 Abraham Tettey
Abraham Tettey Yujie Jiang1
Yujie Jiang1 Ying Li
Ying Li- 1Department of Pharmacology, School of Pharmaceutical Science, Central South University, Changsha, China
- 2Department of Health Management, The Third Xiangya Hospital, Central South University, Changsha, China
- 3Hunan Key Laboratory for Bioanalysis of Complex Matrix Samples, Changsha, China
Pulmonary arterial hypertension (PAH) is a severe disease with a resultant increase of the mean pulmonary arterial pressure, right ventricular hypertrophy and eventual death. Research in recent years has produced various therapeutic options for its clinical management but the high mortality even under treatment remains a big challenge attributed to the complex pathophysiology. Studies from clinical and non-clinical experiments have revealed that the nitric oxide (NO) pathway is one of the key pathways underlying the pathophysiology of PAH. Many of the essential drugs used in the management of PAH act on this pathway highlighting its significant role in PAH. Meanwhile, several novel compounds targeting on NO pathway exhibits great potential to become future therapy medications. Furthermore, the NO pathway is found to interact with other crucial pathways. Understanding such interactions could be helpful in the discovery of new drug that provide better clinical outcomes.
Introduction
Pulmonary arterial hypertension (PAH) is a fatal disease characterized by an increase in pulmonary arterial pressure with subsequent right ventricular failure and death (Rosenkranz, 2015). Generally, the major pathological changes of PAH are vasoconstriction and vascular remodelling of the small pulmonary arteries (Tuder et al., 2009) involving the thickening of the intima-media, smooth muscle cell proliferation, endothelial cell proliferative lesions formation, vascular inflammation and immune dysregulation (Tuder et al., 2007; Price et al., 2012; Humbert et al., 2019). There are different categories in PAH including idiopathic PAH, heritable PAH, drug or toxin-induced PAH and PAH associated with connective tissue disease, human immunodeficiency virus infection, portal hypertension, congenital heart disease and schistosomiasis (Galiè et al., 2016). Idiopathic PAH is the most common type of PAH in western countries with congenital heart disease-related PAH being the most prevalent in Asia (Humbert et al., 2006; Lim et al., 2019).
Currently approved PAH drugs such as prostacyclin analogues, endothelin receptor antagonists and phosphodiesterase-5 (PDE-5) inhibitors aim to control pulmonary vascular tone (Olsson and Hoeper, 2009). Enormous progress has been made in the therapeutic practice of PAH in the past decades but there is still a long way to go because none of these present drugs are curative. They reduce morbidity and only slightly improve survival with no effect on PAH mortality rate.
Many molecular pathways have been linked to the development and treatment of PAH.It has been revealed that the nitric oxide (NO) pathway plays a very essential and central role in regulating vascular tone in the pulmonary circulatory system by interacting with other crucial signaling pathways. Phosphodiesterase-5 (PDE-5) inhibitors and soluble guanylate cyclase stimulators are the current drug classes acting on this pathway. Meanwhile, novel compounds targeting on NO pathway exhibits great potential in development of next generation therapy medications. This review seeks to summarize the research progress of PAH regarding to NO pathway, discuss the critical role of NO pathway in understanding the pathophysiology of PAH and new drug development.
Current Clinical Therapy in Pulmonary Arterial Hypertension
Current PAH therapy focuses on three main pathways. These are the prostacyclin, nitric oxide, and endothelin pathways. Drugs acting on these pathways are summarized in Table 1.
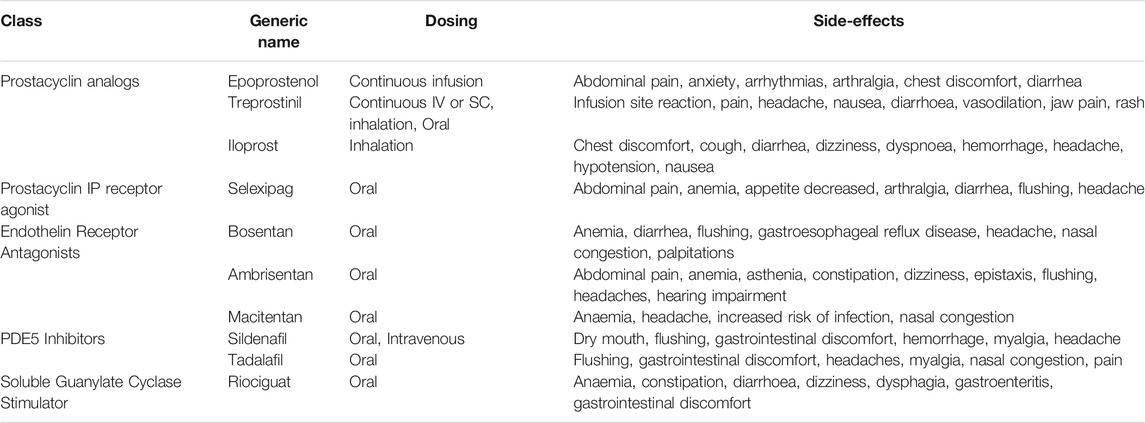
TABLE 1. Classification of drugs used in treating PAH.
Prostanoids
Prostacyclin or prostanoid is produced in the endothelium and it is a potent vasodilator in the pulmonary vasculature (Gryglewski, 2008). Prostacyclin activate the prostacyclin receptor which leads to an increase in cyclic adenosine monophosphate (cAMP) production and consequently, vasodilatation (Coleman et al., 1994). Activating the IP receptor also leads to antithrombotic and antiproliferative effects in the pulmonary vasculature (Wharton et al., 2000; Vane and Corin, 2003). Examples of prostacyclins used in PAH management include epoprostenol and treprostinil. Selexipag is a non-prostanoid IP receptor agonist. Epoprostenol is the only drug shown to improve mortality (Barst et al., 2009) and remains the drug of choice in severe cases (Galiè et al., 2016). It is administered by continuous infusion. The different prostanoids have different routes of administration with varying degrees of efficacy.
Endothelin-1 Receptor Blockers
Endothelin-1 is a potent vasoconstrictor produced by the endothelial cells and it facilitates pulmonary artery smooth muscle cell proliferation (Yanagisawa et al., 1988; Davie et al., 2002). Endothelin-1 binds to two main receptors namely endothelin receptor A (ETA) and endothelin receptor B (ETB). ETA is predominant in vascular smooth muscle cells (VSMC) and facilitates contraction and proliferation of VSMCs in PAH (Rubin et al., 2011). ETB is predominantly expressed in vascular endothelial cells where it enhances vasodilation via the production of prostacyclin and nitric oxide (NO) as well as clearance of ET-1 (Eguchi et al., 1993; Hirata et al., 1993; Seo et al., 1994). Interestingly, ETB is also found in VSMCs where it possesses vasoconstrictive and proliferative properties (Seo et al., 1994). Endothelin-1 receptor blockers prevent endothelin-1 from binding to its receptors thereby abrogating its destructive effects in PAH. Endothelin-1 receptor blockers can be grouped as selective (eg., ambrisentan) and non-selective (eg., Bosentan and macitentan) depending on their endothelin-1 receptor binding properties (Correale et al., 2013). Bosentan is the first orally administered PAH drug, notably it causes abnormal liver function in some patients necessitating monthly liver function tests (Humbert et al., 2007). Ambrisentan and macitentan have lower chances of causing liver damage (Galiè et al., 2005a; Pulido et al., 2013).
Drugs Acting on the Nitric Oxide Pathway
Nitric oxide (NO) is produced by the endothelial cells and serves as a potent vasodilator of the pulmonary circulation through cyclic guanosine monophosphate (cGMP). Phosphodiesterase-5 (PDE5) inhibitors such as sildenafil and tadalafil and the soluble guanylate cyclase stimulator, riociguat act on this pathway. Further explanation of drugs acting on this pathway is in section 4 of this review.
Nitric Oxide Pathway and Its Implication in Pulmonary Arterial Hypertension
Nitric oxide (NO) is a biological molecule that regulates many physiological and pathological processes in the body. It was proposed that the release of a vasodilating factor by the endothelial cells as one of the mechanisms behind acetylcholine-induced vasodilation in vivo (Furchgott and Zarwodski, 1980). The identity of nitric oxide as the endothelial-derived relaxing factor was not known until 1987 when two different studies confirmed it (Ignarro et al., 1987; Palmer et al., 1987).
Nitric oxide is formed from the oxidation of L-arginine to form citrulline and NO in the presence of nitric oxide synthases (NOS) and molecular oxygen (Moncada and Higgs, 1993) as shown in Figure 1. Arginase competes with eNOS for L-arginine by converting L-arginine to urea and L-ornithine (Luiking et al., 2012). NG, NG-dimethyl-L-arginine (ADMA) is known to inhibit this process by competing with L-arginine for eNOS (Zakrzewicz and Eickelberg, 2009). NG, NG-dimethylarginine dimethylaminohydrolases (DDAH1 and DDAH2) are responsible for the degradation of ADMA (Tain and Hsu, 2017). Three isoforms of nitric oxide synthases have been described in mammals. These are neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3) (Nathan and Xie, 1994).
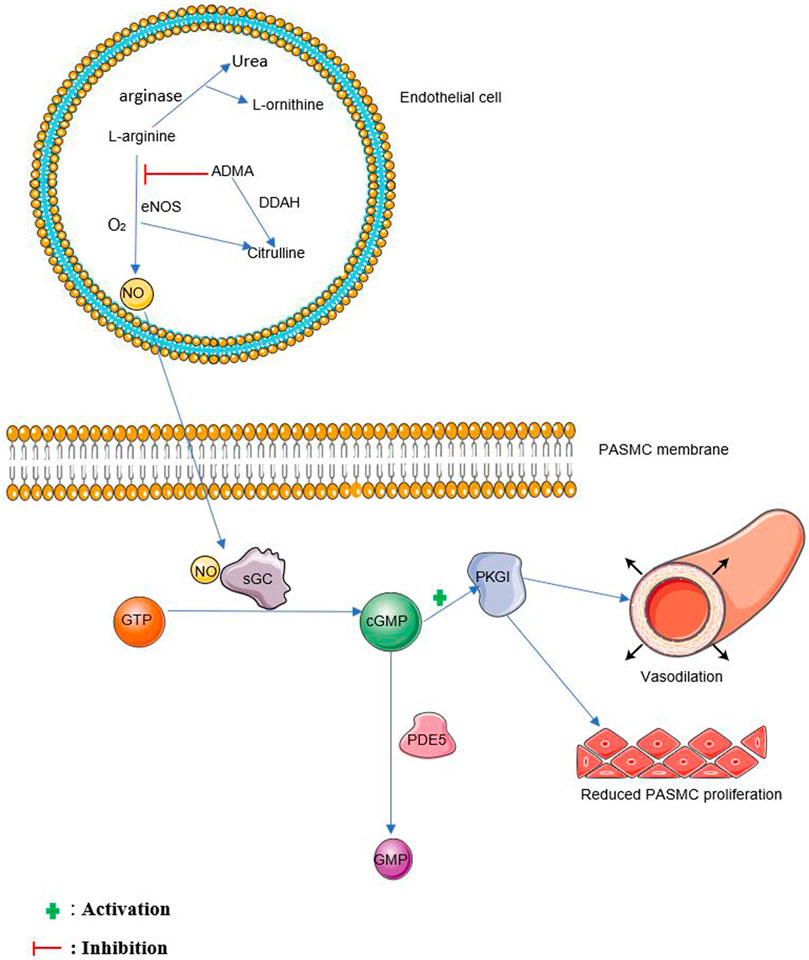
FIGURE 1. The NO pathway. NO, nitric oxide, sGC, soluble guanylate cyclase, GTP, guanosine triphosphate, cGMP, cyclic guanosine monophosphate, PDE5, phosphodiesterase 5, GMP, guanosine monophosphate, PKGI, protein kinase G I. eNOS, endothelial nitric oxide synthase, O2, oxygen, ADMA, Asymmetric dimethylarginine, DDAH, dimethylarginine dimethylaminohydrolase, PASMC, pulmonary arterial smooth muscle cell.
Endothelium-derived NO diffuses into vascular smooth muscle cells and stimulates soluble guanylate cyclase (sGC) to produce cGMP, further activating associated protein kinases such as protein kinase G I (PKGI) which causes vasorelaxation (Sausbier et al., 2000; Surks, 2007). cGMP is broken down mainly by phosphodiesterase 5 (PDE5) (Francis et al., 2000). See Figure 1 for more details.
Conflicting levels of exhaled NO have been reported in PAH patients. Data from studies with patients suffering from idiopathic PAH and scleroderma-/drug-associated PAH indicated a reduction in exhaled NO as compared to healthy patients (Kharitonov et al., 1997; Kaneko et al., 1998; Archer et al., 2012). Other studies also found no significant change in exhaled NO levels in idiopathic PAH and scleroderma-associated PAH when compared to healthy subjects (Riley et al., 1997; Olivieri et al., 2006; Malekmohammad et al., 2019). There was no difference in exhaled NO after 3 months of treatment with endothelin receptor antagonists, guanylate cyclase stimulants, phosphodiesterase type 5 (PDE5) inhibitors and prostanoids in comparison with healthy subjects. However, other diagnostic markers such as 6MWD and N-terminal prohormone of brain natriuretic peptide (NT-proBNP) were significantly correlated to disease severity and treatment response (Malekmohammad et al., 2019). Confounding factors such as exhalation flow rate, measurement technique, the NO analyzer used, nasal NO contamination, age, height and smoking (Borrill et al., 2006; Dweik et al., 2012) makes exhaled NO an unreliable marker that needs standardization to be of diagnostic value in PAH.
The use of NO Plasma metabolites (NOX) as a biomarker and prognostic indicator of PAH is being studied. A study found that patients (age range: 20–57 years) with IPAH had reduced levels of plasma NOx, which correlated inversely with mPAP and patient survival (Zhang et al., 2016). Contradictory studies found elevated levels of NOx in IPAH and congenital heart disease-associated PAH patients with age ranges of 31–77 and 5 days -12 years (Ikemoto et al., 2002; Malinovschi et al., 2011). The patients in the extreme age groups (age averages of 6 and 54 years) seem to show increased levels of NOx. The different results from the various studies could be attributed to the PAH type and ages of the patients studied (Zhang et al., 2016).
Reduced eNOS levels have been reported in PAH patients (Giaid and Saleh, 1995). Another study found an increased expression of eNOS in the plexiform lesions of PAH patients (Berger et al., 2012). The eNOS that is elevated in some PAH patients is likely to be in the uncoupled state causing it to produce more superoxides (NO scavengers) than NO (Klinger et al., 2013). eNOS uncoupling can occur as a result of a decrease in amounts of tetrahydrobiopterin (BH4), a cofactor for eNOS in NO synthesis. There are studies to confirm eNOS uncoupling in pulmonary hypertension models that are BH4-deficient (Khoo et al., 2005; Nandi et al., 2005).
Also, several studies have described elevated levels of ADMA in plasma and serum of IPAH and connective tissue disease (CTD)-associated pulmonary arterial hypertension patients (Kielstein et al., 2005; Fang et al., 2015; Liu et al., 2019). Monocotaline- and hypoxia-induced PAH rat model have been found to have increased ADMA levels with a corresponding decrease in DDAH levels (Millatt et al., 2003; Li et al., 2010). This makes ADMA/DDAH possible diagnostic indicators in PAH
NO signaling remains a very relevant pathway in understanding and treating PAH. Upregulation of NO signaling is crucial in the management of PAH.
Crosstalk With Nitric Oxide
The nitric oxide pathway interacts with several other pathways reiterating its complex role in PAH. Nitric oxide interaction with some of these pathways is illustrated in Figure 2. The nitric oxide pathway has been shown to interact with the bone morphogenetic protein (BMP) pathway. Mutations in the bone morphogenetic protein receptor 2 (BMPR2) gene possess the strongest risk factor in the development of PAH, particularly heritable PAH (Machado et al., 2006). Bone Morphogenetic Protein Receptor II (BMPRII) and its ligands, BMP2 and BMP4 were found to be mediators of endothelial nitric oxide synthase activation (Gangopahyay et al., 2011). BMP2 and BMP4 activates eNOS by enhancing its phosphorylation by protein kinase A in pulmonary arterial endothelial cells (PAEC). Interestingly, knocking down BMPRII in PAEC reduced the ability of BMP2 and BMP4 to stimulate eNOS phosphorylation suggesting a crucial role of BMPRII in eNOS activation (Gangopahyay et al., 2011). Plasma nitric oxide metabolites have also been found to be markedly reduced in idiopathic pulmonary arterial hypertension patients with BMPR2 mutations (Zhang et al., 2016).Protein Kinase G I (PKGI) which is one of the downstream targets in the nitric oxide pathway has been found to crosstalk with the BMP pathway. PKGI contributes to the activation of BMP signaling by BMP4 in Human PASMCs (Yang et al., 2013). PKGI upregulates BMP signaling by phosphorylating the BMPRII receptor and smad proteins. Furthermore, PKGI binds to the phosphorylated smad proteins to form a complex and translocates into the nucleus where it upregulates the transcription of Id1 mRNA (Schwappacher et al., 2013). This then facilitates apoptosis and inhibits PASMC proliferation (Schwappacher et al., 2013; Yang et al., 2013).
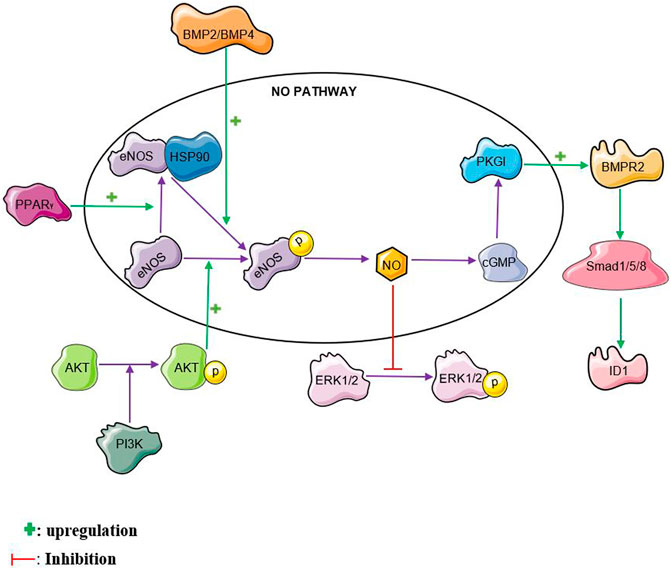
FIGURE 2. NO crosstalk. NO, nitric oxide, BMPR2, bone morphogenetic protein receptor 2 PPARγ, peroxisome proliferator-activated receptor gamma ERK, extracellular signal-regulated kinase, PI3K, phosphatidylinositol 3-kinase, HSP90, heat shock protein 90, BMP2, bone morphogenetic protein 2, BMP4, bone morphogenetic protein 4, ID1, inhibitor of DNA binding 1
The extracellular signal-regulated kinase 1/2 (ERK 1/2) pathway plays a significant role in the pathogenesis of PAH. Activation of the ERK pathway is known to be associated with increased proliferation and reduced apoptosis of PASMCs (Yang et al., 2008; Zhou et al., 2019). NO has been found to induce S-nitrosylation of ERK1/2 leading to reduced ERK phosphorylation and improved MCF-7 cell apoptosis (Feng et al., 2013). This crosstalk could be one of the ways with which NO inhibits proliferation and enhances apoptosis in PASMCs. S-nitrosylation of ERK1/2 by NO in PASMCs is yet to be explored.
The phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway is another major pathway that is implicated PAH. There have been contradictory reports on its role in PAH (Fang et al., 2016; Huang et al., 2017; Zheng et al., 2017). The PI3K/Akt pathway is known to phosphorylate eNOS and increase NO levels in many cell types including human pulmonary arterial endothelial cells (HPAECs) (Kim et al., 2008; Liang et al., 2009; Li et al., 2014). This helps to attenuate monocrotaline-induced pulmonary artery endothelial dysfunction and PAH in rats (Li et al., 2014; Zheng et al., 2017). Relaxation of rat pulmonary artery rings is enhanced through the activation of eNOS by the PI3K/Akt pathway (Zhang et al., 2010).
There is evidence that Peroxisome proliferator-activated receptor gamma (PPARγ) activity is reduced in PASMCs from idiopathic PAH patients (Falcetti et al., 2012). Activation of the Peroxisome proliferator-activated receptor gamma (PPARγ) pathway is known to reduce monocrotaline-and hypoxia-induced PAH in rats (Liu et al., 2012; Legchenko et al., 2018). Increased PPARγ expression has been found to enhance the phosphorylation and activation of eNOS in human pulmonary arterial endothelial cells (Li et al., 2014). Enhanced interaction between heat shock protein 90 and eNOS is one of the ways with which PPARγ facilitates eNOS phosphorylation. PPARγ agonists such as rosiglitazone, ciglitazone and pioglitazone have been found to stimulate eNOS activation through various mechanisms. Rosiglitazone was found to activate eNOS by stimulating heat shock protein (HSP)-90–eNOS interaction, with a subsequent eNOS phosphorylation at Ser1177 (Polikandriotis et al., 2005). Ciglitazone also caused a reduction in human umbilical vein endothelial cells (HUVEC) membrane NADPH-dependent superoxide anion generation leading to a decrease in endothelial superoxide anion generation and oxidative stress with a resulting increase in NO bioavailability (Hwang et al., 2005; Polikandriotis et al., 2005). Rho-kinase, a serine–threonine kinase is known to cause endothelial dysfunction by inactivating eNOS and reducing NO generation (Nohria et al., 2006). Pioglitazone enhances the dephosphorylation on tyrosine residues of the Vav protein, a group of guanosine nucleotide exchange factors (GEFs) involved in the activation of Rho Kinase. The deactivation of Rho Kinase leads to an increase in eNOS activation with a consequent increase in NO production (Wakino et al., 2004). The PPARγ agonist, telmisartan was found to increase the expression of DDAH II thereby enhancing the degradation of ADMA and increasing NO levels in human umbilical vein endothelial cells (Scalera et al., 2008).
Current Drugs ON Nitric Oxide Pathway in Pulmonary Arterial Hypertension
There are two groups of drugs currently used in PAH that act on the nitric oxide pathway, the phosphodiesterase 5 inhibitors (sildenafil and tadalafil) and soluble guanylate cyclase inhibitors (riociguat).
Phosphodiesterase-5 Inhibitors
As the name suggests, PDE5 inhibitors inhibit the PDE5 enzyme responsible for the breakdown of cGMP. This increases the levels of cGMP which activates protein kinase G and consequently leads to vasodilation. PDE5 is very abundant in smooth muscle cells (Rybalkin et al., 2003). Sildenafil was the first PDE5 inhibitor accepted for the management of PAH.
The SUPER-1 trial found 20, 40 and 80 mg doses of sildenafil to increase 6MWD by 45, 46, and 50 m respectively after 12 weeks. These doses also reduced mPAP by 2.1, 2.6 and 4.7 mmHg respectively. The was no significant reduction in clinical worsening (as measured by death, hospitalization for pulmonary hypertension, initiation of prostacyclin and initiation of bosentan) in the different sildenafil dose groups when compared to the placebo. Also, there were no significant dose-dependent changes in exercise capacity making the 20 mg dose the most appropriate for clinical use (Galiè et al., 2005b). A follow-up open-label trial by the SUPER-2 group found the improvement in 6MWD to be maintained after 1 year with a value of 51 m (Rubin et al., 2011). Sildenafil caused mild side effects such as headaches, flushing, and dyspepsia.
Contrary to the SUPER-1 study, the PACES-1 study found the combination of sildenafil with long-term intravenous epoprostenol therapy to improve clinical worsening as compared to intravenous epoprostenol alone after 16 weeks (Simonneau et al., 2008). The proportion of patients that needed a change in epoprostenol dose (an indicator of clinical worsening) was 0.195 in the epoprostenol-only group with a lower proportion (0.062) in the combination group. The sildenafil dose for this study was titrated up from 20 to 40 mg and 80 mg three times daily over 16 weeks depending on patient drug tolerability. An open-label extension study of the PACES-1 trial found the long-term combination of sildenafil and intravenous epoprostenol to be well tolerated with 33% of patients have a maintained or improved 6MWD after 3 years(Simonneau et al., 2014).
Administration of bosentan (125 mg twice daily) to patients on stable sildenafil therapy resulted in no significant difference between the two groups with regards to the primary endpoint (time to the first morbidity/mortality event) after 16 weeks in an event-driven trial. This may be due to limitations of the study such as enrollment of patients with many comorbidities and a high rate of patient dropout, to name but a few. Nonetheless, the addition of bosentan yielded an increase in 6MWD by 7.2 ± 66.0 m in the bosentan group with a reduction of 14.6 ± 80.4 m in the placebo group after 16 weeks. The mean difference in 6MWD between the two groups was 21.8 m. However, this was only a secondary and exploratory endpoint of the study (McLaughlin et al., 2015).
Conversely, a 12 weeks randomized controlled clinical trial found the administration of sildenafil (20 mg three times daily) to patients on stable bosentan therapy offers no significant benefit in terms of the 6MWD as compared to bosentan alone (Vizza et al., 2017). Limitations such as a possible ceiling effect in patients receiving effective bosentan therapy could be responsible for the insignificant difference between the groups.
In a 16 weeks double-blind, placebo-controlled study, 40 mg once daily administration of tadalafil was found to increase 6MWD by 44 m in the bosentan-naive group and by 23 m in patients on bosentan background therapy. These increments were statistically significant in comparison to the placebo group. Tadalafil also reduced the time to clinical worsening and the incidence of clinical worsening (68% relative risk reduction). The most common side-effects were headache, myalgia, and flushing (Galiè et al., 2009a). A 52 weeks extension study confirmed tadalafil to be well-tolerated with a sustained improvement in the 6MWD (Oudiz et al., 2012).
As compared to tadalafil and ambrisentan monotherapies, the combination of tadalafil and ambrisentan resulted in fewer occurrences of clinical failure as defined by the first occurrence of a composite endpoint of death, hospitalization for worsening pulmonary arterial hypertension, disease progression, or unsatisfactory long-term clinical response. Clinical failure occurred in only 18% of patients in the combination group with 34 and 28% occurrences in the ambrisentan only and tadalafil only groups respectively. Side-effects such as headache and nasal congestion occurred more in the combination group than in the monotherapy groups (Galiè et al., 2015a). Because of moderate quality evidence, the 2019 CHEST guideline weakly recommends the initial combination therapy with ambrisentan and tadalafil to improve 6MWD for treatment-naive PAH patients with WHO FC II and III. Also, the addition of tadalafil to improve 6MWD of stable or symptomatic PAH patients on background therapy with ambrisentan is weakly recommended because of low-quality evidence (Klinger et al., 2019).
Vardenafil is also a PDE5 inhibitor. A randomized, double-blind, placebo-controlled study found it to be well tolerated in patients with PAH and also increase 6-MWD by 69 m at a dose of 5 mg twice daily. It also reduced mean pulmonary arterial pressure by 5.3 mmHg at 12 weeks (Jing et al., 2011). Vardenafil was found to significantly decrease mean pulmonary arterial pressure when used for acute vasoreactivity testing in patients with PH (Sandqvist et al., 2015). Vardenafil is not yet approved for the treatment of PAH.
Soluble Guanylate Cyclase Stimulators
Soluble guanylate cyclase is activated by NO to convert guanosine triphosphate (GTP) to cGMP (Stasch et al., 2001). sGC stimulators directly activate soluble guanylate cyclase in smooth muscle cells. sGC stimulators also stabilize the complex formed between NO and sGC to enhance cGMP production (Stasch et al., 2011). Riociguat is the only approved sGC stimulator for treating PAH. It is approved to be used up to a maximum dose of 2.5 mg three times daily. Riociguat increases 6-MWD by a mean of 36 m as compared to placebo after 12 weeks. It also significantly decreases mPAP by 9 ± 11 mmHg from baseline. In the RESPITE trial, riociguat proved to be a viable alternative for patients with PAH not responding to PDE5 inhibitors because of reduced endogenous NO production (Hoeper et al., 2017a). The prospective randomized REPLACE trial further confirmed that patients irresponsive to PDE5 inhibitors are more likely to show clinical improvement upon switching to riociguat therapy (Hoeper et al., 2017b). This has made riociguat a crucial PAH treatment option. Some of its side effects include headache, syncope, and hypotension (Ghofrani et al., 2013a). There was sustained improvement in exercise capacity after 1 year as observed in a long-term extension trial (Rubin et al., 2011). Riociguat is also approved for treating inoperable chronic thromboembolic pulmonary hypertension (Ghofrani et al., 2013b). The combination of riociguat and PDE5 inhibitors is contraindicated due to increased hypotension (Galiè et al., 2015b).
Inhaled Nitric Oxide
Even though inhaled NO has the potential for use in treating PAH, it is mostly used in acute vasoreactivity testing during right heart catheterization. This is to identify acute responders likely to benefit from calcium-channel blockers (CCBs). Acute responders are patients who show a decrease in mPAP of at least 10 mmHg to an absolute level below 40 mmHg with sustained or increased cardiac output (Badesch et al., 2009). These patients are rare with most of them having idiopathic PAH. Such patients have a very good prognosis with CCB use (Rich et al., 2010). Inhaled NO is only approved for the treatment of severe persistent pulmonary hypertension of the newborn (PPHN). There are not enough studies to support the safety and efficacy of the long-term ambulatory use of inhaled NO in PAH. Also, the lack of portable delivery systems and the need for continuous inhalation makes its daily ambulatory use impractical.
Ongoing Pharmaceutical Research ON Nitric Oxide Pathway and Its Implication in Pulmonary Arterial Hypertension
The NO pathway offers various molecular targets that can be exploited to help develop novel drugs to ameliorate the pathophysiological changes that occurs during PAH development. Some of the possible therapeutic targets offered by the NO pathway include arginase, ADMA, DDAH1, eNOS, PDE-5, cGMP, sGC and NO. This section highlights key drug molecules and target that focus mainly on the NO pathway and their implications in PAH.
L-arginine serves as a substrate not just for eNOS but arginase as well. This means an increase in arginase activity leads to a reduction in the availability of L-arginine to be converted to NO by eNOS (Pernow and Jung, 2013). Arginase metabolizes L-arginine to urea and L-ornithine (Luiking et al., 2012) as shown in Figure 1. Arginase exists in two isoforms namely arginase I and arginase II but arginase II has been found to be much more involved in the development of PAH in humans and animals (Jin et al., 2010; Cho et al., 2013). Deletion of arginase II in interleukin-13 overexpressing transgenic mice showed a significant decrease in medial wall thickness suggesting an important role of arginase II in PAH (Cho et al., 2013). PAH patients are known to have high arginase II activity compared to healthy controls with a consequent reduction in NO synthesis (Kao et al., 2015). A recent study found selective arginase II inhibitors L207-0525 and L327-0346 demonstrate a dose-dependent protective activity in monocrotaline-induced PAH rats. Furthermore, combining L207-0525 and L327-0346 with low dose (0.1 mg/kg) tadalafil produced a better protective effect in monocrotakine-induced PAH rats. Therefore, L207-0525 and L327-0346 are potential compounds needing further exploration for PAH treatment (Koklin and Danilenko, 2019).
Nitrates have shown promising signs in the amelioration of PAH in hypoxia- and monocrotaline-induced PAH animals. Both Intraperitoneal and nebulized sodium nitrite reduced pulmonary arterial pressure, right ventricular hypertrophy and vascular remodeling in MCT-induced PAH rats (Zuckerbraun et al., 2010; Pankey et al., 2012). Orally administered sodium nitrate have also shown protective effects in hypoxia-induced PAH mice (Baliga et al., 2012). However, a recent study found oral sodium nitrate does not substantially reduce established MCT-induced PAH in rats (Malikova et al., 2020). The different outcomes in the above studies could be attributed to the difference in disease severity in the PAH models as well as the routes and timing of drug administration (Malikova et al., 2020). Considering the poor response of certain patients to PDE5 inhibitors (PDE5i) due to reduced endogenous nitric oxide production, nitrates could serve as alternative sources of NO in such patients. Also, the impracticability of inhaled nitric oxide therapy further necessitates the need for alternate NO sources. More research has to be done to further explore the potential benefits of nitrates in PAH therapy especially in PDE5i-irresponsive patients.
Oxymatrine, an active alkaloid derived from the traditional Chinese herb Sophora alopecuroides was found to protect against hypoxia- and monocrotaline-induced PAH (Zhang et al., 2014). The mechanism behind the protective effect of Oxymatrine in monocrotaline-induced PAH is likely to be through the reduction of pulmonary ADMA levels even though it did not affect the level of DDAH1 (Dai et al., 2019).
Despite the fact that DDAH1 levels have been found to be reduced in hypoxia-induced PAH, it was uncertain how important of a role DDAH1 dysfunction plays in PAH. Recently, a study on a novel DDAH1 knockout (DDAH1−/−) rat strain model found DDAH1 dysfunction to significantly worsen RVSP and RVHI in DDAH1−/− MCT model compared to the wild type MCT model (Wang et al., 2019). Although no in vitro experiment was carried out to target specific cells that are responsible for the progress of PAH, this study still shows how important DDAH1 dysfunction is in the development of PAH making it a possible PAH treatment target.
Apelin signaling is known to regulate endothelial NOS (eNOS) (Chandra et al., 2011). It has been discovered that the novel cyclic biased agonist of the apelin receptor, MM07 significantly reduces the elevation of right ventricular systolic pressure and hypertrophy induced by monocrotaline (Yang et al., 2019). They also found the elevation of eNOS and its mRNA as one of the mechanisms behind its protective effect in monocrotaline-induced PAH.
Udenafil, an oral phosphodiesterase-5 inhibitor approved for the treatment of erectile dysfunction has been found by a recent double-blind, placebo-controlled phase IIb clinical trial to improve 6-MWD in patients with PAH, especially those with a history of ERA therapy (Chang et al., 2019). The improvement in the 6-MWD with udenafil and ERA combination therapy group was found to be better than that of sildenafil and tadalafil in previous trials (Galiè et al., 2009a; Galiè et al., 2009b; Benza et al., 2018). Adverse side effects in the udenafil were also found to be mild and in the expected range (Chang et al., 2019).
TPN171, a new compound that inhibits PDE5 has been found to significantly reduce mPAP in a rat MCT-PAH model. Its efficacy was comparable to that of the sildenafil control group. It has a long half-life making once-daily dosing possible. The effective dose of TPN171 used in the animal experiment was 1 mg/kg which is lower than that of sildenafil (25 mg/kg). This property could make the occurrence of side-effects less likely if TPN171 is used in a clinical setting (Wang et al., 2019). TPN171 is currently in phase II clinical trial.
Evodiamine is a traditional Chinese medicine for the treatment of cancers (Dong et al., 2012). Evodiamine derivatives (S)-7e and (S)-7 days are newly discovered PDE5 inhibitors with very high selectivity (Zhang et al., 2020). Evodiamine derivative (S)-7 days significantly reduced mPAP and wall thickness in rat MCT-PAH model. Its efficacy is similar to that of sildenafil which was used as a positive control. Additionally, the study found a unique allosteric pocket of PDE5 using evodiamine derivative (S)-7e. Currently approved PDE5 inhibitors only bind to the substrate-binding pocket. This novel allosteric pocket regulates both enzymatic activity and pulmonary hemodynamic function of PDE5 thereby serving as a new therapeutic target for PAH treatment (Zhang et al., 2020).
A new study found PDE10 to be a novel therapeutic target for treating PAH. This study used a highly selective PDE10 inhibitor, 2b to explore the role of PDE10 in PAH. Compound 2b significantly reduced mPAP and RVHI in PAH rats making PDE10 a potential therapeutic target for PAH (Huang et al., 2019).
Studies have found natriuretic peptides to increase cGMP levels (Egom, 2015; Preston et al., 2016). Natriuretic peptides (NP) are inactivated by neprilysin. Natriuretic peptides are known to have antiproliferative effects on PASMCs and also attenuate the development of hypoxia-induced PH (Arjona at al., 1997; Zhao et al., 1999). Furthermore, the beneficial effect of sildenafil was found to be affected by natriuretic peptide activity in mice (Zhao et al., 2003). Also, the infusion of natriuretic peptides in the presence of sildenafil synergistically increased cGMP and reduced RVSP in hypoxia-induced PAH rats (Preston et al., 2016). A recent clinical trial demonstrated that the combination of a neprilysin inhibitor (racecadotril) with a PDE5 inhibitor (sildenafil or tadalafil) acutely increases NP and cGMP levels and improves pulmonary hemodynamics (Hobbs et al., 2019). This indicates that neprilysin inhibitors could have therapeutic use in PAH.
The mechanism of NO/cGMP-induced vasodilation is partly mediated by the activation of voltage-gated K+ (Kv) channels (Cogolludo et al., 2001; Jackson, 2018). Kv1.5 is particularly known to contribute the most to Kv current in PASMCs. Kv7 channels have now been found to play a very significant role in generating Kv current in rat PASMCs (Mondéjar-Parreño et al., 2019). Kv7 channel activation is now thought to be imperative to the electrophysiological and relaxant effects of NO donors and riociguat (Mondéjar-Parreño et al., 2019). This makes activation of Kv7 channels a novel mechanism of action of vasodilators used in managing pulmonary arterial hypertension.
A new sGC stimulator, compound 13a (a pyrazolo [3,4-b] pyridine-3-yl pyrimidine derivative) has recently been found to exhibit similar in vitro vasorelaxation potential as riociguat on rat thoracic aorta rings and rat heart Langendorff preparation. Compound 13a also exhibited good oral bioavailability in male Beagle dogs which could make it a possible treatment candidate for PAH (Li et al., 2019). Compound 2 (a pyrazolo [3,4-b] pyridine derivative) is another novel sGC stimulator found to attenuate PAH (Hu et al., 2020). It reduced the migration of HPASMCs under hypoxic conditions significantly. Furthermore, it significantly improved RVSP, myocardial and vascular remodelling in hypoxia-induced PAH rats (Hu et al., 2020). We should notice that the PDE5 inhibitors block the breakdown of cGMP but these effects are dependent on NO availability and sGC activity. So the sGC stimulator or sGC activator may be effective in patients who have not sufficiently responded to a PDE5 inhibitor. Furthermore, sGC stimulator and sGC activator also show some difference, sGC stimulator acts on mature sGC and keep the activity of sGC, while sGC activator help to activate the damaged sGC.
Conclusion
Great strides have been made in PAH research in recent years but searching for new therapeutical agents remains a big challenge in this field. Current therapy only slows disease progression but does not cure the disease. So far the current clinical practice has strongly suggested that targeting the NO pathway is the strategy with the most potential. Critical molecules in the NO pathway such as DDAH1, PDE10 and cGKI show potential for becoming the next new therapeutic targets in PAH treatment. Compounds or derivatives from plant extracts also have very bright prospect. Furthermore, medicines harboring the ability to enhance NO signal and other key signaling pathways at the same time exhibit better hope to finally cure this thorny disease.
Author Contributions
TT: Writing—Original Draft YJ: Writing—Original Draft XL: Conceptualization YL: Writing—Review&Editing
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Archer, S. L., Djaballah, K., Humbert, M., Weir, E. K., Fartoukh, M., Dall’ava-Santucci, J., et al. (2012). Nitric Oxide Deficiency in Fenfluramine- and Dexfenfluramine-Induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 158, 1061–1067. doi:10.1164/AJRCCM.158.4.9802113
Arjona, A. A., Hsu, C. A., Wrenn, D. S., and Hill, N. S. (1997). Effects of Natriuretic Peptides on Vascular Smooth-Muscle Cells Derived from Different Vascular Beds. Gen. Pharmacol. Vasc. Syst. 28, 387–392. doi:10.1016/S0306-3623(96)00275-3
Badesch, D. B., Champion, H. C., Gomez Sanchez, M. A., Hoeper, M. M., Loyd, J. E., Manes, A., et al. (2009). Diagnosis and Assessment of Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 54, S55–S66. doi:10.1016/J.JACC.2009.04.011
Baliga, R. S., Milsom, A. B., Ghosh, S. M., Trinder, S. L., MacAllister, R. J., Ahluwalia, A., et al. (2012). Dietary Nitrate Ameliorates Pulmonary Hypertension. Circulation 125, 2922–2932. doi:10.1161/CIRCULATIONAHA.112.100586
Barst, R. J., Rubin, L. J., Long, W. A., McGoon, M. D., Rich, S., Badesch, D. B., et al. (2009). A Comparison of Continuous Intravenous Epoprostenol (Prostacyclin) with Conventional Therapy for Primary Pulmonary Hypertension. N. Engl. J. Med. 51, 993. doi:10.1056/NEJM199602013340504
Benza, R. L., Raina, A., Gupta, H., Murali, S., Burden, A., Zastrow, M. S., et al. (2018). Bosentan-Based, Treat-To-Target Therapy in Patients with Pulmonary Arterial Hypertension: Results from the COMPASS-3 Study. Pulm. Circ. 8, 1–13. doi:10.1177/2045893217741480
Berger, R. M. F., Geiger, R., Hess, J., Bogers, A. J. J. C., and Mooi, W. J. (2012). Altered Arterial Expression Patterns of Inducible and Endothelial Nitric Oxide Synthase in Pulmonary Plexogenic Arteriopathy Caused by Congenital Heart Disease. Am. J. Respir. Crit. Care Med. 163, 1493–1499. doi:10.1164/AJRCCM.163.6.9908137
Borrill, Z., Clough, D., Truman, N., Morris, J., Langley, S., and Singh, D. (2006). A Comparison of Exhaled Nitric Oxide Measurements Performed Using Three Different Analysers. Respir. Med. 100, 1392–1396. doi:10.1016/J.RMED.2005.11.018
Chandra, S. M., Razavi, H., Kim, J., Agrawal, R., Kundu, R. K., Perez, V. de. J., et al. (2011). Disruption of the Apelin-APJ System Worsens Hypoxia-Induced Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 31, 814–820. doi:10.1161/ATVBAHA.110.219980
Chang, H.-J., Song, S., Chang, S.-A., Kim, H.-K., Jung, H.-O., Choi, J.-H., et al. (2019). Efficacy and Safety of Udenafil for the Treatment of Pulmonary Arterial Hypertension: a Placebo-Controlled, Double-Blind, Phase IIb Clinical Trial. Clin. Ther. 41, 1499–1507. doi:10.1016/J.CLINTHERA.2019.05.006
Cho, W., Lee, C., Kang, M., Huang, Y., Giordano, F. J., Lee, P. J., et al. (2013). IL-13 Receptor α2-arginase 2 Pathway Mediates IL-13-induced Pulmonary Hypertension. Am. J. Physiol. Lung Cel. Mol. Physiol. 304, L112–L124.
Cogolludo, A. L., Pérez-Vizcaíno, F., Zaragozá-Arnáez, F., Ibarra, M., López-López, G., López-Miranda, V., et al. (2001). Mechanisms Involved in SNP-Induced Relaxation and [Ca2+]I Reduction in Piglet Pulmonary and Systemic Arteries. Br. J. Pharmacol. 132, 959–967. doi:10.1038/SJ.BJP.0703894
Coleman, R. A., Smith, W. L., and Narumiya, S. (1994). International Union of Pharmacology Classification of Prostanoid Receptors: Properties, Distribution, and Structure of the Receptors and Their Subtypes. Pharmacol. Rev. 46.
Correale, M., Totaro, A., Lacedonia, D., Montrone, D., Di Biase, M., Foschino, M. P. B., et al. (2013). Novelty in Treatment of Pulmonary Fibrosis: Pulmonary Hypertension Drugs and Others. Cardiovasc. Hematol. Agents Med. Chem. 11, 169–178. doi:10.2174/187152571131100086
Dai, G., Li, B., Xu, Y., Zeng, Z., and Yang, H. (2019). Oxymatrine Prevents the Development of Monocrotaline-Induced Pulmonary Hypertension via Regulation of the NG, NG-Dimethyl-L-Arginine Metabolism Pathways in Rats. Eur. J. Pharmacol. 842, 338–344. doi:10.1016/J.EJPHAR.2018.11.007
Davie, N., Haleen, S., Upton, P., Polak, J., Yacoub, M., Morrell, N., et al. (2002). ET(A) and ET(B) Receptors Modulate the Proliferation of Human Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Crit. Care Med. 165, 398–405. doi:10.1164/AJRCCM.165.3.2104059
Dong, G., Wang, S., Miao, Z., Yao, J., Zhang, Y., Guo, Z., et al. (2012). New Tricks for an Old Natural Product: Discovery of Highly Potent Evodiamine Derivatives as Novel Antitumor Agents by Systemic Structure–Activity Relationship Analysis and Biological Evaluations. J. Med. Chem. 55, 7593–7613. doi:10.1021/JM300605M
Dweik, R. A., Boggs, P. B., Erzurum, S. C., Irvin, C. G., Leigh, M. W., Lundberg, J. O., et al. (2012). An Official ATS Clinical Practice Guideline: Interpretation of Exhaled Nitric Oxide Levels (FeNO) for Clinical Applications. Am. J. Respir. Crit. Care Med. 184, 602–615. doi:10.1164/RCCM.9120-11ST
Egom, E. E. (2015). BNP and Heart Failure: Preclinical and Clinical Trial Data. J. Cardiovasc. Transl. Res. 8, 149–157. doi:10.1007/s12265-015-9619-3
Eguchi, S., Hirata, Y., and Marumo, F. (1993). Endothelin Subtype B Receptors Are Coupled to Adenylate Cyclase via Inhibitory G Protein in Cultured Bovine Endothelial Cells. J. Cardiovasc. Pharmacol. 22. doi:10.1097/00005344-199322008-00043
Falcetti, E., Hall, S. M., Phillips, P. G., Patel, J., Morrell, N. W., Haworth, S. G., et al. (2012). Smooth Muscle Proliferation and Role of the Prostacyclin (IP) Receptor in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 182, 1161–1170. doi:10.1164/RCCM.201001-0011OC
Fang, X., Chen, X., Zhong, G., Chen, Q., and Hu, C. (2016). Mitofusin 2 Downregulation Triggers Pulmonary Artery Smooth Muscle Cell Proliferation and Apoptosis Imbalance in Rats with Hypoxic Pulmonary Hypertension via the PI3K/Akt and Mitochondrial Apoptosis Pathways. J. Cardiovasc. Pharmacol. 67, 164–174. doi:10.1097/FJC.0000000000000333
Fang, Z., Huang, Y., Tang, L., Hu, X., Shen, X., Tang, J., et al. (2015). Asymmetric Dimethyl-L-Arginine Is a Biomarker for Disease Stage and Follow-Up of Pulmonary Hypertension Associated with Congenital Heart Disease. Pediatr. Cardiol. 36, 1062–1069. doi:10.1007/S00246-015-1127-3
Feng, X., Sun, T., Bei, Y., Ding, S., Zheng, W., Lu, Y., et al. (2013). S-nitrosylation of ERK Inhibits ERK Phosphorylation and Induces Apoptosis. Sci. Rep. 3, 1–6. doi:10.1038/srep01814
Francis, S. H., Turko, I. V., and Corbin, J. D. (2000). Cyclic Nucleotide Phosphodiesterases: Relating Structure and Function. Prog. Nucleic Acid Res. Mol. Biol. 65. doi:10.1016/s0079-6603(00)65001-8
Furchgott, R. F., and Zawadzki, J. V. (1980). The Obligatory Role of Endothelial Cells in the Relaxation of Arterial Smooth Muscle by Acetylcholine. Nat 288, 373–376. doi:10.1038/288373a0
Galié, N., Badesch, D., Oudiz, R., Simonneau, G., McGoon, M. D., Keogh, A. M., et al. (2005a). Ambrisentan Therapy for Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 46, 529–535. doi:10.1016/J.JACC.2005.04.050
Galiè, N., Barberà, J. A., Frost, A. E., Ghofrani, H.-A., Hoeper, M. M., McLaughlin, V. V., et al. (2015a). Initial Use of Ambrisentan Plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 373, 834–844. doi:10.1056/NEJMOA1413687
Galiè, N., Ghofrani, H. A., Torbicki, A., Barst, R. J., Rubin, L. J., Badesch, D., et al. (2009b). Sildenafil Citrate Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 353, 2148–2157. doi:10.1056/NEJMOA050010
Galiè, N., Humbert, M., Vachiery, J.-L., Gibbs, S., Lang, I., Torbicki, A., et al. (2016). 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Rev. Española Cardiol. (English Ed. 69, 177. doi:10.1016/J.REC.2016.01.002
Galiè, N., Müller, K., Scalise, A.-V., and Grünig, E. (2015b). PATENT PLUS: A Blinded, Randomised and Extension Study of Riociguat Plus Sildenafil in Pulmonary Arterial Hypertension. Eur. Respir. J. 45, 1314–1322. doi:10.1183/09031936.00105914
Galiè, N., Brundage, B. H., Ghofrani, H. A., Oudiz, R. J., Simonneau, G., Safdar, Z., et al. (2009a). Tadalafil Therapy for Pulmonary Arterial Hypertension. Circulation 119, 2894–2903. doi:10.1161/CIRCULATIONAHA.108.839274
Gangopahyay, A., Oran, M., Bauer, E. M., Wertz, J. W., Comhair, S. A., Erzurum, S. C., et al. (2011). Bone Morphogenetic Protein Receptor II Is a Novel Mediator of Endothelial Nitric-Oxide Synthase Activation. J. Biol. Chem. 286. doi:10.1074/jbc.M111.274100
Ghofrani, H.-A., D’Armini, A. M., Grimminger, F., Hoeper, M. M., Jansa, P., Kim, N. H., et al. (2013a). Riociguat for the Treatment of Chronic Thromboembolic Pulmonary Hypertension. N. Engl. J. Med. 369, 319–329. doi:10.1056/NEJMOA1209657
Ghofrani, H.-A., Galiè, N., Grimminger, F., Grünig, E., Humbert, M., Jing, Z.-C., et al. (2013b). Riociguat for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 369, 330–340. doi:10.1056/NEJMOA1209655
Giaid, A., and Saleh, D. (1995). Reduced Expression of Endothelial Nitric Oxide Synthase in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 333, 214–221. doi:10.1056/NEJM199507273330403
Hirata, Y., Emori, T., Eguchi, S., Kanno, K., Imai, T., Ohta, K., et al. (1993). Endothelin Receptor Subtype B Mediates Synthesis of Nitric Oxide by Cultured Bovine Endothelial Cells. J. Clin. Invest. 91, 1367–1373. doi:10.1172/JCI116338
Hobbs, A. J., Moyes, A. J., Baliga, R. S., Ghedia, D., Ochiel, R., Sylvestre, Y., et al. (2019). Neprilysin Inhibition for Pulmonary Arterial Hypertension: A Randomized, Double-Blind, Placebo-Controlled, Proof-Of-Concept Trial. Br. J. Pharmacol. 176, 1251–1267. doi:10.1111/BPH.14621
Hoeper, M. M., Ghofrani, H.-A., Benza, R. L., Corris, P. A., Gibbs, J., Klinger, J. R., et al. (2017b). REPLACE: A Prospective, Randomized Trial of Riociguat Replacing Phosphodiesterase 5 Inhibitor Therapy in Patients with Pulmonary Arterial Hypertension Who Are Not at Treatment Goal. Eur. Respir. J. 50, PA2417. doi:10.1183/1393003.CONGRESS-2017.PA2417
Hoeper, M. M., Simonneau, G., Corris, P. A., Ghofrani, H., Klinger, J. R., Langleben, D., et al. (2017a). RESPITE: Switching to Riociguat in Pulmonary Arterial Hypertension Patients with Inadequate Response to Phosphodiesterase-5 Inhibitors. Eur. Respir. J. 50. doi:10.1183/13993003.02425-2016
Hu, L., Li, L., Chang, Q., Fu, S., Qin, J., Chen, Z., et al. (2020). Discovery of Novel Pyrazolo[3,4-B] Pyridine Derivatives with Dual Activities of Vascular Remodeling Inhibition and Vasodilation for the Treatment of Pulmonary Arterial Hypertension. J. Med. Chem. 63, 11215–11234. doi:10.1021/ACS.JMEDCHEM.0C01132
Huang, X., Wu, P., Huang, F., Xu, M., Chen, M., Huang, K., et al. (2017). Baicalin Attenuates Chronic Hypoxia-Induced Pulmonary Hypertension via Adenosine A2A Receptor-Induced SDF-1/CXCR4/PI3K/AKT Signaling. J. Biomed. Sci. 24, 1–14. doi:10.1186/S12929-017-0359-3
Huang, Y.-Y., Yu, Y.-F., Zhang, C., Chen, Y., Zhou, Q., Li, Z., et al. (2019). Validation of Phosphodiesterase-10 as a Novel Target for Pulmonary Arterial Hypertension via Highly Selective and Subnanomolar Inhibitors. J. Med. Chem. 62, 3707–3721. doi:10.1021/ACS.JMEDCHEM.9B00224
Humbert, M., Guignabert, C., Bonnet, S., Dorfmüller, P., Klinger, J. R., Nicolls, M. R., et al. (2019). Pathology and Pathobiology of Pulmonary Hypertension: State of the Art and Research Perspectives. Eur. Respir. J. 53. doi:10.1183/13993003.01887-2018
Humbert, M., Segal, E. S., Kiely, D. G., Carlsen, J., Schwierin, B., and Hoeper, M. M. (2007). Results of European Post-Marketing Surveillance of Bosentan in Pulmonary Hypertension. Eur. Respir. J. 30, 338–344. doi:10.1183/09031936.00138706
Humbert, M., Sitbon, O., Chaouat, A., Bertocchi, M., Habib, G., Gressin, V., et al. (2006). Pulmonary Arterial Hypertension in France: Results from a National Registry. Am. J. Respir. Crit. Care Med. 173, 1023–1030. doi:10.1164/rccm.200510-1668OC
Hwang, J., Kleinhenz, D. J., Lassègue, B., Griendling, K. K., Dikalov, S., Michael Hart, C., et al. (2005). Peroxisome Proliferator-Activated Receptor-Ligands Regulate Endothelial Membrane Superoxide Production. Am. J. Physiol. Cel Physiol 288, 899–905. doi:10.1152/ajpcell
Ignarro, L. J., Buga, G. M., Wood, K. S., Byrns, R. E., and Chaudhuri, G. (1987). Endothelium-Derived Relaxing Factor Produced and Released from Artery and Vein Is Nitric Oxide. Proc. Natl. Acad. Sci. 84, 9265–9269. doi:10.1073/PNAS.84.24.9265
Ikemoto, Y., Teraguchi, M., and Kobayashi, Y. (2002). Plasma Levels of Nitrate in Congenital Heart Disease: Comparison with Healthy Children. Pediatr. Cardiol. 23, 132–136. doi:10.1007/S00246-001-0036-9
Jackson, W. F. (2018). KV Channels and the Regulation of Vascular Smooth Muscle Tone. Microcirculation 25, e12421. doi:10.1111/MICC.12421
Jin, Y., Calvert, T. J., Chen, B., Chicoine, L. G., Joshi, M., Bauer, J. A., et al. (2010). Mice Deficient in Mkp-1 Develop More Severe Pulmonary Hypertension and Greater Lung Protein Levels of Arginase in Response to Chronic Hypoxia. Am. J. Physiol. Heart Circ. Physiol. 298, H1518–H1528.
Jing, Z. C., Yu, Z. X., Shen, J. Y., Wu, B. X., Xu, K. F., Zhu, X. Y., et al. (2011). Vardenafil in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 183, 1723–1729. doi:10.1164/RCCM.201101-0093OC
Kaneko, F. T., Arroliga, A. C., Dweik, R. A., Comhair, S. A., Laskowski, D., Oppedisano, R., et al. (1998). Biochemical Reaction Products of Nitric Oxide as Quantitative Markers of Primary Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 158, 917–923. doi:10.1164/ajrccm.158.3.9802066
Kao, C. C., Wedes, S. H., Hsu, J. W., Bohren, K. M., Comhair, S. A., Jahoor, F., et al. (2015). Arginine Metabolic Endotypes in Pulmonary Arterial Hypertension. Pulm. Circ. 5, 124–134. doi:10.1086/679720
Kharitonov, S. A., Cailes, J. B., Black, C. M., Bois, R. M. du., and Barnes, P. J. (1997). Decreased Nitric Oxide in the Exhaled Air of Patients with Systemic Sclerosis with Pulmonary Hypertension. Thorax 52, 1051–1055. doi:10.1136/THX.52.12.1051
Khoo, J. P., Zhao, L., Alp, N. J., Bendall, J. K., Nicoli, T., Rockett, K., et al. (2005). Pivotal Role for Endothelial Tetrahydrobiopterin in Pulmonary Hypertension. Circulation 111, 2126–2133. doi:10.1161/01.CIR.0000162470.26840.89
Kielstein, J. T., Bode-Böger, S. M., Hesse, G., Martens-Lobenhoffer, J., Takacs, A., Fliser, D., et al. (2005). Asymmetrical Dimethylarginine in Idiopathic Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 25, 1414–1418. doi:10.1161/01.ATV.0000168414.06853.F0
Kim, K. H., Moriarty, K., and Bender, J. R. (2008). Vascular Cell Signaling by Membrane Estrogen Receptors. Steroids 73, 864–869. doi:10.1016/J.STEROIDS.2008.01.008
Klinger, J. R., Abman, S. H., and Gladwin, M. T. (2013). Nitric Oxide Deficiency and Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 188, 639–646. doi:10.1164/RCCM.201304-0686PP
Klinger, J. R., Elliott, C. G., Levine, D. J., Bossone, E., Duvall, L., Fagan, K., et al. (2019). Therapy for Pulmonary Arterial Hypertension in Adults: Update of the CHEST Guideline and Expert Panel Report. Chest 155, 565–586. doi:10.1016/J.CHEST.2018.11.030
Koklin, I. S., and Danilenko, L. M. (2019). Combined Use of Arginase II Inhibitors and Tadalafil for the Correction of Monocrotaline Pulmonary Hypertension. Res. Results Pharmacol. 5 (3), 79. doi:10.3897/RRPHARMACOLOGY.5.39522
Legchenko, E., Chouvarine, P., Borchert, P., Fernandez-Gonzalez, A., Snay, E., Meier, M., et al. (2018). PPARγ Agonist Pioglitazone Reverses Pulmonary Hypertension and Prevents Right Heart Failure via Fatty Acid Oxidation. Sci. Transl. Med. 10, 303. doi:10.1126/SCITRANSLMED.AAO0303
Li, H., Lu, W., Cai, W. W., Wang, P. J., Zhang, N., Yu, C. P., et al. (2014). Telmisartan Attenuates Monocrotaline-Induced Pulmonary Artery Endothelial Dysfunction through A PPAR Gamma-dependent PI3K/Akt/Enos Pathway. Pulm. Pharmacol. Ther. 28, 17–24. doi:10.1016/J.PUPT.2013.11.003
Li, L., Zhang, W., Lin, F., Lu, X., Chen, W., Li, X., et al. (2019). Synthesis and Biological Evaluation of Pyrazolo[3,4-B]Pyridine-3-Yl Pyrimidine Derivatives as sGC Stimulators for the Treatment of Pulmonary Hypertension. Eur. J. Med. Chem. 173, 107–116. doi:10.1016/J.EJMECH.2019.04.014
Li, X. H., Peng, J., Tan, N., Wu, W. H., Li, T. T., Shi, R. Z., et al. (2010). Involvement of Asymmetric Dimethylarginine and Rho Kinase in the Vascular Remodeling in Monocrotaline-Induced Pulmonary Hypertension. Vascul. Pharmacol. 53, 223–229. doi:10.1016/J.VPH.2010.09.002
Liang, C., Ren, Y., Tan, H., He, Z., Jiang, Q., Wu, J., et al. (2009). Rosiglitazone via Upregulation of Akt/Enos Pathways Attenuates Dysfunction of Endothelial Progenitor Cells, Induced by Advanced Glycation End Products. Br. J. Pharmacol. 158, 1865–1873. doi:10.1111/J.1476-5381.2009.00450.X
Lim, Y., Low, T.-T., Chan, S.-P., Teo, T. W., Jang, J.-H. J., Yip, N., et al. (2019). Pulmonary Arterial Hypertension in a Multi-Ethnic Asian Population: Characteristics, Survival and Mortality Predictors from a 14-Year Follow-Up Study. Respirology 24, 162–170. doi:10.1111/RESP.13392
Liu, J., Fu, Q., Jiang, L., Wang, Y., and Yang, Y. (20192019). Clinical Value of Asymmetrical Dimethylarginine Detection in Patients with Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Cardiol. Res. Pract. doi:10.1155/2019/3741909
Liu, Y., Tian, X. Y., Mao, G., Fang, X., Fung, M. L., Shyy, J. Y.-J., et al. (2012). Peroxisome Proliferator-Activated Receptor-γ Ameliorates Pulmonary Arterial Hypertension by Inhibiting 5-Hydroxytryptamine 2B Receptor. Hypertension 60, 1471–1478. doi:10.1161/HYPERTENSIONAHA.112.198887
Luiking, Y. C., Have, G. A. M. T., Wolfe, R. R., and Deutz, N. E. P. (2012). Arginine De Novo and Nitric Oxide Production in Disease States. Am. J. Physiol. Endocrinol. Metab. 303, 1177–1189. doi:10.1152/AJPENDO.00284.2012
Machado, R. D., Aldred, M. A., James, V., Harrison, R. E., Patel, B., Schwalbe, E. C., et al. (2006). Mutations of the TGF-Β Type II Receptor BMPR2 in Pulmonary Arterial Hypertension. Hum. Mutat. 27, 121–132. doi:10.1002/HUMU.20285
Malekmohammad, M., Folkerts, G., Kashani, B. S., Naghan, P. A., Dastenae, Z. H., Khoundabi, B., et al. (20192020). Exhaled Nitric Oxide Is Not a Biomarker for Idiopathic Pulmonary Arterial Hypertension or for Treatment EfficacyEffects of Inorganic Nitrate in a Rat Model of Monocrotaline-Induced Pulmonary Arterial Hypertension. BMC Pulm. Med.Basic Clin. Pharmacol. Toxicol. 19126, 199–7109. doi:10.1186/s12890-019-0954-z10.1111/BCPT.13309
Malinovschi, A., Henrohn, D., Eriksson, A., Lundberg, J. O., Alving, K., and Wikström, G. (2011). Increased Plasma and Salivary Nitrite and Decreased Bronchial Contribution to Exhaled NO in Pulmonary Arterial Hypertension. Eur. J. Clin. Invest. 41, 889–897. doi:10.1111/J.1365-2362.2011.02488.X
McLaughlin, V., Channick, R. N., Ghofrani, H.-A., Lemarié, J.-C., Naeije, R., Packer, M., et al. (2015). Bosentan Added to Sildenafil Therapy in Patients with Pulmonary Arterial Hypertension. Eur. Respir. J. 46, 405–413. doi:10.1183/13993003.02044-2014
Millatt, L. J., Whitley, G. S., Li, D., Leiper, J. M., Siragy, H. M., Carey, R. M., et al. (2003). Evidence for Dysregulation of Dimethylarginine Dimethylaminohydrolase I in Chronic Hypoxia–Induced Pulmonary Hypertension. Circulation 108, 1493–1498. doi:10.1161/01.CIR.0000089087.25930.FF
Moncada, S., and Higgs, A. (1993). The L-Arginine-Nitric Oxide Pathway. N. Engl. J. Med. 329, 2002–2012. doi:10.1056/nejm199312303292706
Mondéjar-Parreño, G., Moral-Sanz, J., Barreira, B., Cruz, A. D. la., Gonzalez, T., Callejo, M., et al. (2019). Activation of Kv7 Channels as a Novel Mechanism for NO/Cgmp-Induced Pulmonary Vasodilation. Br. J. Pharmacol. 176, 2131–2145. doi:10.1111/BPH.14662
Nandi, M., Miller, A., Stidwill, R., Jacques, T. S., Lam, A. A. J., Haworth, S., et al. (2005). Pulmonary Hypertension in a GTP-Cyclohydrolase 1–Deficient Mouse. Circulation 111, 2086–2090. doi:10.1161/01.CIR.0000163268.32638.F4
Nathan, C., and Xie, Q. (1994). Nitric Oxide Synthases: Roles, Tolls, and Controls. Cell 78, 915–918. doi:10.1016/0092-8674(94)90266-6
Nohria, A., Grunert, M. E., Rikitake, Y., Noma, K., Prsic, A., Ganz, P., et al. (2006). Rho Kinase Inhibition Improves Endothelial Function in Human Subjects with Coronary Artery Disease. Circ. Res. 99. doi:10.1161/01.RES.0000251668.39526.c7
Olivieri, M., Talamini, G., Corradi, M., Perbellini, L., Mutti, A., Tantucci, C., et al. (2006). Reference Values for Exhaled Nitric Oxide (Reveno) Study. Respir. Res. 7, 1–6. doi:10.1186/1465-9921-7-94
Olsson, K. M., and Hoeper, M. M. (2009). Novel Approaches to the Pharmacotherapy of Pulmonary Arterial Hypertension. Drug Discov. Today 14, 284–290. doi:10.1016/J.DRUDIS.2008.12.003
Oudiz, R. J., Brundage, B. H., Gali, N., Ghofrani, H. A., Simonneau, G., Botros, F. T., et al. (2012). Tadalafil for the Treatment of Pulmonary Arterial Hypertension: A Double-Blind 52-Week Uncontrolled Extension Study. J. Am. Coll. Cardiol. 60, 768–774. doi:10.1016/J.JACC.2012.05.004
Palmer, R. M. J., Ferrige, A. G., and Moncada, S. (1987). Nitric Oxide Release Accounts for the Biological Activity of Endothelium-Derived Relaxing Factor. Nature 327, 524–526. doi:10.1038/327524a0
Pankey, E. A., Badejo, A. M., Casey, D. B., Lasker, G. F., Riehl, R. A., Murthy, S. N., et al. (2012). Effect of Chronic Sodium Nitrite Therapy on Monocrotaline-Induced Pulmonary Hypertension. Nitric Oxide - Biol. Chem. 27, 1–8. doi:10.1016/j.niox.2012.02.004
Pernow, J., and Jung, C. (2013). Arginase as a Potential Target in the Treatment of Cardiovascular Disease: Reversal of Arginine Steal? Cardiovasc. Res. 98, 334–343. doi:10.1093/CVR/CVT036
Polikandriotis, J. A., Mazzella, L. J., Rupnow, H. L., and Hart, C. M. (2005). Peroxisome Proliferator-Activated Receptor γ Ligands Stimulate Endothelial Nitric Oxide Production through Distinct Peroxisome Proliferator-Activated Receptor γ–Dependent Mechanisms. Arterioscler. Thromb. Vasc. Biol. 25, 1810–1816. doi:10.1161/01.ATV.0000177805.65864.D4
Preston, I. R., Hill, N. S., Gambardella, L. S., Warburton, R. R., and Klinger, J. R. (2016). Synergistic Effects of ANP and Sildenafil on cGMP Levels and Amelioration of Acute Hypoxic Pulmonary Hypertension. Exp. Biol. Med. 229, 920–925. doi:10.1177/153537020422900908
Price, L. C., Wort, S. J., Perros, F., Dorfmüller, P., Huertas, A., Montani, D., et al. (2012). Inflammation in Pulmonary Arterial Hypertension. Chest 141, 210–221. doi:10.1378/CHEST.11-0793
Pulido, T., Adzerikho, I., Channick, R. N., Delcroix, M., Galiè, N., Ghofrani, H.-A., et al. (2013). Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. N. Engl. J. Med. 369, 809–818. doi:10.1056/NEJMOA1213917
Rich, S., Kaufmann, E., and Levy, P. S. (2010). The Effect of High Doses of Calcium-Channel Blockers on Survival in Primary Pulmonary Hypertension. N. Engl. J. Med. 327, 76–81. doi:10.1056/NEJM199207093270203
Riley, M. S., Pórszósz, J., Miranda, J., Engelen, M. P. K. J., Wasserman, K., and Brundage, B. (1997). Exhaled Nitric Oxide during Exercise in Primary Pulmonary Hypertension and Pulmonary Fibrosis. Chest 111, 44–50. doi:10.1378/CHEST.111.1.44
Rosenkranz, S. (2015). Pulmonary Hypertension 2015: Current Definitions, Terminology, and Novel Treatment Options. Clin. Res. Cardiol. 104, 197–207. doi:10.1007/s00392-014-0765-4
Rubin, L. J., Badesch, D. B., Fleming, T. R., Galiè, N., Simonneau, G., Ghofrani, H. A., et al. (2011). Long-term Treatment with Sildenafil Citrate in Pulmonary Arterial Hypertension: The SUPER-2 Study. Chest 140, 1274–1283. doi:10.1378/CHEST.10-0969
Rybalkin, S. D., Yan, C., Bornfeldt, K. E., and Beavo, J. A. (2003). Cyclic GMP Phosphodiesterases and Regulation of Smooth Muscle Function. Circ. Res. 93, 280–291. doi:doi:10.1161/01.RES.0000087541.15600.2B
Sandqvist, A., Henrohn, D., Egeröd, H., Hedeland, M., Wernroth, L., Bondesson, U., et al. (2015). Acute Vasodilator Response to Vardenafil and Clinical Outcome in Patients with Pulmonary Hypertension. Eur. J. Clin. Pharmacol. 71, 1165–1173. doi:10.1007/s00228-015-1914-z
Sausbier, M., Schubert, R., Voigt, V., Hirneiss, C., Pfeifer, A., Korth, M., et al. (2000). Mechanisms of NO/cGMP-dependent Vasorelaxation. Circ. Res. 27, 825–830. doi:10.1161/01.res.87.9.825
Scalera, F., Martens-Lobenhoffer, J., Bukowska, A., Lendeckel, U., Täger, M., and Bode-Böger, S. M. (2008). Effect of Telmisartan on Nitric Oxide–Asymmetrical Dimethylarginine System. Hypertension 51, 696–703. doi:10.1161/HYPERTENSIONAHA.107.104570
Schwappacher, R., Kilic, A., Kojonazarov, B., Lang, M., Diep, T., Zhuang, S., et al. (2013). A Molecular Mechanism for Therapeutic Effects of cGMP-Elevating Agents in Pulmonary Arterial Hypertension. J. Biol. Chem. 288, 16557–16566. doi:10.1074/JBC.M113.458729
Seo, B., Oemar, B. S., Siebenmann, R., von Segesser, L., and Lüscher, T. F. (1994). Both ETA and ETB Receptors Mediate Contraction to Endothelin-1 in Human Blood Vessels. Circulation 89, 1203–1208. doi:10.1161/01.CIR.89.3.1203
Simonneau, G., Rubin, L. J., Galiè, N., Barst, R. J., Fleming, T. R., Frost, A. E., et al. (2008). Addition of Sildenafil to Long-Term Intravenous Epoprostenol Therapy in Patients with Pulmonary Arterial Hypertension: A Randomized Trial. Ann. Intern. Med. 149, 521–530. doi:10.7326/0003-4819-149-8-200810210-00004
Simonneau, G., Rubin, L. J., Galiè, N., Barst, R. J., Fleming, T. R., Frost, A., et al. (2014). Long-Term Sildenafil Added to Intravenous Epoprostenol in Patients with Pulmonary Arterial Hypertension. J. Hear. Lung Transpl. 33, 689–697. doi:10.1016/J.HEALUN.2014.02.019
Stasch, J.-P., Becker, E. M., Alonso-Alija, C., Apeler, H., Dembowsky, K., Feurer, A., et al. (2001). NO-independent Regulatory Site on Soluble Guanylate Cyclase. Nat 410, 212–215. doi:10.1038/35065611
Stasch, J.-P., Pacher, P., and Evgenov, O. V. (2011). Soluble Guanylate Cyclase as an Emerging Therapeutic Target in Cardiopulmonary Disease. Circulation 123, 2263–2273. doi:10.1161/CIRCULATIONAHA.110.981738
Surks, H. K. (2007). cGMP-dependent Protein Kinase I and Smooth Muscle Relaxation: a Tale of Two Isoforms. Circ. Res. 26, 1078–1080. doi:10.1161/CIRCRESAHA.107.165779
Tain, Y. L., and Hsu, C. N. (2017). Toxic Dimethylarginines: Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA). Toxins (Basel). 9, 92. doi:10.3390/toxins9030092
Tuder, R. M., Abman, S. H., Braun, T., Capron, F., Stevens, T., Thistlethwaite, P. A., et al. (2009). Development and Pathology of Pulmonary Hypertension. J. Am. Coll. Cardiol. 54, S3–S9. doi:10.1016/J.JACC.2009.04.009
Tuder, R. M., Marecki, J. C., Richter, A., Fijalkowska, I., and Flores, S. (2007). Pathology of Pulmonary Hypertension. Clin. Chest Med. 28, 23–42. doi:10.1016/J.CCM.2006.11.010
Vane, J., and Corin, R. E. (2003). Prostacyclin: A Vascular Mediator. Eur. J. Vasc. Endovasc. Surg. 26, 571–578. doi:10.1016/S1078-5884(03)00385-X
Vizza, C. D., Jansa, P., Teal, S., Dombi, T., and Zhou, D. (2017). Sildenafil Dosed Concomitantly with Bosentan for Adult Pulmonary Arterial Hypertension in a Randomized Controlled Trial. BMC Cardiovasc. Disord. 17, 1–13. doi:10.1186/S12872-017-0674-3
Wakino, S., Hayashi, K., Kanda, T., Tatematsu, S., Homma, K., Yoshioka, K., et al. (2004). Peroxisome Proliferator-Activated Receptor Ligands Inhibit Rho/Rho Kinase Pathway by Inducing Protein Tyrosine Phosphatase SHP-2. Circ. Res. 95. doi:10.1161/01.RES.0000142313.68389.92
Wang, D., Li, H., Weir, E. K., Xu, Y., Xu, D., and Chen, Y. (2019). Dimethylarginine Dimethylaminohydrolase 1 Deficiency Aggravates Monocrotaline-Induced Pulmonary Oxidative Stress, Pulmonary Arterial Hypertension and Right Heart Failure in Rats. Int. J. Cardiol. 295, 14–20. doi:10.1016/J.IJCARD.2019.07.078
Wang, Z., Jiang, X., Zhang, X., Tian, G., Yang, R., Wu, J., et al. (2019). Pharmacokinetics-Driven Optimization of 4(3H)-Pyrimidinones as Phosphodiesterase Type 5 Inhibitors Leading to TPN171, a Clinical Candidate for the Treatment of Pulmonary Arterial Hypertension. J. Med. Chem. 62, 4979–4990. doi:10.1021/ACS.JMEDCHEM.9B00123
Wharton, J., Davie, N., Upton, P. D., Yacoub, M. H., Polak, J. M., and Morrell, N. W. (2000). Prostacyclin Analogues Differentially Inhibit Growth of Distal and Proximal Human Pulmonary Artery Smooth Muscle Cells. Circulation 102, 3130–3136. doi:10.1161/01.CIR.102.25.3130
Yanagisawa, M., Kurihara, H., Kimura, S., Tomobe, Y., Kobayashi, M., Mitsui, Y., et al. (1988). A Novel Potent Vasoconstrictor Peptide Produced by Vascular Endothelial Cells. Nature 332, 411–415. doi:10.1038/332411a0
Yang, J., Davies, R. J., Southwood, M., Long, L., Yang, X., Sobolewski, A., et al. (2008). Mutations in Bone Morphogenetic Protein Type II Receptor Cause Dysregulation of Id Gene Expression in Pulmonary Artery Smooth Muscle Cells. Circ. Res. 102, 1212–1221. doi:10.1161/CIRCRESAHA.108.173567
Yang, J., Li, X., Al-Lamki, R. S., Wu, C., Weiss, A., Berk, J., et al. (2013). Sildenafil Potentiates Bone Morphogenetic Protein Signaling in Pulmonary Arterial Smooth Muscle Cells and in Experimental Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 33, 34–42. doi:10.1161/ATVBAHA.112.300121
Yang, P., Read, C., Kuc, R. E., Nyimanu, D., Williams, T. L., Crosby, A., et al. (2019). A Novel Cyclic Biased Agonist of the Apelin Receptor, MM07, Is Disease Modifying in the Rat Monocrotaline Model of Pulmonary Arterial Hypertension. Br. J. Pharmacol. 176, 1206–1221. doi:10.1111/BPH.14603
Zakrzewicz, D., and Eickelberg, O. (2009). From Arginine Methylation to ADMA: A Novel Mechanism with Therapeutic Potential in Chronic Lung Diseases. BMC Pulm. Med. 9, 1–7. doi:10.1186/1471-2466-9-5
Zhang, B., Niu, W., Xu, D., Li, Y., Liu, M., Wang, Y., et al. (2014). Oxymatrine Prevents Hypoxia- and Monocrotaline-Induced Pulmonary Hypertension in Rats. Free Radic. Biol. Med. 69, 198–207. doi:10.1016/J.FREERADBIOMED.2014.01.013
Zhang, R., Wang, X.-J., Zhang, H.-D., Sun, X.-Q., Zhao, Q.-H., Wang, L., et al. (2016). Profiling Nitric Oxide Metabolites in Patients with Idiopathic Pulmonary Arterial Hypertension. Eur. Respir. J. 48, 1386–1395. doi:10.1183/13993003.00245-2016
Zhang, S., Liu, Y., Guo, S., Zhang, J., Chu, X., Jiang, C., et al. (2010). Vasoactive Intestinal Polypeptide Relaxes Isolated Rat Pulmonary Artery Rings through Two Distinct Mechanisms. J. Physiol. Sci. 60, 389–397. doi:10.1007/S12576-010-0107-X
Zhang, T., Lai, Z., Yuan, S., Huang, Y.-Y., Dong, G., Sheng, C., et al. (2020). Discovery of Evodiamine Derivatives as Highly Selective PDE5 Inhibitors Targeting a Unique Allosteric Pocket. J. Med. Chem. 63, 9828–9837. doi:10.1021/ACS.JMEDCHEM.0C00983
Zhao, L., Long, L., Morrell, N. W., and Wilkins, M. R. (1999). NPR-A–Deficient Mice Show Increased Susceptibility to Hypoxia-Induced Pulmonary Hypertension. Circulation 99, 605–607. doi:10.1161/01.CIR.99.5.605
Zhao, L., Mason, N. A., Strange, J. W., Walker, H., and Wilkins, M. R. (2003). Beneficial Effects of Phosphodiesterase 5 Inhibition in Pulmonary Hypertension Are Influenced by Natriuretic Peptide Activity. Circulation 107, 234–237. doi:10.1161/01.CIR.0000050653.10758.6B
Zheng, Z., Yu, S., Zhang, W., Peng, Y., Pu, M., Kang, T., et al. (2017). Genistein Attenuates Monocrotaline-Induced Pulmonary Arterial Hypertension in Rats by Activating PI3K/Akt/Enos Signaling. Histol. Histopathol. 32, 35–41. doi:10.14670/HH-11-768
Zhou, C., Chen, Y., Kang, W., Lv, H., Fang, Z., Yan, F., et al. (2019). Mir-455-3p-1 Represses FGF7 Expression to Inhibit Pulmonary Arterial Hypertension through Inhibiting the RAS/ERK Signaling Pathway. J. Mol. Cel. Cardiol. 130, 23–35. doi:10.1016/j.yjmcc.2019.03.002
Zuckerbraun, B. S., Shiva, S., Ifedigbo, E., Mathier, M. A., Mollen, K. P., Rao, J., et al. (2010). Nitrite Potently Inhibits Hypoxic and Inflammatory Pulmonary Arterial Hypertension and Smooth Muscle Proliferation via Xanthine Oxidoreductase-dependent Nitric Oxide Generation. Circulation 121, 98–109. doi:10.1161/CIRCULATIONAHA.109.891077
Keywords: nitric oxide, pulmonary arterial hypertension, phosphodiesterase 5 inhibitor, proliferation, crosstalk
Citation: Tettey A, Jiang Y, Li X and Li Y (2021) Therapy for Pulmonary Arterial Hypertension: Glance on Nitric Oxide Pathway. Front. Pharmacol. 12:767002. doi: 10.3389/fphar.2021.767002
Received: 30 August 2021; Accepted: 25 October 2021;
Published: 12 November 2021.
Edited by:
Xiao-Jian Wang, Fuwai Hospital (CAS), and Peking Union Medical College, ChinaCopyright © 2021 Tettey, Jiang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Li, bHlkaWEwMzEyQGNzdS5lZHUuY24=