INT-777 induces the release of GLP-1 from enteroendocrine L-cells, which can improve liver and pancreatic function in mice, stimulate bile flow, and inhibit macrophage inflammation (Thomas et al., 2009; Pols et al., 2011). In addition to INT-777, which is the most commonly used derivative, other semisynthetic derivatives of BA are also being studied. For example, 7ξ-Me-LCA, 7α-F-LCA and CDC-Sul, which were screened from natural BAs, semisynthetic BA derivatives and steroid hormones, are considered to be highly effective and selective GPBAR1 agonists (Sato et al., 2008). BAR501 was the first UDCA derivative with potent and selective GPBAR1 activity (Festa et al., 2014; De Marino et al., 2019). As a small molecular agonist of GPBAR1, BAR501 has been shown to regulate the M1/M2 phenotype of intestinal macrophages and effectively reduce the expression of inflammatory factors (Biagioli et al., 2017).
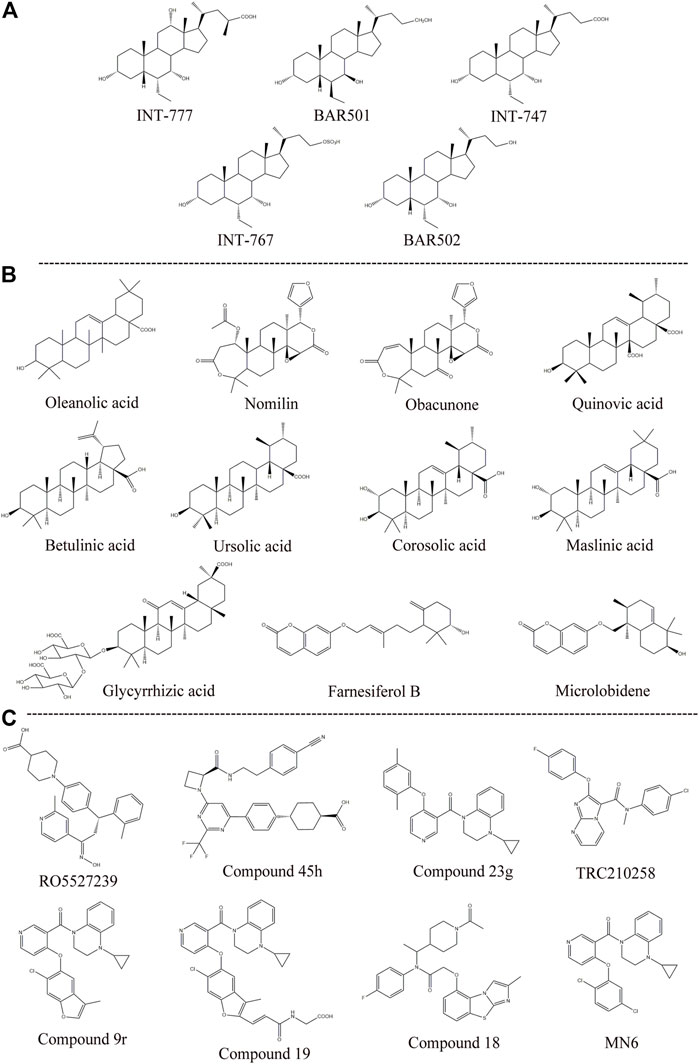
FIGURE 4. Structures of some typical GPBAR1 agonists. (A) Semisynthetic BA derivatives; (B) Natural ligand; (C) Small molecule.
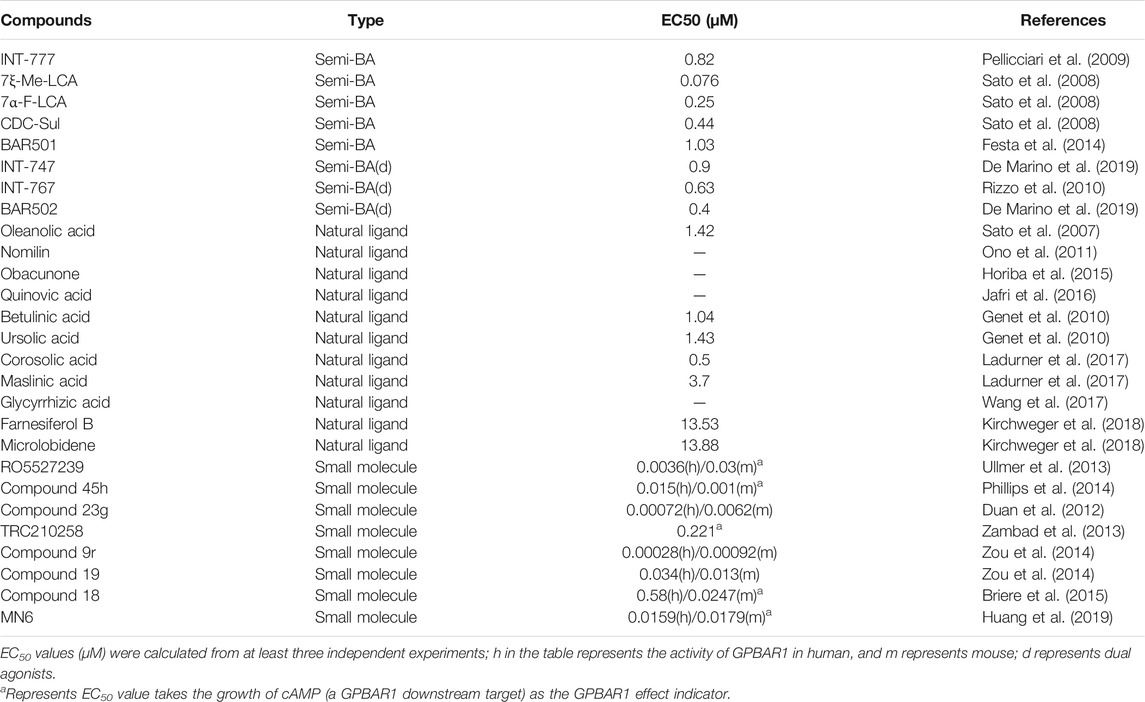
TABLE 1. GPBAR1 agonists and typical GPBAR1/FXR dual agonists.
Apart from specific GPBAR1 agonists, there have been some reports on FXR/GPBAR1 dual agonists, which are also mainly semisynthetic derivatives of BA. We provide a brief summary of several representative dual agonists. The compound INT747, which was first developed in 2002, is a dual ligand of FXR and GPBAR1, but it caused a more intense itch response in clinical trials, so the benefits of this synergistic approach were not realized (Pellicciari et al., 2002). In mice with chronic cholangiopathy, INT-767 can improve liver injury by reducing biliary BA output and promoting HCO3− secretion, showing better hepatoprotection than INT-777 (Baghdasaryan et al., 2011). INT-767 activation of FXR induced GPBAR1 gene expression and increased cAMP and Ca2+ levels, which then stimulated the secretion of GLP-1, thus improving liver glycolipid metabolism (Pathak et al., 2017). INT-767 demonstrated promising drug-like properties in both liver diseases and metabolic diseases and is considered to be an attractive drug candidate (Rizzo et al., 2010). In a recent study, INT767 was reported to inhibit hepatitis B virus (HBV) infection in vivo and in vitro, making it a potential anti-HBV candidate (Ito et al., 2021). BAR502 has been shown to reverse steatohepatitis and liver cirrhosis and protect against liver damage induced by a high-fat diet by promoting adipose tissue browning in NASH (Carino et al., 2017). Furthermore, in mice with cholestasis, BAR502 improved survival and reshaped the BA pool without inducing itching (Cipriani et al., 2015). Compared with GPBAR1 agonists alone, the latter two not only showed high efficacy and selectivity in preclinical trials but also showed great application value by significantly avoiding the risk of pruritus in cholestatic models.
The existing GPBAR1 non-BA agonists are mainly developed by ligands, including natural and synthetic ligands (Gertzen et al., 2015; Figure 4; Table 1). Oleanolic acid is one of the rare natural ligands of GPBAR1, showing robust antidiabetic effects on high-fat-fed mice, matching the potential effect of GPBAR1 to improve metabolic disorders (Sato et al., 2007). Ono et al. showed that nomilin (a naturally occurring limonoid) can activate GPBAR1, which is conducive to metabolic regulation (Ono et al., 2011). Similar natural GPBAR1 agonists also include obacunone (Horiba et al., 2015). Quinovic acid is extracted from the national medicine Fagonia cretica, and Jafri et al. suggested that all quinovic acid compounds activate the GPBAR1 receptor and further stimulate the secretion of GLP-1 (Jafri et al., 2016). The pentacyclic triterpenoid betulinic acid and ursolic acid are considered selective GPBAR1 agonists (Lo et al., 2016, 2017). Furthermore, Ladurner et al. studied the extracts of 19 kinds of plants, and in addition to oleanolic acid and ursolic acid, their results indicated that corosolic acid and maslinic acid also have activating effects on GPBAR1 (Ladurner et al., 2017). Wang et al. suggested that glycyrrhizic acid extracted from licorice root could activate GPBAR1 and promote the secretion of GLP-1 (Wang et al., 2017). Kirchweger et al. found that two sesquiterpene coumarins, farnesiferol B and microlobidene, can effectively activate GPBAR1, and their activity was equivalent to that of the endogenous ligand lycholic acid, which was considered a potent GPBAR1 agonist (Kirchweger et al., 2018).
Moreover, there have been many reports about small molecule synthesis of GPBAR1 agonists in recent years. Ullmer et al. synthesized an orally bioavailable and selective GPBAR1 agonist, RO5527239, which can effectively improve glucose tolerance and stimulate strong and sustained secretion of GLP-1 (Ullmer et al., 2013). Phillips et al. carried out extensive lead optimization from a high-throughput screen hit and ultimately determined that compound 45 h (trifluoromethyl(pyrimidin-2-yl)azetidine-2-carboxamides) was a potent and selective GPBAR1 agonist that plays an effective role in clinical metabolic diseases (Phillips et al., 2014). Similar small molecule agonists also include compound 23 g, TRC210258, compound 9r, compound 19, and compound 18 (Duan et al., 2012; Zambad et al., 2013; Zou et al., 2014; Briere et al., 2015). In recent years, again based on 4-phenoxynicotinamide, researchers synthesized a derivative of 4-phenoxynicotinamide called MN6, which is a potent GPBAR1 agonist. Researchers have suggested that MN6 activates GPBAR1 and further improves insulin sensitivity in skeletal muscles through the cAMP/PKA pathway (Huang et al., 2019). Most of these agonists are based on research achievements in the fields of metabolic disease. How these compounds affect cholestasis liver disease may not be directly known, but current agonist development is still limited to the GPBAR1 exposure range, and more experiments are needed to explore nonsystemic GPBAR1 agonists, which seems to suggest that existing agonists do not have completely specific targets. Of course, in a practical sense, this is the limitation of the status quo of the development of GPBAR1 agonists.
BA is an atypical steroid synthesized from cholesterol. Similar to the developmental trajectory of most biochemicals, BA has not been sufficiently recognized in the medical community since the elucidation of the true chemical structure of BA in the 1930s (Hofmann and Hagey, 2014). With the separation and synthesis of DCA and UDCA and the verification of their therapeutic effects on diseases, BA has been gradually used in the treatment of hepatobiliary diseases for nearly half a century. However, with the development of surgical technology, the lack of activity and side effects of UDCA and CDCA greatly reduce the clinical correlation of BA. The discovery of BA-activated receptors seems to have contributed to a BA renaissance (Fiorucci and Distrutti, 2019). FXR and GPBAR1 are two featured BA receptors. To date, there have been many relevant studies on the former and few on the latter. We summarized the roles and pathways of GPBAR1 in cholestatic liver disease.
The BA pool refers to the total amount of BAs circulating in the enterohepatic circulation, including BAs in the liver, gallbladder and intestines, which is a dynamic and complex composition (Rodrigues et al., 2014). Hydrophobic BAs such as CDCA, LCA, DCA and their conjugates have been proved to have high cytotoxicity. The accumulation and distribution of these hydrophobic BAs are closely related to the progress of cholestatic liver disease. Current research shows that GPBAR1 can regulate the size and hydrophobicity of the BA pool. While cholangiocytes are faced with a large amount of hydrophobic BA-induced injury, GPBAR1 can regulate water homeostasis in biliary epithelial cells and restrict the hydrophobicity of the BA pool through cholehepatic shunting. In addition, GPBAR1 promotes the formation of HCO3− umbrella and enhances the ability of cholangiocytes to resist hydrophobic BAs. Changes in intercholangiocyte tight junctions (TJs) can lead to leakage of the ducts and bile flow back, causing liver damage. GPBAR1 has also been shown to enhance biliary barrier function by affecting the permeability of TJs. GPBAR1 triggers cell proliferation, which is conducive to maintaining the integrity of the biliary tree under cholestatic liver injury. However, due to the wide expression of GPBAR1 in various cells, GPBAR1 also has potential hazards and can promote tumour cell proliferation, and effective inhibition of GPBAR1 may inhibit tumour cell proliferation and metastasis; however, in cholestatic liver disease, because patients with cholestasis can develop CCA at a later stage, the characteristics of GPBAR1 become intriguing. Moreover, existing research suggests that GPBAR1 can inhibit the progression of cholestasis by inhibiting liver inflammation. The liver is rich in a variety of innate immune cells, including natural killer cells, natural killer T cells, neutrophils, macrophages and DCs (Heymann and Tacke, 2016). The latter three cell types have been confirmed to be involved in cholestatic liver injury and interact with GPBAR1 (Li M. et al., 2017). The NF-κB pathway has long been regarded as a typical proinflammatory signalling pathway that can regulate inflammation through a variety of mechanisms such as the production of proinflammatory factors and leukocyte recruitment, and has been considered the Holy Grail of new anti-inflammatory drugs (Lawrence, 2009). Perhaps for this reason, most studies on the anti-inflammatory pathway of the newly discovered BA membrane receptor GPBAR1 have focused on NF-κB. Inevitably, although the general trend in the findings suggests that GPBAR1 activation can effectively inhibit the development of inflammation, it has also been shown that the activation of GPBAR1 induces the proinflammatory effect of inflammasomes. As the most fully characterized inflammasome thus far, the role of the NLRP3 inflammasome in cholestatic liver disease may require further study. Again, with a flurry of research on the gut microbiome, the relationship between the gut microbiome and cholestatic liver disease has been preliminarily established. GPBAR1 can regulate intestinal metabolism and intestinal inflammation, but the mechanism between GPBAR1 and the gut microbiome in cholestatic liver disease remains to be further validated.
While verifying the function of GPBAR1 is critical, the study of effective GPBAR1 agonists is equally important. In studying GPBAR1 agonists, semisynthetic BA derivatives often manifest stronger GPBAR1 agonist functions than natural BAs, which have been widely used in modern experiments. In addition, extracting natural agonists from plants is also an interesting project. The development of botanical medicine has a long history spanning thousands of years, and through integration with contemporary biotechnology, botanical medicine has been constantly breaking through with pharmacological innovation and has had considerable social impact. Compared with the development of natural GPBAR1 agonists, botanical drugs that act directly on diseases may attract more attention, as traditional Chinese herbs both monotherapies and formulations, have shown favourable efficacy in mouse models of cholestasis (Chen et al., 2015; Ma et al., 2015; Zhao et al., 2015; Yan et al., 2017). Whether Chinese herbal medicine also plays a role in the treatment of cholestasis partly through the activation of GPBAR1 may be an interesting line of inquiry.
Pruritus is one of the main symptoms of cholestasis, but its pathogenesis remains elusive. Some studies have suggested that BA-induced pruritus in cholestasis is associated with GPBAR1 receptor activation, suggesting a potential side effect of GPBAR1 in cholestasis treatment (Lieu et al., 2014; Hussain et al., 2019). But there is evidence that BAs, in addition to activating GPBAR1, require co-activation of multiple receptors and mediators, including TRPA1 channels, to cause pruritus (Lieu et al., 2014). In addition, there have also been studies suggesting that BA-induced pruritus may be linked to activation of FXR. Obeticholic acid is considered the most advanced agonist of FXR and has been approved as a second-line treatment for PBC in 2016. However, it often causes severe pruritus in clinical patients (Fiorucci et al., 2020). Therefore, the current research on the cause of pruritus caused by BA is still controversial in many aspects. We may not be able to roughly define the complex relationship between GPBAR1 and pruritus. It is gratifying that the combination of BA agonists with other drugs and the use of BA receptor dual agonists seems to significantly reduce the pruritus in cholestasis. In addition to effectively controlling the pruritus induced by BA, some GPBAR1/FXR dual agonists limits the nonselective proliferation of GPBAR1 since they limits the overexpression of GPBAR1. Both FXR and GPBAR1 are BA-activated receptors that have attracted wide attention. Activation of these receptors leads to different pathway regulation, double activation combines the functions of individual activation, and adaptive generation does not play an additional role in individual activation. These dual agonists tend to show better therapeutic efficacy than single agonists and induce fewer side effects (Wang et al., 2018). Therefore, dual targeting of the BA membrane receptor GPBAR1 and nuclear receptor FXR may be a promising strategy for cholestatic liver disease.
In conclusion, GPBAR1 can protect the liver from the effects of BA overload and effectively control liver inflammation. Then, GPBAR1 can stimulate cell regeneration to maintain the integrity of cholangiocytes and regulate the hydrophobicity of the BA pool. Additionally, GPBAR1 is involved in regulating gut metabolism and inflammation. GPBAR1 appears to form a signalling network between the liver, gallbladder and gut to alleviate cholestatic liver injury.
FZ is the major contributor to this manuscript. FZ wrote the first version of the manuscript, and XX, HW, XD, JW finalized the manuscript. FZ, YL, YJ, and XX downloaded the reference and processed the graph and the table in the manuscript. YJ and WZ extracted the information from the literature. XM and YZ (corresponding author) conceived and coordinated the study, and critically evaluated the data. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (81874365, and 81703725), Sichuan Science and Technology Program (2019YJ0492), Beijing Medical and Health Foundation (YWJKJJHKYJJ-B20645FN), and Chengdu University of TCM Found Grant (QNXZ2018025).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the reviewers and the authors of all references.
Allen, K., Jaeschke, H., and Copple, B. L. (2011). Bile Acids Induce Inflammatory Genes in Hepatocytes: a Novel Mechanism of Inflammation during Obstructive Cholestasis. Am. J. Pathol. 178, 175–186. doi:10.1016/j.ajpath.2010.11.026
Alpini, G., Glaser, S., Robertson, W., Phinizy, J. L., Rodgers, R. E., and Caligiuri, A. (1997a). Bile Acids Stimulate Proliferative and Secretory Events in Large but Not Small Cholangiocytes. Am. J. Physiol. 273, G518–G529. doi:10.1152/ajpgi.1997.273.2.G518
Alpini, G., Glaser, S. S., Ueno, Y., Rodgers, R., Phinizy, J. L., Francis, H., et al. (1999). Bile Acid Feeding Induces Cholangiocyte Proliferation and Secretion: Evidence for Bile Acid-Regulated Ductal Secretion. Gastroenterology 116, 179–186. doi:10.1016/s0016-5085(99)70242-8
Alpini, G., McGill, J. M., and Larusso, N. F. (2002). The Pathobiology of Biliary Epithelia. Hepatology 35, 1256–1268. doi:10.1053/jhep.2002.33541
Alpini, G., Ulrich, C., Roberts, S., Phillips, J. O., Ueno, Y., Podila, P. V., et al. (1997b). Molecular and Functional Heterogeneity of Cholangiocytes from Rat Liver after Bile Duct Ligation. Am. J. Physiol. 272, G289–G297. doi:10.1152/ajpgi.1997.272.2.G289
Alvaro, D., Gigliozzi, A., and Attili, A. F. (2000). Regulation and Deregulation of Cholangiocyte Proliferation. J. Hepatol. 33, 333–340. doi:10.1016/s0168-8278(00)80377-3
Baghdasaryan, A., Claudel, T., Gumhold, J., Silbert, D., Adorini, L., Roda, A., et al. (2011). Dual Farnesoid X receptor/TGR5 Agonist INT-767 Reduces Liver Injury in the Mdr2-/- (Abcb4-/-) Mouse Cholangiopathy Model by Promoting Biliary HCO−₃ Output. Hepatology 54, 1303–1312. doi:10.1002/hep.24537
Baghdasaryan, A., Fuchs, C. D., Österreicher, C. H., Lemberger, U. J., Halilbasic, E., Påhlman, I., et al. (2016). Inhibition of Intestinal Bile Acid Absorption Improves Cholestatic Liver and Bile Duct Injury in a Mouse Model of Sclerosing Cholangitis. J. Hepatol. 64, 674–681. doi:10.1016/j.jhep.2015.10.024
Banales, J. M., Huebert, R. C., Karlsen, T., Strazzabosco, M., LaRusso, N. F., and Gores, G. J. (2019). Cholangiocyte Pathobiology. Nat. Rev. Gastroenterol. Hepatol. 16, 269–281. doi:10.1038/s41575-019-0125-y
Banales, J. M., Masyuk, T. V., Gradilone, S. A., Masyuk, A. I., Medina, J. F., and LaRusso, N. F. (2009). The cAMP Effectors Epac and Protein Kinase a (PKA) Are Involved in the Hepatic Cystogenesis of an Animal Model of Autosomal Recessive Polycystic Kidney Disease (ARPKD). Hepatology 49, 160–174. doi:10.1002/hep.22636
Beuers, U., Hohenester, S., de Buy Wenniger, L. J. M., Kremer, A. E., Jansen, P. L. M., and Elferink, R. P. J. O. (2010). The Biliary HCO(3)(-) Umbrella: a Unifying Hypothesis on Pathogenetic and Therapeutic Aspects of Fibrosing Cholangiopathies. Hepatology 52, 1489–1496. doi:10.1002/hep.23810
Biagioli, M., Carino, A., Cipriani, S., Francisci, D., Marchianò, S., Scarpelli, P., et al. (2017). The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 199, 718–733. doi:10.4049/jimmunol.1700183
Briere, D. A., Ruan, X., Cheng, C. C., Siesky, A. M., Fitch, T. E., Dominguez, C., et al. (2015). Novel Small Molecule Agonist of TGR5 Possesses Anti-diabetic Effects but Causes Gallbladder Filling in Mice. PLoS One 10, e0136873. doi:10.1371/journal.pone.0136873
Cabrera-Rubio, R., Patterson, A. M., Cotter, P. D., and Beraza, N. (2019). Cholestasis Induced by Bile Duct Ligation Promotes Changes in the Intestinal Microbiome in Mice. Sci. Rep. 9, 12324. doi:10.1038/s41598-019-48784-z
Cai, S.-Y., Ouyang, X., Chen, Y., Soroka, C. J., Wang, J., Mennone, A., et al. (2017). Bile Acids Initiate Cholestatic Liver Injury by Triggering a Hepatocyte-specific Inflammatory Response. JCI insight 2, e90780. doi:10.1172/jci.insight.90780
Calmus, Y., and Poupon, R. (2014). Shaping Macrophages Function and Innate Immunity by Bile Acids: Mechanisms and Implication in Cholestatic Liver Diseases. Clin. Res. Hepatol. Gastroenterol. 38, 550–556. doi:10.1016/j.clinre.2014.07.007
Carino, A., Biagioli, M., Marchianò, S., Fiorucci, C., Zampella, A., Monti, M. C., et al. (2019). Ursodeoxycholic Acid Is a GPBAR1 Agonist and Resets Liver/intestinal FXR Signaling in a Model of Diet-Induced Dysbiosis and NASH. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864, 1422–1437. doi:10.1016/j.bbalip.2019.07.006
Carino, A., Cipriani, S., Marchianò, S., Biagioli, M., Santorelli, C., Donini, A., et al. (2017). BAR502, a Dual FXR and GPBAR1 Agonist, Promotes browning of white Adipose Tissue and Reverses Liver Steatosis and Fibrosis. Sci. Rep. 7, 42801. doi:10.1038/srep42801
Chen, H.-L., Wu, S.-H., Hsu, S.-H., Liou, B.-Y., Chen, H.-L., and Chang, M.-H. (2018). Jaundice Revisited: Recent Advances in the Diagnosis and Treatment of Inherited Cholestatic Liver Diseases. J. Biomed. Sci. 25, 75. doi:10.1186/s12929-018-0475-8
Chen, Y., Le, T. H., Du, Q., Zhao, Z., Liu, Y., Zou, J., et al. (2019). Genistein Protects against DSS-Induced Colitis by Inhibiting NLRP3 Inflammasome via TGR5-cAMP Signaling. Int. Immunopharmacol. 71, 144–154. doi:10.1016/j.intimp.2019.01.021
Chen, Z., Ma, X., Zhu, Y., Zhao, Y., Wang, J., Li, R., et al. (2015). Paeoniflorin Ameliorates ANIT-Induced Cholestasis by Activating Nrf2 through an PI3K/Akt-dependent Pathway in Rats. Phytother. Res. 29, 1768–1775. doi:10.1002/ptr.5431
Cipriani, S., Renga, B., D’Amore, C., Simonetti, M., De Tursi, A. A., Carino, A., et al. (2015). Impaired Itching Perception in Murine Models of Cholestasis Is Supported by Dysregulation of GPBAR1 Signaling. PLoS One 10, e0129866. doi:10.1371/journal.pone.0129866
Davis, B. K., Wen, H., and Ting, J. P.-Y. (2011). The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 29, 707–735. doi:10.1146/annurev-immunol-031210-101405
de Aguiar Vallim, T. Q., Tarling, E. J., and Edwards, P. A. (2013). Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab 17, 657–669. doi:10.1016/j.cmet.2013.03.013
De Marino, S., Festa, C., Sepe, V., and Zampella, A. (2019). Chemistry and Pharmacology of GPBAR1 and FXR Selective Agonists, Dual Agonists, and Antagonists. Handb. Exp. Pharmacol. 256, 137–165. doi:10.1007/164_2019_237
Deutschmann, K., Reich, M., Klindt, C., Dröge, C., Spomer, L., Häussinger, D., et al. (2018). Bile Acid Receptors in the Biliary Tree: TGR5 in Physiology and Disease. Biochim. Biophys. Acta. Mol. Basis Dis. 1864, 1319–1325. doi:10.1016/j.bbadis.2017.08.021
Duan, H., Ning, M., Chen, X., Zou, Q., Zhang, L., Feng, Y., et al. (2012). Design, Synthesis, and Antidiabetic Activity of 4-phenoxynicotinamide and 4-Phenoxypyrimidine-5-Carboxamide Derivatives as Potent and Orally Efficacious TGR5 Agonists. J. Med. Chem. 55, 10475–10489. doi:10.1021/jm301071h
Elia, A. E., Lalli, S., Monsurrò, M. R., Sagnelli, A., Taiello, A. C., Reggiori, B., et al. (2016). Tauroursodeoxycholic Acid in the Treatment of Patients with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 23, 45–52. doi:10.1111/ene.12664
Erice, O., Labiano, I., Arbelaiz, A., Santos-Laso, A., Munoz-Garrido, P., Jimenez-Agüero, R., et al. (2018). Differential Effects of FXR or TGR5 Activation in Cholangiocarcinoma Progression. Biochim. Biophys. Acta. Mol. Basis Dis. 1864, 1335–1344. doi:10.1016/j.bbadis.2017.08.016
Festa, C., Renga, B., D’Amore, C., Sepe, V., Finamore, C., De Marino, S., et al. (2014). Exploitation of Cholane Scaffold for the Discovery of Potent and Selective Farnesoid X Receptor (FXR) and G-Protein Coupled Bile Acid Receptor 1 (GP-BAR1) Ligands. J. Med. Chem. 57, 8477–8495. doi:10.1021/jm501273r
Fiorucci, S., Biagioli, M., Sepe, V., Zampella, A., and Distrutti, E. (2020). Bile Acid Modulators for the Treatment of Nonalcoholic Steatohepatitis (NASH). Expert Opin. Investig. Drugs 29, 623–632. doi:10.1080/13543784.2020.1763302
Fiorucci, S., and Distrutti, E. (2019). The Pharmacology of Bile Acids and Their Receptors. Handb. Exp. Pharmacol. 256, 3–18. doi:10.1007/164_2019_238
Francis, H., Glaser, S., Ueno, Y., Lesage, G., Marucci, L., Benedetti, A., et al. (2004). cAMP Stimulates the Secretory and Proliferative Capacity of the Rat Intrahepatic Biliary Epithelium through Changes in the PKA/Src/MEK/ERK1/2 Pathway. J. Hepatol. 41, 528–537. doi:10.1016/j.jhep.2004.06.009
Genet, C., Strehle, A., Schmidt, C., Boudjelal, G., Lobstein, A., Schoonjans, K., et al. (2010). Structure-activity Relationship Study of Betulinic Acid, a Novel and Selective TGR5 Agonist, and its Synthetic Derivatives: Potential Impact in Diabetes. J. Med. Chem. 53, 178–190. doi:10.1021/jm900872z
Gertzen, C. G. W., Spomer, L., Smits, S. H. J., Häussinger, D., Keitel, V., and Gohlke, H. (2015). Mutational Mapping of the Transmembrane Binding Site of the G-Protein Coupled Receptor TGR5 and Binding Mode Prediction of TGR5 Agonists. Eur. J. Med. Chem. 104, 57–72. doi:10.1016/j.ejmech.2015.09.024
Ghonem, N. S., Assis, D. N., and Boyer, J. L. (2015). Fibrates and Cholestasis. Hepatology 62, 635–643. doi:10.1002/hep.27744
Gong, Z., Zhou, J., Zhao, S., Tian, C., Wang, P., Xu, C., et al. (2016). Chenodeoxycholic Acid Activates NLRP3 Inflammasome and Contributes to Cholestatic Liver Fibrosis. Oncotarget 7, 83951–83963. doi:10.18632/oncotarget.13796
Grasset, E., Puel, A., Charpentier, J., Collet, X., Christensen, J. E., Tercé, F., et al. (2017). A Specific Gut Microbiota Dysbiosis of Type 2 Diabetic Mice Induces GLP-1 Resistance through an Enteric NO-dependent and Gut-Brain Axis Mechanism. Cel Metab 25, 1075–1090. doi:10.1016/j.cmet.2017.04.013
Gribble, F. M., and Reimann, F. (2019). Function and Mechanisms of Enteroendocrine Cells and Gut Hormones in Metabolism. Nat. Rev. Endocrinol. 15, 226–237. doi:10.1038/s41574-019-0168-8
Guicciardi, M. E., Trussoni, C. E., LaRusso, N. F., and Gores, G. J. (2020). The Spectrum of Reactive Cholangiocytes in Primary Sclerosing Cholangitis. Hepatology 71, 741–748. doi:10.1002/hep.31067
Guo, C., Chen, W.-D., and Wang, Y.-D. (2016a). TGR5, Not Only a Metabolic Regulator. Front. Physiol. 7, 646. doi:10.3389/fphys.2016.00646
Guo, C., Xie, S., Chi, Z., Zhang, J., Liu, Y., Zhang, L., et al. (2016b). Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 45, 802–816. doi:10.1016/j.immuni.2016.09.008
Haselow, K., Bode, J. G., Wammers, M., Ehlting, C., Keitel, V., Kleinebrecht, L., et al. (2013). Bile Acids PKA-Dependently Induce a Switch of the IL-10/IL-12 Ratio and Reduce Proinflammatory Capability of Human Macrophages. J. Leukoc. Biol. 94, 1253–1264. doi:10.1189/jlb.0812396
Heymann, F., and Tacke, F. (2016). Immunology in the Liver-Ffrom Homeostasis to Disease. Nat. Rev. Gastroenterol. Hepatol. 13, 88–110. doi:10.1038/nrgastro.2015.200
Hofmann, A. F., and Hagey, L. R. (2014). Key Discoveries in Bile Acid Chemistry and Biology and Their Clinical Applications: History of the Last Eight Decades. J. Lipid Res. 55, 1553–1595. doi:10.1194/jlr.R049437
Hogarth, D. K., Sandbo, N., Taurin, S., Kolenko, V., Miano, J. M., and Dulin, N. O. (2004). Dual Role of PKA in Phenotypic Modulation of Vascular Smooth Muscle Cells by Extracellular ATP. Am. J. Physiol. Cel Physiol. 287, C449–C456. doi:10.1152/ajpcell.00547.2003
Hohenester, S., Wenniger, L. M. de. B., Paulusma, C. C., van Vliet, S. J., Jefferson, D. M., Elferink, R. P. O., et al. (2012). A Biliary HCO3- Umbrella Constitutes a Protective Mechanism against Bile Acid-Induced Injury in Human Cholangiocytes. Hepatology 55, 173–183. doi:10.1002/hep.24691
Holter, M. M., Chirikjian, M. K., Govani, V. N., and Cummings, B. P. (2020). TGR5 Signaling in Hepatic Metabolic Health. Nutrients 12:2598. doi:10.3390/nu12092598
Horiba, T., Katsukawa, M., Mita, M., and Sato, R. (2015). Dietary Obacunone Supplementation Stimulates Muscle Hypertrophy, and Suppresses Hyperglycemia and Obesity through the TGR5 and PPARγ Pathway. Biochem. Biophys. Res. Commun. 463, 846–852. doi:10.1016/j.bbrc.2015.06.022
Huang, S., Ma, S., Ning, M., Yang, W., Ye, Y., Zhang, L., et al. (2019). TGR5 Agonist Ameliorates Insulin Resistance in the Skeletal Muscles and Improves Glucose Homeostasis in Diabetic Mice. Metabolism 99, 45–56. doi:10.1016/j.metabol.2019.07.003
Hussain, A. B., Samuel, R., Hegade, V. S., Jones, D. E., and Reynolds, N. J. (2019). Pruritus Secondary to Primary Biliary Cholangitis: a Review of the Pathophysiology and Management with Phototherapy. Br. J. Dermatol. 181, 1138–1145. doi:10.1111/bjd.17933
Ichikawa, R., Takayama, T., Yoneno, K., Kamada, N., Kitazume, M. T., Higuchi, H., et al. (2012). Bile Acids Induce Monocyte Differentiation toward Interleukin-12 Hypo-Producing Dendritic Cells via a TGR5-dependent Pathway. Immunology 136, 153–162. doi:10.1111/j.1365-2567.2012.03554.x
Isaacs-Ten, A., Echeandia, M., Moreno-Gonzalez, M., Brion, A., Goldson, A., Philo, M., et al. (2020). Intestinal Microbiome-Macrophage Crosstalk Contributes to Cholestatic Liver Disease by Promoting Intestinal Permeability in Mice. Hepatology 72, 2090–2108. doi:10.1002/hep.31228
Ito, K., Okumura, A., Takeuchi, J. S., Watashi, K., Inoue, R., Yamauchi, T., et al. (2021). Dual Agonist of Farnesoid X Receptor and Takeda G Protein-Coupled Receptor 5 Inhibits Hepatitis B Virus Infection In Vitro and In Vivo. Hepatology 74, 83–98. doi:10.1002/hep.31712
Jafri, L., Saleem, S., Calderwood, D., Gillespie, A., Mirza, B., and Green, B. D. (2016). Naturally-occurring TGR5 Agonists Modulating Glucagon-like Peptide-1 Biosynthesis and Secretion. Peptides 78, 51–58. doi:10.1016/j.peptides.2016.01.015
Jia, W., Xie, G., and Jia, W. (2018). Bile Acid-Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 15, 111–128. doi:10.1038/nrgastro.2017.119
Jin, P., Deng, S., Tian, M., Lenahan, C., Wei, P., Wang, Y., et al. (2021). INT-777 Prevents Cognitive Impairment by Activating Takeda G Protein-Coupled Receptor 5 (TGR5) and Attenuating Neuroinflammation via cAMP/PKA/CREB Signaling axis in a Rat Model of Sepsis. Exp. Neurol. 335, 113504. doi:10.1016/j.expneurol.2020.113504
Jones, H., Alpini, G., and Francis, H. (2015). Bile Acid Signaling and Biliary Functions. Acta Pharm. Sin. B 5, 123–128. doi:10.1016/j.apsb.2015.01.009
Karlsen, T. H., Franke, A., Melum, E., Kaser, A., Hov, J. R., Balschun, T., et al. (2010). Genome-wide Association Analysis in Primary Sclerosing Cholangitis. Gastroenterology 138, 1102–1111. doi:10.1053/j.gastro.2009.11.046
Katsuma, S., Hirasawa, A., and Tsujimoto, G. (2005). Bile Acids Promote Glucagon-like Peptide-1 Secretion through TGR5 in a Murine Enteroendocrine Cell Line STC-1. Biochem. Biophys. Res. Commun. 329, 386–390. doi:10.1016/j.bbrc.2005.01.139
Kawamata, Y., Fujii, R., Hosoya, M., Harada, M., Yoshida, H., Miwa, M., et al. (2003). A G Protein-Coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 278, 9435–9440. doi:10.1074/jbc.M209706200
Ke, B., Shen, X.-D., Kamo, N., Ji, H., Yue, S., Gao, F., et al. (2013). β-Catenin Regulates Innate and Adaptive Immunity in Mouse Liver Ischemia-Reperfusion Injury. Hepatology 57, 1203–1214. doi:10.1002/hep.26100
Keitel, V., Cupisti, K., Ullmer, C., Knoefel, W. T., Kubitz, R., and Häussinger, D. (2009). The Membrane-Bound Bile Acid Receptor TGR5 Is Localized in the Epithelium of Human Gallbladders. Hepatology 50, 861–870. doi:10.1002/hep.23032
Keitel, V., Donner, M., Winandy, S., Kubitz, R., and Häussinger, D. (2008). Expression and Function of the Bile Acid Receptor TGR5 in Kupffer Cells. Biochem. Biophys. Res. Commun. 372, 78–84. doi:10.1016/j.bbrc.2008.04.171
Keitel, V., and Häussinger, D. (2012). Perspective: TGR5 (Gpbar-1) in Liver Physiology and Disease. Clin. Res. Hepatol. Gastroenterol. 36, 412–419. doi:10.1016/j.clinre.2012.03.008
Keitel, V., and Häussinger, D. (2018). Role of TGR5 (GPBAR1) in Liver Disease. Semin. Liver Dis. 38, 333–339. doi:10.1055/s-0038-1669940
Keitel, V., and Häussinger, D. (2011). TGR5 in the Biliary Tree. Dig. Dis. 29, 45–47. doi:10.1159/000324127
Kirchweger, B., Kratz, J. M., Ladurner, A., Grienke, U., Langer, T., Dirsch, V. M., et al. (2018). In Silico Workflow for the Discovery of Natural Products Activating the G Protein-Coupled Bile Acid Receptor 1. Front. Chem. 6, 242. doi:10.3389/fchem.2018.00242
Koga, K., Takaesu, G., Yoshida, R., Nakaya, M., Kobayashi, T., Kinjyo, I., et al. (2009). Cyclic Adenosine Monophosphate Suppresses the Transcription of Proinflammatory Cytokines via the Phosphorylated C-Fos Protein. Immunity 30, 372–383. doi:10.1016/j.immuni.2008.12.021
Komatsu, H. (2015). Novel Therapeutic GPCRs for Psychiatric Disorders. Int. J. Mol. Sci. 16, 14109–14121. doi:10.3390/ijms160614109
Kumar, D. P., Asgharpour, A., Mirshahi, F., Park, S. H., Liu, S., Imai, Y., et al. (2016). Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet α Cells to Promote Glucose Homeostasis. J. Biol. Chem. 291, 6626–6640. doi:10.1074/jbc.M115.699504
Labib, P. L., Goodchild, G., and Pereira, S. P. (2019). Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer 19, 185. doi:10.1186/s12885-019-5391-0
Ladurner, A., Zehl, M., Grienke, U., Hofstadler, C., Faur, N., Pereira, F. C., et al. (2017). Allspice and Clove as Source of Triterpene Acids Activating the G Protein-Coupled Bile Acid Receptor TGR5. Front. Pharmacol. 8, 468. doi:10.3389/fphar.2017.00468
Lawrence, T. (2009). The Nuclear Factor NF-kappaB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651. doi:10.1101/cshperspect.a001651
Lazaridis, K. N., Strazzabosco, M., and Larusso, N. F. (2004). The Cholangiopathies: Disorders of Biliary Epithelia. Gastroenterology 127, 1565–1577. doi:10.1053/j.gastro.2004.08.006
Lesage, G., Glaser, S. S., Gubba, S., Robertson, W. E., Phinizy, J. L., Lasater, J., et al. (1996). Regrowth of the Rat Biliary Tree after 70% Partial Hepatectomy Is Coupled to Increased Secretin-Induced Ductal Secretion. Gastroenterology 111, 1633–1644. doi:10.1016/s0016-5085(96)70027-6
Li, A.-D., Xie, X.-L., Qi, W., Wang, W.-B., Ma, J.-J., Zhao, D.-Q., et al. (2020). TGR5 Promotes Cholangiocarcinoma by Interacting with Mortalin. Exp. Cel Res. 389, 111855. doi:10.1016/j.yexcr.2020.111855
Li, B., Yang, N., Li, C., Li, C., Gao, K., Xie, X., et al. (2018a). INT-777, a Bile Acid Receptor Agonist, Extenuates Pancreatic Acinar Cells Necrosis in a Mouse Model of Acute Pancreatitis. Biochem. Biophys. Res. Commun. 503, 38–44. doi:10.1016/j.bbrc.2018.05.120
Li, M., Cai, S.-Y., and Boyer, J. L. (2017a). Mechanisms of Bile Acid Mediated Inflammation in the Liver. Mol. Aspects Med. 56, 45–53. doi:10.1016/j.mam.2017.06.001
Li, S., Qiu, M., Kong, Y., Zhao, X., Choi, H.-J., Reich, M., et al. (2018b). Bile Acid G Protein-Coupled Membrane Receptor TGR5 Modulates Aquaporin 2-Mediated Water Homeostasis. J. Am. Soc. Nephrol. 29, 2658–2670. doi:10.1681/ASN.2018030271
Li, Y., Tang, R., Leung, P. S. C., Gershwin, M. E., and Ma, X. (2017b). Bile Acids and Intestinal Microbiota in Autoimmune Cholestatic Liver Diseases. Autoimmun. Rev. 16, 885–896. doi:10.1016/j.autrev.2017.07.002
Liang, H., Matei, N., McBride, D. W., Xu, Y., Zhou, Z., Tang, J., et al. (2021). TGR5 Activation Attenuates Neuroinflammation via Pellino3 Inhibition of caspase-8/NLRP3 after Middle Cerebral Artery Occlusion in Rats. J. Neuroinflammation 18, 40. doi:10.1186/s12974-021-02087-1
Lieu, T., Jayaweera, G., Zhao, P., Poole, D. P., Jensen, D., Grace, M., et al. (2014). The Bile Acid Receptor TGR5 Activates the TRPA1 Channel to Induce Itch in Mice. Gastroenterology 147, 1417–1428. doi:10.1053/j.gastro.2014.08.042
Lo, S.-H., Cheng, K.-C., Li, Y.-X., Chang, C.-H., Cheng, J.-T., and Lee, K.-S. (2016). Development of Betulinic Acid as an Agonist of TGR5 Receptor Using a New In Vitro Assay. Drug Des. Devel. Ther. 10, 2669–2676. doi:10.2147/DDDT.S113197
Lo, S.-H., Li, Y., Cheng, K. C., Niu, C.-S., Cheng, J.-T., and Niu, H.-S. (2017). Ursolic Acid Activates the TGR5 Receptor to Enhance GLP-1 Secretion in Type 1-like Diabetic Rats. Naunyn. Schmiedebergs. Arch. Pharmacol. 390, 1097–1104. doi:10.1007/s00210-017-1409-9
Lv, L.-X., Fang, D.-Q., Shi, D., Chen, D.-Y., Yan, R., Zhu, Y.-X., et al. (2016). Alterations and Correlations of the Gut Microbiome, Metabolism and Immunity in Patients with Primary Biliary Cirrhosis. Environ. Microbiol. 18, 2272–2286. doi:10.1111/1462-2920.13401
Ma, X., Zhao, Y., Zhu, Y., Chen, Z., Wang, J., Li, R., et al. (2015). Paeonia Lactiflora Pall. Protects against ANIT-Induced Cholestasis by Activating Nrf2 via PI3K/Akt Signaling Pathway. Drug Des. Devel. Ther. 9, 5061–5074. doi:10.2147/DDDT.S90030
Malhi, H., and Camilleri, M. (2017). Modulating Bile Acid Pathways and TGR5 Receptors for Treating Liver and GI Diseases. Curr. Opin. Pharmacol. 37, 80–86. doi:10.1016/j.coph.2017.09.008
Maruyama, T., Miyamoto, Y., Nakamura, T., Tamai, Y., Okada, H., Sugiyama, E., et al. (2002). Identification of Membrane-type Receptor for Bile Acids (M-BAR). Biochem. Biophys. Res. Commun. 298, 714–719. doi:10.1016/s0006-291x(02)02550-0
Maruyama, T., Tanaka, K., Suzuki, J., Miyoshi, H., Harada, N., Nakamura, T., et al. (2006). Targeted Disruption of G Protein-Coupled Bile Acid Receptor 1 (Gpbar1/M-Bar) in Mice. J. Endocrinol. 191, 197–205. doi:10.1677/joe.1.06546
Masyuk, A. I., Huang, B. Q., Radtke, B. N., Gajdos, G. B., Splinter, P. L., Masyuk, T. V., et al. (2013). Ciliary Subcellular Localization of TGR5 Determines the Cholangiocyte Functional Response to Bile Acid Signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G1013–G1024. doi:10.1152/ajpgi.00383.2012
Masyuk, T. V., Masyuk, A. I., Lorenzo Pisarello, M., Howard, B. N., Huang, B. Q., Lee, P.-Y., et al. (2017). TGR5 Contributes to Hepatic Cystogenesis in Rodents with Polycystic Liver Diseases through Cyclic Adenosine monophosphate/Gαs Signaling. Hepatology 66, 1197–1218. doi:10.1002/hep.29284
Merlen, G., Bidault-Jourdainne, V., Kahale, N., Glenisson, M., Ursic-Bedoya, J., Doignon, I., et al. (2020a). Hepatoprotective Impact of the Bile Acid Receptor TGR5. Liver Int. Off. J. Int. Assoc. Study Liver 40, 1005–1015. doi:10.1111/liv.14427
Merlen, G., Kahale, N., Ursic-Bedoya, J., Bidault-Jourdainne, V., Simerabet, H., Doignon, I., et al. (2020b). TGR5-dependent Hepatoprotection through the Regulation of Biliary Epithelium Barrier Function. Gut 69, 146–157. doi:10.1136/gutjnl-2018-316975
Meroni, S. B., Galardo, M. N., Rindone, G., Gorga, A., Riera, M. F., and Cigorraga, S. B. (2019). Molecular Mechanisms and Signaling Pathways Involved in Sertoli Cell Proliferation. Front. Endocrinol. (Lausanne). 10, 224. doi:10.3389/fendo.2019.00224
Minagawa, N., Nagata, J., Shibao, K., Masyuk, A. I., Gomes, D. A., Rodrigues, M. A., et al. (2007). Cyclic AMP Regulates Bicarbonate Secretion in Cholangiocytes through Release of ATP into Bile. Gastroenterology 133, 1592–1602. doi:10.1053/j.gastro.2007.08.020
Müller, T. D., Finan, B., Bloom, S. R., D’Alessio, D., Drucker, D. J., Flatt, P. R., et al. (2019). Glucagon-like Peptide 1 (GLP-1). Mol. Metab. 30, 72–130. doi:10.1016/j.molmet.2019.09.010
Nathanson, M. H., Burgstahler, A. D., Mennone, A., and Boyer, J. L. (1996). Characterization of Cytosolic Ca2+ Signaling in Rat Bile Duct Epithelia. Am. J. Physiol. 271, G86–G96. doi:10.1152/ajpgi.1996.271.1.G86
Ono, E., Inoue, J., Hashidume, T., Shimizu, M., and Sato, R. (2011). Anti-obesity and Anti-hyperglycemic Effects of the Dietary Citrus Limonoid Nomilin in Mice Fed a High-Fat Diet. Biochem. Biophys. Res. Commun. 410, 677–681. doi:10.1016/j.bbrc.2011.06.055
Pathak, P., Liu, H., Boehme, S., Xie, C., Krausz, K. W., Gonzalez, F., et al. (2017). Farnesoid X Receptor Induces Takeda G-Protein Receptor 5 Cross-Talk to Regulate Bile Acid Synthesis and Hepatic Metabolism. J. Biol. Chem. 292, 11055–11069. doi:10.1074/jbc.M117.784322
Pathak, P., Xie, C., Nichols, R. G., Ferrell, J. M., Boehme, S., Krausz, K. W., et al. (2018). Intestine Farnesoid X Receptor Agonist and the Gut Microbiota Activate G-Protein Bile Acid Receptor-1 Signaling to Improve Metabolism. Hepatology 68, 1574–1588. doi:10.1002/hep.29857
Péan, N., Doignon, I., Garcin, I., Besnard, A., Julien, B., Liu, B., et al. (2013). The Receptor TGR5 Protects the Liver from Bile Acid Overload during Liver Regeneration in Mice. Hepatology 58, 1451–1460. doi:10.1002/hep.26463
Pellicciari, R., Fiorucci, S., Camaioni, E., Clerici, C., Costantino, G., Maloney, P. R., et al. (2002). 6alpha-ethyl-chenodeoxycholic Acid (6-ECDCA), a Potent and Selective FXR Agonist Endowed with Anticholestatic Activity. J. Med. Chem. 45, 3569–3572. doi:10.1021/jm025529g
Pellicciari, R., Gioiello, A., Macchiarulo, A., Thomas, C., Rosatelli, E., Natalini, B., et al. (2009). Discovery of 6alpha-ethyl-23(S)-methylcholic Acid (S-EMCA, INT-777) as a Potent and Selective Agonist for the TGR5 Receptor, a Novel Target for Diabesity. J. Med. Chem. 52, 7958–7961. doi:10.1021/jm901390p
Perino, A., and Schoonjans, K. (2015). TGR5 and Immunometabolism: Insights from Physiology and Pharmacology. Trends Pharmacol. Sci. 36, 847–857. doi:10.1016/j.tips.2015.08.002
Phillips, D. P., Gao, W., Yang, Y., Zhang, G., Lerario, I. K., Lau, T. L., et al. (2014). Discovery of Trifluoromethyl(pyrimidin-2-Yl)azetidine-2-Carboxamides as Potent, Orally Bioavailable TGR5 (GPBAR1) Agonists: Structure-Activity Relationships, lead Optimization, and Chronic In Vivo Efficacy. J. Med. Chem. 57, 3263–3282. doi:10.1021/jm401731q
Pols, T. W. H., Nomura, M., Harach, T., Lo Sasso, G., Oosterveer, M. H., Thomas, C., et al. (2011). TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cel Metab 14, 747–757. doi:10.1016/j.cmet.2011.11.006
Portincasa, P., Di Ciaula, A., Garruti, G., Vacca, M., De Angelis, M., and Wang, D. Q.-H. (2020). Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 12:3709. doi:10.3390/nu12123709
Rao, J., Yang, C., Yang, S., Lu, H., Hu, Y., Lu, L., et al. (2020). Deficiency of TGR5 Exacerbates Immune-Mediated Cholestatic Hepatic Injury by Stabilizing the β-catenin Destruction Complex. Int. Immunol. 32, 321–334. doi:10.1093/intimm/dxaa002
Razumilava, N., and Gores, G. J. (2014). Cholangiocarcinoma. Lancet (London, England) 383, 2168–2179. doi:10.1016/S0140-6736(13)61903-0
Reich, M., Deutschmann, K., Sommerfeld, A., Klindt, C., Kluge, S., Kubitz, R., et al. (2016). TGR5 Is Essential for Bile Acid-dependent Cholangiocyte Proliferation In Vivo and In Vitro. Gut 65, 487–501. doi:10.1136/gutjnl-2015-309458
Rizzo, G., Passeri, D., De Franco, F., Ciaccioli, G., Donadio, L., Rizzo, G., et al. (2010). Functional Characterization of the Semisynthetic Bile Acid Derivative INT-767, a Dual Farnesoid X Receptor and TGR5 Agonist. Mol. Pharmacol. 78, 617–630. doi:10.1124/mol.110.064501
Rodrigues, A. D., Lai, Y., Cvijic, M. E., Elkin, L. L., Zvyaga, T., and Soars, M. G. (2014). Drug-induced Perturbations of the Bile Acid Pool, Cholestasis, and Hepatotoxicity: Mechanistic Considerations beyond the Direct Inhibition of the Bile Salt export Pump. Drug Metab. Dispos. 42, 566–574. doi:10.1124/dmd.113.054205
Rossen, N. G., Fuentes, S., Boonstra, K., D’Haens, G. R., Heilig, H. G., Zoetendal, E. G., et al. (2015). The Mucosa-Associated Microbiota of PSC Patients Is Characterized by Low Diversity and Low Abundance of Uncultured Clostridiales II. J. Crohns. Colitis 9, 342–348. doi:10.1093/ecco-jcc/jju023
Sand, T. E., Thoresen, G. H., Refsnes, M., and Christoffersen, T. (1992). Growth-regulatory Effects of Glucagon, Insulin, and Epidermal Growth Factor in Cultured Hepatocytes. Temporal Aspects and Evidence for Bidirectional Control by Cyclic AMP. Dig. Dis. Sci. 37, 84–92. doi:10.1007/BF01308347
Sato, H., Genet, C., Strehle, A., Thomas, C., Lobstein, A., Wagner, A., et al. (2007). Anti-hyperglycemic Activity of a TGR5 Agonist Isolated from Olea Europaea. Biochem. Biophys. Res. Commun. 362, 793–798. doi:10.1016/j.bbrc.2007.06.130
Sato, H., Macchiarulo, A., Thomas, C., Gioiello, A., Une, M., Hofmann, A. F., et al. (2008). Novel Potent and Selective Bile Acid Derivatives as TGR5 Agonists: Biological Screening, Structure-Activity Relationships, and Molecular Modeling Studies. J. Med. Chem. 51, 1831–1841. doi:10.1021/jm7015864
Sato, K., Meng, F., Giang, T., Glaser, S., and Alpini, G. (2018). Mechanisms of Cholangiocyte Responses to Injury. Biochim. Biophys. Acta. Mol. Basis Dis. 1864, 1262–1269. doi:10.1016/j.bbadis.2017.06.017
Shi, Y., Su, W., Zhang, L., Shi, C., Zhou, J., Wang, P., et al. (2020). TGR5 Regulates Macrophage Inflammation in Nonalcoholic Steatohepatitis by Modulating NLRP3 Inflammasome Activation. Front. Immunol. 11, 609060. doi:10.3389/fimmu.2020.609060
Shibao, K., Hirata, K., Robert, M. E., and Nathanson, M. H. (2003). Loss of Inositol 1,4,5-trisphosphate Receptors from Bile Duct Epithelia Is a Common Event in Cholestasis. Gastroenterology 125, 1175–1187. doi:10.1016/s0016-5085(03)01201-0
Smith, S. A., Newby, A. C., and Bond, M. (2019). Ending Restenosis: Inhibition of Vascular Smooth Muscle Cell Proliferation by cAMP. Cells 8:1447. doi:doi:10.3390/cells8111447
Sorrentino, G., Perino, A., Yildiz, E., El Alam, G., Bou Sleiman, M., Gioiello, A., et al. (2020). Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration. Gastroenterology 159, 956–968. e8. doi:10.1053/j.gastro.2020.05.067
Tabibian, J. H., O’Hara, S. P., Trussoni, C. E., Tietz, P. S., Splinter, P. L., Mounajjed, T., et al. (2016). Absence of the Intestinal Microbiota Exacerbates Hepatobiliary Disease in a Murine Model of Primary Sclerosing Cholangitis. Hepatology 63, 185–196. doi:10.1002/hep.27927
Tang, R., Wei, Y., Li, Y., Chen, W., Chen, H., Wang, Q., et al. (2018). Gut Microbial Profile Is Altered in Primary Biliary Cholangitis and Partially Restored after UDCA Therapy. Gut 67, 534–541. doi:10.1136/gutjnl-2016-313332
Thomas, C., Gioiello, A., Noriega, L., Strehle, A., Oury, J., Rizzo, G., et al. (2009). TGR5-mediated Bile Acid Sensing Controls Glucose Homeostasis. Cel Metab 10, 167–177. doi:10.1016/j.cmet.2009.08.001
Thomas, C., Pellicciari, R., Pruzanski, M., Auwerx, J., and Schoonjans, K. (2008). Targeting Bile-Acid Signalling for Metabolic Diseases. Nat. Rev. Drug Discov. 7, 678–693. doi:10.1038/nrd2619
Ullmer, C., Alvarez Sanchez, R., Sprecher, U., Raab, S., Mattei, P., Dehmlow, H., et al. (2013). Systemic Bile Acid Sensing by G Protein-Coupled Bile Acid Receptor 1 (GPBAR1) Promotes PYY and GLP-1 Release. Br. J. Pharmacol. 169, 671–684. doi:10.1111/bph.12158
van Nierop, F. S., Scheltema, M. J., Eggink, H. M., Pols, T. W., Sonne, D. P., Knop, F. K., et al. (2017). Clinical Relevance of the Bile Acid Receptor TGR5 in Metabolism. Lancet. Diabetes Endocrinol. 5, 224–233. doi:10.1016/S2213-8587(16)30155-3
Wahlström, A., Sayin, S. I., Marschall, H.-U., and Bäckhed, F. (2016). Intestinal Crosstalk between Bile Acids and Microbiota and its Impact on Host Metabolism. Cel Metab 24, 41–50. doi:10.1016/j.cmet.2016.05.005
Wall, E. A., Zavzavadjian, J. R., Chang, M. S., Randhawa, B., Zhu, X., Hsueh, R. C., et al. (2009). Suppression of LPS-Induced TNF-Alpha Production in Macrophages by cAMP Is Mediated by PKA-AKAP95-P105. Sci. Signal. 2, ra28. doi:10.1126/scisignal.2000202
Wang, L.-Y., Cheng, K. C., Li, Y., Niu, C.-S., Cheng, J.-T., and Niu, H.-S. (2017). Glycyrrhizic Acid Increases Glucagon like Peptide-1 Secretion via TGR5 Activation in Type 1-like Diabetic Rats. Biomed. Pharmacother. 95, 599–604. doi:10.1016/j.biopha.2017.08.087
Wang, X. X., Wang, D., Luo, Y., Myakala, K., Dobrinskikh, E., Rosenberg, A. Z., et al. (2018). FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 29, 118–137. doi:10.1681/ASN.2017020222
Wang, Y.-D., Chen, W.-D., Yu, D., Forman, B. M., and Huang, W. (2011). The G-Protein-Coupled Bile Acid Receptor, Gpbar1 (TGR5), Negatively Regulates Hepatic Inflammatory Response through Antagonizing Nuclear Factor κ Light-Chain Enhancer of Activated B Cells (NF-Κb) in Mice. Hepatology 54, 1421–1432. doi:10.1002/hep.24525
Yan, J., Xie, G., Liang, C., Hu, Y., Zhao, A., Huang, F., et al. (2017). Herbal Medicine Yinchenhaotang Protects against α-naphthylisothiocyanate-induced Cholestasis in Rats. Sci. Rep. 7, 4211. doi:10.1038/s41598-017-04536-5
Yanguas-Casás, N., Barreda-Manso, M. A., Nieto-Sampedro, M., and Romero-Ramírez, L. (2017). TUDCA: An Agonist of the Bile Acid Receptor GPBAR1/TGR5 with Anti-inflammatory Effects in Microglial Cells. J. Cel. Physiol. 232, 2231–2245. doi:10.1002/jcp.25742
Yoneno, K., Hisamatsu, T., Shimamura, K., Kamada, N., Ichikawa, R., Kitazume, M. T., et al. (2013). TGR5 Signalling Inhibits the Production of Pro-inflammatory Cytokines by In Vitro Differentiated Inflammatory and Intestinal Macrophages in Crohn’s Disease. Immunology 139, 19–29. doi:10.1111/imm.12045
Zambad, S. P., Tuli, D., Mathur, A., Ghalsasi, S. A., Chaudhary, A. R., Deshpande, S., et al. (2013). TRC210258, a Novel TGR5 Agonist, Reduces Glycemic and Dyslipidemic Cardiovascular Risk in Animal Models of Diabesity. Diabetes Metab. Syndr. Obes. 7, 1–14. doi:10.2147/DMSO.S50209
Zhao, Y., Ma, X., Wang, J., Wen, R., Jia, L., Zhu, Y., et al. (2015). Large Dose Means Significant Effect-Ddose and Effect Relationship of Chi-Dan-Tui-Huang Decoction on Alpha-Naphthylisothiocyanate-Induced Cholestatic Hepatitis in Rats. BMC Complement. Altern. Med. 15, 104. doi:10.1186/s12906-015-0637-0
Zhong, M. (2010). TGR5 as a Therapeutic Target for Treating Obesity. Curr. Top. Med. Chem. 10, 386–396. doi:10.2174/156802610790980576
Zou, Q., Duan, H., Ning, M., Liu, J., Feng, Y., Zhang, L., et al. (2014). 4-Benzofuranyloxynicotinamide Derivatives Are Novel Potent and Orally Available TGR5 Agonists. Eur. J. Med. Chem. 82, 1–15. doi:10.1016/j.ejmech.2014.05.031
ABAT apical BA transporter
AE2 anion exchanger 2
ALP alkaline phosphatase
AQP2 aquaporin-2
ASBT apical sodium-dependent BA transporter
BA bile acid
BDL bile duct ligation
CA cholic acid
CCA cholangiocarcinoma
CDCA chenodeoxycholic acid
CFTR cystic fibrosis transmembrane conductance regulator
DCA deoxycholic acid
DCs dendritic cells
ECC extrahepatic cholangiocarcinoma
EGFR epidermal growth factor receptor
eNOS endothelial nitric oxide synthase
EPACs exchange proteins activated by cAMP
GCA glycocholic acid
GCDCA glycochenodeoxycholic acid
GDCA glycodeoxycholic acid
GGT gamma-glutamyl transferase
GLCA glycolithocholic acid
GLP1 glucagon-like peptide-1
GPCRs G protein-coupled receptors
GSK3β glycogen synthase kinase 3β
InsP3R inositol 1,4,5-trisphosphate receptor
iNOS inducible nitric oxide synthase
KO knockout
LCA lithocholic acid
LPS lipopolysaccharide
MRP2 multidrug resistance-associated protein 2
MRP4 multidrug resistance-associated protein 4
NASH: non-alcoholic steatohepatitis
NO nitric oxide
PBC primary biliary cirrhosis
PH partial hepatectomy
PKA protein kinase A
PSC primary sclerosing cholangitis
ROS reactive oxygen species
TCA taurocholic acid
TCDCA taurochenodeoxycholic acid
TDCA taurodeoxycholic acid
TJPs tight junction proteins
TLCA taurolithocholic acid
UDCA ursodeoxycholic acid
WT wild-type
Keywords: GPBAR1, bile acids, cholestasis, liver disease, inflammation, GPBAR1 agonists
Citation: Zhang F, Xiao X, Li Y, Wu H, Deng X, Jiang Y, Zhang W, Wang J, Ma X and Zhao Y (2022) Therapeutic Opportunities of GPBAR1 in Cholestatic Diseases. Front. Pharmacol. 12:805269. doi: 10.3389/fphar.2021.805269
Received: 30 October 2021; Accepted: 23 December 2021;
Published: 13 January 2022.
Edited by:
Feng Li, Baylor College of Medicine, United StatesReviewed by:
Stefano Fiorucci, University of Perugia, ItalyCopyright © 2022 Zhang, Xiao, Li, Wu, Deng, Jiang, Zhang, Wang, Ma and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Ma, dG9ieW1heGlhb0AxNjMuY29t; Yanling Zhao, emhhb3lsMjg1NUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.