 Zhengyao Cai
Zhengyao Cai Suxin Yuan
Suxin Yuan Yi Zhong1
Yi Zhong1 Xiaoqiu Tan
Xiaoqiu Tan- 1Department of Cardiology, The Affiliated Hospital of Southwest Medical University, Key Laboratory of Medical Electrophysiology, Ministry of Education and Medical Electrophysiological Key Laboratory of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, Luzhou, China
- 2Department of Rheumatology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
Diabetes mellitus (DM) eventually leads to chronic vascular complications, resulting in cardiovascular diseases. DM-associated endothelial dysfunction (ED) plays an important role in the development of chronic vascular complications. Low endothelial nitric oxide synthase (eNOS) activity, inflammation, and oxidative stress all contribute to ED. The G protein–coupled receptor Takeda G protein–coupled receptor 5 (TGR5) is a membrane receptor for bile acids that plays an important role in the regulation of glucose metabolism. Recent studies have shown that TGR5 is involved in the regulation of various mediators of ED, which suggests that TGR5 may represent a target for the treatment of DM-associated ED. In this review, we summarize the principal mechanisms of DM-associated ED, then propose TGR5 as a novel therapeutic target on the basis of its mechanistic involvement, and suggest potential directions for future research.
Introduction
Diabetes mellitus (DM) is a global public health problem that is associated with a high financial burden for healthcare systems. The progression of DM eventually involves the development of chronic vascular complications, which result in cardiovascular diseases. These cardiovascular complications are the main causes of death in diabetes patients around the world (Naveed et al., 2020). Although clinicians and researchers around the world continue to refine the management of DM, the increase in incidence of diabetic cardiovascular complications continues to outstrip improvements in their prevention and treatment.
There are many potential complications of DM, which have differing mechanisms. DM is considered to be a vascular disease in addition to a metabolic disease (Flyvbjerg, 2010; Arildsen et al., 2019) because vascular complications account for a relatively large proportion of diabetic complications and include diabetic cardiomyopathy, diabetic nephropathy, and diabetic peripheral neuropathy. Endothelial dysfunction (ED) is the initial vascular defect that develops in DM (Legeay et al., 2020; Lespagnol et al., 2020), and it is recognized to be an independent predictor of poor prognosis in patients with microvascular or macrovascular complications of DM (Wiggenhauser and Kroll, 2019; Villano et al., 2020). In this review, we focus on the ED that develops in diabetes, because impairment in endothelial function usually develops before related complications manifest clinically; by reducing ED, we can minimize target organ damage, and by identifying ED, we may diagnose DM in asymptomatic individuals. However, it should be pointed out that DM-related pathology does not always require ED; instead, epithelial cells may be directly affected, such as podocytes and tubules in the diabetic kidney (Pagtalunan et al., 1997).
In patients with DM, the risk of vascular disease has increased by two-to-four times in recent decades (Fox et al., 2004). Macrovascular and microvascular complications related to DM, such as coronary heart disease, stroke, peripheral arterial disease, diabetic retinopathy (DR), and kidney disease (Adler et al., 2003; Yau et al., 2012; Einarson et al., 2018), represent the main health burdens in patients with DM. To date, the management of diabetes has largely focused on the control of hyperglycemia (Triggle and Ding, 2014); however, because the rising burden of DM is mainly related to its vascular complications, it is also important to consider how to protect vascular function in these patients. In fact, although some classic hypoglycemic drugs have been shown to both ameliorate ED and hyperglycemia (Zelniker et al., 2019), there are few specific treatments for the ED associated with DM. Furthermore, in most patients with DM, the goal of controlling cardiovascular disease (CVD) risk factors is not achieved, because hypoglycemic therapy alone does not seem to reduce the incidence of large vessel-related outcomes (Gerstein et al., 2008), and many of the drugs have side effects. For example, metformin, which has been widely used as the first-line therapy for type 2 DM in recent years, can cause gastrointestinal irritation, and other hypoglycemic drugs can cause weight gain and accelerate the loss of pancreatic beta-cells (Maedler et al., 2005; Bennett et al., 2011; Stein et al., 2013; Meza et al., 2019). Therefore, the new frontier in the treatment of DM-associated ED is to identify a hypoglycemic agent that provides endothelial protection and ameliorates ED and is as free of side effects as possible.
Takeda G protein–coupled receptor 5 (TGR5) is a G protein–coupled receptor that is expressed in many organs and tissues, but is also widely expressed in almost all types of endothelial cells (ECs). Recent studies have shown that TGR5 agonists are beneficial in DM and TGR5 has become a promising target for its treatment (Pellicciari et al., 2009; Thomas et al., 2009; Briere et al., 2015). Therefore, in this review, we focus on ED and the role of TGR5 in DM, with the aim of collating evidence for this type of potential targeted therapy for DM.
Diabetes and ED
The endothelium is a layer of squamous epithelial cells that lines the inner surface of vascular systems. ECs form a barrier between blood vessels and tissues, and control the flux of substances in and out of tissues. They provide a metabolic interface between blood and tissue and are therefore essential for the maintenance of vascular homeostasis (Galley and Webster, 2004). The inability of the endothelium to maintain vascular homeostasis is referred to as ED, which is a systemic pathological state that is characterized by alterations to the EC phenotype toward less vasodilatation, and pro-inflammatory and pro-thrombotic states. ED is induced by a number of factors, including turbulent blood flow, shear stress, hypoxia, aging, hyperglycemia, hypercholesterolemia, and hypertension (Gokce et al., 2002; Libby et al., 2002).
ED forms the basis of the chronic microvascular and macrovascular complications of DM. Microvascular complications include DR, nephropathy, and neuropathy, and macrovascular complications affect coronary and peripheral arteries, causing cardiovascular and cerebrovascular diseases and stroke (Suganya et al., 2016). In recent years, great progress has been made in the understanding of the mechanism of EC dysfunction during DM and the difference in its pathogenesis in patients with type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). In T1DM, uncontrolled hyperglycemia and low concentrations of endogenous insulin are the key defects that are involved in the pathogenesis of ED (Joshua et al., 2005), whereas in T2DM, dyslipidemia and insulin resistance play prominent roles (Hamilton et al., 2007). The metabolic milieu in diabetes, which involves hyperglycemia, insulin resistance, hyperinsulinemia, and obesity, induces a wide range of defects, and the principal pathophysiological processes that mediate the development of ED are oxidative stress, endoplasmic reticulum stress, and inflammation (Addabbo et al., 2009; Basha et al., 2012). Endothelial function is impaired in many tissues in diabetes and contributes to the impaired metabolic effects of insulin, as well as diabetic complications, and indicates that the endothelium is a potential target for the therapy of DM.
Mechanism of Diabetic Damage to Endothelia
Furchgott and Zawadzki first described the importance of the intact endothelium for acetylcholine-induced vasodilatation in 1980 (Furchgott and Zawadzki, 1980). The mediator of acetylcholine-induced relaxation was originally designated as endothelium-derived relaxing factor but was eventually identified as NO generated from the amino acid l-arginine by the enzyme eNOS (Palmer et al., 1987; Moncada et al., 1988). eNOS is the main subtype of the enzyme that mediates the physiological generation of NO in ECs. Studies have shown that the reduction in NO production in diabetic patients is involved in the pathogenesis of ED (Förstermann and Münzel, 2006). However, the ED that develops alongside hyperglycemia is not only the result of the reduction in NO production by eNOS but also insulin resistance, oxidative stress, and inflammation (Figure 1). The mechanisms whereby each of these factors promote endothelial injury will be described individually below.
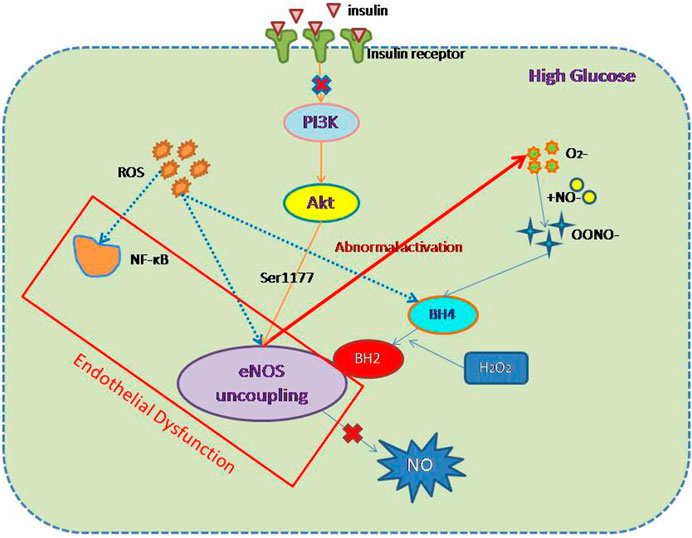
FIGURE 1. Mechanism of endothelial dysfunction associated with diabetes. In DM, cells exist in a high-glucose environment, but the etiology of ED associated with DM can be divided into two components: first, oxidative stress and inflammation; and second the uncoupling of eNOS and a reduction in NO production. The main mediators of oxidative stress and inflammation are ROS and NF-κB. The mechanism of eNOS decoupling involves (1) abnormal activation of eNOS, leading to a reduction in NO production; (2) inhibition of the PI3K/AKT/eNOS pathway as a component of insulin resistance; and (3) ROS activation, leading to a reduction in normal eNOS activation and an increase in the concentration of inhibitory BH2.
eNOS and Endothelial Dysfunction
eNOS is the most important source of NO in ECs (Huang et al., 1995), and the NO generated in this way is crucial for endothelial function (Meza et al., 2019). Therefore, factors that can affect the expression of eNOS also affect the production of NO and can influence the development of ED. In a high-glucose environment, eNOS and endothelial function uncouple, and specifically, abnormal activation of eNOS produces O2− instead of NO. This superoxide quickly combines with NO to form nitrogen-peroxynitrite (ONOO−), which is a highly active oxidant (Stuehr et al., 2001), and reduces the bioavailability NO. It is worth noting that the reaction between NO and superoxide occurs three times faster than the enzyme-catalyzed reduction by superoxide dismutase (Faraci and Didion, 2004), which implies that the cytoplasmic superoxide concentration is an important determinant of ED. In addition, the oxidation of tetrahydrobiopterin (BH4) to dihydrobiopterin (BH2) by ONOO− and H2O2 limits the availability of eNOS substrates and prevents the production of NO (Meza et al., 2019). BH2 cannot function as a cofactor for eNOS, but it competes with the active cofactor BH4. This change in eNOS status is referred to as “eNOS uncoupling” and plays an important role in the development of diabetes-associated ED and CVD (Förstermann and Münzel, 2006; Yang et al., 2009; Karbach et al., 2014).
Insulin resistance also has a significant effect on eNOS production. Under physiological conditions, the delivery of insulin to tissues involves ECs, which regulate capillary recruitment and glucose absorption (Kubota et al., 2011). A large number of previous studies have shown that in insulin resistance, adipose tissue and muscle glucose transporter 4 (GLUT-4) translocation are impaired, blood flow is reduced after a meal, EC function is impaired, and NO production is lower. Insulin signal transduction and glucose uptake are impaired (Kim et al., 2005), which implies that NO production is related to insulin signal transduction. Research by Ardilouze et al. showed that insulin upregulates NOS-dependent vascular activity, thereby increasing total muscle blood flow and capillary recruitment (Ardilouze et al., 2012). This is achieved by insulin binding to its receptor and activating the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/eNOS pathway in the endothelium. The phosphorylation of eNOS at Ser1177 by AKT increases the production of NO (Dimmeler et al., 1999), which results in vasodilation. Insulin resistance is a key defect in T2DM and is therefore also one of the causes of diabetes-related ED. Therefore, the amelioration of insulin resistance should also ameliorate ED.
Multiple lines of evidence have demonstrated an association between greater production of eNOS and an amelioration of diabetic ED. Claybaugh et al. (2014) showed that supplementation of l-arginine (LA), an NO precursor, increases the NO concentration by stimulating the production of eNOS. They found that the glomerular filtration rate of diabetic rats increased when they were administered LA and provided evidence that this effect may be the result of increases in eNOS expression and urinary cGMP, which leads to renal microvascular dilation and the amelioration of ED. Liu (Liu et al., 2014) and Krishnan (Krishnan et al., 2015) have also reached the same conclusion. However, it is worth mentioning that although these findings suggest that increasing the expression of eNOS can ameliorate DM-induced ED, there are some limitations to this approach. eNOS dysfunction is only one of the mechanisms that mediate diabetic ED, and even if an increase in eNOS expression improves endothelial function, hyperglycemia would remain and continue to have adverse effects on endothelia. Therefore, it is still necessary to rigorously prevent and treat DM in order to achieve the maximum impact on ED.
Oxidative Stress and Inflammation
Both oxidative stress and inflammation interfere with insulin signaling (Wen and Duffy, 2017), leading to insulin resistance, DM, and related complications (Baig et al., 2020), and these processes are considered to be important components of the pathogenesis of DM. Persistent oxidative stress and the destruction of normal cellular redox homeostasis typically occur alongside chronic inflammation and the two are highly interrelated; therefore, we will discuss their impact on ECs in diabetes together (Mallard et al., 2020). Vascular ECs are a key site of metabolic dysregulation in diabetes (Khan and Chakrabarti, 2006). In particular, excessive oxidative stress results in the degradation of NO, which causes an imbalance in the effects of vasoconstrictors and vasodilators on the endothelium (Harrison, 1997; Behrendt and Ganz, 2002).
The effects of oxidative stress in ECs are mediated through NADPH oxidase, xanthine oxidase, aldehyde oxidase, and glucose oxidase, which are all enzymes involved in the generation of reactive oxygen species (ROS) (Suganya et al., 2016). ROS are the principal activators of diabetes-related vascular dysfunction (Brownlee, 2001; Brown and Griendling, 2015; Shah and Brownlee, 2016) and have their effects via a number of intracellular signal transduction pathways (El-Daly et al., 2018), through which they alter the phosphorylation and sensitization of eNOS, and also via the oxidation of BH4 to BH2. Thus, ROS can inhibit the production of NO by eNOS through two pathways, which can also form a vicious circle: competition between BH2 and BH4 leads to the dissociation of dimeric eNOS into monomers, which further increases the production of ROS (Wever et al., 1997; Stroes et al., 1998).
ROS also cause redox imbalance through several molecular mechanisms, such as by activating pro-inflammatory signaling pathways and increasing the secretion of pro-inflammatory cytokines (Yaribeygi et al., 2019), which also have deleterious effects on ECs. Previous studies have shown that in streptozotocin-induced diabetic rats, oxidative stress and the production of a series of inflammatory cytokines, including IL-1β, TNF-α, IL-6, and IL-17A, are significantly increased (Zhu et al., 2020), via nuclear factor kappa-B (NF-κB) (Park et al., 2006; Elmarakby and Sullivan, 2012). Therefore, ROS production and the NF-κB pathway represent targets for the treatment of ED.
TGR5 May Represent a Novel Target for the Treatment of Diabetic Endothelial Injury
The pathogenesis of ED in DM involves oxidative stress, chronic inflammation, and low NO production. TGR5 is a G protein–coupled receptor that is present in many organs and tissues and is widely expressed in almost all types of EC. Recent studies have shown that TGR5 mediates beneficial effects in DM and regulates various molecules that mediate DM-associated ED (Pellicciari et al., 2009; Thomas et al., 2009; Briere et al., 2015).
TGR5 and Diabetes
TGR5 (Gpbar1) is a G protein–coupled bile acid receptor that has been widely studied in recent years. It is expressed in various tissues and cell types, such as the gall bladder, intestine, and placenta (Kawamata et al., 2003; Keitel et al., 2009). The roles for TGR5 in regulating metabolic homeostasis have been well documented. Recent studies have shown that TGR5 agonists are beneficial in T2DM and it has become a promising target for the treatment of DM (Pellicciari et al., 2009; Thomas et al., 2009; Briere et al., 2015). Sato et al. found that activation of TGR5 using oleanolic acid reduced the serum glucose and insulin concentrations of high-fat-diet-fed mice (Sato et al., 2007). In addition, Thomas et al. showed that the activation of TGR5 has beneficial effects on metabolism that include resistance to weight gain and improved glucose homeostasis and insulin sensitivity (Thomas et al., 2009). Such improvements in glucose homeostasis and insulin sensitivity implicate TGR5 agonists as potential novel means of ameliorating DM-induced ED.
TGR5 and eNOS
The role of the NO produced by eNOS activity in the maintenance of vascular health and function has been well described (Förstermann and Münzel, 2006). The reinstatement of eNOS activity to improve the bioavailability of NO represents a promising therapeutic strategy, aimed at reducing the incidences of the macrovascular and microvascular complications of DM. Recent studies have shown that TGR5 activation increases eNOS expression through two mechanisms.
First, TGR5 is expressed in ECs (Keitel et al., 2007; Kida et al., 2013). Studies have shown that the activation of TGR5 protects against diabetic nephropathy (Wang et al., 2018). TLCA, TCA, and TCDCA, which are TGR5 agonists, have all been shown to induce eNOS expression and Ser1177 phosphorylation of the enzyme, leading to an increase in NO production (Keitel et al., 2007; Fiorucci et al., 2009). And there are some researchers who have found that increase in eNOS expression is mediated via the “bile-salt-TGR5-cAMP pathway,” which involves binding to the cAMP response element of the human eNOS promoter (Keitel et al., 2007), and the increase of cAMP levels triggered by INT-777 is dose-dependent (Thomas et al., 2009). So the activation of TGR5 may protect against DM-associated ED by inducing an increase in eNOS activation through this pathway. But there is one thing we need to pay attention: Kida et al. (2013) found that TGR5 increased eNOS expression in vascular ECs in a dose-dependent manner in vitro, but care should be taken in extrapolating these results to the endothelium of intact animals, because culture methods cannot fully mimic the complex environment of these cells in blood vessels, nor the complexity of in vivo biochemical and biophysical regulatory mechanisms.
Second, glucagon-like peptide 1 (GLP-1) is an intestinal hormone that has been shown to improve endothelial function (Pratley and Gilbert, 2008; Kolka and Bergman, 2013), and the insulinotropic action of GLP-1 in pancreatic beta-cells has been shown to mediate the glucose lowering effect (Nauck et al., 1997). It has also been shown that GLP-1 is the target of TGR5 and that its secretion is TGR5-dependent (Harach et al., 2012; Briere et al., 2015). The GLP-1 receptor is expressed on ECs and is involved in eNOS activation through a PI3K-dependent pathway (Chai et al., 2012; Lim et al., 2017), which can ultimately be summarized as TGR5-GLP-1-PI3K-eNOS pathway. Further work should aim to clarify the function of this pathway, which may provide new therapeutic targets. Another study also suggests that stimulating effects of TGR5 agonists in the pancreas are mainly due to GLP-1 released from alpha-cells that acts in a paracrine manner on beta-cells (Kumar et al., 2016), improve mass and function of beta-cells in diabetic mouse models (Zheng et al., 2015), resulting in the increase of insulin levels, and then reduce the blood sugar, and at the same time, GLP-1 can protect endothelial function by “GLP-1-PI3K-eNOS” pathway. It is worth noting that the GLP-1 receptor agonists currently on the market do not increase the secretion of endogenous GLP-1, and therefore, they may not prevent certain local effects of disease progression. TGR5 is expressed in many organs and tissues but is principally expressed in the intestine and is found in enteroendocrine L cells, and stimulation of GLP-1 secretion mainly by intestinal L cells is likely to have certain advantages (Knop, 2010; Holst and McGill, 2012). Therefore, TGR5 activation may represent a means of stimulating GLP-1 secretion, as an alternative means of treating DM and ED caused by DM.
As discussed, activation of the bile-salt-TGR5-cAMP and TGR5-GLP-1-PI3K-eNOS pathways induces eNOS expression, which should protect against the ED induced by DM. However, eNOS supplementation does not always protect the endothelium. Krishnan et al. (2015) found that non-diabetic mice which overexpress eNOS show more superoxide production and poor vasorelaxation, and earlier studies by Ohashi (Ohashi et al., 1998) and Yamashita (Yamashita et al., 2000) showed that mice which overexpress eNOS resist the action of vasodilators. However, under specific pathological conditions, the overexpression of eNOS has been shown to be beneficial in specific organs, and we speculate that different disease states are associated with differing vascular phenotypes. Further studies are required to determine which particular disease states would benefit from therapeutic approaches involving eNOS activation.
Effects of TGR5 Activation on Oxidative Stress and Inflammation
As described above, oxidative stress and inflammation are important mechanisms whereby DM leads to endothelial damage. The inhibition of ROS production or inflammatory signaling via the NF-κB pathway represents targets for the treatment of such ED. Previous studies have shown that the activation of TGR5 by INT-777 reduces oxidative stress in human podocytes exposed to a high-glucose environment (Wang et al., 2016). In addition, research by Deng et al. showed that the effect of INT-777 to reduce oxidative stress is achieved via a reduction in the production of ROS and apoptosis in cardiomyocytes exposed to a high-glucose environment (Li et al., 2018; Deng et al., 2019). These studies suggest that TGR5 may ameliorate the DM induced ED by inhibiting the production of ROS. Meanwhile, previous studies have shown that the activation of TGR5 has anti-inflammatory effect on many kind of inflammations through NF-κB signaling pathway. There is evidence that TGR5-induced NO production inhibits the expression of adhesion molecules and the adherence of monocytes, thereby inhibiting NF-κB activity in ECs (Kida et al., 2013). Guo et al. (2015) also found that TGR5 became a negative regulator of gastric inflammation by antagonizing the NF-κB signaling pathway. And in macrophages, activation of TGR5 inhibited NF-κB–mediated inflammatory cytokine production (Pols et al., 2011). Moreover, the study of Wang et al. (2011) found that liver necrosis and inflammation were more severe in TGR5 knockout mice than in wild-type mice in an LPS-induced inflammation model. Therefore, the anti-inflammatory properties of TGR5 may be one of the mechanisms to improve ED caused by DM.
The Identification of a Novel TGR5 Activator for the Treatment of Diabetes-Associated Endothelial Dysfunction
Low eNOS expression, inflammation, and oxidative stress are the principal mechanisms of ED in DM, and the means whereby conventional hypoglycemic drugs protect against ED mainly involve antagonism of these mechanisms (Lu et al., 2019). Many studies have shown that TGR5 plays an important role in DM, inflammation, and oxidative stress, and its agonists may represent novel potential drug substances for the treatment of metabolic and inflammatory disorders (Pols et al., 2011; Lo et al., 2017; Lyu et al., 2019). Therefore, it is important to study the mechanisms whereby TGR5 agonists ameliorate DM-associated ED, to further understand their potential.
Although the use of TGR5 agonists for the treatment of ED in DM holds great promise, a series of questions remain to be resolved. First, TGR5 is expressed in many organs, which may imply that an agonist would be prone to side effects in other organs. For example, Poole et al. (2010) found that the activation of TGR5 reduces gastric and intestinal motility, which can cause nausea and constipation. In addition, the activation of TGR5 delays bile emptying, which was shown to increase the incidence of gallstones over that of TGR5 knockout mice (Keitel et al., 2009; Li et al., 2011). Furthermore, the activation of TGR5 induces hyperexcitability in dorsal root neurons, which results in an itching sensation (Alemi et al., 2013). Finally, there is some in vitro evidence to suggest that TGR5 agonists may induce cancer cell proliferation (Hong et al., 2010; Casaburi et al., 2012; Cao et al., 2013).
Second, even in the same organ, the effects of TGR5 may differ, because of the existence of various distinct binding sites. For example, TGR5 is expressed in the primary cilia of cholangiocytes, where it is coupled to G-α and inhibits cell proliferation, but it is also expressed on the apical plasma membrane, where it is also coupled to G-α proteins but initiates cell proliferation (Masyuk et al., 2013).
Third, TGR5 binds a wide range of ligands, including endogenous and synthetic bile acids and neurosteroids, and also synthetic agonists with a nonsteroidal core (Sato et al., 2008; Martin et al., 2013). Although it is activated by all the known bile acids, the potency of everyone differs from other and depends on the hydrophobicity of its bilane scaffold, and this is reflected in differing effects on the target organ (Sato et al., 2008; Keitel et al., 2020).
Fourth, differing concentrations of TGR5 agonists may have different outcomes. For example, compound 18, a new type of TGR5 agonist, has a significant effect on biliary secretion at a dose as low as 3 mg/kg, whereas doses of compound 18 above 30 mg/kg have a significant effect on GLP-1 secretion (Briere et al., 2015).
Finally, even if an TGR5 agonist is used at the same dose, the results obtained in animal models and humans differ, because of the biological differences between species (Hodge et al., 2013). Therefore, for these reasons, the appropriate choice of TGR5 agonist is critical for its usefulness for the treatment of ED in diabetes.
Nevertheless, in recent years, substantial efforts have been made to identify suitable TGR5 agonists, and there have been some promising results. For example, Zheng et al. (2015) studied the effects of the novel TGR5 agonist WB403 in diabetic mice, and they found that WB403 is not associated with side effects in the gallbladder. Also, in Shan-yao MA et al.'s study, they designed a novel TGR5 agonist called OL3, which combined linagliptin, a DPP-4 inhibitor, with MN6, a novel TGR5 agonist, and they found that OL3 can also lower blood glucose levels without causing gallbladder filling in mice too (Ma et al., 2016); both of the two TGR5 agonists are at a low systemic concentration, and furthermore, WB403’s most significant effects occur in the intestine, which offers new ideas for the design of the best TGR5 agonist for ED caused by DM. And a recently published study also showed that the activation of TGR5 slows the progression of DR in diabetic rats (Zhu et al., 2020), which also confirms that the activation of TGR5 can reduce the ED associated with DM. Thus, although there are many obstacles to the development of TGR5 agonists for the treatment of DM-associated ED, some progress has been made. Nowadays, many researchers found different novel TGR5 agonists which have low side effects (Duan et al., 2015; Finn et al., 2019), future studies should be directed toward the pick of the best TGR5 agonist and evaluation of the safety of TGR5 agonists in both animal study and clinical study. In summary, in order to identify the most appropriate TGR5 agonist, the following issues should be addressed: (1) the biochemical properties of ligands that dictate their distribution and metabolism, such that ligands bind to TGR5 to achieve the target; (2) the design of ligands to render their downstream effects more specifically; and (3) tissue-specific targeting of TGR5 for activation or inhibition.
Discussion
The high prevalence of and numerous risk factors for CVD make it the leading cause of death in the world (Mathers and Loncar, 2006). However, many CVD patients also have DM, which predisposes toward CVD because it is associated with ED (Sun et al., 2019). Hyperglycemia in patients with DM is the principal risk factor for ED-mediated vascular complications, but DM can cause ED through a variety of mechanisms. Low eNOS expression, oxidative stress, and inflammation all cause ED in DM. Because blood from the intestines and the liver is dispersed throughout the body, the vascular endothelium is continuously exposed to bile acids. Therefore, bile acid signaling may affect the physiological functions of ECs. TGR5 is a bile acid receptor that has attracted a lot of attention in recent studies, and studies have shown that its activation has a beneficial effect in DM. This effect of TGR5 activation is mediated not only through a reduction in blood glucose, but also by protecting EC function by increasing eNOS expression, promoting GLP-1 secretion, reducing insulin resistance, and reducing oxidative stress and inflammation (Figure 2). All of these outcomes imply that the activation of TGR5 may represent a therapeutic target for ED in DM. The identification of the optimal TGR5 agonist represents the next research goal.
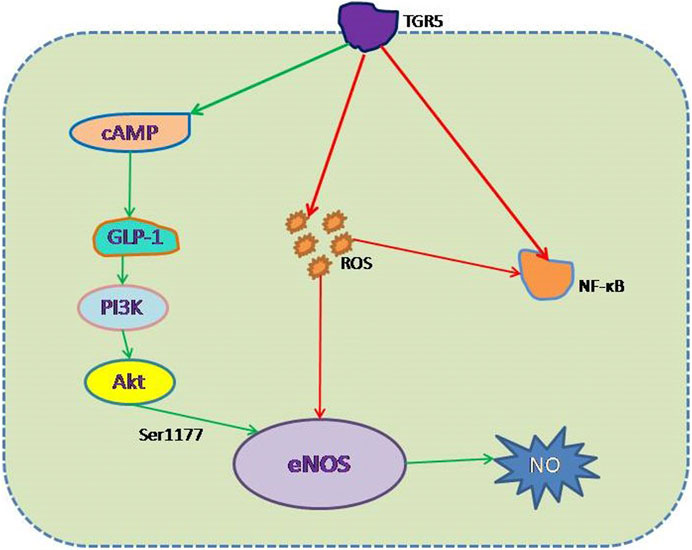
FIGURE 2. Mechanism whereby TGR5 protects against high-glucose-induced endothelial damage. (1) Negative effect (red line): inhibition of the generation of ROS and the activation of NF-κB, thereby reducing eNOS decoupling and NF-κB–mediated ED. (2) Positive effect (green line): increase in eNOS expression via the TGR5-GLP-1-PI3K-eNOS pathway, which mimics GLP-1 action.
Author Contributions
ZC, SY and JF designed the contents of this review article. YZ, JL and XT conducted initial search of literature and prepared the figures. ZC drafted the manuscript. LD and JF revised the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by the fund (KeyME-KeyME-2020-004) of Key Laboratory of Medical Electrophysiology, the fund of Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease of Sichuan Province (xtcx2019-13), Southwest Medical University, and the High-end Talents Recruitment Program (Liu Xinmin group) of Luzhou Municipal People’s Government. The Fund of Affiliated Hospital of Southwest Medical University (17184) and Youth Fund of Southwest Medical University (2019ZQN014).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Addabbo, F., Montagnani, M., and Goligorsky, M. S. (2009). Mitochondria and reactive oxygen species. Hypertension 53, 885–892. doi:10.1161/hypertensionaha.109.130054
Adler, A. I., Stevens, R. J., Manley, S. E., Bilous, R. W., Cull, C. A., Holman, R. R., et al. (2003). Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int 63, 225–232. doi:10.1046/j.1523-1755.2003.00712.x
Alemi, F., Kwon, E., Poole, D. P., Lieu, T., Lyo, V., Cattaruzza, F., et al. (2013). The TGR5 receptor mediates bile acid-induced itch and analgesia. J. Clin. Invest 123, 1513–1530. doi:10.1172/jci64551
Ardilouze, J.-L., Sotorník, R., Dennis, L. A., Fielding, B. A., Frayn, K. N., and Karpe, F. (2012). Failure to increase postprandial blood flow in subcutaneous adipose tissue is associated with tissue resistance to adrenergic stimulation. Diabetes Metab 38, 27–33. doi:10.1016/j.diabet.2011.06.005
Arildsen, L., Andersen, J. V., Waagepetersen, H. S., Nissen, J. B. D., and Sheykhzade, M. (2019). Hypermetabolism and impaired endothelium-dependent vasodilation in mesenteric arteries of type 2 diabetes mellitus db/db mice. Diabetes Vasc. Dis. Res 16, 539–548. doi:10.1177/1479164119865885
Baig, S., Shabeer, M., Parvaresh Rizi, E., et al. (2020). Heredity of type 2 diabetes confers increased susceptibility to oxidative stress and inflammation. BMJ Open Diabetes Res. Care 8, e000945. doi:10.1136/bmjdrc-2019-000945
Basha, B., Samuel, S. M., Triggle, C. R., et al. (2012). Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress?. Exp. Diabetes Res 2012, 481840. doi:10.1155/2012/481840
Behrendt, D., and Ganz, P. (2002). Endothelial function. From vascular biology to clinical applications. Am. J. Cardiol 90, 40L–48L. doi:10.1016/s0002-9149(02)02963-6
Bennett, W. L., Maruthur, N. M., Singh, S., Segal, J. B., Wilson, L. M., Chatterjee, R., et al. (2011). Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann. Intern. Med 154, 602–613. doi:10.7326/0003-4819-154-9-201105030-00336
Briere, D. A., Ruan, X., Cheng, C. C., et al. (2015). Novel small molecule agonist of TGR5 possesses anti-diabetic effects but causes gallbladder filling in mice. PloS one 10, e0136873. doi:10.1371/journal.pone.0136873
Brown, D. I., and Griendling, K. K. (2015). Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res 116, 531–549. doi:10.1161/circresaha.116.303584
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820. doi:10.1038/414813a
Cao, W., Tian, W., Hong, J., Li, D., Tavares, R., Noble, L., et al. (2013). Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am. J. Physiology-Gastrointestinal Liver Physiol 304, G322–G327. doi:10.1152/ajpgi.00263.2012
Casaburi, I., Avena, P., Lanzino, M., Sisci, D., Giordano, F., Maris, P., et al. (2012). Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle 11, 2699–2710. doi:10.4161/cc.21029
Chai, W., Dong, Z., Wang, N., Wang, W., Tao, L., Cao, W., et al. (2012). Glucagon-like peptide 1 recruits microvasculature and increases glucose use in muscle via a nitric oxide-dependent mechanism. Diabetes 61, 888–896. doi:10.2337/db11-1073
Claybaugh, T., Decker, S., McCall, K., Slyvka, Y., Steimle, J., Wood, A., et al. (2014). L-arginine supplementation in type II diabetic rats preserves renal function and improves insulin sensitivity by altering the nitric oxide pathway. Int. J. Endocrinol 2014, 1–7. doi:10.1155/2014/171546
Deng, L., Chen, X., Zhong, Y., et al. (2019). Activation of TGR5 partially alleviates high glucose-induced cardiomyocyte injury by inhibition of inflammatory responses and oxidative stress. Oxid Med. Cel Longev 2019, 6372786. doi:10.1155/2019/6372786
Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R., and Zeiher, A. M. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605. doi:10.1038/21224
Duan, H., Ning, M., Zou, Q., Ye, Y., Feng, Y., Zhang, L., et al. (2015). Discovery of intestinal targeted TGR5 agonists for the treatment of type 2 diabetes. J. Med. Chem 58, 3315–3328. doi:10.1021/jm500829b
Einarson, T. R., Acs, A., Ludwig, C., et al. (2018). Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc. diabetology 17, 83. doi:10.1186/s12933-018-0728-6
El-Daly, M., Pulakazhi Venu, V. K., Saifeddine, M., Mihara, K., Kang, S., Fedak, P. W. M., et al. (2018). Hyperglycaemic impairment of PAR2-mediated vasodilation: prevention by inhibition of aortic endothelial sodium-glucose-co-Transporter-2 and minimizing oxidative stress. Vasc. Pharmacol 109, 56–71. doi:10.1016/j.vph.2018.06.006
Elmarakby, A. A., and Sullivan, J. C. (2012). Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc. Ther 30, 49–59. doi:10.1111/j.1755-5922.2010.00218.x
Faraci, F. M., and Didion, S. P. (2004). Vascular protection. Arterioscler Thromb Vasc Biol 24, 1367–1373. doi:10.1161/01.atv.0000133604.20182.cf
Finn, P. D., Rodriguez, D., Kohler, J., Jiang, Z., Wan, S., Blanco, E., et al. (2019). Intestinal TGR5 agonism improves hepatic steatosis and insulin sensitivity in Western diet-fed mice. Am. J. Physiology-Gastrointestinal Liver Physiol 316, G412–G424. doi:10.1152/ajpgi.00300.2018
Fiorucci, S., Mencarelli, A., Palladino, G., and Cipriani, S. (2009). Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacological Sciences 30, 570–580. doi:10.1016/j.tips.2009.08.001
Flyvbjerg, A. (2010). Diabetic angiopathy, the complement system and the tumor necrosis factor superfamily. Nat. Rev. Endocrinol 6. doi:10.1038/nrendo.2009.266
Förstermann, U., and Münzel, T. (2006). Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113, 1708–1714. doi:10.1161/CIRCULATIONAHA.105.602532
Fox, C. S., Coady, S., Sorlie, P. D., et al. (2004). Trends in cardiovascular complications of diabetes. JAMA 292, 2495–2499. doi:10.1001/jama.292.20.2495
Furchgott, R. F., and Zawadzki, J. V. (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376. doi:10.1038/288373a0
Galley, H. F., and Webster, N. R. (2004). Physiology of the endothelium. Br. J. Anaesth 93, 105–113. doi:10.1093/bja/aeh163
Gerstein, H. C., Gerstein, H. C., Miller, M. E., Byington, R. P., Goff, D. C., Bigger, J. T., et al. (2008). Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med 358, 2545–2559. doi:10.1056/NEJMoa0802743
Gokce, N., Keaney, J. F., Hunter, L. M., Watkins, M. T., Menzoian, J. O., and Vita, J. A. (2002). Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function. Circulation 105, 1567–1572. doi:10.1161/01.cir.0000012543.55874.47
Guo, C., Qi, H., Yu, Y., et al. (2015). The G-protein-coupled bile acid receptor Gpbar1 (TGR5) inhibits gastric inflammation through antagonizing NF-kappaB signaling pathway. Front. Pharmacol 6, 287. doi:10.3389/fphar.2015.00287
Hamilton, S. J., Chew, G. T., and Watts, G. F. (2007). Therapeutic regulation of endothelial dysfunction in type 2 diabetes mellitus. Diabetes Vasc. Dis. Res 4. doi:10.3132/dvdr.2007.026
Harach, T., Pols, T. W., Nomura, M., et al. (2012). TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci. Rep 2, 430. doi:10.1038/srep00430
Harrison, D. G. (1997). Endothelial function and oxidant stress. Clin. Cardiol 20, II–11. doi:10.1002/j.1932-8737.1997.tb00007.x
Hodge, R. J., Lin, J., Vasist Johnson, L. S., Gould, E. P., Bowers, G. D., and Nunez, D. J. (2013). Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 agonist, SB-756050, in type 2 diabetes. Clin. Pharmacol. Drug Dev 2, 213–222. doi:10.1002/cpdd.34
Holst, J. J., and McGill, M. A. (2012). Potential new approaches to modifying intestinal GLP-1 secretion in patients with type 2 diabetes mellitus: focus on bile acid sequestrants. Clin. Drug Investig 32. doi:10.2165/11595370-000000000-00000
Hong, J., Behar, J., Wands, J., Resnick, M., Wang, L. J., DeLellis, R. A., et al. (2010). Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut 59, 170–180. doi:10.1136/gut.2009.188375
Huang, P. L., Huang, Z., Mashimo, H., Bloch, K. D., Moskowitz, M. A., Bevan, J. A., et al. (1995). Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377, 239–242. doi:10.1038/377239a0
Joshua, I. G., Zhang, Q., Falcone, J. C., Bratcher, A. P., Rodriguez, W. E., and Tyagi, S. C. (2005). Mechanisms of endothelial dysfunction with development of type 1 diabetes mellitus: role of insulin and C-peptide. J. Cel. Biochem 96, 1149–1156. doi:10.1002/jcb.20620
Karbach, S., Wenzel, P., Waisman, A., Munzel, T., and Daiber, A. (2014). eNOS uncoupling in cardiovascular diseases - the role of oxidative stress and inflammation. Curr Pharm Des 20, 3579–3594. doi:10.2174/13816128113196660748
Kawamata, Y., Fujii, R., Hosoya, M., Harada, M., Yoshida, H., Miwa, M., et al. (2003). A G protein-coupled receptor responsive to bile acids. J. Biol. Chem 278, 9435–9440. doi:10.1074/jbc.m209706200
Keitel, V., Gertzen, C. G. W., Schäfer, S., et al. (2020). Bile acids and TGR5 (Gpbar1) signaling. mammalian sterols, 81–100. doi:10.1007/978-3-030-39684-8_4
Keitel, V., Cupisti, K., Ullmer, C., Knoefel, W. T., Kubitz, R., and Häussinger, D. (2009). The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50, 861–870. doi:10.1002/hep.23032
Keitel, V., Reinehr, R., Gatsios, P., Rupprecht, C., Görg, B., Selbach, O., et al. (2007). The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology 45, 695–704. doi:10.1002/hep.21458
Khan, Z., and Chakrabarti, S. (2006). Therapeutic targeting of endothelial dysfunction in chronic diabetic complications. Recent Pat Cardiovasc Drug Discov 1, 167–175. doi:10.2174/157489006777442531
Kida, T., Tsubosaka, Y., Hori, M., Ozaki, H., and Murata, T. (2013). Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler Thromb. Vasc. Biol 33, 1663–1669. doi:10.1161/atvbaha.113.301565
Kim, J.-a., Koh, K. K., and Quon, M. J. (2005). The union of vascular and metabolic actions of insulin in sickness and in health. Arterioscler Thromb Vasc Biol 25, 889–891. doi:10.1161/01.atv.0000164044.42910.6b
Knop, F. K. (2010). Bile-induced secretion of glucagon-like peptide-1: pathophysiological implications in type 2 diabetes?. Am. J. Physiology-Endocrinology Metab 299, E10–E13. doi:10.1152/ajpendo.00137.2010
Kolka, C. M., and Bergman, R. N. (2013). The endothelium in diabetes: its role in insulin access and diabetic complications. Rev. Endocr. Metab. Disord 14, 13–19. doi:10.1007/s11154-012-9233-5
Krishnan, M., Janardhanan, P., Roman, L., Reddick, R. L., Natarajan, M., van Haperen, R., et al. (2015). Enhancing eNOS activity with simultaneous inhibition of IKKβ restores vascular function in Ins2Akita+/− type-1 diabetic mice. Lab. Invest 95, 1092–1104. doi:10.1038/labinvest.2015.96
Kubota, T., Kubota, N., Kumagai, H., Yamaguchi, S., Kozono, H., Takahashi, T., et al. (2011). Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cel Metab 13, 294–307. doi:10.1016/j.cmet.2011.01.018
Kumar, D. P., Asgharpour, A., Mirshahi, F., Park, S. H., Liu, S., Imai, Y., et al. (2016). Activation of transmembrane bile acid receptor TGR5 modulates pancreatic islet α cells to promote glucose homeostasis. J. Biol. Chem 291, 6626–6640. doi:10.1074/jbc.m115.699504
Legeay, S., Fautrat, P., Norman, J. B., Antonova, G., Kennard, S., Bruder-Nascimento, T., et al. (2020). Selective deficiency in endothelial PTP1B protects from diabetes and endoplasmic reticulum stress-associated endothelial dysfunction via preventing endothelial cell apoptosis. Biomed. Pharmacother 127, 110200. doi:10.1016/j.biopha.2020.110200
Lespagnol, E., Dauchet, L., Pawlak-Chaouch, M., et al. (2020). Early endothelial dysfunction in type 1 diabetes is accompanied by an impairment of vascular smooth muscle function: a meta-analysis. Front. Endocrinol. (Lausanne) 11, 203. doi:10.3389/fendo.2020.00203
Li, B., Yang, N., Li, C., Li, C., Gao, K., Xie, X., et al. (2018). INT-777, a bile acid receptor agonist, extenuates pancreatic acinar cells necrosis in a mouse model of acute pancreatitis. Biochem. biophysical Res. Commun 503, 38–44. doi:10.1016/j.bbrc.2018.05.120
Li, T., Holmstrom, S. R., Kir, S., Umetani, M., Schmidt, D. R., Kliewer, S. A., et al. (2011). The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol. Endocrinol 25, 1066–1071. doi:10.1210/me.2010-0460
Libby, P., Ridker, P. M., and Maseri, A. (2002). Inflammation and atherosclerosis. Circulation 105, 1135–1143. doi:10.1161/hc0902.104353
Lim, D.-M., Park, K.-Y., Hwang, W.-M., Kim, J.-Y., and Kim, B.-J. (2017). Difference in protective effects of GIP and GLP-1 on endothelial cells according to cyclic adenosine monophosphate response. Exp. Ther. Med 13, 2558–2564. doi:10.3892/etm.2017.4279
Liu, M., Xiang, G., Lu, J., Xiang, L., Dong, J., and Mei, W. (2014). TRAIL protects against endothelium injury in diabetes via Akt-eNOS signaling. Atherosclerosis 237, 718–724. doi:10.1016/j.atherosclerosis.2014.10.013
Lo, S.-H., Li, Y., Cheng, K. C., Niu, C.-S., Cheng, J.-T., and Niu, H.-S. (2017). Ursolic acid activates the TGR5 receptor to enhance GLP-1 secretion in type 1-like diabetic rats. Naunyn-schmiedeberg's Arch. Pharmacol 390, 1097–1104. doi:10.1007/s00210-017-1409-9
Lu, Q., Li, X., Liu, J., et al. (2019). AMPK is associated with the beneficial effects of antidiabetic agents on cardiovascular diseases. Biosci. Rep 39. doi:10.1042/bsr20181995
Lyu, D., Tang, N., Wang, J., Zhang, Y., Chang, J., Liu, Z., et al. (2019). TGR5 agonist INT-777 mitigates inflammatory response in human endometriotic stromal cells: a therapeutic implication for endometriosis. Int. Immunopharmacology 71, 93–99. doi:10.1016/j.intimp.2019.02.044
Ma, S.-y., Ning, M.-m., Zou, Q.-a., Feng, Y., Ye, Y.-l., Shen, J.-h., et al. (2016). OL3, a novel low-absorbed TGR5 agonist with reduced side effects, lowered blood glucose via dual actions on TGR5 activation and DPP-4 inhibition. Acta Pharmacol. Sin 37, 1359–1369. doi:10.1038/aps.2016.27
Maedler, K., Carr, R. D., Bosco, D., Zuellig, R. A., Berney, T., and Donath, M. Y. (2005). Sulfonylurea induced β-cell apoptosis in cultured human islets. J. Clin. Endocrinol. Metab 90, 501–506. doi:10.1210/jc.2004-0699
Mallard, A. R., Hollekim-Strand, S. M., Ingul, C. B., and Coombes, J. S. (2020). High day-to-day and diurnal variability of oxidative stress and inflammation biomarkers in people with type 2 diabetes mellitus and healthy individuals. Redox Rep 25, 64–69. doi:10.1080/13510002.2020.1795587
Martin, R. E., Bissantz, C., Gavelle, O., Kuratli, C., Dehmlow, H., Richter, H. G. F., et al. (2013). 2-Phenoxy-nicotinamides are potent agonists at the bile acid receptor GPBAR1 (TGR5). ChemMedChem 8, 569–576. doi:10.1002/cmdc.201200474
Masyuk, A. I., Huang, B. Q., Radtke, B. N., Gajdos, G. B., Splinter, P. L., Masyuk, T. V., et al. (2013). Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am. J. Physiology-Gastrointestinal Liver Physiol 304, G1013–G1024. doi:10.1152/ajpgi.00383.2012
Mathers, C. D., and Loncar, D. (2006). Projections of global mortality and burden of disease from 2002 to 2030. Plos Med 3, e442. doi:10.1371/journal.pmed.0030442
Meza, C. A., La Favor, J. D., Kim, D-H., et al. (2019). Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS?. Int. J. Mol. Sci 20. doi:10.3390/ijms20153775
Moncada, S., Palmer, R. M., and Higgs, E. A. (1988). The discovery of nitric oxide as the endogenous nitrovasodilator. Hypertension 12, 365–372. doi:10.1161/01.hyp.12.4.365
Nauck, M. A., Niedereichholz, U., Ettler, R., Holst, J. J., Ørskov, C., Ritzel, R., et al. (1997). Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am. J. Physiology-Endocrinology Metab 273, E981–E988. doi:10.1152/ajpendo.1997.273.5.e981
Naveed, A., Farrukh, L., Sana, M. K., Naveed, B., and Randhawa, F. A. (2020). Pharmacological primary prevention of diabetes mellitus type II: a narrative review. Cureus 12, e10033. doi:10.7759/cureus.10033
Ohashi, Y., Kawashima, S., Hirata, K. i., Yamashita, T., Ishida, T., Inoue, N., et al. (1998). Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J. Clin. Invest 102, 2061–2071. doi:10.1172/jci4394
Pagtalunan, M. E., Miller, P. L., Jumping-Eagle, S., Nelson, R. G., Myers, B. D., Rennke, H. G., et al. (1997). Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Invest 99, 342–348. doi:10.1172/jci119163
Palmer, R. M. J., Ferrige, A. G., and Moncada, S. (1987). Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327, 524–526. doi:10.1038/327524a0
Park, J., Choe, S. S., Choi, A. H., Kim, K. H., Yoon, M. J., Suganami, T., et al. (2006). Increase in glucose-6-phosphate dehydrogenase in adipocytes stimulates oxidative stress and inflammatory signals. Diabetes 55, 2939–2949. doi:10.2337/db05-1570
Pellicciari, R., Gioiello, A., Macchiarulo, A., Thomas, C., Rosatelli, E., Natalini, B., et al. (2009). Discovery of 6α-ethyl-23(S)-methylcholic acid (S-emca, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J. Med. Chem 52, 7958–7961. doi:10.1021/jm901390p
Pols, T. W. H., Nomura, M., Harach, T., Lo Sasso, G., Oosterveer, M. H., Thomas, C., et al. (2011). TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cel Metab 14, 747–757. doi:10.1016/j.cmet.2011.11.006
Poole, D. P., Godfrey, C., Cattaruzza, F., et al. (2010). Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil 22. doi:10.1111/j.1365-2982.2010.01487.x
Pratley, R. E., and Gilbert, M. (2008). Targeting incretins in type 2 diabetes: role of GLP-1 receptor agonists and DPP-4 inhibitors. Rev. Diabet Stud 5, 73–94. doi:10.1900/rds.2008.5.73
Sato, H., Genet, C., Strehle, A., Thomas, C., Lobstein, A., Wagner, A., et al. (2007). Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem. biophysical Res. Commun 362, 793–798. doi:10.1016/j.bbrc.2007.06.130
Sato, H., Macchiarulo, A., Thomas, C., Gioiello, A., Une, M., Hofmann, A. F., et al. (2008). Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, Structure−Activity relationships, and molecular modeling studies. J. Med. Chem 51, 1831–1841. doi:10.1021/jm7015864
Shah, M. S., and Brownlee, M. (2016). Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ. Res 118, 1808–1829. doi:10.1161/circresaha.116.306923
Stein, S. A., Lamos, E. M., and Davis, S. N. (2013). A review of the efficacy and safety of oral antidiabetic drugs. Expert Opin. Drug Saf 12, 153–175. doi:10.1517/14740338.2013.752813
Stroes, E., Hijmering, M., van Zandvoort, M., Wever, R., Rabelink, T. J., and van Faassen, E. E. (1998). Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett 438, 161–164. doi:10.1016/s0014-5793(98)01292-7
Stuehr, D., Pou, S., and Rosen, G. M. (2001). Oxygen reduction by nitric-oxide synthases. J. Biol. Chem 276, 14533–14536. doi:10.1074/jbc.r100011200
Suganya, N., Bhakkiyalakshmi, E., Sarada, D. V. L., and Ramkumar, K. M. (2016). Reversibility of endothelial dysfunction in diabetes: role of polyphenols. Br. J. Nutr 116, 223–246. doi:10.1017/s0007114516001884
Sun, H. J., Wu, Z. Y., Nie, X. W., and Bian, J. S. (2019). Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Front. Pharmacol 10, 1568. doi:10.3389/fphar.2019.01568
Thomas, C., Gioiello, A., Noriega, L., Strehle, A., Oury, J., Rizzo, G., et al. (2009). TGR5-mediated bile acid sensing controls glucose homeostasis. Cel Metab 10, 167–177. doi:10.1016/j.cmet.2009.08.001
Triggle, C. R., and Ding, H. (2014). Cardiovascular impact of drugs used in the treatment of diabetes. Ther. Adv. Chronic Dis 5, 245–268. doi:10.1177/2040622314546125
Villano, A., Mencarelli, E., Melita, V., Rizzi, A., Lamendola, P., De Vita, A., et al. (2020). Endothelial dysfunction and cardiovascular outcome in asymptomatic patients with type 2 diabetes: a pilot study. Diabetes Metab. Res. Rev 36, e3215. doi:10.1002/dmrr.3215
Wang, X. X., Edelstein, M. H., Gafter, U., Qiu, L., Luo, Y., Dobrinskikh, E., et al. (2016). G protein-coupled bile acid receptor TGR5 activation inhibits kidney disease in obesity and diabetes. J Am Soc Nephrol 27, 1362–1378. doi:10.1681/asn.2014121271
Wang, X. X., Wang, D., Luo, Y., Myakala, K., Dobrinskikh, E., Rosenberg, A. Z., et al. (2018). FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity. J Am Soc Nephrol 29, 118–137. doi:10.1681/asn.2017020222
Wang, Y.-D., Chen, W.-D., Yu, D., Forman, B. M., and Huang, W. (2011). The G-Protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 54, 1421–1432. doi:10.1002/hep.24525
Wen, L., and Duffy, A. (2017). Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J. Nutr 147, 1468S–1475S. doi:10.3945/jn.116.240754
Wever, R. M. F., van Dam, T., van Rijn, H. J. M., de Groot, F., and Rabelink, T. J. (1997). Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem. biophysical Res. Commun 237, 340–344. doi:10.1006/bbrc.1997.7069
Wiggenhauser, L. M., and Kroll, J. (2019). Vascular damage in obesity and diabetes: highlighting links between endothelial dysfunction and metabolic disease in zebrafish and man. Curr Vasc Pharmacol 17, 476–490. doi:10.2174/1570161116666181031101413
Yamashita, T., Kawashima, S., Ohashi, Y., et al. (2000). Mechanisms of reduced nitric oxide/cGMP-mediated vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. Hypertension 36, 97-102. doi:10.1161/01.hyp.36.1.97
Yang, Y.-M., Huang, A., Kaley, G., and Sun, D. (2009). eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol 297, H1829–H1836. doi:10.1152/ajpheart.00230.2009
Yaribeygi, H., Atkin, S. L., and Sahebkar, A. (2019). A review of the molecular mechanisms of hyperglycemia-induced free radical generation leading to oxidative stress. J. Cel Physiol 234, 1300–1312. doi:10.1002/jcp.27164
Yau, J. W. Y., Rogers, S. L., Kawasaki, R., Lamoureux, E. L., Kowalski, J. W., Bek, T., et al. (2012). Global prevalence and major risk factors of diabetic retinopathy. Diabetes care 35, 556–564. doi:10.2337/dc11-1909
Zelniker, T. A., Wiviott, S. D., Raz, I., Im, K., Goodrich, E. L., Bonaca, M. P., et al. (2019). SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. The Lancet 393, 31–39. doi:10.1016/s0140-6736(18)32590-x
Zheng, C., Zhou, W., Wang, T., et al. (2015). A novel TGR5 activator WB403 promotes GLP-1 secretion and preserves pancreatic beta-cells in type 2 diabetic mice. PLoS One 10, e0134051. doi:10.1371/journal.pone.0134051
Zhu, L., Sha, L., Li, K., et al. (2020). Dietary flaxseed oil rich in omega-3 suppresses severity of type 2 diabetes mellitus via anti-inflammation and modulating gut microbiota in rats. Lipids Health Dis 19, 20. doi:10.1186/s12944-019-1167-4
Keywords: diabetes mellitus, endothelial dysfunction, TGR5, endothelial nitric oxide synthase, inflammation, oxidative stress
Citation: Cai Z, Yuan S, Zhong Y, Deng L, Li J, Tan X and Feng J (2021) Amelioration of Endothelial Dysfunction in Diabetes: Role of Takeda G Protein–Coupled Receptor 5. Front. Pharmacol. 12:637051. doi: 10.3389/fphar.2021.637051
Received: 02 December 2020; Accepted: 22 February 2021;
Published: 28 April 2021.
Edited by:
Francesco Rossi, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Xiaoxin Wang, Georgetown University Medical Center, United StatesTakahisa Murata, The University of Tokyo, Japan
Divya P. Kumar, JSS Academy of Higher Education and Research, India
Copyright © 2021 Cai, Yuan, Zhong, Deng, Li, Tan and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqiu Tan, dGFueGlhb3FpdUBzd211LmVkdS5jbg==; Jian Feng, amVycnlmZW5nQHN3bXUuZWR1LmNu
†These authors have contributed equally to this work