 Michael P. Isles
Michael P. IslesGiven that nanomedicines and follow-on nanosimilars have complex manufacturing processes and heteromolecular structures, the question is being raised in ever increasing frequency as to whether the current European regulation of medicines for human use is robust enough to authorise these medicinal products and their follow-ons. Until this can be achieved, there is a potential for patient safety to be compromised.
The current situation is that nanomedicines have the potential for being assessed under four different types of procedures: the national procedure, the decentralised procedure, the mutual recognition procedure, and the centralised procedure. In this context, it is important to note that a survey published in 2018 reported “…strong regional differences in the regulation of nanomedicines and confirmed the need for a harmonisation of information requirements on nano-specific properties” (Bremer-Hoffmann et al., 2018). Given their complex nature and the fact that each nanomedicine will have unique features, there is currently a lack of guidelines or protocols so that these medicines can be appropriately processed, which will provide a marketing authorisation (MA) that meets the demanding standards of today and thus ensure patient safety (Nanomedicines and Nanosimilars, 2021).
The EU Nanomedicines Regulatory Coalition (Nanomedicines Regulatory Coalition, 2021) currently comprising seven pan-European organisations is therefore advocating for all nanomedicines to be assessed by the EMA Centralised regulatory procedure (Patient Safety and Nanomedicines, 2020).
This is equally true of the off-patent follow-on copy products, or nanosimilars, as they are also called. Within this context, a centralised regulatory process that addresses this is needed at the EU level, and in the absence of a tailored regulatory pathway similar to that of the biosimilars, the European Alliance for Access to Safe Medicines (EAASM) strongly believes that all future nanosimilars should go through the Hybrid Application process (10.3) and not the Generics Application process (10.1). This pathway, if consistently applied and aligned to the draft guidance (European Medicines Agency, 2015) which the EMA has produced for specific types of nanomedicines, would ensure that follow-on copies are therapeutically similar to their originator and therefore improve patient safety.
There will be different manufacturers producing these similar products from different sites with differing manufacturing processes, and so the production of identical replicas of the originator product cannot ever be achieved (Ehmann et al., 2013; Marden et al., 2018). It is for this reason that a thorough clinical valuation must be carried out before an MA can be granted.
Patient harm has occurred when a nanosimilar has not had this rigorous safety and efficacy check established through a clinical trial (Rottembourg et al., 2011a) program.
This article endeavors to lay out the critical success factors that will enable a centralised procedure for nanomedicines and nanosimilars to be achieved.
Methodology
The recommendations of this article have been developed due to extensive desk research (Patient Safety and Nanomedicines, 2020) and in consultation with field experts in one-on-one interviews and through two round-tables which took place in the European Parliament in April 2019 (Event summary, 2019) and November 2020 and which were fully reported.
This has enabled the EAASM to adopt a robust strategy of a continuous extensive advocacy program with all influential stakeholders and the EU Institutions (Nanomedicines Regulatory Coalition, 2021).
This strategy aims to raise the political temperature (Letter to Commissioner for Health and Food Safety Ms. Stella Kyriakides, 2021) so that even more focus can be placed on the regulatory institutions to ensure that a fit-for-purpose regulatory pan-European procedure is adopted as quickly as possible.
The need for a harmonised centralised regulatory procedure is highlighted by three key factors:
1) The plethora of nanomedicines in the pipeline (see Figure 1; Table 1; Van Trier, 2021) which indicates the diversity and complexity of these medicines
2) The evolution of many NBCD marketing authorisations (of which many are nanomedicines and nanosimilars—see Table 2) adapted by Klein et al. (2019) which show the diverse nature of the regulatory routes. This gives rise to different national health agencies assessing these medicines and allows for the marketing of different brand names, which in turn makes PV linkage difficult and thus compromises patient safety.
3) Interchangeability of “similar” medicines requires strong central guidelines and education programs to ensure that hospital- and community-based policies are implemented by doctors, pharmacists, and nurses in a coordinated way.
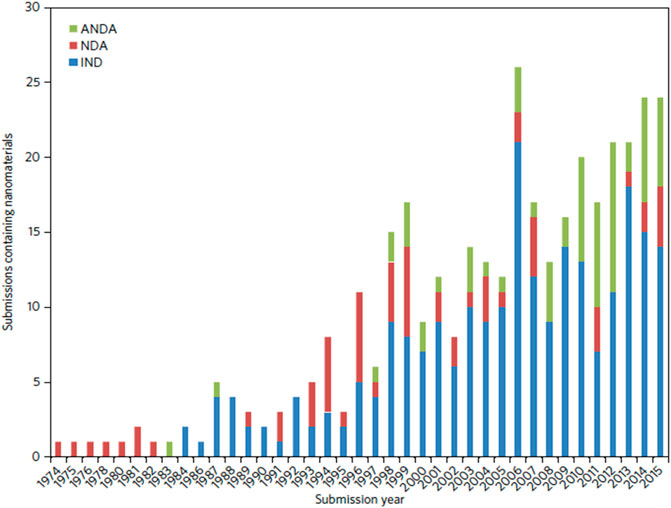
FIGURE 1. Number of nanomaterial product applications submitted to CDER by year. Applications are separated as INDs, NDAs, and ANDAs.
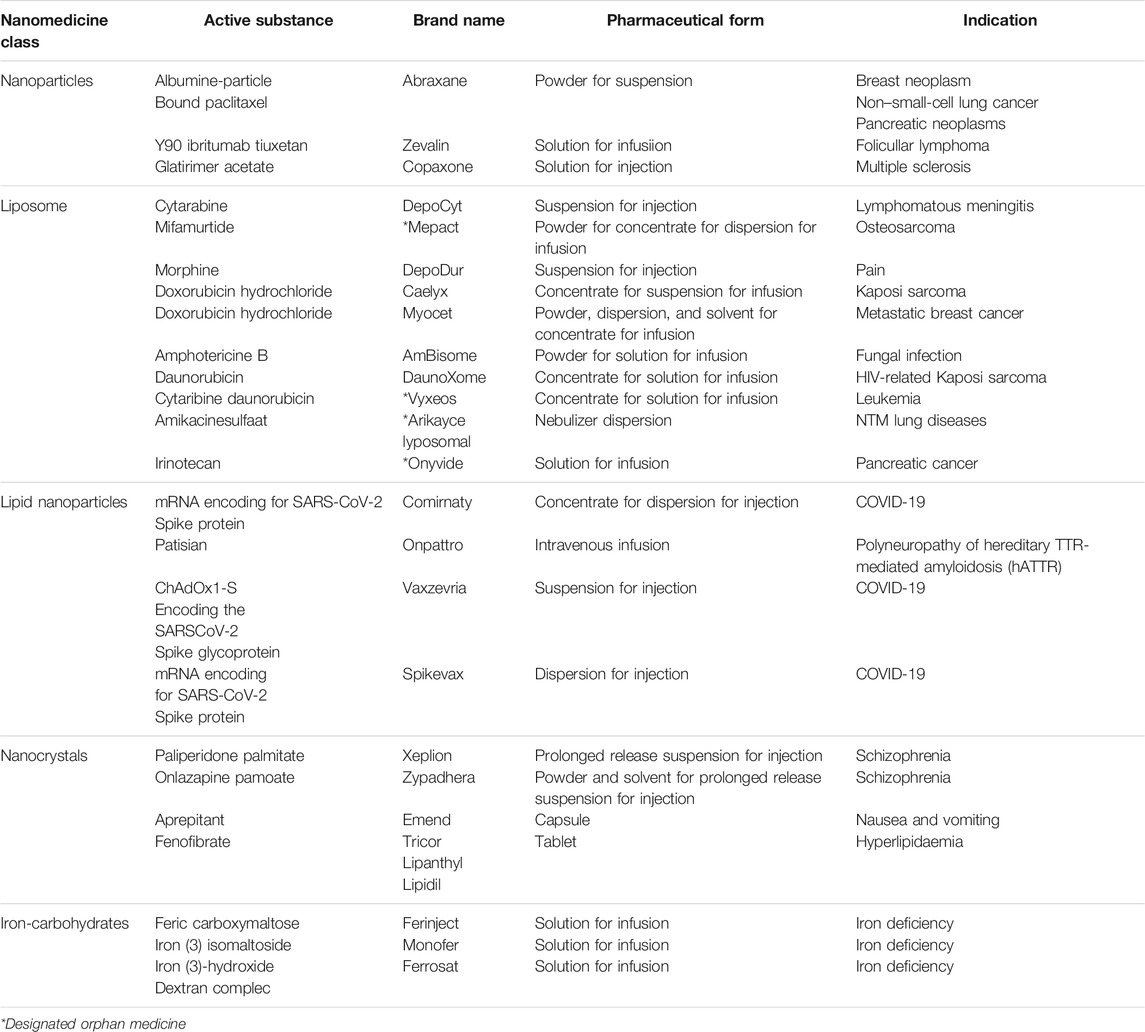
TABLE 1. Overview of the commercially available nanomedicines in the EU (Van Trier, 2021).
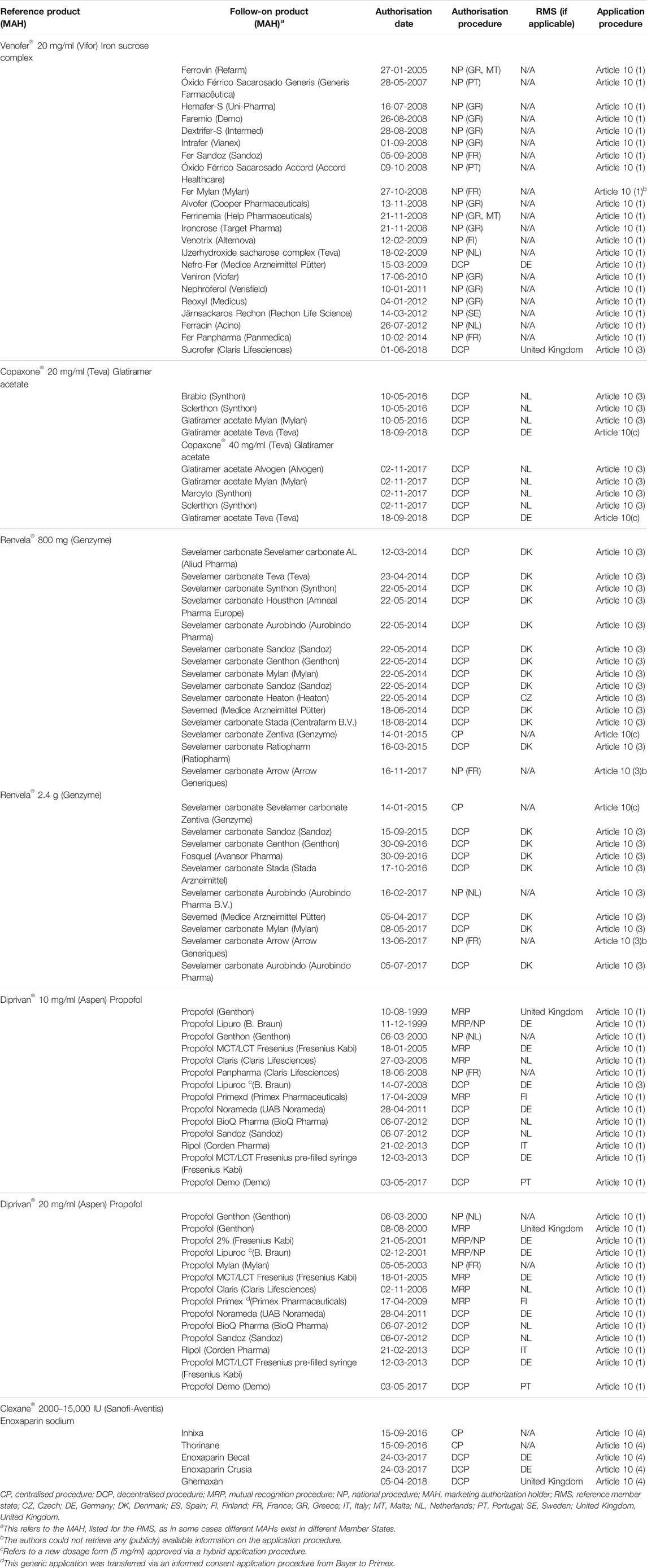
TABLE 2. Overview of NBCD follow-on products approved in the EU via the three abbreviated applications: generic, hybrid, and biosimilar pathways, as well as new applications by originator companies via informed consent, sorted by authorization date since the first approval in 1999 until November 2018.
Nanocolloidal solutions of iron carbohydrates for intravenous applications are another example of frequently used nanomedicines. The first nanotechnology-based intravenous iron product was introduced in the 1950s and is now known as Venofer®. To overcome the high toxicity of iron (II) salts, iron in the form of polynuclear Fe(III)-oxyhydroxide core stabilized by a carbohydrate shell was developed. Intravenously applied Venofer® nanoparticles have been shown to be tolerated at more than 20-fold higher 50% lethal dose (LD 50) levels, compared to iron sulphate solutions in mice (Geisser et al., 1992).
After administration, the iron carbohydrate particles interact with the innate immune system for uptake and release of bioavailable iron (Geisser and Burckhardt, 2011; Koskenkorva-Frank et al., 2013). It is assumed that the characteristics of the nanoparticles affect the fate and disposition in the body (Toblli et al., 2009a; Toblli et al., 2009b; Toblli et al., 2012; Toblli et al., 2015; Toblli et al., 2017). There is a plethora of evidence showing that iron sucrose follow-on products from different manufacturers have different efficacy and safety profiles despite most of them complying with the USP monograph quality requirements (Rottembourg et al., 2011b; Lee et al., 2013; Agüera et al., 2015). Since the structural and functional relationships are not fully understood and, hence, the clinically meaningful critical quality attributes (CQAs) are not fully identified, the manufacturing process defines the product and is crucial for the consistency and quality of the end product and its clinical performance. A robust manufacturing procedure needs to be in place and thoroughly sustained in order to ensure batch-to-batch consistency. Hence, the call for a harmonised centralised regulatory process to ensure the highest safeguards against patient safety issues.
It should be noted that whilst the centralised procedure is already compulsory in a number of situations1, including all those products containing new active substances in, for example, the field of oncology and viral diseases, it does not cover all nanomedicines and nanosimilars. This means that a large number of innovative nanomedicines (including the COVID mRNA) go through the centralised procedure by default. The gap in the system is that for many nanomedicines (i.e., for other indications), it is not yet compulsory for all follow-on/nanosimilars.
As described, for example, by Klein et al., current different routes obtained for marketing approval allows the same nanosimilar to be registered under a variety of brandnames in different countries. This means that when adverse event cases are reported, it is hard to link these patient safety incidences.
As such, nanosimilars would benefit from a mandatory centralised procedure, as this will guarantee consistency in the scientific evaluation of such follow-on products. Another benefit of the centralised procedure is the guarantee of centralised safety monitoring and the obligation for the use of a single brand name throughout the EU. This will facilitate better traceability and adequate identification of product-specific safety issues for nanosimilars.
Results
In 2020, a comprehensive scientific report (Patient Safety and Nanomedicines, 2020) was produced by the EAASM, and a leading politician who sits on the ENVI Committee, namely, MEP Maria da Graca Carvalho (Official website of MEP Petar Vitanov, 2021) (EPP, Portugal), stated in the foreword that (Patient Safety and Nanomedicines, 2021)
“A strong fit-for-purpose regulatory framework is needed, in order to build on all of the current knowledge and expertise. Only then will we be able to have new treatment opportunities that will benefit patients in a timely and safe way.”
An outreach petition encouraging interested parties to join have resulted in the following organizations (Table 3) becoming part of the Nanomedicines Regulatory Coalition, namely, European Alliance for Access to Safe Medicines, European Cancer Patient Coalition, European Liver Patients’ Association, European Parkinson’s Disease Association, European Renal Association, European Specialist Nurses Organization, International Alliance of Patients’ Organizations.

TABLE 3. Nanomedicines Regulatory Coalition.
In addition, the EAASM has held focused interviews with leading MEPs (MEP Cyrus Engerer, 2021) whose major interest is health related and who were co-signatories to a letter sent to the EU Commissioner for Health and Food Safety, Ms. Stella Kyriakides, on 30 June 2021, to which a positive reply was received, and the Commissioner’s statement can be seen in Table 4 2.

TABLE 4. Letter from the EU Commissioner for Health.
The lead rapporteur on the European Parliament INI report, MEP Dolors Montserrat3 (EPP, Spain), charged with challenging the Commission’s legislative proposal, was receptive to the recommendations that nanomedicines should be specifically mentioned in the INI report. The amendments will be voted in the EU Parliament plenary session in Q4 2021, and there is confidence that the inclusion of nanomedicines and nanosimilars will remain and thus be transposed into the EU Pharmaceutical Strategy master policy document that will ultimately result in new legislation.
For the current amendments that are in the Environment, Public Health, and Food Safety (ENVI) Committees’ INI report4 2021/2013/INI 08/11/2021, see Table 5.

TABLE 5. ENVI report -2021/2013/INI.
MEP Petar Vitanov (Official website, 2021) (S&D, Bulgaria) was interviewed by the Parliament Magazine (Parliament Magazine Nanomedicines and Nanosimilars, 2021) and clearly stated the following:
“As an MEP actively involved in health care, and with the progress of the Pharmaceutical Strategy for Europe, it is the right time to set the scene for building a pan-European medical agency consensus so that regulatory weaknesses can be addressed through a robust regulatory pathway and thus provide medicines with the highest quality, safety and efficacy profiles for European patients.”
Following on from two Parliament round-table events, a third follow-up event is tabled for Q3 2021.
Conclusion
In the comprehensive Master Research protocol (Van Trier, 2021) thesis entitled “European stakeholders’ perspectives on the therapeutic opportunities and the regulatory challenges associated with nanomedicines,” the main conclusions under Section 6.3, “The Future of Nanomedicines,” were as follows (see Table 6):

TABLE 6. “European stakeholders’ perspectives on the therapeutic opportunities and the regulatory challenges associated with nanomedicines.” Section 6: “The Future of Nanomedicines.”
This last sentence is a significant cause for concern and so the Nanomedicines Regulatory Coalition under the umbrella of the EAASM intends to continue its strong advocacy program to ensure the following:
• all nanomedicines and nanosimilars be assessed by the EMA Centralised Regulatory Procedure.
• a harmonization of information requirements of regulators in order to correctly characterize nanomedicines
• production of a scientific consensus on definitions for nanomedicines across Europe
• improved education and a fostering of awareness on the complexity and sophistication of nanomedicines among policymakers, prescribers, payers, and patients. This is especially relevant when it centers on issues of interchangeability
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ANDA, Abbreviated New Drug Application; CDER, Center For Drug Evaluation And Research; CQA, critical quality attributes; EAASM, European Alliance for Access to Safe Medicines; EMA, European Medicines Agency; ENVI Committee, the Committee on the Environment, Public Health and Food Safety European Parliament; INI report, own-initiative report of the European Parliament; IND, investigational new drug application; LD, lethal dose; MEP, Member of the European Parliament; NBCD, non-biological complex drugs; NDA, new drug application; USP, United States Pharmacopoeia.
Footnotes
1https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines#scope-of-the-centralised-procedure-section
2https://ec.europa.eu/health/human-use/strategy_en
3https://www.europarl.europa.eu/meps/en/197711/DOLORS_MONTSERRAT/home
4https://www.europarl.europa.eu/doceo/document/A-9-2021-0317_EN.pdf
References
Agüera, M. L., Martin-Malo, A., Alvarez-Lara, M. A., Garcia-Montemayor, V. E., Canton, P., Soriano, S., et al. (2015). Efficiency of Original Versus Generic Intravenous Iron Formulations in Patients on Haemodialysis. PLoS One 10 (10), 8. doi:10.1371/journal.pone.0135967
Bremer-Hoffmann, S., Halamoda-Kenzaoui, B., and Borgos, S. E. (2018). Identification of Regulatory Needs for NanomedicinesJ. Interdiscip. Nanomedicine 3. doi:10.1002/jin2.34
Ehmann, F., Sakai-Kato, K., Duncan, R., Hernán Pérez de la Ossa, D., Pita, R., Vidal, J. M., et al. (2013). Next-Generation Nanomedicines and Nanosimilars: EU Regulators' Initiatives Relating to the Development and Evaluation of Nanomedicines. Nanomedicine (Lond). 8 (5), 849–856. doi:10.2217/nnm.13.68
European Medicines Agency (2015). Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-data-requirements-intravenous-iron-based-nano-colloidal-products-developed_en.pdf.
Event summary (2019). Event Summary: Handling Innovation in NANOMEDICINES: Regulatory Challenges and Opportunities in Modern Healthcare. Available at: https://eaasm.eu/en-gb/2019/04/04/event-summary-handling-innovation-in-nanomedicines-regulatory-challenges-and-opportunities-in-modern-healthcare/.
Geisser, P., Baer, M., and Schaub, E. (1992). Structure/Histotoxicity Relationship of Parenteral Iron Preparations. Arzneimittelforschung 42 (12), 1439–1452.
Geisser, P., and Burckhardt, S. (2011). The Pharmacokinetics and Pharmacodynamics of Iron Preparations. Pharmaceutics 3 (1), 12–33. doi:10.3390/pharmaceutics3010012
Klein, K., Stolk, P., De Bruin, M. L., Leufkens, H. G. M., Crommelin, D. J. A., and De Vlieger, J. S. B. (2019). The EU Regulatory Landscape of Non-Biological Complex Drugs (NBCDs) Follow-On Products: Observations and Recommendations. Eur. J. Pharm. Sci. 133, 228–235. doi:10.1016/j.ejps.2019.03.029
Koskenkorva-Frank, T. S., Weiss, G., Koppenol, W. H., and Burckhardt, S. (2013). The Complex Interplay of Iron Metabolism, Reactive Oxygen Species, and Reactive Nitrogen Species: Insights Into the Potential of Various Iron Therapies to Induce Oxidative and Nitrosative Stress. Free Radic. Biol. Med. 65 (65), 1174–1194. doi:10.1016/j.freeradbiomed.2013.09.001
Lee, E. S., Park, B. R., Kim, J. S., Choi, G. Y., Lee, J. J., and Lee, I. S. (2013). Comparison of Adverse Event Profile of Intravenous Iron Sucrose and Iron Sucrose Similar in Postpartum and Gynecologic Operative Patients. Curr. Med. Res. Opin. 29 (2), 141–147. doi:10.1185/03007995.2012.760444
Letter to Commissioner for Health and Food Safety, Ms. Stella Kyriakides (2021). Entitled Enabling a Centralised Regulatory System for Nanomedicines through the Pharmaceutical Strategy for Europe. Available at: https://eunanomedicinescoalition.eu/wp-content/uploads/2021/08/EAASM-letter-highlighting-the-need-to-fully-harness-the-potential-of-nanomedicines-and-protect-patient-safety-across-Europe.pdf.
Marden, E., Ntai, I., Bass, S., and Flühmann, B. (2018). Reflections on FDA Draft Guidance for Products Containing Nanomaterials: Is the Abbreviated New Drug Application (ANDA) a Suitable Pathway for Nanomedicines? AAPS 20, 92. doi:10.1208/s12248-018-0255-0
MEP Cyrus Engerer (2021). MEP Cyrus Engerer (Malta, Progressive Alliance of Socialists and Democrats), MEP Romana Jerkovic (Croatia, Progressive Alliance of Socialists and Democrats), MEP Petar Vitanov (Bulgaria, Progressive Alliance of Socialists and Democrats), MEP Pietro Fiocchi (Italy, European Conservatives and Reformists Group), MEP Margrete Auken. Denmark: Greens/European Free Alliance.
Nanomedicines and Nanosimilars (2021). Building a Robust Legislative Framework. Available at: https://eunanomedicinescoalition.eu/news/nanomedicines-and-nanosimilars-building-a-robust-legislative-framework/.
Nanomedicines Regulatory Coalition (2021). Nanomedicines Regulatory Coalition. Available at: https://eunanomedicinescoalition.eu/.
Official website of MEP Petar Vitanov (2021). Official Website of MEP Petar Vitanov. Available at: https://www.europarl.europa.eu/meps/en/197844/PETAR_VITANOV/home.
Official website (2021). Official website of MEP Maria da Graça CARVALHO. Available at: https://www.europarl.europa.eu/meps/en/96867/MARIA+DA+GRACA_CARVALHO/home.
Parliament Magazine Nanomedicines and Nanosimilars (2021). Building a Robust Legislative Framework. Available at: https://www.theparliamentmagazine.eu/news/article/nanomedicines-and-nanosimilars-building-a-robust-legislative-framework.
Patient Safety and Nanomedicines (2021). The Need for a Centralised Regulatory Procedure. Available at: https://eunanomedicinescoalition.eu/wp-content/uploads/2020/09/Patient-Safety-and-Nanomedicines-September-2020.pdf.
Rottembourg, J., Kadri, A., Leonard, E., Dansaert, A., and Lafuma, A. (2011a). Do two Intravenous Iron Sucrose Preparations Have the Same Efficacy? Nephrol. Dial. Transplant A Nephrol. Dial. Transpl. 26, 3262–3267. doi:10.1093/ndt/gfr024
Rottembourg, J., Kadri, A., Leonard, E., Dansaert, A., and Lafuma, A. (2011b). Do two Intravenous Iron Sucrose Preparations Have the Same Efficacy? Nephrol. Dial. Transpl. 26 (10), 3262–3267. doi:10.1093/ndt/gfr024
Toblli, J. E., Cao, G., and Angerosa, M. (2015). Nitrosative Stress and Apoptosis in Non-Anemic Healthy Rats Induced by Intravenous Iron Sucrose Similars versus Iron Sucrose Originator. Biometals 28 (2), 279–292. doi:10.1007/s10534-015-9822-3
Toblli, J. E., Cao, G., Giani, J. F., Dominici, F. P., and Angerosa, M. (2017). Markers of Oxidative/Nitrosative Stress and Inflammation in Lung Tissue of Rats Exposed to Different Intravenous Iron Compounds. Drug Des. Devel. Ther. 11, 2251–2263. doi:10.2147/DDDT.S132612
Toblli, J. E., Cao, G., Oliveri, L., and Angerosa, M. (2012). Comparison of Oxidative Stress and Inflammation Induced by Different Intravenous Iron Sucrose Similar Preparations in a Rat Model. Inflamm. Allergy Drug Targets 11 (1), 66–78. doi:10.2174/187152812798889358
Toblli, J. E., Cao, G., Oliveri, L., and Angerosa, M. (2009a). Differences between the Original Iron Sucrose Complex Venofer® and the Iron Sucrose Similar Generis®, and Potential Implications. Port J. Nephrol. Hypert. 23 (1), 53–63.
Toblli, J. E., Cao, G., Oliveri, L., and Angerosa, M. (2009b). Differences Between Original Intravenous Iron Sucrose and Iron Sucrose Similar Preparations. Arzneimittelforschung 59 (4), 176–190. doi:10.1055/s-0031-1296383
Keywords: advocacy program, centralised regulatory procedure, hybrid application, nanomedicines, nanosimilars, follow-on products
Citation: Isles MP (2022) Nanomedicines and Nanosimilars—Why a Robust Centralised Regulatory Framework Is Essential to Enhance Patient Safety. Front. Pharmacol. 12:787239. doi: 10.3389/fphar.2021.787239
Received: 30 September 2021; Accepted: 30 November 2021;
Published: 24 February 2022.
Edited by:
Marc Marie Dooms, University Hospitals Leuven, BelgiumReviewed by:
Jon S. B. De Vlieger, Lygature, NetherlandsRoald Van Der Laan, Insmed Inc., United States
Copyright © 2022 Isles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael P. Isles, bWlrZS5pc2xlc0BlYWFzbS5ldQ==