 Sareshma Sudhesh Dev
Sareshma Sudhesh Dev Syafiq Asnawi Zainal Abidin
Syafiq Asnawi Zainal Abidin Reyhaneh Farghadani
Reyhaneh Farghadani Iekhsan Othman
Iekhsan Othman Rakesh Naidu
Rakesh Naidu- Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, Malaysia
Receptor tyrosine kinases (RTKs) are transmembrane cell-surface proteins that act as signal transducers. They regulate essential cellular processes like proliferation, apoptosis, differentiation and metabolism. RTK alteration occurs in a broad spectrum of cancers, emphasising its crucial role in cancer progression and as a suitable therapeutic target. The use of small molecule RTK inhibitors however, has been crippled by the emergence of resistance, highlighting the need for a pleiotropic anti-cancer agent that can replace or be used in combination with existing pharmacological agents to enhance treatment efficacy. Curcumin is an attractive therapeutic agent mainly due to its potent anti-cancer effects, extensive range of targets and minimal toxicity. Out of the numerous documented targets of curcumin, RTKs appear to be one of the main nodes of curcumin-mediated inhibition. Many studies have found that curcumin influences RTK activation and their downstream signaling pathways resulting in increased apoptosis, decreased proliferation and decreased migration in cancer both in vitro and in vivo. This review focused on how curcumin exhibits anti-cancer effects through inhibition of RTKs and downstream signaling pathways like the MAPK, PI3K/Akt, JAK/STAT, and NF-κB pathways. Combination studies of curcumin and RTK inhibitors were also analysed with emphasis on their common molecular targets.
1 Introduction
In 2020, The International Agency for Research Cancer (IARC) GLOBOCAN reported approximately 19.3 million new cases of cancer and 10 million deaths globally with data from 185 countries/territories. Lung, breast, and prostate cancers were the most commonly diagnosed cancers, while lung, liver, and stomach cancers were the most common causes of cancer death (Ferlay et al., 2021). Most cancer patients undergo combination treatments, for example, surgery combined with chemotherapy or radiotherapy. Chemotherapy alone can also consist of a combination or cocktail of drugs depending on the type and stage of cancer. Common chemotherapeutic drugs can be biochemically classified into alkylating agents (e.g. cisplatin, carboplatin, and etc.), anti-metabolites (e.g. gemcitabine, 5-fluorouracil), anti-tumour antibiotics (e.g. doxorubicin, epirubicin), topoisomerase inhibitors (e.g. etoposide) and tubulin-binding drugs (e.g. vinorelbine, paclitaxel, and doclitaxel) (Dickens and Ahmed, 2018). On the other hand, targeted therapy involves strategies that specifically target characteristic features in cells or proteins that enable cancer.
Receptor tyrosine kinases (RTKs) are a group of membrane-bound receptors that play an important role in the normal function of cells. They act as signal transducers that mediate cell-to-cell communication by phosphorylating tyrosine residues on key intracellular substrate proteins. Essentially, they lie at the centre of complex interconnecting signaling pathways and are actively involved in the maintenance of cellular homeostasis through regulation of cell proliferation, differentiation, metabolism, migration, and etc. (Wheeler and Yarden, 2015). Alteration or abnormal activation of RTKs have been recurrently observed and recognised as a contributing factor in the progression of various cancers (Weigand et al., 2005; Huang et al., 2011; Wang et al., 2011; Ha et al., 2013; Gallant et al., 2015). These observations led to the development of tyrosine kinase inhibitors (TKIs), which is a well-known targeted therapy. A commonly used TKI is the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) against non-small cell lung cancer (NSCLC). These small molecule inhibitors inhibit the tyrosine kinase domain of EGFR (Chan and Hughes, 2014). First- (gefitinib, erlotinib), second- (afatinib, dacomitinib), and third- (osimertinib) generation EGFR TKIs have been developed with slightly different mechanisms aimed at specific activating mutations (Lin et al., 2014). Other examples of TKIs and their targets include sorafenib (VEGFR kinase, RAF, PDGFR), crizotinib (ALK kinase), sunitinib (VEGF, PDGFR), imatinib (PDGFR, ABL kinase), carfilzomib (proteasome), ribociclib (CDK4, CDK6), and others. Despite their perceived efficacy, the use of TKIs are eventually met with the rise of resistance. Tumours either show a lack of response from the beginning of treatment or they slowly develop resistance after exposure to the drug (Simasi et al., 2014). RTKs mediate the emergence of TKI resistance through their oncogenic alterations such as mutations, overexpression, abnormal fusions and autocrine activation loops (Kobayashi et al., 2005; Cepero et al., 2010; Terai et al., 2013; Enrico et al., 2020). This poses a challenge to the clinical use of TKIs against cancer. Hence, recently, many researchers have begun studying the anti-cancer effects of naturally-derived compounds, mainly from the plant species. The idea behind this effort is to find an effective adjuvant that can be administered in combination with existing anti-cancer drugs, thus eliminating the common issue of toxicity associated with combination drug treatments.
Curcumin is a hydrophobic polyphenol extracted from the herb Curcuma longa or commonly known as turmeric. It was first introduced in 1910, but it has recently gained attention due to its potent therapeutic properties (Miłobȩdzka et al., 1910; Boroumand et al., 2018). Curcumin is a diferuloylmethane and its IUPAC name is (1E, 6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione (Giordano and Tommonaro, 2019). Various studies have shown that curcumin has anti-inflammatory (Farhood et al., 2019), anti-proliferative (Teiten et al., 2011), anti-oxidant (Boroumand et al., 2018), anti-microbial (Adamczak et al., 2020), anti-metastatic (Deng et al., 2016), and anti-angiogenic (Shakeri et al., 2019) properties. These properties altogether make curcumin a powerful anti-cancer agent. In India, where turmeric has been widely used as a cooking spice and medication for thousands of years, cancer rates are much lower compared to western countries. The lowest rates of cancer in India include esophagus, colorectal, liver, pancreas, lung, breast, uterine, ovary, prostate, bladder, kidney, renal, and brain cancers as well as non-Hodgkin lymphoma, and leukemia (Hutchins-Wolfbrandt and Mistry, 2011). There is however, a lack of hard evidence proving that turmeric consumption is solely or at least majorly responsible for the reduced cancer rates. Hutchins-Wolfbrandt and Mistry (2011) describe a few studies that have looked into daily turmeric consumption in India and Nepal, however, these studies did not examine how this affected the overall prevalence of cancer. Despite the lack of proven correlations, the availability and rapid expansion of curcumin-related research especially in the last decade, points towards its viability as an anti-cancer agent. Some of the main molecular targets of curcumin include transcription factors, growth factors, inflammatory cytokines, apoptotic proteins, protein kinases, receptors, cell survival proteins, microRNAs, tumour suppressor genes and oncogenes among others (Rahmani et al., 2014; Giordano and Tommonaro, 2019). In cancer, two of the most crucial roles of curcumin involves its ability to inhibit cellular proliferation and induce apoptosis. These features of curcumin target the root cause of cancer which is abnormal cell growth and apoptotic evasion. Several molecular targets of curcumin involving these two hallmarks of cancer are inhibition of growth factors and kinases (TGF-α, EGF, VEGF, FGF, FAK, JAK, MAPKs, mTOR, and etc.) and induction of apoptotic-related proteins (Bax, Bim, Bcl-2, Bcl-XL, and etc.) (Zhou et al., 2011). An essential component regulating these processes are RTKs and curcumin has been found to target RTKs like EGFR, VEGFR, FGFR, PDGFR, and others. Curcumin mainly downregulates RTK expression, inhibits RTK activation, decreases RTK ligands and also inhibits RTK downstream signaling pathways. More recently, several studies have also reported that curcumin enhances the effects of TKIs when administered in combination and in some cases, overcoming resistance altogether. The effects of curcumin are not only mediated through RTKs and involves many other components/molecular targets as mentioned before, however, RTKs seem to be at the core of these processes. Therefore, this current review discussed the role of receptor tyrosine kinases namely epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), platelet-derived growth factor receptor (PDGFR), insulin-like growth factor 1 receptor (IGF-1R), and hepatocyte growth factor receptor (HGFR) in cancer and how curcumin targets these RTK signaling pathways including the mitogen-activated protein kinase (MAPK), the phosphatidylinositol 3-kinases (PI3K)/Akt, the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) and NF-κB pathways. These RTKs were selected because they are well documented targets of curcumin in cancer. Additionally, drug combination studies involving curcumin and tyrosine kinase inhibitors were also reviewed with a particular focus on RTK inhibitors namely those targeting EGFR, VEGFR, and PDGFR.
2 Receptor Tyrosine Kinase Activation
2.1 Receptor Tyrosine Kinase Activation in Normal Cells
Tyrosine kinases can be further divided into receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (NRTKs). Of all the 90 known tyrosine kinases, 58 are RTKs from 20 subfamilies (Table 1) while 32 are NRTKs from 10 subfamilies. All RTKs have a similar basic structure consisting of an amino terminal extracellular domain containing a ligand binding site, a single transmembrane α-helix, an intracellular tyrosine kinase domain, a tyrosine rich carboxy-(C) terminal and juxtamembrane regions (Lemmon and Schlessinger, 2010; Wheeler and Yarden, 2015).
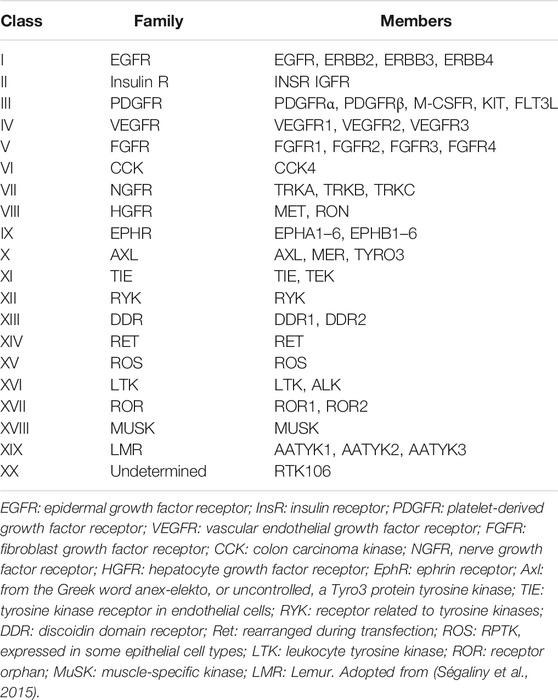
TABLE 1. Classification of RTKs according to family.
RTK activation occurs through the binding of a ligand to the receptor, which then induces receptor dimerization. There are four general modes that have been proposed. These include 1) ligand-mediated dimerization, 2) ligand-mediated dimerization with receptor contacts, 3) ligand-mediated dimerization with receptors contacts and accessory molecules and lastly 4) receptor-mediated dimerization (Lemmon and Schlessinger, 2010). Once ligand-induced dimerization occurs, it activates the intracellular tyrosine kinase domain (TKD) through the transmembrane (TM) domain. The RTK TM dimer interface is very specific and contains essential structural information regarding the positioning of the catalytic domains. Interestingly, studies have found that switching the TM domains between different receptors can still result in constitutive activation as long as the catalytic domains are properly oriented (Cheatham et al., 1993; Petti et al., 1998; Li and Hristova, 2006).
Before TKDs are activated, each TKD is cis-autoinhibited by a specific group of intra-molecular interactions unique to each receptor. RTK activation occurs when this cis-autoinhibition is released after ligand binding and dimerization (Lemmon and Schlessinger, 2010). RTKs can be cis-autoinhibited by their activation loop, juxtamembrane region and C-terminal sequences (Mohammadi et al., 1996; Niu et al., 2002; Till et al., 2002). Transphosphorylation of tyrosine residues in each of these structures are required for activation. Certain TKDs can also be activated allosterically by their partners within a stable dimer. Autophosphorylation of RTKs occurs in several phases, with more tyrosine residues in the cytoplasmic region being autophosphorylated in a precise order (Lemmon and Schlessinger, 2010). The phosphorylated tyrosines or phosphotyrosines then become binding sites that recruit and assemble signaling molecules possessing the Src homology-2 (SH2) and phosphotyrosine-binding (PTB) domains. These specific molecules either bind directly to phosphotyrosine residues, or are indirectly recruited by binding to docking proteins phosphorylated by RTKs (Schlessinger, 2000). Some of these docking proteins include IRS1 (insulin receptor substrate-1), FRS2 and Gab1 (Grb-associated binder). These proteins further activate multiple downstream signaling pathways, with some of the main ones being the MAPK, PI3K, JAK/STAT, and PKC pathways (Lemmon and Schlessinger, 2010; Du and Lovly, 2018). These pathways regulate key processes such as survival, proliferation, differentiation, metabolism and cell-cycle control (Lemmon and Schlessinger, 2010). A more detailed account of these pathways will be included in the following sections.
2.2 Receptor Tyrosine Kinase Activation in Cancer Cells
The abnormal activation of RTKs is a multifaceted process involving not just the RTKs themselves but also partner molecules and their surrounding environments. Their association with diverse groups of cellular components further complicates the mechanics of oncogenic RTK activation. Four main mechanisms leading to aberrant activation have been proposed (Figure 1), which are 1) RTK overexpression, 2) gain-of-function mutations, 3) chromosomal translocations, and 4) autocrine activation. In addition to these basic mechanisms, oncogenic RTK activation can also be influenced by kinase domain duplications, microRNAs, tumour microenvironment changes, negative RTK signaling regulators, protein tyrosine phosphatases, altered endocytic/trafficking genes and also spatial deregulation of RTKs (Casaletto and McClatchey, 2012; Du and Lovly, 2018).
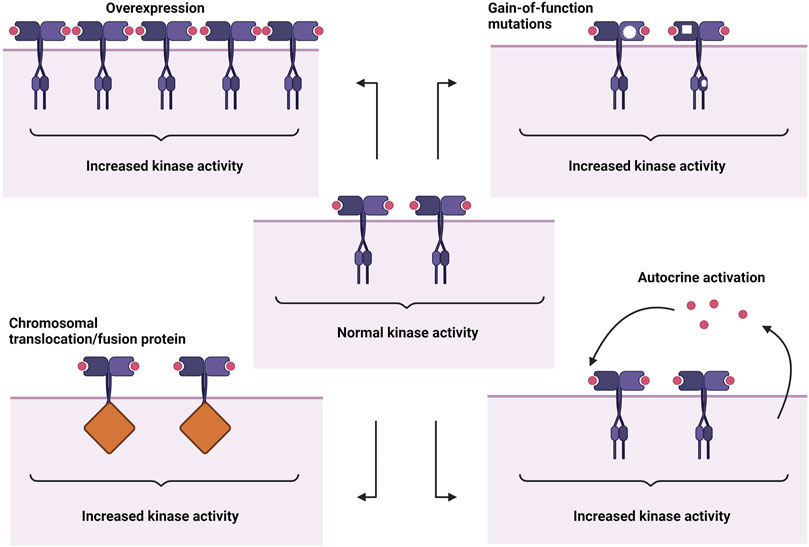
FIGURE 1. Abnormal RTK activation mechanisms. From top left: overexpression, gain-of-function mutations, autocrine activation, chromosomal translocation/fusion protein. Created with BioRender.com.
2.2.1 Epidermal Growth Factor Receptor
EGFR is an extensively studied RTK especially in lung cancer and aptly encapsulates the range of RTK oncogenic alterations. The best well-studied alterations are the EGFR activating mutations that occur in NSCLC. These mutations mainly occur in exons 18, 19, 20, and 21 of the TKD gene (Shigematsu and Gazdar, 2006). Approximately 90% of all EGFR activating mutations involve exon 19 deletions and the L858R point mutation. These mutations allow EGFR activation in the absence of ligand binding and shift the equilibrium between active and inactive states of the TK, enhancing kinase activity (Gazdar, 2009). Mutations also occur in the extracellular domain (ECD) of EGFR in lung (Yu et al., 2017), brain (Idbaih et al., 2009), and colon cancers (Arena et al., 2015). Some of these mutations were found to cause ligand-independent EGFR activation, EGFR amplification and disruption of anti-EGFR mAb binding.
EGFR amplification which commonly occurs due to mutation also occurs in breast (Shao et al., 2011; Park et al., 2014), lung (Morinaga et al., 2008), ovarian (Lassus et al., 2006), and prostate (Schlomm et al., 2007) cancers. The overexpression of EGFR leads to increased surface abundance which stimulates receptor dimerization and subsequent kinase activation (Casaletto and McClatchey, 2012). RTK surface abundance is also influenced by processes involving endocytic machinery and trafficking, whereby alterations of genes/proteins involved in RTK endocytosis can enhanced RTK activation (Bremm et al., 2008; Casaletto and McClatchey, 2012). EGFR gene fusions also occur in lung cancer and the most common fusion is EGFR-RAD51, which is a fusion between the EGFR TKD and RAD51, a DNA damage response protein (Konduri et al., 2016). EGFR-RAD51 can activate MAPK and PI3K/Akt pathways and promote cytokine-independent cell proliferation and colony formation, which are hallmarks of tumour cells (Konduri et al., 2016). Other EGFR fusions in NSCLC include fusions with purine-rich element binding protein B (PURB), septin 14 gene (SEPTIN14) and a recently discovered fusion partner, kinesin family member 5B (KIF5B) (Konduri et al., 2016; Zhu et al., 2019; Xu and Shao, 2020). EGFR can be abnormally activated through kinase domain duplications (KDDs) as well. EGFR-KDDs arise from in-frame tandem duplications of EGFR exons 18–25. Activation of EGFR-KDD occurs through the formation of ligand-independent intra-molecular dimers, which in turn amplifies signaling via ligand-dependent inter-molecular dimers (Du et al., 2021). EGFR-KDD has mostly been studied in lung cancer with regard to clinical outcomes (Wang et al., 2019; Chen et al., 2020). However the first case of EGFR-KDD was reported in a patient with esophageal squamous cell carcinoma, suggesting that it may be linked to hyper-progressive disease (Wang et al., 2020). Several studies also found that EGFR can be activated in an autocrine manner. Autocrine signaling occurs when both the target cell and secreting cell are the same cell. Autocrine signaling has been demonstrated to maintain cancer stem cells and also activate EGFR in tumour cells (Wu et al., 2007; Kim et al., 2012).
2.2.2 Vascular Endothelial Growth Factor Receptor
There are three types of VEGFRs namely VEGFR1, VEGFR2, and VEGFR3 whereas there are five structurally related VEGF ligands including VEGFA, VEGFB, VEGFC, VEGFD, and placenta growth factor (PIGF) (Rapisarda and Melillo, 2012). There are also co-receptors involved in ligand binding called neuropilins (NRPs). Normal activation of VEGFRs generally lead to biological processes like angiogenesis, lymphangiogenesis, migration of endothelial cells, fatty acid uptake, etc. (Rapisarda and Melillo, 2012). VEGFRs and their ligands have been found to be expressed in lung (Tanno et al., 2004; Seto et al., 2006), breast (Filho et al., 2005; Zhao D. et al., 2015), colorectal (Lesslie et al., 2006), prostate (Chen et al., 2004), gastric (Yonemura et al., 2001) cancers. Overexpression is the most common mechanism of abnormal activation in VEGFRs. VEGFR1, and VEGF expressions were found to be elevated in pancreatic cancer cells leading to the activation of the MAPK pathway, which promoted cancer cell growth (Itakura et al., 2000). Meanwhile, an examination of 156 human gastric cancer specimens detected high expressions of VEGFR2, which correlated with poor overall survival (OS) (Lian et al., 2019). Using cDNA construct-transfected cells, they also found that VEGFR2 overexpression accelerated cell proliferation and increased cell invasive properties. VEGFR2 overexpression was also found in ovarian cancer cells (Spannuth et al., 2009), and it was linked to lower E-cadherin expression in breast cancer cells (Yan et al., 2015), suggesting its role in epithelial-mesenchymal transition. However, contradictory results were shown in a human carcinoid cell line whereby downregulation of VEGFR2 correlated to lower E-cadherin expression, suggesting the alternate roles of VEGFR2 in different cancers (Silva et al., 2011). The expression of VEGFR1, VEGFR2, and VEGFR3 has also been demonstrated to vary between the different stages of cervical (Van Trappen et al., 2003), prostate (Grivas et al., 2016) and ovarian (Klasa-Mazurkiewicz et al., 2011) cancers, with VEGFR3 being commonly overexpressed in the later stages. Autocrine activation of VEGFR occurs when VEGF ligands produced by the cancer cells proceed to activate the VEGFRs present on the same cancer cells. This autocrine feed-forward loop has been commonly demonstrated between VEGF:VEGFR2 (Jackson et al., 2002; Chatterjee et al., 2013; Song et al., 2019) and VEGFC:VEGFR3 (Kodama et al., 2008; Matsuura et al., 2009; Chen et al., 2010). Autocrine VEGF signaling also modulates treatment efficacy towards small molecule inhibitors in liver and gastric cancer, whereby higher expressions of VEGFR1/2 within the autocrine loop resulted in higher drug-induced inhibition of cell proliferation and delayed tumour growth (Peng et al., 2014; Lin et al., 2017).
2.2.3 Fibroblast Growth Factor Receptor
There are seven types of FGFRs encoded by four different genes, which are FGFR1, FGFR2, FGFR3, and FGFR4. All of them have two different isoforms produced by alternative splicing except for FGFR4. On the other hand, over twenty FGFs can be grouped into seven families (Porta et al., 2017). FGFR signaling plays an important role during embryonic development and adult life. In cancers containing genetically altered FGFRs, the most frequent alteration can be found in FGFR1 (49%), followed by FGFR3 (23%), FGFR2 (19%), and lastly FGFR4 (7%) (Liu et al., 2021). The most common alteration is the amplification of FGFR genes and based on meta-analysis data, it mainly occurred in lung, breast, and gastric cancers (Chang et al., 2014). FGFR1 amplification was present in approximately 15–18% of lung squamous cell carcinoma patients as shown by three separate studies examining cases from 2000 to 2013 (Heist et al., 2012; Craddock et al., 2013; Monaco et al., 2015). FGFR1-amplified lung and breast cancer cells were shown to have enhanced activation of MAPK and PI3K signaling pathways, increased ligand-dependent signaling, and increased expression of stem cell markers (Turner et al., 2010; Ji et al., 2016). In gastric cancers, FGFR2 amplification was linked to poor progression free survival and overall survival (Matsumoto et al., 2012; Su et al., 2014; Hur et al., 2020), however it was also associated with high sensitivity towards FGFR inhibitors, suggesting the benefits of patient stratification based on FGFR amplification status (Xie et al., 2013; Pearson et al., 2016). Besides FGFR amplification, a comprehensive list of approximately 200 point mutations have also been found in FGFRs (Gallo et al., 2015). Mutations present in all four FGFR receptors were found in breast, colon, brain, lung and head and neck squamous cell carcinomas (Gallo et al., 2015). Acquired resistance to targeted therapies has also been linked to FGFR polymorphisms and gatekeeper mutations like V561M, leading to constitutive activation of FGFR1 (Cowell et al., 2017; Ryan et al., 2019; Mao et al., 2020). FGFR2 and FGFR3 are commonly involved in the formation of oncogenic gene fusions (Porta et al., 2017). The first gene fusion discovered was between FGFR3 and the transforming acidic coiled-coil containing protein (TACC3) forming FGFR3-TACC3 in glioblastoma (Singh et al., 2012; Parker et al., 2013). FGFR3-TACC3 fusions have been observed in lung (Capelletti et al., 2014; Wang et al., 2014), cervical (Carneiro et al., 2015), bladder (Nassar et al., 2018), and nasopharyngeal (Yuan et al., 2014) cancers and usually lead to increased cell proliferation, in vitro transforming abilities, and activation of MAPK and ERK signaling (Nelson et al., 2016). FGFR gene fusions usually involve partners possessing dimerization domains that allow ligand-independent receptor dimerization resulting in constitutive activation (Parker et al., 2014). Other FGFR gene fusions include BAG4-FGFR1, FGFR2-BICC1, FGFR2-CASP7, FGFR2-AFF3, and FGFR3–BAIAP2L1 (Wu et al., 2013).
2.2.4 Platelet-Derived Growth Factor Receptor
There are two platelet-derived growth factors which are PDGFRα and PDGFRβ, also known as PDGFRA and PDGFRB. These two receptors are activated by five PDGFs which include PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD (Fredriksson et al., 2004). Based on The Cancer Genome Atlas (TCGA) data, gene alterations in the PDGF family of ligands and receptors most commonly occur in lung cancer, colon cancer, and glioblastoma (Farooqi and Siddik, 2015). PDGFRB mutations in cancer have not been studied widely however, PDGFRA mutations are frequently observed in gastrointestinal stromal tumours (GISTs), especially in exon 18 (Heinrich et al., 2003; Daniels et al., 2011; Joensuu et al., 2015). PDGFRA and KIT mutations are associated with site and origin of tumours (Penzel et al., 2005), while gain-of-function mutation, V536E, led to increased phosphorylation of ERK and STAT5, causing constitutive receptor activation (Velghe et al., 2014). These mutations can occur on the regulatory domains (extracellular domain and juxtamembrane domain) or the enzymatic domain (tyrosine kinase domain (TKD)), which can lead to ligand-independent receptor dimerization or even kinase activation without receptor dimerization altogether (Lasota and Miettinen, 2006). Overexpression of PDGFRA mRNA has been recently observed in oral squamous cell carcinoma with links to metastasis and reduced patient survival (Ong et al., 2017; Ong et al., 2018). In lung cancer, ovarian cancer and medulloblastomas, PDGFR overexpression was associated with shorter overall survival, co-amplification with other RTKs and potential prognostic value (Lassus et al., 2004; Blom et al., 2010; Tsao et al., 2011).
PDGFR gene fusions are widely observed in hematological malignancies like acute myeloid leukemia, lymphoblastic leukemia and other myeloproliferative neoplasms (MPNs). PDGFRA fuses with five other intracellular proteins and one RTK, whereas PDGFRB fusions occur with twenty-nine other intracellular proteins (Appiah-Kubi et al., 2017). FIP1L1-PDGFRA is the most recurrent PDGFRA fusion gene that was first observed in patients with hypereosinophilic syndrome (Cools et al., 2003). Kinase activation of FIP1L1-PDGFRA is mediated by the disruption of the juxtamembrane domain of PDGFRA (Stover et al., 2006). Other gene fusion partners of PDGFRA include the breakpoint cluster region (BCR) (Yigit et al., 2015), KIF5B (Score et al., 2006), the CDK5 regulatory subunit associated protein 2 (CDK5RAP2) (Walz et al., 2006) and the ETS variant transcription factor 6 (ETV6) (Yoshida et al., 2015). As for PDGFRB, approximately 70 fusions have been identified with most fusion partners normally containing an oligomerization motif that mediates dimerization, causing continuous kinase domain activation in myeloid neoplasms (Campregher et al., 2017; Sheng et al., 2017; Xu et al., 2020). Autocrine PDGFR signaling plays an essential role in cancer progression in ovarian (Matei et al., 2006), breast (Jechlinger et al., 2006), thyroid (Adewuyi et al., 2018), and brain (Lokker et al., 2002) cancers. Both PDGFR and its ligands expressed in these tumours leads to an autocrine loop that fuels activation of downstream PI3K/Akt, MAPK and STAT pathways, in addition to the maintenance of EMT and enhanced metastasis (Jechlinger et al., 2006; Matei et al., 2006; Adewuyi et al., 2018).
2.2.5 Insulin Receptor
The insulin receptor (IR) family consists of IGF-1R, IRA, IRB, IGF-1R/IR (hybrid), and IGF-2R. This review will focus on IGF-1R and IGF-2R, which are more frequently studied. IGF-1R is activated by ligands IGF-1 and IGF-2, while IGF-2R is a non-signaling receptor that mainly functions to clear IGF2 from the cell surface (Chen and Sharon, 2013). IGF-1R is overexpressed in a variety of cancers including colon (Shiratsuchi et al., 2011), pancreatic (Hakam et al., 2003), prostate (Aleksic et al., 2017), lung (Gong et al., 2009; Badzio et al., 2010), and breast (Jones et al., 2007) cancers. High levels of total IGF-1R were associated with higher tumour grade while higher levels of cytoplasmic IGF-1R were linked to a greater risk of post-radiotherapy recurrence in prostate cancer patients (Aleksic et al., 2017). Meanwhile, overexpression of IGF-1R in transgenic mice induces mammary tumour formation through activation of Akt, Erk1/Erk2, and STAT3 (Jones et al., 2007). Moreover, overexpression of IGF-1R also decreases tumour latency time, increases the proliferative genetic signature and enhances migration potential in mammary tumours in addition to protecting cells against stresses of the tumour microenvironment and apoptosis (Resnicoff et al., 1995; Valentinis and Baserga, 1996; Peretz et al., 2002; Ter Braak et al., 2017). IGF-2R has been found to be mutated in 60% of lung squamous cell carcinomas, while levels of IGF-2R appears to be much higher in the malignant stages of endometrial carcinomas (Kong et al., 2000; Pavelić et al., 2007). The existence of an autocrine loop was also found between IL-6 and IGF-1R whereby IL-6 induced expression of itself, forming a positive feedback loop further activating IL-6R, IGF-1R, IGF-1, and IGF-2 in NSCLC (Zheng et al., 2019). In acute myeloid leukemia, autocrine production of IGF-1 was shown to be responsible for the constitutive activation of IGF-1R and PI3K/Akt (Chapuis et al., 2010). Meanwhile, tumour cells have been proposed to secrete IGF-2 which binds to IGF-1R, increasing the rate of cellular proliferation through autocrine/paracrine signaling (Rasmussen and Cullen, 1998; Pavelić et al., 2003).
2.2.6 Hepatocyte Growth Factor Receptor/C-Met
The hepatocyte growth factor receptor (HGFR), also known as MET or c-Met, is encoded by the MET gene, and its ligand is the hepatocyte growth factor (HGF) (Kumar et al., 2018). There is another Met-related RTK called Ron which binds to HGF-like protein/macrophage stimulating-protein (HGFL) (Wagh et al., 2008); however, this review will only focus on c-Met. C-Met has been found to be overexpressed in breast (Zhao et al., 2017), lung (Gumustekin et al., 2012; Awad et al., 2016), ovarian (Sawada et al., 2007), colon (Lee et al., 2018), cervical (Baykal et al., 2003), renal (Miyata et al., 2003), and blood (Jücker et al., 1994) cancers. Overexpression of HGF/c-Met was observed in NSCLC which led to subsequent lymph node invasion mediated by RhoA overexpression (Gumustekin et al., 2012). Mutations were also detected in the TKD of c-Met in this study; however, results suggested that it did not significantly impact RTK activation in NSCLC. In contrast, a more recent study reported MET exon 14 mutations that occurred predominantly in older patients with lung adenocarcinomas. Patients with advanced-stage NSCLC having these mutations also had concurrent MET gene amplification (Awad et al., 2016). A meta-analysis showed that c-Met overexpression correlated to distant metastasis, large tumour size and high histologic grade in breast cancer (Zhao et al., 2017). MET gene fusions have been primarily identified in lung cancer such as the HLA-DRB1-MET (Davies et al., 2017; Blanc-Durand et al., 2020), KIF5B-MET (Gow et al., 2018), MET-UBE2H (Zhu et al., 2018a), and MET-ATXN7L1 (Zhu et al., 2018b). The first case of HLA-DRB1-MET fusion was reported, however further studies are required to elucidate the specific mechanisms of the fusion gene (Davies et al., 2017). Meanwhile, it was also proposed that the MET-UBE2H fusion protein could be a novel resistance mechanism against EGFR-TKI treatment (Zhu et al., 2018a). MET fusions that occur at exon 15 and contain the 3’ MET kinase domain are thought to become activated due to constitutive dimerization of MET (Blanc-Durand et al., 2020).
Autocrine activation of HGF/c-Met signaling has also been described as a novel strategy leading to resistance against multi-kinase inhibitors in heptatocellular carcinoma (Firtina Karagonlar et al., 2016). Resistant cells were shown to have upregulated levels of HGF and activation of c-Met which when inhibited, resulted in lower migration and invasion capabilities (Firtina Karagonlar et al., 2016). Other studies examining hepatocellular carcinoma also found autocrine systems involving the Scatter factor (SF) and HGF/c-Met in metastasis as well as angiogenesis involving VEGF (Xie et al., 2001; Horiguchi et al., 2002). Autocrine activation of the MET receptor was also observed in colorectal cancer and acute myeloid leukemia involving components like β-catenin and co-activation of FGFR1 (Rasola et al., 2007; Kentsis et al., 2012).
3 Receptor Tyrosine Kinase Signaling Pathways Targeted by Curcumin in Cancer
Activation of RTKs leads to a ripple effect whereby multiple signaling cascades are triggered to produce different outcomes. Curcumin has been found to modulate the expression of RTKs, their ligands and components particularly within their downstream MAPK, PI3K/Akt, JAK/STAT, and NF-κB pathways (Figure 2). Curcumin-mediated mechanisms mainly involve inhibition of specific components and in some cases, upregulation as well. Favourable outcomes have been observed in cancer cells following curcumin treatment including enhanced apoptosis, reduced cellular proliferation, reduced angiogenesis and reduced migration. Curcumin appears to act as a tyrosine kinase inhibitor as reviewed by Golonko et al. (2019) and Farghadani and Naidu (2021), similar to the mechanism of TKI drugs but with more pronounced effects. In this section, we take a closer look at how curcumin modulates these signaling pathways.
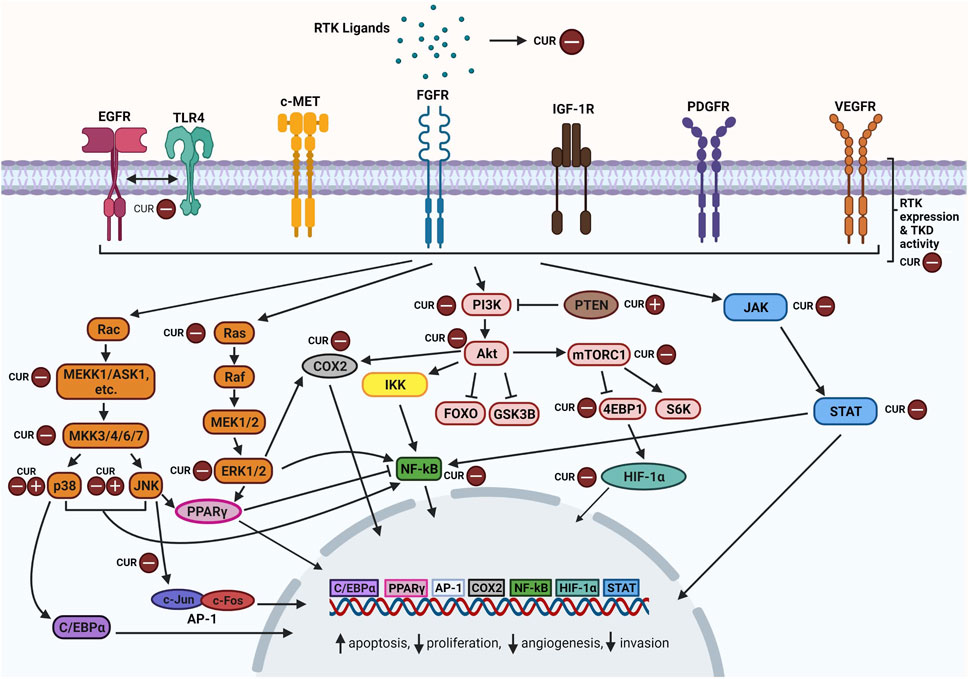
FIGURE 2. Overview of curcumin-inhibition of RTKs and downstream MAPK, PI3K/Akt, JAK/STAT, and NF-κB pathway components. Abbreviations: RTK: Receptor tyrosine kinase; CUR: Curcumin, EGFR: Epidermal growth factor receptor, TLR4: Toll-like receptor 4, c-MET: Mesenchymal epithelial transition factor/Hepatocyte growth factor receptor; FGFR: Fibroblast growth factor receptor; IGF-1R: The insulin-like growth factor 1 receptor; PDGFR: Platelet-derived growth factor receptor; VEGFR: Vascular endothelial growth factor receptor; TKD: Tyrosine kinase domain; Rac: Ras-related C3 botulinum toxin substrate; ASK1: Apoptosis signal-regulating kinase 1; C/EBPα: CCAAT/enhancer-binding protein alpha; JNK: c-Jun N-terminal kinases; AP-1: Activator protein 1; Ras: Rat sarcoma virus protein; Raf: Rapidly accelerated fibrosarcoma protein; MEK: Mitogen-activated protein kinase kinase; ERK: Extracellular signal-regulated kinase; PPARγ: Peroxisome proliferator-activated receptor γ; COX2: Cyclooxygenase-2; PI3K: Phosphoinositide 3-kinase; PTEN: Phosphatase and tensin homolog; Akt: Ak strain transforming; IKK: Inhibitor of nuclear factor kappa B kinase; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; FOXO: Forkhead box transcription factors; GSK3B: Glycogen synthase kinase 3 beta; Mtorc1: Mechanistic target of rapamycin complex 1; 4EBP1: Eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1; S6K: Ribosomal protein S6 kinase; HIF-α: Hypoxia-inducible factor alpha; JAK: Janus kinase; STAT: Signal transducer and activator of transcription. Created with BioRender.com.
3.1 Effects of Curcumin on Receptor Tyrosine Kinase Signaling Pathways in Cancer
3.1.1 Mitogen-Activated Protein Kinase
The mitogen-activated protein kinase (MAPK) pathway is one of the main pathways involved in the regulation of cellular proliferation, differentiation, development, apoptosis, and transformation. Three MAPK families have been well characterised which include extracellular signal-regulated kinase (ERK), Jun kinase (JNK) and p38 kinase. MAP kinases are activated in a cascade fashion following stimulation by growth factors, cytokines, stress and ceramides among others. A MAP kinase cascade is normally a series of activations involving a MAPK kinase kinase (MAPKKK), a MAPK kinase (MAPKK), and a MAP kinase (MAPK) (Zhang and Liu, 2002). The Raf-MEK-ERK is one of the well characterised MAPK signaling pathways. The multistep process that occurs after RTK activation starts with recruitment of adaptor proteins (Grb2, Sos, etc.) followed by activation of c-Raf (MAPKKK), MEK1/2 (MAPKK) and finally ERK1/2 (MAPK). ERK then translocates to the nucleus and phosphorylates transcription factors like the ternary complex factor (TCF) Elk-1, c-Myc, serum response factor accessory protein Sap-1a, Ets1, Tal, and others (Zhang and Liu, 2002). The JNK proteins also known as stress-activated protein kinases (SAPKs) are primarily activated by stress conditions like DNA damage, UV irradiation, and inflammation (Katz et al., 2007). Growth factors like EGF, PDGF, and FGF are less efficient stimulants (Kyriakis and Avruch, 2001). JNKs are directly phosphorylated by MKK4 and MKK7 (MAPKKs) while these MAPKKs are dually phosphorylated by MAPKKKs which include the MEKK family, the mixed-lineage kinase family, the apoptosis signal-regulating kinase family, TAK1 and TPL2 (Davis, 2000). Upon activation, JNKs can activate a range of proteins including the activator protein-1 (AP-1) which is formed through the dimerization of Jun (c-Jun, JunB, and JunD) and Fos (c-Fos, FosB, Fra-1, and Fra-2) proteins. Lastly, the p38 kinase has four isoforms, α, β, γ, and δ (Canovas and Nebreda, 2021). They can be phosphorylated by the MAPKKs, MKK3, MKK4, and MKK6. Prior to this, these MAPKKs are phosphorylated by MAPKKKs such as ASK1, DLK, MEKK3, MEKK4, TAK1, and etc. Overall, growth factors mainly activate the ERK1/2 cascade, partially activate JNK, and rarely activate p38 (Katz et al., 2007). Hence, this review will mainly focus on the ERK pathway.
In cancer, constitutive activation of ERK signaling is normally caused by RTK overexpression and activating mutations in RTKs or components like Ras or B-Raf (Dhillon et al., 2007). Activating mutations in K-Ras and N-Ras have been observed in many cancers and commonly lead to inefficient GTP hydrolysis, leaving Ras in a constantly active, GTP-bound state (Dhillon et al., 2007; Prior et al., 2020). There are three isoforms of Raf namely, Raf-1/C-Raf, B-Raf, and A-Raf which are direct effectors of Ras. B-Raf gene mutations are present most commonly in melanoma (40–70%) and to a lesser extent in thyroid, colorectal and ovarian cancers (Rahman et al., 2013). The missense mutation, V600E, is the most common B-Raf mutation (∼90% of cancers) that results in constitutive activation of the MEK-ERK pathway without external stimuli, causing uncontrolled cellular proliferation (Cantwell-Dorris et al., 2011). C-Raf and A-Raf mutations are quite rare and it was found that C-Raf had a low basal kinase activity, which may explain its weak oncogenic effect (Emuss et al., 2005). MEK1/2 mutations are rare as well and they are mainly influenced by upstream mutations in Ras/Raf. Lastly, mutations in ERK were found to confer resistance to ERK and Raf/MEK inhibitors by disrupting drug binding and maintaining levels of ERK activity in B-raf mutant melanoma cells (Goetz et al., 2014). Oncogenic activation of JNK1 and JNK2 have been found in liver (Chang et al., 2009), pancreatic (Tian et al., 2021), bladder (Pan et al., 2016), and gastric (Mishra et al., 2010) cancers. The JNK pathway also promotes cancer cell survival via autophagy involving Bcl-2, tumour immune evasion, compensatory cell proliferation, and interaction with other signaling components such as NF-κB, p38, and JunD (Wu Q. et al., 2019). As for p38, it also acts as a tumour suppressor and inhibition of p38 mediates Ras-induced transformation (Dhillon et al., 2007). Tumour sizes have been found to be inversely correlated to p38 activity in hepatocellular carcinoma (Iyoda et al., 2003). Oncogenic MAPK signaling activated by RTKs are found in various cancers (Wu et al., 2006; Wang et al., 2012; Tang et al., 2017; Jiang et al., 2020).
In lung cancer cells, two separate studies examined the effects of curcumin on RTKs and their pathway components (Lev-Ari et al., 2006; Lev-Ari et al., 2014). Both studies found that curcumin downregulated expressions of COX-2 and p-ERK1/2 in a dose-dependent manner, however only one study observed downregulation of EGFR (Lev-Ari et al., 2006). EGFR signaling has been shown to induce transcription of COX-2 likely through the activation of MEK/ERK pathway, which explains the simultaneous downregulation of these components by curcumin resulting in decreased survival and enhanced apoptotic effects (Huh et al., 2003; Chi et al., 2016). Curcumin also showed dose-dependent inhibition of MyD88, TLR4, and EGFR in lung cancer cell lines (Zhang et al., 2019). Studies have found that TLR4 requires EGFR to signal and activation of TLR4 has also been linked to the MAPK pathway (Qian et al., 2008; De et al., 2015). It is possible that curcumin indirectly modulates the MAPK pathway by synergistic targeting of EGFR and TLR4. In addition, this study also found that curcumin lowered the expression levels of c-Jun and c-Fos proteins which make up AP-1, a major target of JNK. This led to decreases in other cell cycle proteins like cyclin A1, cyclin B1, cyclin D1, and etc. suggesting curcumin’s role in regulating cell cycle transitions via MAPK signaling activated by RTKs (Zhang et al., 2019). Another study found that curcumin inhibited VEGF and a wide range of downstream MAPK-related components including c-Jun-p, Ras, Grb2, MEKK3, and MKK7, however, levels of JNK and ERK seemed to be upregulated (S. S. Lin et al., 2009). JNK 1 and 2 have been observed to have opposing functions (Yin and Yang) in cellular environments (Wu Q. et al., 2019). Studies have found that JNK1 mediates cell survival while JNK2 contributes to apoptosis however, the opposite has also been observed (Arbour et al., 2002; Liu et al., 2004). Hence, through inhibition of VEGF, curcumin may have indirectly upregulated the pro-apoptotic JNK protein levels since JNK is not a direct target of curcumin (Chen and Tan, 1998; S. S.; Lin et al., 2009). Lastly, the effects of curcumin was examined in an in vivo cancer model involving transgenic mice expressing VEGF-A (Tung et al., 2011). Curcumin significantly downregulated levels of VEGF protein and also mRNA levels of vegf, vegfr2 (kdr), nrp-1, egfr, and erk2. Nrp-1 is the co-receptor of vegfr2 and it was suggested that curcumin-induced downregulation of its downstream pathways, resulted in reduced VEGF expression. This was also evidenced by downregulation of erk2, reaffirming that the MAPK pathway plays a role in curcumin-mediated RTK inhibition (Tung et al., 2011).
In colon cancer, curcumin downregulated the EGFR gene expression by suppressing the early growth response-1 (egr-1) gene and the transactivation activity of Egr-1, a transcription factor that binds to the egfr promoter (Chen et al., 2006). Suppression of egr-1 gene by curcumin was via disruption of ERK signaling which led to a decrease in Elk-1 phosphorylation (Chen et al., 2006). Besides, curcumin also suppresses EGFR expression by activating PPARγ in colon carcinoma cell lines (Chen and Xu, 2005). PPARγ can be inactivated through phosphorylation by ERK and/or JNK. It was found that curcumin-induced inhibition of MAPK activity led to increased activation of PPARγ and subsequent EGFR gene downregulation (Chen and Xu, 2005). Meanwhile, treatment of colon cancer cells with curcumin or dasatinib induced significant reduction of p-EGFR while combination treatment led to much greater reduction of both p-EGFR and p-IGF-1R (Nautiyal et al., 2011). Accordingly, downstream p-ERK1/2 levels were also reduced by a larger magnitude after combination treatment which may have resulted in the reduction of COX-2 levels that was observed as well. In colon cancer cells, after 3 h of exposure to high concentration of curcumin (100 μmol/L), a cDNA microarray analysis showed that levels of MAPK-related genes like MAP3K10 and MAP4K2 and also VEGF and FGFR1 were upregulated (Van Erk et al., 2004). Several other MAPK genes like MAP2K2 and MAPK8 were downregulated after exposure to low concentrations of curcumin (30 μmol/L) for 3 h. The high concentration of curcumin used in this study was found to decrease the cell number and result in floating cells hence, the several unexpected gene expression changes observed could be toxic-related effects of curcumin (Van Erk et al., 2004).
In breast cancer cell lines that overexpress HER-2 (BT-474 and SK-BR-3-h), curcumin downregulated the HER-2 oncoprotein and also the phosphorylation of MAPK in a dose-and time-dependent manner (Lai et al., 2012). Meanwhile, in triple negative breast cancer (TNBC) cells curcumin did not alter the expression of EGFR and ERK1/2 however, it significantly reduced the levels of phosphorylated EGFR and ERK1/2, showing that it specifically inhibits activation of EGFR and its downstream signaling molecules to reduce cell proliferation (Sun et al., 2012). Curcumin was also found to reduce EGFR activation and EGF-induced phosphorylation of ERK1/2 as well as JNK activity in breast cancer cells however, there was a lack of inhibition of p38 (Squires et al., 2003). This provides evidence that curcumin inhibition occurs via RTK signaling pathways since RTKs mainly activate ERK and JNK (partially) and p38 to a much lesser extent.
Using pancreatic cancer cells, it was shown that curcumin reduced hyperglycemia-driven EGF-induced metastatic abilities (Li W. et al., 2019). Under high-glucose conditions (diabetes), which is a risk factor for pancreatic cancer, curcumin suppressed EGF levels and activation of EGFR and ERK, resulting in reduced invasive ability and inhibition of metastatic-related factors (Li W. et al., 2019). Expression of COX-2, EGFR and p-ERK1/2 was also suppressed by curcumin in pancreatic adenocarcinoma cells similar to what was observed in lung adenocarcinoma cells (Lev-Ari et al., 2006). Furthermore, curcumin treatment for 24 h was found to decrease the expression of VEGFR1 and VEGFR2 in HUVECs (Fu et al., 2015). Human umbilical vein endothelial cells (HUVECs) are commonly used to understand tumour angiogenesis due to their major role in vascular homeostasis. Phosphorylation of ERK was also reduced reflecting the ability of curcumin to inhibit growth and migration of endothelial cells via blocking VEGFRs and downstream MAPK signaling pathway. Curcumin also reduced COX-2 expression in VEGF-activated human intestinal microvascular endothelial cells (HIMECs) via inhibition of phosphorylation of MAPK pathway components like p44/42 MAPK, p38 MAPK, and JNK (Binion et al., 2008). In oral cancer, curcumin upregulates the expression of insulin-like growth factor binding protein-5 (IGFBP-5) by increasing the nuclear expression of CCAAT/enhancer-binding protein α (C/EBPα), which is a transcriptional regulator of IGFBP-5 (Chang et al., 2010). This upregulation of IGFBP-5 mediated by activation of p38 by curcumin allowed it to bind to IGF, limiting the activation of IGF-1R and suppressing oral carcinogenesis. Meanwhile, a combination of curcumin and cetuximab decreased levels of phosphorylated EGFR, ERK, JNK, and surprisingly p38 in cisplatin-resistant oral cancer cells which contrasts the curcumin-mediated p38 activation observed by Chang et al. (2010). Accordingly, it has been found that curcumin differentially activates/inhibits p38 in different cancers (Watson et al., 2010; Wang et al., 2013; Tung et al., 2016); however, further research is required to elucidate how RTKs fit in this process. Curcumin treatment also abrogated HGF-induced epithelial-mesenchymal transition (EMT) in oral squamous cell carcinoma and prostate cancer cells by reducing levels of phosphorylated c-Met (HGFR) and inhibiting ERK activation (Hu et al., 2016; Ohnishi et al., 2020). Furthermore, levels of p-ERK, VEGF, and HIF-α were reduced by curcumin in liver cancer cells the same way they were reduced in IGF-1R-knockout liver cancer cells, suggesting that curcumin suppresses tumour progression in an IGF-1R-dependent manner involving the MAPK pathway (Chen et al., 2018). Curcumin also stimulated the expression of PPARγ by interrupting EGF and PDGF signaling in rat hepatic stellate cells (Zhou et al., 2007). This interruption involves repressing phosphorylation of PDGFR-β and EGFR and also reducing p-ERK and p-JNK. This is the second study observing the effects of curcumin on RTKs through PPARγ activation mediated by MAPK-inhibition.
3.1.2 Phosphoinositide 3-Kinase/Akt/Mechanistic Target of Rapamycin
The phosphoinositide 3-kinase (PI3K)–Akt pathway is an ubiquitous signaling network that regulates growth, metabolism, biosynthesis of macromolecules and cellular homeostasis. It is mainly activated by growth factors, insulin, and cytokines. There are three classes of PI3K enzymes, however only class I PI3Ks are involved in cancer (Zhao and Vogt, 2008). Class I PI3Ks have four different isoforms (p110α, β, γ, and δ) which are encoded by PIK3CA, PIK3CB, PIK3CG, and PIK3CD (Fruman et al., 2017). Under normal physiological conditions, PI3K activation at the plasma membrane is followed by phosphorylation of phosphatidylinositol 4, 5-bisphosphate (PtdIns(4,5)P2)
In cancer, there are four common genetic events that drive cancer progression which include 1) PIK3CA activating mutations, 2) PTEN loss-of-function mutations and deletions, 3) gain-of-function mutations in Akt-encoding genes, and lastly 4) amplification of RTKs that activate PI3K signaling (Hoxhaj and Manning, 2020). The PI3K pathway also plays a role in the control of glucose metabolism whereby constitutive activation of Akt promotes aerobic glycolysis and increased glucose uptake through GLUTs in cancer cells (Elstrom et al., 2004; Wieman et al., 2007). It also drives anabolic metabolism in excessively proliferating cells via promoting de novo lipid, nucleotide, and protein synthesis (Faridi et al., 2003; Porstmann et al., 2005; Saha et al., 2014). Moreover, the PI3K-Akt pathway has been found to trigger ROS-producing processes as well however more research in needed to elucidate the exact downstream pathways involved in ROS production in cancer cells (Chen et al., 2003; Hoxhaj and Manning, 2020). Activation of RTKs and downstream PI3K signaling have been implicated in acceleration of tumour growth, malignant transformation, and resistance in different cancers (Zhang et al., 2007; Jung et al., 2015; Starska et al., 2018).
In lung cancer, curcumin has been shown to downregulate EGFR expression by inducing expression of an E1-like ubiquitin-activating enzyme, UBE1L, which represses EGFR protein expression and promotes EGFR internalization (Jiang et al., 2014). Curcumin also reduced Akt phosphorylation and repressed the EGFR/Akt pathway through UBE1L induction. Levels of PI3Kwere also reduced in lung cancer cells following curcumin treatment, which is likely one of the pathways that resulted in the reduced VEGF expression that was also observed (S.-S. Lin et al., 2009). Furthermore, curcumin pre-treatment of lung cancer cells decreased the HGF-induced phosphorylation of c-Met and downstream PI3K signaling components like Akt, mTOR, and S6, leading to EMT inhibition (Jiao et al., 2016).
In colon cancer, curcumin combined with 5-fluorouracil (5-FU) and oxaliplatin (FOLFOX) were found to induce higher levels of apoptosis by reducing both the expression and activation of EGFR, IGF-1R, HER-2, and HER-3 by a greater magnitude then either agent alone (Patel et al., 2008). An analysis of downstream signaling components also found downregulation of expression and activation of Akt and COX-2 after curcumin and FOLFOX combination treatment. COX-2 activation and expression has been linked to Akt phosphorylation in several cancers, hence curcumin may mediate COX-2 activation by targeting RTKs and the downstream PI3K/Akt pathway (St-Germain et al., 2004; Glynn et al., 2010). Similar results were also achieved when curcumin was used to treat FOLFOX-surviving colon cancer cells, highlighting the importance of EGFR/IGF-1R/Akt signaling inhibition by curcumin in chemo-resistant cells (Patel et al., 2010). A combination of curcumin and dasatinib also resulted in the downregulation of the EGFR/IGF-1R/Akt axis, further providing evidence that this may be one of the primary mechanisms of inhibition by curcumin either alone or in synergistic combination with other anti-cancer agents (Nautiyal et al., 2011a). Gene expression analyses carried out on colon cancer cells lines found upregulation of many genes including VEGF, FGFR1, and Akt after exposure to high concentration of curcumin which contrasts the usual downregulation, however it could be toxic-related effects of the fairly high concentration of curcumin used as stated before (Van Erk et al., 2004).
In breast cancer, it was found that curcumin combined with herceptin (trastuzumab) was effective against herceptin-resistant breast cancer cells, likely mediated by the decreased levels of HER-2 oncoprotein and phosphorylated Akt (Lai et al., 2012). In another study, curcumin also inhibited the basal phosphorylation of Akt/PKB in breast cancer cells but not directly, suggesting that it could be due to the decrease in EGF-induced EGFR activation that was observed (Squires et al., 2003). The study by Borah at et al. (2020) aimed to inhibit the Hh/Gli-EGFR signaling pathway in breast cancer by co-delivering curcumin and a Hh/Gli small molecule antagonist GANT61 via polymeric nanoparticles. Based on immunofluorescence studies, they found that the GANT61-curcumin PLGA NPs managed to decrease EGFR protein expression and also PI3K expression, which may contribute to the inhibitory migration potential of breast adenocarcinoma cells.
In liver cancer, curcumin decreased VEGF, PI3K, and Akt expression, with one study suggesting that this effect is mediated via curcumin-inhibition of IGF-1R to suppress angiogenesis (Chen et al., 2018; Pan et al., 2018). Curcumin also reduced the phosphorylated PI3K/Akt levels by inhibiting tyrosine phosphorylation of PDGFRβ and EGFR, which led to activation of PPARγ and subsequent induction of apoptosis (Zhou et al., 2007). It was found that a combination of curcumin and metformin significantly reduced PI3K, p-Akt, and p-mTOR while also significantly increasing expression of PTEN, which is a negative regulator of the PI3K pathway (Zhang et al., 2018). Combination treatments for curcumin and β-phenylethyl isothiocyanate (PEITC) as well as curcumin and docetaxel were found to decrease EGFR expression and activation, PI3K expression, and p-Akt which led to enhanced apoptosis and reduced cell proliferation in prostate cancer cells (Kim et al., 2005; Banerjee et al., 2017). Furthermore, in hyperglycemic-pancreatic and oral cancer cells, curcumin was found to reduce cell proliferation by inhibiting the EGF/EGFR/Akt pathway (Zhen et al., 2014; Li W. et al., 2019). Besides, curcumin also decreased gene expression of EGFR and expression of PI3K (p110α), Akt, and mTOR in tongue and hypopharynx squamous cell carcinoma (SCC), highlighting the therapeutic potential of curcumin-mediated RTK inhibition in preventing head and neck cancer progression (Borges et al., 2020). Meanwhile, another study chemically induced skin carcinogenesis in transgenic mice overexpressing IGF-1 and found that a curcumin diet significantly reduced tumour multiplicity, tumour size, and cell proliferation (Kim et al., 2014). The underlying mechanism leading to these effects was curcumin-mediated inhibition of IGF-1R, insulin receptor substrate-1 (IRS-1), Akt, S6K, and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) phosphorylation in a dose-dependent manner (Kim et al., 2014). Lastly, curcumin downregulated phosphorylation of PI3K-p85, Akt, mTOR, and further downstream effectors 4EBP1 and S6K in bladder cancer. Interestingly, IGF-1 knockdown did not alter the inhibitory effects of curcumin, suggesting that curcumin mainly acts through the IGF-2/IGF-1R pathway and downstream PI3K signaling in bladder cancer (Tian et al., 2017).
3.1.3 Janus Kinase/Signal Transducers and Activators of Transcription
The Janus kinase (JAK)–signal transducer of activators of transcription (STAT) pathway is one of the pathways involved in RTK signal transduction and it can be activated by diverse cytokines, interferons and other related components. It allows direct communication from membrane-to-nucleus through the interaction between four Janus kinases (JAKs)—JAK1, JAK2, JAK3, and TYK2, and seven signal transducers and activators of transcription (STATs)—STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 (O'Shea et al., 2015). Once a ligand binds to the receptor, receptor-associated JAKs are activated and they proceed to cross-phosphorylate each other and also the intracellular tail of their receptors. This creates a docking sites for the recruitment of cytoplasmic STATs. STATs are then activated via JAK-phosphorylation and they translocate to the nucleus to regulate gene expression by binding DNA (O'Shea et al., 2015). Many RTKs engage with the JAK/STAT pathway to promote proliferation and differentiation (Boccaccio et al., 1998; Andl et al., 2004; Masamune et al., 2005).
In cancer, constitutive JAK/STAT activation normally occurs through increased expression of ligands and activating mutations of receptors, JAKs, or STAT themselves (O'Shea et al., 2015). JAK mutations have been found widely in leukemia and many solid tumours that contribute to cancer cell migration, proliferation, and invasion (Walters et al., 2006; Jeong et al., 2008; Stelloo et al., 2016; Xu et al., 2017). STAT3 and STAT5 are also commonly mutated a variety of cancers. Mechanisms leading to their constitutive activation include lack of negative regulation, somatic mutations causing hyperactivation, overstimulation, positive feedback loops and crosstalk with other signaling pathways leading to resistance, poor prognosis, tumour progression, and worse overall survival (Zhang and Lai, 2014; Halim et al., 2020). However, studies have shown that both STAT3 and STAT5 have tumour suppressor roles, reflecting their paradoxical nature (Igelmann et al., 2019). RTKs have also been shown to promote cancer progression and tumour immunosuppression via JAK/STAT pathways (Su et al., 2018; Li P. et al., 2019; Song et al., 2020).
Curcumin was found to inhibit expression of phosphorylated STAT3, JAK1 JAK2, and JAK3 in SCLC cells (Yang et al., 2012). Levels of VEGF were also downregulated after curcumin treatment however this study focused on IL-6-dependent STAT3 activation, hence it is not certain if curcumin mediated inhibition via the RTK signaling pathway. In laryngeal squamous cell carcinoma, curcumin inhibited the expression of JAK2 and phosphorylation of STAT3, which is JAK2-dependent (Hu et al., 2014). Curcumin also inhibited VEGF mRNA and protein expression via the downregulation of this JAK2/STAT3 pathway which likely reduced VEGF-induced activation of VEGFR. However, there is only a handful of studies linking the effects of curcumin to RTKs and the JAK/STAT pathway as most studies only examine how curcumin inhibits JAK/STAT directly or via other signaling pathways.
The available literature on curcumin and JAK/STAT in cancer mainly look at how curcumin inhibits phosphorylation of various JAKs and namely STAT3 and STAT5. In blood cancers, a number of studies found that curcumin downregulated phosphorylation of JAK2, JAK3, TYK2, STAT3, STAT5a, and STAT5b (Rajasingh et al., 2006; Park et al., 2008; Petiti et al., 2019). Meanwhile, it was found that curcumin did not affect the phosphorylation of STAT proteins in chronic leukemia cells but only decreased their nuclear expression (Blasius et al., 2006). Curcumin also reduces migration, proliferation, and invasion directly by modulating levels of phosphorylated JAKs and STATs or indirectly by regulating protein inhibitors of activated STAT-3 (PIAS-3), suppressors of cytokine signaling (SOCS3), and miRNAs involved in JAK/STAT activity in a range of cancers including eye, ovarian, and endometrial cancers (Saydmohammed et al., 2010; Li Y. et al., 2018). Inhibition of the JAK/STAT pathway by curcumin in esophageal cancer cells also increased cell adhesion which is normally reduced in cancer (Zheng et al., 2018). Combination treatment of curcumin and cisplatin managed to inhibit phosphorylation of JAK and STAT3 in ovarian and papillary thyroid cancer cells leading to enhanced proliferation and reduced stemness of potential cancer stem cells (Khan AQ. et al., 2020; Sandhiutami et al., 2021). In osteosarcoma cells, curcumin inhibited the p-JAK2/p-STAT3 pathway which was involved in lung metastasis whereas in lung cancer, curcumin suppressed activation of p-STAT3 both in vitro and in vivo (Alexandrow et al., 2012; Sun et al., 2019). Several studies also examined the potency of curcumin analogues, FLLL31 and FLLL32, which were designed to specifically bind to JAK2 and STAT3 SH2 domains (Lin et al., 2010b). Both analogues were found to effectively suppress p-JAK2 and p-STAT3 and also key apoptotic proteins (Lin et al., 2010a; Lin et al., 2010b; Abuzeid et al., 2011). FLLL32 however showed very little inhibition of RTKs like EGFR, HER2 and Met (Lin et al., 2010b). Another curcumin analogue, L48H37, also decreased the phosphorylation of JAK1, JAK2, JAK3, and STAT3 in osteosarcoma cells (Lu et al., 2020). Gene expression profiling and recent RNA sequencing technology also identified both upregulation and downregulation of JAK/STAT signaling pathway components, with some of these studies also documenting changes in RTK gene expression (Van Erk et al., 2004; Teiten et al., 2009; Zhao W. et al., 2015). Further studies are needed to clarify the link between curcumin, RTKs and the JAK/STAT pathway.
3.1.4 Nuclear Factor Kappa B
The nuclear factor kappa B (NF-κB) consists of a group of transcription factors (TFs) that are responsible for many biological processes like inflammation, cell proliferation, immunity and apoptosis (Zinatizadeh et al., 2021). This family of TFs include five proteins which are RelA, RelB, c-Rel, p100, and p150. These proteins possess the rel homology domain (RHD) that assists in dimerization, binding to DNA and interaction with specific inhibitors (Zinatizadeh et al., 2021). Inhibitors of NF4EBP-κB are from the IκB inhibitor family comprising of IκBα, IκBβ, and IκBε. The association between NF-κB and IκBs form dimers that are retained in the cytoplasm in an inactive state (Dolcet et al., 2005). The phosphorylation of IκBs by IκB-kinases (IKKs) leads to IκB degradation and NF-κB liberation. NF-κB then enters the nucleus and regulates the transcription of a wide array of genes that code for growth factors, cytokines, cell adhesion molecules, pro- and anti-apoptotic proteins (Luo et al., 2005). NF-κB activation can be caused by a variety of signaling pathways including activation of Ras/MAPK, PI3K/Akt, and JAK/STAT which are commonly mediated by RTKs (Dolcet et al., 2005; Zhang et al., 2021).
In cancer, NF-κB activation can lead to apoptosis resistance through the expression of inhibitors of apoptosis (IAPs), members of anti-apoptotic Bcl-2 family and also proteins that disrupt the death receptor apoptotic pathway (Wang et al., 1998; Catz and Johnson, 2001; Kreuz et al., 2001). NF-κB activity also enhances cell cycle progression by inducing expression of key cell cycle proteins like cyclin D1 and invasion-related proteins like matrix metalloproteinases (MMPs) as well as VEGF and COX-2 that are important in tumour growth (Dolcet et al., 2005; Li et al., 2011; Li et al., 2016). Constitutive NF-κB activation has been observed in 66% of colorectal cancer cell lines whereas activating NF-κB mutations commonly occur in hematopoietic tumours (Hassanzadeh, 2011; Xia et al., 2014). Generally, mutations in upstream signaling molecules like MAPK proteins or RTKs themselves lead to constitutive activation of NF-κB in solid tumours (Tilborghs et al., 2017). Similar to many other proteins, NF-κB can act as a tumour promoter or tumour suppressor under different circumstances. As a tumour growth promoter, NF-κB has been found to induce expression of oncogenic microRNAs, promote expression of immune checkpoint proteins like PD-L1 and also act in sync with STAT3 and AP-1 to induce tumour-associated inflammation (Galardi et al., 2011; Asgarova et al., 2018; Ji et al., 2019). Meanwhile, loss or inhibition of NF-κB has been found to increase immortalization of cells and invasion, reflecting its tumour suppressive functions (Vandermark et al., 2012; O’Reilly et al., 2018). Deregulation of RTK/NF-κB signaling has been observed in various cancers (Matušan-Ilijaš et al., 2013; Spirina et al., 2017; Lai et al., 2018).
Curcumin in combination with herceptin decreased levels of NF-κB in a dose-dependent manner in HER-2-overexpressed breast cancer cells, overcoming herceptin resistance (Lai et al., 2012). Curcumin also suppressed osteopontin (OPN)-induced VEGF expression (Chakraborty et al., 2008). OPN is one of the main markers of breast cancer progression. Further analysis found that curcumin inhibited NF-κB activation which led to suppression of OPN-induced VEGF (Chakraborty et al., 2008). This suggests that curcumin may inhibit the VEGF/VEGFR signaling via NF-κB inhibition. In lung cancer, in vivo mice studies also showed that curcumin regulated tumour angiogenesis by decreasing VEGF expression through NF-κB inhibition (Li X. et al., 2018). Two separate studies examined combinations of curcumin with dasatinib and EGF-Receptor Related Protein (ERRP) in colon cancer. Both studies found that curcumin inhibited EGFR, IGF-1R, and NF-κB activity and this effect was more pronounced with combination treatments (Reddy et al., 2006; Nautiyal et al., 2011a). Moreover, curcumin analogues, EF31, and UBS109, were found to induce downregulation of VEGF, HIF-α, and COX-2 as well as inhibit IKKs, NF-κB translocation and NF-κB DNA binding based on in vitro and in vivo cancer studies (Olivera et al., 2012; Nagaraju et al., 2015; Rajitha et al., 2017). However, there is still a lack of studies looking into how curcumin modulates NF-κB via RTK signaling pathways or vice versa.
Similar to the JAK/STAT pathway, a range of studies have examined the direct effect of curcumin on NF-κB signaling. Curcumin was found to inhibit both NF-κB and Wnt signaling in cervical cancer while it also inhibited AP-1, NF-κB, and HPV E6 proteins in HPV-positive oral carcinoma, abolishing HPV transcription (Mishra et al., 2015; Ghasemi et al., 2019). Meanwhile, a phase I/II study on patients with multiple myeloma found that orally administered curcumin had no serious adverse effects and also reduced constitutive NF-κB activation (Vadhan-Raj et al., 2007). Furthermore, curcumin and its analogues have also been combined with cytotoxic drugs like cisplatin and doxorubicin and they were found to downregulate the drug-induced increase of NF-κB in liver and breast cancer (Notarbartolo et al., 2005; Meiyanto et al., 2014). Other curcumin combinations involving tolfenamic acid and Chinese goldthread also inhibited cell proliferation via disruption of NF-κB translocation into the nucleus and NF-κB transcriptional activity respectively (Zhao et al., 2014; Basha et al., 2016).
4 Curcumin-Receptor Tyrosine Kinase Inhibitor Combination
Tyrosine kinase inhibitors (TKIs) are a form of targeted therapy that interfere with the activity of oncogenic tyrosine kinases (TKs). Some of their inhibitory mechanism include competing with ATP for binding sites on the catalytic domain of TKs and decreasing phosphorylation of TKs which lead to inhibition of tumour cell repair, induction of apoptosis, and blockage of G1 phase cell division (Jiao et al., 2018). These small molecule inhibitors are orally active, safe, and effective in tumour inhibition (Arora and Scholar, 2005). As of 2019, the Food and Drug Administration (FDA) has approved 48 protein kinase inhibitors of which 25 target receptor tyrosine kinases (Roskoski, 2019). RTKIs can either be single-targeted or multi-targeted. Single-targeted RTKIs include common ones like gefitinib, erlotinib, and lapatinib that inhibit EGFR and axitinib and lenvatinib that target VEGFR while multi-tyrosine kinase inhibitors include imatinib, sorafenib, sunitinib, pazopanib, and regorafenib which target a mix of RTKs and non-RTKs (Jiao et al., 2018). Certain aspects need to be taken into account when deciding whether to use multiple single kinase inhibitors or a single multi-kinase inhibitor and these include aspects involving efficacy, pharmacokinetics, tumour microenvironment, and resistance (Broekman et al., 2011).
As with the use of most drugs, RTKI use is often followed by the rise of resistance. Mechanisms of resistance against RTKIs include mutations, gene amplification, and RTK overexpression, overexpression of downstream kinases, increased expression of drug efflux pumps, and gene fusion, most of which were mentioned in the section regarding oncogenic RTKs (Broekman et al., 2011; Jiao et al., 2018). RTKI resistance can be either primary (intrinsic) or secondary (acquired) whereby primary resistance is when there is a lack of tumour response to treatment while secondary resistance involves exposure to the RTKI and subsequent selection of resistant tumour cells (Pottier et al., 2020). As a result, combination treatments are becoming the preferred regimen to treat cancers. Many studies are examining the combination RTKIs with chemotherapy drugs, immunotherapy, and radiotherapy (Goldberg et al., 2013; Liang et al., 2018; Khan M. et al., 2020). This in turn presents a new challenge of finding a positive balance between the toxicities caused by increased drug administration and survival benefits. Curcumin is being explored as viable solution to overcome this challenge and can possibly serve as a substitute for certain drugs in combination treatments. A compelling reason for the use of curcumin in drug combinations is that its low toxicity allows doses of up to 12 000 mg a day which are well tolerated in humans (Basnet and Skalko-Basnet, 2011). The combination of curcumin and anti-cancer drugs like RTKIs, can remove a large portion of toxicity induced when two conventional drugs are combined, and indeed studies have found curcumin to reduce chemotherapy- and radiotherapy-induced side effects (Mansouri et al., 2020). Findings from a range of curcumin-RTKI combination studies are summarized in Table 2 and will be further reviewed in the following sections.
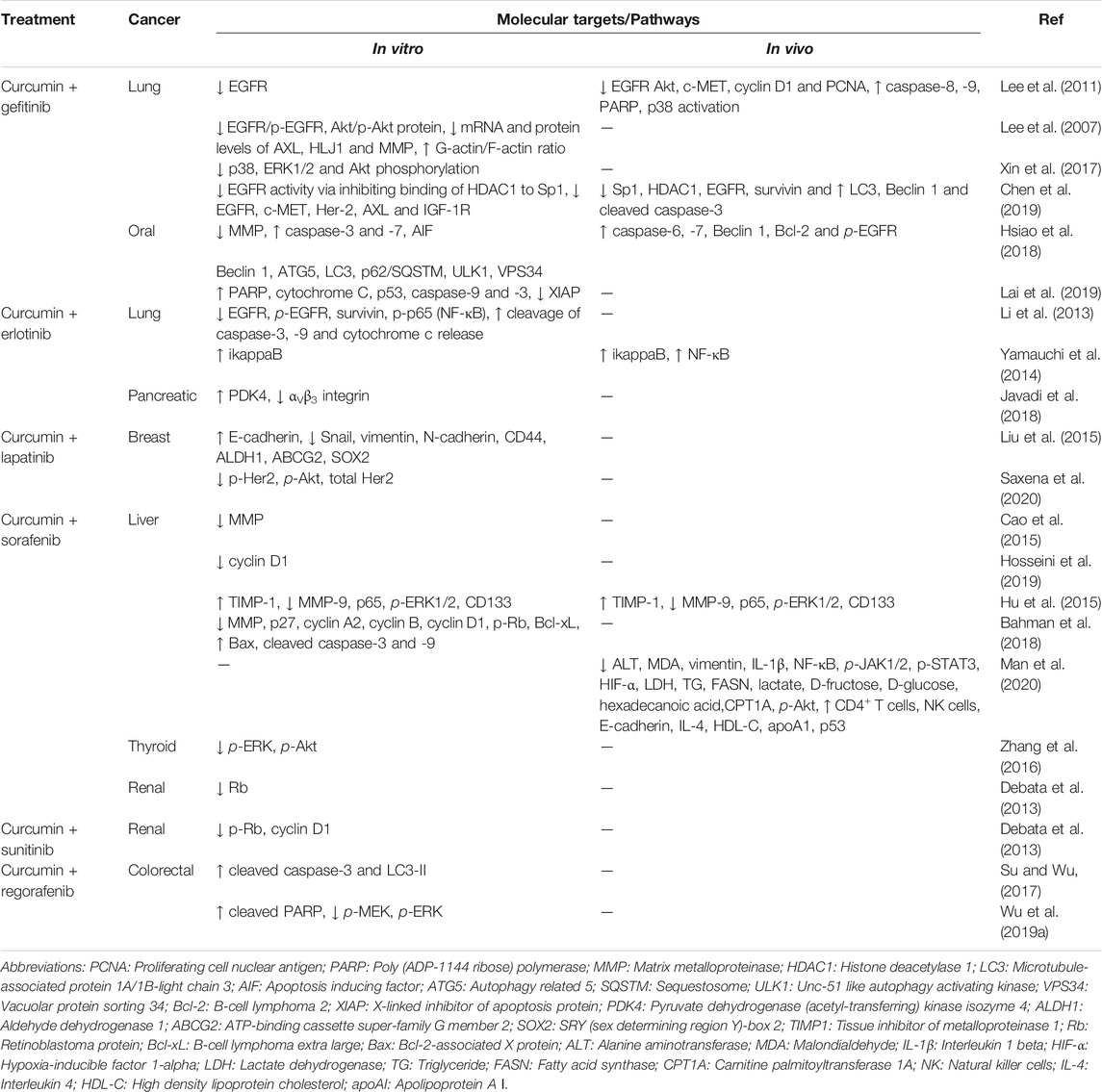
TABLE 2. Summary of curcumin-RTKI combination studies.
4.1 Curcumin and Single-Targeted RTKIs
Curcumin has been studied in combination with a few single-targeted RTKIs mainly EGFR TKIs like gefitinib, erlotinib and lapatinib. In a gefitinib-resistant lung cancer cell line (H1975), a combination of 15 µM of curcumin and 1 µM of gefitinib was found to have the same antiproliferative effect as 20 µM of gefitinib (Lee et al., 2011). In addition to EGFR, c-Met and Akt reduction, this study also found that combination treatment significantly lowered tumour growth on xenograft mice models and more importantly, 60 mg/kg of gefitinib combined with 1 g/kg of curcumin showed comparable results to 120 mg/kg of gefitinib. Side effects of gefitinib like villi damage and gastrointestinal effects were also attenuated by curcumin (Lee et al., 2011). Several other curcumin and gefitinib combination studies also found that curcumin promotes the inhibitory activity of gefitinib through downregulation of EGFR, MAPK, and PI3K signaling pathways (Lee et al., 2007; Xin et al., 2017; Chen et al., 2019). It was further found that curcumin and gefitinib also suppressed Sp1-and HDAC-induced EGFR transcription which led to induction of autophagy (Chen et al., 2019). In human oral cancer SAS cells, curcuminoids (curcumin, demethoxycurcumin or bisdemethoxycurcumin) combined with gefitinib induced certain apoptotic and autophagic proteins and overall led to higher levels of cell death compared to each agent alone (Hsiao et al., 2018). Meanwhile, further in vivo analysis showed that gefitinib combined with curcumin and demethoxycurcumin greatly decreased tumour volume in mice. Curcumin and gefitinib-loaded nanoparticles (NPs) have also been tested in oral cancer SAS cells (Lai et al., 2019). These γ-PGA-Gef/Cur NPs induced cell death through caspase and mitochondria-dependent pathways and low doses of Gef/Cur loaded NPs significantly decreased tumour weight as well. Meanwhile, several curcumin, and erlotinib combination studies were also found to strongly inhibit tumour growth and decrease tumour weight in xenograft mice models (Li et al., 2013; Yamauchi et al., 2014). Co-administration of curcumin and erlotinib was found to reduce cell viability of lung cancer cells via ikappaB elevation (Yamauchi et al., 2014). Additionally, it was also found that a relatively lower dose of curcumin also sensitized erlotinib-resistant NSCLC cells to erlotinib’s cytotoxic effects, reduced expressions of EGFR and also inhibited NF-κB activation (Li et al., 2013). A few studies also employed the use of nano-based delivery systems to combine curcumin and erlotinib. A combination of curcumin and erlotinib-loaded Methoxypoly (ethylene glycol) Poly (caprolactone) (Mpeg-pcl) was found to increase PDK4 gene and decrease αvβ3 integrin expression in colorectal cancer cells (Javadi et al., 2018). These components are involved in erlotinib-resistance, and the addition of curcumin to erlotinib treatment seems to influence associated drug resistance signaling pathways. An erlotinib and curcumin conjugated carrier-free nanoassembly (EPC) was also developed and found to have better tumour-penetrating and anti-migratory properties in addition to the absence of systemic toxicity (Cheng et al., 2020). Lastly, combinations of curcumin and lapatinib were also found to increase lapatinib-induced inhibition of the Her2-Akt pathway, reverse lapatinib resistance and decrease metastatic potential in breast cancer cells (Liu et al., 2015; Saxena et al., 2020). Currently, only one phase I clinical trial has been conducted investigating the combination treatment of curcumin and EGFR-TKIs (gefitinib and erlotinib) (Esfahani et al., 2019). An enhanced bioavailable curcumin formulation was administered together with gefitinib or erlotinib. Overall, no evidence of toxicity was observed and adverse effects, if any, were pre-existing due to TKI therapy. Curcumin was found to improve the quality of life and appeared to be a safe adjuvant to TKI therapy.
4.2 Curcumin and Multi-Targeted RTKIs
Recently, there has been an increase in the number of studies investigating the in vitro combinatorial effects of curcumin and multi-kinase inhibitors, namely sorafenib, sunitinib, and regorafenib. Sorafenib is an orally administered pyridine multi-kinase inhibitor (MKI) that inhibits RTKs like VEGFR, PDGFR, RET, and MAPK signaling components like Raf-1, Braf, Braf mutants, and c-Kit (Wilhelm et al., 2004; Di Gion et al., 2011). As of now, combination treatment of sorafenib and curcumin has mostly been studied in hepatocellular carcinoma (HCC), with most of them involving nanoparticle (NP)-based delivery. These delivery systems include directed self-assembly NPs, pH-sensitive lactosylated NPs, polymeric nanoparticle formulations of curcumin and nanomicelles (Cao et al., 2015; Hu et al., 2015; Hosseini et al., 2019; Bian and Guo, 2020). All these studies found that an NP-based combination of curcumin and sorafenib showed higher cytotoxicity and induced higher apoptosis in HCC than either one alone. Some of them also demonstrated enhanced anti-angiogenic effects (Cao et al., 2015), improved in vivo tissue distribution (Cao et al., 2015; Bian and Guo, 2020), good tolerance (Bian and Guo, 2020), and downregulation of biomarkers/genes involved in cancer progression (Hu et al., 2015). Free drug combination treatments of curcumin and sorafenib also showed promising results like increased apoptosis, disruption of cell cycle progression, and protection of liver function from sorafenib-induced effects (Bahman et al., 2018; Man et al., 2020). This combination also remarkably increased the proportion of CD4+ T cells and natural killer cells and inhibited sorafenib-induced EMT via downregulation of JAK/STAT and NF-κB pathway proteins (Man et al., 2020). MAPK and PI3K pathway components were also reduced by curcumin and sorafenib in thyroid cancer cells, decreasing migration and invasion (Zhang et al., 2016).
Sunitinib is a pyrole multi-kinase inhibitor that mainly inhibits VEGFR and PDGFR, and it is also a first-generation MKI like sorafenib (Mendel et al., 2003; Di Gion et al., 2011). There are only a few studies that have examined curcumin-sunitinib combinations. Combinations of curcumin with erlotinib, sorafenib, and sunitinib were studied in breast cancer cells and it was found that curcumin combined with sunitinib exhibited the highest reduction of cell viability (Chen et al., 2016). This combination was brought forward into in vivo analysis, and the efficacy of combination treatment was higher than mono-therapy; however, no statistical significance was achieved. In addition, bovine serum albumin (BSA)-encapsulated curcumin and sunitinib was found to be more effective than this free drug combination (Chen et al., 2016). These findings inspired an additional study whereby curcumin and sunitinib were co-loaded into BSA-supermagnetic iron oxide nanoparticles (SPIOs) (Chen et al., 2017). This formulation showed the highest amount of tumour inhibition and simultaneously the least amount of toxicity while also efficiently delivering the drugs to the tumour site based on in vitro and in vivo breast cancer models. In renal cancer cells, curcumin combined with sunitinib decreased the IC50 of sunitinib by four-fold; however, this effect was not observed with sorafenib (Debata et al., 2013). This suggests that the therapeutic dose of sunitinib can be reduced when combined with suitable concentrations of curcumin.
Regorafenib is one of the newer orally active MKIs mainly targeting VEGFR and PDGFR (Strumberg and Schultheis, 2012). As such, only two studies have investigated the combination treatment of curcumin and regorafenib in colorectal cancer cells (Su and Wu, 2017; Wu CS. et al., 2019). Curcumin appeared to act like a MEK inhibitor and most likely targets other genes as well, producing a synthetic lethal effect in KRAS-mutant colorectal cancer cells (Wu CS. et al., 2019). The combination of curcumin and regorafenib only showed additive/synergistic effects in KRAS-mutant and not KRAS-wildtype cells, suggesting their possible use in the treatment KRAS-mutant colorectal cancer. Imatinib and dasatinib are first- and second-generation pyrimidine TKIs respectively, and are mainly known to be non-RTKIs,; however, both also target PDGFR (Natoli et al., 2010). Several studies have combined curcumin with both imatinib and dasatinib. Most of the studies found that curcumin enhanced the anti-leukemia effects of imatinib by downregulation of the Bcr/Abl gene (Bae et al., 2005; Gong et al., 2012; Guo et al., 2015). Nanostructured lipid carriers of curcumin and imatinib were also found to have superior effects than imatinib alone (Setareh and Jaleh, 2018; Varshosaz et al., 2021).
Furthermore, a case report stated that curcumin and imatinib successfully treated a patient having c-KIT-positive adenoid cystic carcinoma for the first time, whereby complete anatomic and metabolic response was observed after 24 months (Demiray et al., 2016). On the other hand, curcumin and dasatinib combination treatments have also shown reduced metastatic potential, regression of mice intestinal adenomas and decreased cancer stem cell populations in colon cancer cells (Nautiyal et al., 2011a; Nautiyal et al., 2011b). However, none of the studies combining curcumin and imatinib/dasatinib recorded modulations of PDGFR despite it being a known target of these two drugs. Most of these studies reported changes in downstream signaling pathway components which could possibly be due to upstream regulation of its known target, PDGFR.
5 Conclusion
Curcumin possesses many of the features required to be an ideal anti-cancer therapeutic agent, especially with its enigmatic ability to singularly target a legion of signaling molecules. Further studies revealed that curcumin targets RTKs and their downstream signaling pathways such as MAPK, PI3K/Akt, JAK/STAT, and NF-κB pathways which are involved in essential cellular processes like proliferation, apoptosis, cell cycle progression, and migration. Curcumin-mediated modulation of RTK expression or activation leads to positive outcomes like reduced proliferation, increased apoptosis, and decreased migration. .In many cases, the specific mechanism of action depends on the cellular environment and type of cancer.
Multiple studies have shown that curcumin can overcome resistance and enhance the apoptotic effects of existing TKI drugs. There are still many unanswered questions regarding how curcumin targets RTKs, especially whether or not direct binding occurs. Additional studies are also required to elucidate the effects of curcumin on RTKs along with changes in the JAK/STAT and NF-κB pathways. Many existing studies examine how curcumin targets RTKs or how curcumin targets specific pathways, however, an extensive analysis would require investigating all three components simultaneously (curcumin, RTKs, and signaling pathways) to obtainclearer understanding. In addition, it would be interesting to see how non-RTKs fit into this whole process since they make up many of the essential intracellular components. One of the main limitations of curcumin is its poor bioavailability in cellular environments. Various analogues of curcumin are being developed with superior bioavailability and improved anti-cancer properties. The use of nano-delivery systems is also gaining attention, especially in the delivery of curcumin and chemotherapy drugs. .There is a need for more in vivo and overall toxicity studies involving curcumin and its analogues. Combination treatments of curcumin and TKIs also need to be further studied to build a more substantial basis of evidence to ease curcumin progression into clinical trials. In conclusion, among the many mechanisms employed by curcumin, inhibition of receptor tyrosine kinases appears to be a significant element. It would be crucial to explore the implications for TKI therapy and whether the integration of curcumin and TKIs can improve treatment efficacy.
Author Contributions
Conceptualization, SSD and RN investigation, SSD and RN; writing—original draft preparation, SSD; writing—review and editing, SSD, RN, SA, RF, and IO All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Jeffrey Cheah School of Medicine and Health Sciences, Monash University, Malaysia, for providing the research facilities and support to conduct this study.
References
Abuzeid, W. M., Davis, S., Tang, A. L., Saunders, L., Brenner, J. C., Lin, J., et al. (2011). Sensitization of Head and Neck Cancer to Cisplatin through the Use of a Novel Curcumin Analog. Arch. Otolaryngol. Head Neck Surg. 137, 499–507. doi:10.1001/archoto.2011.63
Adamczak, A., Ożarowski, M., and Karpiński, T. M. (2020). Curcumin, a Natural Antimicrobial Agent with Strain-specific Activity. Pharmaceuticals (Basel) 13, 153. doi:10.3390/ph13070153
Adewuyi, E. E., Deschenes, J., Lopez-Campistrous, A., Kattar, M. M., Ghosh, S., and McMullen, T. P. W. (2018). Autocrine Activation of Platelet-Derived Growth Factor Receptor α in Metastatic Papillary Thyroid Cancer. Hum. Pathol. 75, 146–153. doi:10.1016/j.humpath.2018.01.025
Aleksic, T., Verrill, C., Bryant, R. J., Han, C., Worrall, A. R., Brureau, L., et al. (2017). IGF-1R Associates with Adverse Outcomes after Radical Radiotherapy for Prostate Cancer. Br. J. Cancer 117, 1600–1606. doi:10.1038/bjc.2017.337
Alexandrow, M. G., Song, L. J., Altiok, S., Gray, J., Haura, E. B., and Kumar, N. B. (2012). Curcumin: a Novel Stat3 Pathway Inhibitor for Chemoprevention of Lung Cancer. Eur. J. Cancer Prev. 21, 407–412. doi:10.1097/CEJ.0b013e32834ef194
Andl, C. D., Mizushima, T., Oyama, K., Bowser, M., Nakagawa, H., and Rustgi, A. K. (2004). EGFR-induced Cell Migration Is Mediated Predominantly by the JAK-STAT Pathway in Primary Esophageal Keratinocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G1227–G1237. doi:10.1152/ajpgi.00253.2004
Appiah-Kubi, K., Lan, T., Wang, Y., Qian, H., Wu, M., Yao, X., et al. (2017). Platelet-derived Growth Factor Receptors (PDGFRs) Fusion Genes Involvement in Hematological Malignancies. Crit. Rev. Oncol. Hematol. 109, 20–34. doi:10.1016/j.critrevonc.2016.11.008
Arbour, N., Naniche, D., Homann, D., Davis, R. J., Flavell, R. A., and Oldstone, M. B. (2002). c-Jun NH(2)-terminal Kinase (JNK)1 and JNK2 Signaling Pathways Have Divergent Roles in CD8(+) T Cell-Mediated Antiviral Immunity. J. Exp. Med. 195, 801–810. doi:10.1084/jem.20011481
Arena, S., Bellosillo, B., Siravegna, G., Martínez, A., Cañadas, I., Lazzari, L., et al. (2015). Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 21, 2157–2166. doi:10.1158/1078-0432.Ccr-14-2821
Arora, A., and Scholar, E. M. (2005). Role of Tyrosine Kinase Inhibitors in Cancer Therapy. J. Pharmacol. Exp. Ther. 315, 971–979. doi:10.1124/jpet.105.084145
Asgarova, A., Asgarov, K., Godet, Y., Peixoto, P., Nadaradjane, A., Boyer-Guittaut, M., et al. (2018). PD-L1 Expression Is Regulated by Both DNA Methylation and NF-kB during EMT Signaling in Non-small Cell Lung Carcinoma. Oncoimmunology 7, e1423170. doi:10.1080/2162402x.2017.1423170
Awad, M. M., Oxnard, G. R., Jackman, D. M., Savukoski, D. O., Hall, D., Shivdasani, P., et al. (2016). MET Exon 14 Mutations in Non-small-cell Lung Cancer Are Associated with Advanced Age and Stage-dependent MET Genomic Amplification and C-Met Overexpression. J. Clin. Oncol. 34, 721–730. doi:10.1200/jco.2015.63.4600
Badzio, A., Wynes, M. W., Dziadziuszko, R., Merrick, D. T., Pardo, M., Rzyman, W., et al. (2010). Increased Insulin-like Growth Factor 1 Receptor Protein Expression and Gene Copy Number in Small Cell Lung Cancer. J. Thorac. Oncol. 5, 1905–1911. doi:10.1097/JTO.0b013e3181f38f57
Bae, E. K., Kim, Y.-J., Lee, J.-S., Park, S., Kim, B. K., Yoon, S.-S., et al. (2005). Effect of the Combination of Imatinib Mesylate (Glivec) and Curcumin in Chronic Myeloid Leukemia Cell Line. Cancer Res. 65, 1208.
Bahman, A. A., Abaza, M. S. I., Khoushiash, S. I., and Al-Attiyah, R. J. (2018). Sequence-dependent E-ffect of S-orafenib in C-ombination with N-atural P-henolic C-ompounds on H-epatic C-ancer C-ells and the P-ossible M-echanism of A-ction. Int. J. Mol. Med. 42, 1695–1715. doi:10.3892/ijmm.2018.3725
Banerjee, S., Singh, S. K., Chowdhury, I., Lillard, J. W., and Singh, R. (2017). Combinatorial Effect of Curcumin with Docetaxel Modulates Apoptotic and Cell Survival Molecules in Prostate Cancer. Front. Biosci. (Elite Ed. 9, 235–245. doi:10.2741/e798
Basha, R., Connelly, S. F., Sankpal, U. T., Nagaraju, G. P., Patel, H., Vishwanatha, J. K., et al. (2016). Small Molecule Tolfenamic Acid and Dietary Spice Curcumin Treatment Enhances Antiproliferative Effect in Pancreatic Cancer Cells via Suppressing Sp1, Disrupting NF-kB Translocation to Nucleus and Cell Cycle Phase Distribution. J. Nutr. Biochem. 31, 77–87. doi:10.1016/j.jnutbio.2016.01.003
Basnet, P., and Skalko-Basnet, N. (2011). Curcumin: an Anti-inflammatory Molecule from a Curry Spice on the Path to Cancer Treatment. Molecules 16, 4567–4598. doi:10.3390/molecules16064567
Baykal, C., Ayhan, A., Al, A., Yüce, K., and Ayhan, A. (2003). Overexpression of the C-Met/HGF Receptor and its Prognostic Significance in Uterine Cervix Carcinomas. Gynecol. Oncol. 88, 123–129. doi:10.1016/S0090-8258(02)00073-2
Bian, Y., and Guo, D. (2020). Targeted Therapy for Hepatocellular Carcinoma: Co-delivery of Sorafenib and Curcumin Using Lactosylated pH-Responsive Nanoparticles. Drug Des. Devel Ther. 14, 647–659. doi:10.2147/DDDT.S238955
Binion, D. G., Otterson, M. F., and Rafiee, P. (2008). Curcumin Inhibits VEGF-Mediated Angiogenesis in Human Intestinal Microvascular Endothelial Cells through COX-2 and MAPK Inhibition. Gut 57, 1509–1517. doi:10.1136/gut.2008.152496
Blanc-Durand, F., Alameddine, R., Iafrate, A. J., Tran-Thanh, D., Lo, Y. C., Blais, N., et al. (2020). Tepotinib Efficacy in a Patient with Non-small Cell Lung Cancer with Brain Metastasis Harboring an HLA-DRB1-MET Gene Fusion. Oncologist 25, 916–920. doi:10.1634/theoncologist.2020-0502
Blasius, R., Reuter, S., Henry, E., Dicato, M., and Diederich, M. (2006). Curcumin Regulates Signal Transducer and Activator of Transcription (STAT) Expression in K562 Cells. Biochem. Pharmacol. 72, 1547–1554. doi:10.1016/j.bcp.2006.07.029
Blom, T., Roselli, A., Häyry, V., Tynninen, O., Wartiovaara, K., Korja, M., et al. (2010). Amplification and Overexpression of KIT, PDGFRA, and VEGFR2 in Medulloblastomas and Primitive Neuroectodermal Tumors. J. Neurooncol. 97, 217–224. doi:10.1007/s11060-009-0014-2
Boccaccio, C., Andò, M., Tamagnone, L., Bardelli, A., Michieli, P., Battistini, C., et al. (1998). Induction of Epithelial Tubules by Growth Factor HGF Depends on the STAT Pathway. Nature 391, 285–288. doi:10.1038/34657
Borah, A., Pillai, S. C., Rochani, A. K., Palaninathan, V., Nakajima, Y., Maekawa, T., et al. (2020). GANT61 and Curcumin-Loaded PLGA Nanoparticles for GLI1 and PI3K/Akt-Mediated Inhibition in Breast Adenocarcinoma. Nanotechnology 31, 185102. doi:10.1088/1361-6528/ab6d20
Borges, G. A., Elias, S. T., Amorim, B., de Lima, C. L., Coletta, R. D., Castilho, R. M., et al. (2020). Curcumin Downregulates the PI3K-AKT-mTOR Pathway and Inhibits Growth and Progression in Head and Neck Cancer Cells. Phytother Res. 34, 3311–3324. doi:10.1002/ptr.6780
Boroumand, N., Samarghandian, S., and Hashemy, S. I. (2018). Immunomodulatory, Anti-inflammatory, and Antioxidant Effects of Curcumin. J. Herbmed Pharmacol. 7, 211–219. doi:10.15171/jhp.2018.33
Bremm, A., Walch, A., Fuchs, M., Mages, J., Duyster, J., Keller, G., et al. (2008). Enhanced Activation of Epidermal Growth Factor Receptor Caused by Tumor-Derived E-Cadherin Mutations. Cancer Res. 68, 707–714. doi:10.1158/0008-5472.Can-07-1588
Broekman, F., Giovannetti, E., and Peters, G. J. (2011). Tyrosine Kinase Inhibitors: Multi-Targeted or Single-Targeted? World J. Clin. Oncol. 2, 80–93. doi:10.5306/wjco.v2.i2.80
Campregher, P. V., Halley, N. D. S., Vieira, G. A., Fernandes, J. F., Velloso, E. D. R. P., Ali, S., et al. (2017). Identification of a Novel Fusion TBL1XR1-PDGFRB in a Patient with Acute Myeloid Leukemia Harboring the DEK-Nup214 Fusion and Clinical Response to Dasatinib. Leuk. Lymphoma 58, 2969–2972. doi:10.1080/10428194.2017.1318437
Canovas, B., and Nebreda, A. R. (2021). Diversity and Versatility of P38 Kinase Signalling in Health and Disease. Nat. Rev. Mol. Cel Biol 22, 346–366. doi:10.1038/s41580-020-00322-w
Cantwell-Dorris, E. R., O'Leary, J. J., and Sheils, O. M. (2011). BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 10, 385–394. doi:10.1158/1535-7163.Mct-10-0799
Cao, H., Wang, Y., He, X., Zhang, Z., Yin, Q., Chen, Y., et al. (2015). Codelivery of Sorafenib and Curcumin by Directed Self-Assembled Nanoparticles Enhances Therapeutic Effect on Hepatocellular Carcinoma. Mol. Pharm. 12, 922–931. doi:10.1021/mp500755j
Capelletti, M., Dodge, M. E., Ercan, D., Hammerman, P. S., Park, S. I., Kim, J., et al. (2014). Identification of Recurrent FGFR3-TACC3 Fusion Oncogenes from Lung Adenocarcinoma. Clin. Cancer Res. 20, 6551–6558. doi:10.1158/1078-0432.Ccr-14-1337
Carneiro, B. A., Elvin, J. A., Kamath, S. D., Ali, S. M., Paintal, A. S., Restrepo, A., et al. (2015). FGFR3-TACC3: A Novel Gene Fusion in Cervical Cancer. Gynecol. Oncol. Rep. 13, 53–56. doi:10.1016/j.gore.2015.06.005
Casaletto, J. B., and McClatchey, A. I. (2012). Spatial Regulation of Receptor Tyrosine Kinases in Development and Cancer. Nat. Rev. Cancer 12, 387–400. doi:10.1038/nrc3277
Catz, S. D., and Johnson, J. L. (2001). Transcriptional Regulation of Bcl-2 by Nuclear Factor Kappa B and its Significance in Prostate Cancer. Oncogene 20, 7342–7351. doi:10.1038/sj.onc.1204926
Cepero, V., Sierra, J. R., Corso, S., Ghiso, E., Casorzo, L., Perera, T., et al. (2010). MET and KRAS Gene Amplification Mediates Acquired Resistance to MET Tyrosine Kinase Inhibitors. Cancer Res. 70, 7580–7590. doi:10.1158/0008-5472.Can-10-0436
Chakraborty, G., Jain, S., Kale, S., Raja, R., Kumar, S., Mishra, R., et al. (2008). Curcumin Suppresses Breast Tumor Angiogenesis by Abrogating Osteopontin-Induced VEGF Expression. Mol. Med. Rep. 1, 641–646. doi:10.3892/mmr_00000005
Chan, B. A., and Hughes, B. G. (2014). Targeted Therapy for Non-small Cell Lung Cancer: Current Standards and the Promise of the Future. Transl Lung Cancer Res. 4, 36–54. doi:10.3978/j.issn.2218-6751.2014.05.01
Chang, J., Liu, X., Wang, S., Zhang, Z., Wu, Z., Zhang, X., et al. (2014). Prognostic Value of FGFR Gene Amplification in Patients with Different Types of Cancer: a Systematic Review and Meta-Analysis. PLoS One 9, e105524. doi:10.1371/journal.pone.0105524
Chang, K. W., Hung, P. S., Lin, I. Y., Hou, C. P., Chen, L. K., Tsai, Y. M., et al. (2010). Curcumin Upregulates Insulin-like Growth Factor Binding Protein-5 (IGFBP-5) and C/EBPalpha during Oral Cancer Suppression. Int. J. Cancer 127, 9–20. doi:10.1002/ijc.25220
Chang, Q., Chen, J., Beezhold, K. J., Castranova, V., Shi, X., and Chen, F. (2009). JNK1 Activation Predicts the Prognostic Outcome of the Human Hepatocellular Carcinoma. Mol. Cancer 8, 64. doi:10.1186/1476-4598-8-64
Chapuis, N., Tamburini, J., Cornillet-Lefebvre, P., Gillot, L., Bardet, V., Willems, L., et al. (2010). Autocrine IGF-1/IGF-1R Signaling Is Responsible for Constitutive PI3K/Akt Activation in Acute Myeloid Leukemia: Therapeutic Value of Neutralizing Anti-IGF-1r Antibody. Haematologica 95, 415–423. doi:10.3324/haematol.2009.010785
Chatterjee, S., Heukamp, L. C., Siobal, M., Schöttle, J., Wieczorek, C., Peifer, M., et al. (2013). Tumor VEGF:VEGFR2 Autocrine Feed-Forward Loop Triggers Angiogenesis in Lung Cancer. J. Clin. Invest. 123, 1732–1740. doi:10.1172/jci65385
Cheatham, B., Shoelson, S. E., Yamada, K., Goncalves, E., and Kahn, C. R. (1993). Substitution of the erbB-2 Oncoprotein Transmembrane Domain Activates the Insulin Receptor and Modulates the Action of Insulin and Insulin-Receptor Substrate 1. Proc. Natl. Acad. Sci. U S A. 90, 7336–7340. doi:10.1073/pnas.90.15.7336
Chen, A., and Xu, J. (2005). Activation of PPAR{gamma} by Curcumin Inhibits Moser Cell Growth and Mediates Suppression of Gene Expression of Cyclin D1 and EGFR. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G447–G456. doi:10.1152/ajpgi.00209.2004
Chen, A., Xu, J., and Johnson, A. C. (2006). Curcumin Inhibits Human colon Cancer Cell Growth by Suppressing Gene Expression of Epidermal Growth Factor Receptor through Reducing the Activity of the Transcription Factor Egr-1. Oncogene 25, 278–287. doi:10.1038/sj.onc.1209019
Chen, D., Li, X. L., Wu, B., Zheng, X. B., Wang, W. X., Chen, H. F., et al. (2020). A Novel Oncogenic Driver in a Lung Adenocarcinoma Patient Harboring an EGFR-KDD and Response to Afatinib. Front. Oncol. 10, 867. doi:10.3389/fonc.2020.00867
Chen, H. X., and Sharon, E. (2013). IGF-1R as an Anti-cancer Target-Ttrials and Tribulations. Chin. J. Cancer 32, 242–252. doi:10.5732/cjc.012.10263
Chen, J., De, S., Brainard, J., and Byzova, T. V. (2004). Metastatic Properties of Prostate Cancer Cells Are Controlled by VEGF. Cell Commun Adhes 11, 1–11. doi:10.1080/15419060490471739
Chen, P., Huang, H. P., Wang, Y., Jin, J., Long, W. G., Chen, K., et al. (2019). Curcumin Overcome Primary Gefitinib Resistance in Non-small-cell Lung Cancer Cells through Inducing Autophagy-Related Cell Death. J. Exp. Clin. Cancer Res. 38, 254. doi:10.1186/s13046-019-1234-8
Chen, Q., Powell, D. W., Rane, M. J., Singh, S., Butt, W., Klein, J. B., et al. (2003). Akt Phosphorylates P47phox and Mediates Respiratory Burst Activity in Human Neutrophils. J. Immunol. 170, 5302–5308. doi:10.4049/jimmunol.170.10.5302
Chen, S., Liang, Q., Liu, E., Yu, Z., Sun, L., Ye, J., et al. (2017). Curcumin/sunitinib Co-loaded BSA-Stabilized SPIOs for Synergistic Combination Therapy for Breast Cancer. J. Mater. Chem. B 5, 4060–4072. doi:10.1039/C7TB00040E
Chen, S., Liang, Q., Xie, S., Liu, E., Yu, Z., Sun, L., et al. (2016). Curcumin Based Combination Therapy for Anti-breast Cancer: from In Vitro Drug Screening to In Vivo Efficacy Evaluation. Front. Chem. Sci. Eng. 10, 383–388. doi:10.1007/s11705-016-1574-2
Chen, Y., Jiang, L., She, F., Tang, N., Wang, X., Li, X., et al. (2010). Vascular Endothelial Growth Factor-C Promotes the Growth and Invasion of Gallbladder Cancer via an Autocrine Mechanism. Mol. Cel Biochem 345, 77–89. doi:10.1007/s11010-010-0562-y
Chen, Y., Zhong, W., Chen, B., Yang, C., Zhou, S., and Liu, J. (2018). Effect of Curcumin on Vascular Endothelial Growth Factor in Hypoxic HepG2 Cells via the Insulin-like Growth Factor 1 Receptor Signaling Pathway. Exp. Ther. Med. 15, 2922–2928. doi:10.3892/etm.2018.5783
Chen, Y. R., and Tan, T. H. (1998). Inhibition of the C-Jun N-Terminal Kinase (JNK) Signaling Pathway by Curcumin. Oncogene 17, 173–178. doi:10.1038/sj.onc.1201941
Cheng, C., Sui, B., Wang, M., Hu, X., Shi, S., and Xu, P. (2020). Carrier-Free Nanoassembly of Curcumin-Erlotinib Conjugate for Cancer Targeted Therapy. Adv. Healthc. Mater. 9, e2001128. doi:10.1002/adhm.202001128
Chi, F., Wu, R., Jin, X., Jiang, M., and Zhu, X. (2016). HER2 Induces Cell Proliferation and Invasion of Non-small-cell Lung Cancer by Upregulating COX-2 Expression via MEK/ERK Signaling Pathway. Onco Targets Ther. 9, 2709–2716. doi:10.2147/OTT.S96197
Cools, J., DeAngelo, D. J., Gotlib, J., Stover, E. H., Legare, R. D., Cortes, J., et al. (2003). A Tyrosine Kinase Created by Fusion of the PDGFRA and FIP1L1 Genes as a Therapeutic Target of Imatinib in Idiopathic Hypereosinophilic Syndrome. N. Engl. J. Med. 348, 1201–1214. doi:10.1056/NEJMoa025217
Cowell, J. K., Qin, H., Hu, T., Wu, Q., Bhole, A., and Ren, M. (2017). Mutation in the FGFR1 Tyrosine Kinase Domain or Inactivation of PTEN Is Associated with Acquired Resistance to FGFR Inhibitors in FGFR1-Driven Leukemia/lymphomas. Int. J. Cancer 141, 1822–1829. doi:10.1002/ijc.30848
Craddock, K. J., Ludkovski, O., Sykes, J., Shepherd, F. A., and Tsao, M. S. (2013). Prognostic Value of Fibroblast Growth Factor Receptor 1 Gene Locus Amplification in Resected Lung Squamous Cell Carcinoma. J. Thorac. Oncol. 8, 1371–1377. doi:10.1097/JTO.0b013e3182a46fe9
Daniels, M., Lurkin, I., Pauli, R., Erbstösser, E., Hildebrandt, U., Hellwig, K., et al. (2011). Spectrum of KIT/PDGFRA/BRAF Mutations and Phosphatidylinositol-3-Kinase Pathway Gene Alterations in Gastrointestinal Stromal Tumors (GIST). Cancer Lett. 312, 43–54. doi:10.1016/j.canlet.2011.07.029
Davies, K. D., Ng, T. L., Estrada-Bernal, A., Le, A. T., Ennever, P. R., Camidge, D. R., et al. (2017). Dramatic Response to Crizotinib in a Patient with Lung Cancer Positive for anHLA-DRB1-METGene Fusion. JCO Precision Oncol. 2017, 1–6. doi:10.1200/po.17.00117
Davis, R. J. (2000). Signal Transduction by the JNK Group of MAP Kinases. Cell 103, 239–252. doi:10.1016/s0092-8674(00)00116-1
De, S., Zhou, H., DeSantis, D., Croniger, C. M., Li, X., and Stark, G. R. (2015). Erlotinib Protects against LPS-Induced Endotoxicity Because TLR4 Needs EGFR to Signal. Proc. Natl. Acad. Sci. U S A. 112, 9680–9685. doi:10.1073/pnas.1511794112
Debata, P. R., Begum, S., Mata, A., Genzer, O., Kleiner, M. J., Banerjee, P., et al. (2013). Curcumin Potentiates the Ability of Sunitinib to Eliminate the VHL-Lacking Renal Cancer Cells 786-O: Rapid Inhibition of Rb Phosphorylation as a Preamble to Cyclin D1 Inhibition. Anticancer Agents Med. Chem. 13, 1508–1513. doi:10.2174/18715206113139990093
Demiray, M., Sahinbas, H., Atahan, S., Demiray, H., Selcuk, D., Yildirim, I., et al. (2016). Successful Treatment of C-Kit-Positive Metastatic Adenoid Cystic Carcinoma (ACC) with a Combination of Curcumin Plus Imatinib: A Case Report. Complement. Ther. Med. 27, 108–113. doi:10.1016/j.ctim.2016.06.009
Deng, Y. I., Verron, E., and Rohanizadeh, R. (2016). Molecular Mechanisms of Anti-metastatic Activity of Curcumin. Anticancer Res. 36, 5639–5647. doi:10.21873/anticanres.11147
Dhillon, A. S., Hagan, S., Rath, O., and Kolch, W. (2007). MAP Kinase Signalling Pathways in Cancer. Oncogene 26, 3279–3290. doi:10.1038/sj.onc.1210421
Di Gion, P., Kanefendt, F., Lindauer, A., Scheffler, M., Doroshyenko, O., Fuhr, U., et al. (2011). Clinical Pharmacokinetics of Tyrosine Kinase Inhibitors: Focus on Pyrimidines, Pyridines and Pyrroles. Clin. Pharmacokinet. 50, 551–603. doi:10.2165/11593320-000000000-00000
Dickens, E., and Ahmed, S. (2018). Principles of Cancer Treatment by Chemotherapy. Surgery (Oxford) 36, 134–138. doi:10.1016/j.mpsur.2017.12.002
Dolcet, X., Llobet, D., Pallares, J., and Matias-Guiu, X. (2005). NF-kB in Development and Progression of Human Cancer. Virchows Arch. 446, 475–482. doi:10.1007/s00428-005-1264-9
Du, Z., Brown, B. P., Kim, S., Ferguson, D., Pavlick, D. C., Jayakumaran, G., et al. (2021). Structure-function Analysis of Oncogenic EGFR Kinase Domain Duplication Reveals Insights into Activation and a Potential Approach for Therapeutic Targeting. Nat. Commun. 12, 1382. doi:10.1038/s41467-021-21613-6
Du, Z., and Lovly, C. M. (2018). Mechanisms of Receptor Tyrosine Kinase Activation in Cancer. Mol. Cancer 17, 58. doi:10.1186/s12943-018-0782-4
Elstrom, R. L., Bauer, D. E., Buzzai, M., Karnauskas, R., Harris, M. H., Plas, D. R., et al. (2004). Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 64, 3892–3899. doi:10.1158/0008-5472.Can-03-2904
Emuss, V., Garnett, M., Mason, C., and Marais, R. (2005). Mutations of C-RAF Are Rare in Human Cancer Because C-RAF Has a Low Basal Kinase Activity Compared with B-RAF. Cancer Res. 65, 9719–9726. doi:10.1158/0008-5472.Can-05-1683
Enrico, D., Lacroix, L., Chen, J., Rouleau, E., Scoazec, J. Y., Loriot, Y., et al. (2020). Oncogenic Fusions May Be Frequently Present at Resistance of EGFR Tyrosine Kinase Inhibitors in Patients with NSCLC: A Brief Report. JTO Clin. Res. Rep. 1, 100023. doi:10.1016/j.jtocrr.2020.100023
Esfahani, K., Boodaghians, L., Kasymjanova, G., Agulnik, J. S., Pepe, C., Sakr, L., et al. (2019). A Phase I Open Prospective Cohort Trial of Curcumin Plus Tyrosine Kinase Inhibitors for EGFR-Mutant Advanced Non-small Cell Lung Cancer. Jco 37, e20611. doi:10.1200/JCO.2019.37.15_suppl.e20611
Farghadani, R., and Naidu, R. (2021). Curcumin: Modulator of Key Molecular Signaling Pathways in Hormone-independent Breast Cancer. Cancers (Basel) 13, 3427. doi:10.3390/cancers13143427
Farhood, B., Mortezaee, K., Goradel, N. H., Khanlarkhani, N., Salehi, E., Nashtaei, M. S., et al. (2019). Curcumin as an Anti-inflammatory Agent: Implications to Radiotherapy and Chemotherapy. J. Cel Physiol 234, 5728–5740. doi:10.1002/jcp.27442
Faridi, J., Fawcett, J., Wang, L., and Roth, R. A. (2003). Akt Promotes Increased Mammalian Cell Size by Stimulating Protein Synthesis and Inhibiting Protein Degradation. Am. J. Physiol. Endocrinol. Metab. 285, E964–E972. doi:10.1152/ajpendo.00239.2003
Farooqi, A. A., and Siddik, Z. H. (2015). Platelet-derived Growth Factor (PDGF) Signalling in Cancer: Rapidly Emerging Signalling Landscape. Cell Biochem Funct 33, 257–265. doi:10.1002/cbf.3120
Ferlay, J., Colombet, M., Soerjomataram, I., Parkin, D. M., Piñeros, M., Znaor, A., et al. (2021). Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 149, 778–789. doi:10.1002/ijc.33588
Firtina Karagonlar, Z., Koc, D., Iscan, E., Erdal, E., and Atabey, N. (2016). Elevated Hepatocyte Growth Factor Expression as an Autocrine C-Met Activation Mechanism in Acquired Resistance to Sorafenib in Hepatocellular Carcinoma Cells. Cancer Sci. 107, 407–416. doi:10.1111/cas.12891
Fredriksson, L., Li, H., and Eriksson, U. (2004). The PDGF Family: Four Gene Products Form Five Dimeric Isoforms. Cytokine Growth Factor. Rev. 15, 197–204. doi:10.1016/j.cytogfr.2004.03.007
Fruman, D. A., Chiu, H., Hopkins, B. D., Bagrodia, S., Cantley, L. C., and Abraham, R. T. (2017). The PI3K Pathway in Human Disease. Cell 170, 605–635. doi:10.1016/j.cell.2017.07.029
Fu, Z., Chen, X., Guan, S., Yan, Y., Lin, H., and Hua, Z. C. (2015). Curcumin Inhibits Angiogenesis and Improves Defective Hematopoiesis Induced by Tumor-Derived VEGF in Tumor Model through Modulating VEGF-VEGFR2 Signaling Pathway. Oncotarget 6, 19469–19482. doi:10.18632/oncotarget.3625
Galardi, S., Mercatelli, N., Farace, M. G., and Ciafrè, S. A. (2011). NF-kB and C-Jun Induce the Expression of the Oncogenic miR-221 and miR-222 in Prostate Carcinoma and Glioblastoma Cells. Nucleic Acids Res. 39, 3892–3902. doi:10.1093/nar/gkr006
Gallant, J. N., Sheehan, J. H., Shaver, T. M., Bailey, M., Lipson, D., Chandramohan, R., et al. (2015). EGFR Kinase Domain Duplication (EGFR-KDD) Is a Novel Oncogenic Driver in Lung Cancer that Is Clinically Responsive to Afatinib. Cancer Discov. 5, 1155–1163. doi:10.1158/2159-8290.Cd-15-0654
Gallo, L. H., Nelson, K. N., Meyer, A. N., and Donoghue, D. J. (2015). Functions of Fibroblast Growth Factor Receptors in Cancer Defined by Novel Translocations and Mutations. Cytokine Growth Factor. Rev. 26, 425–449. doi:10.1016/j.cytogfr.2015.03.003
Gazdar, A. F. (2009). Activating and Resistance Mutations of EGFR in Non-small-cell Lung Cancer: Role in Clinical Response to EGFR Tyrosine Kinase Inhibitors. Oncogene 28 (Suppl. 1), S24–S31. doi:10.1038/onc.2009.198
Ghasemi, F., Shafiee, M., Banikazemi, Z., Pourhanifeh, M. H., Khanbabaei, H., Shamshirian, A., et al. (2019). Curcumin Inhibits NF-kB and Wnt/β-Catenin Pathways in Cervical Cancer Cells. Pathol. Res. Pract. 215, 152556. doi:10.1016/j.prp.2019.152556
Giordano, A., and Tommonaro, G. (2019). Curcumin and Cancer. Nutrients 11, 2376. doi:10.3390/nu11102376
Glynn, S. A., Prueitt, R. L., Ridnour, L. A., Boersma, B. J., Dorsey, T. M., Wink, D. A., et al. (2010). COX-2 Activation Is Associated with Akt Phosphorylation and Poor Survival in ER-Negative, HER2-Positive Breast Cancer. BMC Cancer 10, 626. doi:10.1186/1471-2407-10-626
Goetz, E. M., Ghandi, M., Treacy, D. J., Wagle, N., and Garraway, L. A. (2014). ERK Mutations Confer Resistance to Mitogen-Activated Protein Kinase Pathway Inhibitors. Cancer Res. 74, 7079–7089. doi:10.1158/0008-5472.Can-14-2073
Goldberg, S. B., Oxnard, G. R., Digumarthy, S., Muzikansky, A., Jackman, D. M., Lennes, I. T., et al. (2013). Chemotherapy with Erlotinib or Chemotherapy Alone in Advanced Non-small Cell Lung Cancer with Acquired Resistance to EGFR Tyrosine Kinase Inhibitors. Oncologist 18, 1214–1220. doi:10.1634/theoncologist.2013-0168
Golonko, A., Lewandowska, H., Świsłocka, R., Jasińska, U. T., Priebe, W., and Lewandowski, W. (2019). Curcumin as Tyrosine Kinase Inhibitor in Cancer Treatment. Eur. J. Med. Chem. 181, 111512. doi:10.1016/j.ejmech.2019.07.015
Gong, Y., Yao, E., Shen, R., Goel, A., Arcila, M., Teruya-Feldstein, J., et al. (2009). High Expression Levels of Total IGF-1R and Sensitivity of NSCLC Cells In Vitro to an Anti-IGF-1r Antibody (R1507). PLoS One 4, e7273. doi:10.1371/journal.pone.0007273
Gong, Y., Guo, Y., and Niu, T. (2012). Curcumin Potentiates Antitumor Activity of Imatinib via Inhibition of the AKT/mTOR Signaling Pathway and Down-Regulation of Bcr-Abl Gene in Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood 120, 3559. doi:10.1182/blood.V120.21.3559.3559
Gow, C. H., Liu, Y. N., Li, H. Y., Hsieh, M. S., Chang, S. H., Luo, S. C., et al. (2018). Oncogenic Function of a KIF5B-MET Fusion Variant in Non-small Cell Lung Cancer. Neoplasia 20, 838–847. doi:10.1016/j.neo.2018.06.007
Grivas, N., Goussia, A., Stefanou, D., and Giannakis, D. (2016). Microvascular Density and Immunohistochemical Expression of VEGF, VEGFR-1 and VEGFR-2 in Benign Prostatic Hyperplasia, High-Grade Prostate Intraepithelial Neoplasia and Prostate Cancer. Cent. Eur. J Urol 69, 63–71. doi:10.5173/ceju.2016.726
Gumustekin, M., Kargi, A., Bulut, G., Gozukizil, A., Ulukus, C., Oztop, I., et al. (2012). HGF/c-Met Overexpressions, but Not Met Mutation, Correlates with Progression of Non-small Cell Lung Cancer. Pathol. Oncol. Res. 18, 209–218. doi:10.1007/s12253-011-9430-7
Guo, Y., Li, Y., Shan, Q., He, G., Lin, J., and Gong, Y. (2015). Curcumin Potentiates the Anti-leukemia Effects of Imatinib by Downregulation of the AKT/mTOR Pathway and BCR/ABL Gene Expression in Ph+ Acute Lymphoblastic Leukemia. Int. J. Biochem. Cel Biol 65, 1–11. doi:10.1016/j.biocel.2015.05.003
Ha, S. Y., Lee, J., Kang, S. Y., Do, I. G., Ahn, S., Park, J. O., et al. (2013). MET Overexpression Assessed by New Interpretation Method Predicts Gene Amplification and Poor Survival in Advanced Gastric Carcinomas. Mod. Pathol. 26, 1632–1641. doi:10.1038/modpathol.2013.108
Hakam, A., Fang, Q., Karl, R., and Coppola, D. (2003). Coexpression of IGF-1R and C-Src Proteins in Human Pancreatic Ductal Adenocarcinoma. Dig. Dis. Sci. 48, 1972–1978. doi:10.1023/a:1026122421369
Halim, C. E., Deng, S., Ong, M. S., and Yap, C. T. (2020). Involvement of STAT5 in Oncogenesis. Biomedicines 8, 316. doi:10.3390/biomedicines8090316
Hassanzadeh, P. (2011). Colorectal Cancer and NF-Κb Signaling Pathway. Gastroenterol. Hepatol. Bed Bench 4, 127–132.
Heinrich, M. C., Corless, C. L., Duensing, A., McGreevey, L., Chen, C. J., Joseph, N., et al. (2003). PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors. Science 299, 708–710. doi:10.1126/science.1079666
Heist, R. S., Mino-Kenudson, M., Sequist, L. V., Tammireddy, S., Morrissey, L., Christiani, D. C., et al. (2012). FGFR1 Amplification in Squamous Cell Carcinoma of the Lung. J. Thorac. Oncol. 7, 1775–1780. doi:10.1097/JTO.0b013e31826aed28
Horiguchi, N., Takayama, H., Toyoda, M., Otsuka, T., Fukusato, T., Merlino, G., et al. (2002). Hepatocyte Growth Factor Promotes Hepatocarcinogenesis through C-Met Autocrine Activation and Enhanced Angiogenesis in Transgenic Mice Treated with Diethylnitrosamine. Oncogene 21, 1791–1799. doi:10.1038/sj.onc.1205248
Hosseini, S., Chamani, J., Sinichi, M., Bonakdar, A. M., Azad, Z., Ahangari, N., et al. (2019). The Effect of Nanomicelle Curcumin, Sorafenib, and Combination of the Two on the Cyclin D1 Gene Expression of the Hepatocellular Carcinoma Cell Line (HUH7). Iran J. Basic Med. Sci. 22, 1198–1202. doi:10.22038/ijbms.2019.35808.8530
Hoxhaj, G., and Manning, B. D. (2020). The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 20, 74–88. doi:10.1038/s41568-019-0216-7
Hsiao, Y.-T., Kuo, C.-L., Lin, J.-J., Huang, W.-W., Peng, S.-F., Chueh, F.-S., et al. (2018). Curcuminoids Combined with Gefitinib Mediated Apoptosis and Autophagy of Human Oral Cancer SAS Cellsin Vitroand Reduced Tumor of SAS Cell Xenograft Micein Vivo. Environ. Toxicol. 33, 821–832. doi:10.1002/tox.22568
Hu, A., Huang, J. J., Jin, X. J., Li, J. P., Tang, Y. J., Huang, X. F., et al. (2014). Curcumin Suppresses Invasiveness and Vasculogenic Mimicry of Squamous Cell Carcinoma of the Larynx through the Inhibition of JAK-2/STAT-3 Signaling Pathway. Am. J. Cancer Res. 5, 278–288.
Hu, B., Sun, D., Sun, C., Sun, Y. F., Sun, H. X., Zhu, Q. F., et al. (2015). A Polymeric Nanoparticle Formulation of Curcumin in Combination with Sorafenib Synergistically Inhibits Tumor Growth and Metastasis in an Orthotopic Model of Human Hepatocellular Carcinoma. Biochem. Biophys. Res. Commun. 468, 525–532. doi:10.1016/j.bbrc.2015.10.031
Hu, H. J., Lin, X. L., Liu, M. H., Fan, X. J., and Zou, W. W. (2016). Curcumin Mediates Reversion of HGF-Induced Epithelial-Mesenchymal Transition via Inhibition of C-Met Expression in DU145 Cells. Oncol. Lett. 11, 1499–1505. doi:10.3892/ol.2015.4063
Huang, Q., Snyder, D. S., Chu, P., Gaal, K. K., Chang, K. L., and Weiss, L. M. (2011). PDGFRA Rearrangement Leading to Hyper-Eosinophilia, T-Lymphoblastic Lymphoma, Myeloproliferative Neoplasm and Precursor B-Cell Acute Lymphoblastic Leukemia. Leukemia 25, 371–375. doi:10.1038/leu.2010.272
Huh, Y. H., Kim, S. H., Kim, S. J., and Chun, J. S. (2003). Differentiation Status-dependent Regulation of Cyclooxygenase-2 Expression and Prostaglandin E2 Production by Epidermal Growth Factor via Mitogen-Activated Protein Kinase in Articular Chondrocytes. J. Biol. Chem. 278, 9691–9697. doi:10.1074/jbc.M211360200
Hur, J. Y., Chao, J., Kim, K., Kim, S. T., Kim, K. M., Klempner, S. J., et al. (2020). High-level FGFR2 Amplification Is Associated with Poor Prognosis and Lower Response to Chemotherapy in Gastric Cancers. Pathol. Res. Pract. 216, 152878. doi:10.1016/j.prp.2020.152878
Hutchins-Wolfbrandt, A., and Mistry, A. M. (2011). Dietary Turmeric Potentially Reduces the Risk of Cancer. Asian Pac. J. Cancer Prev. 12, 3169–3173.
Idbaih, A., Aimard, J., Boisselier, B., Marie, Y., Paris, S., Crinière, E., et al. (2009). Epidermal Growth Factor Receptor Extracellular Domain Mutations in Primary Glioblastoma. Neuropathol. Appl. Neurobiol. 35, 208–213. doi:10.1111/j.1365-2990.2008.00977.x
Igelmann, S., Neubauer, H. A., and Ferbeyre, G. (2019). STAT3 and STAT5 Activation in Solid Cancers. Cancers (Basel) 11, 1428. doi:10.3390/cancers11101428
Itakura, J., Ishiwata, T., Shen, B., Kornmann, M., and Korc, M. (2000). Concomitant Over-expression of Vascular Endothelial Growth Factor and its Receptors in Pancreatic Cancer. Int. J. Cancer 85, 27–34. doi:10.1002/(sici)1097-0215(20000101)85:1<27:aid-ijc5>3.0.co;2-8
Iyoda, K., Sasaki, Y., Horimoto, M., Toyama, T., Yakushijin, T., Sakakibara, M., et al. (2003). Involvement of the P38 Mitogen-Activated Protein Kinase cascade in Hepatocellular Carcinoma. Cancer 97, 3017–3026. doi:10.1002/cncr.11425
Jackson, M. W., Roberts, J. S., Heckford, S. E., Ricciardelli, C., Stahl, J., Choong, C., et al. (2002). A Potential Autocrine Role for Vascular Endothelial Growth Factor in Prostate Cancer. Cancer Res. 62, 854–859.
Javadi, S., Rostamizadeh, K., Hejazi, J., Parsa, M., and Fathi, M. (2018). Curcumin Mediated Down-Regulation of αV β3 Integrin and Up-Regulation of Pyruvate Dehydrogenase Kinase 4 (PDK4) in Erlotinib Resistant SW480 colon Cancer Cells. Phytother Res. 32, 355–364. doi:10.1002/ptr.5984
Jechlinger, M., Sommer, A., Moriggl, R., Seither, P., Kraut, N., Capodiecci, P., et al. (2006). Autocrine PDGFR Signaling Promotes Mammary Cancer Metastasis. J. Clin. Invest. 116, 1561–1570. doi:10.1172/jci24652
Jeong, E. G., Kim, M. S., Nam, H. K., Min, C. K., Lee, S., Chung, Y. J., et al. (2008). Somatic Mutations of JAK1 and JAK3 in Acute Leukemias and Solid Cancers. Clin. Cancer Res. 14, 3716–3721. doi:10.1158/1078-0432.Ccr-07-4839
Ji, W., Yu, Y., Li, Z., Wang, G., Li, F., Xia, W., et al. (2016). FGFR1 Promotes the Stem Cell-like Phenotype of FGFR1-Amplified Non-small Cell Lung Cancer Cells through the Hedgehog Pathway. Oncotarget 7, 15118–15134. doi:10.18632/oncotarget.7701
Ji, Z., He, L., Regev, A., and Struhl, K. (2019). Inflammatory Regulatory Network Mediated by the Joint Action of NF-kB, STAT3, and AP-1 Factors Is Involved in many Human Cancers. Proc. Natl. Acad. Sci. U S A. 116, 9453–9462. doi:10.1073/pnas.1821068116
Jiang, A. P., Zhou, D. H., Meng, X. L., Zhang, A. P., Zhang, C., Li, X. T., et al. (2014). Down-regulation of Epidermal Growth Factor Receptor by Curcumin-Induced UBE1L in Human Bronchial Epithelial Cells. J. Nutr. Biochem. 25, 241–249. doi:10.1016/j.jnutbio.2013.11.001
Jiang, W., Wang, X., Zhang, C., Xue, L., and Yang, L. (2020). Expression and Clinical Significance of MAPK and EGFR in Triple-Negative Breast Cancer. Oncol. Lett. 19, 1842–1848. doi:10.3892/ol.2020.11274
Jiao, D., Wang, J., Lu, W., Tang, X., Chen, J., Mou, H., et al. (2016). Curcumin Inhibited HGF-Induced EMT and Angiogenesis through Regulating C-Met Dependent PI3K/Akt/mTOR Signaling Pathways in Lung Cancer. Mol. Ther. Oncolytics 3, 16018. doi:10.1038/mto.2016.18
Jiao, Q., Bi, L., Ren, Y., Song, S., Wang, Q., and Wang, Y. S. (2018). Advances in Studies of Tyrosine Kinase Inhibitors and Their Acquired Resistance. Mol. Cancer 17, 36. doi:10.1186/s12943-018-0801-5
Joensuu, H., Rutkowski, P., Nishida, T., Steigen, S. E., Brabec, P., Plank, L., et al. (2015). KIT and PDGFRA Mutations and the Risk of GI Stromal Tumor Recurrence. J. Clin. Oncol. 33, 634–642. doi:10.1200/JCO.2014.57.4970
Jones, R. A., Campbell, C. I., Gunther, E. J., Chodosh, L. A., Petrik, J. J., Khokha, R., et al. (2007). Transgenic Overexpression of IGF-IR Disrupts Mammary Ductal Morphogenesis and Induces Tumor Formation. Oncogene 26, 1636–1644. doi:10.1038/sj.onc.1209955
Jücker, M., Günther, A., Gradl, G., Fonatsch, C., Krueger, G., Diehl, V., et al. (1994). The Met/hepatocyte Growth Factor Receptor (HGFR) Gene Is Overexpressed in Some Cases of Human Leukemia and Lymphoma. Leuk. Res. 18, 7–16. doi:10.1016/0145-2126(94)90003-5
Jung, K. A., Choi, B. H., and Kwak, M. K. (2015). The C-Met/pi3k Signaling Is Associated with Cancer Resistance to Doxorubicin and Photodynamic Therapy by Elevating BCRP/ABCG2 Expression. Mol. Pharmacol. 87, 465–476. doi:10.1124/mol.114.096065
Katz, M., Amit, I., and Yarden, Y. (2007). Regulation of MAPKs by Growth Factors and Receptor Tyrosine Kinases. Biochim. Biophys. Acta 1773, 1161–1176. doi:10.1016/j.bbamcr.2007.01.002
Kentsis, A., Reed, C., Rice, K. L., Sanda, T., Rodig, S. J., Tholouli, E., et al. (2012). Autocrine Activation of the MET Receptor Tyrosine Kinase in Acute Myeloid Leukemia. Nat. Med. 18, 1118–1122. doi:10.1038/nm.2819
Khan, A. Q., Ahmed, E. I., Elareer, N., Fathima, H., Prabhu, K. S., Siveen, K. S., et al. (2020a). Curcumin-Mediated Apoptotic Cell Death in Papillary Thyroid Cancer and Cancer Stem-like Cells through Targeting of the JAK/STAT3 Signaling Pathway. Int. J. Mol. Sci. 21, 438. doi:10.3390/ijms21020438
Khan, M., Zhao, Z., Arooj, S., and Liao, G. (2020b). Impact of Tyrosine Kinase Inhibitors (TKIs) Combined with Radiation Therapy for the Management of Brain Metastases from Renal Cell Carcinoma. Front. Oncol. 10, 1246. doi:10.3389/fonc.2020.01246
Kim, H., Park, J., Tak, K.-H., Bu, S. Y., and Kim, E. (2014). Chemopreventive Effects of Curcumin on Chemically Induced Mouse Skin Carcinogenesis in BK5.Insulin-like Growth Factor-1 Transgenic Mice. In Vitro Cell.Dev.Biol.-Animal 50, 883–892. doi:10.1007/s11626-014-9791-9
Kim, J., Jung, J., Lee, S. J., Lee, J. S., and Park, M. J. (2012). Cancer Stem-like Cells Persist in Established Cell Lines through Autocrine Activation of EGFR Signaling. Oncol. Lett. 3, 607–612. doi:10.3892/ol.2011.531
Kim, J. H., Xu, C., Keum, Y. S., Reddy, B., Conney, A., and Kong, A. N. (2005). Inhibition of EGFR Signaling in Human Prostate Cancer PC-3 Cells by Combination Treatment with Beta-Phenylethyl Isothiocyanate and Curcumin. Carcinogenesis 27, 475–482. doi:10.1093/carcin/bgi272
Klasa-Mazurkiewicz, D., Jarząb, M., Milczek, T., Lipińska, B., and Emerich, J. (2011). Clinical Significance of VEGFR-2 and VEGFR-3 Expression in Ovarian Cancer Patients. Pol. J. Pathol. 62, 31–40.
Kobayashi, S., Boggon, T. J., Dayaram, T., Jänne, P. A., Kocher, O., Meyerson, M., et al. (2005). EGFR Mutation and Resistance of Non-small-cell Lung Cancer to Gefitinib. N. Engl. J. Med. 352, 786–792. doi:10.1056/NEJMoa044238
Kodama, M., Kitadai, Y., Tanaka, M., Kuwai, T., Tanaka, S., Oue, N., et al. (2008). Vascular Endothelial Growth Factor C Stimulates Progression of Human Gastric Cancer via Both Autocrine and Paracrine Mechanisms. Clin. Cancer Res. 14, 7205–7214. doi:10.1158/1078-0432.CCR-08-0818
Konduri, K., Gallant, J. N., Chae, Y. K., Giles, F. J., Gitlitz, B. J., Gowen, K., et al. (2016). EGFR Fusions as Novel Therapeutic Targets in Lung Cancer. Cancer Discov. 6, 601–611. doi:10.1158/2159-8290.CD-16-0075
Kong, F. M., Anscher, M. S., Washington, M. K., Killian, J. K., and Jirtle, R. L. (2000). M6P/IGF2R Is Mutated in Squamous Cell Carcinoma of the Lung. Oncogene 19, 1572–1578. doi:10.1038/sj.onc.1203437
Kreuz, S., Siegmund, D., Scheurich, P., and Wajant, H. (2001). NF-kappaB Inducers Upregulate cFLIP, a Cycloheximide-Sensitive Inhibitor of Death Receptor Signaling. Mol. Cel Biol 21, 3964–3973. doi:10.1128/MCB.21.12.3964-3973.2001
Kumar, R., Jain, A. G., Rashid, M. U., Ali, S., Khetpal, N., Hussain, I., et al. (2018). “HGFR and FGR2: Their Roles in Progression and Metastasis of Esophageal Cancer,” in Role of Tyrosine Kinases in Gastrointestinal Malignancies. Editor G. P. Nagaraju (Singapore: Springer Singapore), 1–14. doi:10.1007/978-981-13-1486-5_1
Kyriakis, J. M., and Avruch, J. (2001). Mammalian Mitogen-Activated Protein Kinase Signal Transduction Pathways Activated by Stress and Inflammation. Physiol. Rev. 81, 807–869. doi:10.1152/physrev.2001.81.2.807
Lai, H.-W., Chien, S.-Y., Kuo, S.-J., Tseng, L.-M., Lin, H.-Y., Chi, C.-W., et al. (2012). The Potential Utility of Curcumin in the Treatment of HER-2-Overexpressed Breast Cancer: AnIn VitroandIn VivoComparison Study with Herceptin. Evidence-Based Complement. Altern. Med. 2012, 1–12. doi:10.1155/2012/486568
Lai, K. C., Chueh, F. S., Hsiao, Y. T., Cheng, Z. Y., Lien, J. C., Liu, K. C., et al. (2019). Gefitinib and Curcumin-Loaded Nanoparticles Enhance Cell Apoptosis in Human Oral Cancer SAS Cells In Vitro and Inhibit SAS Cell Xenografted Tumor In Vivo. Toxicol. Appl. Pharmacol. 382, 114734. doi:10.1016/j.taap.2019.114734
Lai, S. W., Bamodu, O. A., Tsai, W. C., Chang, Y. M., Lee, W. H., Yeh, C. T., et al. (2018). The Therapeutic Targeting of the FGFR1/Src/NF-Κb Signaling axis Inhibits Pancreatic Ductal Adenocarcinoma Stemness and Oncogenicity. Clin. Exp. Metastasis 35, 663–677. doi:10.1007/s10585-018-9919-5
Lasota, J., and Miettinen, M. (2006). KIT and PDGFRA Mutations in Gastrointestinal Stromal Tumors (GISTs). Semin. Diagn. Pathol. 23, 91–102. doi:10.1053/j.semdp.2006.08.006
Lassus, H., Sihto, H., Leminen, A., Joensuu, H., Isola, J., Nupponen, N. N., et al. (2006). Gene Amplification, Mutation, and Protein Expression of EGFR and Mutations of ERBB2 in Serous Ovarian Carcinoma. J. Mol. Med. (Berl) 84, 671–681. doi:10.1007/s00109-006-0054-4
Lassus, H., Sihto, H., Leminen, A., Nordling, S., Joensuu, H., Nupponen, N. N., et al. (2004). Genetic Alterations and Protein Expression of KIT and PDGFRA in Serous Ovarian Carcinoma. Br. J. Cancer 91, 2048–2055. doi:10.1038/sj.bjc.6602252
Lee, J.-Y., Yu, S.-L., Huang, J.-Y., Chen, W.-J., Chen, J. J. W., Yang, P.-C., et al. (2007). Synergistic Antitumor Activity of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Gefitinib and Curcumin in Non-small-cell Lung Cancer. Cancer Res. 67, 4783.
Lee, J. Y., Lee, Y. M., Chang, G. C., Yu, S. L., Hsieh, W. Y., Chen, J. J., et al. (2011). Curcumin Induces EGFR Degradation in Lung Adenocarcinoma and Modulates P38 Activation in Intestine: the Versatile Adjuvant for Gefitinib Therapy. PLoS One 6, e23756. doi:10.1371/journal.pone.0023756
Lee, S. J., Lee, J., Park, S. H., Park, J. O., Lim, H. Y., Kang, W. K., et al. (2018). c-MET Overexpression in Colorectal Cancer: A Poor Prognostic Factor for Survival. Clin. Colorectal Cancer 17, 165–169. doi:10.1016/j.clcc.2018.02.013
Lemmon, M. A., and Schlessinger, J. (2010). Cell Signaling by Receptor Tyrosine Kinases. Cell 141, 1117–1134. doi:10.1016/j.cell.2010.06.011
Lesslie, D. P., Summy, J. M., Parikh, N. U., Fan, F., Trevino, J. G., Sawyer, T. K., et al. (2006). Vascular Endothelial Growth Factor Receptor-1 Mediates Migration of Human Colorectal Carcinoma Cells by Activation of Src Family Kinases. Br. J. Cancer 94, 1710–1717. doi:10.1038/sj.bjc.6603143
Lev-Ari, S., Starr, A., Katzburg, S., Berkovich, L., Rimmon, A., Ben-Yosef, R., et al. (2014). Curcumin Induces Apoptosis and Inhibits Growth of Orthotopic Human Non-small Cell Lung Cancer Xenografts. J. Nutr. Biochem. 25, 843–850. doi:10.1016/j.jnutbio.2014.03.014
Lev-Ari, S., Starr, A., Vexler, A., Karaush, V., Loew, V., Greif, J., et al. (2006). Inhibition of Pancreatic and Lung Adenocarcinoma Cell Survival by Curcumin Is Associated with Increased Apoptosis, Down-Regulation of COX-2 and EGFR and Inhibition of Erk1/2 Activity. Anticancer Res. 26, 4423–4430.
Li, E., and Hristova, K. (2006). Role of Receptor Tyrosine Kinase Transmembrane Domains in Cell Signaling and Human Pathologies. Biochemistry 45, 6241–6251. doi:10.1021/bi060609y
Li, J., Lau, G. K., Chen, L., Dong, S. S., Lan, H. Y., Huang, X. R., et al. (2011). Interleukin 17A Promotes Hepatocellular Carcinoma Metastasis via NF-kB Induced Matrix Metalloproteinases 2 and 9 Expression. PLoS One 6, e21816. doi:10.1371/journal.pone.0021816
Li, P., Huang, T., Zou, Q., Liu, D., Wang, Y., Tan, X., et al. (2019a). FGFR2 Promotes Expression of PD-L1 in Colorectal Cancer via the JAK/STAT3 Signaling Pathway. J. Immunol. 202, 3065–3075. doi:10.4049/jimmunol.1801199
Li, S., Liu, Z., Zhu, F., Fan, X., Wu, X., Zhao, H., et al. (2013). Curcumin Lowers Erlotinib Resistance in Non-small Cell Lung Carcinoma Cells with Mutated EGF Receptor. Oncol. Res. 21, 137–144. doi:10.3727/096504013x13832473330032
Li, S., Zhou, Y., Zheng, X., Wu, X., Liang, Y., Wang, S., et al. (2016). Sphk1 Promotes Breast Epithelial Cell Proliferation via NF-Κb-P65-Mediated Cyclin D1 Expression. Oncotarget 7, 80579–80585. doi:10.18632/oncotarget.13013
Li, W., Wang, Z., Xiao, X., Han, L., Wu, Z., Ma, Q., et al. (2019b). Curcumin Attenuates Hyperglycemia-Driven EGF-Induced Invasive and Migratory Abilities of Pancreatic Cancer via Suppression of the ERK and AKT Pathways. Oncol. Rep. 41, 650–658. doi:10.3892/or.2018.6833
Li, X., Ma, S., Yang, P., Sun, B., Zhang, Y., Sun, Y., et al. (2018a). Anticancer Effects of Curcumin on Nude Mice Bearing Lung Cancer A549 Cell Subsets SP and NSP Cells. Oncol. Lett. 16, 6756–6762. doi:10.3892/ol.2018.9488
Li, Y., Sun, W., Han, N., Zou, Y., and Yin, D. (2018b). Curcumin Inhibits Proliferation, Migration, Invasion and Promotes Apoptosis of Retinoblastoma Cell Lines through Modulation of miR-99a and JAK/STAT Pathway. BMC Cancer 18, 1230. doi:10.1186/s12885-018-5130-y
Lian, L., Li, X. L., Xu, M. D., Li, X. M., Wu, M. Y., Zhang, Y., et al. (2019). VEGFR2 Promotes Tumorigenesis and Metastasis in a Pro-angiogenic-independent Way in Gastric Cancer. BMC Cancer 19, 183. doi:10.1186/s12885-019-5322-0
Liang, H., Liu, X., and Wang, M. (2018). Immunotherapy Combined with Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Non-small-cell Lung Cancer Treatment. Onco Targets Ther. 11, 6189–6196. doi:10.2147/OTT.S178497
Lin, L., Deangelis, S., Foust, E., Fuchs, J., Li, C., Li, P. K., et al. (2010a). A Novel Small Molecule Inhibits STAT3 Phosphorylation and DNA Binding Activity and Exhibits Potent Growth Suppressive Activity in Human Cancer Cells. Mol. Cancer 9, 217. doi:10.1186/1476-4598-9-217
Lin, L., Hutzen, B., Zuo, M., Ball, S., Deangelis, S., Foust, E., et al. (2010b). Novel STAT3 Phosphorylation Inhibitors Exhibit Potent Growth-Suppressive Activity in Pancreatic and Breast Cancer Cells. Cancer Res. 70, 2445–2454. doi:10.1158/0008-5472.CAN-09-2468
Lin, S. S., Lai, K. C., Hsu, S. C., Yang, J. S., Kuo, C. L., Lin, J. P., et al. (2009). Curcumin Inhibits the Migration and Invasion of Human A549 Lung Cancer Cells through the Inhibition of Matrix Metalloproteinase-2 and -9 and Vascular Endothelial Growth Factor (VEGF). Cancer Lett. 285, 127–133. doi:10.1016/j.canlet.2009.04.037
Lin, Y., Wang, X., and Jin, H. (2014). EGFR-TKI Resistance in NSCLC Patients: Mechanisms and Strategies. Am. J. Cancer Res. 4, 411–435.
Lin, Y., Zhai, E., Liao, B., Xu, L., Zhang, X., Peng, S., et al. (2017). Autocrine VEGF Signaling Promotes Cell Proliferation through a PLC-dependent Pathway and Modulates Apatinib Treatment Efficacy in Gastric Cancer. Oncotarget 8, 11990–12002. doi:10.18632/oncotarget.14467
Liu, G., Chen, T., Ding, Z., Wang, Y., Wei, Y., and Wei, X. (2021). Inhibition of FGF-FGFR and VEGF-VEGFR Signalling in Cancer Treatment. Cell Prolif 54, e13009. doi:10.1111/cpr.13009
Liu, J., Minemoto, Y., and Lin, A. (2004). c-Jun N-Terminal Protein Kinase 1 (JNK1), but Not JNK2, Is Essential for Tumor Necrosis Factor Alpha-Induced C-Jun Kinase Activation and Apoptosis. Mol. Cel Biol 24, 10844–10856. doi:10.1128/MCB.24.24.10844-10856.2004
Liu, J., Chen, X., Mao, Y., and Shen, K. (2015). Effect of Curcumin on Lapatinib Sensitivity and Lapatinib Resistance Associated EMT and Stem-like Phenotype in HER2 Positive Breast Cancer. Jco 33, e11594. doi:10.1200/jco.2015.33.15_suppl.e11594
Lokker, N. A., Sullivan, C. M., Hollenbach, S. J., Israel, M. A., and Giese, N. A. (2002). Platelet-derived Growth Factor (PDGF) Autocrine Signaling Regulates Survival and Mitogenic Pathways in Glioblastoma Cells: Evidence that the Novel PDGF-C and PDGF-D Ligands May Play a Role in the Development of Brain Tumors. Cancer Res. 62, 3729–3735.
Longatto Filho, A., Martins, A., Costa, S. M., and Schmitt, F. C. (2005). VEGFR-3 Expression in Breast Cancer Tissue Is Not Restricted to Lymphatic Vessels. Pathol. Res. Pract. 201, 93–99. doi:10.1016/j.prp.2004.11.008
Lu, K. H., Wu, H. H., Lin, R. C., Lin, Y. C., Lu, P. W., Yang, S. F., et al. (2020). Curcumin Analogue L48H37 Suppresses Human Osteosarcoma U2OS and MG-63 Cells' Migration and Invasion in Culture by Inhibition of uPA via the JAK/STAT Signaling Pathway. Molecules 26, 30. doi:10.3390/molecules26010030
Luo, J. L., Kamata, H., and Karin, M. (2005). IKK/NF-kappaB Signaling: Balancing Life and Death-Aa New Approach to Cancer Therapy. J. Clin. Invest. 115, 2625–2632. doi:10.1172/JCI26322
Man, S., Yao, J., Lv, P., Liu, Y., Yang, L., and Ma, L. (2020). Curcumin-enhanced Antitumor Effects of Sorafenib via Regulating the Metabolism and Tumor Microenvironment. Food Funct. 11, 6422–6432. doi:10.1039/c9fo01901d
Mansouri, K., Rasoulpoor, S., Daneshkhah, A., Abolfathi, S., Salari, N., Mohammadi, M., et al. (2020). Clinical Effects of Curcumin in Enhancing Cancer Therapy: A Systematic Review. BMC Cancer 20, 791. doi:10.1186/s12885-020-07256-8
Mao, P., Cohen, O., Kowalski, K. J., Kusiel, J. G., Buendia-Buendia, J. E., Cuoco, M. S., et al. (2020). Acquired FGFR and FGF Alterations Confer Resistance to Estrogen Receptor (ER) Targeted Therapy in ER+ Metastatic Breast Cancer. Clin. Cancer Res. 26, 5974–5989. doi:10.1158/1078-0432.Ccr-19-3958
Masamune, A., Satoh, M., Kikuta, K., Suzuki, N., and Shimosegawa, T. (2005). Activation of JAK-STAT Pathway Is Required for Platelet-Derived Growth Factor-Induced Proliferation of Pancreatic Stellate Cells. World J. Gastroenterol. 11, 3385–3391. doi:10.3748/wjg.v11.i22.3385
Matei, D., Emerson, R. E., Lai, Y. C., Baldridge, L. A., Rao, J., Yiannoutsos, C., et al. (2006). Autocrine Activation of PDGFRalpha Promotes the Progression of Ovarian Cancer. Oncogene 25, 2060–2069. doi:10.1038/sj.onc.1209232
Matsumoto, K., Arao, T., Hamaguchi, T., Shimada, Y., Kato, K., Oda, I., et al. (2012). FGFR2 Gene Amplification and Clinicopathological Features in Gastric Cancer. Br. J. Cancer 106, 727–732. doi:10.1038/bjc.2011.603
Matsuura, M., Onimaru, M., Yonemitsu, Y., Suzuki, H., Nakano, T., Ishibashi, H., et al. (2009). Autocrine Loop between Vascular Endothelial Growth Factor (VEGF)-C and VEGF Receptor-3 Positively Regulates Tumor-Associated Lymphangiogenesis in Oral Squamoid Cancer Cells. Am. J. Pathol. 175, 1709–1721. doi:10.2353/ajpath.2009.081139
Matušan-Ilijaš, K., Damante, G., Fabbro, D., Đorđević, G., Hadžisejdić, I., Grahovac, M., et al. (2013). EGFR Expression Is Linked to Osteopontin and Nf-Κb Signaling in clear Cell Renal Cell Carcinoma. Clin. Transl Oncol. 15, 65–71. doi:10.1007/s12094-012-0889-9
Meiyanto, E., Putri, D. D., Susidarti, R. A., Murwanti, R., SardjimanFitriasari, A., Fitriasari, A., et al. (2014). Curcumin and its Analogues (PGV-0 and PGV-1) Enhance Sensitivity of Resistant MCF-7 Cells to Doxorubicin through Inhibition of HER2 and NF-kB Activation. Asian Pac. J. Cancer Prev. 15, 179–184. doi:10.7314/apjcp.2014.15.1.179
Mendel, D. B., Laird, A. D., Xin, X., Louie, S. G., Christensen, J. G., Li, G., et al. (2003). In Vivo antitumor Activity of SU11248, a Novel Tyrosine Kinase Inhibitor Targeting Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor Receptors: Determination of a Pharmacokinetic/pharmacodynamic Relationship. Clin. Cancer Res. 9, 327–337.
Miłobȩdzka, J., Kostanecki, S. V., and Lampe, V. (1910). Zur Kenntnis des Curcumins. Ber. Dtsch. Chem. Ges. 43, 2163–2170. doi:10.1002/cber.191004302168
Mishra, A., Kumar, R., Tyagi, A., Kohaar, I., Hedau, S., Bharti, A. C., et al. (2015). Curcumin Modulates Cellular AP-1, NF-kB, and HPV16 E6 Proteins in Oral Cancer. Ecancermedicalscience 9, 525. doi:10.3332/ecancer.2015.525
Mishra, P., Senthivinayagam, S., Rangasamy, V., Sondarva, G., and Rana, B. (2010). Mixed Lineage Kinase-3/JNK1 Axis Promotes Migration of Human Gastric Cancer Cells Following Gastrin Stimulation. Mol. Endocrinol. 24, 598–607. doi:10.1210/me.2009-0387
Miyata, Y., Ashida, S., Nakamura, T., Mochizuki, Y., Koga, S., Kanetake, H., et al. (2003). Overexpression of Hepatocyte Growth Factor Receptor in Renal Carcinoma Cells Indirectly Stimulates Tumor Growth In Vivo. Biochem. Biophys. Res. Commun. 302, 892–897. doi:10.1016/S0006-291X(03)00281-X
Mohammadi, M., Schlessinger, J., and Hubbard, S. R. (1996). Structure of the FGF Receptor Tyrosine Kinase Domain Reveals a Novel Autoinhibitory Mechanism. Cell 86, 577–587. doi:10.1016/s0092-8674(00)80131-2
Monaco, S. E., Rodriguez, E. F., Mahaffey, A. L., and Dacic, S. (2015). FGFR1 Amplification in Squamous Cell Carcinoma of the Lung with Correlation of Primary and Metastatic Tumor Status. Am. J. Clin. Pathol. 145, 55–61. doi:10.1093/ajcp/aqv013
Morinaga, R., Okamoto, I., Fujita, Y., Arao, T., Sekijima, M., Nishio, K., et al. (2008). Association of Epidermal Growth Factor Receptor (EGFR) Gene Mutations with EGFR Amplification in Advanced Non-small Cell Lung Cancer. Cancer Sci. 99, 2455–2460. doi:10.1111/j.1349-7006.2008.00962.x
Nagaraju, G. P., Zhu, S., Ko, J. E., Ashritha, N., Kandimalla, R., Snyder, J. P., et al. (2015). Antiangiogenic Effects of a Novel Synthetic Curcumin Analogue in Pancreatic Cancer. Cancer Lett. 357, 557–565. doi:10.1016/j.canlet.2014.12.007
Nassar, A. H., Lundgren, K., Pomerantz, M., Van Allen, E., Harshman, L., Choudhury, A. D., et al. (2018). Enrichment of FGFR3-TACC3 Fusions in Patients with Bladder Cancer Who Are Young, Asian, or Have Never Smoked. JCO Precision Oncol. 2, 1–11. doi:10.1200/po.18.00013
Natoli, C., Perrucci, B., Perrotti, F., Falchi, L., and Iacobelli, S. (2010). Tyrosine Kinase Inhibitors. Curr. Cancer Drug Targets 10, 462–483. doi:10.2174/156800910791517208
Nautiyal, J., Banerjee, S., Kanwar, S. S., Yu, Y., Patel, B. B., Sarkar, F. H., et al. (2011a). Curcumin Enhances Dasatinib-Induced Inhibition of Growth and Transformation of colon Cancer Cells. Int. J. Cancer 128, 951–961. doi:10.1002/ijc.25410
Nautiyal, J., Kanwar, S. S., Yu, Y., and Majumdar, A. P. (2011b). Combination of Dasatinib and Curcumin Eliminates Chemo-Resistant colon Cancer Cells. J. Mol. Signal. 6, 7. doi:10.1186/1750-2187-6-7
Nelson, K. N., Meyer, A. N., Siari, A., Campos, A. R., Motamedchaboki, K., and Donoghue, D. J. (2016). Oncogenic Gene Fusion FGFR3-TACC3 Is Regulated by Tyrosine Phosphorylation. Mol. Cancer Res. 14, 458–469. doi:10.1158/1541-7786.Mcr-15-0497
Niu, X. L., Peters, K. G., and Kontos, C. D. (2002). Deletion of the Carboxyl Terminus of Tie2 Enhances Kinase Activity, Signaling, and Function. Evidence for an Autoinhibitory Mechanism. J. Biol. Chem. 277, 31768–31773. doi:10.1074/jbc.M203995200
Notarbartolo, M., Poma, P., Perri, D., Dusonchet, L., Cervello, M., and D'Alessandro, N. (2005). Antitumor Effects of Curcumin, Alone or in Combination with Cisplatin or Doxorubicin, on Human Hepatic Cancer Cells. Analysis of Their Possible Relationship to Changes in NF-kB Activation Levels and in IAP Gene Expression. Cancer Lett. 224, 53–65. doi:10.1016/j.canlet.2004.10.051
O'Reilly, L. A., Putoczki, T. L., Mielke, L. A., Low, J. T., Lin, A., Preaudet, A., et al. (2018). Loss of NF-Κb1 Causes Gastric Cancer with Aberrant Inflammation and Expression of Immune Checkpoint Regulators in a STAT-1-dependent Manner. Immunity 48, 570–e8. e578. doi:10.1016/j.immuni.2018.03.003
O'Shea, J. J., Schwartz, D. M., Villarino, A. V., Gadina, M., McInnes, I. B., and Laurence, A. (2015). The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 66, 311–328. doi:10.1146/annurev-med-051113-024537
Ohnishi, Y., Sakamoto, T., Zhengguang, L., Yasui, H., Hamada, H., Kubo, H., et al. (2020). Curcumin Inhibits Epithelial-Mesenchymal Transition in Oral Cancer Cells via C-Met Blockade. Oncol. Lett. 19, 4177–4182. doi:10.3892/ol.2020.11523
Olivera, A., Moore, T. W., Hu, F., Brown, A. P., Sun, A., Liotta, D. C., et al. (2012). Inhibition of the NF-Κb Signaling Pathway by the Curcumin Analog, 3,5-Bis(2-Pyridinylmethylidene)-4-Piperidone (EF31): Anti-inflammatory and Anti-cancer Properties. Int. Immunopharmacol 12, 368–377. doi:10.1016/j.intimp.2011.12.009
Ong, H. S., Gokavarapu, S., Tian, Z., Li, J., Xu, Q., Cao, W., et al. (2017). PDGFRA mRNA Is Overexpressed in Oral Cancer Patients as Compared to normal Subjects with a Significant Trend of Overexpression Among Tobacco Users. J. Oral Pathol. Med. 46, 591–597. doi:10.1111/jop.12571
Ong, H. S., Gokavarapu, S., Tian, Z., Li, J., Xu, Q., Zhang, C. P., et al. (2018). PDGFRA mRNA Overexpression Is Associated with Regional Metastasis and Reduced Survival in Oral Squamous Cell Carcinoma. J. Oral Pathol. Med. 47, 652–659. doi:10.1111/jop.12713
Pan, C. W., Liu, H., Zhao, Y., Qian, C., Wang, L., and Qi, J. (2016). JNK2 Downregulation Promotes Tumorigenesis and Chemoresistance by Decreasing P53 Stability in Bladder Cancer. Oncotarget 7, 35119–35131. doi:10.18632/oncotarget.9046
Pan, Z., Zhuang, J., Ji, C., Cai, Z., Liao, W., and Huang, Z. (2018). Curcumin Inhibits Hepatocellular Carcinoma Growth by Targeting VEGF Expression. Oncol. Lett. 15, 4821–4826. doi:10.3892/ol.2018.7988
Park, H. S., Jang, M. H., Kim, E. J., Kim, H. J., Lee, H. J., Kim, Y. J., et al. (2014). High EGFR Gene Copy Number Predicts Poor Outcome in Triple-Negative Breast Cancer. Mod. Pathol. 27, 1212–1222. doi:10.1038/modpathol.2013.251
Park, J., Ayyappan, V., Bae, E. K., Lee, C., Kim, B. S., Kim, B. K., et al. (2008). Curcumin in Combination with Bortezomib Synergistically Induced Apoptosis in Human Multiple Myeloma U266 Cells. Mol. Oncol. 2, 317–326. doi:10.1016/j.molonc.2008.09.006
Parker, B. C., Annala, M. J., Cogdell, D. E., Granberg, K. J., Sun, Y., Ji, P., et al. (2013). The Tumorigenic FGFR3-TACC3 Gene Fusion Escapes miR-99a Regulation in Glioblastoma. J. Clin. Invest. 123, 855–865. doi:10.1172/jci67144
Parker, B. C., Engels, M., Annala, M., and Zhang, W. (2014). Emergence of FGFR Family Gene Fusions as Therapeutic Targets in a Wide Spectrum of Solid Tumours. J. Pathol. 232, 4–15. doi:10.1002/path.4297
Patel, B. B., Gupta, D., Elliott, A. A., Sengupta, V., Yu, Y., and Majumdar, A. P. (2010). Curcumin Targets FOLFOX-Surviving colon Cancer Cells via Inhibition of EGFRs and IGF-1R. Anticancer Res. 30, 319–325.
Patel, B. B., Sengupta, R., Qazi, S., Vachhani, H., Yu, Y., Rishi, A. K., et al. (2008). Curcumin Enhances the Effects of 5-fluorouracil and Oxaliplatin in Mediating Growth Inhibition of colon Cancer Cells by Modulating EGFR and IGF-1R. Int. J. Cancer 122, 267–273. doi:10.1002/ijc.23097
Pavelić, J., Radaković, B., and Pavelić, K. (2007). Insulin-like Growth Factor 2 and its Receptors (IGF 1R and IGF 2R/mannose 6-phosphate) in Endometrial Adenocarcinoma. Gynecol. Oncol. 105, 727–735. doi:10.1016/j.ygyno.2007.02.012
Pavelić, K., Kolak, T., Kapitanović, S., Radosević, S., Spaventi, S., Kruslin, B., et al. (2003). Gastric Cancer: the Role of Insulin-like Growth Factor 2 (IGF 2) and its Receptors (IGF 1R and M6-P/IGF 2R). J. Pathol. 201, 430–438. doi:10.1002/path.1465
Pearson, A., Smyth, E., Babina, I. S., Herrera-Abreu, M. T., Tarazona, N., Peckitt, C., et al. (2016). High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov. 6, 838–851. doi:10.1158/2159-8290.Cd-15-1246
Peng, S., Wang, Y., Peng, H., Chen, D., Shen, S., Peng, B., et al. (2014). Autocrine Vascular Endothelial Growth Factor Signaling Promotes Cell Proliferation and Modulates Sorafenib Treatment Efficacy in Hepatocellular Carcinoma. Hepatology 60, 1264–1277. doi:10.1002/hep.27236
Penzel, R., Aulmann, S., Moock, M., Schwarzbach, M., Rieker, R. J., and Mechtersheimer, G. (2005). The Location of KIT and PDGFRA Gene Mutations in Gastrointestinal Stromal Tumours Is Site and Phenotype Associated. J. Clin. Pathol. 58, 634–639. doi:10.1136/jcp.2004.021766
Peretz, S., Kim, C., Rockwell, S., Baserga, R., and Glazer, P. M. (2002). IGF1 Receptor Expression Protects against Microenvironmental Stress Found in the Solid Tumor. Radiat. Res. 158, 174–180. doi:10.1667/0033-7587(2002)158[0174:irepam]2.0.co;2
Petiti, J., Rosso, V., Lo Iacono, M., Panuzzo, C., Calabrese, C., Signorino, E., et al. (2019). Curcumin Induces Apoptosis in JAK2-Mutated Cells by the Inhibition of JAK2/STAT and mTORC1 Pathways. J. Cel Mol Med 23, 4349–4357. doi:10.1111/jcmm.14326
Petti, L. M., Irusta, P. M., and DiMaio, D. (1998). Oncogenic Activation of the PDGF Beta Receptor by the Transmembrane Domain of P185neu*. Oncogene 16, 843–851. doi:10.1038/sj.onc.1201590
Porstmann, T., Griffiths, B., Chung, Y. L., Delpuech, O., Griffiths, J. R., Downward, J., et al. (2005). PKB/Akt Induces Transcription of Enzymes Involved in Cholesterol and Fatty Acid Biosynthesis via Activation of SREBP. Oncogene 24, 6465–6481. doi:10.1038/sj.onc.1208802
Porta, R., Borea, R., Coelho, A., Khan, S., Araújo, A., Reclusa, P., et al. (2017). FGFR a Promising Druggable Target in Cancer: Molecular Biology and New Drugs. Crit. Rev. Oncol. Hematol. 113, 256–267. doi:10.1016/j.critrevonc.2017.02.018
Pottier, C., Fresnais, M., Gilon, M., Jérusalem, G., Longuespée, R., and Sounni, N. E. (2020). Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers (Basel) 12. doi:10.3390/cancers12030731
Prior, I. A., Hood, F. E., and Hartley, J. L. (2020). The Frequency of Ras Mutations in Cancer. Cancer Res. 80, 2969–2974. doi:10.1158/0008-5472.Can-19-3682
Qian, Y., Deng, J., Geng, L., Xie, H., Jiang, G., Zhou, L., et al. (2008). TLR4 Signaling Induces B7-H1 Expression through MAPK Pathways in Bladder Cancer Cells. Cancer Invest. 26, 816–821. doi:10.1080/07357900801941852
Rahman, M. A., Salajegheh, A., Smith, R. A., and Lam, A. K. (2013). B-raf Mutation: A Key Player in Molecular Biology of Cancer. Exp. Mol. Pathol. 95, 336–342. doi:10.1016/j.yexmp.2013.10.005
Rahmani, A. H., Al Zohairy, M. A., Aly, S. M., and Khan, M. A. (2014). Curcumin: a Potential Candidate in Prevention of Cancer via Modulation of Molecular Pathways. Biomed. Res. Int. 2014, 761608. doi:10.1155/2014/761608
Rajasingh, J., Raikwar, H. P., Muthian, G., Johnson, C., and Bright, J. J. (2006). Curcumin Induces Growth-Arrest and Apoptosis in Association with the Inhibition of Constitutively Active JAK-STAT Pathway in T Cell Leukemia. Biochem. Biophys. Res. Commun. 340, 359–368. doi:10.1016/j.bbrc.2005.12.014
Rajitha, B., Nagaraju, G. P., Shaib, W. L., Alese, O. B., Snyder, J. P., Shoji, M., et al. (2017). Novel Synthetic Curcumin Analogs as Potent Antiangiogenic Agents in Colorectal Cancer. Mol. Carcinog 56, 288–299. doi:10.1002/mc.22492
Rapisarda, A., and Melillo, G. (2012). Role of the VEGF/VEGFR axis in Cancer Biology and Therapy. Adv. Cancer Res. 114, 237–267. doi:10.1016/B978-0-12-386503-8.00006-5
Rasmussen, A. A., and Cullen, K. J. (1998). Paracrine/autocrine Regulation of Breast Cancer by the Insulin-like Growth Factors. Breast Cancer Res. Treat. 47, 219–233. doi:10.1023/a:1005903000777
Rasola, A., Fassetta, M., De Bacco, F., D'Alessandro, L., Gramaglia, D., Di Renzo, M. F., et al. (2007). A Positive Feedback Loop between Hepatocyte Growth Factor Receptor and Beta-Catenin Sustains Colorectal Cancer Cell Invasive Growth. Oncogene 26, 1078–1087. doi:10.1038/sj.onc.1209859
Reddy, S., Rishi, A. K., Xu, H., Levi, E., Sarkar, F. H., and Majumdar, A. P. (2006). Mechanisms of Curcumin- and EGF-Receptor Related Protein (ERRP)-dependent Growth Inhibition of colon Cancer Cells. Nutr. Cancer 55, 185–194. doi:10.1207/s15327914nc5502_10
Resnicoff, M., Abraham, D., Yutanawiboonchai, W., Rotman, H. L., Kajstura, J., Rubin, R., et al. (1995). The Insulin-like Growth Factor I Receptor Protects Tumor Cells from Apoptosis In Vivo. Cancer Res. 55, 2463–2469.
Roskoski, R. (2019). Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors. Pharmacol. Res. 144, 19–50. doi:10.1016/j.phrs.2019.03.006
Ryan, M. R., Sohl, C. D., Luo, B., and Anderson, K. S. (2019). The FGFR1 V561M Gatekeeper Mutation Drives AZD4547 Resistance through STAT3 Activation and EMT. Mol. Cancer Res. 17, 532–543. doi:10.1158/1541-7786.Mcr-18-0429
Saha, A., Connelly, S., Jiang, J., Zhuang, S., Amador, D. T., Phan, T., et al. (2014). Akt Phosphorylation and Regulation of Transketolase Is a Nodal point for Amino Acid Control of Purine Synthesis. Mol. Cel 55, 264–276. doi:10.1016/j.molcel.2014.05.028
Sandhiutami, N. M. D., Arozal, W., Louisa, M., Rahmat, D., and Wuyung, P. E. (2020). Curcumin Nanoparticle Enhances the Anticancer Effect of Cisplatin by Inhibiting PI3K/AKT and JAK/STAT3 Pathway in Rat Ovarian Carcinoma Induced by DMBA. Front. Pharmacol. 11, 603235. doi:10.3389/fphar.2020.603235
Sawada, K., Radjabi, A. R., Shinomiya, N., Kistner, E., Kenny, H., Becker, A. R., et al. (2007). c-Met Overexpression Is a Prognostic Factor in Ovarian Cancer and an Effective Target for Inhibition of Peritoneal Dissemination and Invasion. Cancer Res. 67, 1670–1679. doi:10.1158/0008-5472.Can-06-1147
Saxena, A. R., Ilic, Z., Sripada, V., and Crawford, D. R. (2020). Lower Concentrations of Curcumin Inhibit Her2-Akt Pathway Components in Human Breast Cancer Cells, and Other Dietary Botanicals Potentiate This and Lapatinib Inhibition. Nutr. Res. 78, 93–104. doi:10.1016/j.nutres.2020.05.007
Saydmohammed, M., Joseph, D., and Syed, V. (2010). Curcumin Suppresses Constitutive Activation of STAT-3 by Up-Regulating Protein Inhibitor of Activated STAT-3 (PIAS-3) in Ovarian and Endometrial Cancer Cells. J. Cel Biochem 110, 447–456. doi:10.1002/jcb.22558
Schlessinger, J. (2000). Cell Signaling by Receptor Tyrosine Kinases. Cell 103, 211–225. doi:10.1016/s0092-8674(00)00114-8
Schlomm, T., Kirstein, P., Iwers, L., Daniel, B., Steuber, T., Walz, J., et al. (2007). Clinical Significance of Epidermal Growth Factor Receptor Protein Overexpression and Gene Copy Number Gains in Prostate Cancer. Clin. Cancer Res. 13, 6579–6584. doi:10.1158/1078-0432.Ccr-07-1257
Score, J., Curtis, C., Waghorn, K., Stalder, M., Jotterand, M., Grand, F. H., et al. (2006). Identification of a Novel Imatinib Responsive KIF5B-PDGFRA Fusion Gene Following Screening for PDGFRA Overexpression in Patients with Hypereosinophilia. Leukemia 20, 827–832. doi:10.1038/sj.leu.2404154
Ségaliny, A. I., Tellez-Gabriel, M., Heymann, M. F., and Heymann, D. (2015). Receptor Tyrosine Kinases: Characterisation, Mechanism of Action and Therapeutic Interests for Bone Cancers. J. Bone Oncol. 4, 1–12. doi:10.1016/j.jbo.2015.01.001
Setareh, J., and Jaleh, V. (2018). Co-delivery of Curcumin and Imatinib by Nanostructured Lipid Carriers in the Treatment of Lymphoma. Int. Pharm. Acta 1, 37–38. doi:10.22037/ipa.v1i1.19945
Seto, T., Higashiyama, M., Funai, H., Imamura, F., Uematsu, K., Seki, N., et al. (2006). Prognostic Value of Expression of Vascular Endothelial Growth Factor and its Flt-1 and KDR Receptors in Stage I Non-small-cell Lung Cancer. Lung Cancer 53, 91–96. doi:10.1016/j.lungcan.2006.02.009
Shakeri, A., Ward, N., Panahi, Y., and Sahebkar, A. (2019). Anti-angiogenic Activity of Curcumin in Cancer Therapy: a Narrative Review. Curr. Vasc. Pharmacol. 17, 262–269. doi:10.2174/1570161116666180209113014
Shao, M. M., Zhang, F., Meng, G., Wang, X. X., Xu, H., Yu, X. W., et al. (2011). Epidermal Growth Factor Receptor Gene Amplification and Protein Overexpression in Basal-like Carcinoma of the Breast. Histopathology 59, 264–273. doi:10.1111/j.1365-2559.2011.03921.x
Sheng, G., Zeng, Z., Pan, J., Kou, L., Wang, Q., Yao, H., et al. (2017). Multiple MYO18A-PDGFRB Fusion Transcripts in a Myeloproliferative Neoplasm Patient with T(5;17)(q32;q11). Mol. Cytogenet. 10, 4. doi:10.1186/s13039-017-0306-8
Shigematsu, H., and Gazdar, A. F. (2006). Somatic Mutations of Epidermal Growth Factor Receptor Signaling Pathway in Lung Cancers. Int. J. Cancer 118, 257–262. doi:10.1002/ijc.21496
Shiratsuchi, I., Akagi, Y., Kawahara, A., Kinugasa, T., Romeo, K., Yoshida, T., et al. (2011). Expression of IGF-1 and IGF-1R and Their Relation to Clinicopathological Factors in Colorectal Cancer. Anticancer Res. 31, 2541–2545.
Silva, S. R., Bowen, K. A., Rychahou, P. G., Jackson, L. N., Weiss, H. L., Lee, E. Y., et al. (2011). VEGFR-2 Expression in Carcinoid Cancer Cells and its Role in Tumor Growth and Metastasis. Int. J. Cancer 128, 1045–1056. doi:10.1002/ijc.25441
Simasi, J., Schubert, A., Oelkrug, C., Gillissen, A., and Nieber, K. (2014). Primary and Secondary Resistance to Tyrosine Kinase Inhibitors in Lung Cancer. Anticancer Res. 34, 2841–2850.
Singh, D., Chan, J. M., Zoppoli, P., Niola, F., Sullivan, R., Castano, A., et al. (2012). Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science 337, 1231–1235. doi:10.1126/science.1220834
Song, F., Chen, Q., Rao, W., Zhang, R., Wang, Y., Ge, H., et al. (2019). OVA66 Promotes Tumour Angiogenesis and Progression through Enhancing Autocrine VEGF-VEGFR2 Signalling. EBioMedicine 41, 156–166. doi:10.1016/j.ebiom.2019.02.051
Song, X., Liu, Z., and Yu, Z. (2020). EGFR Promotes the Development of Triple Negative Breast Cancer through JAK/STAT3 Signaling. Cancer Manag. Res. 12, 703–717. doi:10.2147/CMAR.S225376
Spannuth, W. A., Nick, A. M., Jennings, N. B., Armaiz-Pena, G. N., Mangala, L. S., Danes, C. G., et al. (2009). Functional Significance of VEGFR-2 on Ovarian Cancer Cells. Int. J. Cancer 124, 1045–1053. doi:10.1002/ijc.24028
Spirina, L. V., Usynin, Y. A., Yurmazov, Z. A., Slonimskaya, E. M., Kolegova, E. S., and Kondakova, I. V. (2017). Transcription Factors NF-kB, HIF-1, HIF-2, Growth Factor VEGF, VEGFR2 and Carboanhydrase IX mRNA and Protein Level in the Development of Kidney Cancer Metastasis. Mol. Biol. (Mosk) 51, 372–377. doi:10.7868/s0026898417020197
Squires, M. S., Hudson, E. A., Howells, L., Sale, S., Houghton, C. E., Jones, J. L., et al. (2003). Relevance of Mitogen Activated Protein Kinase (MAPK) and Phosphotidylinositol-3-Kinase/protein Kinase B (PI3K/PKB) Pathways to Induction of Apoptosis by Curcumin in Breast Cells. Biochem. Pharmacol. 65, 361–376. doi:10.1016/S0006-2952(02)01517-4
St-Germain, M. E., Gagnon, V., Mathieu, I., Parent, S., and Asselin, E. (2004). Akt Regulates COX-2 mRNA and Protein Expression in Mutated-PTEN Human Endometrial Cancer Cells. Int. J. Oncol. 24, 1311–1324. doi:10.3892/ijo.24.5.1311
Starska, K., Forma, E., Lewy-Trenda, I., Stasikowska-Kanicka, O., Skóra, M., and Bryś, M. (2018). Fibroblast Growth Factor Receptor 1 and 3 Expression Is Associated with Regulatory PI3K/AKT Kinase Activity, as Well as Invasion and Prognosis, in Human Laryngeal Cancer. Cel Oncol (Dordr) 41, 253–268. doi:10.1007/s13402-017-0367-z
Stelloo, E., Versluis, M. A., Nijman, H. W., de Bruyn, M., Plat, A., Osse, E. M., et al. (2016). Microsatellite Instability Derived JAK1 Frameshift Mutations Are Associated with Tumor Immune Evasion in Endometrioid Endometrial Cancer. Oncotarget 7, 39885–39893. doi:10.18632/oncotarget.9414
Stover, E. H., Chen, J., Folens, C., Lee, B. H., Mentens, N., Marynen, P., et al. (2006). Activation of FIP1L1-PDGFRalpha Requires Disruption of the Juxtamembrane Domain of PDGFRalpha and Is FIP1L1-independent. Proc. Natl. Acad. Sci. U S A. 103, 8078–8083. doi:10.1073/pnas.0601192103
Strumberg, D., and Schultheis, B. (2012). Regorafenib for Cancer. Expert Opin. Investig. Drugs 21, 879–889. doi:10.1517/13543784.2012.684752
Su, C., Wang, W., and Wang, C. (2018). IGF-1-induced MMP-11 Expression Promotes the Proliferation and Invasion of Gastric Cancer Cells through the JAK1/STAT3 Signaling Pathway. Oncol. Lett. 15, 7000–7006. doi:10.3892/ol.2018.8234
Su, C. L., and Wu, C. S. (2017). Synthetic Lethality of K-Ras Mutant Human Colorectal Cancer Cells by Phytochemical Curcumin and FDA-Approved Targeted Drug Regorafenib. FASEB J. 31, 630–646.
Su, X., Zhan, P., Gavine, P. R., Morgan, S., Womack, C., Ni, X., et al. (2014). FGFR2 Amplification Has Prognostic Significance in Gastric Cancer: Results from a Large International Multicentre Study. Br. J. Cancer 110, 967–975. doi:10.1038/bjc.2013.802
Sun, X. D., Liu, X. E., and Huang, D. S. (2012). Curcumin Induces Apoptosis of Triple-Negative Breast Cancer Cells by Inhibition of EGFR Expression. Mol. Med. Rep. 6, 1267–1270. doi:10.3892/mmr.2012.1103
Sun, Y., Liu, L., Wang, Y., He, A., Hu, H., Zhang, J., et al. (2019). Curcumin Inhibits the Proliferation and Invasion of MG-63 Cells through Inactivation of the P-JAK2/p-STAT3 Pathway. Onco Targets Ther. 12, 2011–2021. doi:10.2147/OTT.S172909
Tang, L., Yang, J., Chen, J., Yu, J., Zhou, Q., Lu, X., et al. (2017). IGF-1R Promotes the Expression of Cyclin D1 Protein and Accelerates the G1/S Transition by Activating Ras/Raf/MEK/ERK Signaling Pathway. Int. J. Clin. Exp. Pathol. 10, 11652–11658.
Tanno, S., Ohsaki, Y., Nakanishi, K., Toyoshima, E., and Kikuchi, K. (2004). Human Small Cell Lung Cancer Cells Express Functional VEGF Receptors, VEGFR-2 and VEGFR-3. Lung Cancer 46, 11–19. doi:10.1016/j.lungcan.2004.03.006
Teiten, M. H., Eifes, S., Reuter, S., Duvoix, A., Dicato, M., and Diederich, M. (2009). Gene Expression Profiling Related to Anti-inflammatory Properties of Curcumin in K562 Leukemia Cells. Ann. N. Y Acad. Sci. 1171, 391–398. doi:10.1111/j.1749-6632.2009.04890.x
Teiten, M. H., Gaascht, F., Cronauer, M., Henry, E., Dicato, M., and Diederich, M. (2011). Anti-proliferative Potential of Curcumin in Androgen-dependent Prostate Cancer Cells Occurs through Modulation of the Wingless Signaling Pathway. Int. J. Oncol. 38, 603–611. doi:10.3892/ijo.2011.905
Ter Braak, B., Siezen, C. L., Lee, J. S., Rao, P., Voorhoeve, C., Ruppin, E., et al. (2017). Insulin-like Growth Factor 1 Receptor Activation Promotes Mammary Gland Tumor Development by Increasing Glycolysis and Promoting Biomass Production. Breast Cancer Res. 19, 14. doi:10.1186/s13058-017-0802-0
Terai, H., Soejima, K., Yasuda, H., Nakayama, S., Hamamoto, J., Arai, D., et al. (2013). Activation of the FGF2-FGFR1 Autocrine Pathway: a Novel Mechanism of Acquired Resistance to Gefitinib in NSCLC. Mol. Cancer Res. 11, 759–767. doi:10.1158/1541-7786.Mcr-12-0652
Tian, B., Zhao, Y., Liang, T., Ye, X., Li, Z., Yan, D., et al. (2017). Curcumin Inhibits Urothelial Tumor Development by Suppressing IGF2 and IGF2-Mediated PI3K/AKT/mTOR Signaling Pathway. J. Drug Target. 25, 626–636. doi:10.1080/1061186x.2017.1306535
Tian, X., Traub, B., Shi, J., Huber, N., Schreiner, S., Chen, G., et al. (2021). c-Jun N-Terminal Kinase 2 Suppresses Pancreatic Cancer Growth and Invasion and Is Opposed by C-Jun N-Terminal Kinase 1. Cancer Gene Ther. 1–14. doi:10.1038/s41417-020-00290-5
Tilborghs, S., Corthouts, J., Verhoeven, Y., Arias, D., Rolfo, C., Trinh, X. B., et al. (2017). The Role of Nuclear Factor-Kappa B Signaling in Human Cervical Cancer. Crit. Rev. Oncol. Hematol. 120, 141–150. doi:10.1016/j.critrevonc.2017.11.001
Till, J. H., Becerra, M., Watty, A., Lu, Y., Ma, Y., Neubert, T. A., et al. (2002). Crystal Structure of the MuSK Tyrosine Kinase: Insights into Receptor Autoregulation. Structure 10, 1187–1196. doi:10.1016/s0969-2126(02)00814-6
Tsao, A. S., Wei, W., Kuhn, E., Spencer, L., Solis, L. M., Suraokar, M., et al. (2011). Immunohistochemical Overexpression of Platelet-Derived Growth Factor Receptor-Beta (PDGFR-β) Is Associated with PDGFRB Gene Copy Number Gain in Sarcomatoid Non-small-cell Lung Cancer. Clin. Lung Cancer 12, 369–374. doi:10.1016/j.cllc.2011.02.002
Tung, C. L., Jian, Y. J., Chen, J. C., Wang, T. J., Chen, W. C., Zheng, H. Y., et al. (2016). Curcumin Downregulates P38 MAPK-dependent X-ray Repair Cross-Complement Group 1 (XRCC1) Expression to Enhance Cisplatin-Induced Cytotoxicity in Human Lung Cancer Cells. Naunyn Schmiedebergs Arch. Pharmacol. 389, 657–666. doi:10.1007/s00210-016-1235-5
Tung, Y. T., Chen, H. L., Lai, C. W., Shen, C. J., Lai, Y. W., and Chen, C. M. (2011). Curcumin Reduces Pulmonary Tumorigenesis in Vascular Endothelial Growth Factor (VEGF)-overexpressing Transgenic Mice. Mol. Nutr. Food Res. 55, 1036–1043. doi:10.1002/mnfr.201000654
Turner, N., Pearson, A., Sharpe, R., Lambros, M., Geyer, F., Lopez-Garcia, M. A., et al. (2010). FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res. 70, 2085–2094. doi:10.1158/0008-5472.CAN-09-3746
Vadhan-Raj, S., Weber, D. M., Wang, M., Giralt, S. A., Thomas, S. K., Alexanian, R., et al. (2007). Curcumin Downregulates NF-kB and Related Genes in Patients with Multiple Myeloma: Results of a Phase I/II Study. Blood 110, 1177. doi:10.1182/blood.V110.11.1177.1177
Valentinis, B., and Baserga, R. (1996). The IGF-I Receptor Protects Tumor Cells from Apoptosis Induced by High Concentrations of Serum. Biochem. Biophys. Res. Commun. 224, 362–368. doi:10.1006/bbrc.1996.1034
Van Erk, M. J., Teuling, E., Staal, Y. C., Huybers, S., Van Bladeren, P. J., Aarts, J. M., et al. (2004). Time- and Dose-dependent Effects of Curcumin on Gene Expression in Human colon Cancer Cells. J. Carcinog 3, 8. doi:10.1186/1477-3163-3-8
Van Trappen, P. O., Steele, D., Lowe, D. G., Baithun, S., Beasley, N., Thiele, W., et al. (2003). Expression of Vascular Endothelial Growth Factor (VEGF)-C and VEGF-D, and Their Receptor VEGFR-3, during Different Stages of Cervical Carcinogenesis. J. Pathol. 201, 544–554. doi:10.1002/path.1467
Vandermark, E. R., Deluca, K. A., Gardner, C. R., Marker, D. F., Schreiner, C. N., Strickland, D. A., et al. (2012). Human Papillomavirus Type 16 E6 and E 7 Proteins Alter NF-kB in Cultured Cervical Epithelial Cells and Inhibition of NF-kB Promotes Cell Growth and Immortalization. Virology 425, 53–60. doi:10.1016/j.virol.2011.12.023
Varshosaz, J., Jandaghian, S., Mirian, M., and Sajjadi, S. E. (2021). Co-delivery of Rituximab Targeted Curcumin and Imatinib Nanostructured Lipid Carriers in Non-hodgkin Lymphoma Cells. J. Liposome Res. 31, 64–78. doi:10.1080/08982104.2020.1720718
Velghe, A. I., Van Cauwenberghe, S., Polyansky, A. A., Chand, D., Montano-Almendras, C. P., Charni, S., et al. (2014). PDGFRA Alterations in Cancer: Characterization of a Gain-Of-Function V536E Transmembrane Mutant as Well as Loss-Of-Function and Passenger Mutations. Oncogene 33, 2568–2576. doi:10.1038/onc.2013.218
Wagh, P. K., Peace, B. E., and Waltz, S. E. (2008). Met-related Receptor Tyrosine Kinase Ron in Tumor Growth and Metastasis. Adv. Cancer Res. 100, 1–33. doi:10.1016/S0065-230X(08)00001-8
Walters, D. K., Mercher, T., Gu, T. L., O'Hare, T., Tyner, J. W., Loriaux, M., et al. (2006). Activating Alleles of JAK3 in Acute Megakaryoblastic Leukemia. Cancer Cell 10, 65–75. doi:10.1016/j.ccr.2006.06.002
Walz, C., Curtis, C., Schnittger, S., Schultheis, B., Metzgeroth, G., Schoch, C., et al. (2006). Transient Response to Imatinib in a Chronic Eosinophilic Leukemia Associated with Ins(9;4)(q33;q12q25) and a CDK5RAP2-PDGFRA Fusion Gene. Genes Chromosomes Cancer 45, 950–956. doi:10.1002/gcc.20359
Wang, C. Y., Mayo, M. W., Korneluk, R. G., Goeddel, D. V., and Baldwin, A. S. (1998). NF-kappaB Antiapoptosis: Induction of TRAF1 and TRAF2 and C-IAP1 and C-IAP2 to Suppress Caspase-8 Activation. Science 281, 1680–1683. doi:10.1126/science.281.5383.1680
Wang, J., Gui, Z., Deng, L., Sun, M., Guo, R., Zhang, W., et al. (2012). c-Met Upregulates Aquaporin 3 Expression in Human Gastric Carcinoma Cells via the ERK Signalling Pathway. Cancer Lett. 319, 109–117. doi:10.1016/j.canlet.2011.12.040
Wang, J., Li, X., Xue, X., Ou, Q., Wu, X., Liang, Y., et al. (2019). Clinical Outcomes of EGFR Kinase Domain Duplication to Targeted Therapies in NSCLC. Int. J. Cancer 144, 2677–2682. doi:10.1002/ijc.31895
Wang, R., Wang, L., Li, Y., Hu, H., Shen, L., Shen, X., et al. (2014). FGFR1/3 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Non-small Cell Lung Cancer. Clin. Cancer Res. 20, 4107–4114. doi:10.1158/1078-0432.Ccr-14-0284
Wang, W., Wu, M., Liu, M., Yan, Z., Wang, G., Mao, D., et al. (2020). Hyperprogression to Camrelizumab in a Patient with Esophageal Squamous Cell Carcinoma Harboring EGFR Kinase Domain Duplication. J. Immunother. Cancer 8, e000793. doi:10.1136/jitc-2020-000793
Wang, W. Z., Li, L., Liu, M. Y., Jin, X. B., Mao, J. W., Pu, Q. H., et al. (2013). Curcumin Induces FasL-Related Apoptosis through P38 Activation in Human Hepatocellular Carcinoma Huh7 Cells. Life Sci. 92, 352–358. doi:10.1016/j.lfs.2013.01.013
Wang, Z., Longo, P. A., Tarrant, M. K., Kim, K., Head, S., Leahy, D. J., et al. (2011). Mechanistic Insights into the Activation of Oncogenic Forms of EGF Receptor. Nat. Struct. Mol. Biol. 18, 1388–1393. doi:10.1038/nsmb.2168
Watson, J. L., Greenshields, A., Hill, R., Hilchie, A., Lee, P. W., Giacomantonio, C. A., et al. (2010). Curcumin-induced Apoptosis in Ovarian Carcinoma Cells Is P53-independent and Involves P38 Mitogen-Activated Protein Kinase Activation and Downregulation of Bcl-2 and Survivin Expression and Akt Signaling. Mol. Carcinog 49, 13–24. doi:10.1002/mc.20571
Weigand, M., Hantel, P., Kreienberg, R., and Waltenberger, J. (2005). Autocrine Vascular Endothelial Growth Factor Signalling in Breast Cancer. Evidence from Cell Lines and Primary Breast Cancer Cultures In Vitro. Angiogenesis 8, 197–204. doi:10.1007/s10456-005-9010-0
Wheeler, D. L., and Yarden, Y. (2015). Receptor Tyrosine Kinases: Family and Subfamilies. New York, United States: Springer.
Wieman, H. L., Wofford, J. A., and Rathmell, J. C. (2007). Cytokine Stimulation Promotes Glucose Uptake via Phosphatidylinositol-3 kinase/Akt Regulation of Glut1 Activity and Trafficking. Mol. Biol. Cel 18, 1437–1446. doi:10.1091/mbc.e06-07-0593
Wilhelm, S. M., Carter, C., Tang, L., Wilkie, D., McNabola, A., Rong, H., et al. (2004). BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 64, 7099–7109. doi:10.1158/0008-5472.Can-04-1443
Wu, C. S., Wu, S. Y., Chen, H. C., Chu, C. A., Tang, H. H., Liu, H. S., et al. (2019a). Curcumin Functions as a MEK Inhibitor to Induce a Synthetic Lethal Effect on KRAS Mutant Colorectal Cancer Cells Receiving Targeted Drug Regorafenib. J. Nutr. Biochem. 74, 108227. doi:10.1016/j.jnutbio.2019.108227
Wu, Q., Wu, W., Fu, B., Shi, L., Wang, X., and Kuca, K. (2019b). JNK Signaling in Cancer Cell Survival. Med. Res. Rev. 39, 2082–2104. doi:10.1002/med.21574
Wu, W., O'Reilly, M. S., Langley, R. R., Tsan, R. Z., Baker, C. H., Bekele, N., et al. (2007). Expression of Epidermal Growth Factor (EGF)/transforming Growth Factor-Alpha by Human Lung Cancer Cells Determines Their Response to EGF Receptor Tyrosine Kinase Inhibition in the Lungs of Mice. Mol. Cancer Ther. 6, 2652–2663. doi:10.1158/1535-7163.Mct-06-0759
Wu, Y., Hooper, A. T., Zhong, Z., Witte, L., Bohlen, P., Rafii, S., et al. (2006). The Vascular Endothelial Growth Factor Receptor (VEGFR-1) Supports Growth and Survival of Human Breast Carcinoma. Int. J. Cancer 119, 1519–1529. doi:10.1002/ijc.21865
Wu, Y. M., Su, F., Kalyana-Sundaram, S., Khazanov, N., Ateeq, B., Cao, X., et al. (2013). Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 3, 636–647. doi:10.1158/2159-8290.Cd-13-0050
Xia, Y., Shen, S., and Verma, I. M. (2014). NF-κB, an Active Player in Human Cancers. Cancer Immunol. Res. 2, 823–830. doi:10.1158/2326-6066.Cir-14-0112
Xie, L., Su, X., Zhang, L., Yin, X., Tang, L., Zhang, X., et al. (2013). FGFR2 Gene Amplification in Gastric Cancer Predicts Sensitivity to the Selective FGFR Inhibitor AZD4547. Clin. Cancer Res. 19, 2572–2583. doi:10.1158/1078-0432.Ccr-12-3898
Xie, Q., Liu, K. D., Hu, M. Y., and Zhou, K. (2001). SF/HGF-c-Met Autocrine and Paracrine Promote Metastasis of Hepatocellular Carcinoma. World J. Gastroenterol. 7, 816–820. doi:10.3748/wjg.v7.i6.816
Xin, J., Jue, W., Huifen, S., Ran, R., Kai, X., Xiangming, T., et al. (2017). Curcumin Co-treatment Ameliorates Resistance to Gefitinib in Drug-Resistant NCI-H1975 Lung Cancer Cells. J. Traditional Chin. Med. 37, 355–360. doi:10.1016/S0254-6272(17)30071-7
Xu, H., and Shao, C. (2020). KIF5B-EGFR Fusion: A Novel EGFR Mutation in Lung Adenocarcinoma. Onco Targets Ther. 13, 8317–8321. doi:10.2147/OTT.S263994
Xu, X., Lu, Q., Wang, Z., Cai, P., Zeng, Z., Zhang, L., et al. (2021). Identification of a Novel CSNK2A1-PDGFRB Fusion Gene in a Patient with Myeloid Neoplasm with Eosinophilia. Cancer Res. Treat. 53, 889–892. doi:10.4143/crt.2020.1272
Xu, Y., Jin, J., Xu, J., Shao, Y. W., and Fan, Y. (2017). JAK2 Variations and Functions in Lung Adenocarcinoma. Tumour Biol. 39, 1010428317711140. doi:10.1177/1010428317711140
Yamauchi, Y., Izumi, Y., Yamamoto, J., and Nomori, H. (2014). Coadministration of Erlotinib and Curcumin Augmentatively Reduces Cell Viability in Lung Cancer Cells. Phytother Res. 28, 728–735. doi:10.1002/ptr.5056
Yan, J. D., Liu, Y., Zhang, Z. Y., Liu, G. Y., Xu, J. H., Liu, L. Y., et al. (2015). Expression and Prognostic Significance of VEGFR-2 in Breast Cancer. Pathol. Res. Pract. 211, 539–543. doi:10.1016/j.prp.2015.04.003
Yang, C. L., Liu, Y. Y., Ma, Y. G., Xue, Y. X., Liu, D. G., Ren, Y., et al. (2012). Curcumin Blocks Small Cell Lung Cancer Cells Migration, Invasion, Angiogenesis, Cell Cycle and Neoplasia through Janus Kinase-STAT3 Signalling Pathway. PLoS One 7, e37960. doi:10.1371/journal.pone.0037960
Yigit, N., Wu, W. W., Subramaniyam, S., Mathew, S., and Geyer, J. T. (2015). BCR-PDGFRA Fusion in a T Lymphoblastic Leukemia/lymphoma. Cancer Genet. 208, 404–407. doi:10.1016/j.cancergen.2015.04.007
Yonemura, Y., Fushida, S., Bando, E., Kinoshita, K., Miwa, K., Endo, Y., et al. (2001). Lymphangiogenesis and the Vascular Endothelial Growth Factor Receptor (VEGFR)-3 in Gastric Cancer. Eur. J. Cancer 37, 918–923. doi:10.1016/S0959-8049(01)00015-6
Yoshida, M., Tamagawa, N., Nakao, T., Kanashima, H., Ueda, H., Murakami, A., et al. (2015). Imatinib Non-responsive Chronic Eosinophilic Leukemia with ETV6-PDGFRA Fusion Gene. Leuk. Lymphoma 56, 768–769. doi:10.3109/10428194.2014.938330
Yu, S., Zhang, Y., Pan, Y., Cheng, C., Sun, Y., and Chen, H. (2017). The Non-small Cell Lung Cancer EGFR Extracellular Domain Mutation, M277E, Is Oncogenic and Drug-Sensitive. Onco Targets Ther. 10, 4507–4515. doi:10.2147/OTT.S131999
Yuan, L., Liu, Z. H., Lin, Z. R., Xu, L. H., Zhong, Q., and Zeng, M. S. (2014). Recurrent FGFR3-TACC3 Fusion Gene in Nasopharyngeal Carcinoma. Cancer Biol. Ther. 15, 1613–1621. doi:10.4161/15384047.2014.961874
Zhang, H., Bajraszewski, N., Wu, E., Wang, H., Moseman, A. P., Dabora, S. L., et al. (2007). PDGFRs Are Critical for PI3K/Akt Activation and Negatively Regulated by mTOR. J. Clin. Invest. 117, 730–738. doi:10.1172/jci28984
Zhang, H. F., and Lai, R. (2014). STAT3 in Cancer-Friend or Foe? Cancers (Basel) 6, 1408–1440. doi:10.3390/cancers6031408
Zhang, H. H., Zhang, Y., Cheng, Y. N., Gong, F. L., Cao, Z. Q., Yu, L. G., et al. (2018). Metformin Incombination with Curcumin Inhibits the Growth, Metastasis, and Angiogenesis of Hepatocellular Carcinoma In Vitro and In Vivo. Mol. Carcinog 57, 44–56. doi:10.1002/mc.22718
Zhang, J., Yu, J., Xie, R., Chen, W., and Lv, Y. (2016). Combinatorial Anticancer Effects of Curcumin and Sorafenib towards Thyroid Cancer Cells via PI3K/Akt and ERK Pathways. Nat. Prod. Res. 30, 1858–1861. doi:10.1080/14786419.2015.1074229
Zhang, L., Tao, X., Fu, Q., Ge, C., Li, R., Li, Z., et al. (2019). Curcumin Inhibits Cell Proliferation and Migration in NSCLC through a Synergistic Effect on the TLR4/MyD88 and EGFR Pathways. Oncol. Rep. 42, 1843–1855. doi:10.3892/or.2019.7278
Zhang, W., and Liu, H. T. (2002). MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res 12, 9–18. doi:10.1038/sj.cr.7290105
Zhang, X., Hu, B., Sun, Y. F., Huang, X. W., Cheng, J. W., Huang, A., et al. (2021). Arsenic Trioxide Induces Differentiation of Cancer Stem Cells in Hepatocellular Carcinoma through Inhibition of LIF/JAK1/STAT3 and NF-kB Signaling Pathways Synergistically. Clin. Transl Med. 11, e335. doi:10.1002/ctm2.335
Zhao, D., Pan, C., Sun, J., Gilbert, C., Drews-Elger, K., Azzam, D. J., et al. (2015a). VEGF Drives Cancer-Initiating Stem Cells through VEGFR-2/Stat3 Signaling to Upregulate Myc and Sox2. Oncogene 34, 3107–3119. doi:10.1038/onc.2014.257
Zhao, L., and Vogt, P. K. (2008). Class I PI3K in Oncogenic Cellular Transformation. Oncogene 27, 5486–5496. doi:10.1038/onc.2008.244
Zhao, W., Wang, Y., Wang, Y., Gao, N., Han, Z., and Yu, H. (2015b). Potential Anti-cancer Effect of Curcumin in Human Lung Squamous Cell Carcinoma. Thorac. Cancer 6, 508–516. doi:10.1111/1759-7714.12222
Zhao, X., Qu, J., Hui, Y., Zhang, H., Sun, Y., Liu, X., et al. (2017). Clinicopathological and Prognostic Significance of C-Met Overexpression in Breast Cancer. Oncotarget 8, 56758–56767. doi:10.18632/oncotarget.18142
Zhao, Y., Collier, J. J., Huang, E.-C., and Whelan, J. (2014). Turmeric and Chinese Goldthread Synergistically Inhibit Prostate Cancer Cell Proliferation and NF-kB Signaling. Ffhd 4, 312–339. doi:10.31989/ffhd.v4i7.1
Zhen, L., Fan, D., Yi, X., Cao, X., Chen, D., and Wang, L. (2014). Curcumin Inhibits Oral Squamous Cell Carcinoma Proliferation and Invasion via EGFR Signaling Pathways. Int. J. Clin. Exp. Pathol. 7, 6438–6446.
Zheng, B. Z., Liu, T. D., Chen, G., Zhang, J. X., and Kang, X. (2018). The Effect of Curcumin on Cell Adhesion of Human Esophageal Cancer Cell. Eur. Rev. Med. Pharmacol. Sci. 22, 551–560. doi:10.26355/eurrev_201801_14209
Zheng, X., Lu, G., Yao, Y., and Gu, W. (2019). An Autocrine IL-6/IGF-1R Loop Mediates EMT and Promotes Tumor Growth in Non-small Cell Lung Cancer. Int. J. Biol. Sci. 15, 1882–1891. doi:10.7150/ijbs.31999
Zhou, H., Beevers, C. S., and Huang, S. (2011). The Targets of Curcumin. Curr. Drug Targets 12, 332–347. doi:10.2174/138945011794815356
Zhou, Y., Zheng, S., Lin, J., Zhang, Q. J., and Chen, A. (2007). The Interruption of the PDGF and EGF Signaling Pathways by Curcumin Stimulates Gene Expression of PPARgamma in Rat Activated Hepatic Stellate Cell In Vitro. Lab. Invest. 87, 488–498. doi:10.1038/labinvest.3700532
Zhu, Y. C., Wang, W. X., Li, X. L., Xu, C. W., Chen, G., Zhuang, W., et al. (2019). Identification of a Novel Icotinib-Sensitive EGFR-SEPTIN14 Fusion Variant in Lung Adenocarcinoma by Next-Generation Sequencing. J. Thorac. Oncol. 14, e181–e183. doi:10.1016/j.jtho.2019.03.031
Zhu, Y. C., Wang, W. X., Song, Z. B., Zhang, Q. X., Xu, C. W., Chen, G., et al. (2018a). MET-UBE2H Fusion as a Novel Mechanism of Acquired EGFR Resistance in Lung Adenocarcinoma. J. Thorac. Oncol. 13, e202–e204. doi:10.1016/j.jtho.2018.05.009
Zhu, Y. C., Wang, W. X., Xu, C. W., Zhang, Q. X., Du, K. Q., Chen, G., et al. (2018b). Identification of a Novel Crizotinib-Sensitive MET-Atxn7l1 Gene Fusion Variant in Lung Adenocarcinoma by Next Generation Sequencing. Ann. Oncol. 29, 2392–2393. doi:10.1093/annonc/mdy455
Keywords: curcumin, receptor tyrosine kinase, signaling pathway, polyphenol, combination therapy, tyrosine kinase inhibitor
Citation: Sudhesh Dev S, Zainal Abidin SA, Farghadani R, Othman I and Naidu R (2021) Receptor Tyrosine Kinases and Their Signaling Pathways as Therapeutic Targets of Curcumin in Cancer. Front. Pharmacol. 12:772510. doi: 10.3389/fphar.2021.772510
Received: 08 September 2021; Accepted: 01 November 2021;
Published: 15 November 2021.
Edited by:
Maria Teresa Esposito, University of Roehampton London, United KingdomReviewed by:
Bharat B. Aggarwal, University of Texas MD Anderson Cancer Center, United StatesAntonio Giordano, Sbarro Health Research Organization (SHRO), United States
Copyright © 2021 Sudhesh Dev, Zainal Abidin, Farghadani, Othman and Naidu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rakesh Naidu, a2RyYWtlc2huYUBob3RtYWlsLmNvbQ==