
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 04 October 2021
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.751095
This article is part of the Research Topic Emerging Perspectives in Sodium Channelopathies View all 9 articles
 Sophie Nicole1,2*
Sophie Nicole1,2* Philippe Lory1,2
Philippe Lory1,2The voltage-gated sodium channel Nav1.4 is a major actor in the excitability of skeletal myofibers, driving the muscle force in response to nerve stimulation. Supporting further this key role, mutations in SCN4A, the gene encoding the pore-forming α subunit of Nav1.4, are responsible for a clinical spectrum of human diseases ranging from muscle stiffness (sodium channel myotonia, SCM) to muscle weakness. For years, only dominantly-inherited diseases resulting from Nav1.4 gain of function (GoF) were known, i.e., non-dystrophic myotonia (delayed muscle relaxation due to myofiber hyperexcitability), paramyotonia congenita and hyperkalemic or hypokalemic periodic paralyses (episodic flaccid muscle weakness due to transient myofiber hypoexcitability). These last 5 years, SCN4A mutations inducing Nav1.4 loss of function (LoF) were identified as the cause of dominantly and recessively-inherited disorders with muscle weakness: periodic paralyses with hypokalemic attacks, congenital myasthenic syndromes and congenital myopathies. We propose to name this clinical spectrum sodium channel weakness (SCW) as the mirror of SCM. Nav1.4 LoF as a cause of permanent muscle weakness was quite unexpected as the Na+ current density in the sarcolemma is large, securing the ability to generate and propagate muscle action potentials. The properties of SCN4A LoF mutations are well documented at the channel level in cellular electrophysiological studies However, much less is known about the functional consequences of Nav1.4 LoF in skeletal myofibers with no available pertinent cell or animal models. Regarding the therapeutic issues for Nav1.4 channelopathies, former efforts were aimed at developing subtype-selective Nav channel antagonists to block myofiber hyperexcitability. Non-selective, Nav channel blockers are clinically efficient in SCM and paramyotonia congenita, whereas patient education and carbonic anhydrase inhibitors are helpful to prevent attacks in periodic paralyses. Developing therapeutic tools able to counteract Nav1.4 LoF in skeletal muscles is then a new challenge in the field of Nav channelopathies. Here, we review the current knowledge regarding Nav1.4 LoF and discuss the possible therapeutic strategies to be developed in order to improve muscle force in SCW.
Voltage-gated sodium (Na+) channels (Nav) initiate and conduct action potentials (AP) in excitable cells in response to membrane depolarization. The first Na+ channelopathy identified in humans was the hyperkalemic form of periodic paralysis (HYPP or HyperPP, OMIM#170500) 30 years ago (Figure 1): missense mutations in SCN4A, the gene encoding the pore-forming subunit of Nav1.4 channels, were reported as the cause of this familial form of transient muscle weakness. This pioneer demonstration was done using a combination of patch-clamp recordings from HyperPP myotubes, genetic linkage in HyperPP families and screening for mutations in SCN4A (Lehmann-Horn et al., 1983; Fontaine et al., 1990; Ptáček et al., 1991). Strengthening its prototypical role for Nav channels, Nav1.4 is also the first human Nav channel for which the 3D structure has been determined by cryo-electron microscopy (cryoEM) at the atomic resolution (Pan X. et al., 2018). Gain of Function (GoF) of Nav1.4 (i.e., an overactive channel) is now known to cause a spectrum of three clinically delineated dominantly-inherited neuromuscular disorders with overlapping clinical symptoms: sodium channel myotonia (SCM), paramyotonia congenita (PMC) and primary periodic paralyses (PP) (Cannon, 2018). They span a continuum of altered membrane excitability and form the group of muscle Na+ channelopathies, which are ultra-rare diseases with a prevalence estimated to be around 1–2/100,000 (Horga et al., 2013; Stunnenberg et al., 2018a). More than 70 GoF mutations in SCN4A, all missense, have been reported in these diseases (Cannon, 2018; Maggi et al., 2021). A few are de novo, especially those causing neonatal forms of life-threatening myotonia (severe neonatal episodic laryngospasm or SNEL, myotonia permanens) if not treated with Nav blockers (Lion-Francois et al., 2010; Lehmann-Horn et al., 2017).
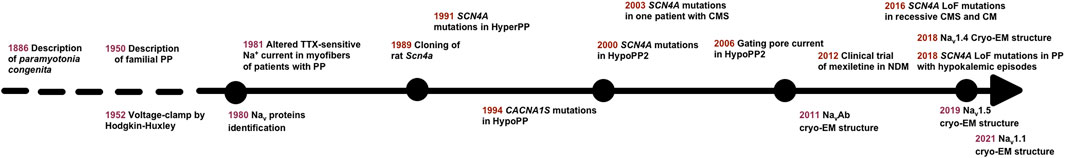
FIGURE 1. Timeline highlighting important events for Nav1.4 channelopathies. PP: periodic paralysis; TTX: tetrodotoxin; HyperPP, hyperkalemic PP; HypoPP, hypokalemic PP; HypoPP2, hypokalemic PP, type 2; CMS, congenital myasthenic syndrome; cryo-EM, cryo-electron microscopy; NDM, non-dystrophic myotonia; LoF, loss of function; CM, congenital myopathy.
The properties of SCN4A GoF mutations on Nav1.4 gating behavior have been well studied using heterologous cell expression systems, mouse models and computer simulations, providing the community with pathophysiological mechanisms. Exhaustive structure-function aspects of Nav1.4 channels, Nav1.4 GoF mutations and related channelopathies are well discussed in recent reviews and are not the scope here (Cannon, 2018; Catterall et al., 2020; Maggi et al., 2021; Mantegazza et al., 2021; Meisler et al., 2021). Briefly, all GoF missense mutations except those resulting in hypokalemic periodic paralyses (HOKPP or HypoPP, type 2 OMIM# 613,345) enhance activation or impair fast inactivation of Nav1.4. That increases Na+ influx in the myofibers, causes repetitive APs and delays muscle relaxation in SCM and PMC. Nav1.4 GoF in HyperPP leads to sustained membrane depolarization, inactivation of Nav1.4 channels and unresponsiveness of myofibers. A distinct mechanism causes the familial forms of HypoPP: the missense mutations favor an inward rectifying cation current through a gating pore that leads to sustained membrane depolarization. The HypoPP2 mutations exert also LoF effects on Nav1.4 gating with unknown physiological impact.
Recessively-inherited hypomorph (reduced function) or null (no function at all) mutations that cause Nav1.4 LoF and muscle weakness have been reported just recently, 25 years after the report of the first SCN4A mutation in HyperPP (Figure 1). These recent works have underlined the lack of efficient therapeutic solutions against Nav1.4 LoF and have drawn attention back to Nav1.4 LoF in HypoPP2. In this review, we describe some molecular aspects of Nav1.4 in skeletal muscles, present the Nav1.4 LoF mutations and discuss possible therapeutic strategies to counteract their deleterious effect on muscle force.
Nav channels are composed of one large, pore-forming α subunit with one or two auxiliary β subunits. Nine Nav isoforms (Nav1.1-Nav1.9) are described in mammals. Each isoform is characterized by its electrophysiological and pharmacological properties, as well as its tissue expression pattern. An additional and atypical Na+ channel isoform, Nax, is not voltage-dependent and arises as a different Na+ channel subfamily (Dolivo et al., 2021). The pore-forming α subunit of Nav forms a functional Na+ channel and the β subunits modulate its trafficking and electrophysiological properties. The α subunit is organized in four homologous transmembrane repeat domains (DI-DIV). Each domain contains six transmembrane segments (S1-S6) that are divided into two main functional modules: the voltage-sensing domain (S1-S4) and the pore module (S5-S6) (Figure 2). The β subunits are multifunctional glycoproteins with one single transmembrane segment, and four isoforms exist in mammals (β1-β4 encoded by SCN1B-SCN4B) (Winters and Isom, 2016).

FIGURE 2. Structure of the pore-forming α subunit of human Nav1.4 and localization of the SCN4A loss-of-function (LoF) mutations in monogenic human disorders. (A) Schematic membrane topology of Nav1.4 α subunit with four domains (DI-DIV), each domain being composed of six transmembrane segments (S1-S6). The voltage-sensor S4 segments are rich in positively-charged amino acid residues (+). The LoF mutations are shown. Hypokalemic periodic paralysis (HypoPP), orange stars; periodic paralysis (PP) with hypokalemic episodes, yellow stars; CMS, green (missense mutation) and red (nonsense or frameshift mutation) circles; CM, blue (missense mutation) and red (nonsense or frameshift mutation) squares. The sites of binding interactions with the transmembrane β1 subunit, and the cytoplasmic ankyrin, calmodulin and syntrophin proteins are indicated. (B) Alignment of amino acid sequences of the four S4 segments of human Nav1.4 with positively-charged residues (in bold). The gating charges are named R1 to R4. The position of the missense mutations causing HypoPP2 (orange), PP with hypokalemic episodes (yellow), CMS (green) and CM (blue) is indicated. Underlined residues cause different phenotypes when substituted: dominant NDM or recessive CM (p.Arg225); de novo HypoPP2, recessive HypoPP2 or CM (p.Arg1135); dominant NDM, glucocorticoid-induced HypoPP, and dominant or recessive PP with hypokalemic episodes (p.Arg1451). To note that the substitutions of p.Arg675 (R3, DIIS4) and p.Arg1135 (R3, DIIIS4) cause HypoPP2 or normokalemic PP with corticosteroid- or thyrotoxicosis-induced hypokalemic episodes of paralysis and depolarization- and not hyperpolarization-activated gating pore current (Vicart et al., 2004; Sokolov et al., 2008; Groome et al., 2014).
Nav channels have three main distinct conformational states: resting (closed), activated (open), inactivated (closed). The kinetics of each transition, i.e. activation (from resting to activated), inactivation (from activated to inactivated) and recovery from inactivation (from inactivated to resting) shape the electrophysiological behavior of Nav channels and critically determine AP frequency. Inactivation includes two distinct components: fast-inactivation and slow-inactivation, fast-inactivation being the main feature of Nav channels. Nav channels activate rapidly (in less than 1 ms) in response to membrane depolarization. The major structural determinants in this process are the S4 transmembrane segments. They are composed of positively charged arginine (or lysine) residues, also called gating charges, occurring in a Arg/Lys-X-X repetitive sequence (Figure 2). Upon depolarization, the S4 helices undergo outward movements by electrostatic interactions of their basic residues with conserved acidic or polar residues of S2 and S3 segments (Catterall et al., 2020). This outward movement drives the conformational shifts of the channel in response to depolarization with opening of the central (also known as α) pore formed by the P-loops of the four S5–S6 segments. The open central pore then conducts a large inward Na+ current that drives the depolarization phase of AP. Within a few milliseconds following their activation, Nav channels undergo fast inactivation, which facilitates membrane repolarization. Fast inactivation is mainly dictated by the movement of DIVS4 during activation (Capes et al., 2013). During this conformational change, the inactivation gate formed by the cytoplasmic linker between DIII and DIV intracellularly blocks the central pore. Prolonged or high-frequency depolarizations drive Nav channels into a slow-inactivated state with time constant ranging from 100 ms to several minutes. Slow inactivation determines long-lasting Nav channels availability, thereby defining the cell firing properties. Its structural basis remains unclear. It probably relies on several sequential conformational changes including constriction of the pore by rearrangement of the ion selectivity filter and S6 segments (Mantegazza et al., 2021).
The force developed by a muscle from a single twitch to its maximal sustained contraction (known as tetanus) resulting from the twitch summation relies on the AP frequency, and by this way on the functional properties of Nav1.4. In the adult myofiber, the AP is generated at the neuromuscular junction (NMJ) located in the middle of the myofiber when the endplate potential (epp), a local depolarization induced by the opening of the post-synaptic nicotinic acetylcholine receptors (nAChRs), reaches the threshold (-50 mV) for AP genesis. To decrease the effective threshold for AP generation and favor the latter, the amplitude of Nav currents is higher at the NMJ than in the extrasynaptic area (80 mA/cm2 against 20 mA/cm2, adult fast-twitch myofibers of mouse Levator auris longus) (Figure 3) (Lupa et al., 1993; Slater, 2003). The high synaptic Na+ current density results from the clustering of Nav channels at the NMJ, more specifically in the depth of the postsynaptic folds while the tops of the folds are enriched in AChRs (Figure 3). The AP generated at the NMJ then propagates longitudinally toward the extremities and radially through the transverse tubules to drive excitation-contraction coupling and myofiber contraction. In the extrasynaptic area, the distribution of Nav channels between the “surface” and transverse tubules sarcolemma is estimated to be in the range between 40:60 and 60:40 (DiFranco and Vergara, 2011).
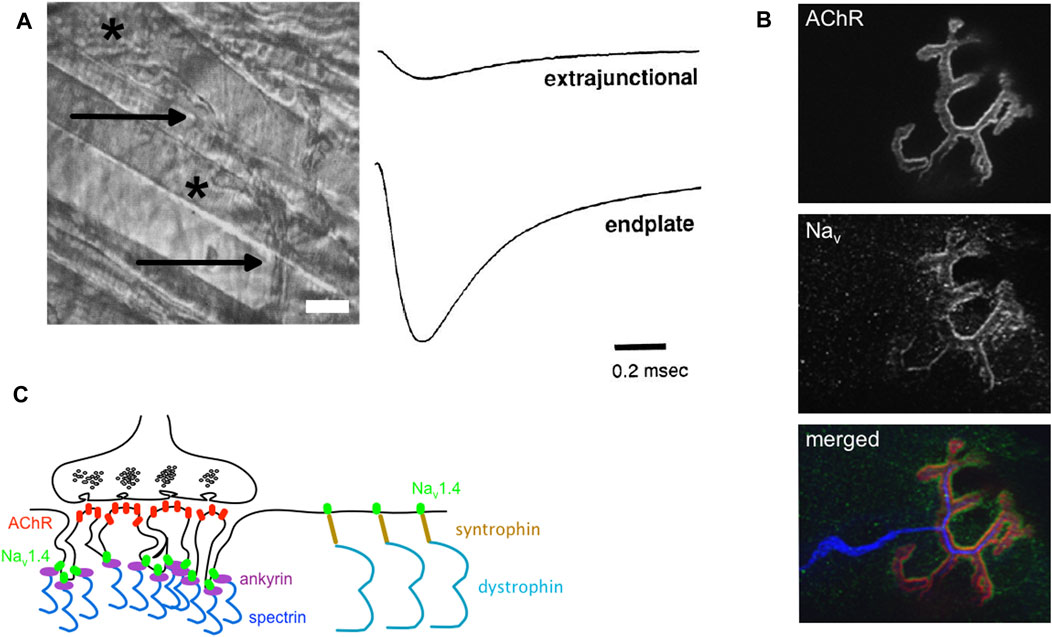
FIGURE 3. Nav channels, Na+ current and neuromuscular junctions. (A) Left: a phase-contrast image of an adult mouse Levator aureus longus (LAL, fast-twitch) muscle. The myelinated nerve (arrows) terminates at neuromuscular junctions (NMJs, stars) (scale bar, 15 μm). Right: examples of Na+ current recordings made with a loose-patch clamp electrode from the extrasynaptic membrane (extrajunctional, top trace) and synaptic (endplate, bottom trace) membrane of a LAL myofiber. The extrasynaptic Na+ current density is equal to 23.3 mA/cm2 and the endplate Na+ current density is equal to 110 mA/cm2 (adapted from (Lupa et al., 1993); Copyright [1993] Society for Neuroscience). (B) Confocal image of fluorescent staining of nAChRs (stained with α bungarotoxin, in red in the merged image), Nav channels (anti-pan antibody binding to all Nav isoforms, in green in the merged image), and nerve (anti-NF 200 and anti-synaptophysin antibodies, in blue in the merged image) on dilacerated Tibialis anterior (fast-twitch) myofibers from an adult mouse (X 63). (C) Schema of one NMJ with its presynaptic (nerve terminal rich in synaptic vesicles, up) and post-synaptic (myofibers with post-synaptic folds, down). AChRs (red) are clustered at the top and Nav channels (Nav1.4 in green) are clustered in the depth of the postsynaptic folds. The synaptic aggregation of Nav1.4 would result from its binding interaction with ankyrins (purple), themselves linked to spectrin (deep blue). Nav1.4 would interact with syntrophin (brown) in the extrasynaptic sarcolemma.
Two Nav currents exist in skeletal myofibers. In innervated adult myofibers of rodents, a tetrodotoxin (TTX)-sensitive Na+ current (IC50 5 nM) generated by Nav1.4 (formerly SkM1) accounts for 98% of the whole inward Na+ current (Fu et al., 2011). In immature neonatal myofibers, this current is low or null compared to a TTX-resistant Na+ current (SkM2, IC50 2000 nM). The TTX-resistant current is mediated by Nav1.5 encoded by SCN5A, which is the main cardiac Nav isoform (Loussouarn et al., 2015). In addition to distinct TTX sensitivity, these two Na+ currents have different electrophysiological properties with higher activation time constants and lower conductance for Nav1.5 current compared to Nav1.4 current (Morel et al., 2010). The switch between Nav1.5 and Nav1.4 occurs between birth and the third postnatal week in rodents and is concomitant to the maturation of NMJs and myofibers (Lupa et al., 1993). The neonatal lethality of children and mice with bi-allelic null mutations in SCN4A demonstrates that Nav1.5 does not compensate for the lack of Nav1.4 in skeletal muscles at birth (Wu et al., 2016; Zaharieva et al., 2016).
Not all myofibers are equal regarding excitability and Nav current properties. Slow-twitch motor units in mammals are active at 10–20 Hz and fast-twitch motor units are active at rates from 40 to 90 Hz to more than 200 Hz for brief periods (Hennig and Lømo, 1985). To assume these high frequencies, the Na+ current density is two-to six-fold higher in fast-than in slow-twitch myofibers (Ruff, 1996; Desaphy et al., 2001). The difference in synaptic versus extrasynaptic Na+ current density is also more important in fast-twitch compared to slow-twitch myofibers (Milton et al., 1992). Slow inactivation of Na+ current is less prominent in slow-twitch fibers (Ruff, 1996). These distinct characteristics probably play a role in the excitability and contractility properties of myofibers such as threshold potential, time to reach contraction threshold or resistance to fatigue.
It is here important to note inter-species differences. For example, the time constants for entry or recovery from slow inactivation are reported to be 4 times faster in human than in rat myofibers (Ruff, 1996). Individual human muscles tend to be a mixture of various fiber types. Whether and when the Nav1.5/Nav1.4 switch occurs in humans is not documented. Human NMJs are smaller and have more extensive postsynaptic folds than rodent NMJs, which may have functional significance since the NMJ size and the spacing and depth of the postsynaptic folds have an important effect on AP genesis (Jones et al., 2017; Slater, 2017). Moreover, proteomics analyses have recently provided evidence that human NMJs have a significantly modified molecular composition compared to mouse NMJs (Jones et al., 2017). These differences must be taken into consideration when interpreting animal-based studies with respect to their applicability to humans.
Protein interactions of Nav channels modulate their trafficking, cellular localization and functional properties. Little is known about these important functional aspects for Nav1.4 compared to other Nav channels (Loussouarn et al., 2015; Eshed-Eisenbach and Peles, 2020). If sequence similarities suggest the possibility that Nav1.4 shares binding partners and regulatory mechanisms with other Nav isoforms, further investigations are required to prove this assumption and to best understand the fine-tuning of Nav1.4 in skeletal myofibers.
One regulatory β subunit, mainly β1 (encoded by SCN1B), associates non-covalently with the Nav1.4 α subunit (Yang et al., 1993). The β2 (SCN2B), β3 (SCN3B) or β4 (SCN4B) subunits might also contribute to the Nav1.4 channel complex since their transcripts are detected in muscle cells (David et al., 2008; Morel et al., 2010). Two extracellular binding sites (DIS5S6 and DIVS5S6) of Nav1.4 are involved in the interaction with β1 (Figure 2) (Makita et al., 1996). Different effects of the interaction of αNav1.4 with β1 on Na+ current have been reported depending on the heterologous cell expression system. They include increased current density, enhanced inactivation with faster kinetics and hyperpolarization shift (Cannon et al., 1993). The differences between cell expression systems may result from distinct β1 glycosylation, which would modify Nav channel gating through the density of the surrounding surface charges (Ferrera and Moran, 2006). No mutations in the genes encoding β subunits are reported to cause disorders with altered skeletal muscle force (Winters and Isom, 2016). It remains to be determined whether this is due to a lower sensitivity of Nav1.4 to functional modulation by β subunits in myofibers compared to other Nav isoforms, to compensation between β isoforms or to the subjective nature of mild variations in muscle force, which may not be considered as “pathogenic” by individuals suffering from them.
The mechanisms leading to the synaptic accumulation of Nav1.4 are still unclear compared to those resulting in the clustering of neuronal channels at axonal initial segments or nodes of Ranvier (Eshed-Eisenbach and Peles, 2020). Synaptic aggregation of Nav1.4 channels would result from their binding interaction with ankyrins (Figure 3). Ankyrins are scaffolding proteins that link membrane-bound proteins to the underlying spectrin-actin cytoskeleton. Ankyrins have a pivotal role in the anchorage of Nav channels to the submembranous cytoskeleton through their binding interaction with a motif of 27 amino acid residues located within the cytoplasmic DII-DIII linker of Nav channels (Figure 2) (Garrido et al., 2003). A recent work has confirmed that ankyrins are required to cluster Nav channels in the depth of synaptic folds since the lack of ankyrins G, R and B induced loss of Nav staining at the NMJ (Zhang et al., 2021). The clustering of Nav channels in the depth of the folds is supposed to result from the physical exclusion of the ankyrin-Nav1.4 complexes from the tops rich in nAChRs during NMJ formation maturation (Flucher and Daniels, 1989; Bailey et al., 2003).
Nav1.4 also interacts with α syntrophin, a peripheral membrane protein localized to the cytosolic face that links a variety of signaling proteins and ion channels to the dystrophin-associated protein complex (Figure 3) (Gee et al., 1998). The C-terminal tail of Nav1.4 possesses a SerLeuVal peptidic sequence that binds the PDZ (for « PSD-95, Dlg1, Zo-1 ») domain of syntrophin (Figure 2). The synaptic immunostaining of syntrophin is distinct from Nav immunostaining at the NMJ, suggesting that their interaction solely takes place in the extrasynaptic region (Bailey et al., 2003). The syntrophin-Nav1.4 interaction could modify the gating properties of Nav1.4 and participate to loss of muscle force in some acquired or inherited myopathic conditions, such as critical illness myopathy or Duchenne muscular dystrophy, a disease acquired by patients in intensive care units and an inherited disorder characterized by progressive muscle degeneration and weakness due to the lack of dystrophin, respectively (Teener and Rich, 2006; Hirn et al., 2008; Kraner et al., 2012).
Calmodulin is one of the rare proteins well known to modulate Nav1.4 gating (Nathan et al., 2021). This intracellular multifunctional protein is ubiquitously expressed and acts as part of a Ca2+ signal transduction pathway by modifying its interactions when bound to Ca2+. Calmodulin interacts with an IQ domain downstream of an EF-hand domain located in the cytoplasmic C-terminal tail of Nav1.4 (Figure 2). The interaction of Nav1.4 with calmodulin facilitates its cell surface expression and mediates its Ca2+-dependent slow inactivation (Biswas et al., 2008; Yoder et al., 2019). The crystal structure of Ca2+-calmodulin bound to Nav1.4 suggests that the Ca2+-dependent inactivation of Nav1.4 would result from the relative reorientation of its EF-hand and IQ domains (Gardill et al., 2019). Increasing the intracellular pool of Ca2+ released from the sarcoplasmic reticulum inhibits Na+ currents in mouse skeletal muscles (Sarbjit-Singh et al., 2020; Liu et al., 2021). These data suggest activity-dependent feedback mechanisms of Nav1.4 activity by intracellular Ca2+ released from the transverse tubules for a fine tuning of muscle contraction during periods of repetitive activity.
To complete this list of Nav1.4 binding interaction, it is worth noting that the α subunits of Nav1.5, Nav1.1, Nav1.2 and Nav1.7 form functional dimers (Clatot et al., 2017; Rühlmann et al., 2020). The dimerization involves a 20 amino acid motif within the DI-DII cytoplasmic loop and is mediated by the 14-3-3 protein. Nav1.4 is the sole Nav channel lacking this motif, suggesting that functional dimerization does not occur for this isoform (Rühlmann et al., 2020).
The human SCN4A gene (24 exons, chromosome 17q23.3) codes for a single mRNA transcript transcribed into a protein of 1,836 amino acids (208 kDa). SCN4A is referred to as the skeletal muscle isoform since its cDNA cloning 30 years ago (Trimmer et al., 1989). A small expression of SCN4A in heart tissue and cardiac myocytes is reported (1.1% of relative Nav channels transcript levels in humans) (Blechschmidt et al., 2008; Kaufmann et al., 2013). The tissue-specific expression of SCN4A most probably explains why Nav1.4 dysfunction only impacts skeletal muscles even if anecdotic reports of cardiac arrhythmia in individuals with dominant SCN4A mutations have been published (Loussouarn et al., 2015).
In rodents, Scn4a expression is developmentally regulated. The amount of Scn4a transcripts increases from birth to 2 weeks after birth while Scn5a (the gene encoding the pore-forming subunit of Nav1.5) expression decreases (Lupa et al., 1993). This developmental period is concomitant to the maturation of the neuromuscular system, especially the NMJ, in rodents (Li et al., 2018). The high density of Nav channels at the NMJ would result in part from the accumulation of Scn4a mRNA at the synapse (Lupa et al., 1993; Awad et al., 2001; Bailey et al., 2003). However, Scn4a expression is insensitive to denervation in the adult myofiber, suggesting that its synaptic expression is not strictly regulated by the nerve terminal (Lupa and Caldwell, 1991; Awad et al., 2001; Carreras et al., 2021). Northern blot analyses have showed that Scn1b expression parallels Scn4a expression during postnatal development and after denervation in rodents, suggesting that common factors regulate their gene expression (Yang et al., 1993). Whether similar modulations of SCN4A and SCN1B gene expression with NMJ development and denervation exist in humans has not been studied.
The specific expression of Scn4a in differentiated myofibers is controlled by at least 4 cis-regulatory promotor elements working together: 1) a core promoter that lacks muscle specificity; 2) a repressor that confers muscle specificity; 3) a muscle-specific positive element, whose accessibility is partially masked by the repressor; and 4) another muscle-specific positive element that confers a 10-fold up-regulation of expression (Hebert et al., 2007; Kraner et al., 1999; Kraner et al., 1998). The cumulative binding of non-specific (Nuclear Factors (NF) I, ZEB/AREB6 and REB) and muscle-specific (myogenin and MRF4) transcription factors on these elements would contribute to the upregulation of Scn4a expression in myofibers. According to the fact that denervation does not modify its expression, no N-box—a response element that promotes synaptic-specific expression in response to the neuronal factors agrin and neuregulins for NMJ formation and maturation (Belotti and Schaeffer, 2020) — is present within the promotor of Scn4a, suggesting the involvement of other regulatory mechanisms. The simplest hypothesis is a higher synaptic amount of Scn4a mRNA due to the high number of myonuclei in this area.
Several transcripts exist for SCN5A but none is specific to skeletal myofibers. Nav1.5 GoF or LoF due to SCN5A mutations cause cardiac arrhythmia (Long QT syndrome (LQTS), type 3 and Brugada syndrome, respectively) but are not reported to cause neuromuscular symptoms. In contrast to Scn4a, Scn5a expression is sensitive to the innervation pattern of the myofibers with downregulation during postnatal development and strong upregulation when the adult myofiber is denervated (Awad et al., 2001; Carreras et al., 2021). The upregulation of Scn5a upon denervation results from the binding of the Gata4 transcription factor, itself upregulated following denervation, to the Scn5a promoter region, and from epigenetic regulations mediated by the H3K27ac and H3K4me3 histones (Carreras et al., 2021).
Familial hypokalemic periodic paralysis (HypoPP) is a rare condition characterized by the episodic occurrence of moderate to severe muscle weakness, which may be focal or generalized, concomitantly to low blood K+ levels (<3,5 mEq/L). The attacks are usually triggered by rest after strenuous exercise, carbohydrate-rich meal, or stress. Each attack has a gradual onset over minutes and resolves spontaneously after few hours or days (Statland et al., 2018). Familial and non-familial forms of HypoPP exist. Non-familial forms are most often secondary to another physiological dysfunction such as thyrotoxicosis, hyperaldosteronism or nephropathic K+ loss. Familial forms are dominantly-inherited and typically develop in the first or second decade of life. Inter-critical electromyographic testing based on compound muscle APs (CMAP) recording demonstrates a decrease of CMAP amplitudes in response to a 5 min-long exercise, which may help to document muscle weakness between episodes (McManis et al., 1986; Fournier et al., 2004). The definitive diagnosis of HypoPP lies on genetic testing with mutations in the genes known to cause HypoPP: CACNA1S, which encodes the pore-forming subunit of the skeletal muscle voltage-gated calcium channel Cav1.1 (HypoPP1), or SCN4A (HypoPP2) (Jurkat-Rott et al., 1994; Ptáček et al., 1994; Jurkat-Rott et al., 2000). Dominant-negative mutations in the KCNJ2 gene, encoding the K+ channel Kir2.1, also cause primary HypoPP in Andersen-Tawil syndrome, a disorder clinically distinct from HypoPP1 and 2 since the neuromuscular phenotype is associated with cardiac arrhythmia and bone deformities (Plaster et al., 2001).
Fourteen SCN4A missense mutations are reported to cause HypoPP2. Eleven substitute the two arginine residues (R1 or R2) nearest the extracellular end of S4 segments in DI, DII and DIII and cause HypoPP2 that are dominantly-inherited (Figure 2 and Table 1). The exception are p.Arg1135His/Cys and p.Arg1451Leu. The p.Arg1135His/Cys mutations substitute the 3rd arginine residue of DIIIS4 and cause de novo (p.Arg1135His) or recessively-inherited (p.Arg1135Cys) HypoPP2 (Groome et al., 2014). P.Arg1451Leu substitutes the 1st arginine residue of DIVS4 and results in HypoPP2 and myotonia in one homozygous individual (Groome et al., 2014; Luo et al., 2018). HypoPP2 mutations that substitute R1 or R2 as well as p.Arg1135His/Cys, but not p.Arg1451Leu, promote a gating pore current (Sokolov et al., 2007; Jurkat-Rott et al., 2009; Wu et al., 2012). The gating pore current (also named omega current) is a cation-selective inward current activated by hyperpolarization through an aqueous pathway created by the neutralization of one of the two outermost positive charges of DIS4, DIIS4 or DIIIS4 in Nav1.4 (Gosselin-Badaroudine et al., 2012). This small inward current explains the paradoxical depolarization characteristics of HypoPP myofibers: they are more frequently depolarized than control myofibers (resting membrane potential (RMP) equal to −55 mV instead of −90 mV) in low extracellular K+ (Rüdel et al., 1984). To simplify complex pathophysiological mechanisms, hypokalemia would be induced by muscle K+ reuptake after exercise or in response to insulin or glucocorticoid (such as cortisol) secretion following carbohydrate-rich meals or stress, respectively. Hypokalemia would modify the cumulated activities of K+ (especially Kir2.1) and Cl- channels, ATPase pumps and Na-K-2Cl (NKCC) transporters in myofibers (Jurkat-Rott et al., 2009; Wu et al., 2011; Mi et al., 2019). These changes would establish the net balance of no ionic current at the depolarized (−55 mV) value in HypoPP2 myofibers because of the gating pore current. Nav1.4 channels would then be mostly inactivated, and the myofibers not excitable (Rüdel et al., 1984; Struyk and Cannon, 2008; Jurkat-Rott et al., 2009). The HypoPP2 mutations could therefore be considered as dominant-negative, the gating pore current altering the central pore current of mutant and wild-type (WT) Nav1.4 channels.
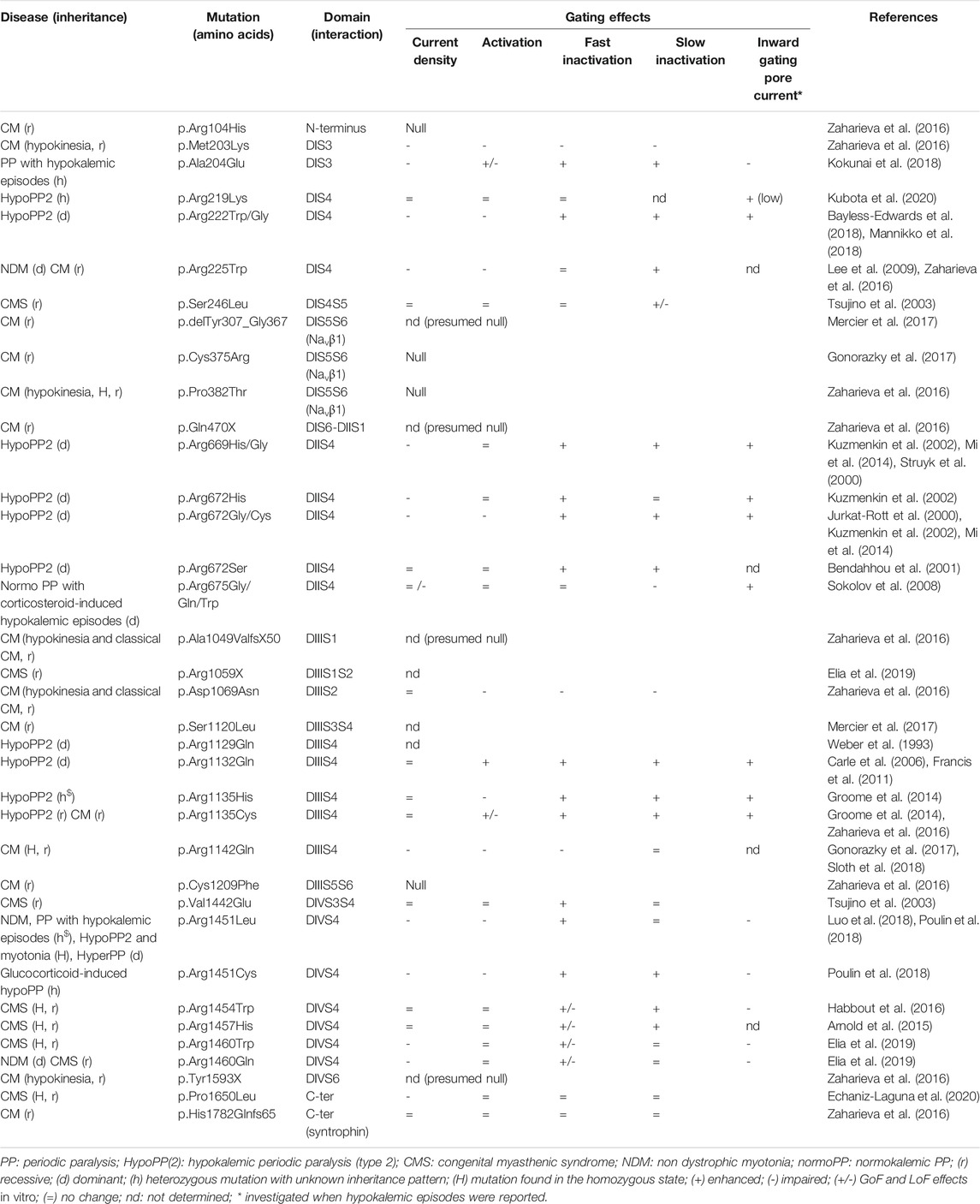
TABLE 1. SCN4A mutations with loss-of-function effects on Nav1.4 gating behavior.
HypoPP2 mutations causing gating pore current have also been reported to cause LoF effects on Nav1.4 gating in heterologous cell expression systems (Bayless-Edwards et al., 2018; Bendahhou et al., 2001; Groome et al., 2014; Jurkat-Rott et al., 2000; Mi et al., 2014; Struyk et al., 2000). The LoF changes include reduced Na+ current density and enhanced fast and slow inactivation (Table 1). Whether they participate to the loss of muscle force in HypoPP2 is an open question. One argument for the physiological significance of these LoF changes is the smaller amplitude and the slower rate of rise of muscle APs in HypoPP2 myofibers compared to control myofibers at normal (−90 mV) RMP (Jurkat-Rott et al., 2000; Groome et al., 2014; Bayless-Edwards et al., 2018). Moreover, introducing one HypoPP2 missense mutation (p.Arg669His) into the mouse Scn4a gene by homologous recombination reproduces LoF changes on muscle APs in addition to the pathogenic gating pore current (Wu et al., 2011). A second argument comes from recent reports of SCN4A missense mutations that do not induce gating pore current in individuals suffering from PP with hypokalemic episodes of muscle weakness (Kokunai et al., 2018; Luo et al., 2018; Poulin et al., 2018). Two individuals are heterozygous for one missense substitution (p.Ala204Glu, p.Arg1451Leu) whereas one is homozygous for one of these two substitutions (p.Arg1451Leu). The recurrence of p.Arg1451Leu in unrelated individuals underlines the link between the functional changes induced by this SCN4A mutation and hypokalemic episodes of paralysis. P.Ala204Glu and p.Arg1451Leu exert GoF effects with enhanced activation. They also exert LoF effects with reduced Na+ current density, slower activation kinetics, accelerated entry into fast inactivation and slower recovery from slow inactivation for mutant channels compared to WT channels (Kokunai et al., 2018; Luo et al., 2018). For one (p.Ala204Glu), LoF changes were more severe in low K+ext with a depolarization shift of activation observed for the mutant but not the WT channels (Kokunai et al., 2018). Recently, a charge-retaining substitution in DIS4 (p.Arg219Lys) has been reported to cause HypoPP2 by a mechanism distinct from gating pore current as the latter was too small to be predicted pathogenic (Kubota et al., 2020). Current density, activation and inactivation of Nav1.4 were unaffected by p.Arg219Lys, but slow inactivation was not investigated. If these works demonstrate that Nav1.4 LoF may result in hypokalemic episodes of muscle paralysis, it cannot be excluded that other genetics and/or environmental factors are important for their expressivity since they were not observed in familial forms.
The demonstration that Nav1.4 LoF causes loss of muscle force has been provided in the last 5 years with the identification and the functional investigation of recessively-inherited SCN4A mutations in children with two forms of congenital muscle weakness: congenital myasthenic syndrome (CMS) and congenital myopathy (CM). Twenty-three recessively-inherited LoF mutations are now described in SCN4A (Figure 2 and Table 1). In contrast to the situation observed for dominantly-inherited Nav1.4 channelopathies, no recurrent SCN4A mutations have been described for recessively-inherited Nav1.4 channelopathies, the reported mutations being private. Both hypomorph (that causes a partial LoF) and null (that causes a complete LoF) mutations are observed.
Congenital myasthenic syndromes (CMS [MIM#608931]) form a clinically and genetically heterogeneous group of inherited disorders with skeletal muscle weakness that worsens with physical exertion (Engel et al., 2015; Nicole et al., 2017). They are caused by defective neurotransmission at the NMJ due to presynaptic (nerve terminal), synaptic cleft or most frequently post-synaptic (muscle) defects. Neurotransmission defects are documented by electromyographic testing showing a decrement of CMAP in response to repetitive nerve stimulation (RNS) at low (3 Hz) frequency or by single fiber electromyography (SFEMG) testing, a technique that detects neuromuscular transmission failure in motor units (Engel et al., 2015; Nicole et al., 2017). Thirty genes are known to cause CMS, which encode proteins that are critical for the formation, the maturation, the maintenance or the function of the NMJ such as post-synaptic nAChRs or proteins required for the synaptic aggregation of these receptors (agrin, Dok-7, rapsyn, MuSK, … ). CMS are most frequently recessively-inherited but dominant forms exist such as the slow-channel CMS that result from GoF mutations in the genes encoding nAChR subunits.
The first report of SCN4A mutations as the cause of fatigable muscle weakness suggestive of CMS was published nearly 20 years ago but the inheritance pattern (dominant or recessive) could not be established (Tsujino et al., 2003). This work reports one individual with abrupt paralytic attacks of respiratory and bulbar muscles in the neonatal period and general muscle weakness worsened by activity later in life. The patient was heterozygous for two SCN4A missense mutations (p.Ser246Leu and p.Val1442Glu). Electromyographic testing showed CMAP decrement at high (10 and 50 Hz) but not at low (2 Hz) RNS. One substitution (p.Val1442Glu) has strong LoF effects with enhanced fast inactivation, predicting that only 13% of Nav1.4 channels are available for activation at the RMP. The second substitution (p.Ser246Leu) was concluded to be benign with mixed enhancing and impairing effects on slow inactivation. The inheritance pattern of the disease remained therefore unclear. The existence of recessively-inherited SCN4A mutations as the cause of fatigable muscle weakness has indeed been proven only recently. Four original articles have reported eight homozygous or compound heterozygous missense mutations in six unrelated individuals (Arnold et al., 2015; Habbout et al., 2016; Elia et al., 2019; Echaniz-Laguna et al., 2020). Symptoms suggestive of CMS with a decrement of CMAP at high (10–50 Hz; 2 out 6 patients tested) but not at low (3 Hz) RNS and jitter at SFEMG (2 out 2) were observed. They were combined to symptoms of PP, CM or even myotonia for p.Arg1460 substitutions. The decrement of CMAP in response to the long-exercise test was reported for 4 out 5 patients, and mild fiber size variabilities on muscle biopsies were observed for 2 out 4 examined.
Six of the eight SCN4A substitutions resulting in CMS are in or close to DIVS4, suggesting that this segment may be a hot-spot for SCN4A mutations causing fatigable muscle weakness (Figure 2). Four neutralize positively-charged residues and exert mixed GoF and LoF effects on fast inactivation (Table 1). They do not induce gating pore current in accordance with previous studies showing that the substitution of a single positively-charged residue in DIVS4 does not induce this current (Gosselin-Badaroudine et al., 2012). All the mutations investigated in patch-clamp experiments are LoF with reduced Na+ current density and/or enhanced fast or slow inactivation (Figure 4). Repetitive pulses at physiological frequencies (10–80 Hz) on heterologous cells induce a decrease of Na+ current amplitude for p.Val1442Glu, p.Arg1454Trp and p.Arg1457His mutant channels but not for WT or p.Ser246Leu channels (Figure 4) (Tsujino et al., 2003; Arnold et al., 2015; Habbout et al., 2016). Decrease of Na+ current in response to pulse trains (60–100 Hz) was on the contrary less pronounced for p.Arg1460Trp/Gln mutant channels than for WT channels (Elia et al., 2019). This may be due to the GoF effects of these variations, and may account for myotonia in individuals heterozygous for p.Arg1460Trp/Gln.
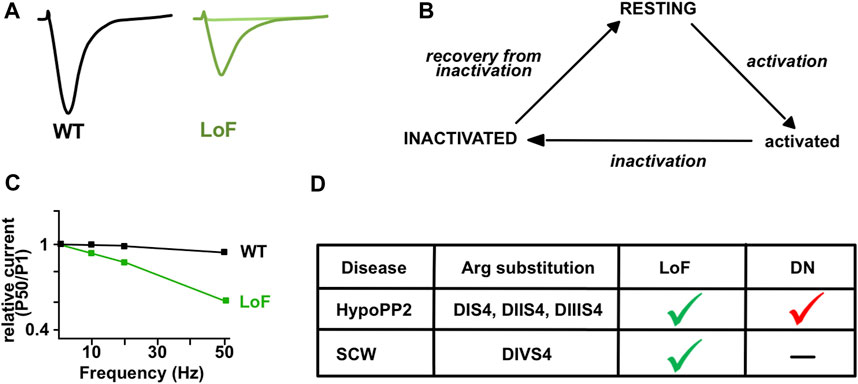
FIGURE 4. Gating of Nav1.4 with sodium channel weakness (SCW) mutations. (A) Illustrative Na+ current traces for wild-type (WT, black) and mutant (LoF, green) Nav1.4 channels obtained in HEK-293 cells. Differences include reduced current density, impaired activation, and enhanced inactivation for hypomorph mutants (dark green). No current is recorded for null mutants (light green). (B) Three-states model of Nav1.4 channels. The resting or inactivated states are favored in SCW. (C) Relative Na+ current amplitude (pulse 50 compared to pulse 1) is observed in response to pulse trains at 10, 20 and 50 Hz for the mutant (LoF, green) but not the WT (black) channels in heterologous cells (adapted from Habbout et al., 2016). (D) Main differences between dominantly-inherited HypoPP2 mutations and recessively-inherited SCW mutations substituting Arginine (Arg) residues in S4 segments of Nav1.4. The gating pore current induces a dominant-negative (DN) effect on central Nav currents in HypoPP2. Loss of function (LoF) occurs in both.
Congenital myopathies (CM) are another group of genetically and clinically heterogeneous diseases with muscle weakness (Gonorazky et al., 2018). There are many different types of CM with more than 30 known causative genes required for skeletal muscle development, structure or function. Most share common features, including lack of muscle tone and weakness. The degree of severity is wide, ranging from severe muscle weakness with in utero or neonatal lethality, to infant- or childhood-onset weakness (Wallgren-Pettersson and Laing, 2010). Several tests are performed to diagnose CM, including blood tests, electromyography, muscle biopsy and genetic testing. Fifteen recessive SCN4A mutations have been reported in nine unrelated kindreds with various severities of muscle weakness ranging from fetal hypokinesia (decreased or absent movements in utero) lethal at birth to “classical” CM for 11 individuals (Zaharieva et al., 2016; Gonorazky et al., 2017; Mercier et al., 2017; Sloth et al., 2018). Electromyographic investigations showed myopathic changes for some but not all individuals with classical CM (5 out 7). Marked fatigability with CMAP decrement at high (10Hz; 1 out 2 patients tested) but not at low (3 Hz) RNS was reported for some patients with “classical” CM (Zaharieva et al., 2016). When done, muscle biopsies showed fiber size variability, some necrotic myofibers and increased nuclear internalization but no specific structural abnormalities.
Mutations causing CM are observed on the entire length of Nav1.4 (Figure 2; Table 1). Four are supposed to be null as they delete several segments. Four missense substitutions (p.Arg104His, p.Cys375Arg, p.Pro382Thr, p.Cys1209Phe) are functionally null as they do not produce any Na+ current in heterologous cells. Two are in one (DIS5S6) binding site for β1 subunit. Whether these missense mutations are null because of defective membrane trafficking or abnormal gating of the mutant channel is unknown. Four substitutions are hypomorph since mutant channels are still functional but have greatly reduced Na+ current density or impaired activation. One (p.Arg1135Cys) is reported to cause a recessively-inherited HypoPP2 phenotype and a gating pore current in vitro (Groome et al., 2014). Another mutation (p.Arg225Trp) has been reported to be heterozygous in one Korean patient with SCM but does not display GoF characteristics in vitro (Lee et al., 2009). One frameshift mutation (p.His1782Glnfs65) does not induce any gating defect in heterologous cells. This frameshift mutation results in the loss of the syntrophin binding site in the C-terminal tail of Nav1.4 and its effect on the membrane expression of Nav1.4 in myofibers remains to be determined. That strengthens the important notion that Nav1.4 gating defects in vitro do not always fit the phenotypic expression. Moreover, the in vitro effects of pulse trains on Na+ current amplitude has not been tested for the SCN4A mutations causing CM.
Sodium Channel Weakness (SCW). LoF mutations that favor non-conducting (resting or inactivated) states of Nav1.4 result in CMS, CM, and unusual forms of PP with hypokalemic episodes that are not due to a gating pore current (Figure 4). If PP, CMS and CM are distinct muscle disorders, the overlapping symptoms observed for individuals with Nav1.4 LoF mutations suggest that they form a clinical spectrum with a continuum of membrane hypoexcitability as proposed for Nav1.4 channelopathies due to GoF mutations (Figure 5) (Cannon, 2018). We propose here to name this spectrum “Sodium Channel Weakness (SCW)” as the clinical mirror of Sodium Channel Myotonia (SCM). A dosage-effect could occur in SCW that would explain the variable severity of muscle weakness: the fewer Nav1.4 would be functional, the more severe would be the muscle weakness. Additional individuals with Nav1.4 LoF mutations must be reported to ascertain this hypothesis important for genetic counseling and therapeutic issues. No specific biomarker (clinical signs, electromyographic testing or muscle biopsies investigations) has been identified for SCW, and the diagnosis of congenital muscle weakness due to Nav1.4 dysfunction requires to perform sequencing of SCN4A and to identify pathogenic variations.
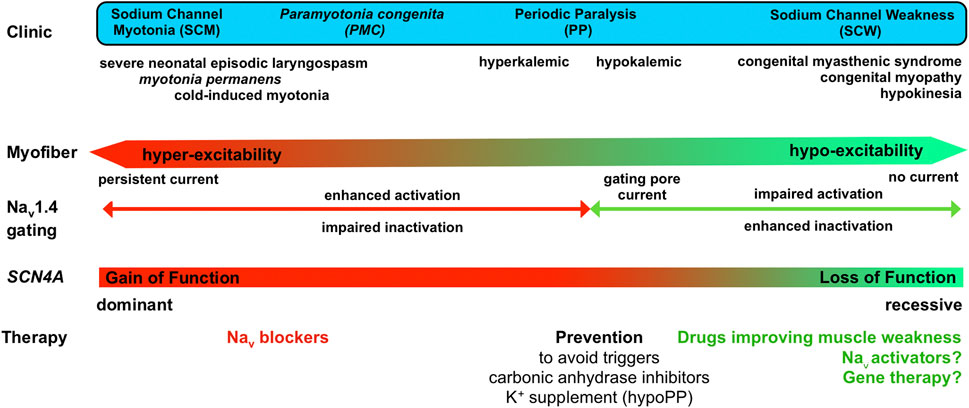
FIGURE 5. Clinical spectrum of Nav1.4 channelopathies with a continuum from membrane hyperexcitability in sodium channel myotonia (SCM) to hypoexcitability in sodium channel weakness (SCW), effect of the mutations on Nav1.4 gating and available as well as potential (?) therapeutic options.
One argument in favor of the dosage-effect hypothesis for SCW is the phenotype of Scn4a knock-out mouse mutants (Wu et al., 2016). Homozygous knock-out mice die at birth. Heterozygous mice have no gross locomotor deficits despite a half-reduced TTX-sensitive Na+ current recorded in dissociated myofibers. Loss of muscle force in response to factors inducing episodes of muscle weakness in PP was not observed in myofibers of heterozygous Scn4a+/- mice despite the reduced Na+ current (Wu et al., 2016). Scn4a haploinsufficiency has nevertheless a functional significance: if the tetanic force developed in response to supramaximal stimulus is like controls, a higher stimulating current density is required to achieve 50% maximal force in Scn4a+/- myofibers. Moreover, force decline is observed for Scn4a+/- myofibers in response to submaximal stimuli with higher sensitivity to partial blockade of neuromuscular transmission with curare (an antagonist of nAChR) compared to controls (Wu et al., 2016). This physiological work demonstrates that a half-reduction of functional Nav1.4 channels in myofibers is not sufficient to induce PP by itself but reduces muscle force in challenging conditions. More recently, the lack of Nav channels clustering at the NMJ in mice knocked-out for ankyrins has also been reported to cause muscle weakness with reduced spontaneous locomotor activity of homozygous mutant mice and CMAP decrements in response to 40 and 60 Hz RNS (Zhang et al., 2021). These two elegant studies confirm that the lack of synaptic Nav1.4 or a half reduction in the whole amount of Nav1.4 in myofibers results in muscle weakness with a fatigability component (Wu et al., 2016; Zhang et al., 2021).
Altogether, these studies suggest the following simplistic pathophysiological mechanism to account for muscle weakness when Nav1.4 is deficient (Figure 6): LoF mutations would reduce the amount of Nav1.4 channels available for activation by reducing their quantity or by altering their biophysical properties. Consequently, the genesis of muscle APs in response to epp and/or their propagation along the sarcolemma would fail more frequently than normal. This would result in a lower muscle AP frequency in the transverse tubules and so in a lower muscle force in response to motoneuronal firing. A fatigable component of the muscle weakness would result, at least in part, from enhanced inactivation properties that reduce Nav1.4 availability for activation with repetitive stimulations.

FIGURE 6. Schematic representation of a simple pathophysiological hypothesis to account for sodium channel weakness (SCW) due to Nav1.4 loss of function (LoF). Repetitive muscle action potentials (myofiber AP) resulting from motoneuronal firing lead to the summation of muscle twitches (contractile activity), in this example up to the maximal sustained contraction (tetanos) in wild-type muscles (WT Nav1.4, black traces). Nav1.4 LoF would reduce the amount of Nav1.4 channels available for activation. This would result in lower muscle AP frequency and muscle force in response to neuronal firing in mutant myofibers (LoF Nav1.4, green). The fatigability would result from a decrease of Nav1.4 availability during neuronal firing, which would progressively reduce muscle AP frequency and muscle force.
To complete this overview of Nav1.4 LoF in human diseases, a case-control genetic study reports more frequent SCN4A missense variants in infants deceased from sudden infant death syndrome (SIDS) than in controls (Mannikko et al., 2018). SIDS is a sudden, unexpected and unexplained death of an apparently healthy baby. The exact cause of SIDS is unknown and is thought to result from a combination of factors. Among the six SCN4A variants observed in a SIDS sample of 278 infants, four have a functional impact on Nav1.4 current: two are GoF and two are LoF (p.Val1442Met in DIVS3S4 and p.Glu1520Lys in DIVS5S6). One (p.Val1442Met) substitutes one residue that induces SCW when substituted by Glu. An independent study performed in another sample of 73 infants with SIDS reported two inherited heterozygous missense variants (p.Lys724Arg in DIIS5S6 and p.Phe103Val in N-terminus) in SCN4A in two infants, but the biophysical characterization of the variant channels has yet to be done (Rochtus et al., 2020). The size of the two studied SIDS samples is low, and robust replicative studies are required to confirm Nav1.4 dysfunction as a susceptibility factor for SIDS. In addition, several challenges are raised by these genetic association studies. The first is that the variant effects should lead to deleterious function in vivo. The second is the question of identifying infants with SCN4A variants and at risk for SIDS with all the bioethical issues raised by genetic testing.
These genetic association studies and the identification of Nav1.4 LoF mutations as the cause of a large clinical spectrum of congenital muscle weakness further underline the influence of modifying factors on the phenotypic expression of SCN4A mutations already reported for GoF mutations (Nicole and Fontaine, 2015). For example, the p.Arg1451Leu substitution results in NDM (cold-induced myotonia), HyperPP, dominant PP with hypokalemic episodes or HypoPP2 with myotonia when homozygous, whereas p.Arg1135Cys results in recessive HypoPP2 or CM (Table 1) (Groome et al., 2014; Zaharieva et al., 2016; Luo et al., 2018; Poulin et al., 2018). The exact identity of the modifying factors is not known, although knowledge of muscle physiology makes ion channels (including trans- and cis- SCN4A variants), proteins important for the fine-tuning of Nav1.4 channels and environmental factors strong candidates.
Several drugs are available as therapeutic tools against Nav1.4 GoF for clinical use in humans (Jitpimolmard et al., 2020; Desaphy et al., 2021). Use-dependent Nav blockers are efficient to reduce myofiber hyperexcitability and improve myotonic symptoms. The first line blocker is mexiletine with conclusive clinical trials and European orphan drug designation (Statland, 2012; Stunnenberg et al., 2018b). Therapeutic management of PP mostly relies on patient education with a life-style aimed to avoid triggering factors. Carbonic anhydrase inhibitors (acetazolamide and dichlorphenamide) also help to prevent paralytic episodes in PP, in addition to K+ supplement in HypoPP. Carbonic anhydrase inhibitors are known to prevent and improve attacks in HypoPP since several years but their mode of action is unclear. Acetazolamide is a diuretic drug used to treat several illnesses. It modulates multiple processes by inducing acidosis and by activating ion channels such as Ca2+-activated K+ (BK) and Cl− channels, and probably acts by this means in PP (Eguchi et al., 2006; Tricarico et al., 2006). Bumetanide, an antagonist of NKCC, is also efficient to prevent hypokalemic-induced weakness in HypoPP1 and 2 mice (Wu et al., 2013a; Wu et al., 2013b). It acts by limiting the intracellular Cl− concentration rise during repetitive APs, which helps to maintain the HypoPP fibers in the normal RMP when extracellular K+ concentration is low. Unfortunately, bumetanide was not efficient in a randomized controlled trial (RCT) pilot study performed on 10 individuals with HypoPP, a negative result that might be due to the small size of the sample. Molecules are also available to improve muscle force in CMS by modulating neuromuscular transmission (Engel, 2018). They include K+ channel blockers (3,4-diaminopyridine), acetylcholinesterase inhibitors (pyridostigmine), agonists of β2 adrenergic receptors (albuterol, ephedrine) for most forms of CMS and blockers of nAChRs for slow-channel CMS (quinidine, quinine, fluoxetine). No generic or specific treatments are available for CM, although several specific, especially gene-based, therapies are either in preclinical development or in early trials (Wallgren-Pettersson and Laing, 2010; Gonorazky et al., 2018).
Some but unfortunately not all individuals with SCW due to hypomorph SCN4A mutations benefited from pyridostigmine with acetazolamide or from acetazolamide alone (Arnold et al., 2015; Habbout et al., 2016; Elia et al., 2019; Echaniz-Laguna et al., 2020). Contrary to severe Nav1.4 GoF conditions that greatly benefit from Nav blockers (mexiletine, carbamazepine), there is no treatment reported to prevent early death linked to SCN4A LoF mutations in SCW (Gay et al., 2008; Lion-Francois et al., 2010).
Several inherited Nav channelopathies are caused by LoF mutations such as Brugada syndrome, a cardiac arrhythmia due in part to Nav1.5 gating defects, and Dravet syndrome, a severe and lifelong form of pediatric epilepsy due to Nav1.1 haploinsufficiency (Loussouarn et al., 2015; Mantegazza et al., 2021). In all, a dosage effect is suggested, driving the therapeutic effort on boosting the mutant Nav channels to eventually improve the clinical consequences of their LoF. The hypomorph nature of several SCN4A LoF mutations and the dosage effect suspected to occur in SCW suggest that such an approach would also be efficient for Nav1.4 channelopathies (Figure 5).
Detailed pharmacological characterizations of Nav activators are sparse compared to inhibitors and the current challenge fostered by the existence of LoF Na+ channelopathies is to identify Nav channels agonists selective enough to be clinically useful. A complementary strategy to target-based drug approach would be to perform phenotype screening in order to identify molecules able to improve muscle weakness when Nav1.4 is deficient (Swinney and Lee, 2020). The last few years have seen the impressive development of gene-based therapy for monogenic diseases with efficient vectors for selective gene delivery in vivo and the use of CRISPR-Cas9 technology for genome editing. Promising preclinical results have been obtained for some Nav channelopathies, which could eventually benefit Nav1.4 channelopathies.
The goal of this approach is to identify molecules that will preferentially or selectively activate Nav1.4 channels. Nav agonists may act by facilitating activation or by impairing inactivation (Figure 4) (Deuis et al., 2017). Different classes of Nav activators are known, including pyrethroid insecticides and natural compounds such as alkaloid-based or peptide toxins from venoms. The high sequence conservation of Nav channels renders challenging the identification of highly subtype-specific modulators, but this is nevertheless possible. For example, AA43279 is one small chemically synthetized compound identified in a screening campaign looking for Nav1.1 activators. AA43279 acts as an inactivation blocker and appears efficient to functionally counteract Nav1.1 haploinsufficiency in a zebrafish model of Dravet syndrome (Frederiksen et al., 2017; Weuring et al., 2020). Due to their serious physiological effects and co-evolution of preys and predators, several Nav modulators including isoform-specific activators, are found in a variety of animal samples. Interestingly, amino acid substitutions may drastically change the toxin affinity for Nav isoforms. This is well exemplified by the natural resistance of poison frog Nav1.4 to alkaloids such as batrachotoxin—a use-dependent activator produced by Phyllobates poison dart frog—to prevent self-intoxication (Tarvin et al., 2016). Some Nav agonists have been successfully used in preclinical studies, rendering peptide toxins useful drug leads for developing novel therapeutic agents. For example, the spider-venom peptide Hm1a binds DIVS1S2 and DIVS3S4 extracellular loops of Nav channels and specifically activates Nav1.1 and Nav1.3 isoforms by inhibiting the VSD gating movement of DIV and hindering both fast and slow inactivation (Osteen et al., 2017; Osteen et al., 2016). Hm1a leads to increased Nav channels availability during high-frequency stimulations and its intracerebroventicular infusion reduced the Dravet syndrome-like phenotype of mice with Nav1.1 haploinsufficiency (Osteen et al., 2016; Richards et al., 2018).
A few Nav channel activators are known to have a great affinity for Nav1.4 such as the sea anemone AFT-II, the α-scorpion toxin OD-1 and the β-scorpion toxins Css4 and Tz1 (Table 2) (Deuis et al., 2017). AFT-II (for Anthopleura fuscoviridis anemone) is a 48-amino acid-long peptide with three disulfide bonds that preferentially binds Nav1.4 and Nav1.5 (Oliveira et al., 2004). It slows the inactivation process with depolarized shift and increased time constants, but its effect on cell excitability has not been explored. OD-1 is an amidated polypeptide of 65 amino acids with four disulfide bonds that efficiently binds Nav1.4, Nav1.6 and Nav1.7 but not Nav1.2, Nav1.3 or Nav1.5 (Durek et al., 2013). The agonist activity of purified OD-1 on Nav1.4 relies on hyperpolarizing shift of activation (Table 2). The Odontobuthus doriae venom, from which OD-1 has been isolated, causes pre- and post-synaptic repetitive firing in response to a single stimulus in mouse nerve-muscle preparations (Jalali et al., 2007). In agreement with its potent agonist effect, OD-1 increases Na+ current peak amplitude and neuronal AP frequency and decreases neuronal AP threshold on rat hippocampal brain slices, but its effect on nerve muscle preparations has not been studied (Lai et al., 2020). Another interesting toxin to explore for SCW is Css4 (for Centruroides suffusus). Css4 is a 65-amino acid-long β-scorpion toxin that activates Nav channels by enhancing activation with an hyperpolarizing shift of its voltage dependence (Cohen et al., 2007). If Css4 is a Nav1.4 agonist, it has no effect on Nav1.5 activation, and substituting three amino acids of Css4 further increases its selectivity for Nav1.4. Interestingly, a genetically-modified form of Css4 counteracts the LoF effects of the HypoPP2 p.Arg672Gly mutation in vitro (Cohen et al., 2007). This modified toxin could be preclinically tested in the knock-in mouse model available for HypoPP2 to determine whether Nav1.4 LoF has a physiological significance in this form of SCW (Wu et al., 2011). Tz1 (for Tityus zulianus) is another β-scorpion toxin (63 amino acids) that induces a hyperpolarized shift of Nav activation with high specificity to Nav1.4 and no effect on Nav1.7 or Nav1.5 (Leipold et al., 2006). Unfortunately, β-scorpion toxins, including Css4 and Tz1, have a bimodal effect on a use-dependent basis since they may reduce Nav conductance, thereby depressing cell excitability. For example, Tz1 reduces Nav1.4 current at 0.1 Hz but increases it at 2 Hz (Cestèle et al., 1998; Leipold et al., 2012). Moreover, its antagonist, but not its agonist, effect is observed on Nav1.5 current at both frequencies (Leipold et al., 2012). Calliotoxin (δ-elapitoxin-Cb1a) is a 57-amino acid-long three-finger peptide from the Calliophis bivirgatus snake. It increases muscle force in chicken nerve-muscle preparations, and enhances peak of NaV1.4 current, shifts the voltage-dependence of its activation to more hyperpolarized potentials, delays its inactivation and causes a persistent Na+ current in heterologous cell expression systems (Yang et al., 2016). However, the activity and selectivity of calliotoxin have not been tested among Nav channels and the boosting effect of this peptide on muscle force may be also due to activation of neuronal Nav channels. Although not natural, 16-amino acids-long peptides corresponding to DI, DII and DIIIS4S5 intracellular linkers increase Na+ current density and shift hNav1.4 activation towards hyperpolarization by allosterically modulating the activation gate and stabilizing the open state (Malak et al., 2020). These synthetic peptides add to the natural toxin arsenal towards identifying compounds able to selectively boost Nav1.4 activity.

TABLE 2. Some Nav1.4 activators reported in the literature. Their effect on Nav1.4 gating behavior in vitro is stated.
If natural toxins and synthetic peptides are promising lead compounds, extensive preclinical research efforts are required to determine whether they may used for developing peptide-based therapeutics to counteract Nav1.4 LoF and improve SCW (Peigneur and Tytgat, 2018). Besides improving Nav1.4 specificity, rational control of the bimodal activity observed for some toxins is necessary to increase and not reduce myofiber excitability. Recent technological breakthroughs will foster these studies, especially CryoEM by facilitating structural pharmacology and the design of Nav subtype-selective profiles (Pan X. et al., 2018; Kong et al., 2019; Noreng et al., 2021). Automated patch-clamp approaches will also help identify additional candidates by performing medium-throughput screening campaigns of toxin and chemical libraries (Pan J. Q. et al., 2018; Obergrussberger et al., 2018). Important preclinical steps for all Nav1.4 activators will be to determine whether they are able 1) to restore a ‘normal’ Nav1.4 current from mutant channels in vitro; 2) to improve muscle force without inducing myotonia or paralysis in preclinical animal models; and 3) are safe enough to be used in humans. These druggability parameters are clues for a therapeutic future of any molecule found to activate Nav1.4 in vitro.
The benefic effect of pyridostigmine and acetazolamide reported for some individuals with SCW illustrates the rationale of such an approach (Tsujino et al., 2003; Habbout et al., 2016; Elia et al., 2019). Phenotypic drug screening has also been fruitful to identify promising lead compounds for other Nav channelopathies. By screening a Food and Drug Administration (FDA)-approved compounds library, clemizole has been identified as efficient to improve the epileptic phenotype resulting from Nav1.1 haploinsufficiency on a zebrafish model for Dravet syndrome (Baraban et al., 2013). Based on the fact that Nav1.6 GoF results in severe epileptic phenotypes, two Nav1.6 inhibitors have also been shown to improve the phenotype in another zebrafish model of Dravet syndrome with Nav1.1 haploinsufficiency (Weuring et al., 2020).
These examples underline the notion that preclinical animal models of SCW are required to successfully perform phenotype drug screening using muscle weakness as a biomarker, as well as for the preclinical investigations of Nav1.4 activators listed above. Some animal models have been developed for Nav1.4 GoF: knock-in mouse lines with HyperPP or HypoPP2, and transgenic zebrafish lines with NDM (Hayward et al., 2008; Wu et al., 2011; Nam et al., 2019). No viable animal model with SCW due to Scn4a LoF has been reported yet. Mice homozygous for a null Scn4a allele die at birth, preventing their use as an efficient model for screening. In the zebrafish, which is a versatile vertebrae model for phenotypic drug screening of small molecule libraries, two Scn4a orthologs exist and are expressed in skeletal muscles (Novak et al., 2006; Chopra et al., 2010). This greatly complexifies the establishment of mutant lines with recessively-inherited Scn4a mutations in this small vertebrate.
The ongoing development of neuromuscular organoids using human induced pluripotent stem cells is another exciting possibility to preclinically model Nav1.4 LoF (Faustino Martins et al., 2020; Mazaleyrat et al., 2020).SCN4A is expressed in the neuromuscular organoids obtained so far, but its contribution to Na+ current and muscle cell excitability must be explored. In addition, the current protocols of differentiation and 3D cell culture have to be further improved since they do not result in NMJ mature enough to be structurally and functionally relevant (Lynch et al., 2021). Such cellular models are nevertheless very promising as they should help deciphering, in patient-derived neuromuscular preparations, the impact of Nav1.4 mutations on excitability and contractility of innervated myofibers and testing novel (bio)pharmacological strategies in Nav1.4 channelopathies.
Gene-based therapies are now biomedical options for devastating monogenic diseases with the development of antisense oligonucleotides (ASO) and recombinant adeno-associated virus (rAAV). More recently, the breakthrough allowed by CRISPR-Cas9 technology has opened a gigantic new area in the field of therapies based on genome engineering. Promising preclinical results using these technologies have been obtained for Nav1.1 and Nav1.5 channelopathies. An ASO-based strategy has been successfully used to enhance the expression of the WT Scn1a allele in a mouse model of Dravet syndrome in order to circumvent Nav1.1 haploinsufficiency (Han et al., 2020). A CRISPR-Cas9 based strategy, which consists in positively and specifically modulating the expression of any gene of interest with a catalytically dead Cas9, has also been successfully used to promote Scn1a expression in mouse models of Dravet syndrome (Colasante et al., 2020; Yamagata et al., 2020). It would be interesting to use these technics to selectively boost SCN5A expression in skeletal myofibers and determine whether its overexpression may compensate Nav1.4 deficiency.
Efficient gene transduction is another way to compensate LoF mutations. AAV vectors are the vectors of choice for efficient gene transduction in vivo with good safety profile, lack of genome integration, long-term expression in non-dividing cells and tissue-specific tropism (Aguti et al., 2018). The length of the cDNAs coding for the α subunits of Nav yet prevents their use, the maximal DNA size encapsulated by rAAV being smaller than 4.7 kb. Development of novel tools, such as trans-splicing events after splitting the coding sequence and packaging it into independent rAAVs, is therefore a prerequisite for efficient delivery of any cDNA of Nav channels. A dual rAAV vector strategy promoting trans-splicing has been successfully used to overexpress one GoF variant of human SCN5A in mouse heart, opening the door to such an approach for efficiently expressing exogenous Nav1.4 in skeletal myofibers (Doisne et al., 2021).
Gene-based strategies are in full expansion with successful examples for devastating neuromuscular disorders such as spinal muscular atrophy with agreements from FDA and European Medical Agency for ASO-based and AAV-based gene therapies (Messina and Sframeli, 2020). We are nevertheless far away to use these approaches for SCW as it requires time to develop potent recombinant vectors and to accurately investigate their therapeutic potential and safety in preclinical models.
These last 5 years have seen the identification of sodium channel weakness (SCW) as a subset of muscle channelopathies. A more complete and accurate clinical spectrum can now be drawn for Nav1.4 channelopathies with a full continuum of myofiber excitability ranging from hyperexcitability in SCM due to GoF changes to unexcitable myofibers in SCW due to LoF changes (Figure 5). Within this spectrum, the quality of life of patients is greatly reduced, and the extreme cases are life-threatening. The identification of LoF mutations in SCW strengthens the need for therapeutic tools able to counteract the loss of muscle force resulting from reduced or lack of Nav1.4 activity. The recent technological breakthroughs in omics technologies, medium-throughput patch-clamp procedures, 3D cultures, induced pluripotent stem cells, and gene-based therapies open exciting possibilities to fill these gaps for the eventual benefit of individuals with Nav1.4 channelopathies in the next years.
SN and PL wrote the manuscript.
This work was supported by Université de Montpellier, CNRS, Inserm, Association Française contre les Myopathies (AFM) - Téléthon (grant numbers 20030 and 23677), Fondation Maladies Rares and Investissements d’Avenir Laboratoire d’Excellence “Ion Channel Science and Therapeutics” (ANR-11-LABX-0015).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aguti, S., Malerba, A., and Zhou, H. (2018). The Progress of AAV-Mediated Gene Therapy in Neuromuscular Disorders. Expert Opin. Biol. Ther. 18, 681–693. doi:10.1080/14712598.2018.1479739
Arnold, W. D., Feldman, D. H., Ramirez, S., He, L., Kassar, D., Quick, A., et al. (2015). Defective Fast Inactivation Recovery of Nav 1.4 in Congenital Myasthenic Syndrome. Ann. Neurol. 77, 840–850. doi:10.1002/ana.24389
Awad, S. S., Lightowlers, R. N., Young, C., Chrzanowska-Lightowlers, Z. M., Lomo, T., and Slater, C. R. (2001). Sodium Channel mRNAs at the Neuromuscular Junction: Distinct Patterns of Accumulation and Effects of Muscle Activity. J. Neurosci. 21, 8456–8463. doi:10.1523/jneurosci.21-21-08456.2001
Bailey, S. J., Stocksley, M. A., Buckel, A., Young, C., and Slater, C. R. (2003). Voltage-gated Sodium Channels and AnkyrinG Occupy a Different Postsynaptic Domain from Acetylcholine Receptors from an Early Stage of Neuromuscular Junction Maturation in Rats. J. Neurosci. 23, 2102–2111. doi:10.1523/jneurosci.23-06-02102.2003
Baraban, S. C., Dinday, M. T., and Hortopan, G. A. (2013). Drug Screening in Scn1a Zebrafish Mutant Identifies Clemizole as a Potential Dravet Syndrome Treatment. Nat. Commun. 4, 2410. doi:10.1038/ncomms3410
Bayless-Edwards, L., Winston, V., Lehmann-Horn, F., Arinze, P., Groome, J. R., and Jurkat-Rott, K. (2018). Nav1.4 DI-S4 Periodic Paralysis Mutation R222W Enhances Inactivation and Promotes Leak Current to Attenuate Action Potentials and Depolarize Muscle Fibers. Sci. Rep. 8, 10372. doi:10.1038/s41598-018-28594-5
Belotti, E., and Schaeffer, L. (2020). Regulation of Gene Expression at the Neuromuscular Junction. Neurosci. Lett. 735, 135163. doi:10.1016/j.neulet.2020.135163
Bendahhou, S., Cummins, T. R., Griggs, R. C., Fu, Y. H., and Ptácek, L. J. (2001). Sodium Channel Inactivation Defects Are Associated with Acetazolamide-Exacerbated Hypokalemic Periodic Paralysis. Ann. Neurol. 50, 417–420. doi:10.1002/ana.1144
Biswas, S., Deschênes, I., Disilvestre, D., Tian, Y., Halperin, V. L., and Tomaselli, G. F. (2008). Calmodulin Regulation of Nav1.4 Current: Role of Binding to the Carboxyl Terminus. J. Gen. Physiol. 131, 197–209. doi:10.1085/jgp.200709863
Blechschmidt, S., Haufe, V., Benndorf, K., and Zimmer, T. (2008). Voltage-gated Na+ Channel Transcript Patterns in the Mammalian Heart Are Species-dependent. Prog. Biophys. Mol. Biol. 98, 309–318. doi:10.1016/j.pbiomolbio.2009.01.009
Cannon, S. C., McClatchey, A. I., and Gusella, J. F. (1993). Modification of the Na+ Current Conducted by the Rat Skeletal Muscle Alpha Subunit by Coexpression with a Human Brain Beta Subunit. Pflugers Arch. 423, 155–157. doi:10.1007/BF00374974
Cannon, S. C. (2018). Sodium Channelopathies of Skeletal Muscle. Handb. Exp. Pharmacol. 246, 309–330. doi:10.1007/164_2017_52
Capes, D. L., Goldschen-Ohm, M. P., Arcisio-Miranda, M., Bezanilla, F., and Chanda, B. (2013). Domain IV Voltage-Sensor Movement Is Both Sufficient and Rate Limiting for Fast Inactivation in Sodium Channels. J. Gen. Physiol. 142, 101–112. doi:10.1085/jgp.201310998
Carle, T., Lhuillier, L., Luce, S., Sternberg, D., Devuyst, O., Fontaine, B., et al. (2006). Gating Defects of a Novel Na+ Channel Mutant Causing Hypokalemic Periodic Paralysis. Biochem. Biophys. Res. Commun. 348, 653–661. doi:10.1016/j.bbrc.2006.07.101
Carreras, D., Martinez-Moreno, R., Pinsach-Abuin, M. L., Santafe, M. M., Gomà, P., Brugada, R., et al. (2021). Epigenetic Changes Governing Scn5a Expression in Denervated Skeletal Muscle. Int. J. Mol. Sci. 22, 2755. doi:10.3390/ijms22052755
Catterall, W. A., Lenaeus, M. J., and Gamal El-Din, T. M. (2020). Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 60, 133–154. doi:10.1146/annurev-pharmtox-010818-021757
Cestèle, S., Qu, Y., Rogers, J. C., Rochat, H., Scheuer, T., and Catterall, W. A. (1998). Voltage Sensor-Trapping: Enhanced Activation of Sodium Channels by Beta-Scorpion Toxin Bound to the S3-S4 Loop in Domain II. Neuron 21, 919–931. doi:10.1016/S0896-6273(00)80606-6
Chopra, S. S., Stroud, D. M., Watanabe, H., Bennett, J. S., Burns, C. G., Wells, K. S., et al. (2010). Voltage-gated Sodium Channels Are Required for Heart Development in Zebrafish. Circ. Res. 106, 1342–1350. doi:10.1161/circresaha.109.213132
Clatot, J., Hoshi, M., Wan, X., Liu, H., Jain, A., Shinlapawittayatorn, K., et al. (2017). Voltage-gated Sodium Channels Assemble and Gate as Dimers. Nat. Commun. 8, 2077. doi:10.1038/s41467-017-02262-0
Cohen, L., Ilan, N., Gur, M., Stühmer, W., Gordon, D., and Gurevitz, M. (2007). Design of a Specific Activator for Skeletal Muscle Sodium Channels Uncovers Channel Architecture. J. Biol. Chem. 282, 29424–29430. doi:10.1074/jbc.M704651200
Colasante, G., Lignani, G., Brusco, S., Di Berardino, C., Carpenter, J., Giannelli, S., et al. (2020). dCas9-based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol. Ther. 28, 235–253. doi:10.1016/j.ymthe.2019.08.018
David, M., Martínez-Mármol, R., Gonzalez, T., Felipe, A., and Valenzuela, C. (2008). Differential Regulation of Na(v)beta Subunits during Myogenesis. Biochem. Biophys. Res. Commun. 368, 761–766. doi:10.1016/j.bbrc.2008.01.138
Desaphy, J. F., Altamura, C., Vicart, S., and Fontaine, B. (2021). Targeted Therapies for Skeletal Muscle Ion Channelopathies: Systematic Review and Steps towards Precision Medicine. J. Neuromuscul. Dis. 8, 357–381. doi:10.3233/JND-200582
Desaphy, J. F., Pierno, S., Léoty, C., George, A. L., De Luca, A., and Camerino, D. C. (2001). Skeletal Muscle Disuse Induces Fibre Type-dependent Enhancement of Na(+) Channel Expression. Brain 124, 1100–1113. doi:10.1093/brain/124.6.1100
Deuis, J. R., Mueller, A., Israel, M. R., and Vetter, I. (2017). The Pharmacology of Voltage-Gated Sodium Channel Activators. Neuropharmacology 127, 87–108. doi:10.1016/j.neuropharm.2017.04.014
DiFranco, M., and Vergara, J. L. (2011). The Na+ Conductance in the Sarcolemma and the Transverse Tubular System Membranes of Mammalian Skeletal Muscle Fibers. J. Gen. Physiol. 138, 393–419. doi:10.1085/jgp.201110682
Doisne, N., Grauso, M., Mougenot, N., Clergue, M., Souil, C., Coulombe, A., et al. (2021). In Vivo Dominant-Negative Effect of an SCN5A Brugada Syndrome Variant. Front. Physiol. 12, 661413. doi:10.3389/fphys.2021.661413
Dolivo, D., Rodrigues, A., Sun, L., Li, Y., Hou, C., Galiano, R., et al. (2021). The Nax (SCN7A) Channel: an Atypical Regulator of Tissue Homeostasis and Disease. Cell. Mol. Life Sci. 78, 5469–5488. doi:10.1007/s00018-021-03854-2
Durek, T., Vetter, I., Wang, C. I., Motin, L., Knapp, O., Adams, D. J., et al. (2013). Chemical Engineering and Structural and Pharmacological Characterization of the α-scorpion Toxin OD1. ACS Chem. Biol. 8, 1215–1222. doi:10.1021/cb400012k
Echaniz-Laguna, A., Biancalana, V., Nadaj-Pakleza, A., Fournier, E., Matthews, E., Hanna, M. G., et al. (2020). Homozygous C-Terminal Loss-Of-Function NaV1.4 Variant in a Patient with Congenital Myasthenic Syndrome. J. Neurol. Neurosurg. Psychiatry 91, 898–900. doi:10.1136/jnnp-2020-323173
Eguchi, H., Tsujino, A., Kaibara, M., Hayashi, H., Shirabe, S., Taniyama, K., et al. (2006). Acetazolamide Acts Directly on the Human Skeletal Muscle Chloride Channel. Muscle Nerve 34, 292–297. doi:10.1002/mus.20585
Elia, N., Palmio, J., Castañeda, M. S., Shieh, P. B., Quinonez, M., Suominen, T., et al. (2019). Myasthenic Congenital Myopathy from Recessive Mutations at a Single Residue in Nav1.4. Neurology 92, e1405–e1415. doi:10.1212/WNL.0000000000007185
Engel, A. G. (2018). Genetic Basis and Phenotypic Features of Congenital Myasthenic Syndromes. Handb. Clin. Neurol. 148, 565–589. doi:10.1016/B978-0-444-64076-5.00037-5
Engel, A. G., Shen, X. M., Selcen, D., and Sine, S. M. (2015). Congenital Myasthenic Syndromes: Pathogenesis, Diagnosis, and Treatment. Lancet Neurol. 14, 420–434. doi:10.1016/S1474-4422(14)70201-7
Eshed‐Eisenbach, Y., and Peles, E. (2020). The Clustering of Voltage‐gated Sodium Channels in Various Excitable Membranes. Develop. Neurobiol. 81, 427–437. doi:10.1002/dneu.22728
Faustino Martins, J. M., Fischer, C., Urzi, A., Vidal, R., Kunz, S., Ruffault, P. L., et al. (2020). Self-organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 27, 498–186.e6. doi:10.1016/j.stem.2019.12.00710.1016/j.stem.2020.08.011
Ferrera, L., and Moran, O. (2006). Beta1-subunit Modulates the Nav1.4 Sodium Channel by Changing the Surface Charge. Exp. Brain Res. 172, 139–150. doi:10.1007/s00221-005-0323-4
Flucher, B. E., and Daniels, M. P. (1989). Distribution of Na+ Channels and Ankyrin in Neuromuscular Junctions Is Complementary to that of Acetylcholine Receptors and the 43 Kd Protein. Neuron 3, 163–175. doi:10.1016/0896-6273(89)90029-9
Fontaine, B., Khurana, T. S., Hoffman, E. P., Bruns, G. A., Haines, J. L., Trofatter, J. A., et al. (1990). Hyperkalemic Periodic Paralysis and the Adult Muscle Sodium Channel Alpha-Subunit Gene. Science 250, 1000–1002. doi:10.1126/science.2173143
Fournier, E., Arzel, M., Sternberg, D., Vicart, S., Laforet, P., Eymard, B., et al. (2004). Electromyography Guides toward Subgroups of Mutations in Muscle Channelopathies. Ann. Neurol. 56, 650–661. doi:10.1002/ana.20241
Francis, D. G., Rybalchenko, V., Struyk, A., and Cannon, S. C. (2011). Leaky Sodium Channels from Voltage Sensor Mutations in Periodic Paralysis, but Not Paramyotonia. Neurology 76, 1635–1641. doi:10.1212/WNL.0b013e318219fb57
Frederiksen, K., Lu, D., Yang, J., Jensen, H. S., Bastlund, J. F., Larsen, P. H., et al. (2017). A Small Molecule Activator of Nav 1.1 Channels Increases Fast-Spiking Interneuron Excitability and GABAergic Transmission In Vitro and Has Anti-convulsive Effects In Vivo. Eur. J. Neurosci. 46, 1887–1896. doi:10.1111/ejn.13626
Fu, Y., Struyk, A., Markin, V., and Cannon, S. (2011). Gating Behaviour of Sodium Currents in Adult Mouse Muscle Recorded with an Improved Two-Electrode Voltage Clamp. J. Physiol. 589, 525–546. doi:10.1113/jphysiol.2010.199430
Gardill, B. R., Rivera-Acevedo, R. E., Tung, C. C., and Van Petegem, F. (2019). Crystal Structures of Ca2+-Calmodulin Bound to NaV C-Terminal Regions Suggest Role for EF-Hand Domain in Binding and Inactivation. Proc. Natl. Acad. Sci. U S A. 116, 10763–10772. doi:10.1073/pnas.1818618116
Garrido, J. J., Fernandes, F., Moussif, A., Fache, M. P., Giraud, P., and Dargent, B. (2003). Dynamic Compartmentalization of the Voltage-Gated Sodium Channels in Axons. Biol. Cel. 95, 437–445. doi:10.1016/S0248-4900(03)00091-1
Gay, S., Dupuis, D., Faivre, L., Masurel-Paulet, A., Labenne, M., Colombani, M., et al. (2008). Severe Neonatal Non-dystrophic Myotonia Secondary to a Novel Mutation of the Voltage-Gated Sodium Channel (SCN4A) Gene. Am. J. Med. Genet. A. 146A, 380–383. doi:10.1002/ajmg.a.32141
Gee, S. H., Madhavan, R., Levinson, S. R., Caldwell, J. H., Sealock, R., and Froehner, S. C. (1998). Interaction of Muscle and Brain Sodium Channels with Multiple Members of the Syntrophin Family of Dystrophin-Associated Proteins. J. Neurosci. 18, 128–137. doi:10.1523/jneurosci.18-01-00128.1998
Gonorazky, H. D., Bönnemann, C. G., and Dowling, J. J. (2018). The Genetics of Congenital Myopathies. Handb Clin. Neurol. 148, 549–564. doi:10.1016/B978-0-444-64076-5.00036-3
Gonorazky, H. D., Marshall, C. R., Al-Murshed, M., Hazrati, L. N., Thor, M. G., Hanna, M. G., et al. (2017). Congenital Myopathy with “corona” Fibres, Selective Muscle Atrophy, and Craniosynostosis Associated with Novel Recessive Mutations in SCN4A. Neuromuscul. Disord. 27, 574–580. doi:10.1016/j.nmd.2017.02.001
Gosselin-Badaroudine, P., Delemotte, L., Moreau, A., Klein, M. L., and Chahine, M. (2012). Gating Pore Currents and the Resting State of Nav1.4 Voltage Sensor Domains. Proc. Natl. Acad. Sci. U S A. 109, 19250–19255. doi:10.1073/pnas.1217990109
Groome, J. R., Lehmann-Horn, F., Fan, C., Wolf, M., Winston, V., Merlini, L., et al. (2014). NaV1.4 Mutations Cause Hypokalaemic Periodic Paralysis by Disrupting IIIS4 Movement during Recovery. Brain 137, 998–1008. doi:10.1093/brain/awu015
Habbout, K., Poulin, H., Rivier, F., Giuliano, S., Sternberg, D., Fontaine, B., et al. (2016). A Recessive Nav1.4 Mutation Underlies Congenital Myasthenic Syndrome with Periodic Paralysis. Neurology 86, 161–169. doi:10.1212/WNL.0000000000002264
Han, Z., Chen, C., Christiansen, A., Ji, S., Lin, Q., Anumonwo, C., et al. (2020). Antisense Oligonucleotides Increase Scn1a Expression and Reduce Seizures and SUDEP Incidence in a Mouse Model of Dravet Syndrome. Sci. Transl. Med. 12, eaaz6100. doi:10.1126/scitranslmed.aaz6100
Hayward, L. J., Kim, J. S., Lee, M. Y., Zhou, H., Kim, J. W., Misra, K., et al. (2008). Targeted Mutation of Mouse Skeletal Muscle Sodium Channel Produces Myotonia and Potassium-Sensitive Weakness. J. Clin. Invest. 118, 1437–1449. doi:10.1172/jci32638
Hebert, S. L., Simmons, C., Thompson, A. L., Zorc, C. S., Blalock, E. M., and Kraner, S. D. (2007). Basic helix-loop-helix Factors Recruit Nuclear Factor I to Enhance Expression of the NaV 1.4 Na+ Channel Gene. Biochim. Biophys. Acta 1769, 649–658. doi:10.1016/j.bbaexp.2007.08.004
Hennig, R., and Lømo, T. (1985). Firing Patterns of Motor Units in normal Rats. Nature 314, 164–166. doi:10.1038/314164a0
Hirn, C., Shapovalov, G., Petermann, O., Roulet, E., and Ruegg, U. T. (2008). Nav1.4 Deregulation in Dystrophic Skeletal Muscle Leads to Na+ Overload and Enhanced Cell Death. J. Gen. Physiol. 132, 199–208. doi:10.1085/jgp.200810024
Horga, A., Raja Rayan, D. L., Matthews, E., Sud, R., Fialho, D., Durran, S. C., et al. (2013). Prevalence Study of Genetically Defined Skeletal Muscle Channelopathies in England. Neurology 80, 1472–1475. doi:10.1212/WNL.0b013e31828cf8d0
Jalali, A., Vatanpour, H., Hosseininasab, Z., Rowan, E. G., and Harvey, A. L. (2007). The Effect of the Venom of the Yellow Iranian Scorpion Odontobuthus Doriae on Skeletal Muscle Preparations In Vitro. Toxicon 50, 1019–1026. doi:10.1016/j.toxicon.2007.05.001
Jitpimolmard, N., Matthews, E., and Fialho, D. (2020). Treatment Updates for Neuromuscular Channelopathies. Curr. Treat. Options Neurol. 22, 34. doi:10.1007/s11940-020-00644-2
Jones, R. A., Harrison, C., Eaton, S. L., Llavero Hurtado, M., Graham, L. C., Alkhammash, L., et al. (2017). Cellular and Molecular Anatomy of the Human Neuromuscular Junction. Cell Rep. 21, 2348–2356. doi:10.1016/j.celrep.2017.11.008
Jurkat-Rott, K., Lehmann-Horn, F., Elbaz, A., Heine, R., Gregg, R. G., Hogan, K., et al. (1994). A Calcium Channel Mutation Causing Hypokalemic Periodic Paralysis. Hum. Mol. Genet. 3, 1415–1419. doi:10.1093/hmg/3.8.1415
Jurkat-Rott, K., Mitrovic, N., Hang, C., Kouzmekine, A., Iaizzo, P., Herzog, J., et al. (2000). Voltage-sensor Sodium Channel Mutations Cause Hypokalemic Periodic Paralysis Type 2 by Enhanced Inactivation and Reduced Current. Proc. Natl. Acad. Sci. U S A. 97, 9549–9554. doi:10.1073/pnas.97.17.9549
Jurkat-Rott, K., Weber, M. A., Fauler, M., Guo, X. H., Holzherr, B. D., Paczulla, A., et al. (2009). K+-dependent Paradoxical Membrane Depolarization and Na+ Overload, Major and Reversible Contributors to Weakness by Ion Channel Leaks. Proc. Natl. Acad. Sci. U S A. 106, 4036–4041. doi:10.1073/pnas.0811277106
Kaufmann, S. G., Westenbroek, R. E., Maass, A. H., Lange, V., Renner, A., Wischmeyer, E., et al. (2013). Distribution and Function of Sodium Channel Subtypes in Human Atrial Myocardium. J. Mol. Cel. Cardiol. 61, 133–141. doi:10.1016/j.yjmcc.2013.05.006
Kokunai, Y., Dalle, C., Vicart, S., Sternberg, D., Pouliot, V., Bendahhou, S., et al. (2018). A204E Mutation in Nav1.4 DIS3 Exerts Gain- and Loss-Of-Function Effects that lead to Periodic Paralysis Combining Hyper- with Hypo-kalaemic Signs. Sci. Rep. 8, 16681. doi:10.1038/s41598-018-34750-8
Kong, D. J., Wang, Y., Wang, H. X., Wang, M. X., Wang, J., and Cheng, M. S. (2019). Molecular Determinants for Ligand Binding at Nav1.4 and Nav1.7 Channels: Experimental Affinity Results Analyzed by Molecular Modeling. Comput. Biol. Chem. 83, 107132. doi:10.1016/j.compbiolchem.2019.107132
Kraner, S. D., Novak, K. R., Wang, Q., Peng, J., and Rich, M. M. (2012). Altered Sodium Channel-Protein Associations in Critical Illness Myopathy. Skelet. Muscle 2, 17. doi:10.1186/2044-5040-2-17
Kraner, S. D., Rich, M. M., Kallen, R. G., and Barchi, R. L. (1998). Two E-Boxes Are the Focal Point of Muscle-Specific Skeletal Muscle Type 1 Na+ Channel Gene Expression. J. Biol. Chem. 273, 11327–11334. doi:10.1074/jbc.273.18.11327
Kraner, S. D., Rich, M. M., Sholl, M. A., Zhou, H., Zorc, C. S., Kallen, R. G., et al. (1999). Interaction between the Skeletal Muscle Type 1 Na+ Channel Promoter E-Box and an Upstream Repressor Element. Release of Repression by Myogenin. J. Biol. Chem. 274, 8129–8136. doi:10.1074/jbc.274.12.8129
Kubota, T., Wu, F., Vicart, S., Nakaza, M., Sternberg, D., Watanabe, D., et al. (2020). Hypokalaemic Periodic Paralysis with a Charge-Retaining Substitution in the Voltage Sensor. Brain Commun. 2, fcaa103. doi:10.1093/braincomms/fcaa103
Kuzmenkin, A., Muncan, V., Jurkat-Rott, K., Hang, C., Lerche, H., Lehmann-Horn, F., et al. (2002). Enhanced Inactivation and pH Sensitivity of Na(+) Channel Mutations Causing Hypokalaemic Periodic Paralysis Type II. Brain 125, 835–843. doi:10.1093/brain/awf071
Lai, M. C., Wu, S. N., and Huang, C. W. (2020). The Specific Effects of OD-1, a Peptide Activator, on Voltage-Gated Sodium Current and Seizure Susceptibility. Int. J. Mol. Sci. 21, 8254. doi:10.3390/ijms21218254
Lee, S. C., Kim, H. S., Park, Y. E., Choi, Y. C., Park, K. H., and Kim, D. S. (2009). Clinical Diversity of SCN4A-Mutation-Associated Skeletal Muscle Sodium Channelopathy. J. Clin. Neurol. 5, 186–191. doi:10.3988/jcn.2009.5.4.186
Lehmann-Horn, F., D'Amico, A., Bertini, E., Lomonaco, M., Merlini, L., Nelson, K. R., et al. (2017). Myotonia Permanens with Nav1.4-G1306E Displays Varied Phenotypes during Course of Life. Acta Myol 36, 125–134.
Lehmann-Horn, F., Rüdel, R., Ricker, K., Lorković, H., Dengler, R., and Hopf, H. C. (1983). Two Cases of Adynamia Episodica Hereditaria: In Vitro Investigation of Muscle Cell Membrane and Contraction Parameters. Muscle Nerve 6, 113–121. doi:10.1002/mus.880060206
Leipold, E., Borges, A., and Heinemann, S. H. (2012). Scorpion β-toxin Interference with NaV Channel Voltage Sensor Gives Rise to Excitatory and Depressant Modes. J. Gen. Physiol. 139, 305–319. doi:10.1085/jgp.201110720
Leipold, E., Hansel, A., Borges, A., and Heinemann, S. H. (2006). Subtype Specificity of Scorpion Beta-Toxin Tz1 Interaction with Voltage-Gated Sodium Channels Is Determined by the Pore Loop of Domain 3. Mol. Pharmacol. 70, 340–347. doi:10.1124/mol.106.024034
Li, L., Xiong, W. C., and Mei, L. (2018). Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 80, 159–188. doi:10.1146/annurev-physiol-022516-034255
Lion-Francois, L., Mignot, C., Vicart, S., Manel, V., Sternberg, D., Landrieu, P., et al. (2010). Severe Neonatal Episodic Laryngospasm Due to De Novo SCN4A Mutations: a New Treatable Disorder. Neurology 75, 641–645. doi:10.1212/WNL.0b013e3181ed9e96
Liu, S. X., Matthews, H. R., and Huang, C. L. (2021). Sarcoplasmic Reticular Ca2+-ATPase Inhibition Paradoxically Upregulates Murine Skeletal Muscle Nav1.4 Function. Sci. Rep. 11, 2846. doi:10.1038/s41598-021-82493-w
Loussouarn, G., Sternberg, D., Nicole, S., Marionneau, C., Le Bouffant, F., Toumaniantz, G., et al. (2015). Physiological and Pathophysiological Insights of Nav1.4 and Nav1.5 Comparison. Front. Pharmacol. 6, 314. doi:10.3389/fphar.2015.00314
Luo, S., Sampedro Castañeda, M., Matthews, E., Sud, R., Hanna, M. G., Sun, J., et al. (2018). Hypokalaemic Periodic Paralysis and Myotonia in a Patient with Homozygous Mutation p.R1451L in NaV1.4. Sci. Rep. 8, 9714. doi:10.1038/s41598-018-27822-2
Lupa, M. T., and Caldwell, J. H. (1991). Effect of Agrin on the Distribution of Acetylcholine Receptors and Sodium Channels on Adult Skeletal Muscle Fibers in Culture. J. Cel. Biol. 115, 765–778. doi:10.1083/jcb.115.3.765
Lupa, M. T., Krzemien, D. M., Schaller, K. L., and Caldwell, J. H. (1993). Aggregation of Sodium Channels during Development and Maturation of the Neuromuscular Junction. J. Neurosci. 13, 1326–1336. doi:10.1523/jneurosci.13-03-01326.1993
Lynch, E., Peek, E., Reilly, M., FitzGibbons, C., Robertson, S., and Suzuki, M. (2021). Current Progress in the Creation, Characterization, and Application of Human Stem Cell-Derived In Vitro Neuromuscular Junction Models. Stem Cel. Rev. Rep. doi:10.1007/s12015-021-10201-2
Maggi, L., Bonanno, S., Altamura, C., and Desaphy, J. F. (2021). Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy. Cells 10, 1521. doi:10.3390/cells10061521
Makita, N., Bennett, P. B., and George, A. L. (1996). Molecular Determinants of Beta 1 Subunit-Induced Gating Modulation in Voltage-dependent Na+ Channels. J. Neurosci. 16, 7117–7127. doi:10.1523/jneurosci.16-22-07117.1996
Malak, O. A., Abderemane-Ali, F., Wei, Y., Coyan, F. C., Pontus, G., Shaya, D., et al. (2020). Up-regulation of Voltage-Gated Sodium Channels by Peptides Mimicking S4-S5 Linkers Reveals a Variation of the Ligand-Receptor Mechanism. Sci. Rep. 10, 5852. doi:10.1038/s41598-020-62615-6
Männikkö, R., Wong, L., Tester, D. J., Thor, M. G., Sud, R., Kullmann, D. M., et al. (2018). Dysfunction of NaV1.4, a Skeletal Muscle Voltage-Gated Sodium Channel, in Sudden Infant Death Syndrome: a Case-Control Study. Lancet 391, 1483–1492. doi:10.1016/S0140-6736(18)30021-7
Mantegazza, M., Cestèle, S., and Catterall, W. A. (2021). Sodium Channelopathies of Skeletal Muscle and Brain. Physiol. Rev. 101, 1633–1689. doi:10.1152/physrev.00025.2020
Mazaleyrat, K., Badja, C., Broucqsault, N., Chevalier, R., Laberthonnière, C., Dion, C., et al. (2020). Multilineage Differentiation for Formation of Innervated Skeletal Muscle Fibers from Healthy and Diseased Human Pluripotent Stem Cells. Cells 9, 1531. doi:10.3390/cells9061531
McManis, P. G., Lambert, E. H., and Daube, J. R. (1986). The Exercise Test in Periodic Paralysis. Muscle Nerve 9, 704–710. doi:10.1002/mus.880090805
Meisler, M. H., Hill, S. F., and Yu, W. (2021). Sodium Channelopathies in Neurodevelopmental Disorders. Nat. Rev. Neurosci. 22, 152–166. doi:10.1038/s41583-020-00418-4
Mercier, S., Lornage, X., Malfatti, E., Marcorelles, P., Letournel, F., Boscher, C., et al. (2017). Expanding the Spectrum of Congenital Myopathy Linked to Recessive Mutations in SCN4A. Neurology 88, 414–416. doi:10.1212/WNL.0000000000003535
Messina, S., and Sframeli, M. (2020). New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 9, 2222. doi:10.3390/jcm9072222
Mi, W., Rybalchenko, V., and Cannon, S. C. (2014). Disrupted Coupling of Gating Charge Displacement to Na+ Current Activation for DIIS4 Mutations in Hypokalemic Periodic Paralysis. J. Gen. Physiol. 144, 137–145. doi:10.1085/jgp.201411199
Mi, W., Wu, F., Quinonez, M., DiFranco, M., and Cannon, S. C. (2019). Recovery from Acidosis Is a Robust Trigger for Loss of Force in Murine Hypokalemic Periodic Paralysis. J. Gen. Physiol. 151, 555–566. doi:10.1085/jgp.201812231
Milton, R. L., Lupa, M. T., and Caldwell, J. H. (1992). Fast and Slow Twitch Skeletal Muscle Fibres Differ in Their Distribution of Na Channels Near the Endplate. Neurosci. Lett. 135, 41–44. doi:10.1016/0304-3940(92)90131-p
Morel, J., Rannou, F., Talarmin, H., Giroux-Metges, M. A., Pennec, J. P., Dorange, G., et al. (2010). Sodium Channel Na(V)1.5 Expression Is Enhanced in Cultured Adult Rat Skeletal Muscle Fibers. J. Membr. Biol. 235, 109–119. doi:10.1007/s00232-010-9262-5
Nam, T. S., Zhang, J., Chandrasekaran, G., Jeong, I. Y., Li, W., Lee, S. H., et al. (2019). A Zebrafish Model of Nondystrophic Myotonia with Sodium Channelopathy. Neurosci. Lett. 714, 134579. doi:10.1016/j.neulet.2019.134579
Nathan, S., Gabelli, S. B., Yoder, J. B., Srinivasan, L., Aldrich, R. W., Tomaselli, G. F., et al. (2021). Structural Basis of Cytoplasmic NaV1.5 and NaV1.4 Regulation. J. Gen. Physiol. 153, e202012722. doi:10.1085/jgp.202012722
Nicole, S., Azuma, Y., Bauché, S., Eymard, B., Lochmüller, H., and Slater, C. (2017). Congenital Myasthenic Syndromes or Inherited Disorders of Neuromuscular Transmission: Recent Discoveries and Open Questions. J. Neuromuscul. Dis. 4, 269–284. doi:10.3233/JND-170257
Nicole, S., and Fontaine, B. (2015). Skeletal Muscle Sodium Channelopathies. Curr. Opin. Neurol. 28, 508–514. doi:10.1097/WCO.0000000000000238
Noreng, S., Li, T., and Payandeh, J. (2021). Structural Pharmacology of Voltage-Gated Sodium Channels. J. Mol. Biol. 433, 166967. doi:10.1016/j.jmb.2021.166967
Novak, A. E., Taylor, A. D., Pineda, R. H., Lasda, E. L., Wright, M. A., and Ribera, A. B. (2006). Embryonic and Larval Expression of Zebrafish Voltage-Gated Sodium Channel Alpha-Subunit Genes. Dev. Dyn. 235, 1962–1973. doi:10.1002/dvdy.20811
Obergrussberger, A., Goetze, T. A., Brinkwirth, N., Becker, N., Friis, S., Rapedius, M., et al. (2018). An Update on the Advancing High-Throughput Screening Techniques for Patch Clamp-Based Ion Channel Screens: Implications for Drug Discovery. Expert Opin. Drug Discov. 13, 269–277. doi:10.1080/17460441.2018.1428555
Oliveira, J. S., Redaelli, E., Zaharenko, A. J., Cassulini, R. R., Konno, K., Pimenta, D. C., et al. (2004). Binding Specificity of Sea Anemone Toxins to Nav 1.1-1.6 Sodium Channels: Unexpected Contributions from Differences in the IV/S3-S4 Outer Loop. J. Biol. Chem. 279, 33323–33335. doi:10.1074/jbc.M404344200
Osteen, J. D., Herzig, V., Gilchrist, J., Emrick, J. J., Zhang, C., Wang, X., et al. (2016). Selective Spider Toxins Reveal a Role for the Nav1.1 Channel in Mechanical Pain. Nature 534, 494–499. doi:10.1038/nature17976
Osteen, J. D., Sampson, K., Iyer, V., Julius, D., and Bosmans, F. (2017). Pharmacology of the Nav1.1 Domain IV Voltage Sensor Reveals Coupling between Inactivation Gating Processes. Proc. Natl. Acad. Sci. U S A. 114, 6836–6841. doi:10.1073/pnas.1621263114
Pan, J. Q., Baez-Nieto, D., Allen, A., Wang, H.-R., and Cottrell, J. R. (2018). “Developing High-Throughput Assays to Analyze and Screen Electrophysiological Phenotypes,” in Phenotypic Screening, Methods in Molecular Biology. Editor B Wagner (New York, NY: Springer New York), 235–252. doi:10.1007/978-1-4939-7847-2_18
Pan, X., Li, Z., Zhou, Q., Shen, H., Wu, K., Huang, X., et al. (2018). Structure of the Human Voltage-Gated Sodium Channel Nav1.4 in Complex with β1. Science 362, eaau2486. doi:10.1126/science.aau2486
Peigneur, S., and Tytgat, J. (2018). Toxins in Drug Discovery and Pharmacology. Toxins (Basel) 10, 126. doi:10.3390/toxins10030126
Plaster, N. M., Tawil, R., Tristani-Firouzi, M., Canún, S., Bendahhou, S., Tsunoda, A., et al. (2001). Mutations in Kir2.1 Cause the Developmental and Episodic Electrical Phenotypes of Andersen's Syndrome. Cell 105, 511–519. doi:10.1016/S0092-8674(01)00342-7
Poulin, H., Gosselin-Badaroudine, P., Vicart, S., Habbout, K., Sternberg, D., Giuliano, S., et al. (2018). Substitutions of the S4DIV R2 Residue (R1451) in NaV1.4 lead to Complex Forms of Paramyotonia Congenita and Periodic Paralyses. Sci. Rep. 8, 2041. doi:10.1038/s41598-018-20468-0
Ptácek, L. J., George, A. L., Griggs, R. C., Tawil, R., Kallen, R. G., Barchi, R. L., et al. (1991). Identification of a Mutation in the Gene Causing Hyperkalemic Periodic Paralysis. Cell 67, 1021–1027. doi:10.1016/0092-8674(91)90374-8
Ptácek, L. J., Tawil, R., Griggs, R. C., Engel, A. G., Layzer, R. B., Kwieciński, H., et al. (1994). Dihydropyridine Receptor Mutations Cause Hypokalemic Periodic Paralysis. Cell 77, 863–868. doi:10.1016/0092-8674(94)90135-x
Richards, K. L., Milligan, C. J., Richardson, R. J., Jancovski, N., Grunnet, M., Jacobson, L. H., et al. (2018). Selective NaV1.1 Activation Rescues Dravet Syndrome Mice from Seizures and Premature Death. Proc. Natl. Acad. Sci. U S A. 115, E8077–E8085. doi:10.1073/pnas.1804764115
Rochtus, A. M., Goldstein, R. D., Holm, I. A., Brownstein, C. A., Pérez-Palma, E., Haynes, R., et al. (2020). The Role of Sodium Channels in Sudden Unexpected Death in Pediatrics. Mol. Genet. Genomic Med. 8, e1309. doi:10.1002/mgg3.1309
Rüdel, R., Lehmann-Horn, F., Ricker, K., and Küther, G. (1984). Hypokalemic Periodic Paralysis: In Vitro Investigation of Muscle Fiber Membrane Parameters. Muscle Nerve 7, 110–120. doi:10.1002/mus.880070205
Ruff, R. L. (1996). Sodium Channel Slow Inactivation and the Distribution of Sodium Channels on Skeletal Muscle Fibres Enable the Performance Properties of Different Skeletal Muscle Fibre Types. Acta Physiol. Scand. 156, 159–168. doi:10.1046/j.1365-201X.1996.189000.x
Rühlmann, A. H., Körner, J., Hausmann, R., Bebrivenski, N., Neuhof, C., Detro-Dassen, S., et al. (2020). Uncoupling Sodium Channel Dimers Restores the Phenotype of a Pain-Linked Nav 1.7 Channel Mutation. Br. J. Pharmacol. 177, 4481–4496. doi:10.1111/bph.15196
Sarbjit-Singh, S. S., Matthews, H. R., and Huang, C. L. (2020). Ryanodine Receptor Modulation by Caffeine Challenge Modifies Na+ Current Properties in Intact Murine Skeletal Muscle Fibres. Sci. Rep. 10, 2199. doi:10.1038/s41598-020-59196-9
Slater, C. R. (2003). Structural Determinants of the Reliability of Synaptic Transmission at the Vertebrate Neuromuscular Junction. J. Neurocytol. 32, 505–522. doi:10.1023/b:neur.0000020607.17881.9b
Slater, C. (2017). The Structure of Human Neuromuscular Junctions: Some Unanswered Molecular Questions. Ijms 18, 2183. doi:10.3390/ijms18102183
Sloth, C. K., Denti, F., Schmitt, N., Bentzen, B. H., Fagerberg, C., Vissing, J., et al. (2018). Homozygosity for SCN4A Arg1142Gln Causes Congenital Myopathy with Variable Disease Expression. Neurol. Genet. 4, e267. doi:10.1212/nxg.0000000000000267
Sokolov, S., Scheuer, T., and Catterall, W. A. (2008). Depolarization-activated Gating Pore Current Conducted by Mutant Sodium Channels in Potassium-Sensitive Normokalemic Periodic Paralysis. Proc. Natl. Acad. Sci. U S A. 105, 19980–19985. doi:10.1073/pnas.0810562105
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating Pore Current in an Inherited Ion Channelopathy. Nature 446, 76–78. doi:10.1038/nature05598
Statland, J. M., Bundy, B. N., Wang, Y., Rayan, D. R., Trivedi, J. R., Sansone, V. A., et al. (2012). Mexiletine for Symptoms and Signs of Myotonia in Nondystrophic Myotonia: a Randomized Controlled Trial. JAMA 308, 1357–1365. doi:10.1001/jama.2012.12607
Statland, J. M., Fontaine, B., Hanna, M. G., Johnson, N. E., Kissel, J. T., Sansone, V. A., et al. (2018). Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve 57, 522–530. doi:10.1002/mus.26009
Struyk, A. F., and Cannon, S. C. (2008). Paradoxical Depolarization of Ba2+- Treated Muscle Exposed to Low Extracellular K+: Insights into Resting Potential Abnormalities in Hypokalemic Paralysis. Muscle Nerve 37, 326–337. doi:10.1002/mus.20928
Struyk, A. F., Scoggan, K. A., Bulman, D. E., and Cannon, S. C. (2000). The Human Skeletal Muscle Na+ Channel Mutation R669H Associated with Hypokalemic Periodic Paralysis Enhances Slow Inactivation. J. Neurosci. 20, 8610–8617. doi:10.1523/jneurosci.20-23-08610.2000
Stunnenberg, B. C., Raaphorst, J., Deenen, J. C. W., Links, T. P., Wilde, A. A., Verbove, D. J., et al. (2018a). Prevalence and Mutation Spectrum of Skeletal Muscle Channelopathies in the Netherlands. Neuromuscul. Disord. 28, 402–407. doi:10.1016/j.nmd.2018.03.006
Stunnenberg, B. C., Raaphorst, J., Groenewoud, H. M., Statland, J. M., Griggs, R. C., Woertman, W., et al. (2018b). Effect of Mexiletine on Muscle Stiffness in Patients with Nondystrophic Myotonia Evaluated Using Aggregated N-of-1 Trials. JAMA 320, 2344–2353. doi:10.1001/jama.2018.18020
Swinney, D. C., and Lee, J. A. (2020). Recent Advances in Phenotypic Drug Discovery. F1000Res 9, F1000. Faculty Rev-944. doi:10.12688/f1000research.25813.1
Tarvin, R. D., Santos, J. C., O'Connell, L. A., Zakon, H. H., and Cannatella, D. C. (2016). Convergent Substitutions in a Sodium Channel Suggest Multiple Origins of Toxin Resistance in Poison Frogs. Mol. Biol. Evol. 33, 1068–1081. doi:10.1093/molbev/msv350
Teener, J. W., and Rich, M. M. (2006). Dysregulation of Sodium Channel Gating in Critical Illness Myopathy. J. Muscle Res. Cel. Motil. 27, 291–296. doi:10.1007/s10974-006-9074-5
Tricarico, D., Mele, A., and Conte Camerino, D. (2006). Carbonic Anhydrase Inhibitors Ameliorate the Symptoms of Hypokalaemic Periodic Paralysis in Rats by Opening the Muscular Ca2+-Activated-K+ Channels. Neuromuscul. Disord. 16, 39–45. doi:10.1016/j.nmd.2005.10.005
Trimmer, J. S., Cooperman, S. S., Tomiko, S. A., Zhou, J. Y., Crean, S. M., Boyle, M. B., et al. (1989). Primary Structure and Functional Expression of a Mammalian Skeletal Muscle Sodium Channel. Neuron 3, 33–49. doi:10.1016/0896-6273(89)90113-x
Tsujino, A., Maertens, C., Ohno, K., Shen, X. M., Fukuda, T., Harper, C. M., et al. (2003). Myasthenic Syndrome Caused by Mutation of the SCN4A Sodium Channel. Proc. Natl. Acad. Sci. U S A. 100, 7377–7382. doi:10.1073/pnas.1230273100
Vicart, S., Sternberg, D., Fournier, E., Ochsner, F., Laforet, P., Kuntzer, T., et al. (2004). New Mutations of SCN4A Cause a Potassium-Sensitive Normokalemic Periodic Paralysis. Neurology 63, 2120–2127. doi:10.1212/01.wnl.0000145768.09934.ec
Wallgren-Pettersson, C., and Laing, N. (2010). “The Congenital Myopathies,” in Disorders of Voluntary Muscle. Editors G Karpati, D Hilton-Jones, K Bushby, and RC Griggs (Cambridge: Cambridge University Press), 282–298.
Weber, F., Jurkat-Rott, K., and Lehmann-Horn, F. (1993). “Hyperkalemic Periodic Paralysis,”. Editors M. P Adam, H H Ardinger, R. A Pagon, S. E Wallace, L. J. H Bean, K Stephenset al. (Seattle (WA): GeneReviews®). Available at: https://www-ncbi-nlm-nih-gov.ezproxy.u-pec.fr/books/NBK1338/(Accessed July 5, 2021).
Weuring, W. J., Singh, S., Volkers, L., Rook, M. B., van 't Slot, R. H., Bosma, M., et al. (2020). NaV1.1 and NaV1.6 Selective Compounds Reduce the Behavior Phenotype and Epileptiform Activity in a Novel Zebrafish Model for Dravet Syndrome. PLoS ONE 15, e0219106. doi:10.1371/journal.pone.0219106
Winters, J. J., and Isom, L. L. (2016). Developmental and Regulatory Functions of Na(+) Channel Non-pore-forming β Subunits. Curr. Top. Membr. 78, 315–351. doi:10.1016/bs.ctm.2016.07.003
Wu, F., Mi, W., Burns, D. K., Fu, Y., Gray, H. F., Struyk, A. F., et al. (2011). A Sodium Channel Knockin Mutant (NaV1.4-R669H) Mouse Model of Hypokalemic Periodic Paralysis. J. Clin. Invest. 121, 4082–4094. doi:10.1172/JCI57398
Wu, F., Mi, W., and Cannon, S. C. (2013a). Beneficial Effects of Bumetanide in a CaV1.1-R528H Mouse Model of Hypokalaemic Periodic Paralysis. Brain 136, 3766–3774. doi:10.1093/brain/awt280
Wu, F., Mi, W., and Cannon, S. C. (2013b). Bumetanide Prevents Transient Decreases in Muscle Force in Murine Hypokalemic Periodic Paralysis. Neurology 80, 1110–1116. doi:10.1212/WNL.0b013e3182886a0e
Wu, F., Mi, W., Fu, Y., Struyk, A., and Cannon, S. C. (2016). Mice with an NaV1.4 Sodium Channel Null Allele Have Latent Myasthenia, without Susceptibility to Periodic Paralysis. Brain 139, 1688–1699. doi:10.1093/brain/aww070
Wu, F., Mi, W., Hernández-Ochoa, E. O., Burns, D. K., Fu, Y., Gray, H. F., et al. (2012). A Calcium Channel Mutant Mouse Model of Hypokalemic Periodic Paralysis. J. Clin. Invest. 122, 4580–4591. doi:10.1172/JCI66091
Yamagata, T., Raveau, M., Kobayashi, K., Miyamoto, H., Tatsukawa, T., Ogiwara, I., et al. (2020). CRISPR/dCas9-based Scn1a Gene Activation in Inhibitory Neurons Ameliorates Epileptic and Behavioral Phenotypes of Dravet Syndrome Model Mice. Neurobiol. Dis. 141, 104954. doi:10.1016/j.nbd.2020.104954
Yang, D. C., Deuis, J. R., Dashevsky, D., Dobson, J., Jackson, T. N., Brust, A., et al. (2016). The Snake with the Scorpion's Sting: Novel Three-Finger Toxin Sodium Channel Activators from the Venom of the Long-Glanded Blue Coral Snake (Calliophis Bivirgatus). Toxins (Basel) 8, 303. doi:10.3390/toxins8100303
Yang, J. S., Bennett, P. B., Makita, N., George, A. L., and Barchi, R. L. (1993). Expression of the Sodium Channel Beta 1 Subunit in Rat Skeletal Muscle Is Selectively Associated with the Tetrodotoxin-Sensitive Alpha Subunit Isoform. Neuron 11, 915–922. doi:10.1016/0896-6273(93)90121-7
Yoder, J. B., Ben-Johny, M., Farinelli, F., Srinivasan, L., Shoemaker, S. R., Tomaselli, G. F., et al. (2019). Ca2+-dependent Regulation of Sodium Channels NaV1.4 and NaV1.5 Is Controlled by the post-IQ Motif. Nat. Commun. 10, 1514. doi:10.1038/s41467-019-09570-7
Zaharieva, I. T., Thor, M. G., Oates, E. C., van Karnebeek, C., Hendson, G., Blom, E., et al. (2016). Loss-of-function Mutations in SCN4A Cause Severe Foetal Hypokinesia or 'classical' Congenital Myopathy. Brain 139, 674–691. doi:10.1093/brain/awv352
Keywords: sodium channel, skeletal muscle, loss of function, therapeutics, congenital myasthenic syndrome (CMS), congenital myopathy (CM)
Citation: Nicole S and Lory P (2021) New Challenges Resulting From the Loss of Function of Nav1.4 in Neuromuscular Diseases. Front. Pharmacol. 12:751095. doi: 10.3389/fphar.2021.751095
Received: 31 July 2021; Accepted: 16 September 2021;
Published: 04 October 2021.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Michael Craig Sanguinetti, The University of Utah, United StatesCopyright © 2021 Nicole and Lory. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Nicole, c29waGllLm5pY29sZUBpbnNlcm0uZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.