 M. Ekholm
M. Ekholm T. Kahan
T. Kahan- Karolinska Institutet, Department of Clinical Sciences, Danderyd Hospital, Division of Cardiovascular Medicine, Stockholm, Sweden
Atherosclerosis is considered a disease caused by a chronic inflammation, associated with endothelial dysfunction, and several mediators of inflammation are up-regulated in subjects with atherosclerotic disease. Healthy, intact endothelium exhibits an antithrombotic, protective surface between the vascular lumen and vascular smooth muscle cells in the vessel wall. Oxidative stress is an imbalance between anti- and prooxidants, with a subsequent increase of reactive oxygen species, leading to tissue damage. The renin-angiotensin-aldosterone system is of vital importance in the pathobiology of vascular disease. Convincing data indicate that angiotensin II accelerates hypertension and augments the production of reactive oxygen species. This leads to the generation of a proinflammatory phenotype in human endothelial and vascular smooth muscle cells by the up-regulation of adhesion molecules, chemokines and cytokines. In addition, angiotensin II also seems to increase thrombin generation, possibly via a direct impact on tissue factor. However, the mechanism of cross-talk between inflammation and haemostasis can also contribute to prothrombotic states in inflammatory environments. Thus, blocking of the renin-angiotensin-aldosterone system might be an approach to reduce both inflammatory and thrombotic complications in high-risk patients. During COVID-19, the renin-angiotensin-aldosterone system may be activated. The levels of angiotensin II could contribute to the ongoing inflammation, which might result in a cytokine storm, a complication that significantly impairs prognosis. At the outbreak of COVID-19 concerns were raised about the use of angiotensin converting enzyme inhibitors and angiotensin receptor blocker drugs in patients with COVID-19 and hypertension or other cardiovascular comorbidities. However, the present evidence is in favor of continuing to use of these drugs. Based on experimental evidence, blocking the renin-angiotensin-aldosterone system might even exert a potentially protective influence in the setting of COVID-19.
Introduction
Cardiovascular disease is the most common cause of death in economically developed countries, and is expected to remain a leading cause globally due to an increasing prevalence in developing countries (Mathers and Loncar, 2006). Given the high morbidity and mortality burden in cardiovascular disease, it is important for societies and their health care systems to improve strategies to decrease the incidence of cardiovascular disease (Leal et al., 2006). Traditional risk factors cardiovascular disease include hypertension, hypercholesterolemia, diabetes mellitus, obesity, tobacco smoking, age, male sex, and family history. These risk factors contribute to endothelial dysfunction, and inflammation plays a key role in early-stage endothelial dysfunction and oxidative stress in the vascular wall, which are hallmarks of subclinical atherosclerosis (Rafieian-Kopaei et al., 2014; Castellon and Bogdanova, 2016).
The renin-angiotensin-aldosterone system (RAAS) is of vital importance in the pathobiology of vascular disease. Angiotensin (Ang) II promotes atherosclerosis (Nickenig, 2002) and has important impact on vascular inflammation and haemostasis. Ang II has been shown to cause oxidative stress, induce endothelial dysfunction and to generate a proinflammatory phenotype in human vascular smooth muscle cell (VSMC)s by stimulating up-regulation of the adhesion molecules, chemokines and cytokines (Schieffer et al., 2000; Brasier et al., 2002). Ang II is also pivotal in vascular remodeling, as it induces the expression of a number of growth factors (Satoh et al., 2001). In addition, Ang II modulates vascular cell migration and growth (Yaghini et al., 2010), decreases VSMC apoptosis (Li et al., 2006), alters extracellular matrix modulation composition (Wesselman and De Mey, 2002), and Ang II has been shown to initiate and accelerate hypertension, endothelial dysfunction and atherosclerosis (Weiss et al., 2001). Conversely, inhibition of the RAAS components decreases experimental atherosclerosis (Rader and Daugherty, 2008; Lu et al., 2012), and death from cardiovascular disease in humans (Yusuf et al., 2000). Thus, it has been proposed that RAAS inhibition may have anti-atherosclerotic effects beyond the effects of the blood pressure reduction (Yusuf et al., 2000).
Ang II also has an impact on haemostasis and may increase thrombin generation. Inflammatory stimuli can prime the coagulation system through several mechanisms, and the cross-talk between inflammation and haemostasis helps to explain prothrombotic states in inflammatory environments (Verhamme and Hoylaerts, 2009). The coagulation cascade in inflammatory states is mainly mediated by tissue factor (TF) (Cimmino et al., 2011). Under physiological conditions TF is not expressed or is in an inactive state in circulatory or endothelial cells (Rao et al., 2012). However, various inflammatory signals, like cytokines, can induce TF activation in endothelial cells (Szotowski et al., 2005). Ang II may increase TF expression in vessels, also platelet activation by circulating Ang II may have an important contributing role in the generation of thrombin (Burger et al., 2011; Cordazzo et al., 2013).
There are multiple effects of Ang II on the atherosclerotic process, as summarized in Figure 1. This review, however, will focus on the RAAS and events associated with inflammation, oxidative stress, endothelial dysfunction. Second, we discuss the influence of the RAAS on the haemostatic system via cross-talk between inflammation and the haemostatic system, and through a direct effect of Ang II on thrombosis. Finally, the relationship to the corona virus disease 2019 (COVID-19) to the RAAS, and the known complications of this infection, are discussed.
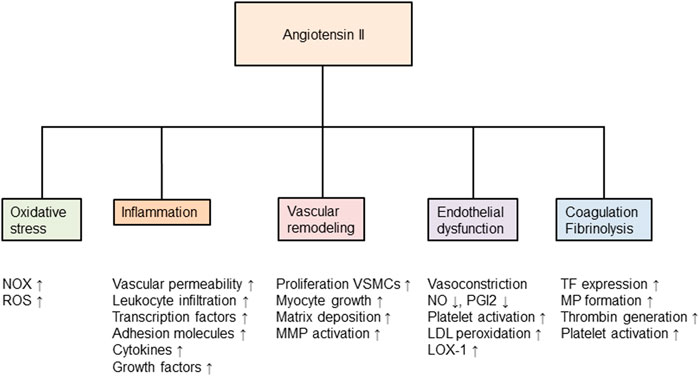
FIGURE 1. Angiotensin II and its effects in the development of atherosclerosis. Nox, nicotinamide-adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species; MMP, matrix metalloproteinase; VSMCs, vascular smooth muscle cells; NO, nitric oxide; PGI2, prostaglandin I2 (also called prostacyclin); LDL, low-density lipoprotein; LOX, lectin-like oxidized low-density lipoprotein receptor; TF, tissue factor; MP, microparticles and tPA, tissue plasminogen activator. Modified from (Volpe, 2012).
The Endothelium
The Importance of Healthy and Intact Endothelium
The endothelium has an important function in preserving a physiological structure and function. Endothelial cells form a monolayer that produces factors that regulate vascular tone, inflammation, haemostasis, vascular cell growth and death, angiogenesis, the migration of leukocytes, and prevents platelet adhesion (Gawaz et al., 2005). Vascular tone is dependent on a delicate balance between vascular dilators, such as NO, prostaglandin I2 (also called prostacyclin); and endothelium derived hyperpolarizing factor and vascular constrictors, such as Ang II, endothelin-1, and thromboxane A2 (Sandoo et al., 2010). Also, VSMCs are affected by endothelial cells and other factors, and VSMCs can themselves release cytokines and growth-regulatory mediators, which in turn have an impact on vessel phenotype and growth.
In inflammatory conditions, endothelial cells release procoagulant and antifibrinolytic mediators, like von Willebrand factor, thromboxane A2 and plasminogen activator inhibitor-1 (PAI-1). Activated endothelial cells express TF and adhesion molecules, which are of vital importance in mediating the interaction of leukocytes and platelets with endothelial cells, and thus activating the coagulant system and promoting an inflammatory response. The cytokines interleukin (IL)-1, IL-6, IL-8, and tumor necrosis factor α (TNF-α), and also the chemokine monocyte chemoattractant protein-1 (MCP-1), and the growth factor transforming growth factor-β1 (TGF-β1), have all been shown to play a key role in mediating the procoagulant changes in endothelial dysfunction (van der Poll et al., 1994; Ceciliani et al., 2002).
Inflammation and Oxidative Stress
The Inflammatory Process
Several mediators of inflammation are up-regulated in subjects with atherosclerotic disease (Kalsch et al., 2007). For diagnostic use, the cytokine IL-6, and C-reactive protein (CRP), have generated considerable attention. CRP is generated by hepatic cells and is modulated by IL-6, but also by TNF-α and IL-1β (Calabró et al., 2003), thereby contributing to the up-regulation of MCP-1 and selectins, such as P- and E-selectin and the cell adhesion molecules intracellular adhesion molecule -1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). CRP attenuates the synthesis of endothelial NO (Verma et al., 2002), and causes augmented PAI-1 (Devaraj et al., 2003). Increased concentrations of acute phase reactants like CRP, IL-6, leukocyte count and fibrinogen are all associated with an increased risk of cardiovascular disease (Ridker et al., 1998; Ridker et al., 2000; Kaptoge et al., 2012). Also, phospholipase A2, which is implicated in the oxidation of low-density lipoprotein (LDL) and subsequent oxidative stress and inflammation, can predict atherosclerotic disease (Di Angelantonio et al., 2012).
The infiltration of inflammatory cells is associated with the secretion of mediators like TGF-β1 and highly pro-inflammatory interleukins like IL-1β and IL-18 (Dinarello, 2018). This stimulates excessive extracellular matrix fibroblasts to differentiate into active myofibroblasts (Wynn, 2008). A key mediator to this process is TGF-β1, and its release can be promoted by Ang II (Wynn, 2008; Meng et al., 2016). The major sources of TGF-β1 are macrophages, monocytes, neutrophils, myofibroblasts, and epithelial cells (Wynn and Ramalingam, 2012), and during states with prolonged activation of Ang II and TGF-β1 a chronic inflammation contributes to adverse effects and diseases like chronic heart failure (Gullestad et al., 2012). In addition, Ang II, TGF-β1, and IL-1β also supress activity of matrix metalloproteinases, which contributes to scar tissue and stiffening (Porter and Turner, 2009).
Inflammasomes have an important role in cardiovascular disease, in particular the subtype nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domains-containing protein 3 (NLRP3) (Pinar et al., 2020). This inflammasome complex consists of three intracellular subunits, the NLRP3 sensor protein, the apoptosis-associated speck-like protein (ASC), and pro-caspase-1. Upon activation NLRP3 first recruits ASC and then pro-caspase-1. This in turn leads to pro-caspase-1 being cleaved to active caspase-1, which cleaves pro- IL-1β and pro-IL-18 to mature inflammatory cytokines (Man and Kanneganti, 2015). Active caspase-1 in turn cleaves gasdermin-D to an N-terminal fragment that can signal transduce promoting inflammation. NLRP3 can be activated by a wide range of danger signals, such as mitochondrial dysfunction, extracellular adenosine triphosphate and damaged nucleic acids, but also by Ang II and TGF-β1 trigger a number of pattern recognition receptors (Suthahar et al., 2017).
The Cell Reduction-Oxidation Reaction State
Oxidative stress is an imbalance between anti- and prooxidants, with a subsequent increase of ROS, leading to tissue damage. Thus, the cell redox state is an important factor of endothelial cell biology. Traditional risk factors for cardiovascular disease can initiate endothelial dysfunction by changing the cell redox state and, consequently, the oxidative stress in the vascular wall. Increased generation of superoxide anion, O2ˉ and, subsequently, oxidative stress results in enhanced catabolism of NO, endothelial dysfunction and impaired vasodilatation. Reactive oxygen species (ROS) also has the ability to reduce the activity of NO synthase (NOS) and to increase the breakdown of NO. Also, NO is a potent endogenous inhibitor of VSMC migration and growth (Sarkar et al., 1996) and, in higher concentrations, impairs up-regulation of adhesion molecules and cytokines (Qian and Fulton, 2012).
The transcription of the pleiotropic nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-ҡB) has a pivotal role in endothelial up-regulation of cytokines and adhesion molecules. In the nucleus NF-ҡB binds to target genes coding for proinflammatory and proatherogenic proteins, and promotes gene transcription (Moynagh, 2005). NO is a powerful inhibitor of activation of NF-ҡB (Grumbach et al., 2005). It is to be noted that NO and the superoxide anion reacts to form the powerful oxidant peroxynitrite, ONOOˉ, that can damage endothelial cells. Reactive oxygen species also lowers the availability of tetrahydrobiopterin. If this occurs, the oxygenase function of NOS is replaced by its reductase function and ROS are produced instead of NO, which increases NF-ҡB activity, and the expression of cytokines (Mittal et al., 2014). Thus, the ratio between ROS and NO regulates the redox state and is of vital importance for proper function of the vascular endothelium. An imbalance between NO and ROS increases the risk for vasospasm, VSMC proliferation, and can cause an imbalance between tissue plasminogen activator (tPA) and PAI-1, which may predispose a prothrombotic state.
Tissue Factor
Coagulation Cascade
The TF pathway (or the extrinsic pathway) and the contact pathway (or the intrinsic pathway), divides the coagulation cascade into two parts. The TF pathway is the major inducer of the coagulation cascade of the two under healthy conditions. The TF pathway is initiated when TF is bound to FVII-FVIIa. The TF-FVIIa complex cleaves FX into FXa, and FIX into FIXa, respectively. FXa then forms the prothrombinase complex when associated with FVa and Ca2+ on activated platelets, resulting in thrombin formation (Grover and Mackman, 2018). The reaction is much faster in the presence of negatively charged surface phospholipids (i.e., phoshatidylserine) on activated platelet membranes. Microparticles may also accelerate thrombin generation due to a procoagulant phosphatidylserine-rich surface that supports thrombin generation (Davizon and Lopez, 2009).
Tissue Factor Circulates in Three Pools
TF is present in the extravascular space (subendothelial tissue) and not exposed to flowing blood, except during plaque rupture. However, TF is also present in circulating blood, mainly in three pools. The most important source of TF is monocytes, expressed as membrane-anchored and membrane-bound. There is no evidence that granulocytes express TF; rather they seem to acquire TF from monocytes (Egorina et al., 2008). Controversial is the expression of TF in platelets. Human platelets appear not to express TF when activated (Osterud and Olsen, 2013). Platelets might store TF in α-granules or acquire TF from TF-containing microparticles (Bouchard et al., 2010; Camera et al., 2010). On the other hand, activated platelets generate microparticles that seem to express TF on their surface and contain negatively charged phospholipids, two very important factors for coagulation reactions (Mallat et al., 2000). The second source of circulating TF is different cell-derived microparticles, which are considered to be key players in atherothrombosis. The third pool, probably of minor importance, is a spliced form of TF, which is soluble and circulates in the blood (Bogdanov et al., 2003).
Blood-borne TF means that the coagulation cascade may be activated without contact between the blood and the extravascular space (i.e., plaque rupture). This appears to be an important contribution in intravascular thrombosis (Balasubramanian et al., 2002). Blood-borne TF implicates that TF apparent on the cell surface under normal conditions is in an inactive, cryptic, state. To be functionally active and to exhibit a procoagulant activity, TF has to undergo an activation step, decryption. The importance of the blood-borne TF is still controversial, and the molecular differences between these states remain to be clarified (Rao et al., 2012).
Inflammation Activates and Decrypts Tissue Factor
The coagulation cascade in inflammatory states is mainly mediated by TF (Cimmino et al., 2011). Various inflammatory signals, like IL-6, IL-1, or TNF-α, can induce TF activation in monocytes and macrophages and endothelial cells. TF expression is mostly dependent of IL-6, and inhibition of IL-6 can completely block thrombin generation (van der Poll et al., 1994). IL-6, but also IL-1β and IL-8 has been shown to be relevant for activation of coagulation (Bester and Pretorius, 2016). Also, CRP seems to have a role in coagulation as CRP can induce TF activation via a NF-κB pathway in both endothelial cells and VSMCs (Cirillo et al., 2005). In addition, Other mediators like oxidized LDL (Cimmino et al., 2019) and oxygen free radicals (Cimmino et al., 2015) can enhance TF activation. This enables active TF to be exposed to blood and bind to FVII. The complex of TF and FVIIa then contributes to the conversion of FIX and FX into the active proteases FXa and FXa, thereby generating thrombin. TF also possesses a function as a signaling receptor, where TF binding of FVIIa triggers VSMC proliferation (Cirillo et al., 2004).
Cross-Talk Between Inflammation and Coagulation
The Effects of Inflammation on Coagulation
Evidence suggests that inflammation and haemostasis are tightly interrelated in a bidirectional way, i.e., inflammation may lead to activation of the haemostatic system, while the haemostatic system has an impact on the inflammatory activity (Verhamme and Hoylaerts, 2009). This helps to explain prothrombotic states in inflammatory environments. Several trials show the importance of the cytokines such as IL-6, IL-1β, IL-8, TNF-α, and MCP-1 in the activation of coagulation by inflammation, (van der Poll et al., 2001; Levi et al., 2012).
The Effects of Coagulation on Inflammation
Coagulation factors can induce vascular inflammation by their binding to protease activated receptor (PAR)s, which are present in endothelial cells, leukocytes, platelets, fibroblasts, and VSMCs (Coughlin, 2000). An exceptional characteristic of PARs is that these receptors carry their own ligand, which is unmasked until the receptor is cleaved. PARs are G protein-coupled receptors, and up to date, four receptors have been identified (PAR 1–4). Thrombin is the most essential player in activation of PARs, and can activate PAR-1, PAR-3 as well as PAR-4. Factor (F)Xa transmits activation of PAR-1 to PAR-3, while TF-FVIIa transmits activation of PAR-2 (Coughlin, 2000). PAR activation up-regulates inflammatory molecules, like cytokines, chemokines, growth factors and adhesion molecules. Experiments in healthy human subjects have shown a 4-fold increase in plasma concentrations of IL-8 and IL-6 by recombinant FVIIa (de Jonge et al., 2003). Activation of PARs transforms endothelial cells into a proinflammatory phenotype, causing vascular permeability and local accumulation of platelets and leukocytes (Alberelli and De Candia, 2014). In VSMCs, PAR activation mediates contraction, proliferation, migration, hypertrophy and modulation of the extracellular matrix, thereby contributing to atherosclerosis and hypertension.
Thrombin, Platelets and Protease-Activated Receptors
The Impact of Thrombin
Figure 2 summarizes the role of thrombin in the coagulation system. The main action of thrombin is to convert fibrinogen to fibrin monomers, the end result of the coagulation cascade. However, thrombin also exercises an anticoagulant role by binding to thrombomodulin at the intact endothelium and to promote activation of the protein C pathway. Activated protein C then inactivates FVa and FVIIIa, two essential cofactors for FXa and FIXa, thereby down-regulating thrombin generation (Esmon, 2003). Thrombin also has a central role in the inflammatory response and stimulates different cell types in the vasculature and in blood, including endothelial cells, VSMCs, and platelets. The effects of thrombin are mediated by its counter-receptors, PARs.
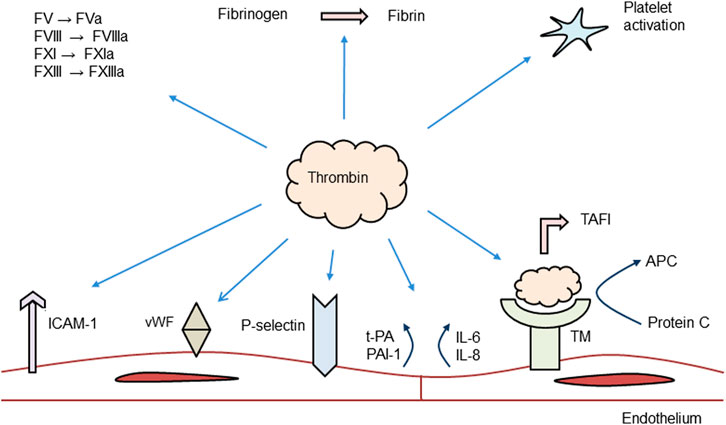
FIGURE 2. Role of thrombin in the coagulation system. ICAM, intracellular cell adhesion molecule; PAI, plasminogen activator inhibitor; vWF, von Willebrand factor; IL, interleukin; TAFI, thrombin fibrinolysis activatable inhibitor; TM, thrombomodulin; APC, activated protein C; and F, coagulation factor. Modified from (Davidson, 2013).
The Impact of Platelets
Human platelets express PAR-1 and PAR-4. PAR-1 seems to be the main platelet receptor for thrombin (Duvernay et al., 2017). Thrombin is the most effective platelet activator and causes platelets to change shape, secrete a number of substances, mobilize P-selectin and cluster of differentiation 40 ligand (CD40L), leading to activation of αIIbβ3 and, ultimately, aggregations of platelets (Brass, 2003). Thus, thrombin signaling in platelets contributes significantly to haemostasis and thrombosis (Duvernay et al., 2017).
IL-1β is a pivotal mediator in the cytokine cascade and also an important activator of endothelial cells, by inducing the cytokines IL-6, IL-8, and chemokine MCP-1 (Gawaz et al., 2002). Platelet IL-1β also induces up-regulation of adhesion molecules like ICAM-1 and the vitronectin receptor (αvβ3), contributing to adhesion of neutrophils and monocytes to endothelial cells (Gawaz et al., 2000). CD40L is also a key player in platelet activation. CD40L and its receptor CD40 are expressed in cells including platelets, endothelial cells, VSMCs, T lymphocytes and macrophages (Daub et al., 2020). Activated, platelets rapidly (within seconds) express CD40L, and the interaction of CD40L and CD40 on endothelial cells up-regulate the cytokines IL-1, IL-8 and IL-6, MCP-1 and adhesion molecules, as well as increasing TF expression (Antoniades et al., 2009). When platelets and endothelial cells are activated, P-selectin is translocated to the cell surface where it functions as a platelet-selectin glycoprotein ligand-1 receptor, expressed on leukocytes (and in small amounts on platelets). The subsequent P-selectin and platelet-selectin glycoprotein ligand-1 interaction in turn increases the release of cytokines and chemokines from neutrophils and monocytes. The interaction also stimulates the up-regulation of adhesion molecules and TF on endothelial cells and leukocytes (Polgar et al., 2005). Thrombin also promotes the release of endothelial cell microparticles, which seems to be critical in vascular pathophysiology (Sapet et al., 2006). Once trace amounts of thrombin have been generated, this can activate FV, FVIII, FXI, and FXIII.
The Shift of the Endothelium
In the healthy endothelium, thrombin activates PAR-1 and stimulates the production of NO and prostaglandin I2, leading to vasodilatation and fibrinolysis by the release of tPA (Gudmundsdottir et al., 2006). During inflammation and endothelial dysfunction PAR expression is increased, priming the response of endothelial cells to thrombin, and shifting endothelial cells to a proinflammatory phenotype, inducing synthesis and release of PAI-1, contributing to impaired fibrinolysis (Hirano, 2007). In pathophysiological conditions, PAR-1 activation causes morphological changes, an increased vascular leakage, release of proinflammatory cytokines, and up-regulation of adhesion molecules (Rahman et al., 1999). In particular the synthesis of IL-6 seems to mediate the transition of the inflammatory process in the vessels from an acute to a chronic phase (Marin et al., 2001). Activation of PAR-1 also mobilizes P-selectin and von Willebrand factor from Weibel-Palade bodies, promoting endothelial cell rolling and subsequently firm adhesion of both platelets and leukocytes.
In healthy arteries PARs are preferably expressed in endothelial cells, while their expression in VSMCs is limited. In hypertension and in atherosclerotic vessels PAR-1 is up-regulated in VSMCs (Hirano, 2007). This implies that PARs on VSMCs have a more prominent role under pathological conditions. In conditions associated with endothelial dysfunction, PARs in VSMCs mediate contraction, proliferation, migration, hypertrophy and the production of extracellular matrix modulation (Hirano, 2007).
Altogether, in healthy conditions thrombin and PAR-1 activation causes endothelium dependent vasodilatation. In contrast, under pathological conditions, thrombin induces a procoagulatory and proinflammatory state.
The RAAS and Vascular Complications
The Components of the RAAS
All RAAS peptides are derived from angiotensinogen, which is synthesized in an abundance in the liver, but angiotensinogen messenger ribonucleic acid (mRNA) has also been detected in many other tissues (Dickson and Sigmund, 2006; Kumar et al., 2012). Figure 3 summarizes the different RAAS components. In the classical pathway, the rate limiting enzyme renin is produced in the kidney (juxtaglomerular cells), and is released in response to vasodilation, low sodium, and by beta1-adrenoceptor stimulation. In the blood stream, renin cleaves angiotensinogen into Ang I, which is processed by the membrane bound exopeptidase angiotensin converting enzyme (ACE), to produce Ang II, the predominant peptide of the RAAS (Putnam et al., 2012; Nehme et al., 2019).
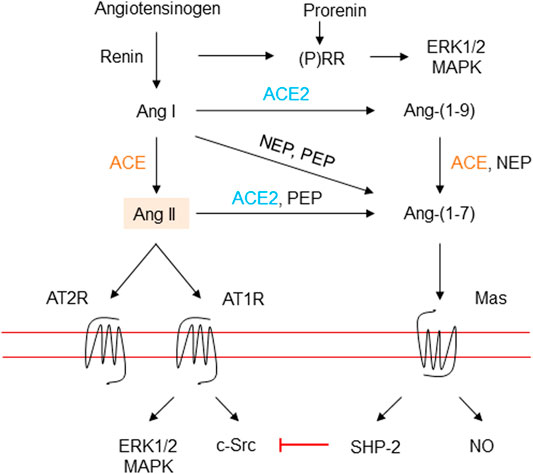
FIGURE 3. The renin-angiotensin-aldosterone system. Renin is secreted due to various stimuli, and then cleaves angiotensinogen into the inactive decapeptide Ang I. Renin and prorenin can also interact with (pro) renin receptors to activate the MAP kinases ERK1/2 and p38 pathways. ACE cleaves Ang I into the octapeptide Ang II, or to Ang-(1–7) by ACE2 and probably ACE. ACE2 may also produce Ang-(1–7) from Ang II. ACE also inactivates bradykinin into inactive fragments. Importantly, Ang II may be generated directly from angiotensinogen through non-ACE pathways. Ang II activates AT1R, a G protein-coupled receptor. Vasoconstriction and stimulation of aldosterone tend to elevate blood pressure. Ang II also activates AT2R, a G protein-coupled receptor, which can antagonize the effects of activation of the AT1R. Ang, angiotensin; ACE, angiotensin converting enzyme; MAPK, mitogen-activated protein kinase; extracellular signal regulated kinase, ERK; angiotensin II type 1 receptor, AT1R; angiotensin II type 2 receptor, AT2R; (P)RR, (pro)renin receptor; c-Src, cellular Src kinase, a non-receptor tyrosine kinase; SHP-2, Src-homology 2 domain-containing phosphatase 2; NO, nitric oxide; NEP, neutral endopeptidase, and PEP, prolyl endopeptidase.
The components of the RAAS have dual roles, as the circulating hormone can also be present in several tissues to act as a potential local system (Dell'Italia, 2011; Nehme et al., 2019). A local or tissue RAAS, Ang II production from independent regulation of angiotensinogen in tissue compartments and locally synthesized enzymes may form a local RAAS operating independently of the circulatory RAAS (De Mello and Frohlich, 2011; Sparks et al., 2014). Ang II can then be produced inside cells and consequently the hormone can bind to its receptors in adjacent cells, thus acting in an autocrine/paracrine manner (Kumar et al., 2012). Thus, the local RAAS in distinct tissues acts via autocrine and paracrine mechanisms to exacerbate the effects of circulating RAAS and/or works independently to induce a response within tissues.
Local Ang II formation is also dependent on other enzymes such as chymase, tonin, and D and G cathepsin. Chymase, which appears the most important, belongs to the serine proteases and is stored in secretory granules in mast cells. Under normal conditions mature mast cells do not circulate in the bloodstream. They are present throughout the body, and are found in mucosal and epithelial tissues and in all vascularized tissues (Krystel-Whittemore et al., 2016). When activated, mast cells undergo degranulation with biological actions in various inflammatory settings (Galli et al., 2005; Wernersson and Pejler, 2014), and large amounts of chymase cleave many peptides and proteins, including Ang I, pro-matrix metalloproteinase-9, pro-IL-1β, pro-IL-18, and IL-6 (Urata et al., 1990; Mizutani et al., 1991; Tchougounova et al., 2005; Fu et al., 2017). Thus, detrimental impact of mast cells has been reported in settings like atherosclerosis, cardiac dysfunction, and abdominal aortic aneurysms (Sun et al., 2007; Wei et al., 2010; Kovanen and Bot, 2017; Tomimori et al., 2019). Chymase has been shown to predominate over ACE activity in human heart, accounting for extremely high total Ang II formation in the human heart, as compared to other species (Balcells et al., 1997). Local formation of Ang II is increased in aged hearts, and chymase is primarily responsible for this increase (Froogh et al., 2017). Accordingly, chronic ACE inhibition did not repress Ang II levels in the cardiac interstitial fluid while combined chymase and ACE inhibition improved cardiac function, decreased adverse cardiac remodeling, and improved survival after myocardial infarction (Wei et al., 2010). In line with these findings, knockout of ACE resulted in revoked formation of Ang II in the circulation, while the formation of Ang II in the heart was not suppressed (Wei et al., 2002). Others have shown protective effects of chymase inhibition in various cardiac conditions like myocardial ischemia-reperfusion injury, inflammation after acute myocardial ischemia/reperfusion, cardiac fibrosis, cardiac function after left ventricular repair, in cardiac function and survival after myocardial infarction, and in preventing cardiac fibrosis and improving diastolic dysfunction in the progression of heart failure (Jin et al., 2003; Matsumoto et al., 2003; Kanemitsu et al., 2008; Oyamada et al., 2011; Maeda et al., 2012; Hooshdaran et al., 2017). Although, chymase inhibitors have been shown to function in animal models, these drugs have failed in early clinical testing, possibly due to differences in chymase genes and hydrolytic activity of chymase isoforms influencing the action of mast cells proteases (Ahmad and Ferrario, 2018). Important to note is also that the findings of chymase have largely been obtained in homogenized tissues, which means that the physiological relevance in humans can be difficult to assess. The challenge is to clarify the mechanisms of intra- and extracellular chymase activation. At present, several newly developed chymase inhibitors are evaluated in pre-clinical studies (Dell'Italia et al., 2018).
The ACE2 – Ang-(1–7) – Mas Axis
Ang-(1–7) is derived from Ang II through the influence of angiotensin ACE2, and exerts its effect via the G-protein-coupled Mas receptor (Santos et al., 2003). The axis of ACE2 – Ang-(1–7) – Mas opposes effects of the ACE – Ang II – Ang II type 1 receptor (AT1R) axis and has been proposed to have a protective effect (Ferreira et al., 2012), and increased Ang-(1–7) has been associated with a favourable phenotype, with attenuated inflammation in atherosclerotic plaques (Fraga-Silva et al., 2014). Ang-(1–7) induces vasodilatation via activation of NO, and decreases fibrosis, thereby enhancing the effects of ACE inhibitors on Ang II (Bader, 2013). Ang-(1–7) may mediate anti-inflammatory and anti-thrombotic effects via activation of NO and inhibition of ROS, derived from nicotinamide-adenine dinucleotide phosphate oxidase (Nox) (Sampaio et al., 2007; Gwathmey et al., 2010). Although animal experiments suggest the ACE2 – Ang-(1–7) – Mas axis to be an important counterregulatory arm within the RAAS (Santos et al., 2018), there are only few and conflicting data available in man. Gaidarov et al. found no evidence that Ang (1–7) interacts with or initiates signaling through individual expressed recombinant MAS1, but can potently antagonize AT1R signaling (Gaidarov et al., 2018). In addition, Ang-(1–7) has been shown to cause vasodilation in the forearm circulation in normotensive and hypertensive subjects (Sasaki et al., 2001) and to potentiate the vasodilatory effect of bradykinin, possibly through NO release, in forearm resistance vessels (Ueda et al., 2001); however others observed no vasodilatory effects by Ang-(1–7) in the human forearm (Wilsdorf et al., 2001).
The Angiotensin II Type 1 and 2 Receptors
Ang II primarily exerts its influence through the AT1R and AT2R. The expression of the AT2R is limited in adults, but is increased after vascular injury and after myocardial or renal tissue injury (Matavelli and Siragy, 2015). The main effects of Ang II by activation of AT1R include vasoconstriction by VSMC stimulation, sodium retention in the kidneys, aldosterone release, cell proliferation with vascular and cardiac hypertrophy and fibrosis, inflammation, oxidative stress, and increased activity of the inflammasome NLRP3, promoting inflammation (Marcus et al., 2013; Whaley-Connell et al., 2013; Wen et al., 2016). In that aspect, AT1R antagonists may reduce inflammation and myocardial fibrosis after acute myocardial infarction by signaling pathways of NF-κB and TGF-β1 (Song et al., 2014), and supress increases in myocardial mRNA expression of proinflammatory cytokines (IL-6, IL-1β), MCP-1 and matrix metalloproteinases-2 and -9 in chronic heart failure (Sukumaran et al., 2010). Also, the AT1R antagonist telmisartan reduced rat cardiac myofibroblast secretion of matrix metalloproteinases-9 during stimulation with Ang II and IL-1β, while an AT2R antagonist, had no effect (Okada et al., 2010). Clinical studies have confirmed that ACE inhibitors and AT1R blockade can modify cardiac remodeling (Malmqvist et al., 2001; Dell'Italia, 2011; Ferrario, 2016), and the ACE inhibitor perindopril reduced the levels of IL-1β in patients with stable coronary disease and in essential hypertension (Madej et al., 2009; Krysiak and Okopień, 2012), implicating that ACE inhibitors may attenuate inflammasome activity.
AT2R-mediated effects generally oppose those effects mediated by AT1R, and include vasodilatation and anti-inflammatory effects in VSMCs, but also antiproliferative effects in vessels and the myocardium (Qi et al., 2012). The selective AT2R agonist compound 21 reduces proliferation, inflammation, remodeling, and fibrosis (Wang et al., 2017; Sumners et al., 2019). The anti-inflammatory effects by compound 21 reduces expression of cytokines such as IL-1β and TGF-1β, suggesting a reduced effect of the inflammasome NLRP3 to prevent abdominal aortic aneurysm progression in rat (Lange et al., 2018). Also, experimental myocardial infarction in Wistar rats and levels of cardiac IL-1β, IL-6, and IL-2 were associated with antiapoptotic and anti-inflammatory mechanisms by compound 21 (Kaschina et al., 2008), and compound 21 prevented vascular inflammation in vitro and in vivo (Sampson et al., 2016).
Aldosterone is produced in the adrenal cortex and acts on sodium reabsorption in the kidney. Ang II stimulates aldosterone synthesis by activation of the adrenal AT1R (Rautureau et al., 2011); further common stimuli are high plasma levels of potassium and adrenocorticotropic hormone. Aldosterone is implicated in vascular inflammation, oxidative stress, fibrosis, remodeling and endothelial dysfunction, particularly in the presence of salt (Park and Schiffrin, 2002). Conversely, mineralocorticoid receptor blockers reduce the effects on vascular remodeling (Virdis et al., 2002). In VSMCs and in endothelial cells, aldosterone exerts its effects via mitogen-activated protein kinase (also known as extracellular signal regulated kinase) and cellular Src kinase (a non-receptor tyrosine kinase), and participates in epidermal growth factor receptor transactivation (Mazak et al., 2004; Nakano et al., 2005). There is cross-talk between aldosterone and Ang II in VSMCs (Min et al., 2007). Aldosterone induced increase in oxidative stress in VSMCs may have a negative impact on endothelial function through a reduction in NO bioavailability (Nakano et al., 2005). Vascular inflammation in endothelial cells is promoted by aldosterone induced expression of ICAM-1 and adhesion of leukocytes in a mineralocorticoid receptor dependent manner (Caprio et al., 2008).
Angiotensin II and Oxidative Stress
Angiotensin II Generates a Proinflammatory Phenotype
A cascade of intracellular signaling responses is initiated when Ang II binds to the AT1R to cause oxidative stress and reduce NO activity. Ang II is a powerful activator of vascular Nox, which induces the production of ROS (H₂O₂) from endothelial cells and VSMCs (Santillo et al., 2015) to act as a second messenger to stimulate multiple signaling molecules (Touyz et al., 2003; Nguyen Dinh Cat et al., 2013). Several isoforms of Nox have been identified (Lambeth, 2004). Vascular endothelial cells express Nox 1, 2, 4, and 5 (Damiano et al., 2013), while Nox 2 is present in VSMCs of resistance arteries (Lassègue and Clempus, 2003), and Nox 1 and 4 are the main isoforms in VSMCs of large arteries (Hilenski et al., 2004). The isoforms Nox 2 and 4 are widely expressed in cardiomyocytes (Byrne et al., 2003; Murray et al., 2013). Nox 1 may play a major role in Ang II mediated impairment of kidney function by increasing oxidative stress (Zimnol et al., 2020). Reactive oxygen species also increase intracellular calcium and activate NF-ҡB and activating protein-1. These molecules participate in migration and cell-growth, and in the expression of inflammatory genes and extracellular matrix modulation. Ang II also activates ras homolog gene family, member A, which is important in vascular contraction and growth (Loirand and Pacaud, 2010).
In man, systemic Ang II infusion increases circulating IL-6 in healthy subjects and patients with familial combined hyperlipidemia and familial hypercholesterolemia (Ekholm et al., 2009; Ekholm et al., 2015) (Figure 4). IL-6 may have reached even higher levels at local areas in vessels, which may represent a local increase of trans-signalling, and a proinflammatory effect (Rose-John, 2012). Thus, circulating Ang II may have proinflammatory effects also in man.
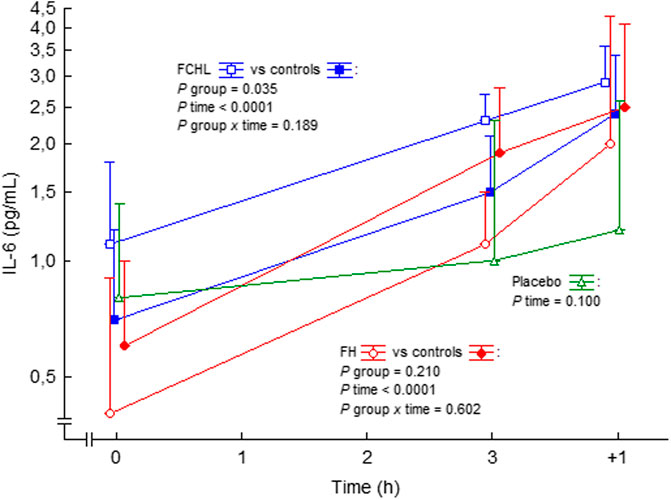
FIGURE 4. IL-6 in plasma before, during and after intravenous 3 h steady state angiotensin II infusion in man. Data are presented as median values and interquartile ranges. Statistical evaluation was made by repeated measures ANOVA. PAI-1, plasminogen activator inhibitor-1; FCHL, familial combined hyperlipidemia, and FH, familial hypercholesterolemia. FCHL, blue line and unfilled squares (n = 16); FCHL-control, blue line and filled squares (n = 16); FH, red line and unfilled circles (n = 16), and FH-control, red line and filled circles (n = 16). The effect of physiological saline infusion in placebo experiments, green line and unfilled triangles (n = 8), is also shown. Data from (Ekholm et al., 2009; Ekholm et al., 2015).
Angiotensin II and Tissue Factor
Angiotensin II Up-Regulates Tissue Factor
There is evidence that Ang II induces synthesis of TF, and that the promotor gene of TF is under control of the key redox-sensitive transcription factor NF-kB (Mackman, 1997; Felmeden et al., 2003; He et al., 2006; Li et al., 2009). Ang II binding to AT1R causes activation of NF-kB and upregulates TF synthesis in human monocytes (He et al., 2006), rat aortic endothelial cells (Nishimura et al., 1997), VSMCs (Taubman et al., 1993), and human glomerular endothelial cells (Felmeden et al., 2003). In addition, platelet activation by circulating Ang II may have an important contributing role in the generation of thrombin (Larsson et al., 2000). This is supported by findings in vitro and in vivo that Ang II stimulates formation of procoagulant microparticles from endothelial cells and mononuclear cells (Burger et al., 2011; Cordazzo et al., 2013).
The Effects of Blocking the RAAS on Tissue Factor
Some clinical effects of ACE inhibitor therapy may possibly be caused by interrupting Nox-derived ROS, and studies have shown antioxidant effects of AT1R blockers. Thus, direct inhibition of Nox and others ROS modulators has emerged as an attractive strategy to improve endothelial dysfunction and vascular damage in hypertensive patients (Cifuentes-Pagano et al., 2014). Blocking the RAAS with ACE inhibitors (idrapril, fosinopril, or captopril) downregulated TF synthesis in monocytes (Soejima et al., 1999; Napoleone et al., 2000), while the ACE inhibitor enalapril and the AT1R blocker losartan reduced TF activity and NF-kB translocation into the nuclei in human cytokine-activated endothelial cells (Nestoridi et al., 2008), and losartan inhibited TF activity (Napoleone et al., 2000), TF mRNA synthesis in rat aortic endothelial cells (Nishimura et al., 1997) and in human glomerular endothelial cells (Nestoridi et al., 2008). In man, AT1R blockade (losartan, irbesartan, candesartan) all reduced blood pressure and TF activity, as compared to placebo, indicating inhibition of TF synthesis (Koh et al., 2004). Moreover, the direct renin inhibiter aliskiren reduced TF synthesis and function in a comparable manner to the ACE inhibitor zofenopril and the AT1R blocker olmesartan in cytokine stimulated human umbilical vein endothelial cells (Del Fiorentino et al., 2010). These results reinforce other results (Nussberger et al., 2008), and suggest that RAAS activation, via cross-talk between an inflamed endothelium and coagulation, contributes to a procoagulant phenotype (Levi et al., 2012).
The Influence of Platelet Activation
Platelet activation in hypertension is important as it induces neurohumoral (sympatho-adrenal and RAAS) overactivity, endothelial dysfunction and dysfunctional NO biosynthesis, and platelet degranulation secondary to increased shear stress (Gkaliagkousi et al., 2010). Platelet activation, TF and hypertension may be linked by P-selectin. The P-selectin molecule, an integral membrane protein that is stored in platelet α-granules (Blair and Flaumenhaft, 2009) or in endothelial cells Weibel-Palade bodies, is not expressed at the cell surface in absence of inflammatory stimuli. However, during stimuli P-selectin is translocated to the plasma membrane, where it functions as an adhesion molecule, which mediates the interaction of platelets and endothelial cells with circulating leukocytes (Blair and Flaumenhaft, 2009). P-selectin also has a role as a signaling molecule, capable of inducing activation of NF-ҡB and to upregulate the expression of TF (Polgar et al., 2005). Thus, platelet activation in hypertension may per se, lead to increased P-selectin and, in turn, to increased TF activation.
The Effect of Blocking the RAAS on Platelet Activation
Both ACE inhibitors and AT1R blockers have impact on the RAAS. However, these drugs have important differences between them, as ACE inhibitors reduce Ang II formation, while AT1R blockers increase Ang II (Schwieler et al., 2013). However, little is known about the impact of these drugs on platelet activity. A review by Blann et al. concluded that blocking the actions of Ang II by ACE inhibitors or AT1R blockers should theoretically have direct antiplatelet effects, but the authors concluded that there was little agreement on this effect in the clinical setting, whichever drug was used (Blann et al., 2003). Thus, ACE inhibitors increased platelet reactivity by enhanced PAR-1 expression on platelets (ref). In addition. endogenous thrombin potential was reduced by treatment with ACE inhibitors but not by ATR1 blockers (Helten et al., 2020). Similar results were obtained with ACE inhibitors and ATR1 blockers in patients with acute coronary syndromes treated with dual antiplatelet therapy (Tscharre et al., 2020).
The RAAS and Thrombin Generation in Man
The Effects on Thrombin Generation by RAAS Blockade in Humans
Treatment with the ACE inhibitor ramipril decreased thrombin generation, as compared to placebo, in essential hypertension (Ekholm et al., 2002), (Figure 5). Thrombin-antithrombin (TAT) complex levels, a marker of thrombin generation in vivo, were attenuated after 6 weeks of treatment, and the effects were retained after 6 months of therapy. This attenuated thrombin generation by ramipril treatment could be of importance for the reduction in thromboembolic events seen during treatment with ramipril in patients at high cardiovascular risk (Yusuf et al., 2000). However, the antithrombotic effects of ramipril might have been due to the reduction in blood pressure per se.
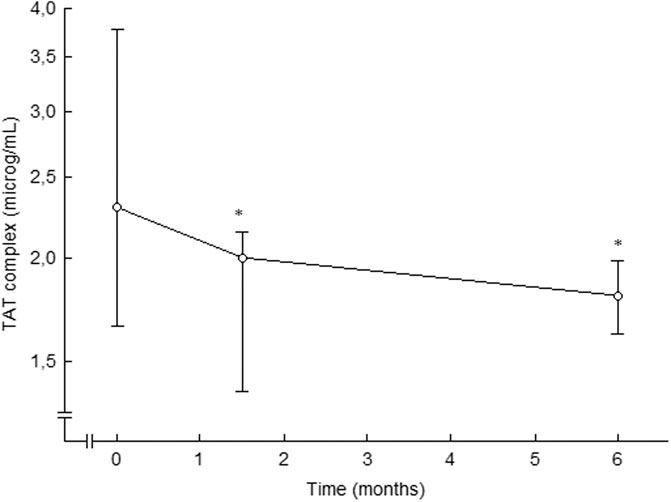
FIGURE 5. Plasma levels of TAT complex in patients with essential hypertension during placebo (0 months) and after 6 weeks and 6 months of ramipril therapy. Data are presented as median and interquartile ranges, n = 15. Statistical evaluation was made by non-parametric test, Wilcoxon sign test. Significant differences are given as; *p < 0.05, compared to placebo. TAT, thrombin-antithrombin. From (Ekholm et al., 2002).
This was further assessed comparing ramipril treatment with the alpha 1-adrenoceptor blocker doxazosin. Ramipril reduced thrombin generation beyond the effects on blood pressure reduction alone (Ekholm et al., 2018) (Figure 6). TAT complex was unaffected in the doxazosin group, the reductions in blood pressure between the groups were comparable, and there was no relation between changes in TAT complex and changes in blood pressure by treatment. Thus, the antithrombin effects of ramipril are not likely to be related to reduction in blood pressure. Our findings of reduced plasma levels of TAT complex following treatment with ramipril is in line with a decreased expression and activity of TF by ACE inhibition, resulting in an attenuated thrombin generation in plasma. This confirmes our previous findings (Ekholm et al., 2002) and offers one possible mechanism by which ACE inhibitors may reduce atherothrombotic complications in high-risk cardiovascular patients (Yusuf et al., 2000; Sleight et al., 2001). Possibly the reduced levels of TAT complex reflect an attenuated thrombin generation which, at least partly, is due to a reduced expression of TF on microparticles originating from endothelial cells and/or blood cells.
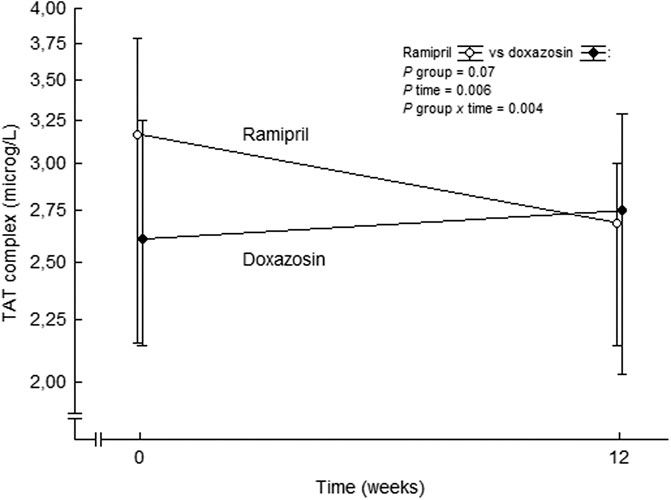
FIGURE 6. TAT complex levels at baseline and after 3 months of double-blind treatment with ramipril or doxazosin in patients with essential hypertension. Data are presented as mean values ± SD, n = 28 for the ramipril group and n = 22 for the doxazosin group. Statistical evaluation was made by multivariate analysis of variance. TAT, thrombin-antithrombin. From (Ekholm et al., 2018).
The Effects on Thrombin Generation During Angiotensin II Infusion in Humans
We have reported unchanged F1+2 and TAT complex values during systemic intravenous 3 h infusion of Ang II in subjects with familial combined hyperlipidemia, familial hypercholesterolemia and control subjects (Ekholm et al., 2009; Ekholm et al., 2015). This is in contrast to a previously reported increase in TAT complex and a tendency to increased F1+2 during short-time Ang II infusion in healthy males (Larsson et al., 2000). However, F1+2 exhibits a diurnal decrease during morning hours (Kapiotis et al., 1997; Ekholm et al., 2016) and these possible effects of diurnal variations of these markers were initially not taken into account by us (Ekholm et al., 2009; Ekholm et al., 2015). The absence of the expected diurnal decreases in F1+2 concentrations during morning hours, during stimulation by Ang II, may be considered as a relative increase in thrombin generation, similar to findings in healthy subjects (Larsson et al., 2000) and in familial combined hyperlipidemia patients (Ekholm et al., 2009). Indeed, in an analysis post hoc TAT complex actually increased in a similar way in controls and in familial combined hyperlipidemia during the ongoing Ang II infusion (Figure 7). Others have shown a circadian variability of TAT complex, with the highest levels at 8 am, and the lowest at 8 pm (Budkowska et al., 2019) with no variation during daytime (Jafri et al., 1992). Taken together, circulating Ang II seems to increase thrombin generation.
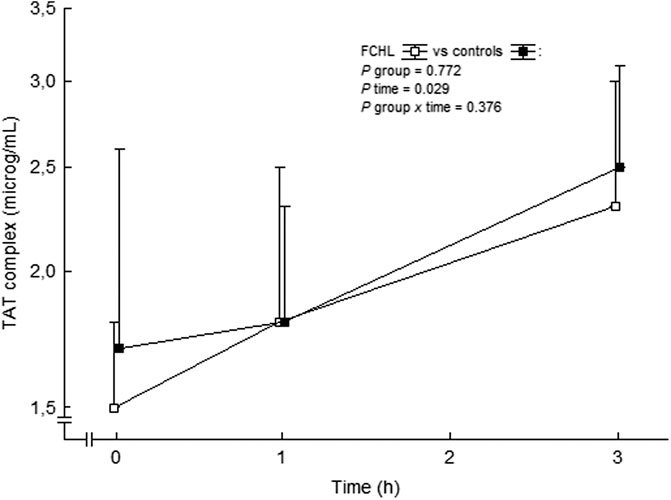
FIGURE 7. TAT complex in plasma before and during the ongoing angiotensin II infusion. Data are presented as median values and interquartile ranges. Statistical evaluation was made by repeated measures ANOVA. TAT, thrombin-antithrombin and FCHL, familial combined hyperlipidemia. FCHL □ (n = 16) and FCHL-control ■ (n = 16). From (Ekholm et al., 2009).
Inflammation can prime the coagulation system, and a proposed mechanism is TF expression by IL-6 (van der Poll et al., 1994). IL-6 increased during systemic Ang II infusion. Possibly Ang II may stimulate activation of TF, at least in part due to increased concentrations of IL-6. Platelet activation by circulating Ang II (Larsson et al., 2000) may have an important contributing role in the generation of thrombin.
Fibrinolysis and the RAAS
The Modulation of the Fibrinolytic System
The fibrinolytic system aims to dissolve and remove clots within the circulation. The zymogen plasminogen is released from the liver and is accumulated in fibrin-rich clots. Upon activation plasminogen is cleaved to plasmin by a variety of enzymes. The main regulator of fibrinolysis is tPA released from endothelial cells. The major inhibitor of tPA and urokinase-P is PAI-1. When PAI-1 binds to plasminogen activators, inactivation of these activators take place, and consequently the activity of the fibrinolysis is supressed.
The ratio between tPA and PAI-1 modulates the fibrinolytic activity (Vaughan, 2002). The immediate response during inflammatory stimuli is a transient increase in the secretion of tPA from Weibel-Palade bodies in endothelial cells (Huber et al., 2002). This increase in fibrinolytic activation is followed by a delayed suppression of tPA production and a sustained increase in PAI-1, resulting in a suppression of fibrinolytic activity. The major regulators of PAI-1 activity at inflammatory sites seem to be cytokines, such as IL-6, IL-1β, and TNF-α (van der Poll et al., 2001; Aso, 2007). CRP has also been shown to stimulate the expression of PAI-1 (Devaraj et al., 2003). Alpha granules in platelets also contain PAI-1 that can be released upon activation, which increases PAI-1 and thereby contributes to suppression of fibrinolysis in inflammatory states.
PAI-1 appears in human blood in three different forms, active, latent (representing an inactive form) and complexed to tPA or urokinase-PA. Concentration of PAI-1 exceeds tPA by a 4:1 ratio and PAI-1 binding to tPA or urokinase-PA occurs in a ratio of 1:1, thereby effectively limiting fibrinolysis. When PAI-1 levels are increased the tPA half-life is considerably shortened, and a negative correlation is present between PAI-1 and plasmin-antiplasmin (PAP) complexes. The tPA/PAI-1 complex is stable, and eliminated from the circulation by the liver. TNF-α is important in the induction of the fibrinolytic responses, while IL-6 have been shown to be most relevant for coagulation activation (Levi et al., 1997), but also enzymes like kallikrein, FXIa and FXIIa may convert plasminogen into plasmin. Plasmin acts by degrading fibrin into d-dimers. The main inhibitor of fibrinolysis and tPA is PAI-1, while α2-antiplasmin and α2-macroglobulin inhibits plasmin. High levels of PAI-1 and tPA have been shown to predict development of a first cardiovascular event (Thögersen et al., 1998). PAI-1 has a diurnal variation with its highest concentrations at 8 am and the lowest at 2 pm (Budkowska et al., 2019).
The Influence of the RAAS on the Fibrinolytic System
The RAAS may influence the fibrinolytic system since Ang II stimulates the production of PAI-1 by endothelial cells and VSMCs, and bradykinin (which is degraded by the ACE) stimulates the production of tPA (Brown and Vaughan, 2000). Ang II can increase protein activities of TF and PAI-1 (Nishimura et al., 1997). Ang II has also been observed to increase PAI-1 and tPA activator messenger RNA (van Leeuwen et al., 1994). Additional experiments have demonstrated that Ang IV increased PAI-1 expression, and the response exhibited a fast time and dose dependence (Kerins et al., 1995). Blocking the RAAS by ACE inhibition after myocardial infarction has been shown to improve the fibrinolytic balance (Boman et al., 2002). Ang II infusion increases the expression of messenger RNA PAI-1 in the rat (Nakamura et al., 2000).
The Effects on the Fibrinolytic System During Angiotensin II Infusion in Humans
In a small study with normotensive and hypertensive subjects, Ang II infusion resulted in an increase in PAI-1 antigen, whereas no changes occurred regarding tPA antigen (Ridker et al., 1993). In conflict, Ang II did not involve any changes in PAI-1 antigen or activity in healthy subjects in other studies (Larsson et al., 1999; Lottermoser et al., 2004). We observed no effects on PAI-1 activity during Ang II infusion during 3 h in control subjects, patients with familial combined hyperlipidemia, or with familial hypercholesterolemia (Ekholm et al., 2015; Ekholm et al., 2016), (Figure 8). Thus, Ang II may induce PAI-1 release in the long-term setting, possibly by a shift of the endothelial cells to a proinflammatory phenotype, but we could not find any short-term impact of Ang II on the concentrations of PAI-1. In addition, we observed that Ang II infusion also seems to induce a progressive increase in tPA activity in healthy volunteers (Ekholm et al., 2016). The response to Ang II stimulation in patients with chronic diseases such as cardiovascular disease or diabetes mellitus, which implies endothelial dysfunction, is not known.
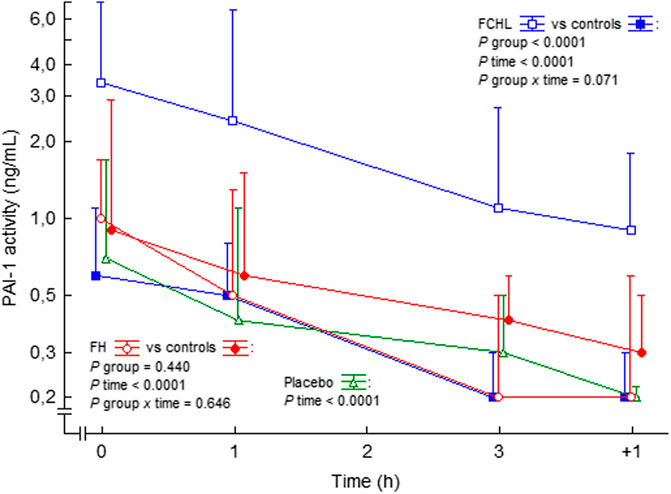
FIGURE 8. PAI-1 activity in plasma before, during and after intravenous 3 h steady state angiotensin II infusion in man. Data are presented as median values and interquartile ranges. Statistical evaluation was made by repeated measures ANOVA. PAI-1, plasminogen activator inhibitor-1; FCHL, familial combined hyperlipidemia; FH, familial hypercholesterolemia. FCHL, blue line and unfilled squares (n = 16); FCHL-control, blue line and filled squares (n = 16); FH, FH, red line and unfilled circles (n = 16), and FH-control, red line and filled circles (n = 16). The effect of physiological saline infusion in placebo experiments, green line and unfilled triangles (n = 8), is also shown. Data from (Ekholm et al., 2009; Ekholm et al., 2015; Ekholm et al., 2016).
The Interaction Between Angiotensin II and COVID-19
The ACE2 Receptor
The outbreak of the COVID-19 pandemic in 2019 affects individuals worldwide. The ACE2 receptor, together with the transmembrane protease serine 2, has a central role for the internalization of the virus (Liu et al., 2020a; Guzik et al., 2020). The expression of ACE2 is not limited to the respiratory system, being present also in other tissues like the gut, heart, kidney, as well as arterial and venous endothelial cells in all organs studied (Hamming et al., 2004). There is clear-cut evidence that endothelial cells are prone to acquire severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (Varga et al., 2020). Preclinical studies have shown that after binding of SARS-CoV-1 and -2 to its receptor, ACE2 activates RAAS leading to downregulation of the expression of ACE2, which in turn results in excessive production of Ang II (Imai et al., 2005; Huang et al., 2014; Wang et al., 2020). Therefore, ACE2 seems to play a dual role in COVID-19. Initially, it acts as a receptor for SARS-CoV-2 entry, then, in the context of SARS-CoV-2 infection, ACE2 is downregulated, which may increase Ang II (Wang et al., 2020).
SARS-CoV-2 Infections May Cause Inflammation and Vascular Thrombi
Recent data suggests that SARS-CoV-2 infections include vascular dysfunction, inflammation, and hypercoagulability, with involvement in multiple organs (Leisman et al., 2020). ACE2 is particularly highly expressed in pericytes, in addition to type II alveolar epithelial cells, and high expression of ACE2 in pericytes, including high levels in the heart, could lead to development of vascular dysfunction (Chen et al., 2020b; Shi et al., 2020). Vascular smooth muscle cells have both ACE2 receptor and the transmembrane protease serine 2, and lung tissue and other affected organs have revealed evidence of vascular inflammation along with vascular thrombi (Varga et al., 2020). Also, distal vasculitis with acro-ischaemic lesions have been observed at the distal aspects of toes and fingers in patients with COVID-19, and these cutaneous vasculitis lesions have been assessed as skin manifestation of SARS-CoV-2 infection (Fernandez-Nieto et al., 2020; Marzano et al., 2020; Zhang et al., 2020).
Cytokines are produced by immune cells like macrophages, dendritic cells, natural killer cells and the adaptive T and B lymphocytes. During inflammation activated macrophages will release cytokines, including IL-1β and IL-6, which stimulate the expression of adhesion molecules leading to endothelial activation and infiltration och inflammatory cells (Guzik et al., 2020). Activated endothelial cells release cytokines that may contribute to the development of microcirculatory lesions (Nencioni et al., 2009), and the dysfunctional endothelium will eventually become pro-adhesive and pro-coagulant (Boisramé-Helms et al., 2013). When SARS-CoV-2 is present in endothelial smooth muscle cells, this process is further enhanced.
COVID-19 Infection May Cause Detrimental RAAS Activation
Patients with COVID-19 are exposed to stress, which results in RAAS activation and subsequent increased Ang II levels, which are positively associated to viral load and lung injury (Huang et al., 2014; Liu et al., 2020b). Thus, the levels of Ang II could cause an inflammatory onset with accompanying increased oxidative stress. In some patients COVID-19 is accompanied by an excessive inflammatory reaction with the release of a large amount of pro-inflammatory cytokines in an event known as a cytokine storm. Studies have suggested that the cytokine storm correlated directly with lung injury, multi-organ failure, and unfavourable prognosis of severe COVID-19 (Chen et al., 2020a; Gao et al., 2020; Ruan et al., 2020). The cytokine IL-6 has a central role in this process, and increased markers of this cytokines predict poor outcome in patients with severe COVID-19 (Cao, 2020).
During severe COVID-19 infection patients suffer from thrombotic complications to a large extent, which may partly be explained by cross-talk between inflammation and coagulation (Verhamme and Hoylaerts, 2009). When generation of thrombin occurs, this prothrombotic state in turn has a spectrum of complicating effects with endothelial dysfunction and activation of platelets, which further contributes to a prothrombotic and proinflammatory state. Localized macrophages can release pro-coagulant factors such as plasminogen activators, and with the retreat of ACE2 and activation of Ang II, the production of downstream of PAI-I is stimulated (Nishimura et al., 1997). COVID-19 patients with acute respiratory failure exhibits hypercoagulability due to hyperfibrinogenemia (Spiezia et al., 2020). This combination further accelerates vascular inflammation, and enhances a prothrombotic state, which may predispose patient to microinfarcts within multiple organs and consequently multi-organ injury and failure. A recent post mortem report of patients with COVID-19 acute respiratory distress syndrome identified severe vascular injury, including alveolar microthrombi that were nine times more prevalent than found in post mortem studies of patients with influenza dito (Ackermann et al., 2020).
Blocking the RAAS or Not During COVID-19 Infection?
Drugs that block the RAAS may play a role in abrogating the inflammatory response, vasoconstriction and thrombotic complications that causes clinical deterioration in patients with COVID-19. We have shown that infusion of Ang II rapidly increases the levels of the cytokine IL-6 in humans (Ekholm et al., 2009; Ekholm et al., 2015), and stimulates thrombin generation. Conversely, ramipril decreased thrombin generation in hypertensive patients beyond the effects on blood pressure reduction alone (Ekholm et al., 2002; Ekholm et al., 2018). In this context, our findings would speak in favor of supressing the RAAS during COVID-19. In contrast, experimental studies have demonstrated that inhibition of the RAAS may result in a compensatory increase in tissue levels of ACE2 (Ferrario et al., 2005). Compensatory effects on ACE may lead to the RAAS being tipped towards the detrimental ACE – Ang II – AT1R axis and away from the protective ACE2 – Ang-(1–7) – Mas axis, leading to suggestions that these drugs may be detrimental in patients exposed to SARS-CoV-2 (Danser et al., 2020). However, there is no clear evidence that ACE inhibitors or AT1 receptor blockers lead to upregulation of ACE2 in human tissues (Danser et al., 2020).
A retrospective cohort study included 18.472 patients tested for COVID-19 found no association between ACE inhibitors or AT1R blockade use and a positive COVID-19 test (Mehta et al., 2020). A cohort study including 8.3 million people concluded that ACE inhibitors and AT1R blockers were associated with reduced risk of COVID-19 disease (Hippisley-Cox et al., 2020) and a meta-analysis that enrolled ten studies and 9.890 hypertensive patients strongly supported the recommendation to continue ACE inhibitors or AT1R blockade for all patients (Flacco et al., 2020). In a meta-analysis that included 25 observational studies neither ACE inhibitors nor AT1R blockers were associated with increased odds ratio for SARS-CoV-2 infection, admission to hospital, severe critical illness, admission to intensive care unit, or SARS-CoV-2 related death. In addition, the authors concluded that ACE inhibitors might be marginally protective regarding SARS-CoV-2 related death compared with AT1R blockade (Patoulias et al., 2020). More recently, a systematic review and meta-analysis of 31 cohort studies with outcome data for 87,951 patients, of whom 27% were on ACE inhibitor or AT1R blocker therapy, and three population based case control studies found no association between the use of RAAS blocking drugs and mortality, severe disease and no differential effect between ACE inhibitor or AT1R blocker therapy and outcome (Bavishi et al., 2021) However, current knowledge on this issue may be considered preliminary until confirmed in properly designed prospective randomized controlled studies. Such studies are ongoing.
Conclusion
The cause of the increased thrombin generation in hypertension and cardiovascular disease remains unclear. However, inflammatory stimuli can prime the coagulation system. Since inflammation participates in vascular remodeling and atherosclerosis, and Ang II has an important role for inflammation in the vessels, Ang II may contribute to vascular dysfunction in hypertension. One possible mechanism could be that Ang II may stimulate activation of TF. Platelet activation by circulating Ang II may also contribute to the generation of thrombin. Antihypertensive treatment with ACE inhibitor may reduce thrombin generation beyond the effects on blood pressure reduction alone. Thus, drugs blocking the RAAS may reduce atherothrombotic complications beyond their effects to reduce blood pressure. In addition, available preliminary experimental and clinical evidence suggests that blocking the RAAS may prevent complications in COVID-19, and blocking RAAS might even exert a potentially protective influence in the setting of COVID-19. Thus, current evidence does not favor the discontinuation of RAAS blocking drugs in patients with COVID-19 and hypertension or other cardiovascular comorbidities.
Author Contributions
All Authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
The research has been supported by grants from the Medical Council of Southeast Sweden (FORSS 806891), and the Swedish Heart-Lung Foundation (20050535 and 20130467).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackermann, M., Verleden, S. E., Kuehnel, M., Haverich, A., Welte, T., Laenger, F., et al. (2020). Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 383 (2), 120–128. doi:10.1056/NEJMoa2015432
Ahmad, S., and Ferrario, C. M. (2018). Chymase Inhibitors for the Treatment of Cardiac Diseases: a Patent Review (2010-2018). Expert Opin. Ther. Patents 28 (11), 755–764. doi:10.1080/13543776.2018.1531848
Alberelli, M. A., and De Candia, E. (2014). Functional Role of Protease Activated Receptors in Vascular Biology. Vasc. Pharmacol. 62 (2), 72–81. doi:10.1016/j.vph.2014.06.001
Antoniades, C., Bakogiannis, C., Tousoulis, D., Antonopoulos, A. S., and Stefanadis, C. (2009). The CD40/CD40 Ligand System. J. Am. Coll. Cardiol. 54 (8), 669–677. doi:10.1016/j.jacc.2009.03.076
Aso, Y. (2007). Plasminogen Activator Inhibitor (PAI)-1 in Vascular Inflammation and Thrombosis. Front. Biosci. 12, 2957–2966. doi:10.2741/2285
Bader, M. (2013). ACE2, Angiotensin-(1-7), and Mas: the Other Side of the coin. Pflugers Arch. - Eur. J. Physiol. 465 (1), 79–85. doi:10.1007/s00424-012-1120-0
Balasubramanian, V., Grabowski, E., Bini, A., and Nemerson, Y. (2002). Platelets, Circulating Tissue Factor, and Fibrin Colocalize in Ex Vivo Thrombi: Real-Time Fluorescence Images of Thrombus Formation and Propagation under Defined Flow Conditions. Blood 100 (8), 2787–2792. doi:10.1182/blood-2002-03-0902
Balcells, E., Meng, Q. C., Johnson, W. H., Oparil, S., and Dell’Italia, L. J. (1997). Angiotensin II Formation from ACE and Chymase in Human and Animal Hearts: Methods and Species Considerations. Am. J. Physiology-Heart Circulatory Physiol. 273 (4), H1769–H1774. doi:10.1152/ajpheart.1997.273.4.H1769
Bavishi, C., Whelton, P. K., Mancia, G., Corrao, G., and Messerli, F. H. (2021). Renin-angiotensin-system Inhibitors and All-Cause Mortality in Patients with COVID-19: a Systematic Review and Meta-Analysis of Observational Studies. J. Hypertens. 39 (4), 784–794. doi:10.1097/hjh.0000000000002784
Bester, J., and Pretorius, E. (2016). Effects of IL-1β, IL-6 and IL-8 on Erythrocytes, Platelets and Clot Viscoelasticity. Sci. Rep. 6 (1), 32188. doi:10.1038/srep32188
Blair, P., and Flaumenhaft, R. (2009). Platelet α-granules: Basic Biology and Clinical Correlates. Blood Rev. 23 (4), 177–189. doi:10.1016/j.blre.2009.04.001
Blann, A. D., Nadar, S., and Lip, G. Y. H. (2003). Pharmacological Modulation of Platelet Function in Hypertension. Hypertension 42 (1), 1–7. doi:10.1161/01.Hyp.0000077901.84467.E1
Bogdanov, V. Y., Balasubramanian, V., Hathcock, J., Vele, O., Lieb, M., and Nemerson, Y. (2003). Alternatively Spliced Human Tissue Factor: a Circulating, Soluble, Thrombogenic Protein. Nat. Med. 9 (4), 458–462. doi:10.1038/nm841
Boisramé-Helms, J., Kremer, H., Schini-Kerth, V., and Meziani, F. (2013). Endothelial Dysfunction in Sepsis. Curr. Vasc. Pharmacol. 11 (2), 150–160. doi:10.2174/1570161111311020005
Boman, K. O., Jansson, J. H., Nyhlén, K. A., and Nilsson, T. K. (2002). Improved Fibrinolysis after One Year of Treatment with Enalapril in Men and Women with Uncomplicated Myocardial Infarction. Thromb. Haemost. 87 (2), 311–316.
Bouchard, B. A., Mann, K. G., and Butenas, S. (2010). No Evidence for Tissue Factor on Platelets. Blood 116 (5), 854–855. doi:10.1182/blood-2010-05-285627
Brasier, A. R., Recinos, A., and Eledrisi, M. S. (2002). Vascular Inflammation and the Renin-Angiotensin System. Arterioscler. Thromb. Vasc. Biol. 22 (8), 1257–1266. doi:10.1161/01.atv.0000021412.56621.a2
Brass, L. F. (2003). Thrombin and Platelet Activation. Chest 124 (3 Suppl. l), 18s–25s. doi:10.1378/chest.124.3_suppl.18s
Brown, N. J., and Vaughan, D. E. (2000). Prothrombotic Effects of Angiotensin. Adv. Intern. Med. 45, 419–429.
Budkowska, M., Lebiecka, A., Marcinowska, Z., Woźniak, J., Jastrzębska, M., and Dołęgowska, B. (2019). The Circadian Rhythm of Selected Parameters of the Hemostasis System in Healthy People. Thromb. Res. 182, 79–88. doi:10.1016/j.thromres.2019.08.015
Burger, D., Montezano, A. C., Nishigaki, N., He, Y., Carter, A., and Touyz, R. M. (2011). Endothelial Microparticle Formation by Angiotensin II Is Mediated via Ang II Receptor Type I/NADPH Oxidase/Rho Kinase Pathways Targeted to Lipid Rafts. Arterioscler. Thromb. Vasc. Biol. 31 (8), 1898–1907. doi:10.1161/atvbaha.110.222703
Byrne, J. A., Grieve, D. J., Bendall, J. K., Li, J.-M., Gove, C., Lambeth, J. D., et al. (2003). Contrasting Roles of NADPH Oxidase Isoforms in Pressure-Overload Versus Angiotensin II–Induced Cardiac Hypertrophy. Circ. Res. 93 (9), 802–805. doi:10.1161/01.RES.0000099504.30207.F5
Calabró, P., Willerson, J. T., and Yeh, E. T. (2003). Inflammatory Cytokines Stimulated C-Reactive Protein Production by Human Coronary Artery Smooth Muscle Cells. Circulation 108 (16), 1930–1932. doi:10.1161/01.Cir.0000096055.62724.C5
Camera, M., Brambilla, M., Toschi, V., and Tremoli, E. (2010). Tissue Factor Expression on Platelets Is a Dynamic Event. Blood 116 (23), 5076–5077. doi:10.1182/blood-2010-09-307306
Cao, X. (2020). COVID-19: Immunopathology and its Implications for Therapy. Nat. Rev. Immunol. 20 (5), 269–270. doi:10.1038/s41577-020-0308-3
Caprio, M., Newfell, B. G., la Sala, A., Baur, W., Fabbri, A., Rosano, G., et al. (2008). Functional Mineralocorticoid Receptors in Human Vascular Endothelial Cells Regulate Intercellular Adhesion Molecule-1 Expression and Promote Leukocyte Adhesion. Circ. Res. 102 (11), 1359–1367. doi:10.1161/circresaha.108.174235
Castellon, X., and Bogdanova, V. (2016). Chronic Inflammatory Diseases and Endothelial Dysfunction. Aging Dis. 7 (1), 81–89. doi:10.14336/ad.2015.0803
Ceciliani, F., Giordano, A., and Spagnolo, V. (2002). The Systemic Reaction during Inflammation: the Acute-phase Proteins. Protein Pept. Lett. 9 (3), 211–223. doi:10.2174/0929866023408779
Chen, G., Wu, D., Guo, W., Cao, Y., Huang, D., Wang, H., et al. (2020a). Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J. Clin. Invest. 130 (5), 2620–2629. doi:10.1172/jci137244
Chen, N., Zhou, M., Dong, X., Qu, J., Gong, F., Han, Y., et al. (2020b). Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: a Descriptive Study. The Lancet 395 (10223), 507–513. doi:10.1016/s0140-6736(20)30211-7
Cifuentes-Pagano, E., Meijles, D. N., and Pagano, P. J. (2014). The Quest for Selective Nox Inhibitors and Therapeutics: Challenges, Triumphs and Pitfalls. Antioxid. Redox Signaling 20 (17), 2741–2754. doi:10.1089/ars.2013.5620
Cimmino, G., Cirillo, P., Conte, S., Pellegrino, G., Barra, G., Maresca, L., et al. (2019). Oxidized Low-Density Lipoproteins Induce Tissue Factor Expression in T-Lymphocytes via Activation of Lectin-like Oxidized Low-Density Lipoprotein Receptor-1. Cardiovasc. Res. 116 (6), 1125–1135. doi:10.1093/cvr/cvz230
Cimmino, G., Cirillo, P., Ragni, M., Conte, S., Uccello, G., and Golino, P. (2015). Reactive Oxygen Species Induce a Procoagulant State in Endothelial Cells by Inhibiting Tissue Factor Pathway Inhibitor. J. Thromb. Thrombolysis 40 (2), 186–192. doi:10.1007/s11239-015-1199-1
Cimmino, G., D’Amico, C., Vaccaro, V., D’Anna, M., and Golino, P. (2011). The Missing Link between Atherosclerosis, Inflammation and Thrombosis: Is it Tissue Factor? Expert Rev. Cardiovasc. Ther. 9 (4), 517–523. doi:10.1586/erc.11.40
Cirillo, P., Calì, G., Golino, P., Calabrò, P., Forte, L., Rosa, S. D., et al. (2004). Tissue Factor Binding of Activated Factor VII Triggers Smooth Muscle Cell Proliferation via Extracellular Signal-Regulated Kinase Activation. Circulation 109 (23), 2911–2916. doi:10.1161/01.cir.0000129312.43547.08
Cirillo, P., Golino, P., Calabro, P., Cali, G., Ragni, M., Derosa, S., et al. (2005). C-reactive Protein Induces Tissue Factor Expression and Promotes Smooth Muscle and Endothelial Cell Proliferation. Cardiovasc. Res. 68 (1), 47–55. doi:10.1016/j.cardiores.2005.05.010
Cordazzo, C., Neri, T., Petrini, S., Lombardi, S., Balìa, C., Cianchetti, S., et al. (2013). Angiotensin II Induces the Generation of Procoagulant Microparticles by Human Mononuclear Cells via an Angiotensin Type 2 Receptor-Mediated Pathway. Thromb. Res. 131 (4), e168–e174. doi:10.1016/j.thromres.2013.01.019
Coughlin, S. R. (2000). Thrombin Signalling and Protease-Activated Receptors. Nature 407 (6801), 258–264. doi:10.1038/35025229
Damiano, S., Petrozziello, T., Ucci, V., Amente, S., Santillo, M., and Mondola, P. (2013). Cu-Zn Superoxide Dismutase Activates Muscarinic Acetylcholine M1 Receptor Pathway in Neuroblastoma Cells. Mol. Cell Neurosci. 52, 31–37. doi:10.1016/j.mcn.2012.11.001
Danser, A. H. J., Epstein, M., and Batlle, D. (2020). Renin-Angiotensin System Blockers and the COVID-19 Pandemic. Hypertension 75 (6), 1382–1385. doi:10.1161/hypertensionaha.120.15082
Daub, S., Lutgens, E., Münzel, T., and Daiber, A. (2020). CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease-The Pros and Cons for Cardioprotection. Int. J. Mol. Sci. 21 (22), 8533. doi:10.3390/ijms21228533
Davidson, S. J. (2013). “Inflammation and Acute Phase Proteins in Haemostasis,” in Acute Phase Proteins. Editor S. Janciauskiene (IntechOpen). doi:10.5772/20408
Davizon, P., and López, J. A. (2009). Microparticles and Thrombotic Disease. Curr. Opin. Hematol. 16 (5), 334–341. doi:10.1097/MOH.0b013e32832ea49c
de Jonge, E., Friederich, P. W., Vlasuk, G. P., Rote, W. E., Vroom, M. B., Levi, M., et al. (2003). Activation of Coagulation by Administration of Recombinant Factor VIIa Elicits Interleukin 6 (IL-6) and IL-8 Release in Healthy Human Subjects. Clin. Vaccin. Immunol 10 (3), 495–497. doi:10.1128/cdli.10.3.495-497.2003
De Mello, W. C., and Frohlich, E. D. (2011). On the Local Cardiac Renin Angiotensin System. Basic and Clinical Implications. Peptides 32 (8), 1774–1779. doi:10.1016/j.peptides.2011.06.018
Del Fiorentino, A., Cianchetti, S., Celi, A., and Pedrinelli, R. (2010). Aliskiren, a Renin Inhibitor, Downregulates TNF-α-Induced Tissue Factor Expression in HUVECS. J. Renin Angiotensin Aldosterone Syst. 11 (4), 243–247. doi:10.1177/1470320310379449
Dell'Italia, L. J., Collawn, J. F., and Ferrario, C. M. (2018). Multifunctional Role of Chymase in Acute and Chronic Tissue Injury and Remodeling. Circ. Res. 122 (2), 319–336. doi:10.1161/circresaha.117.310978
Dell'Italia, L. J. (2011). Translational success Stories: Angiotensin Receptor 1 Antagonists in Heart Failure. Circ. Res. 109 (4), 437–452. doi:10.1161/circresaha.110.238550
Devaraj, S., Xu, D. Y., and Jialal, I. (2003). C-Reactive Protein Increases Plasminogen Activator Inhibitor-1 Expression and Activity in Human Aortic Endothelial Cells. Circulation 107 (3), 398–404. doi:10.1161/01.cir.0000052617.91920.fd
Di Angelantonio, E., Di Angelantonio, E., Gao, P., Pennells, L., Kaptoge, S., Caslake, M., et al. (2012). Lipid-related Markers and Cardiovascular Disease Prediction. Jama 307 (23), 2499–2506. doi:10.1001/jama.2012.6571
Dickson, M. E., and Sigmund, C. D. (2006). Genetic Basis of Hypertension. Hypertension 48 (1), 14–20. doi:10.1161/01.Hyp.0000227932.13687.60
Dinarello, C. A. (2018). Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 281 (1), 8–27. doi:10.1111/imr.12621
Duvernay, M. T., Temple, K. J., Maeng, J. G., Blobaum, A. L., Stauffer, S. R., Lindsley, C. W., et al. (2017). Contributions of Protease-Activated Receptors PAR1 and PAR4 to Thrombin-Induced GPIIbIIIa Activation in Human Platelets. Mol. Pharmacol. 91 (1), 39–47. doi:10.1124/mol.116.106666
Egorina, E. M., Sovershaev, M. A., Olsen, J. O., and Østerud, B. (2008). Granulocytes Do Not Express but Acquire Monocyte-Derived Tissue Factor in Whole Blood: Evidence for a Direct Transfer. Blood 111 (3), 1208–1216. doi:10.1182/blood-2007-08-107698
Ekholm, M., Jekell, A., Wallén, N. H., Gigante, B., and Kahan, T. (2018). Effects of Angiotensin-Converting Enzyme Inhibition and Alpha 1-Adrenergic Receptor Blockade on Inflammation and Hemostasis in Human Hypertension. J. Cardiovasc. Pharmacol. 71 (4), 240–247. doi:10.1097/fjc.0000000000000565
Ekholm, M., Kahan, T., Jörneskog, G., Brinck, J., and Wallén, N. H. (2015). Haemostatic and Inflammatory Alterations in Familial Hypercholesterolaemia, and the Impact of Angiotensin II Infusion. J. Renin Angiotensin Aldosterone Syst. 16 (2), 328–338. doi:10.1177/1470320315575848
Ekholm, M., Kahan, T., Jörneskog, G., Bröijersen, A., and Wallén, N. H. (2009). Angiotensin II Infusion in Man Is Proinflammatory but Has No Short-Term Effects on Thrombin Generation In Vivo. Thromb. Res. 124 (1), 110–115. doi:10.1016/j.thromres.2008.12.040
Ekholm, M., Kahan, T., Jörneskog, G., Bröijersén, A., and Wallén, N. H. (2016). Infusion of Angiotensin II Increases Fibrinolysis in Healthy Individuals but Not in Patients with Familial Combined Hyperlipidemia. Blood Coagul. Fibrinolysis 27 (1), 113–116. doi:10.1097/mbc.0000000000000393
Ekholm, M., Wallén, N. H., Johnsson, H., Eliasson, K., and Kahan, T. (2002). Long-term Angiotensin-Converting Enzyme Inhibition with Ramipril Reduces Thrombin Generation in Human Hypertension. Clin. Sci. (Lond) 103 (2), 151–155. doi:10.1042/cs1030151
Esmon, C. T. (2003). The Protein C Pathway. Chest 124 (3 Suppl. l), 26s–32s. doi:10.1378/chest.124.3_suppl.26s
Felmeden, D. C., Spencer, C. G. C., Chung, N. A. Y., Belgore, F. M., Blann, A. D., Beevers, D. G., et al. (2003). Relation of Thrombogenesis in Systemic Hypertension to Angiogenesis and Endothelial Damage/dysfunction (A Substudy of the Anglo-Scandinavian Cardiac Outcomes Trial [ASCOT]). Am. J. Cardiol. 92 (4), 400–405. doi:10.1016/s0002-9149(03)00657-x
Fernandez-Nieto, D., Jimenez-Cauhe, J., Suarez-Valle, A., Moreno-Arrones, O. M., Saceda-Corralo, D., Arana-Raja, A., et al. (2020). Characterization of Acute Acral Skin Lesions in Nonhospitalized Patients: A Case Series of 132 Patients during the COVID-19 Outbreak. J. Am. Acad. Dermatol. 83, e61–e63. doi:10.1016/j.jaad.2020.04.093
Ferrario, C. M. (2016). Cardiac Remodelling and RAS Inhibition. Ther. Adv. Cardiovasc. Dis. 10 (3), 162–171. doi:10.1177/1753944716642677
Ferrario, C. M., Jessup, J., Chappell, M. C., Averill, D. B., Brosnihan, K. B., Tallant, E. A., et al. (2005). Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 111 (20), 2605–2610. doi:10.1161/circulationaha.104.510461
Ferreira, A. J., Bader, M., and Santos, R. A. (2012). Therapeutic Targeting of the Angiotensin-Converting Enzyme 2/Angiotensin-(1-7)/Mas cascade in the Renin-Angiotensin System: a Patent Review. Expert Opin. Ther. Patents 22 (5), 567–574. doi:10.1517/13543776.2012.682572
Flacco, M. E., Acuti Martellucci, C., Bravi, F., Parruti, G., Cappadona, R., Mascitelli, A., et al. (2020). Treatment with ACE Inhibitors or ARBs and Risk of Severe/lethal COVID-19: a Meta-Analysis. Heart 106 (19), 1519–1524. doi:10.1136/heartjnl-2020-317336
Fraga-Silva, R. A., Savergnini, S. Q., Montecucco, F., Nencioni, A., Caffa, I., Soncini, D., et al. (2014). Treatment with Angiotensin-(1-7) Reduces Inflammation in Carotid Atherosclerotic Plaques. Thromb. Haemost. 111 (4), 736–747. doi:10.1160/th13-06-0448
Froogh, G., Pinto, J. T., Le, Y., Kandhi, S., Aleligne, Y., Huang, A., et al. (2017). Chymase-dependent Production of Angiotensin II: an Old Enzyme in Old Hearts. Am. J. Physiology-Heart Circulatory Physiol. 312 (2), H223–H231. doi:10.1152/ajpheart.00534.2016
Fu, Z., Thorpe, M., Alemayehu, R., Roy, A., Kervinen, J., de Garavilla, L., et al. (2017). Highly Selective Cleavage of Cytokines and Chemokines by the Human Mast Cell Chymase and Neutrophil Cathepsin G. J. Immunol. 198 (4), 1474–1483. doi:10.4049/jimmunol.1601223
Gaidarov, I., Adams, J., Frazer, J., Anthony, T., Chen, X., Gatlin, J., et al. (2018). Angiotensin (1-7) Does Not Interact Directly with MAS1, but Can Potently Antagonize Signaling from the AT1 Receptor. Cell Signal. 50, 9–24. doi:10.1016/j.cellsig.2018.06.007
Galli, S. J., Nakae, S., and Tsai, M. (2005). Mast Cells in the Development of Adaptive Immune Responses. Nat. Immunol. 6 (2), 135–142. doi:10.1038/ni1158
Gao, Y., Li, T., Han, M., Li, X., Wu, D., Xu, Y., et al. (2020). Diagnostic Utility of Clinical Laboratory Data Determinations for Patients with the Severe COVID‐19. J. Med. Virol. 92 (7), 791–796. doi:10.1002/jmv.25770
Gawaz, M., Page, S., Massberg, S., Nothdurfter, C., Weber, M., Fisher, C., et al. (2002). Transient Platelet Interaction Induces MCP-1 Production by Endothelial Cells via I Kappa B Kinase Complex Activation. Thromb. Haemost. 88 (2), 307–314.
Gawaz, M., Brand, K., Dickfeld, T., Pogatsa-Murray, G., Page, S., Bogner, C., et al. (2000). Platelets Induce Alterations of Chemotactic and Adhesive Properties of Endothelial Cells Mediated through an Interleukin-1-dependent Mechanism. Implications for Atherogenesis. Atherosclerosis 148 (1), 75–85. doi:10.1016/s0021-9150(99)00241-5
Gawaz, M., Langer, H., and May, A. E. (2005). Platelets in Inflammation and Atherogenesis. J. Clin. Invest. 115 (12), 3378–3384. doi:10.1172/jci27196
Gkaliagkousi, E., Passacquale, G., Douma, S., Zamboulis, C., and Ferro, A. (2010). Platelet Activation in Essential Hypertension: Implications for Antiplatelet Treatment. Am. J. Hypertens. 23 (3), 229–236. doi:10.1038/ajh.2009.247
Grover, S. P., and Mackman, N. (2018). Tissue Factor. Arterioscler Thromb. Vasc. Biol. 38 (4), 709–725. doi:10.1161/atvbaha.117.309846
Grumbach, I., Chen, W., Mertens, S., and Harrison, D. (2005). A Negative Feedback Mechanism Involving Nitric Oxide and Nuclear Factor Kappa-B Modulates Endothelial Nitric Oxide Synthase Transcription. J. Mol. Cell Cardiol. 39 (4), 595–603. doi:10.1016/j.yjmcc.2005.06.012
Gudmundsdóttir, I. J., Megson, I. L., Kell, J. S., Ludlam, C. A., Fox, K. A., Webb, D. J., et al. (2006). Direct Vascular Effects of Protease-Activated Receptor Type 1 Agonism In Vivo in Humans. Circulation 114 (15), 1625–1632. doi:10.1161/circulationaha.106.638478
Gullestad, L., Ueland, T., Vinge, L. E., Finsen, A., Yndestad, A., and Aukrust, P. (2012). Inflammatory Cytokines in Heart Failure: Mediators and Markers. Cardiology 122 (1), 23–35. doi:10.1159/000338166
Guzik, T. J., Mohiddin, S. A., Dimarco, A., Patel, V., Savvatis, K., Marelli-Berg, F. M., et al. (2020). COVID-19 and the Cardiovascular System: Implications for Risk Assessment, Diagnosis, and Treatment Options. Cardiovasc. Res. 116, 1666–1687. doi:10.1093/cvr/cvaa106
Gwathmey, T. M., Pendergrass, K. D., Reid, S. D., Rose, J. C., Diz, D. I., and Chappell, M. C. (2010). Angiotensin-(1-7)-angiotensin-converting Enzyme 2 Attenuates Reactive Oxygen Species Formation to Angiotensin II within the Cell Nucleus. Hypertension 55 (1), 166–171. doi:10.1161/hypertensionaha.109.141622
Hamming, I., Timens, W., Bulthuis, M., Lely, A., Navis, G., and van Goor, H. (2004). Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 203 (2), 631–637. doi:10.1002/path.1570
He, M., He, X., Xie, Q., Chen, F., and He, S. (2006). Angiotensin II Induces the Expression of Tissue Factor and its Mechanism in Human Monocytes. Thromb. Res. 117 (5), 579–590. doi:10.1016/j.thromres.2005.04.033
Helten, C., Mourikis, P., Dannenberg, L., M’Pembele, R., Trojovsky, K., Ayhan, A., et al. (2020). A Novel Mechanism of ACE Inhibition-Associated Enhanced Platelet Reactivity: Disproof of the ARB-MI Paradox? Eur. J. Clin. Pharmacol. 76 (9), 1245–1251. doi:10.1007/s00228-020-02915-y
Hilenski, L. L., Clempus, R. E., Quinn, M. T., Lambeth, J. D., and Griendling, K. K. (2004). Distinct Subcellular Localizations of Nox1 and Nox4 in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 24 (4), 677–683. doi:10.1161/01.ATV.0000112024.13727.2c
Hippisley-Cox, J., Young, D., Coupland, C., Channon, K. M., Tan, P. S., Harrison, D. A., et al. (2020). Risk of Severe COVID-19 Disease with ACE Inhibitors and Angiotensin Receptor Blockers: Cohort Study Including 8.3 Million People. Heart 106 (19), 1503–1511. doi:10.1136/heartjnl-2020-317393
Hirano, K. (2007). The Roles of Proteinase-Activated Receptors in the Vascular Physiology and Pathophysiology. Arterioscler. Thromb. Vasc. Biol. 27 (1), 27–36. doi:10.1161/01.ATV.0000251995.73307.2d
Hooshdaran, B., Kolpakov, M. A., Guo, X., Miller, S. A., Wang, T., Tilley, D. G., et al. (2017). Dual Inhibition of Cathepsin G and Chymase Reduces Myocyte Death and Improves Cardiac Remodeling after Myocardial Ischemia Reperfusion Injury. Basic Res. Cardiol. 112 (6), 62. doi:10.1007/s00395-017-0652-z
Huang, F., Guo, J., Zou, Z., Liu, J., Cao, B., Zhang, S., et al. (2014). Angiotensin II Plasma Levels Are Linked to Disease Severity and Predict Fatal Outcomes in H7N9-Infected Patients. Nat. Commun. 5, 3595. doi:10.1038/ncomms4595
Huber, D., Cramer, E. M., Kaufmann, J. E., Meda, P., Massé, J.-M., Kruithof, E. K. O., et al. (2002). Tissue-type Plasminogen Activator (t-PA) Is Stored in Weibel-Palade Bodies in Human Endothelial Cells Both In Vitro and In Vivo. Blood 99 (10), 3637–3645. doi:10.1182/blood.v99.10.3637
Imai, Y., Kuba, K., Rao, S., Huan, Y., Guo, F., Guan, B., et al. (2005). Angiotensin-converting Enzyme 2 Protects from Severe Acute Lung Failure. Nature 436 (7047), 112–116. doi:10.1038/nature03712
Jafri, S. M., VanRollins, M., Ozawa, T., Mammen, E. F., Goldberg, A. D., and Goldstein, S. (1992). Circadian Variation in Platelet Function in Healthy Volunteers. Am. J. Cardiol. 69 (9), 951–954. doi:10.1016/0002-9149(92)90799-5
Jin, D., Takai, S., Yamada, M., Sakaguchi, M., Kamoshita, K., Ishida, K., et al. (2003). Impact of Chymase Inhibitor on Cardiac Function and Survival after Myocardial Infarction. Cardiovasc. Res. 60 (2), 413–420. doi:10.1016/s0008-6363(03)00535-2
Kälsch, T., Elmas, E., Nguyen, X. D., Suvajac, N., Klüter, H., Borggrefe, M., et al. (2007). Endotoxin-induced Effects on Platelets and Monocytes in an In Vivo Model of Inflammation. Basic Res. Cardiol. 102 (5), 460–466. doi:10.1007/s00395-007-0667-y
Kanemitsu, H., Takai, S., Tsuneyoshi, H., Yoshikawa, E., Nishina, T., Miyazaki, M., et al. (2008). Chronic Chymase Inhibition Preserves Cardiac Function after Left Ventricular Repair in Rats. Eur. J. Cardio-Thoracic Surg. 33 (1), 25–31. doi:10.1016/j.ejcts.2007.09.040
Kapiotis, S., Jilma, B., Quehenberger, P., Ruzicka, K., Handler, S., and Speiser, W. (1997). Morning Hypercoagulability and Hypofibrinolysis. Circulation 96 (1), 19–21. doi:10.1161/01.cir.96.1.19
Kaptoge, S., Kaptoge, S., Di Angelantonio, E., Pennells, L., Wood, A. M., White, I. R., et al. (2012). C-reactive Protein, Fibrinogen, and Cardiovascular Disease Prediction. N. Engl. J. Med. 367 (14), 1310–1320. doi:10.1056/NEJMoa1107477
Kaschina, E., Grzesiak, A., Li, J., Foryst-Ludwig, A., Timm, M., Rompe, F., et al. (2008). Angiotensin II Type 2 Receptor Stimulation. Circulation 118 (24), 2523–2532. doi:10.1161/circulationaha.108.784868
Kerins, D. M., Hao, Q., and Vaughan, D. E. (1995). Angiotensin Induction of PAI-1 Expression in Endothelial Cells Is Mediated by the Hexapeptide Angiotensin IV. J. Clin. Invest. 96 (5), 2515–2520. doi:10.1172/jci118312
Koh, K. K., Chung, W.-J., Ahn, J. Y., Han, S. H., Kang, W. C., Seo, Y.-H., et al. (2004). Angiotensin II Type 1 Receptor Blockers Reduce Tissue Factor Activity and Plasminogen Activator Inhibitor Type-1 Antigen in Hypertensive Patients: a Randomized, Double-Blind, Placebo-Controlled Study. Atherosclerosis 177 (1), 155–160. doi:10.1016/j.atherosclerosis.2004.07.008
Kovanen, P. T., and Bot, I. (2017). Mast Cells in Atherosclerotic Cardiovascular Disease - Activators and Actions. Eur. J. Pharmacol. 816, 37–46. doi:10.1016/j.ejphar.2017.10.013
Krysiak, R., and Okopień, B. (2012). Different Effects of Perindopril and Enalapril on Monocyte Cytokine Release in Coronary Artery Disease Patients with normal Blood Pressure. Pharmacol. Rep. 64 (6), 1466–1475. doi:10.1016/s1734-1140(12)70944-1
Krystel-Whittemore, M., Dileepan, K. N., and Wood, J. G. (2016). Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 6 (620), 1–12. doi:10.3389/fimmu.2015.00620
Kumar, R., Thomas, C. M., Yong, Q. C., Chen, W., and Baker, K. M. (2012). The Intracrine Renin-Angiotensin System. Clin. Sci. (Lond) 123 (5), 273–284. doi:10.1042/cs20120089
Lambeth, J. D. (2004). NOX Enzymes and the Biology of Reactive Oxygen. Nat. Rev. Immunol. 4 (3), 181–189. doi:10.1038/nri1312
Lange, C., Sommerfeld, M., Namsolleck, P., Kintscher, U., Unger, T., and Kaschina, E. (2018). AT 2 R (Angiotensin AT2 Receptor) Agonist, Compound 21, Prevents Abdominal Aortic Aneurysm Progression in the Rat. Hypertension 72 (3), e20–e29. doi:10.1161/hypertensionaha.118.11168
Larsson, P. T., Schwieler, J. H., Wallen, N. H., and Hjemdahl, P. (1999). Acute Effects of Angiotensin II on Fibrinolysis in Healthy Volunteers. Blood Coagul. Fibrinolysis 10 (1), 19–24. doi:10.1097/00001721-199901000-00003
Larsson, P. T., Schwieler, J. H., and Wallén, N. H. (2000). Platelet Activation during Angiotensin II Infusion in Healthy Volunteers. Blood Coagul. Fibrinolysis 11 (1), 61–69. doi:10.1097/00001721-200011010-00007
Lassègue, B., and Clempus, R. E. (2003). Vascular NAD(P)H Oxidases: Specific Features, Expression, and Regulation. Am. J. Physiology-Regulatory, Integr. Comp. Physiol. 285 (2), R277–R297. doi:10.1152/ajpregu.00758.2002
Leal, J., Luengo-Fernández, R., Gray, A., Petersen, S., and Rayner, M. (2006). Economic burden of Cardiovascular Diseases in the Enlarged European Union. Eur. Heart J. 27 (13), 1610–1619. doi:10.1093/eurheartj/ehi733
Leisman, D. E., Deutschman, C. S., and Legrand, M. (2020). Facing COVID-19 in the ICU: Vascular Dysfunction, Thrombosis, and Dysregulated Inflammation. Intensive Care Med. 46, 1105–1108. doi:10.1007/s00134-020-06059-6
Levi, M., van der Poll, T., ten Cate, H., and van Deventer, S. J. (1997). The Cytokine-Mediated Imbalance between Coagulant and Anticoagulant Mechanisms in Sepsis and Endotoxaemia. Eur. J. Clin. Invest. 27 (1), 3–9. doi:10.1046/j.1365-2362.1997.570614.x
Levi, M., van der Poll, T., and Schultz, M. (2012). Infection and Inflammation as Risk Factors for Thrombosis and Atherosclerosis. Semin. Thromb. Hemost. 38 (5), 506–514. doi:10.1055/s-0032-1305782
Li, Y.-D., Ye, B.-Q., Zheng, S.-X., Wang, J.-T., Wang, J.-G., Chen, M., et al. (2009). NF-κB Transcription Factor P50 Critically Regulates Tissue Factor in Deep Vein Thrombosis. J. Biol. Chem. 284 (7), 4473–4483. doi:10.1074/jbc.M806010200
Li, Y., Song, Y.-H., Mohler, J., and Delafontaine, P. (2006). ANG II Induces Apoptosis of Human Vascular Smooth Muscle via Extrinsic Pathway Involving Inhibition of Akt Phosphorylation and Increased FasL Expression. Am. J. Physiology-Heart Circulatory Physiol. 290 (5), H2116–H2123. doi:10.1152/ajpheart.00551.2005
Liu, P. P., Blet, A., Smyth, D., and Li, H. (2020a). The Science Underlying COVID-19. Circulation 142 (1), 68–78. doi:10.1161/circulationaha.120.047549
Liu, Y., Yang, Y., Zhang, C., Huang, F., Wang, F., Yuan, J., et al. (2020b). Clinical and Biochemical Indexes from 2019-nCoV Infected Patients Linked to Viral Loads and Lung Injury. Sci. China Life Sci. 63 (3), 364–374. doi:10.1007/s11427-020-1643-8
Loirand, G., and Pacaud, P. (2010). The Role of Rho Protein Signaling in Hypertension. Nat. Rev. Cardiol. 7 (11), 637–647. doi:10.1038/nrcardio.2010.136
Lottermoser, K., Hertfelder, H. J., Gohlke, P., Vetter, H., and Düsing, R. (2004). Short‐Term Effects of Exogenous Angiotensin II on Plasma Fibrinolytic Balance in Normal Subjects. Clin. Exp. Hypertens. 26 (1), 13–26. doi:10.1081/ceh-120027328
Lu, H., Balakrishnan, A., Howatt, D. A., Wu, C., Charnigo, R., Liau, G., et al. (2012). Comparative Effects of Different Modes of Renin Angiotensin System Inhibition on Hypercholesterolaemia-Induced Atherosclerosis. Br. J. Pharmacol. 165 (6), 2000–2008. doi:10.1111/j.1476-5381.2011.01712.x
Mackman, N. (1997). Regulation of the Tissue Factor Gene. Thromb. Haemost. 78 (1), 747–754. doi:10.1055/s-0038-1657623
Madej, A., Buldak, L., Basiak, M., Szkrobka, W., Dulawa, A., and Okopien, B. (2009). The Effects of 1 Month Antihypertensive Treatment with Perindopril, Bisoprolol or Both on the Ex Vivo Ability of Monocytes to Secrete Inflammatory Cytokines. Cp 47 (11), 686–694. doi:10.5414/cpp47686
Maeda, Y., Inoguchi, T., Takei, R., Hendarto, H., Ide, M., Inoue, T., et al. (2012). Chymase Inhibition Prevents Myocardial Fibrosis through the Attenuation of NOX4-Associated Oxidative Stress in Diabetic Hamsters. J. Diabetes Investig. 3 (4), 354–361. doi:10.1111/j.2040-1124.2012.00202.x
Mallat, Z., Benamer, H., Hugel, B., Benessiano, J., Steg, P. G., Freyssinet, J.-M., et al. (2000). Elevated Levels of Shed Membrane Microparticles with Procoagulant Potential in the Peripheral Circulating Blood of Patients with Acute Coronary Syndromes. Circulation 101 (8), 841–843. doi:10.1161/01.cir.101.8.841
Malmqvist, K., Kahan, T., Edner, M., Held, C., Hägg, A., Lind, L., et al. (2001). Regression of Left Ventricular Hypertrophy in Human Hypertension with Irbesartan. J. Hypertens. 19 (6), 1167–1176. doi:10.1097/00004872-200106000-00023
Man, S. M., and Kanneganti, T.-D. (2015). Regulation of Inflammasome Activation. Immunol. Rev. 265 (1), 6–21. doi:10.1111/imr.12296
Marcus, Y., Shefer, G., and Stern, N. (2013). Adipose Tissue Renin-Angiotensin-Aldosterone System (RAAS) and Progression of Insulin Resistance. Mol. Cell Endocrinol. 378 (1-2), 1–14. doi:10.1016/j.mce.2012.06.021
Marin, V., Montero-Julian, F. A., Grès, S., Boulay, V., Bongrand, P., Farnarier, C., et al. (2001). The IL-6-Soluble IL-6Rα Autocrine Loop of Endothelial Activation as an Intermediate Between Acute and Chronic Inflammation: an Experimental Model Involving Thrombin. J. Immunol. 167 (6), 3435–3442. doi:10.4049/jimmunol.167.6.3435
Marzano, A. V., Genovese, G., Fabbrocini, G., Pigatto, P., Monfrecola, G., Piraccini, B. M., et al. (2020). Varicella-like Exanthem as a Specific COVID-19-Associated Skin Manifestation: Multicenter Case Series of 22 Patients. J. Am. Acad. Dermatol. 83, 280–285. doi:10.1016/j.jaad.2020.04.044
Matavelli, L. C., and Siragy, H. M. (2015). AT2 Receptor Activities and Pathophysiological Implications. J. Cardiovasc. Pharmacol. 65 (3), 226–232. doi:10.1097/fjc.0000000000000208
Mathers, C. D., and Loncar, D. (2006). Projections of Global Mortality and burden of Disease from 2002 to 2030. Plos Med. 3 (11), e442. doi:10.1371/journal.pmed.0030442
Matsumoto, T., Wada, A., Tsutamoto, T., Ohnishi, M., Isono, T., and Kinoshita, M. (2003). Chymase Inhibition Prevents Cardiac Fibrosis and Improves Diastolic Dysfunction in the Progression of Heart Failure. Circulation 107 (20), 2555–2558. doi:10.1161/01.Cir.0000074041.81728.79
Mazak, I., Fiebeler, A., Muller, D. N., Park, J.-K., Shagdarsuren, E., Lindschau, C., et al. (2004). Aldosterone Potentiates Angiotensin II-Induced Signaling in Vascular Smooth Muscle Cells. Circulation 109 (22), 2792–2800. doi:10.1161/01.cir.0000131860.80444.ab
Mehta, N., Kalra, A., Nowacki, A. S., Anjewierden, S., Han, Z., Bhat, P., et al. (2020). Association of Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 5 (9), 1020–1026. doi:10.1001/jamacardio.2020.1855
Meng, X.-m., Nikolic-Paterson, D. J., and Lan, H. Y. (2016). TGF-β: the Master Regulator of Fibrosis. Nat. Rev. Nephrol. 12 (6), 325–338. doi:10.1038/nrneph.2016.48
Min, L., Mogi, M., Iwanami, J., Li, J., Sakata, A., Fujita, T., et al. (2007). Cross-talk between Aldosterone and Angiotensin II in Vascular Smooth Muscle Cell Senescence. Cardiovasc. Res. 76 (3), 506–516. doi:10.1016/j.cardiores.2007.07.008
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P., and Malik, A. B. (2014). Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signaling 20 (7), 1126–1167. doi:10.1089/ars.2012.5149
Mizutani, H., Schechter, N., Lazarus, G., Black, R. A., and Kupper, T. S. (1991). Rapid and Specific Conversion of Precursor Interleukin 1 Beta (IL-1 Beta) to an Active IL-1 Species by Human Mast Cell Chymase. J. Exp. Med. 174 (4), 821–825. doi:10.1084/jem.174.4.821
Murray, T. V. A., Smyrnias, I., Shah, A. M., and Brewer, A. C. (2013). NADPH Oxidase 4 Regulates Cardiomyocyte Differentiation via Redox Activation of C-Jun Protein and the Cis-Regulation of GATA-4 Gene Transcription. J. Biol. Chem. 288 (22), 15745–15759. doi:10.1074/jbc.M112.439844
Nakamura, S., Nakamura, I., Ma, L., Vaughan, D. E., and Fogo, A. B. (2000). Plasminogen Activator Inhibitor-1 Expression Is Regulated by the Angiotensin Type 1 Receptor In Vivo1. Kidney Int. 58 (1), 251–259. doi:10.1046/j.1523-1755.2000.00160.x
Nakano, S., Kobayashi, N., Yoshida, K., Ohno, T., and Matsuoka, H. (2005). Cardioprotective Mechanisms of Spironolactone Associated with the Angiotensin-Converting Enzyme/epidermal Growth Factor Receptor/extracellular Signal-Regulated Kinases, NAD(P)H Oxidase/lectin-like Oxidized Low-Density Lipoprotein Receptor-1, and Rho-Kinase Pathways in Aldosterone/salt-Induced Hypertensive Rats. Hypertens. Res. 28 (11), 925–936. doi:10.1291/hypres.28.925
Napoleone, E., Di Santo, A., Camera, M., Tremoli, E., and Lorenzet, R. (2000). Angiotensin-converting Enzyme Inhibitors Downregulate Tissue Factor Synthesis in Monocytes. Circ. Res. 86 (2), 139–143. doi:10.1161/01.res.86.2.139
Nehme, A., Zouein, F. A., Zayeri, Z. D., and Zibara, K. (2019). An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 6 (2), 14. doi:10.3390/jcdd6020014
Nencioni, A., Trzeciak, S., and Shapiro, N. I. (2009). The Microcirculation as a Diagnostic and Therapeutic Target in Sepsis. Intern. Emerg. Med. 4 (5), 413–418. doi:10.1007/s11739-009-0297-5
Nestoridi, E., Kushak, R. I., Tsukurov, O., Grabowski, E. F., and Ingelfinger, J. R. (2008). Role of the Renin Angiotensin System in TNF-α and Shiga-Toxin-Induced Tissue Factor Expression. Pediatr. Nephrol. 23 (2), 221–231. doi:10.1007/s00467-007-0636-6
Nguyen Dinh Cat, A., Montezano, A. C., Burger, D., and Touyz, R. M. (2013). Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxid. Redox Signaling 19 (10), 1110–1120. doi:10.1089/ars.2012.4641
Nickenig, G. (2002). Central Role of the AT1-Receptor in Atherosclerosis. J. Hum. Hypertens. 16 (Suppl. 3), S26–S33. doi:10.1038/sj.jhh.1001436
Nishimura, H., Tsuji, H., Masuda, H., Nakagawa, K., Nakahara, Y., Kitamura, H., et al. (1997). Angiotensin II Increases Plasminogen Activator Inhibitor-1 and Tissue Factor mRNA Expression without Changing that of Tissue Type Plasminogen Activator or Tissue Factor Pathway Inhibitor in Cultured Rat Aortic Endothelial Cells. Thromb. Haemost. 77 (6), 1189–1195. doi:10.1055/s-0038-1656136
Nussberger, J., Aubert, J.-F., Bouzourene, K., Pellegrin, M., Hayoz, D., and Mazzolai, L. (2008). Renin Inhibition by Aliskiren Prevents Atherosclerosis Progression. Hypertension 51 (5), 1306–1311. doi:10.1161/hypertensionaha.108.110932
Okada, M., Yamawaki, H., and Hara, Y. (2010). Angiotensin 11 Enhances Interleukin-1.BETA.-Induced MMP-9 Secretion in Adult Rat Cardiac Fibroblasts. J. Vet. Med. Sci. 72 (6), 735–739. doi:10.1292/jvms.09-0582
Østerud, B., and Olsen, J. O. (2013). Human Platelets Do Not Express Tissue Factor. Thromb. Res. 132 (1), 112–115. doi:10.1016/j.thromres.2013.04.010
Oyamada, S., Bianchi, C., Takai, S., Chu, L. M., and Sellke, F. W. (2011). Chymase Inhibition Reduces Infarction and Matrix Metalloproteinase-9 Activation and Attenuates Inflammation and Fibrosis after Acute Myocardial Ischemia/reperfusion. J. Pharmacol. Exp. Ther. 339 (1), 143–151. doi:10.1124/jpet.111.179697
Park, J. B., and Schiffrin, E. L. (2002). Cardiac and Vascular Fibrosis and Hypertrophy in Aldosterone-Infused Rats: Role of Endothelin-1. Am. J. Hypertens. 15 (2 Pt 1), 164–169. doi:10.1016/s0895-7061(01)02291-9
Patoulias, D., Katsimardou, A., Stavropoulos, K., Imprialos, K., Kalogirou, M.-S., and Doumas, M. (2020). Renin-Angiotensin System Inhibitors and COVID-19: a Systematic Review and Meta-Analysis. Evidence for Significant Geographical Disparities. Curr. Hypertens. Rep. 22 (11), 90. doi:10.1007/s11906-020-01101-w
Pinar, A. A., Scott, T. E., Huuskes, B. M., Tapia Cáceres, F. E., Kemp-Harper, B. K., and Samuel, C. S. (2020). Targeting the NLRP3 Inflammasome to Treat Cardiovascular Fibrosis. Pharmacol. Ther. 209, 107511. doi:10.1016/j.pharmthera.2020.107511
Polgar, J., Matuskova, J., and Wagner, D. D. (2005). The P-Selectin, Tissue Factor, Coagulation Triad. J. Thromb. Haemost. 3 (8), 1590–1596. doi:10.1111/j.1538-7836.2005.01373.x
Porter, K. E., and Turner, N. A. (2009). Cardiac Fibroblasts: at the Heart of Myocardial Remodeling. Pharmacol. Ther. 123 (2), 255–278. doi:10.1016/j.pharmthera.2009.05.002
Putnam, K., Shoemaker, R., Yiannikouris, F., and Cassis, L. A. (2012). The Renin-Angiotensin System: a Target of and Contributor to Dyslipidemias, Altered Glucose Homeostasis, and Hypertension of the Metabolic Syndrome. Am. J. Physiology-Heart Circulatory Physiol. 302 (6), H1219–H1230. doi:10.1152/ajpheart.00796.2011
Qi, Y., Li, H., Shenoy, V., Li, Q., Wong, F., Zhang, L., et al. (2012). Moderate Cardiac-Selective Overexpression of Angiotensin II Type 2 Receptor Protects Cardiac Functions from Ischaemic Injury. Exp. Physiol. 97 (1), 89–101. doi:10.1113/expphysiol.2011.060673
Qian, J., and Fulton, D. J. R. (2012). Exogenous, but Not Endogenous Nitric Oxide Inhibits Adhesion Molecule Expression in Human Endothelial Cells. Front. Physio. 3, 3. doi:10.3389/fphys.2012.00003
Rader, D. J., and Daugherty, A. (2008). Translating Molecular Discoveries into New Therapies for Atherosclerosis. Nature 451 (7181), 904–913. doi:10.1038/nature06796
Rafieian-Kopaei, M., Setorki, M., Doudi, M., Baradaran, A., and Nasri, H. (2014). Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int. J. Prev. Med. 5 (8), 927–946.
Rahman, A., Anwar, K. N., True, A. L., and Malik, A. B. (1999). Thrombin-induced P65 Homodimer Binding to Downstream NF-Kappa B Site of the Promoter Mediates Endothelial ICAM-1 Expression and Neutrophil Adhesion. J. Immunol. 162 (9), 5466–5476.
Rao, L. V. M., Kothari, H., and Pendurthi, U. R. (2012). Tissue Factor Encryption and Decryption: Facts and Controversies. Thromb. Res. 129 (Suppl. 2), S13–S17. doi:10.1016/j.thromres.2012.02.021
Rautureau, Y., Paradis, P., and Schiffrin, E. L. (2011). Cross-talk between Aldosterone and Angiotensin Signaling in Vascular Smooth Muscle Cells. Steroids 76 (9), 834–839. doi:10.1016/j.steroids.2011.02.015
Ridker, P. M., Cushman, M., Stampfer, M. J., Tracy, R. P., and Hennekens, C. H. (1998). Plasma Concentration of C-Reactive Protein and Risk of Developing Peripheral Vascular Disease. Circulation 97 (5), 425–428. doi:10.1161/01.cir.97.5.425
Ridker, P. M., Gaboury, C. L., Conlin, P. R., Seely, E. W., Williams, G. H., and Vaughan, D. E. (1993). Stimulation of Plasminogen Activator Inhibitor In Vivo by Infusion of Angiotensin II. Evidence of a Potential Interaction between the Renin-Angiotensin System and Fibrinolytic Function. Circulation 87 (6), 1969–1973. doi:10.1161/01.cir.87.6.1969
Ridker, P. M., Rifai, N., Stampfer, M. J., and Hennekens, C. H. (2000). Plasma Concentration of Interleukin-6 and the Risk of Future Myocardial Infarction Among Apparently Healthy Men. Circulation 101 (15), 1767–1772. doi:10.1161/01.cir.101.15.1767
Rose-John, S. (2012). IL-6 Trans-signaling via the Soluble IL-6 Receptor: Importance for the Pro-inflammatory Activities of IL-6. Int. J. Biol. Sci. 8 (9), 1237–1247. doi:10.7150/ijbs.4989
Ruan, Q., Yang, K., Wang, W., Jiang, L., and Song, J. (2020). Clinical Predictors of Mortality Due to COVID-19 Based on an Analysis of Data of 150 Patients from Wuhan, China. Intensive Care Med. 46 (5), 846–848. doi:10.1007/s00134-020-05991-x
Sampaio, W. O., Souza dos Santos, R. A., Faria-Silva, R., da Mata Machado, L. T., Schiffrin, E. L., and Touyz, R. M. (2007). Angiotensin-(1-7) through Receptor Mas Mediates Endothelial Nitric Oxide Synthase Activation via Akt-dependent Pathways. Hypertension 49 (1), 185–192. doi:10.1161/01.HYP.0000251865.35728.2f
Sampson, A. K., Irvine, J. C., Shihata, W. A., Dragoljevic, D., Lumsden, N., Huet, O., et al. (2016). Compound 21, a Selective Agonist of Angiotensin AT2receptors, Prevents Endothelial Inflammation and Leukocyte Adhesionin Vitroandin Vivo. Br. J. Pharmacol. 173 (4), 729–740. doi:10.1111/bph.13063
Sandoo, A., Veldhuijzen van Zanten, J. J. C. S., Metsios, G. S., Carroll, D., and Kitas, G. D. (2010). The Endothelium and its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 4, 302–312. doi:10.2174/1874192401004010302
Santillo, M., Colantuoni, A., Mondola, P., Guida, B., and Damiano, S. (2015). NOX Signaling in Molecular Cardiovascular Mechanisms Involved in the Blood Pressure Homeostasis. Front. Physiol. 6, 194. doi:10.3389/fphys.2015.00194
Santos, R. A. S., e Silva, A. C. S., Maric, C., Silva, D. M. R., Machado, R. P., de Buhr, I., et al. (2003). Angiotensin-(1-7) Is an Endogenous Ligand for the G Protein-Coupled Receptor Mas. Pnas 100 (14), 8258–8263. doi:10.1073/pnas.1432869100
Santos, R. A. S., Sampaio, W. O., Alzamora, A. C., Motta-Santos, D., Alenina, N., Bader, M., et al. (2018). The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 98 (1), 505–553. doi:10.1152/physrev.00023.2016
Sapet, C., Simoncini, S., Loriod, B., Puthier, D., Sampol, J., Nguyen, C., et al. (2006). Thrombin-induced Endothelial Microparticle Generation: Identification of a Novel Pathway Involving ROCK-II Activation by Caspase-2. Blood 108 (6), 1868–1876. doi:10.1182/blood-2006-04-014175
Sarkar, R., Meinberg, E. G., Stanley, J. C., Gordon, D., and Clinton Webb, R. (1996). Nitric Oxide Reversibly Inhibits the Migration of Cultured Vascular Smooth Muscle Cells. Circ. Res. 78 (2), 225–230. doi:10.1161/01.RES.78.2.225
Sasaki, S., Higashi, Y., Nakagawa, K., Matsuura, H., Kajiyama, G., and Oshima, T. (2001). Effects of Angiotensin-(1-7) on Forearm Circulation in Normotensive Subjects and Patients with Essential Hypertension. Hypertension 38 (1), 90–94. doi:10.1161/01.hyp.38.1.90
Satoh, C., Fukuda, N., Hu, W.-Y., Nakayama, M., Kishioka, H., and Kanmatsuse, K. (2001). Role of Endogenous Angiotensin II in the Increased Expression of Growth Factors in Vascular Smooth Muscle Cells from Spontaneously Hypertensive Rats. J. Cardiovasc. Pharmacol. 37 (1), 108–118. doi:10.1097/00005344-200101000-00013
Schieffer, B., Schieffer, E., Hilfiker-Kleiner, D., Hilfiker, A., Kovanen, P. T., Kaartinen, M., et al. (2000). Expression of Angiotensin II and Interleukin 6 in Human Coronary Atherosclerotic Plaques. Circulation 101 (12), 1372–1378. doi:10.1161/01.cir.101.12.1372
Schwieler, J. H., Kahan, T., Wallén, N. H., Nussberger, J., and Hjemdahl, P. (2013). Inhibition of the Renin-Angiotensin System Does Not Reduce Platelet Activity at Rest or during Stress in Hypertension. J. Hypertens. 31 (8), 1676–1682. doi:10.1097/HJH.0b013e3283617310
Shi, Y., Wang, Y., Shao, C., Huang, J., Gan, J., Huang, X., et al. (2020). COVID-19 Infection: the Perspectives on Immune Responses. Cell Death Differ 27 (5), 1451–1454. doi:10.1038/s41418-020-0530-3
Sleight, P., Yusuf, S., Pogue, J., Tsuyuki, R., Diaz, R., and Probstfield, J. (2001). Blood-pressure Reduction and Cardiovascular Risk in HOPE Study. The Lancet 358 (9299), 2130–2131. doi:10.1016/s0140-6736(01)07186-0
Soejima, H., Ogawa, H., Yasue, H., Kaikita, K., Takazoe, K., Nishiyama, K., et al. (1999). Angiotensin-converting Enzyme Inhibition Reduces Monocyte Chemoattractant Protein-1 and Tissue Factor Levels in Patients with Myocardial Infarction. J. Am. Coll. Cardiol. 34 (4), 983–988. doi:10.1016/s0735-1097(99)00318-6
Song, Z., Bai, J., Zhang, L., Sun, X., Zhang, M., Zheng, H., et al. (2014). Effects of Telmisartan on Inflammation and Fibrosis after Acute Myocardial Infarction in Rats. Zhonghua yi xue za zhi 94 (33), 2628–2633.
Sparks, M. A., Crowley, S. D., Gurley, S. B., Mirotsou, M., and Coffman, T. M. (2014). Classical Renin-Angiotensin System in Kidney Physiology. Compr. Physiol. 4 (3), 1201–1228. doi:10.1002/cphy.c130040
Spiezia, L., Boscolo, A., Poletto, F., Cerruti, L., Tiberio, I., Campello, E., et al. (2020). COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 120, 998–1000. doi:10.1055/s-0040-1710018
Sukumaran, V., Watanabe, K., Veeraveedu, P. T., Thandavarayan, R. A., Gurusamy, N., Ma, M., et al. (2010). Telmisartan, an Angiotensin-II Receptor Blocker Ameliorates Cardiac Remodeling in Rats with Dilated Cardiomyopathy. Hypertens. Res. 33 (7), 695–702. doi:10.1038/hr.2010.67
Sumners, C., Peluso, A. A., Haugaard, A. H., Bertelsen, J. B., and Steckelings, U. M. (2019). Anti‐fibrotic Mechanisms of Angiotensin AT2‐receptor Stimulation. Acta Physiol. 227 (1), e13280. doi:10.1111/apha.13280
Sun, J., Sukhova, G. K., Wolters, P. J., Yang, M., Kitamoto, S., Libby, P., et al. (2007). Mast Cells Promote Atherosclerosis by Releasing Proinflammatory Cytokines. Nat. Med. 13 (6), 719–724. doi:10.1038/nm1601
Suthahar, N., Meijers, W. C., Silljé, H. H. W., and de Boer, R. A. (2017). From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 14 (4), 235–250. doi:10.1007/s11897-017-0343-y
Szotowski, B., Antoniak, S., Poller, W., Schultheiss, H.-P., and Rauch, U. (2005). Procoagulant Soluble Tissue Factor Is Released From Endothelial Cells in Response to Inflammatory Cytokines. Circ. Res. 96 (12), 1233–1239. doi:10.1161/01.RES.0000171805.24799.fa
Taubman, M. B., Marmur, J. D., Rosenfield, C. L., Guha, A., Nichtberger, S., and Nemerson, Y. (1993). Agonist-mediated Tissue Factor Expression in Cultured Vascular Smooth Muscle Cells. Role of Ca2+ Mobilization and Protein Kinase C Activation. J. Clin. Invest. 91 (2), 547–552. doi:10.1172/jci116234
Tchougounova, E., Lundequist, A., Fajardo, I., Winberg, J.-O., Åbrink, M., and Pejler, G. (2005). A Key Role for Mast Cell Chymase in the Activation of Pro-matrix Metalloprotease-9 and Pro-matrix Metalloprotease-2. J. Biol. Chem. 280 (10), 9291–9296. doi:10.1074/jbc.M410396200
Thögersen, A. M., Jansson, J. H., Boman, K., Nilsson, T. K., Weinehall, L., Huhtasaari, F., et al. (1998). High Plasminogen Activator Inhibitor and Tissue Plasminogen Activator Levels in Plasma Precede a First Acute Myocardial Infarction in Both Men and Women: Evidence for the Fibrinolytic System as an Independent Primary Risk Factor. Circulation 98 (21), 2241–2247. doi:10.1161/01.cir.98.21.2241
Tomimori, Y., Manno, A., Tanaka, T., Futamura-Takahashi, J., Muto, T., and Nagahira, K. (2019). ASB17061, a Novel Chymase Inhibitor, Prevented the Development of Angiotensin II-Induced Abdominal Aortic Aneurysm in Apolipoprotein E-Deficient Mice. Eur. J. Pharmacol. 856, 172403. doi:10.1016/j.ejphar.2019.05.032
Touyz, R. M., Cruzado, M., Tabet, F., Yao, G., Salomon, S., and Schiffrin, E. L. (2003). Redox-dependent MAP Kinase Signaling by Ang II in Vascular Smooth Muscle Cells: Role of Receptor Tyrosine Kinase Transactivation. Can. J. Physiol. Pharmacol. 81 (2), 159–167. doi:10.1139/y02-164
Tscharre, M., Wadowski, P. P., Weikert, C., Pultar, J., Eichelberger, B., Panzer, S., et al. (2020). Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers in Acute Coronary Syndrome: Implications for Platelet Reactivity? Cardiovasc. Drugs Ther. [Epub ahead of print]. doi:10.1007/s10557-020-07128-0
Ueda, S., Masumori-Maemoto, S., Wada, A., Ishii, M., Brosnihan, K. B., and Umemura, S. (2001). Angiotensin(1-7) Potentiates Bradykinin-Induced Vasodilatation in Man. J. Hypertens. 19 (11), 2001–2009. doi:10.1097/00004872-200111000-00010
Urata, H., Kinoshita, A., Misono, K. S., Bumpus, F. M., and Husain, A. (1990). Identification of a Highly Specific Chymase as the Major Angiotensin II-Forming Enzyme in the Human Heart. J. Biol. Chem. 265 (36), 22348–22357. doi:10.1016/s0021-9258(18)45712-2
van der Poll, T., de Jonge, E., and Levi, M. (2001). Regulatory Role of Cytokines in Disseminated Intravascular Coagulation. Semin. Thromb. Hemost. 27 (6), 639–652. doi:10.1055/s-2001-18868
van der Poll, T., Levi, M., Hack, C. E., ten Cate, H., van Deventer, S. J., Eerenberg, A. J., et al. (1994). Elimination of Interleukin 6 Attenuates Coagulation Activation in Experimental Endotoxemia in Chimpanzees. J. Exp. Med. 179 (4), 1253–1259. doi:10.1084/jem.179.4.1253
van Leeuwen, R. T., Kol, A., Andreotti, F., Kluft, C., Maseri, A., and Sperti, G. (1994). Angiotensin II Increases Plasminogen Activator Inhibitor Type 1 and Tissue-type Plasminogen Activator Messenger RNA in Cultured Rat Aortic Smooth Muscle Cells. Circulation 90 (1), 362–368. doi:10.1161/01.cir.90.1.362
Varga, Z., Flammer, A. J., Steiger, P., Haberecker, M., Andermatt, R., Zinkernagel, A. S., et al. (2020). Endothelial Cell Infection and Endotheliitis in COVID-19. The Lancet 395 (10234), 1417–1418. doi:10.1016/s0140-6736(20)30937-5
Vaughan, D. E. (2002). Angiotensin and Vascular Fibrinolytic Balance. Am. J. Hypertens. 15 (1 Pt 2), 3S–8S. doi:10.1016/s0895-7061(01)02273-7
Verhamme, P., and Hoylaerts, M. F. (2009). Hemostasis and Inflammation: Two of a Kind? Thromb. J. 7, 15. doi:10.1186/1477-9560-7-15
Verma, S., Wang, C.-H., Li, S.-H., Dumont, A. S., Fedak, P. W. M., Badiwala, M. V., et al. (2002). A Self-Fulfilling Prophecy. Circulation 106 (8), 913–919. doi:10.1161/01.cir.0000029802.88087.5e
Virdis, A., Neves, M. F., Amiri, F., Viel, E., Touyz, R. M., and Schiffrin, E. L. (2002). Spironolactone Improves Angiotensin-Induced Vascular Changes and Oxidative Stress. Hypertension 40 (4), 504–510. doi:10.1161/01.hyp.0000034738.79310.06
Volpe, M. (2012). Should All Patients at High Cardiovascular Risk Receive Renin-Angiotensin System Blockers? Qjm 105 (1), 11–27. doi:10.1093/qjmed/hcr190
Wang, K., Gheblawi, M., and Oudit, G. Y. (2020). Angiotensin Converting Enzyme 2. Circulation 142, 426–428. doi:10.1161/circulationaha.120.047049
Wang, Y., Del Borgo, M., Lee, H. W., Baraldi, D., Hirmiz, B., Gaspari, T. A., et al. (2017). Anti-fibrotic Potential of AT2 Receptor Agonists. Front. Pharmacol. 8, 564. doi:10.3389/fphar.2017.00564
Wei, C.-C., Hase, N., Inoue, Y., Bradley, E. W., Yahiro, E., Li, M., et al. (2010). Mast Cell Chymase Limits the Cardiac Efficacy of Ang I-Converting Enzyme Inhibitor Therapy in Rodents. J. Clin. Invest. 120 (4), 1229–1239. doi:10.1172/jci39345
Wei, C.-C., Tian, B., Perry, G., Meng, Q. C., Chen, Y.-F., Oparil, S., et al. (2002). Differential ANG II Generation in Plasma and Tissue of Mice with Decreased Expression of the ACE Gene. Am. J. Physiology-Heart Circulatory Physiol. 282 (6), H2254–H2258. doi:10.1152/ajpheart.00191.2001
Weiss, D., Kools, J. J., and Taylor, W. R. (2001). Angiotensin II-Induced Hypertension Accelerates the Development of Atherosclerosis in apoE-Deficient Mice. Circulation 103 (3), 448–454. doi:10.1161/01.cir.103.3.448
Wen, Y., Liu, Y., Tang, T., Lv, L., Liu, H., Ma, K., et al. (2016). NLRP3 Inflammasome Activation Is Involved in Ang II-Induced Kidney Damage via Mitochondrial Dysfunction. Oncotarget 7 (34), 54290–54302. doi:10.18632/oncotarget.11091
Wernersson, S., and Pejler, G. (2014). Mast Cell Secretory Granules: Armed for Battle. Nat. Rev. Immunol. 14 (7), 478–494. doi:10.1038/nri3690
Wesselman, J. P. M., and De Mey, J. G. R. (2002). Angiotensin and Cytoskeletal Proteins: Role in Vascular Remodeling. Curr. Sci. Inc 4 (1), 63–70. doi:10.1007/s11906-002-0055-9
Whaley-Connell, A., Habibi, J., Rehmer, N., Ardhanari, S., Hayden, M. R., Pulakat, L., et al. (2013). Renin Inhibition and AT1R Blockade Improve Metabolic Signaling, Oxidant Stress and Myocardial Tissue Remodeling. Metabolism 62 (6), 861–872. doi:10.1016/j.metabol.2012.12.012
Wilsdorf, T., Gainer, J. V., Murphey, L. J., Vaughan, D. E., and Brown, N. J. (2001). Angiotensin-(1-7) Does Not Affect Vasodilator or TPA Responses to Bradykinin in Human Forearm. Hypertension 37 (4), 1136–1140. doi:10.1161/01.hyp.37.4.1136
Wynn, T. A., and Ramalingam, T. R. (2012). Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat. Med. 18 (7), 1028–1040. doi:10.1038/nm.2807
Wynn, T. (2008). Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 214 (2), 199–210. doi:10.1002/path.2277
Yaghini, F. A., Song, C. Y., Lavrentyev, E. N., Ghafoor, H. U. B., Fang, X. R., Estes, A. M., et al. (2010). Angiotensin II-Induced Vascular Smooth Muscle Cell Migration and Growth Are Mediated by Cytochrome P450 1B1-dependent Superoxide Generation. Hypertension 55 (6), 1461–1467. doi:10.1161/hypertensionaha.110.150029
Yusuf, S., Yusuf, S., Sleight, P., Pogue, J., Bosch, J., Davies, R., et al. (2000). Effects of an Angiotensin-Converting-Enzyme Inhibitor, Ramipril, on Cardiovascular Events in High-Risk Patients. N. Engl. J. Med. 342 (3), 145–153. doi:10.1056/nejm200001203420301
Zhang, Y., Cao, W., Xiao, M., Li, Y. J., Yang, Y., Zhao, J., et al. (2020). Clinical and Coagulation Characteristics in 7 Patients with Critical COVID-2019 Pneumonia and Acro-Ischemia. Zhonghua Xue Ye Xue Za Zhi 41, 302–307. doi:10.3760/cma.j.issn.0253-2727.2020.008
Keywords: hypertension, haemostasis, coagulation, renin-angiotensin-aldosterone system, angiotensin II, antihypertensive treatment
Citation: Ekholm M and Kahan T (2021) The Impact of the Renin-Angiotensin-Aldosterone System on Inflammation, Coagulation, and Atherothrombotic Complications, and to Aggravated COVID-19. Front. Pharmacol. 12:640185. doi: 10.3389/fphar.2021.640185
Received: 10 December 2020; Accepted: 07 June 2021;
Published: 17 June 2021.
Edited by:
Ewa Krystyna Szczepanska-Sadowska, Medical University of Warsaw, PolandReviewed by:
Tracey Gaspari, Monash University, AustraliaJan Danser, Erasmus Medical Center, Netherlands
Ichiro Sakuma, Hokko Memorial Hospital, Japan
Luis Miguel Ruilope, University Hospital October 12, Spain
Copyright © 2021 Ekholm and Kahan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Ekholm, bWlrYWVsLmVraG9sbUBraS5zZQ==