 Lia Farahi
Lia Farahi Satyesh K. Sinha
Satyesh K. Sinha Aldons J. Lusis
Aldons J. Lusis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 26 November 2021
Sec. Inflammation Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.785220
This article is part of the Research TopicStreaming Inflammation: From Damage to Healing and ResilienceView all 12 articles
Atherosclerosis is a chronic inflammatory disease that may ultimately lead to local proteolysis, plaque rupture, and thrombotic vascular disease, resulting in myocardial infarction, stroke, and sudden cardiac death. Circulating monocytes are recruited to the arterial wall in response to inflammatory insults and differentiate into macrophages which make a critical contribution to tissue damage, wound healing, and also regression of atherosclerotic lesions. Within plaques, macrophages take up aggregated lipoproteins which have entered the vessel wall to give rise to cholesterol-engorged foam cells. Also, the macrophage phenotype is influenced by various stimuli which affect their polarization, efferocytosis, proliferation, and apoptosis. The heterogeneity of macrophages in lesions has recently been addressed by single-cell sequencing techniques. This article reviews recent advances regarding the roles of macrophages in different stages of disease pathogenesis from initiation to advanced atherosclerosis. Macrophage-based therapies for atherosclerosis management are also described.
Atherosclerosis (AS) is associated with both metabolic dysfunction and chronic inflammatory processes. AS is initiated by endothelial dysfunction caused by factors such as high plasma cholesterol, hypertension, diabetes, leukocytosis, cigarette smoking, and low shear stress (Xu et al., 2019). A particularly central event is the subendothelial accumulation of low-density lipoproteins (LDL) and very-low-density lipoproteins (VLDL), which are then subjected to aggregation, oxidation, and enzymatic modifications. The resulting oxidized phospholipids (OxPLs), then contribute to inflammatory processes that promote the activation of endothelial cells (ECs) to express several types of leukocyte adhesion molecules (Marchio et al., 2019). This, in turn, results in leukocyte recruitment, including blood monocytes, neutrophils, lymphocytes, and also platelets to the vessel wall. The monocytes differentiate into macrophages (Mφs) which then proliferate in response to Mφ colony-stimulating factor (M-CSF) and other factors (Robbins et al., 2013b; Sinha et al., 2021). Local proliferation dominates lesional Mφ accumulation in AS. Monocyte recruitment can not fully account for lesional Mφ accumulation in established AS and lesional Mφ can replenish either through the continuous recruitment of circulating monocytes or through some other processes. Mφ self-renewal was identified as a therapeutic target for cardiovascular diseases (Robbins et al., 2013a). Mφs can polarize and reprogram their functional phenotypes in response to different stimuli, and thus display distinct functions related to tissue homeostasis and inflammation (Park, 2021). Mφs take up the aggregated and oxidized LDL (Ox-LDL) leading to their transformation into cholesterol engorged foam cells. Such uptake of modified lipoproteins is mediated primarily by scavenger receptors (SRs) expressed on Mφs. Besides Mφs, vascular smooth muscle cells (VSMCs) can also transdifferentiate into lesional Mφ-like cells through cholesterol loading (Rong et al., 2003). Advanced plaques are characterized by subendothelial deposition of lipids, necrotic cell debris, calcium phosphate crystals, fibrosis, and inflammatory immune cells including Mφs, T and B lymphocytes, neutrophils, dendritic cells (Canet-Soulas et al., 2021). Lesional T cells can recognize local antigens, leading to activation, clonal expansion, and cytokine production (Robertson and Hansson, 2006). Leukocytosis, an increase in the numbers of circulating leukocytes, is associated with cardiovascular (CV) diseases. Some leukocytes are atherogenic and sustain oxidative stress (OS) and inflammation after myocardial infarction (MI) while others are atheroprotective and help it resolve (Swirski and Nahrendorf, 2013b). For example, CD4+ T cell deficiency results in a delay in the switch of Ly6Chi into Ly6CLow monocytes and impaired healing. The inflammatory Ly-6Chi phenotypes are required during the early response to ischemic injury, but if they persist in the infarct too long, the reparative functions of Ly-6Clow monocytes are impaired (Nahrendorf et al., 2007; Leuschner et al., 2012). The progression of an atherosclerotic lesion including the formation of early fatty streak lesions, fibrous cap, necrotic core, and thrombus have been displayed in Figure 1 (Virmani et al., 2002; Chinetti-Gbaguidi et al., 2015). In this review, we provide an overview of the critical role of Mφs in the pathogenesis of AS, including recent findings relating to clonal hematopoiesis, single-cell sequencing, and senescence. Mφ-based therapies for the management of AS are also discussed.
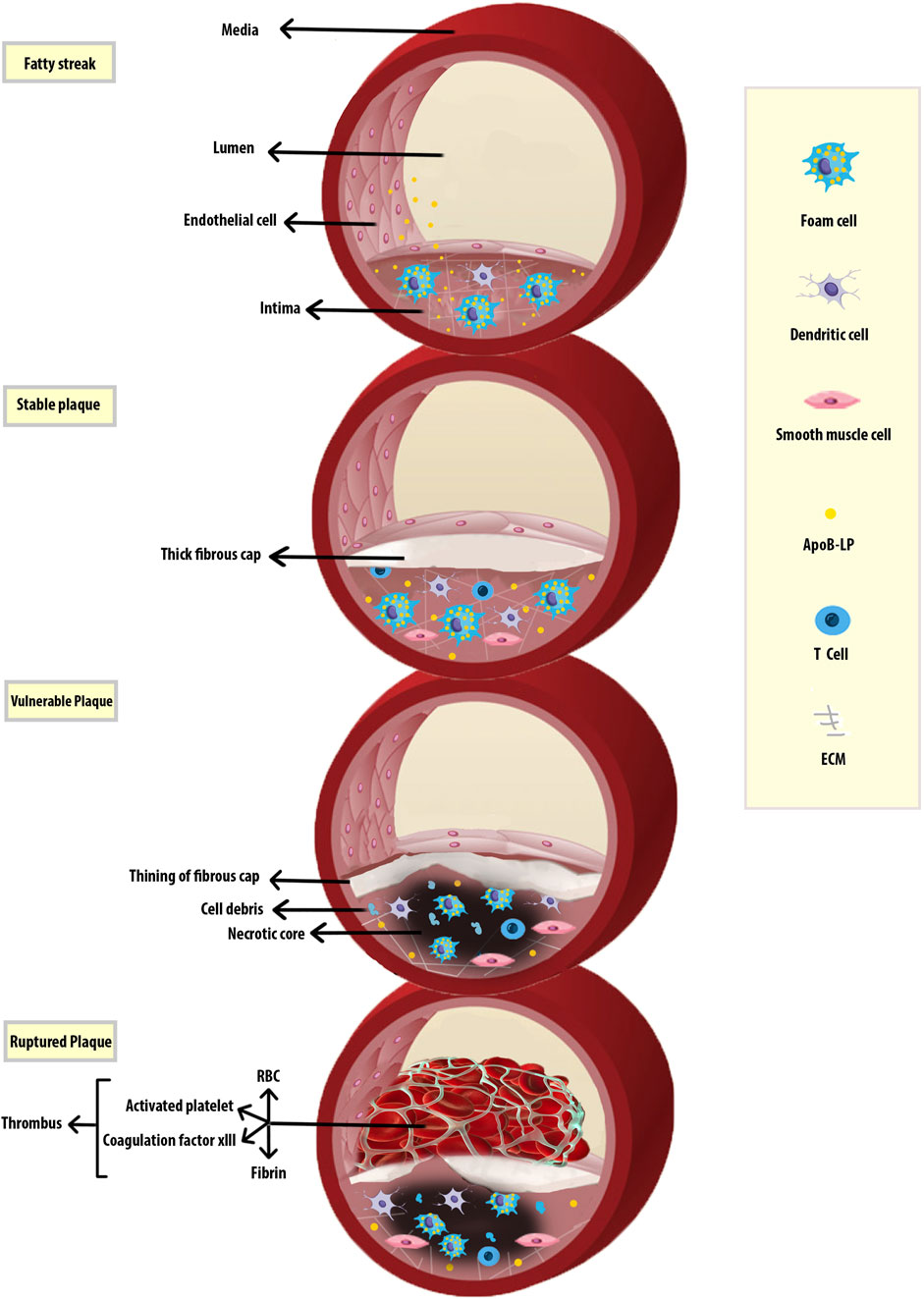
FIGURE 1. Progression of Atherosclerosis. Early fatty streak lesions are characterized by subendothelial accumulation of ApoB-LPs, the main protein constituent of lipoproteins such as VLDL and LDL which promote the recruitment of Mφs. As the atherosclerotic lesion progresses, ApoB-LP retention is amplified. Vulnerable plaques are characterized by the accumulation of ACs and defective efferocytosis, resulting in the lipid-filled necrotic core. A thinning fibrous cap reduces plaque stability and makes them susceptible to rupture and thrombus formation. Apolipoprotein B-containing lipoproteins (ApoB-LPs).
Monocytes originate from hematopoietic stem cell progenitors in the bone marrow. They have also been found to reside in the spleen as a secondary reservoir. Figure 2A depicts the differentiation of blood monocytes from bone marrow progenitors (Teh et al., 2019). The heterogeneous population of monocytes is classified into classical, intermediate, and non-classical subtypes based on the differential expression of CD14 and CD16 (Figure 2B). The characteristics of monocyte subsets are summarized in Table 1 (Kapellos et al., 2019). Evidence supports a causal role of atherogenic lipoproteins, especially LDL, in the pathogenesis of AS.
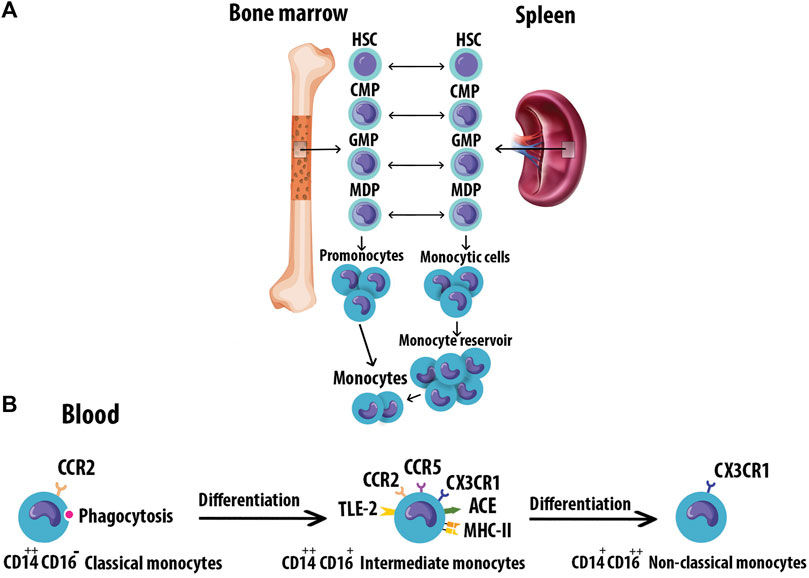
FIGURE 2. Monocyte origin and heterogeneity. (A) Blood monocytes originate from HSC-derived progenitors with myeloid restricted differentiation potential. Successive commitment steps in the bone marrow include CMPs, GMPs, and MDPs. MDPs give rise to monocytes. (B) Human monocytes are classified into three subtypes based on the differential expression of CD14 and CD16. Monocytes mature in the bone marrow and are subsequently released into the circulation. Progressively, classical monocytes CD14++CD16 give rise to non-classical monocytes CD14+CD16++ through an intermediate step of CD14++CD16+ monocytes. Hematopoietic stem cell (HSC), Common myeloid progenitors (CMPs), Granulocyte-Mφ precursors (GMPs), Mφ/Dendritic cell progenitor cells (MDPs).

TABLE 1. Characterization of monocyte subsets.
The subendothelial deposition of LDL is likely a key driver of monocyte-ECs adhesion, an early event of AS by activating the overlying EC (Alderson et al., 1986). Factors such as high-fat diets, smoking, and radiation exposure may increase the risk of long-term OS which manifests itself in excessive reactive oxygen species (ROS) generation and LDL oxidation (Poznyak et al., 2021). In lesions, LDL is oxidized to form Ox-LDL which is pro-inflammatory and appears to contribute to AS initiation and progression (Que et al., 2018). For example, Ox-LDL promotes the expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecules (ICAM-1) by the ECs which attract the leukocytes into the vessel wall (Milioti et al., 2008). Others have suggested that native or aggregated LDL particles are the key drivers of atherogenesis (Boyle, 2005; Libby, 2021).
Activated lesional ECs and platelets express P- and E-selectin while L-selectin is expressed on leukocytes. The major ligand for selectin is P-selectinglycoprotein ligand 1 (PSGL1) which is expressed on leukocytes and binds to all three selectins. The interaction between selectins and PSGL-1 decelerates fast-flowing leukocytes from the central bloodstream and enables circulating leukocytes to adhere to the activated endothelium. Differential expression or glycosylation of PSGL-1 in leukocytes may result in selective recruitment of monocytes or lymphocytes to atherosclerotic lesions (Huo and Xia, 2009). In addition to inflammatory cells, pro-inflammatory mediators such as cytokines and interleukins are also known to contribute to atherogenesis (Kaperonis et al., 2006).
The adhesion molecules, VCAM1 and ICAM1, are overexpressed on activated ECs (Thayse et al., 2020). Integrins mediate firm leukocyte adhesion on ECs through ligation of monocytes or lymphocytes with VCAM1 or ICAM1 by engaging integrin (Ley et al., 2007). PSGL-1 also interacts with chemokine ligand CCL 21 or CCL19 and enhances chemotactic CD4+ T cells to the vulnerable plaques. Activated CD4+ T lymphocytes secrete interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), pro-inflammatory cytokines enhancing immune responses during atherogenesis (Veerman et al., 2007). IFN-β also increases Mφ accumulation in the plaques and accelerates lesion formation (Goossens et al., 2010). Furthermore, activated platelets, a major component of inflammatory lesions, are phagocytosed by monocytes, inducing secretion of pro-inflammatory chemokines such as CCL5. Thus, the presence of activated platelets on the inflamed endothelium may exacerbate pro-inflammatory Mφ activation (Scull et al., 2010). Human IgG1 against a specific Ox-LDL antigenic epitope promoted the regression of atherosclerotic lesions by inhibiting Mφ recruitment and increasing lipid efflux (Schiopu et al., 2007). Monocyte recruitment is also mediated in part by C-C chemokine receptors (CCR)2, CCR5, and CX3C chemokine receptor 1 (CX3CR1) (Tacke et al., 2007).
Monocytes migrate into the intima by chemotactic activity and differentiate into Mφs. Mφs can bind to the circulating lipids through several SRs, such as SR-A1, CD36 (SR-B2), and the lectin-type oxidized LDL receptor 1 (LOX1/SR-E1) that mediate uptake of modified LDL (Syväranta et al., 2014).
SR-A1 is mostly on Mφs but also present on VSMCs and ECs experiencing OS. SR-A1 binds to the modified LDL and is involved in the subendothelial translocation of LDL (Apostolov et al., 2009). The c-Jun N-terminal kinase 2 (JNK2)-dependent phosphorylation of SR-A promotes the uptake of lipids in Mφs and thus mediates foam cell formation (Ricci et al., 2004). Deletion of SR-A reduces plaque inflammation and progression toward more advanced necrotic lesions, but it does not abrogate lipid uptake and Mφ foam cell formation in ApoE−/− mice (Manning-Tobin et al., 2009). SR-A1 expression in aortic VSMCs also leads to foam cell formation and enhanced apoptosis in transfected VSMCs (Lehtolainen et al., 2000). SR-A1 triggers endocytosis through two clathrin- and caveolae-dependent routes. Uptake of modified LDL by SR-A primarily goes through clathrin-dependent pathway. SR-A-induced apoptosis needs endocytosis by the caveola route, which activates p38 MAPK and JNK2 signaling (Zhu et al., 2011). NF-κB also regulates the expression of SR-A1 which can be stimulated by pro-inflammatory cytokines (Chistiakov et al., 2017).
CD36 has a high affinity for Ox-LDL, and Ox-LDL/CD36 interactions inhibit cell polarization and Mφ migration (Park et al., 2012). CD36 is upregulated in Mφ-derived foam cells in plaques. Advanced glycation end products (AGEs) are also recognized by CD36 as overexpression of this receptor facilitated AGE uptake in CHO cells (Ohgami et al., 2001). CD36 is also involved in inflammatory processes, including AS, negative regulation of microvascular angiogenesis, lipid metabolism, and clearance of ACs (Febbraio et al., 2001). CD36 showed a conserved role in lipid sensorial recognition. Ox-LDL binds CD36 and triggers TLR4/TLR6 complex to promote pro-inflammatory signaling (Hoebe et al., 2005). Myricetin, a natural flavonoid, contributes to decreased accumulation of cholesterol in Mφ foam cells by inhibition of CD36-mediated Ox-LDL uptake in Ldlr−/− mice, resulting in a reduction of atherosclerotic lesions (Meng et al., 2019).
LOX-1 augments the uptake of Ox-LDL by Mφs and ECs. Thus, LOX-1 is implicated in atherogenic deposition of lipids and foam cell formation. The expression of LOX-1 is low under normal physiological conditions, but inflammatory modulators, including Ox-LDL, LPS, mitochondrial ROS, angiotensin II, sheer stress, pro-inflammatory cytokines, AGEs, and conditions such as high blood pressure, dyslipidemia, and diabetes mellitus, upregulate LOX-1 in AS (Kattoor et al., 2019). In addition, Ox-LDL and pro-inflammatory molecules like TNF-α upregulate the LOX-1 expression in VSMCs and facilitate VSMCs transformation to foam cells, a major source of plaque foam cells in human atherogenesis (Kattoor et al., 2019). LOX-1 also contributes to endothelial dysfunction, Mφ differentiation, apoptosis, the proliferation and migration of VSMCs, foam cell formation, platelet activation, as well as plaque instability, and subsequent plaque rupture (Xu et al., 2013).
The uptake of lipids by Mφs in lesions leads to the formation of cholesterol engorged foam cells (Figure 3). Foam cells are the first sign of initial atherogenesis, although they also play an important role in lesion development and advanced plaques (Chistiakov et al., 2017). The endothelin-1 receptor antagonist directly reduced Mφ lipid accumulation and AS in Ldlr−/− mice, indicating the role of endothelin-1 in foam-cell formation (Babaei et al., 2000). The cholesteryl esters in modified lipoproteins are hydrolyzed in lysosomes to free cholesteryl, which is then re-esterified by acetyl-CoA acetyltransferase (Chang et al., 2009) and the accumulating cholesteryl esters are stored as cytoplasmic lipid droplets. Increased cholesterol ester accumulation promotes the development of atherosclerotic lesions, unstable plaque with cap rupture, and thrombosis (Yu et al., 2013). Neutral cholesteryl ester hydrolases are responsible for the removal of the ester group from cholesteryl esters to liberate FC which is then effluxed from cells to HDL through ABCA1, ABCG1, and SR-B1 (Chistiakov et al., 2016). Mφ cholesterol uptake and efflux have been shown in Figure 4.
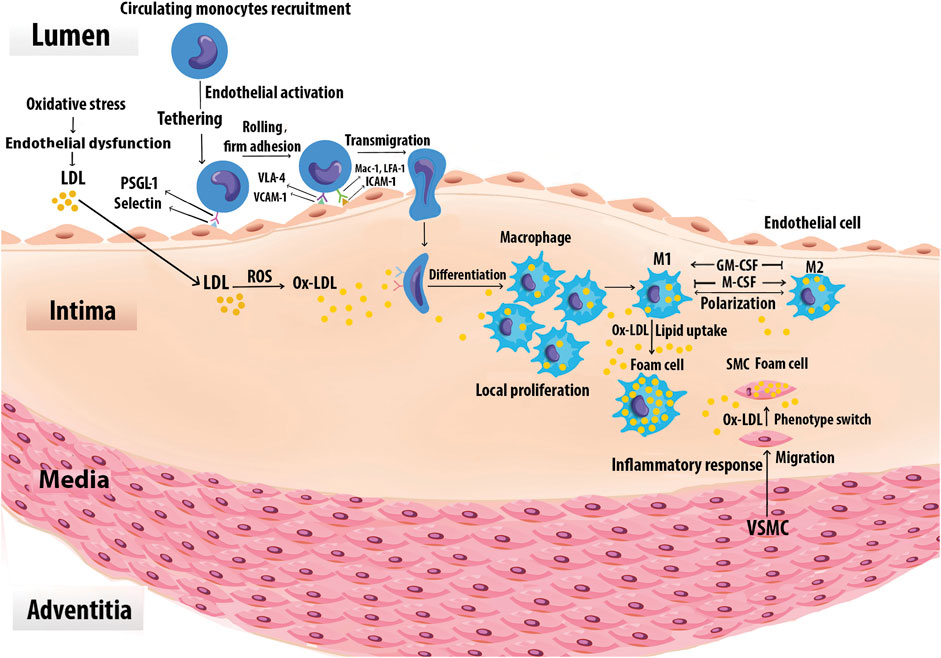
FIGURE 3. Foam cell formation. Monocytes are recruited to the vascular arginase wall in response to Ox-LDL. Specific adhesion molecules such as the selectins, VCAM-1, and ICAM-1 are expressed on the surface of activated vascular ECs, mediate monocyte adhesion. Once adherent, the monocytes enter the intima and differentiate into Mφs. The differentiation process may be mediated by GM-CSF and M-CSF. Local Mφ proliferation contributes to lesion growth. Upon extensive uptake of Ox-LDL via SRs, Mφs are ultimately turned into foam cells. Chemoattractants, growth factors, and cytokines also promote SMC proliferation, uptake of Ox-LDL, and eventually conversion to foam cells. Foam cells derived from SMCs together with those derived from Mφs generate the fatty lesion.
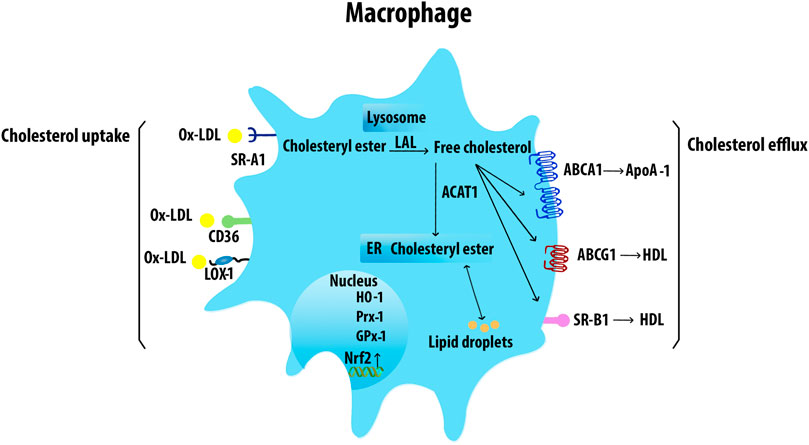
FIGURE 4. Macrophage cholesterol uptake and efflux. Mφs uptake VLDL and Ox-LDL via SRs including SR-A1, CD36, and LOX-1. The internalized LDL is esterified by acetyl-coenzyme A acetyltransferases and stored in lipid droplets. The ester group is removed from cholesteryl by neutral cholesteryl ester hydrolase through lysosomal acid lipase (LAL) to release FC. ABCA1 transporter mediates the FC efflux from Mφs with ApoA-1. ABCG1 and SR-B1 also efflux FC to mature HDL. Acetyl-CoA acetyltransferase 1 (ACAT1).
ABCA1 activity in arterial Mφs plays a major role in the prevention of foam cell formation by mediating the active transfer of cellular phospholipid and FC to apolipoprotein A-1 (ApoA-1), the major apoprotein in HDL (Phillips, 2018). Multiple factors can affect the expression of ABCA1. Extra-virgin olive oil, cineole, quercetin, apelin-13, S-allylcysteine, and TLR2, increase ABCA1 expression, whereas unsaturated FA, miR-26, and IL-12 in synergy with IL-18 inhibit ABCA1 expression.
ABCG1 exports FC or cellular phospholipid to HDL but not to lipid-free ApoA-1. ABCG1 localizes in the plasma membrane, late endosomes, and ER network (Neufeld et al., 2014). Fucosterol, resveratrol, extra-virgin olive oil, and cineole increase ABCG1 expression and significantly reduce cholesterol deposits in the arteries.
SR-B1 promotes FC efflux to mature HDL particles via passive diffusion. The conserved function of SR-B1 across species indicates the importance of its regulation for cholesterol efflux. SR-B1-deficient mice exhibit an increase in accumulation of cholesterol-rich HDL that is accompanied by reduced cholesterol in secreted bile and increased susceptibility to AS (Van Eck et al., 2003). In contradistinction, overexpression of SR-B1 in mice accelerates the metabolism of HDL (Kozarsky et al., 1997). SR-B1 also mediates efferocytosis and reduces atherosclerotic necrosis and inflammation. Deficiency of Mφ SR-BI promoted defective efferocytosis signaling via the Src/PI3K/Rac1 signaling, leading to increased plaque size, necrosis, and inflammation (Tao et al., 2015). Multiple factors were identified to regulate SR-B1 expression. Resveratrol and 13-hydroxy linoleic acid increase the expression of SR-B1, ABCA1, and ABCG1. Caffeic acid and ferulic acid appear to have an antiatherogenic effect by increasing the expression of SR-B1 and ABCG1. Omega-3 fatty acids also induce the activation of SR-B1, ABCA1, and ABCG5, enhancing cholesterol efflux from Mφs (Yu et al., 2013). Risk factors such as infections and fatty diets can change the gut microbiota habitat and increase the levels of circulating LPS which reduces ABCA1/ABCG1 and SR-B1 expression and cholesterol efflux from Mφs (Yu et al., 2013).
Monocytes/Mφs play a central role in innate immune responses, expressing a variety of receptors that modulate their activation. The expression of monocyte and Mφ markers has been shown in Figure 5A. Mφ polarization refers to a process by which Mφs can differentiate into distinct functional phenotypes in response to microenvironmental stimuli, such as cytokines, chemokines, growth factors, and pathogen-derived molecules (Murray, 2017). M-CSF in early lesions induces anti-inflammatory M2 (M2a, M2b, M2c, and M2d) Mφs, whereas granulocyte M-CSF (GM-CSF) expression upon AS development promotes polarization of pro-inflammatory M1 phenotypes (Colin et al., 2014). Mφ activation states and markers associated with distinct activation phenotypes have been shown in Figure 5B. The dynamic plasticity of Mφs is achieved by transcriptional regulation and thus, specific genes are associated with each type of Mφs (Gerrick et al., 2018). The most abundant T cells in atherosclerotic plaques are Th1 cells and memory CD4+ T cells. The Mφ-directed immune response includes both M1/Th1 and M2/Th2 responses. Th1 and Th2 cells in lesions release Mφ-polarizing factors that affect the balancing of M1 and M2 Mφ phenotypes (Bartlett et al., 2019). Both M1 and M2 Mφs contribute to plaque establishment, while M1 and Th1 cells promote plaque destabilization (Bartlett et al., 2019). M2 Mφs were reported to be found at early stages of AS but showed a switch to M1 phenotypes in advanced lesions of ApoE−/− mice (Khallou-Laschet et al., 2010). The mixture of Mφ subsets likely exists simultaneously within atherosclerotic aortas in vivo, contributing to the progression and persistence of atherosclerotic lesions (Butcher and Galkina, 2012). Gene signatures of Mφ activation are highly robust in predicting inflammation, disease susceptibility, and outcomes, suggesting that immune diversity is a valuable parameter in translational research (Buscher et al., 2017). M1/M2 Mφ subpopulations fail to reflect the full complexity of microenvironments in the plaque. A wide range of cytokines and growth factors are present in AS that could affect the phenotype and polarisation state of Mφs. Atherosclerotic plaques contain other Mφ phenotypes include metabolically activated (MMe) Mφs, oxidized (Mox) Mφs, hemoglobin-related (HA-mac, M(Hb), and Mhem) Mφs, M4 Mφs, lipid metabolism-related trigger receptor expressed on myeloid cells 2 (TREM2, also called lipid-associated Mφs), and neuroimmunological Mφs including nerve-associated Mφs (NAM), and sympathetic neuron-associated Mφs (SAM) (Nagenborg et al., 2017; Kolter et al., 2020b; Porsch et al., 2020). The main properties and functions of polarized Mφ subtypes are summarized in Table 2.
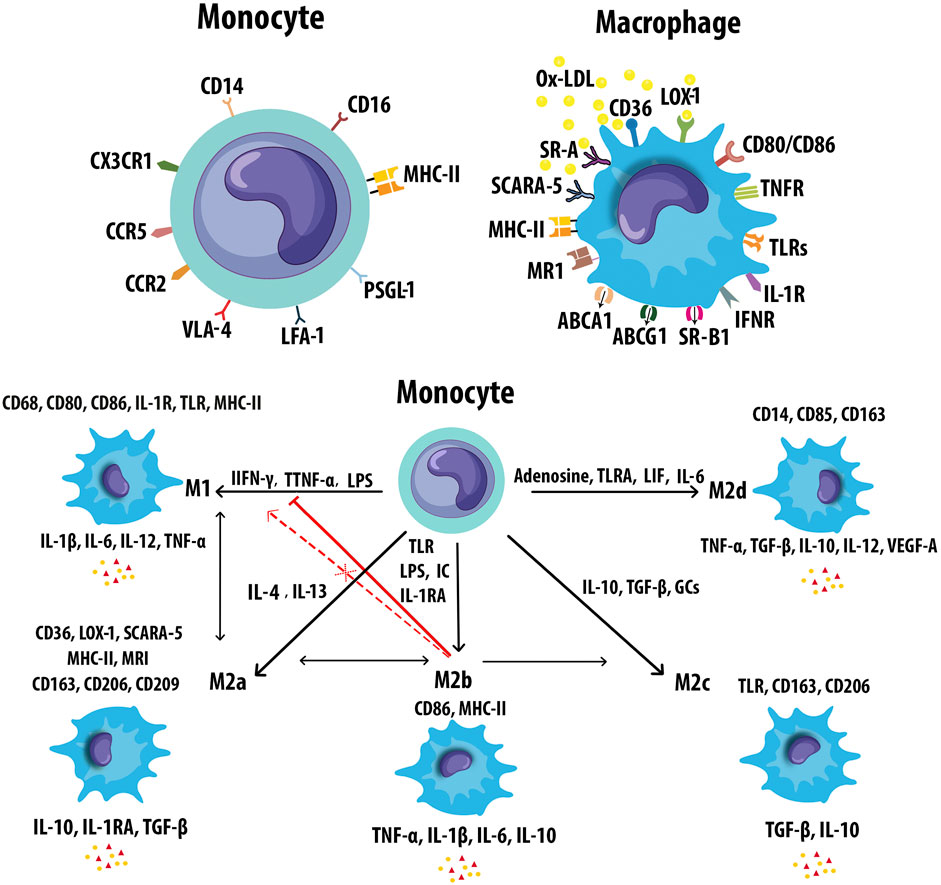
FIGURE 5. Markers of monocyte and macrophages. (A) Monocytes and macrophages express an ample variety of receptors that modulate monocyte and Mφ activation. (B) Mφs can polarize to M1 or M2 (M2a, M2b, M2c, and M2d) in response to different inducers and secrete pro- and anti-inflammatory cytokines respectively. M2b Mφs can convert to the other M2 subtypes in response to activated factors. M2b Mφs inhibit the conversion from monocyte to M1 Mφs. M2b also cannot repolarize to M1 during exposure to M1 inducer. Leukemia inhibitory factor (LIF), mannose receptor (MR).
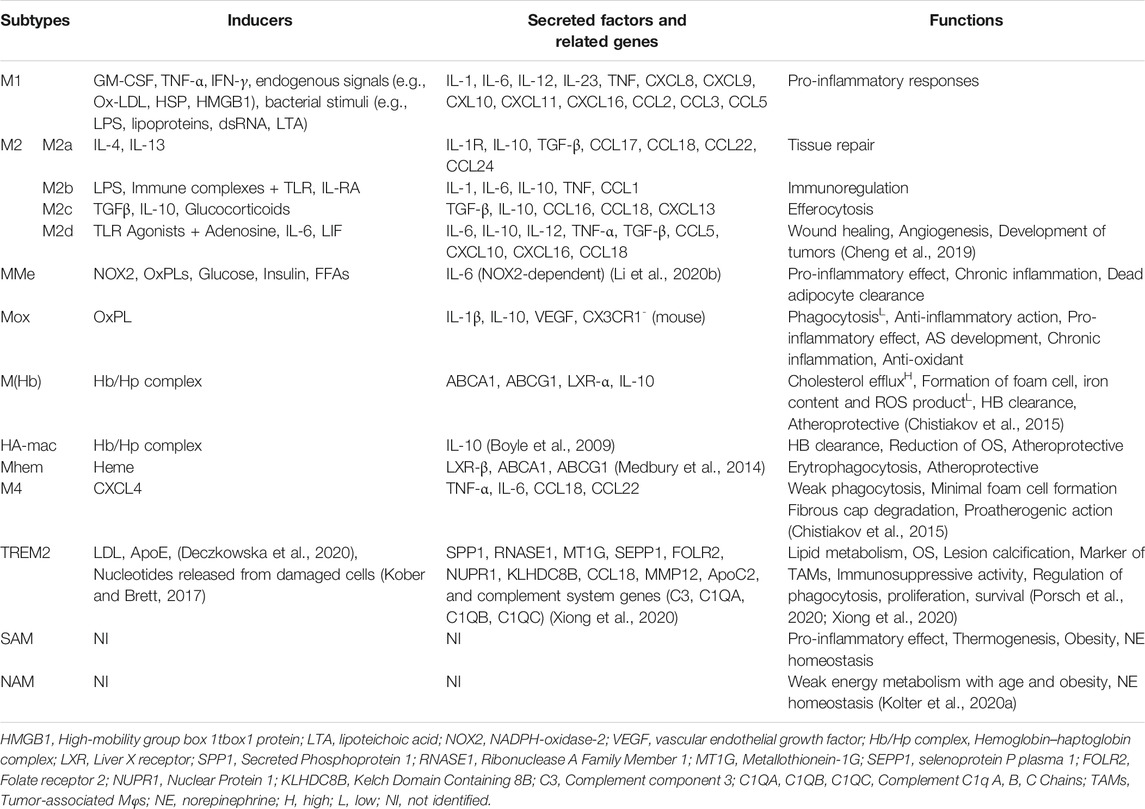
TABLE 2. Activating stimuli, properties, and functions of polarized macrophage subtypes.
Th1 cytokines including IFN-γ, TNF-α, GM-CSF, and also bacterial stimuli such as LPS polarize Mφs toward the M1 phenotype (Nikonova et al., 2020). M1 cells secrete high levels of pro-inflammatory factors including interleukin-1β (IL-1β), IL-6, IL-12, IL-23, low levels of IL-10, and chemokines such as CXCL-9, CXCL-10, and CXCL-11. M1 Mφ polarization is a tightly controlled process including a set of signaling pathways. Toll-like receptor 4 (TLR4) can be stimulated by LPS or other microbial ligands. This, in turn, promotes activation of interferon regulatory factor-3, activator protein 1 (AP-1), and NF-κB. Myeloid differentiation factor 88 (MyD88), one of the adaptors in responding to TLR4 activation, triggers NF-κB pathway (p65 and p50). This pathway regulates inflammatory genes including pro-inflammatory cytokines such as TNF-α, IL-6, and IL-12. MyD88 activates AP-1 via MAPK signaling. Enhanced AP-1 expression is also mediated by cytokine receptors. Signal transducer and activator of transcription 1 (STAT1) is activated by IFN-γ receptor which regulates the expression of specific genes such as MHC-II, IL-12, nitric oxide synthase 2, and the suppressor of cytokine signaling to promote the M1 Mφ polarization (Figure 6) (Wang et al., 2014b). Mφs express Akt1, Akt2, and Akt3 isoforms which are vital for cell proliferation, migration, and survival (Babaev et al., 2014). Akt1 and Akt2 play opposing roles in Mφ polarization. Akt1 deficiency inducing M1 and Akt2 ablation resulting in M2 phenotype (Arranz et al., 2012). Deficiency of Akt2 suppressed the ability of Mφ to undergo M1 polarization reducing the formation of both early and advanced AS in Ldlr−/− mice (Babaev et al., 2014). While deletion of Akt1 resulted in enhanced AS and occlusive coronary artery disease in ApoE−/− mice (Fernández-Hernando et al., 2007). Mice with reduced mitochondrial oxidative phosphorylation (OxPhos), showed increased M1-like Mφ polarization and decreased Th2 cytokine responsiveness (Jung et al., 2018). Activated M1 Mφs also produce ROS and nitric oxide (NO) through NADPH oxidase system, which may regulate the phagocytosis process of Mφ, disrupting normal cell metabolism, inducing apoptosis, and ultimately causing chronic tissue damage and plaque formation (Xu et al., 2019). M1 Mφs are enriched in lipids and implicated in initiating and sustaining inflammation, atherosclerotic lesion enlargement, and promoting unstable plaques. M1 Mφs are also the most abundant cells in the lesions of infarction and coronary artery disease (CAD) patients (Barrett, 2020).
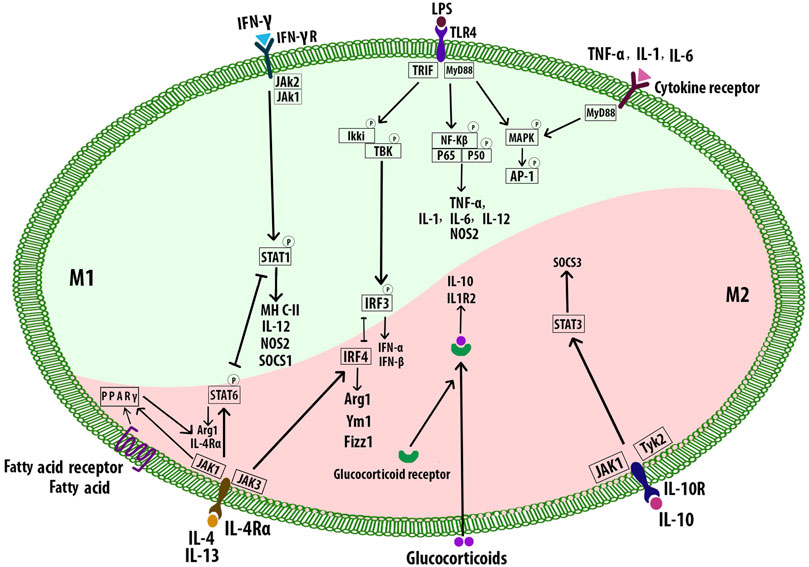
FIGURE 6. Macrophage polarization signaling pathways. M1 phenotypes express transcription factors such as NF-κB, STAT1, AP-1, and IRF3. M2 subtypes are stimulated by the Th2 cytokines IL-4 and IL-13 which bind to the receptor IL-4Rα and cause the activation of STAT6, IRF4, and PPAR-γ. The intracellular glucocorticoids also promote the transcription of anti-inflammatory genes. IL-10 activates STAT3 and subsequent upregulation of SOCS3. Signal transducer and activator of transcription 1 (STAT1), interferon regulatory factor -3 (IRF3), nitric oxide synthase 2 (NOS2), suppressor of cytokine signaling 3 (SOCS3), arginase 1 (Arg1).
Alternatively, activated M2 Mφs accumulate at the site of injury and release mediators that suppress inflammation in plaques, clear apoptotic cells (ACs), promote allergy, angiogenesis, tissue repair, fibrosis, and plaque stability (Wynn and Vannella, 2016; Barrett, 2020; Miki et al., 2021). M2 phenotypes are stimulated by the Th2 cytokines including IL-4 and IL-13 which bind to the receptor IL-4Rα and promote the M2 polarization through several pathways such as JAK1 and JAK3 signaling which further result in the activation of STAT6, IRF4, and peroxisome proliferator-activated receptor γ (PPAR-γ). STAT6, IRF4, and PPARγ regulate many of the genes such as Arg1, Fizz1, and Ym1 associated with mouse M2 Mφs. The fatty acid receptor also activates PPARγ. Inhibition of PPARγ with its antagonist blocked convallatoxin-induced M2 Mφ polarization and enhanced inflammatory responses (Wang et al., 2014a; Zhang et al., 2021). Convallatoxin is a natural cardiac glycoside that protects against AS. Intracellular glucocorticoids bind the glucocorticoid receptor resulting in the nuclear translocation of the complex. The complex binds DNA, promoting the transcription of anti-inflammatory genes such as IL-10 and IL1R2. Alternatively, the complex can also interact with other transcription factors such as NF-κB or AP-1. IL-10 can be produced by all leukocytes, and IL-10 receptor ligation leads to activation of the transcription factor STAT3 that upregulates the expression of SOCS3 which in turn mediates the suppression of pro-inflammatory cytokine signaling (Figure 6) (Wang et al., 2014a). Akt activation is required for M2 activation (Covarrubias et al., 2015). Several signals such as IL-10, and TGF-β promote M2 polarization via PI3K/Akt signaling (Park et al., 2011; Gong et al., 2012). Akt2−/− Mφs attributed to the reduced level of miR-155, which targets C/EBPβ, a key regulator of Arg1 expression and M2 polarization (Arranz et al., 2012). Different stimuli have been associated with the differentiation of M2 subtypes. IL-4, IL-13, and also high levels of CD206 and IL-1 receptor antagonist induce M2a which has anti-inflammatory and tissue remodeling properties. M2b is affected by immune complexes in combination with IL-1β and LPS, and it is involved in immunoregulation. IL-10 and TGFβ or glucocorticoids drive M2c, which has a role in efferocytosis and tissue remodeling. M2b and M2c share regulatory functions, characterized by the secretion of IL-10, and suppression of pro-inflammatory cytokines. IL-6, leukemia inhibitory factor adenosine, and TLR signals induce M2d with angiogenesis, and tumor growth effects (De Paoli et al., 2014; Liberale et al., 2017; Raggi et al., 2017; Abdelaziz et al., 2020). M2 cells also induce the expression of specific chemokines CCL24, CCL17, and CCL22 (M2a), CCL1 (M2b), CCL16, and CCL18 (M2c) (Mantovani et al., 2004). Overall, M2-derived chemokines and cytokines recruit tissue repair cells. Reducing Mφ-inflammation by modulating inflammatory cytokine secretion can promote plaque regression (Lin et al., 2021). M2 Mφs can affect plaque regression via anti-inflammatory effects (Moore et al., 2013) and promote wound healing and tissue repair through collagen formation (Spiller et al., 2014) and removal of dead cells by efferocytosis (Chang et al., 2015). Thus, regulating Mφ polarization to the M2 phenotype could be a therapeutic strategy to prevent the progression of AS or even hasten regression of plaques (Bi et al., 2019). In mouse models of AS regression, the plaque content of M2 markers increased, while M1 markers decreased. Lesions enriched in M2 Mφ are characterized by less inflammation and more stabilizing material, increased content of collagen, and a reduction in the cholesterol content in the plaques (Feig et al., 2011). Administration of IL-13, which is an M2 polarizing factor, protected from AS by increasing lesional collagen content and reducing VCAM-1-dependent monocyte recruitment in Ldlr−/− mice (Cardilo-Reis et al., 2012). AS regression after lipid-lowering is dependent on the recruitment of Ly6Chi inflammatory monocytes and their STAT6-dependent polarization to the M2 subtype in mice. The local accumulation of Mφs are highly efficient in clearing ACS. M2 subtype Mφ localized in areas of neovascularization, outside the lipid core, and can phagocytose apoptotic M1 phenotype Mφ, contributing to the resolution of inflammation and inhibition of necrotic core formation within lesions (Chinetti-Gbaguidi et al., 2015).
Mφ differentiation into MMe and Mox phenotypes in adipose tissue (AT) is induced by cytokines and OxPLs derived from Ox-LDL which play important roles in the development of chronic inflammation (Haka et al., 2016). NADPH-oxidase-2 is the main driver of MMe Mφs functions (Coats et al., 2017). The Mox phenotype is associated with the expression of surface markers including Srnx-1 and Txnrd-1 and it is abundant in advanced plaques comprising CD68+ cells (Kadl et al., 2010; Leitinger and Schulman, 2013).
HA-mac and M(Hb) are atheroprotective phenotypes that are induced by a hemoglobin-haptoglobin complex, implicated in hemoglobin clearance in hemorrhagic sites. M(Hb) Mφs regulate intracellular lipid balance by increasing cholesterol efflux mediators, thereby preventing the formation of foam cells (Chistiakov et al., 2015). Since Mφ intracellular iron levels may drive cholesterol efflux in M(Hb) cells, the manipulation of Mφ iron levels can be useful to retard AS development by upregulation of Mφ cholesterol efflux (Habib and Finn, 2014). Following endocytosis of hemoglobin-haptoglobin complex, heme is released from RBCs which stimulates Mφ polarization to human atheroprotective Mhem subtype implicated in erythrophagocytosis. The Mhem Mφ suppressed OS, lipid accumulation, and foam cell formation, sharing properties with M (Hb) (Skuratovskaia et al., 2020).
M4 Mφs are induced by CXCL-4 in human atherosclerotic plaques. M4 Mφs have pro-inflammatory and proatherogenic properties which may develop in arterial thrombosis (Skuratovskaia et al., 2020). M4 Mφs are unable to efficiently phagocytose Ox-LDL, with minimal formation of foam cells, and also may not function as efferocytes within plaques (Butcher and Galkina, 2012). They can be involved in fibrous cap degradation, and plaque rupture by producing the enzyme MMP12. M4 Mφ also seems to be irreversible and can not switch like M1, M2, or hemorrhage-associated phenotypes (Skuratovskaia et al., 2020).
TREM2 is a main determinant of adipose tissue homeostasis and a modulatory receptor involved in the regulation of phagocytosis, proliferation, and cell survival. TREM2 Mφs correspond to foamy lipid-laden Mφs accumulating in atherosclerotic plaques. Hematopoietic TREM2 is overexpressed on a subset of AS-associated aortic Mφs and is associated with lipid metabolic processes, OS, and lesion calcification while downregulating pro-inflammatory genes (Porsch et al., 2020).
Neuroimmunological Mφs have been reported in different tissues such as the sciatic nerve, adipose tissue, intestine, skin, and microglia which are implicated in thermogenesis, norepinephrine homeostasis, and obesity (Kolter et al., 2020a). Obesity may increase the risk of OS, and subsequent oxidation of LDL particles and atherogenesis (Poznyak et al., 2021). Neuroimmunological Mφs may have therapeutic potential for individuals with obesity (Skuratovskaia et al., 2020).
Bacterial and viral mediators are modulators of Mφ lipid metabolism and AS. ECs and Mφs in atherosclerotic plaques upregulate the expression of TLR in response to microbial antigens followed by inflammatory signals leading to AS. There is evidence that TLR3/4 ligands inhibit cholesterol efflux from Mφs leading to increased susceptibility to AS (Castrillo et al., 2003). Porphyromonas gingivalis induces its uptake by Mφs and stimulates the formation of foam cells, the hallmark of early atherogenesis (Giacona et al., 2004). Similar mechanisms have been observed for Chlamydophila pneumoniae infection where surface antigens such as LPS and heat shock protein (HSP) participate in this process. LPS has been shown to promote Mφs development into foam cells and chlamydial HSP60 may induce LDL oxidation on the lesion site (Milioti et al., 2008). The role of H. pylori, in Mφs differentiation into foam cells in a TLR-2- and TLR-4-dependent way has also been reported (Li B. et al., 2020). During H. pylori infection, Mφs are typically polarized to M1 phenotype which induces pro-inflammatory cytokines and promotes AS (Quiding-Järbrink et al., 2010; Vijayvergiya and Vadivelu, 2015).
Transcriptome analysis suggests that human cytomegalovirus reprograms monocyte differentiation toward pro-inflammatory M1 Mφs following infection. The upregulation of monocyte migration is necessary for the hematogenous spread of the virus and as a consequence, could promote AS associated with human cytomegalovirus infection (Smith et al., 2004; Chan et al., 2008). The enterovirus receptor CXADR is upregulated in Mφs in atherosclerotic plaques. CXADR expression was associated with M1 Mφ polarization and foam cells formation, suggesting a mechanism by which enterovirus may infect the cells in atherosclerotic lesions (Nilchian et al., 2020).
Mφs release cytokines such as IL-1, IL-6, IL-8, IL-12, IL-18, soluble CD40L (sCD40L), and, TNF in response to various inflammatory stimuli. These factors exhibit a variety of effects, discussed below, and appear to induce inflammation-mediated increases in vascular permeability (Arango Duque and Descoteaux, 2014; Jansen et al., 2016).
IL-1 signaling is proatherogenic and implicated in atherothrombosis. The isoforms IL-1α and IL-1β use a shared IL-1R1. Mice deficient in IL-1β show reduced atherosclerotic disease severity (Arango Duque and Descoteaux, 2014). Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) measured the efficacy of IL-1β inhibition in reducing CV event rates (Ridker et al., 2011). Targeting IL-1β by monoclonal antibodies inhibited atherosclerotic plaque formation and the progression of AS (Bhaskar et al., 2011). IL-1β induces angiogenesis by recruitment of myeloid and endothelial lineage cells (Carmi et al., 2009). Deficiency of IL-1α inhibited early lesion formation and inflammation in the ApoE−/− mice (Kamari et al., 2011).
IL-6 is associated with multiple inflammatory disorders. IL-6 can bind to the membrane-bound IL-6R or soluble IL-6R. The CANTOS trial showed that modulation of the IL-6 signaling after taking canakinumab, a human anti-IL-1β monoclonal antibody, is associated with decreased CV events independent of lipid-lowering (Ridker et al., 2018). Senescence-associated IL-6 is upregulated in several tissues and may accelerate atherogenesis due to aging-related alterations. Thus, the blockade of IL-6 might be an effective strategy to reduce AS in old people (Tyrrell and Goldstein, 2020). The MIRACL study showed that the levels of high-sensitivity C-reactive protein (hs-CRP), serum amyloid A protein (SAA), and IL-6 inflammatory markers were related to the risk of stroke (Kinlay et al., 2008). A phase II clinical trial demonstrating ziltivekimab, a fully human monoclonal antibody targeting the IL-6 ligand, markedly reduced multiple biomarkers of systemic inflammation and thrombosis including hsCRP, fibrinogen, SAA, sPLA2, and Lp(a). Thus, the direct inhibition of IL-6 might have the potential to maximize anti-inflammatory atherosclerotic benefits (Ridker et al., 2021). Since IL-6 is a pleiotropic cytokine, depending on the target cell type, it can exhibit both pro- and anti-inflammatory properties. IL-6 contributes to pro-inflammatory responses such as Mφ and neutrophil chemotaxis, induction of chemokine, adhesion molecule production, and promotes vascular endothelial growth factor production and conversely anti-inflammatory functions such as inhibition of IL-1 production, and induction of IL-1R antagonist indicating that IL-6 can act to reduce inflammation and protect the CV system. IL-6 promotes the development of CV disease through activation of ECs, induction of VSMC proliferation, pro-thrombotic effects on platelets, and accumulation of Mφ lipid (Reiss et al., 2017). A high serum level of IL-6 in intermediate CV risk patients referred for coronary angiography is predictive of CAD (Wainstein et al., 2017).
IL-8 is a proatherogenic cytokine produced by Mφs. Upregulation of IL-8 in atherosclerotic plaques promotes recruitment of monocytes by mediating the rolling of monocytes to adhere firmly to the vascular EC (Apostolopoulos et al., 1996; Gerszten et al., 1999). In addition, elevated serum levels of IL-8 are increased in patients with unstable angina (Romuk et al., 2002). High serum levels of IL-8 are related to an increased risk of all-cause mortality independent of the underlying cause (Velásquez et al., 2019).
IL-12 is formed mainly by plaque Mφs and stimulates the differentiation of CD4+ T cells into Th1 cells through the IL-12Rβ2 chain. Th1 cells can further activate Mφs and subsequently the inflammatory cascade (Kleemann et al., 2008). The administration of IL-12 resulted in enhanced lesion size in ApoE−/− mice (Davenport and Tipping, 2003). ApoE−/− mice lacking TLR4 or MyD88 showed a decrease in AS that was associated with a significant reduction in the circulating levels of IL-12 (Michelsen et al., 2004). Exposure of non-stimulated CD4+CD28− T cells to IL-12 induced recruitment of T cells into the atherosclerotic plaque in human atheroma-SCID mouse chimeras (Zhang et al., 2006). These results suggest that IL-12 is a proatherogenic and pro-inflammatory cytokine.
IL-18 acts as a pro-inflammatory cytokine by mediating the production of IL-1β, IL-8, and the expression of ICAM-1 and VCAM-1 (Dinarello et al., 2013). IL-18 accelerated AS by upregulation of CD36 and MMP-9 expression via NF-κB pathway in ApoE−/− mice. NF-κB blockade inhibited IL-18 signaling through downregulation of IL-18, IL-18Rα, CD36, and MMP-9, and upregulation of LXR-α resulted in protection against IL-18-induced AS (Bhat et al., 2015). The proatherogenic effect of IL-18 in the absence of T cells is accompanied by elevation of IFN-γ and CXCL16 expression (Tenger et al., 2005). High serum concentrations of IL-18 increase the risk of future coronary heart disease (Jefferis et al., 2011).
CD40 is expressed mostly on the M1 phenotype (Jansen et al., 2016). Plasma CD40 levels are associated with the severity of carotid AS and an increased risk for future CV events. Intra-plaque levels of sCD40 and sCD40L are also associated with vulnerability and remodeling (Shami et al., 2020). The co-stimulatory CD40 and CD40L are implicated in the regulation of the inflammatory response during AS development. Targeting non-classical CD40L-Mac1 interactions and the inflammatory CD40-TRAF6 signaling may have the potential to reduce the residual inflammatory risk that drives AS (Bosmans et al., 2020).
TNF activates ECs to induce the expression of multiple adhesion molecules and promotes secretion of a variety of inflammatory cytokines and chemokines to enhance the recruitment of activated leukocytes into the lesions (McKellar et al., 2009). TNF-α is a pleiotropic cytokine and functions through its two main receptors including TNF receptors 1 and 2 (TNFR1 and TNFR2). Pro-inflammatory signaling, apoptosis, and degenerative cascades are reported to mediate through TNFR1 while TNF-α signaling through TNFR2 activation is anti-inflammatory and cytoprotective, leading to cell proliferation, differentiation, angiogenesis, and tissue repair (Subedi et al., 2020). TNF-α is implicated in vascular dysfunction. The increase of TNF-α expression by AGEs, their receptors, LOX-1, and NF-κB signaling may induce ROS production leading to endothelial dysfunction in CV disease (Zhang et al., 2009). TNF-α enhances the progression of lesions towards an advanced phenotype by stimulating necrosis and decreasing the incidence of apoptosis in transgenic mice (Boesten et al., 2005). Anti-TNF-α therapy and drugs like thalidomide that inhibit TNF-α production may reduce CV events and inhibit the early development of AS (Chew et al., 2003).
IL-10 exhibits anti-inflammatory properties that may be protective against AS (Mallat et al., 1999). IL-10 appears to impair HIV-related CD4+ T cells and might be a potential target for the treatment of AS in HIV (Fourman et al., 2020). An inverse correlation between AS severity and IL-10+ B cells was found in Ldlr−/− mice, while increased cholesterol may mask the protective effects of IL-10+ B cells (Douna et al., 2019). AS was attenuated through increased expression of IL-10 in programmed cell death protein 4 (PDCD4)-deficient mice. Thus, PDCD4 could affect AS development by regulating the expression of IL-10 (Jiang et al., 2016).
TGF-β is a pleiotropic cytokine that can be both atheroprotective and atherogenic. Deficiency of growth differentiation factor 15 (GDF15), a member of TGF-β family, attenuated lesion formation in Ldlr−/− mice in a TGFβRII-dependent manner that reduced Mφ chemotaxis (de Jager et al., 2011). Suppression of endothelial TGF-β signaling reduced inflammation and vascular permeability in hyperlipidemic mice and attenuated disease progression (Chen et al., 2019). TGF-β can also elicit atherogenic effects through its actions on VSMCs in early plaque lesions (Low et al., 2019). An inverse relationship between serum TGF-β1 levels and advanced AS has been observed (Grainger et al., 1995). Inhibition of TGF-β signaling promoted the development of atherosclerotic lesions with unstable plaque phenotype, increased inflammatory cells, and decreased collagen content in ApoE−/− mice (Mallat et al., 2001). TGF-β1 increased cholesterol efflux and attenuated Mφ foam cell formation in ApoE−/− mice (Panousis et al., 2001). Deletion of Tgfb1 resulted in reduced VSMC differentiation, accelerated lesion formation, and increased inflammation in heterozygous mice, indicating that TGF-β can protect against AS (Low et al., 2019).
Mφ apoptosis, impaired Mφ efferocytosis, and secondary Mφ necrosis have been implicated in necrotic core and vulnerable plaque formation (Gonzalez and Trigatti, 2017). While the most common form of Mφ death is apoptosis, other programmed cell death modalities in Mφ include immune-reactive cell death (pyroptosis), necroptosis, mitochondrial-dependent cell death (parthanatos), and iron-dependent cell death (ferroptosis) (Yan et al., 2020) (Figure 7).
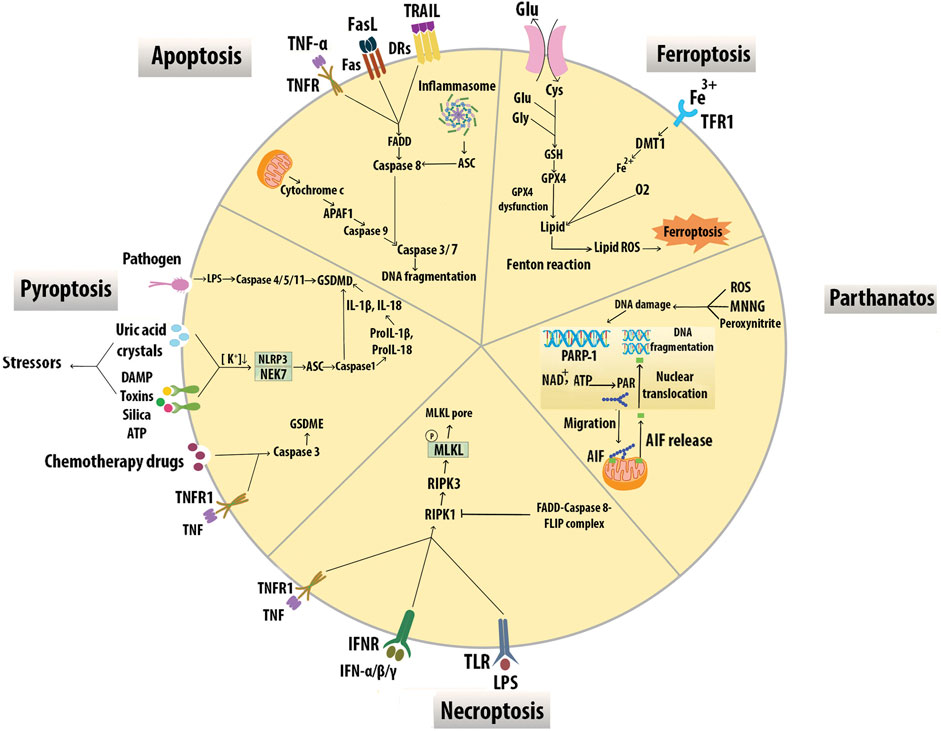
FIGURE 7. Modes of macrophage death. Different modes of cell death including apoptosis, pyroptosis, necroptosis, parthanatos, and ferroptosis have unique activating stimuli and present different signaling pathways and distinct physiological outcomes. Inhibitors of the divalent metal transporter 1 (DMT1).
Apoptosis can be triggered by the activation of a mitochondrial pathway by cellular stress or through the activation of death receptors (DRs) at the cell surface. The mitochondrial intermembrane protein, cytochrome c, is released into the cytosol and triggers apoptotic protease-activating factor 1 (APAF1), which in turn activates the serine protease caspase 9. Active caspase 9 stimulates the executioner caspases caspase 3 and caspase 7, leading to DNA fragmentation. Death receptors include TNFR1, FAS receptor, and TNF-related apoptosis-inducing ligand (TRAIL). Receptor ligation promotes the recruitment of adaptor proteins, including FAS-associated death domain protein (FADD), which bind and activate caspase 8. Caspase 8 activates the executioner caspases results in cell death. Inflammasomes can also trigger caspase 8 via apoptosis-associated protein (ASC) leading to DNA damage-induced apoptosis (Boada-Romero et al., 2020). Mφ SR-A has a protective effect in early lesions but proatherosclerotic roles in advanced plaques for induction of Mφ apoptosis and efferocytosis. (Devries-Seimon et al., 2005). STAT1 appears to be an essential component of ER stress/SRA-induced Mφ apoptosis in advanced atheromata (Lim et al., 2008). Overexpression of IL-10 by T cells was reported to suppress STAT1 activity and protect from excessive cell death and plaque necrosis (Pinderski et al., 2002). Transcription factor Zhx2 deficiency promoted Mφ apoptosis and exhibited a reduction in lesion size and resistance to AS in mice. Evidence including BM transplantation studies also supports this effect of Zhx2 which is mediated in part by monocytes/Mφs (Erbilgin et al., 2018). A prolonged increase of CHOP levels induces apoptosis through cytoplasmic Ca2+ metabolism and suppression of prosurvival molecules like Bcl-2 (Tabas, 2010). In early lesions, efferocytosis prevents cellular necrosis and induces anti-inflammatory pathways through TGF-β and the activation of NF-κB signaling to promote cell survival (Henson et al., 2001). In contrast, ER stress in foam cells induces inflammatory pathways within the lesion by suppressing NF-κB signaling and activating JNK, activator protein-1, ROS, and spliced X-box binding protein 1, which promote apoptosis (Xu et al., 2019).
Pyroptosis results in the death of Mφs in response to bacterial infection, accompanied by activation of inflammasomes and maturation of pro-inflammatory cytokines IL-1β and IL-18. Gasdermin D (GSDMD) pores are the effectors of pyroptosis. Cytosolic LPS binds caspase 4/5/11 to trigger their cleavage of GSDMD and subsequent plasma membrane permeabilization leading to pyroptosis. Inflammasome sensor proteins, such as NLRP3, recognize cellular stressors, including those from bacteria, viruses, toxins, ATP, uric acid crystals, silica, and damage-associated molecular pattern (DAMPs). These stressors activate NLRP3 indirectly through potassium efflux, which leads to NEK7 binding NLRP3 to trigger its oligomerization. NLRP3 subsequently activates caspase-1 via the adaptor protein ASC and cleaves GSDMD resulting in pyroptotic cell death. Active caspase 1 also proteolytically processes IL-1β and IL-18 into their active forms, which are subsequently released from pyroptotic cells (Frank and Vince, 2019). Chemotherapy drugs induce pyroptosis through caspase 3 cleavage of Gasdermin E (GSDME) (Wang et al., 2017). Cleavage of GSDME by caspase 3 may also switch TNF-induced apoptosis to pyroptosis (Rogers et al., 2017). AS risk factors could activate NLRP3 inflammasomes in ECs and Mφs. NLRP3 inflammasome-mediated pyroptosis in the atherosclerotic plaques is correlated with plaque rupture and vascular inflammation, suggesting that NLRP3 inflammasome and related pyroptosis play an important role in the pathogenesis of AS (Zeng et al., 2019).
Necroptosis is activated by death receptors including TNFR1, IFNR, and TLR3/4. Necroptosis is initiated through the activation of RIPK1, which binds and activates RIPK3. RIPK3-mediated phosphorylation of the mixed-lineage kinase domain-like protein (MLKL) results in membrane lysis. This process is inhibited by the activation of caspase 8 and its apoptotic inhibitor FLIP which cleaves RIPK1 to prevent necroptosis (Boada-Romero et al., 2020). The expression of necroptosis mediators RIPK3 and MLKL is upregulated in atherosclerotic plaques, especially in vulnerable plaques supporting the role of RIPK3-mediated Mφ necroptosis in the development of AS (Karunakaran et al., 2016; Tian et al., 2016).
Parthanatos in plaque Mφs is triggered by DNA damaging stimuli such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), peroxynitrite, and ROS-dependent activation of poly ADP-ribose polymerase (PARP)1. The PAR polymer is synthesized by PARP1 in response to DNA breaks, migrates to the mitochondria, and releases apoptosis-inducing factor (AIF) which interacts with Mφ migration inhibitory factor and degrades DNA (Robinson et al., 2019). PARP1 inhibition may affect endothelial function, lipid metabolism, foam cell formation, switch from necrosis to apoptosis, and thus play a major role in atherogenesis (Xu et al., 2014).
Ferroptosis is associated with iron-dependent accumulation of lipid hydroperoxides during cell death. Cysteine (Cys2) can enter the cytoplasm via xCT and synthesizes glutathione (GSH) with glutamate (Glu), and glycine (Gly). Glutathione peroxidase 4 (GPX4) is an antioxidant enzyme that removes oxidative modifications from lipids. When GPX4 dysfunction, lipid transforms into lipid ROS with O2 and Fe2+ via the Fenton reaction (Zuo et al., 2020). Lipid peroxidation, intraplaque hemorrhages, and iron deposition are hallmarks of advanced human plaques, which is indirect evidence for the initiation of Mφ ferroptosis and plaque destabilization. Overexpression of GPX4 decreased lipid peroxidation and inhibited plaque development in ApoE−/− mice indicating the possible role of ferroptosis in CV disease (Guo et al., 2008).
Efferocytosis is the process by which cells undergoing apoptosis are cleared by Mφs, thereby reducing inflammation and maintaining tissue homeostasis (Yan et al., 2020). Disposal of the dying cells requires a variety of signal molecules through which phagocytes recognize and engulf ACs. Find-me signals, such as lysophosphatidylcholine (LPC), ATP/UTP, CX3CL1, and sphingosine-1-phosphate (S1P) are released by ACs, which promote attracting phagocytes to the sites of death. Phagocytes sense these signals via receptors including G2A, P2Y2, CX3CLR, and S1PRs respectively. (Figure 8A). Bridging molecules such as apoE and MFGE8 connect phagocytes to ACs. Afterward, ACs exposed to a variety of signals on their surfaces, interacting with receptors on the phagocytic membrane through Eat-me signals, and efficiently process the AC constituents to maintain homeostasis. The most common Eat-me signal, phosphatidylserine (PS), can interact with a variety of receptors on the surface of phagocytes, such as BAI1 and αvβ3 integrin. Other Eat-me signals, such as calreticulin and ICAM3 modulate the identification and engulfment of ACS via the receptors LRP1 and CD14 respectively (Figure 8B). Healthy cells display don’t-eat-me signals such as CD47, CD31, and CD24, on their surface which binds to receptors SIRP a, CD31, and Siglec-10, respectively expressed on phagocytes to avoid efferocytosis (Figure 8C). Programmed cell removal can be countermanded by anti-phagocytic don’t-eat-me signals such as cell surface expression of CD47. Mouse model studies showed that administration of CD47-blocking antibodies increased intraplaque efferocytosis efficiency and inhibited AS (Kojima et al., 2016). Defective phagocytosis of apoptotic foam cells has several consequences that promote the progression of chronic and non-resolving inflammatory diseases such as advanced AS (Figure 8D) (Wang et al., 2020). Angiotensin Ⅱ is a multifunctional hormone that plays a major role in the development of AS. Angiotensin Ⅱ promotes MerTK shedding via AT1R/ROS/p38 MAPK/ADAM17 pathway in Mφs, which leads to defective efferocytosis, accumulation of ACs, and progression of AS (Zhang et al., 2019). Mφs in atherosclerotic plaques express angiotensin Ⅱ type 1 receptor. This receptor of bone marrow-derived Mφs worsens the renal injury-induced AS by shifting the Mφ phenotype to M1 and decreasing the M2 phenotype which leads to impaired efferocytosis and enhanced necrosis (Yamamoto et al., 2011) Efferocytosis signaling molecules have been shown in Table 3.
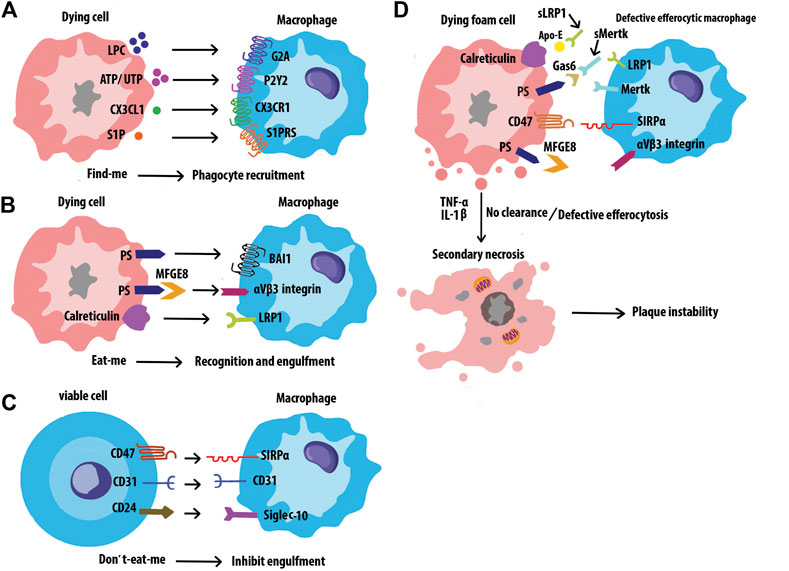
FIGURE 8. Interactions of efferocytosis signaling. Find-me signals recruit phagocytes to the sites of cell death. The dying cells will express Eat-me signals to facilitate interactions with phagocytes. Non-ACs send a Don’t-eat-me signal to avoid efferocytosis when exposed to phagocytes. Defective efferocytosis leading to inefficient clearance of ACs and subsequent necrosis and inflammation in plaques.
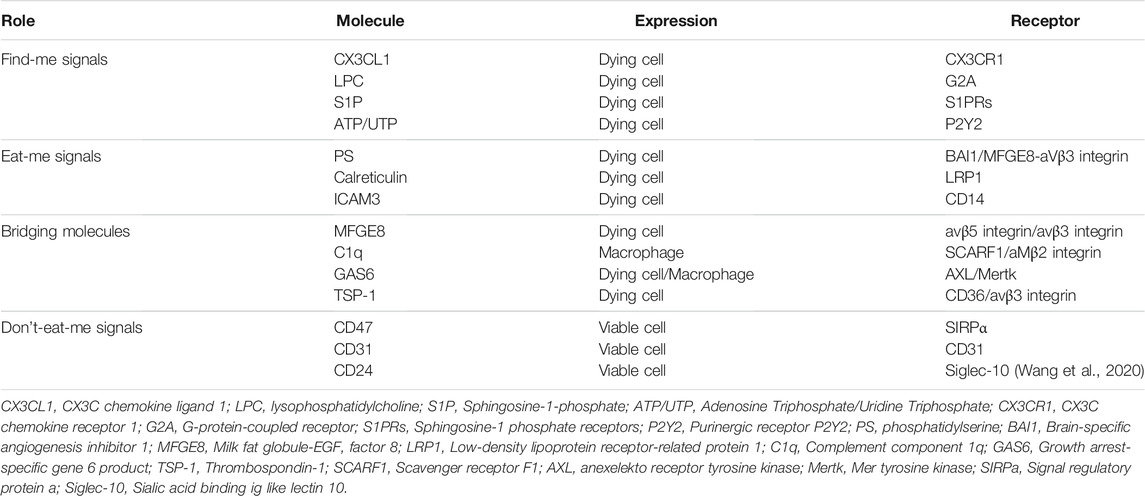
TABLE 3. Efferocytosis signaling molecules.
Atherosclerotic lesions typically consist of a lipid core which is covered by a fibrous cap. Matrix composition, thickness, cellularity, the collagen content of fibrous caps, and chronic inflammatory infiltrate are important factors in plaque stability. Vulnerable plaques are associated with active inflammation and a thin fibrous cap (Sodhi and Brown, 2018). Mφs secrete proteolytic enzymes, especially matrix metalloproteinase (MMP)-2 and MMP-9, causing degradation of ECM proteins such as collagen and elastin leading, to fibrous cap thinning and destabilization of advanced plaques. Thus, MMP-9 inhibitors have the potential to stabilize vulnerable plaques (Gough et al., 2006). Lesional Mφs induced apoptosis of VSMCs by TNF-α, the Fas apoptotic pathway, and NO production and appear to promote rupture-prone plaques (Boyle et al., 2003).
A key feature of unstable plaques is the formation of necrotic cores. If efferocytosis is insufficient, dead M1 Mφs accumulate and undergo postapoptotic necrosis, leading to the formation of a necrotic core. The thin fibrous cap and large necrotic core make lesions susceptible to rupture, leading to thrombosis, heart attack, or stroke (Virmani et al., 2002; Chinetti-Gbaguidi et al., 2015).
Normal circulating blood cells consist of a polyclonal mixture descended from thousands of hematopoietic stem cells (HSC). During aging, or in blood cell cancers, individual HSC clones can become relatively abundant. In 2017–2018, several groups reported that clonal hematopoiesis (CH), defined as expanded somatic blood cell clones in the absence of hematologic abnormalities, dramatically accelerates AS and increases the risk of MI (Fuster et al., 2017; Jaiswal et al., 2017; Sano et al., 2018). These clones were associated with various driver gene mutations such as the epigenetic modifiers TET2 and DNMT3A, suggesting that the mutations not only enhanced cell proliferation but also conferred pro-inflammatory or other properties to the leukocytes. Several subsequent studies on CH and AS have now been reported and they generally support the pro-inflammatory effects of certain driver mutations (Bick et al., 2020; Fidler et al., 2021).
The possibility that these observations with clonal hematopoiesis are due in part to increased hematopoiesis rather than pro-inflammatory clones was raised by Heyde et al. (Heyde et al., 2021). It has long been known that elevated levels of blood leukocytes, a phenomenon termed leukocytosis, predicts CV events, and certain studies have suggested that the presence of a vicious cycle in which events such as MI and AS risk factors promote leukocytosis (Swirski and Nahrendorf, 2013a). Previous studies had shown that the hematopoietic system is activated in AS in a variety of species including mice and humans. The authors showed, using fundamental evolutional dynamics, that this would predict the expansion of clones harboring both advantageous (in terms of increased proliferation) and neutral mutations. The authors quantitatively examined HSC proliferation in atherosclerotic mouse models as well as human subjects using incorporation of bromodeoxyuridine into DNA or the expression of the proliferation marker Ki67. In both atherosclerotic mice and patients with AS, HSC proliferation was markedly increased, roughly 2-fold, as compared to controls. It is noteworthy that smoking, a strong risk factor for AS, promotes CH and leukocytosis. A particularly exciting connection with HSC proliferation and clonal hematopoiesis in AS relates to the role of chronic stress. Sleep modulates hematopoiesis and protects against AS. Previously, McAlpine et al. (McAlpine et al., 2019) showed that sleep fragmentation in mice suppressed hypothalamic secretion of the wake-promoting peptide hypocretin, resulting in elevated M-CSF expression by pre-neutrophils in bone marrow that, in turn, increased blood monocytes and AS. The authors confirm in the present study that sleep fragmentation increases HSC proliferation. Thus, the relative importance in AS of driver mutations such as TET2, as compared to increased hematopoiesis and leukocytosis, is uncertain. Since both are potential therapeutic targets, the answer is of considerable interest.
Single-cell sequencing technologies can be applied to sequence DNA, RNA, open chromatin, or methylated DNA in single-cells in the context of normal or diseased tissues (Williams et al., 2020). The single-cell sequencing studies reported thus far have relied primarily on RNA sequencing (scRNAseq). In some cases, the studies have been complemented by time of flight mass cytometry analyses. Over the past 3 years, several groups have used scRNAseq to examine leukocyte heterogeneity in various mouse models of AS (Cochain et al., 2018; Kim et al., 2018; Winkels et al., 2018; Lin et al., 2019; Zernecke et al., 2020) and in human carotid AS (Fernandez et al., 2019) and these studies have been recently reviewed (Willemsen and de Winther, 2020; Fernandez and Giannarelli, 2021). ScRNAseq analyses of mouse atherosclerotic lesions identified three main clusters of the Mφs in AS, resident-like, inflammatory, and TREM2hi (Willemsen and de Winther, 2020). The resident-like Mφs, some of which are found in the adventitia, can proliferate and resemble an M2-like phenotype. The inflammatory Mφs are the major component of the Mφ population within the intima of the plaque. They appear to be the main cellular drivers of lesional inflammation and are enriched in both M1-associated genes and Mox-associated transcription factor NRF2. They are mainly non-foamy (Kim et al., 2018). The third subtype, TREM2hi Mφs, are foamy lipid-laden Mφs enriched for lipid metabolic processes, regulation of cholesterol efflux, OS and cellular catabolic processes and. have an M2-like phenotype. In a comprehensive meta-analysis of mouse studies, Ley’s group (Zernecke et al., 2020) confirmed these known Mφ subsets and identified a new Mφ subset resembling peritoneal cavity Mφs. Thus far only one study has examined human lesion (Fernandez et al., 2019). In that study, carotid plaques of clinically symptomatic disease (stroke) were compared to asymptomatic disease and scRNAseq was complemented with mass cytometry. Mφs from the plaques exhibited alternatively activated phenotypes, some of which were associated with plaque vulnerability.
Accumulating evidence from both human and mouse studies suggests that cellular senescence contributes to the pathogenesis of AS. The senescent cells can be recognized by the irreversible loss of proliferative capacity, resistance to cell death, and the production of a bioactive secretome, known as the senescence-associated secretory phenotype (SASP). The main components of SASP include inflammatory cytokines, immune modulators, growth factors, and proteases (Childs et al., 2017). Recent findings in mouse models show that among cells with elevated p16INK4a and senescence-associated beta-galactosidase (SA-β-Gal) expression (common biomarkers of senescence) are Mφs (Prattichizzo et al., 2016; Liu et al., 2019). In mouse models of AS foamy senescent Mφs expressing inflammatory cytokines/chemokines accumulate in the subendothelial space in the early stages of disease while in advanced lesions there is evidence senescent Mφs promote plaque instability (Childs et al., 2016). Yang et al. found that microRNA-216a (miR-216a) promotes Mφ senescence as characterized by increased SA-β-GAL activity and p53 and p16 expression, and it has been reported that patients with vulnerable coronary plaques have elevated levels of plasma miR-216a (Yang et al., 2019). A recent study demonstrated that the senescent cells promote thinning of the fibrous cap, and the clearance of these cells by a synolitic agent (ABT263) helped maintain the fibrous caps in mice with advanced lesions. The senescent cells antagonize insulin-like growth factor (IGF)-1 through the secretion of IGF-binding protein-3, thereby inhibiting innate smooth muscle cell repair (Childs et al., 2021). Acting through the phosphatidylinositide 3 kinase-AKT-mTOR-p53 signal pathway, CD9 upregulation initiates cellular senescence while knocking it down in senescent cells reduces senescence (Brosseau et al., 2018; Cho et al., 2020). In human atherosclerotic lesions, most Mφs in plaques exhibited CD9 immunoreactivity (Nishida et al., 2000). In hyperlipidemic ApoE−/− mice, selective delivery of the anti-senescence drug rosuvastatin to the atherosclerotic plaques using nanoparticles alleviated the progression of AS (Kim et al., 2021). Recent studies indicate that certain immune cells, in particular iNKT cells, can function to remove senescent cells (Kale et al., 2020; Arora et al., 2021).
Mφ dynamics play a role in all stages of AS and represent potential drug targets. A Mφ-biomimetic drug delivery system, in which ROS responsive nanoparticles were coated with Mφ membrane was developed. The Mφ membrane may sequester key pro-inflammatory cytokines to decrease local inflammation. The combination of pharmacotherapy and inflammatory cytokines sequestration offered by this platform led to improved therapeutic efficacy in AS (Gao et al., 2020).
Mφ death is an important feature of advanced plaques and is used as a therapeutic target in AS. Ox-LDL within advanced atherosclerotic plaque is directly responsible for the upregulation of RIP3, which is required to induce necroptosis in Mφs and lesion progression. Mφ necroptosis can be inhibited by necrostatin-1 to reduce lesion size and necrotic core formation for diagnostic and therapeutic interventions in AS (Karunakaran et al., 2016). Mφ pyroptosis is associated with the activation of NLRP3 inflammasome which has been linked to CV risk factors such as obesity and is an important regulator of CV inflammation. Thus, the therapeutic approaches targeting the activation of NLRP3 inflammasome and pyroptosis offer good prospects for the treatment of AS (Zeng et al., 2019). PARP inhibition attenuated plaque development and promoted plaque stability, likely through a reduction in the expression of inflammatory factors in ApoE−/− mice. Thus, PARP1 inhibitors such as 3-aminobenzamide, INO-1001, methoxyflavones, PJ34, and DPQ may prove beneficial for the treatment of AS (Oumouna-Benachour et al., 2007; Martinet et al., 2019). Ferrostatin, an inhibitor of Mφ ferroptosis, suffers from inherent stability but anti-ferroptosis analog drugs with improved potency such as liproxstatins and anti-oxidants can inhibit ferroptotic cell death (Hofmans et al., 2016; Martinet et al., 2019).
Lipid deposition in the arterial wall is an initial step in AS, which promotes an inflammatory response. However, it was recently reported that the SR-B1 in ECs binds plasma LDL and mediates the delivery of LDL into arteries and its engulfment by artery wall Mφs to form foam cells. Thus, inhibition of SR-B1 in ECs might reduce lipid deposition with the potential of anti-inflammatory therapy in AS (Huang et al., 2019).
Photobiomodulation therapy has a protective role on AS through promoting the ABCA1-medicated cholesterol efflux in Mφ to inhibit foam cells formation (Yin et al., 2021). Proefferocytic therapy which specifically targets the necrotic core can potentially be used for the treatment of advanced AS (Kojima et al., 2017). 2-hydroxybenzylamine treatment reduced inflammation and plaque apoptotic cells but promoted efferocytosis and features of stable plaques in hypercholesterolemic Ldlr−/− mice supporting its potential as a therapeutic approach for atherosclerotic cardiovascular disease (Tao et al., 2020). An atheroprotective strategy that uses plaque targeting to deliver a proresolving mediator may stabilize advanced atherosclerotic lesions. Thus, defective inflammation resolution may have a role in advanced AS (Fredman et al., 2015).
Inhibition of local proliferation of Mφs is key in plaque regression in response to cholesterol-lowering. Thus, Mφ proliferation was identified as the predominant turnover determinant and a target for induction of plaque regression (Härdtner et al., 2020). HDL nanoparticles are used to target atherosclerotic Mφs. Nanoparticle-based delivery of simvastatin was used to inhibit plaque Mφ proliferation. This resulted in the rapid reduction of plaque inflammation when it was combined with oral statin treatment. Thus, pharmacologically inhibiting plaque Mφ proliferation by nanotherapy can effectively suppress plaque inflammation and reduce AS (Tang et al., 2015).
Defective autophagy in Mφs contributes to impaired cholesterol metabolism and defective efferocytosis leading. Mφs treated with anti-miR-33 showed increased autophagy, lipid droplet catabolism, and enhanced efferocytosis to reduce plaque necrosis (Ouimet et al., 2017).
AS is an inflammation-driven disease and Mφs play a central role in the pathogenesis of AS and controlling inflammation in all stages. Understanding monocytes differentiation into either pro- or anti-inflammatory Mφs within lesions and how Mφs affect plaque initiation and progression is of potential importance for the treatment of AS. Several strategies including anti-inflammatory approaches, depolarizing Mφs, modulation of Mφs, survival, and enhancing efferocytosis have been proposed as possible therapies. Targeting inflammation may be a promising therapeutic option for reducing AS. However, anti-inflammatory strategies may elicit undesirable effects such as infection (Moriya, 2019). Since the complex environmental stimuli within atherosclerotic plaques in vivo affects monocyte-derived Mφs, studying the effects of signaling crosstalk on inflammatory responses can be helpful to understand Mφ activation in the lesion and the development of therapeutic interventions. Another aspect that complicates therapeutic inhibition of Mφ death in atherosclerotic plaques is the crosstalk between cell death mechanisms (Nikoletopoulou et al., 2013). There is a balanced interaction between different types of cell death so that blocking one type of death may stimulate cells to initiate another death pathway. For instance, inhibition of caspases by the pan-caspase inhibitor zVAD promotes apoptosis but may facilitate the necroptosis program downstream of TNFR. Several main mediators of different types of cell death have been identified but more investigation is still needed (Chen et al., 2018). Cholesterol homeostasis in Mφs involves a dynamic balance between cholesterol uptake and efflux (Remmerie and Scott, 2018). These processes are regulated by signaling systems that are also affected by the events such as Mφ pro-inflammatory activation, phagocytosis stimulation, and autophagy induction. The signaling networks regulating these events in Mφs involve specific proteins that might be used as potential therapeutic targets in AS management. The divergent functions of Mφs, including contributions to inflammation, healing, regeneration, and remodeling arise due to the numerous types of Mφs, which can quickly adapt their phenotype in response to the microenvironment changes. Insufficient understanding of Mφ functions represents a major obstacle in Mφ targeting. For instance, M1 Mφs impair wound healing in some situations (Mirza et al., 2014) while promoting regeneration in others (Novak et al., 2014). M2 Mφs have also been associated with both tissue regeneration (Godwin et al., 2013) and fibrosis (Furukawa et al., 2015). In addition, data from cultured Mφs and animal models may not completely reflect the process in human atherosclerotic lesions. In this regard, Useful mouse models of plaque instability have been developed (Chen et al., 2013) Of course, translational studies are needed to confirm the observations made in preclinical murine models.
LF prepared the draft and designed the figures. SS edited and contributed to the manuscript. AJL reviewed, revised, and overall supervised the whole work.
This work was supported in part by NIH grants HL144651, DK117850, HL147883, HL148577, HL148110, HL152176, and DA-Army Medical Research Grant W81XWH2110115.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelaziz, M. H., Abdelwahab, S. F., Wan, J., Cai, W., Huixuan, W., Jianjun, C., et al. (2020). Alternatively Activated Macrophages; a Double-Edged Sword in Allergic Asthma. J. Transl Med. 18 (1), 58. doi:10.1186/s12967-020-02251-w
Alderson, L. M., Endemann, G., Lindsey, S., Pronczuk, A., Hoover, R. L., and Hayes, K. C. (1986). LDL Enhances Monocyte Adhesion to Endothelial Cells In Vitro. Am. J. Pathol. 123 (2), 334–342.
Apostolopoulos, J., Davenport, P., and Tipping, P. G. (1996). Interleukin-8 Production by Macrophages from Atheromatous Plaques. Arterioscler Thromb. Vasc. Biol. 16 (8), 1007–1012. doi:10.1161/01.atv.16.8.1007
Apostolov, E. O., Shah, S. V., Ray, D., and Basnakian, A. G. (2009). Scavenger Receptors of Endothelial Cells Mediate the Uptake and Cellular Proatherogenic Effects of Carbamylated LDL. Arterioscler Thromb. Vasc. Biol. 29 (10), 1622–1630. doi:10.1161/atvbaha.109.189795
Arango Duque, G., and Descoteaux, A. (2014). Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 5, 491. doi:10.3389/fimmu.2014.00491
Arora, S., Thompson, P. J., Wang, Y., Bhattacharyya, A., Apostolopoulou, H., Hatano, R., et al. (2021). Invariant Natural Killer T Cells Coordinate Removal of Senescent Cells. Med 2 (8), 938–950.e8. doi:10.1016/j.medj.2021.04.014
Arranz, A., Doxaki, C., Vergadi, E., Martinez de la Torre, Y., Vaporidi, K., Lagoudaki, E. D., et al. (2012). Akt1 and Akt2 Protein Kinases Differentially Contribute to Macrophage Polarization. Proc. Natl. Acad. Sci. U S A. 109 (24), 9517–9522. doi:10.1073/pnas.1119038109
Babaei, S., Picard, P., Ravandi, A., Monge, J. C., Lee, T. C., Cernacek, P., et al. (2000). Blockade of Endothelin Receptors Markedly Reduces Atherosclerosis in LDL Receptor Deficient Mice: Role of Endothelin in Macrophage Foam Cell Formation. Cardiovasc. Res. 48 (1), 158–167. doi:10.1016/s0008-6363(00)00169-3
Babaev, V. R., Hebron, K. E., Wiese, C. B., Toth, C. L., Ding, L., Zhang, Y., et al. (2014). Macrophage Deficiency of Akt2 Reduces Atherosclerosis in Ldlr Null Mice. J. Lipid Res. 55 (11), 2296–2308. doi:10.1194/jlr.M050633
Barrett, T. J. (2020). Macrophages in Atherosclerosis Regression. Atvb 40 (1), 20–33. doi:10.1161/atvbaha.119.312802
Bartlett, B., Ludewick, H. P., Misra, A., Lee, S., and Dwivedi, G. (2019). Macrophages and T Cells in Atherosclerosis: a Translational Perspective. Am. J. Physiol. Heart Circ. Physiol. 317 (2), H375–H386. doi:10.1152/ajpheart.00206.2019
Bhaskar, V., Yin, J., Mirza, A. M., Phan, D., Vanegas, S., Issafras, H., et al. (2011). Monoclonal Antibodies Targeting IL-1 Beta Reduce Biomarkers of Atherosclerosis In Vitro and Inhibit Atherosclerotic Plaque Formation in Apolipoprotein E-Deficient Mice. Atherosclerosis 216 (2), 313–320. doi:10.1016/j.atherosclerosis.2011.02.026
Bhat, O. M., Kumar, P. U., Giridharan, N. V., Kaul, D., Kumar, M. J., and Dhawan, V. (2015). Interleukin-18-induced Atherosclerosis Involves CD36 and NF-Κb Crosstalk in Apo E-/- Mice. J. Cardiol. 66 (1), 28–35. doi:10.1016/j.jjcc.2014.10.012
Bi, Y., Chen, J., Hu, F., Liu, J., Li, M., and Zhao, L. (2019). M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 6724903. doi:10.1155/2019/6724903
Bick, A. G., Pirruccello, J. P., Griffin, G. K., Gupta, N., Gabriel, S., Saleheen, D., et al. (2020). Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 141 (2), 124–131. doi:10.1161/CIRCULATIONAHA.119.044362
Boada-Romero, E., Martinez, J., Heckmann, B. L., and Green, D. R. (2020). The Clearance of Dead Cells by Efferocytosis. Nat. Rev. Mol. Cel Biol 21 (7), 398–414. doi:10.1038/s41580-020-0232-1
Boesten, L. S., Zadelaar, A. S., van Nieuwkoop, A., Gijbels, M. J., de Winther, M. P., Havekes, L. M., et al. (2005). Tumor Necrosis Factor-Alpha Promotes Atherosclerotic Lesion Progression in APOE*3-Leiden Transgenic Mice. Cardiovasc. Res. 66 (1), 179–185. doi:10.1016/j.cardiores.2005.01.001
Bosmans, L. A., Bosch, L., Kusters, P. J. H., Lutgens, E., and Seijkens, T. T. P. (2020). The CD40-Cd40l Dyad as Immunotherapeutic Target in Cardiovascular Disease. J. Cardiovasc. Transl Res. 14 (1), 1–10. doi:10.1007/s12265-020-09994-3
Boyle, J. J., Harrington, H. A., Piper, E., Elderfield, K., Stark, J., Landis, R. C., et al. (2009). Coronary Intraplaque Hemorrhage Evokes a Novel Atheroprotective Macrophage Phenotype. Am. J. Pathol. 174 (3), 1097–1108. doi:10.2353/ajpath.2009.080431
Boyle, J. J. (2005). Macrophage Activation in Atherosclerosis: Pathogenesis and Pharmacology of Plaque Rupture. Curr. Vasc. Pharmacol. 3 (1), 63–68. doi:10.2174/1570161052773861
Boyle, J. J., Weissberg, P. L., and Bennett, M. R. (2003). Tumor Necrosis Factor-Alpha Promotes Macrophage-Induced Vascular Smooth Muscle Cell Apoptosis by Direct and Autocrine Mechanisms. Arterioscler Thromb. Vasc. Biol. 23 (9), 1553–1558. doi:10.1161/01.atv.0000086961.44581.b7
Brosseau, C., Colas, L., Magnan, A., and Brouard, S. (2018). CD9 Tetraspanin: a New Pathway for the Regulation of Inflammation? Front. Immunol. 9, 2316. doi:10.3389/fimmu.2018.02316
Buscher, K., Ehinger, E., Gupta, P., Pramod, A. B., Wolf, D., Tweet, G., et al. (2017). Natural Variation of Macrophage Activation as Disease-Relevant Phenotype Predictive of Inflammation and Cancer Survival. Nat. Commun. 8 (1), 16041–16110. doi:10.1038/ncomms16041
Butcher, M. J., and Galkina, E. V. (2012). Phenotypic and Functional Heterogeneity of Macrophages and Dendritic Cell Subsets in the Healthy and Atherosclerosis-Prone Aorta. Front. Physiol. 3, 44. doi:10.3389/fphys.2012.00044
Canet-Soulas, E., Bessueille, L., Mechtouff, L., and Magne, D. (2021). The Elusive Origin of Atherosclerotic Plaque Calcification. Front Cel Dev Biol 9, 622736. doi:10.3389/fcell.2021.622736
Cardilo-Reis, L., Gruber, S., Schreier, S. M., Drechsler, M., Papac-Milicevic, N., Weber, C., et al. (2012). Interleukin-13 Protects from Atherosclerosis and Modulates Plaque Composition by Skewing the Macrophage Phenotype. EMBO Mol. Med. 4 (10), 1072–1086. doi:10.1002/emmm.201201374
Carmi, Y., Voronov, E., Dotan, S., Lahat, N., Rahat, M. A., Fogel, M., et al. (2009). The Role of Macrophage-Derived IL-1 in Induction and Maintenance of Angiogenesis. J. Immunol. 183 (7), 4705–4714. doi:10.4049/jimmunol.0901511
Castrillo, A., Joseph, S. B., Vaidya, S. A., Haberland, M., Fogelman, A. M., Cheng, G., et al. (2003). Crosstalk between LXR and Toll-like Receptor Signaling Mediates Bacterial and Viral Antagonism of Cholesterol Metabolism. Mol. Cel 12 (4), 805–816. doi:10.1016/s1097-2765(03)00384-8
Chan, G., Bivins-Smith, E. R., Smith, M. S., Smith, P. M., and Yurochko, A. D. (2008). Transcriptome Analysis Reveals Human Cytomegalovirus Reprograms Monocyte Differentiation toward an M1 Macrophage. J. Immunol. 181 (1), 698–711. doi:10.4049/jimmunol.181.1.698
Chang, H. Y., Lee, H. N., Kim, W., and Surh, Y. J. (2015). Docosahexaenoic Acid Induces M2 Macrophage Polarization through Peroxisome Proliferator-Activated Receptor γ Activation. Life Sci. 120, 39–47. doi:10.1016/j.lfs.2014.10.014
Chang, T. Y., Li, B. L., Chang, C. C., and Urano, Y. (2009). Acyl-coenzyme A:cholesterol Acyltransferases. Am. J. Physiol. Endocrinol. Metab. 297 (1), E1–E9. doi:10.1152/ajpendo.90926.2008
Chen, P. Y., Qin, L., Li, G., Wang, Z., Dahlman, J. E., Malagon-Lopez, J., et al. (2019). Endothelial TGF-β Signalling Drives Vascular Inflammation and Atherosclerosis. Nat. Metab. 1 (9), 912–926. doi:10.1038/s42255-019-0102-3
Chen, Q., Kang, J., and Fu, C. (2018). The independence of and Associations Among Apoptosis, Autophagy, and Necrosis. Signal. Transduct Target. Ther. 3 (1), 18–11. doi:10.1038/s41392-018-0018-5
Chen, Y. C., Bui, A. V., Diesch, J., Manasseh, R., Hausding, C., Rivera, J., et al. (2013). A Novel Mouse Model of Atherosclerotic Plaque Instability for Drug Testing and Mechanistic/therapeutic Discoveries Using Gene and microRNA Expression Profiling. Circ. Res. 113 (3), 252–265. doi:10.1161/CIRCRESAHA.113.301562
Cheng, H., Wang, Z., Fu, L., and Xu, T. (2019). Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 9, 421. doi:10.3389/fonc.2019.00421
Chew, M., Zhou, J., Daugherty, A., Eriksson, T., Ellermann-Eriksen, S., Hansen, P. R., et al. (2003). Thalidomide Inhibits Early Atherogenesis in apoE-Deficient Mice. APMIS Suppl. Suppl (109), 113–116.
Childs, B. G., Baker, D. J., Wijshake, T., Conover, C. A., Campisi, J., and Van Deursen, J. M. (2016). Senescent Intimal Foam Cells Are Deleterious at All Stages of Atherosclerosis. Science 354 (6311), 472–477. doi:10.1126/science.aaf6659
Childs, B. G., Gluscevic, M., Baker, D. J., Laberge, R. M., Marquess, D., Dananberg, J., et al. (2017). Senescent Cells: an Emerging Target for Diseases of Ageing. Nat. Rev. Drug Discov. 16 (10), 718–735. doi:10.1038/nrd.2017.116
Childs, B. G., Zhang, C., Shuja, F., Sturmlechner, I., Trewartha, S., Velasco, R. F., et al. (2021). Senescent Cells Suppress Innate Smooth Muscle Cell Repair Functions in Atherosclerosis. Nat. Aging 1 (8), 1–17. doi:10.1038/s43587-021-00089-5
Chinetti-Gbaguidi, G., Colin, S., and Staels, B. (2015). Macrophage Subsets in Atherosclerosis. Nat. Rev. Cardiol. 12 (1), 10–17. doi:10.1038/nrcardio.2014.173
Chistiakov, D. A., Bobryshev, Y. V., and Orekhov, A. N. (2015). Changes in Transcriptome of Macrophages in Atherosclerosis. J. Cel Mol Med 19 (6), 1163–1173. doi:10.1111/jcmm.12591
Chistiakov, D. A., Bobryshev, Y. V., and Orekhov, A. N. (2016). Macrophage-mediated Cholesterol Handling in Atherosclerosis. J. Cel Mol Med 20 (1), 17–28. doi:10.1111/jcmm.12689
Chistiakov, D. A., Melnichenko, A. A., Myasoedova, V. A., Grechko, A. V., and Orekhov, A. N. (2017). Mechanisms of Foam Cell Formation in Atherosclerosis. J. Mol. Med. (Berl) 95 (11), 1153–1165. doi:10.1007/s00109-017-1575-8
Cho, J. H., Kim, E. C., Son, Y., Lee, D. W., Park, Y. S., Choi, J. H., et al. (2020). CD9 Induces Cellular Senescence and Aggravates Atherosclerotic Plaque Formation. Cell Death Differ 27 (9), 2681–2696. doi:10.1038/s41418-020-0537-9
Coats, B. R., Schoenfelt, K. Q., Barbosa-Lorenzi, V. C., Peris, E., Cui, C., Hoffman, A., et al. (2017). Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Rep 20 (13), 3149–3161. doi:10.1016/j.celrep.2017.08.096
Cochain, C., Vafadarnejad, E., Arampatzi, P., Pelisek, J., Winkels, H., Ley, K., et al. (2018). Single-cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 122 (12), 1661–1674. doi:10.1161/CIRCRESAHA.117.312509
Colin, S., Chinetti-Gbaguidi, G., and Staels, B. (2014). Macrophage Phenotypes in Atherosclerosis. Immunol. Rev. 262 (1), 153–166. doi:10.1111/imr.12218
Covarrubias, A. J., Aksoylar, H. I., and Horng, T. (2015). “Control of Macrophage Metabolism and Activation by mTOR and Akt Signaling,” in Seminars in Immunology (Elsevier), 286–296.
Davenport, P., and Tipping, P. G. (2003). The Role of Interleukin-4 and Interleukin-12 in the Progression of Atherosclerosis in Apolipoprotein E-Deficient Mice. Am. J. Pathol. 163 (3), 1117–1125. doi:10.1016/s0002-9440(10)63471-2
de Jager, S. C., Bermúdez, B., Bot, I., Koenen, R. R., Bot, M., Kavelaars, A., et al. (2011). Growth Differentiation Factor 15 Deficiency Protects against Atherosclerosis by Attenuating CCR2-Mediated Macrophage Chemotaxis. J. Exp. Med. 208 (2), 217–225. doi:10.1084/jem.20100370
De Paoli, F., Staels, B., and Chinetti-Gbaguidi, G. (2014). Macrophage Phenotypes and Their Modulation in Atherosclerosis. Circ. J. 78 (8), 1775–1781. doi:10.1253/circj.cj-14-0621
Deczkowska, A., Weiner, A., and Amit, I. (2020). The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 181 (6), 1207–1217. doi:10.1016/j.cell.2020.05.003
Devries-Seimon, T., Li, Y., Yao, P. M., Stone, E., Wang, Y., Davis, R. J., et al. (2005). Cholesterol-induced Macrophage Apoptosis Requires ER Stress Pathways and Engagement of the Type A Scavenger Receptor. J. Cel Biol 171 (1), 61–73. doi:10.1083/jcb.200502078
Dinarello, C. A., Novick, D., Kim, S., and Kaplanski, G. (2013). Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 4, 289. doi:10.3389/fimmu.2013.00289
Douna, H., Amersfoort, J., Schaftenaar, F. H., Kroon, S., van Puijvelde, G. H. M., Kuiper, J., et al. (2019). Bidirectional Effects of IL-10+ Regulatory B Cells in Ldlr-/- Mice. Atherosclerosis 280, 118–125. doi:10.1016/j.atherosclerosis.2018.11.019
Erbilgin, A., Seldin, M. M., Wu, X., Mehrabian, M., Zhou, Z., Qi, H., et al. (2018). Transcription Factor Zhx2 Deficiency Reduces Atherosclerosis and Promotes Macrophage Apoptosis in Mice. Arterioscler Thromb. Vasc. Biol. 38 (9), 2016–2027. doi:10.1161/ATVBAHA.118.311266
Febbraio, M., Hajjar, D. P., and Silverstein, R. L. (2001). CD36: a Class B Scavenger Receptor Involved in Angiogenesis, Atherosclerosis, Inflammation, and Lipid Metabolism. J. Clin. Invest. 108 (6), 785–791. doi:10.1172/jci14006
Feig, J. E., Parathath, S., Rong, J. X., Mick, S. L., Vengrenyuk, Y., Grauer, L., et al. (2011). Reversal of Hyperlipidemia with a Genetic Switch Favorably Affects the Content and Inflammatory State of Macrophages in Atherosclerotic Plaques. Circulation 123 (9), 989–998. doi:10.1161/circulationaha.110.984146
Fernández-Hernando, C., Ackah, E., Yu, J., Suárez, Y., Murata, T., Iwakiri, Y., et al. (2007). Loss of Akt1 Leads to Severe Atherosclerosis and Occlusive Coronary Artery Disease. Cell Metab 6 (6), 446–457. doi:10.1016/j.cmet.2007.10.007
Fernandez, D. M., Rahman, A. H., Fernandez, N. F., Chudnovskiy, A., Amir, E. D., Amadori, L., et al. (2019). Single-cell Immune Landscape of Human Atherosclerotic Plaques. Nat. Med. 25 (10), 1576–1588. doi:10.1038/s41591-019-0590-4
Fernandez, D. M., and Giannarelli, C. (2021). Immune Cell Profiling in Atherosclerosis: Role in Research and Precision Medicine. Nat. Rev. Cardiol., 1–16. doi:10.1038/s41569-021-00589-2
Fidler, T. P., Xue, C., Yalcinkaya, M., Hardaway, B., Abramowicz, S., Xiao, T., et al. (2021). The AIM2 Inflammasome Exacerbates Atherosclerosis in Clonal Haematopoiesis. Nature 592 (7853), 296–301. doi:10.1038/s41586-021-03341-5
Fourman, L. T., Saylor, C. F., Cheru, L., Fitch, K., Looby, S., Keller, K., et al. (2020). Anti-Inflammatory Interleukin 10 Inversely Relates to Coronary Atherosclerosis in Persons with Human Immunodeficiency Virus. J. Infect. Dis. 221 (4), 510–515. doi:10.1093/infdis/jiz254
Frank, D., and Vince, J. E. (2019). Pyroptosis versus Necroptosis: Similarities, Differences, and Crosstalk. Cel Death Differ 26 (1), 99–114. doi:10.1038/s41418-018-0212-6
Fredman, G., Kamaly, N., Spolitu, S., Milton, J., Ghorpade, D., Chiasson, R., et al. (2015). Erratum for the Research Article: "Targeted Nanoparticles Containing the Proresolving Peptide Ac2-26 Protect against Advanced Atherosclerosis in Hypercholesterolemic Mice" by G. Fredman, N. Kamaly, S. Spolitu, J. Milton, D. Ghorpade, R. Chiasson, G. Kuriakose, M. Perretti, O. Farokzhad, I. Tabas. Sci. Transl Med. 7 (275), 277er2-275ra220. doi:10.1126/scitranslmed.aaa106510.1126/scitranslmed.aaa9877
Furukawa, S., Moriyama, M., Tanaka, A., Maehara, T., Tsuboi, H., Iizuka, M., et al. (2015). Preferential M2 Macrophages Contribute to Fibrosis in IgG4-Related Dacryoadenitis and Sialoadenitis, So-Called Mikulicz's Disease. Clin. Immunol. 156 (1), 9–18. doi:10.1016/j.clim.2014.10.008
Fuster, J. J., MacLauchlan, S., Zuriaga, M. A., Polackal, M. N., Ostriker, A. C., Chakraborty, R., et al. (2017). Clonal Hematopoiesis Associated with TET2 Deficiency Accelerates Atherosclerosis Development in Mice. Science 355 (6327), 842–847. doi:10.1126/science.aag1381
Gao, C., Huang, Q., Liu, C., Kwong, C. H. T., Yue, L., Wan, J. B., et al. (2020). Treatment of Atherosclerosis by Macrophage-Biomimetic Nanoparticles via Targeted Pharmacotherapy and Sequestration of Proinflammatory Cytokines. Nat. Commun. 11 (1), 2622–2714. doi:10.1038/s41467-020-16439-7
Gerrick, K. Y., Gerrick, E. R., Gupta, A., Wheelan, S. J., Yegnasubramanian, S., and Jaffee, E. M. (2018). Transcriptional Profiling Identifies Novel Regulators of Macrophage Polarization. PLoS One 13 (12), e0208602. doi:10.1371/journal.pone.0208602
Gerszten, R. E., Garcia-Zepeda, E. A., Lim, Y. C., Yoshida, M., Ding, H. A., Gimbrone, M. A., et al. (1999). MCP-1 and IL-8 Trigger Firm Adhesion of Monocytes to Vascular Endothelium under Flow Conditions. Nature 398 (6729), 718–723. doi:10.1038/19546
Giacona, M. B., Papapanou, P. N., Lamster, I. B., Rong, L. L., D'Agati, V. D., Schmidt, A. M., et al. (2004). Porphyromonas Gingivalis Induces its Uptake by Human Macrophages and Promotes Foam Cell Formation In Vitro. FEMS Microbiol. Lett. 241 (1), 95–101. doi:10.1016/j.femsle.2004.10.009
Godwin, J. W., Pinto, A. R., and Rosenthal, N. A. (2013). Macrophages Are Required for Adult Salamander Limb Regeneration. Proc. Natl. Acad. Sci. U S A. 110 (23), 9415–9420. doi:10.1073/pnas.1300290110
Gong, D., Shi, W., Yi, S. J., Chen, H., Groffen, J., and Heisterkamp, N. (2012). TGFβ Signaling Plays a Critical Role in Promoting Alternative Macrophage Activation. BMC Immunol. 13 (1), 31–10. doi:10.1186/1471-2172-13-31
Gonzalez, L., and Trigatti, B. L. (2017). Macrophage Apoptosis and Necrotic Core Development in Atherosclerosis: a Rapidly Advancing Field with Clinical Relevance to Imaging and Therapy. Can. J. Cardiol. 33 (3), 303–312. doi:10.1016/j.cjca.2016.12.010
Goossens, P., Gijbels, M. J., Zernecke, A., Eijgelaar, W., Vergouwe, M. N., van der Made, I., et al. (2010). Myeloid Type I Interferon Signaling Promotes Atherosclerosis by Stimulating Macrophage Recruitment to Lesions. Cel Metab 12 (2), 142–153. doi:10.1016/j.cmet.2010.06.008
Gough, P. J., Gomez, I. G., Wille, P. T., and Raines, E. W. (2006). Macrophage Expression of Active MMP-9 Induces Acute Plaque Disruption in apoE-Deficient Mice. J. Clin. Invest. 116 (1), 59–69. doi:10.1172/JCI25074
Grainger, D. J., Kemp, P. R., Metcalfe, J. C., Liu, A. C., Lawn, R. M., Williams, N. R., et al. (1995). The Serum Concentration of Active Transforming Growth Factor-Beta Is Severely Depressed in Advanced Atherosclerosis. Nat. Med. 1 (1), 74–79. doi:10.1038/nm0195-74
Guo, Z., Ran, Q., Roberts, L. J., Zhou, L., Richardson, A., Sharan, C., et al. (2008). Suppression of Atherogenesis by Overexpression of Glutathione Peroxidase-4 in Apolipoprotein E-Deficient Mice. Free Radic. Biol. Med. 44 (3), 343–352. doi:10.1016/j.freeradbiomed.2007.09.009
Härdtner, C., Kornemann, J., Krebs, K., Ehlert, C. A., Jander, A., Zou, J., et al. (2020). Inhibition of Macrophage Proliferation Dominates Plaque Regression in Response to Cholesterol Lowering. Basic Res. Cardiol. 115 (6), 78–19. doi:10.1007/s00395-020-00838-4
Habib, A., and Finn, A. V. (2014). The Role of Iron Metabolism as a Mediator of Macrophage Inflammation and Lipid Handling in Atherosclerosis. Front. Pharmacol. 5, 195. doi:10.3389/fphar.2014.00195
Haka, A. S., Barbosa-Lorenzi, V. C., Lee, H. J., Falcone, D. J., Hudis, C. A., Dannenberg, A. J., et al. (2016). Exocytosis of Macrophage Lysosomes Leads to Digestion of Apoptotic Adipocytes and Foam Cell Formation. J. Lipid Res. 57 (6), 980–992. doi:10.1194/jlr.M064089
Henson, P. M., Bratton, D. L., and Fadok, V. A. (2001). Apoptotic Cell Removal. Curr. Biol. 11 (19), R795–R805. doi:10.1016/s0960-9822(01)00474-2
Heyde, A., Rohde, D., McAlpine, C. S., Zhang, S., Hoyer, F. F., Gerold, J. M., et al. (2021). Increased Stem Cell Proliferation in Atherosclerosis Accelerates Clonal Hematopoiesis. Cell 184 (5), 1348–e22. e1322. doi:10.1016/j.cell.2021.01.049
Hoebe, K., Georgel, P., Rutschmann, S., Du, X., Mudd, S., Crozat, K., et al. (2005). CD36 Is a Sensor of Diacylglycerides. Nature 433 (7025), 523–527. doi:10.1038/nature03253
Hofmans, S., Vanden Berghe, T., Devisscher, L., Hassannia, B., Lyssens, S., Joossens, J., et al. (2016). Novel Ferroptosis Inhibitors with Improved Potency and ADME Properties. J. Med. Chem. 59 (5), 2041–2053. doi:10.1021/acs.jmedchem.5b01641
Huang, L., Chambliss, K. L., Gao, X., Yuhanna, I. S., Behling-Kelly, E., Bergaya, S., et al. (2019). SR-B1 Drives Endothelial Cell LDL Transcytosis via DOCK4 to Promote Atherosclerosis. Nature 569 (7757), 565–569. doi:10.1038/s41586-019-1140-4
Huo, Y., and Xia, L. (2009). P-selectin Glycoprotein Ligand-1 Plays a Crucial Role in the Selective Recruitment of Leukocytes into the Atherosclerotic Arterial wall. Trends Cardiovasc. Med. 19 (4), 140–145. doi:10.1016/j.tcm.2009.07.006
Jaiswal, S., Natarajan, P., Silver, A. J., Gibson, C. J., Bick, A. G., Shvartz, E., et al. (2017). Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 377 (2), 111–121. doi:10.1056/NEJMoa1701719
Jansen, M. F., Hollander, M. R., van Royen, N., Horrevoets, A. J., and Lutgens, E. (2016). CD40 in Coronary Artery Disease: a Matter of Macrophages? Basic Res. Cardiol. 111 (4), 38. doi:10.1007/s00395-016-0554-5
Jefferis, B. J., Papacosta, O., Owen, C. G., Wannamethee, S. G., Humphries, S. E., Woodward, M., et al. (2011). Interleukin 18 and Coronary Heart Disease: Prospective Study and Systematic Review. Atherosclerosis 217 (1), 227–233. doi:10.1016/j.atherosclerosis.2011.03.015
Jiang, Y., Gao, Q., Wang, L., Guo, C., Zhu, F., Wang, B., et al. (2016). Deficiency of Programmed Cell Death 4 Results in Increased IL-10 Expression by Macrophages and Thereby Attenuates Atherosclerosis in Hyperlipidemic Mice. Cell Mol Immunol 13 (4), 524–534. doi:10.1038/cmi.2015.47
Jung, S. B., Choi, M. J., Ryu, D., Yi, H. S., Lee, S. E., Chang, J. Y., et al. (2018). Reduced Oxidative Capacity in Macrophages Results in Systemic Insulin Resistance. Nat. Commun. 9 (1), 1551–1615. doi:10.1038/s41467-018-03998-z
Kadl, A., Meher, A. K., Sharma, P. R., Lee, M. Y., Doran, A. C., Johnstone, S. R., et al. (2010). Identification of a Novel Macrophage Phenotype that Develops in Response to Atherogenic Phospholipids via Nrf2. Circ. Res. 107 (6), 737–746. doi:10.1161/circresaha.109.215715
Kale, A., Sharma, A., Stolzing, A., Desprez, P. Y., and Campisi, J. (2020). Role of Immune Cells in the Removal of Deleterious Senescent Cells. Immun. Ageing 17 (1), 16–19. doi:10.1186/s12979-020-00187-9
Kamari, Y., Shaish, A., Shemesh, S., Vax, E., Grosskopf, I., Dotan, S., et al. (2011). Reduced Atherosclerosis and Inflammatory Cytokines in Apolipoprotein-E-Deficient Mice Lacking Bone Marrow-Derived Interleukin-1α. Biochem. Biophys. Res. Commun. 405 (2), 197–203. doi:10.1016/j.bbrc.2011.01.008
Kapellos, T. S., Bonaguro, L., Gemünd, I., Reusch, N., Saglam, A., Hinkley, E. R., et al. (2019). Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 10, 2035. doi:10.3389/fimmu.2019.02035
Kaperonis, E. A., Liapis, C. D., Kakisis, J. D., Dimitroulis, D., and Papavassiliou, V. G. (2006). Inflammation and Atherosclerosis. Eur. J. Vasc. Endovasc Surg. 31 (4), 386–393. doi:10.1016/j.ejvs.2005.11.001
Karunakaran, D., Geoffrion, M., Wei, L., Gan, W., Richards, L., Shangari, P., et al. (2016). Targeting Macrophage Necroptosis for Therapeutic and Diagnostic Interventions in Atherosclerosis. Sci. Adv. 2 (7), e1600224. doi:10.1126/sciadv.1600224
Kattoor, A. J., Goel, A., and Mehta, J. L. (2019). LOX-1: Regulation, Signaling and its Role in Atherosclerosis. Antioxidants (Basel) 8 (7), 218. doi:10.3390/antiox8070218
Khallou-Laschet, J., Varthaman, A., Fornasa, G., Compain, C., Gaston, A. T., Clement, M., et al. (2010). Macrophage Plasticity in Experimental Atherosclerosis. PLoS One 5 (1), e8852. doi:10.1371/journal.pone.0008852
Kim, K., Shim, D., Lee, J. S., Zaitsev, K., Williams, J. W., Kim, K. W., et al. (2018). Transcriptome Analysis Reveals Nonfoamy rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 123 (10), 1127–1142. doi:10.1161/CIRCRESAHA.118.312804
Kinlay, S., Schwartz, G. G., Olsson, A. G., Rifai, N., Szarek, M., Waters, D. D., et al. (2008). Inflammation, Statin Therapy, and Risk of Stroke after an Acute Coronary Syndrome in the MIRACL Study. Arterioscler Thromb. Vasc. Biol. 28 (1), 142–147. doi:10.1161/ATVBAHA.107.151787
Kleemann, R., Zadelaar, S., and Kooistra, T. (2008). Cytokines and Atherosclerosis: a Comprehensive Review of Studies in Mice. Cardiovasc. Res. 79 (3), 360–376. doi:10.1093/cvr/cvn120
Kober, D. L., and Brett, T. J. (2017). TREM2-Ligand Interactions in Health and Disease. J. Mol. Biol. 429 (11), 1607–1629. doi:10.1016/j.jmb.2017.04.004
Kojima, Y., Volkmer, J. P., McKenna, K., Civelek, M., Lusis, A. J., Miller, C. L., et al. (2016). CD47-blocking Antibodies Restore Phagocytosis and Prevent Atherosclerosis. Nature 536 (7614), 86–90. doi:10.1038/nature18935
Kojima, Y., Weissman, I. L., and Leeper, N. J. (2017). The Role of Efferocytosis in Atherosclerosis. Circulation 135 (5), 476–489. doi:10.1161/circulationaha.116.025684
Kolter, J., Kierdorf, K., and Henneke, P. (2020a). Origin and Differentiation of Nerve-Associated Macrophages. J. Immunol. 204 (2), 271–279. doi:10.4049/jimmunol.1901077
Kolter, J., Kierdorf, K., and Henneke, P. (2020b). Origin and Differentiation of Nerve-Associated Macrophages. J. Immunol. 204 (2), 271–279. doi:10.4049/jimmunol.1901077
Kozarsky, K. F., Donahee, M. H., Rigotti, A., Iqbal, S. N., Edelman, E. R., and Krieger, M. (1997). Overexpression of the HDL Receptor SR-BI Alters Plasma HDL and Bile Cholesterol Levels. Nature 387 (6631), 414–417. doi:10.1038/387414a0
Lehtolainen, P., Takeya, M., and Ylä-Herttuala, S. (2000). Retrovirus-mediated, Stable Scavenger-Receptor Gene Transfer Leads to Functional Endocytotic Receptor Expression, Foam Cell Formation, and Increased Susceptibility to Apoptosis in Rabbit Aortic Smooth Muscle Cells. Arterioscler Thromb. Vasc. Biol. 20 (1), 52–60. doi:10.1161/01.atv.20.1.52
Leitinger, N., and Schulman, I. G. (2013). Phenotypic Polarization of Macrophages in Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 33 (6), 1120–1126. doi:10.1161/ATVBAHA.112.300173
Leuschner, F., Rauch, P. J., Ueno, T., Gorbatov, R., Marinelli, B., Lee, W. W., et al. (2012). Rapid Monocyte Kinetics in Acute Myocardial Infarction Are Sustained by Extramedullary Monocytopoiesis. J. Exp. Med. 209 (1), 123–137. doi:10.1084/jem.20111009
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the Site of Inflammation: the Leukocyte Adhesion cascade Updated. Nat. Rev. Immunol. 7 (9), 678–689. doi:10.1038/nri2156
Li, B., Xia, Y., and Hu, B. (2020a). Infection and Atherosclerosis: TLR-dependent Pathways. Cell Mol Life Sci 77 (14), 2751–2769. doi:10.1007/s00018-020-03453-7
Li, Y., Yun, K., and Mu, R. (2020b). A Review on the Biology and Properties of Adipose Tissue Macrophages Involved in Adipose Tissue Physiological and Pathophysiological Processes. Lipids Health Dis. 19 (1), 164–169. doi:10.1186/s12944-020-01342-3
Libby, P. (2021). The Changing Landscape of Atherosclerosis. Nature 592 (7855), 524–533. doi:10.1038/s41586-021-03392-8
Liberale, L., Dallegri, F., Montecucco, F., and Carbone, F. (2017). Pathophysiological Relevance of Macrophage Subsets in Atherogenesis. Thromb. Haemost. 117 (01), 7–18. doi:10.1160/TH16-08-0593
Lim, W. S., Timmins, J. M., Seimon, T. A., Sadler, A., Kolodgie, F. D., Virmani, R., et al. (2008). Signal Transducer and Activator of Transcription-1 Is Critical for Apoptosis in Macrophages Subjected to Endoplasmic Reticulum Stress In Vitro and in Advanced Atherosclerotic Lesions In Vivo. Circulation 117 (7), 940–951. doi:10.1161/circulationaha.107.711275
Lin, J. D., Nishi, H., Poles, J., Niu, X., Mccauley, C., Rahman, K., et al. (2019). Single-cell Analysis of Fate-Mapped Macrophages Reveals Heterogeneity, Including Stem-like Properties, during Atherosclerosis Progression and Regression. JCI insight 4 (4). doi:10.1172/jci.insight.124574
Lin, P., Ji, H.-H., Li, Y.-J., and Guo, S.-D. (2021). Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosciences 8, 324. doi:10.3389/fmolb.2021.679797
Liu, J. Y., Souroullas, G. P., Diekman, B. O., Krishnamurthy, J., Hall, B. M., Sorrentino, J. A., et al. (2019). Cells Exhibiting strong P16 INK4a Promoter Activation In Vivo Display Features of Senescence. Proc. Natl. Acad. Sci. U S A. 116 (7), 2603–2611. doi:10.1073/pnas.1818313116
Low, E. L., Baker, A. H., and Bradshaw, A. C. (2019). TGFβ, Smooth Muscle Cells and Coronary Artery Disease: a Review. Cell Signal 53, 90–101. doi:10.1016/j.cellsig.2018.09.004
Mallat, Z., Gojova, A., Marchiol-Fournigault, C., Esposito, B., Kamaté, C., Merval, R., et al. (2001). Inhibition of Transforming Growth Factor-Beta Signaling Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Mice. Circ. Res. 89 (10), 930–934. doi:10.1161/hh2201.099415
Mallat, Z., Heymes, C., Ohan, J., Faggin, E., Lesèche, G., and Tedgui, A. (1999). Expression of Interleukin-10 in Advanced Human Atherosclerotic Plaques: Relation to Inducible Nitric Oxide Synthase Expression and Cell Death. Arterioscler Thromb. Vasc. Biol. 19 (3), 611–616. doi:10.1161/01.atv.19.3.611
Manning-Tobin, J. J., Moore, K. J., Seimon, T. A., Bell, S. A., Sharuk, M., Alvarez-Leite, J. I., et al. (2009). Loss of SR-A and CD36 Activity Reduces Atherosclerotic Lesion Complexity without Abrogating Foam Cell Formation in Hyperlipidemic Mice. Arterioscler Thromb. Vasc. Biol. 29 (1), 19–26. doi:10.1161/atvbaha.108.176644
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 25 (12), 677–686. doi:10.1016/j.it.2004.09.015
Marchio, P., Guerra-Ojeda, S., Vila, J. M., Aldasoro, M., Victor, V. M., and Mauricio, M. D. (2019). Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxidative Medicine and Cellular Longevity 2019.
Martinet, W., Coornaert, I., Puylaert, P., and De Meyer, G. R. Y. (2019). Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 10, 306. doi:10.3389/fphar.2019.00306
McAlpine, C. S., Kiss, M. G., Rattik, S., He, S., Vassalli, A., Valet, C., et al. (2019). Sleep Modulates Haematopoiesis and Protects against Atherosclerosis. Nature 566 (7744), 383–387. doi:10.1038/s41586-019-0948-2
McKellar, G. E., McCarey, D. W., Sattar, N., and McInnes, I. B. (2009). Role for TNF in Atherosclerosis? Lessons from Autoimmune Disease. Nat. Rev. Cardiol. 6 (6), 410–7. doi:10.1038/nrcardio.2009.57
Medbury, H. J., Williams, H., and Fletcher, J. P. (2014). Clinical Significance of Macrophage Phenotypes in Cardiovascular Disease. Clin. Transl Med. 3 (1), 63–10. doi:10.1186/s40169-014-0042-1
Meng, Z., Wang, M., Xing, J., Liu, Y., and Li, H. (2019). Myricetin Ameliorates Atherosclerosis in the Low-Density-Lipoprotein Receptor Knockout Mice by Suppression of Cholesterol Accumulation in Macrophage Foam Cells. Nutr. Metab. (Lond) 16 (1), 25–29. doi:10.1186/s12986-019-0354-7
Michelsen, K. S., Wong, M. H., Shah, P. K., Zhang, W., Yano, J., Doherty, T. M., et al. (2004). Lack of Toll-like Receptor 4 or Myeloid Differentiation Factor 88 Reduces Atherosclerosis and Alters Plaque Phenotype in Mice Deficient in Apolipoprotein E. Proc. Natl. Acad. Sci. U S A. 101 (29), 10679–10684. doi:10.1073/pnas.0403249101
Miki, H., Pei, H., Gracias, D. T., Linden, J., and Croft, M. (2021). Clearance of Apoptotic Cells by Lung Alveolar Macrophages Prevents Development of House Dust Mite-Induced Asthmatic Lung Inflammation. J. Allergy Clin. Immunol. 147 (3), 1087–1092. doi:10.1016/j.jaci.2020.10.005
Milioti, N., Bermudez-Fajardo, A., Penichet, M. L., and Oviedo-Orta, E. (2008). Antigen-induced Immunomodulation in the Pathogenesis of Atherosclerosis. Clin. Dev. Immunol. 2008, 723539. doi:10.1155/2008/723539
Mirza, R. E., Fang, M. M., Weinheimer-Haus, E. M., Ennis, W. J., and Koh, T. J. (2014). Sustained Inflammasome Activity in Macrophages Impairs Wound Healing in Type 2 Diabetic Humans and Mice. Diabetes 63 (3), 1103–1114. doi:10.2337/db13-0927
Moore, K. J., Sheedy, F. J., and Fisher, E. A. (2013). Macrophages in Atherosclerosis: a Dynamic Balance. Nat. Rev. Immunol. 13 (10), 709–721. doi:10.1038/nri3520
Moreno Velásquez, I., Gajulapuri, A., Leander, K., Berglund, A., de Faire, U., and Gigante, B. (2019). Serum IL8 Is Not Associated with Cardiovascular Events but with All-Cause Mortality. BMC Cardiovasc. Disord. 19 (1), 34–38. doi:10.1186/s12872-019-1014-6
Moriya, J. (2019). Critical Roles of Inflammation in Atherosclerosis. J. Cardiol. 73 (1), 22–27. doi:10.1016/j.jjcc.2018.05.010
Murray, P. J. (2017). Macrophage Polarization. Annu. Rev. Physiol. 79, 541–566. doi:10.1146/annurev-physiol-022516-034339
Nagenborg, J., Goossens, P., Biessen, E. A. L., and Donners, M. M. P. C. (2017). Heterogeneity of Atherosclerotic Plaque Macrophage Origin, Phenotype and Functions: Implications for Treatment. Eur. J. Pharmacol. 816, 14–24. doi:10.1016/j.ejphar.2017.10.005
Nahrendorf, M., Swirski, F. K., Aikawa, E., Stangenberg, L., Wurdinger, T., Figueiredo, J. L., et al. (2007). The Healing Myocardium Sequentially Mobilizes Two Monocyte Subsets with Divergent and Complementary Functions. J. Exp. Med. 204 (12), 3037–3047. doi:10.1084/jem.20070885
Neufeld, E. B., O'Brien, K., Walts, A. D., Stonik, J. A., Demosky, S. J., Malide, D., et al. (2014). Cellular Localization and Trafficking of the Human ABCG1 Transporter. Biology (Basel) 3 (4), 781–800. doi:10.3390/biology3040781
Nikoletopoulou, V., Markaki, M., Palikaras, K., and Tavernarakis, N. (2013). Crosstalk between Apoptosis, Necrosis and Autophagy. Biochim. Biophys. Acta 1833 (12), 3448–3459. doi:10.1016/j.bbamcr.2013.06.001
Nikonova, A., Khaitov, M., Jackson, D. J., Traub, S., Trujillo-Torralbo, M. B., Kudlay, D. A., et al. (2020). M1-like Macrophages Are Potent Producers of Anti-viral Interferons and M1-Associated Marker-Positive Lung Macrophages Are Decreased during Rhinovirus-Induced Asthma Exacerbations. EBioMedicine 54, 102734. doi:10.1016/j.ebiom.2020.102734
Nilchian, A., Plant, E., Parniewska, M. M., Santiago, A., Rossignoli, A., Skogsberg, J., et al. (2020). Induction of the Coxsackievirus and Adenovirus Receptor in Macrophages during the Formation of Atherosclerotic Plaques. J. Infect. Dis. 222 (12), 2041–2051. doi:10.1093/infdis/jiaa418
Nishida, M., Miyagawa, J., Yamashita, S., Higashiyama, S., Nakata, A., Ouchi, N., et al. (2000). Localization of CD9, an Enhancer Protein for Proheparin-Binding Epidermal Growth Factor-like Growth Factor, in Human Atherosclerotic Plaques: Possible Involvement of Juxtacrine Growth Mechanism on Smooth Muscle Cell Proliferation. Arterioscler Thromb. Vasc. Biol. 20 (5), 1236–1243. doi:10.1161/01.atv.20.5.1236
Novak, M. L., Weinheimer-Haus, E. M., and Koh, T. J. (2014). Macrophage Activation and Skeletal Muscle Healing Following Traumatic Injury. J. Pathol. 232 (3), 344–355. doi:10.1002/path.4301
Ohgami, N., Nagai, R., Ikemoto, M., Arai, H., Kuniyasu, A., Horiuchi, S., et al. (2001). Cd36, a Member of the Class B Scavenger Receptor Family, as a Receptor for Advanced Glycation End Products. J. Biol. Chem. 276 (5), 3195–3202. doi:10.1074/jbc.M006545200
Ouimet, M., Ediriweera, H., Afonso, M. S., Ramkhelawon, B., Singaravelu, R., Liao, X., et al. (2017). microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 37 (6), 1058–1067. doi:10.1161/atvbaha.116.308916
Oumouna-Benachour, K., Hans, C. P., Suzuki, Y., Naura, A., Datta, R., Belmadani, S., et al. (2007). Poly(ADP-ribose) Polymerase Inhibition Reduces Atherosclerotic Plaque Size and Promotes Factors of Plaque Stability in Apolipoprotein E-Deficient Mice: Effects on Macrophage Recruitment, Nuclear Factor-kappaB Nuclear Translocation, and Foam Cell Death. Circulation 115 (18), 2442–2450. doi:10.1161/circulationaha.106.668756
Panousis, C. G., Evans, G., and Zuckerman, S. H. (2001). TGF-beta Increases Cholesterol Efflux and ABC-1 Expression in Macrophage-Derived Foam Cells: Opposing the Effects of IFN-Gamma. J. Lipid Res. 42 (5), 856–863. doi:10.1016/s0022-2275(20)31648-5
Park, H. J., Lee, S. J., Kim, S. H., Han, J., Bae, J., Kim, S. J., et al. (2011). IL-10 Inhibits the Starvation Induced Autophagy in Macrophages via Class I Phosphatidylinositol 3-kinase (PI3K) Pathway. Mol. Immunol. 48 (4), 720–727. doi:10.1016/j.molimm.2010.10.020
Park, S. H. (2021). Regulation of Macrophage Activation and Differentiation in Atherosclerosis. J. Lipid Atheroscler. 10, 251–267. doi:10.12997/jla.2021.10.3.251
Park, Y. M., Drazba, J. A., Vasanji, A., Egelhoff, T., Febbraio, M., and Silverstein, R. L. (2012). Oxidized LDL/CD36 Interaction Induces Loss of Cell Polarity and Inhibits Macrophage Locomotion. Mol. Biol. Cel 23 (16), 3057–3068. doi:10.1091/mbc.E11-12-1051
Pham, L. M., Kim, E. C., Ou, W., Phung, C. D., Nguyen, T. T., Pham, T. T., et al. (2021). Targeting and Clearance of Senescent Foamy Macrophages and Senescent Endothelial Cells by Antibody-Functionalized Mesoporous Silica Nanoparticles for Alleviating Aorta Atherosclerosis. Biomaterials 269, 120677. doi:10.1016/j.biomaterials.2021.120677
Phillips, M. C. (2018). Is ABCA1 a Lipid Transfer Protein? J. Lipid Res. 59 (5), 749–763. doi:10.1194/jlr.R082313
Pinderski, L. J., Fischbein, M. P., Subbanagounder, G., Fishbein, M. C., Kubo, N., Cheroutre, H., et al. (2002). Overexpression of Interleukin-10 by Activated T Lymphocytes Inhibits Atherosclerosis in LDL Receptor-Deficient Mice by Altering Lymphocyte and Macrophage Phenotypes. Circ. Res. 90 (10), 1064–1071. doi:10.1161/01.res.0000018941.10726.fa
Porsch, F., Kiss, M. G., Goederle, L., Hendrikx, T., Hladik, A., Knapp, S., et al. (2020). Haematopoetic TREM2 Deficiency Modulates Atherosclerosis and Lipid Metabolism. Atherosclerosis 315, e58–e59. doi:10.1016/j.atherosclerosis.2020.10.180
Poznyak, A. V., Nikiforov, N. G., Markin, A. M., Kashirskikh, D. A., Myasoedova, V. A., Gerasimova, E. V., et al. (2021). Overview of OxLDL and its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 11, 2248. doi:10.3389/fphar.2020.613780
Prattichizzo, F., Bonafè, M., Olivieri, F., and Franceschi, C. (2016). Senescence Associated Macrophages and "Macroph-Aging": Are They Pieces of the Same Puzzle? Aging (Albany NY) 8 (12), 3159–3160. doi:10.18632/aging.101133
Que, X., Hung, M. Y., Yeang, C., Gonen, A., Prohaska, T. A., Sun, X., et al. (2018). Oxidized Phospholipids Are Proinflammatory and Proatherogenic in Hypercholesterolaemic Mice. Nature 558 (7709), 301–306. doi:10.1038/s41586-018-0198-8
Quiding-Järbrink, M., Raghavan, S., and Sundquist, M. (2010). Enhanced M1 Macrophage Polarization in Human helicobacter Pylori-Associated Atrophic Gastritis and in Vaccinated Mice. PLoS One 5 (11), e15018. doi:10.1371/journal.pone.0015018
Raggi, F., Pelassa, S., Pierobon, D., Penco, F., Gattorno, M., Novelli, F., et al. (2017). Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 8, 1097. doi:10.3389/fimmu.2017.01097
Reiss, A. B., Siegart, N. M., and De Leon, J. (2017). Interleukin-6 in Atherosclerosis: Atherogenic or Atheroprotective? Clin. Lipidol. 12 (1), 14–23.
Remmerie, A., and Scott, C. L. (2018). Macrophages and Lipid Metabolism. Cell Immunol 330, 27–42. doi:10.1016/j.cellimm.2018.01.020
Ricci, R., Sumara, G., Sumara, I., Rozenberg, I., Kurrer, M., Akhmedov, A., et al. (2004). Requirement of JNK2 for Scavenger Receptor A-Mediated Foam Cell Formation in Atherogenesis. Science 306 (5701), 1558–1561. doi:10.1126/science.1101909
Ridker, P. M., Libby, P., MacFadyen, J. G., Thuren, T., Ballantyne, C., Fonseca, F., et al. (2018). Modulation of the Interleukin-6 Signalling Pathway and Incidence Rates of Atherosclerotic Events and All-Cause Mortality: Analyses from the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 39 (38), 3499–3507. doi:10.1093/eurheartj/ehy310
Ridker, P. M., Thuren, T., Zalewski, A., and Libby, P. (2011). Interleukin-1β Inhibition and the Prevention of Recurrent Cardiovascular Events: Rationale and Design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart J. 162 (4), 597–605. doi:10.1016/j.ahj.2011.06.012
Ridker, P. M., Devalaraja, M., Baeres, F. M. M., Engelmann, M. D. M., Hovingh, G. K., Ivkovic, M., et al. (2021). IL-6 Inhibition with Ziltivekimab in Patients at High Atherosclerotic Risk (RESCUE): a Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. The Lancet 397 (10289), 2060–2069. doi:10.1016/s0140-6736(21)00520-1
Robbins, C. S., Hilgendorf, I., Weber, G. F., Theurl, I., Iwamoto, Y., Figueiredo, J. L., et al. (2013a). Local Proliferation Dominates Lesional Macrophage Accumulation in Atherosclerosis. Nat. Med. 19 (9), 1166–1172. doi:10.1038/nm.3258
Robbins, C. S., Hilgendorf, I., Weber, G. F., Theurl, I., Iwamoto, Y., Figueiredo, J. L., et al. (2013b). Local Proliferation Dominates Lesional Macrophage Accumulation in Atherosclerosis. Nat. Med. 19 (9), 1166–72. doi:10.1038/nm.3258
Robertson, A. K., and Hansson, G. K. (2006). T Cells in Atherogenesis: for Better or for Worse? Arterioscler Thromb. Vasc. Biol. 26 (11), 2421–2432. doi:10.1161/01.ATV.0000245830.29764.84
Robinson, N., Ganesan, R., Hegedűs, C., Kovács, K., Kufer, T. A., and Virág, L. (2019). Programmed Necrotic Cell Death of Macrophages: Focus on Pyroptosis, Necroptosis, and Parthanatos. Redox Biol. 26, 101239. doi:10.1016/j.redox.2019.101239
Rogers, C., Fernandes-Alnemri, T., Mayes, L., Alnemri, D., Cingolani, G., and Alnemri, E. S. (2017). Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/pyroptotic Cell Death. Nat. Commun. 8, 14128. doi:10.1038/ncomms14128
Romuk, E., Skrzep-Poloczek, B., Wojciechowska, C., Tomasik, A., Birkner, E., Wodniecki, J., et al. (2002). Selectin-P and Interleukin-8 Plasma Levels in Coronary Heart Disease Patients. Eur. J. Clin. Invest. 32 (9), 657–661. doi:10.1046/j.1365-2362.2002.01053.x
Rong, J. X., Shapiro, M., Trogan, E., and Fisher, E. A. (2003). Transdifferentiation of Mouse Aortic Smooth Muscle Cells to a Macrophage-like State after Cholesterol Loading. Proc. Natl. Acad. Sci. U S A. 100 (23), 13531–13536. doi:10.1073/pnas.1735526100
Sano, S., Oshima, K., Wang, Y., Katanasaka, Y., Sano, M., and Walsh, K. (2018). CRISPR-mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 123 (3), 335–341. doi:10.1161/CIRCRESAHA.118.313225
Schiopu, A., Frendéus, B., Jansson, B., Söderberg, I., Ljungcrantz, I., Araya, Z., et al. (2007). Recombinant Antibodies to an Oxidized Low-Density Lipoprotein Epitope Induce Rapid Regression of Atherosclerosis in Apobec-1(-/-)/low-Density Lipoprotein Receptor(-/-) Mice. J. Am. Coll. Cardiol. 50 (24), 2313–2318. doi:10.1016/j.jacc.2007.07.081
Scull, C. M., Hays, W. D., and Fischer, T. H. (2010). Macrophage Pro-inflammatory Cytokine Secretion Is Enhanced Following Interaction with Autologous Platelets. J. Inflamm. (Lond) 7, 53. doi:10.1186/1476-9255-7-53
Shami, A., Edsfeldt, A., Shore, A. C., Natali, A., Khan, F., Nilsson, J., et al. (2020). CD40 Levels in Plasma Are Associated with Cardiovascular Disease and in Carotid Plaques with a Vulnerable Plaque Phenotype and Remodelling. Eur. Heart J. 41 (Suppl. ment_2), ehaa946–3782. doi:10.1093/ehjci/ehaa946.3782
Sinha, S. K., Miikeda, A., Fouladian, Z., Mehrabian, M., Edillor, C., Shih, D., et al. (2021). Local M-CSF (Macrophage colony-stimulating Factor) Expression Regulates Macrophage Proliferation and Apoptosis in Atherosclerosis. Arteriosclerosis, Thromb. Vasc. Biol. 41 (1), 220–233.
Skuratovskaia, D., Vulf, M., Khaziakhmatova, O., Malashchenko, V., Komar, A., Shunkin, E., et al. (2020). Tissue-Specific Role of Macrophages in Noninfectious Inflammatory Disorders. Biomedicines 8 (10), 400. doi:10.3390/biomedicines8100400
Smith, M. S., Bentz, G. L., Smith, P. M., Bivins, E. R., and Yurochko, A. D. (2004). HCMV Activates PI(3)K in Monocytes and Promotes Monocyte Motility and Transendothelial Migration in a PI(3)K-dependent Manner. J. Leukoc. Biol. 76 (1), 65–76. doi:10.1189/jlb.1203621
Sodhi, N., and Brown, D. L. (2018). Pathophysiology of Acute Coronary Syndromes: Plaque Rupture and Atherothrombosis. Philadelphia: Cardiac Intensive Care-E-Book, 68.
Spiller, K. L., Anfang, R. R., Spiller, K. J., Ng, J., Nakazawa, K. R., Daulton, J. W., et al. (2014). The Role of Macrophage Phenotype in Vascularization of Tissue Engineering Scaffolds. Biomaterials 35 (15), 4477–4488. doi:10.1016/j.biomaterials.2014.02.012
Subedi, L., Lee, S. E., Madiha, S., Gaire, B. P., Jin, M., Yumnam, S., et al. (2020). Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases. Int. J. Mol. Sci. 21 (3), 764. doi:10.3390/ijms21030764
Swirski, F. K., and Nahrendorf, M. (2013a). Leukocyte Behavior in Atherosclerosis, Myocardial Infarction, and Heart Failure. Science 339 (6116), 161–166. doi:10.1126/science.1230719
Syväranta, S., Alanne-Kinnunen, M., Oörni, K., Oksjoki, R., Kupari, M., Kovanen, P. T., et al. (2014). Potential Pathological Roles for Oxidized Low-Density Lipoprotein and Scavenger Receptors SR-AI, CD36, and LOX-1 in Aortic Valve Stenosis. Atherosclerosis 235 (2), 398–407. doi:10.1016/j.atherosclerosis.2014.05.933
Tabas, I. (2010). The Role of Endoplasmic Reticulum Stress in the Progression of Atherosclerosis. Circ. Res. 107 (7), 839–850. doi:10.1161/circresaha.110.224766
Tacke, F., Alvarez, D., Kaplan, T. J., Jakubzick, C., Spanbroek, R., Llodra, J., et al. (2007). Monocyte Subsets Differentially Employ CCR2, CCR5, and CX3CR1 to Accumulate within Atherosclerotic Plaques. J. Clin. Invest. 117 (1), 185–194. doi:10.1172/JCI28549
Tang, J., Lobatto, M. E., Hassing, L., Van Der Staay, S., Van Rijs, S. M., Calcagno, C., et al. (2015). Inhibiting Macrophage Proliferation Suppresses Atherosclerotic Plaque Inflammation. Sci. Adv. 1 (3), e1400223. doi:10.1126/sciadv.1400223
Tao, H., Huang, J., Yancey, P. G., Yermalitsky, V., Blakemore, J. L., Zhang, Y., et al. (2020). Scavenging of Reactive Dicarbonyls with 2-hydroxybenzylamine Reduces Atherosclerosis in Hypercholesterolemic Ldlr-/- Mice. Nat. Commun. 11 (1), 4084–4115. doi:10.1038/s41467-020-17915-w
Tao, H., Yancey, P. G., Babaev, V. R., Blakemore, J. L., Zhang, Y., Ding, L., et al. (2015). Macrophage SR-BI Mediates Efferocytosis via Src/PI3K/Rac1 Signaling and Reduces Atherosclerotic Lesion Necrosis. J. Lipid Res. 56 (8), 1449–1460. doi:10.1194/jlr.M056689
Teh, Y. C., Ding, J. L., Ng, L. G., and Chong, S. Z. (2019). Capturing the Fantastic Voyage of Monocytes through Time and Space. Front. Immunol. 10, 834. doi:10.3389/fimmu.2019.00834
Tenger, C., Sundborger, A., Jawien, J., and Zhou, X. (2005). IL-18 Accelerates Atherosclerosis Accompanied by Elevation of IFN-Gamma and CXCL16 Expression Independently of T Cells. Arterioscler Thromb. Vasc. Biol. 25 (4), 791–796. doi:10.1161/01.atv.0000153516.02782.65
Thayse, K., Kindt, N., Laurent, S., and Carlier, S. (2020). VCAM-1 Target in Non-invasive Imaging for the Detection of Atherosclerotic Plaques. Biology (Basel) 9 (11), 368. doi:10.3390/biology9110368
Tian, F., Yao, J., Yan, M., Sun, X., Wang, W., Gao, W., et al. (2016). 5-Aminolevulinic Acid-Mediated Sonodynamic Therapy Inhibits RIPK1/RIPK3-dependent Necroptosis in THP-1-Derived Foam Cells. Sci. Rep. 6 (1), 21992–22013. doi:10.1038/srep21992
Tyrrell, D. J., and Goldstein, D. R. (2020). Ageing and Atherosclerosis: Vascular Intrinsic and Extrinsic Factors and Potential Role of IL-6. Nat. Rev. Cardiol. 18, 1–11. doi:10.1038/s41569-020-0431-7
Van Eck, M., Twisk, J., Hoekstra, M., Van Rij, B. T., Van der Lans, C. A., Bos, I. S., et al. (2003). Differential Effects of Scavenger Receptor BI Deficiency on Lipid Metabolism in Cells of the Arterial wall and in the Liver. J. Biol. Chem. 278 (26), 23699–23705. doi:10.1074/jbc.M211233200
Veerman, K. M., Williams, M. J., Uchimura, K., Singer, M. S., Merzaban, J. S., Naus, S., et al. (2007). Interaction of the Selectin Ligand PSGL-1 with Chemokines CCL21 and CCL19 Facilitates Efficient Homing of T Cells to Secondary Lymphoid Organs. Nat. Immunol. 8 (5), 532–539. doi:10.1038/ni1456
Vijayvergiya, R., and Vadivelu, R. (2015). Role of Helicobacter pylori Infection in Pathogenesis of Atherosclerosis. World J. Cardiol. 7 (3), 134–143. doi:10.4330/wjc.v7.i3.134
Virmani, R., Burke, A. P., Kolodgie, F. D., and Farb, A. (2002). Vulnerable Plaque: the Pathology of Unstable Coronary Lesions. J. Interv. Cardiol. 15 (6), 439–446. doi:10.1111/j.1540-8183.2002.tb01087.x
Wainstein, M. V., Mossmann, M., Araujo, G. N., Gonçalves, S. C., Gravina, G. L., Sangalli, M., et al. (2017). Elevated Serum Interleukin-6 Is Predictive of Coronary Artery Disease in Intermediate Risk Overweight Patients Referred for Coronary Angiography. Diabetol. Metab. Syndr. 9, 67. doi:10.1186/s13098-017-0266-5
Wang, L., Li, H., Tang, Y., and Yao, P. (2020). Potential Mechanisms and Effects of Efferocytosis in Atherosclerosis. Front. Endocrinol. (Lausanne) 11, 585285. doi:10.3389/fendo.2020.585285
Wang, N., Liang, H., and Zen, K. (2014a). Molecular Mechanisms that Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 5, 614. doi:10.3389/fimmu.2014.00614
Wang, Y., Gao, W., Shi, X., Ding, J., Liu, W., He, H., et al. (2017). Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 547 (7661), 99–103. doi:10.1038/nature22393
Willemsen, L., and de Winther, M. P. (2020). Macrophage Subsets in Atherosclerosis as Defined by Single-Cell Technologies. J. Pathol. 250 (5), 705–714. doi:10.1002/path.5392
Williams, J. W., Winkels, H., Durant, C. P., Zaitsev, K., Ghosheh, Y., and Ley, K. (2020). Single Cell RNA Sequencing in Atherosclerosis Research. Circ. Res. 126 (9), 1112–1126. doi:10.1161/CIRCRESAHA.119.315940
Winkels, H., Ehinger, E., Vassallo, M., Buscher, K., Dinh, H. Q., Kobiyama, K., et al. (2018). Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 122 (12), 1675–1688. doi:10.1161/CIRCRESAHA.117.312513
Wynn, T. A., and Vannella, K. M. (2016). Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44 (3), 450–462. doi:10.1016/j.immuni.2016.02.015
Xiong, D., Wang, Y., and You, M. (2020). A Gene Expression Signature of TREM2hi Macrophages and γδ T Cells Predicts Immunotherapy Response. Nat. Commun. 11 (1), 5084–5112. doi:10.1038/s41467-020-18546-x
Xu, H., Jiang, J., Chen, W., Li, W., and Chen, Z. (2019). Vascular Macrophages in Atherosclerosis. J. Immunol. Res. 2019, 4354786. doi:10.1155/2019/4354786
Xu, S., Bai, P., Little, P. J., and Liu, P. (2014). Poly(ADP-ribose) Polymerase 1 (PARP1) in Atherosclerosis: from Molecular Mechanisms to Therapeutic Implications. Med. Res. Rev. 34 (3), 644–675. doi:10.1002/med.21300
Xu, S., Ogura, S., Chen, J., Little, P. J., Moss, J., and Liu, P. (2013). LOX-1 in Atherosclerosis: Biological Functions and Pharmacological Modifiers. Cel Mol Life Sci 70 (16), 2859–2872. doi:10.1007/s00018-012-1194-z
Yamamoto, S., Yancey, P. G., Zuo, Y., Ma, L. J., Kaseda, R., Fogo, A. B., et al. (2011). Macrophage Polarization by Angiotensin II-type 1 Receptor Aggravates Renal Injury-Acceleration of Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 31 (12), 2856–2864. doi:10.1161/atvbaha.111.237198
Yan, G., Elbadawi, M., and Efferth, T. (2020). Multiple Cell Death Modalities and Their Key Features. World Acad. Sci. J. 2 (2), 39–48.
Yang, S., Li, J., Chen, Y., Zhang, S., Feng, C., Hou, Z., et al. (2019). MicroRNA-216a Promotes M1 Macrophages Polarization and Atherosclerosis Progression by Activating Telomerase via the Smad3/NF-Κb Pathway. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (7), 1772–1781. doi:10.1016/j.bbadis.2018.06.016
Yin, Q., Chang, H., Shen, Q., and Xing, D. (2021). Photobiomodulation Therapy Promotes the ATP-Binding Cassette Transporter A1-dependent Cholesterol Efflux in Macrophage to Ameliorate Atherosclerosis. J. Cel Mol Med 25 (11), 5238–5249. doi:10.1111/jcmm.16531
Yu, X. H., Fu, Y. C., Zhang, D. W., Yin, K., and Tang, C. K. (2013). Foam Cells in Atherosclerosis. Clin. Chim. Acta 424, 245–252. doi:10.1016/j.cca.2013.06.006
Zeng, C., Wang, R., and Tan, H. (2019). Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int. J. Biol. Sci. 15 (7), 1345–1357. doi:10.7150/ijbs.33568
Zernecke, A., Winkels, H., Cochain, C., Williams, J. W., Wolf, D., Soehnlein, O., et al. (2020). Meta-analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ. Res. 127 (3), 402–426. doi:10.1161/CIRCRESAHA.120.316903
Zhang, H., Park, Y., Wu, J., Chen, Xp., Lee, S., Yang, J., et al. (2009). Role of TNF-Alpha in Vascular Dysfunction. Clin. Sci. (Lond) 116 (3), 219–230. doi:10.1042/cs20080196
Zhang, X., Niessner, A., Nakajima, T., Ma-Krupa, W., Kopecky, S. L., Frye, R. L., et al. (2006). Interleukin 12 Induces T-Cell Recruitment into the Atherosclerotic Plaque. Circ. Res. 98 (4), 524–531. doi:10.1161/01.RES.0000204452.46568.57
Zhang, Y., Wang, Y., Zhou, D., Zhang, L. S., Deng, F. X., Shu, S., et al. (2019). Angiotensin II Deteriorates Advanced Atherosclerosis by Promoting MerTK Cleavage and Impairing Efferocytosis through the AT1R/ROS/p38 MAPK/ADAM17 Pathway. Am. J. Physiol. Cel Physiol 317 (4), C776–C787. doi:10.1152/ajpcell.00145.2019
Zhang, Y., Shi, X., Han, J., Peng, W., Fang, Z., Zhou, Y., et al. (2021). Convallatoxin Promotes M2 Macrophage Polarization to Attenuate Atherosclerosis through PPARγ-Integrin αvβ5 Signaling Pathway. Dddt 15, 803–812. doi:10.2147/dddt.s288728
Zhu, X. D., Zhuang, Y., Ben, J. J., Qian, L. L., Huang, H. P., Bai, H., et al. (2011). Caveolae-dependent Endocytosis Is Required for Class A Macrophage Scavenger Receptor-Mediated Apoptosis in Macrophages. J. Biol. Chem. 286 (10), 8231–8239. doi:10.1074/jbc.M110.145888
Keywords: atherosclerosis, inflammation, macrophages, lipid, plaque, biomarker
Citation: Farahi L, Sinha SK and Lusis AJ (2021) Roles of Macrophages in Atherogenesis. Front. Pharmacol. 12:785220. doi: 10.3389/fphar.2021.785220
Received: 28 September 2021; Accepted: 04 November 2021;
Published: 26 November 2021.
Edited by:
Pallavi R. Devchand, University of Calgary, CanadaReviewed by:
Thomas A. Angelovich, RMIT University, AustraliaCopyright © 2021 Farahi, Sinha and Lusis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aldons J. Lusis, amx1c2lzQG1lZG5ldC51Y2xhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.