
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 24 June 2021
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 12 - 2021 | https://doi.org/10.3389/fphar.2021.680349
This article is part of the Research Topic Cardio-Protection and Heart Repair: New Drugs, Targets and Approaches View all 59 articles
 Xuefeng Cao2Zheng Zhang3Yu Wang4Weichao Shan5Ruiting Wang6Shufang Mao1Shi Ding1Chong Pang1Baoqun Li1Jian Zhou1Xiaoyan Guo1Na Guo1Cui Li1Jing Liang7Wenya Ma8*Yu Liu9*
Xuefeng Cao2Zheng Zhang3Yu Wang4Weichao Shan5Ruiting Wang6Shufang Mao1Shi Ding1Chong Pang1Baoqun Li1Jian Zhou1Xiaoyan Guo1Na Guo1Cui Li1Jing Liang7Wenya Ma8*Yu Liu9* Liang Zhao1*
Liang Zhao1*Cardiac hypertrophy is a common pathological process of various cardiovascular diseases, which is often accompanied with structural and electrical remodeling, and can even lead to sudden cardiac death. However, its molecular mechanism still remains largely unknown. Here, we induced cardiomyocyte hypertrophy by angiotensin II (Ang II), and found that miR-27a-3p and hypertrophy-related genes were up-regulated. Further studies showed that miR-27a-3p-inhibitor can alleviate myocardial hypertrophy and electrical remodeling. Moreover, luciferase assay confirmed that miR-27a-3p could regulate the expression of downstream Hoxa10 at the transcriptional level by targeting at its 3′UTR. At the same time, the protein expression of Hoxa10 was significantly reduced in Ang II-treated cardiomyocytes. Furthermore, overexpression of Hoxa10 can reverse myocardial hypertrophy and electrical remodeling induced by Ang II in cardiomyocytes. Finally, we found that Hoxa10 positively regulated the expression of potassium channel protein Kv4.3 which was down-regulated in hypertrophic cardiomyocytes. Taken together, our results revealed miR-27a-3p/Hoxa10/Kv4.3 axis as a new mechanism of Ang II-induced cardiomyocyte hypertrophy, which provided a new target for clinical prevention and treatment of cardiac hypertrophy and heart failure.
Myocardial hypertrophy is a common pathological process of cardiovascular diseases such as hypertension. Long-term myocardial hypertrophy leads to cardiac function decompensation, aggravates cardiac electrical remodeling, greatly increases the susceptibility to arrhythmia (Thompson, 2009; Dey et al., 2018), and even leads to heart failure and sudden cardiac death (SCD) (Coyne et al., 2001). Epidemiological investigation has confirmed that the incidence of arrhythmia, heart failure and sudden cardiac death in patients with myocardial hypertrophy is much higher than normal cohorts (Berger et al., 2002; John et al., 2012). However, current medication or surgery therapy cannot effectively improve the long-term survival rate of patients with myocardial hypertrophy. Therefore, the pathogenesis of cardiac hypertrophic has increasingly become a focus and difficulty in the field of cardiovascular diseases, and it is also of great importance to reveal the mechanism of myocardial remodeling and new targets for clinical treatment of cardiac hypertrophy (Gatzoulis et al., 2012). Although more and more researchers are interested in the study of myocardial hypertrophy and electrical remodeling, the molecular mechanisms of myocardial hypertrophy and electrical remodeling still remain largely unknown (Bacharova, 2019).
Micro-RNA (microRNA, miRNA) is a kind of non-coding RNA with approximately 22 nucleotides, which can inhibit the translation of target gene mRNAs by incomplete complementary binding to the 3′UTR region of target gene mRNAs, and then degrade the expression of mRNA (Yang et al., 2007). A large number of studies have shown that miRNAs play an important role in the occurrence and development of heart diseases, especially in cardiomyopathy such as myocardial hypertrophy and arrhythmia (Wang et al., 2008; Samidurai et al., 2018). For example, muscle-specific microRNA miR-1 has been shown to promote the development of myocardial ischemia and apoptosis in mice. MiRNAs such as miR-101 was linked to cardiac fibrosis and fibroblast transdifferentiation via targeting c-Jun (Pan et al., 2012). Although several studies have revealed that miR-27a-3p is associated with several cardiovascular diseases, its roles in myocardial hypertrophy and electrical remodeling still needs to be further clarified (Chen et al., 2012).
In this study, we observed that the expression of miR-27a-3p was up-regulated in cardiac hypertrophy. MiR-27a-3p-inhibitor could inhibit myocardial hypertrophy and electrical remodeling induced by Ang II. Further studies showed that miR-27a-3p binded to 3′UTR of Hoxa10 which was down-regulated in cardiac hypertrophy. The overexpression of Hoxa10 could reverse cardiac hypertrophy and electrical remodeling by targeting Kv4.3 protein. Therefore, our results revealed for the first time that the miR-27a-3p/Hoxa10/Kv4.3 axis is involved in the pathogenesis of myocardial hypertrophy and electrical remodeling, and these results provided a new therapeutic strategy for myocardial hypertrophy and electrical remodeling clinically.
Sprague-Dawley rats (1–3 days, SPF grade, Certification No. 11400700368598) were obtained from the Beijing Huafukang Biological Technology Co. Ltd. (Beijing, China). The experimental protocols were approved by the Ethics Committee of the Ethics of Animal Experiments of Chengde Medical College (Approval ID: CDMULAC-20191031-009). The animal experiments were approved by the Research Ethics Committee of Chengde Medical College, and were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Cardiomyocytes were obtained from 1 to 3-day-old neonatal rats as previously described, with a few minor modifications (Cai et al., 2018). Briefly, after being digested in 0.25% trypsin solution, myocytes were isolated by selective adhesion of myocytes at a 2 h pre-plating interval. Cardiomyocytes were cultured in DMEM supplemented with 1% penicillin and streptomycin, 10% fetal bovine serum and 0.1 mM 5-bromo-2-deoxyuridine in a 37°C incubator with 5% CO2. MiR-27a-3p-mimics (5′-UUCACAGUGGCUAAGUUCCGC-3′), miR-27a-3p-inhibitor (5′-GCGGAACUUAGCCACUGUGAA-3′), Hoxa10-overexpress and corresponding NC were synthesized by Shanghai Sangon Biotechnology, and transiently transfected with liposome3000 transfection reagent (Invitrogen, Carlsbad, CA, United States) and serum-free medium. Eight hours after transfection, a new serum-free medium with or without Ang II (10 μM) was used instead. After 48 h of culture, the cells were collected for protein/total RNA extraction (Ma et al., 2018).
After treatments, the cardiomyocytes were washed with PBS for three times, then the protein lysate was added and stored at −80°C. Place cells on the ice to dissolve, scrape off the cells with a protein scraper and centrifuge at 13,500 rpm/min × 15 min, 4°C. Collect the supernatant protein solution. Determination of protein concentration by BCA method, proteins were separated by SDS-PAGE and transferred to a PVDF membrane. After blocking with 5% fat-free milk at room temperature for 120 min, the membranes were incubated overnight with the corresponding primary antibody at 4°C, including anti-Kv4.3 (1:1,000, CST), anti-Hoxa10 (1:500, Sigma), anti-ANP (1:1,000, SANTA), anti-BNP (1:1,000, SANTA), anti-MHC (1:1,000, SANTA) and anti-GAPDH (1:10,000, ABclonal). After being washed with TBST by three times, each time 10 min, the membranes were incubated with secondary antibodies (1:8,000, ABclonal) for 2 h at room temperature and washed with TBST again. The proteins were scanned using Odyssey version 1.2 (LI-COR Biosciences, Lincoln, NE, United States) and Image Studio software.
Total RNA samples from different groups of cardiomyocytes were extracted and treated with DNA enzyme 1, and then routine quantitative RT-PCR (qRT-PCR) was used for detection. Real-time quantitative RT-PCR was determined by Taqman probe method (ABI 7500 fast). In the determination of mRNA, GAPDH was used as the internal reference, and U6 was used as the internal reference for microRNAs. Compared with the control group, the related expressions of microRNAs and mRNAs under different treatments were calculated.
Cardiomyocytes in different treatment groups were washed with PBS for three times, and 15 min was fixed with 4% paraformaldehyde at 37°C. Get rid of paraformaldehyde, wash the cells with PBS, then add a mixed solution (0.4% Triton + 10 mg BSA +1 ml PBS) and place 90 min at room temperature. After that, goat serum blocked 30 min at 37°C, then α-actinin was added and incubated at 4°C for 48 h. Finally, after incubated with fluorescent second antibody for 1 h, the cells were washed by PBS for three times, and the morphological changes of cells were observed by fluorescence microscope.
GraphPad Prism software was used for statistical analysis. Values were expressed as the mean ± SEM. Differences between two groups were analyzed using t-test. The significance of the difference between groups in review were analyzed by one-way analysis of variance (ANOVA). A value of *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 were considered statistically significant difference for all analyses.
Firstly, the cardiomyocytes from rats were extracted and exposed to Ang II (10 μM) for 48 h to established cardiac hypertrophy cell model in vitro. Then the expressions of myocardial hypertrophy-related genes and miRNAs in cardiomyocytes were detected. Results showed that the expressions of myocardial hypertrophy-related genes and proteins including ANP, BNP and β-MHC were markedly up-regulated in Ang II-treated cardiomyocytes compared with control group (Figures 1A,B,C,E). Immunofluorescence results showed that the area of cardiomyocytes in Ang II treatment group was significantly larger than the control group (Figure 1F), indicating that Ang II induced cardiac hypertrophy cell model in vitro was successfully established. At the same time, we also found that the expression of miR-27a-3p was up-regulated in cardiomyocytes after Ang II treatment compared with control group (Figure 1D). This result suggested that miR-27a-3p might be involved in myocardial hypertrophy induced by Ang II.
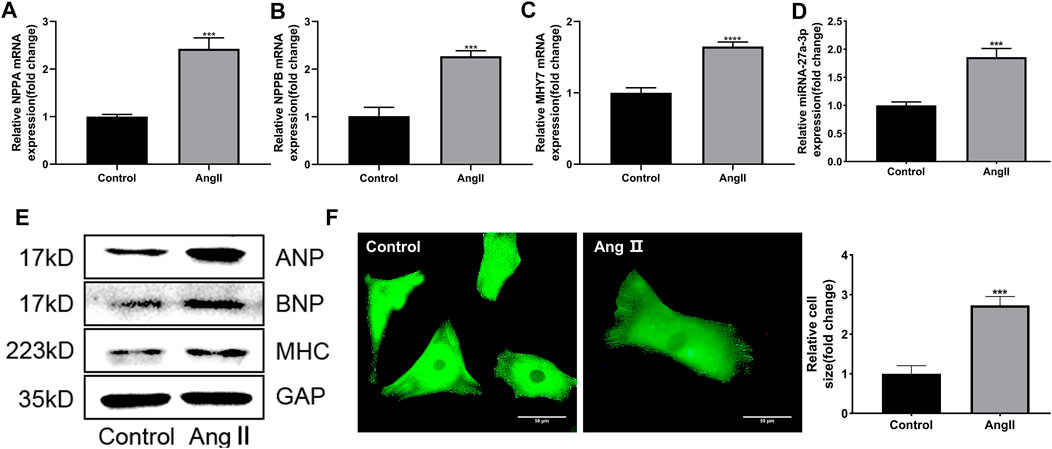
FIGURE 1. Ang II induced cardiac hypertrophy in neonatal rat cardiomyocytes. (A–C) The expressions of cardiac hypertrophy related genes NPPA, NPPB and MHY7 were increased after Ang II treatment. (D) The expression of miR-27a-3p increased after Ang II treatment. (E) The protein expressions of cardiac hypertrophy related genes ANP, BNP and β-MHC were increased after Ang II treatment. (F) Morphological changes after Ang II treatment in neonatal rat cardiomyocytes. Scale bar, 50 μm. All the values are the mean of three independent experiments (n = 3). ****p < 0.0001, ***p < 0.001 vs. control.
To clarify whether miR-27a-3p was involved in cardiac hypertrophy, miR-27a-3p-inhibitor, a specific antagonist of miR-27a-3p, was used in the following experiments. Four hours after the transfection of miR-27a-3p-inhibitor, the serum-free culture medium was replaced and co-cultured with Ang II (10 μM) for 48 h. PCR results showed that the expressions of NPPA, NPPB, and MHY7 in miR-27a-3p-inhibitor + Ang II group was significantly lower than Ang II group, while KCND3 was higher than Ang II group (Figures 2A–D), and the protein expressions showed similar tendencies (Figure 2F). The α-actinin staining showed that the area of cardiomyocytes in miR-27a-3p-inhibitor + Ang II group was significantly smaller than Ang II group (Figure 2E). These results indicated that the inhibition of miR-27a-3p mitigated the process of myocardial hypertrophy and structural remodeling induced by Ang II.
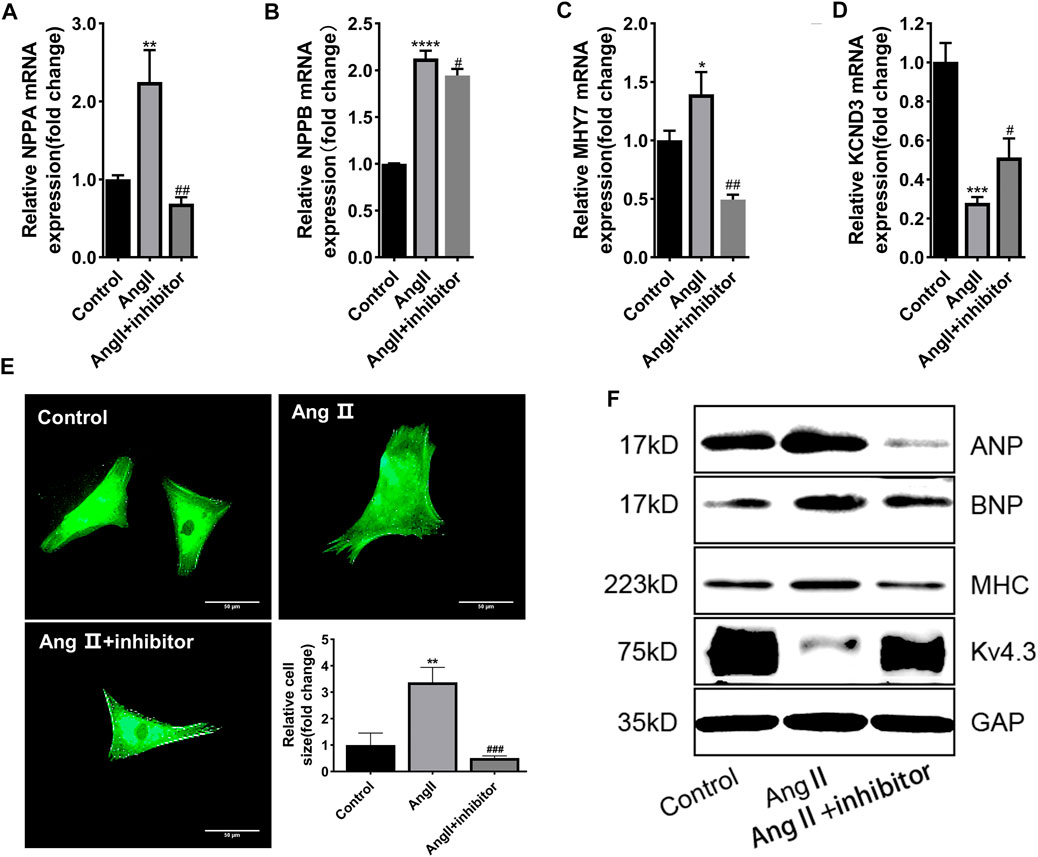
FIGURE 2. MiR-27a-3p is involved in the process of myocardial hypertrophy induced by Ang II. (A–C) The expressions of cardiac hypertrophy related genes NPPA, NPPB, and MHY7 were decreased after Ang II + miR-27a-3p inhibitor treatment compared with the Ang II treatment. (D) The expression of KCND3 decreased after Ang II treatment, while increased in Ang II + miR-27a-3p inhibitor treatment. (E) Morphological changes after Ang II and miR-27a-3p inhibitor treatment in neonatal rat cardiomyocytes. Scale bar, 50 μm. (F) The protein expressions of cardiac hypertrophy related genes ANP, BNP and β-MHC were decreased, while the expression of Kv4.3 increased after Ang II + miR-27a-3p inhibitor treatment. All the values are the mean of three independent experiments (n = 3). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05 vs. control. ###p < 0.001, ##p < 0.01, #p < 0.05 vs. Ang II.
In addition, we detected the effects of miR-27a-3p-mimics and inhibitor on cultured rat ventricular cardiomyocytes. PCR results showed that the expression of miR-27a-3p in miR-27a-3p-mimics group was significantly increased compared with control group, while the expression of miR-27a-3p in miR-27a-3p-inhibitor group was significantly decreased compared with miR-27a-3p-mimics group and the control group (Figure 3A). Furthermore, the expressions of NPPA, NPPB, and MHY7 were significantly up-regulated after miR-27a-3p-mimics treatment, which were effectively alleviated by miR-27a-3p-mimics and miR-27a-3p-inhibitor co-treatment (Figures 3B–D). These results suggested that the overexpression of miR-27a-3p owned the potential to promote cardiomyocyte hypertrophy.
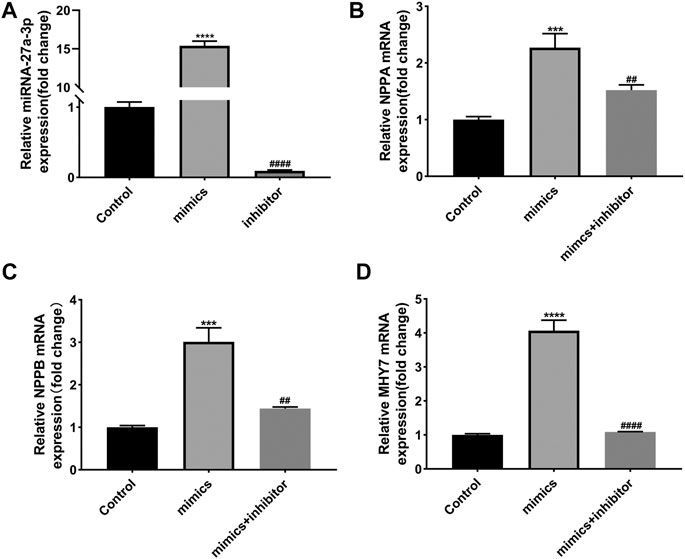
FIGURE 3. Effects of miR-27a-3p-mimics and inhibitor on cardiomyocytes. (A) The expression of miR-27a-3p in miR-27a-3p-mimics and miR-27a-3p-inhibitor treatment group. (B–D) The expressions of NPPA, NPPB, and MHY7 in miR-27a-3p-mimics treatment group and miR-27a-3p-mimics + miR-27a-3p-inhibitor co-treatment group. All the values are the mean of three independent experiments (n = 3). ****p < 0.0001, ***p < 0.001 vs. control, ###p < 0.0001, ##p < 0.01 vs. mimics.
The results above proved that miR-27-3p was involved in hypertrophic growth of cardiomyocytes, but its mechanism still needs to be elucidated. Recent study revealed that potassium channel gene KCND3 acts as an important mediator in cardiomyocyte hypertrophy (Wang et al., 2019). Consistently, we found that the expressions of Kv4.3 protein and KCND3 gene were down-regulated in Ang II-induced cardiomyocyte hypertrophy (Figures 4A,B). Furthermore, miR-27-3p-mimics transfection induced a reduction expression of Kv4.3 channel protein in cardiomyocytes. On the contrary, miR-27-3p-mimics and miR-27-3p-inhibitor co-treatment attenuated the reduction of Kv4.3 protein expression in hypertrophic cardiomyocytes, and PCR results showed the similar tendencies (Figures 4C,D). These results indicated that miR-27-3p regulated cardiomyocyte hypertrophy by affecting the expression of Kv4.3 channel protein. However, no binding sites of miR-27-3p in the 3′UTR region of KCND3 gene were found.
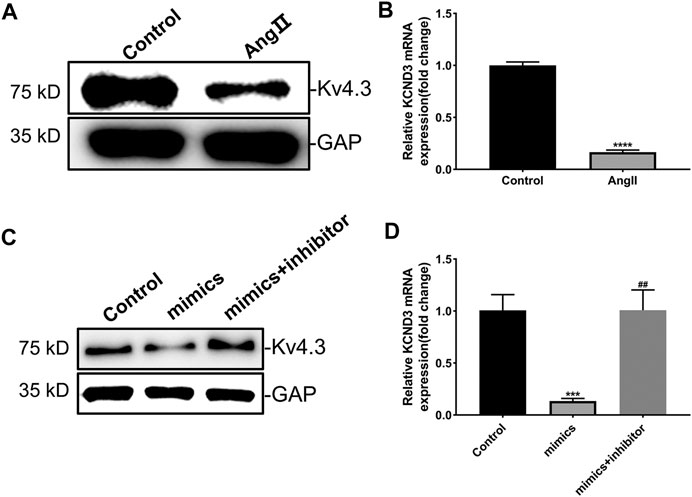
FIGURE 4. MiR-27a-3p negatively regulates the expression of Kv4.3 in rat ventricular cardiomyocytes. (A–B) The expressions of Kv4.3 channel protein and KCND3 gene were down-regulated in Ang II-induced cardiomyocyte hypertrophy. (C–D) MiR-27a-3p-inhibitor can abrogate the changes in the expression of KCND3/Kv4.3 induced by miR-27a-3p-mimics. All the values are the mean of three independent experiments (n = 3). ****p < 0.0001, ***p < 0.001 vs. control, ##p < 0.01 vs. mimics.
Next, we further searched the downstream targets of miR-27a-3p in the pathogenesis of cardiac hypertrophy and electrical remodeling. The TargetScan prediction showed that there was a binding sequence between miR-27a-3p and Hoxa10-mRNA 3′UTR region (Figure 5A). As shown in Figure 5B, we designed wild and mutant types of plasmids of Hoxa10 3′UTR region and transfected into HEK293T cells. Luciferase assay showed that the fluorescence intensity of miR-27a-3p group was significantly lower than the NC group in wild type, but there was no change in mutant type (Figure 5C). In addition, compared with NC group, the mRNA expression of Hoxa10 was significantly inhibited by miR-27a-3p-mimics in cardiomyocytes (Figure 5D). These results confirmed that there was a binding between miR-27a-3p and Hoxa10-mRNA 3′UTR region. Furthermore, miR-27a-3p-mimics significantly inhibited the expression of Hoxa10, while this process could be alleviated by miR-27a-3p-mimics and miR-27a-3p-inhibitor co-treatment. Besides, miR-27a-3p-inhibitor can also alleviate the decreased expression of Hoxa10 induced by Ang II treatment (Figure 5E). These results suggested that miR-27a-3p could negatively regulate the expression of Hoxa10.

FIGURE 5. MiR-27a-3p can negatively regulate the expression of Hoxa10. (A) The binding sequence of miR-27a-3p and 3′UTR region of Hoxa10 was predicted by TargetScan. (B) Wt 3′UTR and Mut 3′UTR of Hxoa10 were constructed. (C) Luciferase assay confirmed that miR-27a-3p had transcriptional regulation effect on Hoxa10. (D) The expression of Hoxa10 was decreased in miR-27a-3p-mimics treatment group compared with the control and NC group. (E) Western blot was applied to detect the protein expression of Hoxa10 after treatment with miR-27a-3p-mimics and miR-27a-3p-inhibitor, as well as the Ang II and Ang II + miR-27a-3p inhibitor treatment. All the values are the mean of three independent experiments (n = 3). ****p < 0.0001, ***p < 0.001 vs. NC.
In order to confirm whether Hoxa10 was involved in myocardial hypertrophy process. The cardiomyocytes were cultured with Hoxa10-overexpress for 8 h, then the serum-free medium was replaced, and the cells were co-cultured with Ang II for 48 h. PCR results showed that the mRNA expression of Hoxa10 in Hoxa10-overexpress group was significantly higher than that in NC group (Figure 6A). Compared with NC group, the expression of cardiac hypertrophy related genes including NPPA, NPPB, and MHY7 in NC + Ang II group was significantly up-regulated, while KCND3 was down-regulated (Figures 6B–E). As shown in Figure 6F, the protein expressions showed similar tendencies. Moreover, the protein expression of Hoxa10 was also down-regulated synchronously in NC + Ang II group. While this process could be inhibited by Hoxa10-overexpress (Figure 6B–F). The α-actinin staining showed that the area of cardiomyocytes in Ang II + Hoxa10-overexpress group was significantly smaller in NC + Ang II group, indicating that Hoxa10-overexpress reversed the process of myocardial hypertrophy induced by Ang II (Figure 6G). To sum up, these results suggested that by targeting the miR-27a-3p/Hoxa10/Kv4.3 axis could effectively inhibit the process of myocardial hypertrophy and cardiomyocyte hypertrophy, which provided us a new therapeutic strategy for myocardial hypertrophy and electrical remodeling.

FIGURE 6. Hoxa10 reverses the process of myocardial hypertrophy and Kv4.3 reduction induced by Ang II. (A) The expression of Hoxa10 in Hoxa10-overexpress group was significantly increased compared with the NC group. (B–D) The expression of cardiac hypertrophy related genes NPPA, NPPB, and MHY7 in NC + Ang II group was significantly up-regulated compared with the NC group, while down-regulated after treatment with Hoxa10 overexpression. (E) The expression of KCND3 in NC + Ang II group was significantly down-regulated compared with the NC group, while up-regulated after Hoxa10 overexpression treatment. (F) The protein expressions of ANP, BNP, β-MHC, and Kv4.3 showed similar tendencies as PCR results. The protein expression of Hoxa10 was down-regulated in NC + Ang II group, while up-regulated in Hoxa10-overexpress treatment group (G) Morphological changes after the overexpression of Hoxa10 showed that the area of cardiomyocytes in Ang II + Hoxa10-overexpress group was significantly smaller than that in NC + Ang II group. Scale bar, 50 μm. All the values are the mean of three independent experiments (n = 3). ***p < 0.001, **p < 0.01, *p < 0.05 vs. NC, ###p < 0.001, ##p < 0.01, #p < 0.05 vs. NC + Ang II.
This study confirmed that Ang II can lead to cardiomyocyte hypertrophy and electrophysiological remodeling. This process is related to the up-regulation of miR-27a-3p and the down-regulation of Hoxa10. We found that miR-27a-3p-inhibitor could mitigate the abnormal mRNA and protein expressions of NPPA/NPPB/MHY7/KNCD3 and ANP/BNP/β-MHC/Kv4.3 induced by Ang II. Further study confirmed that miR-27a-3p-mimics could inhibit the expression of Hoxa10. Overexpression of Hoxa10 could reverse the myocardial hypertrophy process induced by Ang II. Collectively, miR-27a-3p and Hoxa10 are involved in cardiac hypertrophy and electrical remodeling induced by Ang II, and the reversal of this process was closely related to targeting miR-27a-3p, Hoxa10 and Kv4.3.
Recent studies have shown that HOX is involved in the regulation of embryonic development, cell differentiation, cell cycle, apoptosis and other biological processes (Sanchez-Herrero, 2013). A large number of reports have pointed out that Hox is related to the occurrence and development of a variety of tumors, such as breast cancer, colorectal cancer, gastric cancer, lung cancer, ovarian cancer and so on (Grier et al., 2005). Although some studies have found that Hoxa10 was closely associated with the occurrence and development of cardiovascular diseases, there are no reports about its implication in myocardial hypertrophy and electrical remodeling (Ng et al., 2016; Souilhol et al., 2020). Our research demonstrated that the overexpression of Hoxa10 can reverse the expressions of hypertrophy-related genes and electrical remodeling, which is the first time to report the correlation between Hoxa10 and myocardial hypertrophy at home and abroad.
Previous studies have shown that miRNA plays an important role in the occurrence and development of heart disease, especially in cardiomyopathy such as myocardial hypertrophy and arrhythmia (Austin et al., 2019; Colpaert and Calore, 2019). Some abnormally expressed miRNA can regulate the target genes through incomplete complementary binding with the 3′UTR of the target mRNA. Previous studies have shown that the application of arsenic trioxide injection in the treatment of acute promyelocytic leukemia is often accompanied by cardiac side effects, which seriously limits the clinical application of arsenic trioxide (Bao et al., 2019). miR-1 and miR-133 are important targets for cardiac side effects such as arrhythmia and sudden cardiac death caused by arsenic trioxide (Shan et al., 2013). In addition, the expression of miR-133 and miR-590 in the atrium of smokers with atrial fibrillation and nicotine-induced atrial fibrillation was down-regulated, and the protein expressions of target genes TGF-β1 and TGF-β RII was up-regulated, resulting in increased collagen production of atrial fibroblasts, which in turn induced atrial fibrosis and atrial fibrillation (Shan et al., 2009). These reports confirmed that miRNA is closely related to the pathophysiological process of cardiovascular system. In recent years, the role of miR-27a-3p in cardiovascular diseases has become more and more prominent. Researches showed that miR-27a-3p can not only reduce cardiomyocyte injury induced by hypoxia/reoxygenation and lipopolysaccharide (Lozano-Velasco et al., 2015; Zhang et al., 2019), but also promote vascular maturation and angiogenesis by inhibiting SEMA6A and SEMA6D (Zhou et al., 2011; Urbich et al., 2012; Demolli et al., 2017). In addition, it has been reported that miR-27a-3p is closely related to ventricular formation, obesity and cardiac effects in mice (Chinchilla et al., 2011; Sassoon et al., 2016). Our research confirmed that miR-27a-3p could bind to the 3′UTR region of Hoxa10, regulate its transcription and inhibit the protein expression, which further led to cardiomyocyte hypertrophy and electrical remodeling. We firstly demonstrated that miR-27a-3p/Hoxa10 axis was involved in myocardial hypertrophy and electrical remodeling.
The limitations of our study are as followings: first of all, we only discussed myocardial hypertrophy and electrical remodeling from the miR-27a-3p/Hoxa10/Kv4.3 axis, but did not further study myocardial regeneration and myocardial fibrosis. Secondly, we only detected the change of miR-27a-3p/Hoxa10/Kv4.3 in vitro, but did not verify the changes in vivo. Due to the reason of time, we did not further carry out this study.
In summary, our study suggested that miR-27a-3p/Hoxa10/Kv4.3 axis regulated myocardial hypertrophy and electrical remodeling caused by Ang II, thus providing new therapeutic strategies for myocardial hypertrophy and arrhythmias clinically, as well as opening up new fields and expanding new horizons for researchers.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The experimental protocols were approved by the Ethics Committee of the Ethics of Animal Experiments of Chengde Medical College (Approval ID: CDMULAC20191031-009). The animal experiments were approved by the Research Ethics Committee of Chengde Medical College, and were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
XC and ZZ performed the experiments, revised the manuscript and integrated the figures. WS and YW performed the experiments, and produced the original draft of the manuscript. RW and SM helped with the cell biology experiments. CP, BL, and JZ helped with the experiments of molecular biology. XG, NG, JL, CL, and SD also helped with the cell biology and molecular biology experiments, and analyzed the data. WM, YL, and LZ conceived the idea, designed the study and provided the funding support. All authors contributed to and approved the final manuscript.
This work was supported by the Youth Program of National Natural Science Foundation of China (No. 81700310), the Hebei Province Science Foundation for Youths (No. H2018406029), the Natural Science Foundation of Heilongjiang Province (No. H2017025), the Postdoctoral Science Foundation of China (No. 2018T110318), the Hebei Provincial Department of Science and Technology and Technology Innovation Guidance Project-Science and Technology Work Conference, Research Project of basic Scientific Research Business fee of Chengde Medical College (No. KY2020001), the Key Subjects (Pathology and Pathophysiology) at Colleges and Universities of Hebei Province, Innovation and Entrepreneurship training Program for College students in Chengde Medical College (NO. 2019032), and the Chengde Science and Technology support Plan Project (No. 201904A099).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Austin, K. M., Trembley, M. A., Chandler, S. F., Sanders, S. P., Saffitz, J. E., Abrams, D. J., et al. (2019). Molecular Mechanisms of Arrhythmogenic Cardiomyopathy. Nat. Rev. Cardiol. 16 (9), 519–537. doi:10.1038/s41569-019-0200-7
Bacharova, L. (2019). Missing Link between Molecular Aspects of Ventricular Arrhythmias and QRS Complex Morphology in Left Ventricular Hypertrophy. Int. J. Mol. Sci. 21 (1), 48. doi:10.3390/ijms21010048
Bao, Z., Han, Z., Zhang, B., Yu, Y., Xu, Z., Ma, W., et al. (2019). Arsenic Trioxide Blocked Proliferation and Cardiomyocyte Differentiation of Human Induced Pluripotent Stem Cells: Implication in Cardiac Developmental Toxicity. Toxicol. Lett. 309, 51–58. doi:10.1016/j.toxlet.2019.03.008
Berger, R., Huelsman, M., Strecker, K., Bojic, A., Moser, P., Stanek, B., et al. (2002). B-type Natriuretic Peptide Predicts Sudden Death in Patients with Chronic Heart Failure. Circulation 105 (20), 2392–2397. doi:10.1161/01.cir.0000016642.15031.34
Cai, B., Ma, W., Ding, F., Zhang, L., Huang, Q., Wang, X., et al. (2018). The Long Noncoding RNA CAREL Controls Cardiac Regeneration. J. Am. Coll. Cardiol. 72 (5), 534–550. doi:10.1016/j.jacc.2018.04.085
Chen, W.-J., Yin, K., Zhao, G.-J., Fu, Y.-C., and Tang, C.-K. (2012). The Magic and Mystery of microRNA-27 in Atherosclerosis. Atherosclerosis 222 (2), 314–323. doi:10.1016/j.atherosclerosis.2012.01.020
Chinchilla, A., Lozano, E., Daimi, H., Esteban, F. J., Crist, C., Aranega, A. E., et al. (2011). MicroRNA Profiling during Mouse Ventricular Maturation: a Role for miR-27 Modulating Mef2c Expression. Cardiovasc. Res. 89 (1), 98–108. doi:10.1093/cvr/cvq264
Colpaert, R. M. W., and Calore, M. (2019). MicroRNAs in Cardiac Diseases. Cells 8 (7), 737. doi:10.3390/cells8070737
Coyne, J. C., Rohrbaugh, M. J., Shoham, V., Sonnega, J. S., Nicklas, J. M., and Cranford, J. A. (2001). Prognostic Importance of Marital Quality for Survival of Congestive Heart Failure. Am. J. Cardiol. 88 (5), 526–529. doi:10.1016/s0002-9149(01)01731-3
Demolli, S., Doddaballapur, A., Devraj, K., Stark, K., Manavski, Y., Eckart, A., et al. (2017). Shear Stress-Regulated miR-27b Controls Pericyte Recruitment by Repressing SEMA6A and SEMA6D. Cardiovasc. Res. 113 (6), 681–691. doi:10.1093/cvr/cvx032
Dey, S., DeMazumder, D., Sidor, A., Foster, D. B., and O’Rourke, B. (2018). Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 123 (3), 356–371. doi:10.1161/circresaha.118.312708
Gatzoulis, K. A., Archontakis, S., Dilaveris, P., Tsiachris, D., Arsenos, P., et al. (2012). Ventricular Arrhythmias: from the Electrophysiology Laboratory to Clinical Practice. Part II: Potentially Malignant and Benign Ventricular Arrhythmias. Hellenic J. Cardiol. 53 (3), 217–233. doi:10.1007/s11886-014-0510-7
Grier, D., Thompson, A., Kwasniewska, A., McGonigle, G., Halliday, H., and Lappin, T. (2005). The Pathophysiology ofHOX Genes and Their Role in Cancer. J. Pathol. 205 (2), 154–171. doi:10.1002/path.1710
John, R. M., Tedrow, U. B., Koplan, B. A., Albert, C. M., Epstein, L. M., Sweeney, M. O., et al. (2012). Ventricular Arrhythmias and Sudden Cardiac Death. The Lancet 380 (9852), 1520–1529. doi:10.1016/s0140-6736(12)61413-5
Lozano-Velasco, E., Galiano-Torres, J., Jodar-Garcia, A., Aranega, A. E., and Franco, D. (2015). miR-27 and miR-125 Distinctly Regulate Muscle-Enriched Transcription Factors in Cardiac and Skeletal Myocytes. Biomed. Res. Int. 2015, 391306. doi:10.1155/2015/391306
Ma, W., Ding, F., Wang, X., Huang, Q., Zhang, L., Bi, C., et al. (2018). By Targeting Atg7 MicroRNA-143 Mediates Oxidative Stress-Induced Autophagy of C-Kit+ Mouse Cardiac Progenitor Cells. EBioMedicine 32, 182–191. doi:10.1016/j.ebiom.2018.05.021
Ng, E. S., Azzola, L., Bruveris, F. F., Calvanese, V., Phipson, B., Vlahos, K., et al. (2016). Differentiation of Human Embryonic Stem Cells to HOXA+ Hemogenic Vasculature that Resembles the Aorta-Gonad-Mesonephros. Nat. Biotechnol. 34 (11), 1168–1179. doi:10.1038/nbt.3702
Pan, Z., Sun, X., Shan, H., Wang, N., Wang, J., Ren, J., et al. (2012). MicroRNA-101 Inhibited Postinfarct Cardiac Fibrosis and Improved Left Ventricular Compliance via the FBJ Osteosarcoma Oncogene/Transforming Growth Factor-Β1 Pathway. Circulation 126 (7), 840–850. doi:10.1161/circulationaha.112.094524
Samidurai, A., Kukreja, R. C., and Das, A. (2018). Emerging Role of mTOR Signaling-Related miRNAs in Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2018, 6141902. doi:10.1155/2018/6141902
Sanchez-Herrero, E. (2013). Hox Targets and Cellular Functions. Scientifica (Cairo). 2013, 738257. doi:10.1155/2013/738257
Sassoon, D. J., Goodwill, A. G., Noblet, J. N., Conteh, A. M., Herring, B. P., McClintick, J. N., et al. (2016). Obesity Alters Molecular and Functional Cardiac Responses to Ischemia/reperfusion and Glucagon-like Peptide-1 Receptor Agonism. Basic Res. Cardiol. 111 (4), 43. doi:10.1007/s00395-016-0563-4
Shan, H., Zhang, Y., Cai, B., Chen, X., Fan, Y., Yang, L., et al. (2013). Upregulation of microRNA-1 and microRNA-133 Contributes to Arsenic-Induced Cardiac Electrical Remodeling. Int. J. Cardiol. 167 (6), 2798–2805. doi:10.1016/j.ijcard.2012.07.009
Shan, H., Zhang, Y., Lu, Y., Zhang, Y., Pan, Z., Cai, B., et al. (2009). Downregulation of miR-133 and miR-590 Contributes to Nicotine-Induced Atrial Remodelling in Canines. Cardiovasc. Res. 83 (3), 465–472. doi:10.1093/cvr/cvp130
Souilhol, C., Serbanovic-Canic, J., Fragiadaki, M., Chico, T. J., Ridger, V., Roddie, H., et al. (2020). Endothelial Responses to Shear Stress in Atherosclerosis: a Novel Role for Developmental Genes. Nat. Rev. Cardiol. 17 (1), 52–63. doi:10.1038/s41569-019-0239-5
Thompson, B. S. (2009). Sudden Cardiac Death and Heart Failure. AACN Adv. Crit. Care 20 (4), 356–365. doi:10.1097/nci.0b013e3181b82379
Urbich, C., Kaluza, D., Frömel, T., Knau, A., Bennewitz, K., Boon, R. A., et al. (2012). MicroRNA-27a/b Controls Endothelial Cell Repulsion and Angiogenesis by Targeting Semaphorin 6A. Blood 119 (6), 1607–1616. doi:10.1182/blood-2011-08-373886
Wang, Y., Keskanokwong, T., and Cheng, J. (2019). Kv4.3 Expression Abrogates and Reverses Norepinephrine-Induced Myocyte Hypertrophy by CaMKII Inhibition. J. Mol. Cell Cardiol. 126, 77–85. doi:10.1016/j.yjmcc.2018.11.011
Wang, Z., Luo, X., Lu, Y., and Yang, B. (2008). miRNAs at the Heart of the Matter. J. Mol. Med. 86 (7), 771–783. doi:10.1007/s00109-008-0341-3
Yang, B., Lin, H., Xiao, J., Lu, Y., Luo, X., Li, B., et al. (2007). The Muscle-specific microRNA miR-1 Regulates Cardiac Arrhythmogenic Potential by Targeting GJA1 and KCNJ2. Nat. Med. 13 (4), 486–491. doi:10.1038/nm1569
Zhang, X. L., An, B. F., and Zhang, G. C. (2019). MiR‐27 Alleviates Myocardial Cell Damage Induced by Hypoxia/reoxygenation via Targeting TGFBR1 and Inhibiting NF‐κB Pathway. Kaohsiung J. Med. Sci. 35 (10), 607–614. doi:10.1002/kjm2.12092
Keywords: cardiac hypertrophy, electrical remodeling, HOXA10, miRNA-27a-3p, Ang II
Citation: Cao X, Zhang Z, Wang Y, Shan W, Wang R, Mao S, Ding S, Pang C, Li B, Zhou J, Guo X, Guo N, Li C, Liang J, Ma W, Liu Y and Zhao L (2021) MiR-27a-3p/Hoxa10 Axis Regulates Angiotensin II-Induced Cardiomyocyte Hypertrophy by Targeting Kv4.3 Expression. Front. Pharmacol. 12:680349. doi: 10.3389/fphar.2021.680349
Received: 14 March 2021; Accepted: 13 April 2021;
Published: 24 June 2021.
Edited by:
Hua Zhu, The Ohio State University, United StatesReviewed by:
Shanzhi Wang, University of Arkansas at Little Rock, United StatesCopyright © 2021 Cao, Zhang, Wang, Shan, Wang, Mao, Ding, Pang, Li, Zhou, Guo, Guo, Li, Liang, Ma, Liu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Zhao, c2hpamlhbnpoZW5nemFpZmVpQDE2My5jb20=; Yu Liu, cmFpbmZhbGwxOTgyQDE2My5jb20=; Wenya Ma, bXdlbnlhMTk4OUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.