 Dan Qiao1†
Dan Qiao1† Ziwei Zhang2†
Ziwei Zhang2† Yali Zhang2Qian Chen2
Yali Zhang2Qian Chen2 Yujun Chen2
Yujun Chen2 Yingjue Tang2Xiong Sun3Zhipeng Tang2*Yancheng Dai1*
Yingjue Tang2Xiong Sun3Zhipeng Tang2*Yancheng Dai1*- 1Department of Gastroenterology, Shanghai Traditional Chinese Medicine-Integrated Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Institute of Digestive Diseases, LongHua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Department of Gastroenterology, Shanghai PuTuo District People’s Hospital Affiliated to Tongji University, Tongji University School of Medicine, Shanghai, China
Ulcerative colitis (UC) is a chronic nonspecific inflammation that mainly affects the mucosa and submucosa of the rectum and colon. Numerous studies have shown that endoplasmic reticulum stress (ERS)-induced autophagy plays a vital role in the pathogenesis of UC. ERS is the imbalance of internal balance caused by misfolded or unfolded proteins accumulated in the endoplasmic reticulum (ER).Excessive ERS triggers the unfolded protein response (UPR), an increase in inositol-requiring enzyme 1, and a Ca2+ overload, which activates the autophagy pathway. Autophagy is an evolutionarily conserved method of cellular self-degradation. Dysregulated autophagy causes inflammation, disruption of the intestinal barrier, and imbalance of intestinal homeostasis, therefore increasing the risk of colonic diseases. This review summarizes the pathogenesis of ERS, UPR, and ERS-related autophagy in UC, providing potential new targets and more effective treatment options for UC.
Introduction
Ulcerative colitis (UC) is a chronic, nonspecific inflammatory disease of the rectum and colon, whose etiology is unexplained. The main symptoms of UC include hematochezia, diarrhea, abdominal pain and tenesmus (Seyedian et al., 2019; Naseer et al., 2020). UC, one of the refractory diseases of the digestive system with recurrent episodes of intestinal inflammation, is common in Western countries, with a prevalence rate of 100∼200/100,000 in Europe and North America. The number of cases reported in China has also increased significantly in recent years (Li et al., 2017; Ng et al., 2018; Feuerstein et al., 2019). UC patients tend to be relatively young, which decreases social productivity and personal quality of life (Kaplan, 2015).
Destruction of the intestinal mucosal barrier caused by the interaction of genetics, infection, immunity, and environmental pollution is the core event leading to the pathogenesis and progression of UC. Accordingly, as a major component of the intestinal mucosal barrier, damage to intestinal epithelial cells (IECs) may play a decisive role in this event (Ren et al., 2019; Yan et al., 2020). IECs, including microfold (M) cells, enteroendocrine cells, absorptive epithelial cells, goblet cells, and Paneth cells, respond to various types of immune cells, and regulate epithelial barrier function and gut microbiota (Okumura and Takeda, 2017; Soderholm and Pedicord, 2019). IECs, with a well-developed structure of the endoplasmic reticulum (ER), are one of the most metabolically exubera-nt cell types. Sustained and severe endoplasmic reticulum stress (ERS) induces autophagy through the unfolded protein reaction (UPR) in IECs, which causes inflammation. Excessive ERS can also disrupt the intestinal mucosal barrier, and ultimately lead to UC (Hosomi et al., 2015; Iida et al., 2017; Ma et al., 2017). This review is a systematic appraisal of the current literature to provide a better understanding of the role of the pathogenesis of ERS, UPR, and ERS-related autophagy in UC.
ERS in UC
The ER is one of the largest cellular organelles, and has a complex structure (Lu et al., 2020). It is the main site of protein synthesis, folding, lipid synthesis, carbohydrate metabolism, and calcium storage (Schwarz and Blower, 2016; Stevenson et al., 2016). ERS is driven by the accumulation of unfolded and misfolded proteins in the ER (Liu and Green, 2019).
During ERS, the cell activates a response to changes in protein folding, which is called the UPR (Hetz, 2012). Moreover, the other pathway is ER-associated degradation (ERAD), which maintains ER (Olzmann et al., 2013). However, persistent ERS and UPR can induce cell death (Bernales et al., 2006). To date, the UPR is initiated by three kinds of ER transmembrane sensors, including inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Ron and Walter, 2007). The interaction between the heavy-chain-binding protein (BiP) and adenosine nucleotides mainly participates in these three processes (Pobre et al., 2019). Under ERS conditions, BiP dissociation activates IRE1, PERK, or ATF6, and initiates cascades of the UPR and downstream signals (Ma et al., 2017). Above all, three UPR signaling pathways (IRE1, PERK, and ATF6) are involved in the pathogenesis of UC.
IRE1 Pathway
IRE1 is a key factor in the severity and duration of UPR (Pincus et al., 2010). Mammalian IRE1 contains two subtypes: IRE1α and IRE1β. IRE1α is widely expressed in the human body, while IRE1β is mainly expressed in the gastrointestinal tract and pulmonary epithelial cells (Tirasophon et al., 1998; Wang et al., 1998). X-Box-binding protein 1 (XBP1) in mammals is a crucial transcriptional activator in this process. The increase in protein load in the ER activates XBP1 (Calfon et al., 2002), which can relieve ERS. Simultaneously, IRE1 can also bind to and activate tumor necrosis factor receptor–associated factor 2 (TRAF2), which is a binding protein that binds plasma membrane receptors to c-Jun N-terminal kinase (JNK), and then activates JNK (Urano et al., 2000). Regulated IRE1-dependent decay (RIDD) has dual functions: maintaining homeostasis under low ERS and inducing apoptosis by excessive ERS (Pluquet et al., 2013).
IRE1β is expressed in goblet cells, which secrete mucoprotein 2 (MUC2). MUC2 is more easily degraded by pathogens in the colon environment during UC, suggesting that MUC2 acts as a protective mucin in UC. The level of MUC2 increases sharply in IRE1β−/− mice, indicating that IRE1β can degrade MUC2 and maintain the stability of MUC2 in the intestine (Tsuru et al., 2013). Bertolotti found that IRE1β−/- mice developed colitis several days earlier than wild-type mice with dextran sulfate sodium (DSS) induced UC, indicating that IRE1β−/− mice had a marked susceptibility to DSS (Chassaing et al., 2014). IRE1α gene deletion induced the apoptosis of IECs, which destroyed the intestinal mucosal barrier and led to spontaneous colitis (Zhang et al., 2015). Therefore, IRE1 is an essential signal in the pathogenesis of UC, and its absence leads to spontaneous colitis.
MicroRNAs are also involved in UPR/ERS through IREIα, an ER transmembrane kinase-endoribonuclease (RNase). Activation of IRE1α caused decay of select microRNAs (miRs -17, -34a, -96, -125b), which inhibit translation of Caspase-2 mRNA generally. IRE1α regulated Caspase-2 translation via downregulating select anti- Caspase-2 miRNAs by cleaving select pre-miRNAs to prevent proper DICER processing of their mature forms. Thus, IRE1α cleaves select microRNAs to prevent the translation of proapoptotic Caspase-2 during ERS (Upton et al., 2012). Besides that, IRE1α induces thioredoxin-interacting protein (TXNIP) to activate the NLRP3 inflammasome and promote cell death during ERS. However, TXNIP mRNA stability during ER stress is controlled by a specific micro-RNA, miR-17. miR-17 levels decline speedily under ERS. IRE1α increases TXNIP mRNA stability by reducing miR-17. And TXNIP protein activates the NLRP3, causing Caspase-1 cleavage and interleukin 1β (IL-1β) secretion. Therefore, microRNAs indirectly regulates signaling hubs to control cell death during ERS (Lerner et al., 2012).
Transcription of the XBP1u gene regulates the ERS-mediated UPR signaling pathway. Studies found that ERS increased and goblet cells decreased in Xbp1−/− mice, which decreased MUC 2 secretion and enhanced susceptibility to experimental colitis. The expression of tumor necrosis factor (TNF-a) and C/EBP homologous protein (CHOP) increased, while the antimicrobial ability decreased (Kaser et al., 2008). Briefly, IRE1β, IRE1α, and XBP1 in the IRE1 pathway are associated with a protective effect on UC by degrading MUC2 secreted by goblet cells, protecting the intestinal mucosal barrier, improving the sensitivity of mice to DSS, maintaining the homeostasis of the intestinal environment and inhibiting the inflammation process.
PERK Pathway
During ERS, PERK oligomerization and autophosphorylation activate eukaryotic initiation factor 2α (eIF2α) kinase and alleviate ERS by augmenting the UPR (Ma et al., 2002; Verfaillie et al., 2012). Moreover, the expression of ATF4 was induced by phosphorylation of eIF2α (Vattem and Wek, 2004). Subsequently, the expression of CHOP increased and induced apoptosis. (Rozpedek et al., 2016). X-linked inhibitor of apoptosis protein (XIAP) is a potent inhibitor of cysteinyl aspartate specific proteinase (Caspase) activity (Eckelman et al., 2006). ATF4 promotes the degradation of XIAP, while the PERK signaling pathway downregulates XIAP synthesis through two modes: 1) reduction of the synthesis of XIAP through the phosphorylation of eIF2α and 2) degradation of XIAP through ATF4 activation (Hiramatsu et al., 2014). In addition, ATF4 induces the expression of ATF5, which can promote apoptosis (Teske et al., 2013). The repression of PERK signaling blocks the expression of most genes by overcoming ERS, and leads to cell death (Mcquiston and Diehl, 2017).
The extension of eIF2α phosphorylation results in an increase in ATF4 and CHOP expression. Moreover, high expression of CHOP mediates apoptosis in epithelial cells, which promotes the progression of UC (Waldschmitt et al., 2014). Furthermore, CHOP also stimulates MAC-1 to promote macrophage infiltration and induce reactive oxygen species (ROS) production in macrophages by upregulating endoplasmic reticulum oxidoreductin 1α (ERO-1α) (Namba et al., 2009). eIF2α phosphorylation activates nuclear factor kappa-B (NF-κB) signaling, therefore activating more inflammatory factors and disrupting the intestinal mucosal barrier (Deng et al., 2004). Okazaki found that inhibiting the dephosphorylation of eIF2α could inhibit the PERK signaling pathway and alleviate DSS-induced colitis (Okazaki et al., 2014). Specifically, the PERK pathway induced the expression of CHOP and ATF5, and degraded XIAP through ATF4. This mechanism promoted apoptosis and raised proinflammatory cytokines, which disrupted intestinal epithelial function and affected the development of UC.
ATF6 Pathway
The ATF6 transcription factor contains two subtypes: ATF6α and ATF6β. In the process of ERS, ATF6, released from the ER membrane, is cleaved by proteases in the Golgi apparatus and transferred to the nucleus. In the nucleus, ATF6 can bind specific DNA and initiate a series of signals to maintain ER homeostasis (Wang et al., 2000; Sato et al., 2011). In addition, ATF6 also regulates the expression of CHOP, which in turn regulates ERS (Yang et al., 2020). ATF6α induces the phosphorylation of Akt and activates the NF-κB pathway (Yamazaki et al., 2009). The absence of the SIP ATF6-processing enzyme and mutation of the S1P-encoding gene (Mbtps1) resulted in increased susceptibility to DSS-induced colitis (Brandl et al., 2009). The inhibition of ATF6α signaling can significantly generate the expression of IL-8 and TNF-α proinflammatory cytokines (Stengel et al., 2020). Therefore, the ATF6 pathway can enhance the expression of inflammatory cytokines and aggravate intestinal inflammation by activating the NF-κB signaling pathway and by expressing genetic mutations, which exacerbate the development of UC.
ERS, Immune Response and UC
ERS is responsible for the development of UC through a variety of immune responses. Related studies have found that IL-22, dendritic cells (DCs), and nucleotide-binding oligomerization domain (NOD) exhibit potential effects on the intestinal immune response. IL-22, combined with IL-17A, regulate transcription during ERS and promote apoptosis in IECs. The IL-22-ERS axis is vital in the pathogenesis of chronic colitis and might provide a new therapeutic target for future treatment (Powell et al., 2020). Interestingly, William found that ERS stimulated cloned human colorectal gland cells to produce more IL-8, and activated dendritic cells to become proinflammatory cells. This indicated that there was a previously unknown mechanism between epithelial ERS and immune activation in inflammatory bowel disease (IBD) (Rees et al., 2020). In addition, NOD-like receptors (NLRs) were pattern recognition receptors (Caruso et al., 2014). Marijke found that after thapsigarnin treatment, the NOD1/2 level and IL-6 production increased sharply. IL-6 production was significantly decreased in bone marrow–derived macrophages (BMDGs) of dithiothreitol-induced NOD1/2−/− mice compared with wild-type mice. In the NLR family, NOD1 and NOD2 induce ERS to produce more IL-6 through the IRE1α/tumor necrosis factor receptor–associated factor 2 (TRAF2) pathway, causing intestinal inflammation (Keestra-Gounder et al., 2016). Recent studies on ERS, the immune response and UC have shown that ERS can induce apoptosis in IECs and promote the expression of proinflammatory cytokines by regulating the expression of IL-22, DCs, and NOD during the immune response, thereby causing intestinal inflammation and accelerating the development of UC.
Consequently, the mechanism of ERS causing UC can be summarized as follows: 1) regulation of ERS susceptibility genes, 2) induction of apoptosis in IECs, 3) intestinal mucosal barrier dysfunction, and 4) production of pro-inflammatory cytokines, which induces intestinal inflammation and the occurrence of UC (Figure 1).
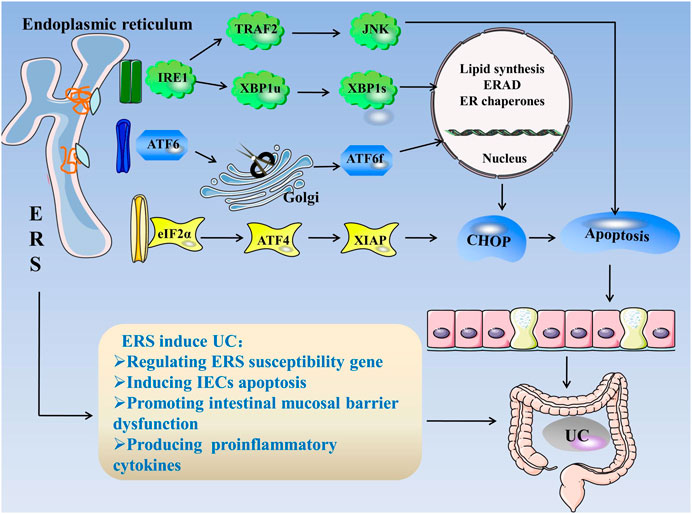
FIGURE 1. ERS in UC. When unfolded or misfolded proteins accumulate excessively in the ER, ERS occurs and the UPR is initiated. The UPR consists of three pathways: IRE1, PERK, and ATF6. When the cell is in steady state, the three stress-related proteins bind to GRP78. When ERS occurs, GRP78 dissociates from three kinds of receptors and activates the IRE1, PERK, and ATF6 pathways. 1) IRE1 cleaves XBP1u into more stable XBP1s, and IRE1 can bind to TRAF2 to activate JNK. The factors related to ER folding, lipid biosynthesis, and ERAD are regulated during the IRE1 reaction. 2) PERK activates eIF2α through autophosphorylation, eIF2α activates ATF4, and ATF4 induces CHOP expression, which is a cytokine that promotes apoptosis. 3) ATF6 is cleaved in the Golgi apparatus and binds to specific DNA to regulate CHOP. ERS regulates ERS susceptibility genes, induces IEC apoptosis, destroys the intestinal mucosal barrier, and produces proinflammatory cytokines, leading to UC. Abbreviations: ATF, activating transcription factor; CHOP, C/EBP homologous protein; ER, endoplasmic reticulum; ERS, endoplasmic reticulum stress; ERAD, endoplasmic reticulum associated degradation; eIF2α, eukaryotic initiation factor 2α; GRP78, glucose-regulated Protein 78; IECs, intestinal epithelial cells; IRE1α, inositol-requiring enzyme 1; JNK, c-Jun N-terminal kinase; PERK, protein kinase R-like endoplasmic reticulum kinase; TRAF2, tumor necrosis factor receptor–associated factor 2; XBP1, X-box-binding protein 1; UC, ulcerative colitis; UPR, unfolded protein response.
Autophagy in UC
Autophagy is an evolutionarily conserved process, whose main function is to degrade endogenous biological macromolecules for recycling (Ravanan et al., 2017). In the case of nutritional deficiency, autophagy is rapidly induced by self-digestion to maintain cell vitality, and core anabolic functions are promoted under conditions of adequate nutrition (Kaur and Debnath, 2015; Yang et al., 2019). Three kinds of autophagy are found in mammals: macroautophagy, microautophagy and chaperone-mediated autophagy (Dikic and Elazar, 2018). Recent studies have reported that autophagy is regulated by autophagy-related genes (ATG) (Li and Zhang, 2019), mammalian target of rapamycin (mTOR) (Munson and Ganley, 2015), adenosine 5′-monophosphate-activated protein kinase (AMPK) (Li and Chen, 2019), Ca2+ (Hu et al., 2019) and NOD2 (Negroni et al., 2016).
ATG is involved in the formation of multiple autophagosomes, starting with the activation of the unc-51 like autophagy activating kinase 1 (ULK1) complex (Kabeya et al., 2000). Similar to the mammalian homolog of Atg8, microtubule-associated protein light chain 3 (LC3) has two forms: LC3I and LC3-II. The level of LC3-II reflects the number of autophagosomes. Recently, LC3 is a classic indicator of autophagy in mammals (Tanida et al., 2008). Paiva demonstrated that the level of LC3II is higher in UC. A decreased number of cells exhibiting colocalized LC3/p62, which was verified by immunofluorescence, indicates that autophagy is involved in the pathogenesis of UC (Paiva et al., 2018). In DSS-induced colitis, the number of Lgr5+ stem cells, the LC3II/I ratio and the level of p62 increase, which is aggravated by activating autophagy in Lgr5+ stem cells (Xie et al., 2020). Ardali found that the level of ATG5 closely relates to autophagy in the stool of UC patients and is significantly higher than that in healthy people (Ardali et al., 2020). This finding indicates that autophagy plays a central role in the pathogenesis of UC and might be used as a diagnostic marker for UC in the future.
After activation, the ULK1 mammalian autophagy complex binds to vesicles and phosphorylates ATG9. Next, at the amplification stage, ATG8 (LC3) combines with phosphatidylethanolamine (PE) to form an ATG8–PE complex, which promotes the elongation of the autophagic membrane, and the closure and formation of autophagosomes and autophagosome lysosomes (Lin and Hurley, 2016; Goodwin et al., 2017; Yu et al., 2017; Levine and Kroemer, 2019; Turco et al., 2020). ATG9A decreases in DSS-induced colitis, and the overexpression of ATG9A improves autophagy induced by rapamycin (Xu et al., 2018). The aforementioned studies indicate that ATG9 and ATG5 autophagy-related genes might be important genes in the pathogenesis of UC, thus assisting in clinical diagnosis.
mTOR forms the catalytic subunits of two different protein complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2), which play an important role in protein synthesis, lipid and glucose metabolism and other physiological functions (Saxton and Sabatini, 2017). The initiation of autophagy requires the Beclin1–VPS34 core complex, and mTORC1 negatively regulates autophagy by inhibiting the ULK1 and VSP34 complex (Rabanal-Ruiz et al., 2017). Death-associated protein 1 (DAP1) has been identified as a novel substrate of mTOR. DAP1 negatively regulates autophagy by inducing apoptosis and reducing the number of autophagosomes with PERK-eIF2α (Yahiro et al., 2014). mTOR can also regulate autophagy and further regulate UC in various ways. In DSS-induced colitis, deficiency of meteorin-like protein (METRNL), secreted by IECs, deteriorated UC partially by inhibiting autophagy through the AMPK-mTOR-p70S6K pathway. METRNL deficiency aggravated UC by inhibiting autophagy, suggesting that UC can be attenuated by activation of autophagy (Zhang et al., 2020). Hypoxia inactivates mTOR and degrades p62 and LC3, reducing inflammation by restoring autophagy (Cosin-Roger et al., 2017). Zhou found that boosting mTOR-dependent autophagy through the NF-κB pathway quenches intestinal inflammation. The mTOR inhibitor AZD8055 (ATP-competitive mTOR inhibitor) alleviates experimental colitis in mice (Zhou et al., 2018). Therefore, the inhibition of mTOR may help alleviate the symptoms of UC by activating autophagy.
MicroRNAs (miRNAs) are noncoding RNAs that are indirectly involved in autophagy and through inhibition of Beclin1 (Chen et al., 2017). Schaefer found that the expression of miRNAs in the blood and tissues of the UC group and the expression of miR-19a, miR-21, miR-31, and miR-101 were significantly increased (Schaefer et al., 2015). This indicates that miRNAs are involved in the pathogenesis of UC and that specific miRNAs distinguish UC from other diseases. Wang found that miRNAs regulate NF-κB or mTOR signaling to modulate autophagy in intestinal cells by releasing anti- or proinflammatory factors (Wang et al., 2018). Based on these studies, miRNAs regulate autophagy in a variety of ways, including disruption of the intestinal mucosa and changes in intestinal permeability to aggravate or improve UC, but the specific role of miRNAs still needs to be verified.
Vitamin D receptor (VDR) is closely related to autophagy. VDR plays a vital role in IECs by reducing apoptosis and enhancing autophagy through Beclin-1 (Lu et al., 2019). Zhang found that VDR deficiency promoted the release of the tight junction protein Claudin-2, which enhanced the permeability of the intestinal mucosal barrier and further accelerated the progression of UC (Zhang et al., 2018). Yongyan Shi found that necroptotic apoptosis was one of the pathogeneses of IBD. VDR inhibits necroptotic apoptosis, alleviates inflammation, and suppresses the induction of colitis by preventing receptor interacting serine/threonine kinase 3 (RIPK3) from binding to RIPK1 (Shi et al., 2020). Jot and others showed that VDR also protects the intestines through the VDR-gut microbiota axis and reduces the susceptibility of DSS-induced colitis (Ooi et al., 2013). These studies all indicate that VDR enhances autophagy and alleviates colitis through different signaling mechanisms.
In summary, autophagy can restore the intestinal mucosal barrier and alleviate UC by regulating expression of susceptibility genes, modifying intestinal microbes, inhibiting proinflammatory cytokines, and suppressing the immune response (Figure 2).
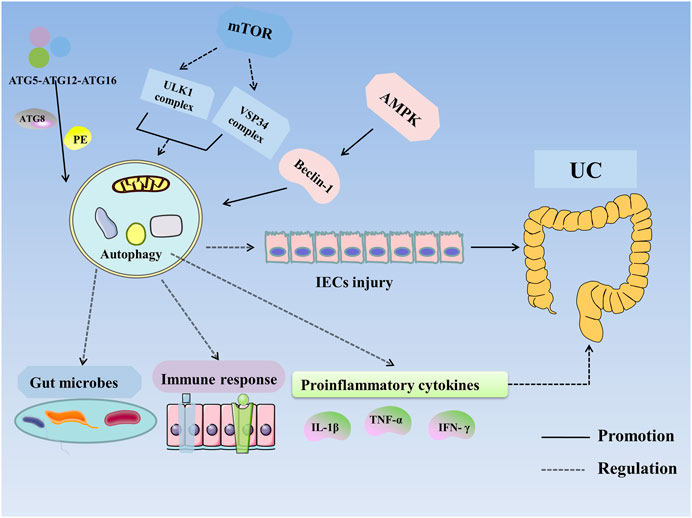
FIGURE 2. Autophagy in UC. Autophagy is regulated by ATG, mTOR, AMPK, and NOD2. ATG participates in the formation of the ULK1 complex and initiates autophagy. The ATG12–ATG5–ATG16 complex recruits autophagosome membranes and combines ATG8 (LC3) with phosphatidylethanolamine (PE) to form an ATG8–PE complex, leading to the formation of autophagosomes and autophagolysosomes. mTOR negatively regulates autophagy by inhibiting the ULK1 complex, VSP34 complex, and DAP1. AMPK activates ULK1 and phosphorylates Beclin-1 to activate autophagy. VDR enhances autophagy through Beclin-1. Autophagy improves UC by altering the expression of susceptibility genes, improving intestinal microbes and the immune response and inhibiting the expression of proinflammatory cytokines. Abbreviations: AMPK, 5′-monophosphate-activated protein kinase; ATG, autophagy associated gene; IECs, intestinal epithelial cells; IFN-γ, Interferon-γ; IL-1β, interleukin-1β; mTOR, mammalian target of rapamycin; PE, phosphatidylethanolamine; TNF-α, tumor necrosis factor-α; UC, Ulcerative colitis.
ERS and Autophagy
Under oxidative stress, energy deficiency, Ca2+ depletion, increased mRNA translation, metabolic changes and inflammatory stimulation, cell homeostasis is destroyed and ERS is activated (Luo and Cao, 2015). Several mediators released under ERS can directly induce the formation of autophagosomes and initiate autophagy (Kaser and Blumberg, 2011). Autophagy plays two major functions: on the one hand, moderate autophagy maintains the stability and survival of cells; on the other hand, excessive autophagy causes cell damage and even apoptosis (Kaur and Debnath, 2015). ERS induces the transformation of LC3 from LC3-I to LC3-II and the formation of autophagy (Ogata et al., 2006; Wu et al., 2021).
Recent studies have confirmed the connection between ERS and autophagy, and ERS induces autophagy through various pathways. For example, PERK regulates ATF4 and CHOP transcription factors, influences autophagosome formation, and further affects autophagy (Rouschop et al., 2010). B cells controlled by Trk-fused gene (TFG) are more sensitive to ERS and contain more LC3 and an increased number and size of autophagosomes (Steinmetz et al., 2020). ERS upregulates death-related protein kinase 1 (DAPK1) through ATF6, mediates mAtg9 trafficking, and activates autophagy (Zhou et al., 2016). In addition, ATF4 induces the expression of DNA damage response 1 (REDD1) during ERS. The upregulated expression of REDD1 in UC is correlated with autophagy induction by inhibiting mTOR (Kimball and Jefferson, 2012; Angelidou et al., 2018).
ERS can induce autophagy not only through the UPR pathway, but also by Akt signal transduction. For instance, ROS mediate Akt inactivation and increase the expression of ERS-related molecules such as CHOP and XBP1, leading to apoptosis. ERS can induce autophagy and reduce inflammation by inhibiting the phosphatidylinositol (3-kinase PI3K)/serine-threonine protein kinase (Akt)/mammalian target of rapamycin (mTOR) pathway (Xue et al., 2017; Chung et al., 2019). This indicates that ERS and autophagy can be activated by inhibiting the Akt pathway. ERS induced an increase in CHOP expression and a decrease in B-cell lymphoma-2 (Bcl-2) expression, and activated autophagy by releasing Beclin-1 through the PERK/CHOP/Bcl-2/Beclin-1 pathway (Liu et al., 2014; Ning et al., 2019). Corazzari found that ERS activates the Tribbles homolog 3 (TRB3) axis and the IRE1/TRAF2/apoptosis signal regulating kinase-1 (ASK1)/JNK signaling axis to induce autophagy (Corazzari et al., 2015). IRE1/XBP1 and IRE1/JNK1 both induce autophagy by activating Beclin-1 (Rather et al., 2020).
In general, ERS induces autophagy through multiple pathways: 1) the IRE1/TRAF2/ASK1/JNK/Bcl-2 (Vps34) signaling axis; 2) the IRE1/XBP1/Beclin-1 signaling axis; 3) the Akt/CHOP/Beclin-1 signaling axis; 4) the ATF4/REDD1/mTOR signaling axis; and 5) the PERK/CHOP/Bcl-2/Beclin-1 signaling axis. Whether ERS-induced autophagy plays a role in more signaling axes, for example, ATF6–XBP1–ULK1 and ATF4–ATG7–Beclin-1, needs to be further explored.
ERS, Autophagy and UC
The interaction between ERS and autophagy can synergistically affect the development of UC. Additionally, IECs, as a target of ERS and autophagy, play a vital role in the pathogenesis of UC (Adolph et al., 2013).
On the one hand, ERS regulates autophagy. Recent reports indicate that the mechanism by which ERSregulates autophagy is complex. Lopes found that ERS activated the ATF6–DAPK1 signal in IECs and enhanced autophagic killing of bacteria (Lopes et al., 2018). The expression of REDD1 in intestinal neutrophils is closely related to the severity of UC. REDD1 activates neutrophil autophagy by inhibiting mTOR phosphorylation, and autophagy positively regulates neutrophil extracellular traps (NETs). Intestinal neutrophil expression through NETs promotes the release of IL-1β, which mediates inflammation and further exacerbates UC. The REDD1/autophagy/NETs/IL-1β pathway plays an important role in the initiation and propagation of UC. Therapy targeting IL-1β could be beneficial for active UC (Angelidou et al., 2018). The expression of glucose-regulated protein (GRP)78 in the colon is increased in UC, especially in the inflamed intestinal mucosa (Shkoda et al., 2007). Tréton found that unspliced and spliced XBP1, GRP78 and GRP94 and ER degradation in the colonic mucosa of UC patients is associated with elevated levels of the active form of ATF6 (p50ATF6α) and impairment of the eIF2α–ATF4–CHOP pathway (Tréton et al., 2011). Stengel found that ATF6α, as an intermediate signaling molecule that regulates its upstream regulators and downstream XBP1 and Atg16L1, participates in the crosstalk between ERS and autophagy of IECs in IBD (Stengel et al., 2020). ERS activates autophagy through multiple signals in IECs and aggravates UC.
On the other hand, autophagy regulates ERS. Moderate autophagy can inhibit ERS, increase the number of goblet cells and mucin secretion, protect intestinal epithelial mucosal barrier function, and relieve the symptoms of UC. In DSS-induced colitis, the inhibition of triggering receptor expressed on myeloid cells 1 (TREM1) induced macroautophagy and chaperone-mediated autophagy, which compensates for the UPR to reduce ERS (Kökten et al., 2018). In addition, the stimulation of NOD and TNF receptors in IECs promote ATG16L1 stabilization via conserved helix-loop-helix ubiquitous kinase (IKKα)-dependent phosphorylation of ATG16L1 at serine 218, thereby protecting ATG16L1 against caspase-3-dependent degradation and limiting ERS activation (Diamanti et al., 2017). In fact, compared with Atg16L1[ΔIEC] or Xbp1[ΔIEC] mice, mice lacking Atg16L1 and Xbp1 (Atg16l1[ΔIEC]Xbp1[ΔIEC]) develop more severe colitis. However, all Atg16L1[ΔIEC], Xbp1[ΔIEC] and Atg16L1[ΔIEC]Xbp1[ΔIEC] mice show increased accumulation and excessive activation of ER to nucleus signaling 1 (ERN1), due to the deficiency of ERN1 degradation caused by autophagy (Tschurtschenthaler et al., 2017).
In general, moderate ERS can maintain intestinal homeostasis, but when ERS is too strong, key molecules such as IRE1, PERK and ATF6 which produce proinflammatory cytokines and destroy the intestinal mucosal barrier to induce UC, are lacking. Autophagy can inhibit the expression of proinflammatory cytokines and improve the immune response and can also reduce UC in other ways, but excessive autophagy increases cell apoptosis and aggravates UC (Figure 3).
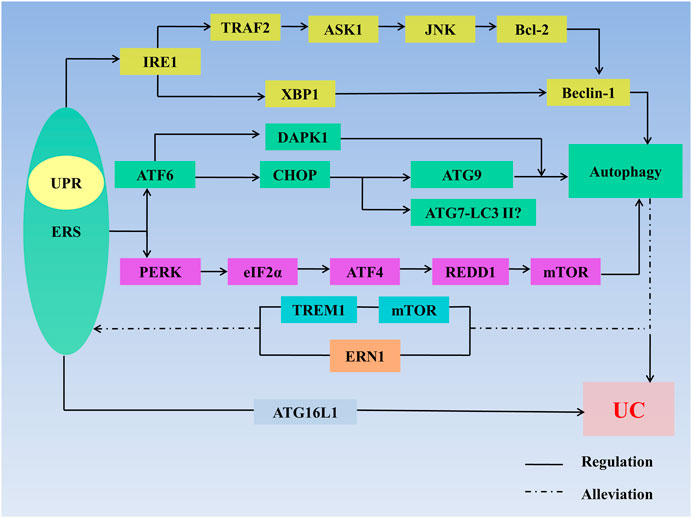
FIGURE 3. ERS, autophagy, and UC. ERS regulates autophagy through multiple pathways: 1) IRE1/TRAF2/ASK1/JNK/Bcl-2/Beclin-1 signaling axis; 2) lIRE1/XBP1/Beclin-1 signaling axis; 3) ATF6/CHOP/ATG9 signaling axis; 4) ATF6/DAPK1 signaling axis; 5) ATF6/DAPK1 signaling axis; 6)ATF6/CHOP/ATG7-LC3II signaling axis; 7) ATF6/CHOP/ATG9 signaling axis; and 8) PERK/elf2α/ATF4/REDD1/mTOR signaling axis. ERS regulates UC through defects in the ATG16L1 gene. Autophagy stimulates mTOR receptors in IECs to inhibit TREM1 and through regulating ERN1 to alleviate ERS. Autophagy also regulates UC. The interaction between ERS and autophagy synergistically affects the development of UC. Abbreviations: AMPK, 5′-monophosphate-activated protein kinase; ASK 1, apoptosis signal regulating kinase-1; ATF, activating transcription factor; ATG, autophagy associated gene; Bcl-2, B-cell lymphoma-2; CHOP, C/EBP homologous protein; DAPK1, death-related protein kinase 1; eIF2α, eukaryotic initiation factor 2α; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; ERN1, endoplasmic reticulum to nucleus signaling 1; ERS, endoplasmic reticulum stress; LC3Ⅱ, microtubleassociated protein light chain 3II; mTOR, mammalian target of rapamycin; IRE1, inositol-requiring enzyme1; JNK, c-Jun N-terminal kinase; PERK, protein kinase R-like endoplasmic reticulum kinase; TRAF2, tumor necrosis factor receptor–associated factor 2; TREM1, triggering receptor expressed on myeloid cells 1; XBP1, X-Box-binding protein 1; UC, ulcerative colitis; UPR, unfolded protein response.
The inhibition of autophagy can be used as a therapeutic strategy in UC. Many drugs can inhibit autophagy. The mTOR inhibitor 2-chloro-N-(6-cyanopy-ridin-3-yl) propanamide alleviated UC symptoms in mice by inhibiting autophagy (Bhonde et al., 2008). Curcumin reduced the number of autophagosomes by regulating autophagy and improved the symptoms of DSS-induced colitis in mice (Yue et al., 2019). Nicotine increased the expression of LC3II/LC3I and Beclin-1 through the AMPK–mTOR–p70S6K pathway, reduced p62 levels, and relieved experimental colitis by regulating autophagy (Gao et al., 2020). Oral administration of Na2SO3 and NaHSO3 (3:1) produces SO2.SO2 suppresses autophagy by reducing oxidative stress and reducing the expression of the proinflammatory cytokines TNF-α, IL-1β, and IL-6, as well as downregulating Beclin1 expression. Therefore SO2 alleviates the pathological manifestations of colitis in rats by anti-inflammatory, antioxidation and autophagy inhibition (Banerjee et al., 2019). Galangin pretreatment increases autophagy-related protein expression and promotes the formation of autophagosomes, which can be used to prevent UC (Xuan et al., 2020). Melatonin alleviates colitis-related colon cancer by reducing autophagy, as revealed by the expression of autophagy-related markers (Trivedi et al., 2016). These drugs all reduce UC inflammation by regulating the expression of autophagy-related proteins.
ERS, Autophagy Related Drugs for UC
Autophagy deficiency is closely related to the pathogenesis of UC. Some therapeutic drugs work by improving autophagy, which makes autophagy a new therapeutic target for the treatment of UC. Drugs that modulate both ERS and autophagy need further investigation (Hooper et al., 2016). Berberine (BBR) inhibits the IRE1/XBP1 pathway and JNK activation and reduces the expression of proinflammatory cytokines and ERS. Hence, BBR might be one of the targeted therapeutic agents for UC (Hao et al., 2012). Lactobacillus paracasei–derived extracellular vesicles (LpEVs) attenuate DSS-induced colitis by reducing the expression of pro-inflammatory cytokines IL-1α, IL-1β, IL-2, and TNF-α proinflammatory cytokines and promoting the expression of anti-inflammatory cytokines, including IL-10 and TGFβ. LpEVs increase IRE1 and PERK phosphorylation, ATF6 cleavage, and CHOP expression by activating ERS to reduce LPS-induced intestinal inflammation and maintain intestinal homeostasis (Choi et al., 2020). Low-dose naltrexone reduces the level of ERS, restores the intestinal mucosal barrier, and improves the severity of IBD (Lie et al., 2018). Lycium barbarum leaves contain a variety of compounds and prevent inflammation through the IRE1–XBP1–dependent ER stress pathway, which might be linked to its antioxidant activity, making the leaves a beneficial food for human health (Lee et al., 2021). Dodecapeptide (LR12) is an inhibitor of triggering receptor expressed on myeloid cells-1 (TREM-1). Compared with healthy mice, the mice in the DSS[+]/LR12[+] group were similar and had no signs of ulcerations. However, DSS[+]/LR12[−]mice developed ulcers with inflammatory cell infiltration, and increased endoscopic scores. LR12 reduced ERS and restored impaired autophagy, indicating that TREM-1 inhibition alleviates the symptoms of colitis, mitigates endoscopic tissue damage, and prevents UC (Kökten et al., 2018). Hooper found that azathioprine induces autophagy by regulating mTORC1 signal transduction and the UPR sensor PERK, thus contributing to its efficacy in treating IBD (Hooper et al., 2019). The aforementioned drugs can reduce or prevent UC by inhibiting ERS.
Conclusion and Prospects
The role of ERS and autophagy in IBD has received increasing attention. This study summarizes the relationship between ERS and autophagy in UC. Moderate ERS can maintain the balance of the intestinal environment, but excessive ERS can induce intestinal inflammation by regulating ERS susceptibility genes, inducing intestinal epithelial cell apoptosis and intestinal mucosal barrier dysfunction, and producing proinflammatory cytokines, leading to the occurrence of UC. However, ERS can activate autophagy, which alleviates UC by inhibiting proinflammatory cytokines and immune responses and improving the intestinal microorganisms, but excessive autophagy still aggravates UC. The interaction between ERS and autophagy can synergistically affect the development of UC. Exploring the important mechanism of ERS-induced autophagy in the pathogenesis of UC can contribute to the understanding of the pathogenesis of UC and provide an effective method for the treatment of UC in the future. However, the specific regulatory mechanism of the ERS–autophagy signaling axis needs further investigation. For example, it is of interest to determine whether there are more stress- and autophagy-related pathways; the specific role and exact function of each protein, molecule, and enzyme in the respective pathways and to identify more autophagy-related genes and their regulatory mechanism in UC. The activation of key molecules in ERS can activate autophagy through multiple pathways. However, the following issues remain unaddressed: whether inhibition of one of the key molecules can affect multiple pathways and will produce side effects; if the effect of single target treatment is not significant, whether multitarget treatment can be used; how each signaling pathway works in multitarget therapy; whether the therapeutic drugs confirmed in current studies that can regulate ERS-autophagy in mice can play the same role in other organisms; and whether the combination of targeted autophagy and targeted ERS agents can exert better effects. There are many drugs for treating UC, but some patients have not been cured. Hence, it is urgent to develop novel drugs for the treatment of UC. A better understanding of the relationship among ERS-induced autophagy in UC, regulation of ERS-autophagy, alleviation of IEC damage, restoration of intestinal mucosal barrier function, and maintenance of intestinal homeostasis can provide potential new targets and more effective therapy for UC.
Author Contributions
DQ and ZZ wrote and revised the manuscript. ZT and YD designed the research. YZ, QC revised the manuscript. YC, YT, and XS edited the manuscript. All authors contributed to the manuscript conception development, data collection and analysis, and discussion on the manuscript writing and revising.
Funding
This study was supported by the National Natural Science Foundation of China (grant numbers 81873253, 81573892, and 81704009); Key clinical specialty construction project supported by Hongkou District Health Committee (grant numbers HKZK2020A01) and Xinglin scholar program of Shanghai University of Traditional Chinese Medicine, The document of the Shanghai University of Traditional Chinese Medicine 2020 No. 23.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adolph, T. E., Tomczak, M. F., Niederreiter, L., Ko, H. J., Böck, J., Martinez-Naves, E., et al. (2013). Paneth Cells as a Site of Origin for Intestinal Inflammation. Nature 503 (7475), 272–276. doi:10.1038/nature12599
Angelidou, I., Chrysanthopoulou, A., Mitsios, A., Arelaki, S., Arampatzioglou, A., Kambas, K., et al. (2018). REDD1/Autophagy Pathway Is Associated with Neutrophil-Driven IL-1β Inflammatory Response in Active Ulcerative Colitis. J. Immunol. 200 (12), 3950–3961. doi:10.4049/jimmunol.1701643
Ardali, R., Kazemipour, N., Nazifi, S., Bagheri Lankarani, K., Razeghian Jahromi, I., and Sepehrimanesh, M. (2020). Pathophysiological Role of Atg5 in Human Ulcerative Colitis. Intest Res. 18 (4), 421–429. doi:10.5217/ir.2019.00120
Banerjee, S., Ghosh, S., Sinha, K., Chowdhury, S., and Sil, P. C. (2019). Sulphur Dioxide Ameliorates Colitis Related Pathophysiology and Inflammation. Toxicology 412, 63–78. doi:10.1016/j.tox.2018.11.010
Bernales, S., Papa, F. R., and Walter, P. (2006). Intracellular Signaling by the Unfolded Protein Response. Annu. Rev. Cel Dev. Biol. 22, 487–508. doi:10.1146/annurev.cellbio.21.122303.120200
Bhonde, M. R., Gupte, R. D., Dadarkar, S. D., Jadhav, M. G., Tannu, A. A., Bhatt, P., et al. (2008). A Novel mTOR Inhibitor Is Efficacious in a Murine Model of Colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 295 (6), G1237–G1245. doi:10.1152/ajpgi.90537.2008
Brandl, K., Rutschmann, S., Li, X., Du, X., Xiao, N., Schnabl, B., et al. (2009). Enhanced Sensitivity to DSS Colitis Caused by a Hypomorphic Mbtps1 Mutation Disrupting the ATF6-Driven Unfolded Protein Response. Proc. Natl. Acad. Sci. U S A. 106 (9), 3300–3305. doi:10.1073/pnas.0813036106
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the XBP-1 mRNA. Nature 415 (6867), 92–96. doi:10.1038/415092a
Caruso, R., Warner, N., Inohara, N., and Núñez, G. (2014). NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 41 (6), 898–908. doi:10.1016/j.immuni.2014.12.010
Chassaing, B., Aitken, J. D., Malleshappa, M., and Vijay-Kumar, M. (2014). Dextran Sulfate Sodium (DSS)-induced Colitis in Mice. Curr. Protoc. Immunol. 104, 15.25.1–15.25.14. doi:10.1002/0471142735.im1525s104
Chen, J., Yu, Y., Li, S., Liu, Y., Zhou, S., Cao, S., et al. (2017). MicroRNA-30a Ameliorates Hepatic Fibrosis by Inhibiting Beclin1-Mediated Autophagy. J. Cel. Mol. Med. 21 (12), 3679–3692. doi:10.1111/jcmm.13278
Choi, J. H., Moon, C. M., Shin, T. S., Kim, E. K., Mcdowell, A., Jo, M. K., et al. (2020). Lactobacillus Paracasei-Derived Extracellular Vesicles Attenuate the Intestinal Inflammatory Response by Augmenting the Endoplasmic Reticulum Stress Pathway. Exp. Mol. Med. 52 (3), 423–437. doi:10.1038/s12276-019-0359-3
Chung, Y. P., Yen, C. C., Tang, F. C., Lee, K. I., Liu, S. H., Wu, C. C., et al. (2019). Methylmercury Exposure Induces ROS/Akt Inactivation-Triggered Endoplasmic Reticulum Stress-Regulated Neuronal Cell Apoptosis. Toxicology 425, 152245. doi:10.1016/j.tox.2019.152245
Corazzari, M., Rapino, F., Ciccosanti, F., Giglio, P., Antonioli, M., Conti, B., et al. (2015). Oncogenic BRAF Induces Chronic ER Stress Condition Resulting in Increased Basal Autophagy and Apoptotic Resistance of Cutaneous Melanoma. Cell Death Differ. 22 (6), 946–958. doi:10.1038/cdd.2014.183
Cosin-Roger, J., Simmen, S., Melhem, H., Atrott, K., Frey-Wagner, I., Hausmann, M., et al. (2017). Hypoxia Ameliorates Intestinal Inflammation through NLRP3/mTOR Downregulation and Autophagy Activation. Nat. Commun. 8 (1), 98. doi:10.1038/s41467-017-00213-3
Deng, J., Lu, P. D., Zhang, Y., Scheuner, D., Kaufman, R. J., Sonenberg, N., et al. (2004). Translational Repression Mediates Activation of Nuclear Factor Kappa B by Phosphorylated Translation Initiation Factor 2. Mol. Cel. Biol. 24 (23), 10161–10168. doi:10.1128/MCB.24.23.10161-10168.2004
Diamanti, M. A., Gupta, J., Bennecke, M., De Oliveira, T., Ramakrishnan, M., Braczynski, A. K., et al. (2017). IKKα Controls ATG16L1 Degradation to Prevent ER Stress during Inflammation. J. Exp. Med. 214 (2), 423–437. doi:10.1084/jem.20161867
Dikic, I., and Elazar, Z. (2018). Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cel Biol. 19 (6), 349–364. doi:10.1038/s41580-018-0003-4
Eckelman, B. P., Salvesen, G. S., and Scott, F. L. (2006). Human Inhibitor of Apoptosis Proteins: Why XIAP Is the Black Sheep of the Family. EMBO Rep. 7 (10), 988–994. doi:10.1038/sj.embor.7400795
Feuerstein, J. D., Moss, A. C., and Farraye, F. A. (2019). Ulcerative Colitis. Mayo Clin. Proc. 94 (7), 1357–1373. doi:10.1016/j.mayocp.2019.01.018
Gao, Q., Bi, P., Luo, D., Guan, Y., Zeng, W., Xiang, H., et al. (2020). Nicotine-induced Autophagy via AMPK/mTOR Pathway Exerts Protective Effect in Colitis Mouse Model. Chem. Biol. Interact. 317, 108943. doi:10.1016/j.cbi.2020.108943
Goodwin, J. M., Dowdle, W. E., Dejesus, R., Wang, Z., Bergman, P., Kobylarz, M., et al. (2017). Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 20 (10), 2341–2356. doi:10.1016/j.celrep.2017.08.034
Hao, X., Yao, A., Gong, J., Zhu, W., Li, N., and Li, J. (2012). Berberine Ameliorates Pro-inflammatory Cytokine-Induced Endoplasmic Reticulum Stress in Human Intestinal Epithelial Cells In Vitro. Inflammation 35 (3), 841–849. doi:10.1007/s10753-011-9385-6
Hetz, C. (2012). The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and beyond. Nat. Rev. Mol. Cel Biol. 13 (2), 89–102. doi:10.1038/nrm3270
Hiramatsu, N., Messah, C., Han, J., Lavail, M. M., Kaufman, R. J., and Lin, J. H. (2014). Translational and Posttranslational Regulation of XIAP by eIF2α and ATF4 Promotes ER Stress-Induced Cell Death during the Unfolded Protein Response. Mol. Biol. Cel. 25 (9), 1411–1420. doi:10.1091/mbc.E13-11-0664
Hooper, K. M., Barlow, P. G., Stevens, C., and Henderson, P. (2016). Inflammatory Bowel Disease Drugs: A Focus on Autophagy. J. Crohns Colitis 11 (1), 118–127. doi:10.1093/ecco-jcc/jjw127
Hooper, K. M., Casanova, V., Kemp, S., Staines, K. A., Satsangi, J., Barlow, P. G., et al. (2019). The Inflammatory Bowel Disease Drug Azathioprine Induces Autophagy via mTORC1 and the Unfolded Protein Response Sensor PERK. Inflamm. Bowel Dis. 25 (9), 1481–1496. doi:10.1093/ibd/izz039
Hosomi, S., Kaser, A., and Blumberg, R. S. (2015). Role of Endoplasmic Reticulum Stress and Autophagy as Interlinking Pathways in the Pathogenesis of Inflammatory Bowel Disease. Curr. Opin. Gastroenterol. 31 (1), 81–88. doi:10.1097/MOG.0000000000000144
Hu, Y. X., Han, X. S., and Jing, Q. (2019). Ca(2+) Ion and Autophagy. Adv. Exp. Med. Biol. 1206, 151–166. doi:10.1007/978-981-15-0602-4_7
Iida, T., Onodera, K., and Nakase, H. (2017). Role of Autophagy in the Pathogenesis of Inflammatory Bowel Disease. World J. Gastroenterol. 23 (11), 1944–1953. doi:10.3748/wjg.v23.i11.1944
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing. EMBO J. 19 (21), 5720–5728. doi:10.1093/emboj/19.21.5720
Kaplan, G. G. (2015). The Global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 12 (12), 720–727. doi:10.1038/nrgastro.2015.150
Kaser, A., and Blumberg, R. S. (2011). Autophagy, Microbial Sensing, Endoplasmic Reticulum Stress, and Epithelial Function in Inflammatory Bowel Disease. Gastroenterology 140 (6), 1738–1747. doi:10.1053/j.gastro.2011.02.048
Kaser, A., Lee, A. H., Franke, A., Glickman, J. N., Zeissig, S., Tilg, H., et al. (2008). XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell 134 (5), 743–756. doi:10.1016/j.cell.2008.07.021
Kaur, J., and Debnath, J. (2015). Autophagy at the Crossroads of Catabolism and Anabolism. Nat. Rev. Mol. Cel Biol. 16 (8), 461–472. doi:10.1038/nrm4024
Keestra-Gounder, A. M., Byndloss, M. X., Seyffert, N., Young, B. M., Chávez-Arroyo, A., Tsai, A. Y., et al. (2016). NOD1 and NOD2 Signalling Links ER Stress with Inflammation. Nature 532 (7599), 394–397. doi:10.1038/nature17631
Kimball, S. R., and Jefferson, L. S. (2012). Induction of REDD1 Gene Expression in the Liver in Response to Endoplasmic Reticulum Stress Is Mediated through a PERK, eIF2α Phosphorylation, ATF4-dependent cascade. Biochem. Biophys. Res. Commun. 427 (3), 485–489. doi:10.1016/j.bbrc.2012.09.074
Kökten, T., Gibot, S., Lepage, P., D'Alessio, S., Hablot, J., Ndiaye, N. C., et al. (2018). TREM-1 Inhibition Restores Impaired Autophagy Activity and Reduces Colitis in Mice. J. Crohns Colitis 12 (2), 230–244. doi:10.1093/ecco-jcc/jjx129
Lee, S. R., An, M.-Y., Hwang, H.-J., Yoon, J.-G., and Cho, J. A. (2021). Antioxidant Effect of Lycium Barbarum Leaf through Inflammatory and Endoplasmic Reticulum Stress Mechanism. Antioxidants 10 (1), 20. doi:10.3390/antiox10010020
Lerner, A. G., Upton, J. P., Praveen, P. V., Ghosh, R., Nakagawa, Y., Igbaria, A., et al. (2012). IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 16 (2), 250–264. doi:10.1016/j.cmet.2012.07.007
Levine, B., and Kroemer, G. (2019). Biological Functions of Autophagy Genes: A Disease Perspective. Cell 176 (1-2), 11–42. doi:10.1016/j.cell.2018.09.048
Li, W., and Zhang, L. (2019). Regulation of ATG and Autophagy Initiation. Adv. Exp. Med. Biol. 1206, 41–65. doi:10.1007/978-981-15-0602-4_2
Li, X., Song, P., Li, J., Tao, Y., Li, G., Li, X., et al. (2017). The Disease Burden and Clinical Characteristics of Inflammatory Bowel Disease in the Chinese Population: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 14 (3), 238. doi:10.3390/ijerph14030238
Li, Y., and Chen, Y. (2019). AMPK and Autophagy. Adv. Exp. Med. Biol. 1206, 85–108. doi:10.1007/978-981-15-0602-4_4
Lie, M. R. K. L., van der Giessen, J., Fuhler, G. M., de Lima, A., Peppelenbosch, M. P., van der Ent, C., et al. (2018). Low Dose Naltrexone for Induction of Remission in Inflammatory Bowel Disease Patients. J. Transl. Med. 16 (1), 55. doi:10.1186/s12967-018-1427-5
Lin, M. G., and Hurley, J. H. (2016). Structure and Function of the ULK1 Complex in Autophagy. Curr. Opin. Cel Biol. 39, 61–68. doi:10.1016/j.ceb.2016.02.010
Liu, K., Shi, Y., Guo, X., Wang, S., Ouyang, Y., Hao, M., et al. (2014). CHOP Mediates ASPP2-Induced Autophagic Apoptosis in Hepatoma Cells by Releasing Beclin-1 from Bcl-2 and Inducing Nuclear Translocation of Bcl-2. Cell Death Dis. 5 (7), e1323. doi:10.1038/cddis.2014.276
Liu, X., and Green, R. M. (2019). Endoplasmic Reticulum Stress and Liver Diseases. Liver Res. 3 (1), 55–64. doi:10.1016/j.livres.2019.01.002
Lopes, F., Keita, Å. V., Saxena, A., Reyes, J. L., Mancini, N. L., Al Rajabi, A., et al. (2018). ER-stress Mobilization of Death-Associated Protein Kinase-1-dependent Xenophagy Counteracts Mitochondria Stress-Induced Epithelial Barrier Dysfunction. J. Biol. Chem. 293 (9), 3073–3087. doi:10.1074/jbc.RA117.000809
Lu, M., van Tartwijk, F. W., Lin, J. Q., Nijenhuis, W., Parutto, P., Fantham, M., et al. (2020). The Structure and Global Distribution of the Endoplasmic Reticulum Network Are Actively Regulated by Lysosomes. Sci. Adv. 6 (51), eabc7209. doi:10.1126/sciadv.abc7209
Lu, R., Zhang, Y. G., Xia, Y., and Sun, J. (2019). Imbalance of Autophagy and Apoptosis in Intestinal Epithelium Lacking the Vitamin D Receptor. FASEB J. 33 (11), 11845–11856. doi:10.1096/fj.201900727R
Luo, K., and Cao, S. S. (2015). Endoplasmic Reticulum Stress in Intestinal Epithelial Cell Function and Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2015, 1–6. doi:10.1155/2015/328791
Ma, K., Vattem, K. M., and Wek, R. C. (2002). Dimerization and Release of Molecular Chaperone Inhibition Facilitate Activation of Eukaryotic Initiation Factor-2 Kinase in Response to Endoplasmic Reticulum Stress. J. Biol. Chem. 277 (21), 18728–18735. doi:10.1074/jbc.M200903200
Ma, X., Dai, Z., Sun, K., Zhang, Y., Chen, J., Yang, Y., et al. (2017). Intestinal Epithelial Cell Endoplasmic Reticulum Stress and Inflammatory Bowel Disease Pathogenesis: An Update Review. Front. Immunol. 8, 1271. doi:10.3389/fimmu.2017.01271
Mcquiston, A., and Diehl, J. A. (2017). Recent Insights into PERK-dependent Signaling from the Stressed Endoplasmic Reticulum. F1000Res 6, 1897. doi:10.12688/f1000research.12138.1
Munson, M. J., and Ganley, I. G. (2015). MTOR, PIK3C3, and Autophagy: Signaling the Beginning from the End. Autophagy 11 (12), 2375–2376. doi:10.1080/15548627.2015.1106668
Namba, T., Tanaka, K., Ito, Y., Ishihara, T., Hoshino, T., Gotoh, T., et al. (2009). Positive Role of CCAAT/Enhancer-Binding Protein Homologous Protein, a Transcription Factor Involved in the Endoplasmic Reticulum Stress Response in the Development of Colitis. Am. J. Pathol. 174 (5), 1786–1798. doi:10.2353/ajpath.2009.080864
Naseer, M., Poola, S., Ali, S., Samiullah, S., and Tahan, V. (2020). Prebiotics and Probiotics in Inflammatory Bowel Disease (IBD): Where Are We Now and where Are We Going?Funders. Curr. Clin. Pharmacol. 15 (3), 216–233. doi:10.2174/1574884715666200312100237
Negroni, A., Colantoni, E., Vitali, R., Palone, F., Pierdomenico, M., Costanzo, M., et al. (2016). NOD2 Induces Autophagy to Control AIEC Bacteria Infectiveness in Intestinal Epithelial Cells. Inflamm. Res. 65 (10), 803–813. doi:10.1007/s00011-016-0964-8
Ng, S. C., Shi, H. Y., Hamidi, N., Underwood, F. E., Tang, W., Benchimol, E. I., et al. (2018). Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st century: a Systematic Review of Population-Based Studies. Lancet 390 (10114), 2769–2778. doi:10.1016/S0140-6736(17)32448-0
Ning, B., Zhang, Q., Wang, N., Deng, M., and Fang, Y. (2019). β-Asarone Regulates ER Stress and Autophagy via Inhibition of the PERK/CHOP/Bcl-2/Beclin-1 Pathway in 6-OHDA-Induced Parkinsonian Rats. Neurochem. Res. 44 (5), 1159–1166. doi:10.1007/s11064-019-02757-w
Ogata, M., Hino, S., Saito, A., Morikawa, K., Kondo, S., Kanemoto, S., et al. (2006). Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress. Mol. Cel. Biol. 26 (24), 9220–9231. doi:10.1128/MCB.01453-06
Okazaki, T., Nishio, A., Takeo, M., Sakaguchi, Y., Fukui, T., Uchida, K., et al. (2014). Inhibition of the Dephosphorylation of Eukaryotic Initiation Factor 2α Ameliorates Murine Experimental Colitis. Digestion 90 (3), 167–178. doi:10.1159/000366414
Okumura, R., and Takeda, K. (2017). Roles of Intestinal Epithelial Cells in the Maintenance of Gut Homeostasis. Exp. Mol. Med. 49 (5), e338. doi:10.1038/emm.2017.20
Olzmann, J. A., Kopito, R. R., and Christianson, J. C. (2013). The Mammalian Endoplasmic Reticulum-Associated Degradation System. Cold Spring Harb. Perspect. Biol. 5 (9), a013185. doi:10.1101/cshperspect.a013185
Ooi, J. H., Li, Y., Rogers, C. J., and Cantorna, M. T. (2013). Vitamin D Regulates the Gut Microbiome and Protects Mice from Dextran Sodium Sulfate-Induced Colitis. J. Nutr. 143 (10), 1679–1686. doi:10.3945/jn.113.180794
Paiva, N. M., Pascoal, L. B., Negreiros, L. M. V., Portovedo, M., Coope, A., Ayrizono, M. L. S., et al. (2018). Ileal Pouch of Ulcerative Colitis and Familial Adenomatous Polyposis Patients Exhibit Modulation of Autophagy Markers. Sci. Rep. 8 (1), 2619. doi:10.1038/s41598-018-20938-5
Pincus, D., Chevalier, M. W., Aragón, T., van Anken, E., Vidal, S. E., El-Samad, H., et al. (2010). BiP Binding to the ER-Stress Sensor Ire1 Tunes the Homeostatic Behavior of the Unfolded Protein Response. Plos Biol. 8 (7), e1000415. doi:10.1371/journal.pbio.1000415
Pluquet, O., Dejeans, N., Bouchecareilh, M., Lhomond, S., Pineau, R., Higa, A., et al. (2013). Posttranscriptional Regulation of PER1 Underlies the Oncogenic Function of IREα. Cancer Res. 73 (15), 4732–4743. doi:10.1158/0008-5472.CAN-12-3989
Pobre, K. F. R., Poet, G. J., and Hendershot, L. M. (2019). The Endoplasmic Reticulum (ER) Chaperone BiP Is a Master Regulator of ER Functions: Getting by with a Little Help from ERdj Friends. J. Biol. Chem. 294 (6), 2098–2108. doi:10.1074/jbc.REV118.002804
Powell, N., Pantazi, E., Pavlidis, P., Tsakmaki, A., Li, K., Yang, F., et al. (2020). Interleukin-22 Orchestrates a Pathological Endoplasmic Reticulum Stress Response Transcriptional Programme in Colonic Epithelial Cells. Gut 69 (3), 578–590. doi:10.1136/gutjnl-2019-318483
Rabanal-Ruiz, Y., Otten, E. G., and Korolchuk, V. I. (2017). mTORC1 as the Main Gateway to Autophagy. Essays Biochem. 61 (6), 565–584. doi:10.1042/EBC20170027
Rather, R. A., Bhagat, M., and Singh, S. K. (2020). Oncogenic BRAF, Endoplasmic Reticulum Stress, and Autophagy: Crosstalk and Therapeutic Targets in Cutaneous Melanoma. Mutat. Res. 785, 108321. doi:10.1016/j.mrrev.2020.108321
Ravanan, P., Srikumar, I. F., and Talwar, P. (2017). Autophagy: The Spotlight for Cellular Stress Responses. Life Sci. 188, 53–67. doi:10.1016/j.lfs.2017.08.029
Rees, W. D., Stahl, M., Jacobson, K., Bressler, B., Sly, L. M., Vallance, B. A., et al. (2020). Enteroids Derived from Inflammatory Bowel Disease Patients Display Dysregulated Endoplasmic Reticulum Stress Pathways, Leading to Differential Inflammatory Responses and Dendritic Cell Maturation. J. Crohns Colitis 14 (7), 948–961. doi:10.1093/ecco-jcc/jjz194
Ren, M. T., Gu, M. L., Zhou, X. X., Yu, M. S., Pan, H. H., Ji, F., et al. (2019). Sirtuin 1 Alleviates Endoplasmic Reticulum Stress-Mediated Apoptosis of Intestinal Epithelial Cells in Ulcerative Colitis. World J. Gastroenterol. 25 (38), 5800–5813. doi:10.3748/wjg.v25.i38.5800
Ron, D., and Walter, P. (2007). Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cel Biol. 8 (7), 519–529. doi:10.1038/nrm2199
Rouschop, K. M., van den Beucken, T., Dubois, L., Niessen, H., Bussink, J., Savelkouls, K., et al. (2010). The Unfolded Protein Response Protects Human Tumor Cells during Hypoxia through Regulation of the Autophagy Genes MAP1LC3B and ATG5. J. Clin. Invest. 120 (1), 127–141. doi:10.1172/JCI40027
Rozpedek, W., Pytel, D., Mucha, B., Leszczynska, H., Diehl, J. A., and Majsterek, I. (2016). The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 16 (6), 533–544. doi:10.2174/1566524016666160523143937
Sato, Y., Nadanaka, S., Okada, T., Okawa, K., and Mori, K. (2011). Luminal Domain of ATF6 Alone Is Sufficient for Sensing Endoplasmic Reticulum Stress and Subsequent Transport to the Golgi Apparatus. Cell Struct. Funct. 36 (1), 35–47. doi:10.1247/csf.10010
Saxton, R. A., and Sabatini, D. M. (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168 (6), 960–976. doi:10.1016/j.cell.2017.02.004
Schaefer, J. S., Attumi, T., Opekun, A. R., Abraham, B., Hou, J., Shelby, H., et al. (2015). MicroRNA Signatures Differentiate Crohn's Disease from Ulcerative Colitis. BMC Immunol. 16 (1), 5. doi:10.1186/s12865-015-0069-0
Schwarz, D. S., and Blower, M. D. (2016). The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell Mol. Life Sci. 73 (1), 79–94. doi:10.1007/s00018-015-2052-6
Seyedian, S. S., Nokhostin, F., and Malamir, M. D. (2019). A Review of the Diagnosis, Prevention, and Treatment Methods of Inflammatory Bowel Disease. J. Med. Life 12 (2), 113–122. doi:10.25122/jml-2018-0075
Shi, Y., Cui, X., Sun, Y., Zhao, Q., and Liu, T. (2020). Intestinal Vitamin D Receptor Signaling Ameliorates Dextran Sulfate Sodium-Induced Colitis by Suppressing Necroptosis of Intestinal Epithelial Cells. FASEB J. 34 (10), 13494–13506. doi:10.1096/fj.202000143RRR
Shkoda, A., Ruiz, P. A., Daniel, H., Kim, S. C., Rogler, G., Sartor, R. B., et al. (2007). Interleukin-10 Blocked Endoplasmic Reticulum Stress in Intestinal Epithelial Cells: Impact on Chronic Inflammation. Gastroenterology 132 (1), 190–207. doi:10.1053/j.gastro.2006.10.030
Soderholm, A. T., and Pedicord, V. A. (2019). Intestinal Epithelial Cells: at the Interface of the Microbiota and Mucosal Immunity. Immunology 158 (4), 267–280. doi:10.1111/imm.13117
Steinmetz, T. D., Schlötzer-Schrehardt, U., Hearne, A., Schuh, W., Wittner, J., Schulz, S. R., et al. (2020). TFG Is Required for Autophagy Flux and to Prevent Endoplasmic Reticulum Stress in CH12 B Lymphoma Cells. Autophagy, 1–19. doi:10.1080/15548627.2020.1821546
Stengel, S. T., Fazio, A., Lipinski, S., Jahn, M. T., Aden, K., Ito, G., et al. (2020). Activating Transcription Factor 6 Mediates Inflammatory Signals in Intestinal Epithelial Cells upon Endoplasmic Reticulum Stress. Gastroenterology 159 (4), 1357. doi:10.1053/j.gastro.2020.06.088
Stevenson, J., Huang, E. Y., and Olzmann, J. A. (2016). Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Annu. Rev. Nutr. 36, 511–542. doi:10.1146/annurev-nutr-071715-051030
Tanida, I., Ueno, T., and Kominami, E. (2008). LC3 and Autophagy. Methods Mol. Biol. 445, 77–88. doi:10.1007/978-1-59745-157-4_4
Teske, B. F., Fusakio, M. E., Zhou, D., Shan, J., Mcclintick, J. N., Kilberg, M. S., et al. (2013). CHOP Induces Activating Transcription Factor 5 (ATF5) to Trigger Apoptosis in Response to Perturbations in Protein Homeostasis. Mol. Biol. Cel. 24 (15), 2477–2490. doi:10.1091/mbc.E13-01-0067
Tirasophon, W., Welihinda, A. A., and Kaufman, R. J. (1998). A Stress Response Pathway from the Endoplasmic Reticulum to the Nucleus Requires a Novel Bifunctional Protein Kinase/endoribonuclease (Ire1p) in Mammalian Cells. Genes Dev. 12 (12), 1812–1824. doi:10.1101/gad.12.12.1812
Tréton, X., Pédruzzi, E., Cazals-Hatem, D., Grodet, A., Panis, Y., Groyer, A., et al. (2011). Altered Endoplasmic Reticulum Stress Affects Translation in Inactive Colon Tissue from Patients with Ulcerative Colitis. Gastroenterology 141 (3), 1024–1035. doi:10.1053/j.gastro.2011.05.033
Trivedi, P. P., Jena, G. B., Tikoo, K. B., and Kumar, V. (2016). Melatonin Modulated Autophagy and Nrf2 Signaling Pathways in Mice with Colitis-Associated colon Carcinogenesis. Mol. Carcinog 55 (3), 255–267. doi:10.1002/mc.22274
Tschurtschenthaler, M., Adolph, T. E., Ashcroft, J. W., Niederreiter, L., Bharti, R., Saveljeva, S., et al. (2017). Defective ATG16L1-Mediated Removal of IRE1α Drives Crohn's Disease-like Ileitis. J. Exp. Med. 214 (2), 401–422. doi:10.1084/jem.20160791
Tsuru, A., Fujimoto, N., Takahashi, S., Saito, M., Nakamura, D., Iwano, M., et al. (2013). Negative Feedback by IRE1β Optimizes Mucin Production in Goblet Cells. Proc. Natl. Acad. Sci. U S A. 110 (8), 2864–2869. doi:10.1073/pnas.1212484110
Turco, E., Fracchiolla, D., and Martens, S. (2020). Recruitment and Activation of the ULK1/Atg1 Kinase Complex in Selective Autophagy. J. Mol. Biol. 432 (1), 123–134. doi:10.1016/j.jmb.2019.07.027
Upton, J. P., Wang, L., Han, D., Wang, E. S., Huskey, N. E., Lim, L., et al. (2012). IRE1α Cleaves Select microRNAs during ER Stress to Derepress Translation of Proapoptotic Caspase-2. Science 338 (6108), 818–822. doi:10.1126/science.1226191
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 287 (5453), 664–666. doi:10.1126/science.287.5453.664
Vattem, K. M., and Wek, R. C. (2004). Reinitiation Involving Upstream ORFs Regulates ATF4 mRNA Translation in Mammalian Cells. Proc. Natl. Acad. Sci. U S A. 101 (31), 11269–11274. doi:10.1073/pnas.0400541101
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK Is Required at the ER-Mitochondrial Contact Sites to Convey Apoptosis after ROS-Based ER Stress. Cel Death Differ. 19 (11), 1880–1891. doi:10.1038/cdd.2012.74
Waldschmitt, N., Berger, E., Rath, E., Sartor, R. B., Weigmann, B., Heikenwalder, M., et al. (2014). C/EBP Homologous Protein Inhibits Tissue Repair in Response to Gut Injury and Is Inversely Regulated with Chronic Inflammation. Mucosal Immunol. 7 (6), 1452–1466. doi:10.1038/mi.2014.34
Wang, S., Huang, Y., Zhou, C., Wu, H., Zhao, J., Wu, L., et al. (2018). The Role of Autophagy and Related MicroRNAs in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2018, 7565076–7565110. doi:10.1155/2018/7565076
Wang, X. Z., Harding, H. P., Zhang, Y., Jolicoeur, E. M., Kuroda, M., and Ron, D. (1998). Cloning of Mammalian Ire1 Reveals Diversity in the ER Stress Responses. EMBO J. 17 (19), 5708–5717. doi:10.1093/emboj/17.19.5708
Wang, Y., Shen, J., Arenzana, N., Tirasophon, W., Kaufman, R. J., and Prywes, R. (2000). Activation of ATF6 and an ATF6 DNA Binding Site by the Endoplasmic Reticulum Stress Response. J. Biol. Chem. 275 (35), 27013–27020. doi:10.1074/jbc.M003322200
Wu, P., Tian, T., Zhao, J., Song, Q., Wu, X., Guo, Y., et al. (2021). IRE1α-JNK Pathway-Mediated Autophagy Promotes Cell Survival in Response to Endoplasmic Reticulum Stress during the Initial Phase of Hepatic Steatosis. Life Sci. 264, 118668. doi:10.1016/j.lfs.2020.118668
Xie, J., Li, L., Deng, S., Chen, J., Gu, Q., Su, H., et al. (2020). Slit2/Robo1 Mitigates DSS-Induced Ulcerative Colitis by Activating Autophagy in Intestinal Stem Cell. Int. J. Biol. Sci. 16 (11), 1876–1887. doi:10.7150/ijbs.42331
Xu, Y., Yang, J., Li, F., Lian, G., and Ouyang, M. (2018). MiR-29a Inhibited Intestinal Epithelial Cells Autophagy Partly by Decreasing ATG9A in Ulcerative Colitis. Anticancer Drugs 29 (7), 652–659. doi:10.1097/CAD.0000000000000636
Xuan, H., Ou, A., Hao, S., Shi, J., and Jin, X. (2020). Galangin Protects against Symptoms of Dextran Sodium Sulfate-Induced Acute Colitis by Activating Autophagy and Modulating the Gut Microbiota. Nutrients 12 (2), 347. doi:10.3390/nu12020347
Xue, J. F., Shi, Z. M., Zou, J., and Li, X. L. (2017). Inhibition of PI3K/AKT/mTOR Signaling Pathway Promotes Autophagy of Articular Chondrocytes and Attenuates Inflammatory Response in Rats with Osteoarthritis. Biomed. Pharmacother. 89, 1252–1261. doi:10.1016/j.biopha.2017.01.130
Yahiro, K., Tsutsuki, H., Ogura, K., Nagasawa, S., Moss, J., and Noda, M. (2014). DAP1, a Negative Regulator of Autophagy, Controls SubAB-Mediated Apoptosis and Autophagy. Infect. Immun. 82 (11), 4899–4908. doi:10.1128/IAI.02213-14
Yamazaki, H., Hiramatsu, N., Hayakawa, K., Tagawa, Y., Okamura, M., Ogata, R., et al. (2009). Activation of the Akt-NF-kappaB Pathway by Subtilase Cytotoxin through the ATF6 branch of the Unfolded Protein Response. J. Immunol. 183 (2), 1480–1487. doi:10.4049/jimmunol.0900017
Yan, S., Yingchao, L., Zhangliu, W., Xianli, R., Si, L., Siyi, N., et al. (2020). Effect of Berberine from Coptis Chinensis on Apoptosis of Intestinal Epithelial Cells in a Mouse Model of Ulcerative Colitis: Role of Endoplasmic Reticulum Stress. Evid. Based Complement. Alternat. Med. 2020, 3784671. doi:10.1155/2020/3784671
Yang, H., Niemeijer, M., van de Water, B., and Beltman, J. B. (2020). ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 23 (2), 100860. doi:10.1016/j.isci.2020.100860
Yang, J., Zhou, R., and Ma, Z. (2019). Autophagy and Energy Metabolism. Adv. Exp. Med. Biol. 1206, 329–357. doi:10.1007/978-981-15-0602-4_16
Yu, L., Chen, Y., and Tooze, S. A. (2017). Autophagy Pathway: Cellular and Molecular Mechanisms. Autophagy 14 (2), 207–215. doi:10.1080/15548627.2017.1378838
Yue, W., Liu, Y., Li, X., Lv, L., Huang, J., and Liu, J. (2019). Curcumin Ameliorates Dextran Sulfate Sodium-Induced Colitis in Mice via Regulation of Autophagy and Intestinal Immunity. Turk J. Gastroenterol. 30 (3), 290–298. doi:10.5152/tjg.2019.18342
Zhang, H. S., Chen, Y., Fan, L., Xi, Q. L., Wu, G. H., Li, X. X., et al. (2015). The Endoplasmic Reticulum Stress Sensor IRE1α in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J. Biol. Chem. 290 (24), 15327–15336. doi:10.1074/jbc.M114.633560
Zhang, S. L., Li, Z. Y., Wang, D. S., Xu, T. Y., Fan, M. B., Cheng, M. H., et al. (2020). Aggravated Ulcerative Colitis Caused by Intestinal Metrnl Deficiency Is Associated with Reduced Autophagy in Epithelial Cells. Acta Pharmacol. Sin. 41 (6), 763–770. doi:10.1038/s41401-019-0343-4
Zhang, Y.-g., Lu, R., Xia, Y., Zhou, D., Petrof, E., Claud, E. C., et al. (2018). Lack of Vitamin D Receptor Leads to Hyperfunction of Claudin-2 in Intestinal Inflammatory Responses. Inflamm. Bowel Dis. 25, 97. doi:10.1093/ibd/izy292
Zhou, M., Xu, W., Wang, J., Yan, J., Shi, Y., Zhang, C., et al. (2018). Boosting mTOR-dependent Autophagy via Upstream TLR4-MyD88-MAPK Signalling and Downstream NF-κB Pathway Quenches Intestinal Inflammation and Oxidative Stress Injury. EBioMedicine 35, 345–360. doi:10.1016/j.ebiom.2018.08.035
Keywords: autophagy, endoplasmic reticulum stress, ulcerative colitis, unfolded protein, unfolded protein response
Citation: Qiao D, Zhang Z, Zhang Y, Chen Q, Chen Y, Tang Y, Sun X, Tang Z and Dai Y (2021) Regulation of Endoplasmic Reticulum Stress-Autophagy: A Potential Therapeutic Target for Ulcerative Colitis. Front. Pharmacol. 12:697360. doi: 10.3389/fphar.2021.697360
Received: 19 April 2021; Accepted: 01 September 2021;
Published: 13 September 2021.
Edited by:
Sung Hwan Ki, Chosun University, South KoreaReviewed by:
Rohit Gundamaraju, University of Tasmania, AustraliaShuhei Hosomi, Osaka City University, Japan
Copyright © 2021 Qiao, Zhang, Zhang, Chen, Chen, Tang, Sun, Tang and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhipeng Tang, emhpcGVuZ3RhbmdAc29odS5jb20=; Yancheng Dai, ZGFpeWFuY2hlbmcyMDA1QDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship