 Alexandrina Campos-Martins1
Alexandrina Campos-Martins1 Bruno Bragança
Bruno Bragança Paulo Correia-de-Sá
Paulo Correia-de-Sá Ana Patrícia Fontes-Sousa
Ana Patrícia Fontes-Sousa
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 20 August 2021
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.724320
This article is part of the Research TopicNovel Perspectives For The Therapy And Prevention Of Heart FailureView all 13 articles
Heart failure with preserved ejection fraction (HFpEF) roughly represents half of the cardiac failure events in developed countries. The proposed ‘systemic microvascular paradigm’ has been used to explain HFpHF presentation heterogeneity. The lack of effective treatments with few evidence-based therapeutic recommendations makes HFpEF one of the greatest unmet clinical necessities worldwide. The endogenous levels of the purine nucleoside, adenosine, increase significantly following cardiovascular events. Adenosine exerts cardioprotective, neuromodulatory, and immunosuppressive effects by activating plasma membrane-bound P1 receptors that are widely expressed in the cardiovascular system. Its proven benefits have been demonstrated in preclinical animal tests. Here, we provide a comprehensive and up-to-date critical review about the main therapeutic advantages of tuning adenosine signalling pathways in HFpEF, without discounting their side effects and how these can be seized.
Heart failure (HF) is a clinical syndrome characterized by alterations in the cardiac structure and/or function; it is associated with a poor quality of life, high rates of hospitalizations, and significant mortality (Ponikowski et al., 2016). HF with preserved ejection fraction (HFpEF) is a subclass of HF defined by left ventricular ejection fraction equal to or above 50% (Ponikowski et al., 2016). Nowadays, it represents up to half of all cases of HF in the developed world, which at least in part may be explained by the current lifestyle and/or prolongation of the life expectancy (Dunlay et al., 2017). HFpEF has a “heterogeneous” presentation often coursing with unspecific clinical symptoms (e.g., exercise intolerance and dyspnoea), challenging its diagnosis and clinical management (Ponikowski et al., 2016). Multiple cardiac-related systemic comorbidities drive the molecular and hemodynamic mechanisms implicated in this disease. Emerging data support systemic microvascular inflammation as an enduring aggravation factor (Paulus and Tschope, 2013). These findings contrast with HF with reduced ejection fraction (left ventricular ejection fraction under 40%), where cardiomyocytes loss and their replacement by fibrotic tissue is the most frequent pathological hallmark (Simmonds et al., 2020). In this case, the systolic dysfunction results in neurohormonal activation of the renin-angiotensin-aldosterone system along with sympathetic stimulation, supporting the indication for using angiotensin-converting enzyme inhibitors, mineralocorticoid receptors antagonists and ß-blockers as disease-modifying drugs (Ponikowski et al., 2016). Conversely, there is still no proven therapy to impact the course of HFpEF, thus representing one of the biggest challenges of cardiovascular medicine nowadays (Iliesiu and Hodorogea, 2018). Therefore, unraveling the dysfunctional signalling pathways associated with HFpEF may shed some light to propose novel pharmacological therapies to this unmet clinical condition.
The purine nucleoside adenosine was first identified in 1929 when Drury and Szent-Gyorgyi successfully extracted a rhythm-influencing adenylic substance from the mammalian heart and other tissues (Drury and Szent-Gyorgyi, 1929). Only in 1972, Geoff Burnstock (born: May 10, 1929, died: June 2, 2020) coined the term purinergic signalling referring to the extracellular effects of adenosine 5′-triphosphate (ATP). Following Burnstock’s pioneering work on the role of ATP-sensitive P2 purinoceptors, its metabolite adenosine soon became recognized as an extracellular signalling molecule through the activation of plasma membrane-bound P1 receptors family that are expressed in every organ systems in the body. In the cardiovascular system, adenosine is considered a “retaliatory metabolite” because it originates from the catabolism of ATP released from stressed cells, thus contributing to decrease cellular energy consumption while favoring the blood tissue supply (Newby et al., 1990). Given this, adenosine-mediated signals have been implicated in many pathophysiological processes. Nowadays, adenosine receptor ligands are being extensively investigated as promising druggable compounds, some of which are undergoing clinical trials (Borah et al., 2019). However, besides the native nucleoside, only a limited number of adenosine-related drugs are approved for clinical use (Borah et al., 2019).
Current knowledge points towards adenosine as a ubiquitous signalling mediator of countless physiological processes throughout the body via the activation of 4 G protein-coupled receptors known as P1 receptors [adenosine receptors (ARs): A1AR, A2AAR, A2BAR, A3AR] (Borea et al., 2018). The A1AR and A3AR were first characterized as negatively coupled to adenylate cyclase (AC) through Gi and Go proteins binding, which normally decrease intracellular cyclic adenosine 5′-monophosphate (cAMP) levels. Contrariwise, both high-affinity A2AAR and low-affinity A2BAR couple to Gs proteins and stimulate AC leading to intracellular cAMP accumulation. Despite this simplistic view of the canonical coupling of P1 receptors to the AC/cAMP pathway, evidence has been gathered demonstrating that A1AR, A2BAR, and A3AR may also activate the phospholipase C beta (PLC-β) isoform. Stimulation of PLC releases inositol 1,4,5-trisphosphate from the plasma membrane and trigger intracellular calcium (Ca2+) mobilization; both intracellular Ca2+ recruitment and diacylglycerol (DAG) production synergize to stimulate Ca2+-dependent protein kinase C (PKC) and/or other Ca2+-associated downstream pathways (Figure 1) (Borea et al., 2018). Interestingly, the cardiovascular (and many other) effects of adenosine are mediated by a multiplicity of more recently discovered intracellular signalling cascades, such as exchange protein directly activated by cAMP (EPAC), phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt), mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinases (Erk), glycogen synthase kinase 3β (see From Cardiovascular Effects of Adenosine to Cardioprotection and Figure 2). Due to their relevance to the current knowledge, involvement of these pathways to adenosine-mediated effects will be further discussed in the following sections.
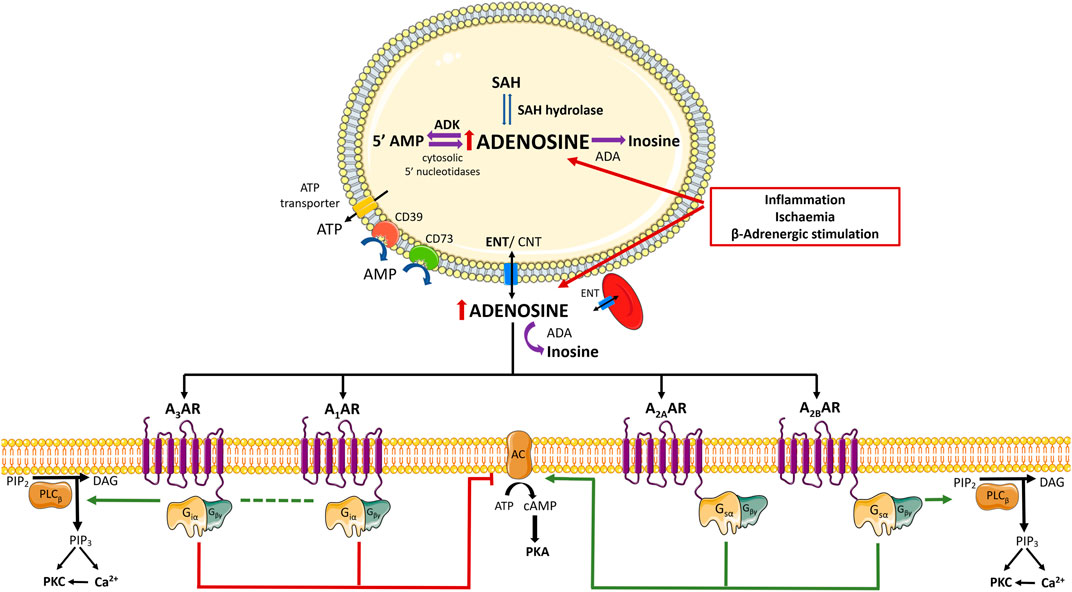
FIGURE 1. Schematic representation of adenosine biosynthesis and complex signalling pathways induced by adenosine receptors activation. Adenosine is a purine nucleoside continuously generated from the catabolism of adenine nucleotides via a cascade of nucleotidases. Regarding the fate of intracellular adenosine, it may (preferentially) re-enter the purines savage pathway by intracellular phosphorylation via adenosine kinase (ADK) or it is inactivated to inosine by adenosine deaminase (ADA). The main source of adenosine results from the hydrolysis of S-adenosylhomocysteine (SAH) by SAH hydrolase. Under low oxygen availability and/or during high energy working loads, recruitment from ATP hydrolysis increases favouring intracellular adenosine accumulation, which promotes its diffusion to the extracellular medium via equilibrative nucleoside transporters (ENTs). Extracellular adenosine may also result from the extracellular breakdown of released adenine nucleotides, namely adenosine 5′-triphosphate (ATP), ADP (adenosine 5′-diphosphate), and AMP (adenosine 5′-monophosphate), by a cascade of ecto-nucleotidases bound to the plasma membrane. CD39 dephosphorylates ATP directly into AMP; the rate limiting enzyme of the ecto-nucleotidase cascade is ecto-5′-nucleotidase/CD73, which dephosphorylates AMP to adenosine and inorganic phosphate. Extracellular adenosine levels are tightly regulated by cellular uptake via ENTs and/or by deamination into inosine by ADA. Adenosine activates 4 G protein-coupled receptor (GPCRs) known as P1 receptors (adenosine receptors (ARs): A1AR, A2AAR, A2BAR, A3AR). In brief, A1AR and A3AR are negatively coupled to adenylate cyclase (AC) through binding to Gi and Go proteins, resulting in decreased intracellular cyclic AMP (cAMP) levels. Both A2AAR and A2BAR are coupled to Gs proteins and stimulate AC leading to increases in cAMP accumulation. Despite the canonical positive and negative coupling to the AC/cAMP system, A1AR, A2BAR, and A3AR are also entitled to activate phospholipase C-beta (PLC- β) isoform, resulting in increased inositol 1,4,5-trisphosphate (IP3) and intracellular Ca2+ mobilization; intracellular Ca2+ and diacylglycerol (DAG) production contribute to stimulate Ca2+-dependent protein kinase C (PKC) and/or downstream Ca2+-dependent pathways. Adapted from (Borea et al., 2018). Green arrows and red bars indicate the effects induced or blocked by adenosine receptor activation, respectively. Illustration used elements from Servier Medical Art (http://smart.servier.com). AC, Adenosine cyclase; ADA, Adenosine deaminase; ADP, 5′adenosine diphosphate; ADK, Adenosine kinase; AMP, 5′-adenosine monophosphate; AR, Adenosine receptor; ATP, 5′-adenosine triphosphate; cAMP, cyclic AMP; CNT, Concentrative nucleoside transporter; DAG, diacylglycerol; ENT, Equilibrative nucleoside transporter; GCPR, G protein-coupled receptor; IP3, inositol 1,4,5-trisphosphate; PKA, protein kinase A; PKC, protein kinase C; PLC- ß, Phospholipase C- beta; SAH, S-adenosylhomocysteine.
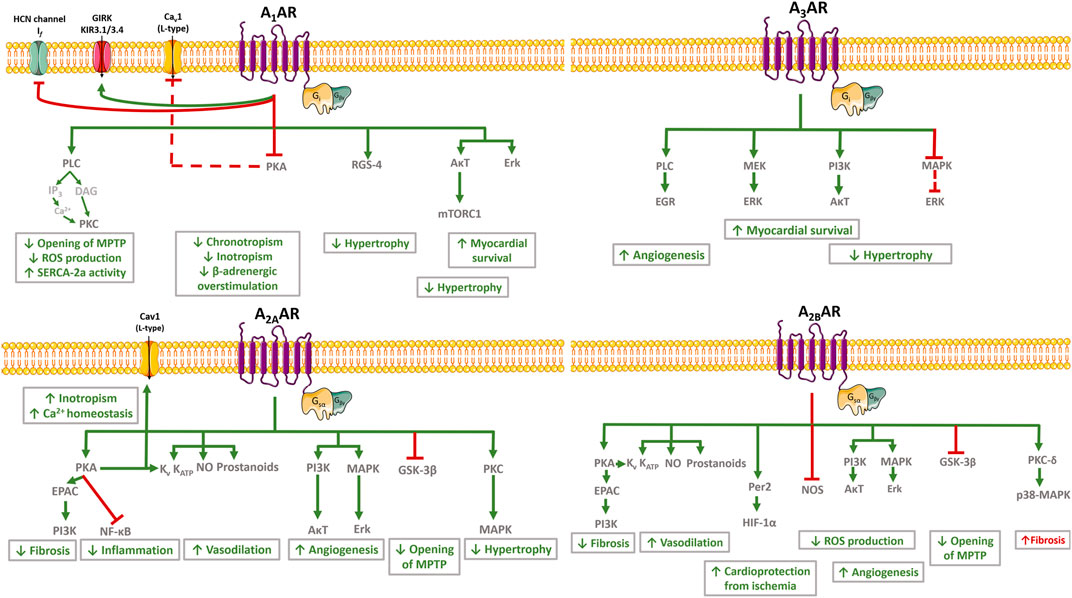
FIGURE 2. Schematic representation of cardiovascular effects of adenosine and related signalling pathways. In the cardiovascular system, adenosine exerts protective effects to control neuronal output (anti-adrenergic effect) and inflammation, while decreasing cardiac metabolism, myocardial contractility, impulse generation and conduction, and the coronary tone. These effects depend on the cellular type involved. The A1AR activation mediates cardioprotective effects by reversing cardiac hypertrophy/remodelling, improving mitochondrial function, enhancing SERCA2a activity and improving Ca2+ handling. The A1AR activation is also associated with an anti-ischemic effect by decreasing catecholamine release and by counteracting ß-adrenergic dysfunction. Activation of A2AAR attenuates cardiac inflammation, fibrosis and hypertrophy, while favoring vasodilation and angiogenesis. Despite the A2BAR-related vasodilation, angiogenesis and protection from ischemic preconditioning, conflicting evidence exist regarding the role of this receptor in fibrosis and related cardiac ischemic remodelling. Activation of A3AR is also associated with controversial results; this receptor mediates cardioprotective effects by increasing angiogenesis and myocardial survival during reperfusion, but it may be deleterious by aggravating cardiomyocytes hypertrophy. Green arrows and red bars indicate effects induced or blocked by adenosine receptors activation, respectively. The red dashed line indicates that the effect is indirectly blocked by adenosine receptors activation. Illustration used elements from Servier Medical Art (http://smart.servier.com) AR, Adenosine receptor; AκT, Protein kinase B; Cav1 channel, L-type calcium channel subunit 1; DAG, diacylglycerol; EGR, Early Growth Response; EPAC, Exchange protein directly activated by cAMP; ERK, Extracellular signal-regulated protein kinase; GIRK and KIR3.1/3.4, G protein coupled inwardly rectifying K+ channels; GSK-3, Glycogen synthase kinase-3; HCN channels, Hyperpolarization-activated cyclic nucleotide-gated channel; HIF-1α, Hypoxia-inducible factor-alpha; IP3, Inositol 1,4,5-trisphosphate; If , “funny” hyperpolarization-activated current; KATP, ATP-sensitive K+ channel; KV, voltage-gated K+ channels; MAPK, Mitogen activated protein kinase; MEK, Mitogen-activated protein kinase; MPTP, Mitochondrial permeability transition pores; mTORC1, Rapamycin complex 1; NF-κB, Nuclear factor-κB; NO, Nitric oxide; Per2, Period 2; PI3K, Phosphoinositol-3 kinase; PKA, Protein kinase A; PKC, Protein kinase C; PLC, Phospholipase C; ROS, Reactive oxygen species; SERCA2a, Sarco/endoplasmic reticulum Ca2+-ATPase 2a.
Adenosine is a purine nucleoside continuously generated from the catabolism of adenine nucleotides via a cascade of nucleotidases, including the rate-limiting enzyme 5′-nucleotidase, which dephosphorylates AMP to adenosine both intra- and extracellularly (Figure 1) (Rubio et al., 1973). Regarding the fate of intracellular adenosine, it may (preferentially) re-enter the purines salvage pathway through intracellular phosphorylation via adenosine kinase (ADK) (Km = 2 µM). Alternatively, the nucleoside can be inactivated to inosine by adenosine deaminase (ADA) (Km = 17–45 µM) when the phosphorylation pathway is overloaded or inhibited (Borea et al., 2018). Another possible source of adenosine comes from the hydrolysis of S-adenosylhomocysteine by S-adenosylhomocysteine hydrolase, being this the main pathway responsible for the intracellular levels of adenosine in conditions of adequate oxygen supply (Schrader et al., 1981). Under low oxygen supply conditions and/or during high energy working loads, ATP hydrolysis increases resulting in intracellular adenosine accumulation. As a consequence, adenosine diffuses to the extracellular milieu via equilibrative nucleoside transporters (ENTs), mostly ENT1 and ENT2 encoded by SLC29A1 and SLC29A2 genes, respectively (Governo et al., 2005; Dip, 2009). NBTI-sensitive (ENT1) and insensitive (ENT2) transporters are expressed in the vascular endothelium, erythrocytes, inflammatory cells, and cardiomyocytes (Eltzschig et al., 2005; Morote-Garcia et al., 2009). The sensitivity of both transporters to dipyridamole allowed this drug to be introduced in the market as a coronary vasodilator more than half a century ago, which is still used nowadays as antithrombotic and vasodilator with promising anti-oxidant properties (Ciacciarelli et al., 2015). Transport of adenosine across the plasma membrane can also occur through concentrative nucleoside transporters encoded by SLC28A1, SLC28A2, and SLC28A3 genes; these proteins concentrate adenosine (and other purines and pyrimidines) inside the cells via a Na+-nucleoside cotransporter (Pastor-Anglada and Perez-Torras, 2018). While ENTs may have a major role in maintaining nucleoside homeostasis, concentrative nucleoside transporters may contribute to adenosine sensing and signal transduction inside the cells (transceptor function).
As aforementioned, extracellular adenosine may result from the release of the nucleoside via ENTs and/or through the extracellular breakdown of released adenine nucleotides, namely ATP, ADP, and AMP, by a cascade of ecto-nucleotidases bound to the plasma membrane. Besides originating from damaged cells, adenine nucleotides may be released from intact cells by vesicular exocytosis, as well as via other mechanisms involving plasma membrane “pores”, namely ionotropic P2 purinoceptors, ABC proteins and hemichannels containing connexins and/or pannexins (Lazarowski et al., 2011).
Four members of the ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase) family (NTPDase1, 2, 3, and 8) and two members of the ecto-nucleotide pyrophosphatases/phosphodiesterases (E-NPP) family (NPP1 and NPP3) are located at the plasma membrane and hydrolyze extracellular nucleotides (Yegutkin, 2008). NTPDase1 (CD39 or apyrase) dephosphorylates ATP directly into AMP, with minimal accumulation of ADP. NTPDase2 (ATPase) is a preferential nucleoside triphosphatase that hydrolysis ADP 10 to 15 times less efficiently than ATP, leading to minimal AMP accumulation. NTPDase3 and NTPDase8 are functional intermediates between NTPDase1 and NTPDase2. NTPDases are considered potential therapeutic targets due to their role in coagulation, immune responses, vascular inflammation, and cancer (Vieira et al., 2014). Interestingly, NPP1 and NPP3 phosphorylated product (e.g., AMP) has a higher binding affinity to this enzyme than substrates do, leading to the inhibition of nucleoside 5′-monophosphate release (Vieira et al., 2014). The rate-limiting enzyme of the ecto-nucleotidase cascade is ecto-5′-nucleotidase/CD73, which dephosphorylates AMP to adenosine and inorganic phosphate. This enzyme is abundantly expressed in immune (e.g., T and B lymphocytes) and mesenchymal originated cells (Bono et al., 2015).
Extracellular adenosine levels are tightly regulated by cellular uptake via ENTs and/or by deamination into inosine by ADA; this enzyme can be found either on the cell surface (ecto-ADA) or as a soluble form after cleavage of its anchor to the plasma membrane (exo-ADA). Low levels of adenosine are normally found in biological fluids. Nevertheless, the extracellular concentration of the nucleoside increases in stressed cells following ischemia/reperfusion, inflammation, neuronal activation, and tissue damage (Sumi et al., 2010; Grenz et al., 2011; Idzko et al., 2014). Adenosine in the plasma has a short half-life due to its rapid inactivation by blood cells and by the vascular endothelium (Meyskens and Williams, 1971). Notwithstanding this, high plasma levels of adenosine have been detected in heart failure, both in humans (Funaya et al., 1997; Asakura et al., 2007) and animals (Correia-de-Sa et al., 2015). Moreover, the ecto-5′-nucleotidase/CD73 activity increases in the plasma and ventricular myocardium of chronic HF patients (Fujita et al., 2008). Together, plasma adenosine and ecto-5′-nucleotidase/CD73 activity may be reliable markers for the diagnosis of severity grade and follow-up of HF, even though adenosine levels are normally underestimated due to its extensive inactivation in the blood stream.
The A1AR is the most expressed adenosine receptor subtype found in the intact myocardium and isolated cardiomyocytes, being particularly abundant in atria. The second most abundant receptor in the heart is the A2AAR, followed by A2BAR and A3AR, which exhibit low expression levels in the heart (Headrick et al., 2013). Nevertheless, one should remember that this pattern is mostly influenced by receptors expressed in cardiomyocytes, which occupy up to 85% of the mammalian heart volume (Zhou and Pu, 2016). Looking more deeply into the distribution of adenosine receptors in cardiac cells, this expression profile changes with A2BAR and A2AAR being more abundant in endothelial cells and cardiac fibroblasts (Headrick et al., 2013). Other tissues and organs influencing the cardiac function are also enriched in adenosine receptors. Large amounts of A1AR are found in kidneys, adipose tissue, pancreas, and brain. The A2AAR is highly expressed in peripheral immune cells (particularly in leucocytes), platelets, smooth muscle fibers and endothelial cells (Headrick et al., 2013).
The activity of the low-affinity A2BAR has been physiologically neglected mostly because higher amounts of adenosine are required to activate this receptor even in cells where its expression is high (Sun and Huang, 2016). However, this assumption is changing as A2BAR over-functioning has been gathered in several pathological conditions, including hypoxia, inflammation, and cell stress, thus providing support for a meaningful role of this receptor in health and disease (Bessa-Goncalves et al., 2018). Considering the retaliatory nature of adenosine, the A2BAR can be seen as a “dormant” receptor that “wakes up” following cells and tissue injury (Bessa-Goncalves et al., 2018). The presence of the A3AR in cardiomyocytes, vascular smooth muscle cells, and immune cells have been implicated in its cardioprotective role against ischemia, as well as in the control of blood vessels tone and remodelling (Headrick et al., 2013).
In the cardiovascular system, adenosine exerts additional protective effects to control neuronal output and inflammation, while decreasing cardiac metabolism, myocardial contractility, impulse generation and conduction, and the coronary tone; the nucleoside is also involved in the control of adrenergic responsiveness and blood pressure (Figure 2) (Headrick et al., 2013).
Stimulation of A1AR induces negative chronotropism and dromotropism, as this receptor is highly expressed in the heart conduction system (sinoatrial and atrioventricular nodes, and the His-Purkinje network) (Headrick et al., 2013). In this regard, while adenosine exerts direct inhibitory effects on chronotropism and dromotropism in atrial cardiomyocytes, it also counteracts ß-adrenergic effects on impulse generation and contractility. The A1AR-induced hyperpolarization of the supraventricular tissue is accomplished by favoring outward potassium (K+) currents through opening G protein-coupled inwardly rectifying K+ channels (GIRK or KIR3.1/3.4), while counteracting adrenergic effects through inhibition of Ca2+ influx and/or attenuation of hyperpolarization-activated cyclic nucleotide-gated channel four mediating the pacemaker “funny” (If) currents (Belardinelli and Isenberg, 1983; Belardinelli et al., 1995). These effects lead to bradycardia and atrial hypocontractility. In this respect, our group demonstrated that adenosine-induced negative atrial inotropism may be partially counteracted by multiple downstream intracellular pathways ending up to increase the time available for Ca2+ influx through Cav1 (L-type) channels (Braganca et al., 2016). Taken together, the electrophysiological properties of the A1AR justify the use of adenosine in the treatment of supraventricular tachycardia and to control the rate of ventricular contractions during atrial fibrillation (Savelieva and Camm, 2008; Lim et al., 2009).
Adenosine counteracts the sympathetic drive operated by catecholamines on cardiac ß-adrenoceptors by decreasing impulse generation and contractility via a mechanism involving both pre- and post-junctional effects. The positive inotropic action of ß-adrenoceptors may be directly controlled by adenosine via inhibition of cAMP production and protein kinase A (PKA) activation (Dobson, 1983; Romano and Dobson, 1990). The A1AR-mediated cardioprotection extends to intracellular organelles, namely mitochondria through a PKC-mediated reduction of the permeability of mitochondrial transition pores, stabilization of mitochondrial membrane potential and inhibition of hypoxia-induced production of reactive oxygen species (ROS), which ends up in the opening of KATP mitochondrial channels and cytoprotection (Xiang et al., 2010a). Activation of A1AR also increases epidermal growth factor receptor activation through the action of myocardial survival kinases Erk 1/2, Aκt (Williams-Pritchard et al., 2011). Overall, these properties confer myocardial protection under ischemic pre-conditioning and ischemia/reperfusion situations (Reichelt et al., 2005; Morrison et al., 2006; Greene et al., 2016).
While myocardial actions of adenosine typically focus on activation of the most abundant A1AR, increasing data also suggest the involvement of the A2AAR in myocardial contractile performance (Dobson and Fenton, 1997) via PKA-dependent increases in intracellular Ca2+ (Dobson et al., 2008). The A2AAR counteracts the antiadrenergic effects of the A1AR at the intracellular signaling level, a phenomenon that is further amplified by the formation of A1AR:A2AAR heteromers (Fenton and Dobson, 2007). Both A2AAR (Iwamoto et al., 1994; Shryock et al., 1998) and A2BAR (Kusano et al., 2010) have vasodilatory effects mediated by cAMP and PKA activation in vascular smooth muscle cells (Iwamoto et al., 1994). Involvement of Kv and KATP channels (Berwick et al., 2010), nitric oxide (NO) (Li et al., 1995; Li et al., 1998), and prostanoids (Faria et al., 2006) production have also been demonstrated. Coupling of A2AAR and A2BAR to such downstream signaling pathways entitles adenosine to regulate coronary blood flow and to protect against chronic myocardial ischemia following endothelial dysfunction. Long-term stimulation of these receptors also fosters mitogenic signaling pathways resulting in vascular overgrowth and angiogenesis; these pathways include A2AAR and A2BAR mediated-PI3K/Aκt activation and MAPK/Erk activation by A2AAR (Ahmad et al., 2013; Du et al., 2015).
Mounting evidence has been gathered suggesting that A2AAR activation is cardioprotective and immunosuppressant during early reperfusion after myocardial infarction (MI). The selective A2AAR agonist, CGS 21680, decreased neutrophils adhesion to the endothelium and subsequent myocardial infiltration, together with a reduction of superoxide production, in a canine model of ischemia and reperfusion (Jordan et al., 1997). Moreover, another A2AAR agonist, ATL146e, attenuated resident mast cells degranulation in a mouse model of myocardial ischemia (Rork et al., 2008). Cardioprotection is also accomplished because activation of A2AAR prevents the infarct-boosting effect of interferon-gamma (IFN-γ) produced by CD4+ T cells, as this cytokine favors reperfusion injury by activating pro-inflammatory macrophages (Yang et al., 2006). Although preconditioning is hardly feasible in clinical settings, clinical trials are ongoing to test whether A2AAR agonists can be used in coronary artery disease and MI (Borah et al., 2019). In this context, it is worth noting that both adenosine and the selective A2AAR agonist, regadenoson, have been clinically approved for myocardial perfusion imaging (Cury et al., 2014).
To our knowledge, A2BAR is the only adenosine receptor subtype upregulated in ischemic hearts of both mice and humans (Gile and Eckle, 2016). Activation of the A2BAR controls Period 2 protein, a metabolic master-switch of myocardial adaptation to ischemia. This protein stabilizes the hypoxia-inducible factor 1α and, thereby, the transduction of glycolytic enzymes (Eckle et al., 2012) to optimize oxygen consumption and to protect the myocardium from infarction reperfusion (Gile and Eckle, 2016). Moreover, both A2BAR and A2AAR are associated to inhibition of mitochondrial glycogen synthase kinase 3β (GSK-3β) phosphorylation, thus preventing the mitochondrial permeability transition pore opening, which is a critical step to afford cardioprotection during myocardial reperfusion (Xi et al., 2009). Inhibition of ROS production by mitochondria broadens the cardioprotective effects of the A2BAR in cardiac ischemia by involving multiple PKC-mediated signalling pathways affecting nitric oxide synthase (NOS), PI3K/Aκt, and Erk 1/2 enzymatic activities (Kuno et al., 2007; Yang et al., 2011; Seo et al., 2015).
Only a few studies have been conducted to elucidate the role of the A3AR in cardiac pathophysiology, most probably because this receptor subtype has low expression levels in cardiac tissues. This situation may also occur because the A3AR has unpredictable effects in rodent models, as it behaves like a low affinity receptor for adenosine contrary to the high affinity for the nucleoside shown in human tissues (Headrick et al., 2013). Current data suggest that the A3AR limits injury processes occurring in the ischemic myocardium, while exerting an anti-inflammatory action during cardiac reperfusion (Ge et al., 2010). Surprisingly, A3AR agonists cause a biphasic hemodynamic response that is characterized, initially, by indirect activation of high affinity A2AAR followed by subsequent A3AR-mediated effects that prevail after A2AAR become desensitized (Tian et al., 2015a). Both in cardiomyocytes and in intact hearts of rats subjected to ischemia and reperfusion, A3AR agonists reduced the infarct size. This effect is related to the upregulation of pro-survival signaling pathways, such as mitogen-activated protein kinase 1/2- Erk 1/2 and PI3K/Aκt, which are known to decrease the activity of caspase-3 used as a biomarker of cellular apoptosis (Hussain et al., 2014). The cardioprotection associated with the A3AR activation may also be due to the induction of human coronary smooth muscle cells proliferation via a mechanism involving phospholipase C and downstream transcriptional factors activation, like the early growth response element 2/3 (Hinze et al., 2012).
HFpEF is associated with systemic inflammation in its genesis that is evidenced by increased circulating levels of pro-inflammatory markers, such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) (Kalogeropoulos et al., 2010). These findings support the “systemic microvascular paradigm” regarding HFpEF in which comorbidities-induced systemic inflammation predisposes and perpetuates dysfunction of the microvasculature (Figure 3) (Paulus and Tschope, 2013). The most important comorbidities associated with this systemic inflammatory state are diabetes mellitus (DM), overweight/obesity, hypertension, chronic obstructive pulmonary disease, and chronic kidney disease (Paulus and Tschope, 2013). Thus, the paradigm of HFpEF has been shifted from the traditional “overload model” to the revolutionary “microvascular hypothesis”, where patients are more easily identified by elevated body mass index rather than by elevated blood arterial pressure (Rozenbaum et al., 2019).
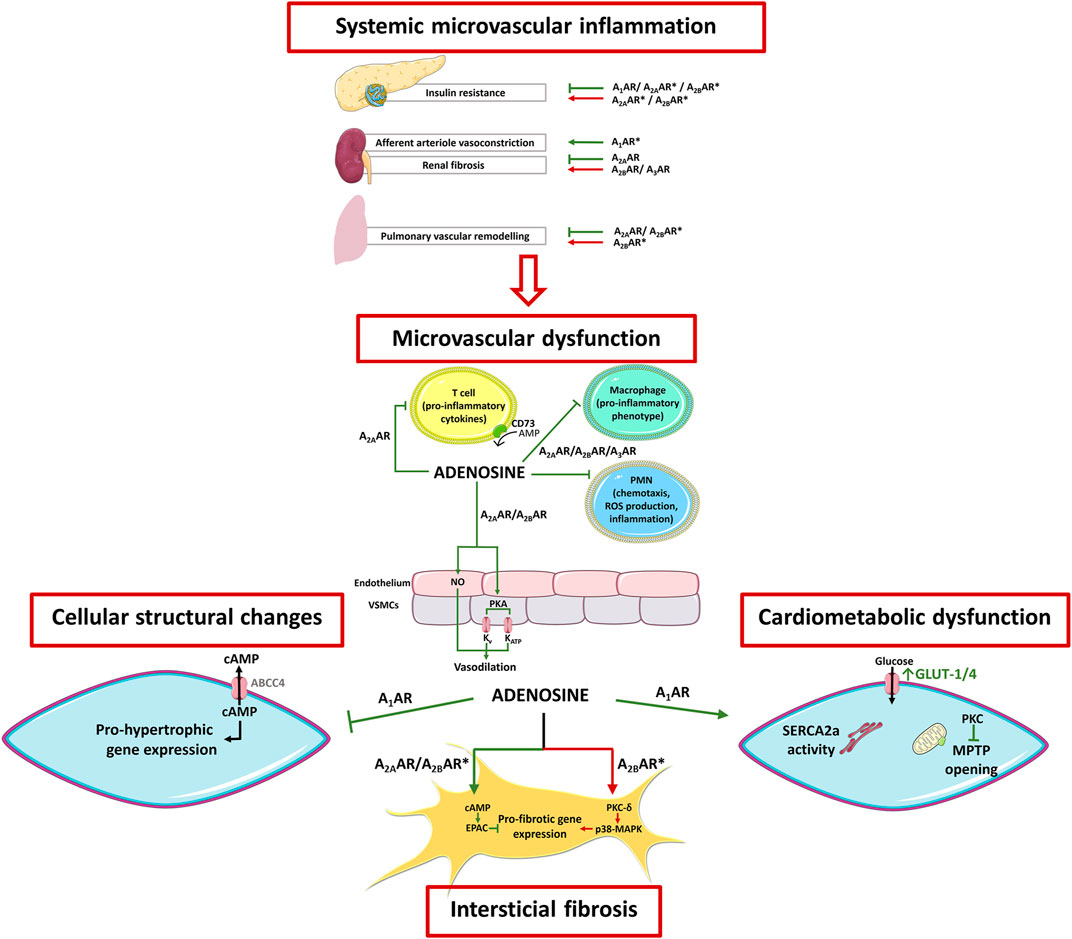
FIGURE 3. Schematic representation of adenosine receptors involvement in HFpEF pathophysiology. HFpEF is a complex clinical syndrome where comorbidities-induced systemic inflammation predispose and perpetuate microvascular dysfunction, as well cardiac structural and metabolic abnormalities. Overall, adenosine counteracts most of the pathophysiological features of this syndrome. These include 1) cardiac inflammation and microvascular dysfunction (via A2AR activation), 2) myocardial structural abnormalities (via A2AR and A1AR activation), and 3) energy metabolism and calcium handling (via A1AR activation). Conflicting evidence, however, exist regarding A2BAR-mediated cardioprotection, as this receptor has been implicated in both pro- and anti-fibrotic effects in the heart and lungs. Adenosine receptors also play important roles in cardiometabolic comorbidities related to HFpEF. Activation of A1AR improves the metabolic profile and induces renal afferent arteriole vasoconstriction, which can be protective when preservation of the glomerular architecture and function is needed. Stimulation of A2AAR and the A2BAR reduce lipolysis, but this beneficial effect may be partially counteracted by their action on skeletal muscles that contribute to insulin resistance. Activation of A2AAR counteracts renal damage due to its ability to reduce fibrosis, independently of A3AR and A2BAR are active or not. The arrows and bars indicate the effects induced or blocked by adenosine receptor activation, respectively. Green = beneficial effect; Red = deleterious effect; Asterisk (*) = contradictory/conflicting data. Adapted from Lam et al., 2018 and Headrick et al., 2013. Illustration used elements from Servier Medical Art (http://smart.servier.com) ABCC4, ATP-binding cassette sub-family C member 4; AR, Adenosine receptor; cAMP, cyclic adenosine monophosphate; EPAC, Exchange factor directly activated by cAMP; FFA, Free fatty acid; GLUT, Glucose transporter; MAPK, Mitogen activated protein kinase; MPTP, Mitochondrial permeability transition pores; NO, Nitric oxide; PKA, Protein kinase A; PKC, Protein kinase C; PMN, Polymorphonuclear leukocytes; SERCA2a, Sarco/endoplasmic reticulum Ca2+-ATPase 2a; a-SMA, Alpha-smooth muscle actin; VSMC, Vascular smooth muscle cell.
Supporting the concept of cardiac inflammation, left ventricular endomyocardial biopsies from patients with HFpEF show increases in the expression of biomarkers of inflammation and fibrosis, such as vascular cell adhesion molecule-1 (VCAM-1), CD3, CD11, and CD45-positive myocardial leucocytes, transforming growth factor-β (TGF-β), types I and III collagen species and extracellular matrix deposition (Westermann et al., 2011). Unbalanced cardiac inflammation causes several of the pathological features seen in HFpEF, such as endothelial dysfunction, fibrosis, concentric hypertrophy, and cardiometabolic functional abnormalities (Lam et al., 2018). Cardiac remodeling lead to left ventricular diastolic dysfunction (Westermann et al., 2011), which ultimately may cause unspecified clinical manifestations related to abnormal haemodynamics (e.g., dyspnoea and fatigue) that are characteristic of HFpEF (Lam et al., 2018).
The pleiotropic actions of adenosine in several organic systems put this retaliatory metabolite in good position to participate in most (if not all) the pathophysiological mechanisms associated with HFpEF. These include the control of systemic microvascular inflammation and related multi-organ comorbidities, in parallel to the nucleoside effects on cardiovascular function and myocardial remodelling. Adenosine regulates the activity of the immune system via self-contained signalling pathways aimed at promoting tissues integrity by resolving inflammatory insults (Figure 3), although a pro-inflammatory activity has also been described (reviewed in (Antonioli et al., 2014; Antonioli et al., 2019)).
Activation of the A2AAR is paramount to the anti-inflammatory effect of adenosine; the coupling of the A2AAR to the canonical AC/cAMP/PKA pathway downstream decreases nuclear factor-kappa B signalling and the release of pro-inflammatory cytokines (Sands et al., 2004). Adenosine signalling in neutrophils either stimulates the expression of adhesion molecules via A1AR or decreases adhesion of these cells to the vascular endothelium, as well as the production of superoxide radicals by A2AAR, A2BAR and A3AR activation. Anti-inflammatory (M2) macrophages overexpress A2AAR, A2BAR and A3AR, which activation attenuates the production of pro-inflammatory mediators (TNF-α, IL-6, IL-12, NO, and macrophage inflammatory protein-1α). Activation of A2AAR and A2BAR also favors 1) the release of the anti-inflammatory cytokine IL-10 by monocytes and macrophages (Antonioli et al., 2014; Antonioli et al., 2019), and 2) the switch from M1 (pro-inflammatory) to M2 (anti-inflammatory) macrophage phenotypes (Grinberg et al., 2009). Adenosine modulates lymphocyte functions on adaptive immunity, mainly through A2AAR activation and related suppressive effects, as it inhibits both IL-4 and IFN-γ production by naïve CD4+ T cells and Th1 and Th2 cells (Antonioli et al., 2014; Antonioli et al., 2019). Taken together, increased extracellular adenosine levels caused by inflammatory processes (Grenz et al., 2011; Idzko et al., 2014) emerges as a putative therapeutic target against inflammatory diseases with encouraging results in preclinical settings (Antonioli et al., 2014). The analogy to clinical data concerning the adenosine actions in rheumatoid arthritis (Silverman et al., 2008) and cancer immunotherapy (Antonioli et al., 2017), also supports this idea. In this context, adenosine mediates the anti-inflammatory effects of methotrexate, a gold standard treatment for patients with rheumatic diseases. By inhibiting certain enzymatic pathways, methotrexate induces adenosine release allowing the nucleoside to exert its anti-inflammatory effects via A2AAR and A3AR activation (Chan and Cronstein, 2010).
Mice submitted to transverse aortic constriction (TAC), a model of HFpEF, show significant cardiac inflammation that is initiated by transient myeloid cells infiltration followed by a chronic increase of myocardial T cells (Table 1) (Quast et al., 2017). Noteworthy, chemotaxis and activation of myocardial T cells are under the immunosuppressive control of adenosine formed by ecto-5′-nucleotidase/CD73 (Eltzschig et al., 2004) and A2AAR activation (Hamad et al., 2012). Indeed, the production of inflammatory cytokines (IL-3, IL-6, IL-13, IL-17, macrophage inflammatory proteins 1α and 1β) associated with cardiac fibrosis and decreased contractility is exaggerated by TAC in mice lacking CD73 on T cells, as well as in global CD73 mutants (Quast et al., 2017). These animals exhibit compensatory increases in the enzymatic machinery participating in adenosine formation (e.g. CD39, pyrophosphatases ENPP1 and ENPP3, and CD38) and overexpress A2AAR in activated T cells infiltrating the injured heart (Quast et al., 2017). Additionally, overexpression of A2AAR attenuates cardiac inflammation and remodelling, thereby attenuating cardiac dysfunction (Table 1) (Hamad et al., 2012). These data provide compelling evidence that deficient adenosine formation by T cells lacking CD73 worsens the HFpEF. This raised the hypothesis that increasing extracellular adenosine formation from the extracellular catabolism of released adenine nucleotides and/or direct activation of the A2AAR subtype may overcome HFpEF by reducing myocardial damage caused by pro-inflammatory cytokines and maladaptive cardiac remodelling (Hamad et al., 2012; Quast et al., 2017). Thus, maintenance of ecto-5′-nucleotidase/CD73 activity may be crucial to define the pattern of extracellular ATP-derived adenosine formation favoring A2AAR activation to prevent the pathological changes observed in HFpEF, as also predicted in other cell systems (Oliveira et al., 2015).

TABLE 1. Results of adenosine receptors agonism/antagonism in heart failure with preserved ejection fraction and pulmonary hypertension. Different agonists and antagonists were used to assess adenosine effects on cardiovascular diseases. The grey area corresponds to studies carried out in human patients.
The current trend is that coronary microvascular dysfunction may be a cornerstone of the complex molecular pathways driving the clinical manifestations of HFpEF (D’Amario et al., 2019). Coronary microvascular dysfunction is inevitably associated with a compromised vasodilatory response and consequently to the existence of myocardial focal ischemic lesions contributing to diastolic dysfunction of the left ventricle (LV) (Tschope et al., 2005). This feature is further compromised by systemic comorbidities and local inflammatory states ending up into structural and functional abnormalities of the heart and blood vessels (D’Amario et al., 2019). Diastolic dysfunction, the hallmark of HFpEF, is related to coronary microvascular dysfunction, independently from the presence of coronary artery disease (D’Amario et al., 2019).
Experimental models of diabetes, obesity and metabolic syndromes predict that systemic inflammation unbalances the equilibrium between endothelium-derived relaxing factors (e.g., NO) and endothelium-derived vasoconstrictors (e.g., endothelin-1), thus increasing stiffness and vasoconstriction of large vessels at rest (Paulus and Tschope, 2013). Endothelial activation favors the expression of adhesion molecules, trans-endothelial migration of circulating leucocytes, and release of pro-inflammatory cytokines and ROS, thus contributing to perpetuate local inflammation (Westermann et al., 2011). Moreover, vascular inflammation decreases NO bioavailability (Griendling et al., 2000; Westermann et al., 2011) resulting in downstream reduction of soluble guanylate cyclase activity, low levels of cyclic GMP and decreased activation of protein kinase G, which is normally associated with titin hypophosphorylation, increased rigidity and myocardial diastolic dysfunction (Paulus and Tschope, 2013).
The vascular endothelium is a key player linking metabolic disorders and HFpEF. Excessive production of ROS by endothelial Nox2 activation has been associated with other cardiovascular diseases, such as hypertension and atherosclerosis, which are common comorbidities of HFpEF (Landmesser et al., 2002). In vitro blockage of the A2AAR effectively inhibits ROS production by endothelial Nox2 and attenuates angiotensin II (AngII)-induced oxidative stress and endothelial dysfunction. This occurs through inhibition of MAPK activation and, consequently, by reducing p47phox phosphorylation and binding to Nox2 (Thakur et al., 2010). Taking together, these findings suggest a potential role for A2AAR antagonism to counteract AngII-induced Nox2 activation in endothelial cells (Thakur et al., 2010). However, contradictory results urge depending on the vascular territory considered; the A2AAR inactivation displays a protective role in reducing oxidative damage in neurodegenerative diseases (Pugliese et al., 2009) and in the heart (Ribe et al., 2008), whereas its inactivation increased tracheal ROS production from Nox2 (Nadeem et al., 2009).
As a matter of fact, the A2AAR activation is reported to play a major role in endothelium homeostasis, culminating in increased endothelial production of NO, and subsequent vasodilation in most vascular beds (Figure 3) (Li et al., 1998). Nevertheless, it has been shown that A2AAR inactivation preserved the endothelium-dependent vasodilation to acetylcholine, while reducing ROS production; this might be explained by the very low Nox2 expression in vascular smooth muscle cells, the predominant cellular component in the vessel wall (Thakur et al., 2010). Even though the A2AAR is mainly expressed in endothelial cells, its activation operates both endothelium-dependent and -independent relaxation of blood vessels. Controversy still exists on the receptor subtype (A2AAR and/or A2BAR) predominating on endothelial cells (Chiang et al., 1994; Iwamoto et al., 1994; Li et al., 1998). Endothelial dysfunction in small penile vessels underlying erectile dysfunction may be taken as an early surrogate of cardiovascular disease severity (Thompson et al., 2005). In this context, adenosine regulates smooth muscle tone in human corpora cavernosa through the activation of high-affinity A2AAR and low-affinity A2BAR located on smooth muscle fibers and endothelial cells, respectively, (Faria et al., 2006). Interestingly, corpora cavernosa from men with vasculogenic impotence is partially resistant to adenosine relaxation due to endothelial A2BAR dysfunction, while keeping almost unaltered relaxation of cavernosal vessels via A2AAR on the smooth muscle layer (Faria et al., 2006). Insufficient adenosine formation linked to deficits in CD73 enzymatic activity has been associated with age-dependent endothelial dysfunction and NO production deficits in mice (Mierzejewska et al., 2019), which may strengthen the production of the pro-inflammatory cytokine, IL-6, and endothelial adhesion molecules, like intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 (VCAM-1) (Mierzejewska et al., 2019).
Another hallmark of coronary microvascular dysfunction in patients with HFpEF is coronary microvascular rarefaction (Mohammed et al., 2015). Thus, adenosine-induced angiogenesis via A2AAR, A2BAR, and A3AR activation may be relevant in this scenario (Hinze et al., 2012; Ahmad et al., 2013; Du et al., 2015). In addition, it has been reported that activation of the A1AR subtype increases capillary density and oxygen diffusion distance in dogs submitted to transverse aortic constriction (Table 1) (Sabbah et al., 2013a). Interestingly, increased intracellular adenosine levels in human endothelial cells lacking ADK lead to hypomethylation in DNA promotor regions of pro-angiogenic genes (Xu et al., 2017b). Epigenetic upregulation of pro-angiogenic genes favor endothelial proliferation in vitro and ischemia induced-angiogenesis in vivo (Xu et al., 2017b).
Besides its antithrombotic properties, the P2Y12R antagonist, ticagrelor, has been increasingly used in cardiometabolic diseases like DM, most probably because of its pleiotropic/anti-inflammatory effects, which at least some of them are shared with adenosine (Wernly et al., 2021). Interestingly, ticagrelor restores defective ATP release and inhibits adenosine uptake by red blood cells directly impacting the actions of these two purines (or its metabolites) on immunocytes and endothelial cells (Wernly et al., 2021), which might be useful to counteract the development of atherosclerosis and related metabolic syndrome. In this regard, adenosine receptors (namely the A2AAR subtype) counteract the development of atherosclerosis by providing endothelial/vascular protection. This effect is attributed to the nucleoside anti-inflammatory properties, as well as to its ability to modulate the cholesterol transport in macrophages and the hepatic metabolism of lipids (reviewed in (Koupenova et al., 2012)). Consistent with these findings, the THEMIS-PCI (The Effect of Ticagrelor on Health Outcomes in Diabetes Mellitus Patients-Percutaneous Coronary Intervention) sub-study revealed a reduction of major adverse cardiac events in diabetic patients submitted to percutaneous coronary intervention and treated with ticagrelor plus aspirin compared to aspirin monotherapy (Bhatt et al., 2019). A more comprehensive understanding about the mechanisms underlying ticagrelor-induced endothelial dysfunction improvement is needed to support indication of this drug for cardiometabolic diseases (Wernly et al., 2021).
Structural abnormalities in HFpEF involve cardiomyocyte hypertrophy and interstitial fibrosis (Paulus and Tschope, 2013; Lam et al., 2018; Simmonds et al., 2020). Under pathological stressful conditions, unopposed actions of pro-fibrotic and pro-hypertrophic paracrine and neurohormonal signals further aggravate cardiac chambers stiffness and impaired relaxation. Diastolic dysfunction and inherent increased LV filling pressure foster secondary left atria remodelling and dysfunction, further aggravating HFpEF symptoms and long-term prognosis (Paulus and Tschope, 2013; Lam et al., 2018; Simmonds et al., 2020). Cardiac fibroblasts are the predominant interstitial cell type in the adult mammalian heart (Souders et al., 2009). Thus, it is not surprising that cardiac fibrosis and associated decrease in myocardial compliance represents a common pathological hallmark in the failing heart (Paulus and Tschope, 2013; Simmonds et al., 2020).
Among all adenosine receptors, A2AAR emerges by its cAMP-dependent anti-fibrotic action (Sassi et al., 2014). Separating the contribution of both A2AAR and A2BAR to fibrotic processes has been difficult due to the lack of selective drugs used in the past (Vecchio et al., 2017). As aforementioned, the cardiac anti-inflammatory/anti-fibrotic role of A2AAR has been translated into disease models of HFpEF (Hamad et al., 2012; Quast et al., 2017) and ischemia/reperfusion (Jordan et al., 1997; Yang et al., 2006; Rork et al., 2008). Activation of the A2AAR decreased the expression of fibrosis-related genes and cardiac fibrosis, while improving cardiac remodelling and function, in the deoxycorticosterone acetate-salt induced hypertensive mice model of cardiac remodelling (Table 1) (Zhou et al., 2020). One may, therefore, hypothesize that the anti-fibrotic effect of adenosine via A2AAR activation is operated either, directly, by inhibiting growth and/or differentiation of cardiac fibroblast, or, indirectly, through nucleoside-induced immunosuppressive effects, thereby preventing pro-fibrotic cells chemotaxis and the release of inflammatory mediators. In addition to these possibilities, adenosine deamination to inosine by third-party ADA cell providers (e.g., inflammatory cells) may also play a role in the anti-fibrotic effect of adenosine, as we recently demonstrated in human subcutaneous fibroblasts (Herman-de-Sousa et al., 2020). Thus, clarification of the mechanism and receptor involved in the anti-fibrotic role of adenosine in myocardial fibrosis is warranted given to the fact it may exert pro-fibrotic effects in other organs, such as the liver (Chan et al., 2006) and the skin (Herman-de-Sousa et al., 2020), while the opposite is verified with the A2BAR which inhibits fibrosis in the heart (see below) and promotes fibrosis of the lung (reviewed in (Bessa-Goncalves et al., 2018)).
The low-affinity A2BAR is the most abundant receptor in rat cardiac fibroblasts (Epperson et al., 2009). Therefore, it is not surprising that the anti-fibrotic effect of the A2BAR is relatively consensual, at least in vitro (Chen et al., 2004), while questions may arise about the endogenous amounts of the nucleoside required to activate this low-affinity receptor under both normal and pathological conditions. Nevertheless, it has been shown that intracellular cAMP accumulation prevents TGF-β-, AngII-, and endothelin-1-induced collagen synthesis and a-smooth muscle actin expression by activated cardiac myofibroblasts via EPAC and PI3K dependent pathways (Delaunay et al., 2019). Likewise, fibroblast proliferation, collagen synthesis, a-smooth muscle actin expression and myofibroblast differentiation were all attenuated by cAMP-coupled A2BAR activation, thus counteracting pro-fibrotic stimuli induced by endothelin-1 and AngII (Figure 3) (Delaunay et al., 2019) via a mechanism involving cAMP and EPAC downstream pathways (Phosri et al., 2017; Phosri et al., 2018). The cAMP/EPAC/PI3K/Akt signalling pathway was also involved in A2BAR-mediated suppression of fibroblasts growth and a-smooth muscle actin-expressing myofibroblast differentiation induced by endothelin-1 (Phosri et al., 2017). In a canine in vivo model of HF it was found that the EPAC1 content in fibroblasts of left atria exhibiting fibrotic remodelling was significantly downregulated (Surinkaew et al., 2019). Taking together, data suggest A2BAR coupling to the cAMP/EPAC pathway may provide potential targets to prevent/overcome cardiac fibrosis that may be required to rescue myocardial compliance.
Even though A2BAR agonists exhibit cardiac anti-fibrotic effects in vitro, these findings were not consistently translated into in vivo data, most probably due to the dual action of this receptor in cardiac inflammation (Vecchio et al., 2017). Available data suggests that A2BAR activation before ischemia followed by reperfusion may have a cardioprotective effect, most likely because of the inhibition of pro-inflammatory/pro-fibrotic cytokines release via PI3K/Akt pathway (Eckle et al., 2007b; Tian et al., 2015b; Ni et al., 2018). Moreover, a more complex interplay between A1AR and A2BAR subtypes is also possible, given to the fact that apparently the A1AR-operated cardiac protection relies on A2BAR overexpression by a PKC-dependent pathway during ischemia and preconditioning (Kuno et al., 2007). Nevertheless, in contrast to the A2AAR, there is no consensus about the putative beneficial roles of the A2BAR on the remodeling phase after myocardial ischemia (Sun and Huang, 2016), with studies demonstrating a protective role of either A2BAR blockage (Toldo et al., 2012; Zhang et al., 2014) or activation (Wakeno et al., 2006). Blockage of A2BAR after MI occurrence seems to attenuate PKC-δ/p38-MAPK-mediated secretion of pro-inflammatory and pro-fibrotic mediators, like TGF-β, TNF-α, and IL-6 (Feng et al., 2010; Toldo et al., 2012; Zhang et al., 2014). On the other hand, deletion of A2BAR did not change fibrosis in the remodelling phase after myocardial ischemia, probably due to opposing pro- and anti-inflammatory stimuli (Alter et al., 2019). In fact, the injured heart depends mostly on the low-affinity A2BAR probably due to functional inactivation of the A2AAR through dimerization (Hinz et al., 2018). It is worth noting that A2BAR may couple to both Gs and Gq proteins, as well as to other G proteins; its preferential coupling to a certain G protein dependents on multiple factors, including the cell type, receptor expression levels, and chemical nature of the agonist - “biased receptor” function (reviewed in (Bessa-Goncalves et al., 2018)). Thus, clarification of the precise mechanisms involved in pro-vs. anti-fibrotic effects of the A2BAR in the heart is still needed to correctly infer any benefit derived from the manipulation of the activity of this receptor in patients with HFpEF.
Overload pressure, oxidative stress and tissue injury, as well as the influence of neurohormonal and inflammatory mediators, like AngII, endothelin-1, catecholamines, growth factors and TNF-α, promote ventricular hypertrophy (reviewed in (Nakamura and Sadoshima, 2018)). While cardiac hypertrophy may be considered a physiologically adaptive response to external insults, over time it becomes pathological and facilitates progression to HF (Nakamura and Sadoshima, 2018; Simmonds et al., 2020).
The myocardial adenosine content highly increases in hypertrophied hearts (Funaya et al., 1997; Asakura et al., 2007). The nucleoside counteracts the activity of many neurohumoral factors involved in cardiac hypertrophy, namely endothelin-1 (Stowe et al., 1997), renin-angiotensin-aldosterone system (Taddei et al., 1992) and TNF-α (Wagner et al., 1998), and noradrenaline release from sympathetic nerves (Burgdorf et al., 2001). Considering these features, it was hypothesized that adenosine may exert an anti-hypertrophic action and, in this sense, might counteract cardiac dysfunction (Liao et al., 2003). Indeed, inhibition of adenosine uptake with dipyridamole increased myocardial adenosine levels, which significantly ameliorated LV filling and pulmonary congestion, while attenuating ß-adrenergic dysfunction in rats with pressure overload cardiac hypertrophy (Table 1) (Chung et al., 1998). Conversely, inhibition of adenosine formation from released adenine nucleotides in CD73 deficient mice promoted myocardial hypertrophy, LV dilation and LV dysfunction (Table 1) after aortic constriction, but not in healthy animals (Xu et al., 2008).
The A1AR seems to play a chief role in the putative anti-hypertrophic action of adenosine in the heart (Liao et al., 2003; Sabbah et al., 2013a; Puhl et al., 2016). The enzymatically stable A1AR agonists, 2-chloroadenosine (CADO) and N6-cyclopentyladenosine (CPA), significantly reduced the plasma concentrations of noradrenaline, renin, and brain natriuretic peptic in a mice model of transverse aortic constriction-induced HFpEF (Table 1) (Liao et al., 2003). When used in vivo, these drugs also effectively attenuated cardiomyocytes hypertrophy and fibrosis in interstitial and perivascular spaces through A1AR activation, resulting in improved cardiac function and reduced pulmonary congestion (Liao et al., 2003). Both CADO and CPA attenuated protein synthesis induced by activation of Gq-protein-coupled receptors for AngII, endothelin-1, and noradrenaline, as well as the PKA-mediated hypertrophic actions of isoproterenol in neonatal rat cardiomyocytes (Liao et al., 2003). Interestingly, in vivo stimulation of α1-adrenoceptor with phenylephrine upregulated A1ARs expression. Activation of overexpressed A1AR with CPA counteracted the hypertrophic phenotype caused by phenylephrine, but the same was not verified concerning cardiac hypertrophy/fibrosis caused by AngII and IGF1 (Table 1) (Puhl et al., 2016). These findings suggest that different signalling pathways operate maladaptive cardiac responses to distinct neurohumoral agents, namely AngII and phenylephrine (Puhl et al., 2016).
Adenosine counteraction of cAMP accumulation induced by ß-adrenoceptors stimulation (anti-adrenergic effect) has been proposed to explain the anti-hypertrophic effect of the nucleoside (Figure 3) (Meyer et al., 2001). Besides this, adenosine by positively modulating G protein signaling 4 may counteract Gq protein-induced hypertrophy, which may contribute to the anti-hypertrophic effect of the nucleoside against α1-adrenoceptor agonists (Liao et al., 2003). A1AR-receptor-mediated interference on intracellular Ca2+ handling may also play a role against myocytes hypertrophy probably by downregulating the calcineurin pathway (Meyer et al., 2001). In this context, sustained ß-adrenergic activation increases interstitial fibrosis and myocyte hypertrophy, two features of myocardial remodelling and HF (Prijic and Buchhorn, 2014). Upon ß-adrenergic stimulation, cardiomyocytes let intracellular cAMP flow to the extracellular space via the ATP-binding cassette sub-family C member 4 transporter where it may act as an important paracrine cardioprotective signalling molecule. Once in the extracellular space, cAMP may be enzymatically converted to AMP by ecto-phosphodiesterases, which will then be dephosphorylated to adenosine by ecto-5′nucleotidase/CD73. In this sense, one may hypothesize that extracellular adenosine yields may partially counteract myocardial hypertrophy due to ß-adrenoceptors overactivation through activation of A1AR negatively coupled to the AC while delivering anti-fibrotic signals to cardiac fibroblasts through A2AAR activation (Figure 3) (Sassi et al., 2014). Thus, the anti-adrenergic effect of adenosine may serve as an endogenous local ß-blocker by limiting intracellular cAMP formation with inherent beneficial cardiac effects. In line with this view, capadesonon, a partial A1AR agonist, decreased myocardial hypertrophy and fibrosis, while increasing capillary density and oxygen supply, which significantly improved LV function in dogs with advanced HF (Table 1) (Sabbah et al., 2013a). All together these pieces of evidence suggest that A1AR may be a potential target to prevent transition from compensated myocardial hypertrophy to irreversible HF.
In addition to the A1AR-mediated anti-hypertrophic effect of adenosine, A2AAR overexpression or agonist activation attenuated cardiomyocyte hypertrophy (Hamad et al., 2012; Zhou et al., 2020) and decreased hypertrophy-associated genes encoding ß-myosin heavy chain and atrial natriuretic peptide along with their transcription activator protein GATA-4 in mice with pressure-induced HF secondary to transverse aortic constriction (Hamad et al., 2012). These cardioprotective A2AAR-mediated effects are associated with attenuation of the inflammatory response via MAPK- and PKC-operated pathways independently of PKA activation (Hamad et al., 2012).
Considering the cardioprotective role of adenosine against chronic cardiac pressure overload, unexpected results were obtained using A3AR knockout (KO) mice. These animals exhibited partial resistance to transverse aortic constriction-induced LV hypertrophy, fibrosis, and oxidative stress (Table 1) (Lu et al., 2008). Hemodynamically, A3AR KO mice exhibited higher ejection fractions and smaller LV end-systolic diameters; the plasma levels of the atrial natriuretic peptide in these animals were also lower compared to wild type controls confirming less cardiac hypertrophy. Authors interpreted these findings by suggesting that A3AR activation might potentiate the inflammatory response triggered by cardiac pressure overload, thus favoring LV hypertrophy and cardiac dysfunction (Lu et al., 2008). In this context, it was interesting to verify that selective blockage of A3AR with MRS1911 potentiated the anti-hypertrophic effect of CADO in cardiomyocytes treated with phenylephrine, strengthening the idea that activation of the A3AR might have a deleterious effect in cardiac remodelling following chronic pressure overload (Lu et al., 2008). The effects obtained with the A3AR antagonist were remarkably similar to those verified upon reducing the activation of MAPK and PI3K-Aκt signalling pathways (Lu et al., 2008), which are often activated in response to oxidative stress and inflammation (Nakamura and Sadoshima, 2018). Confirmation of the protective role of A3AR blockage on the inflammatory component attenuating pressure overload cardiac remodelling requires the use of more potent and selective drugs. It is also worth noting that the adenosine deamination metabolite, inosine, may exert opposite effects to adenosine in the heart via A3AR receptors, which activation is strengthened when tissues become infiltrated by ADA-bearing inflammatory cells (Herman-de-Sousa et al., 2020). This concept raises new lines of research using multi-target drugs designed to increase local adenosine levels while decreasing inosine formation (e.g., ADA inhibitors with blocking properties of adenosine cellular uptake, like the lymphocytotoxic drug 2′-deoxycoformycin or pentostatin) applied in combination with enzymatically stable adenosine receptor agonists and/or antagonists (Herman-de-Sousa et al., 2020).
There is evidence that adenosine may also exert cardioprotective actions in the heart via non-receptor-mediated mechanisms. In support of a role for intracellular adenosine metabolism in regulating hypertrophy, it has been demonstrated that ADK inhibitors, such as iodotubercidin and ABT-702, completely reversed the anti-hypertrophic actions of adenosine and its enzymatically stable analogue, CADO, in phenylephrine-induced hypertrophic neonatal rat cardiomyocytes (Fassett et al., 2011); data identified ADK as an important mediator of adenosine attenuation of cardiomyocyte hypertrophy acting, at least in part, through inhibition of Raf signalling to mTOR/p70S6k. The role of ADK in the anti-hypertrophic effect of adenosine has also been demonstrated in vivo in a KO mice model of cardiac pressure overload induced by transverse aortic constriction (Table 1) (Fassett et al., 2019). ADK enzymatic activity dampened cardiac growth signaling (by mTOR complex 1 and Erk), as well as the excessive microtubule stabilization/detyrosination (Fassett et al., 2019). Overall, these data points towards a novel ADK-dependent adenosine receptor-independent mechanism to protect against adverse hypertrophy remodelling and excessive cardiomyocyte microtubule stabilization.
Mechanisms underlying impaired myocardial energetics in HF can be divided into three subgroups: 1) abnormal mitochondrial structure and function; 2) change in substrate utilization; and 3) intracellular Ca2+ overload (Birkenfeld et al., 2019).
The failing heart is characterized by abnormal mitochondrial structure and function, including hyperplasia and reduced organelle size, inadequate organelle respiration, reduced mitochondrial membrane potential, and opening membrane permeability pores. Therefore, ATP synthesis is reduced because of impaired electron transport chain and excessive ROS generation by mitochondria (Birkenfeld et al., 2019). Association between A1AR activity and mitochondrial dysfunction has been observed in HF (Figure 3) (Sabbah et al., 2013a). Treatment with the partial A1AR agonist, capadesonon, caused a near normalization of mitochondrial dysfunction in animal models of HF (Table 1) (Sabbah et al., 2013a). Capadesonon decreased the opening rate of mitochondrial permeability transition pores resulting in reduced apoptosis and normalization of citrate synthase expression, used as a marker of unaltered mitochondrial function (Sabbah et al., 2013a). Likewise, the A1AR agonist, 2-choloro-N6-cyclopentyladenosine, reduced the permeability of mitochondrial transition pores in rat cardiomyocytes submitted to hypoxia through a mechanism involving PKC activation, stabilization of mitochondrial membrane potential and inhibition of ROS production, culminating with the opening of KATP mitochondrial channels (Xiang et al., 2010b). The low-affinity A2BAR has also been associated with the reduction of the production of mitochondrial ROS via a mechanism involving PI3K, Erk, and NOS in adult rabbit cardiomyocytes (Yang et al., 2011).
Due to abnormal mitochondrial structure and function with consequent impairment of the electron transport chain, there is a switch in the cellular energetics in the failing heart from a preferential metabolism of fatty acids to glucose. This shift in the cellular energetics to anaerobiosis in order to spare oxygen consumption is potentiated by A1AR activation. In fact, this receptor has been shown to restore the plasma membrane expression of glucose transporters 1 and four to normal levels (Table 1) (Sabbah et al., 2013a) and to decrease the levels of free fatty acids (Staehr et al., 2013; Cui et al., 2021).
Intracellular Ca2+ overload in HF causes abnormal function of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA)2a, which normally pumps Ca2+ from the cytosol into the sarcoplasmic reticulum during diastole; as a consequence, active muscle relaxation is impaired and ventricular cardiomyocyte stiffness becomes an issue (Lam et al., 2018). Activation of A1AR with capadesonon ameliorated SERCA2a function, which by restoring the buffering Ca2+ capacity of the sarcoplasmatic reticulum indirectly facilitates the diastolic relaxation of cardiomyocytes, while also restraining intracellular Ca2+ overload before the next contraction (Table 1) (Sabbah et al., 2013a). On the other hand, although the activation of the A2AAR increases intracellular Ca2+ transients during systolic activity, they rapidly subside during diastole in a mouse model of cardiac pressure overload due to transverse aortic constriction (Hamad et al., 2012), through a PKA dependent mechanism (Table 1) (Hamad et al., 2012).
Diabetes mellitus (DM) is frequently associated with the occurrence and severity of HF; roughly 30–40% of patients with HFpEF are also affected by this endocrinopathy (Campbell et al., 2012). According to the clinical trial RELAX (Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure), diabetic HFpEF patients were younger, mostly males, and affected by other comorbidities, namely obesity, hypertension, renal dysfunction, pulmonary disease, and vascular disease (Lindman et al., 2014).
The role of adenosine in insulin resistance and obesity has been recently reviewed by our group (de Oliveira et al., 2020). Adenosine, via A1AR activation, suppresses lipolysis and plasma free fatty acids; increases lipogenesis and adipogenesis; it also regulates insulin sensitivity and glucose tolerance, indirectly interfering with myocardial substrate handling (Dhalla et al., 2007; Johansson et al., 2008). Moreover, it has been recently shown that adenosine, via ADA acting on RNA 1, may control appetite signaling and modulation along with limiting obesity and insulin resistance (Table 2) (Cui et al., 2021). ADA acting on RNA 1-deficient heterozygous mice fed with a high-fat diet (HFD) for 12 weeks exhibited a lean phenotype compared to wild-type controls which became obese within the same time period; the lean phenotype was associated with less severe dyslipidaemia and insulin resistance parameters, along with decreased food intake, decreased gastric ghrelin expression, and attenuated reduction of serum peptide YY levels (Cui et al., 2021). Altogether these findings suggest that ADA acting on RNA 1 may contribute to diet-induced obesity by modulating genes related to appetite control, such as ghrelin (“hunger hormone”) and peptide YY (“satiety hormone”) (Cui et al., 2021).

TABLE 2. Results of adenosine receptors agonism/antagonism in diabetes mellitus/obesity and chronic kidney disease. Different agonists/antagonists were used to assess its effects on HFpEF comorbidities. The grey area corresponds to studies carried out in human patients.
In mice with streptozotocin-induced diabetes, activation of the A1AR with CPA decreases plasma glucose, cholesterol, and triglyceride levels, and improves glucose utilization in peripheral tissues by increasing its uptake and glycogen synthesis by target cells (Table 2) (Cheng et al., 2000). In this context, A1AR agonists have been proposed for the treatment of type 2 DM and obesity (see Pros and Cons of Adenosine A1Receptor Manipulation in HFpEF).
Activation of the low-affinity A2BAR has been given special attention as a putative therapeutic target for obesity due to its ability to inhibit adipogenesis, increase lipolysis, improve insulin signaling and decrease white adipose tissue mass (Gharibi et al., 2012; Peleli et al., 2015). Activation of A2AAR increases lipolysis; this receptor is by far more expressed in the brown adipose tissue compared to the white adipose tissue, which might explain why it favors thermogenesis (de Oliveira et al., 2020). These findings, together with the anti-inflammatory potential of A2AAR activation, put this receptor in good position to counteract insulin resistance (de Oliveira et al., 2020). Previous results using the deoxycorticosterone acetate-salt induced hypertensive mice model showed that A2AAR activation promotes the release of fibroblast growth factor 21 by brown adipocytes, which presence seems to be crucial to combat hypertensive cardiac remodelling (Table 1) (Zhou et al., 2020). Confounding data, however, emerged from studies where the chronic consumption of caffeine reduced the risk of insulin resistance and type 2 DM (van Dam et al., 2006). It is worth noting that caffeine is the most widely psychoactive substance consumed worldwide and it acts mainly through the antagonism of adenosine receptors under moderate consumption of caffeine-containing beverages. A recent study carried out in Wistar rats of both gender fed either with a normalized chow or with a high-sucrose diet showed for the first time that chronic adenosine receptor blockage with caffeine favors insulin resistance in control animals, while restoring insulin sensitivity in animals treated with the high-sucrose diet (Table 1) (Sacramento et al., 2020). These authors also made clear that whole-body insulin sensitivity is under the control of both A2AAR and/or A2BAR given to the fact that selective blockage of each of these receptors rescued insulin sensitivity in skeletal muscles, both in males and females. Concerning the adipose tissue, chronic A1AR antagonism decreased fat accumulation, whereas the opposite was observed upon blockage of A2AAR and/or A2BAR; in this respect, gender differences were found with A1AR blockage being more effective in females, while antagonism of the A2AR was more powerful in males. Overall, these findings indicate that the effect of adenosine against insulin resistance via A2AAR and/or A2BAR is more prominent in skeletal muscles rather than in the adipose tissue where the A1AR seems to predominate (Sacramento et al., 2020).
Accumulating evidence suggest a role for adenosine in endothelial homeostasis maintenance. Due to its high affinity for the nucleoside, ADK is a key intracellular enzyme that is responsible for maintaining low intracellular adenosine levels while consistently driving the nucleoside to the purine nucleotides salvage pathway (Boison, 2013). Deletion of ADK, leading to increased intracellular adenosine accumulation and subsequent outflow of the nucleoside from cells, was found to increase ß-cell replication, thereby protecting against HFD-induced glucose intolerance (Navarro et al., 2017). Endothelial ADK was significantly upregulated in mice submitted to HFD; conversely, endothelial-specific ADK deficiency protected mice from HFD-induced insulin resistance and metabolic syndrome. This may be due to increased adenosine translocation to the extracellular milieu where it can activate plasma-membrane bound receptors, including endothelial A2BAR, which downstream signals contribute to phosphorylate NOS 3 fostering NO production and NO-related protective anti-inflammatory effects due to vasodilation and angiogenesis (Table 2) (Xu et al., 2019). The anti-inflammatory potential of ADK inhibition is also associated with reduced leucocyte adhesion to the endothelium, further contributing to regulation of glucose homeostasis and insulin sensitivity (Xu et al., 2017a).
Due to population aging and incorrect lifestyle habits the prevalence of HFpEF and chronic kidney disease is dramatically increasing (Fang, 2016; Lofman et al., 2017). HFpEF has been considered a “vicious cycle” disorder of kidney and cardiovascular function disequilibrium, as either HFpEF or chronic kidney disease may accelerate the pathological progression of each other. Both disease conditions have not only overlapping comorbidities, including association with hypertension, diabetes and obesity, but also share common underlying features, like hypervolemia, systemic inflammation, and endothelial dysfunction (Fang, 2016).
Adenosine has been reported to reduce urinary protein excretion (Patinha et al., 2014), an earlier marker of underlying kidney injury causing renal dysfunction (Katz et al., 2014), by acting at the kidney level (reviewed in (Pandey et al., 2021)). Specifically, adenosine controls renin release, glomerular filtration rate, tubuloglomerular feedback mechanism and vascular tone through all four P1 receptor subtypes.
The A1AR plays an important role in renal physiology, as it controls afferent arteriole vasoconstriction, reabsorption of sodium in the proximal tubule and the tubuloglomerular feedback. Through inhibition of renin release from justaglomerular cells along with renal arteriole vasodilation, adenosine (via A1AR) reduces renal blood flow lowering the glomerular filtration pressure (Pandey et al., 2021), which is protective against kidney damage caused by metabolic diseases, such as diabetes and hypertension, often complicating HFpEF (Lofman et al., 2017; Pandey et al., 2021). The salutary effects of sodium-glucose cotransporter-2 (SGLT2) inhibitors, like empagliflozin, on diabetes-related hyperglycaemia, obesity and hypertension may be at least in part mediated by the reduction of glomerular hyperfiltration via activation of A1AR receptors and TGF-mediated afferent arteriolar vasoconstriction, independently of their ability to lower the plasma glucose levels (Kidokoro et al., 2019). The use of SGLT2 inhibitors provides a new perspective to the cardio- and reno-protective role of A1AR activation, as it decreases the intraglomerular pressure and prevents hyperfiltration, which may counteract chronic kidney disease progression and impaired renal function, indirectly ameliorating the cardiovascular outcome (Kidokoro et al., 2019). Moreover, renal activation of A1AR reduces the metabolic demand, tubular cell apoptosis and inflammatory reactions in mice models of ischemia/reperfusion (Table 2) (Lee et al., 2004; Xiong et al., 2018), thus strengthening the reno-protective effects of A1AR agonists.
The ability of A1AR antagonists to induce natriuresis without compromising the glomerular filtration rate suggests that this approach may be beneficial to control volume overload disorders, such as HF (Gottlieb et al., 2002), yet controversy still exists in the literature regarding this issue (see details in Pros and Cons of Adenosine A1Receptor Manipulation in HFpEF). Interestingly, long-term dual blockage of A1AR/A2BAR with the orally-available drug tonapofylline (BG9928) reduced glomerulosclerosis in ZSF1 rats used as an animal model of HFpEF related to metabolic syndrome (Tofovic et al., 2016). Diabetic nephropathy is associated with reduced nitrergic vascular innervation, NO bioavailability and VEGF-NO uncoupling leading to 1) excessive endothelial cell proliferation, 2) stimulation of macrophage chemotaxis and 3) vascular smooth muscle cells growth, which ultimately causes glomerular hypertrophy and proteinuria (Nakagawa, 2007). Taking these findings together, it may well be that the renoprotective effect of BG9928 in preventing glomerulosclerosis may be undertaken by the A2BAR antagonism (Tofovic et al., 2016). Indeed, isolated renal fibroblasts treated with the A2BAR agonist, BAY-606583, increased the release of pro-inflammatory and pro-fibrotic mediators (Wilkinson et al., 2016). On the other hand, blockage of the A2BAR with MRS1754 significantly decreased albuminuria and improved the renal function in streptozotocin-induced diabetic mice; the antagonism of overexpressed A2BAR normalized VEGF-A upregulation in these animals while enhancing NO production and decreasing the occurrence of glomerulosclerosis lesions (Table 2) (Patel and Thaker, 2014).
In a recent study performed using a rat model of HF secondary to renal injury, the mineralocorticoid receptor antagonist, spironolactone, significantly up-regulated A2AAR expression, inhibited endothelial-to-mesenchymal transition and attenuated cardiorenal fibrosis, both in vivo and in vitro (Table 2) (Chen et al., 2019). These outcomes suggest the A2AAR as a potential therapeutic target for the cardiorenal syndrome (Chen et al., 2019). In addition to the anti-fibrotic role of the A2AAR, its ability to dilate the efferent arteriole may be renoprotective by reducing the glomerular pressure (Pandey et al., 2021). Long-term administration of the selective A2AAR agonist, CGS21680, reduces diabetes-induced glomerular hyperfiltration in streptozotocin-treated diabetic mice through a mechanism depending on NO production (Table 2) (Persson et al., 2015b). The anti-inflammatory role of A2AAR activation was reflected by a decrease in the number of infiltrating immune cells and by the reduction of urinary pro-inflammatory cytokines along with reduced albuminuria (Awad et al., 2006). These results were corroborated by a different research group using the same animal model, who proved that selective A2AAR activation with CGS21680 prevents proteinuria and glomerular damage by activating T regulatory cells, which results in reduced macrophage infiltration and TNF-α secretion (Table 2) (Persson et al., 2015a). Likewise, inhibition of ADK leading to adenosine overflow to the extracellular milieu had a protective role against renal failure in streptozotocin-induced diabetic mice by decreasing renal inflammation and oxidative stress lesions, as well as by restoring glomerular filtration and permeability (Table 2) (Pye et al., 2014). These effects were consistent with the reduction in renal macrophage infiltration, nuclear factor-κB phosphorylation and monocyte chemoattractant protein-1 excretion, along with increased endothelial NOS expression and MAPK phosphorylation (Pye et al., 2014).
In a model of hypertensive-diabetic nephropathy using spontaneously hypertensive rats treated with streptozotocin, the enzymatically-stable adenosine analogue, CADO, improved glucose metabolism (decreased hyperglycaemia and glycosuria), renal function (decreased proteinuria), and renal fibrosis (decreased glomerular collagen deposition), along with decreased renal oxidative stress (Table 2) (Patinha et al., 2020); authors implicated renal A2AAR activation in these findings, given that overexpression of this receptor, but of A1AR and A2BAR, was observed in superficial glomeruli and proximal and distal tubules of these animals’ kidneys (Patinha et al., 2020). The hypertension-inducing adenosine receptor antagonist, 1,3-dipropyl-8-sulfophenylxanthine, aggravated renal fibrosis, but did not alter the global metabolic status and renal function. This probably results from the selective preservation of tonic A3AR activation, given that downregulation of this receptor is considered protective against renal fibrosis (Patinha et al., 2020). Taken together, these results emphasize the protective role of adenosine in hypertensive-diabetic nephropathy through dynamic expression of its receptors, which may be fine-tuned by A2AAR upregulation and A3AR downregulation, thus prompting for a novel therapeutic target for this disease conditions (Patinha et al., 2020).
A3AR overexpression may be another target to abrogate progression of diabetic nephropathy in db/db leptin receptor-deficient mice, which is the most widely used animal model of type 2 DM normally presenting increased body weight, impaired glucose tolerance, and LV hypertrophy associated with HFpEF, but no significant changes in blood pressure. Chronic administration of LJ-2698 (a novel orally-active and highly selective A3AR antagonist) to these animals was more efficient in preventing diabetic nephropathy progression than the widely used AT1 receptor antagonist, losartan (Table 2) (Dorotea et al., 2018). Moreover, inhibition of renal lipid accumulation with increases in PGC1α, a key regulator of mitochondrial biogenesis, were obtained upon treating these animals with either LJ-2698 or losartan, further supporting the renoprotective effects of selective A3AR antagonism (Dorotea et al., 2018). Whether the combination of A3AR and AT1 receptor antagonists exerts synergic renoprotective effects requires investigation in future studies (Dorotea et al., 2018).
Left heart disease-induced pulmonary hypertension (PH), the WHO group 2 of PH, tend to be more symptomatic and to have worse prognosis when compared to patients with left heart disease alone (Levine et al., 2019). Specifically, PH associated with diastolic dysfunction, also referred as PH secondary to HFpEF (PH-HFpEF) is the most common endophenotype attributable to left heart disease (Levine et al., 2019). Diastolic dysfunction coupled with left atria changes in HFpEF result in a passive backward transmission of increased LV filling pressure with consequent increases in pulmonary congestion (Levine et al., 2019). As aforementioned, studies in animal models of HFpEF induced by cardiac pressure overload revealed a putative beneficial role of adenosine in improving cardiac structure and function alterations and, thereby, attenuating cardiac dysfunction (Liao et al., 2003; Sabbah et al., 2013b) while decreasing pulmonary congestion (Liao et al., 2003). Major benefits of adenosine in this endeavor result from A1AR and A2AAR activation (Hamad et al., 2012; Quast et al., 2017; Zhou et al., 2020) associated or not with A3AR antagonism (Lu et al., 2008), which may also ameliorate symptoms and prognosis of HFpEF caused by cardiac pressure overload.
Adenosine levels in the pulmonary circulation are pathologically low in patients with WHO group 1 pulmonary arterial hypertension (PAH) as a consequence of local endothelial dysfunction and increased inactivation by ADA, possibly indicating that adenosine A1AR and A2AAR activation deficits unbalanced by inosine-mediated A3AR tone (Herman-de-Sousa et al., 2020) may contribute to maladaptive lung disease (Saadjian et al., 1999). In contrast to other vascular beds where adenosine has potent vasodilatory actions, in the pulmonary circulation, the nucleoside exerts dual opposing effects depending on the vascular tone (Cheng et al., 1996). Under low pressure conditions, adenosine fosters vasoconstriction through A1AR activation, whereas upon increasing the vascular tone, as observed in PH, the A2AR subtypes acquire more relevance to promote vasodilatation.
Among all adenosine receptor subtypes, the A2AAR gathered the most attention concerning PAH treatment. This assumption is based on the fact that A2AAR KO mice exhibit structural and functional abnormalities similar to PAH (Xu et al., 2011; Shang et al., 2015) and overexpress RhoA and ROCK proteins, which are known to be involved in pulmonary vascular remodelling and PAH pathophysiology (Table 1) (Shang et al., 2015). In mice with PAH induced by monocrotaline, activation of A2AAR reduced pulmonary endothelial dysfunction and vascular remodelling, while increasing pulmonary vasodilation and the LV stroke volume (Table 1) (Alencar et al., 2013; Alencar et al., 2014). The way adenosine builds its positive influence on RV hypertrophy and dysfunction may be indirectly related to the counteracting effects of the A2AAR on increased pulmonary resistance (Table 1) (Alencar et al., 2014) via NO-mediated synergism (Alencar et al., 2018; see Pros and Cons of Adenosine A1Receptor Manipulation in HFpEF). Additionally, the anti-inflammatory actions of A2AAR agonists may improve cardiopulmonary homeostasis, as discussed above.
Immunolocalization studies demonstrated infiltration of the RV myocardium by A2BAR -positive fibroblasts and macrophages in rats with monocrotaline-induced PAH (Braganca et al., 2015). This result is compatible with adenosine being able to downregulate pro-inflammatory and pro-fibrotic stimuli, thus contributing to mechanical adaptation of RV in response to cardiac pressure overload (Braganca et al., 2015). Chronic obstructive pulmonary disease, a highly prevalent pulmonary comorbidity in patients with HFpEF, is also linked to PH development. Interestingly, chronic obstructive pulmonary disease patients with PH exhibit increased pulmonary vascular remodelling and mastocyte-induced airway hyperactivity associated with A2BAR overexpression (Table 1) (Karmouty-Quintana et al., 2013). Notwithstanding this, the role of the low affinity A2BAR in cardiopulmonary pathophysiology lacks comprehensive studies before inferring any beneficial effects from targeting this receptor to improve RV failure secondary to PAH (reviewed in (Bessa-Goncalves et al., 2018)). Besides controversial studies claiming deleterious effects of A2BAR in cardiopulmonary pathophysiology, others (fewer) emphasize some beneficial effects from A2BAR activation (Bessa-Goncalves et al., 2018).
Considering that most studies available so far about the role of the A2BAR in cardiopulmonary function rely on animal models of PAH, care must be taken before extrapolating the therapeutic effects of this receptor in PH-HFpEF. While the vast majority of PH-HFpEF patients develop HF secondary to pulmonary venous congestion (isolated post-capillary PH) (Levine et al., 2019), studies have shown that many patients with HFpEF also present with coexisting pulmonary vascular disease due to a metabolic syndrome-induced low grade inflammatory status (Ranchoux et al., 2019). Together with the control of actual lifestyle trend leading to increased prevalence of cardiometabolic diseases, development of novel therapeutic strategies towards combined pre- and post-capillary PH is an unmet clinical need (Ranchoux et al., 2019). Through its cardiovascular protective effects and role in metabolic diseases, together with aforementioned effects in PAH, adenosine signalling stands as a putative novel target to manage PH-HFpEF, providing that data from preclinical studies translate correctly to clinical trials in humans.
Adenosine handling, metabolism and signalling are implicated in cardiac remodelling and progression to HF (Table 3). Indeed, increased cardiac and plasma adenosine levels are hallmarks of severity in patients with HF, which might result from decreased cellular energy charge, shift to anaerobic metabolism, plasma membrane damage and decreased ADA activity (Funaya et al., 1997; Asakura et al., 2007). Excessive adenosine endogenous tone may lead to downregulation of adenosine receptor genes, namely A2AAR, A2BAR, and A3AR, in the failing heart (Funaya et al., 1997; Asakura et al., 2007). Increased endogenous levels of adenosine may reflect an adaptive mechanism to counteract adenosine signalling impairment in failing hearts (Funaya et al., 1997; Asakura et al., 2007). This is confirmed because increases in the myocardial concentration of adenosine resulting from inhibition of the nucleoside cellular uptake are protective against cardiac remodeling and ß-adrenergic dysfunction, thereby counteracting progression to HF (Chung et al., 1998).

TABLE 3. Pros and cons of targeting adenosine receptors, enzymes and metabolism in molecular pathways underlying heart failure with preserved ejection fraction and cardioprotection.
As already outlined, all adenosine receptor subtypes may be involved, in one way or another, in the pathophysiology of HFpEF and related comorbidities (Figures 2, 3). Data indicate that A1AR provides cardioprotective effects due to reversal of cardiac hypertrophy/remodelling, improvement of mitochondrial function, enhancement of SERCA2a activity and Ca2+ handling, increases in capillary density, modifications in substrate utilization, and enhancements in skeletal muscle performance. In addition, A1AR activation has been associated to anti-ischemic properties by decreasing catecholamine release and ß-adrenergic overactivation (Albrecht-Kupper et al., 2012; Greene et al., 2016). Activation of A1AR also improves the metabolic profile (de Oliveira et al., 2020) and attenuates renal metabolic demand and glomerular filtration pressure (Pandey et al., 2021). It is, therefore, not surprising that the most expressed adenosine receptor in the heart, the A1AR, has been the most widely studied receptor in cardiovascular, renal and metabolic diseases, both at preclinical and clinical levels (Greene et al., 2016; Borah et al., 2019; Jacobson et al., 2019; Shah et al., 2019) (for details, see Pros and Cons of Adenosine A1Receptor Manipulation in HFpEF). Notwithstanding this, the anti-adrenergic cardioprotective effects of the A1AR may be partially counteracted by A2AAR activation (Chandrasekera et al., 2010), as detected in cardiac pressure overload (Meyer et al., 2001) and hypertensive animals (Tang et al., 1998).
Data from preclinical studies ascribed cardioprotective roles to the A2AAR subtype. Activation of A2AAR attenuates cardiac, renal and pulmonary damage due to its ability to counteract inflammation, in addition to its vasodilation and angiogenic properties; interestingly, this receptor also mediates increases in cardiac inotropism without interfering with Ca2+ homeostasis (Headrick et al., 2013). Positive cardiac repercussions against hypertensive remodelling resulting from A2AAR activation may be owe to amelioration of insulin resistance associated with its effects on brown adipocytes and inflammation (de Oliveira et al., 2020; Zhou et al., 2020). Controversy still exists regarding the impact of the A2AAR on the whole body insulin sensitivity, since antagonism of this receptor in skeletal muscles attenuated whole-body insulin resistance (Sacramento et al., 2020). Like that observed for the A1AR subtype, the anti-adrenergic effect of the A2AAR is also impaired in hypertensive animals (Tang et al., 1998). The chronic use of selective A2AAR agonists is also not free of cardiovascular side effects, like arterial hypotension and tachycardia. To avoid residual actions on other adenosine receptor subtypes, a new era of nucleoside-5′-monophosphates as prodrugs of selective A2AAR activators have been designed, which require ecto-5′-nucleotidase/CD73 activation in close proximity to receptor sites providing that the adenosine forming enzyme is available and functional (El-Tayeb et al., 2009; Flogel et al., 2012).
Conflicting evidence exists regarding A2BAR-mediated cardioprotection, as well as its pulmonary effects (Table 3) (Pye et al., 2014). While activation of A2BAR may counteract fibrosis (Phosri et al., 2017; Phosri et al., 2018; Delaunay et al., 2019) and ischemic preconditioning (Eckle et al., 2007a) in the heart, other studies suggest that this low affinity adenosine receptor may be deleterious in ischemic cardiac remodelling (Toldo et al., 2012; Zhang et al., 2014), as well as in renal fibrosis (Patel and Thaker, 2014; Tofovic et al., 2016). Like the A2AAR, the action of the A2BAR in DM and obesity is also controversial (Sacramento et al., 2020). The same occurs regarding the least expressed A3AR in the heart (Table 3) (Borea et al., 2018). The A3AR may be cardioprotective against reperfusion (Ge et al., 2010; Hussain et al., 2014; Tian et al., 2015a) by promoting angiogenesis (Hinze et al., 2012), but it may aggravate pathological structural changes in the heart (Lu et al., 2008) and kidney (Patinha et al., 2020).
Manipulation of endogenous adenosine inactivation through cellular uptake and/or ADA may be a valuable strategy to increase the extracellular concentration of the nucleoside and, thereby, its receptors activation. Inhibition of ENT by dipyridamole attenuates cardiac remodelling and LV dysfunction, prevents ß-adrenergic dysfunction and induces coronary vasodilation (Drury and Szent-Gyorgyi, 1929; Chung et al., 1998). Downregulation of ADA activity counteracts diet-induced obesity and insulin resistance through decreases in food intake (Cui et al., 2021).
Regulating intracellular adenosine concentration by manipulating ADK, a key intracellular enzyme that is responsible for maintaining low intracellular adenosine levels, thus favoring extracellular adenosine uptake by the cells, may also be a putative strategy to increase the extracellular levels of the nucleoside (Table 3). As a matter of fact, inhibition of ADK may be cardioprotective due to increases in the extracellular levels of the adenosine, which stimulates angiogenesis, promotes insulin sensitivity, and attenuates inflammation and oxidative stress (Pye et al., 2014; Xu et al., 2017b; Xu et al., 2019); contrariwise, when this enzyme is fully operative reduction of the extracellular levels of the nucleoside may be deleterious as it abrogates the anti-hypertrophic effect of adenosine (Fassett et al., 2011; Fassett et al., 2019).
As previously mentioned, the A1AR has been the most widely studied adenosine receptor in the cardiovascular system, both at preclinical and clinical levels. Indeed, activation of A1AR exerts cardioprotective properties that go beyond the preservation of cardiac structure and metabolism, encompassing regulatory metabolic benefits. In this context, several A1AR agonists have been tested in clinical trials for type 2 DM. The first clinical trials using A1AR full agonists, ARA and GR79236, were successful in improving insulin sensitivity, but failed their endpoints due to undesirable cardiovascular side effects (Jacobson et al., 2019). As a matter of fact, the clinical use of A1AR full agonists may present some limitations related to off-target side effects, either cardiac (bradycardia up to atrioventricular block, negative dromotropy and inotropy) and extra-cardiac (functional depression of the central nervous system and reduction of glomerular filtration rate due to vasoconstriction of afferent arteriole) (Albrecht-Kupper et al., 2012; Greene et al., 2016). Another potential limitation of chronic administration of A1AR full agonists is receptor desensitization (Albrecht-Kupper et al., 2012). To overcome these limitations partial A1AR agonists have been produced and tested in clinical trials. These compounds exhibit a more favorable haemodynamic profile with minimal side effects in terms of heart rate, atrioventricular conduction, blood pressure and renal function, along with no evidence of adenosine-mediated negative inotropism (Albrecht-Kupper et al., 2012; Greene et al., 2016), which is in agreement with our prediction that adenosine acting via the A1AR is a chronoselective atrial depressant (Braganca et al., 2016). One of such compounds, GS-9667 (previously known as CVT-3619, a partial agonist that selectively binds to A1AR), reduced plasma free fatty acids and was well-tolerated both in healthy non-obese and obese subjects, without showing any signs of receptor desensitization or rebound problems on suspension (Table 2) (Staehr et al., 2013). The antilipidemic effect of this compound is not surprising because A1AR are highly more abundant in adipocytes than in the atrioventricular node (Wu et al., 2001; Liang et al., 2002).
Controversy still exists regarding the putative cardiorenal protection associated with A1AR modulation. The ability of A1AR antagonists to induce natriuresis without compromising the glomerular filtration rate suggests that this approach may be beneficial to control volume overload disorders, such as HF (Gottlieb et al., 2002). The selective A1AR antagonist, rolofylline, increased diuresis in patients with acute and chronic HF, with enhancements of the renal plasma flow also observed in the latter (Table 2) (Dittrich et al., 2007; Givertz et al., 2007). However, in the large phase III clinical trial PROTECT (Placebo-Controlled Randomized Study of the Selective A1 Adenosine Receptor Antagonist Rolofylline for Patients Hospitalized with Acute Decompensated Heart Failure and Volume Overload to Assess Treatment Effect on Congestion and Renal Function), rolofylline reduced fluid retention, but did not prevent the worsening of renal dysfunction in acute HF patients (Voors et al., 2011). This failure may be attributed to disproportionate diuresis associated with escalating doses of rolofylline, which might have offset the ability of this drug to preserve renal filtration rate and kidney blood supply (Voors et al., 2011). In fact, despite the putative benefits in controlling hypertension and volume overload, inhibition of the A1AR activity may be detrimental for glomerular architecture and function, as it increases intraglomerular pressure by antagonizing preglomerular vasoconstriction operated by A1AR (Tofovic et al., 2016). Thus, considering its cardioprotective and metabolic roles, as well as its ability to reduce intraglomerular pressure, A1AR agonists may stand as promising medications for treatment of the chronic cardiorenal syndrome.
Partial A1AR agonists were also tested in clinical trials for HF in patients presenting reduced or preserved ejection fraction (Voors et al., 2018). Data from two small pilot studies demonstrate that the novel orally-available partial A1AR agonist, neladesonon, seems to be safe with no atrioventricular, neurological, and renal side effects in patients with HF with reduced ejection fraction (Voors et al., 2017), but no beneficial early changes were observed in the cardiac function (Table 1) (Voors et al., 2017). Data from a phase II double-blinded placebo-controlled clinical trial (PANACHE - The Partial AdeNosine A1 receptor agonist in patients with Chronic Heart failure and preserved Ejection fraction), designed to explore the effects of neladesonon on exercise capacity among 300 patients with HFpEF, has been recently provided (Table 1) (Shah et al., 2019). Neladesonon did not have a significant impact on exercise capacity. This was interpreted as consequence of a lower than normal prevalence of coronary artery disease in HFpEF patients, together with a higher number of patients using ß-blockers that may abrogate the beneficial antiadrenergic effects of A1AR agonists (Shah et al., 2019). Although the results were disappointing, a low incidence of major adverse effects was observed in patients under neladesonon treatment (Shah et al., 2019).
The aforementioned data suggest that before translating the putative preclinical benefits of manipulating the adenosinergic signals to the therapeutic armamentarium, one must keep in mind that adenosine production and/or inactivation, receptor subtype expression dynamics, and downstream intracellular messenger pathways might change in a moment-to-moment basis depending on the tissue, pathological situation and stage of disease condition. Another pitfall in our current knowledge is due to the relatively complex pathophysiology of HFpEF owing to a limited number of animal models that reflect entire features of the human pathology; this constrain, further challenges translation of preclinical data to clinical settings (Conceicao et al., 2016). Notwithstanding this, the beauty of targeting the adenosinergic system is that it can differentially control not only heart structure and function abnormalities, but also most of the co-morbidities concurring with HFpEF in terms of precipitating causes and/or aggravating factors.
Taken together, these preclinical and clinical evidence shed some lights on the adenosine role in molecular mechanisms and comorbidities related to HFpEF. Adenosine was found to counteract cardiac pathophysiological mechanisms implicated in this clinical syndrome, namely cardiac inflammation and microvascular dysfunction, especially via A2ARs, in addition to extracellular and cellular structural abnormalities, and energy metabolism and Ca2+ handling, being these effects more under A1AR and A2AR control. Consequently, adenosine also counteracts cardiac remodelling and related diastolic dysfunction, the pathological hallmark of HFpEF clinical syndrome. Considering the new paradigm of a systemic microvascular dysfunction as a perpetuating factor for myocardial dysfunction, adenosine receptors, enzymatic machinery and downstream intracellular messenger pathways surge as promising treatment targets, in part because they are also implicated in a wide range of pathophysiological processes related to HF comorbidities, such as diabetes and metabolic syndrome, along with kidney and pulmonary dysfunction.
AC, AF, PC, and BB designed the work. AC collected the data. AC, AF, PC, and BB wrote the article. AF, PC, and BB critically analysed all data in the article.
This work was supported by Fundação para a Ciência e a Tecnologia (grant number: UIDB/04308/2020). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the article. BB is in receipt of a PhD studentship from FCT (FEDER funding SFRH/BD/104114/2014).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Figures 1–3 use templates from Servier Medical Art.
Ahmad, A., Schaack, J. B., White, C. W., and Ahmad, S. (2013). Adenosine A2A Receptor-dependent Proliferation of Pulmonary Endothelial Cells Is Mediated through Calcium Mobilization, PI3-Kinase and ERK1/2 Pathways. Biochem. Biophys. Res. Commun. 434 (3), 566–571. doi:10.1016/j.bbrc.2013.03.115
Albrecht-Küpper, B. E., Leineweber, K., and Nell, P. G. (2012). Partial Adenosine A1 Receptor Agonists for Cardiovascular Therapies. Purinergic Signal. 8 (Suppl. 1), 91–99. doi:10.1007/s11302-011-9274-3
Alencar, A. K., Carvalho, F. I., Silva, A. M., Martinez, S. T., Calasans-Maia, J. A., Fraga, C. M., et al. (2018). Synergistic Interaction between a PDE5 Inhibitor (Sildenafil) and a New Adenosine A2A Receptor Agonist (LASSBio-1359) Improves Pulmonary Hypertension in Rats. PLoS One 13 (4), e0195047. doi:10.1371/journal.pone.0195047
Alencar, A. K. N., Pereira, S. L., da Silva, F. E., Mendes, L. V. P., Cunha, V. d. M. N., Lima, L. M., et al. (2014). N-acylhydrazone Derivative Ameliorates Monocrotaline-Induced Pulmonary Hypertension through the Modulation of Adenosine AA2R Activity. Int. J. Cardiol. 173 (2), 154–162. doi:10.1016/j.ijcard.2014.02.022
Alencar, A. K. N., Pereira, S. L., Montagnoli, T. L., Maia, R. C., Kümmerle, A. E., Landgraf, S. S., et al. (2013). Beneficial Effects of a Novel Agonist of the Adenosine A2Areceptor on Monocrotaline-Induced Pulmonary Hypertension in Rats. Br. J. Pharmacol. 169 (5), 953–962. doi:10.1111/bph.12193
Alter, C., Ding, Z., Flögel, U., Scheller, J., and Schrader, J. (2019). A2bR-dependent Signaling Alters Immune Cell Composition and Enhances IL-6 Formation in the Ischemic Heart. Am. J. Physiol.-Heart Circ. Physiol. 317 (1), H190–h200. doi:10.1152/ajpheart.00029.2019
Antonioli, L., Csóka, B., Fornai, M., Colucci, R., Kókai, E., Blandizzi, C., et al. (2014). Adenosine and Inflammation: What's New on the Horizon?. Drug Discov. Today 19 (8), 1051–1068. doi:10.1016/j.drudis.2014.02.010
Antonioli, L., Fornai, M., Blandizzi, C., Pacher, P., and Haskó, G. (2019). Adenosine Signaling and the Immune System: When a Lot Could Be Too Much. Immunol. Lett. 205, 9–15. doi:10.1016/j.imlet.2018.04.006
Antonioli, L., Novitskiy, S. V., Sachsenmeier, K. F., Fornai, M., Blandizzi, C., and Haskó, G. (2017). Switching off CD73: a Way to Boost the Activity of Conventional and Targeted Antineoplastic Therapies. Drug Discov. Today 22 (11), 1686–1696. doi:10.1016/j.drudis.2017.06.005
Asakura, M., Asanuma, H., Kim, J., Liao, Y., Nakamaru, K., Fujita, M., et al. (2007). Impact of Adenosine Receptor Signaling and Metabolism on Pathophysiology in Patients with Chronic Heart Failure. Hypertens. Res. 30 (9), 781–787. doi:10.1291/hypres.30.781
Awad, A. S., Huang, L., Ye, H., Duong, E. T. A., Bolton, W. K., Linden, J., et al. (2006). Adenosine A2Areceptor Activation Attenuates Inflammation and Injury in Diabetic Nephropathy. Am. J. Physiol.-Renal Physiol. 290 (4), F828–F837. doi:10.1152/ajprenal.00310.2005
Belardinelli, L., and Isenberg, G. (1983). Isolated Atrial Myocytes: Adenosine and Acetylcholine Increase Potassium Conductance. Am. J. Physiol.-Heart Circ. Physiol. 244 (5), H734–H737. doi:10.1152/ajpheart.1983.244.5.H734
Belardinelli, L., Shryock, J. C., Song, Y., Wang, D., and Srinivas, M. (1995). Ionic Basis of the Electrophysiological Actions of Adenosine on Cardiomyocytes. FASEB j. 9 (5), 359–365. doi:10.1096/fasebj.9.5.7896004
Berwick, Z. C., Payne, G. A., Lynch, B., Dick, G. M., Sturek, M., and Tune, J. D. (2010). Contribution of Adenosine A2A and A2B Receptors to Ischemic Coronary Dilation: Role of KV and KATP Channels. Microcirc. (N.Y) 17 (8), 600–607. doi:10.1111/j.1549-8719.2010.00054.x
Bessa-Gonçalves, M., Bragança, B., Martins-Dias, E., Correia-de-Sá, P., and Fontes-Sousa, A. P. (2018). Is the Adenosine A 2B 'biased' Receptor a Valuable Target for the Treatment of Pulmonary Arterial Hypertension?. Drug Discov. Today 23 (6), 1285–1292. doi:10.1016/j.drudis.2018.05.005
Bhatt, D. L., Steg, P. G., Mehta, S. R., Leiter, L. A., Simon, T., Fox, K., et al. (2019). Ticagrelor in Patients with Diabetes and Stable Coronary Artery Disease with a History of Previous Percutaneous Coronary Intervention (THEMIS-PCI): a Phase 3, Placebo-Controlled, Randomised Trial. Lancet 394 (10204), 1169–1180. doi:10.1016/s0140-6736(19)31887-2
Birkenfeld, A. L., Jordan, J., Dworak, M., Merkel, T., and Burnstock, G. (2019). Myocardial Metabolism in Heart Failure: Purinergic Signalling and Other Metabolic Concepts. Pharmacol. Ther. 194, 132–144. doi:10.1016/j.pharmthera.2018.08.015
Boison, D. (2013). Adenosine Kinase: Exploitation for Therapeutic Gain. Pharmacol. Rev. 65 (3), 906–943. doi:10.1124/pr.112.006361
Bono, M. R., Fernández, D., Flores-Santibáñez, F., Rosemblatt, M., and Sauma, D. (2015). CD73 and CD39 Ectonucleotidases in T Cell Differentiation: Beyond Immunosuppression. FEBS Lett. 589 (22), 3454–3460. doi:10.1016/j.febslet.2015.07.027
Borah, P., Deka, S., Mailavaram, R. P., and Deb, P. K. (2019). P1 Receptor Agonists/Antagonists in Clinical Trials - Potential Drug Candidates of the Future. Cpd 25 (26), 2792–2807. doi:10.2174/1381612825666190716111245
Borea, P. A., Gessi, S., Merighi, S., Vincenzi, F., and Varani, K. (2018). Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 98 (3), 1591–1625. doi:10.1152/physrev.00049.2017
Bragança, B., Oliveira-Monteiro, N., Ferreirinha, F., Lima, P. A., Faria, M., Fontes-Sousa, A. P., et al. (2016). Ion Fluxes through KCa2 (SK) and Cav1 (L-type) Channels Contribute to Chronoselectivity of Adenosine A1 Receptor-Mediated Actions in Spontaneously Beating Rat Atria. Front. Pharmacol. 7, 45. doi:10.3389/fphar.2016.00045
Bragança, B., Rodrigues, T., Gonçalves, M., Ferreirinha, F., Correia-de-Sá, P., and Fontes-Sousa, A. P. (2015). Unveiling the Potential Benefit of Adenosine A2B Receptor Blockage to Counteract Right Cardiac Overload in Rat Pulmonary Arterial Hypertension. Eur. Heart J. 36 (Suppl. 1), 497. doi:10.1093/eurheartj/ehv399
Burgdorf, C., Richardt, D., Kurz, T., Seyfarth, M., Jain, D., Katus, H. A., et al. (2001). Adenosine Inhibits Norepinephrine Release in the Postischemic Rat Heart: the Mechanism of Neuronal Stunning. Cardiovasc. Res. 49 (4), 713–720. doi:10.1016/s0008-6363(00)00309-6
Campbell, R. T., Jhund, P. S., Castagno, D., Hawkins, N. M., Petrie, M. C., and McMurray, J. J. V. (2012). What Have We Learned about Patients with Heart Failure and Preserved Ejection Fraction from DIG-PEF, CHARM-Preserved, and I-PRESERVE?. J. Am. Coll. Cardiol. 60 (23), 2349–2356. doi:10.1016/j.jacc.2012.04.064
Chan, E. S. L., and Cronstein, B. N. (2010). Methotrexate-how Does it Really Work?. Nat. Rev. Rheumatol. 6 (3), 175–178. doi:10.1038/nrrheum.2010.5
Chan, E. S. L., Montesinos, M. C., Fernandez, P., Desai, A., Delano, D. L., Yee, H., et al. (2006). Adenosine A2Areceptors Play a Role in the Pathogenesis of Hepatic Cirrhosis. Br. J. Pharmacol. 148 (8), 1144–1155. doi:10.1038/sj.bjp.0706812
Chandrasekera, P. C., McIntosh, V. J., Cao, F. X., and Lasley, R. D. (2010). Differential Effects of Adenosine A2a and A2b Receptors on Cardiac Contractility. Am. J. Physiol.-Heart Circ. Physiol. 299 (6), H2082–H2089. doi:10.1152/ajpheart.00511.2010
Chen, X., Ge, W., Dong, T., Hu, J., Chen, L., Fan, X., et al. (2019). Spironolactone Inhibits Endothelial-Mesenchymal Transition via the Adenosine A2A Receptor to Reduce Cardiorenal Fibrosis in Rats. Life Sci. 224, 177–186. doi:10.1016/j.lfs.2019.01.017
Chen, Y., Epperson, S., Makhsudova, L., Ito, B., Suarez, J., Dillmann, W., et al. (2004). Functional Effects of Enhancing or Silencing Adenosine A2b Receptors in Cardiac Fibroblasts. Am. J. Physiol.-Heart Circ. Physiol. 287 (6), H2478–H2486. doi:10.1152/ajpheart.00217.2004
Cheng, D. Y., DeWitt, B. J., Suzuki, F., Neely, C. F., and Kadowitz, P. J. (1996). Adenosine A1 and A2 Receptors Mediate Tone-dependent Responses in Feline Pulmonary Vascular Bed. Am. J. Physiol.-Heart Circ. Physiol. 270 (1 Pt 2), H200–H207. doi:10.1152/ajpheart.1996.270.1.H200
Cheng, J.-T., Chi, T.-C., and Liu, I.-M. (2000). Activation of Adenosine A1 Receptors by Drugs to Lower Plasma Glucose in Streptozotocin-Induced Diabetic Rats. Auton. Neurosci. 83 (3), 127–133. doi:10.1016/s0165-1838(00)00106-5
Chiang, P., Wu, S., Tsai, E., Wu, C., Shen, M., Huang, C., et al. (1994). Adenosine Modulation of Neurotransmission in Penile Erection. Br. J. Clin. Pharmacol. 38 (4), 357–362. doi:10.1111/j.1365-2125.1994.tb04366.x
Chung, E. S., Perlini, S., Aurigemma, G. P., Fenton, R. A., Dobson, J. G., and Meyer, T. E. (1998). Effects of Chronic Adenosine Uptake Blockade on Adrenergic Responsiveness and Left Ventricular Chamber Function in Pressure Overload Hypertrophy in the Rat. J. Hypertens. 16 (12 Pt 1), 1813–1822. doi:10.1097/00004872-199816120-00015
Ciacciarelli, M., Zerbinati, C., Violi, F., and Iuliano, L. (2015). Dipyridamole: a Drug with Unrecognized Antioxidant Activity. Ctmc 15 (9), 822–829. doi:10.2174/1568026615666150220111942
Conceição, G., Heinonen, I., Lourenço, A. P., Duncker, D. J., and Falcão-Pires, I. (2016). Animal Models of Heart Failure with Preserved Ejection Fraction. Neth. Heart J. 24 (4), 275–286. doi:10.1007/s12471-016-0815-9
Correia-de-Sá, M., Magalhães-Cardoso, M. T., Lobo, L., Pedro, B., Faria, M., Correia-de-Sá, P., et al. (2015). Biomarker Sensitivity of Plasma Adenosine Levels to Predict Heart Failure in Dogs. Auton. Neurosci. 192, 75. doi:10.1016/j.autneu.2015.07.071
Cui, X.-B., Fei, J., Chen, S., Edwards, G. L., and Chen, S.-Y. (2021). ADAR1 Deficiency Protects against High-Fat Diet-Induced Obesity and Insulin Resistance in Mice. Am. J. Physiol.-Endocrinol. Metab. 320 (1), E131–e138. doi:10.1152/ajpendo.00175.2020
Cury, R. C., Kitt, T. M., Feaheny, K., Akin, J., and George, R. T. (2014). Regadenoson-stress Myocardial CT Perfusion and Single-Photon Emission CT: Rationale, Design, and Acquisition Methods of a Prospective, Multicenter, Multivendor Comparison. J. Cardiovasc. Comput. Tomogr. 8 (1), 2–12. doi:10.1016/j.jcct.2013.09.004
D'Amario, D., Migliaro, S., Borovac, J. A., Restivo, A., Vergallo, R., Galli, M., et al. (2019). Microvascular Dysfunction in Heart Failure with Preserved Ejection Fraction. Front. Physiol. 10, 1347. doi:10.3389/fphys.2019.01347
de Oliveira, M., Mathias, L. S., de Sibio, M. T., Noronha-Matos, J. B., Costa, M. A., Nogueira, C. R., et al. (2020). Pitfalls and Challenges of the Purinergic Signaling Cascade in Obesity. Biochem. Pharmacol. 182, 114214. doi:10.1016/j.bcp.2020.114214
Delaunay, M., Osman, H., Kaiser, S., and Diviani, D. (2019). The Role of Cyclic AMP Signaling in Cardiac Fibrosis. Cells 9 (1), 69. doi:10.3390/cells9010069
Dhalla, A. K., Wong, M. Y., Voshol, P. J., Belardinelli, L., and Reaven, G. M. (2007). A1 Adenosine Receptor Partial Agonist Lowers Plasma FFA and Improves Insulin Resistance Induced by High-Fat Diet in Rodents. Am. J. Physiol.-Endocrinol. Metab. 292 (5), E1358–E1363. doi:10.1152/ajpendo.00573.2006
Dip, R. G. (2009). Adenosine Receptor Modulation: Potential Implications in Veterinary Medicine. Vet. J. 179 (1), 38–49. doi:10.1016/j.tvjl.2007.08.005
Dittrich, H. C., Gupta, D. K., Hack, T. C., Dowling, T., Callahan, J., and Thomson, S. (2007). The Effect of KW-3902, an Adenosine A1 Receptor Antagonist, on Renal Function and Renal Plasma Flow in Ambulatory Patients with Heart Failure and Renal Impairment. J. Card. Fail. 13 (8), 609–617. doi:10.1016/j.cardfail.2007.08.006
Dobson, J. G. (1983). Mechanism of Adenosine Inhibition of Catecholamine-Induced Responses in Heart. Circ. Res. 52 (2), 151–160. doi:10.1161/01.res.52.2.151
Dobson, J. G., Shea, L. G., and Fenton, R. A. (2008). Adenosine A2A and β-adrenergic Calcium Transient and Contractile Responses in Rat Ventricular Myocytes. Am. J. Physiol.-Heart Circ. Physiol. 295 (6), H2364–H2372. doi:10.1152/ajpheart.00927.2008
Dobson, J., and Fenton, R. A. (1997). Adenosine A2 Receptor Function in Rat Ventricular Myocytes. Cardiovasc. Res. 34 (2), 337–347. doi:10.1016/S0008-6363(97)00023-0
Dorotea, D., Cho, A., Lee, G., Kwon, G., Lee, J., Sahu, P. K., et al. (2018). Orally Active, Species-independent Novel A3 Adenosine Receptor Antagonist Protects against Kidney Injury in Db/db Mice. Exp. Mol. Med. 50 (4), 1–14. doi:10.1038/s12276-018-0053-x
Drury, A. N., and Szent-Györgyi, A. (1929). The Physiological Activity of Adenine Compounds with Especial Reference to Their Action upon the Mammalian Heart1. J. Physiol. 68 (3), 213–237. doi:10.1113/jphysiol.1929.sp002608
Du, X., Ou, X., Song, T., Zhang, W., Cong, F., Zhang, S., et al. (2015). Adenosine A2B Receptor Stimulates Angiogenesis by Inducing VEGF and eNOS in Human Microvascular Endothelial Cells. Exp. Biol. Med. (Maywood) 240 (11), 1472–1479. doi:10.1177/1535370215584939
Dunlay, S. M., Roger, V. L., and Redfield, M. M. (2017). Epidemiology of Heart Failure with Preserved Ejection Fraction. Nat. Rev. Cardiol. 14 (10), 591–602. doi:10.1038/nrcardio.2017.65
Eckle, T., Hartmann, K., Bonney, S., Reithel, S., Mittelbronn, M., Walker, L. A., et al. (2012). Adora2b-elicited Per2 Stabilization Promotes a HIF-dependent Metabolic Switch Crucial for Myocardial Adaptation to Ischemia. Nat. Med. 18 (5), 774–782. doi:10.1038/nm.2728
Eckle, T., Krahn, T., Grenz, A., Köhler, D., Mittelbronn, M., Ledent, C., et al. (2007b). Cardioprotection by Ecto-5′-Nucleotidase (CD73) and A2B Adenosine Receptors. Circulation 115 (12), 1581–1590. doi:10.1161/CIRCULATIONAHA.106.669697
Eckle, T., Krahn, T., Grenz, A., Köhler, D., Mittelbronn, M., Ledent, C., et al. (2007a). Cardioprotection by Ecto-5′-Nucleotidase (CD73) and A 2B Adenosine Receptors. Circulation 115 (12), 1581–1590. doi:10.1161/circulationaha.106.669697
El-Tayeb, A., Iqbal, J., Behrenswerth, A., Romio, M., Schneider, M., Zimmermann, H., et al. (2009). Nucleoside-5′-monophosphates as Prodrugs of Adenosine A2A Receptor Agonists Activated by Ecto-5′-Nucleotidase†Contribution to Celebrate the 100th Anniversary of the Division of Medicinal Chemistry of the American Chemical Society. J. Med. Chem. 52 (23), 7669–7677. doi:10.1021/jm900538v
Eltzschig, H. K., Abdulla, P., Hoffman, E., Hamilton, K. E., Daniels, D., Schönfeld, C., et al. (2005). HIF-1-dependent Repression of Equilibrative Nucleoside Transporter (ENT) in Hypoxia. J. Exp. Med. 202 (11), 1493–1505. doi:10.1084/jem.20050177
Eltzschig, H. K., Thompson, L. F., Karhausen, J., Cotta, R. J., Ibla, J. C., Robson, S. C., et al. (2004). Endogenous Adenosine Produced during Hypoxia Attenuates Neutrophil Accumulation: Coordination by Extracellular Nucleotide Metabolism. Blood 104 (13), 3986–3992. doi:10.1182/blood-2004-06-2066
Epperson, S. A., Brunton, L. L., Ramirez-Sanchez, I., and Villarreal, F. (2009). Adenosine Receptors and Second Messenger Signaling Pathways in Rat Cardiac Fibroblasts. Am. J. Physiol.-Cell Physiol. 296 (5), C1171–C1177. doi:10.1152/ajpcell.00290.2008
Fang, J. C. (2016). Heart Failure with Preserved Ejection Fraction. Circulation 134 (6), 435–437. doi:10.1161/circulationaha.116.022249
Faria, M., Magalhães-Cardoso, T., Lafuente-de-Carvalho, J.-M., and Correia-de-Sá, P. (2006). Corpus Cavernosum from Men with Vasculogenic Impotence Is Partially Resistant to Adenosine Relaxation Due to Endothelial A2B Receptor Dysfunction. J. Pharmacol. Exp. Ther. 319 (1), 405–413. doi:10.1124/jpet.106.107821
Fassett, J. T., Hu, X., Xu, X., Lu, Z., Zhang, P., Chen, Y., et al. (2011). Adenosine Kinase Regulation of Cardiomyocyte Hypertrophy. Am. J. Physiol.-Heart Circ. Physiol. 300 (5), H1722–H1732. doi:10.1152/ajpheart.00684.2010
Fassett, J., Xu, X., Kwak, D., Zhu, G., Fassett, E. K., Zhang, P., et al. (2019). Adenosine Kinase Attenuates Cardiomyocyte Microtubule Stabilization and Protects against Pressure Overload-Induced Hypertrophy and LV Dysfunction. J. Mol. Cell Cardiol. 130, 49–58. doi:10.1016/j.yjmcc.2019.03.015
Feng, W., Song, Y., Chen, C., Lu, Z. Z., and Zhang, Y. (2010). Stimulation of Adenosine A2B Receptors Induces Interleukin-6 Secretion in Cardiac Fibroblasts via the PKC-δ-P38 Signalling Pathway. Br. J. Pharmacol. 159 (8), 1598–1607. doi:10.1111/j.1476-5381.2009.00558.x
Fenton, R. A., and Dobson, J. G. (2007). Adenosine A1 and A2A Receptor Effects on G-Protein Cycling in β-adrenergic Stimulated Ventricular Membranes. J. Cel. Physiol. 213 (3), 785–792. doi:10.1002/jcp.21149
Flögel, U., Burghoff, S., van Lent, P. L. E. M., Temme, S., Galbarz, L., Ding, Z., et al. (2012). Selective Activation of Adenosine A2A Receptors on Immune Cells by a CD73-dependent Prodrug Suppresses Joint Inflammation in Experimental Rheumatoid Arthritis. Sci. Transl. Med. 4 (146), 146ra108. doi:10.1126/scitranslmed.3003717
Fujita, M., Asakura, M., Sanada, S., Funaya, H., Tsukamoto, O., Komamura, K., et al. (2008). Activation of Ecto-5′-Nucleotidase in the Blood and Hearts of Patients with Chronic Heart Failure. J. Card. Fail. 14 (5), 426–430. doi:10.1016/j.cardfail.2008.01.011
Funaya, H., Kitakaze, M., Node, K., Minamino, T., Komamura, K., and Hori, M. (1997). Plasma Adenosine Levels Increase in Patients with Chronic Heart Failure. Circulation 95 (6), 1363–1365. doi:10.1161/01.cir.95.6.1363
Ge, Z.-D., der Hoeven, D. v., Maas, J. E., Wan, T. C., and Auchampach, J. A. (2010). A3 Adenosine Receptor Activation during Reperfusion Reduces Infarct Size through Actions on Bone Marrow-Derived Cells. J. Mol. Cell Cardiol. 49 (2), 280–286. doi:10.1016/j.yjmcc.2010.01.018
Gharibi, B., Abraham, A. A., Ham, J., and Evans, B. A. J. (2012). Contrasting Effects of A1 and A2b Adenosine Receptors on Adipogenesis. Int. J. Obes. 36 (3), 397–406. doi:10.1038/ijo.2011.129
Givertz, M. M., Massie, B. M., Fields, T. K., Pearson, L. L., and Dittrich, H. C. (2007). The Effects of KW-3902, an Adenosine A1-Receptor Antagonist,on Diuresis and Renal Function in Patients with Acute Decompensated Heart Failure and Renal Impairment or Diuretic Resistance. J. Am. Coll. Cardiol. 50 (16), 1551–1560. doi:10.1016/j.jacc.2007.07.019
Gottlieb, S. S., Brater, D. C., Thomas, I., Havranek, E., Bourge, R., Goldman, S., et al. (2002). BG9719 (CVT-124), an A 1 Adenosine Receptor Antagonist, Protects against the Decline in Renal Function Observed with Diuretic Therapy. Circulation 105 (11), 1348–1353. doi:10.1161/hc1102.105264
Governo, R. J. M., Deuchars, J., Baldwin, S. A., and King, A. E. (2005). Localization of the NBMPR-Sensitive Equilibrative Nucleoside Transporter, ENT1, in the Rat Dorsal Root Ganglion and Lumbar Spinal Cord. Brain Res. 1059 (2), 129–138. doi:10.1016/j.brainres.2005.08.017
Greene, S. J., Sabbah, H. N., Butler, J., Voors, A. A., Albrecht-Küpper, B. E., Düngen, H.-D., et al. (2016). Partial Adenosine A1 Receptor Agonism: a Potential New Therapeutic Strategy for Heart Failure. Heart Fail. Rev. 21 (1), 95–102. doi:10.1007/s10741-015-9522-7
Grenz, A., Homann, D., and Eltzschig, H. K. (2011). Extracellular Adenosine: a Safety Signal that Dampens Hypoxia-Induced Inflammation during Ischemia. Antioxid. Redox Signal. 15 (8), 2221–2234. doi:10.1089/ars.2010.3665
Griendling, K. K., Sorescu, D., and Ushio-Fukai, M. (2000). NAD(P)H Oxidase. Circ. Res. 86 (5), 494–501. doi:10.1161/01.res.86.5.494
Grinberg, S., Hasko, G., Wu, D., and Leibovich, S. J. (2009). Suppression of PLCβ2 by Endotoxin Plays a Role in the Adenosine A2A Receptor-Mediated Switch of Macrophages from an Inflammatory to an Angiogenic Phenotype. Am. J. Pathol. 175 (6), 2439–2453. doi:10.2353/ajpath.2009.090290
Hamad, E. A., Zhu, W., Chan, T. O., Myers, V., Gao, E., Li, X., et al. (2012). Cardioprotection of Controlled and Cardiac-specific Over-expression of A2A-Adenosine Receptor in the Pressure Overload. PLoS One 7 (7), e39919. doi:10.1371/journal.pone.0039919
Headrick, J. P., Ashton, K. J., Rose'meyer, R. B., and Peart, J. N. (2013). Cardiovascular Adenosine Receptors: Expression, Actions and Interactions. Pharmacol. Ther. 140 (1), 92–111. doi:10.1016/j.pharmthera.2013.06.002
Herman-de-Sousa, C., Pinheiro, A. R., Paramos-de-Carvalho, D., Costa, M. A., Ferreirinha, F., Magalhães-Cardoso, T., et al. (2020). Opposing Effects of Adenosine and Inosine in Human Subcutaneous Fibroblasts May Be Regulated by Third Party ADA Cell Providers. Cells 9 (3), 651. doi:10.3390/cells9030651
Hinz, S., Navarro, G., Borroto-Escuela, D., Seibt, B. F., Ammon, Y.-C., de Filippo, E., et al. (2018). Adenosine A2A Receptor Ligand Recognition and Signaling Is Blocked by A2B Receptors. Oncotarget 9 (17), 13593–13611. doi:10.18632/oncotarget.24423
Hinze, A. V., Mayer, P., Harst, A., and von Kügelgen, I. (2012). Adenosine A3 Receptor-Induced Proliferation of Primary Human Coronary Smooth Muscle Cells Involving the Induction of Early Growth Response Genes. J. Mol. Cell Cardiol. 53 (5), 639–645. doi:10.1016/j.yjmcc.2012.08.003
Hussain, A., Gharanei, A. M., Nagra, A. S., and Maddock, H. L. (2014). Caspase Inhibition via A3 Adenosine Receptors: a New Cardioprotective Mechanism against Myocardial Infarction. Cardiovasc. Drugs Ther. 28 (1), 19–32. doi:10.1007/s10557-013-6500-y
Idzko, M., Ferrari, D., and Eltzschig, H. K. (2014). Nucleotide Signalling during Inflammation. Nature 509 (7500), 310–317. doi:10.1038/nature13085
Ilieşiu, A. M., and Hodorogea, A. S. (2018). Treatment of Heart Failure with Preserved Ejection Fraction. Adv. Exp. Med. Biol. 1067, 67–87. doi:10.1007/5584_2018_149
Iwamoto, T., Umemura, S., Toya, Y., Uchibori, T., Kogi, K., Takagi, N., et al. (1994). Identification of Adenosine A2 Receptor-cAMP System in Human Aortic Endothelial Cells. Biochem. Biophys. Res. Commun. 199 (2), 905–910. doi:10.1006/bbrc.1994.1314
Jacobson, K. A., Tosh, D. K., Jain, S., and Gao, Z.-G. (2019). Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cel. Neurosci. 13, 124. doi:10.3389/fncel.2019.00124
Johansson, S. M., Lindgren, E., Yang, J.-N., Herling, A. W., and Fredholm, B. B. (2008). Adenosine A1 Receptors Regulate Lipolysis and Lipogenesis in Mouse Adipose Tissue - Interactions with Insulin. Eur. J. Pharmacol. 597 (1), 92–101. doi:10.1016/j.ejphar.2008.08.022
Jordan, J. E., Zhao, Z. Q., Sato, H., Taft, S., and Vinten-Johansen, J. (1997). Adenosine A2 Receptor Activation Attenuates Reperfusion Injury by Inhibiting Neutrophil Accumulation, Superoxide Generation and Coronary Endothelial Adherence. J. Pharmacol. Exp. Ther. 280 (1), 301–309.
Kalogeropoulos, A., Georgiopoulou, V., Psaty, B. M., Rodondi, N., Smith, A. L., Harrison, D. G., et al. (2010). Inflammatory Markers and Incident Heart Failure Risk in Older Adults. J. Am. Coll. Cardiol. 55 (19), 2129–2137. doi:10.1016/j.jacc.2009.12.045
Karmouty-Quintana, H., Weng, T., Garcia-Morales, L. J., Chen, N.-Y., Pedroza, M., Zhong, H., et al. (2013). Adenosine A2B Receptor and Hyaluronan Modulate Pulmonary Hypertension Associated with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cel Mol Biol 49 (6), 1038–1047. doi:10.1165/rcmb.2013-0089OC
Katz, D. H., Burns, J. A., Aguilar, F. G., Beussink, L., and Shah, S. J. (2014). Albuminuria Is Independently Associated with Cardiac Remodeling, Abnormal Right and Left Ventricular Function, and Worse Outcomes in Heart Failure with Preserved Ejection Fraction. JACC: Heart Fail. 2 (6), 586–596. doi:10.1016/j.jchf.2014.05.016
Kidokoro, K., Cherney, D. Z. I., Bozovic, A., Nagasu, H., Satoh, M., Kanda, E., et al. (2019). Evaluation of Glomerular Hemodynamic Function by Empagliflozin in Diabetic Mice Using In Vivo Imaging. Circulation 140 (4), 303–315. doi:10.1161/circulationaha.118.037418
Koupenova, M., Johnston-Cox, H., and Ravid, K. (2012). Regulation of Atherosclerosis and Associated Risk Factors by Adenosine and Adenosine Receptors. Curr. Atheroscler. Rep. 14 (5), 460–468. doi:10.1007/s11883-012-0263-y
Kuno, A., Critz, S. D., Cui, L., Solodushko, V., Yang, X.-M., Krahn, T., et al. (2007). Protein Kinase C Protects Preconditioned Rabbit Hearts by Increasing Sensitivity of Adenosine A2b-dependent Signaling during Early Reperfusion. J. Mol. Cell Cardiol. 43 (3), 262–271. doi:10.1016/j.yjmcc.2007.05.016
Kusano, Y., Echeverry, G., Miekisiak, G., Kulik, T. B., Aronhime, S. N., Chen, J. F., et al. (2010). Role of Adenosine A2 Receptors in Regulation of Cerebral Blood Flow during Induced Hypotension. J. Cereb. Blood Flow Metab. 30 (4), 808–815. doi:10.1038/jcbfm.2009.244
Lam, C. S. P., Voors, A. A., de Boer, R. A., Solomon, S. D., and van Veldhuisen, D. J. (2018). Heart Failure with Preserved Ejection Fraction: from Mechanisms to Therapies. Eur. Heart J. 39 (30), 2780–2792. doi:10.1093/eurheartj/ehy301
Landmesser, U., Cai, H., Dikalov, S., McCann, L., Hwang, J., Jo, H., et al. (2002). Role of P47 Phox in Vascular Oxidative Stress and Hypertension Caused by Angiotensin II. Hypertension 40 (4), 511–515. doi:10.1161/01.hyp.0000032100.23772.98
Lazarowski, E. R., Sesma, J. I., Seminario-Vidal, L., and Kreda, S. M. (2011). Molecular Mechanisms of Purine and Pyrimidine Nucleotide Release. Adv. Pharmacol. 61, 221–261. doi:10.1016/b978-0-12-385526-8.00008-4
Lee, H. T., Gallos, G., Nasr, S. H., and Emala, C. W. (2004)., 15. Ae, 102–111. doi:10.1097/01.Asn.0000102474.6861310.1097/01.asn.0000102474.68613.aeA1 Adenosine Receptor Activation Inhibits Inflammation, Necrosis, and Apoptosis after Renal Ischemia-Reperfusion Injury in MiceJ. Am. Soc. Nephrol.1
Levine, A. R., Simon, M. A., and Gladwin, M. T. (2019). Pulmonary Vascular Disease in the Setting of Heart Failure with Preserved Ejection Fraction. Trends Cardiovasc. Med. 29 (4), 207–217. doi:10.1016/j.tcm.2018.08.005
Li, J.-m., Fenton, R. A., Wheeler, H. B., Powell, C. C., Peyton, B. D., Cutler, B. S., et al. (1998). Adenosine A2aReceptors Increase Arterial Endothelial Cell Nitric Oxide. J. Surg. Res. 80 (2), 357–364. doi:10.1006/jsre.1998.5439
Li, J. M., Fenton, R. A., Cutler, B. S., and Dobson, J. G. (1995). Adenosine Enhances Nitric Oxide Production by Vascular Endothelial Cells. Am. J. Physiology-Cell Physiol. 269 (2 Pt 1), C519–C523. doi:10.1152/ajpcell.1995.269.2.C519
Liang, H.-X., Belardinelli, L., Ozeck, M. J., and Shryock, J. C. (2002). Tonic Activity of the Rat Adipocyte A1-Adenosine Receptor. Br. J. Pharmacol. 135 (6), 1457–1466. doi:10.1038/sj.bjp.0704586
Liao, Y., Takashima, S., Asano, Y., Asakura, M., Ogai, A., Shintani, Y., et al. (2003). Activation of Adenosine A 1 Receptor Attenuates Cardiac Hypertrophy and Prevents Heart Failure in Murine Left Ventricular Pressure-Overload Model. Circ. Res. 93 (8), 759–766. doi:10.1161/01.Res.0000094744.88220.62
Lim, S. H., Anantharaman, V., Teo, W. S., and Chan, Y. H. (2009). Slow Infusion of Calcium Channel Blockers Compared with Intravenous Adenosine in the Emergency Treatment of Supraventricular Tachycardia. Resuscitation 80 (5), 523–528. doi:10.1016/j.resuscitation.2009.01.017
Lindman, B. R., Dávila-Román, V. G., Mann, D. L., McNulty, S., Semigran, M. J., Lewis, G. D., et al. (2014). Cardiovascular Phenotype in HFpEF Patients with or without Diabetes. J. Am. Coll. Cardiol. 64 (6), 541–549. doi:10.1016/j.jacc.2014.05.030
Löfman, I., Szummer, K., Dahlström, U., Jernberg, T., and Lund, L. H. (2017). Associations with and Prognostic Impact of Chronic Kidney Disease in Heart Failure with Preserved, Mid-range, and Reduced Ejection Fraction. Eur. J. Heart Fail. 19 (12), 1606–1614. doi:10.1002/ejhf.821
Lu, Z., Fassett, J., Xu, X., Hu, X., Zhu, G., French, J., et al. (2008). Adenosine A 3 Receptor Deficiency Exerts Unanticipated Protective Effects on the Pressure-Overloaded Left Ventricle. Circulation 118 (17), 1713–1721. doi:10.1161/circulationaha.108.788307
Meyer, T. E., Chung, E. S., Perlini, S., Norton, G. R., Woodiwiss, A. J., Lorbar, M., et al. (2001). Antiadrenergic Effects of Adenosine in Pressure Overload Hypertrophy. Hypertension 37 (3), 862–868. doi:10.1161/01.hyp.37.3.862
Meyskens, F. L., and Williams, H. E. (1971). Adenosine Metabolism in Human Erythrocytes. Biochim. Biophys. Acta (Bba) - Nucleic Acids Protein Synth. 240 (2), 170–179. doi:10.1016/0005-2787(71)90654-x
Mierzejewska, P., Zabielska, M. A., Kutryb-Zajac, B., Tomczyk, M., Koszalka, P., Smolenski, R. T., et al. (2019). Impaired L-Arginine Metabolism marks Endothelial Dysfunction in CD73-Deficient Mice. Mol. Cel Biochem 458 (1-2), 133–142. doi:10.1007/s11010-019-03537-4
Mohammed, S. F., Hussain, S., Mirzoyev, S. A., Edwards, W. D., Maleszewski, J. J., and Redfield, M. M. (2015). Coronary Microvascular Rarefaction and Myocardial Fibrosis in Heart Failure with Preserved Ejection Fraction. Circulation 131 (6), 550–559. doi:10.1161/circulationaha.114.009625
Morote-Garcia, J. C., Rosenberger, P., Nivillac, N. M., Coe, I. R., and Eltzschig, H. K. (2009). Hypoxia-inducible Factor-dependent Repression of Equilibrative Nucleoside Transporter 2 Attenuates Mucosal Inflammation during Intestinal Hypoxia. Gastroenterology 136 (2), 607–618. doi:10.1053/j.gastro.2008.10.037
Morrison, R. R., Teng, B., Oldenburg, P. J., Katwa, L. C., Schnermann, J. B., and Mustafa, S. J. (2006). Effects of Targeted Deletion of A1 Adenosine Receptors on Postischemic Cardiac Function and Expression of Adenosine Receptor Subtypes. Am. J. Physiol.-Heart Circ. Physiol. 291 (4), H1875–H1882. doi:10.1152/ajpheart.00158.2005
Nadeem, A., Ponnoth, D. S., Ansari, H. R., Batchelor, T. P., Dey, R. D., Ledent, C., et al. (2009). A2A Adenosine Receptor Deficiency Leads to Impaired Tracheal Relaxation via NADPH Oxidase Pathway in Allergic Mice. J. Pharmacol. Exp. Ther. 330 (1), 99–108. doi:10.1124/jpet.109.151613
Nakagawa, T. (2007). Uncoupling of the VEGF-Endothelial Nitric Oxide axis in Diabetic Nephropathy: an Explanation for the Paradoxical Effects of VEGF in Renal Disease. Am. J. Physiol.-Renal Physiol. 292 (6), F1665–F1672. doi:10.1152/ajprenal.00495.2006
Nakamura, M., and Sadoshima, J. (2018). Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 15 (7), 387–407. doi:10.1038/s41569-018-0007-y
Navarro, G., Abdolazimi, Y., Zhao, Z., Xu, H., Lee, S., Armstrong, N. A., et al. (2017). Genetic Disruption of Adenosine Kinase in Mouse Pancreatic β-Cells Protects against High-Fat Diet-Induced Glucose Intolerance. Diabetes 66 (7), 1928–1938. doi:10.2337/db16-0816
Newby, A., Worku, Y., Meghji, P., Nakazawa, M., and Skladanowski, A. (1990). Adenosine: A Retaliatory Metabolite or Not?. Physiology 5 (2), 67–70. doi:10.1152/physiologyonline.1990.5.2.67
Ni, Y., Liang, D., Tian, Y., Kron, I. L., French, B. A., and Yang, Z. (2018). Infarct-Sparing Effect of Adenosine A2B Receptor Agonist Is Primarily Due to its Action on Splenic Leukocytes via a PI3K/Akt/IL-10 Pathway. J. Surg. Res. 232, 442–449. doi:10.1016/j.jss.2018.06.042
Oliveira, L., Correia, A., Cristina Costa, A., Guerra-Gomes, S., Ferreirinha, F., Magalhães-Cardoso, M. T., et al. (2015). Deficits in Endogenous Adenosine Formation by Ecto-5′-Nucleotidase/CD73 Impair Neuromuscular Transmission and Immune Competence in Experimental Autoimmune Myasthenia Gravis. Mediators Inflamm. 2015, 1–16. doi:10.1155/2015/460610
Pandey, S., Aggarwal, D., Gupta, K., Kumari, A., Sen, P., Singh, R., et al. (2021). “Adenosine an Old Player with New Possibilities in Kidney Diseases”: Preclinical Evidences and Clinical Perspectives. Life Sci. 265, 118834. doi:10.1016/j.lfs.2020.118834
Pastor-Anglada, M., and Pérez-Torras, S. (2018). Emerging Roles of Nucleoside Transporters. Front. Pharmacol. 9, 606. doi:10.3389/fphar.2018.00606
Patel, L., and Thaker, A. (2014). The Effects of Adenosine A2Breceptor Inhibition on VEGF and Nitric Oxide axis-mediated Renal Function in Diabetic Nephropathy. Ren. Fail. 36 (6), 916–924. doi:10.3109/0886022x.2014.900404
Patinha, D., Abreu, C., Carvalho, C., Cunha, O. M., Mota, M., Afonso, J., et al. (2020). Adenosine A2A and A3 Receptors as Targets for the Treatment of Hypertensive-Diabetic Nephropathy. Biomedicines 8 (11), 529. doi:10.3390/biomedicines8110529
Patinha, D., Afonso, J., Sousa, T., Morato, M., and Albino-Teixeira, A. (2014). Activation of Adenosine Receptors Improves Renal Antioxidant Status in Diabetic Wistar but Not SHR Rats. Upsala J. Med. Sci. 119 (1), 10–18. doi:10.3109/03009734.2013.851748
Paulus, W. J., and Tschöpe, C. (2013). A Novel Paradigm for Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 62 (4), 263–271. doi:10.1016/j.jacc.2013.02.092
Peleli, M., Hezel, M., Zollbrecht, C., Persson, A. E. G., Lundberg, J. O., Weitzberg, E., et al. (2015). In Adenosine A2B Knockouts Acute Treatment with Inorganic Nitrate Improves Glucose Disposal, Oxidative Stress, and AMPK Signaling in the Liver. Front. Physiol. 6 (222). doi:10.3389/fphys.2015.00222
Persson, P., Friederich-Persson, M., Fasching, A., Hansell, P., Inagi, R., and Palm, F. (2015a). Adenosine A2a Receptor Stimulation Prevents Proteinuria in Diabetic Rats by Promoting an Anti-inflammatory Phenotype without Affecting Oxidative Stress. Acta Physiol. 214 (3), 311–318. doi:10.1111/apha.12511
Persson, P., Hansell, P., and Palm, F. (2015b). Reduced Adenosine A2a Receptor–Mediated Efferent Arteriolar Vasodilation Contributes to Diabetes-Induced Glomerular Hyperfiltration. Kidney Int. 87 (1), 109–115. doi:10.1038/ki.2014.219
Phosri, S., Arieyawong, A., Bunrukchai, K., Parichatikanond, W., Nishimura, A., Nishida, M., et al. (2017). Stimulation of Adenosine A2B Receptor Inhibits Endothelin-1-Induced Cardiac Fibroblast Proliferation and α-Smooth Muscle Actin Synthesis through the cAMP/Epac/PI3K/Akt-Signaling Pathway. Front. Pharmacol. 8, 428. doi:10.3389/fphar.2017.00428
Phosri, S., Bunrukchai, K., Parichatikanond, W., Sato, V. H., and Mangmool, S. (2018). Epac Is Required for Exogenous and Endogenous Stimulation of Adenosine A2B Receptor for Inhibition of Angiotensin II-Induced Collagen Synthesis and Myofibroblast Differentiation. Purinergic Signal. 14 (2), 141–156. doi:10.1007/s11302-017-9600-5
Ponikowski, P., Voors, A. A., Anker, S. D., Bueno, H., Cleland, J. G. F., Coats, A. J. S., et al. (2016). 2016 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 37 (27), 2129–2200. doi:10.1093/eurheartj/ehw128
Prijic, S., and Buchhorn, R. (2014). Mechanisms of Beta-Blockers Action in Patients with Heart Failure. Rrct 9 (2), 58–60. doi:10.2174/1574887109666140908125402
Pugliese, A., Traini, C., Cipriani, S., Gianfriddo, M., Mello, T., Giovannini, M., et al. (2009). The Adenosine A2Areceptor Antagonist ZM241385 Enhances Neuronal Survival after Oxygen-Glucose Deprivation in Rat CA1 Hippocampal Slices. Br. J. Pharmacol. 157 (5), 818–830. doi:10.1111/j.1476-5381.2009.00218.x
Puhl, S.-L., Kazakov, A., Müller, A., Fries, P., Wagner, D. R., Böhm, M., et al. (2016). Adenosine A1receptor Activation Attenuates Cardiac Hypertrophy and Fibrosis in Response to α1-adrenoceptor Stimulationin Vivo. Br. J. Pharmacol. 173 (1), 88–102. doi:10.1111/bph.13339
Pye, C., Elsherbiny, N. M., Ibrahim, A. S., Liou, G. I., Chadli, A., Al-Shabrawey, M., et al. (2014). Adenosine Kinase Inhibition Protects the Kidney against Streptozotocin-Induced Diabetes through Anti-inflammatory and Anti-oxidant Mechanisms. Pharmacol. Res. 85, 45–54. doi:10.1016/j.phrs.2014.05.004
Quast, C., Alter, C., Ding, Z., Borg, N., and Schrader, J. (2017). Adenosine Formed by CD73 on T Cells Inhibits Cardiac Inflammation and Fibrosis and Preserves Contractile Function in Transverse Aortic Constriction-Induced Heart Failure. Circ. Heart Fail. 10 (4). doi:10.1161/circheartfailure.116.003346
Ranchoux, B., Nadeau, V., Bourgeois, A., Provencher, S., Tremblay, É., Omura, J., et al. (2019). Metabolic Syndrome Exacerbates Pulmonary Hypertension Due to Left Heart Disease. Circ. Res. 125 (4), 449–466. doi:10.1161/circresaha.118.314555
Reichelt, M. E., Willems, L., Molina, J. G., Sun, C.-X., Noble, J. C., Ashton, K. J., et al. (2005). Genetic Deletion of the A 1 Adenosine Receptor Limits Myocardial Ischemic Tolerance. Circ. Res. 96 (3), 363–367. doi:10.1161/01.Res.0000156075.00127.C3
Ribé, D., Sawbridge, D., Thakur, S., Hussey, M., Ledent, C., Kitchen, I., et al. (2008). Adenosine A2A Receptor Signaling Regulation of Cardiac NADPH Oxidase Activity. Free Radic. Biol. Med. 44 (7), 1433–1442. doi:10.1016/j.freeradbiomed.2007.12.035
Romano, F., and Dobson, J. G. (1990). Adenosine Modulates β-adrenergic Signal Transduction in guinea-pig Heart Ventricular Membranes. J. Mol. Cell Cardiol. 22 (12), 1359–1370. doi:10.1016/0022-2828(90)90981-7
Rork, T. H., Wallace, K. L., Kennedy, D. P., Marshall, M. A., Lankford, A. R., and Linden, J. (2008). Adenosine A2Areceptor Activation Reduces Infarct Size in the Isolated, Perfused Mouse Heart by Inhibiting Resident Cardiac Mast Cell Degranulation. Am. J. Physiology-Heart Circulatory Physiol. 295 (5), H1825–H1833. doi:10.1152/ajpheart.495.2008
Rozenbaum, Z., Topilsky, Y., Khoury, S., Pereg, D., and Laufer-Perl, M. (2019). Association of Body Mass index and Diastolic Function in Metabolically Healthy Obese with Preserved Ejection Fraction. Int. J. Cardiol. 277, 147–152. doi:10.1016/j.ijcard.2018.08.008
Rubio, R., Berne, R., and Dobson Jg, J. G. (1973). Sites of Adenosine Production in Cardiac and Skeletal Muscle. Am. J. Physiology-Legacy Content 225 (4), 938–953. doi:10.1152/ajplegacy.1973.225.4.938
Saadjian, A., Paganelli, F., Gaubert, M. L., Levy, S., and Guieu, R. P. (1999). Adenosine Plasma Concentration in Pulmonary Hypertension. Cardiovasc. Res. 43 (1), 228–236. doi:10.1016/s0008-6363(99)00059-0
Sabbah, H. N., Gupta, R. C., Kohli, S., Wang, M., Rastogi, S., Zhang, K., et al. (2013a). Chronic Therapy with a Partial Adenosine A1-Receptor Agonist Improves Left Ventricular Function and Remodeling in Dogs with Advanced Heart Failure. Circ. Heart Fail. 6(3), 563–571. doi:10.1161/CIRCHEARTFAILURE.112.000208
Sabbah, H. N., Gupta, R. C., Kohli, S., Wang, M., Rastogi, S., Zhang, K., et al. (2013b). Chronic Therapy with a Partial Adenosine A1-Receptor Agonist Improves Left Ventricular Function and Remodeling in Dogs with Advanced Heart Failure. Circ. Heart Fail. 6 (3), 563–571. doi:10.1161/circheartfailure.112.000208
Sacramento, J. F., Martins, F. O., Rodrigues, T., Matafome, P., Ribeiro, M. J., Olea, E., et al. (2020). A2 Adenosine Receptors Mediate Whole-Body Insulin Sensitivity in a Prediabetes Animal Model: Primary Effects on Skeletal Muscle. Front. Endocrinol. 11, 262. doi:10.3389/fendo.2020.00262
Sands, W. A., Martin, A. F., Strong, E. W., and Palmer, T. M. (2004). Specific Inhibition of Nuclear Factor-κb-dependent Inflammatory Responses by Cell Type-specific Mechanisms upon A2AAdenosine Receptor Gene Transfer. Mol. Pharmacol. 66 (5), 1147–1159. doi:10.1124/mol.104.001107
Sassi, Y., Ahles, A., Truong, D.-J. J., Baqi, Y., Lee, S.-Y., Husse, B., et al. (2014). Cardiac Myocyte-Secreted cAMP Exerts Paracrine Action via Adenosine Receptor Activation. J. Clin. Invest. 124 (12), 5385–5397. doi:10.1172/jci74349
Savelieva, I., and Camm, J. (2008). Anti-arrhythmic Drug Therapy for Atrial Fibrillation: Current Anti-arrhythmic Drugs, Investigational Agents, and Innovative Approaches. Europace 10 (6), 647–665. doi:10.1093/europace/eun130
Schrader, J., Schütz, W., and Bardenheuer, H. (1981). Role of S-Adenosylhomocysteine Hydrolase in Adenosine Metabolism in Mammalian Heart. Biochem. J. 196 (1), 65–70. doi:10.1042/bj1960065
Seo, S.-w., Koeppen, M., Bonney, S., Gobel, M., Thayer, M., Harter, P. N., et al. (2015). Differential Tissue-specific Function of Adora2b in Cardioprotection. J.I. 195 (4), 1732–1743. doi:10.4049/jimmunol.1402288
Shah, S. J., Voors, A. A., McMurray, J. J. V., Kitzman, D. W., Viethen, T., Bomfim Wirtz, A., et al. (2019). Effect of Neladenoson Bialanate on Exercise Capacity Among Patients with Heart Failure with Preserved Ejection Fraction. JAMA 321 (21), 2101–2112. doi:10.1001/jama.2019.6717
Shang, P., He, Z.-Y., Chen, J.-F., Huang, S.-Y., Liu, B.-H., Liu, H.-X., et al. (2015). Absence of the Adenosine A2A Receptor Confers Pulmonary Arterial Hypertension through RhoA/ROCK Signaling Pathway in Mice. J. Cardiovasc. Pharmacol. 66 (6), 569–575. doi:10.1097/fjc.0000000000000305
Shryock, J. C., Snowdy, S., Baraldi, P. G., Cacciari, B., Spalluto, G., Monopoli, A., et al. (1998). A 2A -Adenosine Receptor Reserve for Coronary Vasodilation. Circulation 98 (7), 711–718. doi:10.1161/01.cir.98.7.711
Silverman, M. H., Strand, V., Markovits, D., Nahir, M., Reitblat, T., Molad, Y., et al. (2008). Clinical Evidence for Utilization of the A3 Adenosine Receptor as a Target to Treat Rheumatoid Arthritis: Data from a Phase II Clinical Trial. J. Rheumatol. 35 (1), 41–48.
Simmonds, S. J., Cuijpers, I., Heymans, S., and Jones, E. A. V. (2020). Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 9 (1), 242. doi:10.3390/cells9010242
Souders, C. A., Bowers, S. L. K., and Baudino, T. A. (2009). Cardiac Fibroblast. Circ. Res. 105 (12), 1164–1176. doi:10.1161/circresaha.109.209809
Staehr, P. M., Dhalla, A. K., Zack, J., Wang, X., Ho, Y. L., Bingham, J., et al. (2013). Reduction of Free Fatty Acids, Safety, and Pharmacokinetics of Oral GS-9667, an A1Adenosine Receptor Partial Agonist. J. Clin. Pharmacol. 53 (4), 385–392. doi:10.1002/jcph.9
Stowe, D. F., O'Brien, W. C., Chang, D., Knop, C. S., and Kampine, J. P. (1997). Reversal of Endothelin-Induced Vasoconstriction by Endothelium-dependent and -independent Vasodilators in Isolated Hearts and Vascular Rings. J. Cardiovasc. Pharmacol. 29 (6), 747–754. doi:10.1097/00005344-199706000-00007
Sumi, Y., Woehrle, T., Chen, Y., Yao, Y., Li, A., and Junger, W. G. (2010). Adrenergic Receptor Activation Involves ATP Release and Feedback through Purinergic Receptors. Am. J. Physiol.-Cell Physiol. 299 (5), C1118–C1126. doi:10.1152/ajpcell.00122.2010
Sun, Y., and Huang, P. (2016). Adenosine A2B Receptor: From Cell Biology to Human Diseases. Front. Chem. 4, 37. doi:10.3389/fchem.2016.00037
Surinkaew, S., Aflaki, M., Takawale, A., Chen, Y., Qi, X.-Y., Gillis, M.-A., et al. (2019). Exchange Protein Activated by Cyclic-Adenosine Monophosphate (Epac) Regulates Atrial Fibroblast Function and Controls Cardiac Remodelling. Cardiovasc. Res. 115 (1), 94–106. doi:10.1093/cvr/cvy173
Taddei, S., Arzilli, F., Arrighi, P., and Salvetti, A. (1992). Dipyridamole Decreases Circulating Renin-Angiotensin System Activity in Hypertensive Patients. Am. J. Hypertens. 5 (1), 29–31. doi:10.1093/ajh/5.1.29
Tang, X.-L., Wang, H.-X., Cho, C. H., and Wong, T. M. (1998). Reduced Responsiveness of [Ca2+]i to Adenosine A1- and A2-Receptor Stimulation in the Isoproterenol-Stimulated Ventricular Myocytes of Spontaneously Hypertensive Rats. J. Cardiovasc. Pharmacol. 31 (4), 493–498. doi:10.1097/00005344-199804000-00004
Thakur, S., Du, J., Hourani, S., Ledent, C., and Li, J.-M. (2010). Inactivation of Adenosine A2A Receptor Attenuates Basal and Angiotensin II-Induced ROS Production by Nox2 in Endothelial Cells. J. Biol. Chem. 285 (51), 40104–40113. doi:10.1074/jbc.M110.184606
Thompson, I. M., Tangen, C. M., Goodman, P. J., Probstfield, J. L., Moinpour, C. M., and Coltman, C. A. (2005). Erectile Dysfunction and Subsequent Cardiovascular Disease. Jama 294 (23), 2996–3002. doi:10.1001/jama.294.23.2996
Tian, Y., Marshall, M., French, B. A., Linden, J., and Yang, Z. (2015a). The Infarct-Sparing Effect of IB-MECA against Myocardial Ischemia/reperfusion Injury in Mice Is Mediated by Sequential Activation of Adenosine A3 and A2A Receptors. Basic Res. Cardiol. 110 (2), 16. doi:10.1007/s00395-015-0473-x
Tian, Y., Piras, B. A., Kron, I. L., French, B. A., and Yang, Z. (2015b). Adenosine 2B Receptor Activation Reduces Myocardial Reperfusion Injury by Promoting Anti-inflammatory Macrophages Differentiation via PI3K/Akt Pathway. Oxid. Med. Cell Longev. 2015, 1–8. doi:10.1155/2015/585297
Tofovic, S. P., Salah, E. M., Smits, G. J., Whalley, E. T., Ticho, B., Deykin, A., et al. (2016). Dual A1/A2B Receptor Blockade Improves Cardiac and Renal Outcomes in a Rat Model of Heart Failure with Preserved Ejection Fraction. J. Pharmacol. Exp. Ther. 356 (2), 333–340. doi:10.1124/jpet.115.228841
Toldo, S., Zhong, H., Mezzaroma, E., Van Tassell, B. W., Kannan, H., Zeng, D., et al. (2012). GS-6201, a Selective Blocker of the A2BAdenosine Receptor, Attenuates Cardiac Remodeling after Acute Myocardial Infarction in the Mouse. J. Pharmacol. Exp. Ther. 343 (3), 587–595. doi:10.1124/jpet.111.191288
Tschöpe, C., Bock, C. T., Kasner, M., Noutsias, M., Westermann, D., Schwimmbeck, P. L., et al. (2005). High Prevalence of Cardiac Parvovirus B19 Infection in Patients with Isolated Left Ventricular Diastolic Dysfunction. Circulation 111 (7), 879–886. doi:10.1161/01.Cir.0000155615.68924.B3
van Dam, R. M., Willett, W. C., Manson, J. E., and Hu, F. B. (2006). Coffee, Caffeine, and Risk of Type 2 Diabetes: a Prospective Cohort Study in Younger and Middle-Aged U.S. Women. Diabetes Care 29 (2), 398–403. doi:10.2337/diacare.29.02.06.dc05-1512
Vecchio, E. A., White, P. J., and May, L. T. (2017). Targeting Adenosine Receptors for the Treatment of Cardiac Fibrosis. Front. Pharmacol. 8, 243. doi:10.3389/fphar.2017.00243
Vieira, C., Magalhães-Cardoso, M. T., Ferreirinha, F., Silva, I., Dias, A. S., Pelletier, J., et al. (2014). Feed-forward Inhibition of CD73 and Upregulation of Adenosine Deaminase Contribute to the Loss of Adenosine Neuromodulation in Postinflammatory Ileitis. Mediators Inflamm. 2014, 1–19. doi:10.1155/2014/254640
Voors, A. A., Dittrich, H. C., Massie, B. M., DeLucca, P., Mansoor, G. A., Metra, M., et al. (2011). Effects of the Adenosine A1 Receptor Antagonist Rolofylline on Renal Function in Patients with Acute Heart Failure and Renal Dysfunction. J. Am. Coll. Cardiol. 57 (19), 1899–1907. doi:10.1016/j.jacc.2010.11.057
Voors, A. A., Düngen, H.-D., Senni, M., Nodari, S., Agostoni, P., Ponikowski, P., et al. (2017). Safety and Tolerability of Neladenoson Bialanate, a Novel Oral Partial Adenosine A1 Receptor Agonist, in Patients with Chronic Heart Failure. J. Clin. Pharmacol. 57 (4), 440–451. doi:10.1002/jcph.828
Voors, A. A., Shah, S. J., Bax, J. J., Butler, J., Gheorghiade, M., Hernandez, A. F., et al. (2018). Rationale and Design of the Phase 2b Clinical Trials to Study the Effects of the Partial Adenosine A1-Receptor Agonist Neladenoson Bialanate in Patients with Chronic Heart Failure with Reduced (PANTHEON) and Preserved (PANACHE) Ejection Fraction. Eur. J. Heart Fail. 20 (11), 1601–1610. doi:10.1002/ejhf.1295
Wagner, D. R., McTiernan, C., Sanders, V. J., and Feldman, A. M. (1998). Adenosine Inhibits Lipopolysaccharide-Induced Secretion of Tumor Necrosis Factor-α in the Failing Human Heart. Circulation 97 (6), 521–524. doi:10.1161/01.cir.97.6.521
Wakeno, M., Minamino, T., Seguchi, O., Okazaki, H., Tsukamoto, O., Okada, K.-i., et al. (2006). Long-Term Stimulation of Adenosine A2b Receptors Begun after Myocardial Infarction Prevents Cardiac Remodeling in Rats. Circulation 114(18), 1923–1932. doi:10.1161/CIRCULATIONAHA.106.630087
Wernly, B., Erlinge, D., Pernow, J., and Zhou, Z. (2021). Ticagrelor: a Cardiometabolic Drug Targeting Erythrocyte-Mediated Purinergic Signaling?. Am. J. Physiol.-Heart Circ. Physiol. 320 (1), H90–h94. doi:10.1152/ajpheart.00570.2020
Westermann, D., Lindner, D., Kasner, M., Zietsch, C., Savvatis, K., Escher, F., et al. (2011). Cardiac Inflammation Contributes to Changes in the Extracellular Matrix in Patients with Heart Failure and normal Ejection Fraction. Circ. Heart Fail. 4 (1), 44–52. doi:10.1161/circheartfailure.109.931451
Wilkinson, P. F., Farrell, F. X., Morel, D., Law, W., and Murphy, S. (2016). Adenosine Signaling Increases Proinflammatory and Profibrotic Mediators through Activation of a Functional Adenosine 2B Receptor in Renal Fibroblasts. Ann. Clin. Lab. Sci. 46 (4), 339–345.
Williams-Pritchard, G., Knight, M., Hoe, L. S., Headrick, J. P., and Peart, J. N. (2011). Essential Role of EGFR in Cardioprotection and Signaling Responses to A1 Adenosine Receptors and Ischemic Preconditioning. Am. J. Physiol.-Heart Circ. Physiol. 300 (6), H2161–H2168. doi:10.1152/ajpheart.00639.2010
Wu, L., Belardinelli, L., Zablocki, J. A., Palle, V., and Shryock, J. C. (2001). A Partial Agonist of the A1-Adenosine Receptor Selectively Slows AV Conduction in guinea Pig Hearts. Am. J. Physiol.-Heart Circ. Physiol. 280 (1), H334–H343. doi:10.1152/ajpheart.2001.280.1.H334
Xi, J., McIntosh, R., Shen, X., Lee, S., Chanoit, G., Criswell, H., et al. (2009). Adenosine A2A and A2B Receptors Work in Concert to Induce a strong protection against Reperfusion Injury in Rat Hearts. J. Mol. Cell Cardiol. 47 (5), 684–690. doi:10.1016/j.yjmcc.2009.08.009
Xiang, F., Huang, Y.-s., Zhang, D.-x., Chu, Z.-g., Zhang, J.-p., and Zhang, Q. (2010a). Adenosine A1receptor Activation Reduces Opening of Mitochondrial Permeability Transition Pores in Hypoxic Cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 37 (3), 343–349. doi:10.1111/j.1440-1681.2009.05300.x
Xiang, F., Huang, Y.-s., Zhang, D.-x., Chu, Z.-g., Zhang, J.-p., and Zhang, Q. (2010b). Adenosine A1receptor Activation Reduces Opening of Mitochondrial Permeability Transition Pores in Hypoxic Cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 37 (3), 343–349. doi:10.1111/j.1440-1681.2009.05300.x
Xiong, B., Li, M., Xiang, S., and Han, L. (2018). A1AR-mediated Renal protection against Ischemia/reperfusion Injury Is Dependent on HSP27 Induction. Int. Urol. Nephrol. 50 (7), 1355–1363. doi:10.1007/s11255-018-1797-x
Xu, J., Yang, Q., Zhang, X., Liu, Z., Cao, Y., Wang, L., et al. (2019). Endothelial Adenosine Kinase Deficiency Ameliorates Diet-Induced Insulin Resistance. J. Endocrinol. 242 (2), 159–172. doi:10.1530/joe-19-0126
Xu, M. H., Gong, Y. S., Su, M. S., Dai, Z. Y., Dai, S. S., Bao, S. Z., et al. (2011). Absence of the Adenosine A2A Receptor Confers Pulmonary Arterial Hypertension and Increased Pulmonary Vascular Remodeling in Mice. J. Vasc. Res. 48 (2), 171–183. doi:10.1159/000316935
Xu, X., Fassett, J., Hu, X., Zhu, G., Lu, Z., Li, Y., et al. (2008). Ecto-5′-Nucleotidase Deficiency Exacerbates Pressure-Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Hypertension 51 (6), 1557–1564. doi:10.1161/hypertensionaha.108.110833
Xu, Y., Wang, Y., Yan, S., Yang, Q., Zhou, Y., Zeng, X., et al. (2017a). Regulation of Endothelial Intracellular Adenosine via Adenosine Kinase Epigenetically Modulates Vascular Inflammation. Nat. Commun. 8 (1), 943. doi:10.1038/s41467-017-00986-7
Xu, Y., Wang, Y., Yan, S., Zhou, Y., Yang, Q., Pan, Y., et al. (2017b). Intracellular Adenosine Regulates Epigenetic Programming in Endothelial Cells to Promote Angiogenesis. EMBO Mol. Med. 9 (9), 1263–1278. doi:10.15252/emmm.201607066
Yang, X., Xin, W., Yang, X.-M., Kuno, A., Rich, T. C., Cohen, M. V., et al. (2011). A2Badenosine Receptors Inhibit Superoxide Production from Mitochondrial Complex I in Rabbit Cardiomyocytes via a Mechanism Sensitive toPertussistoxin. Br. J. Pharmacol. 163 (5), 995–1006. doi:10.1111/j.1476-5381.2011.01288.x
Yang, Z., Day, Y.-J., Toufektsian, M.-C., Xu, Y., Ramos, S. I., Marshall, M. A., et al. (2006). Myocardial Infarct-Sparing Effect of Adenosine A 2A Receptor Activation Is Due to its Action on CD4 + T Lymphocytes. Circulation 114 (19), 2056–2064. doi:10.1161/circulationaha.106.649244
Yegutkin, G. G. (2008). Nucleotide- and Nucleoside-Converting Ectoenzymes: Important Modulators of Purinergic Signalling cascade. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1783 (5), 673–694. doi:10.1016/j.bbamcr.2008.01.024
Zhang, H., Zhong, H., Everett, T. H., Wilson, E., Chang, R., Zeng, D., et al. (2014). Blockade of A2B Adenosine Receptor Reduces Left Ventricular Dysfunction and Ventricular Arrhythmias 1 Week after Myocardial Infarction in the Rat Model. Heart Rhythm 11 (1), 101–109. doi:10.1016/j.hrthm.2013.10.023
Zhou, P., and Pu, W. T. (2016). Recounting Cardiac Cellular Composition. Circ. Res. 118(3), 368–370. doi:10.1161/CIRCRESAHA.116.308139
Keywords: adenosine, adenosine receptor, preserved ejection fraction heart failure, cardiac comorbidities, cardiac fibrosis and hypertrophy, endothelial dysfunction
Citation: Campos-Martins A, Bragança B, Correia-de-Sá P and Fontes-Sousa AP (2021) Pharmacological Tuning of Adenosine Signal Nuances Underlying Heart Failure With Preserved Ejection Fraction. Front. Pharmacol. 12:724320. doi: 10.3389/fphar.2021.724320
Received: 12 June 2021; Accepted: 04 August 2021;
Published: 20 August 2021.
Edited by:
Eliot Ohlstein, Drexel University, United StatesReviewed by:
Zhuoming Li, Sun Yat-Sen University, ChinaCopyright © 2021 Campos-Martins, Bragança, Correia-de-Sá and Fontes-Sousa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paulo Correia-de-Sá, ZmFybWFjb2xAaWNiYXMudXAucHQ=; Ana Patrícia Fontes-Sousa, YXBzb3VzYUBpY2Jhcy51cC5wdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.