
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 27 August 2021
Sec. Pharmacology of Anti-Cancer Drugs
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.720619
This article is part of the Research Topic Clinical Therapeutic Development Against Cancers Resistant to Targeted Therapies View all 31 articles
 Shi-Long Jiang1,2Zhi-Bin Wang1,2Tao Zhu1,2Ting Jiang3Jiang-Feng Fei4
Shi-Long Jiang1,2Zhi-Bin Wang1,2Tao Zhu1,2Ting Jiang3Jiang-Feng Fei4 Chong Liu1,2
Chong Liu1,2 Chao Luo1,5Yan Cheng3*
Chao Luo1,5Yan Cheng3* Zhao-Qian Liu1,2*
Zhao-Qian Liu1,2*Vemurafenib, a BRAF V600E inhibitor, provides therapeutic benefits for patients with melanoma, but the frequent emergence of drug resistance remains a challenge. An understanding of the mechanisms underlying vemurafenib resistance may generate novel therapeutic strategies for patients with melanoma. Here, we showed that eIF3a, a translational regulatory protein, was an important mediator involved in vemurafenib resistance. eIF3a was expressed at significantly lower levels in vemurafenib-resistant A375 melanoma cells (A375R) than in parental A375 cells. Overexpression of eIF3a enhanced the sensitivity to BRAF inhibitors by reducing p-ERK levels. Furthermore, eIF3a controlled ERK activity by regulating the expression of the phosphatase PPP2R1B via a translation mechanism, thus determining the sensitivity of melanoma cells to vemurafenib. In addition, a positive correlation between eIF3a and PPP2R1B expression was also observed in tumor samples from the Human Protein Atlas and TCGA databases. In conclusion, our studies reveal a previously unknown molecular mechanism of BRAF inhibitor resistance, which may provide a new strategy for predicting vemurafenib responses in clinical treatment.
Melanoma is an aggressive cancer with a rapidly increasing incidence (Bellmann et al., 2020). Approximately 60% of melanoma patients harbored BRAF kinase mutation (BRAFV600E), which activates the MAPK pathway and contributes to the immortalization of cancer (Betancourt et al., 2019). Selective BRAF inhibitors (BRAFis), such as vemurafenib, have been approved for clinical use and significantly improved the progression-free survival (PFS) and overall survival (OS) of patients with melanoma carrying BRAF mutations compared with the traditional chemotherapy dacarbazine (Chapman et al., 2011; Kim and Cohen, 2016; Chapman et al., 2017). However, the universal emergence of resistance after vemurafenib treatment limits its application in the clinic (Sosman et al., 2012; Sullivan and Flaherty, 2013; Samatar and Poulikakos, 2014; Garbe and Eigentler, 2018). Multiple mechanisms involved in vemurafenib resistance have been identified (Sullivan and Flaherty, 2013); however, these mechanisms do not fully characterize the causes of vemurafenib resistance, and effective targets and strategies for overcoming clinical vemurafenib resistance are lacking, encouraging further research (Rizos et al., 2014).
Eukaryotic translation initiation factor 3a (eIF3a), the largest subunit of the eIF3 complex, plays an important role in interacting with and recruiting mRNA to the ribosome (Zhang et al., 2007; Saletta et al., 2010; Wang et al., 2016). eIF3a expression is associated with physiological and pathological processes by regulating the cell cycle, apoptosis, differentiation and fibrosis (Dong et al., 2009; Zhang Y. F. et al., 2015; Wang et al., 2016; Yin et al., 2018). Many researchers have reported an important role for eIF3a in the occurrence and development of tumors, and elevated eIF3a expression favors the maintenance of the tumor malignant phenotype (Yin et al., 2011a; Jiang et al., 2020). Recent reports suggested that eIF3a could mediate glycolytic metabolism, and the level of anti-eIF3A autoantibody in serum may represent a potential diagnostic marker for hepatocellular carcinoma (Heo et al., 2019; Miao et al., 2019). Moreover, many studies have suggested that eIF3a affects patient prognosis and treatment responses; for example, patients with cancer presenting high eIF3a levels experience better relapse-free and overall survival than those with low eIF3a expression (Dellas et al., 1998; Chen and Burger, 1999). Previous studies, including our own study, supported the hypothesis that eIF3a knockdown reduces the cellular response to cisplatin by regulating the expression of DNA repair proteins in lung cancer (Yin et al., 2011a), nasopharyngeal carcinoma (Liu et al., 2011), and ovarian cancer (Zhang Y. et al., 2015). Tumia R et al. also reported that high eIF3a expression increases radiotherapy and chemotherapy responses in patients with breast, gastric, lung, and ovarian cancer (Tumia et al., 2020), further suggesting that eIF3a may affect patient responses to treatments.
Here, this study reported for the first time that eIF3a expression is positively correlated with vemurafenib sensitivity in melanoma cells, and this effect of eIF3a is mediated by regulating the translation of the protein phosphatase PPP2R1B, which inhibits ERK phosphorylation. Furthermore, the relationship between eIF3a and PPP2R1B expression was detected in samples from patients with cancer. These findings not only reveal a novel role for eIF3a in vemurafenib resistance and present eIF3a/PPP2R1B/ERK as a new regulatory pathway in cancer but also may provide a novel biomarker for predicting the vemurafenib response.
The human malignant melanoma cell lines A375 and SK-28 were purchased from ATCC (Manassas, VA, United States). Vemurafenib-resistant A375 cells (A375R) were established in our lab. HEK293T cells were maintained in our lab. A375, SK-28, and HEK293T cells were cultured in high-glucose DMEM. A375R cells were cultured in DMEM supplemented with 2 μM vemurafenib. All cell culture media were supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin. All cell lines were maintained at 37°C in a humidified atmosphere containing 5% CO2/95% air. All cells were confirmed negative for mycoplasma using a mycoplasma detection kit.
Vemurafenib, dabrafenib and L-mimosine were purchased from Selleck (Shanghai, China). The ERK inhibitor SCH772984 was purchased from TOPSCIENCE (Shanghai, China). The anti-eIF3a antibody (cat. no. ab128996, 1:1,000) and anti-PPP2R1B antibody (cat. no. ab154815, 1:1,000) were purchased from Abcam. The anti-ERK antibody (cat. no. 4695, 1:1,000) and anti-p-ERK antibody (Thr202/Tyr204) (cat. no. 4370, 1:1,000) were purchased from Cell Signaling Technologies. The anti-puromycin antibody (cat. no. EQ0001, 1:1,000) was purchased from Kerafast. The anti-Flag antibody (cat. no. M185, 1:1,000) and anti-HA antibody (cat. no. M180, 1:1,000) were purchased from MBL. β-action (1:10,000) and β-Tubulin (1:10,000) antibodies were purchased from Proteintech. An enhanced chemiluminescence (ECL) kit was purchased from Cytiva. CCK8 was purchased from Bimake (Shanghai, China).
The siRNAs targeting eIF3a and PPP2R1B were purchased from RiboBio (Guangzhou, China). The PPP2R1B plasmid was purchased from Gene (Shanghai, China). Transfection of siRNAs was performed according to the manufacturer’s protocol. Briefly, cells in the exponential phase of growth were plated in six-well tissue culture plates and then transfected with siRNAs using Lipofectamine RNAimax (Invitrogen) reagent and OPTI-MEM medium. The plasmid was transfected using the Lipofectamine 2000 (Invitrogen) reagent according to the manufacturer’s protocol.
After treatment, the cells were lysed on ice for 30 min in RIPA buffer supplemented with protease inhibitor and phosphatase inhibitor cocktails A and B (Biotool), followed by centrifugation at 12,000 × g for 15 min at 4°C. The protein concentration of the supernatant was determined using a BCA assay (Beyotime Biotechnology, Shanghai, China). Proteins were resolved on SDS-PAGE gels and then transferred to PVDF membranes (0.22 µm, Merck Millipore). After blocking with skim milk, the PVDF membranes were incubated with the respective antibodies in 5% BSA at 4°C overnight, followed by an incubation with a secondary antibody at room temperature for 1 h. The protein signals were detected using an enhanced chemiluminescence kit.
Cells were plated in 6-well tissue culture plates (1,000 cells per well) and exposed to the indicated treatment at 37°C in a humidified atmosphere containing 5% CO2/95% air. At the end of the incubation, cells were fixed with 4% paraformaldehyde, stained with crystal violet for 30 min, and washed with PBS, and then the colonies were photographed and counted.
Gene expression data from GSE118239 were extracted from the NCBI Gene Expression Omnibus (GEO) database. The Gene Set Enrichment Analysis (GSEA) was completed using the R package, “clusterProfiler”.
Cell viability was measured using CCK8 (Bimake, Shanghai, China) reagent. Briefly, 2 × 103 cells/well were plated in each well of 96-well plates and treated with various drug concentrations for the indicated durations. After treatment, 10 μl of CCK8 reagent were added to each well and incubated for 1 h. The absorbance was read at a 450 nm wavelength.
Total RNA was isolated from cells using TRIzol reagent (Takara), and 1 μg of total RNAs was reverse transcribed using the PrimeScript RT Reagent Kit (Perfect real time) (Takara). Real-time PCR was performed using SYBR Premix Ex Tap (Takara) and run on an LC480 instrument. For the quantification of gene expression, the 2−ΔΔCt method was used. The actin expression level was used for normalization.
Cells were transfected with the eIF3a siRNA or a control siRNA. After transfection, the cells were washed twice with ice-cold PBS, fixed with 4% paraformaldehyde for 30 min and treated with 0.5% Triton-100X for 10 min. The cells were blocked at room temperature for 60 min and incubated with a p-ERK antibody overnight at 4°C. Cells were then incubated with a secondary antibody at room temperature for 1 h. The cell nuclei were stained with DAPI for 2 min. Finally, p-ERK levels were observed with a fluorescence microscope.
To analyze PPP2R1B translation activity, eIF3a-silenced 293T cells were cotransfected with the PPP2R1B-containing luciferase reporter plasmid. Cells were harvested, and the luciferase activities were measured using the Promega Luciferase Assay System (Promega, Madison, Wisconsin).
The RIP assay was performed according to the manufacturer’s instructions using an EZMagna RIP Kit (Millipore, Billerica, MA). Briefly, 293T cells lysates were incubated with anti-eIF3a antibodies or anti-IgG at 4°C overnight. Next, the expression of the PPP2R1B mRNA was determined using qRT-PCR.
Protein synthesis activity was assessed using puromycin labeling. Briefly, cells were transfected with the eIF3a siRNA or non-targeting RNA. After transfection, the cells were incubated with puromycin (10 mg/ml) for 30 min. Cells were lysed at 4°C, and the protein concentration was determined using a BCA assay. Equal amounts of protein were resolved on SDS-PAGE gels and then transferred to PVDF membranes. Signals were detected with an anti-puromycin antibody.
All experiments were performed at least three times. For measurements of CCK8 assays, statistical analyses were performed using Student’s t-test with GraphPad Prism software. p values <0.05 were considered statistically significant.
To explore the potential function of eIF3a in cancer therapy, gene set enrichment analysis (GSEA) was used for pathway enrichment analysis. The KEGG analysis showed that the KRAS signaling pathway was significantly enriched after knockdown of eIF3a (GSE118239) (Figure 1A). Reactivation of RAS is associated with vemurafenib resistance (Boussemart et al., 2014), therefore, we hypothesize that eIF3a may be involved in vemurafenib resistance. To test this hypothesis, we first compared the levels of eIF3a in A375 and A375R cells, which is less responsive to vemurafenib than A375 cells (Figure 1B) and observed a significant downregulation of eIF3a in A375R cells (Figure 1C). Furthermore, siRNA-mediated knockdown of eIF3a decreased the responses of human melanoma cells to vemurafenib, as evidenced by the results of the CCK8 assay and colony formation assay (Figures 1D,E). Furthermore, overexpression of eIF3a rendered A375R cells more sensitive to vemurafenib (Figure 1F). To further support the idea that eIF3a is associated with vemurafenib sensitivity, A375 and SK-28 cells were transfected with different amounts of siRNA, followed by vemurafenib treatment. Figures 1G,H showed that the level of eIF3a was positively correlated with vemurafenib sensitivity. These data suggest that the reduced expression of eIF3a in melanoma may result in vemurafenib resistance.
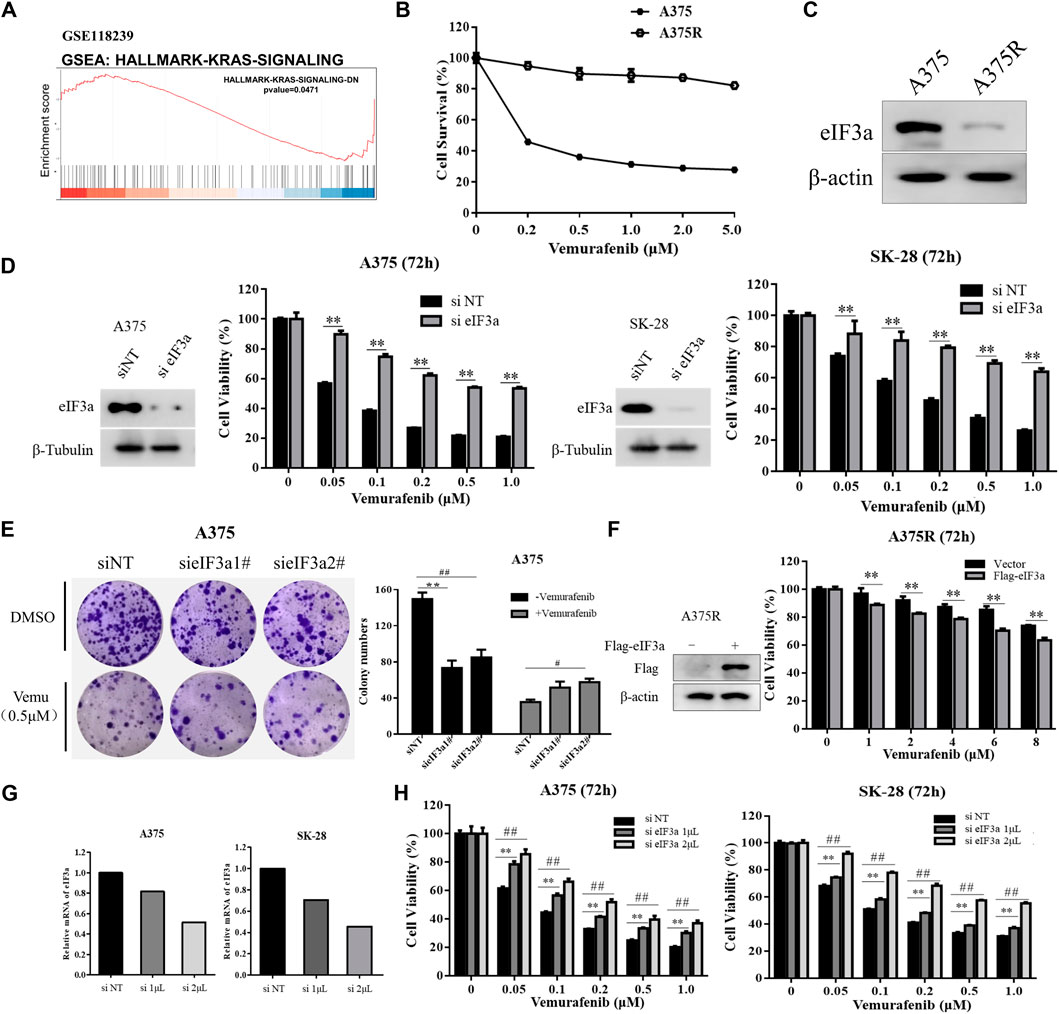
FIGURE 1. eIF3a is associated with vemurafenib sensitivity. (A) Enrichment plots from the gene set enrichment analysis (GSEA) of eIF3a in the KRAS signaling pathway. (B) A375 and A375R cells were treated with a series of vemurafenib concentrations for 72 h, cell survival was measured using the CCK8 assay. (C) The expression of eIF3a in A375 and A375R cells was measured using western blotting. (D) A375 and SK-28 cells were transfected with the non-targeting RNA or eIF3a siRNA and then treated with vemurafenib for 72 h, cell viability was measured by the CCK8 assay. **p < 0.01, t-test. (E) A375 cells were transfected with eIF3a siRNA and treated with vemurafenib, cell proliferation was measured by the colony formation assay. */#p < 0.05, **/##p < 0.01, t-test. (F) A375R cells transfected with the empty vector or Flag-eIF3a plasmid and then treated with vemurafenib for 72 h, cell viability was measured by the CCK8 assay. **p < 0.01, t-test. (G,H) A375 and SK-28 cells were transfected with a non-targeting siRNA or an eIF3a siRNA with the indicated concentrations, then treated with vemurafenib for 72 h, cell viability was measured by the CCK8 assay. **/##p < 0.01, t-test.
In addition, we also evaluated the effect of eIF3a on the antitumor activity of dabrafenib, another BRAF inhibitor approved by the FDA to treat melanoma (Puszkiel et al., 2019). Consistent with the results obtained with vemurafenib, eIF3a silencing also reduced the cytotoxicity of dabrafenib (Figures 2A,B), and eIF3a expression was positively correlated with the toxicity of dabrafenib (Figures 2C,D). These experiments confirmed the involvement of eIF3a in regulating the sensitivity of human melanoma cells to dabrafenib.
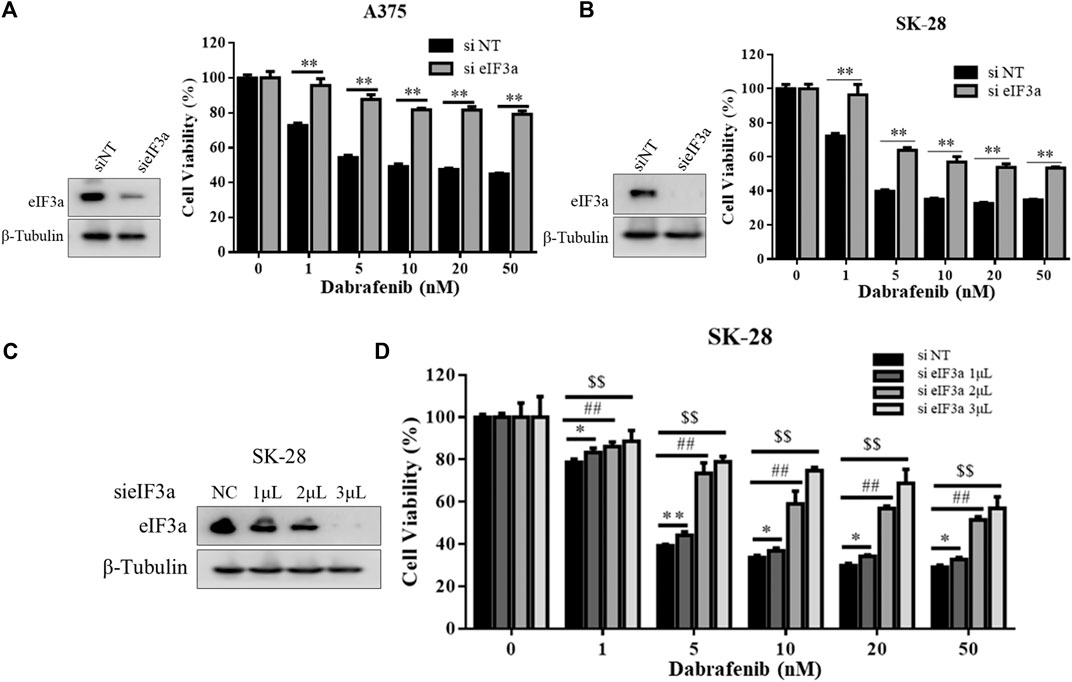
FIGURE 2. eIF3a was also associated with dabrafenib sensitivity. (A,B) A375 and SK-28 cells were transfected with the non-targeting RNA or eIF3a siRNA and then treated with dabrafenib for 72 h, cell viability was measured by the CCK8 assay. **p < 0.01, t-test. (C) SK-28 cells were transfected with a non-targeting siRNA or an eIF3a siRNA with the indicated concentrations, the expression of eIF3a was measured by western blotting. (D) SK-28 cells were transfected with the indicated concentrations of a non-targeting siRNA or an eIF3a siRNA, and then treated with dabrafenib for 72 h, cell viability was measured using the CCK8 assay. */#/$p < 0.05, **/##/$$p < 0.01, t-test.
Next, we investigated the molecular mechanism by which eIF3a regulates sensitivity to vemurafenib. Figure 1A shows that inhibiting eIF3a activated the RAS signaling pathway. It’s well known that ERK is the classical downstream target of the RAS signaling pathway, and the aberrant activation of ERK plays a central role in vemurafenib resistance (Davies et al., 2002). Consistent with this finding, we also verified that the A375R cells harbored higher p-ERK and lower eIF3a levels than the A375 cells (Figure 3A). Therefore, we next determined whether eIF3a regulates the sensitivity of melanoma cells to vemurafenib by modulating ERK activity. As expected, inhibiting eIF3a with either siRNA or L-mimosine, a small-molecule inhibitor of eIF3a, significantly increased ERK phosphorylation in melanoma cells (Figures 3B,C). Figures 3D,E further shows the negative correlation between the expression of eIF3a and p-ERK. Immunofluorescence staining was also used to detect p-ERK levels after silencing eIF3a (Supplementary Figures S1A,B). Moreover, the regulation of p-ERK by eIF3a was also observed in non-small cell lung cancer and breast cancer cell lines (Supplementary Figures S1C,D). Ectopic expression of eIF3a resulted in a reduction in ERK phosphorylation (Figure 3F). We reintroduced eIF3a into eIF3a knockdown cells by transfecting an eIF3a overexpression plasmid and then measured p-ERK levels to further validate the effect of eIF3a on ERK activation. As shown in Figure 3G, p-ERK levels were increased in the cells with eIF3a knockdown, and eIF3a overexpression blocked the silencing expression. Furthermore, knockdown of eIF3a in A375 cells mitigated the suppressive effects of vemurafenib on p-ERK levels, revealing that the vemurafenib-induced MAP kinase signaling blockade was attenuated by silencing eIF3a (Figure 3H). And by combination with an ERK inhibitor, the insensitivity to vemurafenib after knockdown of eIF3a was overcome (Figure 3I). Based on these results, the loss of eIF3a contributed to ERK activation, in turn conferring BRAF inhibitor resistance.
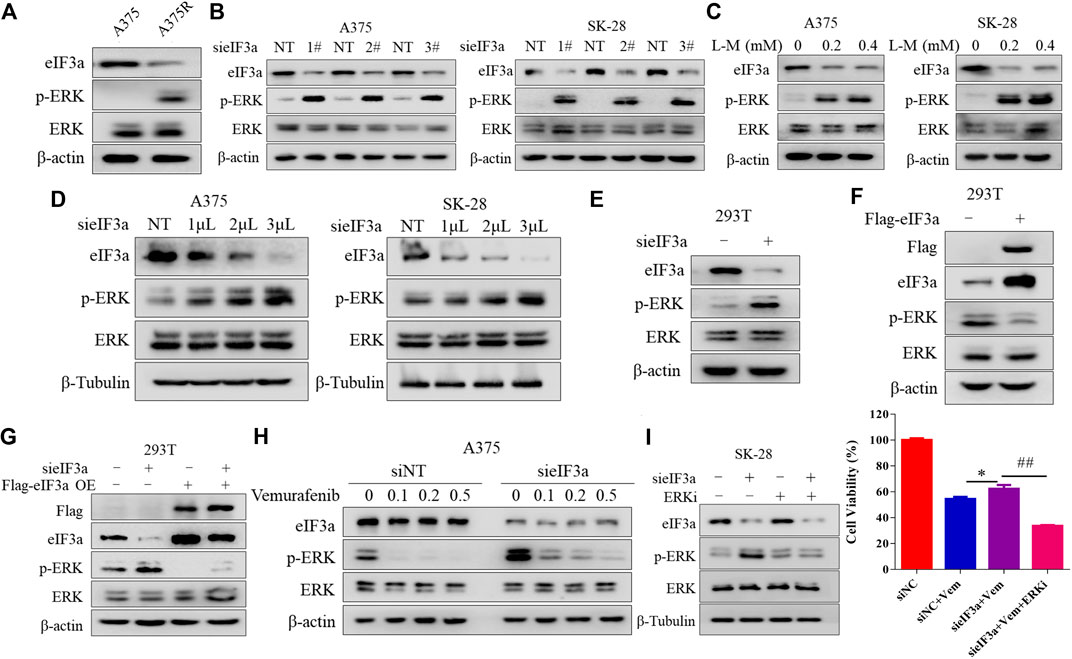
FIGURE 3. eIF3a alters the sensitivity of melanoma cells to vemurafenib by controlling ERK signaling. (A) The levels of eIF3a, p-ERK and ERK in A375 and A375R cells were measured using western blotting. (B) A375 and SK-28 cells were transfected with a non-targeting siRNA or an eIF3a siRNA, the levels of eIF3a, p-ERK and ERK were measured using western blotting. (C) A375 or SK-28 cells were treated with the indicated concentrations of the eIF3a inhibitor L-mimosine, and the levels of eIF3a, p-ERK and ERK were measured using western blotting. (D) A375 and SK-28 cells were transfected with the indicated concentrations of a non-targeting siRNA or an eIF3a siRNA, and the levels of eIF3a, p-ERK and ERK were measured using western blotting. (E) HEK293T cells were transfected with a non-targeting siRNA or an eIF3a siRNA, and the levels of eIF3a, p-ERK and ERK were measured using western blotting. (F) HEK293T cells were transfected with a control plasmid or a Flag-eIF3a plasmid, and the levels of eIF3a, p-ERK and ERK were measured using western blotting. (G) HEK293T cells were transfected with a non-targeting siRNA or an eIF3a siRNA, followed by transfection with the Flag-eIF3a plasmid. The levels of Flag, eIF3a, p-ERK and ERK were measured using western blotting. (H) A375 cells were transfected with the non-targeting RNA or eIF3a siRNA and then treated with vemurafenib. The levels of eIF3a, p-ERK and ERK were measured using western blotting. (I) SK-28 cells were transfected with a non-targeting siRNA or an eIF3a siRNA, and then treated with vemurafenib alone or in combination with an ERK inhibitor. Cell viability was measured using the CCK8 assay. *p < 0.05, ##p < 0.01, t-test.
We next sought to investigate the molecular mechanism underlying eIF3a knockdown-mediated ERK activation. The MAPK pathway is dephosphorylated by numerous protein phosphatases, for example, reductions in the levels of dual specificity phosphatase 4 (DUSP4) and dual specificity phosphatase 6 (DUSP6) result in activation of the MAP kinase pathway (Kondoh and Nishida, 2007; Kidger and Keyse, 2016; Gupta et al., 2019). Therefore, we wondered whether a phosphatase was involved in regulating eIF3a knockdown-mediated ERK activation. We first measured the expressions of protein phosphatases after silencing eIF3a using mass spectrometry to assess this hypothesis. Among the phosphatases examined, the expression of PPP2R1B, a regulatory subunit of the PP2A complex (Hamano et al., 2011), was decreased in eIF3a-knockdowned cells compared with that in control cells (Figure 4A).
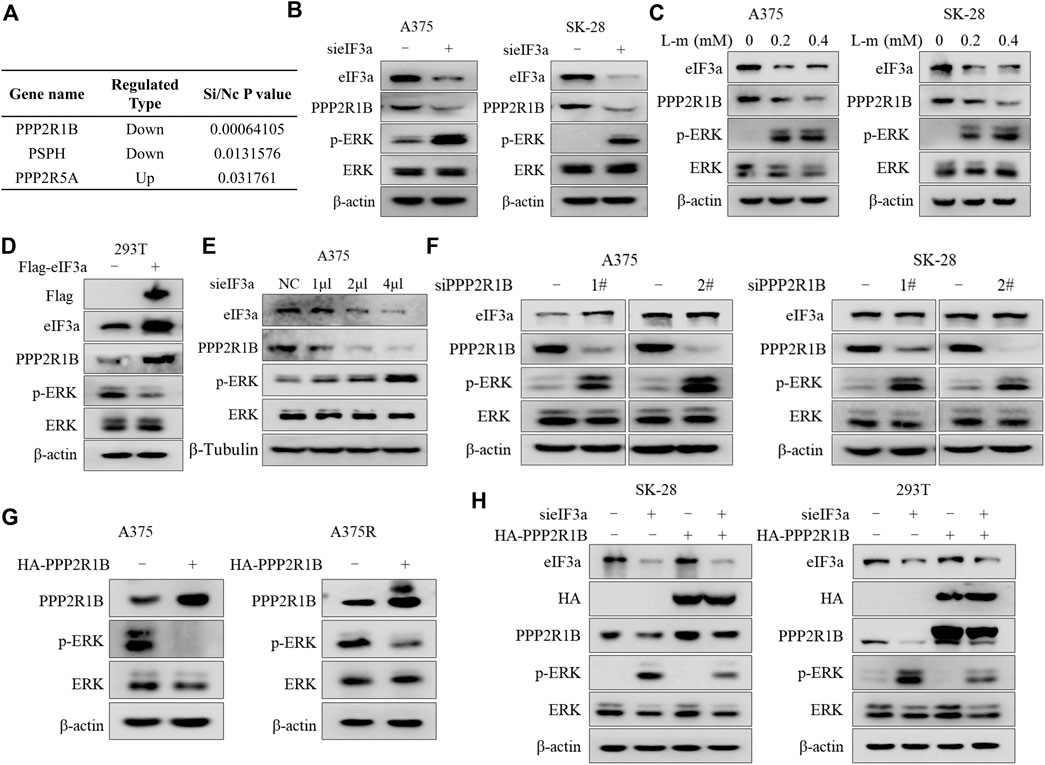
FIGURE 4. eIF3a suppresses ERK activity by upregulating the expression of PPP2R1B. (A) Differential expression of three protein phosphatases was observed after silencing eIF3a using mass spectrometry. (B) A375 and SK-28 cells were transfected with a non-targeting siRNA or an eIF3a siRNA, and the levels of eIF3a, PPP2R1B, p-ERK and ERK were measured using western blotting. (C) A375 and SK-28 cells were treated with the indicated concentrations of the eIF3a inhibitor L-mimosine, and the levels of eIF3a, PPP2R1B, p-ERK and ERK were measured using western blotting. (D) HEK293T cells were transfected with a control plasmid or a Flag-eIF3a plasmid, and the levels of Flag, eIF3a, PPP2R1B, p-ERK and ERK were measured using western blotting. (E) A375 cells were transfected with the indicated concentrations of a non-targeting siRNA or an eIF3a siRNA, and the levels of eIF3a, PPP2R1B and p-ERK were measured using western blotting. (F) A375 and SK-28 cells were transfected with a non-targeting siRNA or PPP2R1B siRNA, and the levels of eIF3a, PPP2R1B, p-ERK and ERK were measured using western blotting. (G) A375 and A375R cells were transfected with a control plasmid or a HA-PPP2R1B plasmid, and the levels of PPP2R1B, p-ERK and ERK were measured using western blotting. (H) SK-28 or HEK293T cells were transfected with a non-targeting siRNA or an eIF3a siRNA, followed by transfection with a HA-PPP2R1B plasmid. The levels of eIF3a, HA, PPP2R1B, p-ERK and ERK were measured using western blotting.
Consistent with the proteomics results, knockdown or inhibition of eIF3a with an siRNA or small-molecule inhibitor decreased PPP2R1B expression and increased p-ERK levels (Figures 4B,C), and overexpression of eIF3a resulted in PPP2R1B elevation (Figure 4D). Figure 4E further demonstrated that eIF3a could regulate PPP2R1B expression. In addition, the regulatory effect of eIF3a on PPP2R1B protein levels was also determined in non-small cell lung cancer cells and breast cancer cells (Supplementary Figure S2). Next, we examined whether a regulatory effect existed between PPP2R1B and ERK. As shown in Figure 4F, PPP2R1B silencing caused a significant increase in p-ERK levels, and overexpression of PPP2R1B induced a remarkable reduction in p-ERK levels (Figure 4G). We overexpressed PPP2R1B after silencing eIF3a to further determine whether PPP2R1B mediates the regulatory effect of eIF3a on ERK activity. Figure 4H shows that reintroduction of PPP2R1B reversed the upregulation of p-ERK induced by eIF3a deficiency.
We further investigated the functional role of PPP2R1B in vemurafenib resistance, and found that PPP2R1B knockdown reduced the sensitivity of A375 cells to vemurafenib, whereas overexpression of PPP2R1B significantly enhanced the antitumor effect of vemurafenib (Supplementary Figures S3A,B). Furthermore, ectopic expression of PPP2R1B in eIF3a-silenced cells partially restored the sensitivity to vemurafenib (Supplementary Figure S3C). These findings support the hypothesis that eIF3a controls the action of ERK by regulating PPP2R1B expression, thus determining the sensitivity of melanoma cells to vemurafenib.
Next, we wanted to explore how eIF3a affects PPP2R1B expression. We examined the PPP2R1B mRNA level after knockdown or overexpression of eIF3a. Silencing or overexpression of eIF3a did not change the PPP2R1B mRNA level (Figures 5A,B), indicating that eIF3a regulated PPP2R1B at the posttranscriptional level. Because eIF3a is a translation regulator, we monitored the role of eIF3a in ongoing translational regulation by measuring protein synthesis using puromycin labeling. Figure 5C showed that the translational activity was reduced after eIF3a knockdown, indicating that eIF3a is indeed involved in protein synthesis in cells. In addition, the PPP2R1B mRNA could bind to the eIF3a protein (Figure 5D), and the reporter gene with PPP2R1B showed significant lower activities in the eIF3a knockdown cells compared with control cells (Figure 5E). Furthermore, HCQ, an autophagy inhibitor, or MG132, a proteasome inhibitor, had no effect on the downregulation of PPP2R1B in the cells with eIF3a knockdown (Figures 5F,G). Based on these results, we deduced that eIF3a regulated the translation of PPP2R1B.
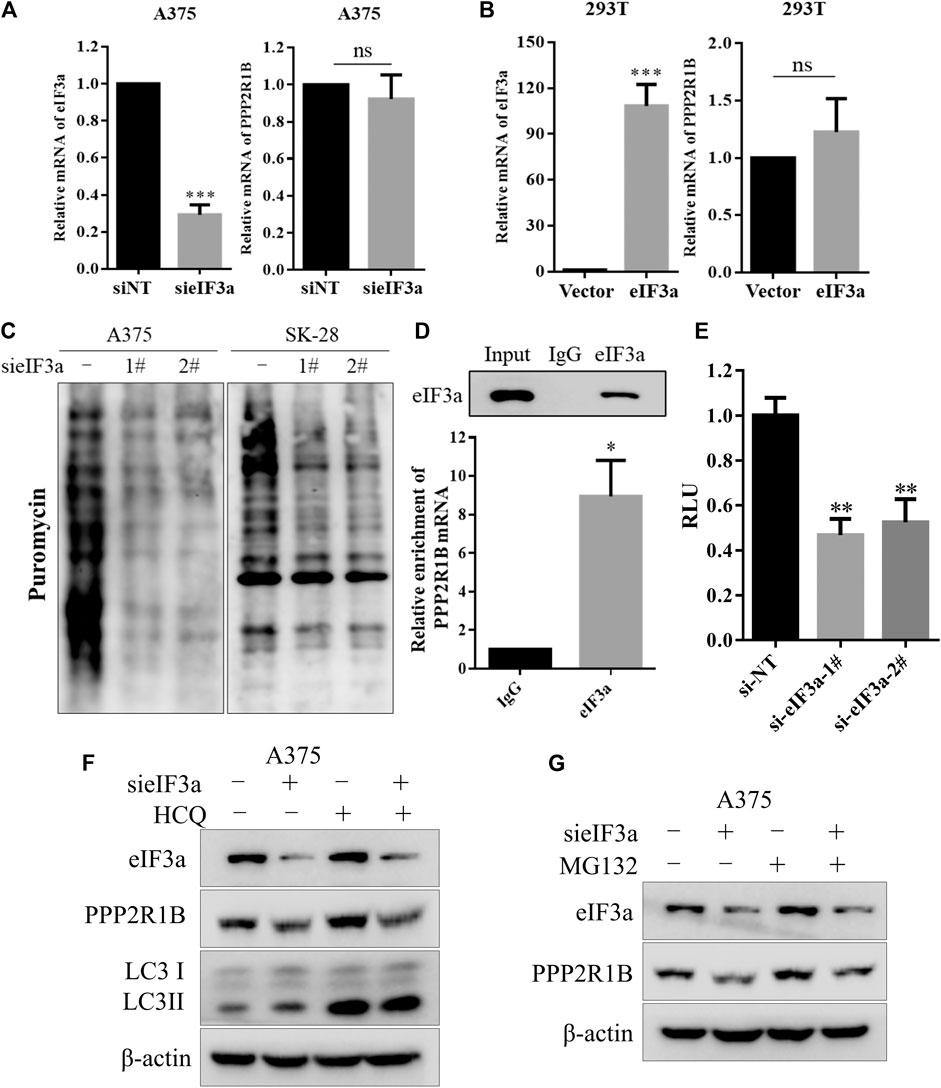
FIGURE 5. eIF3a upregulates the expression of PPP2R1B by promoting its translation. (A) A375 cells were transfected with a non-targeting siRNA or an eIF3a siRNA, and levels of the eIF3a and PPP2R1B mRNAs were measured using qRT-PCR. (B) HEK293T cells were transfected with a control plasmid or a Flag-eIF3a plasmid, and the expression of the eIF3a and PPP2R1B mRNAs was measured using qRT-PCR. (C) A375 or SK-28 cells were transfected with a non-targeting siRNA or an eIF3a siRNA, followed by treatment with puromycin for 30 min. Puromycin incorporation was measured using western blotting. (D) RIP assay indicating that the PPP2R1B mRNA bound to the eIF3a protein. (E) Luciferase assay showing significantly lower activity of the PPP2R1B reporter construct after eIF3a knockdown in HEK293T cells. (F) A375 cells were transfected with a non-targeting siRNA or an eIF3a siRNA, followed by treatment with 20 μM HCQ for 24 h. The levels of eIF3a, PPP2R1B and LC3 were measured using western blotting. (G) A375 cells were transfected with a nontargeting siRNA or an eIF3a siRNA, followed by treatment with 20 μM MG132 for 4 h. The levels of eIF3a and PPP2R1B were measured using western blotting.
To further validate the association between eIF3a and PPP2R1B in human melanoma patient samples, we first assessed protein expression levels of eIF3a and PPP2R1B by the Human Protein Atlas (https://www.proteinatlas.org/). The results showed that the expression of eIF3a was positively correlated with PPP2R1B expression in patients with melanoma (Figure 6A and Supplementary Figure S4). We further analyzed the correlation between eIF3a and PPP2R1B expression in TCGA database. Consistently, the strong positive correlation between eIF3a and PPP2R1B expression was observed in melanoma, lung cancer and breast cancer (Figures 6B–D). In addition, we also analyzed the relationship between eIF3a and PPP2R1B expression in primary, metastatic and uveal melanoma using the TIMER web server (https://cistrome.shinyapps.io/timer/), and found that eIF3a expression was positively correlated with PPP2R1B expression (Figure 6E).
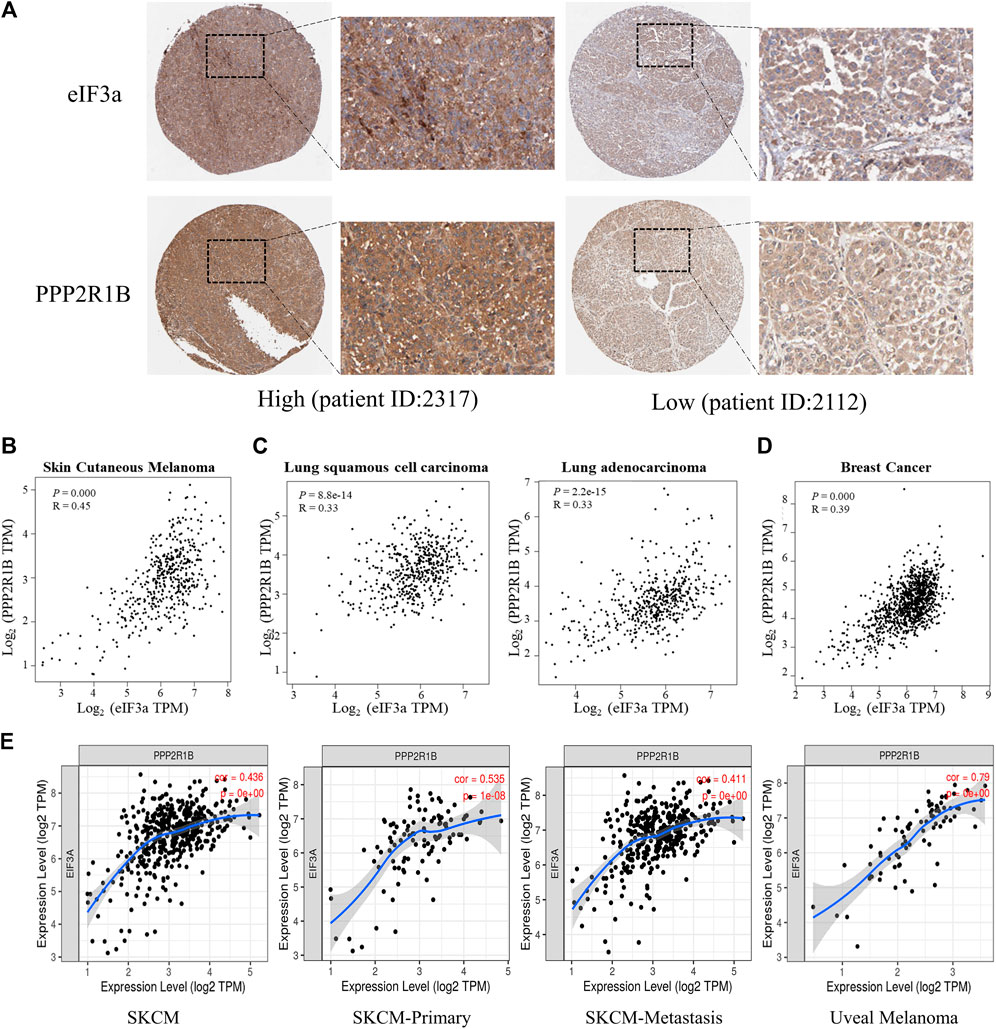
FIGURE 6. The association of eIF3a and PPP2R1B is validated in samples from patients with melanoma. (A) IHC analyses of eIF3a and PPP2R1B levels in melanoma tissues from the HPA database. (B) Pearson’s correlation analyses of eIF3a and PPP2R1B mRNA levels in melanoma (data from TCGA). (C) Pearson’s correlation analyses of eIF3a and PPP2R1B mRNA levels in lung cancer. (D) Pearson’s correlation analyses of eIF3a and PPP2R1B mRNA levels in breast cancer. (E) Pearson’s correlation analyses of the eIF3a and PPP2R1B mRNA levels in primary, metastatic, and uveal melanoma. TPM represents transcripts per million.
Acquired resistance to vemurafenib is a major hurdle in the management of melanoma; therefore, studies exploring the mechanism underlying vemurafenib resistance and developing more effective therapeutic strategies for patients with melanoma carrying the BRAFV600E are imperative. In this study, we revealed for the first time the potentially important role of eIF3a in targeted therapy. We found that eIF3a was downregulated in vemurafenib-resistant A375 cells, and its depletion significantly decreased the response of melanoma cells to BRAF inhibitors. Furthermore, eIF3a modulated the sensitivity of melanoma cells to vemurafenib by promoting the translation of PPP2R1B, and PPP2R1B further resulted in decreased ERK activity by dephosphorylating this kinase, indicating that the eIF3a/PPP2R1B/ERK axis was a key mediator of melanoma resistance to vemurafenib (Figure 7). The positive relationship between eIF3a and PPP2R1B expression was also observed in melanoma tissues. These results suggested that eIF3a may be a predictor to evaluate the efficacy of BRAF inhibitors.
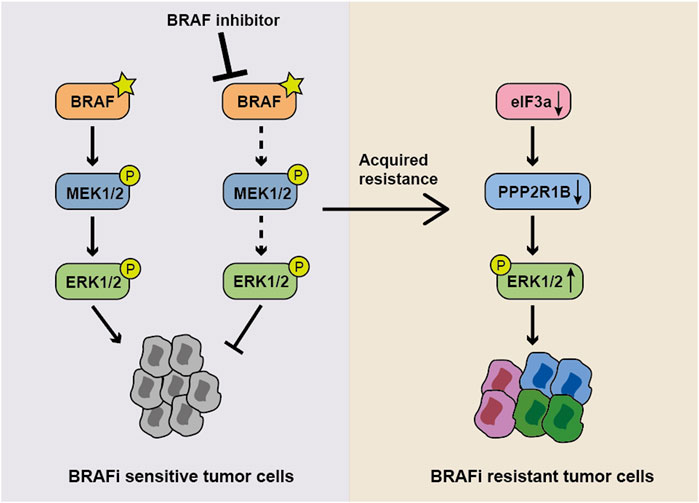
FIGURE 7. Model showing the role of the eIF3a-PPP2R1B-ERK axis in regulating BRAF inhibitor resistance. In normal BRAFV600E mutant cells, the BRAF inhibitor suppresses ERK activity and cell proliferation. When eIF3a is depleted, PPP2R1B translation is inhibited, resulting in the persistence of p-ERK and the subsequent occurrence of drug resistance.
As a key regulator in cancer, eIF3a is expressed at high levels in multiple cancers and is relevant to the sensitivity of DNA damage-induced therapy, such as platinum agents (Zhang Y. et al., 2015) and ionizing radiation (Tumia et al., 2020). As shown in our previous study, eIF3a increases the sensitivity of ovarian cancer and non-small cell lung cancer to platinum-based chemotherapy, and the patients with a high level of eIF3a have a better prognosis (Yin et al., 2011b; Zhang Y. et al., 2015).
ERK activation is critical for cell proliferation and differentiation, and even alters the response of cancer cells to chemotherapies and targeted therapies (Marshall, 1995). In addition, ERK regulates the metabolic processes of cancer cell, such as glucose metabolism and fatty acid metabolism, and promotes cancer cell invasion and migration (Lee et al., 2020). As a critical mechanism significantly affecting cancer pathogenesis, the regulation of ERK has been extensively investigated. Levels of activated ERK are regulated by multiple proteins or miRNAs, including BOP1, VRK3, and miR-30 (Han and Kim, 2020; Wang et al., 2020). Therefore, a better understanding of ERK-mediated drug resistance may be helpful for in-depth studies of the antitumor mechanism. Here, we showed that reduced eIF3a expression caused ERK activation, as evidence by increased ERK phosphorylation, thus leading to vemurafenib resistance, which may provide novel strategy to rescue drug sensitivity for anticancer drug development.
We performed a proteomics analysis using mass spectrometry to identify eIF3a interacting proteins and illustrate the regulatory mechanism of eIF3a in ERK activation. The expression of PPP2R1B, a phosphatase, was decreased in cells subjected to eIF3a knockdown. Notably, eIF3a knockdown or overexpression decreased or increased the expression of the PPP2R1B protein, respectively. In addition, consistent with other phosphatases of ERK, PPP2R1B knockdown in melanoma cell lines led to increased ERK phosphorylation and mediated the negative regulatory effect of eIF3a on vemurafenib sensitivity. As a tumor suppressor, mutations and alterations in PPP2R1B have been found in human cancers, including colon cancer, lung cancer and cervical cancer, and is involved in chemotherapy sensitivity (Wang et al., 1998; Tamaki et al., 2004; Yeh et al., 2007). Previous studies have reported that silencing PPP2R1B enhances 5-FU resistance (Zhang et al., 2015), and PPP2R1B overexpression increases the sensitivity of tongue squamous cell carcinoma and esophageal cancer cells to docetaxel and cisplatin, respectively by acting as an AKT phosphatase (Hamano et al., 2011; Cui et al., 2020). In the present study, PPP2R1B influenced vemurafenib sensitivity by inhibiting ERK phosphorylation, and ectopic expression of PPP2R1B restored cell sensitivity to vemurafenib. Thus, ERK may be a novel protein target of PPP2R1B phosphatase, and PPP2R1B may be a potential target for developing novel strategies against vemurafenib resistance.
Last, we also observed strong correlations between eIF3a and PPP2R1B expression between tumor tissues from HPA and TCGA databases, further supporting our findings in cells. In addition, the correlation between eIF3a and PPP2R1B was also observed in lung cancer and breast cancer tissue, suggesting that the mechanism is universal. Due to the importance of vemurafenib in clinical applications, studies investigating the regulatory axis of eIF3a-PPP2R1B-ERK are crucial and worthwhile. Moreover, our findings highlight eIF3a as a promising biomarker for melanoma or other cancers to predict the therapeutic effect of vemurafenib.
Taken together, our results revealed that the eIF3a-PPP2R1B-ERK axis may contribute to vemurafenib resistance, which provided new insights into not only vemurafenib resistance but also the prediction of the response to vemurafenib treatment.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Z-QL and YC designed, conceived the study, and revised the manuscript. S-LJ, Z-BW, and TZ performed the experiments and analyzed data. S-LJ drafted the manuscript. CLu, TJ, CLi, and J-FF provided technological support in the experiments.
This work was supported by the National Key Research and Development Program of China (2016YFC1306900), National Natural Science Foundation of China (81874327), Key Research and Development Program of Hunan Province (2019SK2251), Innovation and Research Project of Development and Reform Committee of Hunan Province (2019-875), Project Program of National Clinical Research Center for Geriatric Disorders (Xiangya Hospital, 2020LNJJ02), and Science and Technology Program of Changsha (kh2003010).
JFF was employed by Sinocare Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.720619/full#supplementary-material
eIF3a, eukaryotic translation initiation factor 3a; ERK, p44/42 MAPK (Erk1/2); PPP2R1B, protein phosphatase 2 scaffold subunit Abeta; RIP, RNA immunoprecipitation; TCGA, the cancer genome atlas; GEO, gene expression omnibus; qPCR, quantitative real-time PCR.
Bellmann, L., Cappellano, G., Schachtl-Riess, J. F., Prokopi, A., Seretis, A., Ortner, D., et al. (2020). A TLR7 Agonist Strengthens T and NK Cell Function during BRAF-Targeted Therapy in a Preclinical Melanoma Model. Int. J. Cancer 146 (5), 1409–1420. doi:10.1002/ijc.32777
Betancourt, L. H., Szasz, A. M., Kuras, M., Rodriguez Murillo, J., Sugihara, Y., Pla, I., et al. (2019). The Hidden Story of Heterogeneous B-Raf V600E Mutation Quantitative Protein Expression in Metastatic Melanoma-Association with Clinical Outcome and Tumor Phenotypes. Cancers (Basel) 11 (12), 1981. doi:10.3390/cancers11121981
Boussemart, L., Malka-Mahieu, H., Girault, I., Allard, D., Hemmingsson, O., Tomasic, G., et al. (2014). eIF4F Is a Nexus of Resistance to Anti-BRAF and Anti-MEK Cancer Therapies. Nature. 513 (7516), 105–109. doi:10.1038/nature13572
Chapman, P. B., Hauschild, A., Robert, C., Haanen, J. B., Ascierto, P., Larkin, J., et al. (2011). Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 364 (26), 2507–2516. doi:10.1056/NEJMoa1103782
Chapman, P. B., Robert, C., Larkin, J., Haanen, J. B., Ribas, A., Hogg, D., et al. (2017). Vemurafenib in Patients with BRAFV600 Mutation-Positive Metastatic Melanoma: Final Overall Survival Results of the Randomized BRIM-3 Study. Ann. Oncol. 28 (10), 2581–2587. doi:10.1093/annonc/mdx339
Chen, G., and Burger, M. M. (1999). p150 Expression and its Prognostic Value in Squamous-Cell Carcinoma of the Esophagus. Int. J. Cancer 84 (2), 95–100. doi:10.1002/(sici)1097-0215(19990420)84:2<95::aid-ijc1>3.0.co;2-n
Cui, J., Wang, H., Zhang, X., Sun, X., Zhang, J., and Ma, J. (2020). Exosomal miR-200c Suppresses Chemoresistance of Docetaxel in Tongue Squamous Cell Carcinoma by Suppressing TUBB3 and PPP2R1B. Aging (Albany NY) 12 (8), 6756–6773. doi:10.18632/aging.103036
Davies, H., Bignell, G. R., Cox, C., Stephens, P., Edkins, S., Clegg, S., et al. (2002). Mutations of the BRAF Gene in Human Cancer. Nature 417 (6892), 949–954. doi:10.1038/nature00766
Dellas, A., Torhorst, J., Bachmann, F., Banziger, R., Schultheiss, E., and Burger, M. M. (1998). Expression of P150 in Cervical Neoplasia and its Potential Value in Predicting Survival. Cancer 83 (7), 1376–1383. doi:10.1002/(sici)1097-0142(19981001)83:7<1376::aid-cncr15>3.0.co;2-1
Dong, Z., Liu, Z., Cui, P., Pincheira, R., Yang, Y., Liu, J., et al. (2009). Role of eIF3a in Regulating Cell Cycle Progression. Exp. Cel Res 315 (11), 1889–1894. doi:10.1016/j.yexcr.2009.03.009
Garbe, C., and Eigentler, T. K. (2018). Vemurafenib. Recent Results Cancer Res. 211, 77–89. doi:10.1007/978-3-319-91442-8_6
Gupta, R., Bugide, S., Wang, B., Green, M. R., Johnson, D. B., and Wajapeyee, N. (2019). Loss of BOP1 Confers Resistance to BRAF Kinase Inhibitors in Melanoma by Activating MAP Kinase Pathway. Proc. Natl. Acad. Sci. U S A. 116 (10), 4583–4591. doi:10.1073/pnas.1821889116
Hamano, R., Miyata, H., Yamasaki, M., Kurokawa, Y., Hara, J., Moon, J. H., et al. (2011). Overexpression of miR-200c Induces Chemoresistance in Esophageal Cancers Mediated through Activation of the Akt Signaling Pathway. Clin. Cancer Res. 17 (9), 3029–3038. doi:10.1158/1078-0432.CCR-10-2532
Han, S. H., and Kim, K. T. (2020). RNF144a Induces ERK-dependent Cell Death under Oxidative Stress via Downregulation of Vaccinia-Related Kinase 3. J. Cel Sci 133 (21), jcs247304. doi:10.1242/jcs.247304
Heo, C. K., Hwang, H. M., Lee, H. J., Kwak, S. S., Yoo, J. S., Yu, D. Y., et al. (2019). Serum Anti-eif3a Autoantibody as a Potential Diagnostic Marker for Hepatocellular Carcinoma. Sci. Rep. 9 (1), 11059. doi:10.1038/s41598-019-47365-4
Jiang, S. L., Mo, J. L., Peng, J., Lei, L., Yin, J. Y., Zhou, H. H., et al. (2020). Targeting Translation Regulators Improves Cancer Therapy. Genomics. 113 (1 Pt 2), 1247–1256. doi:10.1016/j.ygeno.2020.11.011
Kidger, A. M., and Keyse, S. M. (2016). The Regulation of Oncogenic Ras/ERK Signalling by Dual-Specificity Mitogen Activated Protein Kinase Phosphatases (MKPs). Semin. Cel Dev Biol 50, 125–132. doi:10.1016/j.semcdb.2016.01.009
Kim, A., and Cohen, M. S. (2016). The Discovery of Vemurafenib for the Treatment of BRAF-Mutated Metastatic Melanoma. Expert Opin. Drug Discov. 11 (9), 907–916. doi:10.1080/17460441.2016.1201057
Kondoh, K., and Nishida, E. (2007). Regulation of MAP Kinases by MAP Kinase Phosphatases. Biochim. Biophys. Acta 1773 (8), 1227–1237. doi:10.1016/j.bbamcr.2006.12.002
Lee, S., Rauch, J., and Kolch, W. (2020). Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 21 (3), 1102. doi:10.3390/ijms21031102
Liu, R. Y., Dong, Z., Liu, J., Yin, J. Y., Zhou, L., Wu, X., et al. (2011). Role of eIF3a in Regulating Cisplatin Sensitivity and in Translational Control of Nucleotide Excision Repair of Nasopharyngeal Carcinoma. Oncogene. 30 (48), 4814–4823. doi:10.1038/onc.2011.189
Marshall, C. J. (1995). Specificity of Receptor Tyrosine Kinase Signaling: Transient versus Sustained Extracellular Signal-Regulated Kinase Activation. Cell. 80 (2), 179–185. doi:10.1016/0092-8674(95)90401-8
Miao, B., Wei, C., Qiao, Z., Han, W., Chai, X., Lu, J., et al. (2019). eIF3a Mediates HIF1alpha-dependent Glycolytic Metabolism in Hepatocellular Carcinoma Cells through Translational Regulation. Am. J. Cancer Res. 9 (5), 1079–1090.
Puszkiel, A., Noe, G., Bellesoeur, A., Kramkimel, N., Paludetto, M. N., Thomas-Schoemann, A., et al. (2019). Clinical Pharmacokinetics and Pharmacodynamics of Dabrafenib. Clin. Pharmacokinet. 58 (4), 451–467. doi:10.1007/s40262-018-0703-0
Rizos, H., Menzies, A. M., Pupo, G. M., Carlino, M. S., Fung, C., Hyman, J., et al. (2014). BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clin. Cancer Res. 20 (7), 1965–1977. doi:10.1158/1078-0432.CCR-13-3122
Saletta, F., Suryo Rahmanto, Y., and Richardson, D. R. (2010). The Translational Regulator eIF3a: the Tricky eIF3 Subunit!. Biochim. Biophys. Acta 1806 (2), 275–286. doi:10.1016/j.bbcan.2010.07.005
Samatar, A. A., and Poulikakos, P. I. (2014). Targeting RAS-ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 13 (12), 928–942. doi:10.1038/nrd4281
Sosman, J. A., Kim, K. B., Schuchter, L., Gonzalez, R., Pavlick, A. C., Weber, J. S., et al. (2012). Survival in BRAF V600-Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 366 (8), 707–714. doi:10.1056/NEJMoa1112302
Sullivan, R. J., and Flaherty, K. T. (2013). Resistance to BRAF-Targeted Therapy in Melanoma. Eur. J. Cancer 49 (6), 1297–1304. doi:10.1016/j.ejca.2012.11.019
Tamaki, M., Goi, T., Hirono, Y., Katayama, K., and Yamaguchi, A. (2004). PPP2R1B Gene Alterations Inhibit Interaction of PP2A-Abeta and PP2A-C Proteins in Colorectal Cancers. Oncol. Rep. 11 (3), 655–659. doi:10.3892/or.11.3.655
Tumia, R., Wang, C. J., Dong, T., Ma, S., Beebe, J., Chen, J., et al. (2020). eIF3a Regulation of NHEJ Repair Protein Synthesis and Cellular Response to Ionizing Radiation. Front Cel Dev Biol 8, 753. doi:10.3389/fcell.2020.00753
Wang, S. S., Esplin, E. D., Li, J. L., Huang, L., Gazdar, A., Minna, J., et al. (1998). Alterations of the PPP2R1B Gene in Human Lung and colon Cancer. Science 282 (5387), 284–287. doi:10.1126/science.282.5387.284
Wang, S. Q., Liu, Y., Yao, M. Y., and Jin, J. (2016). Eukaryotic Translation Initiation Factor 3a (eIF3a) Promotes Cell Proliferation and Motility in Pancreatic Cancer. J. Korean Med. Sci. 31 (10), 1586–1594. doi:10.3346/jkms.2016.31.10.1586
Wang, Y., Jie, L., Gong, H., Li, Y., Xie, A., Li, Y., et al. (2020). miR-30 Inhibits Proliferation of Trophoblasts in Preeclampsia Rats Partially Related to MAPK/ERK Pathway. Exp. Ther. Med. 20 (2), 1379–1384. doi:10.3892/etm.2020.8866
Yeh, L. S., Hsieh, Y. Y., Chang, J. G., Chang, W. W., Chang, C. C., and Tsai, F. J. (2007). Mutation Analysis of the Tumor Suppressor Gene PPP2R1B in Human Cervical Cancer. Int. J. Gynecol. Cancer 17 (4), 868–871. doi:10.1111/j.1525-1438.2007.00880.x
Yin, J. Y., Dong, Z., Liu, Z. Q., and Zhang, J. T. (2011a). Translational Control Gone Awry: a New Mechanism of Tumorigenesis and Novel Targets of Cancer Treatments. Biosci. Rep. 31 (1), 1–15. doi:10.1042/BSR20100077
Yin, J. Y., Shen, J., Dong, Z. Z., Huang, Q., Zhong, M. Z., Feng, D. Y., et al. (2011b). Effect of eIF3a on Response of Lung Cancer Patients to Platinum-Based Chemotherapy by Regulating DNA Repair. Clin. Cancer Res. 17 (13), 4600–4609. doi:10.1158/1078-0432.CCR-10-2591
Yin, J. Y., Zhang, J. T., Zhang, W., Zhou, H. H., and Liu, Z. Q. (2018). eIF3a: A New Anticancer Drug Target in the eIF Family. Cancer Lett. 412, 81–87. doi:10.1016/j.canlet.2017.09.055
Zhang, L., Pan, X., and Hershey, J. W. (2007). Individual Overexpression of Five Subunits of Human Translation Initiation Factor eIF3 Promotes Malignant Transformation of Immortal Fibroblast Cells. J. Biol. Chem. 282 (8), 5790–5800. doi:10.1074/jbc.M606284200
Zhang, Y., Talmon, G., and Wang, J. (2015). MicroRNA-587 Antagonizes 5-FU-Induced Apoptosis and Confers Drug Resistance by Regulating PPP2R1B Expression in Colorectal Cancer. Cell Death Dis 6, e1845. doi:10.1038/cddis.2015.200
Zhang, Y., Yu, J. J., Tian, Y., Li, Z. Z., Zhang, C. Y., Zhang, S. F., et al. (2015). eIF3a Improve Cisplatin Sensitivity in Ovarian Cancer by Regulating XPC and p27Kip1 Translation. Oncotarget 6 (28), 25441–25451. doi:10.18632/oncotarget.4555
Keywords: melanoma, vemurafenib resistance, eIF3a, ERK, PPP2R1B
Citation: Jiang S-L, Wang Z-B, Zhu T, Jiang T, Fei J-F, Liu C, Luo C, Cheng Y and Liu Z-Q (2021) The Downregulation of eIF3a Contributes to Vemurafenib Resistance in Melanoma by Activating ERK via PPP2R1B. Front. Pharmacol. 12:720619. doi: 10.3389/fphar.2021.720619
Received: 04 June 2021; Accepted: 12 August 2021;
Published: 27 August 2021.
Edited by:
Yongchuan Gu, The University of Auckland, New ZealandReviewed by:
Oscar Gee Wan Wong, The University of Hong Kong, Hong Kong, SAR ChinaCopyright © 2021 Jiang, Wang, Zhu, Jiang, Fei, Liu, Luo, Cheng and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhao-Qian Liu, enFsaXVAY3N1LmVkdS5jbg==; Yan Cheng, eWFuY2hlbmdAY3N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.