 Emine Eren-Koçak
Emine Eren-Koçak Turgay Dalkara
Turgay Dalkara- 1Institute of Neurological Sciences and Psychiatry, Hacettepe University, Ankara, Turkey
- 2Department of Psychiatry, Medical Faculty, Hacettepe University, Ankara, Turkey
Migraine and major depression are debilitating disorders with high lifetime prevalence rates. Interestingly these disorders are highly comorbid and show significant heritability, suggesting shared pathophysiological mechanisms. Non-homeostatic function of ion channels and neuroinflammation may be common mechanisms underlying both disorders: The excitation-inhibition balance of microcircuits and their modulation by monoaminergic systems, which depend on the expression and function of membrane located K+, Na+, and Ca+2 channels, have been reported to be disturbed in both depression and migraine. Ion channels and energy supply to synapses not only change excitability of neurons but can also mediate the induction and maintenance of inflammatory signaling implicated in the pathophysiology of both disorders. In this respect, Pannexin-1 and P2X7 large-pore ion channel receptors can induce inflammasome formation that triggers release of pro-inflammatory mediators from the cell. Here, the role of ion channels involved in the regulation of excitation-inhibition balance, synaptic energy homeostasis as well as inflammatory signaling in migraine and depression will be reviewed.
Introduction
Migraine and depression are comorbid diseases (Amiri et al., 2019; Karsan and Goadsby, 2021). Migraine is a risk factor for depression, whereas migraine attack prevalence and intensity increase during depression. Despite this well-established epidemiological data, the biological basis of this comorbidity is unclear. Emerging data from animal models and human imaging studies suggest that neuroinflammation could be a common pathway in the pathophysiology of both disorders (Mueller and Schwarz, 2007; Richards et al., 2018; Albrecht et al., 2019; Hadjikhani et al., 2020; Afridi and Suk, 2021). However, how neuroinflammatory signaling is initiated without an injury to the brain tissue is unknown. A non-homeostatic synaptic transmission has been suggested as an initiator of neuroinflammatory signaling (Dalkara and Kilic, 2013; Kilic et al., 2018; Afridi and Suk, 2021; Petit et al., 2021). In this regard, some ion channels and activation of cellular stress sensors such as pannexin1 (Panx1) and P2X7 channels and the downstream inflammatory cascade may play a role. Here, we review the recent evidence suggesting that a non-homeostatic ion channel activity or a mismatch between synaptic energy supply and glutamatergic transmission can activate the inflammatory pathway in migraine and depression. Since we limit the scope of the review to these potentially common central mechanisms in both disorders, we will not be able to cover recent developments regarding the success of CGRP antagonists as novel anti-migraine drugs and their implications for peripheral nociceptive mechanisms of migraine headache.
Lessons Learned From Familial Migraine Mutations
Most of our knowledge about the potential role of ion channels and glutamate in migraine comes from the discovery of mutant genes causing familial hemiplegic migraine (FHM) (Moskowitz et al., 2004; Russell and Ducros, 2011; Pietrobon and Moskowitz, 2014; Dalkara and Moskowitz, 2017; De Boer et al., 2019; Pietrobon and Brennan, 2019). FHM patients suffer from typical migraine with aura episodes along with transient hemiplegic or hemiparetic attacks (Russell and Ducros, 2011). Some patients may also experience confusion, memory loss, seizures and coma episodes and, rarely, persistent deficits like ataxia. However, these severe phenotypes are typical of FHM1 and 2 but not of FHM3 (Mantegazza, 2018) and, other than episodic attacks, majority of FHM 1 and 2 patients are healthy except being more sensitive to head trauma. Hemiplegic attacks and an autosomal dominant inheritance pattern distinguish them from non-familial, common forms of migraine (Sutherland H. G. et al., 2019). Although they are rare and the mutations have yet been detected in less than half of the cases (Sutherland et al., 2020), the three genes identified so far have provided insight into how a migraine attack can be initiated in an otherwise healthy brain (Moskowitz et al., 2004; Pietrobon and Brennan, 2019). Mutations in CACNA1A, ATP1A2, and SCN1A genes, which encode a P/Q-type voltage-gated calcium channel (CaV2.1), an astrocytic Na/K pump (α2 Na/K-ATPase) and the α1 subunit of voltage-gated NaV1.1 sodium channel, account for FHM1, FHM2 and FHM3, respectively (Figure 1). Of these, CACNA1A, ATP1A2 mutations have been comprehensively studied with regard to their effects on cortical excitability (for review, (Pietrobon and Brennan, 2019). In vitro and in vivo evidence suggests that CACNA1A mutations can lead to enhanced glutamate release during excitatory synaptic activity because gain-of-function mutations on the pore-forming α1-subunit of the presynaptic P/Q type calcium channels enable them to open at more hyperpolarized membrane potentials than normal, allowing more calcium influx to the terminal during action potential trains (Tottene et al., 2009). Interestingly, the P/Q type calcium channels on terminals of GABAergic neurons are not affected by these mutations (Vecchia et al., 2014). This causes a shift in cortical excitation/inhibition balance towards excitation (Vecchia and Pietrobon, 2012; Tottene et al., 2019), which is thought to account for the vulnerability to cortical spreading depolarization (CSD), the putative cause of migraine aura and, for the hyperexcitability seen in FHM1 brain (Takizawa et al., 2020a). Given the presence of glutamatergic synapses driving the feed-forward and feed-back inhibitory neurons, one may expect an increased inhibitory control as well. However, this is probably not the case owing to the fact that short-term depression develops faster in these glutamatergic synapses than does in glutamatergic synapses terminating on principal neurons (Tottene et al., 2019). Although the net effect of the excitation/inhibition balance might be different in various neuronal circuits depending on the organization of neuronal interconnections and release kinetics of synapses, including pain-transmitting/processing networks, it has been clearly shown that the CSD threshold is decreased in transgenic knock-in mice harboring human CACNA1A mutations (Van Den Maagdenberg et al., 2004). In fact, the threshold was found to be even lower in S218L compared to R192Q knock-in mouse in parallel with the clinical severity of FHM symptoms in patients carrying these mutations (Van Den Maagdenberg et al., 2010). However, some FHM1 and FHM2 patients also suffer from epileptic seizures suggesting that excitation/inhibition imbalance has also potential to generate epileptic discharges (Marconi et al., 2003; Lebas et al., 2008; Costa et al., 2014).
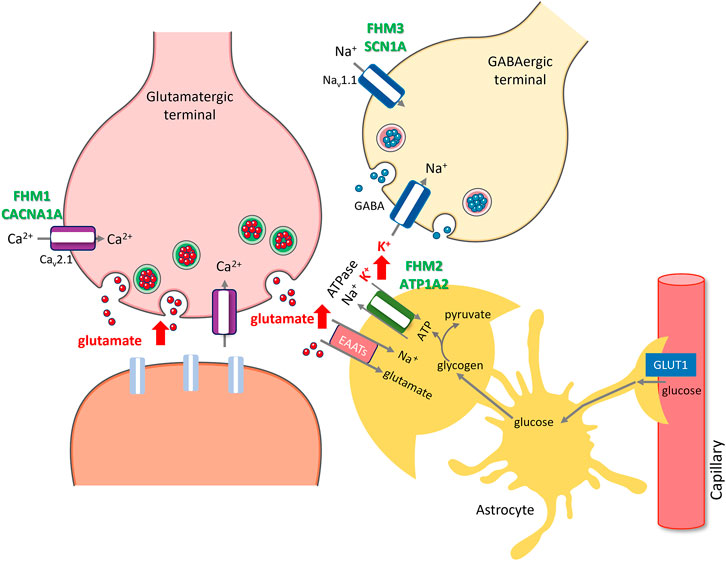
FIGURE 1. Rises in extracellular glutamate and K+ caused by ion channel and transporter mutations or transcriptional changes induced by migraine triggers create susceptibility to migraine with aura. Facilitation of excitatory synaptic transmission by either promoting glutamate release (red arrows) due to gain of function mutations in P/Q type presynaptic calcium channels (purple) encoded by CACNA1A in FHM1 or decreasing its uptake along with K+ (red arrow) due to mutations in alpha2 subunit of Na+/K + ATPase (green) encoded by ATP1A2 in FHM2, increases susceptibility to CSD and migraine with aura. Glutamate released during synaptic activity is taken up to astrocyte processes (amber) by excitatory amino acid transporters (pink, EAATs), which is driven by the Na + gradient created by Na+/K + ATPase, hence, glutamate and K+ uptakes are coupled. Excess K+ released from overactive GABAergic terminals (red arrow) due to gain of function mutations in NaV 1.1 type sodium channels (blue) encoded by SCN1A in FHM3 also creates susceptibility to CSD and migraine with aura. Glycogen granules in peri-synaptic astrocyte processes instantly provide glycosyl units to meet the rapidly escalating energy demand during excitatory synaptic transmission (e.g., by Na+/K + ATPase), whereas glucose transported from circulation to astrocytes via GLUT1 is primarily used in replenishing glycogen. Insufficient glycogen breakdown due to transcriptional changes induced by migraine triggers such as sleep deprivation, which promote glycogen synthesis while reducing glycogen breakdown, hinders glutamate and K+ uptake as seen in FHM2, creating susceptibility to CSD and migraine in non-familial common migraine. The illustration is prepared by Dr. Zeynep Kaya. Cellular templates are copied from Servier Medical Art (smart.servier.com).
Cortical spreading depolarization is considered the electrophysiological correlate of migraine aura (Takizawa et al., 2020a). First discovered by Leao in 1944 on the rabbit brain, it is caused by intense depolarization of neurons and astrocytes, propagating along the cortical gray matter at a speed of 2–6 mm per minute (Leao, 1944). As it spreads, it depresses the ongoing electrical activity (e.g., the EEG as originally observed), hence, named spreading depression by Leao. However, cortical spreading depolarization is increasingly used instead of cortical spreading depression because of the confusion the term “depression” causes. Leao was the first to note the similarity between the propagation rate of visual aura in migraine and CSD (Leao, 1947). However, it only became possible to show the presence of CSD in migraineur brain in the past 4 decades with advances in imaging technologies that enabled detection of the CSD-induced cerebral blood flow changes (Olesen et al., 1982; Cao et al., 1999; Hadjikhani et al., 2001).
High extracellular levels of glutamate and K+ are thought to be responsible for ignition and propagation of CSD (Pietrobon and Moskowitz, 2014). Rising extracellular glutamate levels along with K+ concentration exceeding 15 mM appear to be necessary for synchronized depolarization of an aggregate of neurons involving, at least partly, the NR2 type NMDA receptors (Tang et al., 2014; Takizawa et al., 2020a; Bu et al., 2020). During CSD, K+ levels further rise to 30–60 mM, which contribute to spread of the depolarizing wave to neighboring gray matter (Pietrobon and Moskowitz, 2014). Accordingly, application of glutamate, NMDA or high K+ to the cortex all ignites CSD. Therefore, increased glutamate release in FHM1 knock-in mice is consistent with the enhanced susceptibility to CSD (Pietrobon and Brennan, 2019). Indeed, foci of glutamate “plumes” spontaneously bursting in the cortex of awake ATP1A2 knock-in mice, a model of FHM2, have recently been demonstrated by expressing a fluorescent glutamate reporter in the cortex (Parker et al., 2021). The same study also showed that a surge of glutamate plumes preceded the onset of CSD as hypothesized before.
Astrocytic Na/K pump (α2 Na/K-ATPase) clears K+ spilling out of the synaptic cleft during synaptic activity (Cholet et al., 2002). The transmembrane Na+ gradient it creates as it takes up K+ is essential for uptake of glutamate by astrocyte processes around excitatory synapses (Cholet et al., 2001; Petit et al., 2021). Accordingly, ATP1A2 hypofunction caused by FHM2 mutations leads to reduced K+ and glutamate uptake as demonstrated both in vitro and in knock-in mice, in vivo (Capuani et al., 2016; Parker et al., 2021). As with CACNA1A mutations, GABA release is not affected in FHM2 because α2 Na/K-ATPase is not appreciably expressed on astrocyte processes around GABAergic terminals (Cholet et al., 2002; Capuani et al., 2016). Consistent with these findings, FHM2 knock-in mice have a low CSD induction threshold and spontaneously generate the glutamate plums mentioned above (Leo et al., 2011; Parker et al., 2021).
Although the pathophysiological phenotype for FHM1 and FHM2 converge on the glutamatergic synapse, this is not the case for SCN1A gene mutations underlying FHM3 (Desroches et al., 2019; Chever et al., 2021; Lemaire et al., 2021). SCN1A encodes the α1 subunit of the neuronal voltage-gated sodium channel Nav1.1, which contributes to the action potential firing on primarily GABAergic interneurons. Indeed, hundreds of loss-of-function SCN1A mutations have been reported in various epileptic syndromes caused by reduced GABAergic inhibition (Hedrich et al., 2014; Sutherland HG. et al., 2019; Mantegazza et al., 2021). In FHM3, however, mutations are gain-of-function, causing increased firing of GABAergic interneurons. Increased GABAergic activity may sound at odds with CSD generation but recent studies have shed light on the mechanism of CSD initiation by documenting that hyperactive GABAergic interneurons discharging at high frequencies can cause significant elevations in extracellular K+ (over 12 mM), hence, increase CSD susceptibility as discussed above (Desroches et al., 2019; Chever et al., 2021; Lemaire et al., 2021). Supporting this view, knock-in mice bearing L263V or L1649Q human mutation exhibit a low CSD induction threshold (Jansen et al., 2020; Auffenberg et al., 2021) as well as spontaneous CSDs (Jansen et al., 2020) and hyperexcitable GABAergic neurons (Auffenberg et al., 2021).
Another gain of function change in glutamatergic transmission secondary to impairment of frequency-dependent presynaptic adaptation has been reported for mutations in casein kinase 1 delta, which cause a non-hemiplegic form of familial migraine along with disrupted sleep phases (Brennan et al., 2013). Knock-in mice harboring this mutation have also a reduced CSD induction threshold (Suryavanshi et al., 2019). Knock-in mouse models have been instrumental to study various aspects of migraine although they basically model familial migraines. In addition to enhanced susceptibility to CSD, these mice also exhibit heightened sensory perceptions such as hypersensitivity to light, consistent with sensory abnormalities reported by patients (Russell and Ducros, 2011; Chanda et al., 2013; Brennan and Pietrobon, 2018). They also seem to suffer from headaches and periorbital allodynia (Langford et al., 2010; Chanda et al., 2013). Mutations associated with severe clinical phenotypes additionally lead to spontaneous seizures or ataxia in FHM 1 and 2 knock-in mice (Van Den Maagdenberg et al., 2010; Kros et al., 2018).
Non-Familial Common Migraine
The mutations detected in familial migraine are usually not found in non-familial common migraine cases although migraine has been established to have a complex polygenic genetic heritability estimated to be as high as 60% (Sutherland H. G. et al., 2019). Meta analysis of several GWAS studies encompassing 59,674 patients and 316,078 controls reveled 44 susceptibility loci (Gormley et al., 2016). Most of them are associated with vascular and neuronal function, ion channels and circadian rhythm (Sutherland H. G. et al., 2019). Three of them are near ion channels (TRPM8 and KCNK5) or an ion transporter (sodium/potassium/calcium exchanger3 SLC24A3). TRPM8 is expressed on type C- and A-delta nociceptors and is activated by cold temperatures (Dussor and Cao, 2016). Although migraine is generally initiated by intrinsic brain mechanisms, it can also be triggered by some volatile irritants such as umbellulone emanating from the leaves of headache tree (umbellularia californica), which is thought to cause headache by stimulating TRPA1 channels on meningeal nociceptors (Edelmayer et al., 2012). KCNK5 belongs to the superfamily of potassium channel proteins containing two pore-forming P domains as does KCNK18 (Reyes et al., 1998). Although the neuronal function of KCNK5 is unclear, TRESK potassium channels encoded by KCNK18 regulates neuronal excitability in the dorsal root and trigeminal ganglia (Enyedi and Czirjak, 2015). Supporting a role for two-pore-domain potassium channels in migraine, KCNK18 mutations cause non-hemiplegic familial migraine with aura. F139Wfsx24 KCNK18 frame-shift mutation has been shown to result in hyperexcitability of cultured trigeminal ganglion neurons as well as allodynia in rodent migraine models (isosorbide dinitrate injection) by a negative dominant effect on TREK1 and 2, two other two-pore-domain potassium channels with which TRESK channels form functional heterodimers (Liu et al., 2013; Royal et al., 2019). Suggesting a suppressive role of TRESK over trigeminal ganglia excitability, KCNK18 knockout mice reportedly exhibit mechanical and thermal hyperalgesia in response to systemic glyceryl trinitrate treatment (a migraine model) (Pettingill et al., 2019) and exaggerated nocifensive behaviors in response to dural application of inflammatory soup (Guo et al., 2019). Of note, these families with KCNK18 mutations exhibit migraine with aura, suggesting that reduced control over neuronal hyperexcitability due to hypofunction of TRESK channels in cortical neurons may also create susceptibility to CSD.
Initiation of Inflammatory Signaling That Causes Headache
CSD has been shown to activate neuronal pannexin1 channels and initiate the downstream inflammatory signaling following formation of the inflammasome complex (Karatas et al., 2013; Ghaemi et al., 2016; Takizawa et al., 2016; Chen et al., 2017; Eising et al., 2017; Ghaemi et al., 2018; Bu et al., 2020; Takizawa et al., 2020b). Inflammasome formation causes release of pro-inflammatory mediators such IL1-ß and HMGB1 from neurons, which triggers translocation of inflammatory transcription factor NF-kappaB to the nucleus in astrocytes (Karatas et al., 2013; Takizawa et al., 2016; Dehghani et al., 2021). NF-kappaB induces hundreds of transcripts including tens of inflammatory mediators such as iNOS, COX2, and cytokines (https://www.bu.edu/nf-kb/gene-resources/target-genes/). This is thought to cause release of prostanoids, NO, cytokines and other algesic mediators to CSF and, hence, to activate meningeal nociceptors and inflammatory cells, culminating in a sterile meningeal inflammation that can sustain migraine headache for hours to days (Karatas et al., 2013; Erdener and Dalkara, 2014; Dalkara and Moskowitz, 2017). With a single CSD, microglia do not switch to a pro-inflammatory state, which is only seen a few days after exposure to multiple CSDs (Takizawa et al., 2017), suggesting that aura (CSD)-triggered parenchymal inflammatory signaling is mainly mediated by astrocytes, whereas microglia may take part in inflammatory signaling in patients suffering from frequent migraine with aura attacks. Supporting this formulation, a recent PET/MRI study using a sensitive inflammatory tracer (11C-PBR28) taken up by active glia, has documented the presence of parenchymal as well as meningeal inflammatory signaling in 13 patients suffering from repeated migraine with aura attacks in the past month (Albrecht et al., 2019; Hadjikhani et al., 2020).
Interestingly, in the absence CSD, it has been shown that parenchymal inflammatory signaling pathway can be initiated via activation of neuronal Panx1 channels, this time, by migraine triggers such as sleep deprivation (Kilic et al., 2018). Sleep deprivation induces transcriptional changes favoring glycogen synthesis over its breakdown (Petit and Magistretti, 2016; Petit et al., 2021). Insufficient glycogen breakdown within astrocyte processes around glutamatergic terminals can lead to Panx1 channel activation due to inadequate glutamate and K+ clearance. This is because glycogen-derived ATP is preferentially used for glutamate and K+ uptake even in the presence of sufficient glucose owing to more favorable kinetics of the enzymes involved in metabolizing the glycosyl units liberated from glycogen over glucose, whereas glucose is quickly metabolized to replenish glycogen (Petit et al., 2021). Thus, extracellular glutamate and K+ accumulation during intense glutamatergic synaptic activity create a favorable extracellular milieu, first, for activation of Panx1 channels, and then, for CSD ignition upon further rise in extracellular glutamate and K+ (Kilic et al., 2018; Petit et al., 2021). This proposed mechanism could explain why migraine with and without aura attacks exist in the same person and how CSD can emerge in the absence of monogenic mutations but with transcriptional changes triggering the same mechanisms in non-familial migraine.
Master Switch of Inflammatory Signaling: Pannexin Channels
Pannexins are large-pore membrane channels similar to connexins but, unlike connexins, they do not form gap junctions (Macvicar and Thompson, 2010). Panx1 and Panx2 are widely expressed in the central nervous system on all main cell types (Yeung et al., 2020). In vitro studies have disclosed that Panx1 can open in three different states; a low conductance Cl-selective opening at positive membrane potentials (in vivo function unknown); large conductance, non-selective opening allowing calcium, ATP and molecules up to 900 Dalton to pass through; and a persistent opening due to caspase-mediated cleavage of the C-terminal domain, leading to cell death (Dahl, 2018). Panx1 channels can be physiologically activated in large-pore state by increases in extracellular K+, glutamate and intracellular Ca2+ concentration, NMDA and P2X7 receptor stimulation, swelling (membrane stretch), c-Jun N-terminal kinases and Src family of tyrosine kinases (Whyte-Fagundes and Zoidl, 2018). Most of the Panx1 activating conditions are present during CSD as well as when glycogen breakdown, hence, K+ and glutamate uptake is reduced as discussed above (Karatas et al., 2013; Kilic et al., 2018; Petit et al., 2021). Opening of neuronal Panx 1 channels can be monitored in vivo and in vitro by using membrane-impermeant fluorescent dyes like propidium iodide or YoPro-1 (Macvicar and Thompson, 2010), which flux into cells through Panx 1 channels opened in large conductance state (Karatas et al., 2013; Bu et al., 2020) (Figure 2). CSD-induced dye uptake to neurons was suppressed with several Panx1 inhibitors such as carbenoxolone, probenecid, 10Panx peptide and inhibition of Panx1 expression by siRNA, supporting the view that the dye fluxed in neurons through Panx1 channels (Karatas et al., 2013; Bu et al., 2020). Large pore opening of P2X7 receptors can also mediate the dye influx (Bhaskaracharya et al., 2014). However, recent transcriptomics studies consistently show that P2X7 receptors are not expressed in adult neurons unlike macrophages where they are closely coupled with Panx1 channels (Illes et al., 2017; Kaczmarek-Hajek et al., 2018). Further supporting opening of Panx1 channels with CSD or insufficient glycogen breakdown, formation of the inflammasome complex and activation of caspase-1 along with HMGB1 release have also been demonstrated (Karatas et al., 2013; Takizawa et al., 2016; Kilic et al., 2018). These advances open the exciting possibility of developing Panx1 inhibitors as migraine prophylactic drugs; especially considering some Panx1 inhibitors such as carbenoxolone, probenecid and mefloquine are already clinically registered medicines (Yeung et al., 2020). Similarly, agents acting on downstream steps in the inflammatory cascade including inhibitors of inflammasome, caspase-1 or interleukins may be promising drug targets for migraine treatment. Successes of anakinra in relieving severe migraine headaches seen in cyropyrin-associated periodic syndromes characterized by IL1-ß over production due to inflammasome mutations (Parker et al., 2016) and, the relief obtained by NSAIDs in treating common migraine headaches support this view.
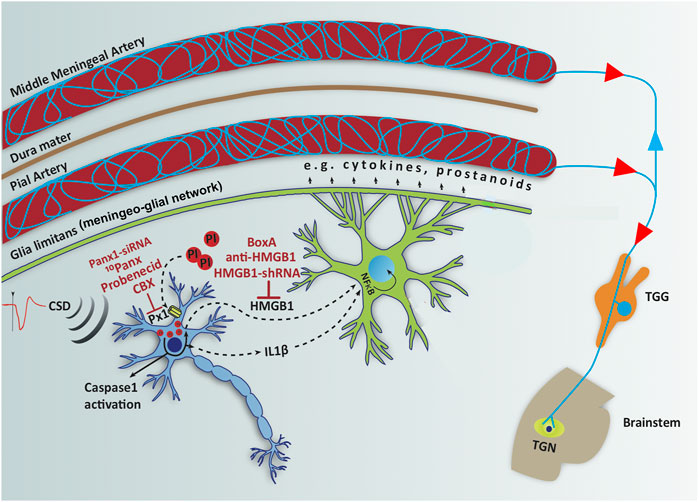
FIGURE 2. Opening neuronal Panx1 channels after CSD initiates a neuroinflammatory signaling cascade, characterized by inflammasome formation and caspase-1 activation in neurons followed by release of HMGB1 and IL-1β, which trigger NF-κB nuclear translocation in astrocytes. Red labels show agents used to inhibit each step in the inflammatory cascade and red circles represent propidium iodide (PI) influx through open Panx1 (Px1) large-pore channels. NF-κB induces transcription of several pro-inflammatory genes including cytokines and prostanoids, which are thought to be released to CSF through glia limitans and meningeo-glial network and activate the nociceptive nerves around pial and dural vessels, causing headache. Modified from Karatas et al., 2013 with permission.
Ion Channel Dysfunctions in Stress and Depression
As proposed for migraine, changes in the excitation/inhibition (E: I) balance resulting in increased excitability have also been implicated in major depression (Fee et al., 2017). Ion channels are important regulators of excitability, network activity as well as plasticity. Changes in ion channel attributes alter GABAergic and glutamatergic neuron excitability and firing, hence, modify the E: I balance in microcircuits. A recent transcriptomic analysis revealed that out of 1,153 differentially expressed genes (DEGs) in the hippocampi of chronically stressed rats, a rodent model of depression; 46 DEGs were related to potassium channels, calcium channels, sodium channels, and chloride channels on plasma membrane (Ren et al., 2021). This finding supports the hypothesis that changes in the expression or function of ion channels may disrupt the E: I balance throughout the brain regions relevant for depression, and contribute to the development of depression. Indeed, a recent paper reported that global inactivation of γ2 subunit gene of GABA-A receptors on somatostatin interneurons (SST-IN) disinhibited effects of SST-IN on pyramidal cells, resulting in an anxiolytic and antidepressant phenotype (Fuchs et al., 2017). On the other hand, ionotropic P2X7 receptors can additionally trigger formation of the inflammasome complex, activating interleukin-1ß and downstream inflammatory signaling, as do Panx1 channels. Indeed, several lines of evidence implicate the involvement of P2X7 receptors and inflammatory signaling, including inflammasome formation in the pathophysiology of stress and depression (Dowlati et al., 2010; Iwata et al., 2016; Wohleb et al., 2016; Yue et al., 2017). Accordingly, in the second part of the article, we will review the potential roles of purinergic P2X7 receptors along with hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, M-type K+ (KCNQ) channels, TWIK-related K+ channels (TREK) and Ca+2 channels in the pathophysiology of depression, dysfunction of which are also implicated in migraine.
P2X7 Receptor
The purinergic P2X7 receptor (P2X7R) is a ligand gated ion channel activated by binding of adenosine triphosphate (ATP) (Khakh and North, 2006). ATP is released from the presynaptic terminal as a co-transmitter as well as from astrocytes (Bezzi and Volterra, 2001; Burnstock, 2007). Its release is increased in response to physical stressors and cellular injury as part of an adaptive response. Psychological stressors like acute restraint stress, in which restriction of the movement of animals leads to stress, also have been shown to increase extracellular ATP concentrations in rat hippocampus and prefrontal cortex (PFC) (Iwata et al., 2016), which then can activate low affinity P2X7R (Donnelly-Roberts et al., 2009). Mice susceptible to chronic social defeat stress (CSDS), on the other hand, displayed lower ATP concentrations in hippocampus and PFC (Cao et al., 2013). In line with the latter finding, intracerebroventircular administration of ATP or its nonhydrolyzable analog, ATP-γ-S, for 7-days or 28-days reverses social avoidance caused by CSDS and anhedonia caused by chronic unpredictable mild stress (CUMS) (Cao et al., 2013) (Table 1). Inhibition of ATP release selectively from astrocytes by genetic knockout of inositol 1,4,5- trisphosphate (IP3) receptor type 2 (IP3R2−/−) lead to increased behavioral despair and anhedonia (Cao et al., 2013) (Table 1). These findings suggest that increased ATP release from astrocytes in the hippocampus and PFC in response to stressors is an important mediator in developing adaptive responses to stress.
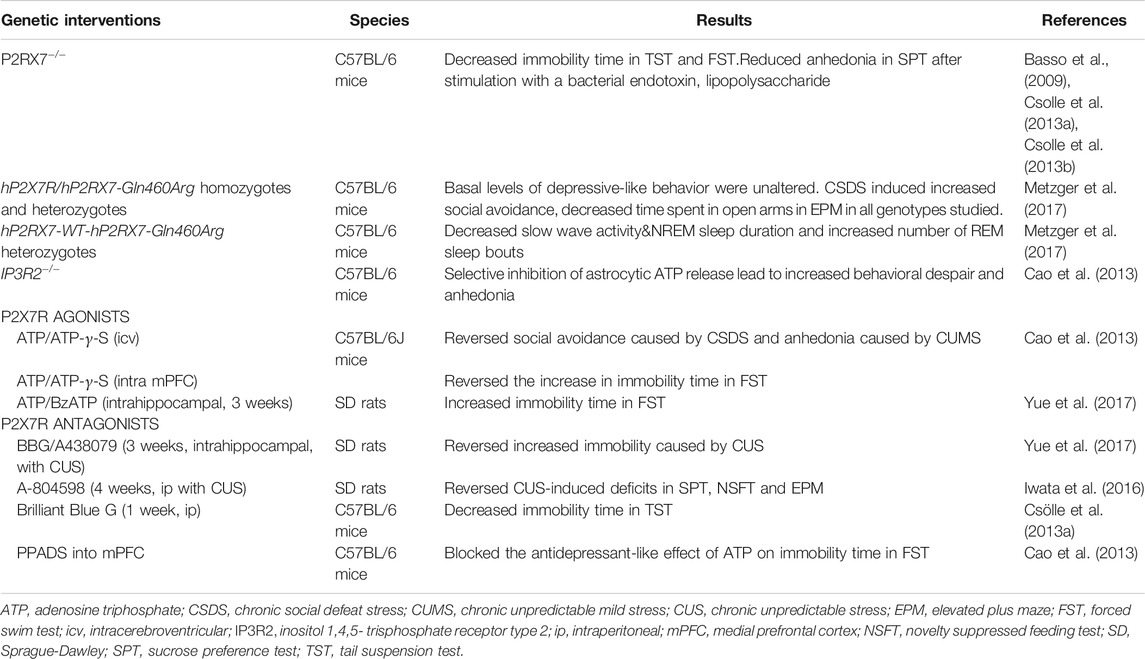
TABLE 1. Behavioral effects of interventions to P2X7R expression and functions.
However, the story is more complex: Intra-PFC, but not intrahippocampal injections of ATP or ATP-γ-S reversed the depression-like behavior (Cao et al., 2013); whereas chronic (3weeks) intrahippocampal injections of ATP or BzATP, a P2XR agonist, induced depression-like behavior to a similar extent as chronic unpredictable stress (CUS) (Table 1). Long-term blockade of P2X7R for 3–4 weeks by systemic (intraperitoneal, i.p.) or intrahippocampal injections of selective P2XR7 antagonists Brilliant Blue G (BBG), A438079, A-804598, reversed depressogenic behavior, specifically behavioral despair, anhedonia and anxiety-like behavior caused by CUMS in rats (Iwata et al., 2016; Yue et al., 2017) (Table 1). Subacute (1 week) but not acute systemic treatment of mice with BBG also caused a reduction in behavioral despair (Csolle et al., 2013a) (Table 1). Further complicating understanding the role of P2X7R in depression, preinfusions of a non-selective P2XR antagonist, pyridoxal phosphate-6-azophenyl-2′-4′-disulphonic acid, and P2X2R shRNA into the mPFC blocked the antidepressant-like effect of ATP (Cao et al., 2013) (Table 1). These findings altogether suggest that; 1) ATP and P2XR signaling exhibit an inverted U-shaped relationship in mood regulation; i.e., both very low and very high doses of ATP and P2XR signaling are involved in the development of depression-like behavior whereas moderate doses are necessary to maintain euthymia (Figure 3); 2) effects of ATP and P2XR signaling are region-specific.
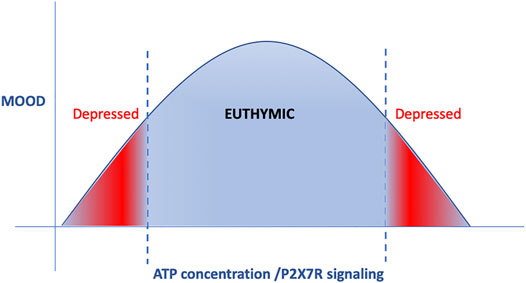
FIGURE 3. ATP and P2XR signaling displays a reverse U-curve. Both very low and very high doses of ATP and P2XR signaling are involved in the development of depression-like behavior, whereas moderate doses are necessary to maintain euthymic mood.
Genetic tools, in which control of the “dose effect” is far limited compared to pharmacological approaches, have confirmed the important role of P2XR signaling in the pathophysiology of depression but the net consequnce of their effect was also model-dependent. Studies using P2RX7−/− KO mice reported a decrease in behavioral despair, i.e. an antidepressant effect (Basso et al., 2009; Csolle et al., 2013a; Csolle et al., 2013b) (Table 1). In human genetic studies, polymorphisms of P2RX7 the gene encoding P2X7R has been reported to be associated with increased risk/severity of mood disorders (Lucae et al., 2006; Mcquillin et al., 2009; Halmai et al., 2013; Vereczkei et al., 2019). As a more precise model of the human condition, a humanized transgenic mouse line was generated in which exon 2–13 of murine P2X7R was substituted by either human wild-type (WT) hP2X7R or hP2X7R-Gln460Arg variant. Intriguingly, the ion channel function of hP2X7R-Gln460Arg variant was not different than that of the WT, but it was impaired when hP2X7R-Gln460Arg variant was co-expressed with WT P2X7R (Aprile-Garcia et al., 2016). Contrary to P2x7R KO mice, mice homozygous or heterozygous for hP2X7R-Gln460Arg allele displayed no change in depression-like behavior at basal conditions, but they showed increased depression- and anxiety-like behavior following social defeat stress (Metzger et al., 2017) (Table 1). These findings indicate that P2X7R polymorphism creates vulnerability to stress and depression and supports the involvement of gene X environment interactions in the development of mood disorders. Interestingly sleep quality was disturbed in heterozygous mice (WT hP2X7R/hP2X7R-Gln460Arg), but not in homozygous mice (hP2X7R-Gln460Arg/hP2X7R-Gln460Arg) (Metzger et al., 2017). Specifically, slow wave activity and NREM sleep duration were reduced and the number of REM sleep bouts was increased in heterozygous mice (Metzger et al., 2017) (Table 1), which is similar to sleep disturbances seen in depressive patients (Nutt et al., 2008). These findings have led to a randomized, placebo-controlled, double blinded clinical trial in order to evaluate the antidepressant efficacy of P2X7R antagonists in the treatment of major depression (ClinicalTrials.gov Identifier: NCT04116606).
Potential Mechanisms Underlying P2X7R’s Effects on Mood Regulation
P2X7R mediates several physiological neural functions, disturbances of which may be involved in the pathophysiology of depression:
1. P2X7R increases the release of neurotransmitters noradrenaline (NA), serotonin (5-HT), glutamate and GABA (Sperlagh et al., 2002; Papp et al., 2004; Alloisio et al., 2008; Marcoli et al., 2008; Goloncser et al., 2017). These neurotransmitters long implicated in the pathophysiology of depression also modulate astrocytic ATP release by altering intracellular Ca+2 levels (Hirschfeld, 2000; Arcuino et al., 2002; Luscher et al., 2011; Marpegan et al., 2011; Duman et al., 2019; Fogaca and Duman, 2019). Indeed, selective inhibition of ATP release from astrocytes by genetic knockout of IP3R2 mediating intra-astrocytic Ca+2 rise induces depression-like behavior (Cao et al., 2013), which may be related with the resulting dysregulation of the release of neurotransmitters.
2. P2X7R regulates synaptic plasticity. Chronic stress has been reported to decrease synaptic plasticity, which is thought to be involved in depression-like phenotype by limiting adaptive responses. In P2RX7−/− KO mice, whole genome microarray analysis disclosed changes in expression of genes involved in synaptic plasticity (Csolle et al., 2013a). P2X7R ligand ATP has been shown to alter synaptic plasticity by several mechanisms, including glutamate release and regulation of NMDA receptor expression and function (Pankratov et al., 2002; Sperlagh et al., 2002; Csolle et al., 2013b; Guo et al., 2015; Otrokocsi et al., 2017). ATP and BzATP application evoked glutamate efflux in hippocampal slices obtained from WT mice, which was abolished in hippocampal slices of P2RX7−/− mice (Sperlagh et al., 2002; Csolle et al., 2013b). There is an ongoing debate on the presence of P2X7R in adult neurons (Illes et al., 2017; Miras-Portugal et al., 2017). P2X7R was proposed to modulate presynaptic release of glutamate, however, recent transcriptomic studies report that neurons isolated from adult rodents do not express P2X7R unlike the in vitro preparations prepared from young or embryonic animals (Illes et al., 2017; Kaczmarek-Hajek et al., 2018). Therefore, the glutamate efflux induced by ATP and P2X7R agonists in adult animals can also be attributed to the astrocytic release (Parpura et al., 1994; Savtchouk and Volterra, 2018). The basal and stress-induced expression of the NR2B subunit of NMDA receptor was found to be upregulated in P2RX7−/− mice both in vitro and in vivo (Csolle et al., 2013b; Otrokocsi et al., 2017), possibly as a compensatory response to decreased glutamate release. As P2X7R ion channel has considerable Ca+2 permeability, when expressed at the synapse, P2X7R can modulate synaptic plasticity. Indeed, in hippocampal slices prepared from 3-week old rats, ATP blocked induction of long-term potentiation (LTP) with low frequency stimulation at the CA3-CA1 synapse by inhibiting NMDA-mediated currents through Ca+2-dependent inactivation of NMDA receptors. At higher frequencies of stimulation, owing to desensitization of P2X receptors, ATP loses its inhibitory effect on LTP (Pankratov et al., 2002). In accordance with these observations inhibition or desensitization of P2XR with pyridoxal phosphate-6-azophenyl-2′-4′-disulphonic acid or nonhydrolyzable ATP analog α,β-methylene ATP facilitated LTP at the CA3-CA1 synapse (Pankratov et al., 2002).
3. P2X7R can induce neuroinflammation. Binding of ATP to P2X7R trigger NLRP3 inflammasome cascade resulting in the activation of caspase-1, which converts pro-IL-1ß and pro-IL-18 to their active forms: IL-1ß and IL18 (Guo et al., 2015). As mentioned above, psychological stressors increase extracellular ATP levels, which reportedly lead to activation of caspase-1 and IL-1ß release by binding to P2X7R on microglia (Iwata et al., 2016; Yue et al., 2017). This may be related to the increased peripheral levels of cytokines, acute phase proteins and chemokines detected in depressed patients (Levine et al., 1999; Lanquillon et al., 2000; Alesci et al., 2005; Pace et al., 2006; Dowlati et al., 2010; Milenkovic et al., 2019). Indeed, increased microglial activity in the prefrontal cortex, anterior cingulate cortex and insula of major depression patients was visualized by positron emission tomography (Setiawan et al., 2015).
The role of microglia in sculpturing synapses is well documented. Microglial processes in close contact with dendritic spines can function in both the elimination and formation of spines, suggesting that they play important roles in synaptic plasticity (Paolicelli et al., 2011; Zhan et al., 2014; Miyamoto et al., 2016; Weinhard et al., 2018; Cangalaya et al., 2020). Stress is known to cause a decrease in dendritic length and branching in apical dendrites of layer II/III and layer V pyramidal neurons of the medial prefrontal cortex as well as apical dendrites of CA3 pyramidal neurons (Mcewen, 1999; Mcewen, 2001; Cook and Wellman, 2004; Liston et al., 2006; Radley et al., 2006; Czeh et al., 2008; Goldwater et al., 2009). On the other hand, chronic stress increases the length of apical dendrites in the ventral orbital subregion of the orbitofrontal cortex as well as the dendritic arborization and spine density in the basolateral amygdala spiny neurons (Vyas et al., 2003; Dias-Ferreira et al., 2009). Recent work showed that microglia activated after chronic unpredictable stress were involved in pruning of spines both in the Layer-1 of mPFC and hippocampus (Milior et al., 2016; Wohleb et al., 2018). Interestingly, deletion of P2X7R gene inhibited the decrease in synapse numbers in dorsal dentate gyrus (DG) following inescapable footshock, a rodent model of depression (Otrokocsi et al., 2017), indicating a possible role of P2X7R in pruning of synapses, which needs to be addressed in future studies.
In conclusion, as with ATP release, ATP-P2X-mediated microglial activation may exert depressant or anti-depressant effect possibly depending on the intensity of the stimulus and brain region, which may yield contrasting results in in vitro and in vivo experiments. Further studies are needed to document the prevailing mechanisms involved under in situ conditions as well as in patients.
Hyperpolarization-Activated Cyclic Nucleotide-gated Channels and M-type K+ (KCNQ) Channels
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are nonselective cation channels located mainly at dendrites, but also present at axons and the neuronal soma. They open at membrane voltages more negative than −40 mV and when HCN channels open, membrane resistance decreases (Biel et al., 2009; Kase and Imoto, 2012). Thus, HCN channels can counteract both membrane hyperpolarization and depolarization allowing both neuronal excitation as well as inhibition (Kase and Imoto, 2012; Benarroch, 2013). Four subtypes of HCN’s have been identified: HCN1 to HCN4. HCN1 and HCN2 were reported to be highly expressed in brain regions known to be involved in the pathophysiology of major depression, namely prefrontal cortex, hippocampus, ventral tegmental area (VTA) and nucleus accumbens (NAc) (Notomi and Shigemoto, 2004).
HCN’s interact with cyclic adenosine monophosphate (cAMP), which modulates voltage dependence of their activation kinetics and facilitate their opening. Signaling through several neuromodulators like noradrenaline and dopamine, whose dysfunction may contribute to the pathophysiology of depression, alter cAMP levels in the dendrites and thus HCN channel functioning, especially of HCN 2, the subtype most strongly modulated by cAMP (Biel et al., 2009). HCN also interacts with tetratricopeptide repeat-containing Rab8b interacting protein (TRIP8b), which regulates trafficking of HCN to dendrites (Biel et al., 2009).
Global knockout of HCN1, HCN2, or TRIP8b genes (leading to a decrease in both HCN1 and HCN2 protein levels) in mice resulted in a reduction in behavioral despair, an antidepressant-like effect (Lewis et al., 2011) (Table 2). Studies showed that HCNs regulate neuronal excitability and plasticity in response to stress, depending on the brain region and cell type that they are expressed in, as well as the duration and type of the stressor. For example, VTA dopamine (DA) neurons in the brain reward circuit has important role in regulation of the stress response. Evidence indicates that there are disturbances in the brain reward circuitry in major depression, which is associated with anhedonia, one of the 2 cardinal symptoms of major depression (Nestler and Carlezon, 2006; Russo and Nestler, 2013; Nestler, 2015; Hoflich et al., 2019). Recent evidence showed that the social stress susceptible mice display increased VTA DA neuronal firing and increase in HCN mediated I(h) currents (Cao et al., 2010; Chaudhury et al., 2013), whereas mice exposed to chronic unpredictable mild stress show a reduction in I(h) and DA neuronal firing (Zhong et al., 2018) (Table 2). In NAc, one of the major targets of VTA DA neurons, both HCN 2 expression and function [I(h)] were reduced in cholinergic interneurons (ChIN) in two different mice depression models, namely chronic social defeat stress and p11 knockout mice (Cheng et al., 2019) (Figure 4). Because decreased activity of ChIN in NAc was found to be related to depressive behavior, the possible involvement of HCN2 channels in regulating ChIN activity was studied by pharmacological and genetic interventions. Decreased tonic firing of ChIN by pharmacological blockade of HCN2 channels (ZD7288) suggest that dysfunction of HCN channels participates in the diminished ChIN firing rate observed in depressive mice (Table 2; Figure 4). Intriguingly, selective overexpression of HCN2 in both NAc ChIN and VTA DA neurons resulted in antidepressant-like effects (Friedman et al., 2014; Zhong et al., 2018; Cheng et al., 2019) (Table 2; Figure 4). Selective knockdown of HCN2 in VTA DA neurons by shRNA increased anxiety-like and depression-like behaviors (Zhong et al., 2018). Intriguingly, knockdown of HCN1 in CA1 region of dorsal hippocampus resulted in anxiolytic and antidepressant effects (Kim et al., 2012) (Table 2). Whether this effect was associated with the difference in HCN subtype or the brain region studied warrants further research investigating the role of HCN channels in other brain regions relevant to depression.
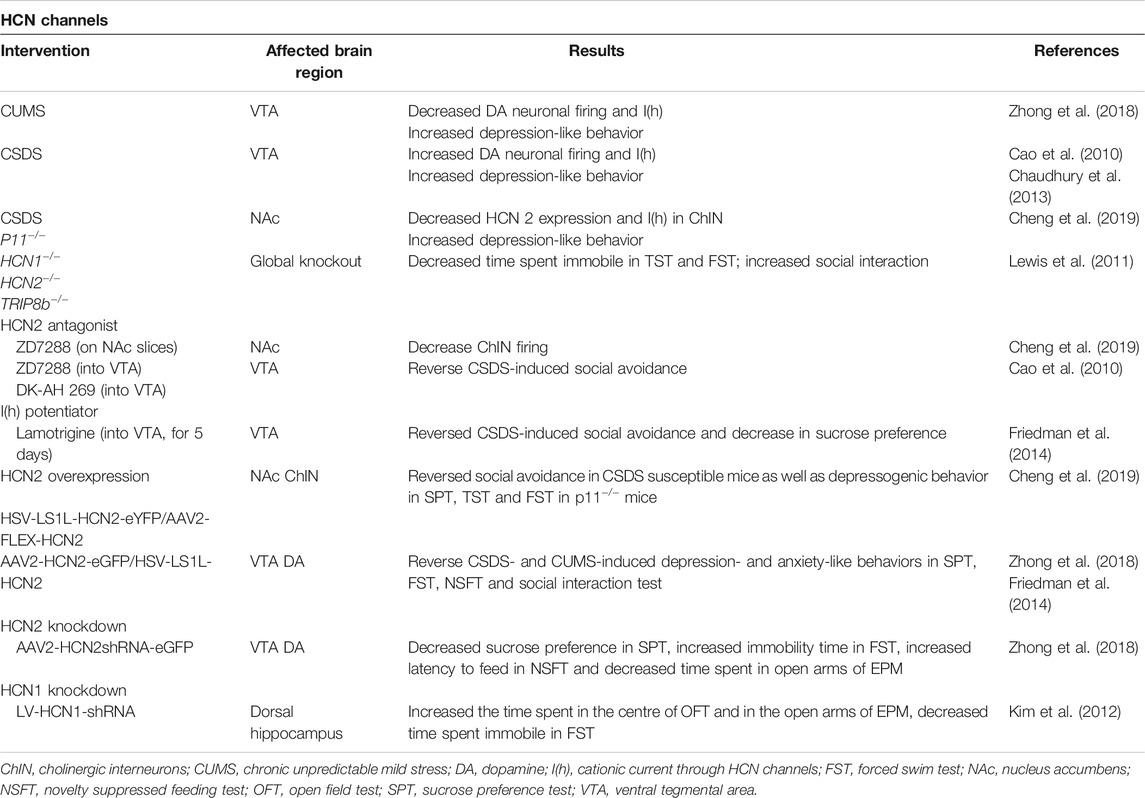
TABLE 2. Effects of modulating HCN2 expression and function on depression-like behavior.
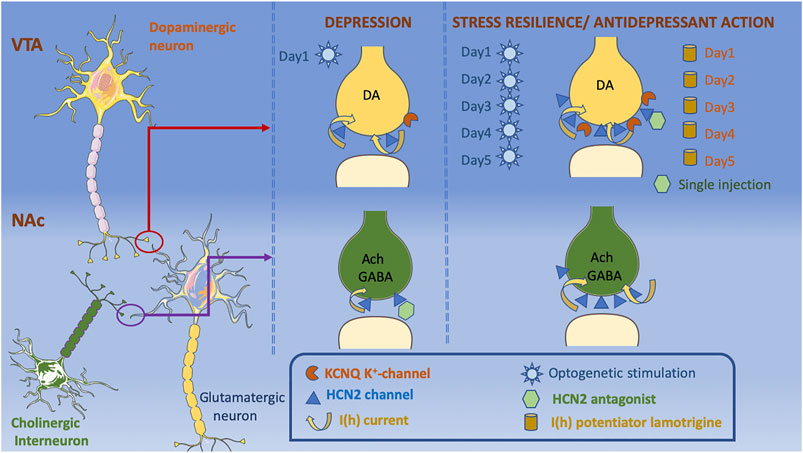
FIGURE 4. The decrease in the expression of HCN2 channels in NAc on both dopaminergic terminals projecting from VTA and local ChIN terminals is depressogenic. Decrease in I(h) currents in ChIN terminals is associated with depression-like phenotype, whereas an increase in I(h) currents on the VTA dopaminergic terminals is observed in both stress susceptible and stress resilient mice. K+ channels are increased only in the stress resilient mice counterbalancing the effects of increased I(h) currents. Application of an HCN2 antagonist into NAc causes a depressogenic phenotype, whereas it has antidepressant effects when applied into VTA. Lamotrigine administration for 5-days also reduce depressogenic behaviors. Acute optogenetic stimulation of VTA dopaminergic neurons increase depression-like behaviors, whereas chronic optogenetic stimulation of VTA dopaminergic neurons shows antidepressant effects. Cellular templates are copied from Servier Medical Art (smart.servier.com).
HCN2 channels’ ability to modulate VTA DA neurons depend on the duration of neuronal excitation: Acute phasic optogenetic stimulation of VTA DA neurons projecting to NAc (20 Hz, 10 min) during a subthreshold social defeat stress increases spontaneous and evoked activity that lasts for 8–12 h after stimulation in parallel with increased depression-like behaviors in social interaction and sucrose preference tests (Chaudhury et al., 2013) (Figure 4). Repetitive optogenetic stimulation of VTA DA neurons for 5 days (20 Hz, 20 min), on the other hand, reduced DA neuron firing rate and displayed antidepressant-like effects in social interaction, forced swim and sucrose preference tests (Friedman et al., 2014) (Figure 4). In line with these observations, both acute injections of HCN channel blockers (ZD7288 or DK-AH 269) into VTA (Cao et al., 2010) and repeated infusions of the I(h) potentiator, lamotrigine into VTA for 5 days showed antidepressant-like effects (Friedman et al., 2014) (Table 2; Figure 4). The group treated with repeated lamotrigine displayed normalization of the hyperactivity of VTA DA neurons (Friedman et al., 2014). A more detailed look at the mechanisms underlying these opposite effects of acute vs repeated manipulations of HCN channels and DA neuron activity showed that I(h) currents were also amplified in social stress resilient mice whose DA neuronal firing rate was comparable to that of controls (Friedman et al., 2014) (Figure 4). Recordings in brain slices of VTA DA neuron demonstrated an increase in K+ currents and reduction in firing rate in resilient mice (Friedman et al., 2014). Altogether, these findings suggest that increase in K+ currents is a homeostatic plasticity mechanism that stabilizes VTA DA neuronal activity (Figure 4). In support of this proposition, I(h) was found to have a depolarizing effect on the peak voltage of weak EPSPs, whereas it exerted a hyperpolarizing effect on the peak voltage of stronger, but still subthreshold, EPSPs (George et al., 2009). George et al. (2009) showed that blockade of the delayed-rectifier M-type K(+) current resulted in shift of dual I(h) influence on EPSPs to only an excitatory effect, suggesting an interaction between M-type K(+) current and I(h) (George et al., 2009).
Intriguingly, M-type K+ (KCNQ) channels have been linked to depression-like behavior and identified as a novel antidepressant drug target (Friedman et al., 2016; Li et al., 2017; Tan et al., 2020; Ren et al., 2021) (Figure 4). RNA sequencing analysis revealed that genes encoding K+ channels are differentially expressed in chronically stressed rats (Ren et al., 2021). Transcript levels of KCNQ4 channels that are selectively expressed in VTA DA neurons were negatively correlated with firing rate of VTA DA neurons (Li et al., 2017). Supporting the involvement of KCNQ channels in the pathophysiology of depression, pharmacological activators of KCNQ channels normalized the hyperactivity of VTA DA neurons and alleviated stress-induced depression-like behaviors (Friedman et al., 2016; Li et al., 2017; Ren et al., 2021). A recent open-label clinical study conducted in 18 major depression subjects reported that 10 weeks of treatment with KCNQ channel opener, retigabine, significantly reduced depressive symptoms and decreased functional connectivity between ventral caudate and mid-cingulate and posterior cingulate cortices (Tan et al., 2020).
In contrast to well documented mutations of HCN and KCNQ channel genes in a spectrum of epileptic diseases (Nappi et al., 2020; Rivolta et al., 2020), genetic studies in depressed patients have not conclusively acknowledged a convincing association between single nucleotide polymorphisms (SNPs) in HCN or KCNQ channel genes and depression. Polymorphisms in HCN4 gene were reported to be associated with a broad spectrum of mood disorders including major depression, bipolar affective disorder and obsessive compulsive disorder (Kelmendi et al., 2011). A study with a larger sample failed to replicate these findings and reported no association between any HCN channel genes studied (HCN1-4) and depression (Mcintosh et al., 2012). Despite the lack of evidence from human genetic studies supporting the involvement of SNPs in HCN or KCNQ channel genes, the above summarized findings in animals suggest that both HCN channels and KCNQ channels are important players of the physiological adaptations to stress, perturbations of which can lead to depression.
Voltage-Gated Calcium Channels and Two-Pore Domain K+ Channels
Voltage-gated calcium channels and two-pore domain K+ channels, the role of which are discussed in the context of migraine in the previous section are also involved in the pathophysiology of mood disorders: Genetic variations in CACNA1C gene, which encodes for the alpha subunit of L-type calcium channels (Cav1.2) as well as CACNA1E, which encodes for the alpha 1E subunit of R-type calcium channel (CaV2.3 channel) were reported in mood disorders (Ferreira et al., 2008; Psychiatric, 2011; Nurnberger et al., 2014; Howard et al., 2019). Animal studies have confirmed the involvement of CACNA1C in the pathophysiolgy of depression: Following CUMS, Cav1.2 levels were found to be increased in the PFC (Bavley et al., 2017). The depression-like behaviors were decreased in both CACNA1C+/− heterozygous and forebrain specific CACNA1C KO mice at baseline and after chronic stress (Dao et al., 2010; Bavley et al., 2017; Kabir et al., 2017; Dedic et al., 2018). In the NAc, on the other hand, CSDS decreased Cav1.2 channel expression in stress susceptible mice and selective KO of CACNA1C in the NAc resulted in an increase in susceptibility to subthreshold social defeat stress as well as a reduction in both the time spent sniffing female urine soaked cotton tips and the time spent in open arms of elevated plus maze (Terrillion et al., 2017). In line with these observations, alterations in calcium signaling has long been implicated in the pathophysiology of mood disorders and a recent metaanalysis of 21 studies reported that both basal and stimulated Ca+2 levels were found to be higher in platelets and lymphocytes of bipolar affective disorder patients (Harrison et al., 2019). The Ca+2 channel blocking properties of mood stabilizer drugs, lithium, valproic acid, carbamazepine and lamotrigine as well as efficacy of verapamil, a Ca+2 channel blocker as a mood stabilizing agent support the role of Ca+2 in the pathophysiology of mood disorders and point to the promising potential of BBB-permeable Ca+2 channel blockers in the treatment of mood disordes (for detailed reviews please see (Cipriani et al., 2016; Dubovsky, 2019; Harrison et al., 2020). Intriguingly, mood stabilizers valproic acid, lamotrigine and carbamazepine also block voltage-gated Na+ channels, raising the possibility that their actions might, in part, be attributed to Na+ channel blockade (Stahl, 2004). However voltage-gated Na+ channels have not been implicated so far in the pathophysiology of depression (Mantegazza et al., 2021).
Among two-pore domain K+ channels, the involvement of TWIK-related K+ channel-1 (TREK-1) in depression pathophysiology is the most well documented (for detailed reviews see (Borsotto et al., 2015; Djillani et al., 2019)). They are involved in the regulation of plasma membrane potential and excitability similar to HCN channels (Lesage, 2003; Talley et al., 2003). SNPs in kcnk2, gene encoding TREK-1 channel, were reported in major depression patients, and some SNPs were found to be associated with antidepressant response and resistance to treatment (Perlis et al., 2008; Liou et al., 2009). TREK-1 inhibitors reversed behavioral despair and decreased sucrose preference induced by CUMS in rats when given intraperitoneously (Ye et al., 2015; Djillani et al., 2017). Deletion of TREK-1 channels resulted in depression-resistant phenotype (Heurteaux et al., 2006). Interestingly selective serotonin uptake inhibitors, fluoxetine, paroxetine and citalopram were reported to block both TREK-1 and TREK-2 mediated currents in vitro (Kennard et al., 2005; Kim et al., 2017). Mood stabilizers, lithium, valproic acid and carbamazepine, on the other hand, increased TREK-1 mediated currents without affecting TREK-2 mediated currents (Kim et al., 2017). The therapeutic use of SSRIs in the treatment of both depression and migraine patients may be in part related to their effects on TREK-mediated currents.
In conclusion, mutations detected in familial cases of migraine and polymorphisms creating susceptibility to depression as well as genetic manipulations in animal models of depression have provided novel insight to the pathophysiology of these disorders. Since subtle changes in, for instance, ATP release or K+ channel activity may lead to variable consequences within a microcircuit, and hence, in network activity depending on the microcircuit structure and brain region, experimental studies (especially those using isolated preparations) have yielded to varying, sometimes contradictory, results. Therefore, identification of overlapping genetic variants in non-familial forms of migraine and depression can provide further insight to understand the phenotypic traits and role of ion channels, E:I balance, metabolic coupling of astrocytic glycogen to glutamatergic synaptic activity and neuroinflammation in migraine as well as depression, both of which display significant heritability and comorbidity. These findings can also help development of novel therapies for both conditions.
Author Contributions
TD wrote the sections regarding ion channel dysfunctions in migraine (Lessons Learned From Familial Migraine Mutations, Non-Familial Common Migraine, Initiation of Inflammatory Signaling That Causes Headache, and Master Switch of Inflammatory Signaling: Pannexin Channels). EE-K wrote the sections regarding ion channel dysfunctions in depression (P2X7 Receptor, Potential Mechanisms Underlying P2X7R’s Effects on Mood Regulation?) Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and M-type K+ (KCNQ) channels and Voltage-gated calcium channels and two-pore domain K+ channels. Figures 3 and 4 were prepared by EE-K. Figure 1 was prepared by Zeynep Kaya and Figure 2 was adapted from Karatas et al. (2013)
Funding
TD's research is supported by Turkish Academy of Sciences and TUBITAK (120C122).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Afridi, R., and Suk, K. (2021). Neuroinflammatory Basis of Depression: Learning from Experimental Models. Front. Cell Neurosci. 15, 691067. doi:10.3389/fncel.2021.691067
Albrecht, D. S., Mainero, C., Ichijo, E., Ward, N., Granziera, C., Zürcher, N. R., et al. (2019). Imaging of Neuroinflammation in Migraine With Aura: A [11C]PBR28 PET/MRI Study. Neurology. 92, e2038–e2050. doi:10.1212/WNL.0000000000007371
Alesci, S., Martinez, P. E., Kelkar, S., Ilias, I., Ronsaville, D. S., Listwak, S. J., et al. (2005). Major Depression Is Associated With Significant Diurnal Elevations in Plasma Interleukin-6 Levels, a Shift of its Circadian Rhythm, and Loss of Physiological Complexity in its Secretion: Clinical Implications. J. Clin. Endocrinol. Metab. 90, 2522–2530. doi:10.1210/jc.2004-1667
Alloisio, S., Cervetto, C., Passalacqua, M., Barbieri, R., Maura, G., Nobile, M., et al. (2008). Functional Evidence for Presynaptic P2X7 Receptors in Adult Rat Cerebrocortical Nerve Terminals. FEBS Lett. 582, 3948–3953. doi:10.1016/j.febslet.2008.10.041
Amiri, S., Behnezhad, S., and Azad, E. (2019). Migraine Headache and Depression in Adults: a Systematic Review and Meta-Analysis. Neuropsychiatr. 33, 131–140. doi:10.1007/s40211-018-0299-5
Aprile-Garcia, F., Metzger, M. W., Paez-Pereda, M., Stadler, H., Acuna, M., Liberman, A. C., et al. (2016). Co-Expression of Wild-Type P2X7R With Gln460Arg Variant Alters Receptor Function. PLoS One. 11, e0151862. doi:10.1371/journal.pone.0151862
Arcuino, G., Lin, J. H., Takano, T., Liu, C., Jiang, L., Gao, Q., et al. (2002). Intercellular Calcium Signaling Mediated by Point-Source Burst Release of ATP. Proc. Natl. Acad. Sci. U S A. 99, 9840–9845. doi:10.1073/pnas.152588599
Auffenberg, E., Hedrich, U. B., Barbieri, R., Miely, D., Groschup, B., Wuttke, T. V., et al. (2021). Hyperexcitable Interneurons Trigger Cortical Spreading Depression in an Scn1a Migraine Model. J. Clin. Invest. 21, e142202. doi:10.1172/jci142202
Basso, A. M., Bratcher, N. A., Harris, R. R., Jarvis, M. F., Decker, M. W., and Rueter, L. E. (2009). Behavioral Profile of P2X7 Receptor Knockout Mice in Animal Models of Depression and Anxiety: Relevance for Neuropsychiatric Disorders. Behav. Brain Res. 198, 83–90. doi:10.1016/j.bbr.2008.10.018
Bavley, C. C., Fischer, D. K., Rizzo, B. K., and Rajadhyaksha, A. M. (2017). Cav1.2 Channels Mediate Persistent Chronic Stress-Induced Behavioral Deficits That Are Associated With Prefrontal Cortex Activation of the p25/Cdk5-Glucocorticoid Receptor Pathway. Neurobiol. Stress. 7, 27–37. doi:10.1016/j.ynstr.2017.02.004
Benarroch, E. E. (2013). HCN Channels: Function and Clinical Implications. Neurology. 80, 304–310. doi:10.1212/WNL.0b013e31827dec42
Bezzi, P., and Volterra, A. (2001). A Neuron-Glia Signalling Network in the Active Brain. Curr. Opin. Neurobiol. 11, 387–394. doi:10.1016/s0959-4388(00)00223-3
Bhaskaracharya, A., Phuong, D. U., Jalilian, I., Spildrejorde, M., Skarratt, K. K., Fuller, S. J., et al. (2014). Probenecid Blocks Human P2X7 Receptor-Induced Dye Uptake via a Pannexin-1 Independent Mechanism. Plos One. 9, e93058. doi:10.1371/journal.pone.0093058
Biel, M., Wahl-Schott, C., Michalakis, S., and Zong, X. (2009). Hyperpolarization-Activated Cation Channels: From Genes to Function. Physiol. Rev. 89, 847–885. doi:10.1152/physrev.00029.2008
Borsotto, M., Veyssiere, J., Moha Ou Maati, H., Devader, C., Mazella, J., and Heurteaux, C. (2015). Targeting Two-Pore Domain K(+) Channels TREK-1 and TASK-3 for the Treatment of Depression: a New Therapeutic Concept. Br. J. Pharmacol. 172, 771–784. doi:10.1111/bph.12953
Brennan, K. C., Bates, E. A., Shapiro, R. E., Zyuzin, J., Hallows, W. C., Huang, Y., et al. (2013). Casein Kinase Iδ Mutations in Familial Migraine and Advanced Sleep Phase. Sci. Transl Med. 5 (183ra56), 1–11. doi:10.1126/scitranslmed.3005784
Brennan, K. C., and Pietrobon, D. (2018). A Systems Neuroscience Approach to Migraine. Neuron. 97, 1004–1021. doi:10.1016/j.neuron.2018.01.029
Bromet, E., Andrade, L. H., Hwang, I., Sampson, N. A., Alonso, J., De Girolamo, G., et al. (2011). Cross-National Epidemiology of DSM-IV Major Depressive Episode. BMC Med. 9, 90. doi:10.1186/1741-7015-9-90
Bu, F., Nie, L., Quinn, J. P., and Wang, M. (2020). Sarcoma Family Kinase-Dependent Pannexin-1 Activation After Cortical Spreading Depression Is Mediated by NR2A-Containing Receptors. Int. J. Mol. Sci. 21, 1269. doi:10.3390/ijms21041269
Burnstock, G. (2007). Physiology and Pathophysiology of Purinergic Neurotransmission. Physiol. Rev. 87, 659–797. doi:10.1152/physrev.00043.2006
Cangalaya, C., Stoyanov, S., Fischer, K. D., and Dityatev, A. (2020). Light-Induced Engagement of Microglia to Focally Remodel Synapses in the Adult Brain. Elife. 9, e58435. doi:10.7554/elife.58435
Cao, J. L., Covington, H. E., Friedman, A. K., Wilkinson, M. B., Walsh, J. J., Cooper, D. C., et al. (2010). Mesolimbic Dopamine Neurons in the Brain Reward Circuit Mediate Susceptibility to Social Defeat and Antidepressant Action. J. Neurosci. 30, 16453–16458. doi:10.1523/JNEUROSCI.3177-10.2010
Cao, X., Li, L. P., Wang, Q., Wu, Q., Hu, H. H., Zhang, M., et al. (2013). Astrocyte-Derived ATP Modulates Depressive-Like Behaviors. Nat. Med. 19, 773–777. doi:10.1038/nm.3162
Cao, Y., Welch, K. M., Aurora, S., and Vikingstad, E. M. (1999). Functional MRI-BOLD of Visually Triggered Headache in Patients With Migraine. Arch. Neurol. 56, 548–554. doi:10.1001/archneur.56.5.548
Capuani, C., Melone, M., Tottene, A., Bragina, L., Crivellaro, G., Santello, M., et al. (2016). Defective Glutamate and K+ Clearance by Cortical Astrocytes in Familial Hemiplegic Migraine Type 2. EMBO Mol. Med. 8, 967–986. doi:10.15252/emmm.201505944
Chanda, M. L., Tuttle, A. H., Baran, I., Atlin, C., Guindi, D., Hathaway, G., et al. (2013). Behavioral Evidence for Photophobia and Stress-Related Ipsilateral Head Pain in Transgenic Cacna1a Mutant Mice. Pain. 154, 1254–1262. doi:10.1016/j.pain.2013.03.038
Chaudhury, D., Walsh, J. J., Friedman, A. K., Juarez, B., Ku, S. M., Koo, J. W., et al. (2013). Rapid Regulation of Depression-Related Behaviours by Control of Midbrain Dopamine Neurons. Nature 493, 532–536. doi:10.1038/nature11713
Chen, S. P., Qin, T., Seidel, J. L., Zheng, Y., Eikermann, M., Ferrari, M. D., et al. (2017). Inhibition of the P2X7-PANX1 Complex Suppresses Spreading Depolarization and Neuroinflammation. Brain. 140, 1643–1656. doi:10.1093/brain/awx085
Cheng, J., Umschweif, G., Leung, J., Sagi, Y., and Greengard, P. (2019). HCN2 Channels in Cholinergic Interneurons of Nucleus Accumbens Shell Regulate Depressive Behaviors. Neuron. 101, 662–e5. doi:10.1016/j.neuron.2018.12.018
Chever, O., Zerimech, S., Scalmani, P., Lemaire, L., Pizzamiglio, L., Loucif, A., et al. (2021). Initiation of Migraine-Related Cortical Spreading Depolarization by Hyperactivity of GABAergic Neurons and NaV1.1 Channels. J. Clin. Invest. 7, 142203. doi:10.1172/jci142203
Cholet, N., Pellerin, L., Magistretti, P. J., and Hamel, E. (2002). Similar Perisynaptic Glial Localization for the Na+,K+-ATPase Alpha 2 Subunit and the Glutamate Transporters GLAST and GLT-1 in the Rat Somatosensory Cortex. Cereb. Cortex. 12, 515–525. doi:10.1093/cercor/12.5.515
Cholet, N., Pellerin, L., Welker, E., Lacombe, P., Seylaz, J., Magistretti, P., et al. (2001). Local Injection of Antisense Oligonucleotides Targeted to the Glial Glutamate Transporter GLAST Decreases the Metabolic Response to Somatosensory Activation. J. Cereb. Blood Flow Metab. 21, 404–412. doi:10.1097/00004647-200104000-00009
Cipriani, A., Saunders, K., Attenburrow, M. J., Stefaniak, J., Panchal, P., Stockton, S., et al. (2016). A Systematic Review of Calcium Channel Antagonists in Bipolar Disorder and Some Considerations for Their Future Development. Mol. Psychiatry. 21, 1324–1332. doi:10.1038/mp.2016.86
Cook, S. C., and Wellman, C. L. (2004). Chronic Stress Alters Dendritic Morphology in Rat Medial Prefrontal Cortex. J. Neurobiol. 60, 236–248. doi:10.1002/neu.20025
Costa, C., Prontera, P., Sarchielli, P., Tonelli, A., Bassi, M. T., Cupini, L. M., et al. (2014). A Novel ATP1A2 Gene Mutation in Familial Hemiplegic Migraine and Epilepsy. Cephalalgia. 34, 68–72. doi:10.1177/0333102413498941
Csölle, C., Andó, R. D., Kittel, Á., Gölöncsér, F., Baranyi, M., Soproni, K., et al. (2013a). The Absence of P2X7 Receptors (P2rx7) on Non-Haematopoietic Cells Leads to Selective Alteration in Mood-Related Behaviour With Dysregulated Gene Expression and Stress Reactivity in Mice. Int. J. Neuropsychopharmacol. 16, 213–233. doi:10.1017/S1461145711001933
Csölle, C., Baranyi, M., Zsilla, G., Kittel, A., Gölöncsér, F., Illes, P., et al. (2013b). Neurochemical Changes in the Mouse Hippocampus Underlying the Antidepressant Effect of Genetic Deletion of P2X7 Receptors. PLoS One. 8, e66547. doi:10.1371/journal.pone.0066547
Czéh, B., Perez-Cruz, C., Fuchs, E., and Flügge, G. (2008). Chronic Stress-Induced Cellular Changes in the Medial Prefrontal Cortex and Their Potential Clinical Implications: Does Hemisphere Location Matter? Behav. Brain Res. 190, 1–13. doi:10.1016/j.bbr.2008.02.031
Dahl, G. (2018). The Pannexin1 Membrane Channel: Distinct Conformations and Functions. FEBS Lett. 592, 3201–3209. doi:10.1002/1873-3468.13115
Dalkara, T., and Kiliç, K. (2013). How Does Fasting Trigger Migraine? A Hypothesis. Curr. Pain Headache Rep. 17, 368. doi:10.1007/s11916-013-0368-1
Dalkara, T., and Moskowitz, M. A. (2017). From Cortical Spreading Depression to Trigeminovascular Activation in Migraine Neurobiological Basis of Migraine. Hoboken, New Jersey: John Wiley & Sons, 267–284. doi:10.1002/9781118967225.ch16
Dao, D. T., Mahon, P. B., Cai, X., Kovacsics, C. E., Blackwell, R. A., Arad, M., et al. (2010). Mood Disorder Susceptibility Gene CACNA1C Modifies Mood-Related Behaviors in Mice and Interacts With Sex to Influence Behavior in Mice and Diagnosis in Humans. Biol. Psychiatry. 68, 801–810. doi:10.1016/j.biopsych.2010.06.019
De Boer, I., Van Den Maagdenberg, A. M. J. M., and Terwindt, G. M. (2019). Advance in Genetics of Migraine. Curr. Opin. Neurol. 32, 413–421. doi:10.1097/wco.0000000000000687
Dedic, N., Pöhlmann, M. L., Richter, J. S., Mehta, D., Czamara, D., Metzger, M. W., et al. (2018). Cross-Disorder Risk Gene CACNA1C Differentially Modulates Susceptibility to Psychiatric Disorders During Development and Adulthood. Mol. Psychiatry. 23, 533–543. doi:10.1038/mp.2017.133
Dehghani, A., Phisonkunkasem, T., Ozcan, S. Y., Dalkara, T., Van Den Maagdenberg, A. M. J. M., Tolner, E. A., et al. (2021). Widespread Brain Parenchymal HMGB1 and NF-KB Neuroinflammatory Responses Upon Cortical Spreading Depolarization in Familial Hemiplegic Migraine Type 1 Mice. Neurobiol. Dis. 156, 105424. doi:10.1016/j.nbd.2021.105424
Desroches, M., Faugeras, O., Krupa, M., and Mantegazza, M. (2019). Modeling Cortical Spreading Depression Induced by the Hyperactivity of Interneurons. J. Comput. Neurosci. 47, 125–140. doi:10.1007/s10827-019-00730-8
Dias-Ferreira, E., Sousa, J. C., Melo, I., Morgado, P., Mesquita, A. R., Cerqueira, J. J., et al. (2009). Chronic Stress Causes Frontostriatal Reorganization and Affects Decision-Making. Science. 325, 621–625. doi:10.1126/science.1171203
Djillani, A., Pietri, M., Mazella, J., Heurteaux, C., and Borsotto, M. (2019). Fighting Against Depression With TREK-1 Blockers: Past and Future. A Focus on Spadin. Pharmacol. Ther. 194, 185–198. doi:10.1016/j.pharmthera.2018.10.003
Djillani, A., Pietri, M., Moreno, S., Heurteaux, C., Mazella, J., and Borsotto, M. (2017). Shortened Spadin Analogs Display Better TREK-1 Inhibition, In Vivo Stability and Antidepressant Activity. Front. Pharmacol. 8, 643. doi:10.3389/fphar.2017.00643
Donnelly-Roberts, D. L., Namovic, M. T., Han, P., and Jarvis, M. F. (2009). Mammalian P2X7 Receptor Pharmacology: Comparison of Recombinant Mouse, Rat and Human P2X7 Receptors. Br. J. Pharmacol. 157, 1203–1214. doi:10.1111/j.1476-5381.2009.00233.x
Dowlati, Y., Herrmann, N., Swardfager, W., Liu, H., Sham, L., Reim, E. K., et al. (2010). A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry. 67, 446–457. doi:10.1016/j.biopsych.2009.09.033
Dubovsky, S. L. (2019). Applications of Calcium Channel Blockers in Psychiatry: Pharmacokinetic and Pharmacodynamic Aspects of Treatment of Bipolar Disorder. Expert Opin. Drug Metab. Toxicol. 15, 35–47. doi:10.1080/17425255.2019.1558206
Duman, R. S., Sanacora, G., and Krystal, J. H. (2019). Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron. 102, 75–90. doi:10.1016/j.neuron.2019.03.013
Dussor, G., and Cao, Y. Q. (2016). TRPM8 and Migraine. Headache. 56, 1406–1417. doi:10.1111/head.12948
Edelmayer, R. M., Le, L. N., Yan, J., Wei, X., Nassini, R., Materazzi, S., et al. (2012). Activation of TRPA1 on Dural Afferents: a Potential Mechanism of Headache Pain. Pain. 153, 1949–1958. doi:10.1016/j.pain.2012.06.012
Eising, E., Shyti, R., 't Hoen, P. A. C., Vijfhuizen, L. S., Huisman, S. M. H., Broos, L. A. M., et al. (2017). Cortical Spreading Depression Causes Unique Dysregulation of Inflammatory Pathways in a Transgenic Mouse Model of Migraine. Mol. Neurobiol. 54, 2986–2996. doi:10.1007/s12035-015-9681-5
Enyedi, P., and Czirják, G. (2015). Properties, Regulation, Pharmacology, and Functions of the K₂p Channel, TRESK. Pflugers Arch. 467, 945–958. doi:10.1007/s00424-014-1634-8
Erdener, S. E., and Dalkara, T. (2014). Modelling Headache and Migraine and its Pharmacological Manipulation. Br. J. Pharmacol. 171, 4575–4594. doi:10.1111/bph.12651
Fee, C., Banasr, M., and Sibille, E. (2017). Somatostatin-Positive Gamma-Aminobutyric Acid Interneuron Deficits in Depression: Cortical Microcircuit and Therapeutic Perspectives. Biol. Psychiatry. 82, 549–559. doi:10.1016/j.biopsych.2017.05.024
Ferreira, M. A., O'donovan, M. C., Meng, Y. A., Jones, I. R., Ruderfer, D. M., Jones, L., et al. (2008). Collaborative Genome-Wide Association Analysis Supports a Role for ANK3 and CACNA1C in Bipolar Disorder. Nat. Genet. 40, 1056–1058. doi:10.1038/ng.209
Fogaça, M. V., and Duman, R. S. (2019). Cortical GABAergic Dysfunction in Stress and Depression: New Insights for Therapeutic Interventions. Front. Cel Neurosci. 13, 87. doi:10.3389/fncel.2019.00087
Friedman, A. K., Juarez, B., Ku, S. M., Zhang, H., Calizo, R. C., Walsh, J. J., et al. (2016). KCNQ Channel Openers Reverse Depressive Symptoms via an Active Resilience Mechanism. Nat. Commun. 7, 11671. doi:10.1038/ncomms11671
Friedman, A. K., Walsh, J. J., Juarez, B., Ku, S. M., Chaudhury, D., Wang, J., et al. (2014). Enhancing Depression Mechanisms in Midbrain Dopamine Neurons Achieves Homeostatic Resilience. Science. 344, 313–319. doi:10.1126/science.1249240
Fuchs, T., Jefferson, S. J., Hooper, A., Yee, P. H., Maguire, J., and Luscher, B. (2017). Disinhibition of Somatostatin-Positive GABAergic Interneurons Results in an Anxiolytic and Antidepressant-Like Brain State. Mol. Psychiatry. 22, 920–930. doi:10.1038/mp.2016.188
George, M. S., Abbott, L. F., and Siegelbaum, S. A. (2009). HCN Hyperpolarization-Activated Cation Channels Inhibit EPSPs by Interactions With M-type K(+) Channels. Nat. Neurosci. 12, 577–584. doi:10.1038/nn.2307
Ghaemi, A., Alizadeh, L., Babaei, S., Jafarian, M., Khaleghi Ghadiri, M., Meuth, S. G., et al. (2018). Astrocyte-Mediated Inflammation in Cortical Spreading Depression. Cephalalgia. 38, 626–638. doi:10.1177/0333102417702132
Ghaemi, A., Sajadian, A., Khodaie, B., Lotfinia, A. A., Lotfinia, M., Aghabarari, A., et al. (2016). Immunomodulatory Effect of Toll-Like Receptor-3 Ligand Poly I:C on Cortical Spreading Depression. Mol. Neurobiol. 53, 143–154. doi:10.1007/s12035-014-8995-z
Goldwater, D. S., Pavlides, C., Hunter, R. G., Bloss, E. B., Hof, P. R., Mcewen, B. S., et al. (2009). Structural and Functional Alterations to Rat Medial Prefrontal Cortex Following Chronic Restraint Stress and Recovery. Neuroscience. 164, 798–808. doi:10.1016/j.neuroscience.2009.08.053
Gölöncsér, F., Baranyi, M., Balázsfi, D., Demeter, K., Haller, J., Freund, T. F. F., et al. (2017). Regulation of Hippocampal 5-HT Release by P2X7 Receptors in Response to Optogenetic Stimulation of Median Raphe Terminals of Mice. Front. Mol. Neurosci. 10, 325. doi:10.3389/fnmol.2017.00325
Gormley, P., Anttila, V., Winsvold, B. S., Palta, P., Esko, T., Pers, T. H., et al. (2016). Meta-Analysis of 375,000 Individuals Identifies 38 Susceptibility Loci for Migraine. Nat. Genet. 48, 856–866. doi:10.1038/ng.3598
Guo, H., Callaway, J. B., and Ting, J. P. (2015). Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 21, 677–687. doi:10.1038/nm.3893
Guo, Z., Qiu, C. S., Jiang, X., Zhang, J., Li, F., Liu, Q., et al. (2019). TRESK K+ Channel Activity Regulates Trigeminal Nociception and Headache. eNeuro. 6, ENEURO.0236-19.2019. doi:10.1523/ENEURO.0236-19.2019
Hadjikhani, N., Albrecht, D. S., Mainero, C., Ichijo, E., Ward, N., Granziera, C., et al. (2020). Extra-Axial Inflammatory Signal in Parameninges in Migraine With Visual Aura. Ann. Neurol. 87, 939–949. doi:10.1002/ana.25731
Hadjikhani, N., Sanchez Del Rio, M., Wu, O., Schwartz, D., Bakker, D., Fischl, B., et al. (2001). Mechanisms of Migraine Aura Revealed by Functional MRI in Human Visual Cortex. Proc. Natl. Acad. Sci. U S A. 98, 4687–4692. doi:10.1073/pnas.071582498
Halmai, Z., Dome, P., Vereczkei, A., Abdul-Rahman, O., Szekely, A., Gonda, X., et al. (2013). Associations between Depression Severity and Purinergic Receptor P2RX7 Gene Polymorphisms. J. Affect Disord. 150, 104–109. doi:10.1016/j.jad.2013.02.033
Harrison, P. J., Tunbridge, E. M., Dolphin, A. C., and Hall, J. (2020). Voltage-Gated Calcium Channel Blockers for Psychiatric Disorders: Genomic Reappraisal. Br. J. Psychiatry. 216, 250–253. doi:10.1192/bjp.2019.157
Harrison, P. J., Hall, N., Mould, A., Al-Juffali, N., and Tunbridge, E. M. (2019). Cellular Calcium in Bipolar Disorder: Systematic Review and Meta-Analysis. Mol. Psychiatry. doi:10.1038/s41380-019-0622-y
Hedrich, U. B., Liautard, C., Kirschenbaum, D., Pofahl, M., Lavigne, J., Liu, Y., et al. (2014). Impaired Action Potential Initiation in GABAergic Interneurons Causes Hyperexcitable Networks in an Epileptic Mouse Model Carrying a Human Na(V)1.1 Mutation. J. Neurosci. 34, 14874–14889. doi:10.1523/JNEUROSCI.0721-14.2014
Heurteaux, C., Lucas, G., Guy, N., El Yacoubi, M., Thümmler, S., Peng, X. D., et al. (2006). Deletion of the Background Potassium Channel TREK-1 Results in a Depression-Resistant Phenotype. Nat. Neurosci. 9, 1134–1141. doi:10.1038/nn1749
Hirschfeld, R. M. (2000). History and Evolution of the Monoamine Hypothesis of Depression. J. Clin. Psychiatry. 61 (Suppl. 6), 4–6.
Höflich, A., Michenthaler, P., Kasper, S., and Lanzenberger, R. (2019). Circuit Mechanisms of Reward, Anhedonia, and Depression. Int. J. Neuropsychopharmacol. 22, 105–118. doi:10.1093/ijnp/pyy081
Howard, D. M., Adams, M. J., Clarke, T. K., Hafferty, J. D., Gibson, J., Shirali, M., et al. (2019). Genome-wide Meta-Analysis of Depression Identifies 102 Independent Variants and Highlights the Importance of the Prefrontal Brain Regions. Nat. Neurosci. 22, 343–352. doi:10.1038/s41593-018-0326-7
Illes, P., Khan, T. M., and Rubini, P. (2017). Neuronal P2X7 Receptors Revisited: Do They Really Exist? J. Neurosci. 37, 7049–7062. doi:10.1523/JNEUROSCI.3103-16.2017
Iwata, M., Ota, K. T., Li, X. Y., Sakaue, F., Li, N., Dutheil, S., et al. (2016). Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol. Psychiatry. 80, 12–22. doi:10.1016/j.biopsych.2015.11.026
Jansen, N. A., Dehghani, A., Linssen, M. M. L., Breukel, C., Tolner, E. A., and Van Den Maagdenberg, A. M. J. M. (2020). First FHM3 Mouse Model Shows Spontaneous Cortical Spreading Depolarizations. Ann. Clin. Transl Neurol. 7, 132–138. doi:10.1002/acn3.50971
Kabir, Z. D., Lee, A. S., Burgdorf, C. E., Fischer, D. K., Rajadhyaksha, A. M., Mok, E., et al. (2017). Cacna1c in the Prefrontal Cortex Regulates Depression-Related Behaviors via REDD1. Neuropsychopharmacol. 42, 2032–2042. doi:10.1038/npp.2016.271
Kaczmarek-Hajek, K., Zhang, J., Kopp, R., Grosche, A., Rissiek, B., Saul, A., et al. (2018). Re-Evaluation of Neuronal P2X7 Expression Using Novel Mouse Models and a P2X7-Specific Nanobody. Elife. 7, e36217. doi:10.7554/elife.36217
Karatas, H., Erdener, S. E., Gursoy-Ozdemir, Y., Lule, S., Eren-Koçak, E., Sen, Z. D., et al. (2013). Spreading Depression Triggers Headache by Activating Neuronal Panx1 Channels. Science. 339, 1092–1095. doi:10.1126/science.1231897
Karsan, N., and Goadsby, P. J. (2021). Migraine Is More Than Just Headache: Is the Link to Chronic Fatigue and Mood Disorders Simply Due to Shared Biological Systems? Front. Hum. Neurosci. 15, 646692. doi:10.3389/fnhum.2021.646692
Kase, D., and Imoto, K. (2012). The Role of HCN Channels on Membrane Excitability in the Nervous System. J. Signal. Transduct. 2012, 619747. doi:10.1155/2012/619747
Kelmendi, B., Holsbach-Beltrame, M., Mcintosh, A. M., Hilt, L., George, E. D., Kitchen, R. R., et al. (2011). Association of Polymorphisms in HCN4 with Mood Disorders and Obsessive Compulsive Disorder. Neurosci. Lett. 496, 195–199. doi:10.1016/j.neulet.2011.04.026
Kennard, L. E., Chumbley, J. R., Ranatunga, K. M., Armstrong, S. J., Veale, E. L., and Mathie, A. (2005). Inhibition of the Human Two-Pore Domain Potassium Channel, TREK-1, by Fluoxetine and its Metabolite Norfluoxetine. Br. J. Pharmacol. 144, 821–829. doi:10.1038/sj.bjp.0706068
Khakh, B. S., and North, R. A. (2006). P2X Receptors as Cell-Surface ATP Sensors in Health and Disease. Nature. 442, 527–532. doi:10.1038/nature04886
Kilic, K., Karatas, H., Dönmez-Demir, B., Eren-Kocak, E., Gursoy-Ozdemir, Y., Can, A., et al. (2018). Inadequate Brain Glycogen or Sleep Increases Spreading Depression Susceptibility. Ann. Neurol. 83, 61–73. doi:10.1002/ana.25122
Kim, C. S., Chang, P. Y., and Johnston, D. (2012). Enhancement of Dorsal Hippocampal Activity by Knockdown of HCN1 Channels Leads to Anxiolytic- and Antidepressant-like Behaviors. Neuron. 75, 503–516. doi:10.1016/j.neuron.2012.05.027
Kim, E. J., Lee, D. K., Hong, S. G., Han, J., and Kang, D. (2017). Activation of TREK-1, but Not TREK-2, Channel by Mood Stabilizers. Int. J. Mol. Sci. 18, 2460. doi:10.3390/ijms18112460
Kros, L., Lykke-Hartmann, K., and Khodakhah, K. (2018). Increased Susceptibility to Cortical Spreading Depression and Epileptiform Activity in a Mouse Model for FHM2. Scientific Rep. 8, 16959. doi:10.1038/s41598-018-35285-8
Langford, D. J., Bailey, A. L., Chanda, M. L., Clarke, S. E., Drummond, T. E., Echols, S., et al. (2010). Coding of Facial Expressions of Pain in the Laboratory Mouse. Nat. Methods. 7, 447–449. doi:10.1038/nmeth.1455
Lanquillon, S., Krieg, J. C., Bening-Abu-Shach, U., and Vedder, H. (2000). Cytokine Production and Treatment Response in Major Depressive Disorder. Neuropsychopharmacology. 22, 370–379. doi:10.1016/S0893-133X(99)00134-7
Leao, A. A. (1947). Further Observations on the Spreading Depression of Activity in the Cerebral Cortex. J. Neurophysiol. 10, 409–414. doi:10.1152/jn.1947.10.6.409
Leao, A. A. P. (1944). Spreading Depression of Activity in the Cerebral Cortex. J. Neurophysiol. 7, 359–390. doi:10.1152/jn.1944.7.6.359
Lebas, A., Guyant-Maréchal, L., Hannequin, D., Riant, F., Tournier-Lasserve, E., and Parain, D. (2008). Severe Attacks of Familial Hemiplegic Migraine, Childhood Epilepsy and ATP1A2 Mutation. Cephalalgia. 28, 774–777. doi:10.1111/j.1468-2982.2008.01603.x
Lemaire, L., Desroches, M., Krupa, M., Pizzamiglio, L., Scalmani, P., and Mantegazza, M. (2021). Modeling NaV1.1/SCN1A Sodium Channel Mutations in a Microcircuit With Realistic Ion Concentration Dynamics Suggests Differential GABAergic Mechanisms Leading to Hyperexcitability in Epilepsy and Hemiplegic Migraine. Plos Comput. Biol. 17, e1009239. doi:10.1371/journal.pcbi.1009239
Leo, L., Gherardini, L., Barone, V., De Fusco, M., Pietrobon, D., Pizzorusso, T., et al. (2011). Increased Susceptibility to Cortical Spreading Depression in the Mouse Model of Familial Hemiplegic Migraine Type 2. Plos Genet. 7, e1002129. doi:10.1371/journal.pgen.1002129
Lesage, F. (2003). Pharmacology of Neuronal Background Potassium Channels. Neuropharmacology. 44, 1–7. doi:10.1016/s0028-3908(02)00339-8
Levine, J., Barak, Y., Chengappa, K. N., Rapoport, A., Rebey, M., and Barak, V. (1999). Cerebrospinal Cytokine Levels in Patients With Acute Depression. Neuropsychobiology. 40, 171–176. doi:10.1159/000026615
Lewis, A. S., Vaidya, S. P., Blaiss, C. A., Liu, Z., Stoub, T. R., Brager, D. H., et al. (2011). Deletion of the Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel Auxiliary Subunit TRIP8b Impairs Hippocampal Ih Localization and Function and Promotes Antidepressant Behavior in Mice. J. Neurosci. 31, 7424–7440. doi:10.1523/JNEUROSCI.0936-11.2011
Li, L., Sun, H., Ding, J., Niu, C., Su, M., Zhang, L., et al. (2017). Selective Targeting of M-type Potassium Kv 7.4 Channels Demonstrates Their Key Role in the Regulation of Dopaminergic Neuronal Excitability and Depression-Like Behaviour. Br. J. Pharmacol. 174, 4277–4294. doi:10.1111/bph.14026
Liou, Y. J., Chen, T. J., Tsai, S. J., Yu, Y. W., Cheng, C. Y., and Hong, C. J. (2009). Support for the Involvement of the KCNK2 Gene in Major Depressive Disorder and Response to Antidepressant Treatment. Pharmacogenet Genomics. 19, 735–741. doi:10.1097/FPC.0b013e32832cbe61
Liston, C., Miller, M. M., Goldwater, D. S., Radley, J. J., Rocher, A. B., Hof, P. R., et al. (2006). Stress-induced Alterations in Prefrontal Cortical Dendritic Morphology Predict Selective Impairments in Perceptual Attentional Set-Shifting. J. Neurosci. 26, 7870–7874. doi:10.1523/JNEUROSCI.1184-06.2006
Liu, P., Xiao, Z., Ren, F., Guo, Z., Chen, Z., Zhao, H., et al. (2013). Functional Analysis of a Migraine-Associated TRESK K+ Channel Mutation. J. Neurosci. 33, 12810–12824. doi:10.1523/JNEUROSCI.1237-13.2013
Lucae, S., Salyakina, D., Barden, N., Harvey, M., Gagné, B., Labbé, M., et al. (2006). P2RX7, a Gene Coding for a Purinergic Ligand-Gated Ion Channel, Is Associated with Major Depressive Disorder. Hum. Mol. Genet. 15, 2438–2445. doi:10.1093/hmg/ddl166
Luscher, B., Shen, Q., and Sahir, N. (2011). The GABAergic Deficit Hypothesis of Major Depressive Disorder. Mol. Psychiatry. 16, 383–406. doi:10.1038/mp.2010.120
Macvicar, B. A., and Thompson, R. J. (2010). Non-Junction Functions of Pannexin-1 Channels. Trends Neurosci. 33, 93–102. doi:10.1016/j.tins.2009.11.007
Mantegazza, M., Cestèle, S., and Catterall, W. A. (2021). Sodium Channelopathies of Skeletal Muscle and Brain. Physiol. Rev. 101, 1633–1689. doi:10.1152/physrev.00025.2020
Mantegazza, M. (2018). Epilepsy: Advances in Genetics and Pathophysiology. Neurosci. Lett. 667, 1–3. doi:10.1016/j.neulet.2017.10.026
Marcoli, M., Cervetto, C., Paluzzi, P., Guarnieri, S., Alloisio, S., Thellung, S., et al. (2008). P2X7 Pre-Synaptic Receptors in Adult Rat Cerebrocortical Nerve Terminals: a Role in ATP-Induced Glutamate Release. J. Neurochem. 105, 2330–2342. doi:10.1111/j.1471-4159.2008.05322.x
Marconi, R., De Fusco, M., Aridon, P., Plewnia, K., Rossi, M., Carapelli, S., et al. (2003). Familial Hemiplegic Migraine Type 2 Is Linked to 0.9Mb Region on Chromosome 1q23. Ann. Neurol. 53, 376–381. doi:10.1002/ana.10464
Marpegan, L., Swanstrom, A. E., Chung, K., Simon, T., Haydon, P. G., Khan, S. K., et al. (2011). Circadian Regulation of ATP Release in Astrocytes. J. Neurosci. 31, 8342–8350. doi:10.1523/JNEUROSCI.6537-10.2011
Mcewen, B. S. (1999). Stress and Hippocampal Plasticity. Annu. Rev. Neurosci. 22, 105–122. doi:10.1146/annurev.neuro.22.1.105
Mcewen, B. S. (2001). Plasticity of the hippocampus: Adaptation to Chronic Stress and Allostatic Load. Ann. N. Y Acad. Sci. 933, 265–277. doi:10.1111/j.1749-6632.2001.tb05830.x
Mcintosh, A. M., Simen, A. A., Evans, K. L., Hall, J., Macintyre, D. J., Blackwood, D., et al. (2012). Genetic Variation in Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels and its Relationship With Neuroticism, Cognition and Risk of Depression. Front. Genet. 3, 116. doi:10.3389/fgene.2012.00116
Mclaughlin, K. A., Conron, K. J., Koenen, K. C., and Gilman, S. E. (2010). Childhood Adversity, Adult Stressful Life Events, and Risk of Past-Year Psychiatric Disorder: a Test of the Stress Sensitization Hypothesis in a Population-Based Sample of Adults. Psychol. Med. 40, 1647–1658. doi:10.1017/S0033291709992121
Mcquillin, A., Bass, N. J., Choudhury, K., Puri, V., Kosmin, M., Lawrence, J., et al. (2009). Case-Control Studies Show That a Non-Conservative Amino-Acid Change From a Glutamine to Arginine in the P2RX7 Purinergic Receptor Protein Is Associated with Both Bipolar- and Unipolar-Affective Disorders. Mol. Psychiatry. 14, 614–620. doi:10.1038/mp.2008.6
Metzger, M. W., Walser, S. M., Dedic, N., Aprile-Garcia, F., Jakubcakova, V., Adamczyk, M., et al. (2017). Heterozygosity for the Mood Disorder-Associated Variant Gln460Arg Alters P2X7 Receptor Function and Sleep Quality. J. Neurosci. 37, 11688–11700. doi:10.1523/JNEUROSCI.3487-16.2017
Milenkovic, V. M., Stanton, E. H., Nothdurfter, C., Rupprecht, R., and Wetzel, C. H. (2019). The Role of Chemokines in the Pathophysiology of Major Depressive Disorder. Int. J. Mol. Sci. 20, 2283. doi:10.3390/ijms20092283
Milior, G., Lecours, C., Samson, L., Bisht, K., Poggini, S., Pagani, F., et al. (2016). Fractalkine Receptor Deficiency Impairs Microglial and Neuronal Responsiveness to Chronic Stress. Brain Behav. Immun. 55, 114–125. doi:10.1016/j.bbi.2015.07.024
Miras-Portugal, M. T., Sebastián-Serrano, Á., de Diego García, L., and Díaz-Hernández, M. (2017). Neuronal P2X7 Receptor: Involvement in Neuronal Physiology and Pathology. J. Neurosci. 37, 7063–7072. doi:10.1523/JNEUROSCI.3104-16.2017
Miyamoto, A., Wake, H., Ishikawa, A. W., Eto, K., Shibata, K., Murakoshi, H., et al. (2016). Microglia Contact Induces Synapse Formation in Developing Somatosensory Cortex. Nat. Commun. 7, 12540. doi:10.1038/ncomms12540
Moskowitz, M. A., Bolay, H., and Dalkara, T. (2004). Deciphering Migraine Mechanisms: Clues From Familial Hemiplegic Migraine Genotypes. Ann. Neurol. 55, 276–280. doi:10.1002/ana.20035
Mueller, N., and Schwarz, M. J. (2007). The Immune-Mediated Alteration of Serotonin and Glutamate: towards an Integrated View of Depression. Mol. Psychiatry. 12, 988–1000. doi:10.1038/sj.mp.4002006
Nappi, P., Miceli, F., Soldovieri, M. V., Ambrosino, P., Barrese, V., and Taglialatela, M. (2020). Epileptic Channelopathies Caused by Neuronal Kv7 (KCNQ) Channel Dysfunction. Pflugers Arch. 472, 881–898. doi:10.1007/s00424-020-02404-2
Nestler, E. J., and Carlezon, W. A. (2006). The Mesolimbic Dopamine Reward Circuit in Depression. Biol. Psychiatry. 59, 1151–1159. doi:10.1016/j.biopsych.2005.09.018
Nestler, E. J. (2015). Role of the Brain's Reward Circuitry in Depression: Transcriptional Mechanisms. Int. Rev. Neurobiol. 124, 151–170. doi:10.1016/bs.irn.2015.07.003
Notomi, T., and Shigemoto, R. (2004). Immunohistochemical Localization of Ih Channel Subunits, HCN1-4, in the Rat Brain. J. Comp. Neurol. 471, 241–276. doi:10.1002/cne.11039
Nurnberger, J. I., Koller, D. L., Jung, J., Edenberg, H. J., Foroud, T., Guella, I., et al. (2014). Identification of Pathways for Bipolar Disorder: a Meta-Analysis. JAMA Psychiatry. 71, 657–664. doi:10.1001/jamapsychiatry.2014.176
Nutt, D., Wilson, S., and Paterson, L. (2008). Sleep Disorders as Core Symptoms of Depression. Dialogues Clin. Neurosci. 10, 329–336. doi:10.31887/DCNS.2008.10.3/dnutt
Olesen, J., Lauritzen, M., Tfelt-Hansen, P., Henriksen, L., and Larsen, B. (1982). Spreading Cerebral Oligemia in Classical- and Normal Cerebral Blood Flow in Common Migraine. Headache. 22, 242–248. doi:10.1111/j.1526-4610.1982.hed2206242.x
Otrokocsi, L., Kittel, Á., and Sperlágh, B. (2017). P2X7 Receptors Drive Spine Synapse Plasticity in the Learned Helplessness Model of Depression. Int. J. Neuropsychopharmacol. 20, 813–822. doi:10.1093/ijnp/pyx046
Pace, T. W., Mletzko, T. C., Alagbe, O., Musselman, D. L., Nemeroff, C. B., Miller, A. H., et al. (2006). Increased Stress-Induced Inflammatory Responses in Male Patients with Major Depression and Increased Early Life Stress. Am. J. Psychiatry. 163, 1630–1633. doi:10.1176/ajp.2006.163.9.1630
Pankratov, Y. V., Lalo, U. V., and Krishtal, O. A. (2002). Role for P2X Receptors in Long-Term Potentiation. J. Neurosci. 22, 8363–8369. doi:10.1523/jneurosci.22-19-08363.2002
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic Pruning by Microglia Is Necessary for normal Brain Development. Science. 333, 1456–1458. doi:10.1126/science.1202529
Papp, L., Vizi, E. S., and Sperlágh, B. (2004). Lack of ATP-Evoked GABA and Glutamate Release in the hippocampus of P2X7 Receptor-/- Mice. Neuroreport. 15, 2387–2391. doi:10.1097/00001756-200410250-00017
Parker, P. D., Suryavanshi, P., Melone, M., Sawant-Pokam, P. A., Reinhart, K. M., Kaufmann, D., et al. (2021). Non-canonical Glutamate Signaling in a Genetic Model of Migraine with Aura. Neuron. 109, 611–628. doi:10.1016/j.neuron.2020.11.018
Parker, T., Keddie, S., Kidd, D., Lane, T., Maviki, M., Hawkins, P. N., et al. (2016). Neurology of the Cryopyrin-Associated Periodic Fever Syndrome. Eur. J. Neurol. 23, 1145–1151. doi:10.1111/ene.12965
Parpura, V., Basarsky, T. A., Liu, F., Jeftinija, K., Jeftinija, S., and Haydon, P. G. (1994). Glutamate-mediated Astrocyte-Neuron Signalling. Nature. 369, 744–747. doi:10.1038/369744a0
Perlis, R. H., Moorjani, P., Fagerness, J., Purcell, S., Trivedi, M. H., Fava, M., et al. (2008). Pharmacogenetic Analysis of Genes Implicated in Rodent Models of Antidepressant Response: Association of TREK1 and Treatment Resistance in the STAR*D Study. Neuropsychopharmacol. 33, 2810–2819. doi:10.1038/npp.2008.6
Petit, J. M., Eren-Koçak, E., Karatas, H., Magistretti, P., and Dalkara, T. (2021). Brain Glycogen Metabolism: A Possible Link Between Sleep Disturbances, Headache and Depression. Sleep Med. Rev. 59, 101449. doi:10.1016/j.smrv.2021.101449
Petit, J. M., and Magistretti, P. J. (2016). Regulation of Neuron-Astrocyte Metabolic Coupling Across the Sleep-Wake Cycle. Neuroscience. 323, 135–156. doi:10.1016/j.neuroscience.2015.12.007
Pettingill, P., Weir, G. A., Wei, T., Wu, Y., Flower, G., Lalic, T., et al. (2019). A Causal Role for TRESK Loss of Function in Migraine Mechanisms. Brain. 142, 3852–3867. doi:10.1093/brain/awz342
Pietrobon, D., and Brennan, K. C. (2019). Genetic Mouse Models of Migraine. J. Headache Pain. 20, 79. doi:10.1186/s10194-019-1029-5
Pietrobon, D., and Moskowitz, M. A. (2014). Chaos and Commotion in the Wake of Cortical Spreading Depression and Spreading Depolarizations. Nat. Rev. Neurosci. 15, 379–393. doi:10.1038/nrn3770
Psychiatric, G. C. B. D. W. G. (2011). Large-scale Genome-wide Association Analysis of Bipolar Disorder Identifies a New Susceptibility Locus Near ODZ4. Nat. Genet. 43, 977–983. doi:10.1038/ng.943
Radley, J. J., Rocher, A. B., Miller, M., Janssen, W. G., Liston, C., Hof, P. R., et al. (2006). Repeated Stress Induces Dendritic Spine Loss in the Rat Medial Prefrontal Cortex. Cereb. Cortex. 16, 313–320. doi:10.1093/cercor/bhi104
Ren, J., Guo, J., Zhu, S., Wang, Q., Gao, R., Zhao, C., et al. (2021). The Role of Potassium Channels in Chronic Stress-Induced Brain Injury. Biol. Pharm. Bull. 44, 169–180. doi:10.1248/bpb.b20-00504
Reyes, R., Duprat, F., Lesage, F., Fink, M., Salinas, M., Farman, N., et al. (1998). Cloning and Expression of a Novel pH-Sensitive Two Pore Domain K+ Channel From Human Kidney. J. Biol. Chem. 273, 30863–30869. doi:10.1074/jbc.273.47.30863
Richards, E. M., Zanotti-Fregonara, P., Fujita, M., Newman, L., Farmer, C., Ballard, E. D., et al. (2018). PET Radioligand Binding to Translocator Protein (TSPO) Is Increased in Unmedicated Depressed Subjects. Ejnmmi Res. 8, 57. doi:10.1186/s13550-018-0401-9
Rivolta, I., Binda, A., Masi, A., and Difrancesco, J. C. (2020). Cardiac and Neuronal HCN Channelopathies. Pflugers Arch. 472, 931–951. doi:10.1007/s00424-020-02384-3
Royal, P., Andres-Bilbe, A., Ávalos Prado, P., Verkest, C., Wdziekonski, B., Schaub, S., et al. (2019). Migraine-Associated TRESK Mutations Increase Neuronal Excitability Through Alternative Translation Initiation and Inhibition of TREK.. Neuron. 101, 232–e6. doi:10.1016/j.neuron.2018.11.039
Russell, M. B., and Ducros, A. (2011). Sporadic and Familial Hemiplegic Migraine: Pathophysiological Mechanisms, Clinical Characteristics, Diagnosis, and Management. Lancet Neurol. 10, 457–470. doi:10.1016/S1474-4422(11)70048-5
Russo, S. J., and Nestler, E. J. (2013). The Brain Reward Circuitry in Mood Disorders. Nat. Rev. Neurosci. 14, 609–625. doi:10.1038/nrn3381
Savtchouk, I., and Volterra, A. (2018). Gliotransmission: Beyond Black-And-White. J. Neurosci. 38, 14–25. doi:10.1523/JNEUROSCI.0017-17.2017
Setiawan, E., Wilson, A. A., Mizrahi, R., Rusjan, P. M., Miler, L., Rajkowska, G., et al. (2015). Role of Translocator Protein Density, a Marker of Neuroinflammation, in the Brain During Major Depressive Episodes. JAMA Psychiatry. 72, 268–275. doi:10.1001/jamapsychiatry.2014.2427
Sperlágh, B., Köfalvi, A., Deuchars, J., Atkinson, L., Milligan, C. J., Buckley, N. J., et al. (2002). Involvement of P2X7 Receptors in the Regulation of Neurotransmitter Release in the Rat hippocampus. J. Neurochem. 81, 1196–1211. doi:10.1046/j.1471-4159.2002.00920.x
Stahl, S. M. (2004). Anticonvulsants as Mood Stabilizers and Adjuncts to Antipsychotics: Valproate, Lamotrigine, Carbamazepine, and Oxcarbazepine and Actions at Voltage-Gated Sodium Channels. J. Clin. Psychiatry. 65, 738–739. doi:10.4088/jcp.v65n0601
Suryavanshi, P., Pokam, P. S., and Brennan, K. C. (2019). Altered Synaptic Adaptation and Gain in Sensory Circuits of the Casein Kinase 1 delta (CK1dT44A) Mouse Model of Migraine. bioRxiv
Sutherland, H. G., Albury, C. L., and Griffiths, L. R. (2019). Advances in Genetics of Migraine. J. Headache Pain. 20, 72. doi:10.1186/s10194-019-1017-9
Sutherland, H. G., Maksemous, N., Albury, C. L., Ibrahim, O., Smith, R. A., Lea, R. A., et al. (2020). Comprehensive Exonic Sequencing of Hemiplegic Migraine-Related Genes in a Cohort of Suspected Probands Identifies Known and Potential Pathogenic Variants. Cells. 9, 2368. doi:10.3390/cells9112368
Takizawa, T., Ayata, C., and Chen, S. P. (2020a). Therapeutic Implications of Cortical Spreading Depression Models in Migraine. Prog. Brain Res. 255, 29–67. doi:10.1016/bs.pbr.2020.05.009
Takizawa, T., Qin, T., Lopes De Morais, A., Sugimoto, K., Chung, J. Y., Morsett, L., et al. (2020b). Non-Invasively Triggered Spreading Depolarizations Induce a Rapid Pro-Inflammatory Response in Cerebral Cortex. J. Cereb. Blood Flow Metab. 40, 1117–1131. doi:10.1177/0271678X19859381
Takizawa, T., Shibata, M., Kayama, Y., Shimizu, T., Toriumi, H., Ebine, T., et al. (2017). High-Mobility Group Box 1 Is an Important Mediator of Microglial Activation Induced by Cortical Spreading Depression. J. Cereb. Blood Flow Metab. 37, 890–901. doi:10.1177/0271678X16647398
Takizawa, T., Shibata, M., Kayama, Y., Toriumi, H., Ebine, T., Koh, A., et al. (2016). Temporal Profiles of High-Mobility Group Box 1 Expression Levels after Cortical Spreading Depression in Mice. Cephalalgia. 36, 44–52. doi:10.1177/0333102415580100
Talley, E. M., Sirois, J. E., Lei, Q., and Bayliss, D. A. (2003). Two-Pore-Domain (KCNK) Potassium Channels: Dynamic Roles in Neuronal Function. Neuroscientist. 9, 46–56. doi:10.1177/1073858402239590
Tan, A., Costi, S., Morris, L. S., Van Dam, N. T., Kautz, M., Whitton, A. E., et al. (2020). Effects of the KCNQ Channel Opener Ezogabine on Functional Connectivity of the Ventral Striatum and Clinical Symptoms in Patients With Major Depressive Disorder. Mol. Psychiatry. 25, 1323–1333. doi:10.1038/s41380-018-0283-2
Tang, Y. T., Mendez, J. M., Theriot, J. J., Sawant, P. M., López-Valdés, H. E., Ju, Y. S., et al. (2014). Minimum Conditions for the Induction of Cortical Spreading Depression in Brain Slices. J. Neurophysiol. 112, 2572–2579. doi:10.1152/jn.00205.2014
Terrillion, C. E., Francis, T. C., Puche, A. C., Lobo, M. K., and Gould, T. D. (2017). Decreased Nucleus Accumbens Expression of Psychiatric Disorder Risk Gene Cacna1c Promotes Susceptibility to Social Stress. Int. J. Neuropsychopharmacol. 20, 428–433. doi:10.1093/ijnp/pyw112
Tottene, A., Conti, R., Fabbro, A., Vecchia, D., Shapovalova, M., Santello, M., et al. (2009). Enhanced Excitatory Transmission at Cortical Synapses as the Basis for Facilitated Spreading Depression in Ca(v)2.1 Knockin Migraine Mice. Neuron. 61, 762–773. doi:10.1016/j.neuron.2009.01.027
Tottene, A., Favero, M., and Pietrobon, D. (2019). Enhanced Thalamocortical Synaptic Transmission and Dysregulation of the Excitatory-Inhibitory Balance at the Thalamocortical Feedforward Inhibitory Microcircuit in a Genetic Mouse Model of Migraine. J. Neurosci. 39, 9841–9851. doi:10.1523/JNEUROSCI.1840-19.2019
Van Den Maagdenberg, A. M., Pietrobon, D., Pizzorusso, T., Kaja, S., Broos, L. A., Cesetti, T., et al. (2004). A Cacna1a Knockin Migraine Mouse Model With Increased Susceptibility to Cortical Spreading Depression. Neuron. 41, 701–710. doi:10.1016/s0896-6273(04)00085-6
Van Den Maagdenberg, A. M., Pizzorusso, T., Kaja, S., Terpolilli, N., Shapovalova, M., Hoebeek, F. E., et al. (2010). High Cortical Spreading Depression Susceptibility and Migraine-Associated Symptoms in Ca(V)2.1 S218L Mice. Ann. Neurol. 67, 85–98. doi:10.1002/ana.21815
Vecchia, D., and Pietrobon, D. (2012). Migraine: a Disorder of Brain Excitatory-Inhibitory Balance? Trends Neurosci. 35, 507–520. doi:10.1016/j.tins.2012.04.007
Vecchia, D., Tottene, A., Van Den Maagdenberg, A. M., and Pietrobon, D. (2014). Mechanism Underlying Unaltered Cortical Inhibitory Synaptic Transmission in Contrast With Enhanced Excitatory Transmission in CaV2.1 Knockin Migraine Mice. Neurobiol. Dis. 69, 225–234. doi:10.1016/j.nbd.2014.05.035
Vereczkei, A., Abdul-Rahman, O., Halmai, Z., Nagy, G., Szekely, A., Somogyi, A., et al. (2019). Association of Purinergic Receptor P2RX7 Gene Polymorphisms With Depression Symptoms. Prog. Neuropsychopharmacol. Biol. Psychiatry. 92, 207–216. doi:10.1016/j.pnpbp.2019.01.006
Vyas, A., Bernal, S., and Chattarji, S. (2003). Effects of Chronic Stress on Dendritic Arborization in the Central and Extended Amygdala. Brain Res. 965, 290–294. doi:10.1016/s0006-8993(02)04162-8
Weinhard, L., Di Bartolomei, G., Bolasco, G., Machado, P., Schieber, N. L., Neniskyte, U., et al. (2018). Microglia Remodel Synapses by Presynaptic Trogocytosis and Spine Head Filopodia Induction. Nat. Commun. 9, 1228. doi:10.1038/s41467-018-03566-5
Whyte-Fagundes, P., and Zoidl, G. (2018). Mechanisms of Pannexin1 Channel Gating and Regulation. Biochim. Biophys. Acta Biomembr. 1860, 65–71. doi:10.1016/j.bbamem.2017.07.009
Wohleb, E. S., Franklin, T., Iwata, M., and Duman, R. S. (2016). Integrating Neuroimmune Systems in the Neurobiology of Depression. Nat. Rev. Neurosci. 17, 497–511. doi:10.1038/nrn.2016.69
Wohleb, E. S., Terwilliger, R., Duman, C. H., and Duman, R. S. (2018). Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biol. Psychiatry. 83, 38–49. doi:10.1016/j.biopsych.2017.05.026
Ye, D., Li, Y., Zhang, X., Guo, F., Geng, L., Zhang, Q., et al. (2015). TREK1 Channel Blockade Induces an Antidepressant-Like Response Synergizing With 5-HT1A Receptor Signaling. Eur. Neuropsychopharmacol. 25, 2426–2436. doi:10.1016/j.euroneuro.2015.09.007
Yeung, A. K., Patil, C. S., and Jackson, M. F. (2020). Pannexin-1 in the CNS: Emerging Concepts in Health and Disease. J. Neurochem. 154, 468–485. doi:10.1111/jnc.15004
Yue, N., Huang, H., Zhu, X., Han, Q., Wang, Y., Li, B., et al. (2017). Activation of P2X7 Receptor and NLRP3 Inflammasome Assembly in Hippocampal Glial Cells Mediates Chronic Stress-Induced Depressive-Like Behaviors. J. Neuroinflammation. 14, 102. doi:10.1186/s12974-017-0865-y
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., et al. (2014). Deficient Neuron-Microglia Signaling Results in Impaired Functional Brain Connectivity and Social Behavior. Nat. Neurosci. 17, 400–406. doi:10.1038/nn.3641
Keywords: ATP1A2, HCN, KCNQ, CACNA, TREK, Pannnexin-1, P2X7R, neuroinflammation
Citation: Eren-Koçak E and Dalkara T (2021) Ion Channel Dysfunction and Neuroinflammation in Migraine and Depression. Front. Pharmacol. 12:777607. doi: 10.3389/fphar.2021.777607
Received: 15 September 2021; Accepted: 22 October 2021;
Published: 10 November 2021.
Edited by:
Jacques Joubert, University of the Western Cape, South AfricaReviewed by:
Yong Li, Shanghai Jiao Tong University, ChinaMassimo Mantegazza, UMR7275 Institut de Pharmacologie Moléculaire et Cellulaire (IPMC), France
Copyright © 2021 Eren-Koçak and Dalkara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Turgay Dalkara, dGRhbGthcmFAaGFjZXR0ZXBlLmVkdS50cg==