
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 05 January 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.774069
This article is part of the Research TopicChemoinformatics Approaches to Structure- and Ligand-Based Drug Design, Volume IIView all 20 articles
 Ivani Pauli1†
Ivani Pauli1† Celso de O. Rezende Jr.2†
Celso de O. Rezende Jr.2† Brian W. Slafer2
Brian W. Slafer2 Marco A. Dessoy2
Marco A. Dessoy2 Mariana L. de Souza1
Mariana L. de Souza1 Leonardo L. G. Ferreira1Abraham L. M. Adjanohun3Rafaela S. Ferreira3
Leonardo L. G. Ferreira1Abraham L. M. Adjanohun3Rafaela S. Ferreira3 Luma G. Magalhães1Renata Krogh1Simone Michelan-Duarte1Ricardo Vaz Del Pintor4Fernando B. R. da Silva4
Luma G. Magalhães1Renata Krogh1Simone Michelan-Duarte1Ricardo Vaz Del Pintor4Fernando B. R. da Silva4 Fabio C. Cruz4
Fabio C. Cruz4 Luiz C. Dias2*
Luiz C. Dias2* Adriano D. Andricopulo1*
Adriano D. Andricopulo1*Cruzain, the main cysteine protease of Trypanosoma cruzi, plays key roles in all stages of the parasite’s life cycle, including nutrition acquisition, differentiation, evasion of the host immune system, and invasion of host cells. Thus, inhibition of this validated target may lead to the development of novel drugs for the treatment of Chagas disease. In this study, a multiparameter optimization (MPO) approach, molecular modeling, and structure-activity relationships (SARs) were employed for the identification of new benzimidazole derivatives as potent competitive inhibitors of cruzain with trypanocidal activity and suitable pharmacokinetics. Extensive pharmacokinetic studies enabled the identification of metabolically stable and permeable compounds with high selectivity indices. CYP3A4 was found to be involved in the main metabolic pathway, and the identification of metabolic soft spots provided insights into molecular optimization. Compound 28, which showed a promising trade-off between pharmacodynamics and pharmacokinetics, caused no acute toxicity and reduced parasite burden both in vitro and in vivo.
Endemic in Latin America, Chagas disease affects 6–7 million people worldwide and has become an emerging public health problem in nonendemic countries1. Among nonendemic nations, the greatest burden occurs in the United States, which is estimated to have approximately 300,000 cases of the disease (Pérez-Molina and Molina, 2018). Chagas disease kills ∼12,000 people annually, and 70 million people are at risk of infection in the Americas2. Moreover, the disease is an important cause of infectious cardiopathy worldwide, playing a key role in the global prevalence of cardiovascular disease (Bern, 2015; Cucunubá et al., 2016). Chagas disease significantly impacts the productivity of endemic countries, which are estimated to lose more than US $7.2 billion per year because of the disease (GBD DALYs and HALE Collaborators, 2016; Arnal et al., 2019). According to the World Health Organization (WHO), the development of innovative therapeutic approaches is required for this neglected tropical disease (NTD) because of the lack of efficient control measures and the insufficient research and development (R&D) funding. The need for novel therapeutic approaches has become more evident this year, as the WHO released a new roadmap for NTDs for 2021–2030, whose target is to eliminate the epidemics of these diseases by 2030. Chemotherapy for Chagas disease consists of benznidazole (BZ) and nifurtimox, two nitro compounds that have limited efficacy and produce serious adverse reactions that lead up to 40% of patients to discontinue treatment (Rodriques Coura and De Castro, 2002). Given these shortcomings, the development of novel, effective and safe drugs for the treatment of Chagas disease is critically needed.
Cruzain (EC 3.4.22.51), the main cysteine protease of Trypanosoma cruzi, has been broadly explored as a molecular target in Chagas disease drug discovery (Engel et al., 1998; McKerrow, 1999; Jose Cazzulo et al., 2001; Massarico Serafim et al., 2014). This enzyme plays a key role in all stages of the parasite’s life cycle, participating in processes such as nutrition, differentiation, evasion of the host immune system, and invasion of host cells (Ferreira and Andricopulo, 2017). Genetic studies of T. cruzi and the efficacy of cruzain inhibitors in reducing parasite load in vivo have validated the enzyme as a molecular target for the discovery of novel drugs for Chagas disease (Zanatta et al., 2008; Doyle et al., 2011; Ndao et al., 2014). Following these investigations, various classes of cruzain inhibitors, such as nitroalkenes, vinyl sulfones, thiosemicarbazones, and triazoles, have been described in the literature (Rogers et al., 2012; Avelar et al., 2015; Espíndola et al., 2015; Neitz et al., 2015; Latorre et al., 2016). In this work, we describe the design, synthesis, and in vitro and in vivo evaluations of novel benzimidazole derivatives. In addition to improving pharmacodynamic properties, such as binding affinity and potency, we evaluated the pharmacokinetic (PK) profile of newly synthesized and previously described benzimidazoles (Ferreira et al., 2014) by applying a multiparameter optimization (MPO) approach. MPO has increasingly been adopted in the early phases of pharma R&D to exclude pipeline compounds that feature poor PK profiles as early as possible (Eddershaw et al., 2000; Andricopulo and Montanari, 2005; Wang et al., 2007; Wang, 2009; Wang and Skolnik, 2009). This study led to the discovery of potent cruzain inhibitors with trypanocidal activity and innovatively contributed to the identification of compounds with improved safety and PK profiles to be explored for Chagas disease drug discovery.
Pro-cruzain truncated at the C-terminus was expressed and purified using a previously described protocol (Ferreira et al., 2019). Escherichia coli (strain M15) cultures were grown overnight at 37°C and 200 rpm in Luria Bertani (LB) medium supplemented with ampicillin (100 μg/ml) and kanamycin (50 μg/ml). Next, the cultures were diluted 10-fold in fresh LB medium supplemented with 0.5 M NaCl, 0.2% glucose, 1 mM betaine, 0.5 M sorbitol, 100 μg/ml ampicillin, and 50 μg/ml kanamycin and incubated at 37°C and 200 rpm. At an optical density (OD600) of 0.9, the cultures were incubated at 47°C for 20 min to promote the expression of chaperones. Then, the expression of cruzain was induced by adding isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.2 mM, which was followed by overnight incubation of the cultures at 20°C and 200 rpm. Next, the cultures were centrifuged (5,000 rpm, 30 min, 4°C), and the cells were suspended in 50 ml of lysis buffer (300 mM NaCl, 50 mM Tris-HCl, and 1.6 mg/ml lysozyme, pH 8.0) per liter of culture and lysed by sonication (12 cycles of 30 s). This cell lysate was centrifuged (9,000 rpm, 30 min, 4°C), and the supernatant was collected. Cruzain was precipitated by incubation with 35% ammonium sulfate (2 h), and this suspension was centrifuged at 9,000 rpm for 30 min at 4°C. The precipitated cruzain was resuspended in lysis buffer, and the sample was dialyzed to eliminate ammonium sulfate. The soluble fraction of the dialysate was loaded on a Ni−NTA column (Qiagen, Hilden, Germany), and the contaminants were washed using washing buffer (300 mM NaCl, 50 mM Tris-HCl, and 10 mM imidazole, pH 8.0). Cruzain was eluted by applying an increasing imidazole gradient: 25, 50, 75, 100, and 250 mM. The fractions containing cruzain were pooled together and dialyzed against 1.5 L of 0.1 M acetate buffer, pH 5.5, and then concentrated to 0.5 mg/ml. Pro-cruzain was activated by incubation with activation buffer (100 mM sodium acetate, pH 5.5, 10 mM EDTA, 5 mM DTT, and 1 M NaCl) at 37°C. The activation of cruzain was monitored by following the enzymatic activity at 30-min intervals, and the process was observed to stop after approximately 1 h. After activation, the enzyme was diluted 20-fold in binding buffer (20 mM sodium phosphate and 150 mM NaCl, pH 7.2) and added to thiopropyl Sepharose 6B resin (GE Healthcare Life Sciences, Pittsburgh, PA). After overnight incubation at 4°C, the resin was loaded on a column, and cruzain was eluted with binding buffer supplemented with 20 mM DTT. Fractions containing cruzain were pooled together and stored in 0.1 M sodium acetate, pH 5.5, at −80°C.
Cruzain activity was followed by monitoring the cleavage of the fluorogenic substrate Z-Phe-Arg-aminomethyl coumarin (Z-FR-AMC), as previously described (Ferreira et al., 2019), using 96-well flat-bottom black plates and wavelengths of 355 nm for excitation and 460 nm for emission. All cruzain assays were performed in 0.1 M sodium acetate buffer with 5 mM dithiothreitol (DTT) and 0.01% Triton X-100, pH 5.5. The final concentration of cruzain was 1.5 nM, and the substrate concentration was 5.0 μM (Km = 1.6 μM), except in the experiments for Ki determination, in which several concentrations of substrate were used. The cleavage of the substrate was monitored for 5 min, and the activity was calculated based on the initial reaction rates compared with the rate of a DMSO control at 30°C. The IC50 values were independently calculated by considering the rate measurements for at least six inhibitor concentrations, each evaluated in triplicate. To determine the mechanism of cruzain inhibition, eight concentrations of the substrate Z-FR-AMC and four concentrations of the inhibitor were employed, each in triplicate. Kinetic parameters were determined using the SigmaPlot (Systat Software Inc., Erkrath, Germany) enzyme kinetics module. Compounds were tested in two or three independent experiments. All enzyme assays were performed using varying Triton X-100 concentrations (0, 0.01, and 0.1%) (Ferreira et al., 2009). Compound concentrations were 100 μM in the single-dose percentage inhibition assays.
Rhodesain activity was measured using a fluorescence-based assay as previously described (Fonseca et al., 2015). The cleavage rates of the fluorogenic substrate Z-Phe-Arg-aminomethyl coumarin (Z-FR-AMC) were monitored at wavelengths of 340 nm for excitation and 440 nm for emission. All assays were performed in triplicate in a 0.1 M sodium acetate buffer, pH 5.5, with 1 mM beta-mercaptoethanol and 0.01% Triton X-100. The final concentration of rhodesain was 3 nM, and the substrate concentration was 2.5 μM. The cleavage of the substrate was followed by continuous reading for 5 min, and enzyme activity in the presence of 100 μM of each potential inhibitor was calculated based on initial velocity rates compared to DMSO controls. All compounds were tested in triplicate in three independent experiments.
The three-dimensional structures of the cruzain inhibitors were constructed using the standard geometric parameters embedded in SYBYL-X 2.1 (Certara, Princeton, NJ). Each compound was energetically minimized employing the Tripos force field (Clark et al., 1989) and Powell conjugate gradient method (Powell, 1977), with a convergence value of 0.05 kcal/mol.Å, and the Gasteiger-Hückel model was used for charge calculation (Gasteiger and Marsili, 1980). The molecules were docked using GOLD 5.3 (Cambridge Crystallographic Data Centre, Cambridge, United Kingdom) (Jones et al., 1997; Verdonk et al., 2003) against the X-ray structure of cruzain (PDB ID 3KKU, 1.28 Å) (Ferreira et al., 2010). The preparation of the cruzain structure consisted of removing all water molecules and inserting hydrogen atoms. The active site Cys25 was kept negatively charged, and His162 was kept protonated. The binding site was defined as a sphere with a 10 Å radius centered on the Cys25 sulfur atom. The default GOLD parameters were applied for the molecular docking runs, except for the search efficiency, which was changed to its maximum value of 200%. The generated poses were evaluated using the GoldScore scoring function, and the analysis of the binding conformations was visualized using PyMOL 3.1 (Schrödinger, New York, NY) (Lill and Danielson, 2011).
Biological assays against T. cruzi intracellular amastigotes were performed as reported previously using the T. cruzi Tulahuen strain, which is genetically engineered to express the E. coli β-galactosidase gene lacZ (Buckner et al., 1996). β-Galactosidase catalyzes a colorimetric reaction with chlorophenol red β-D-galactopyranoside (CPRG, Sigma Chemical Co., St. Louis, MO) as the substrate. The assays were conducted in 96-well tissue culture plates, and the compounds to be tested were prepared in 100% DMSO. Epimastigotes were maintained in liver infusion tryptone (LIT) enriched with 10% fetal calf serum (FCS), streptomycin, and penicillin at 28°C. Epimastigotes were converted to trypomastigotes by incubation in Grace’s insect medium (Sigma–Aldrich, St. Louis, MO) enriched with 10% FCS at 28°C. Human HFF-1 fibroblasts were seeded at 2 × 103/well in 80 μl of RPMI 1640 without phenol red and incubated overnight at 37°C and 5% CO2. Trypomastigotes were seeded at 1.0 × 104/well in 20 μl of RPMI 1640, and the plates were incubated at 37°C and 5% CO2. The next day, the synthesized compounds were added (50 μl) in 3-fold serial dilutions at concentrations ranging from 0.4 to 300 μM, and the plates were incubated at 37°C and 5% CO2. Each compound concentration was assayed in triplicate. After 120 h, 50 μl of chlorophenol red β-D-galactopyranoside (CPRG, Sigma–Aldrich) and IGEPAL CA-630 (Sigma–Aldrich) at a final concentration of 0.1% were added. The absorbance was measured at a wavelength of 570 nm in an automated microplate reader. The data were transferred to SigmaPlot 10.0 (Systat Software Inc., Erkrath, Germany) to determine the IC50 values. Benznidazole (BZ, Sigma–Aldrich) was used as a positive control, and untreated wells (100% parasite growth) were used as negative controls in all plates. All compounds were tested in three independent assays.
The synthesized compounds were evaluated for their cytotoxicity against HFF-1 cells using the MTS assay (Promega, Madison, WI) (Barltrop et al., 1991) as previously described (Ferreira et al., 2019). HFF-1 fibroblasts were plated at 2 × 103/well in 96-well culture plates in RPMI 1640 without phenol red enriched with 10% FCS and incubated overnight at 37°C and 5% CO2. Next, 7 concentrations (0.1–100 µM) of the compounds were added in 3-fold serial dilutions, each concentration in triplicate, and the plates were incubated for 72 h at 37°C and 5% CO2. Next, 20 µl of MTS was added to each well, and the plates were incubated for an additional 4 h at 37°C and 5% CO2. The absorbance was measured at 490 nm using a spectrophotometer, and the data were transferred to SigmaPlot 10.0 (Systat Software Inc., Erkrath, Germany) to determine the IC50 values. Doxorubicin (Sigma–Aldrich) was used as a positive control, and untreated wells (100% growth) were used as negative controls in all plates. All compounds were tested in two independent assays.
Isolated mouse (BD Gentest, Bedford, MA) and human liver microsomes (XenoTech, Kansas City, KS) (Plant, 2004) were added at a final concentration of 0.25 mg/ml to a solution containing 40 mM dibasic potassium phosphate and 10 mM monobasic potassium phosphate. Stock solutions of compounds at 5 mM were prepared in 100% DMSO. A 50:50 quenching solution of acetonitrile (ACN) and methanol (MeOH) was prepared. An NADPH solution was prepared at 10 mM. The preparations containing the microsomes were added to each well of the incubation plate (450 µl), which was then heated to 37°C for 10 min. The compounds were added to the respective wells of the test plate (2 µl). Then, 300 µl of the microsome preparation was added to each well of the test plate. The test plate was heated under gentle rotation for 5 min at 37°C. Next, 90 µl of the mixture contained in the test plate was added to the incubation plate, making a final volume of 540 µl. Samples were collected at the following incubation times: 0, 5, 10, 15, 20, and 30 min. To the 0 min sample plate, quenching solution (180 µl), NADPH (6 µl), and the incubation plate mixture (54 µl) were added to each well. The sample plate was then sealed, homogenized, and stored at 4°C. Then, 54 µl of the NADPH solution was added to the incubation plate, which was homogenized. Before the collection of each sample at the established times, 45 µl of the quenching solution was added to each well of the corresponding sample plates. After reaching the incubation times, 60 µl of the mixture contained in the incubation plate was added to the corresponding sample plates. The sample plates were then sealed and stored at 4°C. After the collection of the last sample (30 min), all sample plates were centrifuged at 3,800 rpm for 30 min. The supernatant from each well was collected and transferred to clean plates for mass spectrometry. Five to 10 µl of each sample was injected into an AB Sciex Triple Quad 5500 LC-MS/MS instrument.
Stock solutions of compounds at 5 mM were prepared in 100% DMSO. An NADPH solution at 10 mM was prepared in a 50 mM potassium phosphate buffer. A solution at 100 pmol/ml of each of the recombinant CYP450 enzymes (1A2, 2C8, 2C9, 2C19, 2D6, and 3A4) was prepared in 50 mM potassium phosphate buffer (Proctor et al., 2004). A 50:50 quenching solution of ACN and MeOH was prepared. 320 µl of each CYP solution was added to the incubation plate, and 1 µl of each compound was added to the compound plate. Then, 100 µl of each CYP solution was added to the compound plate, and 10 µl from the compound plate was added to the incubation plate. The incubation plate was heated to 37°C with 600 rpm rotation for 10 min. To the 0 min plate, 30 µl of quenching solution, 1 µl of NADPH, and 9 µl of the incubation plate solution were added to each well, and the plate was sealed and stored at 4°C. Next, 10-µl samples were collected from the incubation plate at different incubation times (5, 10, 20, 30, and 60 min). For each time point, a separate plate containing 30 µl of quenching solution and incubation plate solution was used. After collecting the last sample, the plates were centrifuged for 20 min at 3,000 rpm. The supernatant from each well was collected and transferred to new plates for mass spectrometry. Five to 10 µl of each sample was injected into an AB Sciex Triple Quad 5500 LC-MS/MS instrument. The compounds were tested at a final concentration of 5 µM.
Rat and human hepatocytes were used (McGinnity, et al., 2004). Samples were collected at six different time points during the incubation period and analyzed by LC-MS/MS to determine the T½ and the intrinsic clearance. Stock solutions of the CYP inhibitors azamulin (8.75 mM) and 1-ABT (350 mM) were prepared. Next, the test compounds were added to 96-well plates and incubated in Williams medium (Invitrogen, A12176-01) supplemented with 2 mM L-glutamine and 15 mM HEPES to reach a final concentration of 2 µM. Then, compounds (50 µl) were transferred to other plates, one plate for each time point (0, 15, 30, 60, 120, and 240 min), and incubated at 37°C and 5% CO2 for 30 min. Cryopreserved hepatocytes were heated in a wet bath at 37°C and dispensed in InVitroGRO HT medium supplemented with 10% FCS, 0.15 µM hydrocortisone, 0.2 mg/ml BSA, fructose, insulin, and amino acids. The cells were centrifuged at 500 rpm for 5 min. The supernatant was discarded, and the cells were resuspended in 1 ml of incubation medium heated to 37°C. Next, the cells were counted and diluted to 1 × 106/ml in incubation medium. The cell suspension was divided into 3 samples: without CYP inhibitors; with 25 µM azamulin, a CYP3A4 inhibitor; and with 1 mM 1-ABT, an inhibitor of all CYPs. Each hepatocyte sample with 12,500 cells/well was incubated with either azamulin or 1-ABT for 30 min (except the group without CYP inhibitor). To the plates with the test compounds, 12,500 cells/well were added. The plates were incubated in a shaker at 37°C, 5% CO2, and 300 rpm. At the specified time points, the hepatocyte enzymatic activity was interrupted by the addition of 75 µl of cold ACN, and the samples were read by LC-MS/MS.
The permeability of the compounds was assessed using the PAMPA method (Yu et al., 2015). The test compounds were dissolved in DMSO to a concentration of 5 mM. Next, the compounds were diluted in a stock plate in saline phosphate buffer (PBS), pH 6.5, containing 1% DMSO to a concentration of 1 µM. Then, 300 µl of each test compound was added to the donor plate. Afterward, 200 µl of PBS buffer, pH 7.4, was added to the acceptor plate. The donor plate was attached to the acceptor plate. The assembled acceptor-donor plate was then incubated at 37°C for 5 h under gentle agitation. To analyze the concentration of the compounds by mass spectrometry, two analysis plates (one for the donor and another for the acceptor plate) containing 300 μl of MEOH:ACN 50:50 were prepared. To the donor analysis plate, 90 µl of PBS, pH 6.5, and 10 µl of the content of the donor plate were added. For the acceptor analysis plate, 100 µl of the content of the acceptor plate was added. To monitor any potential decomposition/intrinsic instability of the test compounds in solution, samples from the stock plate were subjected to LC-MS/MS. The compounds that presented Pe ≥ 1.5 × 10−6 cm/s were classified as permeable, while compounds that presented Pe < 1.5 × 10−6 cm/s were considered poorly permeable or nonpermeable.
For the eLogD assays (Waring, 2010), each compound (5 μl) from a 5 mM stock solution was diluted in 245 μl of a solution containing buffers A (5% MeOH, 10 mM ammonium acetate, pH 7.4) and B (100% MeOH, pH 7.4) (50:50). Nine control compounds with known eLogD and column retention times (acyclovir, atenolol, antipyrine, fluconazole, metoprolol, carbamazepine, ketoconazole, tolnaftate, and amiodarone; eLogD values ranging from −1.86 to 6.1) were subjected to LC-MS/MS in triplicate before and after the analysis of the test compounds. Retention times were recorded for each control and test compound in a C18 column. The retention time of each control compound was plotted against the respective eLogD values described in the literature (Lombardo et al., 2002; Lombardo et al., 2004; Alelyunas et al., 2010). The resulting linear equation (y = mx + b) was used to calculate the eLogD values of the test compounds, in which x is the retention time in minutes and y is the eLogD value.
The fraction unbound (fu) (Masimirembwa et al., 2003) of the test compounds was determined after incubation with different media, namely, plasma, microsomes, and buffer with 10% FCS. Equilibrium dialysis was performed in a 96-well plate (HT-Dialysis, Gales Ferry, CT) in which each well was divided by a semipermeable membrane (12–14 kDa cutoff). The test compounds diluted in either plasma, microsome suspension, or buffer were added to one side of the membrane. Potassium phosphate buffer (50 mM, pH 7.4) was added to the other side of the membrane. A standard curve was used to calculate the compound concentration (fu) on each side of the membrane. Stock solutions of each compound were prepared in 100% DMSO to obtain a final concentration of 1 mM. The following compound concentrations were used: 1, 2, 20, 200, 2,000 and 5,000 nM. To 500 μl of medium, 0.5 μl of the compound stock solution was added, and this was applied to one side of the well. Each compound was evaluated in triplicate. Next, the plate was assembled, sealed, and incubated at 37°C under rotation (150 rpm) for 4 h. After the incubation period, the plates were subjected to LC-MS/MS.
Stock solutions of compounds at 5 mM were prepared in 100% DMSO for the biotransformation analyses (Obach, 1999). Test solutions of compounds at 1 mM were prepared in H2O/MeOH: 2/1 (v/v). A solution of 10 mM NADPH, a 50:50 quenching solution of ACN and MeOH, and 100 mM phosphate buffer was prepared. Isolated mouse (BD Gentest, Bedford, MA) and human liver microsomes (XenoTech, Kansas City, KS) were dissolved to 2 mg/ml in phosphate buffer. One plate for the 0 min time point and another for the 60 min time point were prepared. To these plates, 178 μl of the microsome solution and 2 μl of the compound stock solution (final test concentration of 5 μM) were added. The plates were incubated at 37°C for 5 min under gentle agitation. Next, 400 μl of the quenching solution was added to the 0 min plate, which was followed by the addition of 20 μl of NADPH. The plate was sealed and kept under refrigeration (4°C). To the 60 min plate, 20 μl of NADPH was added, and the plate was sealed and incubated at 37°C for 60 min under gentle agitation. Next, 400 μl of quenching solution was added to the 60 min plate. The two plates were centrifuged (4°C, 3300 rpm, 30 min), and the supernatant was collected for mass spectrometry.
The in vivo pharmacokinetic profiles of the compounds were determined using male CD1 mice weighing 50 g (Davies and Morris, 1993; Liu and Jia, 2007). The compounds were administered in a single dose orally (0.5 mg/kg) and intravenously (0.5 mg/kg). Stock solutions of the compounds in DMSO were diluted in Tween 80, PEG-400, and D5W (5% dextrose in water) at a ratio of 2:5:20:73 (v/v). The injection volume was 10 ml/kg. The remaining plasma concentration was monitored over time by LC-MS/MS by collecting blood samples (40 μl) at 10, 25, and 50 min and 1, 3, 6, 9, 12, and 24 h after administration of the compound.
For the biotransformation experiments and analysis of metabolites in microsomes, an Ultra-High-Pressure Liquid Chromatography instrument (UHPLC, Thermo Accela, Waltham, MA) (Spaggiari et al., 2014) connected to an automatic sample injector and a 1250 series pump was used. The UHPLC system was connected to a Thermo Fisher (Waltham, MA) LTQ Orbitrap mass spectrometer. For all other analyses, an AB Sciex Triple Quad 5500 coupled to a UHPLC equipped with a UV 1290 diode detector (Agilent Technologies, Santa Clara, CA) and a CTC PAL self-collecting system (LEAP Technologies, Carrboro, NC) was used. Q1 MS positive ion mode (300–500 Da) was used to detect the ions of the parent compounds. The UV detector was operated in spectral mode (250–280 nm). A Hypersil Gold C18 (2.1 mm × 100 mm, 1.9 µm, Thermo Fisher) HPLC column was used. The mobile phases were solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in ACN). The flow was adjusted to 0.55 ml/min, and the injection volume was adjusted to 20 ml. The gradient started with 1% solvent B for 0.4 min, reached 40% (solvent B) in 2.3 min and 95% (solvent B) in 0.67 min, was maintained for 0.5 min, and returned to the initial condition of 1%. This condition was maintained for 1 min before injection of the next sample. The peak area ratio (peak area of the test compounds/peak area of the control compounds) was converted to the percentage of remaining compound, with the 0 min time point ratio set to 100%. T½ and CLint were calculated from the percentage of remaining compound versus the incubation time. From the resulting function, the slope (k) was determined. The equations T1/2 (min) = ln(2)/k and CLint in vitro (μL/min/mg) = k*1000/0.25 were used to determine T½ and CLint.
Thirty-day-old female Swiss mice weighing 20–25 g and procured from the Center for the Development of Experimental Models for Medicine and Biology (CEDEME/UNIFESP) served as the subjects for these experiments. Animals were housed (5–6 per cage) in polypropylene cages and kept under controlled temperature (22–23°C) and humidity on a 12-h light/dark cycle (12 h light, 12 h dark; lights on at 6:30 am). Rodent chow and water were available ad libitum throughout the experiments. The Committee of Ethics in Research of the Universidade Federal de São Paulo approved all the experiments (CEUA n° 5301080816).
Unless stated otherwise, all reactions were performed under an atmosphere of argon with dry solvents and magnetic stirring (detailed organic synthesis methods are in the Supplementary Material). Dichloromethane (DCM) and triethylamine (Et3N) were distilled from CaH2. Tetrahydrofuran (THF) was distilled from sodium/benzophenone. Dimethyl formamide (DMF) was purchased from Aldrich (anhydrous) and used without further purification. Yields refer to homogeneous materials obtained after purification of reaction products by flash column chromatography using silica gel (200–400 mesh) or recrystallization. Analytical thin-layer chromatography was performed on silica gel 60 and GF (5–40 μm thickness) plates, and the plates were treated with a basic potassium permanganate stain or ninhydrin solution, heated and visualized under UV light. Melting points were measured with a Buchi M-565 instrument and are uncorrected. 1H and proton-decoupled 13C NMR spectra were acquired in CDCl3, CD3OD or d6-DMSO at 250 MHz (1H) and 62.5 MHz (13C) (Bruker DPX250), 400 MHz (1H) and 100 MHz (13C) (Bruker AVANCE 400), 500 MHz (1H) and 125 MHz (13C) (Varian Inova 500), or 600 MHz (1H) and 150 MHz (13C) (Bruker AVANCE 600). Chemical shifts (δ) are reported in ppm using residual undeuterated solvent as an internal standard (CDCl3 at 7.26 ppm, CD3OD at 3.31 ppm, d6-DMSO at 2.50 ppm, and TMS at 0.00 ppm for 1H NMR spectra and CDCl3 at 77.16 ppm, CD3OD at 49.0 ppm, d6-DMSO at 39.52 ppm for 13C NMR spectra). Multiplicity data are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, br s = broad singlet, dd = doublet of doublets, dt = doublet of triplets, app d = apparent doublet, app t = apparent triplet, m = multiplet, and br m = broad multiplet. The multiplicity is followed by the coupling constant(s) in Hz and integration. High-resolution mass spectrometry (HRMS) was measured using electrospray ionization (ESI) (Waters xevo Q-tof, Thermo LTQ-FT ultra, or Thermo Q Exactive) or using electron ionization (EI) (GCT Premier Waters). The synthesis and characterization of compounds 1, 17, 18, 31–63 were previously reported (Ferreira et al., 2014).
Phenoxyacetic acids of type I were prepared from the corresponding substituted phenols by nucleophilic substitution with 2-bromoacetic acid or nucleophilic substitution with alkyl 2-bromoacetic ester, followed by ester hydrolysis (Scheme 1A). A subsequent reaction of activated carboxylic acid I with amine II led to the formation of amides 1–4, 6, 8, 10–12, 14, and 16–18. Alcohols 5 and 7 and aniline 9 were prepared by reduction reactions of imides 4 and 6 with sodium borohydride and nitrobenzene derivative 8 using hydrogenation under Pd/C catalysis. Carboxylic acid derivatives 13 and 15 were synthesized by hydrolysis under basic conditions of methyl esters 12 and 14, respectively. N-alkylated compounds 19–29 were synthesized by N-alkylation of the benzimidazole moiety of compounds 1, 17, and 18 with different electrophiles. N-Phenyl derivative 30 was prepared as described in Scheme 1B by an amidation reaction followed by cyclization and dehydration.
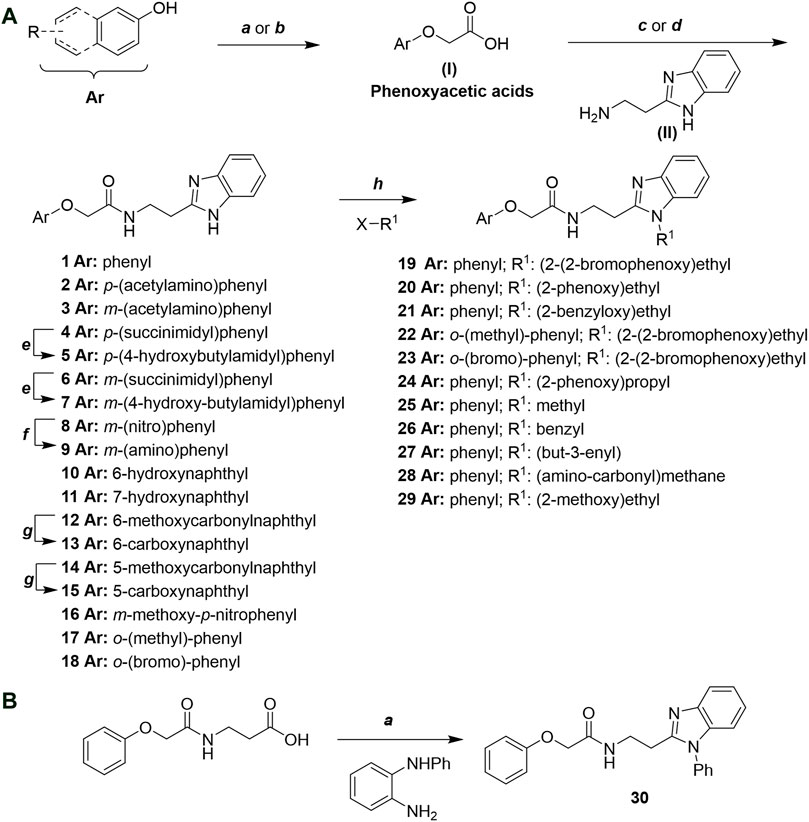
Scheme 1. (A) Reagents and conditions: (a) i) ethyl 2-bromoacetate, K2CO3, DMF, r.t., 4–6 h; ii) NaOH (6 mol. L−1), MeOH, r.t., 30 min; iii) HCl (6 mol. L−1), 0 C, 10 min; (b) i) benzyl 2-bromoacetate, K2CO3, DMF, r.t., 4–6 h; ii) Pd/C (20%), H2(g), EtOAc, MeOH, r.t., 1–2 h; (c) i) oxalyl chloride, DMF, DCM, r.t., 1 h; ii) N-Hydroxysuccinimide, DCM, triethylamine, 0°C, 30 min; iii) II, sodium carbonate, EtOAc, r.t., 1 h; (d) II, EDC, HOBt, trimethylamine, DMF, r.t., 8–15 h; (e) sodium borohydride, MeOH, THF, r.t., 5 h; (f) Pd/C (20%), H2(g), MeOH, r.t., 2 h; (g) i) NaOH (6 mol. L−1), MeOH, r.t., 20 min; ii) HCl (6 mol. L−1), 0°C, 10 min; (h) haloalkyls, 18-crown-6, potassium tert-butoxide, THF, r.t. or 45°C, 13–48 h. (B) Reagents and conditions: (a) i) oxalyl chloride, DMF, DCM, r.t., 30 min, ii) N1-phenylbenzene-1,2-diamine, n-butanol, 110°C, 18 h.
In this work, we designed a series of cruzain inhibitors based on a previously identified benzimidazole derivative (18, Figure 1A) (Ferreira et al., 2010; Ferreira et al., 2014). Considering the lead-like profile of compound 18 and its activity against cruzain and T. cruzi, we selected this compound for a lead optimization program and, for the first time, the pharmacokinetics and the in vivo trypanocidal ability of this molecule and its analogs were investigated. We explored compound 18 by appending diverse substituents at the phenyl and benzimidazole rings to improve both the interaction with cruzain and the PK profile. By adding substituents at the phenyl ring, we aimed to enhance the selectivity for cruzain over other proteases by promoting hydrogen bonding with Glu208, a critical residue located in the S2 subsite of the active site (Figure 1B). Glu208 is absent in most other proteases, including human cathepsins. We additionally focused on increasing the affinity and potency of the compounds by exploring N-substitutions at the benzimidazole and enabling additional interactions with the S1 and S1’ subsites.
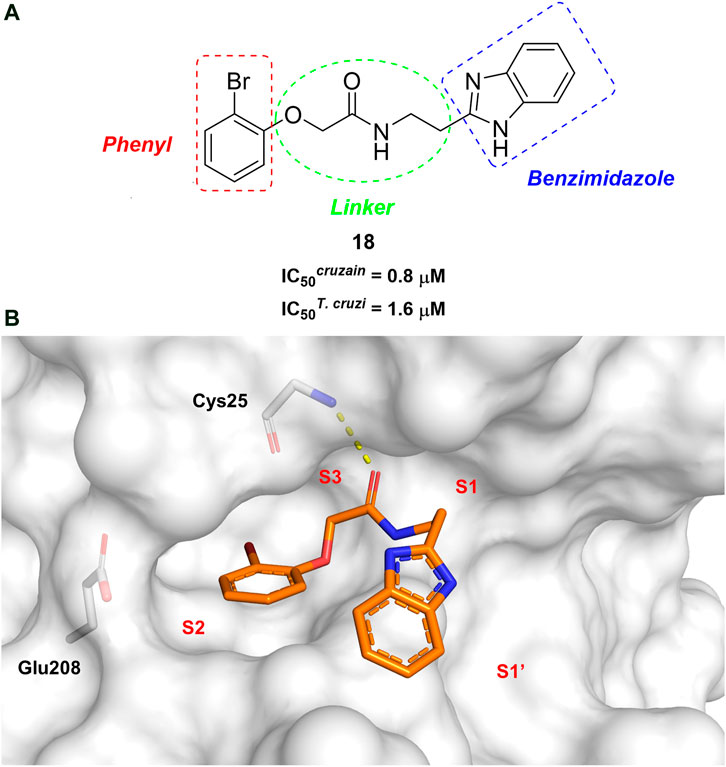
FIGURE 1. (A) Cruzain inhibitor 18 was used as the lead compound for the design of novel benzimidazole derivatives. (B) X-ray structure of compound 18 in complex with cruzain (PDB 3KKU, 1.28 Å). Binding site residues (carbon in gray) and compound 18 (carbon in orange) are shown as sticks. A hydrogen bond is shown as a dashed line. Cruzain subsites are labeled as S1, S1′, S2, and S3.
The structure and activity against cruzain of N-substituted benzimidazoles are summarized in Table 1. Three out of the derivatives that were initially evaluated showed IC50 values below 3 μM. Only compounds lacking the o-bromine at the substituent appended to the benzimidazole core were active against cruzain. No significant variation in the percent inhibition values was observed for different Triton X-100 concentrations (0, 0.01, and 0.1%), demonstrating that the inhibitors do not act as aggregators (Supplementary Table S1).
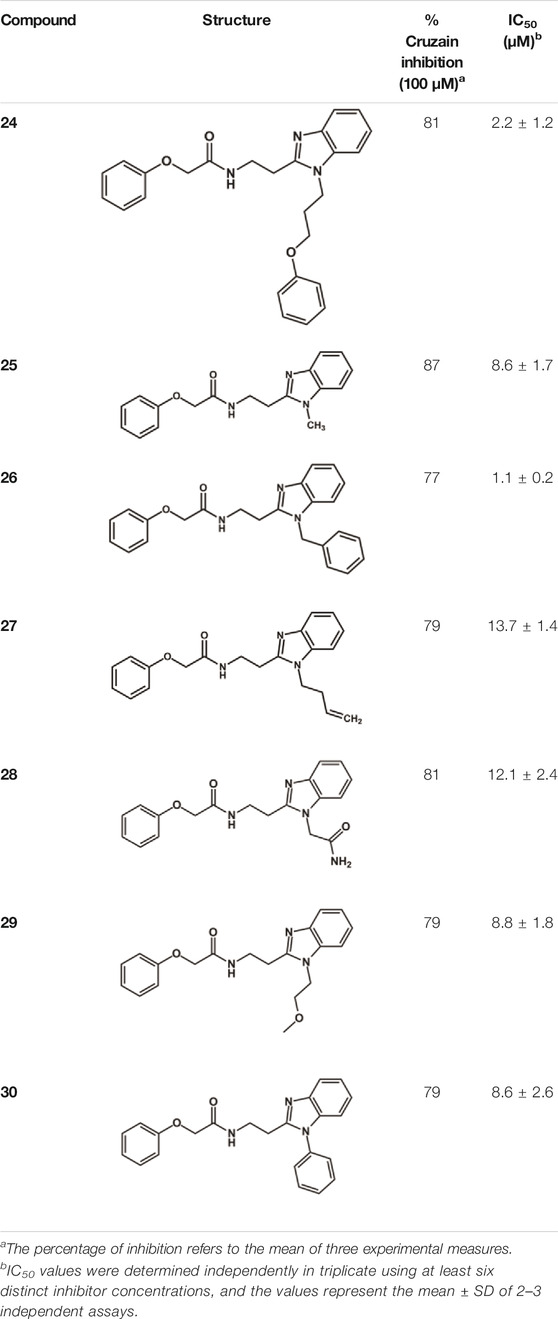
TABLE 1. Structure and activity against cruzain of new N-substituted benzimidazole derivatives.a
The mechanisms of action of compounds 20 and 24 were determined by measuring their remaining enzymatic activity in the presence of distinct concentrations of the substrate and inhibitors. Double reciprocal Lineweaver-Burk plots (Figure 2) showed that unlike the benzimidazole analogs previously described (Ferreira et al., 2014), compounds 20 and 24 act as noncompetitive cruzain inhibitors with a higher affinity for the free enzyme than for the corresponding enzyme-substrate complex. The typical behavior of noncompetitive inhibitors was additionally confirmed in another experiment, in which no significant variation in IC50 values was observed with increasing substrate concentrations at a constant protein concentration (Supplementary Table S2).
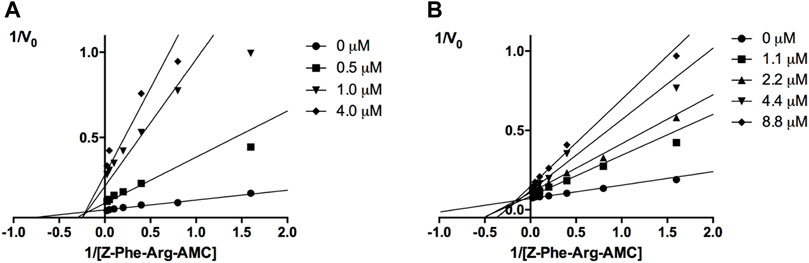
FIGURE 2. Lineweaver-Burk plots for compounds 20 (A) and 24 (B). Each curve represents a different inhibitor concentration.
Next, novel compounds were synthesized to evaluate the effect of growing the N-substituent on the mechanism of action against cruzain. Compound 1 (IC50 = 10.9 μM, Table 1), which, in contrast with lead compound 18, lacks the o-bromine at the phenyl ring, is more than 10-fold less potent than 18 (IC50 = 0.8 μM). Installing a methyl group as the N-substituent also resulted in a decrease in activity (25, IC50 = 8.6 μM). As shown in Figure 3, compounds 1 and 25 act as competitive inhibitors. Growing the N-substituent to a benzyl (26, IC50 = 1.1 μM) enhanced the activity; however, expanding to but-3-enyl (27, IC50 = 13.7 μM) significantly reduced the activity. Interestingly, in contrast with compounds 1 and 25, compounds 26 and 27 act as noncompetitive inhibitors (Figure 3). No significant variation in the IC50 values of 26 and 27 was observed with increasing substrate and constant protein concentrations, further corroborating the noncompetitive inhibition mechanism (Supplementary Table S2).
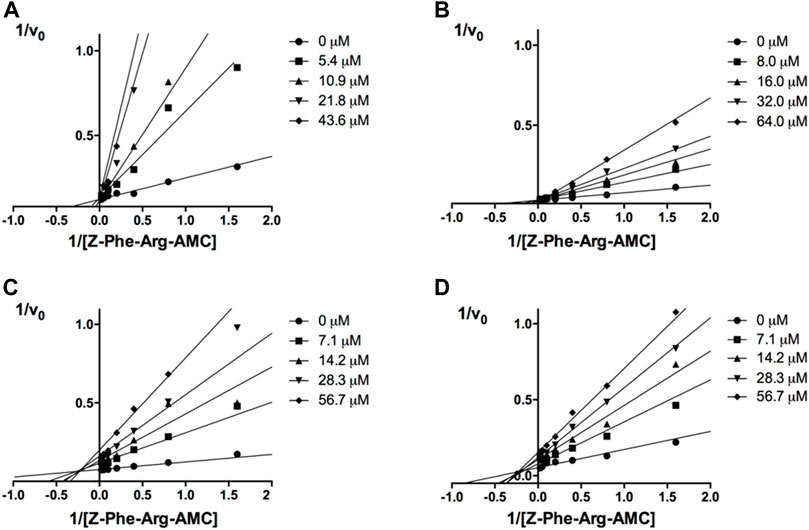
FIGURE 3. Lineweaver-Burk plots for compounds 1 (A); 25 (B); 27 (C); and 26 (D). Each curve represents a different inhibitor concentration.
These results clearly highlight the role played by the N-substituents in the mechanism of cruzain inhibition. The importance of the amine was previously demonstrated by replacing the nitrogen with an oxygen atom, and the activity was lost (Ferreira et al., 2014). Additionally, the distinct N-substituents allowed us to correlate the substituent volume with the mechanism of inhibition. The lack of a substituent (1) or the presence of a methyl (25) results in competitive inhibition, while bulkier groups such as benzyl (26) and but-3-enyl (27) lead to noncompetitive inhibition.
In the next step, we explored substitutions at the phenyl ring. Given that the phenyl ring of lead compound 18 occupies the S2 pocket of the cruzain-binding site, we expanded the phenyl into a naphthyl system and appended different hydrogen bond donors and acceptors to the phenyl ring. The goal was to explore a potential interaction with Glu208. As shown in Table 2, among the 15 synthesized compounds, the three naphthyl analogs with either hydroxyl or ether at the meta or para positions were the most potent: 6-hydroxynaphthyl 10 (IC50 = 3.4 μM), 7-hydroxynaphthyl 11 (IC50 = 2.7 μM), and 6-methoxycarbonyl 12 (IC50 = 2.3 μM). The design concept was corroborated by molecular docking runs, which predicted the formation of a hydrogen bond between the hydroxyl groups of 10 and 11 and Glu208 (Figure 4). To further corroborate the formation of a hydrogen bond with Glu208, we evaluated the activities of six compounds against the enzyme rhodesain, a cysteine protease that has a similar active site to that of cruzain, in which Glu208 is replaced with an alanine residue (Lima et al., 2013). The activities of the compounds against cruzain were significantly more pronounced than their activities against rhodesain, indicating the importance of interactions with Glu208 for inhibition by 10 and 11 (Supplementary Table S3).
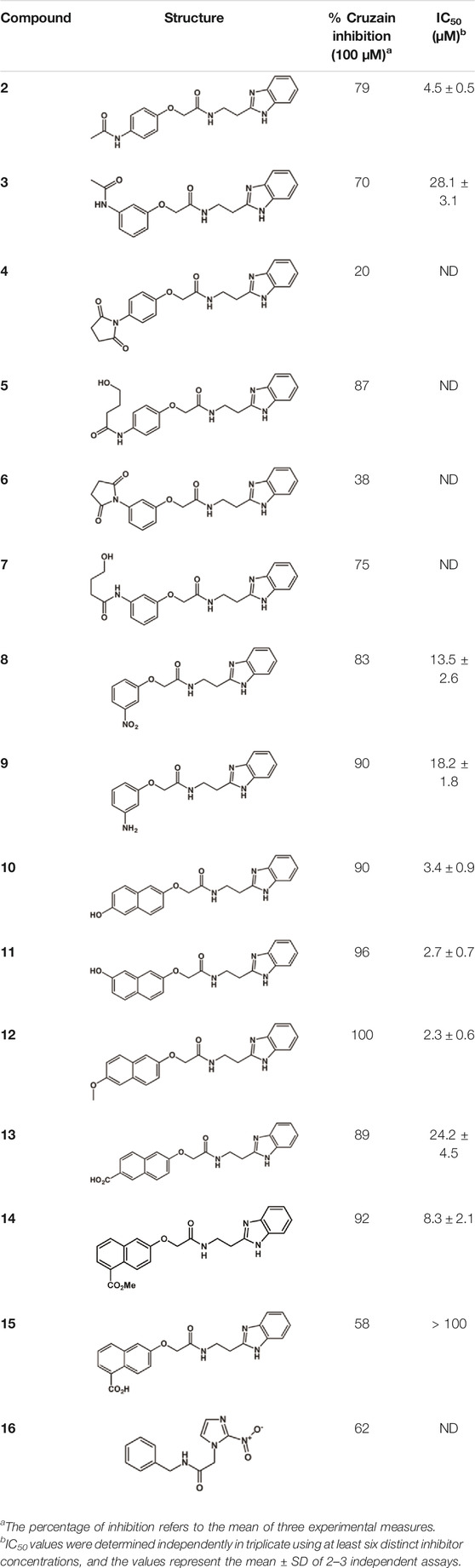
TABLE 2. Structure and activity against cruzain of new benzimidazoles with substituents at the phenyl ring.a
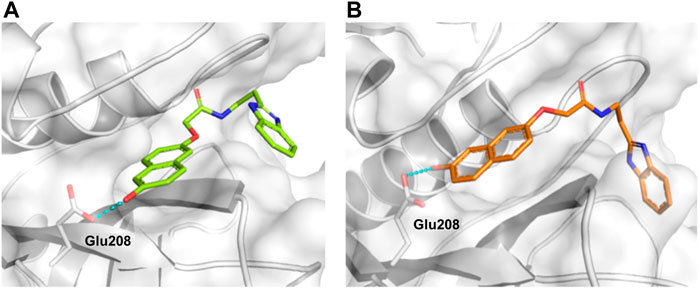
FIGURE 4. Molecular docking predicted the binding conformations of compounds 10 (A) and 11 (B) in complex with cruzain (PDB 3KKU, 1.28 Å), showing the formation of hydrogen bonds (dashed lines) between the hydroxyl groups and Glu208. Binding site residues (carbon in gray) and compounds 10 and 11 (carbon in green and orange, respectively) are shown as sticks.
After the enzyme inhibition studies, active compounds were evaluated for their activity against T. cruzi intracellular amastigotes and PK properties (Table 3). Among the N-substituted analogs, compounds 20 (IC50 = 2.04 μM) and 24 (IC50 = 1.43 μM) were equipotent to the reference drug BZ (IC50 = 1.45 μM). The only inactive compound in this series was the N-methyl analog 25. In general, these compounds are more lipophilic than BZ, as shown by the LogP and eLogD values. Among the molecules in Table 3, six were classified as high-permeability compounds (PAMPA higher than 1.5 × 10−6 cm/s), and nine were classified as low-permeability compounds (PAMPA lower than 1.5 × 10−6 cm/s).
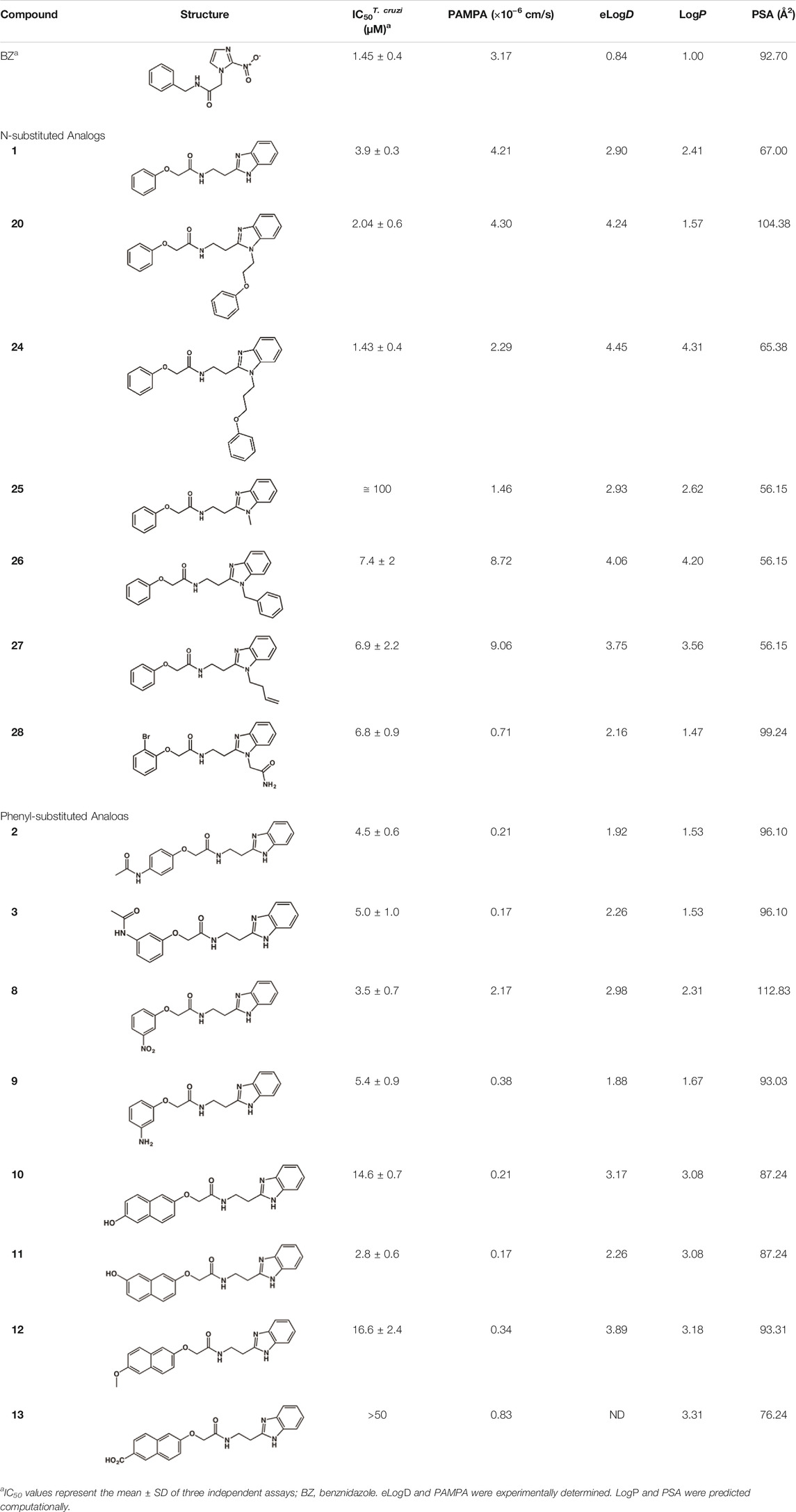
TABLE 3. In vitro activity against T. cruzi and physicochemical properties of a subset of the benzimidazoles.
Most compounds with substituents on the phenyl ring were active against T. cruzi, with IC50 values in the low micromolar range (Table 3). The exception was compound 13, which has a 6-carboxynaphthyl moiety. Compound 13 showed moderate activity against cruzain (IC50 = 24.2 μM) in addition to a LogP value higher than those of the other analogs. The combination of these two properties may be the cause of the lack of trypanocidal activity of this compound.
The benzimidazole derivatives were further evaluated regarding their cytotoxicity against human HFF-1 fibroblasts, which were used as host cells for T. cruzi (Table 4). Selectivity indices (SI), which express the ratio between the IC50 values for HFF-1 cells and T. cruzi, were calculated. Overall, the evaluated compounds exhibited no significant toxicity against human HFF-1 fibroblasts. Three compounds showed SI values comparable to or greater than that of the reference drug BZ (SI > 33): 18 (SI > 61), 17 (SI > 35), and 37 (SI > 34). It is worth noting that compounds 1 (SI > 26) and 8 (SI > 29) also exhibited suitable SI values.
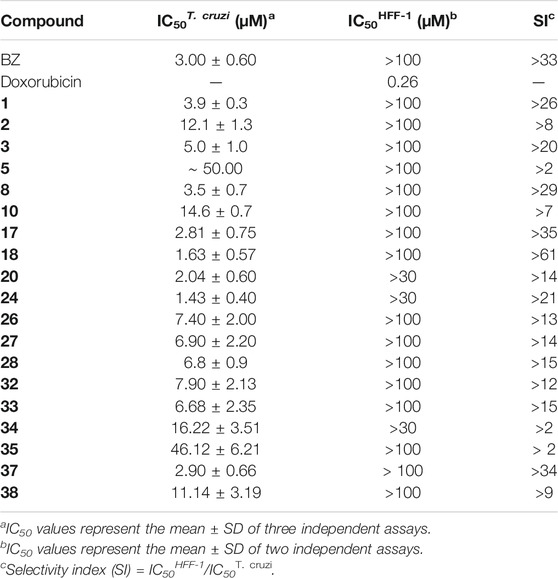
TABLE 4. Biological evaluation of a subset of the benzimidazoles against T. cruzi and human HFF-1 fibroblasts.
A series of 10 benzimidazole derivatives were selected based on their activity against cruzain and T. cruzi to undergo PK studies, including in vitro and in vivo metabolism. Table 5 shows the in vitro results for CLint after incubation with human and mouse microsomes, fu, LogD, and PAMPA. Corrected clearance values (CLint_u) were obtained by calculating the ratio between CLint and fu. It is important to note that only unbound drug molecules are available for clearance, interaction with metabolizing enzymes and transporters, equilibration into tissues, and pharmacological activity. Thus, PK, pharmacodynamics, and toxicity are driven by unbound drug concentrations (Zamek-Gliszczynski, et al., 2011). As such, protein binding (PPB) in plasma, microsomes, and target tissues is routinely evaluated in drug discovery to determine the respective fu values (Wang, et al., 2014). The drug-like space for unbound clearance lies at approximately 10 L/h/kg. As shown in Table 5, all benzimidazole derivatives have CLint_u values much higher than the drug-like reference and that of the reference drug BZ. Compounds 17, 18, 37, which have IC50T. cruzi values comparable to that of BZ, have CLint_u values ranging from ∼10 to 21 times higher than that of BZ, which could undermine the achievement of the bioavailability levels required for biological response. Most compounds listed in Table 5 had PAMPA values higher than 1.5 × 10−6 cm/s and were classified as having good permeability.
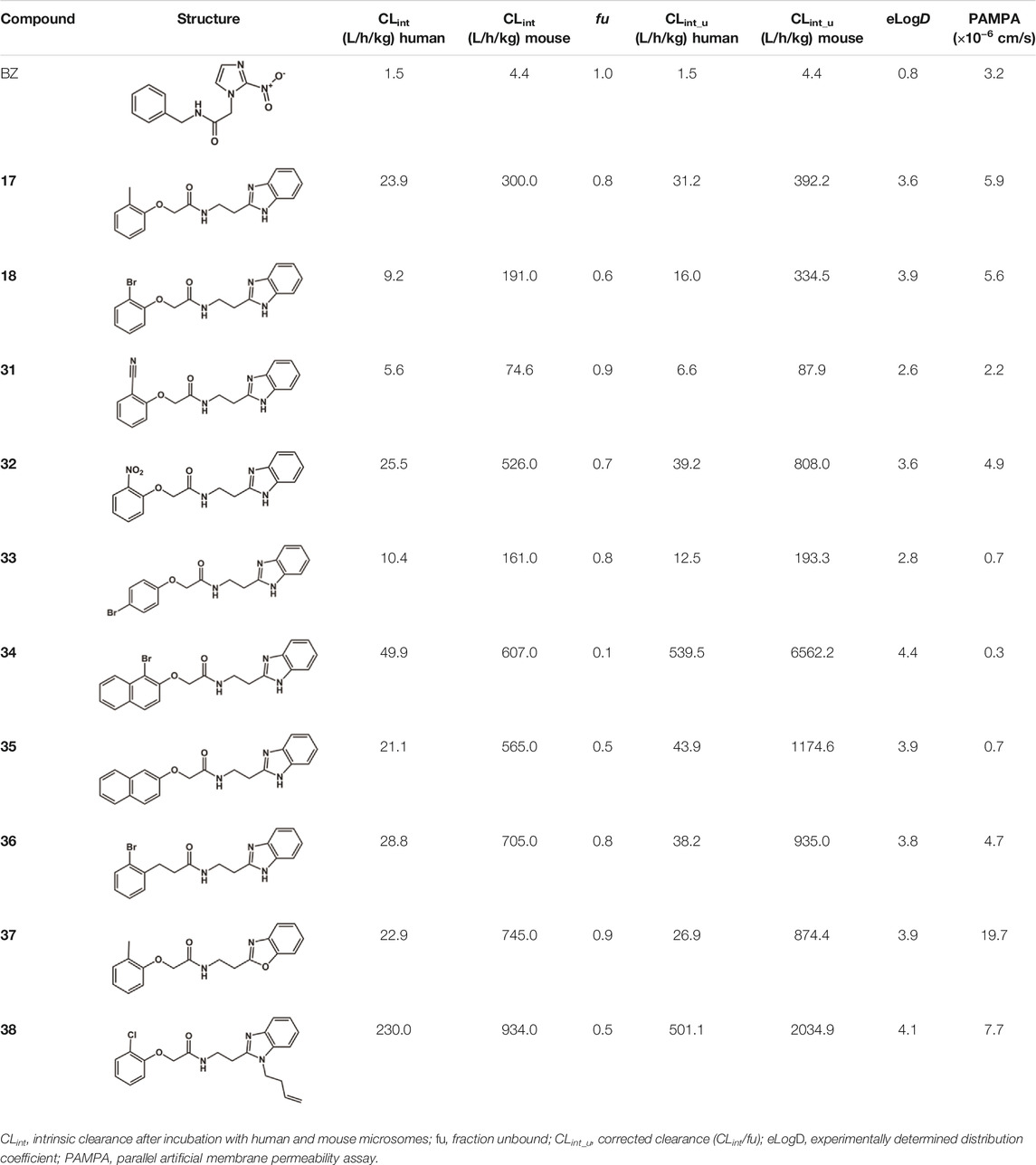
TABLE 5. In vitro PK profile of benzimidazole analogs.
Next, the same set of molecules was evaluated for their in vivo PK profile (Table 6). From this assay, information such as T1/2, plasma clearance (CLp), and bioavailability (F) were obtained. As observed for the in vitro assays, all benzimidazoles had high unbound clearance compared to that of BZ. The in vivo assays reinforced the concept that the high clearance may be the reason for the very low oral bioavailability (F) observed for the benzimidazoles (0–35%) compared to that of BZ (90%).
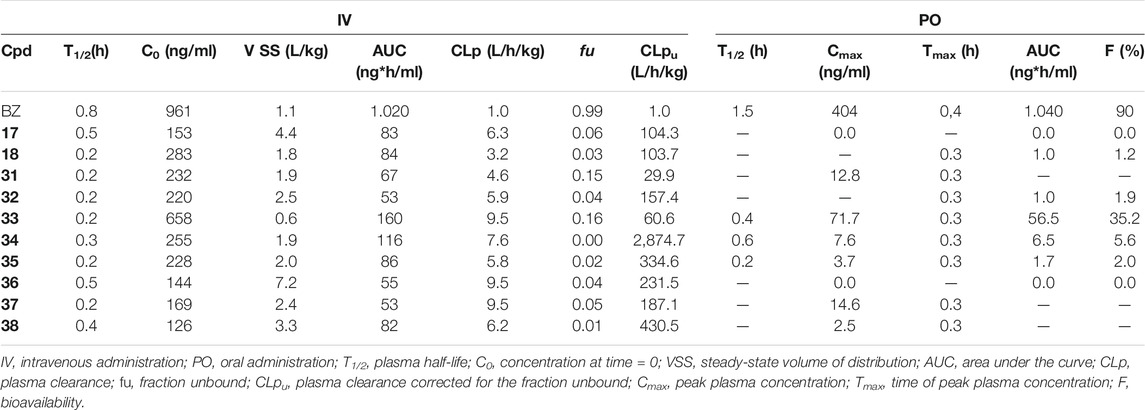
TABLE 6. In vivo PK profiles of selected benzimidazole derivatives.
In vitro experiments are faster and less expensive than in vivo assays. Accessing the in vitro-in vivo correlation (IVIVC) for metabolic stability is important to demonstrate whether one can rely on in vitro studies and keep the use of animals to a minimum for a series of molecules. The lack of IVIVC is also informative, indicating that other metabolic routes are likely to be responsible for the observed in vivo clearance. In our experiments, a positive IVIVC was observed for fu-corrected clearance (Figure 5), which allowed us to rely on in vitro assays for the prediction of in vivo metabolic stability and prioritize compounds for further studies.
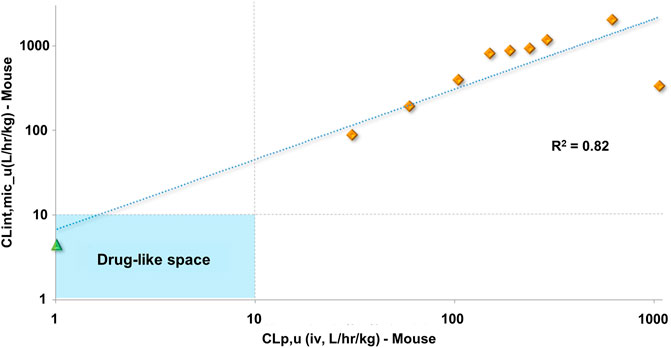
FIGURE 5. Clearance in vitro-in vivo correlation (IVIVC) plot showing a good correlation between the metabolic stability data. Benznidazole (green triangle); benzimidazole analogs (orange diamonds).
All compounds evaluated showed high clearance values, reaching 60–180% of mouse liver blood flow (5.4 L/h/kg), which is a plausible explanation for their low bioavailability (0–35%). To better understand pathways involved in the elimination of the compounds, metabolic stability studies in human hepatocytes, which contain the whole set of human phase I and phase II hepatic metabolizing enzymes, were conducted. This experiment was performed in the absence of CYP inhibitors for the determination of the total clearance (Phase I + Phase II); in the presence of 1-ABT (a CYP450 inhibitor) for the determination of the fraction of the compounds that are metabolized by the remaining Phase I as well as the conjugating Phase II enzymes; and in the presence of azamulin (a CYP3A4 inhibitor) for the identification of the fraction metabolized by this isoform. This last assay provides critical information for prioritizing compounds since CYP3A4 plays a key role in drug-drug interactions (DDIs) and is associated with adverse effects and low efficacy when two or more drugs are taken together. Phase I metabolism, performed mainly by the CYP450 family, was responsible for 56–95% of the metabolism of the compounds (mean = 78.1 ± 12.9%). A much lower contribution to total clearance was observed for all other Phase I enzymes, which were responsible for 5–35% of the metabolism (mean = 21.9 ± 13.6%). The central role played by CYP3A4 became apparent when the CYP3A4 inhibitor azamulin was used in the assay: the resulting clearance values were approximately 40% lower, reaching a minimum value of 31.6% and a maximum value of 59.5% when compared with total clearance values (Supplementary Figure S1).
After identifying the central role of CYP3A4 in the metabolism of this series, the next step was to identify the involved isoforms using recombinant CYP enzymes. CLint was determined based on the residual amount of the compound over time. Additionally, the contribution of each CYP isoform to metabolism was calculated based on their relative abundance in humans. The information generated by this experiment was essential to assess the risk of potential drug-drug interactions (DDIs) for this series of compounds. Molecules eliminated through multiple pathways have reduced DDI potential and are therefore more suitable for advancing to further steps in a drug discovery pipeline. The benzimidazole derivatives are mainly metabolized by CYP3A4 (23–90%) (Supplementary Table S4). Although modest, a contribution from isoforms CYP2D6 and CYP1A2 is observed, featuring an attractive profile from a DDI perspective despite the high clearance values.
All studies on benzimidazoles showed that these molecules are metabolically unstable, and biotransformation mediated by CYP3A4 is the major metabolic route. Therefore, studies to identify the molecular sites of metabolism (SOM) were performed. These studies can enable the blockage of these sites by the inclusion of blocking groups to achieve appropriate levels of metabolic stability. Ideally, these molecular changes should not significantly affect the potency toward the molecular target. The test compounds were then incubated with human and mouse liver microsomes. Most of the metabolites were found to be oxidation products mainly of the linker and benzimidazole moieties (Supplementary Figures S2–S5). Strategies to block these SOMs could include switching the amide position and adding halogen atoms to the linker. At the benzimidazole ring, N substitutions and the addition of halogens could be explored (Supplementary Figure S6).
After completing the PK profile for 10 molecules (set 1), an additional set of 55 compounds (set 2) was evaluated to provide additional information for the establishment of a SAR for metabolic stability. The results for clearance after incubation with human and mouse liver microsomes, eLogD, and PAMPA are summarized in Supplementary Table S5. Some set 2 compounds with lower clearance values than those of set 1 molecules were identified, some of which exhibited clearance values comparable to that of BZ (Supplementary Figure S7).
The clearance values listed in Supplementary Table S5 show that the presence of substituents on the phenyl ring might influence the metabolic stability of the compounds. Substituents at para and meta, for example, led to the most stable compounds with the lowest CLint_u values. Among the compounds with substituents at para, compounds 2, 4 and 5 are highlighted, for which clearance values are in the same range of BZ (CLint_u = 1.50 L/h/kg). In addition, compounds 6 (CLint_u = 3.54 L/h/kg), 7 (CLint_u = 1.50 L/h/kg), and 3 (CLint_u = 6.90 L/h/kg), with substituents at meta, showed drug-like profiles for metabolic stability. Substituents at ortho also led to stable compounds: 31 (CLint_u = 6.6 L/h/kg), 42 (CLint_u = 4.64 L/h/kg), and 43 (CLint_u = 3.94 L/h/kg).
Set 2 compounds did not show significant structural variability in the linker region. Among the few exceptions are compounds 48, in which sulfur replaced the linker oxygen (CLint_u = 11.38 L/h/kg), and 58 (CLint_u = 16.99 L/h/kg) and 49 (CLint_u = 3.28 L/h/kg), in which the position of the phenoxy fragment was modified by the introduction of a methylene group at the linker. At the benzimidazole ring, the introduction of a hydrophilic amide led to high metabolic stability (28, CLint_u = 1.53 L/h/kg). It is important to highlight the influence exerted by the physicochemical nature of the substituents at the phenyl and the benzimidazole on the stability of the compounds (Supplementary Figure S8). The introduction of hydrophobic substituents at the phenyl resulted in high clearance values, such as those observed for compounds 44 (CLint_u = 70.62 L/h/kg), 51 (CLint_u = 65.56 L/h/kg), 52 (CLint_u = 2,122.45 L/h/kg), and 60 (CLint_u = 194.48 L/h/kg). Hydrophobic substituents at the benzimidazole (19, 20, 21, 23, 26, 27, and 30) followed the same trend, with CLint_u values ranging from 70.23 to 3,366.34 L/h/kg. Overall, the clearance values for the benzimidazole derivatives increased with increasing hydrophobicity (Supplementary Figure S9). Seven set 2 compounds (1, 2, 3, 8, 10, 11, and 28) with suitable trypanocidal activity and in vitro clearance underwent in vivo PK studies. Overall, the set 2 compounds exhibited lower CLp_u values compared to those of the set 1 analogs (Supplementary Table S6), with benzimidazoles 2 and 28 showing the most promising profiles (CLp_u of 4.16 and 3.98, respectively). Additionally, similar to the profile observed for the set 1 compounds, a good correlation between in vitro and in vivo clearance was found for the set 2 benzimidazoles (Figure 6).
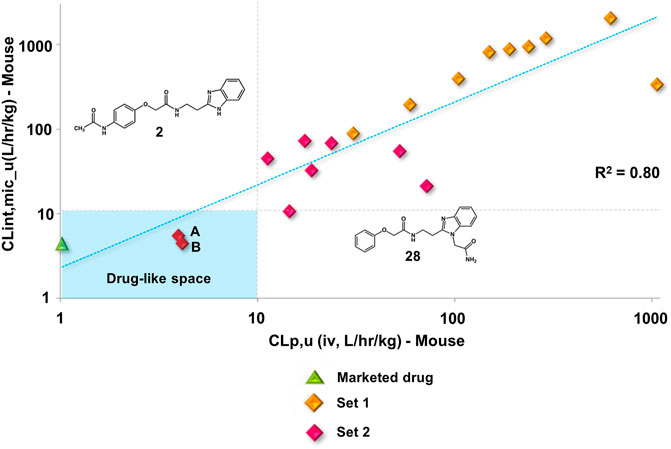
FIGURE 6. Clearance in vitro-in vivo correlation (IVIVC) plot showing a good correlation between the metabolic stability data. Benznidazole (green triangle); set 1 compounds (orange diamonds); set 2 compounds (red diamonds). Compounds 2 (A) and 28 (B).
Compound 28 (IC50T. cruzi = 6.8 µM) was selected for a proof-of-concept study given its suitable balance between pharmacodynamics and PK properties. Initially, we determined the doses that elicited no acute toxicity. The compound, solubilized in 10% DMSO aqueous solution, was administered orally in a single dose of 150 and 300 mg/kg of body weight in female Swiss mice. Parameters related to behavior, autonomic functions, neurological activity, and mortality were assessed as toxicity signs. Soon after the administration of the compound, mice were placed in a circular open-field arena (40 cm diameter) with 50-cm-high walls to assess motor deficits. Mortality and clinical signs associated with toxicity were recorded 0.5, 2, 4, 8, and 24 h after the single-dose administration. After this period, toxicity signs were assessed once a day for two consecutive weeks. The reference drug BZ at 150 mg/kg and vehicle (10% DMSO) were administered as controls. No toxicity signs were observed within the 2 weeks of observation for any of the tested doses. Additionally, no mortality was observed (Figure 7A). One-way ANOVA did not reveal any difference among the groups [F3,4 = 0.15; p = 0.9238], indicating that treatment with 28 at doses of 150 and 300 mg/kg did not cause locomotor deficits.
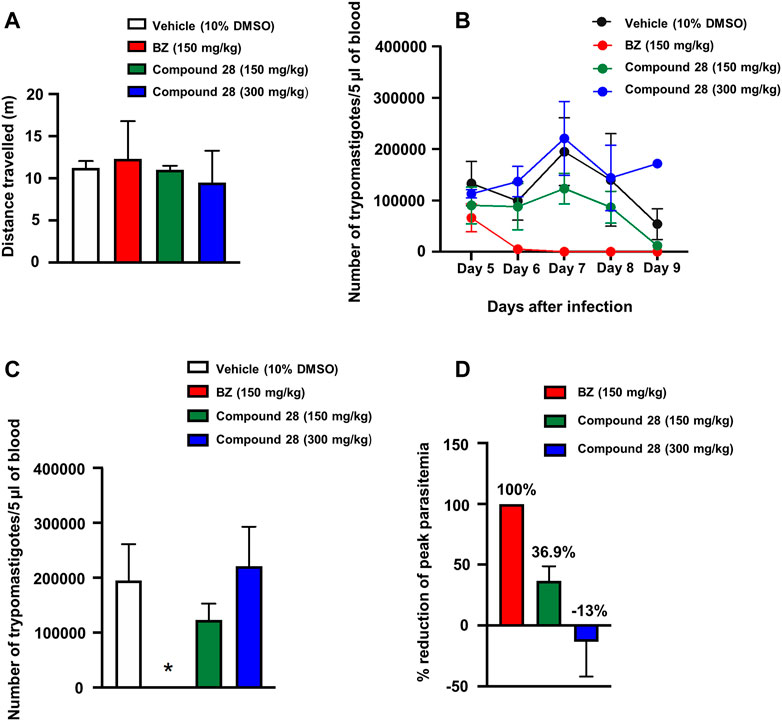
FIGURE 7. Acute toxicity and trypanocidal activity in vivo. (A) Open-field test. Mice were orally treated with vehicle (10% DMSO) or benznidazole (BZ) at doses of 150 mg/kg or 28 at doses of 150 and 300 mg/kg. (B) Parasitemia during T. cruzi infection in mice treated with vehicle, BZ or 28 (150 and 300 mg/kg) expressed as the number of trypomastigotes per 5 μl of blood. The data represent the mean parasitemia ± SEM (4–8 animals per group) for all assays. (C) Peak parasitemia expressed as the number of trypomastigotes per 5 μl of blood in mice treated with vehicle, BZ or 28 (150 and 300 mg/kg) (*p <0.05 when compared to vehicle and other groups). (D) Reduction of peak parasitemia (seventh day of infection) in mice treated with vehicle, BZ or 28 (150 and 300 mg/kg). Vehicle solution: 0.9% NaCl + 10% DMSO.
Considering the favorable acute toxicity results, the in vivo trypanocidal activity of 28 was determined at single doses of 150 and 300 mg/kg for 5 days. Female Swiss mice were infected with T. cruzi (Y strain) (Rodriguez et al., 2014) and treated via gavage with five daily doses of BZ (150 mg/kg of body weight), 28 (150 and 300 mg/kg of body weight) and vehicle (10% DMSO). Treatment started on day five after infection with T. cruzi. The following parameters were evaluated in these experiments: level of parasitemia after the treatment, suppression of peak parasitemia (day seven after the infection), and reduction of parasitemia on the peak day (day seven after infection). Parasitemia was expressed as the number of T. cruzi trypomastigotes per 5 µl of blood and was calculated using the Brener method (Brener, 1962). Repeated ANOVA measures considering the factors treatment and day (repeated measure) showed a main effect of treatment (F3,18 = 12.68; p < 0.05) (Figure 7B). On the seventh day of treatment, when parasitemia reached its peak, one-way ANOVA indicated a treatment effect (F3,16 = 7.47; p = 0.002). Post hoc analyses indicated that the treatment with BZ significantly decreased parasite burden compared with the other treatments (p < 0.05) (Figure 7C). BZ and 28 (150 mg/kg) reduced peak parasitemia by 100 and 36.9%, respectively, compared with the vehicle. At a dose of 300 mg/kg, benzimidazole 28 showed an increase of 13% in the peak parasitemia when compared with the vehicle (Figure 7D). This dose-response effect is likely associated with the modulation of physiological systems that increase the susceptibility of the animals to infection with T. cruzi at high doses of the compound. Further tests with lower doses (75 mg/kg and 37.5 mg/kg) showed no reduction in parasitemia levels (Supplementary Figure S10). The results of the in vivo studies indicate a moderate ability of 28 to suppress peak parasitemia at 150 mg/kg.
Considering that few molecular targets are validated in NTDs (De Rycker et al., 2018) and the relatively unsuitable compounds regarding toxicity and drug-likeness that have been historically explored in the area, the findings reported herein address an important gap in Chagas disease drug discovery. Regardless of the mechanism of action, it is noteworthy that in rare cases a compound succeeds in terms of efficacy in Chagas disease in vivo infection models. This is a major hurdle in the field that can be related to the complex life-cycle biology of T. cruzi, and the many poorly understood aspects of the interplay between the parasite and the host (Libisch et al., 2021). Among the compounds that reached this milestone, we can highlight vinyl sulfone K777, CYP51-inhibitor azoles (including posaconazole), and cruzain-inhibitor triazoles and carbamoyl imidazoles (Ferraz et al., 2007; McKerrow et al., 2009; Brak et al., 2010; de Souza et al., 2020). K777 was a landmark in the field as it was the first compound to show the possibility to enter clinical trials for Chagas disease. However, tolerability issues in dogs and primates during the preclinical phase hampered the progression of this compound toward clinical development. After the failure of K777, it was discussed whether the toxicity issues would be due to the irreversible mechanism of action and resulting lack of selectivity of K777 over other proteases, which could include human proteases. The case of K777 highlights the importance of designing reversible cruzain inhibitors with improved selectivity as are the benzimidazole derivatives investigated in this work. In this study, we adopted the strategy of diversifying the substitution pattern at the phenyl and benzimidazole regions. This approach led to an enhanced interaction with cruzain and, by enabling the formation of a hydrogen bond with Glu208, it improves the selectivity for cruzain over other proteases. Glu208 is part of the S2 subsite in the cruzain active site and is lacking in most other proteases such as human cathepsins. The role played by the formation of a hydrogen bond with Glu208 was investigated by evaluating a set of compounds against rhodesain, a cysteine protease that has an active site that resemble that of cruzain in which an alanine replaces Glu208. The compounds were far more active against cruzain over rhodesain, which indicates the important part played by Glu208 in selectivity toward cruzain.
Another key finding reported in Chagas disease drug design was the identification of CYP51-inhibitor antifungal azoles (Ferraz et al., 2007). These compounds, particularly Posaconazole and E1224 (the ravuconazole prodrug), showed promising suppressive effects in parasite burden in animal models of Chagas disease. However, their failure in clinical trials raised fruitful discussions regarding the mechanism of action of the compounds. Although these azoles displayed a remarkable suppressive effect, they failed in providing sustained parasite clearance when opposed to benznidazole. These studies served to establish the landmark that T. cruzi CYP51 is not a molecular target to be pursued in Chagas disease drug discovery. These previous findings demonstrate the critical importance of target validation and identification of compounds that act by different modes of action, for example, the modulation of cruzain. The compounds studied herein showed a moderate reduction of parasite burden and, therefore, open novel possibilities for future work on this molecular target. Moreover, the benzimidazoles did not demonstrate toxicity in animal studies, which, as seen in the K777 case, can be an issue of cysteine protease inhibitors. The best compound (28) administered orally to mice in a single dose of 150 and 300 mg/kg showed no toxicity signs for any of the doses and, importantly, no mortality was observed.
Another important class of compounds is triazole-based cruzain inhibitors, whose representative analogs showed promising in vivo efficacy (Brak et al., 2010; Neitz et al., 2015). These non-peptidic ketones irreversibly inactivate cruzain by attaching covalently to Cys25, which can raise selectivity issues and, therefore, be a drawback for further development. Regarding the PK profile, optimization of these triazoles resulted in enhanced bioavailability and exposure after oral dosing, although they proved to inhibit CYP3A4, the most important CYP isoform for the elimination of xenobiotics. The best compound identified herein (28), showed a suitable tradeoff among pharmacodynamics and PK properties. Regarding its mechanism of action, inhibitor 28 is a reversible inhibitor and interacts with Glu208, which reduces the probability of inhibition of human proteases. Additionally, the extensive PK studies enabled the identification of permeable, metabolically stable, and bioavailable compounds with high selectivity indices, and that are metabolized mainly by CYP3A4. Incubation of the compounds with isolated recombinant CYPs using CYP3A4 and pan-CYP inhibitors as controls showed that the benzimidazoles do not inhibit CYP3A4. These findings are pivotal in the context of drug-drug interactions, particularly in the case of chagasic patients who need to use different drugs to mitigate the complications of the disease.
An MPO strategy for the optimization of benzimidazole derivatives as antichagasic agents was developed. This strategy relied on the parallel optimization of activity against cruzain and T. cruzi, selectivity, and PK parameters such as metabolic stability and permeability. New compounds were synthesized, and previously synthesized analogs were thoroughly evaluated for PK properties. Newly introduced N-substituents at the benzimidazole ring revealed that increasing bulkiness at this site modifies the mechanism of action toward cruzain from competitive to noncompetitive. These results introduce new and interesting aspects regarding the binding mode and mechanism of action of cruzain inhibitors. Newly designed phenyl-substituted analogs showed increased inhibition of cruzain over rhodesain, demonstrating the key role played by Glu208 in the selective inhibition of cruzain over other proteases. Some of the benzimidazole derivatives showed appropriate metabolic stability and clearance values comparable to those of drug-like molecules. Phase I oxidation reactions catalyzed by CYP3A4 were detected as the main elimination pathway, and the identified sites of metabolism provided insights into the improvement of metabolic stability. Moreover, the analysis of the in vitro trypanocidal and cytotoxicity data revealed a sound selectivity index for the investigated compounds, indicating a low potential for toxicity.
The applied MPO approach enabled the prioritization of compounds considering an appropriate combination of in vitro activity, toxicity, and PK properties. The gathered in vitro data supported in vivo PK studies for representative compounds. A solid IVIVC was obtained, demonstrating the high predictive ability of the in vitro PK models for the corresponding in vivo endpoints. Finally, acute toxicity and efficacy studies were conducted for compound 28, which showed no toxicity signs and a moderate reduction in peak parasitemia at 150 mg/kg. Importantly, the knowledge gathered in this study opens novel opportunities to understand the molecular aspects of cruzain inhibition, enabling the discovery of compounds with a good trade-off between pharmacodynamics and pharmacokinetics.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by The Committee of Ethics in Research of the Universidade Federal de São Paulo (CEUA No. 5301080816) and AbbVie DMPK Department.
IP conceptualization, writing, molecular design and biological evaluation; CORJ: conceptualization, writing, molecular design and organic synthesis; BS: organic synthesis; MD: organic synthesis; MS: molecular design and in vitro experiments; LF: writing, molecular design and in vitro experiments; ALMA: molecular design and in vitro experiments; RF: molecular design and in vitro experiments; LM, RK, and SM-D: in vitro experiments; FS: in vivo experiments; RD: in vivo experiments; FC: in vivo experiments; LD: conceptualization, supervision, organic synthesis and writing; and ADA: conceptualization, supervision, molecular design, fund acquisition and writing.
The National Council for Scientific and Technological Development (CNPq), Brazil, the Coordination for the Improvement of Higher Education Personnel (CAPES), Brazil, the Sao Paulo Research Foundation (FAPESP), Brazil (CEPID-CIBFar grant 13/07600-3, IP grants 11/13789-6 and 14/26324-0) for financial support and post-doctoral fellowship for CORJ, grant 158926/2014-5.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors acknowledge the Abbvie DMPK Department for the support in the DMPK experiments and Luiz Severino da Silva for the support in the in vivo experiments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.774069/full#supplementary-material
1https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis).
2https://www.paho.org/en/topics/chagas-disease.
Alelyunas, Y. W., Pelosi-Kilby, L., Turcotte, P., Kary, M. B., and Spreen, R. C. (2010). A High Throughput Dried DMSO LogD Lipophilicity Measurement Based on 96-Well Shake-Flask and Atmospheric Pressure Photoionization Mass Spectrometry Detection. J. Chromatogr. A. 1217, 1950–1955. doi:10.1016/j.chroma.2010.01.071
Andricopulo, A. D., and Montanari, C. A. (2005). Structure-Activity Relationships for the Design of Small-Molecule Inhibitors. Mini Rev. Med. Chem. 5, 585–593. doi:10.2174/1389557054023224
Arnal, A., Waleckx, E., Rico-Chávez, O., Herrera, C., and Dumonteil, E. (2019). Estimating the Current burden of Chagas Disease in Mexico: A Systematic Review and Meta-Analysis of Epidemiological Surveys from 2006 to 2017. Plos Negl. Trop. Dis. 13, e0006859. doi:10.1371/journal.pntd.0006859
Avelar, L. A., Camilo, C. D., de Albuquerque, S., Fernandes, W. B., Gonçalez, C., Kenny, P. W., et al. (2015). Molecular Design, Synthesis and Trypanocidal Activity of Dipeptidyl Nitriles as Cruzain Inhibitors. Plos Negl. Trop. Dis. 9, e0003916. doi:10.1371/journal.pntd.0003916
Barltrop, J. A., Owen, T. C., Cory, A. H., and Cory, J. G. (1991). 5-(3-Carboxymethoxyphenyl)-2-(4,5-Dimethylthiazolyl)-3-(4-Sulfophenyl)tetrazolium, Inner Salt (MTS) and Related Analogs of 3-(4,5-Dimethylthiazolyl)-2,5-Diphenyltetrazolium Bromide (MTT) Reducing to Purple Water-Soluble Formazans as Cell-Viability Indicators. Bioorg. Med. Chem. Lett. 1, 611–614. doi:10.1016/S0960-894X(01)81162-8
Brak, K., Kerr, I. D., Barrett, K. T., Fuchi, N., Debnath, M., Ang, K., et al. (2010). Nonpeptidic Tetrafluorophenoxymethyl Ketone Cruzain Inhibitors as Promising New Leads for Chagas Disease Chemotherapy. J. Med. Chem. 53, 1763–1773. doi:10.1021/jm901633v
Brener, Z. (1962). Therapeutic Activity and Criterion of Cure on Mice Experimentally Infected with Trypanosoma Cruzi. Rev. Inst. Med. Trop. Sao Paulo 4, 389–396.
Buckner, F. S., Verlinde, C. L., La Flamme, A. C., and Van Voorhis, W. C. (1996). Efficient Technique for Screening Drugs for Activity against Trypanosoma Cruzi Using Parasites Expressing Beta-Galactosidase. Antimicrob. Agents Chemother. 40, 2592–2597. doi:10.1128/AAC.40.11.2592
Clark, M., Cramer, R. D., and Van Opdenbosch, N. (1989). Validation of the General Purpose Tripos 5.2 Force Field. J. Comput. Chem. 10, 982–1012. doi:10.1002/jcc.540100804
Cucunubá, Z. M., Okuwoga, O., Basáñez, M. G., and Nouvellet, P. (2016). Increased Mortality Attributed to Chagas Disease: A Systematic Review and Meta-Analysis. Parasit. Vectors 9, 42. doi:10.1186/s13071-016-1315-x
Davies, B., and Morris, T. (1993). Physiological Parameters in Laboratory Animals and Humans. Pharm. Res. 10, 1093–1095. doi:10.1023/a:1018943613122
De Rycker, M., Baragaña, B., Duce, S. L., and Gilbert, I. H. (2018). Challenges and Recent Progress in Drug Discovery for Tropical Diseases. Nature 559, 498–506. doi:10.1038/s41586-018-0327-4
de Souza, M. L., de Oliveira Rezende Junior, C., Ferreira, R. S., Espinoza Chávez, R. M., Ferreira, L. L. G., Slafer, B. W., et al. (2020). Discovery of Potent, Reversible, and Competitive Cruzain Inhibitors with Trypanocidal Activity: A Structure-Based Drug Design Approach. J. Chem. Inf. Model. 60, 1028–1041. doi:10.1021/acs.jcim.9b00802
Doyle, P. S., Zhou, Y. M., Hsieh, I., Greenbaum, D. C., McKerrow, J. H., and Engel, J. C. (2011). The Trypanosoma Cruzi Protease Cruzain Mediates Immune Evasion. Plos Pathog. 7, e1002139. doi:10.1371/journal.ppat.1002139
Eddershaw, P. J., Beresford, A. P., and Bayliss, M. K. (2000). ADME/PK as Part of a Rational Approach to Drug Discovery. Drug Discov. Todaytoday 5, 409–414. doi:10.1016/s1359-6446(00)01540-3
Engel, J. C., Doyle, P. S., Palmer, J., Hsieh, I., Bainton, D. F., and McKerrow, J. H. (1998). Cysteine Protease Inhibitors Alter Golgi Complex Ultrastructure and Function in Trypanosoma Cruzi. J. Cel Sci. 111, 597–606. doi:10.1242/jcs.111.5.597
Espíndola, J. W., Cardoso, M. V., Filho, G. B., Oliveira E Silva, D. A., Moreira, D. R., Bastos, T. M., et al. (2015). Synthesis and Structure-Activity Relationship Study of a New Series of Antiparasitic Aryloxyl Thiosemicarbazones Inhibiting Trypanosoma Cruzi Cruzain. Eur. J. Med. Chem. 101, 818–835. doi:10.1016/j.ejmech.2015.06.048
Ferraz, M. L., Gazzinelli, R. T., Alves, R. O., Urbina, J. A., and Romanha, A. J. (2007). The Anti-Trypanosoma Cruzi Activity of Posaconazole in a Murine Model of Acute Chagas' Disease Is Less Dependent on Gamma Interferon Than that of Benznidazole. Antimicrob. Agents Chemother. 51, 1359–1364. doi:10.1128/AAC.01170-06
Ferreira, L. G., and Andricopulo, A. D. (2017). Targeting Cysteine Proteases in Trypanosomatid Disease Drug Discovery. Pharmacol. Ther. 180, 49–61. doi:10.1016/j.pharmthera.2017.06.004
Ferreira, R. A. A., Pauli, I., Sampaio, T. S., de Souza, M. L., Ferreira, L. L. G., Magalhães, L. G., et al. (2019). Structure-Based and Molecular Modeling Studies for the Discovery of Cyclic Imides as Reversible Cruzain Inhibitors with Potent Anti-Trypanosoma Cruzi Activity. Front. Chem. 7, 798. doi:10.3389/fchem.2019.00798
Ferreira, R. S., Bryant, C., Ang, K. K., McKerrow, J. H., Shoichet, B. K., and Renslo, A. R. (2009). Divergent Modes of Enzyme Inhibition in a Homologous Structure-Activity Series. J. Med. Chem. 52, 5005–5008. doi:10.1021/jm9009229
Ferreira, R. S., Dessoy, M. A., Pauli, I., Souza, M. L., Krogh, R., Sales, A. I., et al. (2014). Synthesis, Biological Evaluation, and Structure-Activity Relationships of Potent Noncovalent and Nonpeptidic Cruzain Inhibitors as Anti-Trypanosoma Cruzi Agents. J. Med. Chem. 57, 2380–2392. doi:10.1021/jm401709b
Ferreira, R. S., Simeonov, A., Jadhav, A., Eidam, O., Mott, B. T., Keiser, M. J., et al. (2010). Complementarity between a Docking and a High-Throughput Screen in Discovering New Cruzain Inhibitors. J. Med. Chem. 53, 4891–4905. doi:10.1021/jm100488w
Fonseca, N. C., da Cruz, L. F., da Silva Villela, F., do Nascimento Pereira, G. A., de Siqueira-Neto, J. L., Kellar, D., et al. (2015). Synthesis of a Sugar-Based Thiosemicarbazone Series and Structure-Activity Relationship versus the Parasite Cysteine Proteases Rhodesain, Cruzain, and Schistosoma Mansoni Cathepsin B1. Antimicrob. Agents Chemother. 59, 2666–2677. doi:10.1128/AAC.04601-14
Gasteiger, J., and Marsili, M. (1980). Iterative Partial Equalization of Orbital Electronegativity-A Rapid Access to Atomic Charges. Tetrahedron 36, 3219–3228. doi:10.1016/0040-4020(80)80168-2
GBD DALYs and HALE Collaborators (2016). Global, Regional, and National Disability-Adjusted Life-Years (DALYs) for 315 Diseases and Injuries and Healthy Life Expectancy (HALE), 1990-2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 388, 1603–1658. doi:10.1016/S0140-6736(16)31460-X
Jones, G., Willett, P., Glen, R. C., Leach, A. R., and Taylor, R. (1997). Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 267, 727–748. doi:10.1006/jmbi.1996.0897
Jose Cazzulo, J., Stoka, V., and Turk, V. (2001). The Major Cysteine Proteinase of Trypanosoma Cruzi: A Valid Target for Chemotherapy of Chagas Disease. Curr. Pharm. Des. 7, 1143–1156. doi:10.2174/1381612013397528
Latorre, A., Schirmeister, T., Kesselring, J., Jung, S., Johé, P., Hellmich, U. A., et al. (2016). Dipeptidyl Nitroalkenes as Potent Reversible Inhibitors of Cysteine Proteases Rhodesain and Cruzain. ACS Med. Chem. Lett. 7, 1073–1076. doi:10.1021/acsmedchemlett.6b00276
Libisch, M. G., Rego, N., and Robello, C. (2021). Transcriptional Studies on Trypanosoma Cruzi - Host Cell Interactions: A Complex Puzzle of Variables. Front Cel. Infect. Microbiol. 11, 692134. doi:10.3389/fcimb.2021.692134
Lill, M. A., and Danielson, M. L. (2011). Computer-Aided Drug Design Platform Using PyMOL. J. Comput. Aided Mol. Des. 25, 13–19. doi:10.1007/s10822-010-9395-8
Lima, A. P., Reis, F. C., and Costa, T. F. (2013). Cysteine Peptidase Inhibitors in Trypanosomatid Parasites. Curr. Med. Chem. 20, 3152–3173. doi:10.2174/0929867311320250009
Liu, X., and Jia, L. (2007). The Conduct of Drug Metabolism Studies Considered Good Practice (I): Analytical Systems and In Vivo Studies. Curr. Drug Metab. 8, 815–821. doi:10.2174/138920007782798153
Lombardo, F., Obach, R. S., Shalaeva, M. Y., and Gao, F. (2004). Prediction of Human Volume of Distribution Values for Neutral and Basic Drugs. 2. Extended Data Set and Leave-Class-Out Statistics. J. Med. Chem. 47, 1242–1250. doi:10.1021/jm030408h
Lombardo, F., Obach, R. S., Shalaeva, M. Y., and Gao, F. (2002). Prediction of Volume of Distribution Values in Humans for Neutral and Basic Drugs Using Physicochemical Measurements and Plasma Protein Binding Data. J. Med. Chem. 45, 2867–2876. doi:10.1021/jm0200409
Masimirembwa, C. M., Bredberg, U., and Andersson, T. B. (2003). Metabolic Stability for Drug Discovery and Development: Pharmacokinetic and Biochemical Challenges. Clin. Pharmacokinet. 42, 515–528. doi:10.2165/00003088-200342060-00002
Massarico Serafim, R. A., Gonçalves, J. E., de Souza, F. P., de Melo Loureiro, A. P., Storpirtis, S., Krogh, R., et al. (2014). Design, Synthesis and Biological Evaluation of Hybrid Bioisoster Derivatives of N-Acylhydrazone and Furoxan Groups with Potential and Selective Anti-Trypanosoma Cruzi Activity. Eur. J. Med. Chem. 82, 418–425. doi:10.1016/j.ejmech.2014.05.077
McKerrow, J. H. (1999). Development of Cysteine Protease Inhibitors as Chemotherapy for Parasitic Diseases: Insights on Safety, Target Validation, and Mechanism of Action. Int. J. Parasitol. 29, 833–837. doi:10.1016/S0020-7519(99)00044-2
McKerrow, J. H., Doyle, P. S., Engel, J. C., Podust, L. M., Robertson, S. A., Ferreira, R., et al. (2009). Two Approaches to Discovering and Developing New Drugs for Chagas Disease. Mem. Inst. Oswaldo Cruz. 104 (Suppl. 1), 263–269. doi:10.1590/s0074-02762009000900034
Ndao, M., Beaulieu, C., Black, W. C., Isabel, E., Vasquez-Camargo, F., Nath-Chowdhury, M., et al. (2014). Reversible Cysteine Protease Inhibitors Show Promise for a Chagas Disease Cure. Antimicrob. Agents Chemother. 58, 1167–1178. doi:10.1128/AAC.01855-13
Neitz, R. J., Bryant, C., Chen, S., Gut, J., Hugo Caselli, E., Ponce, S., et al. (2015). Tetrafluorophenoxymethyl Ketone Cruzain Inhibitors with Improved Pharmacokinetic Properties as Therapeutic Leads for Chagas' Disease. Bioorg. Med. Chem. Lett. 25, 4834–4837. doi:10.1016/j.bmcl.2015.06.066
Obach, R. S. (1999). Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: An Examination of In Vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos. 27, 1350–1359.
Pérez-Molina, J. A., and Molina, I. (2018). Chagas Disease. The Lancet 391, 82–94. doi:10.1016/S0140-6736(17)31612-4
Plant, N. (2004). Strategies for Using In Vitro Screens in Drug Metabolism. Drug Discov. Today 9, 328–336. doi:10.1016/s1359-6446(03)03019-8
Powell, M. J. D. (1977). Restart Procedures for the Conjugate Gradient Method. Math. Programming 12, 241–254. doi:10.1007/BF01593790
Proctor, N. J., Tucker, G. T., and Rostami-Hodjegan, A. (2004). Predicting Drug Clearance from Recombinantly Expressed CYPs: Intersystem Extrapolation Factors. Xenobiotica 34, 151–178. doi:10.1080/00498250310001646353
Rodriguez, H. O., Guerrero, N. A., Fortes, A., Santi-Rocca, J., Gironès, N., and Fresno, M. (2014). Trypanosoma Cruzi Strains Cause Different Myocarditis Patterns in Infected Mice. Acta Trop. 139, 57–66. doi:10.1016/j.actatropica.2014.07.005
Rodriques Coura, J., and De Castro, S. L. (2002). A Critical Review on Chagas Disease Chemotherapy. Mem. Inst. Oswaldo Cruz. 97, 3–24. doi:10.1590/s0074-02762002000100001
Rogers, K. E., Keränen, H., Durrant, J. D., Ratnam, J., Doak, A., Arkin, M. R., et al. (2012). Novel Cruzain Inhibitors for the Treatment of Chagas' Disease. Chem. Biol. Drug Des. 80, 398–405. doi:10.1111/j.1747-0285.2012.01416.x
Spaggiari, D., Geiser, L., and Rudaz, S. (2014). Coupling Ultra-high-pressure Liquid Chromatography with Mass Spectrometry for Iin-Vvitro Drug-Metabolism Studies. Trac Trends Anal. Chem. 63, 129–139. doi:10.1016/j.trac.2014.06.021
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W., and Taylor, R. D. (2003). Improved Protein-Ligand Docking Using GOLD. Proteins 52, 609–623. doi:10.1002/prot.10465
Wang, H., Zrada, M., Anderson, K., Katwaru, R., Harradine, P., Choi, B., et al. (2014). Understanding and Reducing the Experimental Variability of In Vitro Plasma Protein Binding Measurements. J. Pharm. Sci. 103, 3302–3309. doi:10.1002/jps.24119
Wang, J. (2009). Comprehensive Assessment of ADMET Risks in Drug Discovery. Curr. Pharm. Des. 15, 2195–2219. doi:10.2174/138161209788682514
Wang, J., and Skolnik, S. (2009). Recent Advances in Physicochemical and ADMET Profiling in Drug Discovery. Chem. Biodivers. 6, 1887–1899. doi:10.1002/cbdv.200900117
Wang, J., Urban, L., and Bojanic, D. (2007). Maximising Use of In Vitro ADMET Tools to Predict In Vivo Bioavailability and Safety. Expert Opin. Drug Metab. Toxicol. 3, 641–665. doi:10.1517/17425255.3.5.641
Waring, M. J. (2010). Lipophilicity in Drug Discovery. Expert Opin. Drug Discov. 5, 235–248. doi:10.1517/17460441003605098
Yu, H., Wang, Q., Sun, Y., Shen, M., Li, H., and Duan, Y. (2015). A New PAMPA Model Proposed on the Basis of a Synthetic Phospholipid Membrane. PLoS One 10, e0116502. doi:10.1371/journal.pone.0116502
Zamek-Gliszczynski, M. J., Ruterbories, K. J., Ajamie, R. T., Wickremsinhe, E. R., Pothuri, L., Rao, M. V., et al. (2011). Validation of 96-Well Equilibrium Dialysis with Non-Radiolabeled Drug for Definitive Measurement of Protein Binding and Application to Clinical Development of Highly-Bound Drugs. J. Pharm. Sci. 100, 2498–2507. doi:10.1002/jps.22452
Zanatta, N., Amaral, S. S., Dos Santos, J. M., de Mello, D. L., Fernandes, Lda. S., Bonacorso, H. G., et al. (2008). Convergent Synthesis and Cruzain Inhibitory Activity of Novel 2-(N'-Benzylidenehydrazino)-4-Trifluoromethyl-Pyrimidines. Bioorg. Med. Chem. 16, 10236–10243. doi:10.1016/j.bmc.2008.10.052
Keywords: chagas disease, cruzain, medicinal chemistry, drug design, multiparameter optimization, pharmacokinetics, molecular modeling
Citation: Pauli I, Rezende Jr. CdO, Slafer BW, Dessoy MA, de Souza ML, Ferreira LLG, Adjanohun ALM, Ferreira RS, Magalhães LG, Krogh R, Michelan-Duarte S, Del Pintor RV, da Silva FBR, Cruz FC, Dias LC and Andricopulo AD (2022) Multiparameter Optimization of Trypanocidal Cruzain Inhibitors With In Vivo Activity and Favorable Pharmacokinetics. Front. Pharmacol. 12:774069. doi: 10.3389/fphar.2021.774069
Received: 10 September 2021; Accepted: 22 November 2021;
Published: 05 January 2022.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
Aparecida Donizette Malvezi, State University of Londrina, BrazilCopyright © 2022 Pauli, Rezende Jr., Slafer, Dessoy, de Souza, Ferreira, Adjanohun, Ferreira, Magalhães, Krogh, Michelan-Duarte, Del Pintor, da Silva, Cruz, Dias and Andricopulo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriano D. Andricopulo, YWFuZHJpY29AaWZzYy51c3AuYnI=; Luiz C. Dias, bGRpYXNAdW5pY2FtcC5icg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.