 Alessandro Rimessi
Alessandro Rimessi Veronica A. M. Vitto
Veronica A. M. Vitto Simone Patergnani
Simone Patergnani Paolo Pinton
Paolo Pinton- 1Department of Medical Sciences and Laboratory for Technologies of Advanced Therapies (LTTA), University of Ferrara, Ferrara, Italy
- 2Center of Research for Innovative Therapies in Cystic Fibrosis, University of Ferrara, Ferrara, Italy
Cystic fibrosis (CF) is an autosomal recessive disorder characterized by mutations in the cystic fibrosis transmembrane conductance regulator gene, which causes multifunctional defects that preferentially affect the airways. Abnormal viscosity of mucus secretions, persistent pathogen infections, hyperinflammation, and lung tissue damage compose the classical pathological manifestation referred to as CF lung disease. Among the multifunctional defects associated with defective CFTR, increasing evidence supports the relevant role of perturbed calcium (Ca2+) signaling in the pathophysiology of CF lung disease. The Ca2+ ion is a critical player in cell functioning and survival. Its intracellular homeostasis is maintained by a fine balance between channels, transporters, and exchangers, mediating the influx and efflux of the ion across the plasma membrane and the intracellular organelles. An abnormal Ca2+ profile has been observed in CF cells, including airway epithelial and immune cells, with heavy repercussions on cell function, viability, and susceptibility to pathogens, contributing to proinflammatory overstimulation, organelle dysfunction, oxidative stress, and excessive cytokines release in CF lung. This review discusses the role of Ca2+ signaling in CF and how its dysregulation in airway epithelial and immune cells contributes to hyperinflammation in the CF lung. Finally, we provide an outlook on the therapeutic options that target the Ca2+ signaling to treat the CF lung disease.
Introduction
Cystic fibrosis (CF) is a multiorgan genetic disease associated with mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which preferentially affects the airways causing abnormal infiltration of polymorphonucleated cells, hyperinflammation, and severe lung damage (Riordan, 1993). According to the CF foundation patient registries, more than 70,000 people are living with CF worldwide and about one thousand new cases of CF are diagnosed only in United States each year. The CF is a complex disease, in which the type and severity of symptoms may differ from patient to patient, influencing the individual’s health and the course of disease in a different manner.
The gene’s product is a plasma membrane (PM) ion channel protein located on the apical surface of epithelial cells. Its activation is due by ATP and cAMP-dependent protein kinase A phosphorylations, which extrudes chloride (Cl-) and bicarbonate ions from airway cells (Jacquot et al., 2008). Although CF has long been recognized as an epithelial disease, the channel is expressed also in immune cells (Di et al., 2006; Ng et al., 2016). The most common mutation is the deletion of phenylalanine at position 508 (F508del-CFTR) and the substitution of the amino acid glycine by aspartate at position 551 (G551D-CFTR). The first mutation results in a misfolded protein retained in the endoplasmic reticulum (ER) to be prematurely degraded through the ubiquitin-proteasome pathway. The second is the most prevalent gating mutation, which abolishing the ATP-dependent gating led to a pronounced reduction of channel activity (Pedemonte et al., 2005; Farinha et al., 2013).
Moreover, defective CFTR induces an increased absorption of sodium (Na+) coupled with the absence of Cl- secretion. This electrolytic disorder causes the dehydration of periciliary and mucus layers, leading to mucociliary dysfunction and airway mucus plugging (Boucher et al., 1988). An increased susceptibility to pathogen infections, including Pseudomonas aeruginosa (P. aeruginosa), is associated with defective CFTR, which leads to exaggerated lung inflammatory responses (Bruscia and Bonfield, 2016). In addition, CF patients airways are characterized by abnormal infiltration of neutrophils, which synthesize and release abundant proinflammatory mediators such as interleukin-8 (IL-8) and IL-1β, that contribute to overstimulating the inflammatory responses and worsening the pulmonary injury (Bruscia and Bonfield, 2016).
To improve the lung function, airway clearance techniques and administration of mucus thinner, such as mucolytics, contribute to maintaining the lung clear. Meanwhile, antibiotics and anti-inflammatory drugs fight the infection and consequent hyperinflammation, conditioning the days of CF patients. Recently, a new class of CFTR modulators has been introduced in CF therapy to correct and potentiate the defective CFTR channel. “Correctors” and “potentiators” have garnered much attention in the CF community, although their impact on downstream consequences, such as inflammation, remains debated. The new advances in CF care have drastically ameliorated the quality and duration of life of CF patients.
The calcium ion (Ca2+) is a second messenger, which fulfills a plethora of intracellular functions (Giorgi et al., 2018). In addition to its prominent but ambiguous role in energy metabolism and cell death, Ca2+ is intimately involved in various cellular processes, such as autophagy and inflammation (Carafoli and Krebs, 2016; Marchi et al., 2018; Patergnani et al., 2020a). Therefore, it is not surprising that altered Ca2+ signaling represents a key factor in several inflammatory diseases, including CF. Airway epithelial and immune cells are critically dependent on Ca2+ signaling function and integrity. Consequently, perturbations in Ca2+ signaling have been observed in CF and are caused by intrinsic defects associated with CFTR deficiency and environmental stress related to recurrent bacterial infections, resulting in an exacerbated inflammatory response that favors lung injury (Ribeiro and Boucher, 2010; Antigny et al., 2011a; Rimessi et al., 2015a).
This review analyzes Ca2+ signaling in CF and how its dysregulation contributes to CF lung disease. We summarize the current knowledge and provide an outlook on the therapeutic options to treat the CF lung disease. In particular, we will focus on compounds that target the Ca2+ signaling and potentially would lead to adjusting the inflammatory response, thus suggesting new therapeutic strategies for this pulmonary disease.
Ca2+ Signaling in Airway Epithelial and Immune Cells
The free cytosolic Ca2+ concentration [Ca2+]cyt is very low with concentrations in the order of hundreds of nM. This characteristic is guaranteed by a regulated activity of pumps, exchangers, and channels that reside on the PM and by intracellular organelles, such as ER and Golgi, that act as intracellular Ca2+ store by accumulating ions in the order of hundreds of μM (Figure 1) (Bootman and Bultynck, 2020).
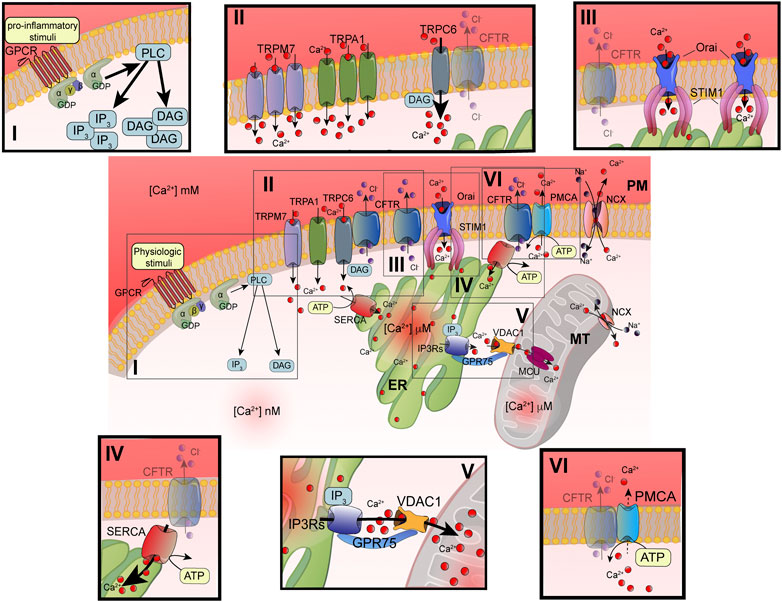
FIGURE 1. Principal defects in Ca2+ signaling associated with defective CFTR channel. Schematic representation of intracellular Ca2+ signaling in airway cells. The cellular Ca2+ homeostasis is regulated by systems of Ca2+-entry and Ca2+-efflux located in plasma membrane and organelles. In the inset are reported the principal molecular systems involved in the abnormal intracellular Ca2+ signaling associated with defective CFTR channel, where the their dysfunction contributes to physiopathology of CF lung disease: I) increased PLC activity, due to GPCR-dependent overstimulation; II) increased TRP-dependent Ca2+-entry, due to enhanced functional activity and/or expression; III) increased Orai insertion with consequent augments in Ca2+-influx; IV) increased SERCA activity; V) increased MCU activity; and VI) reduced Ca2+-efflux, due to altered PMCA activity. ATP, adenosine triphosphate; Ca2+, calcium; CFTR, cystic fibrosis transmembrane conductance regulator; Cl-, chloride; DAG, diacylglycerol; ER, endoplasmic reticulum; GPCR, G protein coupled receptor; GPD, guanosine diphosphate; GRP75, glucose-related protein 75; IP3, inositol 1,4,5-triphosphate; IP3Rs; inositol trisphosphate receptors; MCU, mitochondrial calcium uniporter; NCX, sodium-calcium exchanger; Orai, calcium release activated calcium channel; PLC, phospholipase C; PM, plasma membrane; PMCA, PM-resident Ca2+-ATPase; SERCA, ER-resident Ca2+-ATPase; STIM1; stromal interaction molecule 1; TRPC, transient receptor potential (TRP) channels; VDAC1, voltage-dependent anion-selective channel 1.
The advancements in the definition of Ca2+ signaling have evidenced the high spatiotemporal complexity and asynchronicity of Ca2+ responses. These responses are represented by localized [Ca2+]cyt spikes that gradually propagate into the cell as Ca2+ waves (Berridge et al., 2003). In nonexcitable cells, such as airway epithelial and immune cells, the [Ca2+]cyt spikes are caused by extracellular stimuli, which result in a Ca2+-influx from the extracellular space. Agonists, including some proinflammatory and infectious stimuli (e.g., cytokines, bradykinin, prostaglandins, lipopolysaccharides, bacterial flagellin, and pili), are translated in intracellular Ca2+ signals through their interaction and subsequent activation of membrane receptors, such as Toll-Like Receptors (TLR) and G protein coupled receptors (GPCRs) linked to phospholipase C (PLC) (Wootten et al., 2018). Activating PLC types, such as PLCβ or γ, catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2), giving rise to two second messengers: diacylglycerol (DAG) and 1,4,5-inositol trisphosphate (IP3) (Figure 1i) (Bill and Vines, 2020). DAG may activate transient receptor potential (TRP) canonic (TRPC) channels and classical and novel protein kinase C (PKC) isoforms (Rimessi et al., 2007; Curcic et al., 2019) (Figure 1ii). The DAG-triggering [Ca2+]i spikes occur through the direct binding with TRPC channels in PM, in a PKC-independent manner (Curcic et al., 2019). The subfamily of TRPC channels is composed of seven members (TRPC1-7). They are nonselective ion channels permeable to Na+ and Ca2+. TRPC3, TRPC6, and TRPC7 are the principal contributors of DAG-dependent Ca2+-entry in nonexcitable and excitable cells (Figure 1ii).
IP3 induces a transient increase in [Ca2+]cyt through the binding with IP3 receptors (IP3Rs), which in turn trigger ER Ca2+-release (Foskett et al., 2007) (Figure 1v). The temporal kinetics, the amplitude, and localization of generated Ca2+ spikes are strictly dependent on the nature of stimuli. All three IP3Rs induce local Ca2+ spikes with similar mean amplitudes, temporal characteristics, and spatial extents (Lock et al., 2018). Normally, IP3Rs are localized in cluster positioned near ER-mitochondria and ER-PM junctions, where the stromal interaction molecule (STIM)/Orai (Ca2+-release activated Ca2+ modulator 1) complex accumulates Ca2+ after the ER store depletion (Thillaiappan et al., 2017; Marchi et al., 2018; Taylor and Machaca, 2019). Intraluminal [Ca2+] depletion induces a conformational change in STIM1 and STIM2 isoforms, which translocate in ER-PM interface to bind the PM Orai channel (Figure 1iii). The STIM/Orai complex stimulates the Orai channel opening, giving rise to the store operated Ca2+-entry (SOCE) process from the extracellular space. This Ca2+-influx mechanism replenishes the ER Ca2+ stores and sustains the IP3Rs-dependent phase of increased [Ca2+]cyt (Bodnar et al., 2017). A preferential platform for clustering SOCE channels is the caveolae, PM lipid raft microdomains, where Ca2+ channels and their regulators are grouped to provide the Ca2+-entry also in an IP3R-dependent manner upon the intracellular administration of IP3 (Pani et al., 2008; Pulli et al., 2015).
Most of this free intracellular Ca2+ is bound by cytosolic proteins or organelles, such as mitochondria and lysosomes, which act as Ca2+ buffers (Schwaller, 2020). Another cytosolic Ca2+-binding protein involved in the regulation of intracellular Ca2+ signaling is Calmodulin. It binds Ca2+ through four high affinity binding sites, promoting a direct association and opening of CFTR channel (Bozoky et al., 2017) and regulation of the activity of protein kinases and Ca2+-pumps (Villalobo et al., 2018).
Mitochondria influence the [Ca2+]cyt by inducing a transient sequestering of the Ca2+ released at the ER-mitochondria interfaces (Marchi et al., 2018). In these intimate and dynamic regions between ER and mitochondrial outer membranes (OMMs), called also mitochondria-associated ER membranes (MAMs), a series of specialized molecular bridges control the frequency of interactions, the size and the spacing between the organelles, and changing at front of cellular and functional requests (Simmen and Herrera-Cruz, 2018; Bootman and Bultynck, 2020). Thus, the mitochondrial Ca2+-transfer is firstly favored by the distance from ER and number of mitochondria involved in these interorganelle couplings and secondly by the negative membrane potential in mitochondrial matrix generated by the respiratory chain (Csordas et al., 2010; Rimessi et al., 2015b). The Ca2+ is then transmitted into the matrix by the mitochondrial Ca2+ uniporter (MCU) (Figure 1v) (Baughman et al., 2011; De Stefani et al., 2011). MCU is a transmembrane protein of inner mitochondrial membrane (IMM), assembled as tetramer, which forms a high selective Ca2+-channel with low affinity for the ion. Its activity is regulated by the EF-hand-containing Ca2+-binding proteins mitochondrial calcium uptake 1 (MICU1) and MICU2, which together with other forming-channel elements, such as MCUb and essential MCU regulator (EMRE), constitute the MCU complex (Figure 2) (Marchi and Pinton, 2014). However, before reaching the IMM, Ca2+ must cross the OMM mediating the voltage-dependent anion channels (VDACs), involved also in the transport of adenosines (ATP, ADP) and metabolites, including pyruvate and malate (Shoshan-Barmatz et al., 2010). Three different isoforms of VDAC have been identified: VDAC1-3. Among them, VDAC1 has Ca2+ binding sites and is highly Ca2+ permeable and modulates the accessibility of ion to the mitochondrial intermembrane space (IMS) (Gincel et al., 2001). The mitochondrial Ca2+ is released more slowly back into the cytosol by Na+-dependent exchange mechanisms in excitable and nonexcitable cells via Na+/Ca2+ exchanger (NCX) and Na+/Ca2+/Li+ exchanger (NCLX) (Khananshvili, 2014; Kostic and Sekler, 2019). NCX, located on the OMM, may operate either in forward mode, extruding one Ca2+ ion from mitochondrial intermembrane space vs three Na+ ions in influx from cytosol, or in reverse mode, exchanging Ca2+-influx/Na+-efflux. NCLX, located on the IMM, transports Ca2+ outside the matrix in exchange of either Na+ or Li+ at similar rates (Figures 1, 2). In nonexcitable cells, the mitochondrial Ca2+ is also extruded by H+/Ca2+ exchanger (Nishizawa et al., 2013).
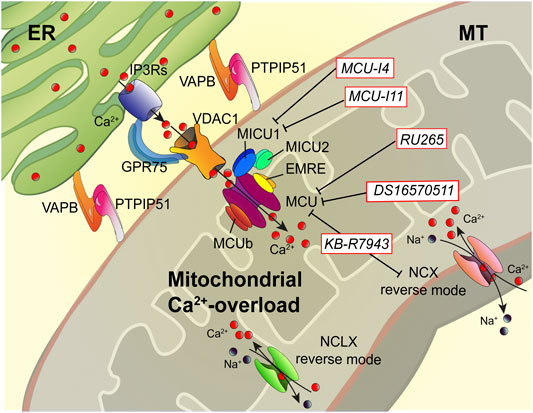
FIGURE 2. Dampening the mitochondrial Ca2+-overload in cystic fibrosis. The dysregulation of Ca2+ signaling in CF causes mitochondrial Ca2+-overload in airway cells during the recurrent pathogen infections, which leads to organelle dysfunction with repercussion on ROS production and inflammatory responses. The mitochondrial Ca2+-overload is mediated by an increased ER-mitochondria Ca2+ transfer through the IP3Rs-VDAC-MCU axis due to the stabilization of VAPB-PTPIP51 tethers. Indeed, the increased ENaC-dependent Na+ absorption due to defective CFTR in CF could stimulate NCX and NCLX exchangers to work in reverse mode triggering intracellular and mitochondrial Ca2+-influx, which may worsen the excessive mitochondrial Ca2+-uptake. To dampen the detrimental Ca2+ accumulation in matrix, a new class of Ca2+ modulator drugs are under investigation; the mitochondrial Ca2+-overload inhibitors act on MCU complex and mitochondrial Ca2+ exchangers in reverse mode to control the amount of Ca2+ imported into the matrix to avoid mitochondrial injury and oxidative stress in CF. Ca2+, calcium; EMRE, essential MCU regulator; ER, endoplasmic reticulum; GRP75, glucose-related protein 75; IP3Rs, inositol trisphosphate receptors; MCU, mitochondrial Ca2+ uniporter; MICU1, mitochondrial calcium uptake protein 1; MICU2, mitochondrial calcium uptake protein 2; MT, mitochondrion; Na+, sodium; NCX, sodium-calcium exchanger; NCLX, mitochondrial Na/Ca exchanger; PTPIP51, protein tyrosine phosphatase interacting protein 51; VAPB, vesicle-associated membrane protein-associated protein B; VDAC1, voltage-dependent anion-selective channel 1.
However, after removing the stimulus, the [Ca2+]cyt is rapidly lowered through the activation of Ca2+-ATPase pumps located on the PM and ER, respectively (Figure 1iv–vi). PM Ca2+-ATPase (PMCA) push out Ca2+ from cell while sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps Ca2+ back into the ER (Domi et al., 2007). These pumps are P-type ATPase, which exchange one (PMCA) or two (SERCA) Ca2+ ions for hydrolyzed ATP (Strehler and Treiman, 2004; Chen et al., 2020a). PMCA presents a high Ca2+-affinity but low Ca2+-transporting rate. In support of the PM Na+/Ca2+ exchangers, a second Ca2+-efflux system with low Ca2+-affinity but high Ca2+-transporting rate contributes to clamping the [Ca2+]cyt at homeostatic levels.
Abnormal Ca2+ Signaling in Cystic Fibrosis and Physiopathological Consequences
To date, increasing evidence highlights the importance of perturbed Ca2+ signaling in CF lung disease’s physiopathology. The abnormal Ca2+ profile observed in CF airway epithelial and immune cells is initially due to intrinsic defects associated with mutated CFTR. It is sustained successively by recurrent pathogen infections and by overstimulation of released proinflammatory mediators, resulting in detrimental lung inflammation (Ribeiro, 2006; Antigny et al., 2011a).
Defective CFTR and Ca2+ Signaling
Ca2+ signals have key roles in the CFTR channel function and in airway immune responses, which are perturbed in CF. Ca2+ signaling controls the CFTR protein expression levels and internalization (Bargon et al., 1992; Patel et al., 2019), while at level of airways, it regulates ciliary beating and secretion of fluid and antimicrobial agents (Salathe, 2007; Waterer, 2012; Lee and Foskett, 2014).
In CF, Ca2+ exacerbates the airway inflammatory responses (Figure 1i). Its dysregulation has been observed in several human CF patient-derived primary cells: airway epithelial cells (Rimessi et al., 2015a), bronchial goblet cells (Roomans, 1986), skin fibroblasts (Shapiro et al., 1978), kidney cells (Katz et al., 1988), and immune cells such as leukocytes, neutrophils, and lymphocytes (Banschbach et al., 1978; Waller et al., 1984; Robledo-Avila et al., 2018). In all of them, the [Ca2+]cyt was increased compared to non-CF cells, demonstrating that functional CFTR regulates the Ca2+ homeostasis conditioning and in turn the interorganelle Ca2+-transfer evoked by stimuli (Rimessi et al., 2015a).
The increased [Ca2+]cyt in CF airways results from an enhanced Ca2+-entry mediated by PM Ca2+-channels and reduced Ca2+-efflux operated by PMCA, influencing the Ca2+ accumulation into the stores (Figures 1i,ii,vi) (Philippe et al., 2015). In particular, the TRP channel family is involved in abnormal Ca2+-entry in CF airway cells (reviewed in (Grebert et al., 2019)) (Figure 1ii). TRPC6-mediated Ca2+-influx was increased in F508del-CFTR and G551D-CFTR airway cells with respect to non-CF cells when exposed to 1-oleoyl-sn-glycerol, a synthetic, cell-permeable compound analogous to DAG, used as an activator of PKC (Antigny et al., 2011b). No difference in TRPC6 expression justified the discrepancy, but it has been observed that the physical interaction between WT-CFTR and TRPC6 channel downregulated the Ca2+-influx in airway epithelial cells, suggesting that the lacked or reduced CFTR expression in PM in CF cells perturbs the functional coupling between the two channels favoring the abnormal Ca2+-entry (Antigny et al., 2011b). However, specific TRPC6-silencing or the CFTR-corrector agent’s administration, VX-770, reduced the abnormal [Ca2+]cyt increment and IL-8 release, indicating that defective CFTR worsens the inflammatory response affecting the TRPC6 activity (Vachel et al., 2013). Similar effects were also observed for TRP vanilloid 4 and 6 channels (TRPV4 and TRPV6), respectively. The intracellular Ca2+ elevation by TRPV4 stimulation led to CFTR channel activation in bronchial epithelial cells in physiological condition (Genovese et al., 2019). The increased Ca2+-influx and the inflammatory response in CF were attenuated by genetic manipulation of TRPV channels and by low temperature-mediated F508del-CFTR rescue in CFTR-deficient cells, mediating reduction of [Ca2+]cyt mitigated release of IL-8, prostaglandin E2 and keratinocyte chemo-attractant (Vachel et al., 2015; Henry et al., 2016).
In addition, an increased expression in PM of nonselective Ca2+ channel TRP subfamily M member 7 (TRPM7) has been observed in both F508del-CFTR- and G551D-CFTR-expressing cells, resulting in higher Ca2+-influx than WT-CFTR-expressing cells (Huguet et al., 2016). A similar increase in expression of TRP ankyrin subtype 1 channel (TRPA1) was observed in human CF respiratory epithelium and in CF airway cells. Here, TRPA1 pharmacological modulation controls the transcription and release of several proinflammatory mediators, including IL-8 and IL-1β, in a Ca2+-dependent manner (Prandini et al., 2016). Orai expression resulted in enhanced CF airway cells with a consequent increase in Ca2+-entry and release of IL-8 (Figure 1iii) (Balghi et al., 2011). The STIM1 migration and Orai activity do not occur only during ER depletion but may also occur upon mitochondrial Ca2+-efflux, as demonstrated in basophilic leukemia, embryonic fibroblast, and kidney cells, with dangerous repercussions on inflammation (Singaravelu et al., 2011; Delmotte et al., 2012).
Abnormal Ca2+-entry is a primary signal associated with defective CFTR that conditions the ER Ca2+-accumulation and mitochondrial Ca2+-overload elicited in CF cells, already due also to increased SERCA activity (Figure 1vi) (Philippe et al., 2015). The functional interaction between WT-CFTR with SERCA2b and PMCA observed in non-CF cells is partially lost in CF cells, due to ER retention of F508del-CFTR mutant, with several implications on the ER and PM Ca2+ channel activities, which potentiate the intraluminal Ca2+ accumulation (Norez et al., 2006). A study identified Calumenin as interactor of CFTR channel, a Ca2+-binding protein located primarily in the ER able to regulate ER Ca2+ homeostasis, interacting with SERCA and the ER Ca2+ channel ryanodine receptors (RyRs) (Vorum et al., 1999; Jung et al., 2006; Sahoo et al., 2009; Teng et al., 2012). The binding between Calumenin and CFTR increased when the channel presented the prevalent gating mutation G551D-CFTR (Teng et al., 2012). Indeed, Calumenin contributed to the ER retention of mutated F508del-CFTR channel, if silenced PM expression and activity of mutated CFTR channel were restored in bronchial epithelial cells (Philippe et al., 2017).
Mitochondrial Ca2+-uptake was increased in primary CF airway epithelial cells, skin fibroblasts, and lymphocytes (Feigal et al., 1982; Waller et al., 1984; Rimessi et al., 2015a), mediated by a greater ER Ca2+-transfer and by perturbed respiratory activity, which stimulated mitochondrial ROS production, mitochondrial injury, and release of mitochondrial damage-associated molecular patterns in CF lung (Figure 1v) (Feigal et al., 1982; Antigny et al., 2011a). Severe mitochondrial dysfunctions in basal condition were restricted to human F508del-CFTR tracheal gland CF-KM4 cell clone. In this cell clone, authors found a reduced mitochondrial Ca2+ uptake consequence of mitochondrial membrane depolarization and perturbed network, resulting from an altered mitochondrial physiology (Antigny et al., 2009).
At demonstration of the presence of an abnormal mitochondrial Ca2+ accumulation, different studies unveiled that, by using corrector agents, such as VX-770 and VX-809, or by rescuing functional F508del-CFTR, it is possible to normalize the mitochondrial [Ca2+] levels with beneficial repercussions on oxidative stress and the levels of proinflammatory mediators released, such as IL-8 and the inflammasome-dependent cytokine IL-1β (Vachel et al., 2013; Rimessi et al., 2015a; Philippe et al., 2015).
Ca2+ Signaling in CF Infection and Inflammation
Airway epithelial cells respond to pathogens, such as P. aeruginosa, through Ca2+-dependent mechanisms to produce proinflammatory mediators to initiate the inflammatory response (Ratner et al., 2001; Fu et al., 2007). Different bacterial constituents, including LPS, promote TRP-dependent Ca2+-entry and ER Ca2+-release via IP3Rs (Buyck et al., 2013). Pili and flagellin interact with TLR2, TLR4, TLR5, and Asialo ganglio-N-tetraosylceramide (Asialo GM1) receptor and induce the expression of IL-8 mediating the activation of NF-kB, which is also phosphorylated by Ca2+-dependent PKC isoforms α and β that respond to intracellular Ca2+ flux following IP3R-dependent ER Ca2+-release (Pinton et al., 2004; Asehnoune et al., 2005; Chun and Prince, 2006). This Ca2+-dependent activation of NF-kB is also sustained by Asialo GM1-dependent nucleotides released from airway epithelial cells that interact with flagellin, which binds the purinergic P2Y receptors active intracellular Ca2+-signaling (Mcnamara et al., 2006; Billet and Hanrahan, 2013).
In CF, the airway epithelial cells respond to the recurrent infection generating abnormal Ca2+ mobilization to produce many cytokines and chemokines, useful to recruit leukocytes to contrast the accumulated bacteria in the airways. This overstimulation determines a Ca2+-dependent hyperinflammation phenotype. CF airway epithelial cells result in hyperresponsiveness to pathogens due to the increased [Ca2+]cyt, which contributes to 1) ER expansion and increased intraluminal [Ca2+]; 2) mitochondrial Ca2+-overload and consequent organelle dysfunction; and 3) an exuberant and more prolonged NF-kB activation, priming the cells to excessive expression and release of proinflammatory mediators (Ribeiro et al., 2005; Tabary et al., 2006; Rimessi et al., 2015a; Rimessi et al., 2020).
The ER Ca2+ store expansion in CF airway cells is due to activation of the inositol-requiring enzyme 1 (IRE1)/X-box binding 1 (XBP-1) pathway, which is not the consequence of misfolded CFTR. Reductions in ER Ca2+-release in CF samples were also obtained by correcting F508del-CFTR trafficking by miglustat (N-butyldeoxynojirimycin) or low temperature (27°C) (Antigny et al., 2008a; Antigny et al., 2008b). Antigny et al. demonstrated that the abnormal ER Ca2+-release in CF gland CF-KM4 clone was due to a dysfunctional IP3Rs, consequence of ER retention of mutant CFTR channel (Antigny et al., 2008b). The importance of IP3Rs in CF was then confirmed by Martins et al. in nasal epithelial cells, where it was demonstrated that the ER retention of F508del-CFTR determined a functional interference with IP3-receptor binding protein IRBIT, which suppresses the activation of IP3Rs by competing with IP3 for binding to the ligand-binding domain (Ando et al., 2006; Martins et al., 2011).
However, the increased flux of newly synthesized proinflammatory mediators into the ER in response to recurrent infections contributes to ER expansion (Ribeiro and Lubamba, 2017). Besides, changes in the intracellular redistribution of ER have been observed in response to pathogens. In this case, the ER moved to the apical level of polarized CF airway epithelial cells to facilitate the GPCR-induced Ca2+ responses (Ribeiro et al., 2005).
Recently, Rimessi et al. demonstrated that P. aeruginosa infection increases ER-mitochondria juxtapositions in CF airway epithelial cells by stabilizing the ER protein vesicle-associated membrane protein-associated protein B (VAPB) and the outer mitochondrial membrane protein tyrosine phosphatase interacting protein 51 (PTPIP51) tethers, favoring the mitochondrial Ca2+ transfer via MCU (Figure 2) (Rimessi et al., 2020). This led to mitochondrial membrane potential loss, ROS production, and organelle dysfunction, inducing persistent mitochondrial Unfolding Protein Response (UPRmt) and NLRP3 inflammasome activation. In turn, these processes downregulated the selective autophagic responses, mitophagy, and xenophagy, resulting in augmented pathogen survival and worsening of inflammatory response (Rimessi et al., 2015a; Rimessi et al., 2020). Thus, the mitochondrial Ca2+-overload in CF airway cells plays a crucial role in the evolution of CF pulmonary inflammation. Preventing the mitochondrial Ca2+-overload, via MCU inhibition, the P. aeruginosa-dependent mitochondrial dysfunction was abrogated in CF airway cells, while the selective autophagic responses were rectified (Rimessi et al., 2020).
A higher predisposition of CF airway cells to NLRP3 inflammasome activation is also due to the dysregulation of ENaC-dependent Na+-influx associated with defective CFTR, which predisposes the cells to K+-efflux, a further activating signal to NLRP3 inflammasome (Scambler et al., 2019).
Excessive Ca2+-dependent IL-8 secretion is critical for CF lung disease development and is responsible for abundant neutrophil recruitment into the lung. IL-8 production is 13-fold higher in CF bronchial cells than non-CF cells and occurs through persistent and prolonged NF-kB activation (Tabary et al., 2000). A Single Nucleotide Polymorphisms (SNP) genetic study from a panel of 135 genes implicated in the signal transduction for neutrophil recruitment, identifying PLC beta-3 (PLCB3) gene on top of the rank, involved in the excessive expression and release of IL-8 during P. aeruginosa infection in F508del-CFTR patients (Bezzerri et al., 2011). The c.2534C > T (p.S845L) PLCB3 is a loss-of-function variant associated with a mild progression of CF lung disease, where its inability to trigger intracellular Ca2+ transient limited the activation of Ca2+-dependent PKCs and NF-kB, reducing the P. aeruginosa-dependent induction of IL-8 transcription and protein release in primary CF patient-derived airway epithelial cells (Rimessi et al., 2018). Similar effects have been observed by inhibiting the Ca2+-dependent PKCα isoform with β-sitosterol, which blocked the P. aeruginosa-triggering IL-8 induction and release in CF airway cells (Lampronti et al., 2017).
The abnormal intracellular Ca2+ signaling associated with defective CFTR also implies the abundant neutrophils recruited into CF lung during the recurrent bacterial infections. An increased [Ca2+]cyt has been measured in human CF neutrophils compared to non-CF, which correlates with a reduced antimicrobial killing capacity due to diminished NADPH oxidase response and impaired secretion of neutrophil extracellular traps (Robledo-Avila et al., 2018). The higher [Ca2+]cyt in CF neutrophils is sustained by increased Ca2+-entry via TRP channels, especially TRPM2 and TRPM7 channels, which when inhibited with 2-aminoethoxydiphenylborane restored the antimicrobial response of CF neutrophils during infection, preventing the intracellular Ca2+-overloading (Heiner et al., 2005; Park et al., 2014; Robledo-Avila et al., 2018).
Targeting Ca2+ Signaling as Alternative Anti-Inflammatory Approach
Understanding the molecular mechanisms that induce hyperinflammation in CF lung through the Ca2+ signaling impairment helps to identify new and alternative therapeutic targets to treat the CF lung disease. Thus, pharmacological Ca2+ signaling-targeting agents aim to control the increased [Ca2+]cyt, ER, and mitochondrial Ca2+-overload in CF airway epithelial and immune cells, which may be considered a new class of anti-inflammatory drugs to prevent the hyperinflammatory response in CF.
TRP Channel Inhibitors
TRPA1 results in a druggable target to control the excessive inflammation in CF. The selective inhibition of TRPA1, by HC03 or A96 antagonist, reduced the induction and release of IL-8, IL-1β, and TNFα in CF patient-derived airway cells during P. aeruginosa infection (Prandini et al., 2016). Recently, the new antagonists of TRPA1 ODM-108, CB-625, CB-189625, and HX-100, which were under investigation in different phases of clinical trials to treat asthma and chronic obstructive pulmonary disease, have been discontinued for pharmacokinetics reasons (Chen and Terrett, 2020). In addition, GRC-17536, which had obtained promising results in preclinical studies on inflammatory animal models, has been suspended from clinical trials (Preti et al., 2012; Mukhopadhyay et al., 2014; Chen and Terrett, 2020). Also, TRPV4 plays a role in the excessive Ca2+-entry in CF. A new inhibitor derived from the TRPV4-inhibitor GSK205, called compound 16-8, has been developed to target simultaneously TRPV4 and TRPA1 channel to block the Ca2+-influx, showing the potentially advantageous property to apply to CF hyperinflammation (Kanju et al., 2016).
TRPC6 is another druggable target to counteract the increased [Ca2+]cyt in CF. BI-749327 is an orally selective TRPC6-inhibitor used to suppress renal inflammatory cell infiltration and fibrosis, ameliorating renal stress-induced disease (Lin et al., 2019). SAR7334 was initially identified as a potent TRPC6-inhibitor, but this agent may inhibit the Ca2+-influx mediated by TRPC3 and TRPC7. SAR7334 attenuated the IL-6 and IL-8 release in human bronchial epithelial cells exposed to ozone (O3), protecting from the O3-induced airway inflammatory response in vivo (Chen et al., 2020b). The nonselectivity of TRPC antagonist SKF-96365 limits the clinical usefulness of this drug although it reduced the LPS-dependent secretion of TNFα and IL-6 in microglia. In contrast, carvacrol, a phenolic monoterpene, through the selective inhibition of TRPM7, reduced the oversecretion of proinflammatory cytokines such as IL-1β, TNFα, and IL-6 in endotoxemic rats (Heo et al., 2015; Gatica et al., 2019).
PLC Inhibitors
PLC activity concurs to abnormal Ca2+ signaling in CF inducing the TRP channels activation and ER Ca2+-release through the recurrent generation of DAG and IP3. Thus, the inhibition of PLC may contribute to attenuating the hyperinflammatory response in CF, reducing the Ca2+-entry and the mitochondrial Ca2+-overload limiting TRP channels activation and ER-mitochondria Ca2+-transfer, respectively. Edelfosine was the first PLC inhibitor identified, decreasing the ER Ca2+-release in tumor cells, but its cytotoxicity limits the clinical usefulness (Berkovic, 1998). U73122 is another PLC inhibitor and is used as an anti-inflammatory agent in different pathological contexts. U73122 may inhibit the LPS- or influenza A virus-induced expression of cytokines IL-1β and TNFα in human promonocyte U937 cells and in mouse primary peritoneal macrophages (Zhu et al., 2015; Zhu et al., 2016). Unfortunately, accumulating reports highlight off-target effects of U73122, including SERCA, Kir3, and Ca2+-activated K+ channels, calling into question its selectivity (Klose et al., 2008; Hollywood et al., 2010). A high-throughput analysis has been performed on 6,280 compounds, identifying three putative PLC activity inhibitors. Unfortunately, these compounds are not optimal to inhibit cellular PLC activity because they present a reduced cell permeability and a limited potency. Nevertheless, these compounds may be useful for development of new drugs to interrupt the abnormal signaling cascades controlled by PLCs, for the treatment of human diseases, including cancer (Huang et al., 2013).
Mitochondrial Ca2+-Overload Inhibitors
The mitochondrial Ca2+-overload inhibitors are pharmacological agents that by preventing the detrimental Ca2+ accumulation in the matrix reduce mitochondrial injury and oxidative stress, which are necessary to amplify the proinflammatory signals and activate NLRP3 inflammasome in CF lung disease (Figure 2) (Rimessi et al., 2015a; Rimessi et al., 2020). Limiting the excessive Ca2+ transport into mitochondria, via MCU, represents the first therapeutic approach with promising results both in vitro and in vivo in CF (Rimessi et al., 2015a; Rimessi et al., 2020). The MCU inhibitor, KB-R7943, reduced lung inflammation in P. aeruginosa-inoculated CF mice. The treated mice showed a reduction of interstitial inflammatory infiltrate with a general reduction in the congestion of lung parenchyma and restoration at the level of interalveolar septa of inflammatory infiltrate clearing conditions (Rimessi et al., 2020). Indeed, KB-R7943 rectified the unbalanced selective autophagic activities, thus restoring mitochondrial quality control and bacterial clearance capacity in CF airway cells. KB-R7943, designed to inhibit NCX in reverse mode, is the first cell-permeable MCU inhibitor available (Iwamoto et al., 1996; Santo-Domingo et al., 2007). Its nonspecificity and toxicity at high concentrations have limited its clinical usefulness, but its multitasking activity in CF on inflammation, mitochondrial stress response, and autophagy could represent an important starting point to develop new drugs to treat CF (Figure 2). A new class of selective and cell-permeable MCU inhibitors is now commercially available, namely, Ru265 and DS16570511, until now used only in vitro, but could have therapeutic implications in CF in the future (Kon et al., 2017; Woods et al., 2019).
The abnormal mitochondrial Ca2+-uptake in CF may be controlled through MCU and mitochondrial NCX reverse targeting. The increased ENaC-dependent Na+ absorption in CF could stimulate NCX and NCLX exchanger to work in reverse mode, triggering intracellular and mitochondrial Ca2+-influx (Berdiev et al., 2009; Verkhratsky et al., 2018).
As an alternative, mitochondrial Ca2+-overload may be prevented by inhibiting MICU1 activity (Figure 2). MCU complex comprises the pore-forming MCU protein, EMRE, and the gatekeepers MICU1 and MICU2, which regulate the MCU activity sensing the changes in [Ca2+]cyt. Recently, two new pharmacological MICU1 inhibitors have been developed, MCU-i4 and MCU-i11, both blocking the IP3-dependent mitochondrial Ca2+-uptake, maintaining the gatekeeping role of their target (Di Marco et al., 2020). Hence, the MICU1 inhibitors should allow a greater fine-tuning modulation of mitochondrial Ca2+-uptake than the known MCU inhibitors. This aspect could be relevant to treat the hyperinflammation in CF lung disease. The last strategy feasible should be to activate the mitochondrial Ca2+-efflux mechanisms, but selective activators or inducer have not been identified.
Some potential pitfalls could emerge about mitochondrial Ca2+-overload inhibitors, concerning the safety and biochemical stability of new molecules in vivo, the limitations in current knowledge (being recently discovered), and the long-term efficacy of mitochondrial Ca2+ signaling modulation that would be expected to alter the cell metabolism. However, no differences in basal oxygen consumption have been observed between WT and MCU-KO mice, suggesting that basal metabolism was not markedly altered in absence of mitochondrial Ca2+ signal (Pan et al., 2013; Murphy et al., 2014).
The enhancement of mitochondrial quality control through the pharmacological modulation of mitochondrial Ca2+ signaling is emerging as alternative anti-inflammatory strategy for the prevention or treatment of mitochondrial-associated disorders, such as CF (Patergnani et al., 2020b). Drugs that directly affect mitochondria, and thus mitochondrial Ca2+ signaling, have been recently used as a main mode of action to treat diseases, such as type 2 diabetes and cancer, as well as upregulating the immune system to clear infection with promising success (Stoker et al., 2019).
Conclusion
Substantial evidence supports the theory that the dysregulation in Ca2+ signaling associated with defective CFTR is essential for the development of the hyperinflammatory phenotype observed in CF lungs. This dysregulation involves different cells leading to multifunctional defects in CF patients. Both airway epithelial and immune cells are affected, with heavy repercussions on cell function, viability, and susceptibility to pathogens, which contribute significantly to the degeneration of pathological conditions of CF lung disease. Targeting the abnormal Ca2+ signaling in CF represents a new and attractive therapeutic strategy useful for reducing the proinflammatory overstimulation, organelle dysfunction, oxidative stress, and cytokines release in the CF lung.
“Correctors” and “potentiators” that are the new frontier in CF therapy, despite their positive impact in the CF community, are debated about their downstream consequences, in particular on inflammation. Evidence shows that P. aeruginosa burden decreased in the first six months of modulator therapy but rebounded thereafter, increasing the inflammatory response (Hisert et al., 2017). In addition, P. aeruginosa has been shown to directly reduce the apical membrane expression of rescued ΔF508CFTR and the following chloride secretion (Rubino et al., 2014). These facts are relevant since the presence of P. aeruginosa infection could per se render the recent CF therapies less effective. Therefore, alternative approaches aimed at activating early anti-inflammatory pathways to prevent organ damage before patients become symptomatic are needed. To date, new class of alternative anti-inflammatory drugs is emerging to prevent the inflammatory signal amplification and tissue degeneration related to chronic inflammation in CF. The cure of the lung pathology of CF patients will rely on the association of drugs acting as “correctors” and “potentiators” on the mutated CFTR protein together with novel anti-inflammatory drugs, such as the Ca2+-modulators, and more active antibacterial drugs against P. aeruginosa.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
AR is supported by the Italian Cystic Fibrosis Research Foundation, grant FFC #20/2015, local funds from the University of Ferrara, FIR-2017, the Italian Ministry of Health (GR-2016-02364602), and the Italian Ministry of Education, University and Research (PRIN Grant 2017XA5J5N). The research was funded by grants from the Italian Cystic Fibrosis Research Foundation, FFC#12/2010 and #19/2014, Italian Association for Cancer Research (AIRC, IG-23670), Telethon (GGP11139B), local funds from the University of Ferrara, and the Italian Ministry of Education, University and Research (PRIN Grant 2017E5L5P3) to PP. PP is grateful to Camilla degli Scrovegni for continuous support. SP is supported by Fondazione Umberto Veronesi.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with some of the authors, AR and PP.
References
Ando, H., Mizutani, A., Kiefer, H., Tsuzurugi, D., Michikawa, T., and Mikoshiba, K. (2006). IRBIT suppresses IP3 receptor activity by competing with IP3 for the common binding site on the IP3 receptor. Mol. Cel. 22, 795–806. doi:10.1016/j.molcel.2006.05.017
Antigny, F., Girardin, N., Raveau, D., Frieden, M., Becq, F., and Vandebrouck, C. (2009). Dysfunction of mitochondria Ca2+ uptake in cystic fibrosis airway epithelial cells. Mitochondrion 9, 232–241. doi:10.1016/j.mito.2009.02.003
Antigny, F., Norez, C., Becq, F., and Vandebrouck, C. (2008a). Calcium homeostasis is abnormal in cystic fibrosis airway epithelial cells but is normalized after rescue of F508del-CFTR. Cell Calcium 43, 175–183. doi:10.1016/j.ceca.2007.05.002
Antigny, F., Norez, C., Becq, F., and Vandebrouck, C. (2011a). CFTR and Ca signaling in cystic fibrosis. Front. Pharmacol. 2, 67. doi:10.3389/fphar.2011.00067
Antigny, F., Norez, C., Cantereau, A., Becq, F., and Vandebrouck, C. (2008b). Abnormal spatial diffusion of Ca2+ in F508del-CFTR airway epithelial cells. Respir. Res. 9, 70. doi:10.1186/1465-9921-9-70
Antigny, F., Norez, C., Dannhoffer, L., Bertrand, J., Raveau, D., Corbi, P., et al. (2011b). Transient receptor potential canonical channel 6 links Ca2+ mishandling to cystic fibrosis transmembrane conductance regulator channel dysfunction in cystic fibrosis. Am. J. Respir. Cel Mol. Biol. 44, 83–90. doi:10.1165/rcmb.2009-0347OC
Asehnoune, K., Strassheim, D., Mitra, S., Yeol Kim, J., and Abraham, E. (2005). Involvement of PKCalpha/beta in TLR4 and TLR2 dependent activation of NF-kappaB. Cell Signal 17, 385–394. doi:10.1016/j.cellsig.2004.08.005
Balghi, H., Robert, R., Rappaz, B., Zhang, X., Wohlhuter-Haddad, A., Evagelidis, A., et al. (2011). Enhanced Ca2+ entry due to Orai1 plasma membrane insertion increases IL-8 secretion by cystic fibrosis airways. FASEB J. 25, 4274–4291. doi:10.1096/fj.11-187682
Banschbach, M. W., Karam, A. G., Love, P. K., and Hilman, B. C. (1978). Cystic fibrosis serum promotes [45Ca] uptake by normal human leukocytes. Biochem. Biophys. Res. Commun. 84, 922–927. doi:10.1016/0006-291x(78)91671-6
Bargon, J., Trapnell, B. C., Chu, C. S., Rosenthal, E. R., Yoshimura, K., Guggino, W. B., et al. (1992). Down-regulation of cystic fibrosis transmembrane conductance regulator gene expression by agents that modulate intracellular divalent cations. Mol. Cel Biol. 12, 1872–1878. doi:10.1128/mcb.12.4.1872
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi:10.1038/nature10234
Berdiev, B. K., Qadri, Y. J., and Benos, D. J. (2009). Assessment of the CFTR and ENaC association. Mol. Biosyst. 5, 123–127. doi:10.1039/b810471a
Berkovic, D. (1998). Cytotoxic etherphospholipid analogues. Gen. Pharmacol. 31, 511–517. doi:10.1016/s0306-3623(98)00081-0
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cel Biol. 4, 517–529. doi:10.1038/nrm1155
Bezzerri, V., D'adamo, P., Rimessi, A., Lanzara, C., Crovella, S., Nicolis, E., et al. (2011). Phospholipase C-beta3 is a key modulator of IL-8 expression in cystic fibrosis bronchial epithelial cells. J. Immunol. 186, 4946–4958. doi:10.4049/jimmunol.1003535
Bill, C. A., and Vines, C. M. (2020). Phospholipase C. Adv. Exp. Med. Biol. 1131, 215–242. doi:10.1007/978-3-030-12457-1_9
Billet, A., and Hanrahan, J. W. (2013). The secret life of CFTR as a calcium-activated chloride channel. J. Physiol. (Lond) 591, 5273–5278. doi:10.1113/jphysiol.2013.261909
Bodnar, D., Chung, W. Y., Yang, D., Hong, J. H., Jha, A., and Muallem, S. (2017). STIM-TRP pathways and microdomain organization: Ca2+ influx channels: the orai-STIM1-TRPC complexes. Adv. Exp. Med. Biol. 993, 139–157. doi:10.1007/978-3-319-57732-6_8
Bootman, M. D., and Bultynck, G. (2020). Fundamentals of cellular calcium signaling: a primer. Cold Spring Harb Perspect. Biol. 12, a038802. doi:10.1101/cshperspect.a038802
Boucher, R. C., Cotton, C. U., Gatzy, J. T., Knowles, M. R., and Yankaskas, J. R. (1988). Evidence for reduced Cl- and increased Na+ permeability in cystic fibrosis human primary cell cultures. J. Physiol. (Lond) 405, 77–103. doi:10.1113/jphysiol.1988.sp017322
Bozoky, Z., Ahmadi, S., Milman, T., Kim, T. H., Du, K., Di Paola, M., et al. (2017). Synergy of cAMP and calcium signaling pathways in CFTR regulation. Proc. Natl. Acad. Sci. USA 114, E2086–E2095. doi:10.1073/pnas.1613546114
Bruscia, E. M., and Bonfield, T. L. (2016). Innate and adaptive immunity in cystic fibrosis. Clin. Chest Med. 37, 17–29. doi:10.1016/j.ccm.2015.11.010
Buyck, J. M., Verriere, V., Benmahdi, R., Higgins, G., Guery, B., Matran, R., et al. (2013). P. aeruginosa LPS stimulates calcium signaling and chloride secretion via CFTR in human bronchial epithelial cells. J. Cyst Fibros 12, 60–67. doi:10.1016/j.jcf.2012.06.007
Carafoli, E., and Krebs, J. (2016). Why calcium? How calcium became the best communicator. J. Biol. Chem. 291, 20849–20857. doi:10.1074/jbc.R116.735894
Chen, H., and Terrett, J. A. (2020). Transient receptor potential ankyrin 1 (TRPA1) antagonists: a patent review (2015–2019). Expert Opin. Ther. Pat. 30, 643–657. doi:10.1080/13543776.2020.1797679
Chen, J., Sitsel, A., Benoy, V., Sepúlveda, M. R., and Vangheluwe, P. (2020a). Primary active Ca2+ transport systems in health and disease. Cold Spring Harb Perspect. Biol. 12, a035113. doi:10.1101/cshperspect.a035113
Chen, Q., Zhou, Y., Zhou, L., Fu, Z., Yang, C., Zhao, L., et al. (2020b). TRPC6-dependent Ca2+ signaling mediates airway inflammation in response to oxidative stress via ERK pathway. Cell Death Dis. 11, 170. doi:10.1038/s41419-020-2360-0
Chun, J., and Prince, A. (2006). Activation of Ca2+-dependent signaling by TLR2. J. Immunol. 177, 1330–1337. 177/2/133010.4049/jimmunol.177.2.1330.
Csordas, G., Várnai, P., Golenár, T., Roy, S., Purkins, G., Schneider, T. G., et al. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cel. 39, 121–132. doi:10.1016/j.molcel.2010.06.029
Curcic, S., Schober, R., Schindl, R., and Groschner, K. (2019). TRPC-mediated Ca2+ signaling and control of cellular functions. Semin. Cel Dev. Biol. 94, 28–39. doi:10.1016/j.semcdb.2019.02.001
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi:10.1038/nature10230
Delmotte, P., Yang, B., Thompson, M. A., Pabelick, C. M., Prakash, Y. S., and Sieck, G. C. (2012). Inflammation alters regional mitochondrial Ca2+ in human airway smooth muscle cells. Am. J. Physiol. Cel Physiol. 303, C244–C256. doi:10.1152/ajpcell.00414.2011
Di, A., Brown, M. E., Deriy, L. V., Li, C., Szeto, F. L., Chen, Y., et al. (2006). CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cel Biol. 8, 933–944. doi:10.1038/ncb1456
Di Marco, G., Vallese, F., Jourde, B., Bergsdorf, C., Sturlese, M., De Mario, A., et al. (2020). A high-throughput screening identifies MICU1 targeting compounds. Cell Rep. 30, 2321–2331.e6. doi:10.1016/j.celrep.2020.01.081
Domi, T., Di Leva, F., Fedrizzi, L., Rimessi, A., and Brini, M. (2007). Functional specificity of PMCA isoforms? Ann. N. Y Acad. Sci. 1099, 237–246. doi:10.1196/annals.1387.043
Farinha, C. M., King-Underwood, J., Sousa, M., Correia, A. R., Henriques, B. J., Roxo-Rosa, M., et al. (2013). Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 20, 943–955. doi:10.1016/j.chembiol.2013.06.004
Feigal, R. J., Tomczyk, M. S., and Shapiro, B. L. (1982). The calcium abnormality in cystic fibrosis mitochondria: relative role of respiration and ATP hydrolysis. Life Sci. 30, 93–98. doi:10.1016/0024-3205(82)90640-3
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658. doi:10.1152/physrev.00035.2006
Fu, Z., Bettega, K., Carroll, S., Buchholz, K. R., and Machen, T. E. (2007). Role of Ca2+ in responses of airway epithelia to Pseudomonas aeruginosa, flagellin, ATP, and thapsigargin. Am. J. Physiol. Lung Cel. Mol. Physiol. 292, L353–L364. doi:10.1152/ajplung.00042.2006
Gatica, S., Eltit, F., Santibanez, J. F., Varela, D., Cabello-Verrugio, C., and Simon, F. (2019). Expression suppression and activity inhibition of TRPM7 regulate cytokine production and multiple organ dysfunction syndrome during endotoxemia: a new target for sepsis. Curr. Mol. Med. 19, 547–559. doi:10.2174/1566524019666190709181726
Genovese, M., Borrelli, A., Venturini, A., Guidone, D., Caci, E., Viscido, G., et al. (2019). TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J. Physiol. 597, 5859–5878. doi:10.1113/JP278784
Gincel, D., Zaid, H., and Shoshan-Barmatz, V. (2001). Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358, 147–155. doi:10.1042/0264-6021:3580147
Giorgi, C., Danese, A., Missiroli, S., Patergnani, S., and Pinton, P. (2018). Calcium dynamics as a machine for decoding signals. Trends Cel Biol. 28, 258–273. doi:10.1016/j.tcb.2018.01.002
Grebert, C., Becq, F., and Vandebrouck, C. (2019). Focus on TRP channels in cystic fibrosis. Cell Calcium 81, 29–37. doi:10.1016/j.ceca.2019.05.007
Heiner, I., Radukina, N., Eisfeld, J., Kühn, F., and Lückhoff, A. (2005). Regulation of TRPM2 channels in neutrophil granulocytes by ADP-ribose: a promising pharmacological target. Naunyn Schmiedebergs Arch. Pharmacol. 371, 325–333. doi:10.1007/s00210-005-1033-y
Henry, C. O., Dalloneau, E., Pérez-Berezo, M. T., Plata, C., Wu, Y., Guillon, A., et al. (2016). In vitro and in vivo evidence for an inflammatory role of the calcium channel TRPV4 in lung epithelium: potential involvement in cystic fibrosis. Am. J. Physiol. Lung Cel. Mol. Physiol. 311, L664–L675. doi:10.1152/ajplung.00442.2015
Heo, D. K., Lim, H. M., Nam, J. H., Lee, M. G., and Kim, J. Y. (2015). Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cel Signal 27, 177–186. doi:10.1016/j.cellsig.2014.11.003
Hisert, K. B., Heltshe, S. L., Pope, C., Jorth, P., Wu, X., Edwards, R. M., et al. (2017). Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 195, 1617–1628. doi:10.1164/rccm.201609-1954OC
Hollywood, M. A., Sergeant, G. P., Thornbury, K. D., and Mchale, N. G. (2010). The PI-PLC inhibitor U-73122 is a potent inhibitor of the SERCA pump in smooth muscle. Br. J. Pharmacol. 160, 1293–1294. doi:10.1111/j.1476-5381.2010.00795.x
Huang, W., Barrett, M., Hajicek, N., Hicks, S., Harden, T. K., Sondek, J., et al. (2013). Small molecule inhibitors of phospholipase C from a novel high-throughput screen. J. Biol. Chem. 288, 5840–5848. doi:10.1074/jbc.M112.422501
Huguet, F., Calvez, M. L., Benz, N., Le Hir, S., Mignen, O., Buscaglia, P., et al. (2016). Function and regulation of TRPM7, as well as intracellular magnesium content, are altered in cells expressing DeltaF508-CFTR and G551D-CFTR. Cell Mol. Life Sci. 73, 3351–3373. doi:10.1007/s00018-016-2149-6
Iwamoto, T., Watano, T., and Shigekawa, M. (1996). A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J. Biol. Chem. 271, 22391–22397. doi:10.1074/jbc.271.37.22391
Jacquot, J., Tabary, O., Le Rouzic, P., and Clement, A. (2008). Airway epithelial cell inflammatory signalling in cystic fibrosis. Int. J. Biochem. Cel Biol 40, 1703–1715. doi:10.1016/j.biocel.2008.02.002
Jung, D. H., Mo, S. H., and Kim, D. H. (2006). Calumenin, a multiple EF-hands Ca2+-binding protein, interacts with ryanodine receptor-1 in rabbit skeletal sarcoplasmic reticulum. Biochem. Biophys. Res. Commun. 343, 34–42. doi:10.1016/j.bbrc.2006.02.115
Kanju, P., Chen, Y., Lee, W., Yeo, M., Lee, S. H., Romac, J., et al. (2016). Small molecule dual-inhibitors of TRPV4 and TRPA1 for attenuation of inflammation and pain. Sci. Rep. 6, 26894. doi:10.1038/srep26894
Katz, S. M., Krueger, L. J., and Falkner, B. (1988). Microscopic nephrocalcinosis in cystic fibrosis. N. Engl. J. Med. 319, 263–266. doi:10.1056/NEJM198808043190502
Khananshvili, D. (2014). Sodium-calcium exchangers (NCX): molecular hallmarks underlying the tissue-specific and systemic functions. Pflugers Arch. 466, 43–60. doi:10.1007/s00424-013-1405-y
Klose, A., Huth, T., and Alzheimer, C. (2008). 1-[6-[[(17beta)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) selectively inhibits Kir3 and BK channels in a phospholipase C-independent fashion. Mol. Pharmacol. 74, 1203–1214. doi:10.1124/mol.108.047837
Kon, N., Murakoshi, M., Isobe, A., Kagechika, K., Miyoshi, N., and Nagayama, T. (2017). DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 3, 17045. doi:10.1038/cddiscovery.2017.45
Kostic, M., and Sekler, I. (2019). Functional properties and mode of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin. Cel Dev. Biol. 94, 59–65. doi:10.1016/j.semcdb.2019.01.009
Lampronti, I., Dechecchi, M. C., Rimessi, A., Bezzerri, V., Nicolis, E., Guerrini, A., et al. (2017). Beta-sitosterol reduces the expression of chemotactic cytokine genes in cystic fibrosis bronchial epithelial cells. Front. Pharmacol. 8, 236. doi:10.3389/fphar.2017.00236
Lee, R. J., and Foskett, J. K. (2014). Ca2+ signaling and fluid secretion by secretory cells of the airway epithelium. Cell Calcium 55, 325–336. doi:10.1016/j.ceca.2014.02.001
Lin, B. L., Matera, D., Doerner, J. F., Zheng, N., Del Camino, D., Mishra, S., et al. (2019). In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. U S A. 116, 10156–10161. doi:10.1073/pnas.1815354116
Lock, J. T., Alzayady, K. J., Yule, D. I., and Parker, I. (2018). All three IP3 receptor isoforms generate Ca2+ puffs that display similar characteristics. Sci. Signal. 11, 11. doi:10.1126/scisignal.aau0344
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. doi:10.1016/j.ceca.2017.05.003
Marchi, S., and Pinton, P. (2014). The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J. Physiol. (Lond) 592, 829–839. doi:10.1113/jphysiol.2013.268235
Martins, J. R., Kongsuphol, P., Sammels, E., Dahimène, S., Aldehni, F., Clarke, L. A., et al. (2011). F508del-CFTR increases intracellular Ca2+ signaling that causes enhanced calcium-dependent Cl(−) conductance in cystic fibrosis. Biochim. Biophys. Acta 1812, 1385–1392. doi:10.1016/j.bbadis.2011.08.008
Mcnamara, N., Gallup, M., Sucher, A., Maltseva, I., Mckemy, D., and Basbaum, C. (2006). AsialoGM1 and TLR5 cooperate in flagellin-induced nucleotide signaling to activate Erk1/2. Am. J. Respir. Cel. Mol. Biol. 34, 653–660. doi:10.1165/rcmb.2005-0441OC
Mukhopadhyay, I., Kulkarni, A., Aranake, S., Karnik, P., Shetty, M., Thorat, S., et al. (2014). Transient receptor potential ankyrin 1 receptor activation in vitro and in vivo by pro-tussive agents: GRC 17536 as a promising anti-tussive therapeutic. PLoS One 9, e97005. doi:10.1371/journal.pone.0097005
Murphy, E., Pan, X., Nguyen, T., Liu, J., Holmström, K. M., and Finkel, T. (2014). Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 449, 384–385. doi:10.1016/j.bbrc.2014.04.144
Ng, H. P., Valentine, V. G., and Wang, G. (2016). CFTR targeting during activation of human neutrophils. J. Leukoc. Biol. 100, 1413–1424. doi:10.1189/jlb.4A0316-130RR
Nishizawa, T., Kita, S., Maturana, A. D., Furuya, N., Hirata, K., Kasuya, G., et al. (2013). Structural basis for the counter-transport mechanism of a H+/Ca2+ exchanger. Science 341, 168–172. doi:10.1126/science.1239002
Norez, C., Antigny, F., Becq, F., and Vandebrouck, C. (2006). Maintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cells. Traffic 7, 562–573. doi:10.1111/j.1600-0854.2006.00409.x
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cel Biol. 15, 1464–1472. doi:10.1038/ncb2868
Pani, B., Ong, H. L., Liu, X., Rauser, K., Ambudkar, I. S., and Singh, B. B. (2008). Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE). J. Biol. Chem. 283, 17333–17340. doi:10.1074/jbc.M800107200
Park, H. S., Hong, C., Kim, B. J., and So, I. (2014). The pathophysiologic roles of TRPM7 channel. Korean J. Physiol. Pharmacol. 18, 15–23. doi:10.4196/kjpp.2014.18.1.15
Patel, W., Moore, P. J., Sassano, M. F., Lopes-Pacheco, M., Aleksandrov, A. A., Amaral, M. D., et al. (2019). Increases in cytosolic Ca2+ induce dynamin- and calcineurin-dependent internalisation of CFTR. Cel. Mol. Life Sci. 76, 977–994. doi:10.1007/s00018-018-2989-3
Patergnani, S., Danese, A., Bouhamida, E., Aguiari, G., Previati, M., Pinton, P., et al. (2020a). Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int. J. Mol. Sci. 21, 8323. doi:10.3390/ijms21218323
Patergnani, S., Vitto, V. A. M., Pinton, P., and Rimessi, A. (2020b). Mitochondrial stress responses and “Mito-Inflammation” in cystic fibrosis. Front. Pharmacol. 11, 581114. doi:10.3389/fphar.2020.581114
Pedemonte, N., Sonawane, N. D., Taddei, A., Hu, J., Zegarra-Moran, O., Suen, Y. F., et al. (2005). Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol. Pharmacol. 67, 1797–1807. doi:10.1124/mol.105.010959
Philippe, R., Antigny, F., Buscaglia, P., Norez, C., Becq, F., Frieden, M., et al. (2015). SERCA and PMCA pumps contribute to the deregulation of Ca2+ homeostasis in human CF epithelial cells. Biochim. Biophys. Acta 1853, 892–903. doi:10.1016/j.bbamcr.2015.01.010
Philippe, R., Antigny, F., Buscaglia, P., Norez, C., Huguet, F., Castelbou, C., et al. (2017). Calumenin contributes to ER-Ca2+ homeostasis in bronchial epithelial cells expressing WT and F508del mutated CFTR and to F508del-CFTR retention. Cell Calcium 62, 47–59. doi:10.1016/j.ceca.2017.01.011
Pinton, P., Leo, S., Wieckowski, M. R., Di Benedetto, G., and Rizzuto, R. (2004). Long-term modulation of mitochondrial Ca2+ signals by protein kinase C isozymes. J. Cel Biol. 165, 223–232. doi:10.1083/jcb.200311061
Prandini, P., De Logu, F., Fusi, C., Provezza, L., Nassini, R., Montagner, G., et al. (2016). Transient receptor potential ankyrin 1 channels modulate inflammatory response in respiratory cells from patients with cystic fibrosis. Am. J. Respir. Cel Mol Biol 55, 645–656. doi:10.1165/rcmb.2016-0089OC
Preti, D., Szallasi, A., and Patacchini, R. (2012). TRP channels as therapeutic targets in airway disorders: a patent review. Expert Opin. Ther. Pat 22, 663–695. doi:10.1517/13543776.2012.696099
Pulli, I., Blom, T., Löf, C., Magnusson, M., Rimessi, A., Pinton, P., et al. (2015). A novel chimeric aequorin fused with caveolin-1 reveals a sphingosine kinase 1-regulated Ca2+ microdomain in the caveolar compartment. Biochim. Biophys. Acta 1853, 2173–2182. doi:10.1016/j.bbamcr.2015.04.005
Ratner, A. J., Bryan, R., Weber, A., Nguyen, S., Barnes, D., Pitt, A., et al. (2001). Cystic fibrosis pathogens activate Ca2+-dependent mitogen-activated protein kinase signaling pathways in airway epithelial cells. J. Biol. Chem. 276, 19267–19275. doi:10.1074/jbc.M007703200
Ribeiro, C. M., and Boucher, R. C. (2010). Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc. Am. Thorac. Soc. 7, 387–394. doi:10.1513/pats.201001-017AW
Ribeiro, C. M., and Lubamba, B. A. (2017). Role of IRE1alpha/XBP-1 in cystic fibrosis airway inflammation. Int. J. Mol. Sci. 18, ijms18010118. doi:10.3390/ijms18010118
Ribeiro, C. M., Paradiso, A. M., Carew, M. A., Shears, S. B., and Boucher, R. C. (2005). Cystic fibrosis airway epithelial Ca2+ i signaling: the mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia. J. Biol. Chem. 280, 10202–10209. doi:10.1074/jbc.M410617200
Ribeiro, C. M. (2006). The role of intracellular calcium signals in inflammatory responses of polarised cystic fibrosis human airway epithelia. Drugs R. D 7, 17–31. doi:10.2165/00126839-200607010-00002
Rimessi, A., Bezzerri, V., Patergnani, S., Marchi, S., Cabrini, G., and Pinton, P. (2015a). Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 6, 6201. doi:10.1038/ncomms7201
Rimessi, A., Bezzerri, V., Salvatori, F., Tamanini, A., Nigro, F., Dechecchi, M. C., et al. (2018). PLCB3 loss of function reduces Pseudomonas aeruginosa-dependent IL-8 release in cystic fibrosis. Am. J. Respir. Cel Mol Biol 59, 428–436. doi:10.1165/rcmb.2017-0267OC
Rimessi, A., Patergnani, S., Bonora, M., Wieckowski, M. R., and Pinton, P. (2015b). Mitochondrial Ca2+ remodeling is a prime factor in oncogenic behavior. Front. Oncol. 5, 143. doi:10.3389/fonc.2015.00143
Rimessi, A., Pozzato, C., Carparelli, L., Rossi, A., Ranucci, S., De Fino, I., et al. (2020). Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci. Adv. 6, eaax9093. doi:10.1126/sciadv.aax9093
Rimessi, A., Rizzuto, R., and Pinton, P. (2007). Differential recruitment of PKC isoforms in HeLa cells during redox stress. Cell Stress Chaperones 12, 291–298. doi:10.1379/csc-211.1
Riordan, J. R. (1993). The cystic fibrosis transmembrane conductance regulator. Annu. Rev. Physiol. 55, 609–630. doi:10.1146/annurev.ph.55.030193.003141
Robledo-Avila, F. H., Ruiz-Rosado, J. D., Brockman, K. L., Kopp, B. T., Amer, A. O., Mccoy, K., et al. (2018). Dysregulated calcium homeostasis in cystic fibrosis neutrophils leads to deficient antimicrobial responses. J. Immunol. 201, 2016–2027. doi:10.4049/jimmunol.1800076
Rubino, R., Bezzerri, V., Favia, M., Facchini, M., Tebon, M., Singh, A. K., et al. (2014). Pseudomonas aeruginosa reduces the expression of CFTR via post-translational modification of NHERF1. Pflugers Arch. 466, 2269–2278. doi:10.1007/s00424-014-1474-6
Sahoo, S. K., Kim, T., Kang, G. B., Lee, J. G., Eom, S. H., and Kim, D. H. (2009). Characterization of calumenin-SERCA2 interaction in mouse cardiac sarcoplasmic reticulum. J. Biol. Chem. 284, 31109–31121. doi:10.1074/jbc.M109.031989
Salathe, M. (2007). Regulation of mammalian ciliary beating. Annu. Rev. Physiol. 69, 401–422. doi:10.1146/annurev.physiol.69.040705.141253
Santo-Domingo, J., Vay, L., Hernández-Sanmiguel, E., Lobatón, C. D., Moreno, A., Montero, M., et al. (2007). The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br. J. Pharmacol. 151, 647–654. doi:10.1038/sj.bjp.0707260
Scambler, T., Jarosz-Griffiths, H. H., Lara-Reyna, S., Pathak, S., Wong, C., Holbrook, J., et al. (2019). ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife 8. doi:10.7554/eLife.49248
Schwaller, B. (2020). Cytosolic Ca2+ buffers are inherently Ca2+ signal modulators. Cold Spring Harb Perspect. Biol. 12, a035543. doi:10.1101/cshperspect.a035543
Shapiro, B. L., Feigal, R. J., Laible, N. J., Biros, M. H., and Warwick, W. J. (1978). Doubling time alpha-aminoisobutyrate transport and calcium exchange in cultured fibroblasts from cystic fibrosis and control subjects. Clin. Chim. Acta 82, 125–131. doi:10.1016/0009-8981(78)90035-9
Shoshan-Barmatz, V., De Pinto, V., Zweckstetter, M., Raviv, Z., Keinan, N., and Arbel, N. (2010). VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285. doi:10.1016/j.mam.2010.03.002
Simmen, T., and Herrera-Cruz, M. S. (2018). Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cel Biol. 53, 61–69. doi:10.1016/j.ceb.2018.04.014
Singaravelu, K., Nelson, C., Bakowski, D., De Brito, O. M., Ng, S. W., Di Capite, J., et al. (2011). Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J. Biol. Chem. 286, 12189–12201. doi:10.17402910.1074/jbc.M110.174029
Stoker, M. L., Newport, E., Hulit, J. C., West, A. P., and Morten, K. J. (2019). Impact of pharmacological agents on mitochondrial function: a growing opportunity? Biochem. Soc. Trans. 47, 1757–1772. doi:10.1042/BST20190280
Strehler, E. E., and Treiman, M. (2004). Calcium pumps of plasma membrane and cell interior. Curr. Mol. Med. 4, 323–335. doi:10.2174/1566524043360735
Tabary, O., Boncoeur, E., De Martin, R., Pepperkok, R., Clément, A., Schultz, C., et al. (2006). Calcium-dependent regulation of NF-(kappa)B activation in cystic fibrosis airway epithelial cells. Cel Signal 18, 652–660. doi:10.1016/j.cellsig.2005.06.004
Tabary, O., Escotte, S., Couetil, J. P., Hubert, D., Dusser, D., Puchelle, E., et al. (2000). High susceptibility for cystic fibrosis human airway gland cells to produce IL-8 through the I kappa B kinase alpha pathway in response to extracellular NaCl content. J. Immunol. 164, 3377–3384. doi:10.4049/jimmunol.164.6.3377
Taylor, C. W., and Machaca, K. (2019). IP3 receptors and store-operated Ca2+ entry: a license to fill. Curr. Opin. Cel. Biol. 57, 1–7. doi:10.1016/j.ceb.2018.10.001
Teng, L., Kerbiriou, M., Taiya, M., Le Hir, S., Mignen, O., Benz, N., et al. (2012). Proteomic identification of calumenin as a G551D-CFTR associated protein. PLoS One 7, e40173. doi:10.1371/journal.pone.0040173
Thillaiappan, N. B., Chavda, A. P., Tovey, S. C., Prole, D. L., and Taylor, C. W. (2017). Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 8, 1505. doi:10.1038/s41467-017-01644-8
Vachel, L., Norez, C., Becq, F., and Vandebrouck, C. (2013). Effect of VX-770 (ivacaftor) and OAG on Ca2+ influx and CFTR activity in G551D and F508del-CFTR expressing cells. J. Cyst Fibros 12, 584–591. doi:10.1016/j.jcf.2013.05.008
Vachel, L., Norez, C., Jayle, C., Becq, F., and Vandebrouck, C. (2015). The low PLC-delta1 expression in cystic fibrosis bronchial epithelial cells induces upregulation of TRPV6 channel activity. Cell Calcium 57, 38–48. doi:10.1016/j.ceca.2014.11.005
Verkhratsky, A., Trebak, M., Perocchi, F., Khananshvili, D., and Sekler, I. (2018). Crosslink between calcium and sodium signalling. Exp. Physiol. 103, 157–169. doi:10.1113/EP086534
Villalobo, A., Ishida, H., Vogel, H. J., and Berchtold, M. W. (2018). Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta Mol. Cel. Res. 1865, 507–521. doi:10.1016/j.bbamcr.2017.12.004
Vorum, H., Hager, H., Christensen, B. M., Nielsen, S., and Honoré, B. (1999). Human calumenin localizes to the secretory pathway and is secreted to the medium. Exp. Cel. Res. 248, 473–481. doi:10.1006/excr.1999.4431
Waller, R. L., Brattin, W. J., and Dearborn, D. G. (1984). Cytosolic free calcium concentration and intracellular calcium distribution in lymphocytes from cystic fibrosis patients. Life Sci. 35, 775–781. doi:10.1016/0024-3205(84)90347-3
Waterer, G. W. (2012). Airway defense mechanisms. Clin. Chest Med. 33, 199–209. doi:10.1016/j.ccm.2012.03.003
Woods, J. J., Nemani, N., Shanmughapriya, S., Kumar, A., Zhang, M., Nathan, S. R., et al. (2019). A selective and cell-permeable mitochondrial calcium uniporter (MCU) inhibitor preserves mitochondrial bioenergetics after hypoxia/reoxygenation injury. ACS Cent. Sci. 5, 153–166. doi:10.1021/acscentsci.8b00773
Wootten, D., Christopoulos, A., Marti-Solano, M., Babu, M. M., and Sexton, P. M. (2018). Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cel Biol. 19, 638–653. doi:10.1038/s41580-018-0049-3
Zhu, L., Yuan, C., Ding, X., Xu, S., Yang, J., Liang, Y., et al. (2016). PLC-gamma1 is involved in the inflammatory response induced by influenza A virus H1N1 infection. Virology 496, 131–137. doi:10.1016/j.virol.2016.06.003
Zhu, L., Yuan, C., Ma, Y., Ding, X., Zhu, G., and Zhu, Q. (2015). Anti-inflammatory activities of phospholipase C inhibitor U73122: inhibition of monocyte-to-macrophage transformation and LPS-induced pro-inflammatory cytokine expression. Int. Immunopharmacol. 29, 622–627. doi:10.1016/j.intimp.2015.09.019
Keywords: inflammatory disease, calcium signaling, lung disease, inflammation, cystic fibrosis, calcium
Citation: Rimessi A, Vitto VAM, Patergnani S and Pinton P (2021) Update on Calcium Signaling in Cystic Fibrosis Lung Disease. Front. Pharmacol. 12:581645. doi: 10.3389/fphar.2021.581645
Received: 09 July 2020; Accepted: 19 January 2021;
Published: 11 March 2021.
Edited by:
Giulio Cabrini, University of Ferrara, ItalyReviewed by:
Magdalena Lebiedzinska, Nencki Institute of Experimental Biology (PAS), PolandGeert Bultynck, KU Leuven, Belgium
Copyright © 2021 Rimessi, Vitto, Patergnani and Pinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Pinton, cGFvbG8ucGludG9uQHVuaWZlLml0
†These authors have contributed equally to this work