
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 16 December 2021
Sec. Neuropharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.708141
 Lihui Wang1,2†
Lihui Wang1,2† Jinjin Cao1†Qianqian Xu1†Xiaomei Lu1Xin Yang1Qiong Song1Shuai Chen1Kechen Du1Renbin Huang2*
Jinjin Cao1†Qianqian Xu1†Xiaomei Lu1Xin Yang1Qiong Song1Shuai Chen1Kechen Du1Renbin Huang2* Chunlin Zou1*
Chunlin Zou1*Diabetes mellitus (DM) is an independent risk factor for cognitive impairment. Although the etiology of diabetic cognitive impairment is complex and multifactorial, the hippocampus neuronal apoptosis is recognized as a main cause of diabetes-induced cognitive impairment. 2-Dodecyl-6-methoxycyclohexa-2,5-diene-1,4-dione (DMDD) was purified from the roots of Averrhoa carambola L. Previous research demonstrated that DMDD was safe and effective in delaying some diabetic complications. However, the efficacy of DMDD to ameliorate diabetic cognitive impairment in type 2 diabetes mice has not been reported. In the present study, the behavioral evaluation was performed by Y maze and novel object recognition in db/db mice. Gene expression profiles were detected using mouse lncRNA microarray analysis in the hippocampi of db/db mice. Changes in the neurodegeneration-associated proteins and the apoptosis-related proteins were determined in both db/db mice and high glucose-treated HT22 cells by Western blotting. We observed that DMDD treatment significantly ameliorated the spatial working memory and object recognition memory impairment in db/db mice. Further study showed that neurodegeneration-associated protein tau was decreased after DMDD treatment in the hippocampi of db/db mice. Eleven lncRNAs and four mRNAs including pro-apoptotic gene Hif3a were significantly differently expressed after DMDD treatment in the hippocampi of db/db mice. The expression of Hif3a, cleaved parp, and caspase 3 proteins was significantly increased in the hippocampi of diabetic db/db mice compared with db/m control mice and then decreased after DMDD treatment. Similar beneficial effects of DMDD were observed in HG-treated HT22 cells. These data indicate that DMDD can alleviate cognitive impairment by inhibiting neuronal apoptosis through decreasing the expression of pro-apoptotic protein Hif3a. In conclusion, our study suggests that DMDD has great potential to be a new preventive and therapeutic compound for diabetic cognitive impairment.
Diabetes mellitus (DM) is a complex metabolic disturbances characterized by chronic hyperglycemia. Epidemiological findings indicate that DM is a strong and independent risk factor for cognitive impairment (Chatterjee et al., 2016), Parkinson’s disease (PD) (Athauda and Foltynie, 2016), and Alzheimer’s disease (AD) (Zilliox et al., 2016). The pathophysiology of cognitive dysfunction in diabetes is multifactorial, including insulin signaling defect, inflammatory pathways, oxidative stress, mitochondrial abnormalities, and Tau signaling (Zilliox et al., 2016; Bharadwaj et al., 2017). Cognitive deficits can occur in the early stages of diabetes, which can be exacerbated by metabolic syndrome (Yaffe et al., 2004). With the prolongation of life span, the need for new methods for preventing the development of neurodegenerative disease in T2DM is becoming more and more urgent.
The link between diabetes and neurodegenerative disease has been well established, but the underlying mechanisms of pathological development remains unclear. Previous studies focused on tauopathies, α-synuclein accumulation (Ashraf et al., 2014), mitochondrial dysfunction, and apoptosis (Cheng et al., 2019). Multiple studies showed that diabetes-related metabolic dysfunction affected tau phosphorylation and resulted in hyperphosphorylated tau accumulates to form neurofibrillary tangles (NFTs) and lead to neuronal death (Zhang et al., 2018; Ho et al., 2020). In addition, α-synuclein accumulation plays a key role in diabetic cognitive impairment. Under pathological conditions, misfolded α-synuclein aggregates into a form of high molecular weight and becomes toxic to neurons. Recent studies have shown that compared with normal controls, patients with diabetes had higher CSF tau and α-synuclein levels (Pagano et al., 2018).
Both hyperphosphorylated tau (Stoothoff et al., 2009) and α-synuclein (Ordonez et al., 2018) can cause mitochondrial dysfunction and eventually lead to neuronal death. Mitochondria are essential for generating ATP through respiration and oxidative phosphorylation, and have a central role in apoptosis. Mitochondrial dysfunction is a key factor of cognitive deficits in neurodevelopmental disorders and neurodegenerative diseases (Sheng and Cai, 2012; Khacho et al., 2019). Growing evidence shows that even before the occurrence of AD symptoms or any detectable cognitive decline, mitochondrial dysfunction already happened (Kulic et al., 2011). Interaction between dysfunctional mitochondria and impaired insulin signaling can finally promote the development of AD in diabetic patients. This suggests that overlapping pathological mechanisms exists in neurodegenerative diseases and diabetes (Craft and Watson, 2004). Hence, antidiabetic agents might have potential benefits in the treatment of diabetic cognitive impairment.
Natural products are the main source of new drug compounds. Studies have shown that natural products and derived compounds can attenuate cognitive impairment and diabetes. 2-Dodecyl-6-methoxycyclohexa-2,5-diene-1,4-dione (DMDD) (Figure 1A) is an active compound, which was purified from the roots of Averrhoa carambola L. Previous studies in our lab have suggested that DMDD had an antidiabetic effect by decreasing AGE expression and downregulating the NFκB–TGFβ1 pathway in type 2 diabetic KKAy mice (Zheng et al., 2013). In addition, DMDD also protected against apoptosis in PC-12 pheochromocytoma cells in vitro and APP/PS1 Alzheimer’s disease mice in vivo (Wei et al., 2018). Although the therapeutic effect of DMDD on diabetes mellitus and Alzheimer’s disease were studied, the therapeutic potential of DMDD on diabetic cognitive impairment and its underlying mechanisms remains unclear. Here, we investigated the neuroprotective effect and possible mechanisms of DMDD on high-glucose-induced HT22 mouse hippocampal neuron injury and on diabetic db/db mice with impaired learning and memory.
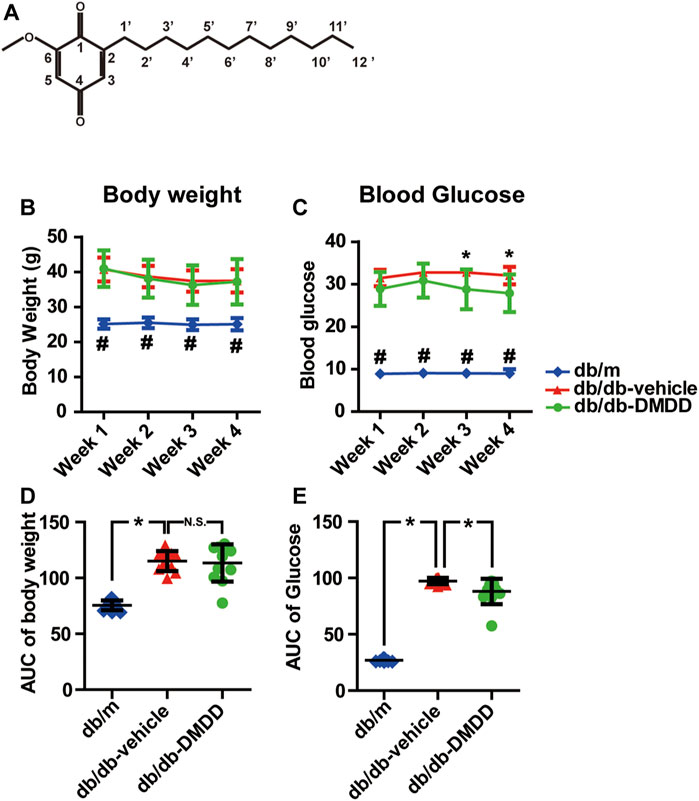
FIGURE 1. 2-Dodecyl-6-methoxycyclohexa-2,5-diene-1,4-dione (DMDD) improved hyperglycemia in db/db mice. (A) Chemical structural diagrams of DMDD. (B) Body weights and (D) area under the curve (AUC) during 4 weeks of drug treatment. (C) Blood glucose and (E) AUC during drug treatment. All data are shown as means ± SD. *p < 0.05 for one-way ANOVA test.
2-Dodecyl-6-methoxycyclohexa-2,5-diene-1,4-dione (DMDD), a cyclohexanedione, was isolated and purified from Averrhoa carambola L., which is cultivated in tropical and subtropical regions. The roots of Averrhoa carambola L. were collected from Lingshan, Guangxi, China, and identified by Prof Maoxiang Lai (Traditional Chinese Medicine Research Institute of Guangxi). For in vitro assay, DMDD was dissolved in 100% DMSO as 20 mM stock solution. DMDD working solution was prepared by diluting appropriate stock solution in cell culture medium. The chemical structure of DMDD is shown in Figure 1A.
Male 9-week-old db/db mice and age-matched heterozygous db/m control mice were bought from the Model Animal Research Center of Nanjing University (Nanjing, China). The mice were housed in a controlled environment (25 ± 2°C and 50 ± 5% relative humidity) with a 12-h/12-h day/night cycle. The mice were given free access to food and water. All animal experimental protocols were approved by the Animal Ethics Committee of Guangxi Medical University.
Db/db mice were randomly assigned to vehicle-treated db/db and DMDD-treated db/db group (10 mice per group) after 1 week of adaption. The wild-type db/m control mice were used as the control group (n = 10). Db/m control group and vehicle-treated db/db group were administrated intragastrically with 1% Tween 80 per day. The DMDD-treated db/db group was administrated intragastrically with 50 mg/ kg of DMDD per day. Drug treatment was performed once daily during the daytime for 28 successive days. Body weight and blood glucose were detected every 2 days. The animal behavior test was carried out before and after the drug treatment. At the end of the study, the mice were euthanized. Then the hippocampus were isolated and kept in liquid nitrogen for Western blotting, real-time PCR, and lncRNA microarray analysis.
Y-maze test, a two-trial spatial memory task was designed to assess spatial memory in mice (Kraeuter et al., 2019). During the first trial (acquisition), the mouse was able to visit two arms (named as start arm and other arm) of the Y-maze for 10 min, and the third arm (named as new arm) was blocked by a door with the same color as the aisle. After the end of the first trial, the mouse was placed in a single cage. After 1 h, the second trial (retrieval) started. The door of the new arm was opened, and the mouse accessed freely all three arms for 5 min (Figure 2A). The number and the duration of visiting each arm were recorded for each trial. During the second trial, every entry different from the last two entries was regarded as a successful alternation. The following formula was used to calculate working memory: spontaneous alternation percentage (%) = [(number of successful alternations)/(total number of arm entries −2)] × 100%. Recognition memory index (%) = (time in the novel arm/time in all arms during the second trial) × 100%.
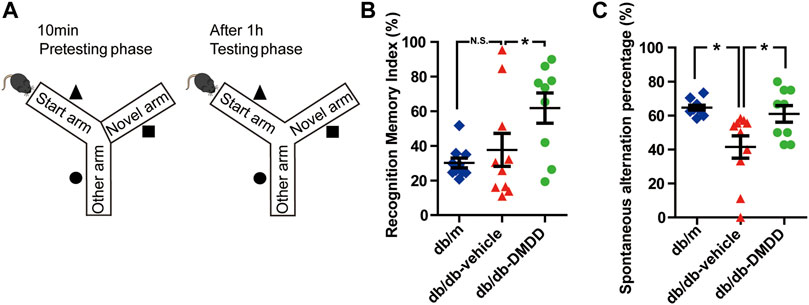
FIGURE 2. DMDD attenuated spatial working memory impairment in db/db mice. (A) Schematic diagram of the Y-maze. Figures representing recognition memory index (B) and spontaneous alternation percentage (C) in db/m-control (n = 10), vehicle-treated db/db (n = 10), and DMDD-treated db/db (n = 9) mice. Representing recognition memory index and spontaneous alternation percentage are shown by scatter plot. All data are shown as means ± SD. *p < 0.05 for one-way ANOVA test.
Novel object recognition task was executed using the following steps(Lueptow, 2017): 1) Habituation: the mouse was allowed to explore freely in the empty test tank for 10 min per day for 2 days. 2) Training: 24 h after habituation, two indistinguishable objects were placed in the symmetric quadrants of the arena, and then the mouse was put in the tank and allowed free exploration for 10 min. 3) Testing: 24 h after training, one of the previous objects was replaced with a new object at the same location. The mouse was again placed in the tank and allowed free exploration for 10 min. As illustrated in Figure 3A, the whole testing phase was recorded and analyzed by a behavioral analyzing system (Shanghai Xinruan Information Technology Co. Ltd). The following formula was used to calculate short-term memory: discrimination index = [(exploration time at novel object - exploration time at old object)/(exploration time at novel object + exploration time at old object)]. Preference index = exploration time at novel object/(exploration time at novel object + exploration time at old object).
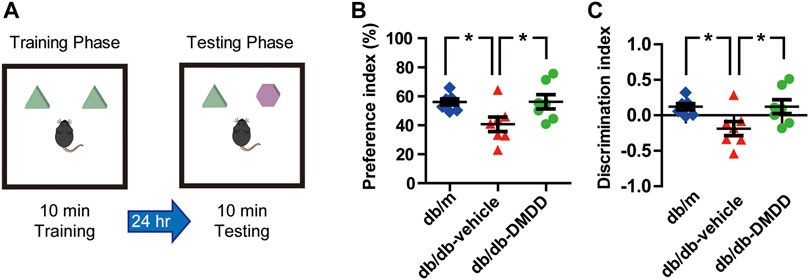
FIGURE 3. DMDD attenuated object recognition memory impairment in db/db mice. (A) Schematic diagram of the object recognition task during the training phase and testing phase. Preference index (B) and discrimination index (C) increased significantly in DMDD-treated db/db group compared with vehicle-treated db/db group during the testing phase. All data are shown as means ± SD. *p < 0.05 for one-way ANOVA test.
The HT22 mouse hippocampal neuron cells were obtained from GuangZhou Jennio Biotech Co., Ltd. The HT22 cell line was genotyped for identity and was tested routinely for Mycoplasma contamination. The HT22 cell was maintained in DMEM medium (Gibco) commonly with 10% FBS (Gibco) at 37°C in cell incubator with 5% CO2. Normal glucose (NG, 25 mM) DMEM medium contained 25 mM glucose. Additional 25 mM glucose was added into NG DMEM to prepare the high-glucose (HG, 50 mM) DMEM medium. Cells were treated with vehicle (DMSO) or DMDD for 48 h after incubation with NG or HG DMEM medium for 24 h.
The HT22 cells were seeded at 1.5 × 103/well in a 96-well cell culture plate and then treated with HG medium as described above. Cell viability was measured by CellTiter 96® AQueous one solution cell proliferation assay system (Promega) according to the instructions of the manufacturer. Cell viability or death rates were calculated by normalizing to the 0 μM DMDD or NG medium group. Apoptosis was detected by Hoechst 33342 (7 mM) and propidium iodide (2 mM) (Invitrogen). Images were captured by Olympus microscope.
Shbio mouse lncRNA array V6.0 (4 × 180K, Shanghai Biotechnology Corporation) is designed for global profiling of mouse lncRNAs and mRNA transcripts and includes 64,221 lncRNA and 26,531 mRNA probes. Total RNA from mice hippocampus tissues and cell lines was extracted using RNeasy mini kit (Qiagen). RNA integrity was checked for an RIN number by an Agilent Bioanalyzer 2,100. For lncRNA microarray analysis, total RNA was amplified and labeled by Labeling Kit (Agilent technologies). Slides were hybridized with labeled cRNA and were scanned by Agilent Microarray Scanner. Microarray data were extracted by Feature Extraction software 12.0 (Agilent technologies) and normalized by Quantile algorithm. To reveal the potential biological processes associated with DMDD regulatory genes, Gene Ontology enrichment analysis and KEGG pathway analysis were performed based on significantly different expression genes and lncRNA target genes using clusterProfiler in R/bioconductor.
Total RNA was reverse transcribed into cDNA using Reverse Transcription Kit (Thermo Fisher Scientific). qRT-PCR was performed using Power SYBR™ Green (Thermo Fisher Scientific) and ABI 7300 detection system (Applied Biosystems). qRT-PCR data were normalized to the expression of housekeeping gene GAPDH, and relative expressions were calculated using the 2−ΔΔCt method. The oligonucleotide primers were shown as follows: Hif3α (sense: 5′-TCGTGCATTCTCATGAGCCC-3′, antisense: 5′-GTTCCTGGGCTGGACAGTTT-3′), Mmp8 (sense: 5′-CCAATGCCTTCCCAGTACCTGA-3′, antisesce: 5′-GGCATTCCTCGAAGACCGGAAT-3′), Timp1 (sense: 5′- CTATAGTGCTGGCTGTGGGG-3′, antisense: 5′-GGACCTGATCCGTCCACAAA-3′), Fam187b (sense: 5′ ACCAACCAGCCGTACTTTGT3′, antisense: 5′-TTGTCAGCTTCCCAGTGGAC-3′). All tests were performed in triplicate, and the data were presented as mean ± SD.
Gene expression datasets GDS810 (Blalock et al., 2004) and GDS2519 (Scherzer et al., 2008) were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and analyzed by GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/). These datasets include AD (GDS810, brain hippocampi samples from nine healthy normal control and 22 ADs at various stages including 7 incipient,8 moderate, and 7 severe AD) and PD (GDS2519, blood samples from 22 healthy control and 50 PDs) with healthy control. These two microarray datasets were detected by the same platform (HG-U133A) Affymetrix Human Genome U133A Array.
The 3D format of DMDD was obtained from the ZINC15 drug database (http://zinc15.docking.org/) (Sterling and Irwin, 2015). The x-ray crystal structure of Hif3α was obtained from the RCSB PDB protein databank (http://www.rcsb.org/). The water molecules and ligand molecules were removed from the crystal structures by Discovery Studio 4.5 Client before the docking simulation began. Docking simulations of DMDD and Hif3α was carried out by PyRx software. Interaction energies were calculated for predicting docking positions and selecting the binding pose that had the lowest binding energy (kcal mol−1). Results were visualized and analyzed using Discovery Studio 4.5 Client.
Cells or hippocampus tissues were homogenized in RIPA buffer (SIGMA). Concentration of protein was measured using BCA Assay Kit (Beyotime). Equivalent protein was separated by polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore). After blocking the membranes with 5% nonfat milk, the membranes were incubated overnight at 4°C with primary antibodies against Tau phospho Serine 199/202 (1:1,000, Cat. AB9674, Millipore), Tau-1 (1:1,000, Cat. MAB3420, Millipore), phospho-α-Synuclein (Ser129) (1:1,000, Cat. MABN826, Millipore), α-Synuclein (1:1,000, Cat. 2,647, Cell signaling), HIF-3 alpha (1:1,000, Cat. NB100-2,529, Novus), and gapdh (1:1,000, Cat. 2,118, Cell signaling) overnight at 4°C. Then membranes were incubated with HRP-conjugated secondary antibody (1:2,000 dilution) and detected using Chemiluminescent Substrate (Thermo Scientific). Band intensity was quantified by ImageJ.
Data were analyzed using GraphPad Prism 5 and SPSS 19.0 software. To identify gene expression patterns, an unsupervised hierarchical cluster analysis using un-centered average linkage was performed using Cluster v3.0 after median-centering genes and arrays. Heat maps were visualized using Tree View software. The data were analyzed by one-way ANOVA followed by Tuckey’s tests. A value of p < 0.05 was considered significant.
Compared with db/m control mice, the blood glucose levels (Figure 1C) and the total area under the curve (AUC) of the blood glucose (Figure 1E) were significantly increased in vehicle-treated db/db mice (p < 0.05). DMDD treatment (50 mg/ kg) significantly reduced hyperglycemia (p < 0.05) in DMDD-treated db/db mice at weeks 3 and 4 (Figure 1C). The AUC of blood glucose was significantly decreased after DMDD treatment (p < 0.05) (Figure 1E). A marked increase in vehicle-treated db/db mice body weight was observed at the first week compared with db/m control mice (Figures 1B,D). However, DMDD treatment had no meaningful effect on body weight in DMDD-treated db/db mice (Figures 1B,D).
The spatial working memory of mice was investigated using Y maze (Figure 2A). As shown in Figure 2C, compared with the db/m-control mice, the spontaneous alternation percentage was significantly decreased (p < 0.05), and DMDD treatment reversed this change (p < 0.05) in vehicle-treated db/db mice. Meanwhile, the percentage of duration in the novel arm (measured as recognition memory index) was significantly higher in DMDD-treated db/db mice than in the vehicle-treated db/db mice (p < 0.05) (Figure 2B). These results suggested that DMDD treatment had significant positive effects on the spontaneous alternation and improved spatial working memory in db/db mice.
We performed novel object recognition (NOR) test to examine the object recognition memory and short-term memory of mice (Figure 3A). During the training phase, no significant difference was found among each group. During the testing phase, preference and discrimination index of vehicle-treated db/db mice was decreased significantly compared with that of db/m-control mice (p < 0.05) (Figures 3B,C), which suggested that vehicle-treated db/db mice had decreased abilities to recognize a novel object. DMDD treatment significantly improved the preference and discrimination index (p < 0.05) (Figures 3B,C). Collectively, our results showed that administration of DMDD attenuated object recognition memory impairment in db/db mice.
Hyperphosphorylation of tau and α-synuclein is an important feature of neurodegenerative disease. We further explored whether DMDD treatment could influence the expression of tau and α-synuclein in the hippocampi of db/db mice. As shown in Figures 4A,B, compared with db/m control mice, tau proteins phosphorylated at Serine 199/202, the p-Tau/Tau ratio, p-α-synuclein, α-synuclein, and oligomers of α-synuclein were markedly increased in the hippocampus of vehicle-treated db/db mice (p < 0.05). The expression of p-tau 199/202, the ratio of p-Tau 199/202/total tau, p-α-synuclein (Ser129), α-synuclein, and oligomers of α-synuclein were increased by 1.35-, 1.50-, 2.05-, 2.10-, and 3.34-fold in db/db mice compared with db/m control mice (p < 0.05). After DMDD treatment, the expression of Tau-1 was significantly decreased (p < 0.05) in db/db mice. Although p-α-synuclein (Ser129, 18, and 46 kDa), α-synuclein, and its oligomers protein levels showed a decreasing trend after DMDD treatment, there was no significant difference between db/db-vehicle and db/db-DMDD group.
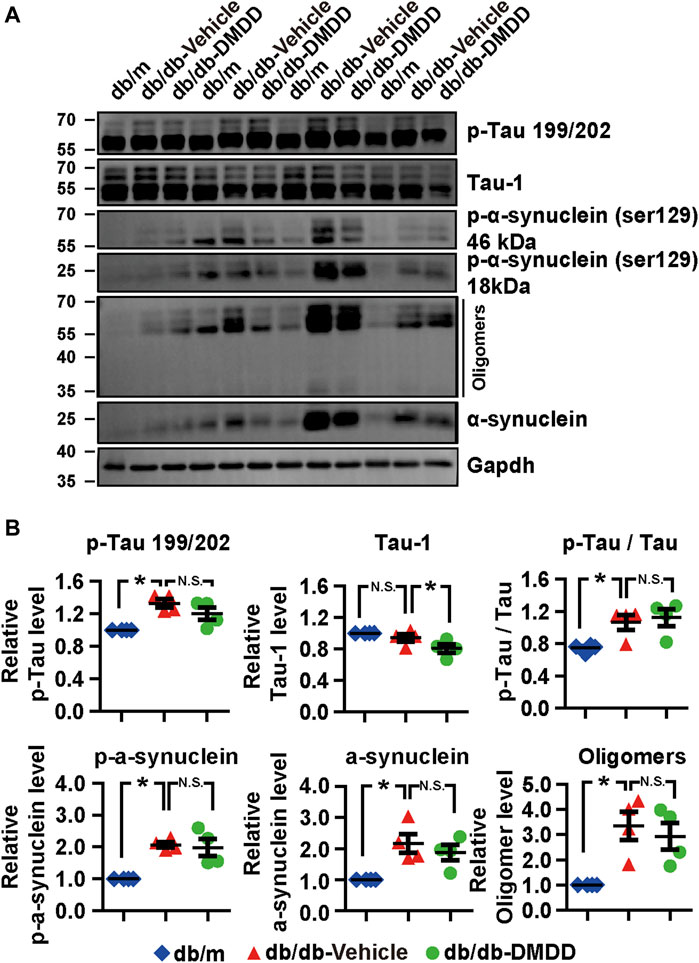
FIGURE 4. Neurodegeneration disease-related protein expression was reduced after DMDD treatment in the type 2 diabetes (T2D) db/db mice hippocampus. (A) The expressions of neurodegeneration disease-related proteins in the hippocampi of db/m-control mice (n = 4), vehicle-treated db/db (n = 4), and DMDD-treated db/db (n = 4) mice were analyzed by Western immunoblotting. (B) The relative expression of proteins was standardized by gapdh and db/m-control group for each sample. Data are expressed as means ± SD of four independent samples, *p < 0.05 for one-way ANOVA test.
In order to reveal the underlying mechanisms and potential targets of DMDD treatment in db/db mice, we performed RNA array on db/db-vehicle (n = 3) and db/db-DMDD (n = 3) mice hippocampal samples to find the genes that changed after DMDD treatment. Results showed that a total of 34 mRNAs and 120 lncRNAs probes had significantly different expression in hippocampi of DMDD treated mice compared with that of db/db-vehicle mice (p < 0.05, |log2 (fold change)| ≥ 0.5). Of these, 12 mRNAs and 59 lncRNAs were upregulated, and 22 mRNAs and 61 lncRNAs were downregulated in DMDD-treated db/db mice (Supplementary Table S1). Further analysis identified 11 lncRNAs and 4 mRNAs as candidate genes (p < 0.05, |log2 (fold change)| ≥ 1.00) (Figures 5A,B). We predicted cis and trans mRNA targets of these 11 lncRNAs by the sequence of lncRNA (Supplementary Table S2). Unfortunately, there was no intersection between the candidate mRNAs and the predicted lncRNA targets. To understand the function of candidate mRNAs (Mmp8, Hif3α, Timp1, and Fam187b), Gene Ontology enrichment analysis was performed and three cellular components were identified, including proteinaceous extracellular matrix (p = 0.0012), extracellular matrix (p = 0.0016), and extracellular space (p = 0.017). Eight biological processes were identified including proteolysis (p = 0.021), apoptotic process (p = 0.027), programmed cell death (p = 0.028), cell death (p = 0.031), death (p = 0.031), catabolic process (p = 0.032), negative regulation of cellular metabolic process (p = 0.043) and negative regulation of macromolecule metabolic process (p = 0.046). Molecular functions were identified including metalloendopeptidase inhibitor activity (p = 0.0018), metalloenzyme inhibitor activity (p = 0.0018), metalloenzyme regulator activity (p = 0.021), metalloendopeptidase activity (p = 0.017), protease binding (p = 0.020), growth factor activity (p = 0.024), and transcription corepressor activity (p = 0.024) (Figure 5C). We validated Mmp8, Hif3α, Timp1, and Fam187b expression in hippocampi of vehicle and DMDD-treated db/db mice by independent real-time PCR. The result showed that Hif3α (p = 0.0033) and Mmp8 (p = 0.0047) were significantly lower in db/db-DMDD mice than in db/db-vehicle mice. Although Timp1 decreased after DMDD treatment, there was no significant difference (p > 0.05) (Figure 5D). The Ct value of Fam187b was too low to detect (Ct value >40). Based on these results, we chose Mmp8 and Hif3α as DMDD potential targets for the following study.
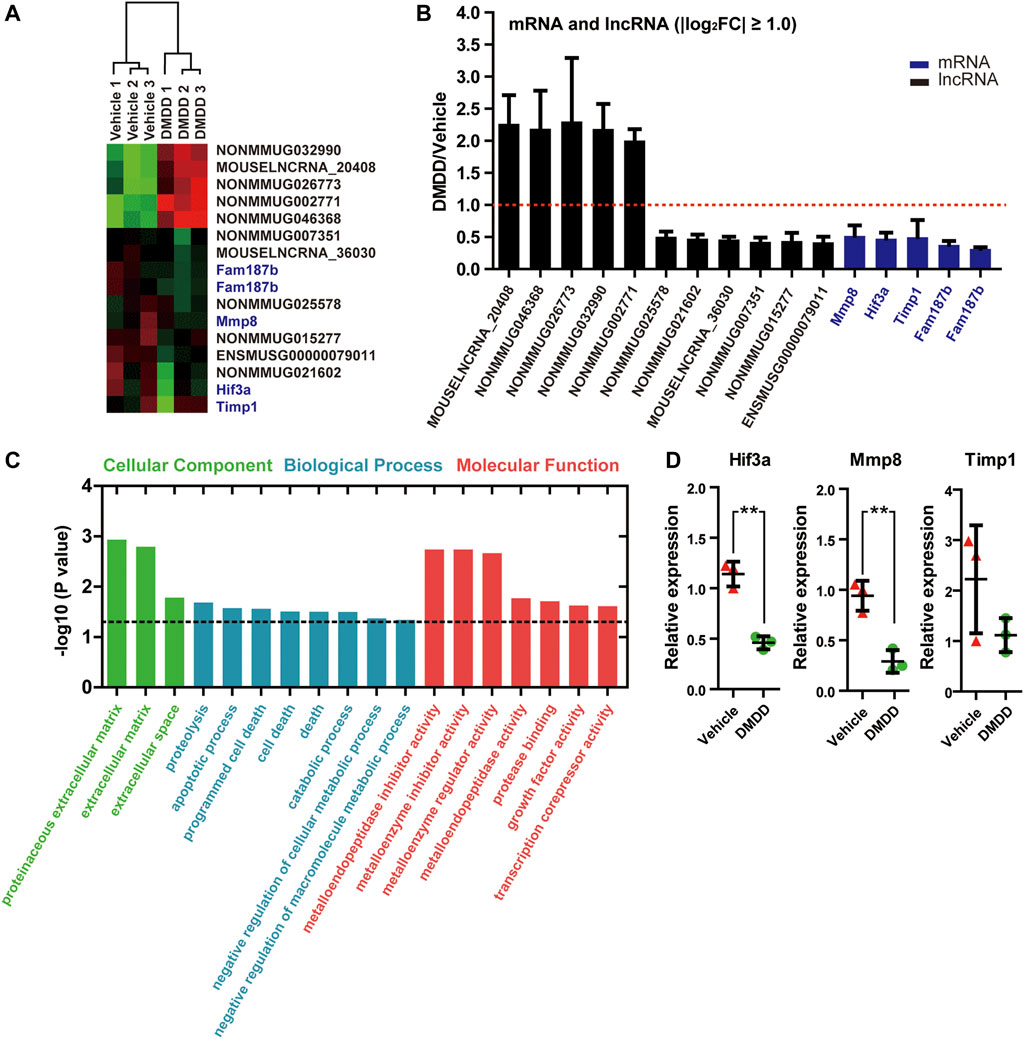
FIGURE 5. mRNAs and lncRNAs regulations in the hippocampi of DMDD-treated db/db mice. (A) Heat maps showing 16 different expressed genes between vehicle (n = 3) and DMDD (n = 3)-treated db/db mice, including 11 lncRNAs and5 mRNAs (fold change >2.0 and p < 0.05). Columns represent samples, rows represent lncRNAs (black) and mRNAs (blue), red means high expression, and green means low expression. (B) Bar graph showing altered mRNAs and lncRNAs in DMDD-treated T2D db/db mice hippocampus (n = 3) using microarray analysis. Vehicle group (n = 3) is represented by red dashed line. (C) Gene Ontology enrichment analysis of the different expressions mRNAs in vehicle (n = 3) and DMDD (n = 3)-treated db/db mice. A value of p equal to 0.05 is represented by a black dashed line. (D) Expressions of hypoxia-inducible factor-3α (Hif3α), Mmp8, and Timp1 were verified in three pairs of vehicle and DMDD-treated db/db mice by qRT-PCR. All data are shown as means ± SD. * p < 0.05, ** p < 0.01 for Student’s t-test.
Due to the fact that T2D and neurodegeneration disease shared the overlapping pathophysiological mechanisms and pathways, we detected Mmp8 and Hif3α mRNA expression value in AD and PD samples by analyzing gene expression datasets in GEO. Two related datasets were downloaded and analyzed, including expression profiling of AD brain hippocampi (GDS810, 9 healthy normal control and 22 ADs at various stages including 7 incipient, 8 moderate, and 7 severe AD) (Blalock et al., 2004) and expression profiling of PD blood (GDS2519, 22 healthy control, and 50 PDs) (Scherzer et al., 2008). The results showed that no significant difference was found for Mmp8 expression between healthy control and AD or PD (p > 0.05). However, Hif3α was significantly increased in AD and PD compared with healthy control (p < 0.05, Figures 6A,C). Remarkably, the expression of Hif3α was significantly higher in severe AD than in incipient AD (p < 0.05, Figure 6B).
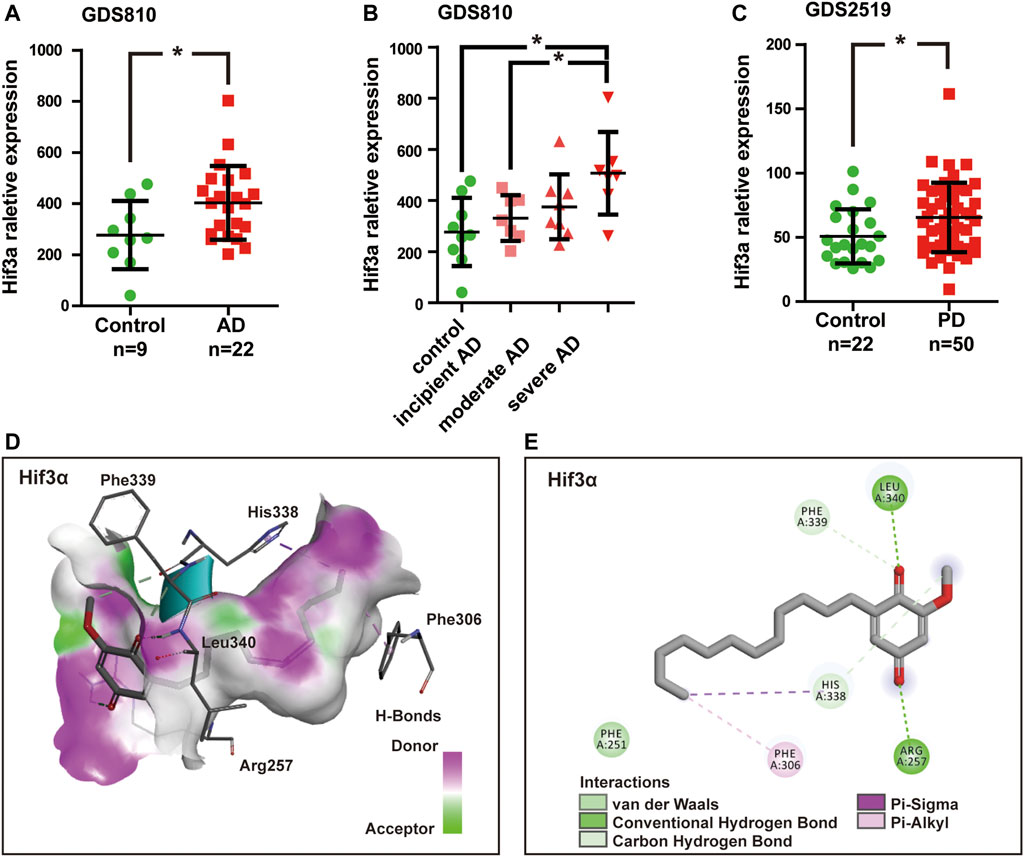
FIGURE 6. Hif3a mRNA expression was significantly increased in Alzheimer’s disease (AD) and Parkinson’s disease (PD). (A) Hif3a expression levels in AD and healthy control hippocampi in GDS810 data set (9 healthy control vs. 22 ADs). (B) Hif3a expression level was significantly higher in severe AD than in incipient AD in GDS810 data set (9 healthy control vs. 22 ADs at various stages including 7 incipient, 8 moderate, and 7 severe ADs). (C) Hif3a expression levels in PD and healthy control blood samples in GDS2519 data set (22 healthy control vs. 50 PDs). (D) DMDD binding with the pocket of Hif3a is composed of hydrogen bonds and the interaction pattern of DMDD with the residues. (E) A 2D diagram between the Hif3a and residues.*p < 0.05 for Student’s t-test.
A molecular docking model was constructed to further explore the mechanism of interaction between DMDD and Hif3α (PDB, 4WN5). Processing of the DMDD included energy minimized. The refinement of structure of DMDD was used for the dock. PyRx was used for the docking studies. The docked conformation corresponding to the lowest binding energy was selected as the most probable binding conformation. The result showed that DMDD could stay in a binding pocket surrounded by Hif3α amino acid Arg257, Phe306, His338, Phe339, and Leu340 (Figures 6D,E). These suggested that Hif3a was not only a potential target of DMDD, but also a diagnostic biomarker of AD and PD.
In order to further confirm the neuroprotection of DMDD, the cell injury model was established by high-glucose treatment in HT22 cells. The protective effects of DMDD were investigated by MTS assay. The result showed that the apparent cytotoxicity could be detected when HT22 cells were cultured for 48 h with high concentrations of DMDD (7.00 μM) (Figures 7A,B). However, there was no obvious difference in cell viability between the DMDD-treated groups of different low concentrations (0.22, 0.44, 0.88, 1.75, and 3.50 μM) and the vehicle group (p > 0.05) (Figure 7B). As shown in Figure 7C, the cell viability of HT22 cells was significantly decreased after high-glucose treatment for 48 h (p < 0.05), and the administration of DMDD at concentrations of 0.44, 0.88, 1.75, and 3.50 μM obviously inhibited the decrease in cell viability induced by high glucose in HT22 cells (p < 0.05) (Figure 7C). In addition, the treatment with 1.75 μM of DMDD reduced Tau-1 protein levels (p < 0.05) and treatment with 3.50 μM of DMDD reduced p-Tau 199/202 protein levels (p < 0.05) compared with the high glucose-treated HT22 cells (Figures 7D,E). These results indicated that DMDD treatment had a protective effect on high glucose-induced neural damage.
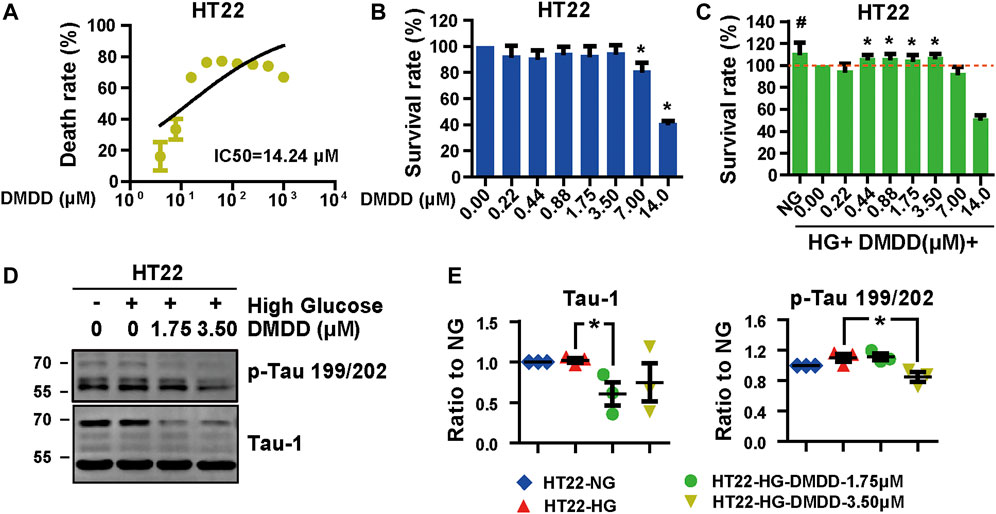
FIGURE 7. DMDD reduced neurodegeneration disease-related protein expression in HT22 cells. (A) The dose-effective curve of DMDD on HT22 cells. (B) DMDD treatment had no obvious cytotoxicity with concentrations under 3.50 μM in HT22 cell. (C) The protection effect of DMDD on the high glucose-treated HT22 cells. (D) AD-related proteins were tested by Western blotting. (E) Relative expression of proteins was standardized by gapdh and HT22-NG group for each sample. Data are expressed as means ± SD, *p < 0.05 for Student’s t-test.
Considering that Hif3a is potential target for DMDD and is also a pro-apoptotic factor (Torii et al., 2011), we performed Hoechst and PI staining to evaluate the potential effect of DMDD on high glucose-induced apoptosis in HT22 cells. The results showed that cell apoptosis and death were significantly increased after high glucose treatment in HT22 cells, whereas DMDD prevented high glucose induced-apoptosis and death in HT22 cells (Figures 8A,B). The expression levels of apoptosis markers further validated the above results. Western blot analysis showed that high glucose upregulated the expression of Hif3a, cleaved parp, Bad, Bax, Caspase 9, cleaved Caspase 9 (37 kDa), and cleaved Caspase3 in HT22 cells (Figure 8C). DMDD treatment inhibited high glucose-induced expression of Hif3a, cleaved parp, Bad, Bax, caspase 9, cleaved caspase 9 (37 kDa), and cleaved caspase 3 in HT22 cells (Figure 8C). Moreover, DMDD exerts a dose-dependent effect on the expression of Hif3a, Bad, Bax, caspase 9, cleaved caspase 9 (37 kDa), and cleaved caspase 3 (Figure 8C). Similar results were also demonstrated in the hippocampi of db/db mice. As shown in Figures 8D,E, there were significantly higher Hif3a, caspase 3, and Bad expression in vehicle-treated db/db group than in db/m group (p < 0.05). However, the expression levels of Hif3a, cleaved Parp, caspase 3, and Bax were significantly reduced in DMDD-treated db/db group compared with db/db-vehicle group (p < 0.05). These results indicated that DMDD treatment decreased pro-apoptotic factor Hif3a expression and protected neurons from high glucose-induced apoptosis in vivo and in vitro.
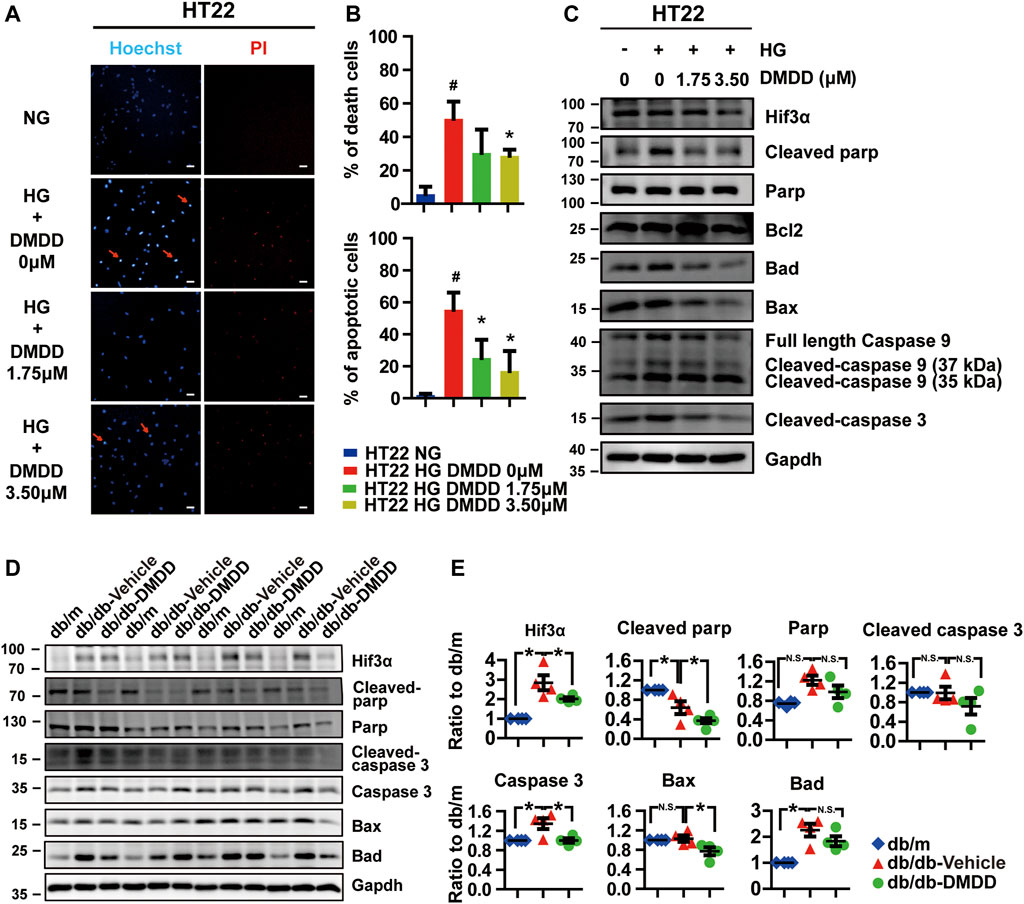
FIGURE 8. DMDD prevented high glucose-induced apoptosis in HT22 cells and hippocampi of T2D db/db mice. (A) Images of Hoechst 33342 and PI staining in HT22 cells pre-incubated with DMDD (0, 1.75, 3.50 μM) for 48 h in normal glucose (NG) or high-glucose (HG) medium. Scale bars, 20 mm. (B) Quantification of dead and apoptotic cells (n = 4). (C) HT22 cells were incubated with DMDD (0, 1.75, 3.50 μM) for 48 h in NG or HG medium. Hif3α and apoptosis-related proteins were detected by Western blot. (D) The expressions of HIF3α and apoptosis-related protein in db/m-control mice (n = 4), vehicle-treated db/db (n = 4), and DMDD-treated db/db (n = 4) mice hippocampus tissues were measured by Western blotting. (E) Relative expression of proteins was standardized by gapdh and db/m-control group for each sample. Data are expressed as means ± SD, * p < 0.05 for one-way ANOVA test.
Type 2 diabetes mellitus (T2DM) is a progressive, chronic disease characterized by hyperglycemia and affects millions of people worldwide. The high incidence of neurodegenerative disease in patients with T2DM is supported by epidemiological data, and it is well recognized that T2DM is an independent risk factor of cognitive impairment (Sims-Robinson et al., 2010). In fact, mounting evidence showed that insulin resistance and deficiency could lead to structural and functional alterations in the brain. This suggests that overlapping pathological mechanisms exist in neurodegenerative diseases and diabetes (Craft and Watson, 2004). Hence, antidiabetic agents might have potential therapeutic benefits for the treatment of diabetic cognitive impairment.
2-Dodecyl-6-methoxycyclohexa-2,5-diene-1,4-dione (DMDD), an active compound was purified from the roots of Averrhoa carambola L. Previous studies have reported that DMDD improves cognitive impairment by protected against neuronal apoptosis in APP/PS1 Alzheimer’s disease mice (Wei et al., 2018). In addition to anticognitive impairment activity, DMDD had an antidiabetic activity by decreasing AGE expression and downregulating key genes in the NFκB–TGFβ1 pathway in KKAy mice. DMDD alleviates diabetic nephropathy by mitigating kidney damage and inflammation via the inhibition of the TLR4/MyD88/NF-κB signaling pathway (Lu et al., 2019). Notably, the NF-κB signaling pathway plays a critical role across many cellular processes, especially neuronal development (Garcia-Garcia et al., 2021). Suppression of the NF-κβ signaling pathway can prevent diabetes-associated cognitive impairment (Kuhad et al., 2009). Although therapeutic actions of DMDD on diabetes mellitus and Alzheimer’s disease were studied, the therapeutic potential of DMDD on diabetic cognitive impairment and its underlying mechanisms remains unclear.
Emerging evidence showed that the pathogenesis of AD, PD, and T2DM are synergistic (Moran et al., 2015; Pagano et al., 2018). Abnormal insulin level in T2DM promotes Lewy body (composed of α-synuclein) formation, tau phosphorylation, and ultimately leads to diabetic cognitive impairment (Sato and Morishita, 2014). Therefore, it is particularly important to choose a suitable animal model for studying diabetic cognitive impairment. Several animal models are used to investigate the effect of spontaneous type 2 diabetes on neurodegenerative disease. Most of them are characterized by leptin impairment. Leptin plays a key role in immunity, regulation of insulin secretion, and hippocampal behavioral deficits (Bjørbaek and Kahn, 2004; Yadav et al., 2013; Platt et al., 2016). Studies showed that the db/db mouse model, which carries mutated leptin receptor, exhibited metabolic syndrome of T2DM (such as hyperglycemia, overweight, dyslipidemia, insulin resistance, and inflammation), also accompanied with cognition impairment symptoms (Yermakov et al., 2019). In the present study, we observed significant hyperglycemia, excessive body weight, spatial working memory impairment, and object recognition memory impairment in db/db mice compared with db/m control mice. DMDD treatment improves hyperglycemia and learning memory impairment in db/db mice. In terms of pathological mechanism, tau and α-synuclein, which are the pathologic characteristics of AD and PD, were significantly upregulated in the hippocampus of db/db mice. DMDD treatment reduced the abnormal protein expressions of Tau-1 and α-synuclein in the hippocampi of db/db mice. These results confirmed that the db/db mice model could be effectively applied to diabetic cognitive impairment study. Furthermore, DMDD protects db/db mice from hyperglycemia and neural damage by decreasing the expression of Tau-1 and α-synuclein.
To date, only a few mRNAs and lncRNAs have been reported to be dysregulated and functionally characterized in diabetic cognitive impairment. Microarray provides a comprehensive assessment of expressed probes and allows identification of mRNAs and lncRNAs with diagnostic, prognostic, and functional potential in diabetic cognitive impairment (Ci et al., 2018; Santiago et al., 2019). To find the potential target genes of DMDD and cell signaling associated with diabetic cognitive impairment treatment, we test the gene expression profile in the hippocampi of db/db mice by RNA microarray. Eleven lncRNAs and four mRNAs had significantly different expressions in the hippocampi of DMDD-treated mice compared with db/db-vehicle mice. In order to identify molecular mechanisms of lncRNA, we predicted mRNA targets of these 11 lncRNAs by the sequence of lncRNA. Unfortunately, there was no intersection between the candidate mRNAs and the predicted lncRNA targets. GO analysis indicated that the potential target mRNAs of DMDD (Mmp8, Hif3α, Timp1, and Fam187b) were correlated with proteolysis, apoptotic process, programmed cell death, protease binding, and other important biological processes. Among them, defective proteolysis (Homma and Fujii, 2020), apoptosis (Li et al., 2020), and cell death (Nguyen et al., 2020) are closely related to neurodegenerative and metabolic diseases. We validated the potential target mRNA expression of DMDD by independent real-time PCR. The results showed that Hif3α and Mmp8 were significantly lower in the hippocampi of DMDD-treated db/db mice than in that of db/db-vehicle mice. These mRNAs could potentially serve as DMDD targets in diabetic cognitive impairment treatment.
Hypoxia-inducible factor-3α (Hif3a) is member of hypoxia-inducible factors (HIFs). As transcriptional factors, the HIF family has a vital role in transcriptional response to hypoxic stress by regulating different target genes. Compared with HIF1A and HIF2A, much less is known about HIF3A. This is, in part, due to the existence of multiple HIF3A variants that made it challenging to elucidate HIF3A functions. Recently, studies have shown that HIF3A expression and methylation are related to adipose tissue dysfunction (Pfeiffer et al., 2016), insulin resistance (Main et al., 2016), and gestational diabetes mellitus (Zhang et al., 2019). In addition, researchers also noted that microglial cells expressed HIF3A under inflammatory conditions, and hypoxia treatment resulted in a marked upregulation of HIF3A mRNA levels in the adult rat cortex, hippocampus, lungs, kidneys, and myocardial tissue (Heidbreder et al., 2003). These pathophysiological events are believed to be key factors contributing to neurodegeneration disease and diabetes mellitus progression. Significantly, a splicing variant of HIF3A, named IPAS (Makino et al., 2002; Torii et al., 2011; Kasai et al., 2017), has recently been found to act as a pro-apoptotic factor in the mitochondria by binding directly to the pro-survival Bcl-2 family and activating caspase 3 in PC12 cells. In addition, a previous study indicated that IPAS might take part in the pathogenesis of both sporadic and familial PD by acting as a substrate of PINK1 and Parkin and mediating neuronal cell death (Torii et al., 2015). In this study, Hif3a was further validated for its expression in two independent AD and PD microarray data sets. Moreover, we found that Hif3α was significantly increased in the hippocampi of AD patients and the blood of PD patients compared with that of healthy control, and related to the severity of AD. Then we conducted molecular docking between DMDD and Hif3α. The result showed that DMDD could stay in a binding pocket surrounded by Hif3α amino acid Arg257, Phe306, His338, Phe339, and Leu340. The mRNA and protein levels of Hif3α were upregulated in the hippocampi of db/db mice and high glucose-treated HT22 cells. It suggested that Hif3a was not only a potential target of DMDD to protect neurons from damage, but also a diagnostic biomarker of neurodegenerative disease.
Apoptosis plays a crucial role in diabetes-induced cognitive and memory impairment. Mitochondrial dysfunction is one of the main causes of apoptosis and is also a key factor of cognitive deficits in neurodevelopmental disorders and neurodegenerative diseases (Cheng et al., 2019). Growing evidence showed that even before the occurrence of AD pathological symptoms or any detectable cognitive decline, there are mitochondrial dysfunctions that happened (Iglesias-Gato et al., 2016). Interaction between dysfunctional mitochondria and impaired insulin signaling can finally promote the development of AD in diabetic patients (Jadiya et al., 2019). When mitochondrial dysfunction happened, antiapoptotic proteins Bcl-2 and Bcl-xL participated in this process by controlling mitochondrial permeability. They exist in the outer mitochondrial membrane and inhibit the release of pro-apoptotic protein cytochrome c from mitochondria. Following death signal, the pro-apoptotic proteins Bad, Bid, Bax, and Bim, which exist in the cytosol, will be transferred to the mitochondria, promote the release of cytochrome c to the cytosol, activate the caspase cascade, and lead to cell death (Pradelli et al., 2010; Ola et al., 2011). Apoptosis caused by mitochondrial dysfunction has been implicated in the pathogenesis of diseases characterized by excessive cell death, such as neurodegenerative diseases and diabetes (Quintanilla et al., 2009). Hif3a, as a pro-apoptotic protein, can block the interaction between Bcl-xL and Bax, and inhibits the antiapoptosis function of Bcl-xL (Torii et al., 2011). In this study, we found that cell apoptosis and death were significantly increased after high glucose treatment, and DMDD prevents high glucose-induced apoptosis and death in HT22 cells. Hif3a and apoptosis-related proteins cleaved parp, Bad, Bax, caspase 9, cleaved caspase 9, and cleaved caspase 3 were upregulated after high-glucose treatment. DMDD treatment inhibited high glucose-induced expression of Hif3a, cleaved parp, Bad, Bax, caspase 9, cleaved caspase 9, and cleaved caspase 3 in vivo and in vitro. These results indicated that DMDD treatment protected neurons from high glucose-induced apoptosis by regulating apoptosis-related protein Hif3α. However, the function and mechanism of Hif3a in the pathogenesis of high glucose-induced cognitive deficits is still unknown, and further research is needed.
We studied the neuroprotective effects of DMDD to high glucose-induced HT22 hippocampal neuron cell injury in vitro and learning memory impairment of db/db mice in vivo. The results indicated that DMDD treatment reduced the abnormal protein expressions of Tau-1 in the hippocampi of db/db mice. Moreover, DMDD treatment inhibited hyperglycemia-induced apoptosis through inhibiting the pro-apoptotic protein Hif3a, cleaved parp, caspase 3, and Bax in hippocampal neurons. Our findings suggest that DMDD has the potential to prevent and treat cognitive impairment in people with diabetes.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174378
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Guangxi Medical University.
LW, JC, and QX carried out the majority of the experiments and wrote the original draft. XL, XY, QS, SC, and KD participated in some of the experiments. RH and CZ designed the project, analyzed the data, and reviewed and edited the manuscript.
This work was supported by the National Natural Science Foundation of China (81670750, 81971191, 81360123, 61627807, 81803564, and 81760665), China Postdoctoral Science Foundation Grant (2018M633619XB), and Natural Science Foundation of Shandong Province, China (ZR2019ZD39).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.708141/full#supplementary-material
Ashraf, G. M., Greig, N. H., Khan, T. A., Hassan, I., Tabrez, S., Shakil, S., et al. (2014). Protein Misfolding and Aggregation in Alzheimer's Disease and Type 2 Diabetes Mellitus. CNS Neurol. Disord. Drug Targets 13, 1280–1293. doi:10.2174/1871527313666140917095514
Athauda, D., and Foltynie, T. (2016). Insulin Resistance and Parkinson's Disease: A New Target for Disease Modification. Prog. Neurobiol. 145–146, 98–120. doi:10.1016/j.pneurobio.2016.10.001
Bharadwaj, P., Wijesekara, N., Liyanapathirana, M., Newsholme, P., Ittner, L., Fraser, P., et al. (2017). The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimers Dis. 59, 421–432. doi:10.3233/JAD-161192
Bjorbaek, C., and Kahn, B. B. (2004). Leptin Signaling in the central Nervous System and the Periphery. Recent Prog. Horm. Res. 59, 305–331. doi:10.1210/rp.59.1.305
Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R., and Landfield, P. W. (2004). Incipient Alzheimer's Disease: Microarray Correlation Analyses Reveal Major Transcriptional and Tumor Suppressor Responses. Proc. Natl. Acad. Sci. U S A. 101, 2173–2178. doi:10.1073/pnas.0308512100
Chatterjee, S., Peters, S. A., Woodward, M., Mejia Arango, S., Batty, G. D., Beckett, N., et al. (2016). Type 2 Diabetes as a Risk Factor for Dementia in Women Compared with Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes care 39, 300–307. doi:10.2337/dc15-1588
Cheng, H., Gang, X., Liu, Y., Wang, G., Zhao, X., and Wang, G. (2019). Mitochondrial Dysfunction Plays a Key Role in the Development of Neurodegenerative Diseases in Diabetes. Am. J. Physiol. Endocrinol. Metab. 318 (5), E750–E764. doi:10.1152/ajpendo.00179.02019
Ci, L. Y., Liu, D. S., Yang, J. Q., Liu, Y. Z., Li, C. L., Zhang, X., et al. (2018). Expression of Long Non-coding RNA and mRNA in the hippocampus of M-ice with T-ype 2 D-iabetes. Mol. Med. Rep. 18, 4960–4968. doi:10.3892/mmr.2018.9504
Craft, S., and Watson, G. S. (2004). Insulin and Neurodegenerative Disease: Shared and Specific Mechanisms. Lancet Neurol. 3, 169–178. doi:10.1016/S1474-4422(04)00681-7
Garcia-Garcia, V. A., Alameda, J. P., Page, A., and Casanova, M. L. (2021). Role of NF-kappaB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 10 (8), 1906. doi:10.3390/cells10081906
Heidbreder, M., Fröhlich, F., Jöhren, O., Dendorfer, A., Qadri, F., and Dominiak, P. (2003). Hypoxia Rapidly Activates HIF-3alpha mRNA Expression. FASEB J. 17, 1541–1543. doi:10.1096/fj.02-0963fje
Ho, K. H., Yang, X., Osipovich, A. B., Cabrera, O., Hayashi, M. L., Magnuson, M. A., et al. (2020). Glucose Regulates Microtubule Disassembly and the Dose of Insulin Secretion via Tau Phosphorylation. Diabetes 69, 1936–1947. doi:10.2337/db19-1186
Homma, T., and Fujii, J. (2020). Emerging Connections between Oxidative Stress, Defective Proteolysis, and Metabolic Diseases. Free Radic. Res. 54, 931–946. doi:10.1080/10715762.2020.1734588
Iglesias-Gato, D., Wikström, P., Tyanova, S., Lavallee, C., Thysell, E., Carlsson, J., et al. (2016). The Proteome of Primary Prostate Cancer. Eur. Urol. 69, 942–952. doi:10.1016/j.eururo.2015.10.053
Jadiya, P., Kolmetzky, D. W., Tomar, D., Di Meco, A., Lombardi, A. A., Lambert, J. P., et al. (2019). Impaired Mitochondrial Calcium Efflux Contributes to Disease Progression in Models of Alzheimer's Disease. Nat. Commun. 10, 3885. doi:10.1038/s41467-019-11813-6
Kasai, S., Richardson, M. J. E., Torii, S., Yasumoto, K. I., Shima, H., Igarashi, K., et al. (2017). Increase in Proapoptotic Activity of Inhibitory PAS Domain Protein via Phosphorylation by MK2. Febs J. 284, 4115–4127. doi:10.1111/febs.14300
Khacho, M., Harris, R., and Slack, R. S. (2019). Mitochondria as central Regulators of Neural Stem Cell Fate and Cognitive Function. Nat. Rev. Neurosci. 20, 34–48. doi:10.1038/s41583-018-0091-3
Kraeuter, A. K., Guest, P. C., and Sarnyai, Z. (2019). The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 1916, 105–111. doi:10.1007/978-1-4939-8994-2_10
Kuhad, A., Bishnoi, M., Tiwari, V., and Chopra, K. (2009). Suppression of NF-Kappabeta Signaling Pathway by Tocotrienol Can Prevent Diabetes Associated Cognitive Deficits. Pharmacol. Biochem. Behav. 92, 251–259. doi:10.1016/j.pbb.2008.12.012
Kulic, L., Wollmer, M. A., Rhein, V., Pagani, L., Kuehnle, K., Cattepoel, S., et al. (2011). Combined Expression of Tau and the Harlequin Mouse Mutation Leads to Increased Mitochondrial Dysfunction, Tau Pathology and Neurodegeneration. Neurobiol. Aging 32, 1827–1838. doi:10.1016/j.neurobiolaging.2009.10.014
Li, J. M., Zhao, Y., Sun, Y., and Kong, L. D. (2020). Potential Effect of Herbal Antidepressants on Cognitive Deficit: Pharmacological Activity and Possible Molecular Mechanism. J. Ethnopharmacol 257, 112830. doi:10.1016/j.jep.2020.112830
Lu, S., Zhang, H., Wei, X., Huang, X., Chen, L., Jiang, L., et al. (2019). 2-dodecyl-6-methoxycyclohexa-2,5-diene-1,4-dione Isolated from Averrhoa carambola L. Root Ameliorates Diabetic Nephropathy by Inhibiting the TLR4/MyD88/NF-Κb Pathway. Diabetes Metab. Syndr. Obes. 12, 1355–1363. doi:10.2147/DMSO.S209436
Lueptow, L. M. (2017). Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. visualized experiments : JoVE 126, 55718. doi:10.3791/55718
Main, A. M., Gillberg, L., Jacobsen, A. L., Nilsson, E., Gjesing, A. P., Hansen, T., et al. (2016). DNA Methylation and Gene Expression of HIF3A: Cross-Tissue Validation and Associations with BMI and Insulin Resistance. Clin. Epigenetics 8, 89. doi:10.1186/s13148-016-0258-6
Makino, Y., Kanopka, A., Wilson, W. J., Tanaka, H., and Poellinger, L. (2002). Inhibitory PAS Domain Protein (IPAS) is a Hypoxia-Inducible Splicing Variant of the Hypoxia-Inducible Factor-3alpha Locus. J. Biol. Chem. 277, 32405–32408. doi:10.1074/jbc.C200328200
Moran, C., Beare, R., Phan, T. G., Bruce, D. G., Callisaya, M. L., Srikanth, V., et al. (2015). Type 2 Diabetes Mellitus and Biomarkers of Neurodegeneration. Neurology 85, 1123–1130. doi:10.1212/WNL.0000000000001982
Nguyen, T. T., Ta, Q. T. H., Nguyen, T. T. D., Le, T. T., and Vo, V. G. (2020). Role of Insulin Resistance in the Alzheimer’s Disease Progression. Neurochem. Res. 45, 1481–1491. doi:10.1007/s11064-020-03031-0
Ola, M. S., Nawaz, M., and Ahsan, H. (2011). Role of Bcl-2 Family Proteins and Caspases in the Regulation of Apoptosis. Mol. Cel Biochem 351, 41–58. doi:10.1007/s11010-010-0709-x
Ordonez, D. G., Lee, M. K., and Feany, M. B. (2018). α-Synuclein Induces Mitochondrial Dysfunction through Spectrin and the Actin Cytoskeleton. Neuron 97, 108–124.e6. doi:10.1016/j.neuron.2017.11.036
Pagano, G., Polychronis, S., Wilson, H., Giordano, B., Ferrara, N., Niccolini, F., et al. (2018). Diabetes Mellitus and Parkinson Disease. Neurology 90, e1654–e1662. doi:10.1212/WNL.0000000000005475
Pfeiffer, S., Krüger, J., Maierhofer, A., Böttcher, Y., Klöting, N., El Hajj, N., et al. (2016). Hypoxia-inducible Factor 3A Gene Expression and Methylation in Adipose Tissue is Related to Adipose Tissue Dysfunction. Sci. Rep. 6, 27969. doi:10.1038/srep27969
Platt, T. L., Beckett, T. L., Kohler, K., Niedowicz, D. M., and Murphy, M. P. (2016). Obesity, Diabetes, and Leptin Resistance Promote Tau Pathology in a Mouse Model of Disease. Neuroscience 315, 162–174. doi:10.1016/j.neuroscience.2015.12.011
Pradelli, L. A., Bénéteau, M., and Ricci, J. E. (2010). Mitochondrial Control of Caspase-dependent and -independent Cell Death. Cell Mol Life Sci 67, 1589–1597. doi:10.1007/s00018-010-0285-y
Quintanilla, R. A., Matthews-Roberson, T. A., Dolan, P. J., and Johnson, G. V. (2009). Caspase-cleaved Tau Expression Induces Mitochondrial Dysfunction in Immortalized Cortical Neurons: Implications for the Pathogenesis of Alzheimer Disease. J. Biol. Chem. 284, 18754–18766. doi:10.1074/jbc.M808908200
Santiago, J. A., Bottero, V., and Potashkin, J. A. (2019). Transcriptomic and Network Analysis Highlight the Association of Diabetes at Different Stages of Alzheimer’s Disease. Front. Neurosci. 13, 1273. doi:10.3389/fnins.2019.01273
Sato, N., and Morishita, R. (2014). Brain Alterations and Clinical Symptoms of Dementia in Diabetes: Aβ/tau-dependent and Independent Mechanisms. Front. Endocrinol. 5, 143. doi:10.3389/fendo.2014.00143
Scherzer, C. R., Grass, J. A., Liao, Z., Pepivani, I., Zheng, B., Eklund, A. C., et al. (2008). GATA Transcription Factors Directly Regulate the Parkinson's Disease-Linked Gene Alpha-Synuclein. Proc. Natl. Acad. Sci. U.S.A. 105, 10907–10912. doi:10.1073/pnas.0802437105
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial Transport in Neurons: Impact on Synaptic Homeostasis and Neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi:10.1038/nrn3156
Sims-Robinson, C., Kim, B., Rosko, A., and Feldman, E. L. (2010). How Does Diabetes Accelerate Alzheimer Disease Pathology. Nat. Rev. Neurol. 6, 551–559. doi:10.1038/nrneurol.2010.130
Sterling, T., and Irwin, J. J. (2015). ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337. doi:10.1021/acs.jcim.5b00559
Stoothoff, W., Jones, P. B., Spires-Jones, T. L., Joyner, D., Chhabra, E., Bercury, K., et al. (2009). Differential Effect of Three-Repeat and Four-Repeat Tau on Mitochondrial Axonal Transport. J. Neurochem. 111, 417–427. doi:10.1111/j.1471-4159.2009.06316.x
Torii, S., Goto, Y., Ishizawa, T., Hoshi, H., Goryo, K., Yasumoto, K., et al. (2011). Pro-apoptotic Activity of Inhibitory PAS Domain Protein (IPAS), a Negative Regulator of HIF-1, through Binding to Pro-survival Bcl-2 Family Proteins. Cell Death Differ 18, 1711–1725. doi:10.1038/cdd.2011.47
Torii, S., Kasai, S., Suzuki, A., Todoroki, Y., Yokozawa, K., Yasumoto, K. I., et al. (2015). Involvement of Inhibitory PAS Domain Protein in Neuronal Cell Death in Parkinson’s Disease. Cell Death Discov 1, 15015. doi:10.1038/cddiscovery.2015.15
Wei, X., Xu, X., Chen, Z., Liang, T., Wen, Q., Qin, N., et al. (2018). Protective Effects of 2-Dodecyl-6-Methoxycyclohexa-2,5 -Diene-1,4-Dione Isolated from Averrhoa carambola L. (Oxalidaceae) Roots on Neuron Apoptosis and Memory Deficits in Alzheimer’s Disease. Cell Physiol Biochem 49, 1064–1073. doi:10.1159/000493289
Yadav, A., Kataria, M. A., Saini, V., and Yadav, A. (2013). Role of Leptin and Adiponectin in Insulin Resistance. Clin. Chim. Acta 417, 80–84. doi:10.1016/j.cca.2012.12.007
Yaffe, K., Blackwell, T., Kanaya, A. M., Davidowitz, N., Barrett-Connor, E., and Krueger, K. (2004). Diabetes, Impaired Fasting Glucose, and Development of Cognitive Impairment in Older Women. Neurology 63, 658–663. doi:10.1212/01.wnl.0000134666.64593.ba
Yermakov, L. M., Griggs, R. B., Drouet, D. E., Sugimoto, C., Williams, M. T., Vorhees, C. V., et al. (2019). Impairment of Cognitive Flexibility in Type 2 Diabetic Db/db Mice. Behav. Brain Res. 371, 111978. doi:10.1016/j.bbr.2019.111978
Zhang, Y., Chen, Y., Qu, H., and Wang, Y. (2019). Methylation of HIF3A Promoter CpG Islands Contributes to Insulin Resistance in Gestational Diabetes Mellitus. Mol. Genet. Genomic Med. 7, e00583. doi:10.1002/mgg3.583
Zhang, Y., Huang, N. Q., Yan, F., Jin, H., Zhou, S. Y., Shi, J. S., et al. (2018). Diabetes Mellitus and Alzheimer's Disease: GSK-3β as a Potential Link. Behav. Brain Res. 339, 57–65. doi:10.1016/j.bbr.2017.11.015
Zheng, N., Lin, X., Wen, Q., KintokoZhang, S., Zhang, S., Huang, J., et al. (2013). Effect of 2-Dodecyl-6-Methoxycyclohexa-2,5-Diene-1,4-Dione, Isolated from Averrhoa carambola L. (Oxalidaceae) Roots, on Advanced Glycation End-Product-Mediated Renal Injury in Type 2 Diabetic KKAy Mice. Toxicol. Lett. 219, 77–84. doi:10.1016/j.toxlet.2013.03.001
Keywords: DMDD, diabetic cognitive impairment, db/db mice, neuronal apoptosis, Hif3a
Citation: Wang L, Cao J, Xu Q, Lu X, Yang X, Song Q, Chen S, Du K, Huang R and Zou C (2021) 2-Dodecyl-6-Methoxycyclohexa-2,5-Diene-1,4-Dione Ameliorates Diabetic Cognitive Impairment Through Inhibiting Hif3α and Apoptosis. Front. Pharmacol. 12:708141. doi: 10.3389/fphar.2021.708141
Received: 11 May 2021; Accepted: 15 November 2021;
Published: 16 December 2021.
Edited by:
William Lee Klein, Northwestern University, United StatesReviewed by:
Vinod Tiwari, Indian Institute of Technology (BHU), IndiaCopyright © 2021 Wang, Cao, Xu, Lu, Yang, Song, Chen, Du, Huang and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renbin Huang, aHVhbmdyZW5iaW41MThAMTYzLmNvbQ==; Chunlin Zou, em91Y2h1bmxpbkBzb2h1LmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.