 Steven R. Botts
Steven R. Botts Jason E. Fish
Jason E. Fish Kathryn L. Howe
Kathryn L. Howe
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 22 December 2021
Sec. Inflammation Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.787541
This article is part of the Research TopicStreaming Inflammation: From Damage to Healing and ResilienceView all 12 articles
Atherosclerosis, the chronic accumulation of cholesterol-rich plaque within arteries, is associated with a broad spectrum of cardiovascular diseases including myocardial infarction, aortic aneurysm, peripheral vascular disease, and stroke. Atherosclerotic cardiovascular disease remains a leading cause of mortality in high-income countries and recent years have witnessed a notable increase in prevalence within low- and middle-income regions of the world. Considering this prominent and evolving global burden, there is a need to identify the cellular mechanisms that underlie the pathogenesis of atherosclerosis to discover novel therapeutic targets for preventing or mitigating its clinical sequelae. Despite decades of research, we still do not fully understand the complex cell-cell interactions that drive atherosclerosis, but new investigative approaches are rapidly shedding light on these essential mechanisms. The vascular endothelium resides at the interface of systemic circulation and the underlying vessel wall and plays an essential role in governing pathophysiological processes during atherogenesis. In this review, we present emerging evidence that implicates the activated endothelium as a driver of atherosclerosis by directing site-specificity of plaque formation and by promoting plaque development through intracellular processes, which regulate endothelial cell proliferation and turnover, metabolism, permeability, and plasticity. Moreover, we highlight novel mechanisms of intercellular communication by which endothelial cells modulate the activity of key vascular cell populations involved in atherogenesis, and discuss how endothelial cells contribute to resolution biology – a process that is dysregulated in advanced plaques. Finally, we describe important future directions for preclinical atherosclerosis research, including epigenetic and targeted therapies, to limit the progression of atherosclerosis in at-risk or affected patients.
Atherosclerosis is a chronic inflammatory process in which the accumulation of cholesterol-laden plaque restricts blood flow within the arterial vasculature. The occlusion of arteries by luminal encroachment of expanding plaque or emboli from plaque rupture underlies a spectrum of cardiovascular diseases (CVDs) including myocardial infarction, ischemic cardiomyopathy, stroke, and peripheral vascular disease. Although CVD has remained a leading cause of morbidity and mortality in high-income countries (Piepoli et al., 2016), an epidemiological shift has occurred in recent decades (Dai et al., 2020; Libby, 2021), where improvements in vaccination and treatment of infectious diseases have led to a notable increase in CVD prevalence within low- and middle-income nations. The prominent and evolving burden of atherosclerotic CVD has stimulated continued interest in the identification of cellular mechanisms that govern its pathogenesis, which may aid in the discovery of novel biomarkers and therapeutic targets for CVD prevention, detection, and treatment.
Endothelial cells (ECs) comprise the vascular endothelium, the inner lining of all blood vessels, which forms the interface between systemic circulation and underlying tissues. The quiescent or non-proliferating endothelium, once considered to be dormant outside the settings of vascular development or disease, is now understood to play an active role in maintaining vascular homeostasis by receiving and generating diverse biochemical (i.e., autocrine, paracrine, and endocrine) and mechanical signals (Cahill and Redmond, 2016; Ricard et al., 2021). The systemic functions of the endothelium are numerous, and include the provision of oxygen and nutrients to tissues, regulation of vascular tone and permeability, maintenance of hemostasis and coagulation, induction of angiogenesis, and coordination of the inflammatory response (Cahill and Redmond, 2016; Ricard et al., 2021). These essential processes are modulated by rich crosstalk between ECs and other vascular cell populations, including smooth muscle cells (SMCs), monocytes, and macrophages, which contribute to normal vascular function in physiological settings. Likewise, dysregulated communication between ECs and other vascular cell types is associated with vascular dysfunction and pathological remodeling in CVDs such as hypertension, atherosclerosis, and aneurysm (Jaipersad et al., 2014; Méndez-Barbero et al., 2021).
The pathophysiology of atherosclerosis begins with the perturbed endothelium and is mediated by a cascade of intra- and intercellular signaling events that shape the behaviour of cells within the vasculature (da Luz et al., 2018). Vascular ECs facilitate the active transport of low-density lipoprotein (LDL) to the subendothelial space through transcytosis pathways (Mundi et al., 2018), and LDL accumulation initiates a vascular inflammatory response. Early in atherosclerosis, the endothelium transitions from a quiescent to an activated state in response to proatherogenic stimuli, including oxidized LDL (oxLDL), proinflammatory cytokines, and disturbed flow (Cahill and Redmond, 2016; da Luz et al., 2018). In turn, the activated endothelium plays a critical role in the recruitment of inflammatory cells including T lymphocytes, neutrophils, and monocytes to the arterial intima, the first of which induces the adaptive immune response, and the latter of which gives rise to intimal macrophages (da Luz et al., 2018; Libby, 2021). Subsequent lipid engulfment by macrophages produces foam cells, which undergo necrosis and apoptosis to form the lipid core of the progressing atherosclerotic lesion. Vascular SMCs that comprise the medial layer of arteries migrate to the intima, form fibrous tissue through the production of collagen and elastin, and can also differentiate into macrophage-like foam cells in the developing plaque (Bennett et al., 2016). In this proatherogenic environment, communication between the endothelium and other vascular cell populations stimulates the release of proinflammatory signals, which augment local inflammation and contribute to sustained plaque progression. Intimal thickening occurs during plaque development and creates a hypoxic intraplaque environment, which stimulates angiogenesis of the vasa vasorum – adventitial blood vessels that supply larger arteries – and promotes neovascularization into the vascular wall (Jaipersad et al., 2014). Progressive thinning of the fibrous cap results from an inflammation-associated decrease in collagen synthesis and increase in degradation, which in combination with erosion of the endothelium, contributes to plaque rupture, thrombosis, and obstruction of the affected vessel (Libby, 2021). Although beyond the scope of this review, the role of endothelial dysfunction in plaque rupture (Bentzon et al., 2014; White et al., 2016), as well as the contribution to pathogenesis and the therapeutic potential of the vasa vasorum in treating atherosclerosis (Xu et al., 2015; Boyle et al., 2017; Sedding et al., 2018), has been previously discussed in detail. The pathophysiological relevance of the arterial endothelium has been similarly outlined (Cahill and Redmond, 2016; da Luz et al., 2018; Libby, 2021), however, the specific and modifiable intra- and intercellular mechanisms that mediate endothelial dysfunction and atherogenesis remain to be elucidated.
In this review, we present emerging studies that implicate the vascular endothelium as a driver of atherosclerotic CVD by directing site-specificity of plaque formation and governing plaque progression through intracellular processes. Furthermore, we highlight recent studies that describe intercellular communication between ECs and key vascular cell types involved in atherogenesis (Figure 1), and discuss how ECs participate in resolution biology during plaque development. Finally, we outline outstanding questions and future directions for atherosclerosis treatments, including epigenetic interventions and the targeted delivery of therapeutics to the activated endothelium to resolve vascular inflammation and limit atherosclerotic plaque progression.
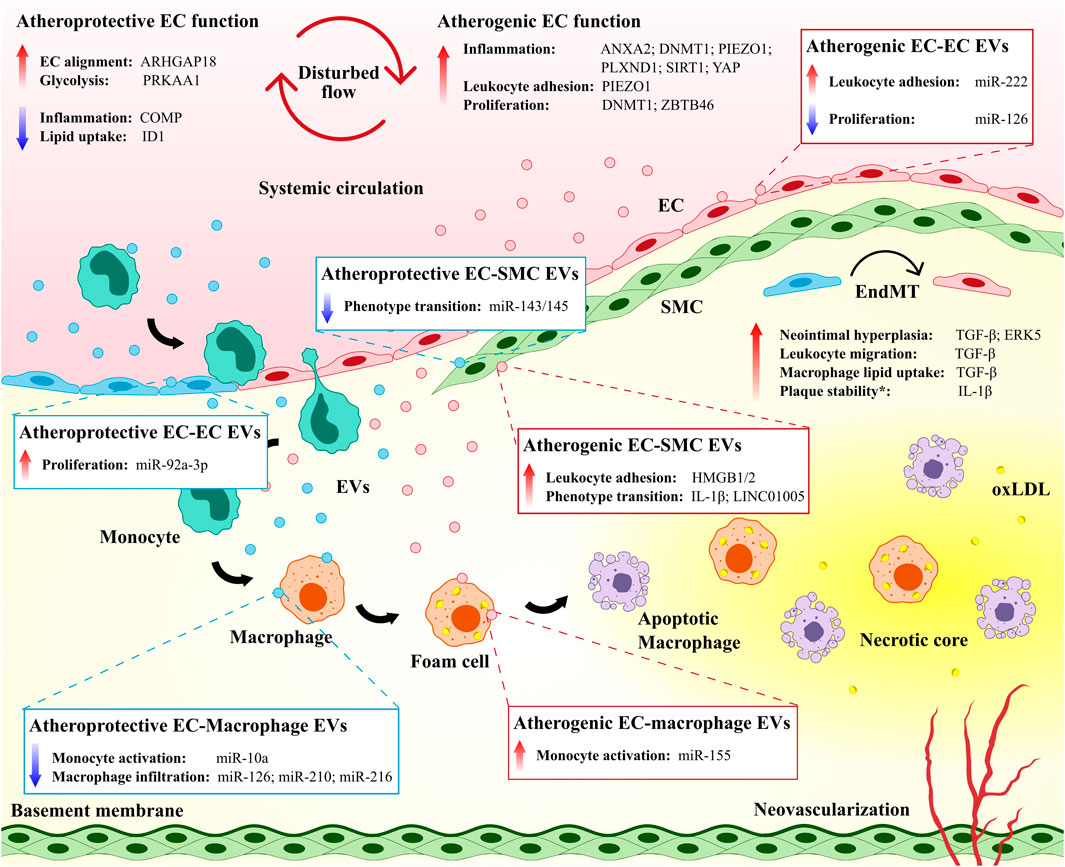
FIGURE 1. The vascular endothelium directs site-specificity for plaque development and governs plaque progression. Both atheroprotective and atherogenic mechanisms are operative in endothelial cells (ECs) exposed to disturbed flow. EC-derived extracellular vesicles (EVs) mediate atheroprotective and atherogenic intercellular communication among ECs and between ECs and other immune and non-immune cell populations. Endothelial-to-mesenchymal transition (EndMT) contributes to the atherosclerotic disease process but may also maintain plaque stability.* ANXA2, annexin A2; ARHGAP18, Rho GTPase activating protein 18; COMP, cartilage oligomeric matrix protein; DNMT1, DNA methyltransferase 1; ERK5, extracellular signal-regulated kinase 5; HMGB1/2, high mobility group box protein 1/2; ID1, inhibitor of DNA binding 1; IL-1β, interleukin 1 beta; oxLDL, oxidized low-density lipoprotein; PLXND1, plexin D1; PRKAA1, protein kinase AMP-activated catalytic subunit alpha 1; SIRT1, sirtuin 1; SMC, smooth muscle cell; TGF-β, transforming growth factor beta; YAP, yes-associated protein; ZBTB46, zinc finger and BTB domain-containing protein 46.
The vascular endothelium is subjected to both tangential (i.e., shear stress) and circumferential forces (i.e., pulsatile stretch) that result from circulating blood flow. ECs at this interface convert mechanical stimuli to biochemical signals through mechanotransduction, which modulates key cellular processes in response to fluctuations in the vascular environment, including proliferation and turnover (Hahn and Schwartz, 2009; Nigro et al., 2011). The development of early atherosclerotic lesions is characteristically localized to regions of the vasculature where laminar blood flow is disturbed (e.g., arterial branch points and lesser curvatures of vessels) (Giddens et al., 1993; VanderLaan et al., 2004), which have previously been associated with decreased expression of endothelial nitric oxide synthase (eNOS) and increased nuclear factor kappa B (NF-κB) activation (Hajra et al., 2000; Collins and Cybulsky, 2001; Won et al., 2007).
In the setting of undisturbed laminar flow, nitric oxide (NO) is produced by elevated levels of eNOS in ECs and diffuses across cell membranes to modulate the activity of vascular cells, including SMCs and leukocytes (Pan, 2009). NO-mediated activation of soluble guanylate cyclase and S-nitrosylation has been observed to inhibit SMC proliferation (Ignarro et al., 2001), suppress EC inflammation (Matsushita et al., 2003; Kang-Decker et al., 2007), and regulate vascular tone, blood flow, and oxygen delivery to tissues (Stamler et al., 1997; Haldar and Stamler, 2013). Hydrogen sulfide, a gaseous signaling molecule and upstream regulator of NO, is similarly produced under laminar flow conditions (Huang et al., 2015) and has a critical role in reducing vascular inflammation (Zanardo et al., 2006), promoting antioxidative activity (Muzaffar et al., 2008), and limiting the formation of foam cells (Zhao et al., 2011). Laminar flow conditions have also been found to promote EC alignment (Hahn and Schwartz, 2009), and foster a reducing environment that limits oxidative stress (Berk, 2008), tumor necrosis factor α- and signal transducer and activator of transcription 3-induced inflammation (Ni et al., 2003; Yamawaki et al., 2003), and EC turnover via apoptosis (Pi et al., 2004). The transcription factor Kruppel-like factor 2 (KLF2) has emerged as a critical transcriptional mediator of vascular homeostasis that is induced under laminar shear stress and increases eNOS expression and represses adhesion proteins such as vascular cell adhesion molecule 1 (VCAM-1) and E-selectin (SenBanerjee et al., 2004; Berk, 2008), which are induced by NF-κB in regions of disturbed flow (Hajra et al., 2000; Collins and Cybulsky, 2001). In contrast to regions of laminar flow, altered hemodynamics in atheroprone regions has been associated with increased oxidative stress (Förstermann et al., 2017), reorganization of cytoskeletal and cell-cell junction proteins (Chong et al., 2013; Pfenniger et al., 2013), and enhanced senescence and turnover in ECs (Xu, 2009; Warboys et al., 2014). Curiously, hydrogen sulfide has been observed to impair dilation of coronary arteries by reducing NO production under disturbed flow (Chai et al., 2015), although hydrogen sulfide administration has also been shown to inhibit leukocyte adhesion to the endothelium in regions of shear stress by upregulating Akt/eNOS signaling (Go et al., 2012).
Recent studies continue to elucidate a plethora of EC regulatory pathways that induce a proinflammatory and proatherogenic phenotype in response to disturbed flow (Qu et al., 2020; Zhao et al., 2020; Xia et al., 2021) (Figure 1). The mechanosensitive cation channel Piezo1, though essential for coordinating vascular morphogenesis under physiological shear stress in embryogenesis and adulthood (Li J. et al., 2014), has been previously linked to altered flow-induced inflammation and leukocyte recruitment to the endothelium. Indeed, pathological activation of Piezo1 has been associated with induction of downstream integrin α5 and NF-κB pathways, which contributes to endothelial inflammation and the progression of murine atherosclerosis (Albarrán-Juárez et al., 2018), and elevation of intracellular calcium, which results in actin disruption and increased monocyte adhesion in cultured ECs (Swain and Liddle, 2020). Likewise, integrin α5β1-associated phosphorylation of yes-associated protein (YAP) by c-Abl kinase (Li B. et al., 2019), the coupling of integrin α5 to annexin A2 (Zhang C. et al., 2020), and the guidance receptor plexin D1, upstream of EC integrins (Mehta et al., 2020), have been identified as novel mechanisms for endothelial activation under oscillatory shear stress. In contrast, several EC processes confer in vivo protection against atherosclerosis under disturbed flow, including inhibition of integrin α5 by cartilage oligomeric matrix protein (Lv et al., 2021), increased glycolysis by protein kinase AMP-activated catalytic subunit alpha 1 (Yang et al., 2018), decreased lipid uptake by the transcription factor inhibitor of DNA binding 1 (Zhang K. et al., 2018), and increased EC alignment by Rho GTPase activating protein 18 (Lay et al., 2019). Many of these preclinical findings hold promise for the development of atherosclerosis treatments through pharmacological modulation of the endothelium (e.g., mitigating atherogenic responses or promoting atheroprotective functions in ECs). However, further investigation is required in vitro to determine the generalizability of endothelial responses between vascular cell models (e.g., shared and distinct behaviors of different EC lines under varying flow conditions) (Maurya et al., 2021), as well as in vivo to identify translatability in preclinical models and long-term safety and efficacy in individuals with CVD.
Emerging evidence has also implicated disturbed blood flow in the dysregulation of EC proliferation and turnover during early atherosclerosis. Under physiological conditions, ECs that become senescent or apoptotic are replaced by the replication of neighboring cells, and more significant disruptions to the endothelium (e.g., during injury) are mitigated by circulating endothelial progenitor cells (Mannarino and Pirro, 2008). Atherosclerosis is characterized by cellular processes that promote EC turnover and induce proliferation of the activated endothelium (Xu, 2007). However, whether this proliferation is protective or detrimental has not been fully resolved. The mechanosensitive transcription factor zinc finger and BTB domain containing 46, for instance, inhibits proliferation in quiescent ECs and is downregulated by disturbed flow in vitro (Wang et al., 2019a). Hemodynamic signaling also impacts epigenetic pathways, and we have reviewed the impact of epigenetics on atherosclerosis elsewhere (Khyzha et al., 2017). For example, altered flow has been shown to induce DNA methyltransferase 1-mediated hypermethylation of the endothelium and its subsequent inhibition limits atherosclerosis via the cell cycle regulator cyclin A (Zhang et al., 2017). Furthermore, laminar flow-induced autophagy and expression of the deacetylase sirtuin 1 (SIRT1) together inhibit Hippo/YAP signaling to attenuate atherosclerotic plaque formation (Yuan P. et al., 2020), and hypermethylation of eNOS promoter elements under chronic disturbed flow has been shown to contribute to repressed eNOS expression in wildtype mice (Ku et al., 2021).
Flow-sensitive microRNAs have also been identified as key modulators of endothelial proliferation and turnover in atherosclerosis (Kumar et al., 2019), including miR-126-5p, which has been shown to contribute to proliferative reserve in ECs and prevents plaque development through upregulation of Notch signaling (Schober et al., 2014). Curiously, increased expression of miR-126 has been found in atheroprone regions of the endothelium (Zhou et al., 2013). Furthermore, physiological flow conditions have also been observed to promote antiproliferative microRNA activity within the endothelium, whereby KLF2-induced miR-23b represses cyclin H to reduce activity of the cyclin-dependent kinase–activating kinase complex and limit cell cycle progression (Wang et al., 2014). The regulatory interplay of antagonistic, flow-sensitive microRNAs may serve as a mechanism for fine-tuning EC proliferation and warrants further characterization in both quiescent and activated endothelial states.
A robust characterization of the transcriptional and epigenetic regulatory elements that contribute to endothelial dysfunction has remained an important challenge for atherosclerosis research and has been recently addressed with next-generation approaches for global EC profiling. Of note, the integration of chromatin immunoprecipitation, chromatin accessibility, and RNA sequencing has enabled identification of diverse DNA regulatory elements associated with disturbed flow and proinflammatory activation (e.g., NF-κB and hypoxia inducible factor 1α, as well as ETS, zinc finger, and activator protein 1 transcription factor families) (Hogan et al., 2017; Bondareva et al., 2019; Alizada et al., 2021). Moreover, combined single-cell RNA sequencing and genome-wide chromatin accessibility assays have been used to profile the genome- and epigenome-wide changes associated with proatherogenic ECs under oscillatory shear stress (Andueza et al., 2020). Likewise, these technologies have allowed for characterization of EC heterogeneity in human atherosclerotic plaque (Depuydt et al., 2020) and identification of coronary artery disease-associated genetic variants in the open chromatin regions of activated ECs (Örd et al., 2021). The future integration of global EC profiling with functional assays will aid in the validation of putative modulators of atherogenesis, which may serve as prospective therapeutic targets for CVD treatment. Notably, real-time monitoring of the vascular endothelium has been enabled with organ-on-a-chip technologies (Sei et al., 2017), allowing for specific and quantifiable testing to elucidate the impact of environmental stimuli on endothelial function. Nevertheless, the complex cellular milieu of the atherosclerotic plaque remains difficult to fully model.
Metabolic pathways prominently contribute to EC phenotypes in health and disease. Despite direct exposure to oxygen in the blood, ECs do not utilize oxidative phosphorylation as a primary means of energy production, perhaps because this might enhance oxidative stress and would hamper angiogenesis in hypoxic environments (Eelen et al., 2018). Instead, glycolysis serves as the primary method of energy delivery for ECs, in which 75–85% of ATP is generated via hexokinase 2-mediated phosphorylation of glucose to glucose-6-phosphate and conversion to lactate (Krützfeldt et al., 1990; De Bock et al., 2013; Yu et al., 2017). Fatty acid oxidation, used as a secondary source of energy by ECs, is modulated by carnitine palmitoyltransferase 1A-mediated shuttling of fatty acids to the mitochondria and ATP production via adenosine monophosphate activated protein kinase signaling (Dagher et al., 2001; Currie et al., 2013). Alternatively, fatty acids can be generated within ECs via fatty acid synthase, which has important functions for EC migration, permeability, and eNOS-mediated activity (Wei et al., 2011; Hagberg et al., 2013). Moreover, the proliferative and vasodilatory capacities of the endothelium can also be regulated by the metabolism of amino acids, in which inhibition of glutaminase-mediated conversion of glutamine to α-ketoglutarate represses angiogenesis (Kim B. et al., 2017), and the eNOS-induced conversion of arginine to NO controls vascular tone (Morris Jr, 2009).
Altered EC metabolism has been identified as both a consequence of, and a contributor to, endothelial dysfunction in atherosclerosis. In the quiescent endothelium, laminar shear stress reduces glucose uptake and glycolytic and mitochondrial activity in ECs via KLF2 (Doddaballapur et al., 2015). Conversely, in the diabetic and proatherogenic environment, elevated levels of circulating glucose induce the production of reactive oxygen species, DNA damage, and the accumulation of advanced glycation end products, which contribute to endothelial dysfunction via NF-κB signaling and increased vascular permeability (Theodorou and Boon, 2018). Disturbed flow similarly induces NF-κB and hypoxia inducible factor 1α expression, EC proliferation, and inflammation via upregulation of glycolytic enzymes (Feng et al., 2017), and can also promote EC activation and atherosclerosis through YAP/tafazzin (TAZ) signaling (Wang K.-C. et al., 2016; Wang et al., 2016 L.). Importantly, YAP/TAZ signaling has been shown to induce EC glycolysis, and glycolytic activity can in turn upregulate the YAP/TAZ pathway (Enzo et al., 2015; Bertero et al., 2016; Kim J. et al., 2017), which has been hypothesized to result in a cyclical and sustained pro-inflammatory response in the perturbed endothelium (Theodorou and Boon, 2018). Emerging preclinical studies propose altered EC metabolism as a therapeutic target for mitigating the development of atherosclerosis, including coenzyme Q10-mediated activation of the AMP-activated protein kinase-YAP-optic atrophy protein 1 pathway to promote mitochondrial function and energy metabolism (Xie et al., 2020), and inhibition of the glycolytic regulator 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 to improve plaque stability in atheroprone mice (Poels et al., 2020). Although repressing glycolysis in the activated endothelium remains a promising strategy for treating patients with atherosclerotic CVD, consideration must be given to the complex interactions between genetics and environment (e.g., age, diet, diabetes, and cardiovascular fitness) that shape individual metabolic profiles to allow for patient-tailored therapies.
In its quiescent state, the vascular endothelium forms a semipermeable barrier between luminal and abluminal environments that allows for selective bidirectional movement of molecules via EC cell-cell junctions, vesicle-mediated transport within ECs, and diffusion between ECs or across endothelial gaps (Cahill and Redmond, 2016). In pioneering studies, LDL and other serum macromolecules were shown to enter the vessel wall using a paracellular route in permeable regions of the vasculature (Weinbaum et al., 1985; Lin et al., 1990). However, more recently, ECs have been implicated in the active transcytosis of LDL via caveolae, scavenger receptor B1, activin receptor-like kinase 1, LDL receptor, and high mobility group box protein 1, which contribute to the proinflammatory accumulation of LDL within the subendothelial space and promote atherogenesis (Zhang X. et al., 2018; Ghaffari et al., 2021). Loss of barrier function also potentiates atherosclerosis by facilitating leukocyte extravasation into the vessel wall through paracellular diapedesis from the vascular lumen, and may promote inflammatory infiltration through the vasa vasorum, contributing to subsequent plaque instability (Mulligan-Kehoe and Simons, 2014; Sluiter et al., 2021). Vascular insults including atherosclerosis, ischemia, and trauma are characterized by the accumulation of pathological proinflammatory mediators, which can induce acute (e.g., due to vascular injury) or chronic disruptions in endothelial permeability (e.g., due to plaque progression). Numerous mechanisms that underlie endothelial barrier disruption have been characterized, including protein kinase C-induced phosphorylation of cell junction proteins, which promotes actin reorganization and increased paracellular flux (Lum and Malik, 1996), and stimulation of myosin light chain kinase by inflammatory factors, which promotes EC retraction via actin-myosin network dynamics (Lum and Malik, 1996). Mediators including histamine, thrombin, and the proinflammatory cytokines interleukin (IL) 1 beta (IL-1β) and tumor necrosis factor α also increase permeability via modulation of tight junctions (e.g., zonula occludens 1 and occludins) and adhesion complexes (Lum and Malik, 1996). Furthermore, prolonged inflammation and oxidative stress can act to disrupt adherens junctions [e.g., vascular endothelial (VE)-cadherin] and gap junctions (e.g., connexins) via decreased NO (Komarova et al., 2017).
Emerging studies continue to uncover novel causal roles for established atherosclerosis mediators in the disruption of endothelial permeability. The mechanosensitive channel Piezo1, previously discussed as a coordinator of vascular structure and activator of integrin α5/NF-κB pathways, also promotes VE-cadherin degradation and increased vascular permeability under altered hemodynamics (Friedrich et al., 2019). Furthermore, the NOD like receptor family pyrin domain containing 3 (NLRP3) inflammasome, discussed in greater detail below as a therapeutic target for inflammation resolution in atherosclerosis, induces a loss of barrier in the diabetic vascular environment (Li X.-X. et al., 2019). Recent findings have also implicated the chemokine C–C motif ligand 8 in promoting atherosclerosis through enhanced permeability via NADPH oxidase 2 and reactive oxygen species (Xue et al., 2021), while the inhibition of insulin-like growth factor-1 signaling has been associated with disrupted endothelial barrier function in vitro and elevated atherosclerotic burden in mice (Higashi et al., 2020). The multifaceted roles of these atherogenic factors offer potential advantages for prospective therapies, which may simultaneously target multiple aspects of the atherosclerotic disease processes to limit plaque development (e.g., combined anti-inflammatory and pro-barrier effects of pharmacological agents that inhibit Piezo1 or NLRP3).
The endothelium is an essential regulator of vascular homeostasis, defined as the balance of vascular injury and repair, and dynamic changes in endothelial phenotype can both potentiate and limit atherogenesis and associated complications (Bäck et al., 2019; Chen P.-Y. et al., 2020). In the setting of chronic inflammatory diseases, including atherosclerosis, ECs undergo complete or partial endothelial-to-mesenchymal transition (EndMT), during which they lose endothelial properties and gain mesenchymal cell characteristics (e.g., extracellular matrix production and contractile function) (Souilhol et al., 2018; Chen P.-Y. et al., 2020). EndMT is driven by proatherogenic stimuli including inflammation and disturbed flow (Chen et al., 2015) and early activation of transforming growth factor beta signaling (Ma et al., 2020), and has garnered interest as an important pathophysiological mechanism for atherosclerotic CVD. Indeed, previous work has linked EndMT to neointimal hyperplasia (Chen et al., 2015; Moonen et al., 2015), increased leukocyte migration (Evrard et al., 2016), lipid uptake by lesional macrophages (Chen et al., 2019), and oxidative stress (Evrard et al., 2016) in the developing plaque (Figure 1). Moreover, statin therapy (Li Y. et al., 2021), histone deacetylase inhibitors (Chen et al., 2021), and microRNA inhibition (Wu et al., 2021) have recently been investigated as potential strategies for limiting EndMT, the latter of which was observed to reduce plaque formation in atherosclerotic mice.
Future study is warranted to address several outstanding research areas in EndMT and atherosclerosis, including the characterization of EndMT as a discrete versus continuous process, as well as the potential beneficial versus detrimental role of EndMT in plaque vulnerability. First, the proatherogenic endothelium has recently been shown to exist in a “metastable or partial” state of EndMT, where perturbed ECs perform both endothelial and mesenchymal functions (Helmke et al., 2019; Fledderus et al., 2021). The metastable nature of EndMT may be further elucidated with next-generation technologies that profile the atherosclerotic plaque and neighboring regions at the single-cell level, as recently used in studies of disturbed flow and endothelial reprogramming (Andueza et al., 2020) and diabetic atherogenesis (Zhao et al., 2021), as well as through the incorporation of computational models that predict endothelial activation and EndMT in various genetic and environmental conditions (Weinstein et al., 2020). Second, although augmentation of EndMT has previously been associated with increased plaque progression (Chen et al., 2015), emerging studies have highlighted a potential protective role of EndMT by maintaining plaque stability in the absence of SMC-derived myofibroblast-like cells (Evrard et al., 2016; Newman et al., 2021). Evidently, a more nuanced understanding of EndMT in the context of plaque development and rupture is required before therapeutic inhibition of EndMT is considered for the treatment of atherosclerotic CVD.
Vascular cell populations including ECs, monocytes, macrophages, and SMCs have well-established roles in driving atherosclerosis. ECs orchestrate the cellular interactions between these cell populations through the local expression of adhesion proteins and secretion of signaling molecules (Raines and Ferri, 2005; Liebner et al., 2006; Okamoto and Suzuki, 2017). The intercellular processes that mediate this pathophysiology have been a focus of previous studies, which have characterized EC-EC and EC-leukocyte communication via adherens junctions (e.g., VE-cadherin), tight junctions (e.g., junctional adhesion molecules), and other adhesion proteins including occludin, claudins, and platelet endothelial cell adhesion molecule (Liebner et al., 2006; Reglero-Real et al., 2016). Furthermore, a diverse group of EC-derived cytokines, chemokines, and other molecules (e.g., NO and endothelin-1) regulate vascular function by controlling proatherogenic processes such as leukocyte activation and SMC proliferation (Raines and Ferri, 2005; Gimbrone and García-Cardeña, 2016; Fledderus et al., 2021). Inflammatory cell recruitment to the activated endothelium is coordinated by the expression of EC adhesion proteins such as E-selectin, VCAM-1, and intercellular adhesion molecule-1 (ICAM-1), which facilitate leukocyte rolling and extravasation into the intimal layer, as well as the chemotactic factor monocyte chemoattractant protein-1, which attracts circulating monocytes, and the mitogen macrophage colony-stimulating factor, which stimulates monocyte proliferation and differentiation to intimal macrophages (Kleemann et al., 2008; Fledderus et al., 2021). Additionally, the proinflammatory polarization of macrophages has been associated with EndMT (Wu et al., 2017) and sprouting angiogenesis, the latter of which supports plaque progression through increased supply of oxygen and nutrients (Camaré et al., 2017; Graney et al., 2020). The developing atherosclerotic lesion is further characterized by intimal hyperplasia and neointimal formation, which are mediated by decreased production of EC-derived NO and a resultant increase in SMC proliferation, in conjunction with EndMT (Fledderus et al., 2021).
In recent years, vascular cell-cell communication mediated by the secretion of extracellular vesicles (i.e., EVs; nano-sized packages of proteins, mRNAs, noncoding RNAs, and lipids) has emerged as an important research area for the pathogenesis and treatment of atherosclerosis (Charla et al., 2020; Chen Y.-T. et al., 2020; Wang H. et al., 2020). Three broad categories of EVs – exosomes (30–150 nm in diameter), microparticles (100 nm–1 μm in diameter), and apoptotic bodies (1–5 μm in diameter) – have been shown to modulate vascular function in atherogenesis via intercellular signaling among ECs, as well as between ECs and other immune and non-immune cell types (He et al., 2018; Li M. et al., 2018; Charla et al., 2020; Peng et al., 2020) (Figure 1). Within the vascular endothelium, EC-derived EVs with distinct microRNA content have been observed to both induce (Jansen et al., 2013; Arderiu et al., 2015) and inhibit angiogenesis (Ou et al., 2011; Liang et al., 2017, 2) via paracrine regulation of nearby ECs, whereby EVs fuse to recipient cells, and the release of EV-derived microRNAs allows for silencing of complementary mRNA targets. Moreover, the uptake of EC-secreted microparticles by neighboring ECs was shown to promote ICAM-1 expression and monocyte adhesion via miR-222 in diabetic conditions (Jansen et al., 2015), and EV-derived apoptotic bodies containing miR-126 were found to reduce macrophage content in plaque and limit atherosclerotic burden (Zernecke et al., 2009). The specific role of the proatherogenic environment in governing EV loading and function remains of interest for future study, and characterization of this environment has begun with exposure of donor ECs to proatherogenic stimuli (i.e., oxLDL and IL-6), which were observed to decrease thrombospondin 1 and increase angiogenesis in recipient ECs via EV-derived miR-92a-3p (Liu Y. et al., 2019).
Beyond the endothelium, EC-EVs regulate both immune and non-immune vascular cell populations involved in the development of atherosclerosis (Li M. et al., 2018; He et al., 2018) and circulating EVs may have systemic effects at distant sites (Bär et al., 2019; Liu et al., 2021). Exosomal microRNAs mediate crosstalk between ECs and macrophages by suppressing (e.g., miR-10a) (Njock et al., 2015) or promoting monocyte activation (e.g., miR-155) (He et al., 2018), reducing macrophage infiltration, and delaying plaque progression (e.g., miR-126, miR-210, and miR-216) (Wang et al., 2019b). In turn, monocyte- and macrophage-derived EVs have been shown to induce EC apoptosis and promote murine atherosclerosis through elevated blood lipids, oxidative stress, and inflammation (Aharon et al., 2008; Li K. et al., 2021), as well as by reducing proliferation and angiogenesis in human coronary artery ECs via miR-503-5p (Wang et al., 2021). Reciprocal communication between cells of the vasculature has been similarly observed in ECs and SMCs, where EC-secreted EVs induce both atheroprone (Boyer et al., 2020; Yuan X. et al., 2020; Zhang Z. et al., 2020) and atheroprotective SMC phenotypes (Hergenreider et al., 2012; Xiang et al., 2021), while SMC-derived EVs govern endothelial migration under stimulation by platelet-derived growth factor (Heo et al., 2020) and can increase endothelial permeability and potentiate atherosclerosis via miR-155 (Zheng et al., 2017).
EC-EVs that are secreted into systemic circulation may serve as biomarkers for CVD and have the potential to govern cell behaviour at distant areas of the vasculature, although definitive evidence for this regulation is lacking (Bär et al., 2019; Liu et al., 2021). Preliminary animal studies have demonstrated that microRNAs secreted by circulating blood cells, and presumably contained within EVs, can modulate SMC activity in atherogenesis (Shan et al., 2015) and have proapoptotic and antiproliferative effects on ECs (Chu et al., 2017). Further investigation is warranted to determine whether circulating EVs are present in sufficient quantities to elicit systemic effects in patients and if their regulatory functions extend to other CVDs (e.g., cardiac fibrosis and ischemic heart disease), as well as to identify the precise mechanisms by which circulating EVs promote vascular dysfunction in recipient cells. Notably, future study is also required to determine whether EVs undergo transcytosis or pass through intercellular or intracellular gaps to the subendothelial space and elicit direct effects on medial cells, or are endocytosed by ECs, which then serve as indirect mediators of altered vascular function. The continued integration of EV profiling in patients with atherosclerosis (i.e., local EVs in plaque; circulating EVs in plasma) with experimental models of EV activity will shed light on the causal role of EVs and EV-contents in the setting of atherosclerotic CVD. Such models will require the transition from in vitro co-culture towards in vivo tracking approaches to allow for a robust, physiological characterization of EV activity, which will be made possible with advances in the visualization of EV release and uptake (Durak-Kozica et al., 2018; Oesterreicher et al., 2020). With respect to the diagnostic potential of vascular EVs, the application of machine learning strategies to predict CVD from circulating EV biomarkers has garnered recent interest (Burrello et al., 2020; Castellani et al., 2020) and will likely play an important role in the development of precision cardiovascular medicine over the coming decades.
Present therapeutic strategies to limit atherosclerosis and plaque rupture are broadly categorized as those which reduce atherosclerotic risk (e.g., lipid-lowering and antihypertensive agents) and those which prevent associated complications (e.g., antithrombotic agents) (Fledderus et al., 2021). Lipid-lowering therapies, including 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitors (i.e., statins) and proprotein convertase subtilisin/kexin type 9 inhibitors, reduce circulating levels of LDL-cholesterol and thus oxLDL in the vessel wall, and decrease the incidence of severe events in CVD (Silverman et al., 2016; Mach et al., 2020), although these agents are not sufficient to prevent plaque formation (Tran-Dinh et al., 2013; Moss and Ramji, 2016). Moreover, a spectrum of anti-atherogenic effects of statins on the endothelium has been previously discussed in detail (Xu et al., 2021), and includes reduced inflammation through NF-κB blockade (Greenwood and Mason, 2007), inhibition of EC apoptosis via Janus kinase 2/signal transducer and activator of transcription 3 signaling (Wang K. et al., 2020), protection against EndMT via Kruppel-like factor 4/miR-483 (He et al., 2017), epigenetic modulation of ECs through histone modification (Mohammadzadeh et al., 2020), and increased NO production via hydrogen sulfide and eNOS (Citi et al., 2021; Xu et al., 2021). Notably, the statin-mediated mechanisms that contribute to elevated eNOS activity include increased eNOS transcription via KLF2 (Parmar et al., 2005), improved eNOS mRNA stability via polyadenylation (Kosmidou et al., 2007), and increased eNOS phosphorylation via phosphatidylinositol 3-kinase/Akt signaling (Kureishi et al., 2000). Other anti-atherogenic treatments have also been observed to mitigate endothelial dysfunction by increasing NO production, such as antihypertensive agents (e.g., angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers) (Silva et al., 2019), antihyperglycemic drugs (e.g., insulin) (Muniyappa et al., 2008), and antioxidants (e.g., streptozotocin) (Varadharaj et al., 2017). In experimental settings, treatments for atherosclerosis have largely focused on antagonizing broad inflammatory pathways (e.g., IL-1β and 5-lipoxygenase) (Tardif et al., 2010; Ridker et al., 2017). Therapeutic agents that specifically target the perturbed endothelium to resolve inflammation and limit atherosclerosis, however, have not yet been fully realized in clinical studies (Fledderus et al., 2021).
The NLRP3 inflammasome is now recognized as a key player in the coordination of vascular inflammation and onset of atherogenesis (Duewell et al., 2010; Jin and Fu, 2019). NLRP3-mediated induction of atherosclerosis has been associated with a host of causal factors including hypoxia (Folco et al., 2014), cholesterol crystals and oxLDL (Duewell et al., 2010), and disturbed flow (Xiao et al., 2013). Activation of NLRP3 subsequently leads to the maturation of proinflammatory cytokines (e.g., IL-1β and IL-18), increases the migration and lipid loading of macrophages (Li X. et al., 2014), and promotes pyroptosis, a proinflammatory form of programmed cell death that contributes to the release of additional inflammatory mediators (Bergsbaken et al., 2009). Therapeutic agents that directly inhibit the NLRP3 inflammasome, including the natural compound arglabin (Abderrazak et al., 2015), colchicine (Fernando et al., 2017), and the small molecule inhibitor MCC950 (van der Heijden et al., 2017), have been shown to ameliorate endothelial inflammation and atherosclerosis in preclinical studies. Moreover, adjunct colchicine therapy has entered clinical trials for repressing vascular inflammation in acute coronary syndrome (Bouabdallaoui et al., 2020) and coronary artery disease (Nidorf et al., 2013; Kajikawa et al., 2019; Hays et al., 2021). Previous trials have employed several outcomes related to improved cardiovascular health, including CVD endpoints, serum inflammatory markers, and proxy measures for endothelial dysfunction including flow-mediated vasodilation and exercise-induced coronary blood flow. Although the latter two of these measures remain the gold standard for clinical assessment of the perturbed endothelium (Flammer et al., 2012; Xu et al., 2021), they rely on endothelial responsiveness to vasodilatory or vasoconstrictive manipulation and do not adequately capture changes in EC phenotype during atherogenesis (e.g., permeability, metabolism, EndMT, or intercellular communication). Emerging studies have begun to address this limitation by applying machine learning techniques to predict CVD events from clinical data, CT imaging, and circulating biomarkers, including matrix metalloproteinase 9 and polymeric immunoglobulin receptor (Tamarappoo et al., 2021), very low-density lipoprotein and leucine (Coelewij et al., 2021), and serum EVs (Burrello et al., 2020; Castellani et al., 2020). Alongside functional assessment of the endothelium, robust models for CVD prediction have the potential to identify subclinical disease (Coelewij et al., 2021), aid in risk stratification, and inform treatment and prevention strategies.
Recent studies have also highlighted the potential of endogenous specialized proresolving mediators (SPMs) as anti-inflammatory therapies for limiting endothelial inflammation in CVD (Fredman and Tabas, 2017; Xu et al., 2021). Resolvin D1 (RvD1) belongs to a family of SPMs formed from metabolic processing of polyunsaturated docosahexaenoic acid and eicosapentaenoic acid, and is synthesized by ECs under proinflammatory conditions including stimulation with oxLDL (Dufour et al., 2018). Treatment of ECs with RvD1 has been observed to repress lipopolysaccharide- and cholesterol crystal-stimulated EC-monocyte interactions in vitro by downregulating NFκB-ICAM-1/VCAM-1 signaling (Chattopadhyay et al., 2018; Pichavaram et al., 2019), and attenuates leukocyte trafficking to the endothelium in mice fed a cholesterol rich diet (Pichavaram et al., 2019). Beyond the resolvin family, the SPMs maresin 1 and lipoxin A4 have likewise been observed to disrupt leukocyte adhesion to activated ECs (Filep et al., 1999; Chatterjee et al., 2014), suggesting that endogenous production or exogenous administration of diverse SPM metabolites may have therapeutic benefits for limiting endothelial activation in the setting of atherosclerosis.
Notably, the potential therapeutic impact of promoting inflammation resolution through efferocytosis (i.e., the clearing of apoptotic cells) is being established with preclinical studies, wherein atherosclerotic plaque progression has been attenuated by proefferocytic cluster of differentiation 47 (CD47)-blocking therapies (Kojima et al., 2016; Flores et al., 2020) or activation of the macrophage cell surface receptor MER proto-oncogene, tyrosine kinase (Thorp et al., 2008; Cai et al., 2018). While these effects appear to be mediated by lesional macrophages, ECs could facilitate the transport of inflammation resolving agents to cells within the developing plaque. Indeed, a proefferocytic and anti-atherosclerotic nanoparticle therapy has been previously hypothesized to function by uptake within circulating monocytes or delivery to macrophages within the intima (Flores et al., 2020), the latter of which would require the monocyte-independent transport of nanoparticles across the endothelium. In this role, ECs may contribute to therapeutic delivery by modulating vascular permeability and trafficking nanoparticles to macrophages via EC-secreted EVs. Clinically, the promise of inflammation resolving strategies has been highlighted in a subset of patients enrolled in a phase 1b-2 trial of the proefferocytic anti-CD47 antibody magrolimab for treating B-cell non-Hodgkin’s lymphoma, where vascular inflammation was significantly reduced in the carotid arteries of patients with CVD after 9 weeks of treatment (Jarr et al., 2021).
Epigenetic interventions allow for broad targeting of the diverse mechanisms underlying endothelial dysfunction and have shown recent promise as anti-atherosclerotic therapies (Fledderus et al., 2021). The methyltransferase enhancer of zeste homologue 2 (EZH2) and histone deacetylase SIRT1 respectively induce and limit endothelial dysfunction in murine models of CVD (Zhou et al., 2011; Lv et al., 2016) and represent novel avenues for epigenetic treatment. EZH2 antagonism and SIRT1 agonism similarly increase NO production (Gracia-Sancho et al., 2010; Kumar et al., 2013) and decrease EC activation (Zhou et al., 2011; Wang J. et al., 2020) and EndMT (Maleszewska et al., 2016; Liu Z.-H. et al., 2019) in experimental models of CVD (Figure 2). More recently, natural antioxidants including epigallocatechin and naringenin have been shown to reduce oxidative damage in ECs via SIRT1 (Li H. et al., 2021; Pai et al., 2021), and an intermittent fasting regimen was found to ameliorate vascular dysfunction in a murine model of diabetes by activating the SIRT1 pathway (Hammer et al., 2021). Small molecule agents that target EZH2 and SIRT1 are currently available or under clinical study in several disease areas (e.g., oncology, dermatology, and nephrology) (Ganesan et al., 2019; Fledderus et al., 2021), and may be investigated as potential anti-atherosclerotic agents by incorporating CVD endpoints into ongoing clinical trials. Beyond their desired therapeutic potential, the unintended effects of genome-wide modification via epigenetic agents also warrant consideration. Future studies may improve upon the specificity of these agents by making use of targeted CRISPR/Cas9 epigenome-editing techniques for epigenetic modifications associated with CVD risk loci (Cano-Rodriguez et al., 2016; Cano-Rodriguez and Rots, 2016; Xu et al., 2018). Alternatively, targeted EC therapies, as discussed below, may be considered for the resolution of vascular inflammation at the site of the activated endothelium in atherosclerosis. Prospective agents for targeted resolution include ICAM-1- or VCAM-1-targeted nanocarriers, which have previously been employed to limit neurovascular (Lutton et al., 2017; Marcos-Contreras et al., 2020) and pulmonary inflammation (Li S. et al., 2018; Park et al., 2021) in experimental models of disease.
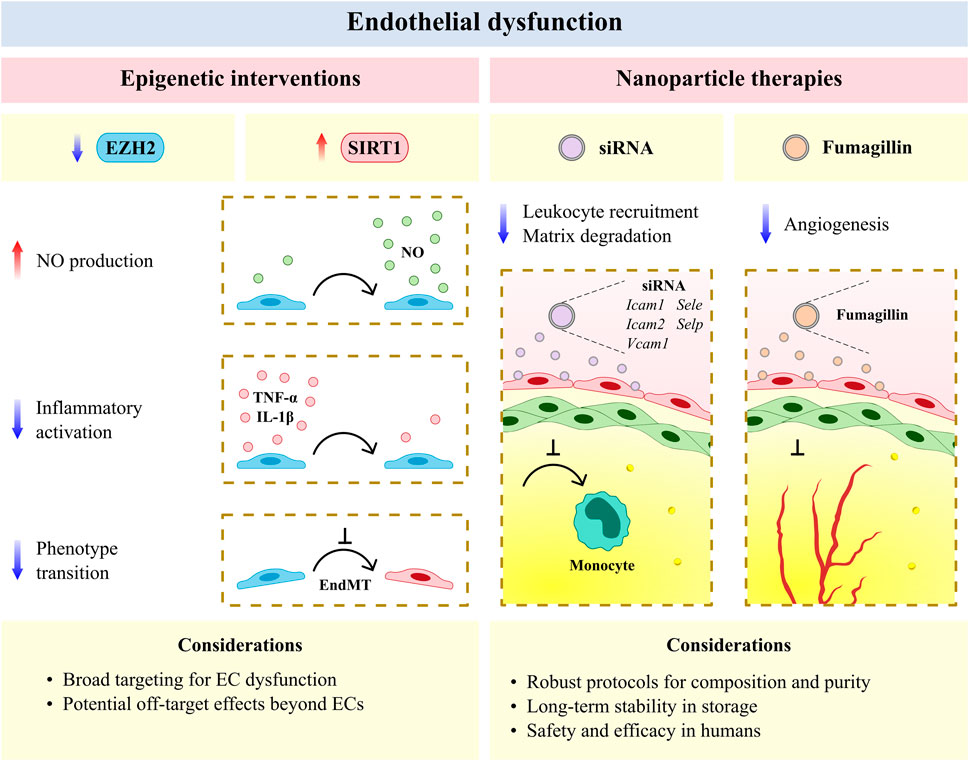
FIGURE 2. The vascular endothelium is an emerging therapeutic target for atherosclerotic cardiovascular disease. Epigenic interventions including enhancer of zeste homologue 2 (EZH2) antagonism and sirtuin 1 (SIRT1) agonism allow for broad targeting of endothelial cell (EC) dysfunction but may have undesirable effects in other cell types and tissues. Anti-inflammatory and anti-angiogenic nanotherapies have reduced off-target effects and increased efficacy per dose, but require rigorous protocols for assessing composition and purity, as well as the development of long-term stability in storage and safety and efficacy in humans with cardiovascular disease. EndMT, endothelial-to-mesenchymal transition; Icam1, intercellular adhesion molecule 1; Icam2, intercellular adhesion molecule 2; IL-1β, interleukin 1 beta; NO, nitric oxide; Sele, selectin E; Selp, selectin P; siRNA, small interfering RNA; TNF-α, tumor necrosis factor alpha; Vcam1, vascular cell adhesion molecule 1.
Advances in the development of nanoparticles (on a scale of <0.1 μm) have likewise afforded new opportunities for anti-atherosclerotic therapies that resolve endothelial dysfunction (Flores et al., 2019; Marchio et al., 2019; Chen L. et al., 2020). Vascular nanotherapies have been broadly classified as those which resolve inflammation and dysfunction in efferocytosis, limit plaque neovascularization and neointimal growth, modulate lipid metabolism, and decrease thrombosis (Flores et al., 2019). Several of these therapies have been shown to target the perturbed endothelium in experimental models of CVD, including the encapsulation of five cell adhesion molecule small interfering RNA, which reduced leukocyte migration to plaque and suppressed post-myocardial infarction inflammation in murine models (Sager et al., 2016). Integrin-targeted nanoparticles containing the antiangiogenic compound fumagillin have likewise been shown to limit neovascularization in mice and demonstrate prolonged activity when combined with statin treatment (Winter et al., 2006; Winter et al., 2008) (Figure 2). More recently, engineered endothelial adrenoreceptor- (Ul Ain et al., 2017), VCAM-1- (Distasio et al., 2021), and P-selectin-targeted nanoparticles (Mocanu et al., 2021), which respectively carried the genes eNOS and IL-10 and receptor for advanced glycation end products-silencing RNA, have been found to localize to ECs and resolve inflammation within the murine atherosclerotic plaque. Nano-selenium particles have also been observed to improve endothelial dysfunction in murine atherosclerosis via Na+/H+ exchanger 1 inhibition (Zhu et al., 2019). Despite the therapeutic advantages offered by nanoparticles (e.g., reduced off-target effects and increased efficacy per dose), several methodological and biological challenges remain for clinical translation (Flores et al., 2019). Importantly, robust protocols are required to determine the composition and purity of nanoparticles derived using various formulations (e.g., lipid or polymeric). Furthermore, the long-term stability of nanoparticles, as well as their safety and selectivity for the activated endothelium in patients with atherosclerosis, must be assessed before these agents are clinically adopted. Nevertheless, vascular nanotherapies remain a promising avenue for the targeted resolution of endothelial dysfunction and reduction of plaque progression in atherosclerotic CVD, with encouraging evidence being generated for the systemic administration of nanoparticle-enveloped small interfering RNA in nonhuman primates to inhibit EC gene expression in multiple organs (Khan et al., 2018).
An emerging body of literature has implicated the vascular endothelium as an essential driver of atherosclerosis, a disease process that contributes to substantial mortality and healthcare burden among aging populations. These discoveries shed light on diverse cellular mechanisms that underlie endothelial activation, dysfunctional cell-cell communication, and perturbed vascular homeostasis in atherogenesis. Moreover, they highlight novel therapeutic targets and delivery methods with significant potential to prevent or limit plaque development in patients with atherosclerotic CVD. Although a broad spectrum of intra- and intercellular processes has been implicated in preclinical models of endothelial dysfunction and atherosclerosis, several methodological challenges remain to effectively translate this research to clinical practice. First, the generalizability of findings between diverse in vitro and in vivo models of endothelial activation and atherosclerosis remains an important consideration to identify strategies that hold significant therapeutic potential for distinct plaque locations and diverse patient groups. Additionally, the safety, efficacy, timing, and sustained response of candidate treatments must be considered, as the atherosclerotic disease process develops over decades and necessitates long-term intervention. A robust integration of experimental techniques (e.g., high-throughput EC profiling with functional validation), data sources (e.g., human samples with experimental models), and cutting-edge computational methods (e.g., machine learning) will be required for the creation of next-generation biomarkers and therapies that effectively mitigate atherosclerotic CVD in vulnerable patients.
SB synthesized literature and drafted the review. JF and KH participated in manuscript editing and supervision. All authors have read and approve the published version of the manuscript.
SB is supported by a University of Toronto MD/PhD Studentship and a Canadian Institutes of Health Research (CIHR) Canada Graduate Scholarship – Doctoral (CGS D). Research in the laboratory of JF is supported by CIHR Project Grants (PJT148487, PJT173489, and PJT175301) and Medicine by Design, which received funding from the Canada First Research Excellence Fund. JF is supported by a Tier 2 Canada Research Chair in Vascular Cell and Molecular Biology from CIHR. KH is supported by a CIHR Project Grant (PJT178006) and the Wylie Scholar Award (Vascular Cures), Blair Early Career Professorship in Vascular Surgery (University of Toronto), Peter Munk Cardiac Centre, and University Health Network.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
BioRender.com was used in the creation of figures for this review.
CD47, cluster of differentiation 47; CVD, cardiovascular disease; EC, endothelial cell; EndMT, endothelial-to-mesenchymal transition; eNOS, endothelial nitric oxide synthase; EV, extracellular vesicle; EZH2, enhancer of zeste homologue 2; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; IL-1β, interleukin 1 beta; KLF2, Kruppel-like factor 2; LDL, low-density lipoprotein; NF-κB, nuclear factor kappa B; NLRP3, NOD like receptor family pyrin domain containing 3; oxLDL, oxidized low-density lipoprotein; RvD1, resolvin D1; SIRT1, sirtuin 1; SMC, smooth muscle cell; SPM, specialized proresolving mediator; TAZ, tafazzin; VCAM-1, vascular cell adhesion molecule 1; VE-cadherin, vascular endothelial-cadherin; YAP, yes-associated protein.
Abderrazak, A., Couchie, D., Mahmood, D. F., Elhage, R., Vindis, C., and Laffargue, M. (2015). Anti-Inflammatory and Antiatherogenic Effects of the NLRP3 Inflammasome Inhibitor Arglabin in ApoE2.Ki Mice Fed a High-Fat Diet. Circulation 131, 1061–1070. doi:10.1161/CIRCULATIONAHA.114.013730
Aharon, A., Tamari, T., and Brenner, B. (2008). Monocyte-derived Microparticles and Exosomes Induce Procoagulant and Apoptotic Effects on Endothelial Cells. Thromb. Haemost. 100, 878–885. doi:10.1160/th07-11-0691
Albarrán-Juárez, J., Iring, A., Wang, S., Joseph, S., Grimm, M., Strilic, B., et al. (2018). Piezo1 and Gq/G11 Promote Endothelial Inflammation Depending on Flow Pattern and Integrin Activation. J. Exp. Med. 215, 2655–2672. doi:10.1084/jem.20180483
Alizada, A., Khyzha, N., Wang, L., Antounians, L., Chen, X., Khor, M., et al. (2021). Conserved Regulatory Logic at Accessible and Inaccessible Chromatin During the Acute Inflammatory Response in Mammals. Nat. Commun. 12, 567. doi:10.1038/s41467-020-20765-1
Andueza, A., Kumar, S., Kim, J., Kang, D.-W., Mumme, H. L., Perez, J. I., et al. (2020). Endothelial Reprogramming by Disturbed Flow Revealed by Single-Cell RNA and Chromatin Accessibility Study. Cell Rep. 33, 108491. doi:10.1016/j.celrep.2020.108491
Arderiu, G., Peña, E., and Badimon, L. (2015). Angiogenic Microvascular Endothelial Cells Release Microparticles Rich in Tissue Factor that Promotes Postischemic Collateral Vessel Formation. Arterioscler. Thromb. Vasc. Biol. 35, 348–357. doi:10.1161/ATVBAHA.114.303927
Bäck, M., Yurdagul, A., Tabas, I., Öörni, K., and Kovanen, P. T. (2019). Inflammation and its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities. Nat. Rev. Cardiol. 16, 389–406. doi:10.1038/s41569-019-0169-2
Bär, C., Thum, T., and de Gonzalo-Calvo, D. (2019). Circulating miRNAs as Mediators in Cell-To-Cell Communication. Epigenomics 11, 111–113. doi:10.2217/epi-2018-0183
Bennett, M. R., Sinha, S., and Owens, G. K. (2016). Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 118, 692–702. doi:10.1161/CIRCRESAHA.115.306361
Bentzon, J. F., Otsuka, F., Virmani, R., and Falk, E. (2014). Mechanisms of Plaque Formation and Rupture. Circ. Res. 114, 1852–1866. doi:10.1161/CIRCRESAHA.114.302721
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 7, 99–109. doi:10.1038/nrmicro2070
Berk, B. C. (2008). Atheroprotective Signaling Mechanisms Activated by Steady Laminar Flow in Endothelial Cells. Circulation 117, 1082–1089. doi:10.1161/CIRCULATIONAHA.107.720730
Bertero, T., Oldham, W. M., Cottrill, K. A., Pisano, S., Vanderpool, R. R., Yu, Q., et al. (2016). Vascular Stiffness Mechanoactivates YAP/TAZ-dependent Glutaminolysis to Drive Pulmonary Hypertension. J. Clin. Invest. 126, 3313–3335. doi:10.1172/JCI86387
Bondareva, O., Tsaryk, R., Bojovic, V., Odenthal-Schnittler, M., Siekmann, A. F., and Schnittler, H.-J. (2019). Identification of Atheroprone Shear Stress Responsive Regulatory Elements in Endothelial Cells. Cardiovasc. Res. 115, 1487–1499. doi:10.1093/cvr/cvz027
Bouabdallaoui, N., Tardif, J.-C., Waters, D. D., Pinto, F. J., Maggioni, A. P., Diaz, R., et al. (2020). Time-to-treatment Initiation of Colchicine and Cardiovascular Outcomes after Myocardial Infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur. Heart J. 41, 4092–4099. doi:10.1093/eurheartj/ehaa659
Boyer, M. J., Kimura, Y., Akiyama, T., Baggett, A. Y., Preston, K. J., Scalia, R., et al. (2020). Endothelial Cell-Derived Extracellular Vesicles Alter Vascular Smooth Muscle Cell Phenotype through High-Mobility Group Box Proteins. J. Extracell. Vesicles 9, 1781427. doi:10.1080/20013078.2020.1781427
Boyle, E. C., Sedding, D. G., and Haverich, A. (2017). Targeting Vasa Vasorum Dysfunction to Prevent Atherosclerosis. Vascul. Pharmacol. 96–98, 5–10. doi:10.1016/j.vph.2017.08.003
Burrello, J., Biemmi, V., Dei Cas, M., Amongero, M., Bolis, S., Lazzarini, E., et al. (2020). Sphingolipid Composition of Circulating Extracellular Vesicles after Myocardial Ischemia. Sci. Rep. 10, 16182. doi:10.1038/s41598-020-73411-7
Cahill, P. A., and Redmond, E. M. (2016). Vascular Endothelium – Gatekeeper of Vessel Health. Atherosclerosis 248, 97–109. doi:10.1016/j.atherosclerosis.2016.03.007
Cai, B., Kasikara, C., Doran, A. C., Ramakrishnan, R., Birge, R. B., and Tabas, I. (2018). MerTK Signaling in Macrophages Promotes the Synthesis of Inflammation Resolution Mediators by Suppressing CaMKII Activity. Sci. Signal. 11, eaar3721. doi:10.1126/scisignal.aar3721
Camaré, C., Pucelle, M., Nègre-Salvayre, A., and Salvayre, R. (2017). Angiogenesis in the Atherosclerotic Plaque. Redox Biol. 12, 18–34. doi:10.1016/j.redox.2017.01.007
Cano-Rodriguez, D., Gjaltema, R. A. F., Jilderda, L. J., Jellema, P., Dokter-Fokkens, J., Ruiters, M. H. J., et al. (2016). Writing of H3K4Me3 Overcomes Epigenetic Silencing in a Sustained but Context-dependent Manner. Nat. Commun. 7, 12284. doi:10.1038/ncomms12284
Cano-Rodriguez, D., and Rots, M. G. (2016). Epigenetic Editing: On the Verge of Reprogramming Gene Expression at Will. Curr. Genet. Med. Rep. 4, 170–179. doi:10.1007/s40142-016-0104-3
Castellani, C., Burrello, J., Fedrigo, M., Burrello, A., Bolis, S., Di Silvestre, D., et al. (2020). Circulating Extracellular Vesicles as Non-invasive Biomarker of Rejection in Heart Transplant. J. Heart Lung Transpl. 39, 1136–1148. doi:10.1016/j.healun.2020.06.011
Chai, Q., Lu, T., Wang, X.-L., and Lee, H.-C. (2015). Hydrogen Sulfide Impairs Shear Stress-Induced Vasodilation in Mouse Coronary Arteries. Pflüg. Arch. - Eur. J. Physiol. 467, 329–340. doi:10.1007/s00424-014-1526-y
Charla, E., Mercer, J., Maffia, P., and Nicklin, S. A. (2020). Extracellular Vesicle Signalling in Atherosclerosis. Cell. Signal. 75, 109751. doi:10.1016/j.cellsig.2020.109751
Chatterjee, A., Sharma, A., Chen, M., Toy, R., Mottola, G., and Conte, M. S. (2014). The Pro-resolving Lipid Mediator Maresin 1 (MaR1) Attenuates Inflammatory Signaling Pathways in Vascular Smooth Muscle and Endothelial Cells. PLOS ONE 9, e113480. doi:10.1371/journal.pone.0113480
Chattopadhyay, R., Mani, A. M., Singh, N. K., and Rao, G. N. (2018). Resolvin D1 Blocks H2O2-Mediated Inhibitory Crosstalk between SHP2 and PP2A and Suppresses Endothelial-Monocyte Interactions. Free Radic. Biol. Med. 117, 119–131. doi:10.1016/j.freeradbiomed.2018.01.034
Chen, L., Jiang, Z., Akakuru, O. U., Yang, L., Li, J., Ma, S., et al. (2020a). Recent Progress in the Detection and Treatment of Atherosclerosis by Nanoparticles. Mater. Today Chem. 17, 100280. doi:10.1016/j.mtchem.2020.100280
Chen, L., Shang, C., Wang, B., Wang, G., Jin, Z., Yao, F., et al. (2021). HDAC3 Inhibitor Suppresses Endothelial-To-Mesenchymal Transition via Modulating Inflammatory Response in Atherosclerosis. Biochem. Pharmacol. 192, 114716. doi:10.1016/j.bcp.2021.114716
Chen, P.-Y., Qin, L., Baeyens, N., Li, G., Afolabi, T., Budatha, M., et al. (2015). Endothelial-to-mesenchymal Transition Drives Atherosclerosis Progression. J. Clin. Invest. 125, 4514–4528. doi:10.1172/JCI82719
Chen, P.-Y., Qin, L., Li, G., Wang, Z., Dahlman, J. E., Malagon-Lopez, J., et al. (2019). Endothelial TGF-β Signalling Drives Vascular Inflammation and Atherosclerosis. Nat. Metab. 1, 912–926. doi:10.1038/s42255-019-0102-3
Chen, P.-Y., Schwartz, M. A., and Simons, M. (2020b). Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front. Cardiovasc. Med. 7, 53. doi:10.3389/fcvm.2020.00053
Chen, Y.-T., Yuan, H.-X., Ou, Z.-J., and Ou, J.-S. (2020c). Microparticles (Exosomes) and Atherosclerosis. Curr. Atheroscler. Rep. 22, 23. doi:10.1007/s11883-020-00841-z
Chong, W., Baker, B. M., Chen, C. S., and Schwartz, M. A. (2013). Endothelial Cell Sensing of Flow Direction. Arterioscler. Thromb. Vasc. Biol. 33, 2130–2136. doi:10.1161/ATVBAHA.113.301826
Chu, M., Wu, R., Qin, S., Hua, W., Shan, Z., Xing, R., et al. (2017). Bone Marrow–Derived MicroRNA‐223 Works as an Endocrine Genetic Signal in Vascular Endothelial Cells and Participates in Vascular Injury from Kawasaki Disease. J. Am. Heart Assoc. 6, e004878. doi:10.1161/JAHA.116.004878
Citi, V., Martelli, A., Gorica, E., Brogi, S., Testai, L., and Calderone, V. (2021). Role of Hydrogen Sulfide in Endothelial Dysfunction: Pathophysiology and Therapeutic Approaches. J. Adv. Res. 27, 99–113. doi:10.1016/j.jare.2020.05.015
Coelewij, L., Waddington, K. E., Robinson, G. A., Chocano, E., McDonnell, T., Farinha, F., et al. (2021). Serum Metabolomic Signatures Can Predict Subclinical Atherosclerosis in Patients with Systemic Lupus Erythematosus. Arterioscler. Thromb. Vasc. Biol. 41, 1446–1458. doi:10.1161/ATVBAHA.120.315321
Collins, T., and Cybulsky, M. I. (2001). NF-κB: Pivotal Mediator or Innocent Bystander in Atherogenesis? J. Clin. Invest. 107, 255–264. doi:10.1172/JCI10373
Currie, E., Schulze, A., Zechner, R., Walther, T. C., and Farese, R. V. (2013). Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 18, 153–161. doi:10.1016/j.cmet.2013.05.017
da Luz, P. L., Chagas, A. C. P., Dourado, P. M. M., and Laurindo, F. R. M. (2018). “Chapter 33 - Endothelium in Atherosclerosis: Plaque Formation and its Complications,” in Endothelium And Cardiovascular Diseases. Editors P. L. Da Luz, P. Libby, A. C. P. Chagas, and F. R. M. Laurindo (Academic Press), 493–512. doi:10.1016/b978-0-12-812348-5.00033-7
Dagher, Z., Ruderman, N., Tornheim, K., and Ido, Y. (2001). Acute Regulation of Fatty Acid Oxidation and AMP-Activated Protein Kinase in Human Umbilical Vein Endothelial Cells. Circ. Res. 88, 1276–1282. doi:10.1161/hh1201.092998
Dai, H., Much, A. A., Maor, E., Asher, E., Younis, A., Xu, Y., et al. (2020). Global, Regional, and National burden of Ischaemic Heart Disease and its Attributable Risk Factors, 1990–2017: Results from the Global Burden of Disease Study 2017. Eur. Heart J. - Qual. Care Clin. Outcomes, qcaa076. doi:10.1093/ehjqcco/qcaa076
De Bock, K., Georgiadou, M., Schoors, S., Kuchnio, A., Wong, B. W., Cantelmo, A. R., et al. (2013). Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 154, 651–663. doi:10.1016/j.cell.2013.06.037
Depuydt, M. A. C., Prange, K. H. M., Slenders, L., Örd, T., Elbersen, D., Boltjes, A., et al. (2020). Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 127, 1437–1455. doi:10.1161/CIRCRESAHA.120.316770
Distasio, N., Salmon, H., Dierick, F., Ebrahimian, T., Tabrizian, M., and Lehoux, S. (2021). VCAM-1-Targeted Gene Delivery Nanoparticles Localize to Inflamed Endothelial Cells and Atherosclerotic Plaques. Adv. Ther. 4, 2000196. doi:10.1002/adtp.202000196
Doddaballapur, A., Michalik, K. M., Manavski, Y., Lucas, T., Houtkooper, R. H., You, X., et al. (2015). Laminar Shear Stress Inhibits Endothelial Cell Metabolism via KLF2-Mediated Repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 35, 137–145. doi:10.1161/ATVBAHA.114.304277
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 464, 1357–1361. doi:10.1038/nature08938
Dufour, D., Khalil, A., Nuyens, V., Rousseau, A., Delporte, C., Noyon, C., et al. (2018). Native and Myeloperoxidase-Oxidized Low-Density Lipoproteins Act in Synergy to Induce Release of Resolvin-D1 from Endothelial Cells. Atherosclerosis 272, 108–117. doi:10.1016/j.atherosclerosis.2018.03.012
Durak-Kozica, M., Baster, Z., Kubat, K., and Stępień, E. (2018). 3D Visualization of Extracellular Vesicle Uptake by Endothelial Cells. Cell. Mol. Biol. Lett. 23, 57. doi:10.1186/s11658-018-0123-z
Eelen, G., de Zeeuw, P., Treps, L., Harjes, U., Wong, B. W., and Carmeliet, P. (2018). Endothelial Cell Metabolism. Physiol. Rev. 98, 3–58. doi:10.1152/physrev.00001.2017
Enzo, E., Santinon, G., Pocaterra, A., Aragona, M., Bresolin, S., Forcato, M., et al. (2015). Aerobic Glycolysis Tunes YAP/TAZ Transcriptional Activity. EMBO J. 34, 1349–1370. doi:10.15252/embj.201490379
Evrard, S. M., Lecce, L., Michelis, K. C., Nomura-Kitabayashi, A., Pandey, G., Purushothaman, K.-R., et al. (2016). Endothelial to Mesenchymal Transition Is Common in Atherosclerotic Lesions and Is Associated with Plaque Instability. Nat. Commun. 7, 1–16. doi:10.1038/ncomms11853
Feng, S., Bowden, N., Fragiadaki, M., Souilhol, C., Hsiao, S., Mahmoud, M., et al. (2017). Mechanical Activation of Hypoxia-Inducible Factor 1α Drives Endothelial Dysfunction at Atheroprone Sites. Arterioscler. Thromb. Vasc. Biol. 37, 2087–2101. doi:10.1161/ATVBAHA.117.309249
Fernando, S., Schwarz, N., Williamson, A., Toledo, D., Zareh, J., Bartolo, B. D., et al. (2017). Anti-Inflammatory Effects of Colchicine on Oxidised Low-Density Lipoproteins and Cholesterol Crystal-Induced Macrophage Activation In Vitro. Heart Lung Circ. 26, S69–S70. doi:10.1016/j.hlc.2017.06.060
Filep, J. G., Zouki, C., Petasis, N. A., Hachicha, M., and Serhan, C. N. (1999). Anti-Inflammatory Actions of Lipoxin A4 Stable Analogs Are Demonstrable in Human Whole Blood: Modulation of Leukocyte Adhesion Molecules and Inhibition of Neutrophil-Endothelial Interactions. Blood 94, 4132–4142. doi:10.1182/blood.V94.12.4132
Flammer, A. J., Anderson, T., Celermajer, D. S., Creager, M. A., Deanfield, J., Ganz, P., et al. (2012). The Assessment of Endothelial Function. Circulation 126, 753–767. doi:10.1161/CIRCULATIONAHA.112.093245
Fledderus, J., Vanchin, B., Rots, M. G., and Krenning, G. (2021). The Endothelium as a Target for Anti-atherogenic Therapy: A Focus on the Epigenetic Enzymes EZH2 and SIRT1. J. Pers. Med. 11, 103. doi:10.3390/jpm11020103
Flores, A. M., Hosseini-Nassab, N., Jarr, K.-U., Ye, J., Zhu, X., Wirka, R., et al. (2020). Pro-efferocytic Nanoparticles Are Specifically Taken up by Lesional Macrophages and Prevent Atherosclerosis. Nat. Nanotechnol. 15, 154–161. doi:10.1038/s41565-019-0619-3
Flores, A. M., Ye, J., Kai-Uwe, J., Niloufar, H. N., Smith Bryan, R., and Leeper Nicholas, J. (2019). Nanoparticle Therapy for Vascular Diseases. Arterioscler. Thromb. Vasc. Biol. 39, 635–646. doi:10.1161/ATVBAHA.118.311569
Folco, E. J., Sukhova, G. K., Quillard, T., and Libby, P. (2014). Moderate Hypoxia Potentiates Interleukin-1β Production in Activated Human Macrophages. Circ. Res. 115, 875–883. doi:10.1161/CIRCRESAHA.115.304437
Förstermann, U., Xia, N., and Li, H. (2017). Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 120, 713–735. doi:10.1161/CIRCRESAHA.116.309326
Fredman, G., and Tabas, I. (2017). Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol. 187, 1211–1221. doi:10.1016/j.ajpath.2017.01.018
Friedrich, E. E., Hong, Z., Xiong, S., Zhong, M., Di, A., Rehman, J., et al. (2019). Endothelial Cell Piezo1 Mediates Pressure-Induced Lung Vascular Hyperpermeability via Disruption of Adherens Junctions. Proc. Natl. Acad. Sci. 116, 12980–12985. doi:10.1073/pnas.1902165116
Ganesan, A., Arimondo, P. B., Rots, M. G., Jeronimo, C., and Berdasco, M. (2019). The Timeline of Epigenetic Drug Discovery: from Reality to Dreams. Clin. Epigenetics 11, 174. doi:10.1186/s13148-019-0776-0
Ghaffari, S., Jang, E., Naderinabi, F., Sanwal, R., Khosraviani, N., Wang, C., et al. (2021). Endothelial HMGB1 Is a Critical Regulator of LDL Transcytosis via an SREBP2-SR-BI Axis. Arterioscler. Thromb. Vasc. Biol. 41, 200–216. doi:10.1161/ATVBAHA.120.314557
Giddens, D. P., Zarins, C. K., and Glagov, S. (1993). The Role of Fluid Mechanics in the Localization and Detection of Atherosclerosis. J. Biomech. Eng. 115, 588–594. doi:10.1115/1.2895545
Gimbrone, M. A., and García-Cardeña, G. (2016). Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 118, 620–636. doi:10.1161/CIRCRESAHA.115.306301
Go, Y.-M., Lee, H.-R., and Park, H. (2012). H2S Inhibits Oscillatory Shear Stress-Induced Monocyte Binding to Endothelial Cells via Nitric Oxide Production. Mol. Cell 34, 449–455. doi:10.1007/s10059-012-0200-5
Gracia-Sancho, J., Villarreal, G., Zhang, Y., and García-Cardeña, G. (2010). Activation of SIRT1 by Resveratrol Induces KLF2 Expression Conferring an Endothelial Vasoprotective Phenotype. Cardiovasc. Res. 85, 514–519. doi:10.1093/cvr/cvp337
Graney, P. L., Ben-Shaul, S., Landau, S., Bajpai, A., Singh, B., Eager, J., et al. (2020). Macrophages of Diverse Phenotypes Drive Vascularization of Engineered Tissues. Sci. Adv. 6, eaay6391. doi:10.1126/sciadv.aay6391
Greenwood, J., and Mason, J. C. (2007). Statins and the Vascular Endothelial Inflammatory Response. Trends Immunol. 28, 88–98. doi:10.1016/j.it.2006.12.003
Hagberg, C., Mehlem, A., Falkevall, A., Muhl, L., and Eriksson, U. (2013). Endothelial Fatty Acid Transport: Role of Vascular Endothelial Growth Factor B. Physiology 28, 125–134. doi:10.1152/physiol.00042.2012
Hahn, C., and Schwartz, M. A. (2009). Mechanotransduction in Vascular Physiology and Atherogenesis. Nat. Rev. Mol. Cell Biol. 10, 53–62. doi:10.1038/nrm2596
Hajra, L., Evans, A. I., Chen, M., Hyduk, S. J., Collins, T., and Cybulsky, M. I. (2000). The NF-κB Signal Transduction Pathway in Aortic Endothelial Cells Is Primed for Activation in Regions Predisposed to Atherosclerotic Lesion Formation. Proc. Natl. Acad. Sci. 97, 9052–9057. doi:10.1073/pnas.97.16.9052
Haldar, S. M., and Stamler, J. S. (2013). S-nitrosylation: Integrator of Cardiovascular Performance and Oxygen Delivery. J. Clin. Invest. 123, 101–110. doi:10.1172/JCI62854
Hammer, S. S., Vieira, C. P., McFarland, D., Sandler, M., Levitsky, Y., Dorweiler, T. F., et al. (2021). Fasting and Fasting-Mimicking Treatment Activate SIRT1/LXRα and Alleviate Diabetes-Induced Systemic and Microvascular Dysfunction. Diabetologia 64, 1674–1689. doi:10.1007/s00125-021-05431-5
Hays, A. G., Schär, M., Barditch-Crovo, P., Bagchi, S., Bonanno, G., Meyer, J., et al. (2021). A Randomized, Placebo-Controlled, Double-Blinded Clinical Trial of Colchicine to Improve Vascular Health in People Living with HIV. AIDS Lond. Engl. 35, 1041–1050. doi:10.1097/QAD.0000000000002845
He, M., Chen, Z., Martin, M., Zhang, J., Sangwung, P., Woo, B., et al. (2017). miR-483 Targeting of CTGF Suppresses Endothelial-To-Mesenchymal Transition. Circ. Res. 120, 354–365. doi:10.1161/CIRCRESAHA.116.310233
He, S., Wu, C., Xiao, J., Li, D., Sun, Z., and Li, M. (2018). Endothelial Extracellular Vesicles Modulate the Macrophage Phenotype: Potential Implications in Atherosclerosis. Scand. J. Immunol. 87, e12648. doi:10.1111/sji.12648
Helmke, A., Casper, J., Nordlohne, J., David, S., Haller, H., Zeisberg, E. M., et al. (2019). Endothelial-to-mesenchymal Transition Shapes the Atherosclerotic Plaque and Modulates Macrophage Function. FASEB J. 33, 2278–2289. doi:10.1096/fj.201801238R
Heo, J., Yang, H. C., Rhee, W. J., and Kang, H. (2020). Vascular Smooth Muscle Cell-Derived Exosomal MicroRNAs Regulate Endothelial Cell Migration under PDGF Stimulation. Cells 9, 639. doi:10.3390/cells9030639
Hergenreider, E., Heydt, S., Tréguer, K., Boettger, T., Horrevoets, A. J. G., Zeiher, A. M., et al. (2012). Atheroprotective Communication between Endothelial Cells and Smooth Muscle Cells through miRNAs. Nat. Cell Biol. 14, 249–256. doi:10.1038/ncb2441
Higashi, Y., Sukhanov, S., Shai, S.-Y., Danchuk, S., Snarski, P., Li, Z., et al. (2020). Endothelial Deficiency of Insulin-like Growth Factor-1 Receptor Reduces Endothelial Barrier Function and Promotes Atherosclerosis in Apoe-Deficient Mice. Am. J. Physiol.-Heart Circ. Physiol. 319, H730–H743. doi:10.1152/ajpheart.00064.2020
Hogan, N. T., Whalen, M. B., Stolze, L. K., Hadeli, N. K., Lam, M. T., Springstead, J. R., et al. (2017). Transcriptional Networks Specifying Homeostatic and Inflammatory Programs of Gene Expression in Human Aortic Endothelial Cells. eLife 6, e22536. doi:10.7554/eLife.22536
Huang, B., Chen, C.-T., Chen, C.-S., Wang, Y.-M., Hsieh, H.-J., and Wang, D. L. (2015). Laminar Shear Flow Increases Hydrogen Sulfide and Activates a Nitric Oxide Producing Signaling cascade in Endothelial Cells. Biochem. Biophys. Res. Commun. 464, 1254–1259. doi:10.1016/j.bbrc.2015.07.115
Ignarro, L. J., Buga, G. M., Wei, L. H., Bauer, P. M., Wu, G., and del Soldato, P. (2001). Role of the Arginine-Nitric Oxide Pathway in the Regulation of Vascular Smooth Muscle Cell Proliferation. Proc. Natl. Acad. Sci. 98, 4202–4208. doi:10.1073/pnas.071054698
Jaipersad, A. S., Lip, G. Y. H., Silverman, S., and Shantsila, E. (2014). The Role of Monocytes in Angiogenesis and Atherosclerosis. J. Am. Coll. Cardiol. 63, 1–11. doi:10.1016/j.jacc.2013.09.019
Jansen, F., Yang, X., Baumann, K., Przybilla, D., Schmitz, T., Flender, A., et al. (2015). Endothelial Microparticles Reduce ICAM-1 Expression in a microRNA-222-dependent Mechanism. J. Cell. Mol. Med. 19, 2202–2214. doi:10.1111/jcmm.12607
Jansen, F., Yang, X., Hoelscher, M., Cattelan, A., Schmitz, T., Proebsting, S., et al. (2013). Endothelial Microparticle–Mediated Transfer of MicroRNA-126 Promotes Vascular Endothelial Cell Repair via SPRED1 and Is Abrogated in Glucose-Damaged Endothelial Microparticles. Circulation 128, 2026–2038. doi:10.1161/CIRCULATIONAHA.113.001720
Jarr, K.-U., Nakamoto, R., Doan, B. H., Kojima, Y., Weissman, I. L., Advani, R. H., et al. (2021). Effect of CD47 Blockade on Vascular Inflammation. N. Engl. J. Med. 384, 382–383. doi:10.1056/NEJMc2029834
Jin, Y., and Fu, J. (2019). Novel Insights into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Heart Assoc. 8, e012219. doi:10.1161/JAHA.119.012219
Kajikawa, M., Higashi, Y., Tomiyama, H., Maruhashi, T., Kurisu, S., Kihara, Y., et al. (2019). Effect of Short-Term Colchicine Treatment on Endothelial Function in Patients with Coronary Artery Disease. Int. J. Cardiol. 281, 35–39. doi:10.1016/j.ijcard.2019.01.054
Kang-Decker, N., Cao, S., Chatterjee, S., Yao, J., Egan, L. J., Semela, D., et al. (2007). Nitric Oxide Promotes Endothelial Cell Survival Signaling through S-Nitrosylation and Activation of Dynamin-2. J. Cell Sci. 120, 492–501. doi:10.1242/jcs.03361
Khan, O. F., Kowalski, P. S., Doloff, J. C., Tsosie, J. K., Bakthavatchalu, V., Winn, C. B., et al. (2018). Endothelial siRNA Delivery in Nonhuman Primates Using Ionizable Low–Molecular Weight Polymeric Nanoparticles. Sci. Adv. 4, eaar8409. doi:10.1126/sciadv.aar8409
Khyzha, N., Alizada, A., Wilson, M. D., and Fish, J. E. (2017). Epigenetics of Atherosclerosis: Emerging Mechanisms and Methods. Trends Mol. Med. 23, 332–347. doi:10.1016/j.molmed.2017.02.004
Kim, B., Li, J., Jang, C., and Arany, Z. (2017a). Glutamine Fuels Proliferation but Not Migration of Endothelial Cells. EMBO J. 36, 2321–2333. doi:10.15252/embj.201796436
Kim, J., Kim, Y. H., Kim, J., Park, D. Y., Bae, H., Lee, D.-H., et al. (2017b). YAP/TAZ Regulates Sprouting Angiogenesis and Vascular Barrier Maturation. J. Clin. Invest. 127, 3441–3461. doi:10.1172/JCI93825
Kleemann, R., Zadelaar, S., and Kooistra, T. (2008). Cytokines and Atherosclerosis: a Comprehensive Review of Studies in Mice. Cardiovasc. Res. 79, 360–376. doi:10.1093/cvr/cvn120
Kojima, Y., Volkmer, J.-P., McKenna, K., Civelek, M., Lusis, A. J., Miller, C. L., et al. (2016). CD47-blocking Antibodies Restore Phagocytosis and Prevent Atherosclerosis. Nature 536, 86–90. doi:10.1038/nature18935
Komarova, Y. A., Kruse, K., Mehta, D., and Malik, A. B. (2017). Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 120, 179–206. doi:10.1161/CIRCRESAHA.116.306534
Kosmidou, I., Moore, J. P., Weber, M., and Searles, C. D. (2007). Statin Treatment and 3′ Polyadenylation of eNOS mRNA. Arterioscler. Thromb. Vasc. Biol. 27, 2642–2649. doi:10.1161/ATVBAHA.107.154492
Krützfeldt, A., Spahr, R., Mertens, S., Siegmund, B., and Piper, H. M. (1990). Metabolism of Exogenous Substrates by Coronary Endothelial Cells in Culture. J. Mol. Cell. Cardiol. 22, 1393–1404. doi:10.1016/0022-2828(90)90984-A
Ku, K. H., Dubinsky, M. K., Sukumar, A. N., Subramaniam, N., Feasson, M. Y. M., Nair, R., et al. (2021). In Vivo Function of Flow-Responsive Cis-DNA Elements of eNOS Gene: A Role for Chromatin-Based Mechanisms. Circulation 144, 365–381. doi:10.1161/CIRCULATIONAHA.120.051078
Kumar, A., Kumar, S., Vikram, A., Hoffman, T. A., Naqvi, A., Lewarchik, C. M., et al. (2013). Histone and DNA Methylation–Mediated Epigenetic Downregulation of Endothelial Kruppel-like Factor 2 by Low-Density Lipoprotein Cholesterol. Arterioscler. Thromb. Vasc. Biol. 33, 1936–1942. doi:10.1161/ATVBAHA.113.301765
Kumar, S., Williams, D., Sur, S., Wang, J.-Y., and Jo, H. (2019). Role of Flow-Sensitive microRNAs and Long Noncoding RNAs in Vascular Dysfunction and Atherosclerosis. Vascul. Pharmacol. 114, 76–92. doi:10.1016/j.vph.2018.10.001
Kureishi, Y., Luo, Z., Shiojima, I., Bialik, A., Fulton, D., Lefer, D. J., et al. (2000). The HMG-CoA Reductase Inhibitor Simvastatin Activates the Protein Kinase Akt and Promotes Angiogenesis in Normocholesterolemic Animals. Nat. Med. 6, 1004–1010. doi:10.1038/79510
Lay, A. J., Coleman, P. R., Formaz-Preston, A., Ting, K. K., Roediger, B., Weninger, W., et al. (2019). ARHGAP18: A Flow‐Responsive Gene that Regulates Endothelial Cell Alignment and Protects against Atherosclerosis. J. Am. Heart Assoc. 8, e010057. doi:10.1161/JAHA.118.010057
Li, B., He, J., Lv, H., Liu, Y., Lv, X., Zhang, C., et al. (2019a). c-Abl Regulates YAPY357 Phosphorylation to Activate Endothelial Atherogenic Responses to Disturbed Flow. J. Clin. Invest. 129, 1167–1179. doi:10.1172/JCI122440
Li, H., Liu, L., Cao, Z., Li, W., Liu, R., Chen, Y., et al. (2021a). Naringenin Ameliorates Homocysteine Induced Endothelial Damage via the AMPKα/Sirt1 Pathway. J. Adv. Res. 34, 137–147. doi:10.1016/j.jare.2021.01.009
Li, J., Hou, B., Tumova, S., Muraki, K., Bruns, A., Ludlow, M. J., et al. (2014a). Piezo1 Integration of Vascular Architecture with Physiological Force. Nature 515, 279–282. doi:10.1038/nature13701
Li, K., Cui, M., Zhang, K., Wang, G., and Zhai, S. (2021b). M1 Macrophages-Derived Extracellular Vesicles Elevate microRNA-185-3p to Aggravate the Development of Atherosclerosis in ApoE-/- Mice by Inhibiting Small Mothers against Decapentaplegic 7. Int. Immunopharmacol. 90, 107138. doi:10.1016/j.intimp.2020.107138
Li, M., Qian, M., Kyler, K., and Xu, J. (2018a). Endothelial–Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front. Cardiovasc. Med. 5, 151. doi:10.3389/fcvm.2018.00151
Li, S., Chen, L., Wang, G., Xu, L., Hou, S., Chen, Z., et al. (2018b). Anti-ICAM-1 Antibody-Modified Nanostructured Lipid Carriers: a Pulmonary Vascular Endothelium-Targeted Device for Acute Lung Injury Therapy. J. Nanobiotechnology 16, 105. doi:10.1186/s12951-018-0431-5
Li, X.-X., Ling, S.-K., Hu, M.-Y., Ma, Y., Li, Y., and Huang, P.-L. (2019b). Protective Effects of Acarbose against Vascular Endothelial Dysfunction through Inhibiting Nox4/NLRP3 Inflammasome Pathway in Diabetic Rats. Free Radic. Biol. Med. 145, 175–186. doi:10.1016/j.freeradbiomed.2019.09.015
Li, X., Zhang, Y., Xia, M., Gulbins, E., Boini, K. M., and Li, P.-L. (2014b). Activation of Nlrp3 Inflammasomes Enhances Macrophage Lipid-Deposition and Migration: Implication of a Novel Role of Inflammasome in Atherogenesis. PLOS ONE 9, e87552. doi:10.1371/journal.pone.0087552
Li, Y., Zhang, Y.-X., Ning, D.-S., Chen, J., Li, S.-X., Mo, Z.-W., et al. (2021c). Simvastatin Inhibits POVPC-Mediated Induction of Endothelial-To-Mesenchymal Cell Transition. J. Lipid Res. 62, 100066. doi:10.1016/j.jlr.2021.100066
Liang, H., Li, S., and Chen, H. (2017). GW28-e0938 Endothelial Microparticles-Mediated Transfer of microRNA-19b,a Novel Messenger in Cell-Cell Communication, Plays a Key Role in Endothelial Migration and Angiogenesis. J. Am. Coll. Cardiol. 70, C34. doi:10.1016/j.jacc.2017.07.118
Libby, P. (2021). The Changing Landscape of Atherosclerosis. Nature 592, 524–533. doi:10.1038/s41586-021-03392-8
Liebner, S., Cavallaro, U., and Dejana, E. (2006). The Multiple Languages of Endothelial Cell-To-Cell Communication. Arterioscler. Thromb. Vasc. Biol. 26, 1431–1438. doi:10.1161/01.ATV.0000218510.04541.5e
Lin, S. J., Jan, K. M., and Chien, S. (1990). Role of Dying Endothelial Cells in Transendothelial Macromolecular Transport. Arteriosclerosis 10, 703–709. doi:10.1161/01.atv.10.5.703
Liu, Q., Piao, H., Wang, Y., Zheng, D., and Wang, W. (2021). Circulating Exosomes in Cardiovascular Disease: Novel Carriers of Biological Information. Biomed. Pharmacother. 135, 111148. doi:10.1016/j.biopha.2020.111148
Liu, Y., Li, Q., Hosen, M. R., Zietzer, A., Flender, A., Levermann, P., et al. (2019a). Atherosclerotic Conditions Promote the Packaging of Functional MicroRNA-92a-3p into Endothelial Microvesicles. Circ. Res. 124, 575–587. doi:10.1161/CIRCRESAHA.118.314010
Liu, Z.-H., Zhang, Y., Wang, X., Fan, X.-F., Zhang, Y., Li, X., et al. (2019b). SIRT1 Activation Attenuates Cardiac Fibrosis by Endothelial-To-Mesenchymal Transition. Biomed. Pharmacother. 118, 109227. doi:10.1016/j.biopha.2019.109227
Lum, H., and Malik, A. B. (1996). Mechanisms of Increased Endothelial Permeability. Can. J. Physiol. Pharmacol. 74, 787–800. doi:10.1139/y96-081
Lutton, E. M., Razmpour, R., Andrews, A. M., Cannella, L. A., Son, Y.-J., Shuvaev, V. V., et al. (2017). Acute Administration of Catalase Targeted to ICAM-1 Attenuates Neuropathology in Experimental Traumatic Brain Injury. Sci. Rep. 7, 3846. doi:10.1038/s41598-017-03309-4
Lv, H., Wang, H., Quan, M., Zhang, C., Fu, Y., Zhang, L., et al. (2021). Cartilage Oligomeric Matrix Protein fine-tunes Disturbed Flow-Induced Endothelial Activation and Atherogenesis. Matrix Biol. 95, 32–51. doi:10.1016/j.matbio.2020.10.003
Lv, Y.-C., Tang, Y.-Y., Zhang, P., Wan, W., Yao, F., He, P.-P., et al. (2016). Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis. PLOS ONE 11, e0157265. doi:10.1371/journal.pone.0157265
Ma, J., Sanchez-Duffhues, G., Goumans, M.-J., and ten Dijke, P. (2020). TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 8, 260. doi:10.3389/fcell.2020.00260
Mach, F., Baigent, C., Catapano, A. L., Koskinas, K. C., Casula, M., Badimon, L., et al. (2020). 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 41, 111–188. doi:10.1093/eurheartj/ehz455
Maleszewska, M., Vanchin, B., Harmsen, M. C., and Krenning, G. (2016). The Decrease in Histone Methyltransferase EZH2 in Response to Fluid Shear Stress Alters Endothelial Gene Expression and Promotes Quiescence. Angiogenesis 19, 9–24. doi:10.1007/s10456-015-9485-2
Mannarino, E., and Pirro, M. (2008). Endothelial Injury and Repair: A Novel Theory for Atherosclerosis. Angiology 59, 69S–72S. doi:10.1177/0003319708320761
Marchio, P., Guerra-Ojeda, S., Vila, J. M., Aldasoro, M., Victor, V. M., and Mauricio, M. D. (2019). Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, e8563845. doi:10.1155/2019/8563845
Marcos-Contreras, O. A., Greineder, C. F., Kiseleva, R. Y., Parhiz, H., Walsh, L. R., Zuluaga-Ramirez, V., et al. (2020). Selective Targeting of Nanomedicine to Inflamed Cerebral Vasculature to Enhance the Blood–Brain Barrier. Proc. Natl. Acad. Sci. 117, 3405–3414. doi:10.1073/pnas.1912012117
Matsushita, K., Morrell, C. N., Cambien, B., Yang, S.-X., Yamakuchi, M., Bao, C., et al. (2003). Nitric Oxide Regulates Exocytosis by S-Nitrosylation of N-Ethylmaleimide-Sensitive Factor. Cell 115, 139–150. doi:10.1016/S0092-8674(03)00803-1
Maurya, M. R., Gupta, S., Li, J. Y.-S., Ajami, N. E., Chen, Z. B., Shyy, J. Y.-J., et al. (2021). Longitudinal Shear Stress Response in Human Endothelial Cells to Atheroprone and Atheroprotective Conditions. Proc. Natl. Acad. Sci. 118, e2023236118. doi:10.1073/pnas.2023236118
Mehta, V., Pang, K.-L., Rozbesky, D., Nather, K., Keen, A., Lachowski, D., et al. (2020). The Guidance Receptor Plexin D1 Is a Mechanosensor in Endothelial Cells. Nature 578, 290–295. doi:10.1038/s41586-020-1979-4
Méndez-Barbero, N., Gutiérrez-Muñoz, C., and Blanco-Colio, L. M. (2021). Cellular Crosstalk between Endothelial and Smooth Muscle Cells in Vascular Wall Remodeling. Int. J. Mol. Sci. 22, 7284. doi:10.3390/ijms22147284
Mocanu, C. A., Fuior, E. V., Voicu, G., Rebleanu, D., Safciuc, F., Deleanu, M., et al. (2021). P-selectin Targeted RAGE-shRNA Lipoplexes Alleviate Atherosclerosis-Associated Inflammation. J. Controlled Release 338, 754–772. doi:10.1016/j.jconrel.2021.09.012
Mohammadzadeh, N., Montecucco, F., Carbone, F., Xu, S., Al-Rasadi, K., and Sahebkar, A. (2020). Statins: Epidrugs with Effects on Endothelial Health? Eur. J. Clin. Invest. 50, e13388. doi:10.1111/eci.13388
Moonen, J.-R. A. J., Lee, E. S., Schmidt, M., Maleszewska, M., Koerts, J. A., Brouwer, L. A., et al. (2015). Endothelial-to-mesenchymal Transition Contributes to Fibro-Proliferative Vascular Disease and Is Modulated by Fluid Shear Stress. Cardiovasc. Res. 108, 377–386. doi:10.1093/cvr/cvv175
Morris, S. M. (2009). Recent Advances in Arginine Metabolism: Roles and Regulation of the Arginases. Br. J. Pharmacol. 157, 922–930. doi:10.1111/j.1476-5381.2009.00278.x
Moss, J. W. E., and Ramji, D. P. (2016). Nutraceutical Therapies for Atherosclerosis. Nat. Rev. Cardiol. 13, 513–532. doi:10.1038/nrcardio.2016.103
Mulligan-Kehoe, M. J., and Simons, M. (2014). Vasa Vasorum in normal and Diseased Arteries. Circulation 129, 2557–2566. doi:10.1161/CIRCULATIONAHA.113.007189
Mundi, S., Massaro, M., Scoditti, E., Carluccio, M. A., van Hinsbergh, V. W. M., Iruela-Arispe, M. L., et al. (2018). Endothelial Permeability, LDL Deposition, and Cardiovascular Risk Factors—A Review. Cardiovasc. Res. 114, 35–52. doi:10.1093/cvr/cvx226
Muniyappa, R., Iantorno, M., and Quon, M. J. (2008). An Integrated View of Insulin Resistance and Endothelial Dysfunction. Endocrinol. Metab. Clin. North. Am. 37, 685–711. doi:10.1016/j.ecl.2008.06.001
Muzaffar, S., Shukla, N., Bond, M., Newby, A. C., Angelini, G. D., Sparatore, A., et al. (2008). Exogenous Hydrogen Sulfide Inhibits Superoxide Formation, NOX-1 Expression and Rac1 Activity in Human Vascular Smooth Muscle Cells. J. Vasc. Res. 45, 521–528. doi:10.1159/000129686
Newman, A. A. C., Serbulea, V., Baylis, R. A., Shankman, L. S., Bradley, X., Alencar, G. F., et al. (2021). Multiple Cell Types Contribute to the Atherosclerotic Lesion Fibrous Cap by PDGFRβ and Bioenergetic Mechanisms. Nat. Metab. 3, 166–181. doi:10.1038/s42255-020-00338-8
Ni, C.-W., Hsieh, H.-J., Chao, Y.-J., and Wang, D. L. (2003). Shear Flow Attenuates Serum-Induced STAT3 Activation in Endothelial Cells. J. Biol. Chem. 278, 19702–19708. doi:10.1074/jbc.M300893200
Nidorf, S. M., Eikelboom, J. W., Budgeon, C. A., and Thompson, P. L. (2013). Low-dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 61, 404–410. doi:10.1016/j.jacc.2012.10.027
Nigro, P., Abe, J.-I., and Berk, B. C. (2011). Flow Shear Stress and Atherosclerosis: a Matter of Site Specificity. Antioxid. Redox Signal. 15, 1405–1414. doi:10.1089/ars.2010.3679
Njock, M.-S., Cheng, H. S., Dang, L. T., Nazari-Jahantigh, M., Lau, A. C., Boudreau, E., et al. (2015). Endothelial Cells Suppress Monocyte Activation through Secretion of Extracellular Vesicles Containing Antiinflammatory microRNAs. Blood 125, 3202–3212. doi:10.1182/blood-2014-11-611046
Oesterreicher, J., Pultar, M., Schneider, J., Mühleder, S., Zipperle, J., Grillari, J., et al. (2020). Fluorescence-Based Nanoparticle Tracking Analysis and Flow Cytometry for Characterization of Endothelial Extracellular Vesicle Release. Int. J. Mol. Sci. 21, 9278. doi:10.3390/ijms21239278
Okamoto, T., and Suzuki, K. (2017). The Role of Gap Junction-Mediated Endothelial Cell–Cell Interaction in the Crosstalk between Inflammation and Blood Coagulation. Int. J. Mol. Sci. 18, 2254. doi:10.3390/ijms18112254
Örd, T., Õunap, K., Stolze, L. K., Aherrahrou, R., Nurminen, V., Toropainen, A., et al. (2021). Single-Cell Epigenomics and Functional Fine-Mapping of Atherosclerosis GWAS Loci. Circ. Res. 129, 240–258. doi:10.1161/CIRCRESAHA.121.318971
Ou, Z.-J., Chang, F.-J., Luo, D., Liao, X.-L., Wang, Z.-P., Zhang, X., et al. (2011). Endothelium-derived Microparticles Inhibit Angiogenesis in the Heart and Enhance the Inhibitory Effects of Hypercholesterolemia on Angiogenesis. Am. J. Physiol.-Endocrinol. Metab. 300, E661–E668. doi:10.1152/ajpendo.00611.2010
Pai, P.-Y., Chou, W.-C., Chan, S.-H., Wu, S.-Y., Chen, H.-I., Li, C.-W., et al. (2021). Epigallocatechin Gallate Reduces Homocysteine-Caused Oxidative Damages through Modulation SIRT1/AMPK Pathway in Endothelial Cells. Am. J. Chin. Med. 49, 113–129. doi:10.1142/S0192415X21500063
Pan, S. (2009). Molecular Mechanisms Responsible for the Atheroprotective Effects of Laminar Shear Stress. Antioxid. Redox Signal. 11, 1669–1682. doi:10.1089/ars.2009.2487
Park, J. H., Jiang, Y., Zhou, J., Gong, H., Mohapatra, A., Heo, J., et al. (2021). Genetically Engineered Cell Membrane–Coated Nanoparticles for Targeted Delivery of Dexamethasone to Inflamed Lungs. Sci. Adv. 7, eabf7820. doi:10.1126/sciadv.abf7820
Parmar, K. M., Nambudiri, V., Dai, G., Larman, H. B., Gimbrone, M. A., and García-Cardeña, G. (2005). Statins Exert Endothelial Atheroprotective Effects via the KLF2 Transcription Factor. J. Biol. Chem. 280, 26714–26719. doi:10.1074/jbc.C500144200
Peng, M., Liu, X., and Xu, G. (2020). Extracellular Vesicles as Messengers in Atherosclerosis. J. Cardiovasc. Transl. Res. 13, 121–130. doi:10.1007/s12265-019-09923-z
Pfenniger, A., Chanson, M., and Kwak, B. R. (2013). Connexins in Atherosclerosis. Biochim. Biophys. Acta BBA - Biomembr. 1828, 157–166. doi:10.1016/j.bbamem.2012.05.011
Pi, X., Yan, C., and Berk, B. C. (2004). Big Mitogen-Activated Protein Kinase (BMK1)/ERK5 Protects Endothelial Cells from Apoptosis. Circ. Res. 94, 362–369. doi:10.1161/01.RES.0000112406.27800.6F
Pichavaram, P., Mani, A. M., Singh, N. K., and Rao, G. N. (2019). Cholesterol Crystals Promote Endothelial Cell and Monocyte Interactions via H2O2-Mediated PP2A Inhibition, NFκB Activation and ICAM1 and VCAM1 Expression. Redox Biol. 24, 101180. doi:10.1016/j.redox.2019.101180
Piepoli, M. F., Hoes, A. W., Agewall, S., Albus, C., Brotons, C., Catapano, A. L., et al. (2016). 2016 European Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 37, 2315–2381. doi:10.1093/eurheartj/ehw106
Poels, K., Schnitzler, J. G., Waissi, F., Levels, J. H. M., Stroes, E. S. G., Daemen, M. J. A. P., et al. (2020). Inhibition of PFKFB3 Hampers the Progression of Atherosclerosis and Promotes Plaque Stability. Front. Cell Dev. Biol. 8, 581641. doi:10.3389/fcell.2020.581641
Qu, D., Wang, L., Huo, M., Song, W., Lau, C.-W., Xu, J., et al. (2020). Focal TLR4 Activation Mediates Disturbed Flow-Induced Endothelial Inflammation. Cardiovasc. Res. 116, 226–236. doi:10.1093/cvr/cvz046
Raines, E. W., and Ferri, N. (2005). Thematic Review Series: The Immune System and Atherogenesis. Cytokines Affecting Endothelial and Smooth Muscle Cells in Vascular Disease. J. Lipid Res. 46, 1081–1092. doi:10.1194/jlr.R500004-JLR200
Reglero-Real, N., Colom, B., Bodkin, J. V., and Nourshargh, S. (2016). Endothelial Cell Junctional Adhesion Molecules. Arterioscler. Thromb. Vasc. Biol. 36, 2048–2057. doi:10.1161/ATVBAHA.116.307610
Ricard, N., Bailly, S., Guignabert, C., and Simons, M. (2021). The Quiescent Endothelium: Signalling Pathways Regulating Organ-specific Endothelial Normalcy. Nat. Rev. Cardiol. 18, 565–580. doi:10.1038/s41569-021-00517-4
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 377, 1119–1131. doi:10.1056/NEJMoa1707914
Sager, H. B., Dutta, P., Dahlman, J. E., Hulsmans, M., Courties, G., Sun, Y., et al. (2016). RNAi Targeting Multiple Cell Adhesion Molecules Reduces Immune Cell Recruitment and Vascular Inflammation after Myocardial Infarction. Sci. Transl. Med. 8, 342ra80. doi:10.1126/scitranslmed.aaf1435
Schober, A., Nazari-Jahantigh, M., Wei, Y., Bidzhekov, K., Gremse, F., Grommes, J., et al. (2014). MicroRNA-126-5p Promotes Endothelial Proliferation and Limits Atherosclerosis by Suppressing Dlk1. Nat. Med. 20, 368–376. doi:10.1038/nm.3487
Sedding, D. G., Boyle, E. C., Demandt, J. A. F., Sluimer, J. C., Dutzmann, J., Haverich, A., et al. (2018). Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front. Immunol. 9, 706. doi:10.3389/fimmu.2018.00706
Sei, Y. J., Ahn, S. I., Virtue, T., Kim, T., and Kim, Y. (2017). Detection of Frequency-dependent Endothelial Response to Oscillatory Shear Stress Using a Microfluidic Transcellular Monitor. Sci. Rep. 7, 10019. doi:10.1038/s41598-017-10636-z
SenBanerjee, S., Lin, Z., Atkins, G. B., Greif, D. M., Rao, R. M., Kumar, A., et al. (2004). KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 199, 1305–1315. doi:10.1084/jem.20031132
Shan, Z., Qin, S., Li, W., Wu, W., Yang, J., Chu, M., et al. (2015). An Endocrine Genetic Signal between Blood Cells and Vascular Smooth Muscle Cells. J. Am. Coll. Cardiol. 65, 2526–2537. doi:10.1016/j.jacc.2015.03.570
Silva, I. V. G., de Figueiredo, R. C., and Rios, D. R. A. (2019). Effect of Different Classes of Antihypertensive Drugs on Endothelial Function and Inflammation. Int. J. Mol. Sci. 20, 3458. doi:10.3390/ijms20143458
Silverman, M. G., Ference, B. A., Im, K., Wiviott, S. D., Giugliano, R. P., Grundy, S. M., et al. (2016). Association between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-Analysis. JAMA 316, 1289–1297. doi:10.1001/jama.2016.13985
Sluiter, T. J., van Buul, J. D., Huveneers, S., Quax, P. H. A., and de Vries, M. R. (2021). Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis. Biomedicines 9, 328. doi:10.3390/biomedicines9040328
Souilhol, C., Harmsen, M. C., Evans, P. C., and Krenning, G. (2018). Endothelial–mesenchymal Transition in Atherosclerosis. Cardiovasc. Res. 114, 565–577. doi:10.1093/cvr/cvx253
Stamler, J. S., Jia, L., Eu, J. P., McMahon, T. J., Demchenko, I. T., Bonaventura, J., et al. (1997). Blood Flow Regulation by S-Nitrosohemoglobin in the Physiological Oxygen Gradient. Science 276, 2034–2037. doi:10.1126/science.276.5321.2034
Swain, S. M., and Liddle, R. A. (2020). Piezo1 Acts Upstream of TRPV4 to Induce Pathological Changes in Endothelial Cells Due to Shear Stress. J. Biol. Chem. 296, 100171. doi:10.1074/jbc.RA120.015059
Tamarappoo, B. K., Lin, A., Commandeur, F., McElhinney, P. A., Cadet, S., Goeller, M., et al. (2021). Machine Learning Integration of Circulating and Imaging Biomarkers for Explainable Patient-specific Prediction of Cardiac Events: A Prospective Study. Atherosclerosis 318, 76–82. doi:10.1016/j.atherosclerosis.2020.11.008
Tardif, J.-C., L'Allier, P. L., Ibrahim, R., Grégoire, J. C., Nozza, A., Cossette, M., et al. (2010). Treatment with 5-Lipoxygenase Inhibitor VIA-2291 (Atreleuton) in Patients with Recent Acute Coronary Syndrome. Circ. Cardiovasc. Imaging 3, 298–307. doi:10.1161/CIRCIMAGING.110.937169
Theodorou, K., and Boon, R. A. (2018). Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 6, 82. doi:10.3389/fcell.2018.00082
Thorp, E., Cui, D., Schrijvers, D. M., Kuriakose, G., and Tabas, I. (2008). Mertk Receptor Mutation Reduces Efferocytosis Efficiency and Promotes Apoptotic Cell Accumulation and Plaque Necrosis in Atherosclerotic Lesions of Apoe-/- Mice. Arterioscler. Thromb. Vasc. Biol. 28, 1421–1428. doi:10.1161/ATVBAHA.108.167197
Tran‐Dinh, A., Diallo, D., Delbosc, S., Varela‐Perez, L. M., Dang, Q. B., Lapergue, B., et al. (2013). HDL and Endothelial protection. Br. J. Pharmacol. 169, 493–511. doi:10.1111/bph.12174
Ul Ain, Q., Chung, H., Chung, J. Y., Choi, J.-H., and Kim, Y.-H. (2017). Amelioration of Atherosclerotic Inflammation and Plaques via Endothelial Adrenoceptor-Targeted eNOS Gene Delivery Using Redox-Sensitive Polymer Bearing L-Arginine. J. Controlled Release 262, 72–86. doi:10.1016/j.jconrel.2017.07.019
van der Heijden, T., Kritikou, E., Venema, W., van Duijn, J., van Santbrink, P. J., Slütter, B., et al. (2017). NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice—Brief Report. Arterioscler. Thromb. Vasc. Biol. 37, 1457–1461. doi:10.1161/ATVBAHA.117.309575
VanderLaan, P. A., Reardon, C. A., and Getz, G. S. (2004). Site Specificity of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 12–22. doi:10.1161/01.ATV.0000105054.43931.f0
Varadharaj, S., Kelly, O. J., Khayat, R. N., Kumar, P. S., Ahmed, N., and Zweier, J. L. (2017). Role of Dietary Antioxidants in the Preservation of Vascular Function and the Modulation of Health and Disease. Front. Cardiovasc. Med. 4, 64. doi:10.3389/fcvm.2017.00064
Wang, H., Xie, Y., Salvador, A. M., Zhang, Z., Chen, K., Li, G., et al. (2020a). Exosomes: Multifaceted Messengers in Atherosclerosis. Curr. Atheroscler. Rep. 22, 57. doi:10.1007/s11883-020-00871-7
Wang, J., Li, P., Xu, X., Zhang, B., and Zhang, J. (2020b). MicroRNA-200a Inhibits Inflammation and Atherosclerotic Lesion Formation by Disrupting EZH2-Mediated Methylation of STAT3. Front. Immunol. 11, 907. doi:10.3389/fimmu.2020.00907
Wang, K.-C., Nguyen, P., Weiss, A., Yeh, Y.-T., Chien, H. S., Lee, A., et al. (2014). MicroRNA-23b Regulates Cyclin-dependent Kinase–Activating Kinase Complex through Cyclin H Repression to Modulate Endothelial Transcription and Growth under Flow. Arterioscler. Thromb. Vasc. Biol. 34, 1437–1445. doi:10.1161/ATVBAHA.114.303473
Wang, K.-C., Yeh, Y.-T., Nguyen, P., Limqueco, E., Lopez, J., Thorossian, S., et al. (2016a). Flow-dependent YAP/TAZ Activities Regulate Endothelial Phenotypes and Atherosclerosis. Proc. Natl. Acad. Sci. 113, 11525–11530. doi:10.1073/pnas.1613121113
Wang, K., Li, B., Xie, Y., Xia, N., Li, M., and Gao, G. (2020c). Statin Rosuvastatin Inhibits Apoptosis of Human Coronary Artery Endothelial Cells through Upregulation of the JAK2/STAT3 Signaling Pathway. Mol. Med. Rep. 22, 2052–2062. doi:10.3892/mmr.2020.11266
Wang, L., Luo, J.-Y., Li, B., Tian, X. Y., Chen, L.-J., Huang, Y., et al. (2016b). Integrin-YAP/TAZ-JNK cascade Mediates Atheroprotective Effect of Unidirectional Shear Flow. Nature 540, 579–582. doi:10.1038/nature20602
Wang, Y., Sun, H.-Y., Kumar, S., Puertadel, M. M., Jo, H., and Rezvan, A. (2019a). ZBTB46 Is a Shear-Sensitive Transcription Factor Inhibiting Endothelial Cell Proliferation via Gene Expression Regulation of Cell Cycle Proteins. Lab. Invest. 99, 305–318. doi:10.1038/s41374-018-0060-5
Wang, Y., Xie, Y., Zhang, A., Wang, M., Fang, Z., and Zhang, J. (2019b). Exosomes: An Emerging Factor in Atherosclerosis. Biomed. Pharmacother. 115, 108951. doi:10.1016/j.biopha.2019.108951
Wang, Y., Xu, Z., Wang, X., Zheng, J., Peng, L., Zhou, Y., et al. (2021). Extracellular-vesicle Containing miRNA-503-5p Released by Macrophages Contributes to Atherosclerosis. Aging 13, 12239–12257. doi:10.18632/aging.103855
Warboys, C. M., de Luca, A., Amini, N., Luong, L., Duckles, H., Hsiao, S., et al. (2014). Disturbed Flow Promotes Endothelial Senescence via a P53-dependent Pathway. Arterioscler. Thromb. Vasc. Biol. 34, 985–995. doi:10.1161/ATVBAHA.114.303415
Wei, X., Schneider, J. G., Shenouda, S. M., Lee, A., Towler, D. A., Chakravarthy, M. V., et al. (2011). De Novo Lipogenesis Maintains Vascular Homeostasis through Endothelial Nitric-Oxide Synthase (eNOS) Palmitoylation. J. Biol. Chem. 286, 2933–2945. doi:10.1074/jbc.M110.193037
Weinbaum, S., Tzeghai, G., Ganatos, P., Pfeffer, R., and Chien, S. (1985). Effect of Cell Turnover and Leaky Junctions on Arterial Macromolecular Transport. Am. J. Physiol. 248, H945–H960. doi:10.1152/ajpheart.1985.248.6.H945
Weinstein, N., Mendoza, L., and Álvarez-Buylla, E. R. (2020). A Computational Model of the Endothelial to Mesenchymal Transition. Front. Genet. 11, 40. doi:10.3389/fgene.2020.00040
White, S. J., Newby, A. C., and Johnson, T. W. (2016). Endothelial Erosion of Plaques as a Substrate for Coronary Thrombosis. Thromb. Haemost. 115, 509–519. doi:10.1160/TH15-09-0765
Winter, P. M., Caruthers, S. D., Zhang, H., Williams, T. A., Wickline, S. A., and Lanza, G. M. (2008). Antiangiogenic Synergism of Integrin-Targeted Fumagillin Nanoparticles and Atorvastatin in Atherosclerosis. JACC Cardiovasc. Imaging 1, 624–634. doi:10.1016/j.jcmg.2008.06.003
Winter, P. M., Neubauer, A. M., Caruthers, S. D., Harris, T. D., Robertson, J. D., Williams, T. A., et al. (2006). Endothelial ανβ3 Integrin–Targeted Fumagillin Nanoparticles Inhibit Angiogenesis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 26, 2103–2109. doi:10.1161/01.ATV.0000235724.11299.76
Won, D., Zhu, S.-N., Chen, M., Teichert, A.-M., Fish, J. E., Matouk, C. C., et al. (2007). Relative Reduction of Endothelial Nitric-Oxide Synthase Expression and Transcription in Atherosclerosis-Prone Regions of the Mouse Aorta and in an In Vitro Model of Disturbed Flow. Am. J. Pathol. 171, 1691–1704. doi:10.2353/ajpath.2007.060860
Wu, K. Q., Muratore, C. S., So, E.-Y., Sun, C., Dubielecka, P. M., Reginato, A. M., et al. (2017). M1 Macrophage–Induced Endothelial-To-Mesenchymal Transition Promotes Infantile Hemangioma Regression. Am. J. Pathol. 187, 2102–2111. doi:10.1016/j.ajpath.2017.05.014
Wu, X., Du, X., Yang, Y., Liu, X., Liu, X., Zhang, N., et al. (2021). Inhibition of miR-122 Reduced Atherosclerotic Lesion Formation by Regulating NPAS3-Mediated Endothelial to Mesenchymal Transition. Life Sci. 265, 118816. doi:10.1016/j.lfs.2020.118816
Xia, L., Zhang, B., Sun, Y., Chen, B., and Yu, Z. (2021). Analysis of Syk/PECAM-1 Signaling Pathway in Low Shear Stress Induced Atherosclerosis Based on Ultrasound Imaging. Comput. Methods Programs Biomed. 201, 105953. doi:10.1016/j.cmpb.2021.105953
Xiang, D., Li, Y., Cao, Y., Huang, Y., Zhou, L., Lin, X., et al. (2021). Different Effects of Endothelial Extracellular Vesicles and LPS-Induced Endothelial Extracellular Vesicles on Vascular Smooth Muscle Cells: Role of Curcumin and its Derivatives. Front. Cardiovasc. Med. 8, 649352. doi:10.3389/fcvm.2021.649352
Xiao, H., Lu, M., Lin, T. Y., Chen, Z., Chen, G., Wang, W.-C., et al. (2013). Sterol Regulatory Element Binding Protein 2 Activation of NLRP3 Inflammasome in Endothelium Mediates Hemodynamic-Induced Atherosclerosis Susceptibility. Circulation 128, 632–642. doi:10.1161/CIRCULATIONAHA.113.002714
Xie, T., Wang, C., Jin, Y., Meng, Q., Liu, Q., Wu, J., et al. (2020). CoenzymeQ10-Induced Activation of AMPK-YAP-OPA1 Pathway Alleviates Atherosclerosis by Improving Mitochondrial Function, Inhibiting Oxidative Stress and Promoting Energy Metabolism. Front. Pharmacol. 11, 1034. doi:10.3389/fphar.2020.01034
Xu, J., Lu, X., and Shi, G.-P. (2015). Vasa Vasorum in Atherosclerosis and Clinical Significance. Int. J. Mol. Sci. 16, 11574–11608. doi:10.3390/ijms160511574
Xu, Q. (2009). Disturbed Flow-Enhanced Endothelial Turnover in Atherosclerosis. Trends Cardiovasc. Med. 19, 191–195. doi:10.1016/j.tcm.2009.12.002
Xu, Q. (2007). Progenitor Cells in Vascular Repair. Curr. Opin. Lipidol. 18, 534–539. doi:10.1097/MOL.0b013e3282a66082
Xu, S., Ilyas, I., Little, P. J., Li, H., Kamato, D., Zheng, X., et al. (2021). Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 73, 924–967. doi:10.1124/pharmrev.120.000096
Xu, X., Tan, X., Tampe, B., Wilhelmi, T., Hulshoff, M. S., Saito, S., et al. (2018). High-fidelity CRISPR/Cas9- Based Gene-specific Hydroxymethylation Rescues Gene Expression and Attenuates Renal Fibrosis. Nat. Commun. 9, 3509. doi:10.1038/s41467-018-05766-5
Xue, S., Tang, H., Zhao, G., Fang, C., Shen, Y., Yan, D., et al. (2021). C–C Motif Ligand 8 Promotes Atherosclerosis via NADPH Oxidase 2/reactive Oxygen Species-Induced Endothelial Permeability Increase. Free Radic. Biol. Med. 167, 181–192. doi:10.1016/j.freeradbiomed.2021.02.022
Yamawaki, H., Lehoux, S., and Berk, B. C. (2003). Chronic Physiological Shear Stress Inhibits Tumor Necrosis Factor–Induced Proinflammatory Responses in Rabbit Aorta Perfused Ex Vivo. Circulation 108, 1619–1625. doi:10.1161/01.CIR.0000089373.49941.C4
Yang, Q., Xu, J., Ma, Q., Liu, Z., Sudhahar, V., Cao, Y., et al. (2018). PRKAA1/AMPKα1-driven Glycolysis in Endothelial Cells Exposed to Disturbed Flow Protects against Atherosclerosis. Nat. Commun. 9, 4667. doi:10.1038/s41467-018-07132-x
Yu, P., Wilhelm, K., Dubrac, A., Tung, J. K., Alves, T. C., Fang, J. S., et al. (2017). FGF-dependent Metabolic Control of Vascular Development. Nature 545, 224–228. doi:10.1038/nature22322
Yuan, P., Hu, Q., He, X., Long, Y., Song, X., Wu, F., et al. (2020a). Laminar Flow Inhibits the Hippo/YAP Pathway via Autophagy and SIRT1-Mediated Deacetylation against Atherosclerosis. Cell Death Dis. 11, 141. doi:10.1038/s41419-020-2343-1
Yuan, X., Bhat, O. M., Samidurai, A., Das, A., Zhang, Y., and Li, P.-L. (2020b). Reversal of Endothelial Extracellular Vesicle-Induced Smooth Muscle Phenotype Transition by Hypercholesterolemia Stimulation: Role of NLRP3 Inflammasome Activation. Front. Cell Dev. Biol. 8, 597423. doi:10.3389/fcell.2020.597423
Zanardo, R. C. O., Brancaleone, V., Distrutti, E., Fiorucci, S., Cirino, G., and Wallace, J. L. (2006). Hydrogen Sulfide Is an Endogenous Modulator of Leukocyte-Mediated Inflammation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 20, 2118–2120. doi:10.1096/fj.06-6270fje
Zernecke, A., Bidzhekov, K., Noels, H., Shagdarsuren, E., Gan, L., Denecke, B., et al. (2009). Delivery of MicroRNA-126 by Apoptotic Bodies Induces CXCL12-dependent Vascular Protection. Sci. Signal. 2, ra81. doi:10.1126/scisignal.2000610
Zhang, C., Zhou, T., Chen, Z., Yan, M., Li, B., Lv, H., et al. (2020a). Coupling of Integrin α5 to Annexin A2 by Flow Drives Endothelial Activation. Circ. Res. 127, 1074–1090. doi:10.1161/CIRCRESAHA.120.316857
Zhang, K., Chen, Y., Zhang, T., Huang, L., Wang, Y., Yin, T., et al. (2018a). A Novel Role of Id1 in Regulating Oscillatory Shear Stress-Mediated Lipid Uptake in Endothelial Cells. Ann. Biomed. Eng. 46, 849–863. doi:10.1007/s10439-018-2000-3
Zhang, X., Sessa, W. C., and Fernández-Hernando, C. (2018b). Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front. Cardiovasc. Med. 5, 130. doi:10.3389/fcvm.2018.00130
Zhang, Y.-P., Huang, Y.-T., Huang, T.-S., Pang, W., Zhu, J.-J., Liu, Y.-F., et al. (2017). The Mammalian Target of Rapamycin and DNA Methyltransferase 1 axis Mediates Vascular Endothelial Dysfunction in Response to Disturbed Flow. Sci. Rep. 7, 14996. doi:10.1038/s41598-017-15387-5
Zhang, Z., Yi, D., Zhou, J., Zheng, Y., Gao, Z., Hu, X., et al. (2020b). Exosomal LINC01005 Derived from Oxidized Low-Density Lipoprotein-Treated Endothelial Cells Regulates Vascular Smooth Muscle Cell Phenotypic Switch. BioFactors 46, 743–753. doi:10.1002/biof.1665
Zhao, G., Lu, H., Liu, Y., Zhao, Y., Zhu, T., Garcia-Barrio, M. T., et al. (2021). Single-Cell Transcriptomics Reveals Endothelial Plasticity during Diabetic Atherogenesis. Front. Cell Dev. Biol. 9, 689469. doi:10.3389/fcell.2021.689469
Zhao, Y., Ren, P., Li, Q., Umar, S. A., Yang, T., Dong, Y., et al. (2020). Low Shear Stress Upregulates CX3CR1 Expression by Inducing VCAM-1 via the NF-κB Pathway in Vascular Endothelial Cells. Cell Biochem. Biophys. 78, 383–389. doi:10.1007/s12013-020-00931-4
Zhao, Z.-Z., Wang, Z., Li, G.-H., Wang, R., Tan, J.-M., Cao, X., et al. (2011). Hydrogen Sulfide Inhibits Macrophage-Derived Foam Cell Formation. Exp. Biol. Med. 236, 169–176. doi:10.1258/ebm.2010.010308
Zheng, B., Yin, W., Suzuki, T., Zhang, X., Zhang, Y., Song, L., et al. (2017). Exosome-Mediated miR-155 Transfer from Smooth Muscle Cells to Endothelial Cells Induces Endothelial Injury and Promotes Atherosclerosis. Mol. Ther. 25, 1279–1294. doi:10.1016/j.ymthe.2017.03.031
Zhou, J., Li, Y.-S., Nguyen, P., Wang, K.-C., Weiss, A., Kuo, Y.-C., et al. (2013). Regulation of Vascular Smooth Muscle Cell Turnover by Endothelial Cell–Secreted MicroRNA-126. Circ. Res. 113, 40–51. doi:10.1161/CIRCRESAHA.113.280883
Zhou, S., Chen, H.-Z., Wan, Y., Zhang, Q.-J., Wei, Y.-S., Huang, S., et al. (2011). Repression of P66Shc Expression by SIRT1 Contributes to the Prevention of Hyperglycemia-Induced Endothelial Dysfunction. Circ. Res. 109, 639–648. doi:10.1161/CIRCRESAHA.111.243592
Keywords: vascular endothelium, endothelial dysfunction, atherosclerosis, disturbed flow, endothelial-to-mesenchymal transition, extracellular vesicles, inflammation resolution, targeted therapy
Citation: Botts SR, Fish JE and Howe KL (2021) Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment. Front. Pharmacol. 12:787541. doi: 10.3389/fphar.2021.787541
Received: 01 October 2021; Accepted: 06 December 2021;
Published: 22 December 2021.
Edited by:
Pallavi R. Devchand, University of Calgary, CanadaReviewed by:
Elisabetta Caiazzo, University of Naples Federico II, ItalyCopyright © 2021 Botts, Fish and Howe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn L. Howe, a2F0aHJ5bi5ob3dlQHVobi5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.