 Linyi Li1,2,3
Linyi Li1,2,3 Yunyun Yang1,2,3Huina Zhang1,2,3Yunhui Du1,2,3Xiaolu Jiao1,2,3Huahui Yu1,2,3Yu Wang1,2,3Qianwen Lv1,2,3Fan Li1,2,3Qiuju Sun1,2,3
Yunyun Yang1,2,3Huina Zhang1,2,3Yunhui Du1,2,3Xiaolu Jiao1,2,3Huahui Yu1,2,3Yu Wang1,2,3Qianwen Lv1,2,3Fan Li1,2,3Qiuju Sun1,2,3 Yanwen Qin1,2,3*
Yanwen Qin1,2,3*- 1The Key Laboratory of Upper Airway Dysfunction-Related Cardiovascular Diseases, Beijing Anzhen Hospital, Beijing Institute of Heart, Lung and Blood Vessel Diseases, Capital Medical University, Beijing, China
- 2The Key Laboratory of Remodeling-Related Cardiovascular Diseases, Beijing Anzhen Hospital, Ministry of Education, Capital Medical University, Beijing, China
- 3Beijing Institute of Heart, Lung and Blood Vessel Disease, Beijing, China
Background: Endothelial barrier dysfunction plays a key role in atherosclerosis progression. The primary pathology of obstructive sleep apnea-hypopnea syndrome is chronic intermittent hypoxia (IH), which induces reactive oxygen species (ROS) overproduction, endothelial barrier injury, and atherosclerosis. Salidroside, a typical pharmacological constituent of Rhodiola genus, has documented antioxidative, and cardiovascular protective effects. However, whether salidroside can improve IH-aggravated endothelial barrier dysfunction and atherosclerosis has not been elucidated.
Methods and results: In normal chow diet-fed ApoE−/− mice, salidroside (100 mg/kg/d, p. o.) significantly ameliorated the formation of atherosclerotic lesions and barrier injury aggravated by 7-weeks IH (21%–5%–21%, 120 s/cycle). In human umbilical vein endothelial cells (HUVECs), exposure to IH (21%–5%–21%, 40 min/cycle, 72 cycles) decreased transendothelial electrical resistance and protein expression of vascular endothelial cadherin (VE-cadherin) and zonula occludens 1. In addition, IH promoted ROS production and activated ras homolog gene family member A (RhoA)/Rho-associated protein kinase (ROCK) pathway. All of these effects of IH were reversed by salidroside. Similar to salidroside, ROCK-selective inhibitors Y26732, and Fasudil protected HUVECs from IH-induced ROS overproduction and endothelial barrier disruption. Furthermore, salidroside increased intracellular cAMP levels, while the PKA-selective inhibitor H-89 attenuated the effects of salidroside on IH-induced RhoA/ROCK suppression, ROS scavenging, and barrier protection.
Conclusion: Our findings demonstrate that salidroside effectively ameliorated IH-aggravated endothelial barrier injury and atherosclerosis, largely through the cAMP/PKA/RhoA signaling pathway.
Introduction
Atherosclerotic cardio- or cerebrovascular diseases are common causes of morbidity and mortality worldwide (Gibbons et al., 2021). Atherosclerosis is the pathological basis of peripheral arterial disease, coronary heart disease, and stroke. In addition to traditional risk factors such as hereditary susceptibility, aging, hypertension, obesity, diabetes, and smoking, obstructive sleep apnea-hypopnea syndrome (OSA) that affect up to 100 million people worldwide has emerged as a new significant and independent risk factor for atherosclerosis (Ali et al., 2014; Souza et al., 2021). OSA is characterized by repetitive episodes of hypopnea or apnea due to upper airway collapse during sleep (Lévy et al., 2015). OSA prevalence in patients with coronary heart disease ranges from 30 to 58% (Bradley and Floras, 2009). OSA causes endothelial dysfunction (Hoyos et al., 2015; Khalyfa et al., 2016), unstable plaque characteristics (Trzepizur et al., 2014; Konishi et al., 2019), and atherosclerosis (Wattanakit et al., 2008; Ma et al., 2016; Zhou et al., 2017). Recently, one of the largest studies in this area to date (n = 2009) found that OSA is independently associated with increased carotid intima-media thickness in a dose-responsive manner (Souza et al., 2021), reinforcing the role of OSA as a potential nontraditional risk factor for atherosclerosis.
Chronic intermittent hypoxia (CIH), a main feature of OSA, is characterized by repeated circulating hypoxemia and reoxidation (Lévy et al., 2015). CIH induces atherosclerosis in murine models and plays a critical role in OSA-associated cardiovascular morbidities (Drager et al., 2013; Gautier-Veyret et al., 2013; Gileles-Hillel et al., 2014). Endothelial barriers are crucial for maintaining vascular homeostasis. In atherosclerosis, endothelial barrier function is impaired, which triggers the formation of atherosclerotic plaques (Sun et al., 2011; Döring et al., 2017; Pi et al., 2018). CIH has been shown to increase vascular permeability and endothelial barrier dysfunction (Makarenko et al., 2014). However, whether CIH destroys vascular endothelial barriers to promote atherosclerosis remains unknown.
CIH can reportedly increase reactive oxygen species (ROS) generation, which subsequently cause vascular endothelial cadherin (VE-cadherin) cleavage, endothelial barrier injury, and dysfunction (Makarenko et al., 2014; Harki et al., 2021). Ras homolog gene family member A (RhoA) and its downstream Rho-associated protein kinase (ROCK) play crucial roles in both oxidative stress and endothelial barrier function (Shimokawa and Satoh, 2015; Komarova et al., 2017; Dee et al., 2019; Cong and Kong, 2020). cAMP/protein kinase A (PKA) signaling is a potent protective pathway to stabilize the endothelial barrier in response to oxidative stress, and an essential negative regulator of RhoA/ROCK (Qiao et al., 2003; Cong and Kong, 2020).
Salidroside is a typical pharmacological constituent found in medicinal plants under the Rhodiola genus. In addition to their anti-fatigue and anti-hypoxia roles in traditional medicine, Rhodiola extract, and salidroside also displayed medicinal properties as anti-cardiovascular diseases agents, which was mainly attributed to their antioxidant effects (Li et al., 2017; Zhao et al., 2021). Rhodiola crenulata exerts protective effects on hypoxia/high glucose-induced endothelial dysfunction and CIH-induced cardiac apoptosis (Lai et al., 2015; Chang et al., 2018; Huang et al., 2020). Rhodiola rosea extract attenuated pulmonary hypertension in chronic hypoxic rats (Kosanovic et al., 2013) and inhibited atherosclerosis formation in high fat diet-fed rabbits (Shen et al., 2008). Salidroside exhibited activity similar to that of Rhodiola extract. Salidroside improved CIH-mediated myocardial cell apoptosis (Zhong et al., 2010; Lai et al., 2014), chronic persistent hypoxia-induced pulmonary hypertension (Chen et al., 2016) and high fat diet-related atherosclerosis (Xing et al., 2015; Bai et al., 2020; Song et al., 2021) in animal models. Moreover, salidroside suppresses oxidative stress-induced HUVECs cell injury (Zhao et al., 2013; Zhu et al., 2016). Our previous study also demonstrated that salidroside could improve homocysteine-induced endothelium-dependent relaxation dysfunction by reducing oxidative stress (Leung et al., 2013). However, the effect of salidroside on endothelial barrier and CIH-aggravated atherosclerosis has not been reported. Wang et al. found that salidroside attenuated inflammatory responses by inhibiting the RhoA/ROCK pathway in vascular smooth muscle cells (Wang et al., 2021). Guan et al. reported that salidroside attenuated hydrogen peroxide-induced cell damage by elevating cAMP levels (Guan et al., 2011). Therefore, in the present study, we hypothesized that salidroside protects the integrity of endothelial barriers and alleviates CIH-aggravated atherosclerosis through effects on cAMP/PKA/RhoA signaling.
Materials and Methods
Reagents
The following drugs were used: salidroside (98% purity; Sigma-Aldrich, St. Louis, MO, United States), H-89 dihydrochloride hydrate (PKA-specific inhibitor; Sigma-Aldrich), Y-27632 dihydrochloride (ROCK-specific inhibitor; Tocris Bioscience, Bristol, United Kingdom), and Fasudil hydrochloride (ROCK-specific inhibitor, Tocris Bioscience). All drugs were freshly dissolved in medium at the beginning of each experiment. The following primary antibodies were purchased from the cited commercial sources: zonula occludens 1 (ZO-1, Invitrogen, Carlsbad, CA, United States), VE-cadherin (Santa Cruz Biotechnology, Santa Cruz, CA, United States), CD31 (Invitrogen), ROCK (Abcam, Cambridge, United Kingdom), RhoA (Invitrogen and Abcam), and GAPDH (Abcam). FITC-conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Abcam.
Experiments in ApoE-/- Mice
All animal handling complied with the standard animal welfare regulations of Capital Medical University (Beijing, China). The Animal Subjects Committee of Capital Medical University approved the animal study protocol. Animals were randomly assigned to the experimental groups. Ten-week-old male ApoE-/- mice were purchased from HFK Bioscience (Beijing, China). All animals were fed a normal chow diet and maintained in a controlled environment with 12-h light/dark cycles, temperature of 22 ± 2°C, and humidity of 50 ± 2%. To deliver CIH, mice were housed in an OxyCycler A84 System (BioSpherix, Redfield, NY, United States). Briefly, a gas control system regulated the room airflow (N2 and O2). A series of programs and flow regulators enabled manipulation of the fraction of inspired O2 from 21 to 5.0% over a 90-s period, followed by rapid reoxygenation to normal air levels via a burst of 100% O2 in the succeeding 30-s period. Hypoxic events occurred at a rate of one event per 120 s throughout the 12-h light period. During the 12-h dark period, CIH animals were maintained in a normoxic environment. Non-CIH mice were exposed to normoxia at all times. Mice were randomly assigned to one of three groups (n = 8 in each group): 1) control, mice not subjected to CIH, N.S., p. o.; 2) CIH, mice subjected to CIH, N.S., p. o.; and 3) CIH plus salidroside (CIH + Sali), mice subjected to CIH and salidroside (100 mg/kg/d), p. o. After 7 weeks, all mice were anesthetized with pentobarbital sodium and arterial blood were collected by cardiac puncture from the left ventricle. Then mice were perfused with ice-cold saline solution and their ascending aortas were collected for Oil Red O staining. Plaque sizes were quantified using Image-Pro Plus software (Media Cybernetic, Tokyo, Japan). Serial cross-sections (8 μm) of the heart throughout the entire aortic valve area were cut with a cryostat (CM 1900; Leica, Wetzlar, Germany). Sections stained with VE-cadherin (1:50; Cat. No. Sc-9989, Santa Cruz), CD31 (1:50; Cat. No. PA5-16301, Invitrogen), ROCK (1:100; Cat. No. 39749, Abcam), or RhoA (1:50; Cat. No. PA5-87403, Invitrogen) were digitally scanned (Pannoramic DESK, P-MIDI, P250; 3DHISTECH, Budapest, Hungary) to obtain images for analysis by a previously described method (Yang et al., 2020). The methods for oral glucose tolerance test, insulin tolerance test, and biochemical analysis are included in the Supplementary Material.
Cell Culture and Treatment
All experiments using human umbilical vein endothelial cells (HUVECs) were approved by the Medical Ethics Committee of Beijing Anzhen Hospital (Approval No. 2017005) and conducted in accordance with guidelines of the Declaration of Helsinki. Written informed consent was obtained from all participating donors. Human umbilical cord veins were donated by the Maternal and Child Care Service Centre in Beijing, China. HUVECs were isolated, purified, and identified as described previously (Qin et al., 2012). Cells were cultured in VascuLife Basal Medium (Lifeline Cell Technology, Frederick, MD) supplemented with 2% fetal bovine serum, 5 ng/ml recombinant human epidermal growth factor, 10 mM l-glutamine, 1 mg/ml hydrocortisone hemisuccinate, 0.75 U/mL heparin sulfate, 0.2% EnGS (Lifeline Cell Technology), 104 U/mL penicillin, and 104 U/mL streptomycin (HyClone, Logan, UT) in humidified incubator with 95% air (v/v) and 5% CO2 (v/v) at 37°C. HUVECs were used from passages 2–5.
The intermittent hypoxia (IH) protocol consisted of alternating cycles of 20-min hypoxia (1% O2 and 5% CO2) and 20-min reoxygenation (21% O2 and 5% CO2), using a BioSpherix OxyCycler C42 system (BioSpherix, Redfield, NY, United States). The total duration of one cycle was 40 min. Cultured HUVECs were pretreated with the indicated concentration of drugs for 2 h and then exposed to the indicated number of cycles of IH.
Measurement of Transendothelial Electrical Resistance
HUVECs were seeded in the top chambers of 24-well Transwell plates (8.0-µm pore size; Cat. No. 35-3097; Becton-Dickinson, Franklin Lakes, NJ, United States) at a density of 1 × 104 cells/well. Volumes of culture media in the top and lower chambers were 300 and 700 μL, respectively. TEER was measured using a Millicell-ERS (MERS00002; Millipore, Burlington, MA, United States), as previously described (Li et al., 2015). Values for blank wells and cell wells represented the blank resistance (R blank) and sample resistance (R Sample), respectively. The following calculation was performed: TEER = (R Sample)–(R blank). The effect of IH on TEER was determined in HUVECs after 9, 18, 36, 72, and 108 cycles of IH exposure. The effect of drugs on permeability was determined in HUVECs pretreated with salidroside (10 µM or 100 µM), H-89 (10 μM), Y27632 (10 µM), or Fasudil (10 µM) for 2 h before exposure to either normoxia or 72 cycles of IH.
Measurement of HUVEC Permeability to FITC-Dextran
HUVECs were seeded in the top chambers of 24-well Transwell plates (0.4-µm pore size polyester membrane inserts; Corning, Corning, NY, United States) at a density of 1 × 104 cells/well. Volumes of culture media in the top and lower chambers were 200 and 700 μL, respectively. Permeability was measured by adding 1 mg/ml FITC-dextran (70 kDa; Sigma-Aldrich) immediately after IH exposure. Following a 20-min incubation with FITC-dextran, 100 µL of medium was aspirated from each of the lower chambers into wells of black 96-well microplates (Nunc, Roskilde, Denmark). The fluorescence value of each well was determined by an EnSpire 2300 Multimode Plate Reader (PerkinElmer, Waltham, MA, United States) at a 490-nm excitation wavelength and 525-nm emission wavelength. The effect of IH on permeability of HUVECs to FITC-dextran was determined after 9, 18, 36, 72, and 108 cycles of IH exposure. The effect of salidroside on permeability was determined in HUVECs pretreated with salidroside (10 µM or 100 µM) for 2 h before exposure to either normoxia or 72 cycles of IH.
Assay of ROS Production
Intracellular ROS was detected by fluorescence microscopy and quantitated by a plate reader using 2ʹ,7ʹ-dichlorofluorescein diacetate (DCFH-DA, Sigma-Aldrich) as the probe, as previously described (Naha et al., 2010). HUVECs were cultured in black 96-well microplates (Nunc) at a density of 4 × 105 cells/mL in 100 µL of medium. After exposure to drugs under normoxia or IH, microplates were washed with 100 µL/well of phosphate-buffered saline (PBS) and then 100 µL of 10 µM DCFH-DA was added to each well. After incubating the microplates at 37°C for 20 min and washing three times with PBS, an EnSpire 2300 plate reader was used to quantify the fluorescence of each well at a 490-nm excitation wavelength and 525-nm emission wavelength. Fluorescence images were obtained with a Ni-U Nikon Upright Microscope equipped with a DS-Ri2 color charge-coupled device (Nikon, Tokyo, Japan). The effect of each drug on ROS production was determined in HUVECs pretreated with salidroside (10 µM or 100 µM), Tiron (10 µM), H-89 (10 µM), Y27632 (10 µM), or Fasudil (10 µM) for 2 h before exposure to either normoxia or 72 cycles of IH.
GST Pull-Down Assay to Evaluate RhoA Activity
After experimental treatments, HUVECs were lysed and cell lysates were collected. The supernatant was centrifuged at 4°C at 700 g for 10 min. The resulting protein was quantified by bicinchoninic acid method, and GST-RBD fusion was added to the total protein samples of each group. After incubation at 4°C for 1 h, protein samples were centrifuged at 1900 g for 3 min. The precipitated protein was GST-bound. Expression of RhoA protein in total and GST-bound samples was detected by western blot using a rabbit monoclonal RhoA antibody (1:1,000; Cat. No. ab187027, Abcam). RhoA protein in the total protein sample was expressed as total RhoA, while RhoA protein in the GST-bound protein sample was expressed as GTP-RhoA. The ratio of GTP-RhoA expression to total RhoA expression reflected the activity level of RhoA in HUVECs.
Measurement of Intracellular cAMP
HUVECs were cultured by seeding 100 µL of cell suspension with a density of 1 × 105 cells/mL in wells of 24-well plates. After exposure to drugs under normoxia or IH, intracellular cAMP was extracted with hydrochloric acid (HCl, 0.1 M). cAMP concentrations were assessed with a cAMP Assay Kit (Cat. No. ab65355, Abcam) according to the manufacturer’s protocol. The effect of drugs on intracellular cAMP was determined in HUVECs pretreated with salidroside (10 µM or 100 µM) for 2 h before exposure to either normoxia or 72 cycles of IH.
Immunostaining for HUVECs
HUVECs were exposed to either normoxia or 72 cycles of IH, and then fixed with methanol for 30 min at −40°C. After blocking with 3% bovine serum albumin for 30 min, cells were incubated overnight with primary antibodies against VE-cadherin (1:100; Cat. No. 36-1900, Invitrogen) and ZO-1 (1:100; Cat. No. 33-9100, Invitrogen). The following day, cells were washed three times with PBS and incubated with FITC-conjugated anti-mouse and TRITC-conjugated anti-rabbit secondary antibodies (1:200) for 1 h at room temperature. Images of immunofluorescence staining were acquired with a Leica TCS SP2 confocal laser-scanning microscope.
Western Blot Analysis
To extract protein from HUVECs, a protein extraction kit containing protease inhibitors and a protein phosphatase inhibitor cocktail was used (Thermo Fisher Scientific). Equal amounts of protein (30 µg/lane) were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Blots were probed overnight at 4°C with primary antibodies (1:1,000), washed with Tris-buffered saline containing Tween 20, and incubated with secondary antibodies (1:10,000; ZSGB-BIO, Beijing, China) for 1 h at room temperature. Finally, blots were washed, incubated with SuperSignal™ WestFemto Maximum Sensitivity Substrate (Thermo Fisher Scientific), and analyzed using a ChemiDoc™ Touch Imaging System (Bio-Rad, Hercules, CA, United States).
Statistical Analysis
Data were analyzed using Prism 5.0 software (GraphPad Software, San Diego, CA) and are presented as mean ± standard deviation (SD). In all cases, the results of at least three independent experiments were used. Statistical comparisons between two groups were performed using Student’s t-test. Multiple comparison tests used one-way analysis of variance with Bonferroni’s procedure. Values of p < 0.05 were considered statistically significant.
Results
Salidroside Alleviated Atherosclerotic Lesion Formation in CIH-Exposed ApoE-/- Mice
Analysis of Oil Red O staining revealed a significant increasein lipid accumulation (p < 0.01) in aortic sinus tissue of CIH mice compared with control mice, as well as a significant decrease (p < 0.05) in CIH + Sali mice compared with CIH mice (Figures 1A,B). Salidroside did not alter fasting blood glucose, oral glucose tolerance, fasting blood triglyceride, fasting total blood cholesterol, or insulin sensitivity levels in CIH-exposed ApoE-/- mice (Supplementary Figures S1A–E). To determine the effect of salidroside on endothelial barriers in CIH-exposed ApoE-/- mice, immunohistochemistry, and immunofluorescence double staining was conducted to measure expression levels of VE-cadherin, CD31, and ROCK in aortic plaques. As shown in Figures 1A,C–E, a significant decrease in VE-cadherin (p < 0.001) and significant increase of ROCK (p < 0.01) were observed in the CIH group compared with the control group. Meanwhile, salidroside significantly increased VE-cadherin (p < 0.001) and significantly attenuated ROCK expression (p < 0.01).
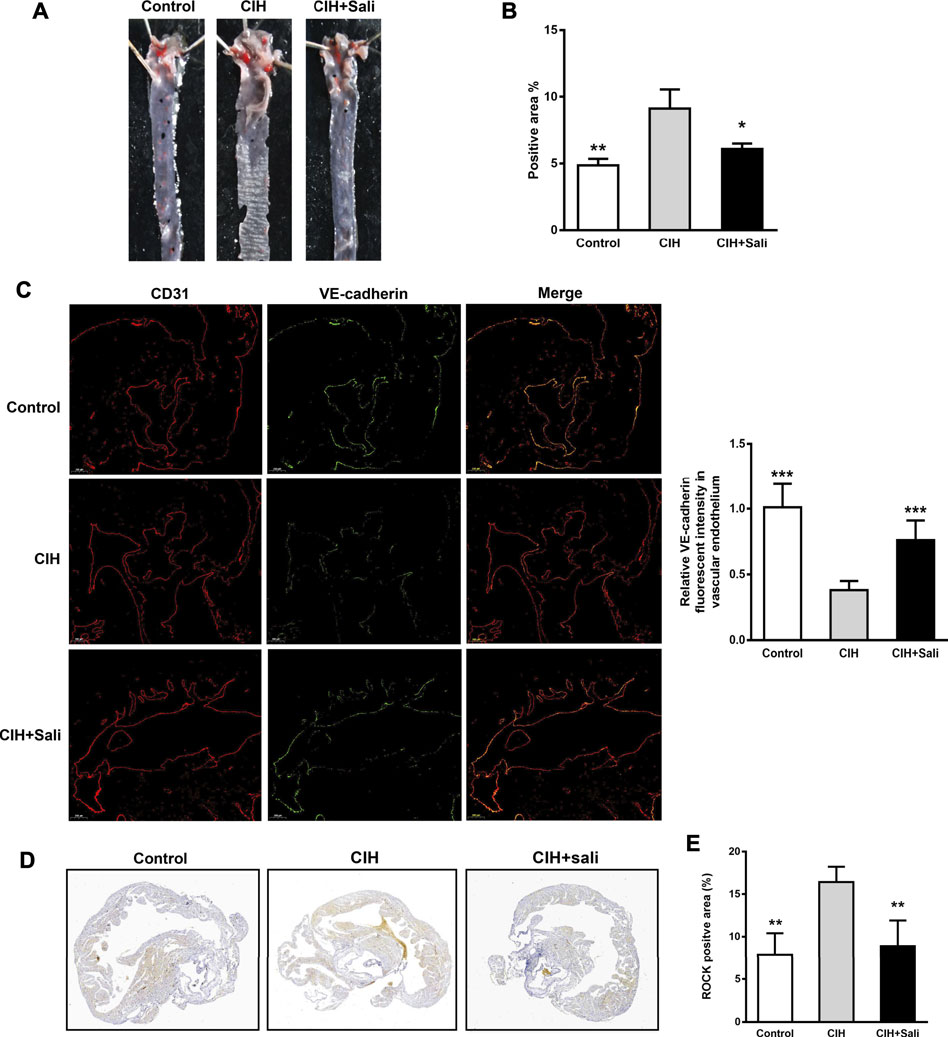
FIGURE 1. Salidroside attenuated CIH-aggravated atherosclerosis in ApoE-/- mice. Male ApoE-/- mice were randomly divided into normoxia (control), CIH, and CIH + Sali groups. After experimental intervention for 7 weeks, mice were sacrificed under anesthesia (A) Representative images of aortic Oil Red O staining (B) Quantitative summary of atherosclerotic lesions in aortas (n = 4) (C) Immunofluorescence staining of VE-cadherin and CD31 and relative fluorescent intensity of VE-cadherin in the endothelium of aortic roots (5 × magnification, n = 3 sections per tissue, at least three analysis sites per slide) (D) Plaques of aortic roots were stained with ROCK (2 × magnification) (E) Quantitative summary of ROCK expression in aortic root plaques (n = 4). CIH, chronic intermittent hypoxia; Sali, salidroside; VE-cadherin, vascular endothelial cadherin; ROCK, Rho-associated protein kinase. All data are presented as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with CIH group.
Salidroside Ameliorated IH-Induced Endothelial Barrier Dysfunction in HUVECs
Endothelial barrier function was assayed by measuring TEER and permeability to FITC-dextran in HUVECs immediately after IH exposure. Compared with the normoxia group, IH exposure significantly decreased TEER (p < 0.001) and increased permeability to FITC-dextran (p < 0.001) of HUVECs in a time-dependent manner, reaching a plateau with 72 cycles of IH (Figures 2A,B). Therefore, the following studies were performed on HUVECs exposed to 72 cycles of IH. Pretreatment of HUVECs with salidroside (10 µM or100 µM) dose-dependently and significantly increased TEER (p < 0.01) and decreased permeability to FITC-dextran (p < 0.01) compared with the IH group (Figures 2C,D).
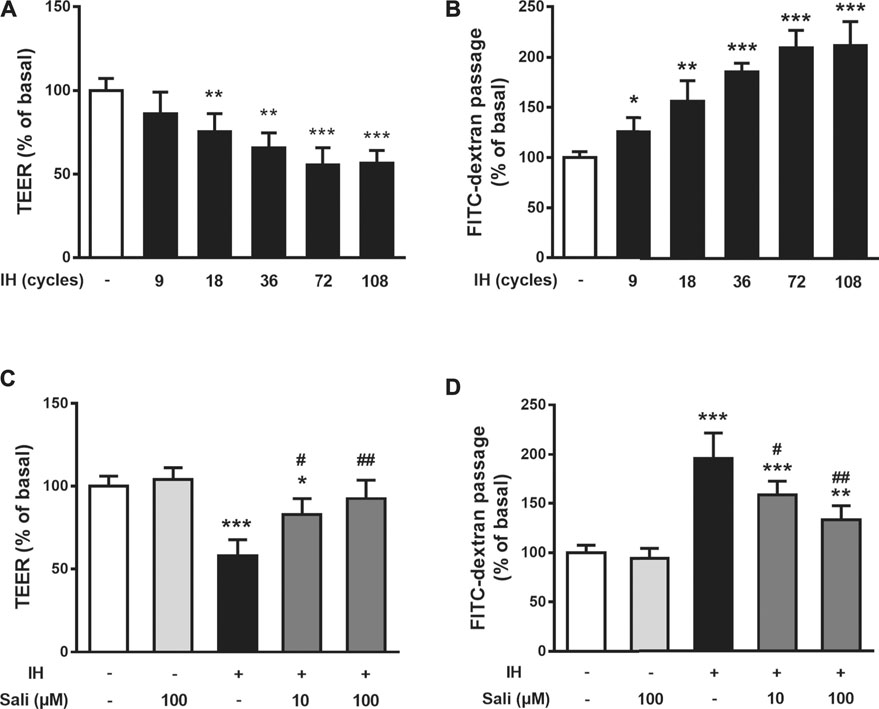
FIGURE 2. Salidroside ameliorated IH-induced endothelial barrier dysfunction in HUVECs. The effects of IH on TEER (A) and permeability to FITC-dextran (B) were determined in HUVECs after 9, 18, 36, 72, 108 cycles of IH exposure. Effects of Sali on TEER (C) and permeability to FITC-dextran (D) were determined in HUVECs pretreated with Sali (10 µM or 100 µM) for 2 h before exposure to either normoxia or 72 cycles of IH. IH, intermittent hypoxia [one cycle includes reducing O2 for a 20-min hypoxia period (5% O2 and 5% CO2) followed by 20-min reoxygenation (21% O2 and 5% CO2)]; Sali, salidroside; TEER, transendothelial electrical resistance. All data are presented as mean ± SD. #p < 0.05 and ##p < 0.01 compared with IH group; *p < 0.05, **p < 0.01, and ***p < 0.001 compared with normoxia group.
Salidroside Ameliorated the Suppressive Effect of IH on VE-Cadherin and ZO-1 Expression in HUVECs
As shown in Figures 3A,B, IH-exposed HUVECs displayed obvious disruptions of VE-cadherin and ZO-1 by immunofluorescence staining that were dose-dependently ameliorated by salidroside pretreatment. Similarly, western blot assay results showed that IH exposure significantly reduced protein expression levels of VE-cadherin (p < 0.001) and ZO-1 (p < 0.001), but this effect was largely reversed by salidroside pretreatment (p < 0.001, Figures 3C–E).
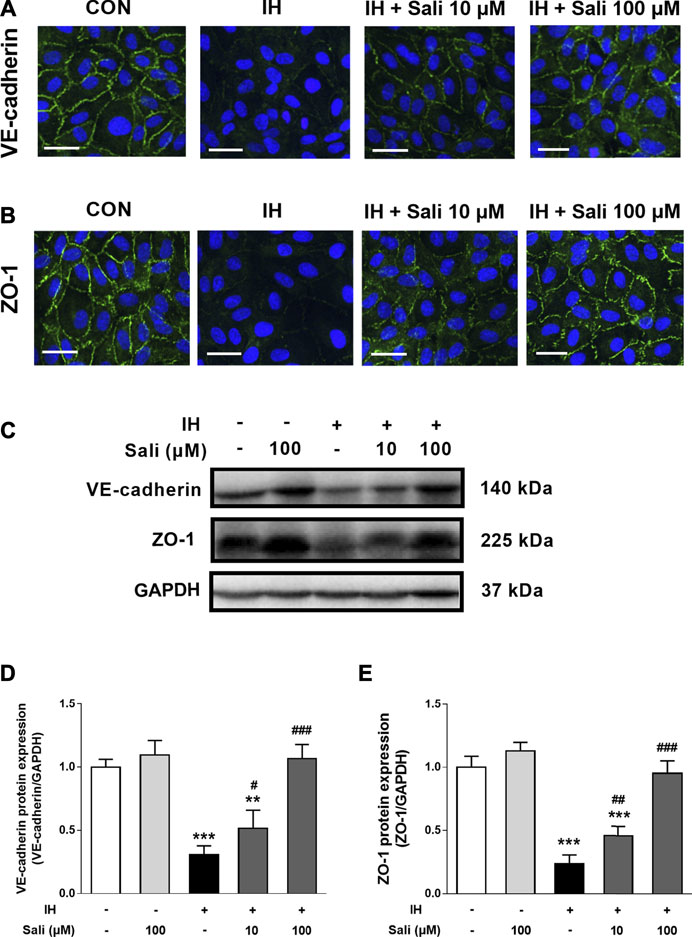
FIGURE 3. Salidroside ameliorated the suppressive effect of IH on VE-cadherin and ZO-1 expression in HUVECs. HUVECs were pretreated with Sali (10 µM or 100 µM) for 2 h before exposure to 72 cycles of IH (A) Immunofluorographs of VE-cadherin (20 × magnification; Bar = 50 µm) and (B) ZO-1 (20 × magnification; Bar = 50 μm) (C) VE-cadherin and ZO-1 protein levels were measured by western blot analysis (D–E) Relative protein levels of VE-cadherin and ZO-1 were quantified by densitometry analysis. IH, intermittent hypoxia; Sali, salidroside; VE-cadherin, vascular endothelial cadherin; ZO-1, zonula occludens 1. All data are presented as mean ± SD. #p < 0.05, ##p < 0.01, and ###p < 0.001 compared with IH group; **p < 0.01 and ***p < 0.001 compared with normoxia group.
Salidroside Attenuated IH-Induced ROS Overproduction in HUVECs
ROS play a major role in IH-related endothelial dysfunction. To determine whether salidroside attenuated IH-induced oxidative stress in HUVECs, intracellular ROS production was measured by a fluorescent method. Immunostaining revealed elevated ROS production in the IH group, which was decreased in groups treated with salidroside (10 and 100 μM) or the ROS scavenger Tiron (Figure 4A). Quantification of fluorescence assay results further showed that exposure to 72 cycles of IH significantly increased ROS production (p < 0.001) in HUVECs, while salidroside could dose-dependently and significantly suppress ROS production (p < 0.001) in IH-exposed HUVECs (Figure 4B).
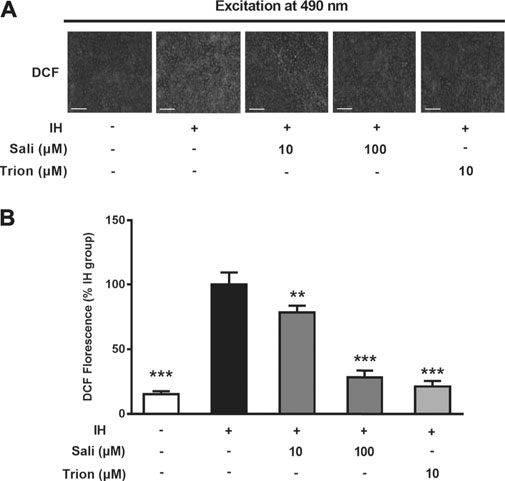
FIGURE 4. Salidroside attenuated IH-induced ROS overproduction in HUVECs. Intracellular ROS levels were determined in HUVECs pretreated with Sali (10 µM or 100 µM) or Tiron (10 µM) for 2 h before exposure to either normoxia or 72 cycles of IH. DCFH-DA was used as a fluorescent probe for ROS. Tiron was used as a positive control (A) Fluorographs of intracellular ROS were acquired with a fluorescence microscope (5 × magnification; Bar = 500 µm) (B) Fluorescence quantification results of intracellular ROS. IH, intermittent hypoxia; Sali, salidroside; DCFH-DA, 2ʹ,7ʹ-dichlorofluorescein diacetate; ROS, reactive oxygen species. All data are presented as mean ± SD. *p < 0.05 and ***p < 0.001 compared with IH group.
Salidroside Elevated Intracellular cAMP in IH-Exposed HUVECs
Endothelial cAMP is one of the most potent endothelial barrier stabilizers. As shown in Figure 5A, IH exposure did not affect cellular cAMP levels in HUVECs. However, salidroside significantly increased cAMP levels up to 2.4-fold compared with the IH group (p < 0.001).
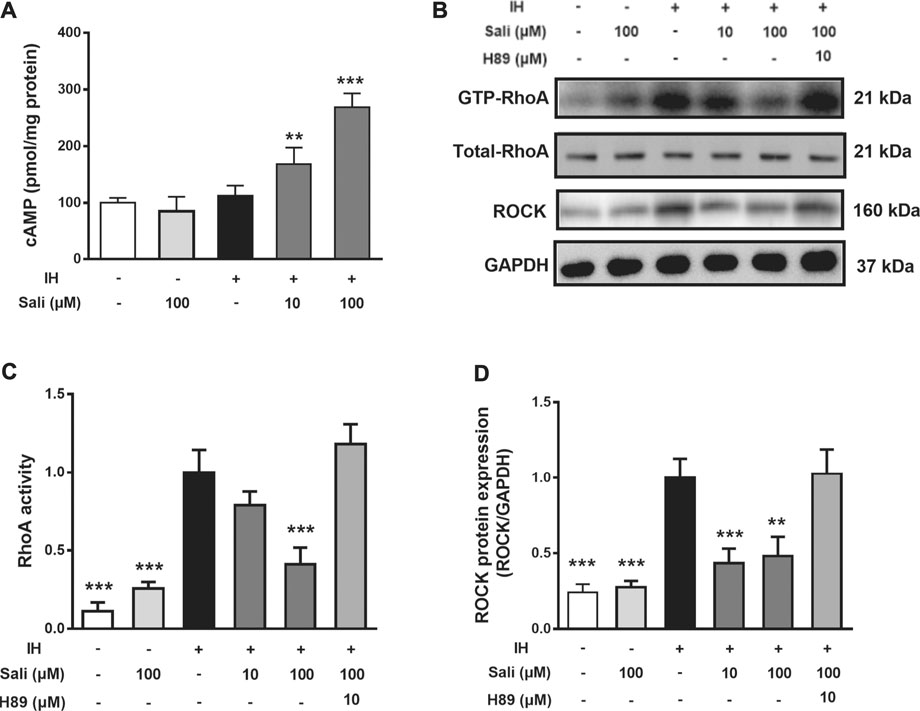
FIGURE 5. Salidroside suppressed IH-induced elevation of RhoA activity and ROCK protein expression in HUVECs through the cAMP/PKA pathway. HUVECs were pretreated with Sali (10 µM or 100 µM) or the PKA inhibitor H-89 for 2 h before exposure to 72 cycles of IH (A) Intracellular cAMP levels, as well as (B) RhoA activity and ROCK protein levels were measured (C–D) Relative levels of RhoA activity and ROCK protein expression were quantified by densitometry analysis. IH, intermittent hypoxia; Sali, salidroside; RhoA, ras homolog gene family member A; ROCK, Rho-associated protein kinase. All data are presented as mean ± SD. *p < 0.05 and ***p < 0.001 compared with IH group.
Salidroside Suppressed IH-Induced Elevation of RhoA Activity and ROCK Protein Expression in HUVECs Through the PKA Pathway
We next examined the effect of salidroside on RhoA activity and ROCK protein expression in HUVECs. Exposure of HUVECs to 72 cycles of IH significantly upregulated RhoA activity (p < 0.001) and ROCK (p < 0.001) expression. Pretreatment of HUVECs with salidroside significantly reduced IH-induced increases in RhoA activity (p < 0.001) and ROCK expression (p < 0.01). The PKA-specific inhibitor H-89 significantly attenuated the effect of salidroside on reducing RhoA/ROCK signaling (p < 0.001, Figures 5B–D).
Salidroside Ameliorated IH-Induced ROS Overproduction and Endothelial Barrier Dysfunction in HUVECs via cAMP/PKA/RhoA Signaling
The ROCK-selective inhibitors Y27632 and Fasudil were employed to further explore the mechanism of salidroside. IH exposure increased ROS production and induced barrier dysfunction in HUVECs, the effects of which were significantly attenuated by salidroside (p < 0.01). Similar to salidroside, pretreatment of HUVECs with Y27632 or Fasudil markedly ameliorated IH-induced ROS generation (p < 0.001) and barrier disruption (p < 0.01 or p < 0.001). Moreover, the inhibitory effects of salidroside on IH-induced ROS accumulation (p < 0.001) and endothelial barrier disruption (p < 0.001) were significantly attenuated by H-89 (Figures 6A,B). Taken together, these results suggest that salidroside ameliorated IH-induced ROS production and barrier dysfunction in HUVECs via the cAMP/PKA/RhoA signaling pathway.
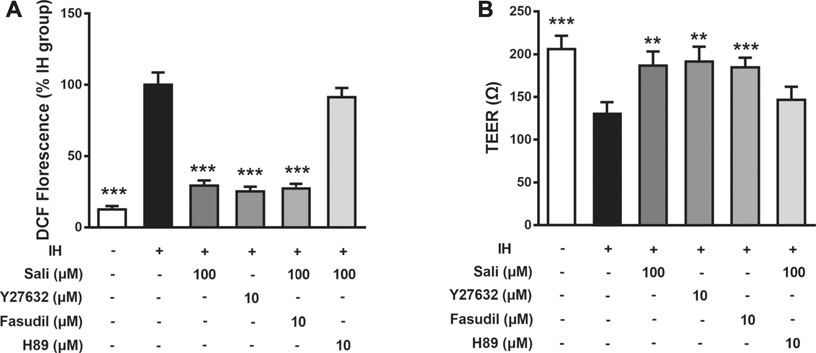
FIGURE 6. Salidroside ameliorated IH-induced ROS overproduction and barrier dysfunction in HUVECs via the cAMP/PKA/RhoA signaling pathway. Effects of drugs on ROS generation (A) and TEER (B) were determined in HUVECs pretreated with Sali (100 µM), H-89 (10 µM), Y27632 (10 µM), or Fasudil (10 µM) for 2 h before exposure to either normoxia or 72 cycles of IH. IH, intermittent hypoxia; Sali, salidroside; ROS, reactive oxygen species; TEER, transendothelial electrical resistance. All data are presented as mean ± SD of three experiments. ***p < 0.001 compared with IH group.
Discussion
The observations of this study can be summarized as follows. Firstly, salidroside ameliorated CIH-induced atherosclerosis progression and barrier injury in normal chow diet-fed ApoE-/- mice. Secondly, salidroside inhibited the deleterious effect of IH on barrier function of HUVECs through effects on cAMP/PKA/RhoA signaling. Furthermore, similar to salidroside, the ROCK inhibitors Y27632 and Fasudil protected HUVECs from IH-induced ROS overproduction and endothelial barrier disruption. This research demonstrates for the first time that salidroside has a protective effect on endothelial barrier function via cAMP/PKA/RhoA signaling. These results also suggest that RhoA/ROCK signaling plays important roles in IH-induced oxidative stress and disruption of endothelial integrity, representing a novel mechanism by which sleep breathing disorders exacerbate atherosclerosis.
Clinical and basic studies have demonstrated a causal relationship between CIH and atherosclerosis. Continuous positive airway pressure (CPAP) treatment, which is mainly used to correct CIH in OSA patients, can reduce intima-media thickness and femoral artery wave velocity to improve the early signs of atherosclerosis (Drager et al., 2007; Chen et al., 2017). In addition, CIH promoted atherosclerotic plaque formation in both normal and high cholesterol diet-fed ApoE-/- mice (Drager et al., 2013; Gautier-Veyret et al., 2013; Gileles-Hillel et al., 2014) However, CPAP cannot improve the incidence or mortality rate of cardiovascular diseases in OSA patients, and compliance with CPAP treatment was shown to be very low in actual practice (McEvoy et al., 2016; Peker et al., 2016). Therefore, potential drugs for OSA-related atherosclerotic cardiovascular diseases remain to be further investigated. Salidroside can improve atherosclerosis in high fat diet-fed ApoE-/- mice (Bai et al., 2020; Song et al., 2021). However, the effect of salidroside on CIH-related atherosclerosis has not been reported. Consistent with previous studies, our results indicate that CIH can significantly promote the progression of atherosclerosis in normal chow diet-fed ApoE-/- mice. However, we are the first to report that salidroside can significantly reduce CIH-aggravated plaque progression.
Endothelial barrier dysfunction is the key factor in triggering and aggravating the formation of atherosclerosis. Previous studies showed that endothelial cells in the intima of human atherosclerotic plaques are wider and the endothelial barrier is destroyed, leading to infiltration of various inflammatory cells and the formation of lipid-rich foam cells, a key pathological change of atherosclerosis (Pi et al., 2018). The fundamental basis for barrier function is predominantly maintained by adherens junctions and tight junctions (Komarova et al., 2017). The adherent junction protein VE-cadherin and tight junction protein ZO-1 are important factors to maintain and regulate endothelial barrier function. CIH can reportedly increase vascular permeability and impair endothelial barrier function by VE-cadherin cleavage (Makarenko et al., 2014; Harki et al., 2021). Consistent with previous studies, exposure of HUVECs to IH decreased TEER, increased permeability to FITC-dextran, and suppressed expression of junction proteins VE-cadherin and ZO-1. In vivo, CIH significantly reduced VE-cadherin expression in the aortas of ApoE-/- mice. We found that salidroside significantly improved IH-induced endothelial barrier injury in both ApoE-/- mice and HUVECs, suggesting that salidroside may alleviate CIH-aggravated atherosclerosis by improving endothelial barrier damage, and endothelial barrier dysfunction may be a mechanism by which IH aggravates atherosclerosis.
Oxidative stress caused by ROS overproduction is a common mechanism of endothelial barrier dysfunction in both OSA and atherosclerotic diseases (Lévy et al., 2015). The RhoA/ROCK pathway plays a crucial role in ROS augmentation and endothelial barrier function (Shimokawa and Satoh, 2015; Komarova et al., 2017; Dee et al., 2019; Cong and Kong, 2020). Accumulating evidence indicates that Rho-kinase inhibitors have beneficial effects for the treatment of cardiovascular diseases (Dai et al., 2018; Lei et al., 2020). Exposure to IH activated RhoA/Rho-kinase in the aorta and mesenteric arteries of animal models, which is required for IH-induced arterial remodeling, and hypertension (de Frutos et al., 2010; Lu et al., 2017; Li et al., 2018). However, it remains unknown whether the RhoA/ROCK pathway is involved in IH-induced endothelial barrier dysfunction and, if so, what mechanisms are involved. In the present study, we observed that IH increased ROCK expression in the aortas of ApoE-/- mice, as well as ROS generation, RhoA activity, and ROCK expression in HUVECs. Furthermore, the ROCK-selective inhibitors Y27632 and Fasudil attenuated IH-induced ROS overproduction and endothelial barrier disruption. These results suggest that the RhoA/ROCK pathway is essential for IH-induced ROS augmentation and endothelial barrier dysfunction. Because ROS also activates the RhoA/ROCK pathway in endothelial cells (Lu et al., 2017; Li et al., 2018), IH-induced ROS overproduction likely activates RhoA/ROCK pathway, thus creating a vicious cycle for ROS augmentation and endothelial barrier breakdown. Salidroside treatment reversed the increase of ROCK expression in aortas of chronic IH-exposed ApoE-/- mice. Moreover, similar to the ROCK-selective inhibitors Y26732 and Fasudil, salidroside reduced RhoA/ROCK activation and protected HUVECs from ROS overproduction and endothelial barrier disruption induced by IH. These results suggest that the molecular mechanism by which salidroside improved IH-mediated endothelial barrier dysfunction and atherosclerosis was be related to the RhoA/ROCK signaling pathway.
An increase in endothelial cAMP is one of the most potent triggers to stabilize the endothelial barrier against almost any barrier-compromising stimulus (Schlegel and Waschke, 2014; Radeva and Waschke, 2018). Furthermore, cAMP/PKA is an essential negative regulator of RhoA/ROCK (Qiao et al., 2003; Cong and Kong, 2020). Our results suggest that salidroside significantly increased the cAMP level of IH-exposed HUVEC cells (2.4-fold), whereas the PKA-specific inhibitor H-89 attenuated the RhoA/ROCK inhibition, ROS clearance, and barrier protection effects of salidroside. These findings indicate that cAMP/PKA activation acts upstream of RhoA/ROCK inhibition to elicit the endothelial barrier-protective effect of salidroside.
Salidroside did not affect the glucolipid metabolism or insulin sensitivity of CIH-exposed ApoE-/- mice. Therefore, the beneficial effects of salidroside were not secondary to the improvement of glucolipid metabolism or insulin resistance. Moreover, the present results indicate that salidroside has only a moderate regulatory effect under pathophysiological conditions. Under normal conditions, barrier-forming junction proteins undergo dynamic regulation of assembly and disassembly. During these processes, proper Rho activity is very important to maintain the endothelial barrier in a healthy state (Terry et al., 2010). The application of salidroside did not inhibit RhoA activity below a physiological level, but instead maintained RhoA activity at a medium activation level in IH-treated HUVECs, which could provide reliable evidence for the safety of salidroside. However, it remains far from clinical application. The study provides a reference for future clinical research.
In summary, salidroside exerted significant barrier-protective and anti-atherosclerosis effects against IH via the cAMP/PKA/RhoA signaling pathway (Figure 7). We propose that this evidence supports potential therapeutic actions of salidroside in applications for OSA-associated vascular system protection.
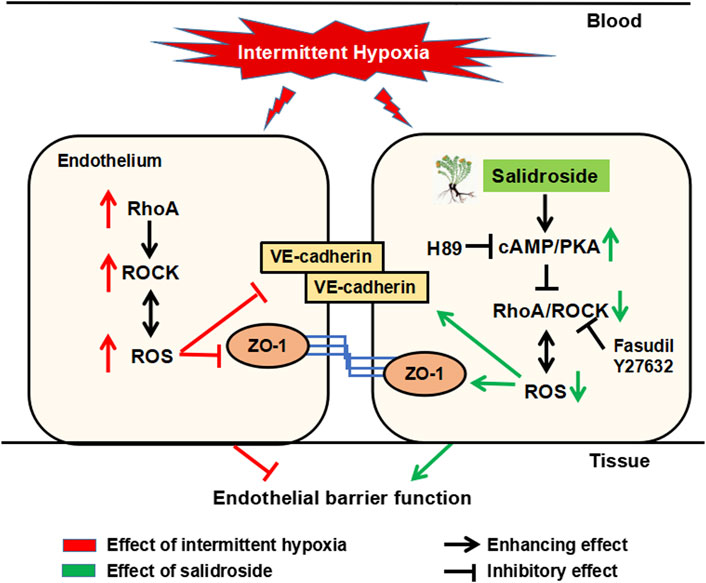
FIGURE 7. Schematic diagram of the potential mechanism by which salidroside protects against IH-induced ROS overproduction and barrier disruption in the endothelium. Salidroside increased intracellular cAMP levels, which subsequently activated PKA in HUVECs. Activated cAMP/PKA suppressed IH-induced elevation of RhoA and ROCK, and thereafter inhibited ROS generation. In turn, reduced ROS production inhibited RhoA/ROCK expression, breaking the vicious cycle of ROS augmentation induced by IH in the endothelium. IH, intermittent hypoxia; ROS, reactive oxygen species; RhoA, ras homolog gene family member A; ROCK, Rho-associated protein kinase; PKA, protein kinase A.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Beijing Anzhen Hospital (Approval No. 2017005). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by The Animal Subjects Committee of Capital Medical University approved the animal study protocol.
Author Contributions
LL designed and performed research, and wrote the paper; YY, HZ, YD, XJ, HY, YW, QL, FL, and QS performed research and analyzed data; YQ designed research.
Funding
This study was supported by the Beijing Talents Fund (No. 2016000021469G193), Natural Science Foundation of China (Grant Nos. 81970224 and 81670331), Beijing Natural Science Foundation (Grant No. 7192030), and the Beijing Billion Talent Project (No. 2017-A-10).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.723922/full#supplementary-material
References
Ali, S. S., Oni, E. T., Warraich, H. J., Blaha, M. J., Blumenthal, R. S., Karim, A., et al. (2014). Systematic Review on Noninvasive Assessment of Subclinical Cardiovascular Disease in Obstructive Sleep Apnea: New Kid on the Block!. Sleep Med. Rev. 18, 379–391. doi:10.1016/j.smrv.2014.01.004
Bai, X., Jia, X., Lu, Y., Zhu, L., Zhao, Y., Cheng, W., et al. (2020). Salidroside-mediated Autophagic Targeting of Active Src and Caveolin-1 Suppresses Low-Density Lipoprotein Transcytosis across Endothelial Cells. Oxid Med. Cel Longev 2020, 9595036. doi:10.1155/2020/9595036
Bradley, T. D., and Floras, J. S. (2009). Obstructive Sleep Apnoea and its Cardiovascular Consequences. Lancet 373, 82–93. doi:10.1016/S0140-6736(08)61622-0
Chang, P. K., Yen, I. C., Tsai, W. C., Chang, T. C., and Lee, S. Y. (2018). Protective Effects of Rhodiola Crenulata Extract on Hypoxia-Induced Endothelial Damage via Regulation of AMPK and ERK Pathways. Int. J. Mol. Sci. 19. doi:10.3390/ijms19082286
Chen, L. D., Lin, L., Lin, X. J., Ou, Y. W., Wu, Z., Ye, Y. M., et al. (2017). Effect of Continuous Positive Airway Pressure on Carotid Intima-media Thickness in Patients with Obstructive Sleep Apnea: A Meta-Analysis. PLoS ONE 12, e0184293. doi:10.1371/journal.pone.0184293
Chen, M., Cai, H., Yu, C., Wu, P., Fu, Y., Xu, X., et al. (2016). Salidroside Exerts Protective Effects against Chronic Hypoxia-Induced Pulmonary Arterial Hypertension via AMPKα1-dependent Pathways. Am. J. Transl Res. 8, 12–27.
Cong, X., and Kong, W. (2020). Endothelial Tight Junctions and Their Regulatory Signaling Pathways in Vascular Homeostasis and Disease. Cell. Signal. 66, 109485. doi:10.1016/j.cellsig.2019.109485
Dai, Y., Luo, W., and Chang, J. (2018). Rho Kinase Signaling and Cardiac Physiology. Curr. Opin. Physiol. 1, 14–20. doi:10.1016/j.cophys.2017.07.005
de Frutos, S., Caldwell, E., Nitta, C. H., Kanagy, N. L., Wang, J., Wang, W., et al. (2010). NFATc3 Contributes to Intermittent Hypoxia-Induced Arterial Remodeling in Mice. Am. J. Physiol. Heart Circ. Physiol. 299, H356–H363. doi:10.1152/ajpheart.00341.2010
Dee, R. A., Mangum, K. D., Bai, X., Mack, C. P., and Taylor, J. M. (2019). Druggable Targets in the Rho Pathway and Their Promise for Therapeutic Control of Blood Pressure. Pharmacol. Ther. 193, 121–134. doi:10.1016/j.pharmthera.2018.09.001
Döring, Y., Noels, H., van der Vorst, E. P. C., Neideck, C., Egea, V., Drechsler, M., et al. (2017). Vascular CXCR4 Limits Atherosclerosis by Maintaining Arterial Integrity: Evidence from Mouse and Human Studies. Circulation 136, 388–403. doi:10.1161/CIRCULATIONAHA.117.027646
Drager, L. F., Bortolotto, L. A., Figueiredo, A. C., Krieger, E. M., and Lorenzi, G. F. (2007). Effects of Continuous Positive Airway Pressure on Early Signs of Atherosclerosis in Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 176, 706–712. doi:10.1164/rccm.200703-500OC
Drager, L. F., Yao, Q., Hernandez, K. L., Shin, M. K., Bevans-Fonti, S., Gay, J., et al. (2013). Chronic Intermittent Hypoxia Induces Atherosclerosis via Activation of Adipose Angiopoietin-like 4. Am. J. Respir. Crit. Care Med. 188, 240–248. doi:10.1164/rccm.201209-1688OC
Gautier-Veyret, E., Arnaud, C., Bäck, M., Pépin, J. L., Petri, M. H., Baguet, J. P., et al. (2013). Intermittent Hypoxia-Activated Cyclooxygenase Pathway: Role in Atherosclerosis. Eur. Respir. J. 42, 404–413. doi:10.1183/09031936.00096512
Gibbons, G. H., Seidman, C. E., and Topol, E. J. (2021). Conquering Atherosclerotic Cardiovascular Disease - 50 Years of Progress. N. Engl. J. Med. 384, 785–788. doi:10.1056/NEJMp2033115
Gileles-Hillel, A., Almendros, I., Khalyfa, A., Zhang, S. X., Wang, Y., and Gozal, D. (2014). Early Intermittent Hypoxia Induces Proatherogenic Changes in Aortic wall Macrophages in a Murine Model of Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 190, 958–961. doi:10.1164/rccm.201406-1149LE
Guan, S., Wang, W., Lu, J., Qian, W., Huang, G., Deng, X., et al. (2011). Salidroside Attenuates Hydrogen Peroxide-Induced Cell Damage through a cAMP-dependent Pathway. Molecules 16, 3371–3379. doi:10.3390/molecules16043371
Harki, O., Tamisier, R., Pépin, J.-L., Bailly, S., Mahmani, A., Gonthier, B., et al. (2021). VE-cadherin Cleavage in Sleep Apnoea: New Insights into Intermittent Hypoxia-Related Endothelial Permeability. Eur. Respir. J., 2004518. doi:10.1183/13993003.04518-2020
Hoyos, C. M., Melehan, K. L., Liu, P. Y., Grunstein, R. R., and Phillips, C. L. (2015). Does Obstructive Sleep Apnea Cause Endothelial Dysfunction? A Critical Review of the Literature. Sleep Med. Rev. 20, 15–26. doi:10.1016/j.smrv.2014.06.003
Huang, L. Y., Yen, I. C., Tsai, W. C., and Lee, S. Y. (2020). Rhodiola Crenulata Suppresses High Glucose-Induced Matrix Metalloproteinase Expression and Inflammatory Responses by Inhibiting ROS-Related HMGB1-TLR4 Signaling in Endothelial Cells. Am. J. Chin. Med. 48, 91–105. doi:10.1142/S0192415X20500056
Khalyfa, A., Kheirandish-Gozal, L., Khalyfa, A. A., Philby, M. F., Alonso-Álvarez, M. L., Mohammadi, M., et al. (2016). Circulating Plasma Extracellular Microvesicle microRNA Cargo and Endothelial Dysfunction in Children with Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 194, 1116–1126. doi:10.1164/rccm.201602-0323OC
Komarova, Y. A., Kruse, K., Mehta, D., and Malik, A. B. (2017). Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 120, 179–206. doi:10.1161/CIRCRESAHA.116.306534
Konishi, T., Kashiwagi, Y., Funayama, N., Yamamoto, T., Murakami, H., Hotta, D., et al. (2019). Obstructive Sleep Apnea Is Associated with Increased Coronary Plaque Instability: an Optical Frequency Domain Imaging Study. Heart Vessels 34, 1266–1279. doi:10.1007/s00380-019-01363-8
Kosanovic, D., Tian, X., Pak, O., Lai, Y. J., Hsieh, Y. L., Seimetz, M., et al. (2013). Rhodiola: an Ordinary Plant or a Promising Future Therapy for Pulmonary Hypertension? a Brief Review. Pulm. Circ. 3, 499–506. doi:10.1086/674303
Lai, M. C., Lin, J. G., Pai, P. Y., Lai, M. H., Lin, Y. M., Yeh, Y. L., et al. (2015). Effects of Rhodiola Crenulata on Mice Hearts under Severe Sleep Apnea. BMC Complement. Altern. Med. 15, 198. doi:10.1186/s12906-015-0698-0
Lai, M. C., Lin, J. G., Pai, P. Y., Lai, M. H., Lin, Y. M., Yeh, Y. L., et al. (2014). Protective Effect of Salidroside on Cardiac Apoptosis in Mice with Chronic Intermittent Hypoxia. Int. J. Cardiol. 174, 565–573. doi:10.1016/j.ijcard.2014.04.132
Lei, S., Peng, F., Li, M. L., Duan, W. B., Peng, C. Q., and Wu, S. J. (2020). LncRNA-SMILR Modulates RhoA/ROCK Signaling by Targeting miR-141 to Regulate Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Physiol. Heart Circ. Physiol. 319, H377–377H391. doi:10.1152/ajpheart.00717.2019
Leung, S. B., Zhang, H., Lau, C. W., Huang, Y., and Lin, Z. (2013). Salidroside Improves Homocysteine-Induced Endothelial Dysfunction by Reducing Oxidative Stress. Evid. Based Complement. Alternat Med. 2013, 679635. doi:10.1155/2013/679635
Lévy, P., Kohler, M., McNicholas, W. T., Barbé, F., McEvoy, R. D., Somers, V. K., et al. (2015). Obstructive Sleep Apnoea Syndrome. Nat. Rev. Dis. Primers 1, 15015. doi:10.1038/nrdp.2015.15
Li, J. R., Zhao, Y. S., Chang, Y., Yang, S. C., Guo, Y. J., and Ji, E. S. (2018). Fasudil Improves Endothelial Dysfunction in Rats Exposed to Chronic Intermittent Hypoxia through RhoA/ROCK/NFATc3 Pathway. PLoS ONE 13, e0195604. doi:10.1371/journal.pone.0195604
Li, L., Hu, J., He, T., Zhang, Q., Yang, X., Lan, X., et al. (2015). P38/MAPK Contributes to Endothelial Barrier Dysfunction via MAP4 Phosphorylation-dependent Microtubule Disassembly in Inflammation-Induced Acute Lung Injury. Sci. Rep. 5, 8895. doi:10.1038/srep08895
Li, Y., Wu, J., Shi, R., Li, N., Xu, Z., and Sun, M. (2017). Antioxidative Effects of Rhodiola Genus: Phytochemistry and Pharmacological Mechanisms against the Diseases. Curr. Top. Med. Chem. 17, 1692–1708. doi:10.2174/1568026617666161116141334
Lu, W., Kang, J., Hu, K., Tang, S., Zhou, X., Xu, L., et al. (2017). The Role of the Nox4-Derived ROS-Mediated RhoA/Rho Kinase Pathway in Rat Hypertension Induced by Chronic Intermittent Hypoxia. Sleep Breath 21, 667–677. doi:10.1007/s11325-016-1449-2
Ma, L., Zhang, J., and Liu, Y. (2016). Roles and Mechanisms of Obstructive Sleep Apnea-Hypopnea Syndrome and Chronic Intermittent Hypoxia in Atherosclerosis: Evidence and Prospective. Oxid Med. Cel Longev 2016, 8215082. doi:10.1155/2016/8215082
Makarenko, V. V., Usatyuk, P. V., Yuan, G., Lee, M. M., Nanduri, J., Natarajan, V., et al. (2014). Intermittent Hypoxia-Induced Endothelial Barrier Dysfunction Requires ROS-dependent MAP Kinase Activation. Am. J. Physiol. Cel Physiol 306, C745–C752. doi:10.1152/ajpcell.00313.2013
McEvoy, R. D., Antic, N. A., Heeley, E., Luo, Y., Ou, Q., Zhang, X., et al. (2016). CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea. N. Engl. J. Med. 375, 919–931. doi:10.1056/NEJMoa1606599
Naha, P. C., Davoren, M., Lyng, F. M., and Byrne, H. J. (2010). Reactive Oxygen Species (ROS) Induced Cytokine Production and Cytotoxicity of PAMAM Dendrimers in J774A.1 Cells. Toxicol. Appl. Pharmacol. 246, 91–99. doi:10.1016/j.taap.2010.04.014
Peker, Y., Glantz, H., Eulenburg, C., Wegscheider, K., Herlitz, J., and Thunström, E. (2016). Effect of Positive Airway Pressure on Cardiovascular Outcomes in Coronary Artery Disease Patients with Nonsleepy Obstructive Sleep Apnea. The RICCADSA Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 194, 613–620. doi:10.1164/rccm.201601-0088OC
Pi, X., Xie, L., and Patterson, C. (2018). Emerging Roles of Vascular Endothelium in Metabolic Homeostasis. Circ. Res. 123, 477–494. doi:10.1161/CIRCRESAHA.118.313237
Qiao, J., Huang, F., and Lum, H. (2003). PKA Inhibits RhoA Activation: a protection Mechanism against Endothelial Barrier Dysfunction. Am. J. Physiol. Lung Cel Mol. Physiol. 284, L972–L980. doi:10.1152/ajplung.00429.2002
Qin, W., Lu, W., Li, H., Yuan, X., Li, B., Zhang, Q., et al. (2012). Melatonin Inhibits IL1β-induced MMP9 Expression and Activity in Human Umbilical Vein Endothelial Cells by Suppressing NF-Κb Activation. J. Endocrinol. 214, 145–153. doi:10.1530/JOE-12-0147
Radeva, M. Y., and Waschke, J. (2018). Mind the gap: Mechanisms Regulating the Endothelial Barrier. Acta Physiol. (Oxf) 222. doi:10.1111/apha.12860
Schlegel, N., and Waschke, J. (2014). cAMP with Other Signaling Cues Converges on Rac1 to Stabilize the Endothelial Barrier- a Signaling Pathway Compromised in Inflammation. Cell Tissue Res 355, 587–596. doi:10.1007/s00441-013-1755-y
Shen, W., Fan, W. H., and Shi, H. M. (2008). Effects of Rhodiola on Expression of Vascular Endothelial Cell Growth Factor and Angiogenesis in Aortic Atherosclerotic Plaque of Rabbits. Zhongguo Zhong Xi Yi Jie He Za Zhi 28, 1022–1025.
Shimokawa, H., and Satoh, K. (2015). Light and Dark of Reactive Oxygen Species for Vascular Function: 2014 ASVB (Asian Society of Vascular Biology). J. Cardiovasc. Pharmacol. 65, 412–418. doi:10.1097/FJC.0000000000000159
Song, T., Wang, P., Li, C., Jia, L., Liang, Q., Cao, Y., et al. (2021). Salidroside Simultaneously Reduces De Novo Lipogenesis and Cholesterol Biosynthesis to Attenuate Atherosclerosis in Mice. Biomed. Pharmacother. 134, 111137. doi:10.1016/j.biopha.2020.111137
Souza, S. P., Santos, R. B., Santos, I. S., Parise, B. K., Giatti, S., Aielo, A. N., et al. (2021). Obstructive Sleep Apnea, Sleep Duration, and Associated Mediators with Carotid Intima-media Thickness: the ELSA-Brasil Study. Arterioscler. Thromb. Vasc. Biol. 41, 1549–1557. doi:10.1161/ATVBAHA.120.315644
Sun, C., Wu, M. H., and Yuan, S. Y. (2011). Nonmuscle Myosin Light-Chain Kinase Deficiency Attenuates Atherosclerosis in Apolipoprotein E-Deficient Mice via Reduced Endothelial Barrier Dysfunction and Monocyte Migration. Circulation 124, 48–57. doi:10.1161/CIRCULATIONAHA.110.988915
Terry, S., Nie, M., Matter, K., and Balda, M. S. (2010). Rho Signaling and Tight junction Functions. Physiology (Bethesda) 25, 16–26. doi:10.1152/physiol.00034.2009
Trzepizur, W., Martinez, M. C., Priou, P., Andriantsitohaina, R., and Gagnadoux, F. (2014). Microparticles and Vascular Dysfunction in Obstructive Sleep Apnoea. Eur. Respir. J. 44, 207–216. doi:10.1183/09031936.00197413
Wang, C., Nan, X., Pei, S., Zhao, Y., Wang, X., Ma, S., et al. (2021). Salidroside and Isorhamnetin Attenuate Urotensin II-induced I-nflammatory R-esponse In V-ivo and In V-itro: Involvement in R-egulating the RhoA/ROCK II P-athway. Oncol. Lett. 21, 292. doi:10.3892/ol.2021.12553
Wattanakit, K., Boland, L., Punjabi, N. M., and Shahar, E. (2008). Relation of Sleep-Disordered Breathing to Carotid Plaque and Intima-media Thickness. Atherosclerosis 197, 125–131. doi:10.1016/j.atherosclerosis.2007.02.029
Xing, S. S., Yang, X. Y., Zheng, T., Li, W. J., Wu, D., Chi, J. Y., et al. (2015). Salidroside Improves Endothelial Function and Alleviates Atherosclerosis by Activating a Mitochondria-Related AMPK/PI3K/Akt/eNOS Pathway. Vascul. Pharmacol. 72, 141–152. doi:10.1016/j.vph.2015.07.004
Yang, Y. Y., Li, L. Y., Jiao, X. L., Jia, L. X., Zhang, X. P., Wang, Y. L., et al. (2020). Intermittent Hypoxia Alleviates β-Aminopropionitrile Monofumarate Induced Thoracic Aortic Dissection in C57BL/6 Mice. Eur. J. Vasc. Endovasc Surg. 59, 1000–1010. doi:10.1016/j.ejvs.2019.10.014
Zhao, C. C., Wu, X. Y., Yi, H., Chen, R., and Fan, G. (2021). The Therapeutic Effects and Mechanisms of Salidroside on Cardiovascular and Metabolic Diseases: an Updated Review. Chem. Biodivers. 18, e2100033. doi:10.1002/cbdv.202100033
Zhao, X., Jin, L., Shen, N., Xu, B., Zhang, W., Zhu, H., et al. (2013). Salidroside Inhibits Endogenous Hydrogen Peroxide Induced Cytotoxicity of Endothelial Cells. Biol. Pharm. Bull. 36, 1773–1778. doi:10.1248/bpb.b13-00406
Zhong, H., Xin, H., Wu, L. X., and Zhu, Y. Z. (2010). Salidroside Attenuates Apoptosis in Ischemic Cardiomyocytes: a Mechanism through a Mitochondria-dependent Pathway. J. Pharmacol. Sci. 114, 399–408. doi:10.1254/jphs.10078fp
Zhou, M., Guo, B., Wang, Y., Yan, D., Lin, C., and Shi, Z. (2017). The Association between Obstructive Sleep Apnea and Carotid Intima-media Thickness: a Systematic Review and Meta-Analysis. Angiology 68, 575–583. doi:10.1177/0003319716665985
Keywords: atherosclerosis, obstructive sleep apnea-hypopnea syndrome, intermittent hypoxia, salidroside, endothelial barrier
Citation: Li L, Yang Y, Zhang H, Du Y, Jiao X, Yu H, Wang Y, Lv Q, Li F, Sun Q and Qin Y (2021) Salidroside Ameliorated Intermittent Hypoxia-Aggravated Endothelial Barrier Disruption and Atherosclerosis via the cAMP/PKA/RhoA Signaling Pathway. Front. Pharmacol. 12:723922. doi: 10.3389/fphar.2021.723922
Received: 11 June 2021; Accepted: 11 August 2021;
Published: 24 August 2021.
Edited by:
Annalisa Chiavaroli, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Maria Luisa Del Moral, University of Jaén, SpainIna Yosifova Aneva, Bulgarian Academy of Sciences, Bulgaria
Copyright © 2021 Li, Yang, Zhang, Du, Jiao, Yu, Wang, Lv, Li, Sun and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanwen Qin, cWlueWFud2VuQHZpcC4xMjYuY29t