 Yinghui Cui
Yinghui Cui Fenglan Chu2
Fenglan Chu2 Hui Dong
Hui Dong Feng Xu
Feng Xu- 1Department of Pediatric Intensive Care Unit, Children’s Hospital of Chongqing Medical University; National Clinical Research Center for Child Health and Disorders; Ministry of Education Key Laboratory of Child Development and Disorders; Chongqing Key Laboratory of Pediatrics, Chongqing, China
- 2Department of Gastroenterology, Xinqiao Hospital, Army Medical University, Chongqing, China
As little is known about the role of calcium (Ca2+) signaling mediating the small intestinal epithelial anion secretion, we aimed to study its regulatory role in secretagogue-stimulated duodenal anion secretion and the underlying molecular mechanisms. Therefore, intestinal anion secretion from native mouse duodenal epithelia was examined with Ussing chambers to monitor PGE2-, 5-HT-, and CCh-induced short-circuit currents (Isc). PGE2 (10 μM) and 5-HT (10 μM) induced mouse duodenal Isc, markedly attenuated by serosal Ca2+-free solution and selective blockers of store-operated Ca2+ channels on the serosal side of the duodenum. Furthermore, PGE2- and 5-HT-induced duodenal Isc was also inhibited by ER Ca2+ chelator TPEN. However, dantrolene, a selective blocker of ryanodine receptors, inhibited PGE2-induced duodenal Isc, while LiCl, an inhibitor of IP3 production, inhibited 5-HT-induced Isc. Moreover, duodenal Isc response to the serosal applications of both PGE2 and 5-HT was significantly attenuated in transient receptor potential vanilloid 4 (TRPV4) knockout mice. Finally, mucosal application of carbachol (100 μM) also induced duodenal Isc via selective activation of muscarinic receptors, which was significantly inhibited in serosal Ca2+-free solution but neither in mucosal Ca2+-free solution nor by nifedipine. Therefore, the serosal TRPV4-constituted SOCE mechanism is likely universal for the most common and important secretagogues-induced and Ca2+-dependent intestinal anion secretion. These findings will enhance our knowledge about gastrointestinal (G.I.) epithelial physiology and the associated G.I. diseases, such as diarrhea and constipation.
Introduction
Intestinal epithelial ion secretion is a critical physiological process in the human gastrointestinal (G.I.) tract. Since water follows ion movement across osmotic gradients, which is primarily generated by chloride ion (Cl−) and bicarbonate (HCO3−) secretion (Barrett and Keely, 2000; Kiela and Ghishan, 2009), the clarification of these processes is essential to delineate the pathophysiology of various diarrheal diseases. The Cl− and HCO3− secretions are under the control of several secretagogues in the G.I. system. Like prostaglandin E2 (PGE2), is a potent chloride secretagogue likely to be active under physiological and pathophysiological circumstances (Weymer et al., 1985; Rajagopal et al., 2014); meantime, PGE2 stimulates duodenal bicarbonate secretion to protect the mucosal epithelium against acid-induced injury in various species (Takeuchi and Amagase, 2018). Furthermore, 5-hydroxytryptamine (5-HT) is also an essential secretagogue of Cl− and HCO3− secretion, and it is released by enterochromaffin (E.C.) cells situated in the intestine epithelium (Brown, 1995). Besides, acetylcholine (ACh) is a primary neurotransmitter in activating intestinal anion secretion.
These secretagogues described above mediate epithelial ion transports via three major second messengers: cAMP, cGMP, and Ca2+ (Murek et al., 2010). Among these messengers, the physiological roles and molecular mechanisms of cAMP- and cGMP-dependent regulation of intestinal ion transports have been well elucidated (Rao et al., 1984; Tuo et al., 2009), while those mediated via calcium signaling remain relatively poorly understood.
It is commonly believed that in non-excitable cells, secretagogues evoke calcium signaling through two necessary processes: the release of Ca2+ from intracellular stores, then an enhanced extracellular Ca2+ entry (Putney, 2007), which was called capacitative or store-operated Ca2+ channels (SOCs) classically. The intracellular store in the endoplasmic reticulum (E.R.) from which Ca2+ is released in two main ways, which is via the ryanodine receptor (RyR) or the inositol trisphosphate receptor (IP3R) (MacMillan et al., 2005). These Ca2+ release-activated Ca2+ channels (CRAC) were first described in mast cells and Jurkat lymphocytes (Hoth and Penner, 1992; Hoth and Penner, 1993). However, detailed underlying mechanisms that secretagogues mediated cytosolic Ca2+ signaling in duodenal anion secretion still need to elucidate (Xie et al., 2014). In addition, while molecular components of SOCE are well defined in immune cells, their molecular identification is still elusive in intestinal epithelial cells.
We previously demonstrated that carbachol (CCh), a stable chemical analog of neurotransmitter ACh, triggered IP3R/ER Ca2+ release, but caffeine triggered RyR/ER Ca2+ release, both of which stimulated serosal store-operated Ca2+ entry (SOCE) mechanism and eventually induced Ca2+-dependent duodenal anion secretion (Yang et al., 2018; Zhang et al., 2019; Zhang et al., 2021) However, it is currently unclear: 1) whether [Ca2+]cyt is also a critical cell signaling for other most common and important secretagogues, such as PGE2 and 5-HT; 2) if serosal SOCE is a universal mechanism for Ca2+-dependent duodenal anion secretion; 3) if so, what molecular components of the SOCE are involved in this process; and 4) if CCh evokes a Ca2+-dependent anion secretion when applied from the mucosal side of the duodenum, although it is well known to stimulate it from the serosal side. Therefore, we aimed to investigate these important issues using native duodenal epithelial tissues in mice as a follow-up study.
Materials and Methods
Animals and Cells
All experiments were adopted with adult male Harlan C-57BL/6 mice (6–8 weeks old; 18–22 g; Chongqing Tengxin Biotechnology Co. Ltd., Chongqing, China) and transient receptor potential vanilloid 4 (TPPV4) deficient (TRPV4 KO) mice which generated from C-57BL/6 mice (6–12 weeks old; 20–25 g; Cyagen bioscience, China). Animal care and experiments conformed with the guidance of the Animal Ethical Committee of the University and were approved by the University Committee on Investigations Involving Animal Subjects. According to the ARRIVE guidelines (Kilkenny et al., 2010), the mice were bred and housed in a standard animal care room at an ambient temperature of 20°C and air humidity of 50–55% on a 12 h: 12 h light-dark cycle with free access to water and food pellets until the time of experiments. Before each experiment, mice’s food and water were deprived for at least 1 h. Mice were sacrificed by cervical dislocation under narcosis with 100% CO2. Animals were assigned randomly to different experimental groups of all studies. Data collection and evaluation of all experiments performed blindly, and the experimenters were unaware of group treatments.
IEC-6, a small intestinal epithelial cell line of rat origin (Thomas and Oates, 2002), was obtained from the American Type Culture Collection (ATCC, Rockville, MD, United States) and routinely cultured in fresh Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% Fetal bovine serum (FBS), glutamine and penicillin/streptomycin every 2 days. After the cells had grown well for experiments, they were replated onto 12 mm round coverslips (Warner Instruments Inc., Hamden, CT, United States) and incubated for at least 24 h before use for [Ca2+]cyt measurement.
Solutions
Solutions to the mucosal side in the Ussing chamber experiments contained the following: 115 mM NaCl, 25 mM sodium-D-gluconate, 5.2 mM potassium-D-gluconate, 1.2 mM CaCl2, 1.2 mM MgCl2, and 10 mM D-mannitol at pH 7.4 when gassed with Oxygen (100% O2) at 37°C. The serosal solution contained the following: 115 mM NaCl, 25 mM NaHCO3, 2.2 mM K2HPO4, 1.2 mM CaCl2, 1.2 mM MgCl2, 0.8 mM KH2PO4, 10 mM D-glucose, and this solution was gassed with carbogen (5% CO2 and 95% O2, v/v) at 37°C and had a pH 7.4. For the Ca2+-free experiments, Ca2+ was omitted, and EGTA (0.5 mM) was added in both mucosal and serosal solutions to prevent potential Ca2+ contamination. The physiological salt solution (PSS) used in digital Ca2+ measurement contained the following: 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 10 mM HEPES, and 10 mM glucose (pH 7.4). For the Ca2+-free PSS solution, Ca2+ was omitted, but 0.5-mM EGTA was added. The osmolalities for all solutions were 300 mOsmol kg−1 of H2O.
Tissue Preparations
Following euthanasia, the mice’s abdomen was opened by a midline incision. Next, we dissected the proximal duodenum 4 cm from the pylorus carefully and immediately but not pulled to avoid damaging the epithelium. Afterward, the duodenum section was incubated in ice-cold iso-osmolar mannitol (300 mM) and indomethacin (1μM) solution 10 min before seromuscular stripping to inhibit possible endogenous PGE2, which is resulting from mucosal injury during experiments, to avoid affecting the basal Isc. Finally, the section is opened longitudinally, with the mesenteric attachment remnant, seromusculature stripped, and divided into four segments. The segment, which is likely to undergo less excision damage, will be situated in the chambers’ aperture (window area, 0.1 cm2).
Ussing Chamber Experiments
Segments were fixed in a modified Ussing chamber bathed with a volume of 3 ml on each side of the mucosa preparation at 37°C and short-circuited by a computer-controlled voltage-clamp device (Voltage-Current Clamp, VCC MC6; Physiologic Instruments, San Diego, CA, United States) under continuous short-circuited conditions. An automatic voltage-clamp measured the transepithelial short-circuit currents (Isc), while μA was used for the original recordings, and μA cm−2 was used for summary data. After 10–15 min of measurements for basal parameters, various agonists or antagonists were added to one side or both sides for 10–20 min, followed by PGE2, 5-HT, and carbachol.
Calcium Image Experiments
[Ca2+]cyt measurement experiments were performed as previously described (Zhang et al., 2019). ICE-6 Cells grown on coverslips were loaded with 5 μM Fura-2/AM in PSS, described above, at room temperature (22–25°C) for 50 min and then washed for 30 min. After that, the coverslips with epithelial cells were mounted in a perfusion chamber on a Nikon microscope stage (Nikon Corp., Tokyo, Japan). The ratio of Fura-2/AM fluorescence with excitation at 340 or 380 nm (F340/380) was followed over time and captured using an intensified charge-coupled device camera (ICCD200) and a MetaFluor imaging system (Universal Imaging Corp., Downingtown, PA, United States).
Materials
Prostaglandin E2, Serotonin hydrochloride(5-HT), SKF-96365, TPEN (N, N, N′, N′- tetrakis (2-pyridylmethyl) ethylenediamine), GSK-7975A, ouabain, HC067047, and GSK1016790A were purchased from MedChemExpress (MCE; Monmouth Junction, NJ, United States). Sigma (Saint Louis, MO, United States) supplied carbamylcholine chloride (CCh), nifedipine, gadolinium chloride, and cyclopiazonic acid (CPA). Tocris Bioscience (Ellisville, MO, United States) supplied 2-Aminoethoxydiphenyl borate (2-APB), while APExBIO Technology LLC (Houston, TX, United States) provided dantrolene. Fura-2 was purchased from Invitrogen (UT, United States), meantime DMEM and FBS were obtained from Hyclone (Logan, UT, United States). Trypsin and penicillin/streptomycin were purchased from Gibco (CA, United States). The other chemicals were obtained from BBI Life Science (Shanghai, China).
Data and Statistical Analysis
Data and statistical analysis yield to the recommendations of Frontiers in Pharmacology. All results are given as means ± standard error of the mean number (n) of investigated tissues. Net peak of duodenal Isc refers to drug-stimulated maximal peak minus basal level. The statistical significance of differences in experimental groups’ means was determined by using Student’s unpaired, two-tailed t-test or one-way ANOVA followed by Dunnett’s post-test. Post hoc tests were run if F achieved p < 0.05 (GraphPad Prism 8.0), and there was no significant variance in inhomogeneity. A probability P-value <0.05 was considered statistically significant.
Results
Prostaglandin E2 Induced Ca2+-Dependent Epithelial Anion Secretion in Duodenum
Since PGE2 is one of the most common and important secretagogues, we conducted Ussing chamber experiments to test its effect on Ca2+-dependent duodenal epithelial anion transports. Because the duodenal epithelium is polarized, with the mucosal side and the serosal side, we tested which side was acted by PGE2. The addition of PGE2 (10 μM) in the serosal induced a transient high Isc peak with a sustained phase following (Figure 1A). However, PGE2 and vehicle (DMSO) mucosal application did not affect the duodenal Isc (Figures 1A,B). Therefore, PGE2 acts on the serosal side of the duodenum exclusively.
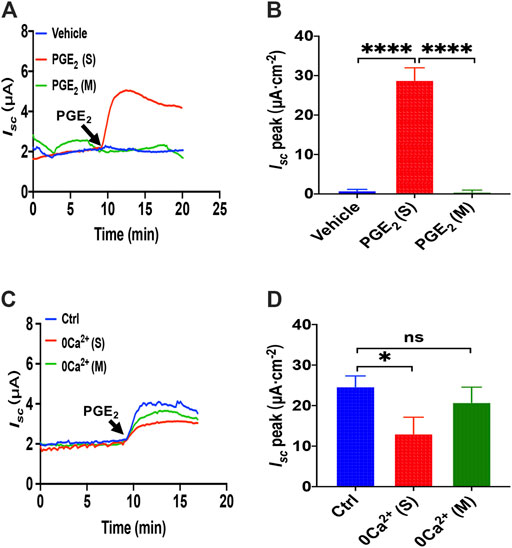
FIGURE 1. PGE2 induced Ca2+-dependent small intestine epithelial anion secretion. (A) Time courses of PGE2 (10 μM) -evoked Isc or vehicle (DMSO) when applied to the serosal (s) or mucosal (m) side of mice duodenal mucosal tissues (n ≥ 6). (B) When added to the serosal or mucosal side, vehicle or PGE2-stimulated Isc peak (n = 6). (C) Time courses of PGE2-evoked Isc after extracellular Ca2+ omission (0 Ca2+) from each side in duodenal mucosal tissues. (D) PGE2-stimulated Isc peak after Ca2+ omission from each side (n ≥ 6). Ctrl represents as the control in which normal extracellular Ca2+ was on both sides. Results are presented as mean ± SE. *p < 0.05, ****p < 0.0001, significantly different from the corresponding control by one-way ANOVA followed by Dunnett’s post-test.
To examine whether extracellular Ca2+ is vital for PGE2-evoked anion secretion, we omitted extracellular Ca2+ on the serosal or mucosal sides of the Ussing chamber. We found that calcium omission of the serosal side weakened the PGE2-evoked Isc peak but not the mucosal side (Figures 1A,B). Therefore, the presence of Ca2+ in the serosal side is critical for PGE2-induced duodenal Isc.
Prostaglandin E2 Induced Ca2+-Dependent Ion Secretion by Serosal Store-Operated Ca2+ Entry Mechanism
To examine if the SOCE mechanism was involved in PGE2-mediated anion transports, we adapted four inhibitors with different chemical structures to block SOCE. Considering 2-APB is a SOCE and an inconsistent IP3R inhibitor (Bootman et al., 2002), we first applied 2-APB to test. We found that 2-APB (100 μM) had no effect on PGE2-stimulated duodenal Isc after mucosal addition while adding in the serosal side significantly attenuated the duodenal Isc (Figures 2A,B). Like 2-APB, SKF-96365, a selective SOCE blocker added in the serosal side but not the mucosal side, also significantly suppressed the duodenal Isc (Figures 2C,D).
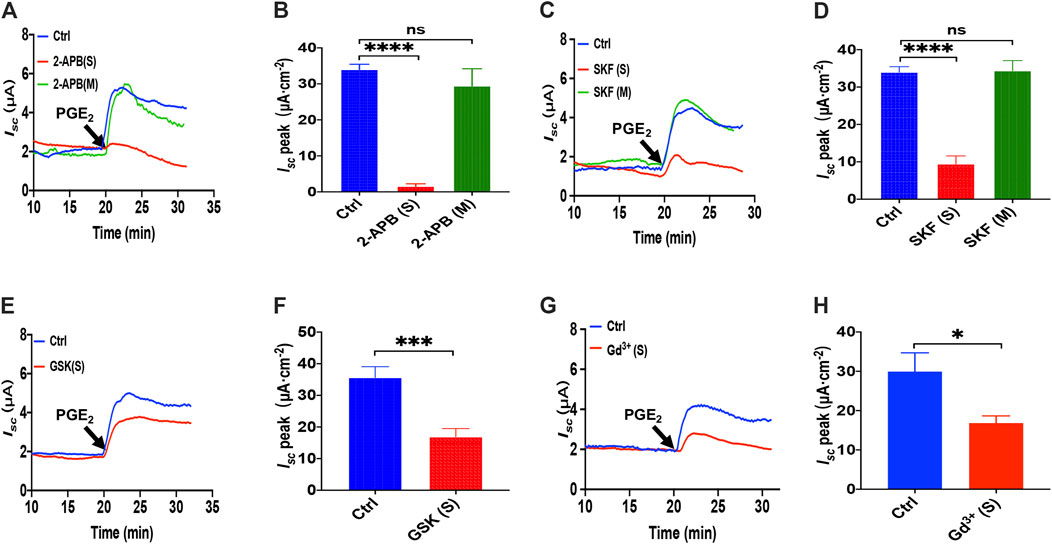
FIGURE 2. PGE2 induced Ca2+-dependent ion secretion by serosal SOCE mechanism in mice duodenum. (A) Time course of PGE2 (10 μM) -evoked Isc after mucosal or serosal application of 2-aminoethoxydiphenyl borate (2-APB, 100 μM, n = 6). (B) PGE2-evoked Isc peak after 2-APB was added to the serosal or mucosal side. (C) Time course of PGE2-evoked Isc after mucosal or serosal application of SKF-96365 (SKF, 30 μM, n = 6). (D) PGE2-evoked Isc peak after SKF-96365 was added to the serosal or mucosal side. (E) Time course of PGE2-evoked duodenal Isc after serosal application of GSK-7975A (GSK, 100 μM, n = 6). (F) PGE2-evoked duodenal Isc peak after serosal application of GSK-7975A. Ctrl represents the control without drug treatment. (G) Representative of the time course of PGE2-stimulated duodenal Isc after serosal addition of GdCl3 (Gd3+, 30 μM, n = 6). (F) Summary of the effect of GdCl3 on PGE2-stimulated duodenal Isc peak after serosal addition. Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences, *p < 0.05, ***p < 0.001, ****p < 0.0001 vs. corresponding control by Student’s unpaired, two-tailed t-test or one-way ANOVA followed Dunnett’s post-test.
Since GSK-7975A is a specific blocker of the CRAC channel (Molnar et al., 2016), we tested if the CRAC channel is SOCE in the duodenal epithelium utilizing it. As shown in Figures 2E,F, GSK-7975A (100 μM) markedly reduced PGE2-stimulated duodenal Isc. Furthermore, considering Gd3+ has been the most widely employed tool for blocking SOCE and CRAC/Orai channel (Bird et al., 2008; Sataloff et al., 2017), we added GdCl3 (30 μM) in the serosal side significantly reduced PGE2-stimulated duodenal Isc (Figures 2G,H). Therefore, PGE2 induced Ca2+-dependent ion secretion by acting on the serosal SOCE mechanism and probably CRAC channels in the duodenal epithelium.
ER Ca2+ Store and Ryanodine Receptors in PGE2-Induced Intestinal Ion Transports
As is know that N, N, N′, N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN) can rapidly and reversibly chelate Ca2+ within E.R. stores without influencing [Ca2+]cyt for its low affinity with Ca2+ (Caroppo et al., 2003), which we applied to investigate further the role of ER Ca2+ store in PGE2-induced duodenal Isc. As shown in Figures 3A,B, serosal addition of TPEN (1 mM) significantly suppressed PGE2-evoked Isc. Considering ryanodine receptors (RyR) can mediate Ca2+ release from E.R., serosal addition of dantrolene (300 μM), a selective RyR antagonist, markedly inhibited PGE2-induced Isc (Figures 3C,D), suggesting E.R. Ca2+ store release dominantly by the serosal side. As the inositol 1,4,5-triphosphate (IP3) also leads to E.R. intracellular Ca2+release through E.R. membrane by IP3 receptors (Lindqvist et al., 1998), we used LiCl (30 mM) that inhibits IP3 production, but LiCl added either on the serosal side or both sides of the tissues did not significantly alter PGE2-induced Isc (Figure 3E). These findings suggest that PGE2 acts via RyR/Ca2+ rather than IP3/Ca2+ in E.R. to induce duodenal epithelial anion transports.
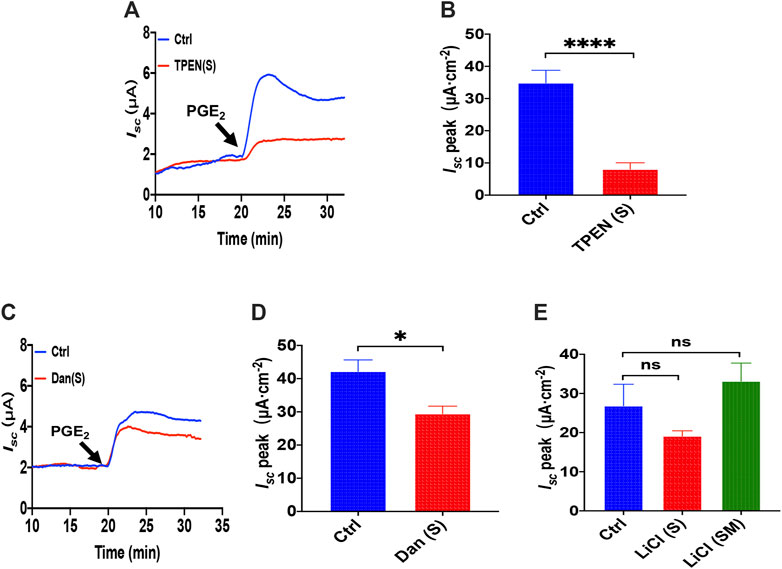
FIGURE 3. The ER Ca2+ store and ryanodine receptors in PGE2 -induced duodenal ion transports. (A,B) PGE2 (10 μM) -evoked Isc after serosal (s) application of TPEN (1 mM, n = 6). (C,D) PGE2-evoked Isc after dantrolene (Dan, 300 μM, n ≥ 6) was added to the serosal (s). (E) PGE2-evoked Isc after LiCl (30 mM, n ≥ 6) was added to the serosal side or both sides (m + s). Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences, *p < 0.05, ****p < 0.0001 vs. corresponding control by Student’s unpaired, two-tailed t-test or one-way ANOVA followed by Dunnett’s post-test.
Prostaglandin E2 Induced Duodenal Ion Secretion by Serosal Transient Receptor Potential Vanilloid 4 Channels and Na+/K+ ATPase
Since that 2-APB, SKF-96365 and GSK-7975A also act on the transient receptor potential V(TRPV) family and that TRPV4 channels are expressed in the G.I. tract (Blackshaw et al., 2010), we tested if TRPV4 channel may represent the molecular constituents of CRAC channels in the process of the PGE2-stimulated duodenal Isc. We all know that GSK1016790A, a highly selective agonist of TRPV4, can activate TRPV4 in diverse cells (Baratchi et al., 2019). However, unlike the effect in cultured cells detected in the ussing chamber at the tissue level, GSK1016790A alone had no effect on the basal Isc (Supplementary Figure S1). Hence we first chose the application of HC067047, a potent and selective TRPV4 antagonist (Xia et al., 2013), to block TRPV4 Channels. As shown in Figures 4A,B, serosal addition of HC067047 (30 μM) suppressed PGE2-stimulated duodenal Isc. Secondly, we compared PGE2-stimulated duodenal Isc between wild-type and TRPV4 KO mice. Duodenal Isc induced by serosal addition of PGE2 was significantly attenuated in TRPV4 KO mice (Figures 4C,D), while HC067047 (30 μM) did not affect PGE2 evoked Isc of TRPV4 KO mice to exclude non-specific effects other than TRPV4 inhibition (Supplementary Figure S2). Thirdly, to further verify TRPV4 involved in the molecular composition of SOCE, we used SOCE and CRAC blocker, Gd3+, to test whether it works on TRPV4 knockout mice. GdCl3 (30 μM) serosal addition did not affect the PGE2-stimulated duodenal Isc of TRPV4 knockout mice. Above all, our findings are suggesting that TRPV4 is the molecular constituent of CRAC channels.
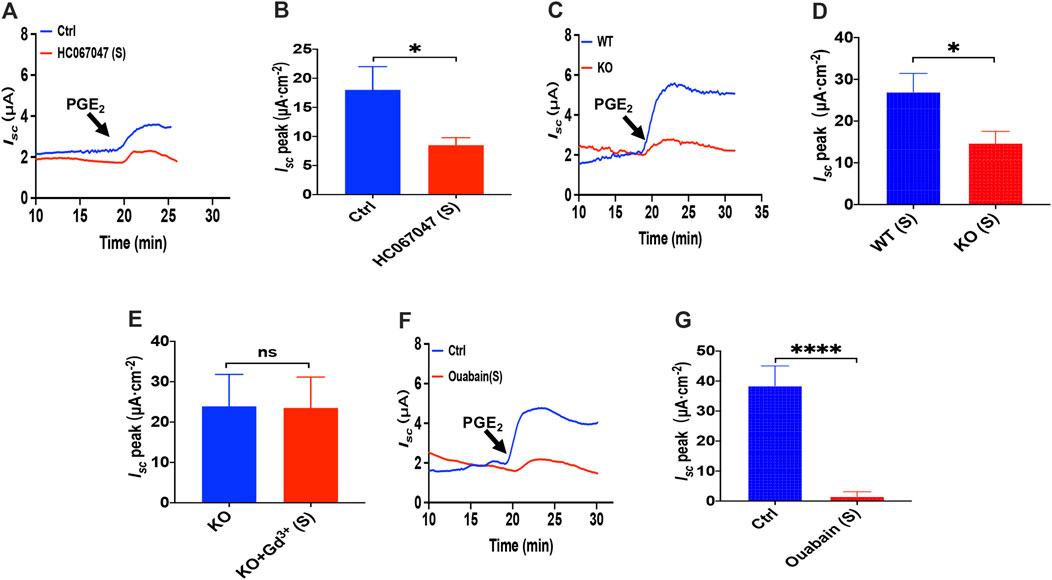
FIGURE 4. PGE2 induced duodenal ion secretion by serosal TRPV4 channels and NKA. (A,B) on PGE2-evoked Isc after HC067047 (30 μM, n = 6) added in serosal (s) side. (C,D) PGE2 (10 μM) -evoked duodenal Isc in wild-type (W.T.) and TRPV4 knockout (K.O.) mice (n = 6). (E) PGE2-evoked Isc after GdCl3 (Gd3+, 30 μM, n ≥ 6) was added to the serosal (s) side of the TRPV4 KO mice duodenum. (F,G) PGE2-stimulated Isc after Ouabain (1 mM, n = 7) was added to the serosal (s) side. Ctrl represents the control without drug treatment. Results are presented as mean ± SE. *p < 0.05, ****p < 0.0001 significantly different from the corresponding control by Student’s unpaired, two-tailed t-test.
Since Cl− movement across the epithelial cells is facilitated by Na+/K+ ATPase (NKA), we tested if it is involved in the process of PGE2-stimulated duodenal Isc. Because NKA is exclusively expressed at the serosal side in the intestinal epithelium (Hammerton et al., 1991; Thorsen et al., 2014), addition of 1 mM ouabain, a selective NKA inhibitor, to the serosal side completely abolished PGE2-induced duodenum Isc (Figures 4F,G), indicating that NKA participates in PGE2-mediated duodenal secretion.
5-HT Induced Ca2+-Dependent Duodenal Epithelial Ion Transports
When another important secretagogue, 5-HT (10 μM), was added to the serosal side, duodenal Isc increased, peaking within 2 min, and then sustained for more than 10 min (Figures 5A,B). However, mucosal application of 5-HT or vehicle (DMSO) did not alter basal Isc. Therefore, to test if Ca2+ is involved in the process of 5-HT-stimulated duodenal ion transports, we omitted extracellular Ca2+ of each side, and then we found that 5-HT- evoked Isc was markedly suppressed in either side of the duodenal tissues (Figures 5C,D). Therefore, 5-HT-stimulated duodenal ion transports are strongly Ca2+-dependent.
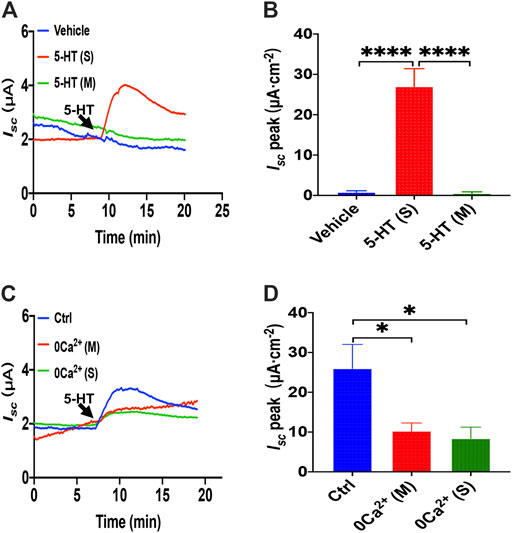
FIGURE 5. 5-HT induced Ca2+-dependent duodenal epithelial ion transports. (A) Time courses of 5-HT (10 μM) -evoked Isc or vehicle (DMSO) when added to serosal (s) or mucosal side. (B) When added to the serosal or mucosal side, vehicle or 5-HT-evoked Isc peak (n = 6). (C) Time courses of 5-HT-evoked Isc after extracellular Ca2+ omission (0 Ca2+) from each side in duodenal mucosal tissues. (D) 5-HT-evoked Isc peak after Ca2+ omission from each side (s or m, n = 6). Ctrl represents as the control in which normal extracellular Ca2+ was on both sides. Results are presented mean ± SE. *p < 0.05, ****p < 0.0001 significantly different from the corresponding control by one-way ANOVA followed by Dunnett’s post-test.
5-HT Induced Ca2+-Dependent Intestinal Ion Secretion by Serosal Store-Operated Ca2+ Entry Mechanism
Because it is still elusive for the mechanisms underlying 5-HT induced Ca2+-dependent duodenal ion secretion, we examined if the above mechanisms similar to PGE2 are involved. First, we utilized three antagonists of SOCE. 2-APB (100 μM) application to the serosal side significantly suppressed 5-HT-evoked Isc, but the mucosal side application did not affect Isc (Figures 6A,B). SKF-96365 (30 μM) markedly decreased the Isc peak from serosal side application but not from the mucosal side of the duodenum (Figures 6C,D). Hence, 5-HT also mediates SOCE mechanisms that act entirely on the duodenal serosal side, consistently with our findings with PGE2 described above.
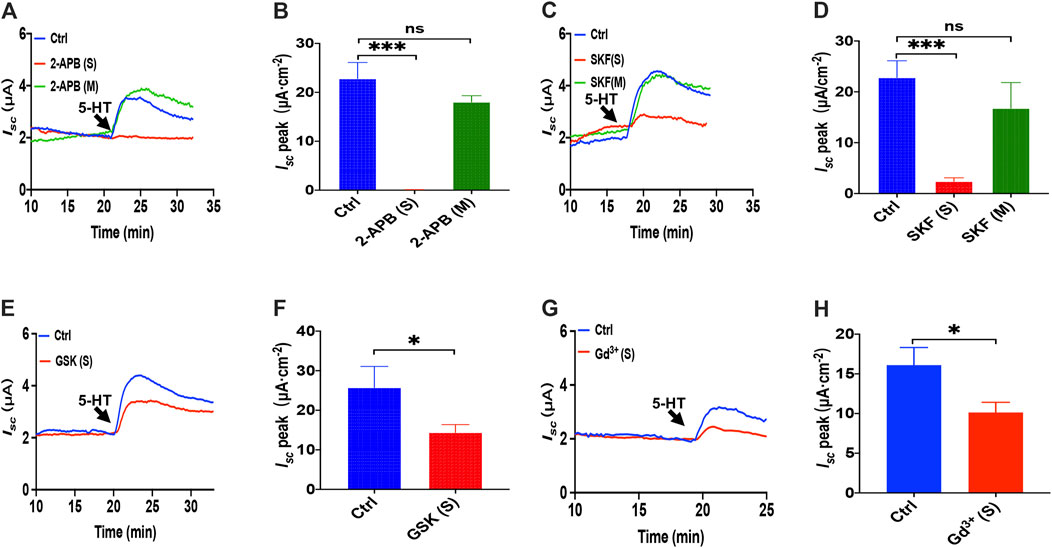
FIGURE 6. 5-HT induced Ca2+-dependent intestinal ion secretion by serosal SOCE mechanism in mice duodenum. (A) Time course of 5-HT-evoked Isc after 2-aminoethoxydiphenyl borate (2-APB, 100 μM, n = 6) added to the serosal or mucosal side. (B) 5-HT-stimulated murine duodenal Isc peak after 2-APB was added to the serosal or mucosal side. (C) Time course of 5-HT-evoked Isc after SKF-96365 (SKF, 30 μM, n = 6) added to the serosal or mucosal side. (D) 5-HT-evoked duodenal Isc peak after SKF-96365 was added to the serosal or mucosal side. (E) Time course of 5-HT-evoked Isc after GSK-7975A (GSK, 100 μM, n = 6) added to the serosal side. (F) 5-HT-evoked Isc peak after GSK-7975A was added to the serosal side. (G-H) 5-HT-evoked Isc after GdCl3 (Gd3+, 30 μM, n = 6) added to the side. Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences, *p < 0.05, ***p < 0.001 vs. corresponding control by Student's unpaired, two-tailed t-test or one-way ANOVA followed Dunnett’s post-test.
We further identified if the CRAC channel is 5-HT-mediated SOCE in the duodenal epithelium by utilizing GSK-7975A and Gd3+. When GSK-7975A (100 μM) was added to the serosal side, 5-HT-stimulated duodenal Iscis markedly inhibited in duodenal tissue (Figures 6E,F). Furthermore, GdCl3 (30 μM) application on the serosal side also significantly reduced 5-HT-stimulated duodenal Isc (Figures 6G,H). Therefore, 5-HT induced Ca2+-dependent intestinal ion secretion by serosal SOCE mechanism and probably CRAC channels.
5-HT Induced Duodenal Ion Secretion via Serosal Transient Receptor Potential Vanilloid 4 Channels
Like PGE2 experiments, we first chose HC067047, a potent and selective TRPV4 antagonist, to block TRPV4 channels. As shown in Figures 7A,B, serosal addition of HC067047 (30 μM) suppressed the 5-HT-stimulated duodenal Isc. Secondly, the duodenal Isc response to serosal application of 5-HT was significantly attenuated in TRPV4 knockout mice (Figures 7C,D), while HC067047 (30 μM) did not affect PGE2 evoked Isc of TRPV4 KO mice to exclude non-specific effects other than TRPV4 inhibition (Supplementary Figure S3). Thirdly, GdCl3 (30 μM) serosal addition did not affect the 5-HT-stimulated duodenal Isc of TRPV4 knockout mice (Figure 7E). Therefore, the present studies suggest that TRPV4 is the molecular constituent of CRAC channels in the duodenum again.
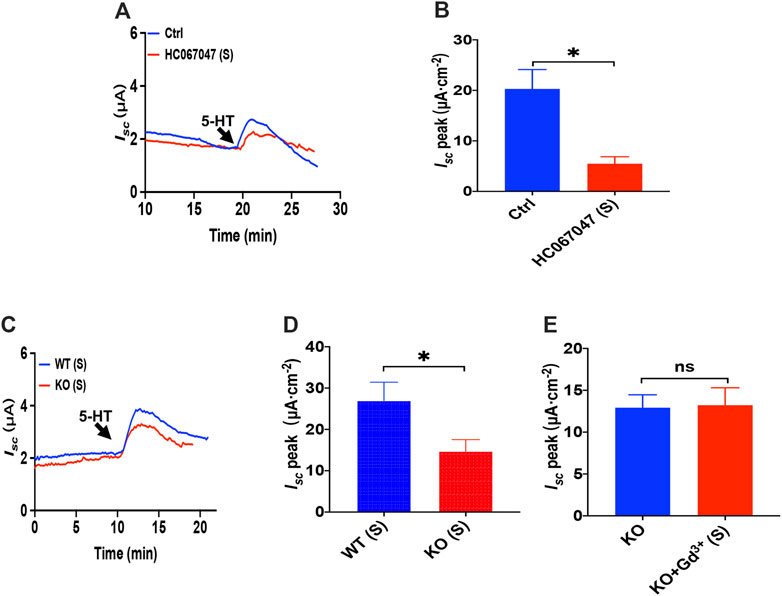
FIGURE 7. 5-HT induced duodenal ion secretion via serosal TRPV4 channels. (A,B) 5-HT-evoked Isc after HC067047 (30 μM, n = 6) was added to the serosal (s) side. (C,D) 5-HT (10 μM) -evoked Isc in wild-type (W.T.) or TRPV4 knockout (K.O.) mice (n = 6). (E) 5-HT-evoked Isc after GdCl (Gd3+, 30 μM, n = 6) was added to the serosal (s) side of the TRPV4 KO mice duodenum. Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences, *p < 0.05 vs. corresponding control by Student’s unpaired, two-tailed t-test.
The ER Ca2+ Store and IP3/Ca2+ Signaling in 5-HT-Induced Intestinal Ion Transports
To investigate how the ER Ca2+ store act in 5-HT-evoked Isc, we added TPEN (1 mM) to serosal side of the duodenum and found that TPEN significantly inhibited 5-HT-stimulated Isc (Figures 8A,B), indicating a vital role of the ER Ca2+ store in this course. Then we used LiCl (30 mM) to inhibit IP3 production but dantrolene (300 μM) to inhibit RyR. Interestingly, we found LiCl but not dantrolene significantly inhibited 5-HT-induced duodenal Isc (Figures 8C–E). Therefore, unlike PGE2, 5-HT induced duodenal epithelial anion transports via IP3/Ca2+ rather than RyR/Ca2+ in the E.R.
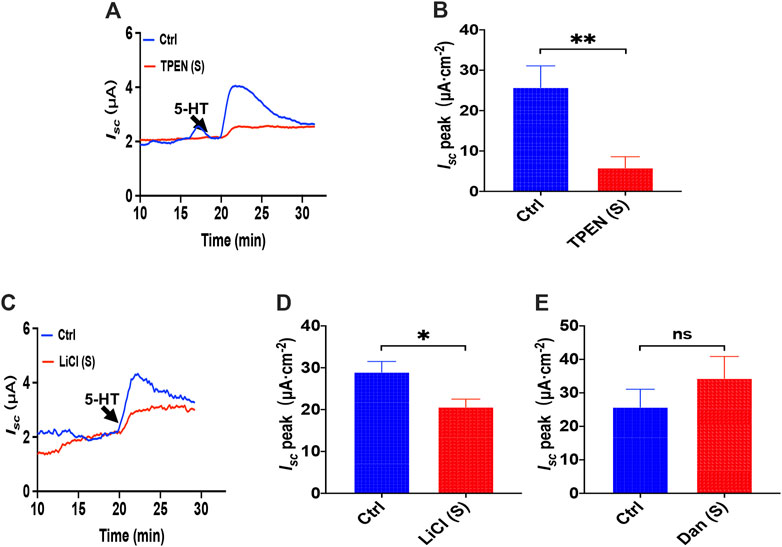
FIGURE 8. The ER Ca2+ store and IP3/Ca2+ signaling in 5-HT-induced intestinal ion transports. (A,B) 5-HT-evoked duodenal Isc after TPEN (1 mM, n = 6) added to serosal (s) side. (C,D) 5-HT-evoked Isc after LiCl (30 mM, n = 6) was added to the serosal (s) side. (E) 5-HT-evoked Isc after dantrolene (Dan, 100 μM, n = 6) was added to the serosal side. Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences, *p < 0.05, **p < 0.01 vs. corresponding control by Student’s unpaired, two-tailed t-test.
Luminal Carbachol Induced Ca2+-Dependent Duodenal Isc Through Serosal Ca2+ Entry
We previously demonstrated a critical role of serosal SOCE mechanism mediated by CCh, one of the most common and important secretagogues in Ca2+-dependent duodenal ion secretion. (Yang et al., 2018); however, it is not known if luminal addition of CCh can induce duodenal ion transports. Unlike PGE2 and 5-HT that act on seroal side of the duodenum exclusively (Figures 1A, 5A), we found that luminal addition of CCh (100 μM) induced a significant duodenal Isc (Figure 9A), although it was only about one-third of that induced by serosal addition (Figure 9B). To verify whether the action of luminal CCh is through the muscarinic receptors, atropine, a muscarinic receptor antagonist, was applied. As shown in Figures 9C,D, atropine (10 μM) added in mucosal side markedly attenuated luminal CCh-induced Isc indicating that luminal CCh evokes duodenal Isc via specific activation of muscarinic receptors expressed on the mucosal side of epithelial cells as well.
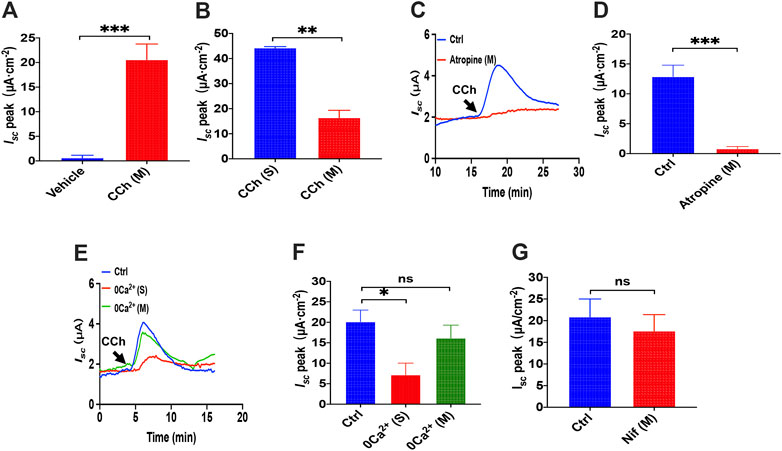
FIGURE 9. luminal CCh induced Ca2+-dependent duodenal Isc through serosal Ca2+ entry. (A) Summary data of vehicle (H2O) or CCh (100 μM) -stimulated Isc peak after mucosal application (n = 6). (B) Comparison between CCh-evoked Isc peak after mucosal and serosal application. (C,D) luminal CCh-induced duodenal Isc after atropine (10 μM, n = 6) added to the mucosal side. Ctrl represents the control without atropine treatment. (E,F) luminal CCh-evoked Isc after extracellular Ca2+ omitted from the serosal or mucosal side (n = 6). Ctrl represents as the control in which normal extracellular Ca2+ was on both sides. (G) luminal CCh-evoked duodenal Isc after mucosal addition of Nifedipine (Nif, 10 μM, n = 6). Ctrl represents the control without Nif treatment. Results are presented as mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001 vs. corresponding control by Student’s unpaired, two-tailed t-test.
To test the extracellular Ca2+ effect in CCh-induced duodenal Isc, we omitted extracellular Ca2+ in each side of the tissues. Figures 9E,F show that luminal CCh-evoked Isc was markedly suppressed when Ca2+ has omitted on the serosal side but not the mucosal side. Furthermore, we verified further that nifedipine (10 μM), an L-type calcium channel blocker, did not inhibit luminal CCh-induced Isc either (Figure 9G). Together, these findings indicate that even luminal CCh induced Ca2+-dependent duodenal Isc through serosal Ca2+ entry, further supporting our previous notion that CCh induced Ca2-dependent duodenal Isc by serosal SOCE mechanism exclusively (Yang et al., 2018).
Luminal Carbachol Induced Ca2+-Dependent Duodenal Isc via Transient Receptor Potential Vanilloid 4-Constituted Store-Operated Ca2+ Entry
As Gd3+ is a potential blocker of SOCE and CRAC/Orai channel, we added GdCl3 (30 μM) in the serosal side to test if it affects the luminal CCh induced Ca2+-dependent duodenal Isc. As shown in Figures 10A,B, Gd3+ significantly reduced luminal CCh-stimulated duodenal Isc, which suggested luminal CCh evokes anion secretion through SOCE mechanisms.
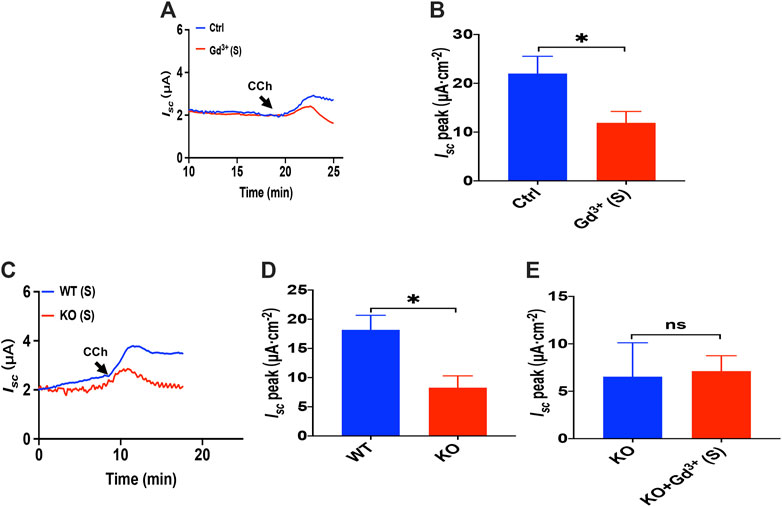
FIGURE 10. Luminal CCh induced Ca2+-dependent duodenal Isc via TRPV4-constituted SOCE. (A,B) luminal CCh(100 μM)-evoked Isc after GdCl3 (30 μM, n = 6) added to the serosal (s) side. (C,D) luminal CCh (100 μM) -evoked Isc in wild-type (W.T.) or TRPV4 K.O. mice (n = 6). (E) luminal-stimulated Isc after GdCl (Gd3+, 30 μM, n = 6) was added to the serosal (s) side of TRPV4 KO mice duodenum. Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences, *p < 0.05 vs. corresponding control by Student’s unpaired, two-tailed t-test.
To verify that TRPV4 is the molecular constituent of CRAC channels in the duodenum, we compared PGE2-stimulated duodenal Isc between wild-type and TRPV4 KO mice. As shown in Figures 10C,D, duodenal Isc induced by serosal addition of luminal CCh was significantly attenuated in TRPV4 KO mice. Meanwhile, GdCl3 (30 μM) serosal addition did not affect luminal CCh-stimulated duodenal Isc of TRPV4 knockout mice (Figure 10E), further indicating that luminal CCh induced Ca2+-dependent duodenal anion secretion via TRPV4-constituted SOCE.
Activator of Store-Operated Ca2+ Entry in the Duodenal Epithelium and TRPV4-Constituted Store-Operated Ca2+ Entry Mechanism in IECs
As we have already known that cyclopiazonic acid (CPA), an ER-Ca2+-ATPase inhibitor (Dolmetsch and Lewis, 1994), can activate SOCE (Bird et al., 2008), TPEN also evoked SOCE by chelating Ca2+ within E.R. (Gwozdz et al., 2012). The addition of CPA (10 μM) in the serosal induced a transient high Isc peak with a sustained phase following (Figure 11A), and the addition of GdCl3 (30 μM) in the serosal side significantly reduced CPA-stimulated duodenal Isc (Figures 11A,B). The addition of TPEN (1 mM) in the serosal induced a transient high Isc peak with a sustained phase following (Figures 11C,E), while the addition of GdCl3 (30 μM) and HC067047 (30 μM) in the serosal side significantly reduced TPEN-stimulated duodenal Isc (Figures 11C–F).
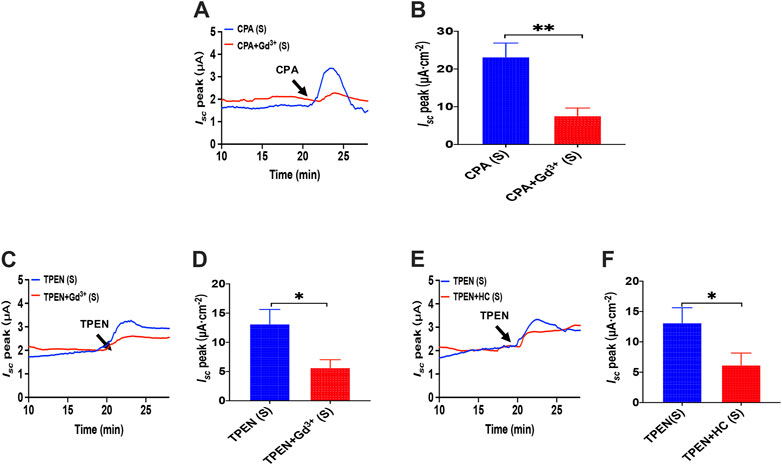
FIGURE 11. Activator of SOCE in the duodenal epithelium. (A,B) serosal CPA(10 μM)-stimulated duodenal Isc after addition of GdCl3 (30 μM) in the serosal side (n = 6). (C,D) TPEN-stimulated duodenal Isc after addition of GdCl3 (30 μM) in the serosal side (n = 6). (E,F) TPEN-stimulated duodenal Isc after adding HC067047 (30 μM) to the serosal side (n = 6). Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences, *p < 0.05, **p < 0.01 vs. corresponding control by Student’s unpaired, two-tailed t-test.
Our former studies, including T29 (Yang et al., 2018) and SCBN (Zhang et al., 2019; Zhang et al., 2021), confirmed that SOCE/ORAC mechanism is in IECs through SOCE blockers or knockdown cells. IEC-6 cells are usually applied as an IEC model to study intestine epithelial anion secretion (Wenzl et al., 1989), which has also been certificated the participation of SOCE mechanism (Chung et al., 2015). Meanwhile, TRPV4 expressed in IEC-6 cells basolateral and GSK1016790A induced [Ca2+]cyt rising were suppressed by pretreatment with RN1734 or extracellular Ca2+ omission in IEC-6 cells (Yamawaki et al., 2014). So we measured [Ca2+]cyt in IEC-6 cells to test the TRPV4 constituent SOCE mechanism. Superfused with Ca2+-free solution (0 Ca), CPA (10 μM), and TPEN (50 μM), first induced a rapid increase in [Ca2+]cyt in IEC-6 cells as a result of ER Ca2+ release (Figures 12A,D). After completion of Ca2+ release from E.R., the restoration of extracellular Ca2+ (2 Ca) caused an enhanced increase in [Ca2+]cyt owing to the SOCE mechanism (Figures 12A,D). As shown in Figures 12B,C,E,F, HC067047 (10 μM), a commonly used TRPV4 blocker in IEC-6, significantly inhibited CPA-and TPEN-induced SOCE, further advocating that TRPV4 participates in the composition of SOCE.
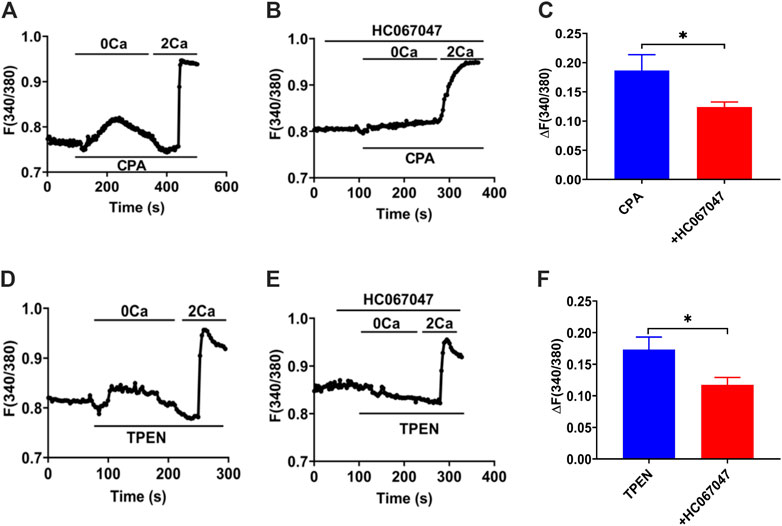
FIGURE 12. TRPV4-constituted SOCE mechanism in IECs. (A)Time courses of CPA (10 μM)-induced Ca2+ signaling in the extracellular Ca2+ omission (0 Ca, left) or not (2 Ca, right) in ICE-6 cells. (B) Time courses of CPA-evoked Ca2+ signaling with HC067047 (30 μM) pretreated in extracellular Ca2+ omission or not. (C) CPA-induced [Ca2+]cyt mobilization with HC067047 (30 μM) pretreated in HEC-6 cells superfused with Ca2+ - containing solution (2 Ca). Results are presented as mean SE (n = 25–35 cells). (D) TPEN (50 μM) -evoked Ca2+ signaling in the extracellular Ca2+ omission (0 Ca, left) or not (2 Ca, right) in ICE-6 cells. (E) TPEN-evoked Ca2+ signaling with HC067047 (30 μM) pretreated in extracellular Ca2+ omission or not. (F) TPEN-induced [Ca2+]cyt mobilization with HC067047 (30 μM) pretreated in HEC-6 cells superfused with Ca2+ - containing solution (2 Ca). Results are presented as mean SE (n = 25–35 cells) *p < 0.05 vs. corresponding control by Student’s unpaired, two-tailed t-test.
Discussion
Albeit it is well recognized that the critical role of calcium signaling in epithelial ion transports of the salivary gland, pancreatic ducts, and colonic epithelia, the detailed regulatory in the small intestinal epithelial and the underlying molecular mechanisms are not fully understood. Our research demonstrates that: 1) serosal PGE2 stimulates anion secretion mainly through RyR/ER Ca2+ release-initiated serosal SOCE mechanism; however, serosal 5-HT and luminal CCh stimulate anion secretion mainly through IP3R/ER Ca2+ release-initiated serosal SOCE mechanisms. 2) CARC may act as the SOCE mechanism in the process of Ca2+-dependent anion secretion. 3) TRPV4 channels may represent the molecular constituents of serosal SOCE/CARC channels to mediate Ca2+-dependent anion secretion. Therefore, our results indicate that Ca2+ signaling is essential for three most common and important secretagogues-induced small intestinal anion secretion, in which serosal TRPV4-constituted SOCE mechanism may play a critical role. Thus, our findings supple a novel insight into the molecular mechanisms of secretagogues-mediated epithelial anion secretion via Ca2+ signaling.
Being a prevalent second messenger, [Ca2+]cyt serve to regulate numerous cellular functions in various mammalian cells (Berridge et al., 2003), and it has been an essential regulator for intestinal epithelial ion secretion (Chew et al., 1998; Flemström and Isenberg, 2001; Jung and Lee, 2014). However, compared with excitable cells in which Ca2+ entry is mainly through VGCC, less is known about Ca2+ entry in non-excitable intestinal epithelial cells since functional VGCC may not be expressed (Parekh and Penner, 1997). In our research, we firstly confirmed the essential role of pure Ca2+ signaling in three secretagogues-induced small intestine anion secretion by using native mice duodenal epithelium, and then we revealed that they induced Ca2+ entry from the serosal side rather than from the mucosal side, which is consistent with our previous study (Yang et al., 2018). This phenomenon may be due to the following facts: 1) most secretagogues are derived from enterochromaffin (E.C.) cells or enteric neurons and transferred from the bloodstream, 2) their corresponding receptors are predominately located on the serosal side of intestinal epithelium, and 3) external Ca2+ concentrations in interstitial fluid of epithelia are maintained relatively consistent under physiological conditions.
It is well known that muscarinic receptors predominately express on the serosal side of the intestinal epithelium. However, we revealed for the first time that luminal CCh also evoked a significant duodenal Isc, which was inhibited by mucosal application of muscarinic receptor antagonist and omission of serosal calcium, suggesting that luminal CCh also activates mucosal muscarinic receptor to media Ca2+ entry from serosal side instead of mucosal side of duodenal epithelium. These new findings are not only mostly consistent with those obtained from our previous study but extend them (Yang et al., 2018). Although the muscarinic receptors in pancreatic acinar cells are localized to the apical side (Ashby et al., 2003), they are still elusive for their localization on the apical side of duodenal epithelial cells and their physiological significance. It was previously reported that bile acids might interact with apical muscarinic receptors on gastric chief cells and intestinal cells (Raufman et al., 2003); however, the detailed localization and significance of apical muscarinic receptors in the G.I. tract need further investigation.
Previous studies demonstrated that the serosal addition of CCh triggers IP3/IP3R/ER Ca2+ release that stimulates serosal SOCE mechanisms and ultimately induces Ca2+-dependent duodenal anion secretion (Yang et al., 2018; Zhang et al., 2019). Here, we further examined if PGE2 and 5-HT induced intestinal anion secretion through serosal SOCE mechanism. First, either omission of extracellular Ca2+ or ER Ca2+ chelation markedly attenuated PGE2- and 5-HT-induced duodenal Isc. Second, relatively selective SOCE blockers with different chemical structures significantly inhibited PGE2- and 5-HT-induced duodenal Isc from serosal side instead of mucosal side of the duodenum. Third, GSK-7975A and Gd3+, selective ICRAC blockers (Bird et al., 2008; Derler et al., 2013; Sataloff et al., 2017), also inhibited PGE2- and 5-HT-evoked Isc from serosal side. Therefore, together with our previous studies, these findings support a universal role of serosal SOCE mechanism in Ca2+-dependent duodenal anion secretion induced by three most common secretagogues.
Furthermore, we reveal a difference between them: 1) dantrolene, a selective RyR antagonist, inhibited PGE2-induced but not 5-HT-induced duodenal Isc; 2) LiCl, an inhibitor of IP3 production attenuated 5-HT-induced but not PGE2-induced Isc. These findings suggest that ER Ca2+ release may be induced via different pathways: PGE2 and 5-HT activate RyR and IP3R, respectively. Although ER Ca2+ release is mainly mediated by two well-known channels RyR and IP3R, they have different Ca2+ affinities (Carreras-Sureda et al., 2018). 5-HT couples to IP3/IP3R signaling pathway in glioma cells, which subsequently cause intracellular Ca2+ release (Noda et al., 2003). In contrast, PGE2 induces Ca2+ release through RyR in bovine adrenal medullary cells (Shibuya et al., 2002). The concentration of dantrolene (300 μM) in our research is relatively high. However, it is consistent with our previous study (Zhang et al., 2019) that serosal application of dantrolene (100 μM) caused significant inhibition of caffeine-induced Isc, and an increase in the concentration of dantrolene (300 μM and 1 mM) dose-dependently enhanced the inhibition. However, whether the high concentration of dantrolene has a non-specific effect needs further investigation in RyR KO mice or cell lines. Moreover, 2-APB is a non-specific IP3R inhibitor, so we chose to examine the involvement of IP3R by inhibition of IP3 with LiCl. Further experiments in K.O. mice or cell lines may be needed to verify IP3R involvement in Isc and SOCE further.
Molecular components of SOCE in intestinal epithelial cells have not been well identified. They are considered as TRPC1 in IEC (Rao et al., 2006) or STIM1/Orai1 in rat colonic epithelium (Onodera et al., 2013) and Caco2 cells (Yang et al., 2018). TRPV4 channels are nonselective cation channels with higher permeability for Ca2+ (Gees et al., 2010) and expressed on the G.I. tract (Balemans et al., 2017). It was previously reported that SOCE could be constituted by TRPV4 alone or together with TRPC1 to form a heteromeric channel (Ma et al., 2011). Therefore, using TRPV4 antagonist and TRPV4-KO mice, we examined if it represents molecular components of SOCE in mouse native duodenum. Indeed, we demonstrate that TRPV4 channels may constitute SOCE to contribute to PGE2- and 5-HT-induced Ca2+-dependent duodenal anion secretion for the first time. In the meantime, we adopted a TRPV4 blocker to suppress SOCE activation in the IEC-6 cell line, which elucidates that TRPV4 participates in SOCE. However, GSK1016790A alone had no effect on the basal Isc in W.T., and K.O. mice, which could be due to the fact that directly opening TRPV4 channel itself may not be sufficient to cause calcium influx in situ because TRPV4 is only one of many components for SOCE activation, including STIM1, Orai, TRPC channels.
Although the role of TRPV4 channels in G.I. disease has been broadly examined (Vergnolle, 2014), their physiological roles in the gut are still elusive, which need further investigation.
In conclusion, we underscore an essential role of Ca2+ signaling mediated anion secretion by TRPV4-constituted serosal SOCE mechanisms, which is universal for the three most common and important secretagogues. Although PGE2 and 5-HT stimulate this mechanism exclusively from serosal side of the duodenum, CCh stimulates it from both sides. We also reveal that 5-HT and CCh trigger ER Ca2+ release to initiate SOCE likely via IP3/IP3R, but PGE2 triggers it likely via RyR. A diagram is depicting, which we find in Figure 13. Complete comprehension of molecular mechanisms underlying secretagogues-mediated intestinal ion transports via Ca2+ signaling will enhance our knowledge of G.I. epithelial physiology and G.I. disease associated with abnormal anion secretion, such as diarrhea and constipation.
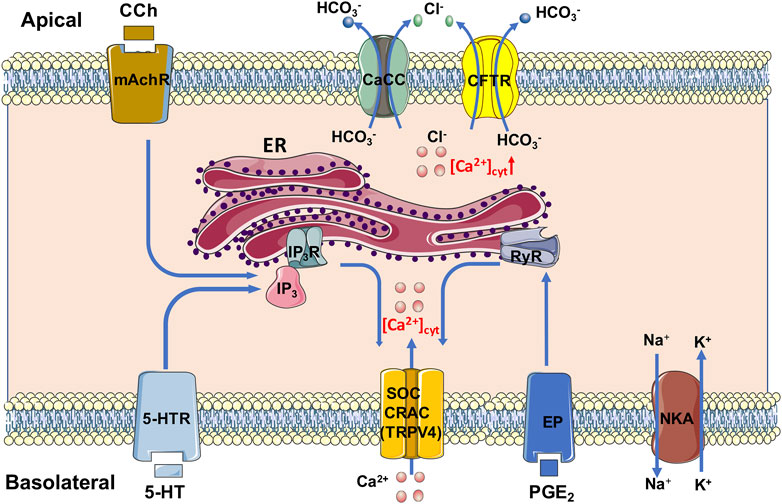
FIGURE 13. Mechanism diagram of secretagogues mediating Ca2+ signaling in duodenal epithelial cells via TRPV4-constituted serosal SOCE mechanism. PGE2 and 5-HT stimulate this mechanism exclusively from serosal side of the duodenum; CCh stimulates it from both sides. 5-HT and CCh trigger the ER Ca2+ release to initiate the SOCE likely via IP3/IP3R, but PGE2 triggers it likely via RyR. CaCC, Ca2+-activated Cl− channels; CFTR, cystic fibrosis transmembrane conductance regulator; CRAC, Ca2+ release-activated Ca2+ channel; ER, endoplasmic reticulum; EP, PGE2 receptor; IP3 and IP3R, inositol 1,4,5-triphosphate and its receptor; mAchR, muscarinic acetylcholine receptor; NKA, Na+/K+ ATPase; RyR, ryanodine receptor; SOC, store-operated channels; TRPV4, transient receptor potential V4; 5-HTR, 5-HT receptor.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Third Military Medical University Committee on Investigations Involving Animal Subjects.
Author Contributions
HD conceived the study, designed most experiments, and finalized the manuscript. YC performed most experiments and data analysis and drafted the manuscript. FX designed, and FC, KY, XC, HW, and GL helped perform some experiments. Finally, all authors reviewed the manuscript.
Funding
This study was supported by research grants from China's National Key Research and Development Program (No. 2016YFC1302200 to H.D.) and the National Natural Science Foundation of China (No. 81873544 to H.D., and No. 81602474 to G.L.).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.684538/full#supplementary-material
SUPPLEMENTARY FIGURE 1. (A) Time courses of GSK1016790A (10 μM)-induced Isc in wild-type (W.T.) and TRPV4 K.O. mice (n = 6), while CCh verifies that the mice tissue is alive. (B) PGE2-evoked Isc after HC067047 (30 μM, n = 6) was added to the serosal (s) side of the TRPV4 knockout (K.O.) mice duodenum. (C) 5-HT-evoked Isc after HC067047 (30 μM, n = 6) was added to the serosal (s) side of the TRPV4 knockout (K.O.) mice duodenum. Ctrl represents the control without drug treatment. Results are presented as mean ± SE. NS, no significant differences vs. corresponding control by Student’s unpaired, two-tailed t-test.
References
Ashby, M. C., Camello-Almaraz, C., Gerasimenko, O. V., Petersen, O. H., and Tepikin, A. V. (2003). Long Distance Communication between Muscarinic Receptors and Ca2+ Release Channels Revealed by Carbachol Uncaging in Cell-Attached Patch Pipette. J. Biol. Chem. 278, 20860–20864. doi:10.1074/jbc.M302599200
Balemans, D., Boeckxstaens, G. E., Talavera, K., and Wouters, M. M. (2017). Transient Receptor Potential Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades Underlying Visceral Hypersensitivity. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G635–G648. doi:10.1152/ajpgi.00401.2016
Baratchi, S., Keov, P., Darby, W. G., Lai, A., Khoshmanesh, K., Thurgood, P., et al. (2019). The TRPV4 Agonist GSK1016790A Regulates the Membrane Expression of TRPV4 Channels. Front. Pharmacol. 10, 1–12. doi:10.3389/fphar.2019.00006
Barrett, K. E., and Keely, S. J. (2000). Chloride Secretion by the Intestinal Epithelium: Molecular Basis and Regulatory Aspects. Annu. Rev. Physiol. 62, 535–572. doi:10.1146/annurev.physiol.62.1.535
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi:10.1038/nrm1155
Bird, G. S., DeHaven, W. I., Smyth, J. T., and Putney, J. W. (2008). Methods for Studying Store-Operated Calcium Entry. Methods 46, 204–212. doi:10.1016/j.ymeth.2008.09.009
Blackshaw, L. A., Brierley, S. M., and Hughes, P. A. (2010). TRP Channels: New Targets for Visceral Pain. Gut 59, 126–135. doi:10.1136/gut.2009.179523
Bootman, M. D., Collins, T. J., Mackenzie, L., Roderick, H. L., Berridge, M. J., and Peppiatt, C. M. (2002). 2‐Aminoethoxydiphenyl Borate (2‐APB) is a Reliable Blocker of Store‐Operated Ca2+ Entry but an Inconsistent Inhibitor of InsP3‐Induced Ca2+ Release. FASEB J. 16, 1145–1150. doi:10.1096/fj.02-0037rev
Brown, D. R. (1995). Mucosal Protection Through Active Intestinal Secretion: Neural and Paracrine Modulation by 5-Hydroxytryptamine. Behav. Brain Res. 73, 193–197. doi:10.1016/0166-4328(96)00095-2
Caroppo, R., Colella, M., Colasuonno, A., DeLuisi, A., Debellis, L., Curci, S., et al. (2003). A Reassessment of the Effects of Luminal [Ca2+] on Inositol 1,4,5-Trisphosphate-Induced Ca2+ Release from Internal Stores. J. Biol. Chem. 278, 39503–39508. doi:10.1074/jbc.M305823200
Carreras-Sureda, A., Pihán, P., and Hetz, C. (2018). Calcium Signaling at the Endoplasmic Reticulum: Fine-Tuning Stress Responses. Cell Calcium 70, 24–31. doi:10.1016/j.ceca.2017.08.004
Chew, C. S., Säfsten, B., and Flemström, G. (1998). Calcium Signaling in Cultured Human and Rat Duodenal Enterocytes. Am. J. Physiol. 275, G296–G304. doi:10.1152/ajpgi.1998.275.2.G296
Chung, H. K., Rathor, N., Wang, S. R., Wang, J.-Y., and Rao, J. N. (2015). RhoA Enhances Store-Operated Ca2+ Entry and Intestinal Epithelial Restitution by Interacting With TRPC1 After Wounding. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G759–G767. doi:10.1152/ajpgi.00185.2015
Derler, I., Schindl, R., Fritsch, R., Heftberger, P., Riedl, M. C., Begg, M., et al. (2013). The Action of Selective CRAC Channel Blockers is Affected by the Orai Pore Geometry. Cell Calcium 53, 139–151. doi:10.1016/j.ceca.2012.11.005
Dolmetsch, R. E., and Lewis, R. S. (1994). Signaling between Intracellular Ca2+ Stores and Depletion-Activated Ca2+ Channels Generates [Ca2+]i Oscillations in T Lymphocytes. J. Gen. Physiol. 103, 365–388. doi:10.1085/jgp.103.3.365
Flemström, G., and Isenberg, J. I. (2001). Gastroduodenal Mucosal Alkaline Secretion and Mucosal Protection. Physiology 16, 23–28. doi:10.1152/physiologyonline.2001.16.1.23
Gees, M., Colsoul, B., and Nilius, B. (2010). The Role of Transient Receptor Potential Cation Channels in Ca2+ Signaling. Cold Spring Harb. Perspect. Biol. 2, a003962. doi:10.1101/cshperspect.a003962
Gwozdz, T., Dutko-Gwozdz, J., Schafer, C., and Bolotina, V. M. (2012). Overexpression of Orai1 and STIM1 Proteins Alters Regulation of Store-Operated Ca2+ Entry by Endogenous Mediators. J. Biol. Chem. 287, 22865–22872. doi:10.1074/jbc.M112.356626
Hammerton, R., Krzeminski, K., Mays, R., Ryan, T., Wollner, D., and Nelson, W. (1991). Mechanism for Regulating Cell Surface Distribution of Na+,K(+)-ATPase in Polarized Epithelial Cells. Science 254, 847–850. doi:10.1126/science.1658934
Hoth, M., and Penner, R. (1993). Calcium Release-Activated Calcium Current in Rat Mast Cells. J. Physiol. 465, 359–386. doi:10.1113/jphysiol.1993.sp019681
Hoth, M., and Penner, R. (1992). Depletion of Intracellular Calcium Stores Activates a Calcium Current in Mast Cells. Nature 355, 353–356. doi:10.1038/355353a0
Jung, J., and Lee, M. G. (2014). Role of Calcium Signaling in Epithelial Bicarbonate Secretion. Cell Calcium 55, 376–384. doi:10.1016/j.ceca.2014.02.002
Kiela, P. R., and Ghishan, F. K. (2009). Ion Transport in the Intestine. Curr. Opin. Gastroenterol. 25, 87–91. doi:10.1097/MOG.0b013e3283260900
Kilkenny, C., Browne, W., Cuthill, I. C., Emerson, M., and Altman, D. G. (2010). Animal Research: Reporting In Vivo Experiments: The ARRIVE Guidelines. Br. J. Pharmacol. 160, 1577–1579. doi:10.1111/j.1476-5381.2010.00872.x
Lindqvist, S., Sharp, P., Johnson, I., Satoh, Y., and Williams, M. (1998). Acetylcholine-Induced Calcium Signaling Along the Rat Colonic Crypt Axis. Gastroenterology 115, 1131–1143. doi:10.1016/s0016-5085(98)70084-8
Ma, X., Cheng, K.-T., Wong, C.-O., O’Neil, R. G., Birnbaumer, L., Ambudkar, I. S., et al. (2011). Heteromeric TRPV4-C1 Channels Contribute to Store-Operated Ca2+ Entry in Vascular Endothelial Cells. Cell Calcium 50, 502–509. doi:10.1016/j.ceca.2011.08.006
MacMillan, D., Chalmers, S., Muir, T. C., and McCarron, J. G. (2005). IP3-Mediated Ca2+ Increases do not Involve the Ryanodine Receptor, but Ryanodine Receptor Antagonists Reduce IP3-Mediated Ca2+ Increases in Guinea-Pig Colonic Smooth Muscle Cells. J. Physiol. 569, 533–544. doi:10.1113/jphysiol.2005.096529
Molnar, T., Yarishkin, O., Iuso, A., Barabas, P., Jones, B., Marc, R. E., et al. (2016). Store-Operated Calcium Entry in Muller Glia Is Controlled by Synergistic Activation of TRPC and Orai Channels. J. Neurosci. 36, 3184–3198. doi:10.1523/JNEUROSCI.4069-15.2016
Murek, M., Kopic, S., and Geibel, J. (2010). Evidence for Intestinal Chloride Secretion. Exp. Physiol. 95, 471–478. doi:10.1113/expphysiol.2009.049445
Noda, M., Yasuda, S., Okada, M., Higashida, H., Shimada, A., Iwata, N., et al. (2003). Recombinant Human Serotonin 5A Receptors Stably Expressed in C6 Glioma Cells Couple to Multiple Signal Transduction Pathways. J. Neurochem. 84, 222–232. doi:10.1046/j.1471-4159.2003.01518.x
Onodera, K., Pouokam, E., and Diener, M. (2013). STIM1-Regulated Ca2+ Influx across the Apical and the Basolateral Membrane in Colonic Epithelium. J. Membr. Biol. 246, 271–285. doi:10.1007/s00232-013-9528-9
Parekh, A. B., and Penner, R. (1997). Store Depletion and Calcium Influx. Physiol. Rev. 77, 901–930. doi:10.1152/physrev.1997.77.4.901
Putney, J. W. (2007). Recent Breakthroughs in the Molecular Mechanism of Capacitative Calcium Entry (With Thoughts on How We Got Here). Cell Calcium 42, 103–110. doi:10.1016/j.ceca.2007.01.011
Rajagopal, M., Thomas, S. V., Kathpalia, P. P., Chen, Y., and Pao, A. C. (2014). Prostaglandin E2 Induces Chloride Secretion through Crosstalk between cAMP and Calcium Signaling in Mouse Inner Medullary Collecting Duct Cells. Am. J. Physiol. Cell Physiol. 306, C263–C278. doi:10.1152/ajpcell.00381.2012
Rao, J. N., Platoshyn, O., Golovina, V. A., Liu, L., Zou, T., Marasa, B. S., et al. (2006). TRPC1 Functions as a Store-Operated Ca2+ Channel in Intestinal Epithelial Cells and Regulates Early Mucosal Restitution after Wounding. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G782–G792. doi:10.1152/ajpgi.00441.2005
Rao, M. C., Nash, N. T., and Field, M. (1984). Differing Effects of cGMP and cAMP on Ion Transport across Flounder Intestine. Am. J. Physiol. 246, C167–C171. doi:10.1152/ajpcell.1984.246.1.C167
Raufman, J.-P., Cheng, K., and Zimniak, P. (2003). Activation of Muscarinic Receptor Signaling by Bile Acids: Physiological and Medical Implications. Dig. Dis. Sci. 48, 1431–1444. doi:10.1023/a:1024733500950
Sataloff, R. T., Johns, M. M., and Kost, K. M. (2017). “Signaling ER Store Depletion to Plasma Membrane Orai Channels,” in Calcium Entry Channels in Non-Excitable Cells. Editors J. A. Kozak, and J. W. Putney (Boca Raton, FL: CRC Press/Taylor & Francis).
Shibuya, I., Tanaka, K., Uezono, Y., Ueta, Y., Toyohira, Y., Yanagihara, N., et al. (2002). Prostaglandin E2 Induces Ca2+ Release from Ryanodine/Caffeine-Sensitive Stores in Bovine Adrenal Medullary Cells via EP1-Like Receptors. J. Neurochem. 73, 2167–2174. doi:10.1046/j.1471-4159.1999.02167.x
Takeuchi, K., and Amagase, K. (2018). Roles of Cyclooxygenase, Prostaglandin E2 and EP Receptors in Mucosal Protection and Ulcer Healing in the Gastrointestinal Tract. Curr. Pharm. Des. 24, 2002–2011. doi:10.2174/1381612824666180629111227
Thomas, C., and Oates, P. S. (2002). IEC-6 Cells are an Appropriate Model of Intestinal Iron Absorption in Rats. J. Nutr. 132, 680–687. doi:10.1093/jn/132.4.680
Thorsen, K., Drengstig, T., and Ruoff, P. (2014). Transepithelial Glucose Transport and Na+/K+ Homeostasis in Enterocytes: An Integrative Model. Am. J. Physiol. Cell Physiol. 307, C320–C337. doi:10.1152/ajpcell.00068.2013
Tuo, B., Wen, G., Zhang, Y., Liu, X., Wang, X., Liu, X., et al. (2009). Involvement of Phosphatidylinositol 3-Kinase in cAMP- and cGMP-Induced Duodenal Epithelial CFTR Activation in Mice. Am. J. Physiol. Cell Physiol. 297, C503–C515. doi:10.1152/ajpcell.00460.2008
Vergnolle, N. (2014). TRPV4: New Therapeutic Target for Inflammatory Bowel Diseases. Biochem. Pharmacol. 89, 157–161. doi:10.1016/j.bcp.2014.01.005
Wenzl, E., Sjaastad, M. D., Weintraub, W. H., and Machen, T. E. (1989). Intracellular pH Regulation in IEC-6 Cells, a Cryptlike Intestinal Cell Line. Am. J. Physiol. 257, G732–G740. doi:10.1152/ajpgi.1989.257.5.g732
Weymer, A., Huott, P., Liu, W., McRoberts, J. A., and Dharmsathaphorn, K. (1985). Chloride Secretory Mechanism Induced by Prostaglandin E1 in a Colonic Epithelial Cell Line. J. Clin. Invest. 76, 1828–1836. doi:10.1172/JCI112175
Xia, Y., Fu, Z., Hu, J., Huang, C., Paudel, O., Cai, S., et al. (2013). TRPV4 Channel Contributes to Serotonin-Induced Pulmonary Vasoconstriction and the Enhanced Vascular Reactivity in Chronic Hypoxic Pulmonary Hypertension. Am. J. Physiol. Cell Physiol. 305, C704–C715. doi:10.1152/ajpcell.00099.2013
Xie, R., Dong, X., Wong, C., Vallon, V., Tang, B., Sun, J., et al. (2014). Molecular Mechanisms of Calcium-Sensing Receptor-Mediated Calcium Signaling in the Modulation of Epithelial Ion Transport and Bicarbonate Secretion. J. Biol. Chem. 289, 34642–34653. doi:10.1074/jbc.M114.592774
Yamawaki, H., Mihara, H., Suzuki, N., Nishizono, H., Uchida, K., Watanabe, S., et al. (2014). Role of Transient Receptor Potential Vanilloid 4 Activation in Indomethacin-Induced Intestinal Damage. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G33–G40. doi:10.1152/ajpgi.00105.2013
Yang, X., Wen, G., Tuo, B., Zhang, F., Wan, H., He, J., et al. (2018). Molecular Mechanisms of Calcium Signaling in the Modulation of Small Intestinal Ion Transports and Bicarbonate Secretion. Oncotarget 9, 3727–3740. doi:10.18632/oncotarget.23197
Zhang, F., Wan, H., Chu, F., Lu, C., Chen, J., and Dong, H. (2021). Small intestinal glucose and sodium absorption through calcium-induced calcium release and store-operated Ca2+ entry mechanisms. Br. J. Pharmacol. 178 (2), 346–362. doi:10.1111/bph.15287
Keywords: calcium signaling, PGE2, 5-HT, SOCE, CRAC, TRPV4, CCH
Citation: Cui Y, Chu F, Yin K, Chen X, Wan H, Luo G, Dong H and Xu F (2021) Role of Serosal TRPV4-Constituted SOCE Mechanism in Secretagogues-Stimulated Intestinal Epithelial Anion Secretion. Front. Pharmacol. 12:684538. doi: 10.3389/fphar.2021.684538
Received: 23 March 2021; Accepted: 24 June 2021;
Published: 14 July 2021.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
James S. K. Sham, Johns Hopkins University, United StatesYasutada Akiba, University of California, Los Angeles, United States
Copyright © 2021 Cui, Chu, Yin, Chen, Wan, Luo, Dong and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Dong, ZGh4cUB0bW11LmVkdS5jbg==; Feng Xu, eHVmZW5nOTg5OUAxNjMuY29t