 Rafael Franco
Rafael Franco Rafael Rivas‐Santisteban
Rafael Rivas‐Santisteban Irene Reyes-Resina3*
Irene Reyes-Resina3*- 1Department Biochemistry and Molecular Biomedicine, School of Biology, University of Barcelona, Barcelona, Spain
- 2Centro de Investigación en Red, Enfermedades Neurodegenerativas (CiberNed), Instituto de Salud Carlos iii, Madrid, Spain
- 3RG Neuroplasticity, Leibniz Institute for Neurobiology, Magdeburg, Germany
- 4Department of Biochemistry and Physiology, School of Pharmacy, University of Barcelona, Barcelona, Spain
Biased signaling is a concept that has arisen in the G protein-coupled receptor (GCPR) research field, and holds promise for the development of new drug development strategies. It consists of different signaling outputs depending on the agonist’s chemical structure. Here we review the most accepted mechanisms for explaining biased agonism, namely the induced fit hypothesis and the key/lock hypothesis, but we also consider how bias can be produced by a given agonist. In fact, different signaling outputs may originate at a given receptor when activated by, for instance, the endogenous agonist. We take advantage of results obtained with adenosine receptors to explain how such mechanism of functional selectivity depends on the context, being receptor-receptor interactions (heteromerization) one of the most relevant and most studied mechanisms for mammalian homeostasis. Considering all the possible mechanisms underlying functional selectivity is essential to optimize the selection of biased agonists in the design of drugs targeting GPCRs.
Introduction
Biased signaling consists of different signaling outputs depending on the agonist chemical structure. The concept has taken hold in the field of G protein-coupled receptor (GPCR) research and has opened up new perspectives for therapeutic drug development. The underlying idea is that a given agonist biased towards a particular signaling may be therapeutic while another agonist biased towards activating an alternative pathway may not be helpful, and may even be harmful.
Biased agonism is an attractive concept to try to get agonist use off the ground in clinical practice. At present, agonists have by far less potential than antagonists. Usually, endogenous agonists approved as therapeutic drugs are used in acute conditions and during short times. In contrast, antagonists may be used in a chronic regime. The classical example is epinephrine that is used as adrenergic agonist to save lives in critical situations (e.g., anaphylaxis) whereas beta-adrenergic blockers/antagonists are used for a variety of diseases in both acute and chronic regimes. In the purine field, adenosine is used in bolus administration to combat paroxysmal tachycardia whereas the adenosine A2AR antagonist, istradefylline (Nouriast™ in Japan; Nourianz™ in the United States), has been approved for chronic use in the therapy of Parkinson’s disease (Pinna et al., 2007; Simola et al., 2008; Jenner et al., 2009; Mizuno and Kondo, 2013; Kondo et al., 2015; Navarro et al., 2015).
Two complementary points of view are needed to underline the mechanisms underlying differential signaling arising from a given GPCR. In a previous paper we already made a distinction between biased signaling and biased functionality (Franco et al., 2018). Here we will provide more information on the possibility that biased signaling arises from different compounds acting on the same receptor but, also, on the possibility that biased signaling arises from the same agonist acting in the same receptor but expressed in a different context. By different context we mean that a given GPCR may be expressed in different cells coupled to different proteins, not only to different G proteins but to other receptors, to scaffolding proteins, etc.
The Most Accepted Mechanism to Explain Biased Signaling
The resolution of the structure of various GPCRs and the molecular dynamics of macromolecules in aqueous solutions give indications as to how GPCR-mediated signaling occurs. Binding of the agonist to the orthosteric site leads to significant structural rearrangements that are transmitted to the coupled G protein and allow signaling (Westfield et al., 2011; Masureel et al., 2018).
It is not necessary to be very specific with the details to explain the basis of the most accepted mechanism underlying biased signaling. In fact, assuming that GPCRs have a loose orthosteric center, the binding of structurally different chemicals to the site can result in different conformations (Figure 1 up). Said different conformations will couple differently to the signaling machinery, thus providing different signaling outputs. The agonist/receptor interaction would be similar to the so-called induced fit in the case of a substrate that interacts with the active site of an enzyme (Urban et al., 2007; Kenakin and Miller, 2010) (Kenakin, 2009; Kenakin, 2011; Kenakin and Christopoulos, 2013).
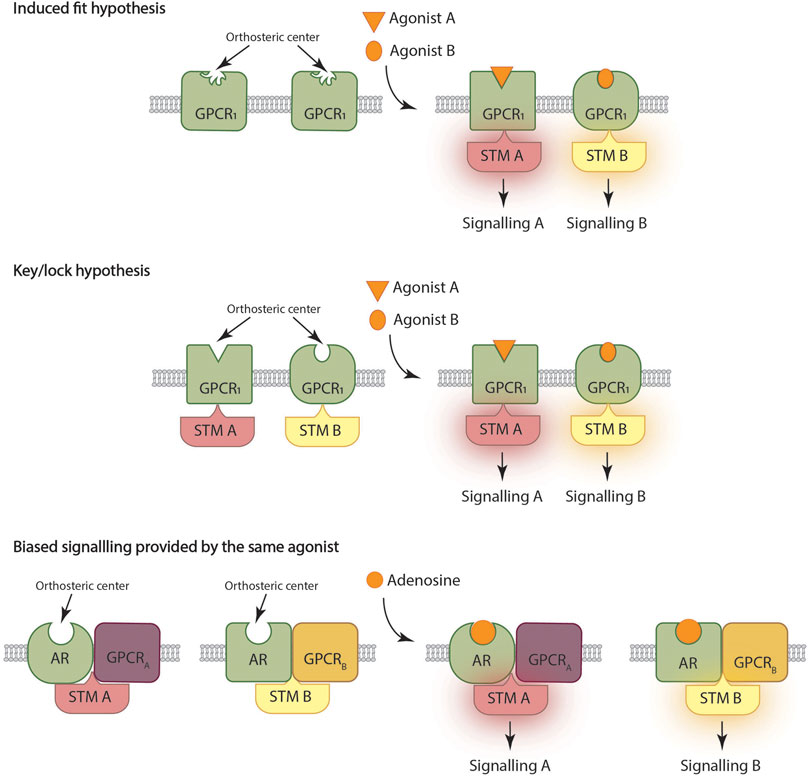
FIGURE 1. Three ways to deliver biased signaling. Top: G protein coupled receptor (GPCR) orthosteric center of the GPCR has a loose conformation that is fixed upon agonist binding, thus allowing coupling and engagement of the signal transduction machinery (STM). Center: There are different conformational states of a given GPCR with structurally different orthosteric sites; each agonist preferentially binds to a given conformation thus preferentially engaging a given STM. Bottom: A given ligand acting on a given GPCR may lead to different signaling outputs depending on the context of the receptor. The case of heteromer formation is exemplified with a given adenosine receptor (AR) able to interact with GPCRA or with GPCRB.
Following the analogy with substrate/enzyme interaction, there is another point of view which is that the cell surface receptor is in different conformational states while waiting for the arrival of the agonist. Similar to the key/lock idea (Figure 1 center), each of these conformational states would have a different lock and each agonist would interact more strongly (i.e., with more affinity) with some conformations than with others (Costa-Neto et al., 2016; Michel and Charlton, 2018).
In summary, in the classical view each agonist favors a specific signal transduction and that this may be due to two conceptually different mechanisms. One is by assuming different receptor states due to pre-coupling to signaling mechanisms and each chemical structure preferentially binding to a given state, thereby preferentially engaging such particular signaling pathway. The second is by assuming the GPCR in a given state that, after agonist-induced conformational changes, would lead to a receptor prone to interact to (and engage) a particular signaling machinery.
The Alternative Mechanism to Approach Biased Signaling. How the Endogenous Agonist May Provide Functional Diversity
Biased agonism fits into a more general framework, called functional selectivity. A given GPCR may provide different signaling outputs depending on the context. In other words, functional selectivity may be afforded using a single agonist. Yet another way to express the idea is that the endogenous agonist (hormone/neurotransmitter) will give rise to different signals depending on the cell/tissue and the general pathophysiological state.
We argue, as suggested elsewhere (Franco et al., 2018), that biased signaling does not require a biased agonist, that is, that the endogenous agonist may engage different signaling pathways depending on the cell context. In short, it would be the functional unit itself, made up of the receptor and the direct receptor/receptor and receptor/protein interactions, which is coupled to a certain signaling machinery. Consequently, cells will respond according to the coupling assigned to the specific structure of the functional unit and the existence, or not, of more than one functional unit.
Below we will present some examples of differential functional selectivity provided by an endogenous agonist (Figure 1 bottom). Let us first describe the classic case discovered by Susan George and her colleagues working with dopamine receptors. According to IUPHAR, the cognate G proteins for the D1 and D2 receptors are, respectively, Gs and Gi (Alexander et al., 2019). However, D1 and D2 can interact to form D1-D2 receptor heteromers that do not couple to Gs/Gi but to Gq. Coupling to Gq allows dopamine to activate not only cAMP- but also calcium-related mechanisms (Lee et al., 2004; Rashid et al., 2007; Perreault et al., 2015; Perreault et al., 2016). The controversy that arose about the appearance of such complexes in primates has been resolved by finding that about 18% of the neurons of the striatum of Macaca fascicularis express the D1-D2 receptor heteromers (Rico et al., 2016). In fact, there are neurons in different parts of the central nervous system that expressing those heteromers provide a long-suspected link between dopaminergic neurotransmission and calcium signals.
Dopamine D1 receptors can also form heteromers with histamine receptors, whose exact role in the central nervous system has yet to be fully clarified. Interestingly, the formation of D1 and the histamine H3 receptor heteromer is required for histamine to activate the mitogen-activated protein kinase (MAPK) signaling pathway. Surprisingly, it appears that D1 receptors within this heteromeric context bind to Gi rather than its cognate G protein, Gs. (Ferrada et al., 2009).
A final example we provide here is related to GPCRs that regulate intraocular pressure. Melatonin receptors form functional complexes with α1-adrenergic receptors, which involve the C-terminal tail of the latter. Surprisingly, activation of α1-adrenergic receptors in this particular heteromeric context does not lead to changes in cytoplasmic levels of Ca2+ but of cAMP. Once again, the heteromeric context leads to a change, from Gq to Gs, in the G protein coupling (see Figure 1 bottom). Glaucoma coursing with elevated intraocular pressure is correlated with a decreased expression of the complexes in stromal cells (Alkozi et al., 2019; Alkozi et al., 2020). Whether this fact is a cause or a consequence of the disease, the melatonin-adrenergic heteromers arise as targets for fighting the disease.
Biased Signaling Under the Prism of Results Derived From Adenosine Receptor Signaling Characterization
Four are the adenosine receptors identified so far in mammals: A1, A2A, A2B, and A3. The cognate G proteins for the A1 and the A3 are of the Gi type and the cognate G proteins for the A2A and the A2B are of the Gs type. Via G protein-mediated signaling or via the ßγ subunits of G proteins, activation of adenosine receptors may activate the mitogen-activated protein kinase (MAPK) pathway. Also ß-arrestin recruitment may lead to receptor internalization and intracellular signaling (see Borea et al., 2018 for review).
Recently, we have performed a classic study of biased agonism using one of the four adenosine receptors, the A2A, expressed in a heterologous system. In addition to identifying two chemical structures, PSB-0777 and LUF-5834, that behaved differently from the rest of the agonists, we noticed that removing part of the receptor’s C-terminal tail does not qualitatively change the results (Navarro et al., 2020). This finding was unexpected as the long C-terminal end of the receptor is potentially interacting with some components of the signaling machinery. Interestingly, removal of the C-terminal tail of the A3 receptor is dispensable for its capability to recruit ß-arrestins (Storme et al., 2018).
It is now suspected that the four adenosine receptors, A1, A2A, A2B, and A3, can interact with each other. The A1-A2A, A1-A3, and A2A-A2B interactions have already been described (Ciruela et al., 2006a; Hill et al., 2014; Hinz et al., 2018; Lillo et al., 2020). The interaction between the adenosine A2A and A1 receptors was identified several years ago (Ciruela et al., 2006a) and the functional role of the complex has been well understood ever since (Ciruela et al., 2006b; Cristóvão-Ferreira et al., 2013; Lin et al., 2020). A recent review on structure and function of adenosine receptor heteromers is available (Franco et al., 2021).
Adenosine leads to marked biased signaling based on the heteromeric context, even considering only interactions between adenosine receptors. On the one hand, signaling mediated by the A3 receptor is blocked if A2AR is co-expressed and A2A-A3 receptor heteromers are formed. A2A receptor antagonists abrogate the blockade, thus providing a novel approach to the development of drugs that target the heteromers of the A2A-A3 receptor. On the other hand, the expression of the A2B receptor blocks signaling through the A2A receptor. This finding raises several questions, as the A2A receptor has a much higher affinity for adenosine than the A2B. The actual physiological significance of this interaction is under close scrutiny, although the A2A-A2B receptor functional complex has already been shown to be relevant in aging and obesity (Gnad et al., 2020).
Remarkably, the A1-A2A receptor heteromer adds an additional dimension to functional selectivity. In fact, the signal is biased not only by the endogenous agonist, but also by its concentration. As we often mention, this complex is an adenosine concentration sensor. At concentrations at which only the A1 receptor is occupied by adenosine, only Gi-mediated signaling is observed, with Gi being the cognate protein of the A1 receptor. In contrast, when the adenosine concentration increases and the A2A receptor is occupied, only Gs-mediated signaling originates in the heteromer, with Gs being the cognate protein of the A2A receptor. The mechanistic molecular basis of such a phenomenon has been fully elucidated and, more importantly, it is the C-terminal tail of A2A that is relevant for blocking the partner (A1) receptor function (Navarro et al., 2016; Navarro et al., 2018).
In summary, biased signaling is produced by the endogenous agonist, adenosine, depending on the context of the target receptor, even depending on the adenosine concentration itself. It should be noted that the panorama of functional diversity that adenosine can cause is not limited to the interaction between adenosine receptors, but extends to the complexes that adenosine receptors establishes with other GPCRs or other proteins (see (Ginés et al., 2001; Agnati et al., 2003; Burgueño et al., 2003; Franco et al., 2005; Ciruela et al., 2006b; Fuxe et al., 2007; Navarro et al., 2014) for review).
Conclusion
The hopes placed on the biased agonism to give an extra boost to the drug discovery are not being fulfilled. For the above reasons, a biased agonist may provide a benefit in a given setting but provide a detrimental effect in other receptor settings and thus not be useful in therapy. Also relevant is how to reliably measure the output of the signaling pathway one wants to target; in fact, different assays claiming to evaluate the same pathway may produce a different result, some suggesting bias, some not. One wonders if it would be more proactive to skip in vitro pharmacological assays and test different agonists for their efficacy and safety in in vivo disease models. To date, trying to decipher the mechanism underlying functional selectivity for a given GPCR is challenging. Without this information, it is virtually impossible to optimize the selection of biased agonists for drug development. Therefore, it seems necessary to carry out an investigation aimed at knowing both 1) what is the signaling pathway to target 2) how to reliably measure the pathway output and 3) what is the status of the target GPCR. Status means identifying the proteins/receptors that interact with the target GPCR and how that macromolecular complex is specifically coupled to the signaling machineries.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
RF planned the contents and discussed them with RR‐S, IR-R, and GN. RF, and GN wrote a first draft. RR‐S, and IR-R made significant additions to the draft. IR-R made the figure. All authors have approved the submitted version.
Funding
This research was funded by the Spanish Ministry of Economy and Competitiveness (grants: SAF2017-84117-R and RTI2018-094204-B-I00; they may include European Regional Development -FEDER- funds) and the Alzheimer’s Association (grant: AARFD-17-503612). The laboratory of the University of Barcelona is considered of excellence (grup consolidat #2017 SGR 1497) by the Regional Catalonian Government, which does not provide any specific funding for reagents or for payment of services or for Open Access fees.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agnati, L. F., Ferré, S., Lluis, C., Franco, R., and Fuxe, K. (2003). Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol. Rev. 55, 509–550. doi:10.1124/pr.55.3.2
Alexander, S. P. H., Christopoulos, A., Davenport, A. P., Kelly, E., Mathie, A., Peters, J. A., et al. (2019). The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 176 (Suppl 1), S21–S141. doi:10.1111/bph.14748
Alkozi, H. A., Navarro, G., Aguinaga, D., Reyes-Resina, I., Sanchez-Naves, J., Pérez de Lara, M. J., et al. (2020). Adreno–melatonin receptor complexes control ion homeostasis and intraocular pressure - their disruption contributes to hypertensive glaucoma. Br. J. Pharmacol. 177, 2090–2105. doi:10.1111/bph.14971
Alkozi, H. A., Navarro, G., Franco, R., and Pintor, J. (2019). Melatonin and the control of intraocular pressure. Prog. Retin. Eye Res. 75, 100798. doi:10.1016/j.preteyeres.2019.100798
Borea, P. A., Gessi, S., Merighi, S., Vincenzi, F., and Varani, K. (2018). Pharmacology of adenosine receptors : the state of the art. Physiol. Rev. 98, 1591–1625. doi:10.1152/physrev.00049.2017
Burgueño, J., Blake, D. J., Benson, M. A., Tinsley, C. L., Esapa, C. T., Canela, E. I., et al. (2003). The adenosine A2A receptor interacts with the actin-binding protein alpha-actinin. J. Biol. Chem. 278, 37545–37552. doi:10.1074/jbc.M302809200
Ciruela, F., Casadó, V., Rodrigues, R., Luján, R., Burgueño, J., Canals, M., et al. (2006a). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 26, 2080–2087. doi:10.1523/JNEUROSCI.3574-05.2006
Ciruela, F., Ferré, S., Casadó, V., Cortés, A., Cunha, R., Lluis, C., et al. (2006b). Heterodimeric adenosine receptors: a device to regulate neurotransmitter release. Cell. Mol. Life Sci. 63, 2427–2431. doi:10.1007/s00018-006-6216-2
Costa-Neto, C. M., Parreiras-E-Silva, L. T., and Bouvier, M. (2016). A pluridimensional view of biased agonism. Mol. Pharmacol. 90, 587–595. doi:10.1124/mol.116.105940
Cristóvão-Ferreira, S., Navarro, G., Brugarolas, M., Pérez-Capote, K., Vaz, S. H., Fattorini, G., et al. (2013). A1R-A2AR heteromers coupled to Gs and G i/o proteins modulate GABA transport into astrocytes. Purinergic Signal 9, 433–449. doi:10.1007/s11302-013-9364-5
Ferrada, C., Moreno, E., Casadó, V., Bongers, G., Cortés, A., Mallol, J., et al. (2009). Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br. J. Pharmacol. 157, 64–75. doi:10.1111/j.1476-5381.2009.00152.x
Franco, R., Aguinaga, D., Jiménez, J., Lillo, J., Martínez-Pinilla, E., and Navarro, G. (2018). Biased receptor functionality versus biased agonism in G-protein-coupled receptors. Biomol. Concepts 9, 143–154. doi:10.1515/bmc-2018-0013
Franco, R., Ciruela, F., Casadó, V., Cortes, A., Canela, E. I., Mallol, J., et al. (2005). Partners for adenosine A1 receptors. J. Mol. Neurosci. 26, 221–231. doi:10.1385/JMN:26:2-3:221
Franco, R., Cordomí, A., Llinas del Torrent, C., Lillo, A., Serrano-Marín, J., Navarro, G., et al. (2021). Structure and function of adenosine receptor heteromers. Cell. Mol. Life Sci. In the Press.
Fuxe, K., Canals, M., Torvinen, M., Marcellino, D., Terasmaa, A., Genedani, S., et al. (2007). Intramembrane receptor-receptor interactions: a novel principle in molecular medicine. J. Neural. Transm. (Vienna) 114, 49–75. doi:10.1007/s00702-006-0589-0
Ginés, S., Ciruela, F., Burgueño, J., Casadó, V., Canela, E. I. I., Mallol, J., et al. (2001). Involvement of caveolin in ligand-induced recruitment and internalization of A(1) adenosine receptor and adenosine deaminase in an epithelial cell line. Mol. Pharmacol. 59, 1314–1323. doi:10.1124/mol.59.5.1314
Gnad, T., Navarro, G., Lahesmaa, M., Reverte-Salisa, L., Copperi, F., Cordomi, A., et al. (2020). Adenosine/A2B receptor signaling ameliorates the effects of aging and counteracts obesity. Cell Metabol. 32, 56–70.e7. doi:10.1016/j.cmet.2020.06.006
Hill, S. J., May, L. T., Kellam, B., and Woolard, J. (2014). Allosteric interactions at adenosine A(1) and A(3) receptors: new insights into the role of small molecules and receptor dimerization. Br. J. Pharmacol. 171, 1102–1113. doi:10.1111/bph.12345
Hinz, S., Navarro, G., Borroto-Escuela, D., Seibt, B. F., Ammon, C., de Filippo, E., et al. (2018). Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget. 9, 13593–13611. doi:10.18632/oncotarget.24423
Jenner, P., Mori, A., Hauser, R., Morelli, M., Fredholm, B. B., and Chen, J. F. (2009). Adenosine, adenosine A 2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 15, 406–413. doi:10.1016/j.parkreldis.2008.12.006
Kenakin, T., and Christopoulos, A. (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216. doi:10.1038/nrd3954
Kenakin, T. (2011). Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 336, 296–302. doi:10.1124/jpet.110.173948
Kenakin, T., and Miller, L. J. (2010). Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304. doi:10.1124/pr.108.000992
Kondo, T., and Mizuno, Y.Japanese Istradefylline Study Group (2015). A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin. Neuropharmacol. 38, 41–46. doi:10.1097/WNF.0000000000000073
Lee, S. P., So, C. H., Rashid, A. J., Varghese, G., Cheng, R., Lança, A. J., et al. (2004). Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J. Biol. Chem. 279, 35671–35678. doi:10.1074/jbc.M401923200
Lillo, A., Martínez-Pinilla, E., Reyes-Resina, I., Navarro, G., and Franco, R. (2020). Adenosine A2a and A3 receptors are able to interact with each other. A further piece in the puzzle of adenosine receptor-mediated signaling. Int. J. Mol. Sci. 21, 1–14. doi:10.3390/ijms21145070
Lin, Y., Xu, J., Gao, J., Huang, Y., Wang, Q., Xie, S., et al. (2020). “Real-time calcium imaging by the interaction of adenosine receptors in living HEK293T cells,” in Optics in health care and biomedical optics X. Editors Q. Luo, X. Li, Y. Gu, and D. Zhu (Bellingham, WA: SPIE), 6. doi:10.1117/12.2573547
Masureel, M., Zou, Y., Picard, L. P., van der Westhuizen, E., Mahoney, J. P., Rodrigues, J. P. G. L. M., et al. (2018). Structural insights into binding specificity, efficacy and bias of a β 2 AR partial agonist. Nat. Chem. Biol. 14, 1059–1066. doi:10.1038/s41589-018-0145-x
Michel, M. C., and Charlton, S. J. (2018). Biased agonism in drug discovery-is it too soon to choose a path? Mol. Pharmacol. 93, 259–265. doi:10.1124/mol.117.110890
Mizuno, Y., and Kondo, T. (2013). Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 28, 1138–1141. doi:10.1002/mds.25418
Navarro, G., Borroto-Escuela, D. O., Fuxe, K., and Franco, R. (2014). Potential of caveolae in the therapy of cardiovascular and neurological diseases. Front. Physiol. 5, 370. doi:10.3389/fphys.2014.00370
Navarro, G., Borroto-Escuela, D. O., Fuxe, K., and Franco, R. (2015). Purinergic signaling in Parkinson’s disease. Relevance for treatment. Neuropharmacology 104, 161–168. doi:10.1016/j.neuropharm.2015.07.024
Navarro, G., Cordomí, A., Brugarolas, M., Moreno, E., Aguinaga, D., Pérez-Benito, L., et al. (2018). Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 16 (1), 24. doi:10.1186/s12915-018-0491-x
Navarro, G., Cordomí, A., Zelman-Femiak, M., Brugarolas, M., Moreno, E., Aguinaga, D., et al. (2016). Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with Gi and Gs. BMC Biol. 14, 26. doi:10.1186/s12915-016-0247-4
Navarro, G., Gonzalez, A., Campanacci, S., Rivas-Santisteban, R., Reyes-Resina, I., Casajuana-Martin, N., et al. (2020). Experimental and computational analysis of biased agonism on full-length and a C-terminally truncated adenosine A2A receptor. Comput. Struct. Biotechnol. J. 18, 2723–2732. doi:10.1016/j.csbj.2020.09.028
Perreault, M. L., Hasbi, A., Shen, M. Y. F., Fan, T., Navarro, G., Fletcher, P. J., et al. (2016). Disruption of a dopamine receptor complex amplifies the actions of cocaine. Eur. Neuropsychopharmacol. 26, 1366–1377. doi:10.1016/j.euroneuro.2016.07.008
Perreault, M. L., Shen, M. Y. F., Fan, T., and George, S. R. (2015). Regulation of c-fos expression by the dopamine D1-D2 receptor heteromer. Neuroscience 285, 194–203. doi:10.1016/j.neuroscience.2014.11.017
Pinna, A., Pontis, S., Borsini, F., and Morelli, M. (2007). Adenosine A2A receptor antagonists improve deficits in initiation of movement and sensory motor integration in the unilateral 6-hydroxydopamine rat model of Parkinson’s disease. Synapse 61, 606–614. doi:10.1002/syn.20410
Rashid, A. J., So, C. H., Kong, M. M. C., Furtak, T., El-Ghundi, M., Cheng, R., et al. (2007). D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. U. S. A. 104, 654–659. doi:10.1073/pnas.0604049104
Rico, A. J., Dopeso-Reyes, I. G., Martínez-Pinilla, E., Sucunza, D., Pignataro, D., Roda, E., et al. (2016). Neurochemical evidence supporting dopamine D1–D2 receptor heteromers in the striatum of the long-tailed macaque: changes following dopaminergic manipulation. Brain Struct. Funct. 222, 1767–1784. doi:10.1007/s00429-016-1306-x
Simola, N., Fenu, S., Baraldi, P. G., Tabrizi, M. A., and Morelli, M. (2008). Blockade of globus pallidus adenosine A2A receptors displays antiparkinsonian activity in 6-hydroxydopamine-lesioned rats treated with D 1 or D2 dopamine receptor agonists. Synapse 62, 345–351. doi:10.1002/syn.20504
Storme, J., Cannaert, A., Van Craenenbroeck, K., and Stove, C. P. (2018). Molecular dissection of the human A3 adenosine receptor coupling with β-arrestin2. Biochem. Pharmacol. 148, 298–307. doi:10.1016/j.bcp.2018.01.008
Urban, J. D., Clarke, W. P., von Zastrow, M., Nichols, D. E., Kobilka, B., Weinstein, H., et al. (2007). Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320, 1–13. doi:10.1124/jpet.106.104463
Keywords: cAMP, MAPK pathway, adenylyl cyclase, GPCR, tetramer, heteromer, receptor-receptor interactions, functional selectivity
Citation: Franco R, Rivas‐Santisteban R, Reyes-Resina I and Navarro G (2021) The Old and New Visions of Biased Agonism Through the Prism of Adenosine Receptor Signaling and Receptor/Receptor and Receptor/Protein Interactions. Front. Pharmacol. 11:628601. doi: 10.3389/fphar.2020.628601
Received: 12 November 2020; Accepted: 21 December 2020;
Published: 29 January 2021.
Edited by:
Francesco Caciagli, University of Studies G d'Annunzio Chieti and Pescara, ItalyCopyright © 2021 Franco, Rivas-Santisteban, Reyes-Resina and Navarro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Franco, cmZyYW5jbzEyM0BnbWFpbC5jb20=; Irene Reyes Resina, aXJlbmUucmV5ZXNyZXNpbmFAbGluLW1hZ2RlYnVyZy5kZQ==
†These authors have contributed equally to this work