
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 29 January 2021
Sec. Experimental Pharmacology and Drug Discovery
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.606656
This article is part of the Research TopicRecent Advances in G Protein-Coupled Receptor Signalling: Impact of Intracellular Location, Environment and Biased AgonismView all 9 articles
 Emma T. van der Westhuizen1*
Emma T. van der Westhuizen1* K. H. Christopher Choy1
K. H. Christopher Choy1 Celine Valant1
Celine Valant1 Simon McKenzie-Nickson1
Simon McKenzie-Nickson1 Sophie J. Bradley2Andrew B. Tobin2Patrick M. Sexton1
Sophie J. Bradley2Andrew B. Tobin2Patrick M. Sexton1 Arthur Christopoulos1*
Arthur Christopoulos1*The M1 and M4 muscarinic acetylcholine receptors (mAChRs) are highly pursued drug targets for neurological diseases, in particular for Alzheimer’s disease and schizophrenia. Due to high sequence homology, selective targeting of any of the M1-M5 mAChRs through the endogenous ligand binding site has been notoriously difficult to achieve. With the discovery of highly subtype selective mAChR positive allosteric modulators in the new millennium, selectivity through targeting an allosteric binding site has opened new avenues for drug discovery programs. However, some hurdles remain to be overcome for these promising new drug candidates to progress into the clinic. One challenge is the potential for on-target side effects, such as for the M1 mAChR where over-activation of the receptor by orthosteric or allosteric ligands can be detrimental. Therefore, in addition to receptor subtype selectivity, a drug candidate may need to exhibit a biased signaling profile to avoid such on-target adverse effects. Indeed, recent studies in mice suggest that allosteric modulators for the M1 mAChR that bias signaling toward specific pathways may be therapeutically important. This review brings together details on the signaling pathways activated by the M1 and M4 mAChRs, evidence of biased agonism at these receptors, and highlights pathways that may be important for developing new subtype selective allosteric ligands to achieve therapeutic benefit.
Acetylcholine (ACh) is a neurotransmitter that plays a vital role in central nervous system (CNS) function. ACh is synthesized from the nutrient, choline, by the enzyme, cholineacetyltransferease and subsequently stored in intracellular vesicles in cholinergic projection neurons and cholinergic interneurons (Amenta and Tayebati, 2008). The cell bodies of cholinergic neurons reside in eight distinct clusters, which are named Ch1-Ch8. These clusters send projections to innervate distinct regions of the brain, as depicted in Figure 1 (Thiele 2013; Allaway and Machold, 2017; Li et al., 2018). Besides the main clusters of cholinergic projection neurons, cholinergic signaling also occurs in other local networks of neurons. One such cluster is located within the basal ganglia (striatum, caudate-putamen and globus pallidus), which is without external cholinergic inputs. Another cluster is found in the cortex and contains many cholinergic interneurons (Lanciego et al., 2012; Li et al., 2018). The central cholinergic signaling system contributes to many critical brain functions, including arousal, attention, learning and memory, sensory perception, motor function, sleep, nociception, motivation, reward, mood, psychosis and neuroplasticity (Thiele 2013; Hangya et al., 2015). At the cellular level, ACh regulates neuronal functions such as cell excitability and firing, neurotransmitter release and synaptic plasticity through its actions at pre- and post-synaptic acetylcholine receptors (Picciotto et al., 2012; Thiele 2013).
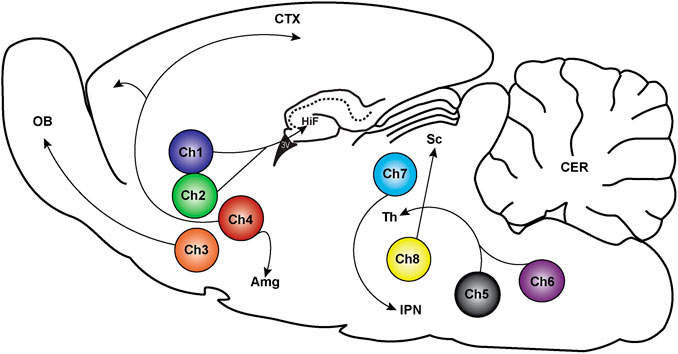
FIGURE 1. Location of cholinergic neurons and their projections in the rodent brain. Cholinergic neurons exist in eight distinct clusters labeled Ch1-Ch8. Ch1 and Ch2 are located in the medial septum and the diagonal band of Broca and send projections to the hippocampus (HiF), medial cortex, and thalamic nuclei (Th). The Ch3 cluster is in the diagonal band of Broca with projections to the olfactory bulb (OB). Ch4 neurons are in the nucleus basalis magnocellularis contains, which project to the cortex (CTX) and the amygdala (Amg). Ch5 and Ch6 are in the pedunculopontine tegmental nucleus and laterodorsal tegmental nucleus, respectively, which project to the thalamus (Th), pontine reticular formation, ventral midbrain, ventral tegmental area and to the substantia nigra. Ch7 neurons are in the habenula with projections to the interpeduncular nucleus (IPN). Ch8 are in the parabigeminal nucleus that project to the superior colliculus (Sc) (Thiele 2013; Li et al., 2018).
ACh signaling is triggered through activation of two major classes of cell-surface receptor proteins, the nicotinic ACh receptors (nAChRs; ligand-gated ion channels) and the muscarinic AChRs (mAChRs; G protein-coupled receptors; GPCRs). Both the nAChRs and the mAChRs are expressed in the CNS and in the periphery (Gotti et al., 2009; Chatzidaki and Millar, 2015) and participate in neuronal signal transduction events. The nAChRs are located both pre- and post-synaptically on neurons. They are important for fast synaptic transmission, allowing the passage of sodium, potassium and calcium ions (Gotti et al., 2009). Within the mAChR family, there are five subtypes that are aptly named M1–M5. All five subtypes of mAChRs are expressed in both the CNS and within peripheral tissues.
Quantitative immunoprecipitation, immunocytochemistry and electron microscopy techniques identified high expression levels of M1 mAChR in the striatum, prefrontal cortex, hippocampus and many neocortical regions (Buckley et al., 1988; Levey et al., 1991; Mrzljak et al., 1993; Vilaro et al., 1993; Flynn et al., 1995; Levey, 1996). The M1 mAChR is predominantly post-synaptic, playing important roles in the control of glutamatergic neurotransmission (Buckley et al., 1988; Levey et al., 1991; Levey, 1996). The M2 and M4 mAChRs are co-expressed with the M1 mAChR in forebrain regions, albeit at much lower levels (Buckley et al., 1988; Levey et al., 1991; Levey, 1996). The M4 mAChR is the most abundant subtype in the striatum, caudate and putamen (Buckley et al., 1988; Levey et al., 1991; Levey, 1996), with both pre- and post-synaptic expression, regulating dopamine release in the striatum (Foster et al., 2016; Moehle et al., 2017; Klawonn et al., 2018; Nair et al., 2019). The M2 mAChR is the predominant subtype in the basal forebrain, and is a pre-synaptic autoreceptor that sends signals to stop the release of ACh into synaptic clefts (Levey et al., 1991; Levey, 1996). In contrast, the M3 and M5 subtypes are expressed at low levels within the brain (Levey et al., 1991; Levey, 1996). Although all of these subtypes play important functional roles in central cholinergic signaling, the focus for this review will be on the M1 and M4 mAChRs, which have recently emerged as promising drug targets for the treatment of cognitive and behavioral symptoms of neurodegenerative and neurodevelopmental disorders.
Increasing evidence indicates that an imbalance in cholinergic signaling is a major contributor to the prevailing symptoms of many neurological diseases, including Alzheimer's disease, Parkinson's disease, schizophrenia, depression and bipolar disorder; either directly or as a result of modulating other important neurotransmitters (Langmead et al., 2008; Carruthers et al., 2015; van Enkhuizen et al., 2015; Foster et al., 2016; Nair et al., 2019).
Alzheimer's disease is a neurodegenerative disease that presents as a progressive decline in memory. It is characterised by the accumulation of amyloid-β protein plaques in neurons and hyperphosphorylation of microtubule associated Tau proteins (Kar et al., 2004; Thathiah and De Strooper, 2011; Hartl et al., 2015). Accumulation of protein aggregates in neurons, results in the dysregulation of multiple neurotransmitter systems. The cholinergic system is one of the key transmitter systems that are affected, with degeneration of the cholinergic neurons of the basal forebrain reducing the amount of ACh available for neurotransmission. In post-mortem Alzheimer's disease brain tissue, the expression levels of the M2 mAChRs are decreased but there are no significant changes to the expression level of the M1 mAChRs when compared to normal aged brain tissue (Nordberg and Winblad, 1986; Flynn et al., 1991; Hampel et al., 2018); an important consideration for therapeutic development. Treatment for Alzheimer's disease is currently achieved with the use of acetylcholinesterase inhibitors, which prevent the degradation of ACh in the synapse, thus directly increasing cholinergic signaling (Lleo, 2007). Unfortunately, this treatment is only modestly effective for treating the cognitive symptoms and does not treat the underlying pathology of the disease itself, only delaying the symptomatic progression of the disease. Acetylcholinesterase inhibitors are associated with dose-limiting gastrointestinal side effects, including nausea, vomiting and diarrhea, due to the global increase in ACh throughout the periphery (Lockhart et al., 2009; Harvey, 2012). As an alternative to increasing ACh levels, directly targeting the mAChRs with agonist ligands could increase neuronal stimulation to improve cognitive functions. This hypothesis was indeed tested in human clinical trials with the rigid ACh analogue, AF102B (cevimeline) or with the M1/M4 mAChR-preferring agonist, xanomeline (Fisher et al., 1996; Bodick et al., 1997). Treatment with either cevimeline or xanomeline significantly improved the cognitive abilities of Alzheimer's disease patients when compared with placebo controls (Fisher et al., 1996; Bodick et al., 1997), proving that direct activation of the mAChRs is a feasible drug targeting strategy. However, like the acetylcholinesterase inhibitors, cevimeline and xanomeline both display similar gastrointestinal side effects and can cause syncope in patients (Fisher et al., 1996; Bodick et al., 1997). Although directly targeting the mAChRs remains a possible approach for developing new therapies for cognitive enhancement, the use of mAChR agonists is currently limited by the side effects mediated by activation of peripherally expressed mAChRs.
Cleavage of the amyloid precursor protein (APP) by secretase enzymes produces several amyloid-β peptides (Kar et al., 2004). Disturbances in APP processing increase the production of toxic amyloid-β proteins that form plaques on neurons, disrupt neurotransmission and cause neuronal cell death (Harkany et al., 1995; Kelly et al., 1996; Kar et al., 2004; Hartl et al., 2015). Interestingly, these amyloid-β proteins directly affect the coupling of the M1 mAChR to G proteins (Janickova et al., 2013). In [3H]-N-methyl-scopolamine radioligand binding experiments, high (G protein-coupled state) and low (G protein-uncoupled state) affinity binding sites can be defined by increasing concentrations of the agonist, carbachol in M1 mAChR expressing Chinese hamster ovary (CHO) cells. Following a four day pre-treatment of the cells with amyloid-β(1-42), the high affinity binding site is lost, suggesting that the peptide disrupts G protein coupling to the M1 mAChR (Janickova et al., 2013). Carbachol-stimulated GTPase activity, IP3 production and intracellular calcium release are also inhibited in M1 mAChR expressing CHO cells and rat neocortical cultures following a 24–96 h pre-treatment with amyloid-β(1-40/42) (Kelly et al., 1996; Janickova et al., 2013). Thus the amyloid-β peptides can uncouple the M1 mAChR from G proteins and affect Gαq-mediated signaling events. This is an important point to consider for the development of M1 mAChR selective ligands for the treatment of Alzheimer's disease. In M1 mAChR knockout mice, amyloid-β peptide production is increased, while reintroduction of the M1 mAChR into neuronal cultures from these mice reverses the changes in amyloid-β peptide production (Davis et al., 2010a). In addition, activation of the M1 mAChR with agonists increases the production of soluble APP and decreases the production of amyloid-β, suggesting that direct activation of this receptor may also yield disease modifying benefits (Fisher et al., 2003).
Dopamine abnormalities consisting of low dopamine levels in the cortex and high dopamine levels in the striatum are classically proposed to cause the main symptoms of schizophrenia (Weinberger, 1987; Kesby et al., 2018). Positron emission tomography imaging shows that people with schizophrenia have increased synaptic dopamine levels, release higher levels of dopamine in response to amphetamines and have increased basal dopamine synthesis capacity (Kesby et al., 2018). The altered dopamine levels contribute to three distinct symptom domains: positive symptoms (delusions, hallucinations), negative symptoms (lack of emotional expression, low motivation and social withdrawal) and cognitive symptoms (impaired learning, memory, attention and executive function) (Kesby et al., 2018). Interestingly, activation of the M4 mAChR in the striatum controls the release of dopamine through a mechanism involving 2-arachidonoylglycerol and glutamate release, acting upon the cannabinoid receptors (CB2) and the glutamate receptors (mGluR1), respectively (Foster et al., 2016; Yohn et al., 2018). In striatal projection neurons, the M4 mAChR is highly co-expressed with the D1 dopamine receptor (Jeon et al., 2010). When oxotremorine, a mAChR agonist, is administered to these neurons, decreases in dopamine-stimulated cAMP signaling are observed (Nair et al., 2019). This occurs via crosstalk between the receptors, where dopamine activates cAMP signaling via the Gαs-coupled D1 dopamine receptor, and ACh inhibits cAMP production via the Gαi/o-coupled M4 mAChR. This complex interplay between the different neurotransmitter systems highlights how modulation of one system can perturb the network of neurotransmission in the brain.
Studies using post-mortem brain tissues indicate that there is a reduction in the expression levels of both the M1 and the M4 mAChRs in the caudate, putamen, hippocampus, cingulate cortex and the prefrontal cortex in schizophrenic brains compared to non-schizophrenic brains (Raedler et al., 2007; Dean et al., 2016). This suggests that a decrease in signaling from both of these receptors may contribute to symptoms of schizophrenia. A role for the M4 mAChR is further supported by evidence from human clinical trials, where the M1/M4-preferring agonist, xanomeline, improved the positive, negative and cognitive symptoms in schizophrenic patients (Shekhar et al., 2008). While these results are encouraging, an unfavourable, peripheral side effect profile has prevented xanomeline progressing further into the clinic. In 2018, Karuna therapeutics created ‘KarXT’, a combination therapy of xanomeline and trospium (a peripherally restricted mAChR antagonist) (Brannan et al., 2020). In KarXT, trospium specifically blocks the peripheral actions of xanomeline, while allowing xanomeline to provide therapeutic efficacy in the CNS. In phase IIb clinical trials, KarXT successfully reduced positive, negative and cognitive symptoms in schizophrenic patients, but some anti-muscarinic side effects such as constipation, nausea, dry mouth, dyspepsia and vomiting were still observed (Brannan et al., 2020). While these clinical results are very promising, they also suggest that further improvements could be made to reduce the adverse effects of mAChR targeting treatments.
Schizophrenia is a syndrome that is likely composed of multiple etiologies, but presents with similar symptoms. Thus, in the path forward for treatments, the syndrome should be broken up into distinct biological problems that can be solved with the correctly tailored treatment (Jablensky, 2006). [3H]-pirenzipine binding in human post-mortem brain slices identified a subset (∼25%) of schizophrenic patients that have a reduction of approximately 75% M1 mAChR expression in the Brodmann's area nine of the pre-frontal cortex when compared to non-schizophrenic controls (Salah-Uddin et al., 2009; Scarr et al., 2009; Dean et al., 2016; Scarr et al., 2018). Patients with this reduced M1 mAChR expression profile have been classified as a distinct subset of patients with ‘muscarinic receptor-deficit syndrome’ (MRDS) schizophrenia. Gene expression microarray data indicate that 65 genes are distinctly altered in the MRDS group; these genes are important for controlling cell movement and cell signaling pathways, upstream of M1 mAChR (Scarr et al., 2018). This finding is important, because treatment of this subset of schizophrenic patients with M1 mAChR-selective ligands may fail. Indeed, in [3H]-N-methyl-scopolamine binding experiments using post-mortem brain tissues from MRDS and non-schizophrenics, an M1 mAChR-selective ligand, benzyl quinolone carboxylic acid (BQCA), had much weaker effects in the MRDS tissues than in the non-schizophrenic control tissues (Dean et al., 2016; Hopper et al., 2019). Thus, targeting an alternative receptor, such as the M4 mAChR, may be more beneficial for this subgroup of schizophrenic patients.
Important information about the role that the M1 and M4 mAChRs play in the brain can be gained through the use of receptor knockout and chemogenetically modified receptor mice. Knockout of the gene of interest often results in biochemical and behavioral changes that can then be associated with the function of that particular gene. An alternative mouse model uses a mutant mAChR that has two orthosteric binding site mutations, which cause the endogenous ligand ACh to lose activity at the receptor. This mutant receptor, known as a "Designer Receptor Exclusively Activated by Designer Drugs" (DREADD) can however, be activated by the otherwise pharmacologically inactive compound, clozapine-N-oxide (Roth, 2016).
Knockout of either M1 or M4 mAChRs produces hyperactive mice relative to wild-type controls, suggesting that both mAChRs play a role in the control of locomotor activity (Gomeza et al., 1999; Miyakawa et al., 2001; Wess, 2004; Koshimizu et al., 2012). It should be noted, however, that the hyperactivity effect is greater in the M1 knockout mouse than in the M4 knockout mouse (Wess, 2004). The M1 DREADD mice behave similarly to the M1 knockout mice, with respect to locomotor activity levels; however, the hyper-locomotor activity is reversed when the M1 DREADD is activated following administration of clozapine-N-oxide (Bradley et al., 2020). This hyperactivity may be linked to increases in dopamine release, since increases in striatal extracellular dopamine were observed by in vivo quantitative microdialysis in the M1 mAChR knockout mice (Gerber et al., 2001; Zhang et al., 2002). In a striatal specific M4 mAChR knockout, dopamine efflux increases in the nucleus accumbens, which increases cAMP signaling through the D1 dopamine receptor (Jeon et al., 2010). These results suggest that both M1 and M4 mAChR subtypes play a role in the control of central dopamine signaling, which can be linked to increased locomotor activity in mice.
Dopamine transporter knockout mice have a reduction in dopamine reuptake from the synapse, resulting in an accumulation of synaptic dopamine levels and a characteristic hyperactivity phenotype (Gainetdinov et al., 1999; Carpenter et al., 2012; Nair et al., 2019). Thus, measurement of locomotor activity is a commonly used behavioral assay to assess antipsychotic medications. Ligand-induced hyperactivity, using amphetamine, MK-801 or phencyclidine, is typically reduced by compounds that are efficient antipsychotics. Muscarinic agonists acting at either the M1 or the M4 mAChRs decrease the level of synaptic dopamine; therefore, increasing cholinergic activity at both the M1 and M4 mAChR subtypes may be helpful in treating the psychotic symptoms of schizophrenia. Intriguingly, M1 mAChR-mediated locomotor activity is linked to Gαq-mediated signaling events; in mice that express a phosphodeficient M1 mAChR (G protein-signalling pathway biased) the locomotor activity was equivalent to the wild type mice when monitored over a 24 h period (Bradley et al., 2020). In addition, monitoring another Gαq-linked signaling event, ligand-induced IP1 accumulation in the striatum, can predict the behavioral effects of ligands toward reversing amphetamine-induced locomotor activity (Popiolek et al., 2016). Thus M1 mAChR-selective compounds that activate Gαq-IP3 pathways may exhibit good antipsychotic efficacy.
Initial characterisation of M1 mAChR knockout mice in the elevated plus maze, a mouse model of anxiety, indicated that the M1 mAChR knockout mice and wild type mice both spent a similar amount of time in the open arms, suggesting that M1 mAChR knockout is not associated with anxiety (Miyakawa et al., 2001). However, in more recent studies using the same behavioral paradigm, M1 mAChR knockout, M1 DREADD and M1 mAChR-phosphodeficient mice spent less time exploring the open arms than the wild-type controls (Bradley et al., 2020). This latter study suggests that the loss of the M1 mAChR does indeed produce mice with greater anxiety. Interestingly, the amount of time the M1 DREADD mice spent exploring the open arms was restored to that of the control mice upon administration of clozapine-N-oxide, further supporting a role for the M1 mAChR in anxiety (Bradley et al., 2020). The M1 mAChR driven anxiety was also apparent in the M1 mAChR-phosphodeficient mouse, suggesting that the anxiety occurs downstream of G protein-dependent signaling pathways for the M1 mAChR (Bradley et al., 2020). The only study that explored anxiety responses in M4 mAChR knockout mice to date revealed that M4 mAChR knockout mice have decreased burying responses when tested in a shock-probe burying model, indicating that they also have increased anxiety levels (Degroot and Nomikos, 2006).
Knockout of the M4 mAChR generates mice with only subtle physiological changes when they are compared to wild type mice, indicating that this is a very mild receptor knockout (Gomeza et al., 1999; Bymaster et al., 2001; Bymaster et al., 2003; Koshimizu et al., 2012). M4 mAChR knockout mice display abnormal social behavior, with less contact observed when compared to wild type mice (Koshimizu et al., 2012). This contrasts with the results in the M1 mAChR knockout mice, where increases in social contacts are observed compared to wild type mice, which may be due to the increase in locomotor activity of these mice rather than a reduction in anxiety (Miyakawa et al., 2001). The M4 mAChR may be involved in psychotic symptoms because the M4 mAChR knockout mice have increased sensitivity to psychomimetic drugs, with increased startle responses observed when M4 mAChR knockout mice are given phencyclidine (an NMDA receptor antagonist) compared with wild type controls (Felder et al., 2001; Bubser et al., 2014). The pre-pulse inhibition of the startle reflex behavioral paradigm is another test used to assess antipsychotic drugs for efficacy. When the M4 mAChR knockout mice are pre-treated with phencyclidine, these animals have increased disruption of pre-pulse inhibition, suggesting that the M4 mAChR may be involved in psychosis (Felder et al., 2001; Wess 2004). When M4 mAChR knockout mice were tested in the Morris water maze or by touchscreen discrimination tasks, they performed equally well as wild type controls, suggesting that the M4 mAChR has little involvement in cognition (Koshimizu et al., 2012; Bubser et al., 2014). Together these results suggest that the M4 mAChR is important in psychosis, social behavior and anxiety but is less important in cognition. Thus, M4 mAChR-targeting drugs may be particularly helpful for treating the positive and negative symptoms of schizophrenia.
Historically, it has proven extremely challenging to selectively target one mAChR subtype over the other mAChR subtypes because the residues lining the orthosteric ACh binding site of all five mAChR subtypes are absolutely conserved. Figure 2 shows the alignment for the amino acid residues of the M1-M5 mAChRs. This absolute conservation of the residues within the orthosteric site has thus hindered the design and synthesis of highly subtype selective mAChR ligands. It is thought that activation of the peripheral mAChRs by non-selective orthosteric ligands is one of the main reasons for the failure of many mAChR-based drug candidates for the treatment of neurological disorders.
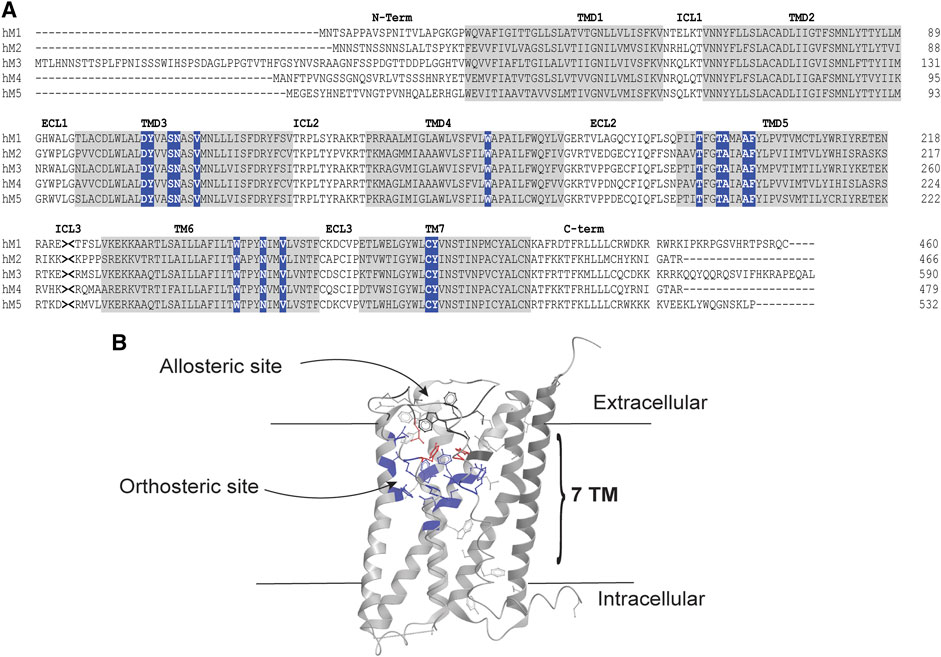
FIGURE 2. Orthosteric and allosteric sites of the mAChRs. (A) Amino acid sequence alignment of the human M1-M5 mAChRs. The amino (N)-terminal tail, Intracellular loops (ICL), extracellular loops (ECL) and carboxy (C)-terminal tails are shown as black letters on a white background. Transmembrane domains (TMD) are shown as black letters on a gray background. Orthosteric site residues are white letters on a blue background. The intracellular loop three was truncated, as indicated by >< for presentation of the alignment. Alignment was performed using clustal omega. (B) X-ray crystal structure of the human M4 mAChR (RCSB PDB number 5DSG), showing the location of orthosteric and allosteric sites. Orthosteric site residues are highlighted in blue, allosteric site residues are highlighted in black and residues that contribute to both binding pockets are highlighted in red.
The orthosteric site lies deep within the transmembrane helices of the mAChRs, defined by amino acid residues in the transmembrane spanning helices 3, 4, 5, 6, and 7, as shown in Figure 2B (Thal et al., 2016). Interestingly, it appears that all mAChRs have an allosteric binding site that is located in an extracellular vestibule above the orthosteric site (Haga et al., 2012; Kruse et al., 2013; Thal et al., 2016). Allosteric sites recognize structurally distinct ligands to modulate the activity of co-bound orthosteric ligands (Christopoulos and Mitchelson, 1995; Christopoulos et al., 1997; Lanzafame et al., 1997; Christopoulos et al., 1998). Gallamine was one of the earliest and best-studied allosteric ligands for the mAChRs, and remains an exemplar molecule for GPCR allostery in general (Clark and Mitchelson, 1976). Through receptor mutagenesis and structural biology studies, many residues that are important for binding allosteric ligands into the allosteric site of the M1 and M4 mAChRs have been identified in this region (Nawaratne et al., 2010; Leach et al., 2011; Abdul-Ridha et al., 2014; Keov et al., 2014; Thal et al., 2016). The important residues for this site include those in the top of transmembrane helices 2, 6, and 7 as well as in extracellular loop 2 (Nawaratne et al., 2010; Leach et al., 2011; Abdul-Ridha et al., 2014; Keov et al., 2014; Thal et al., 2016). Excitingly, the amino acid residues of this extracellular allosteric site of the mAChRs show greater diversity between the different subtypes, thus providing the framework for designing mAChR subtype selective allosteric ligands. In fact, the allosteric sites of the M1 and M4 mAChRs have successfully been targeted by rationally designed synthetic allosteric ligands, with (now) a large number of subtype selective allosteric ligands available as pharmacological tools (Ma et al., 2009; Kuduk et al., 2010; Kuduk et al., 2011; Salovich et al., 2012; Le et al., 2013; Mistry et al., 2013; Croy et al., 2014; Huynh et al., 2015; Davoren et al., 2016a; Mistry et al., 2016a; Wood et al., 2016a; Davoren et al., 2016b; Mistry et al., 2016b; Wood et al., 2016b; Wood et al., 2017a; Wood et al., 2017b; Davoren et al., 2017; Long et al., 2017; Tarr et al., 2017; Bertron et al., 2018; Beshore et al., 2018; Dallagnol et al., 2018; Engers et al., 2019a; Engers et al., 2019b; Chopko et al., 2019; Jorg et al., 2019; Poslusney et al., 2019; Schubert et al., 2019; Temple et al., 2019; Temple et al., 2020a; Temple et al., 2020b). Since the orthosteric and allosteric sites are topographically distinct, two ligands can bind one receptor simultaneously. Upon binding to the receptor, the allosteric ligand can alter the pharmacological properties of the co-bound orthosteric ligand. This alteration in the pharmacological profile of the orthosteric ligand is defined as an ‘allosteric interaction’ (Christopoulos et al., 2014), and can be identified as either a change of binding affinity (KA) or signaling efficacy (τA) of the orthosteric ligand at the receptor.
Several different types of allosteric modulators have been identified thus far, depending on their effect on the co-bound orthosteric ligand and their potential direct effect on receptor-mediated signaling. Positive allosteric modulators (PAMs) are ligands that enhance the pharmacological properties of the orthosteric ligand. Negative allosteric modulators (NAMs) are ligands that diminish the affinity and/or efficacy of the orthosteric ligand. Neutral allosteric ligands (NALs) are ligands that bind to the allosteric site, but do not alter the properties of the co-bound orthosteric ligand (May et al., 2007; Christopoulos et al., 2014). In addition to their potential modulatory effects on orthosteric ligand binding and/or signaling, allosteric ligands can also have a direct effect on the receptor, and trigger a signaling response, in a not too dissimilar manner to orthosteric agonists, but from the allosteric site specifically. These ligands are called ‘PAM-agonists’ as they potentiate the effects of the orthosteric ligand and simultaneously have a direct effect on the receptor. This is in contrast to “pure” PAMs, which only potentiate the effect of the orthosteric ligand but have no direct effect on the receptor’s signaling capabilities on their own. The discovery of allosteric modulators of mAChRs has rejuvenated drug discovery programs, due to their significant advantages over orthosteric drugs. They can achieve subtype selectivity, maintain normal spatial and temporal profiles of physiological signaling and can be devoid of agonist activity, a key feature for targeting mAChRs in the CNS.
There are a variety of biochemical and functional approaches for experimentally determining and validating allosteric interactions (Christopoulos, 2014; Christopoulos et al., 2014). The majority involve the determination of the effect of increasing concentrations of an allosteric modulator on the binding or signaling of an orthosteric ligand. In the case of a PAM, the allosteric effect may result in an increase in orthosteric ligand affinity, which would manifest as an increase in the dissociation constant (KA) and the functional potency (EC50) of the orthosteric ligand, and/or an increase in ligand efficacy, which would manifest functionally as an increase in agonist potency for full agonists or an increase in agonist maximal effect (Emax) for partial agonists. In contrast, a NAM would have the opposite effects on orthosteric ligand affinity and/or efficacy, thus reducing agonist affinity, potency and/or Emax depending on the type of NAM effect and the intrinsic efficacy of the orthosteric ligand (Figure 3). If the allosteric modulator is a NAL, then there will be no change in the affinity or potency of the orthosteric ligand at equilibrium, although the NAL may still result in changes on orthosteric agonist binding kinetics as well as competing with PAMs or NAMs that bind to the same allosteric site (Christopoulos et al., 2014). Additional effects can be observed, depending on the properties of the allosteric modulator. For example, if the allosteric ligand is a PAM-agonist, it can activate the receptor in the absence of an orthosteric ligand in addition to allosteric modulatory effects (Figure 3).
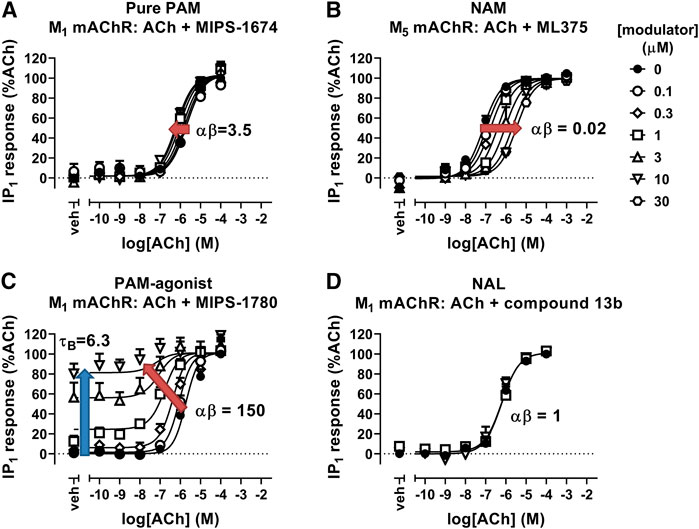
FIGURE 3. Allosteric modulation of mAChRs. (A) The PAM, MIPS-1674, increases the potency (αβ) of ACh toward IP1 accumulation at the M1 mAChR. (B) The NAM, ML375, decreases the potency of ACh at the M5 mAChR in an IP1 accumulation assay. (C) The PAM-agonist, MIPS-1780, increases the potency (αβ) of ACh toward IP1 accumulation, but also has its own allosteric agonist effect (τB). (D) The NAL, compound 13b, has no effect on the potency of ACh toward IP1 accumulation at the M1 mAChR. Data are from van der Westhuizen et al. (2018), Berrizzi et al. (2016), and Jorg et al. (2020). Data were fit with the simplified operational model of allostery and agonism (Aurelio et al., 2009), to quantify the cooperativity (αβ) and intrinsic efficacy (τB) parameters.
To quantify the effects that an allosteric modulator can exert on an orthosteric ligand, a number of mechanistic and operational models have been developed for analysis of experimental data. The most common mechanistic model that describes allosteric effects on orthosteric ligand affinity is the allosteric ternary complex model (ATCM; Stockton et al., 1982; Ehlert, 1988; Christopoulos and Kenakin, 2002). The ATCM quantifies the affinity of the allosteric modulator for the allosteric site (KB) and the effect the modulator has on the affinity of the co-bound orthosteric ligand (binding cooperativity; α), in addition to orthosteric ligand affinity (KA). Values of α > 1 denote positive cooperativity, values of α < 1 but > 0 denote negative cooperativity, and α values equal to one denote neutral binding cooperativity. For signaling assays, the ATCM has been incorporated into an operational model that also allows for quantification of allosteric effects on orthosteric agonist efficacy (β) as well as direct orthosteric (τA) and allosteric (τB) agonism (Price et al., 2005; Leach et al., 2007). In operational terms, β values of >1 denote positive efficacy modulation, values of β < 1 but > 0 denote negative efficacy modulation, and β values equal to one denote no modulation of orthosteric signaling efficacy. Of note, in situations where the orthosteric ligand is a full agonist in both the absence and presence of modulator, the operational model can be simplified to allow for derivation of an overall combined modulatory effect, quantified through the composite parameter, αβ (Aurelio et al., 2009). Application of these allosteric models has been successful in determining structure activity relationships for allosteric modulators of multiple GPCRs including the adenosine A1 (Ferguson et al., 2008; Aurelio et al., 2009; Aurelio et al., 2010; Aurelio et al., 2011; Valant et al., 2012a) the mAChRs (Mistry et al., 2013; Mistry et al., 2016b; Dallagnol et al., 2018; Jorg et al., 2019; Jorg et al., 2020), the mGlu receptors (Mueller et al., 2012; Gregory et al., 2013; Turlington et al., 2013) and the GLP1-R (Wootten et al., 2012; Wootten et al., 2013b; Hager et al., 2017).
The first M1 mAChR PAM with high subtype-selectivity was presented by Merck with the discovery of BQCA (Ma et al., 2009). BQCA increases the binding affinity and functional potency of ACh and carbachol at the M1 mAChR overexpressed in CHO cells or primary cortical neurons (Ma et al., 2009; Canals et al., 2012; Abdul-Ridha et al., 2013). In vivo animal studies also showed that BQCA reverses scopolamine-induced memory loss, decreases amphetamine-induced hyperlocomotion in rodents (Ma et al., 2009; Chambon et al., 2012) and can act synergistically with sub-threshold doses of antipsychotics (Choy et al., 2016). Notably, it also increases APP processing (Shirey et al., 2009) and extends the lifespan of terminally-sick mice with neurodegenerative disease (Bradley et al., 2017), suggesting that M1 PAMs have the potential to improve cognition and to modify the underlying cause of Alzheimer's disease. However, this compound was not progressed into clinical trials due to its poor solubility, limited brain penetration and high plasma protein binding properties (Kuduk et al., 2011). Subsequently, there have been substantial efforts to develop novel M1 mAChR PAMs with improved physicochemical properties (Mistry et al., 2013; Davie et al., 2014; Kuduk et al., 2014; Davoren et al., 2016a; Mistry et al., 2016a; Davoren et al., 2016b; Mistry et al., 2016b; Panarese et al., 2016; Davoren et al., 2017; Flohr et al., 2017; Bertron et al., 2018; Beshore et al., 2018; Dallagnol et al., 2018; Engers et al., 2019b; Jorg et al., 2019; Mandai et al., 2019; Jorg et al., 2020)
High M4 mAChR-subtype selectivity was first described for the PAM, LY2033298 (Chan et al., 2008). This PAM increased the binding affinity and potency of ACh in CHO cells expressing the human M4 mAChR, however, LY2033298 was also noted to have some activity at the M2 mAChR (Chan et al., 2008; Valant et al., 2012b). In addition, LY2033298 did not potentiate ACh at the rodent M4 mAChR to the same extent as at the human M4 mAChR, indicating that there is species variability in the modulatory responses at the M4 mAChR (Chan et al., 2008; Suratman et al., 2011). When administered in vivo, LY2033298 has weak to modest effects. However, when LY2033298 is co-administered with a sub-effective dose of oxotremorine, it reduced conditioned avoidance responses and reversed apo-morphine induced pre-pulse inhibition (Chan et al., 2008; Leach et al., 2010). This compound provided important proof of concept that the M4 mAChR can be allosterically targeted and has served as a basis for the development of novel compounds through medicinal chemistry efforts (Salovich et al., 2012; Huynh et al., 2013; Le et al., 2013; Byun et al., 2014; Huynh et al., 2015; Wood et al., 2016a; Wood et al., 2016b; Wood et al., 2017a; Bewley et al., 2017; Wood et al., 2017b; Tarr et al., 2017; Engers et al., 2019a; Schubert et al., 2019; Temple et al., 2019; Temple et al., 2020a; Temple et al., 2020b).
Achieving receptor subtype selectivity was an important milestone in the development of potentially therapeutic mAChR ligands. Unfortunately, most subtype-selective PAMs that have been disclosed to date are still plagued by both central and peripheral adverse side effects, particularly at the M1 mAChR. One of the most worrying CNS-based side effects of M1 mAChR-selective compounds is their ability to trigger epileptic like seizures (Turski et al., 1989; Davoren et al., 2016a; Bradley et al., 2020). M1 mAChR-selective PAMs, such as PF06767832 and MK-7622, have high intrinsic agonism toward calcium signaling in transfected cell systems, and trigger seizures in rodents. It is hypothesised that this on-target effect is due to the strong allosteric agonism (τB) of these ligands (Davoren et al., 2016a; Beshore et al., 2018; Moran et al., 2019). This adverse effect is not observed in M1 mAChR knockout mice administered orthosteric agonists, confirming that the seizure events are indeed driven by the M1 mAChR (Hamilton et al., 1997). Therefore, focusing the development of next generation selective PAMs on compounds with minimal intrinsic allosteric efficacy and/or modest positive cooperativity may eliminate this and other on-target side effects while retaining the therapeutic benefit of allosteric potentiation of the ACh response.
GPCRs are highly dynamic cell-surface proteins that can activate multiple signaling pathways through recruitment and activation of different families of G proteins, cellular kinases and scaffold proteins. Excitingly, there is a large body of evidence suggesting that recruitment and activation of the different transducer proteins can occur in a ligand-dependent and in a cell-type-dependent manner (Smith et al., 2018; Wootten et al., 2018). It is now clear that structurally distinct ligands can stabilise different receptor conformations. Whilst some GPCR ligands are capable of stabilising a large set of receptor conformations, allowing the receptor to couple to the full range of transducers to transmit the signals from the extracellular environment to intracellular proteins, others can only stabilise a subset of conformations. Such ligands are called biased ligands, because they can direct the signal that emanates from the receptor to one or several particular signaling pathways over all the signaling pathways that are available to the GPCR (Smith et al., 2018; Wootten et al., 2018). This ability to selectively activate certain pathways at the detriment of others has made the concept of bias agonism extremely attractive for development of drugs targeting the M1 and M4 mAChRs. If some of the side effects observed with mAChR agonists and allosteric modulators are due to on-target driven side effects, then developing selective biased agonists or modulators for these receptors may be another method to reduce the adverse effects of drugs.
Biased agonists often display a reversal of efficacy and/or potency in concentration-response curves at different signaling pathways relative to a reference compound. Such effects have been well documented for many GPCRs (Smith et al., 2018; Wootten et al., 2018). Relative agonist activity values are then calculated for a range of different ligands and for a range of different pathways then compared to a reference agonist that ideally activates all known coupled pathways. There are several methods that have been proposed to quantify bias at GPCRs (Ehlert, 2008; Rajagopal et al., 2011; Kenakin, 2012; Kenakin et al., 2012; Kenakin and Christopoulos, 2013). These models essentially condense comparisons between a test and reference ligand's affinity, potency and efficacy into a single parameter, which is then used to compare the effects of ligands on different pathways relative to the reference agonist. Due to the difficulties in achieving subtype selectivity with orthosteric agonists at the mAChRs, engendering biased allosteric modulation is a viable strategy for achieving subtype-selective biased signaling at mAChRs. Allosterically-mediated bias can be experimentally observed by changes in the efficacy parameter (β), where an allosteric modulator can impose positive modulation of an orthosteric agonist toward one pathway, but may yield negative modulation of the same co-bound orthosteric agonist on a distinct signaling pathway (Goupil et al., 2010; Wootten et al., 2013a). More subtly, it can also manifest as a variation in the cooperativity that an allosteric ligand may exert on an orthosteric ligand, or as a change in the functional affinity (KB) of allosteric ligands for cellularly-compartmentalised receptor conformations linked to different signaling pathways (Davey et al., 2012; Wootten et al., 2013a).
Excitingly, the physiological relevance of biased signaling was recently shown using phospho-deficient and chemogenetically modified M1 mAChRs mouse models (Bradley et al., 2020). This study suggested that mAChR ligands that induce M1 mAChR phosphorylation are critical for reducing adverse effects, such as salivation and gastrointestinal disturbances. In contrast, mAChR ligands that displayed weaker ability to phosphorylate M1 mAChR, thus biased toward G protein signaling pathways, produced greater central and peripheral adverse effects (Bradley et al., 2020). When all the phosphorylation sites of the M1 mAChR were mutated to alanine to create an M1-phophodeficient mAChR, activation of Gαq-mediated signaling events remained intact, but recruitment of β-arrestin was decreased by ∼50% (Bradley et al., 2020). Excitingly, M1-phosphodeficient mAChR transgenic mice exhibited similar locomotor activity over 24 h compared to wild type mice, but displayed increased anxiety-like behaviors and cognitive deficits in a Y-maze test, similar to M1 mAChR knockout mice (Bradley et al., 2020). Together, these ground-breaking studies suggest that the regulation of learning and memory processes and anxiety-related behaviors, are directly linked to M1 mAChR phosphorylation and subsequent downstream signaling pathways, such as β-arrestin recruitment, whereas, adverse effects such as salivation, seizures, and hyperactivity are linked to Gαq, IP3 and calcium signaling pathways (Bradley et al., 2020). M1 mAChR-selective positive allosteric modulators that stabilize mAChR conformations linked to receptor phosphorylation and β-arrestin signaling pathways may be strong drug candidates to improve cognition in Alzheimer's disease. In contrast, given that the locomotor activity is linked to G protein signaling events, drugs that exhibit strong phosphorylation and β-arrestin signaling pathways, yet retain some degree of signaling via G proteins may be superior for treating some symptom domains of schizophrenia.
Although there are a wide range of ligands available for the mAChRs, only a handful of studies have systematically tested for biased agonists and allosteric modulators at the M1 and M4 mAChRs. The M1 mAChR couples to Gαq/11, Gαi/o and Gαs proteins, to increase calcium release, inhibit and stimulate cAMP, respectively. Early studies that characterised mAChR agonist profiles across different receptor subtypes and signaling pathways suggested that pilocarpine, oxotremorine, arecoline, cevimeline, McN-A-343 and cis-AF30, were Gαq-biased ligands that did not activate Gαs and cAMP signaling at the M1 mAChR in CHO cells (Table 1; Gurwitz et al., 1994). However, subsequent studies have demonstrated that pilocarpine and arecoline are weak partial agonists for the cAMP signaling pathway (Thomas et al., 2008). Interestingly, the non-selective agonist, cis-dioxolane, increases arachidonic acid release and cAMP accumulation without activating IP3 signaling (Figure 4, Table 1) (Gurwitz et al., 1994), a finding that has not been confirmed by other studies but could represent a potentially interesting bias profile. At the M1 mAChR expressed in CHO cells, five agonists were investigated for biased signaling toward Gαq activation, Gαi1/2 activation, Gαs activation, IP3 accumulation and cAMP accumulation (Table 1) (Thomas et al., 2008). The non-subtype selective mAChR agonists, oxotremorine-M, pilocarpine and arecoline activated Gαq, Gαi1/2, IP3 and cAMP pathways. Whereas, the M1-selective bitopic ligands, AC-42 and 77-LH-28–1 activated Gαq, IP3 and cAMP pathways but did not activate Gαi1/2, suggesting that the allosteric ligands were biased toward Gαs and Gαq-coupled pathways (Figure 4, Table 1) (Thomas et al., 2008; Davis et al., 2009).
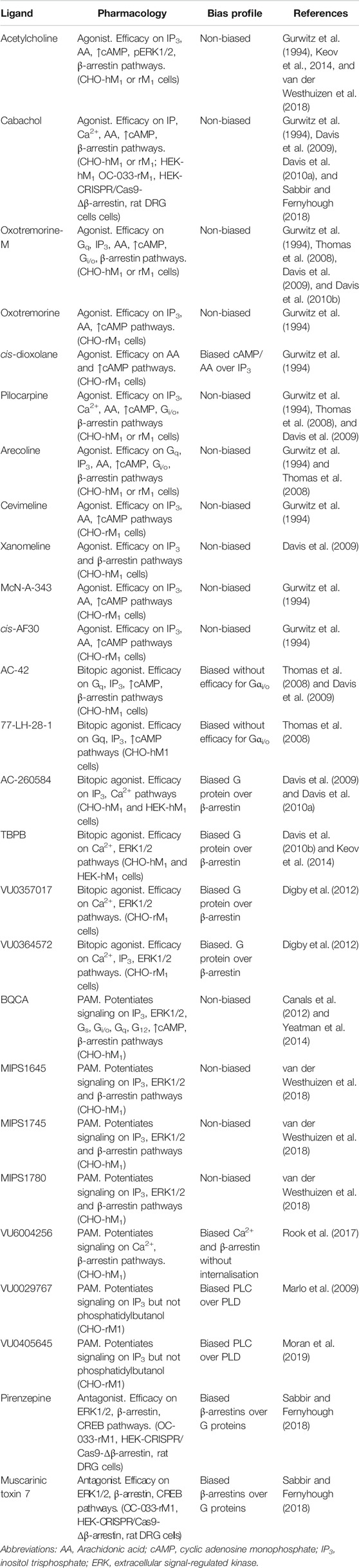
TABLE 1. Ligands screened, pharmacology assessed and biased profiles determined for a range of orthosteric, bitopic and allosteric ligands at the M1 mAChR. Different combinations of ligands and pathways have been used to assess a range of mAChR ligands for biased signaling. Biased signaling has been assessed in recombinant Chinese hamster ovary (CHO) or Human embryonic kidney (HEK) cells stably expressing either the rat (rM1) or human (hM1) mAChRs, OC-033 mouse tumor cell lines or Sprague-Dawley rat dorsal root ganglia (DRG) neurons.
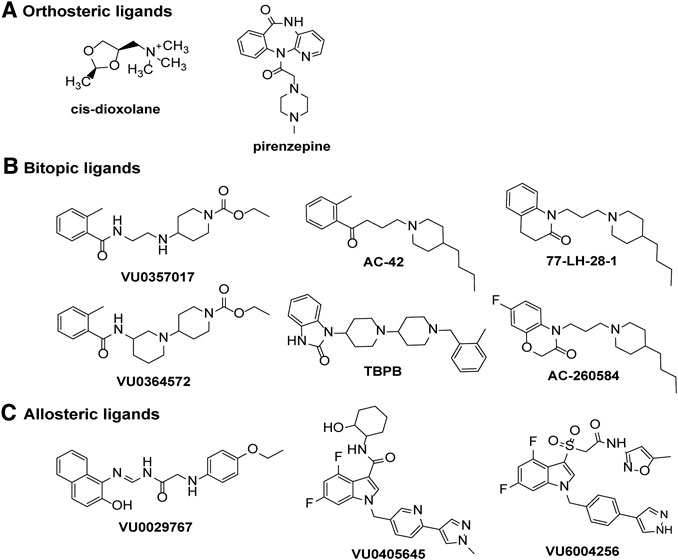
FIGURE 4. Chemical structures of ligands displaying signaling bias at the M1 or M4 mAChRs. Biased signaling profiles are reported for (A) orthosteric agonists or antagonists, (B) bitopic ligands or (C) allosteric ligands at the M1 and M4 mAChRs.
At the M4 mAChR, to date, seven agonists have been systematically and quantitatively assayed for potential biased signaling across measures of cAMP inhibition and cAMP accumulation in CHO cells stably expressing the M4 mAChR and for IP3 accumulation in human embryonic kidney (HEK) cells co-expressing Gα15 and the M4 mAChR (Figueroa et al., 2009). There was no evidence of agonist-directed signaling for oxotremorine-M, carbachol, McN-A-343, S-aceclidine, R-aceclidine, arecoline or pilocarpine at any of the pathways tested (Table 2). A similar study assessed methacholine, oxotremorine-M, arecoline, bethanacol, oxotremorine or pilocarpine for bias in modulating cAMP inhibition vs. cAMP accumulation at the M4 mAChR, but found no quantitative differences in relative responses (Table 2) (Mistry et al., 2005). Due to the lack of apparent bias for mAChR agonists in these well-documented signaling assays, further studies looking for biased orthosteric agonists have not been pursued.
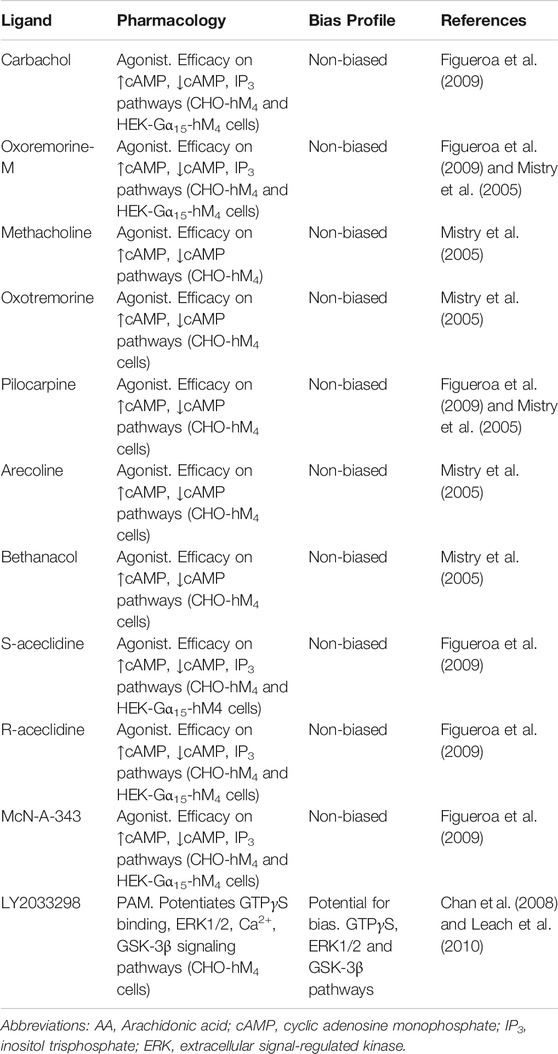
TABLE 2. Ligands screened, pharmacology assessed and biased profiles determined for a range of orthosteric, bitopic and allosteric ligands at the M4 mAChR. Different combinations of ligands and pathways have been used to assess a range of mAChR ligands for biased signaling. Biased signaling has been assessed in recombinant Chinese hamster ovary (CHO) or Human embryonic kidney (HEK) cells stably expressing the human (hM4) mAChRs.
In CHO cells expressing the M1 mAChR, carbachol increases calcium levels, ERK1/2 phosphorylation, β-arrestin 2 translocation and causes receptor internalization (Davis et al., 2010b). In the same cell system, AC260584 and TBPB (Figure 4), which are M1 mAChR bitopic ligands that span both the orthosteric and allosteric sites, increased calcium levels and ERK1/2 phosphorylation, but did not translocate β-arrestin or cause receptor internalization (Table 1) (Davis et al., 2010b). Similarly, the bitopic ligands, VU0357017 and VU0364572 (Figure 4), also increased calcium and ERK1/2 phosphorylation in CHO cells overexpressing M1 mAChR, but they did not recruit β-arrestin (Table 1) (Digby et al., 2012). These results suggest that traditional orthosteric ligands, such as carbachol, activate all signaling pathways available to the receptor, whereas compounds that interact with the allosteric site, can direct their signal activation away from β-arrestin recruitment, receptor internalization and subsequent downstream signaling pathways. In vivo work with VU0357017 and VU0364572 showed that both compounds improved hippocampal dependent memory but failed to decrease amphetamine-induced hyper-locomotion in rats (Digby et al., 2012). A result also suggesting a link between G protein activation at the M1 mAChR and hyperlocomotion responses in rats, however, further work to confirm such a link is required.
Pharmacologically distinct effects on different measures of cellular function are not restricted to mAChR agonists, with mAChR antagonists now linked to selective effects on intracellular signaling. Prolonged treatment (1–1.5 h) of M1 mAChR expressing cell lines with the mAChR antagonists muscarinic toxin 7 (MT7) or pirenzepine (Figure 4), increases ERK1/2 phosphorylation in acidic fractions (pH ∼3) and CREB phosphorylation in several different cell lines; an effect not replicated with the agonist carbachol (Table 1) (Sabbir and Fernyhough, 2018). In Δβ-arrestin 1/2 CRISPR/Cas-9 knockout HEK293 cells, CREB and ERK1/2 phosphorylation by pirenzepine and MT7 is lost, suggesting that this pathway requires β-arrestins (Sabbir and Fernyhough, 2018). Activation of this β-arrestin-ERK1/2-CREB signaling pathway increases neurite outgrowth in cultured primary dorsal root ganglion neurons (Sabbir and Fernyhough, 2018), suggesting that this signaling pathway has some physiological relevance. Further work to confirm this result and to explore whether other mAChR antagonists exhibit the same ERK1/2 phosphorylation effects may yield some interesting biased compounds that could be further developed for therapeutic benefit.
With the recent advances in allosteric drug discovery, some mAChR allosteric modulators have been assessed for the degree to which they can induce bias by altering the pattern of signaling of orthosteric ligands. BQCA potentiates the responses to carbachol on cAMP production, ERK1/2 phosphorylation, IP1 accumulation, Gαs activation, Gαi1/2 activation, Gαq activation, Gα12 activation and β-arrestin recruitment without altering the relative activation of the different pathways (Table 1) (Canals et al., 2012; Yeatman et al., 2014). Similarly, the structurally diverse M1 PAMs, MIPS1674, MIPS1745 and MIPS1780 induce equivalent potentiation of ACh-mediated IP1 accumulation, ERK1/2 phosphorylation, and β-arrestin recruitment (van der Westhuizen et al., 2018). Mechanistically, this is consistent with enhancement of affinity as the driver of observed cooperativity for these compounds (van der Westhuizen et al., 2018). When BQCA was assessed at the M1 DREADD, it weakly potentiated the effects of the DREADD ligand clozapine-N-oxide, in measures of calcium mobilisation, IP1, ERK1/2 and cAMP signaling (Abdul-Ridha et al., 2013). Of note, although BQCA was a PAM of the potency of clozapine-N-oxide on the cAMP pathway, it was a NAM of the efficacy of clozapine-N-oxide on the cAMP pathway. In contrast, when ACh was used as the agonist BQCA was a PAM for both potency and efficacy on the cAMP pathway (Abdul-Ridha et al., 2013). Thus, BQCA is a selective biased modulator of clozapine-N-oxide at the M1 DREADD. The mechanistic simplicity of the cooperativity between BQCA and ACh at M1 mAChRs, at all pathways investigated to date, makes this compound a good "reference" modulator for future studies.
In contrast to BQCA, biased modulation has been observed with the PAMs VU6004256, VU0029767 and VU0405645 (Figure 4) at the M1 mAChR (Table 1) (Marlo et al., 2009; Rook et al., 2017; Moran et al., 2019). In the case of VU6004256, comparison of this PAM with PF06764427 revealed that both PAMs potentiated ACh-mediated calcium fluxes and β-arrestin recruitment, however, PF-06764427 promoted M1 mAChR internalisation whereas VU6004256 did not (Rook et al., 2017). Further studies revealed that the PAMs VU0090157 and VU0453595 activate both phospholipase C (PLC) and phospholipase D (PLD) pathways, whereas VU0029767 and VU0405645 coupled only to PLC pathways but not PLD pathways (Marlo et al., 2009; Moran et al., 2019). Furthermore, VU0453595 potentiated carbachol-mediated field excitatory postsynaptic potentials (fEPSPs) in mouse prefrontal cortex containing coronal slices, but VU0405645 did not, suggesting that compounds that select for PLC pathways over PLD pathways can actually be detrimental to the formation of long term depression in the prefrontal cortex (Moran et al., 2019).
At the M4 mAChR, LY2033298 has a robust allosteric agonist profile toward G protein activation, ERK1/2 phosphorylation and the GSK-3β signaling pathway. Whilst LY2033298 displayed various degree of agonist activity in most signaling assays, no allosteric agonism for the calcium signaling pathway was detected (Table 2) (Chan et al., 2008; Leach et al., 2010). In receptor internalization assays, LY2033298 had a small degree of allosteric agonist activity but a large degree of allosteric potentiation, more so than for the any other signaling assays investigated. Whilst the potential for bias was not directly quantified in these studies, the results hinted at the potential for biased modulation at the M4 mAChR. With the development of many new M4 mAChR PAMs, further work to explore the potential for biased modulation of the M4 mAChR may yield new and exciting results in this field.
Like most GPCRs, the M1 and M4 mAChRs couple to a wide array of signal transduction pathways, as can be seen in Figures 5, 6. These include those linked to phospholipase C (PLC), calcium release, phospholipase D (PLD), phospholipase A2(PLA2), cAMP increases and decreases, and mitogen-activated protein kinase pathways (Felder, 1995; Nathanson, 2000). Some of these pathways have well-described roles in learning, memory and synaptic plasticity, and are implicated in the symptomology of neurological diseases, while others require further characterisation to identify potential avenues for the development of new biased ligands for the mAChRs.
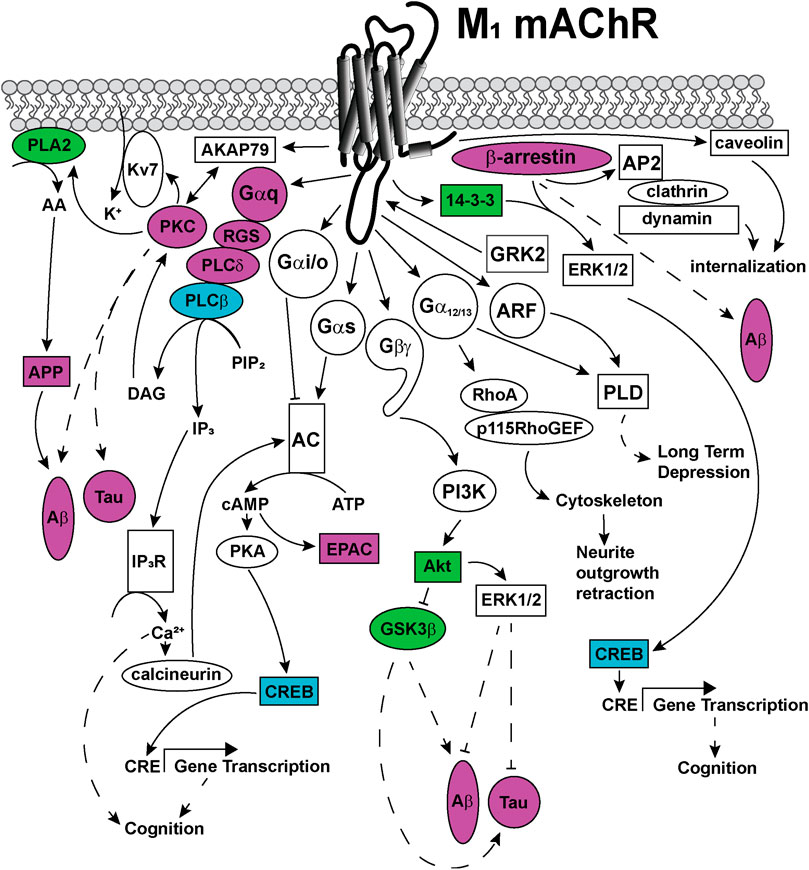
FIGURE 5. Signaling pathways coupled to the M1 mAChR. Upon activation, the M1 mAChR principally couples to Gαq proteins to activate phospholipase C (PLC) enzymes to catalyze the membrane lipid substrate, phosphatidyl inositol 4,5-bisphosphate (PIP2) into inositol trisphophate (IP3) and diacylglycerol (DAG). DAG activates protein kinase C (PKC), which is anchored to the M1 mAChR via a direct interaction with the scaffold protein, A-kinase anchoring protein (AKAP)-79. This AKAP-PKC complex activates the potassium M-channel (Kv7/KCNQ), permitting the influx of potassium (K+) into neurons to regulate neuronal excitability. IP3 activates the IP3 receptor (IP3R) on the sarcoplasmic reticulum, releasing calcium (Ca2+) from the intracellular stores to the cytosol. Ca2+ activates intracellular signaling proteins, such as calcineurin, which can in turn activate some adenylate cyclase (AC) isoforms. PKC activates phospholipase A (PLA)2, which increases cytosolic arachidonic acid (AA), releasing amyloid precursor proteins (APP) and driving the formation of toxic amyloid-β (Aβ) plaques in Alzheimer's disease. The M1 mAChR also couples to Gαs and Gαi/o proteins, which can directly activate or inhibit AC, respectively. Activated AC converts cytosolic adenosine triphosphate (ATP) to the second messenger, cyclic adenosine monophosphate (cAMP). cAMP can in turn, activate signaling proteins such as protein kinase A (PKA) or exchange protein directly activated by cAMP (EPAC). PKA phosphorylates and activates cAMP response element binding protein (CREB), which increases gene transcription from cAMP response elements (CRE). Genes with CRE promoter regions are linked to improved cognition. The M1 mAChR couples to Gα12/13 proteins, which activate Ras homolog family member A (RhoA), Rho guanine nucleotide exchange factor 1 (p115RhoGEF), resulting in cytoskeletal rearrangements and neurite outgrowth retraction. Phospholipase D (PLD) is activated by either Gα12/13 proteins or the small GTPase adenosine diphosphate-ribosylation factor (ARF) proteins, to induce long-term depression. The Gβγ-subunits are also linked to signal transduction, through activation of phosphoinosotide 3-kinase (PI3K) and protein kinase B (Akt). Akt activates extracellular signal-regulated kinase (ERK) 1/2 which is linked to decreases in Aβ levels and decreases in Tau protein phosphorylation. Akt inhibits the activity of glycogen synthase kinase (GSK) 3β, a kinase that is associated with increases in Aβ and Tau hyperphosphorylation in Alzheimer's disease. M1 mAChR signaling is regulated by phosphorylation of the receptor at serine and threonine residues located in intracellular loops and carboxy-terminal tail, by kinases including GRK2. GRK2 phophorylation promotes the recruitment of β-arrestin scaffold proteins that promote M1 mAChR internalisation via clathrin-coated pits through interactions with the adaptor complex, AP2 protein, clathrin and dynamin. Additional M1 mAChR internalization may occur via recruitment of caveolins, through clathrin-independent pathways. Recruitment of scaffold proteins such as, β-arrestin and 14-3-3 are also linked to ERK1/2 activation and increases in transcription from CRE elements. Dashed arrows indicate pathways requiring further work to characterise the signaling proteins involved in the pathway. Solid arrows represent experimentally determined interactions. Proteins that are disrupted in Alzheimer's disease are colored in purple, those disrupted in schizophrenia are colored in blue and those disrupted in both diseases are colored in green.
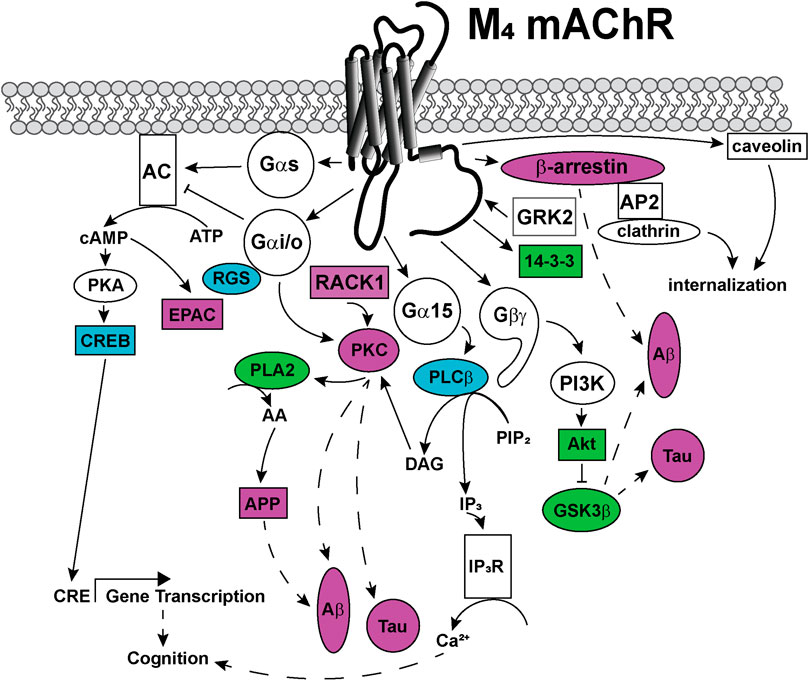
FIGURE 6. Signaling pathways coupled to the M4 mAChR. Upon activation, the M4 mAChR is principally coupled to Gαi/o proteins that inhibit adenylate cyclase (AC) to decrease cytosolic cyclic adenosine monophosphate (cAMP) levels. The M4 mAChR also couples to Gαs proteins, when activated by high (100 μM) concentrations of agonists. Gαs directly activates adenylate cyclase to convert cytosolic adenosine triphosphate (ATP) to cAMP, a second messenger that can activate protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC). PKA phosphorylates and activates cAMP response element binding protein (CREB), which increases gene transcription from cAMP response elements (CRE). Phospholipase A (PLA)2 is activated by the M4 mAChR via a mechanism involving Gαi/o and protein kinase C (PKC). PLA2 increases intracellular arachidonic acid (AA) levels, which promotes amyloid precursor protein (APP) release, amyloid-β (Aβ) plaque formation and Tau protein hyperphosphorylation in Alzheimer's disease. Phosphoinositol 3-kinase (PI3K) is activated by Gβγ-subunits, and PI3K activates protein kinase B (Akt), which in turn inhibits glycogen synthase kinase (GSK) 3β affecting the production of toxic Aβ plaques and Tau hyperphosphorylation. Receptor for activated C-kinase (RACK) one is recruited to the M4 mAChR and scaffolds PKC, which may be involved in Aβ plaque formation and Tau hyperphosphorylation. Gα15 coupling links M4 mAChR activation with increases in intracellular IP3 levels and related signaling responses. The M4 mAChR is phosphorylated by GRK2 and recruits β-arrestin to terminate G protein-mediated signaling events. M4 mAChR cell surface expression levels are regulated by internalization, either by recruitment of β-arrestins and internalization via clathrin-coated pits or by caveolin-dependent mechanisms. Dashed arrows indicate pathways requiring further work to characterise the signaling proteins involved in the pathway. Solid arrows represent experimentally determined interactions. Proteins that are disrupted in Alzheimer's disease are colored in purple, those disrupted in schizophrenia are colored in blue and those disrupted in both diseases are colored in green.
The M1 mAChR is principally coupled to Gαq/11 linking the receptor to phospholipase C (PLC), inositol trisphosphate (IP3), diacylglycerol (DAG), protein kinase C (PKC) and calcium signaling pathways (Figure 5). The Gαq-PLC signaling pathway is also linked to the activation of cAMP, via a mechanism involving IP3-calcium release and calmodulin (Felder et al., 1989). [35S]GTPγS assays in hippocampal and cortical cultures revealed that direct activation of the Gαq/11 proteins was abolished in the M1 mAChR knockout mouse (Porter et al., 2002; Wess, 2004), confirming a role for Gαq for the M1 mAChR. In M1 mAChR knockout mouse primary cortical neurons, inositol phosphate (PI) hydrolysis was also dramatically reduced (Hamilton and Nathanson, 2001; Wess, 2004), linking Gαq and PI signaling pathways to the M1 mAChR in areas of the brain that are important in cognition. Activation of this signaling pathway may be particularly relevant for improving cognition, because influxes of calcium into neurons increase the activity of calcium-dependent adenylate cyclases, which are linked to generating short term memory, coding for memories that last minutes to days to weeks (Kandel, 2012).
Targeting Gαq/11 signaling pathways may also be relevant for treating the symptoms of schizophrenia. Gene expression microarray data comparing genes from schizophrenic and non-schizophrenic patients identified decreases in PLCβ1 in the dorsolateral prefrontal cortex of schizophrenic patients (McOmish et al., 2008a). In addition, PLCβ1 knockout mice display abnormal cortical development, are hyperactive, have reduced pre-pulse inhibition responses and have diminished spatial memory in the Morris water maze test (McOmish et al., 2008a). Therefore, in schizophrenia the decreases in PLCβ1 may influence the downstream signaling pathways by way of decreased IP3 production, decreased calcium release and decreases in cognition.
The Gαq/11-mediated signaling pathway may also be relevant to treating Alzheimer's disease. In the prefrontal cortex of Alzheimer’s disease brains, there is decreased activity of PKC and glycogen synthase kinase (GSK) 3β (Tsang et al., 2007; Medeiros et al., 2011). The reduction in PKC and GSK3β activity induces Tau protein hyper-phosphorylation and amyloid-β processing (Medeiros et al., 2011). Receptors for activated C kinase 1 (RACK1) is reported to directly interact with the M1 mAChR (Borroto-Escuela et al., 2011a), and the expression of RACK1 is also decreased in the cortex of Alzheimer's disease post-mortem brain tissues compared to healthy aged brain tissues (Battaini et al., 1999). This loss in RACK1 may contribute to the decrease in PKC activity by disrupting the subcellular localisation of PKC. Overexpression of amyloid-β in cortical neuron cultures disrupts the membrane distribution of RACK1 (Liu et al., 2011), thus it is likely that disruption of the RACK1-PKC complex by amyloid-β contributes to the pathology of Alzheimer's disease. The neurofibrillary tangles within the neurons of Alzheimer's disease brains also accumulate phospholipase Cδ (Shimohama et al., 1991), which likely hinders the functioning of this PLC. Furthermore, the duration of signaling from the G protein is regulated by direct interactions between the Gα subunits and regulator of G proteins signaling (RGS) proteins. The M1 mAChR directly interacts with RGS2 and RGS8 (Bernstein et al., 2004; Itoh et al., 2006; Borroto-Escuela et al., 2011a), where both RGS2 and RGS8 can interact with Gαq and the third intracellular loop of the M1 mAChR to switch-off Gαq signaling (Bernstein et al., 2004; Itoh et al., 2006). Interestingly, as observed with other proteins involved in the Gαq signaling pathway, RGS2 levels are decreased in Alzheimer’s patients (Hadar et al., 2016). Together, these findings suggest that the pathological increases in amyloid-β and neurofibrillary tangles that are seen in Alzheimer's disease reduce Gαq-mediated signaling events, which may have implications for the treatment of the disease.
Although the expression level of the M1 mAChR is unchanged in Alzheimer’s disease brains when compared to age matched controls, the functionality of the M1 mAChR is possibly compromised (Jope et al., 1994; Bradley et al., 2017). Several studies have explored the binding and signaling properties of M1 mAChRs in post mortem tissue samples from Alzheimer's disease and healthy aged brains. In radioligand binding studies, high and low affinity binding components are often observed with increasing concentrations of competing agonists, which are associated with the G protein-coupled and uncoupled states of the receptor, respectively. In Alzheimer's disease post-mortem tissues there is a loss in the high affinity binding component for carbachol when either [3H]-N-methylscopolamine or [3H]-pirenzepine are used as the radioligand, suggesting disruption in coupling to Gα proteins (Ferrari-DiLeo and Flynn, 1993). Furthermore, carbachol or oxotremorine M-stimulated PIP2 hydrolysis is decreased in cortical membranes of Alzheimer's disease patients, and can be blocked with the M1 mAChR antagonist, pirenzepine (Ferrari-DiLeo and Flynn, 1993; Jope et al., 1994). The functionality of PLC activity from Alzheimer's disease brains was unchanged compared to that in unaffected brain; therefore, it is likely due to inefficient activation of the PLC by Gαq rather than due to a deficiency in PLC itself (Ferrari-DiLeo and Flynn, 1993; Jope et al., 1994). More recently, direct assessment of G protein activation by [35S]-GTPγS binding assays, demonstrated that G protein activation was the same in Alzheimer's disease tissues compared to aged matched controls (Bradley et al., 2017). Thus G protein activation is equivalent in healthy and diseased brain tissues. Further work to explore the coupling of the M1 mAChR with G proteins and the efficiency of Gα activation is required to determine the nature of the disruption of M1 mAChR signal transduction in Alzheimer’s disease.
Potassium M-currents occur through the Kv7 (KCNQ) potassium channels, and regulate a slow ingress of potassium ions into neurons, to regulate the excitability of neurons (Passmore et al., 2012). In M1 mAChR knockout mice, inhibition of the potassium M-current was observed in sympathetic neurons (Hamilton et al., 1997). Further work examining the potassium M-current in dentate gyrus granule cells and hippocampal CA1 pyramidal cells indicate that the M1 mAChR may enhance rather than supress M-currents in different neuronal populations (Carver and Shapiro, 2019). This enhancement of the M-current is proposed to occur via phosphatidylinositol 4,5-bisphosphate (PIP2) synthesis from phosphatidylinositol 4-phosphate or phosphatidylinositol 5-phosphate in the dentate gyrus granule cells (Carver and Shapiro, 2019). In CA1 pyramidal neurons, the M-current was suppressed via a mechanism involving the depletion of PIP2 (Carver and Shapiro, 2019). The M1 mAChR directly interacts with AKAP79 (Borroto-Escuela et al., 2011a), which anchors PKC to the receptor. The M1 mAChR-AKAP79-PKC complex phosphorylates and activates potassium channels to regulate the M-current in post-synaptic cells (Hoshi et al., 2010). This mechanism is important for regulating neuronal excitability. Together, these results suggest that the M1 mAChR is involved in cell-type specific regulation of neuronal excitability state through the regulation of potassium channels.
Extracellular regulated kinases (ERK) are members of the mitogen activated protein kinase (MAPK) family. ERK1/2 are the most abundant isoforms of ERK expressed in the brain (Mazzucchelli et al., 2002). ERK1 and ERK2 knockout mouse studies directly link ERK1/2 activation with enhanced cognition (Mazzucchelli et al., 2002; Satoh et al., 2007). The mechanism underlying this improvement in memory may occur via ERK1/2 phosphorylation and subsequent activation of CREB, which is linked to increases in memory formation (Mazzucchelli et al., 2002; Kandel, 2012; Xia and Storm, 2012).
When ACh levels are increased in rats with the acetylcholinesterase inhibitor, physostigmine, enhanced levels of ERK1/2 phosphorylation are observed in the hippocampus and the cortex (Rosenblum et al., 2000). Physostigmine-induced ERK1/2 phosphorylation in the cortex and hippocampus are blocked with the administration of atropine, suggesting that this response occurs via activation of mAChRs (Rosenblum et al., 2000; Xia and Storm, 2012). In whole anesthetized rats, long term potentiation (LTP) is induced by tetanic stimulation in the dentate gyrus (Rosenblum et al., 2000). This LTP is blocked in animals that are pre-treated with the mitogen activated protein kinase kinase (MEK) inhibitor, PD98059 (Rosenblum et al., 2000). Together these results suggest that activation of the ERK1/2 signaling pathway in the brain is involved in the formation of memory and that ERK1/2 activation can occur via the mAChRs in the rat brain. In the M1 mAChR knockout mouse, ERK1/2 signaling was not evident in primary cortical neurons or CA1 hippocampal neurons from newborn pups (Berkeley et al., 2001). This suggests that activation of ERK1/2 downstream of the M1 mAChR is important, and may represent an ideal pathway to activate to improve cognition.
ERK1/2 activation by the M1 mAChR likely involves both G protein-dependent and β-arrestin-dependent pathways. In primary cortical cultures or African green monkey kidney (COS7) cells over-expressing the M1 mAChR, carbachol-stimulated ERK1/2 phosphorylation is blocked by PI3K (LY294002) and Src (PP1) inhibitors, indicating a role for both of these kinases in the M1 mAChR-mediated ERK1/2 activation pathway (Rosenblum et al., 2000). When the same cells were treated with the calcium chelators, BAPTA-AM, EGTA or a PKC inhibitor (BIM1), ERK1/2 phosphorylation was still observed (Rosenblum et al., 2000). Other studies suggest that the M1 mAChR activates ERK1/2 by Gαq-PKC dependent manner in CHO or COS7 cells (Hawes et al., 1995), or via Gαo in a pertussis toxin-sensitive manner in CHO cells (van Biesen et al., 1996). M1 mAChR activation of ERK1/2 via a PI3K-dependent pathway may be important because activation of a PI3K-Akt-ERK1/2-dependent pathway is important for protecting against Alzheimer's disease (Rai et al., 2019). Given that these pathways may contribute to the symptoms of neurological diseases, selective targeting of ERK1/2 pathways may also be an important pathway to target with biased modulators.
Less is known about the mechanism of ERK1/2 activation by the M4 mAChR. However, parallels may be drawn from the M2 mAChR, which is also a Gαi/o coupled receptor. In COS7 cells over-expressing the M2 mAChR, carbachol increases ERK1/2 phosphorylation, which is blocked with a PI3K inhibitor (wortmannin) and the Gβγ subunit inhibitor (βARK-ct) (Lopez-Ilasaca et al., 1997). In M4 mAChR knockout mice, ERK1/2 signaling was unaffected; suggesting that activation of ERK1/2 is not a critical pathway downstream of the M4 mAChR (Berkeley et al., 2001; Wess, 2004). Allosteric modulation of the M4 mAChR with VU0152100 inhibits D1 dopamine receptor-induced ERK1/2 activation in rat striatum and medial prefrontal cortex (Xue et al., 2015). Thus, the M4 mAChR exerts an inhibitory effect toward the ERK1/2 signaling pathway via crosstalk with the D1 dopamine receptor. Western blots of post-mortem brain tissue of patients with schizophrenia and mood disorders determined that in the prefrontal cortex, there are decreased protein expression levels of B-raf, MEK1, MEK2, RSK1, CREB, and Rap1, all members of the MAPK signaling pathway (Yuan et al., 2010; Funk et al., 2012). This indicates that in schizophrenia and mood disorders, an under-stimulation of ERK1/2 signaling pathway may be involved in the symptomology of the disease, further supporting the notion that activation of the ERK1/2 pathway may be beneficial for treating neurological disorders.
Interestingly, the M1 mAChR is reported to directly interact with Gα12/13 proteins (Borroto-Escuela et al., 2011a). Gα12/13 proteins activate Rho proteins, which are involved in the rearrangement of the actin cytoskeleton and regulate membrane trafficking (Ridley et al., 1992; Ridley and Hall, 1992; Ridley 2001; Suzuki et al., 2009). Activation of Gα13 is linked to the retraction of neurite outgrowths in adult sensory neurons, an effect that is reversed with the M1 mAChR antagonists, pirenzepine and muscarinic toxin 7 (Sabbir and Fernyhough, 2018). Gα12/13 proteins are also linked to the activation of PLD (Plonk et al., 1998). Although a direct link between the M1 mAChR activation of Gα12/13 and subsequent PLD activation is lacking, such a link has been established for the related M3 mAChR, which activates PLD via Gα12/13 proteins in HEK293 cells (Rumenapp et al., 2001). Given the link between Gα12/13, PLD activation and the important role of PLD in long-term depression in the prefrontal cortex, further work to determine whether or not the M1 mAChR biased allosteric modulators can selectively activate Gα12/13 signaling pathways may provide vital information regarding the direction for the development of future M1 mAChR targeted therapeutics.
Mass spectrometry experiments suggest that the M1 mAChR interacts with ADP ribosylation factors (ARF) 1, 3, 5 and 6, which are small Ras-family, GTP binding proteins (Borroto-Escuela et al., 2011a). Although these interacting partners have been identified for the M1 mAChR, there are no subsequent studies to date confirming these interactions with the M1 mAChR. Studies looking at the related M3 mAChR demonstrate that ARF 1 and 6 directly interact with the M3 mAChR to activate PLD (Mitchell et al., 2003). This interaction requires the asparagine residue of the NPxxY motif at the end of transmembrane 7 (Borroto-Escuela et al., 2011b). Given the importance of the PLD pathway in long-term depression in the prefrontal cortex and the potential for biased allosteric modulation of this pathway, ARFs may be an important family of proteins to monitor for novel mAChR ligand characterisation. Although the M1 and M3 mAChR are both Gαq-coupled related receptors, further work to confirm direct interactions between the M1 mAChR and ARFs is required. In addition, links between ARFs and PLD signaling at the M1 mAChR may provide a new avenue to exploit for biased allosteric modulators.
Because the M4 mAChR is a Gαi/o-coupled receptor, one of its best characterised second messenger responses is the inhibition of adenylate cyclase, thus decreasing intracellular cAMP levels. There are five subtypes of Gαi/o proteins, and the M4 mAChR specifically couples to the Gαi2 and GαoA and GαoB alpha subunits (Migeon et al., 1994; Migeon et al., 1995; Borroto-Escuela et al., 2011a). Mass spectrometry data also identified a possible interaction between the M4 mAChR and Gαi3 (Borroto-Escuela et al., 2011a), however, the functional consequences of this interaction remain to be confirmed. The M4 mAChR also interacts with RGS4, which is reported to be selective for regulating Gαi activity (Roy et al., 2003; Borroto-Escuela et al., 2011a). Interestingly, DNA microarray analysis demonstrated a significant reduction in RGS4 expression levels in the prefrontal cortex of schizophrenic patients (Mirnics et al., 2001) and RGS4 expression is dysregulated in the PLCβ1 knockout mouse (McOmish et al., 2008b). This may provide a hint that the Gαi/o signaling pathway is dysregulated in schizophrenia; however a direct link between the M4 mAChR-RGS4 and schizophrenia remains to be established. Interestingly, although activation of the M4 mAChR is normally linked to decreases in forskolin-stimulated cAMP levels, activation with high agonist concentrations can increase intracellular cAMP levels in CHO cells (Migeon and Nathanson, 1994; Migeon et al., 1995). Chronic activation of the M4 mAChR with the agonist McN-A-343 (100 μM; 18 h) also increases cAMP levels in CHO cells (Nevo et al., 1998). This possibly occurs through a “super-activation” mechanism involving Gβγ subunit signaling of the Gαi/o proteins, which has been observed for mAChRs (Sunahara et al., 1996). This super-activation mechanism may be therapeutically relevant, as the lifetime of synthetic agonist ligands may impact the direction of cAMP levels in neurons. This is an important consideration for the design of synthetic M4 mAChR ligands to avoid overstimulation of receptors.
Some studies have also reported that the M1 mAChR can decrease cAMP signaling by coupling to Gαi/o proteins. Direct activation of Gαi1/2, determined using [35S]-GTPγS binding assays, is observed for several orthosteric and bitopic mAChR ligands in CHO cells overexpressing M1 mAChR (Thomas et al., 2008). Of note, interactions between the M1 mAChR and the Gαi/o proteins were not observed by mass spectrometry (Borroto-Escuela et al., 2011a). However, this may be explained by the faster dissociation rates of the Gαi/o proteins from the M1 mAChR when compared with the dissociation rates of the Gαq proteins (Ilyaskina et al., 2018). Further work is required to determine the potential physiological relevance of M1 mAChR coupling to Gαi/o proteins and to inhibition of cAMP signaling.
The PI3K-Akt-GSK3β pathway is downregulated in schizophrenia (Karam et al., 2010; Emamian, 2012; Enriquez-Barreto and Morales, 2016; McGuire et al., 2017). Decreased levels of Akt and decreased inhibition of GSK3β are observed in the cortex and peripheral lymphocytes of human schizophrenic patients relative to controls (Emamian et al., 2004). The reduction of Akt expression levels may be linked to certain single nucleotide polymorphisms identified in a subset of schizophrenic patients, which cause a reduction in the transcription or translation of the AKT1 gene (Emamian et al., 2004). Increased activity of Akt is observed in Alzheimer's disease brain tissues, resulting in an increase in the phosphorylation of the downstream proteins GSK3β(Ser9) (inhibits activity), Tau(Ser214) and mTor(Ser2448) (Pei et al., 2003; Griffin et al., 2005). Carbachol stimulation of an endogenously expressed mAChR in PC12 cells is linked to Akt phosphorylation (Wu and Wong, 2006). The carbachol-stimulated Akt phosphorylation is inhibited by pertussis toxin, suggesting that it occurs downstream of a Gαi/o-coupled mAChR (Wu and Wong, 2006). Few studies have investigated the GSK3β phosphorylation by mAChR ligands, however, this would be a pathway to try to target for the treatment of both Alzheimer's disease and schizophrenia.
cAMP is a second messenger that activates PKA, exchange protein activated by cAMP (EPAC) and cAMP response element binding protein (CREB). When CREB is activated by PKA, mitogen-activated protein kinase (MAPK) or calcium/calmodulin-stimulated protein kinase (CaMK), CREB binds to cAMP response element promoter regions of DNA to increase gene transcription. This in turn, increases protein expression, increases synaptic strength and promotes long term memory storage (Davis et al., 1996; Montminy, 1997; Pittenger et al., 2006; Kandel, 2012). Long term memory can be blocked by overexpression of a phosphodeficient CREB(S133A) mutant in rat dorsolateral striatum (Pittenger et al., 2006; Brightwell et al., 2008). Thus, biased ligands that selectively enhanced the cAMP-CREB pathway could have potential to improve long term memory in patients with neurological disorders.
The M1 and M4 mAChRs are also reported to increase cAMP levels by coupling to Gαs proteins in CHO cells (Burford and Nahorski, 1996; Canals et al., 2012). cAMP accumulation is observed in cells pre-treated with pertussis toxin (Gαi/o inhibitor) and is blocked in cells treated with a Gαs antiserum (Migeon et al., 1995; Burford and Nahorski, 1996). In addition, when the AC activator, forskolin, is co-administered with mAChR agonists, an increase in cAMP level is observed (Burford and Nahorski, 1996). Increases in cAMP in M1 mAChR expressing CHO cells are reported with both orthosteric and allosteric agonists (Thomas et al., 2008). There is, however, a lack of evidence showing a direct interaction between the M1 mAChR and Gαs. Data from mass spectrometry suggests that Gαs interacts with the M4 mAChR but not the M1 mAChR (Borroto-Escuela et al., 2011a). Furthermore, at the M1 mAChR, [35S]-GTPγS assays using a Gαs antibody to pull down activated Gαs proteins, did not detect Gαs activation (Thomas et al., 2008), and Forster resonance energy transfer (FRET) studies did not detect an interaction between the M1 mAChR and Gαs (Ilyaskina et al., 2018). Together, these results may suggest that the M1 mAChR couples extremely weakly to Gαs, and may be a product of the overexpression of the M1 mAChR in a recombinant system. In CHO cells, recombinantly expressing the M4 mAChR, increases in cAMP levels are routinely reported; an effect that is reversed by overexpressing Gαi2 or Gαo proteins (Migeon et al., 1995). Additionally, the M4 mAChR may activate cAMP through the Gβγ subunits depending on which AC isoforms are present in the cell (Varga et al., 1998). Interestingly, high agonist concentrations are required to increase cAMP levels at both the M1 and M4 mAChRs (Migeon et al., 1995; Burford and Nahorski, 1996), which makes the physiological relevance of cAMP signaling by the mAChRs questionable. Thus, further work to identify the mechanism and physiological relevance of Gαs activation by the M1 and M4 mAChRs is required.
Changes in levels of cAMP pathway-associated proteins may be important in producing some symptoms of schizophrenia. In post-mortem schizophrenic brain tissues, changes in the expression level of proteins associated with the cAMP signaling pathway are indeed observed. Rap2, CREB and phosphodiesterase (PDE) 4B are either upregulated or downregulated, depending on the region of the brain that is investigated (Millar et al., 2005; Funk et al., 2012). Since schizophrenia is primarily considered a disorder of dopamine signaling, much of the evidence linking the cAMP pathway with schizophrenia is described within the context of D1 or D2 dopamine receptor activation (Wang et al., 2018). There is currently no direct evidence linking the mAChRs with cAMP activation to improve the symptoms of schizophrenia.
β-arrestin is a scaffold protein that can bind to members of the MAPK pathway, including cRaf, MEK and ERK1/2 (Song et al., 2009). Through scaffolding the relevant proteins together, β-arrestin activates the ERK1/2 signaling pathway. Following activation of Gαq protein, the M1 mAChR is regulated by phosphorylation at serine and threonine residues in the intracellular loop three and carboxy-terminal tail by intracellular kinases. Phosphorylation of the M1 mAChR at Ser228/Ser273 by G protein-coupled receptor kinase (GRK) 2 results in translocation of β-arrestin 2 to the receptor, which in turn scaffolds the MAPK and results in ERK1/2 activation (Jung et al., 2017). Similar studies interrogating β-arrestin-ERK1/2 activation have not yet been performed for the M4 mAChR, but would be important for understanding M4 mAChR signaling. Since M1 mAChR-Gαq pathways may be compromised in Alzheimer's disease, ERK1/2 activation to improve cognition could be achieved with β-arrestin-biased modulators. This could also be beneficial in reducing salivation and gastrointestinal disturbances that appear to occur downstream of Gαq proteins in the M1 mAChR phosphodeficient mice (Bradley et al., 2020).
Most GPCRs are regulated by phosphorylation on their intracellular loops and carboxy-terminal tails by cellular kinases. These phosphorylation events uncouple GPCRs from G protein signaling and trigger receptor internalisation and downstream trafficking events. Intracellular scaffold proteins are subsequently recruited to phosphorylated GPCRs to assemble cellular proteins into specific signaling complexes. GRKs that phosphorylate GPCRs are one of the most studied GPCR kinases. The M1 and M4 mAChRs are phosphorylated in the third intracellular loop by GRK2 (Haga et al., 1996; Borroto-Escuela et al., 2011a; Jung et al., 2017). In the case of the M1 mAChR, GRK2 directly interacts with Gαq to inhibit phosphoinositide signaling events (Willets et al., 2005), whilst for the M4 mAChR, GRK2 overexpression increases the rate of internalisation of the receptor (Holroyd et al., 1999; Vogler et al., 1999; Reiner and Nathanson, 2008; Borroto-Escuela et al., 2011a). Phosphorylation of GPCRs by GRKs increases β-arrestin translocation from the cytosol to the plasma membrane. The M1 mAChR rapidly recruits β-arrestin with a preference for β-arrestin 2 over β-arrestin 1 in RBL-2H3 or CHO cells expressing systems (Santini et al., 2000; Canals et al., 2012; Yeatman et al., 2014). Interestingly, both orthosteric and allosteric agonists promote β-arrestin recruitment to and internalisation of the M1 mAChR.
β-arrestin 2 is important in memory as evidenced by impaired fear conditioning responses in β-arrestin 2 knockout mice (Li et al., 2009). This suggests that β-arrestin 2 is important for amygdala based fear associative memory (Li et al., 2009). β-arrestin 1 and 2 are also implicated in Alzheimer’s disease, because the levels of both β-arrestin 1 and 2 are elevated in autopsied brains of Alzheimer's disease patients, when compared to age matched controls (Thathiah et al., 2013; Jiang et al., 2013). Overexpression of β-arrestin 2 in HEK 293-APP695 cells increased the secretion of amyloid-β, which was blocked when the cells were pre-treated with a γ-secretase inhibitor, L-685458 (Thathiah et al., 2013). The secretion of amyloid-β is also reduced in cells where the β-arrestin 2 gene is silenced, and in the β-arrestin 2 knockout mouse (Thathiah et al., 2013). Interestingly, there was no effect on the APP, with no change in expression levels with overexpression or silencing of β-arrestin 2 (Thathiah et al., 2013). β-arrestin 2 signaling may also be an important pathway to target for novel anti-schizophrenic therapies. In mouse models of schizophrenia (phencyclidine treated or NMDA receptor NR1 subunit knockdown mice), selective activation of β-arrestin 2 pathways with D2 dopamine receptor biased agonists reduced hyperlocomotion, restored pre-pulse inhibition, improved novel object recognition, improved social behaviors and reduced seizures (Park et al., 2016). Therefore, selectively activating β-arrestin 2-dependent signaling pathways may be beneficial in treating schizophrenia.
The M1 and M4 mAChRs are reported to interact with the adapter 14-3-3 proteins (Borroto-Escuela et al., 2011a). These proteins are most abundantly expressed in the brain and are reported to modulate GPCR trafficking and assemble signaling complexes, such as ERK1/2 signaling complexes (Li et al., 2016; Yuan et al., 2019). The M1 mAChR recruits 14-3-3ε-pLuc following stimulation with carbachol (Yuan et al., 2019). For the M4 mAChR, validation and characterisation of the potential interaction with 14-3-3 requires further investigation. Characterising mAChR ligands toward 14-3-3 recruitment may be of physiological relevance because 14-3-3 proteins are dysregulated in neurological diseases such as Alzheimer’s disease and schizophrenia (Foote and Zhou, 2012; Graham et al., 2019). This largely unexplored scaffold protein may therefore represent an important novel signaling pathway to target for the development of biased allosteric modulators.
The M1 and M4 mAChRs are reported to modulate arachidonic acid (AA) levels via activation of PLA2. Activation of PLA2 by the M1 mAChR increases AA release via a PKC-dependent pathway in mouse striatal neurons (Tence et al., 1994), suggesting that this may lead to physiologically relevant signaling. Activation of CHO cells overexpressing the M1 mAChR with carbachol increases release of the APP (Emmerling et al., 1993). This secretion of APP is inhibited with the PLA2 inhibitors, quinacrine, manoalide and scalaradial, suggesting that M1 mAChR-stimulated APP secretion occurs via a mechanism involving PLA2 (Emmerling et al., 1993). Inhibition of PLA2 signaling may represent an important signaling pathway to target for the treatment of Alzheimer's disease, to reduce the amount of APP that is released from neurons and decrease the burden of accumulation of toxic amyloid plaques. This is supported by evidence from a mouse model of Alzheimer's disease (Transgenic human APP mouse), where increased PLA2 activity and AA levels were observed (Sanchez-Mejia et al., 2008). In this mouse model, PLA2 was activated by amyloid-β (Sanchez-Mejia et al., 2008). Knockdown of PLA2 Group IVA reduced hippocampal AA levels and improved cognition in the Morris water maze test (Sanchez-Mejia et al., 2008). Thus, ligands that can inhibit PLA2 signaling via the M1 mAChR could have disease modifying utility that could be harnessed for the treatment of Alzheimer's disease.
The M4 mAChR can weakly increase AA release when activated by carbachol in CHO cells over-expressing the M4 mAChR (Felder et al., 1991). Carbachol stimulation of the M4 mAChR also potentiated the adenosine triphosphate (ATP)-mediated release of AA (Felder et al., 1991). In cells pre-treated with pertussis toxin or staurosporine, the AA release was blocked, suggesting that it occurs downstream of PKC and Gαi/o proteins (Felder et al., 1991). Interestingly, increased PLA2 activity and AA levels are observed by magnetic resonance imaging of schizophrenic patient brains (Smesny et al., 2010). Thus, compounds that reduce PLA2 activity via the M4 mAChR may be beneficial in the treatment of schizophrenia.
The M1 and M4 mAChRs have the potential to be targeted by new medications to treat the symptoms of neurological disorders, such as Alzheimer's disease and schizophrenia. The challenge in successfully targeting these receptors lays in the selectivity of the ligands and their associated side effect profiles. With the development and characterisation of many new selective compounds for the mAChRs has come the realisation that not all side effects are due to activation of other receptor subtypes, either centrally or peripherally. Instead, some of the side effects are also driven through on-target overstimulation. Biased allosteric modulators offer the potential to overcome these hurdles, through selectively targeting a given mAChR subtype and subsets of its signal transduction pathways within the cell. However, a greater understanding of the pathways and their involvement in the diseases themselves and in the symptoms of the diseases are needed to determine the path forward for developing novel biased allosteric modulators.
In Alzheimer's disease, the M1 mAChR is the primary drug target. Studies in genetically modified mice suggest that biasing the M1 mAChR to pathways occurring after GRK2 phosphorylation of the receptor would provide relief from adverse effects such as salivation and hyperactivity. Since the M1 mAChR is uncoupled from the Gαq proteins in Alzheimer's disease, agonists biased toward β-arrestin pathways may be the most beneficial pathways to target. M1 mAChR-β-arrestin-biased modulators may link the receptor to ERK1/2 signaling pathways and improve cognition. Given that overexpression of β-arrestin 2 is linked to increases in amyloid-β secretion, it will be important to avoid overstimulating this pathway. Activation of PLD signaling pathways may also be beneficial, since PLD activation is linked to improved cognition. This could be achieved with ARF-biased or Gα12/13-biased ligands, relative to the native repertoire of signaling. However, considering that the Gα12/13 pathway is linked to neurite retraction, ligands with bias away from Gα12/13 recruitment may be preferred. Compounds that improve M1 mAChR coupling to Gαq and away from PLA2 may yield Alzheimer's disease modifying benefits, by decreasing APP secretion, amyloid-β production and Tau hyperphosphorylation. Additional benefits may also be seen through targeting the M1 mAChR-AKAP79 interaction, by increasing potassium channel activity and cognition.
In treating schizophrenia, the M4 mAChR is an attractive drug target, as it is potentially linked to improvements in the positive, negative and cognitive symptoms of the disease. However, the pathways that would be beneficial to target for the M4 mAChR are less clear. Based on the currently available evidence, M4 mAChR biased allosteric modulators directed toward Gβγ-PI3K-ERK1/2 or β-arrestin-ERK1/2 pathways may improve cognition. Further work is required to identify and characterise the signaling pathways that are activated downstream of the M4 mAChR to identify additional pathways that may be beneficial to activate. Treatment of schizophrenic symptoms via the M1 mAChR, may require the development of ligands with a more balanced profile of potentiation. This is because the M1 mAChR-Gαq pathways are linked with anti-psychotic and anti-anxiety potential. Furthermore, activation of ERK1/2 and PLD signaling pathways via the M1 mAChR may improve memory and increase neuronal connectivity, thus directing signaling to specific pathways may lead to the successful development of a new generation of anti-schizophrenic treatments with reduced adverse effects.
EW wrote the manuscript and prepared the figures. KC, SM-N, and SB contributed sections regarding mouse studies. CV contributed information for the allostery sections. AT, PS, and AC contributed to the writing of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
This work was supported by a Wellcome Trust Collaborative Research Award (201529/Z/16/Z), a National and Medical Research Council of Australia program grant (APP1150083) and an Australia Research Council of Australia discovery project (DP190102950). PS is a senior principal research fellow of the National health and medical research council of Australia. SB is a senior lecturer at the University of Glasgow, and supported by a Medical Research Council industrial collaboration agreement (MR/P019366/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdul-Ridha, A., Lane, J. R., Sexton, P. M., Canals, M., and Christopoulos, A. (2013). Allosteric modulation of a chemogenetically modified G protein-coupled receptor. Mol. Pharmacol. 83, 521–530. doi:10.1124/mol.112.083006
Abdul-Ridha, A., López, L., Keov, P., Thal, D. M., Mistry, S. N., Sexton, P. M., et al. (2014). Molecular determinants of allosteric modulation at the M1 muscarinic acetylcholine receptor. J. Biol. Chem. 289, 6067–6079. doi:10.1074/jbc.M113.539080
Allaway, K. C., and Machold, R. (2017). Developmental specification of forebrain cholinergic neurons. Dev. Biol. 421, 1–7. doi:10.1016/j.ydbio.2016.11.007
Amenta, F., and Tayebati, S. K. (2008). Pathways of acetylcholine synthesis, transport and release as targets for treatment of adult-onset cognitive dysfunction. Curr. Med. Chem. 15, 488–498. doi:10.2174/092986708783503203
Aurelio, L., Valant, C., Flynn, B. L., Sexton, P. M., Christopoulos, A., and Scammells, P. J. (2009). Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J. Med. Chem. 52, 4543–4547. doi:10.1021/jm9002582
Aurelio, L., Valant, C., Flynn, B. L., Sexton, P. M., White, J. M., Christopoulos, A., et al. (2010). Effects of conformational restriction of 2-amino-3-benzoylthiophenes on A(1) adenosine receptor modulation. J. Med. Chem. 53, 6550–6559. doi:10.1021/jm1008538
Aurelio, L., Christopoulos, A., Flynn, B. L., Scammells, P. J., Sexton, P. M., and Valant, C. (2011). The synthesis and biological evaluation of 2-amino-4,5,6,7,8,9-hexahydrocycloocta[b]thiophenes as allosteric modulators of the A1 adenosine receptor. Bioorg. Med. Chem. Lett. 21, 3704–3707. doi:10.1016/j.bmcl.2011.04.080
Battaini, F., Pascale, A., Lucchi, L., Pasinetti, G. M., and Govoni, S. (1999). Protein kinase C anchoring deficit in postmortem brains of Alzheimer’s disease patients. Exp. Neurol. 159, 559–564. doi:10.1006/exnr.1999.7151
Berizzi, A. E., Gentry, P. R., Rueda, P., Den Hoedt, S., Sexton, P. M., Langmead, C. J., et al. (2016). Molecular mechanisms of action of M5 muscarinic acetylcholine receptor allosteric modulators. Mol. Pharmacol. 90, 427–436. doi:10.1124/mol.116.104182
Berkeley, J. L., Gomeza, J., Wess, J., Hamilton, S. E., Nathanson, N. M., and Levey, A. I. (2001). M1 muscarinic acetylcholine receptors activate extracellular signal-regulated kinase in CA1 pyramidal neurons in mouse hippocampal slices. Mol. Cell. Neurosci. 18, 512–524. doi:10.1006/mcne.2001.1042
Bernstein, L. S., Ramineni, S., Hague, C., Cladman, W., Chidiac, P., Levey, A. I., et al. (2004). RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J. Biol. Chem. 279, 21248–21256. doi:10.1074/jbc.M312407200
Bertron, J. L., Cho, H. P., Garcia-Barrantes, P. M., Panarese, J. D., Salovich, J. M., Nance, K. D., et al. (2018). The discovery of VU0486846: steep SAR from a series of M1 PAMs based on a novel benzomorpholine core. Bioorg. Med. Chem. Lett. 28, 2175–2179. doi:10.1016/j.bmcl.2018.05.009
Beshore, D. C., Di Marco, C. N., Chang, R. K., Greshock, T. J., Ma, L., Wittmann, M., et al. (2018). MK-7622: a first-in-Class M1 positive allosteric modulator development candidate. ACS Med. Chem. Lett. 9, 652–656. doi:10.1021/acsmedchemlett.8b00095
Bewley, B. R., Spearing, P. K., Weiner, R. L., Luscombe, V. B., Zhan, X., Chang, S., et al. (2017). Discovery of a novel, CNS penetrant M4 PAM chemotype based on a 6-fluoro-4-(piperidin-1-yl)quinoline-3-carbonitrile core. Bioorg. Med. Chem. Lett. 27, 4274–4279. doi:10.1016/j.bmcl.2017.08.043
Brannan, S., Sawchak, S., Miller, A., Paul, S. M., and Breier, A. (2020). Efficacy and safety of xanomeline, a M1/M4 receptor preferring agonist, plus trospium, a peripheral muscarinic antagonist, in schizophrenia: phase 2 clinical trial results. Biol. Psychiatry. 87, S169. doi:10.1016/j.biopsych.2020.02.446
Bodick, N. C., Offen, W. W., Levey, A. I., Cutler, N. R., Gauthier, S. G., Satlin, A., et al. (1997). Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol. 54, 465–473. doi:10.1001/archneur.1997.00550160091022
Borroto-Escuela, D. O., Correia, P. A., Romero-Fernandez, W., Narvaez, M., Fuxe, K., Ciruela, F., et al. (2011a). Muscarinic receptor family interacting proteins: role in receptor function. J. Neurosci. Methods. 195, 161–169. doi:10.1016/j.jneumeth.2010.11.025
Borroto-Escuela, D. O., Romero-Fernandez, W., García-Negredo, G., Correia, P. A., Garriga, P., Fuxe, K., et al. (2011b). Dissecting the conserved NPxxY motif of the M3 muscarinic acetylcholine receptor: critical role of Asp-7.49 for receptor signaling and multiprotein complex formation. Cell. Physiol. Biochem. 28, 1009–1022. doi:10.1159/000335788
Bradley, S. J., Bourgognon, J. M., Sanger, H. E., Verity, N., Mogg, A. J., White, D. J., et al. (2017). M1 muscarinic allosteric modulators slow prion neurodegeneration and restore memory loss. J. Clin. Invest. 127, 487–499. doi:10.1172/JCI87526
Bradley, S. J., Molloy, C., Valuskova, P., Dwomoh, L., Scarpa, M., Rossi, M., et al. (2020). Biased M1-muscarinic-receptor-mutant mice inform the design of next-generation drugs. Nat. Chem. Biol. 16, 240–249. doi:10.1038/s41589-019-0453-9
Brightwell, J. J., Smith, C. A., Neve, R. L., and Colombo, P. J. (2008). Transfection of mutant CREB in the striatum, but not the hippocampus, impairs long-term memory for response learning. Neurobiol. Learn. Mem. 89, 27–35. doi:10.1016/j.nlm.2007.09.004
Bubser, M., Bridges, T. M., Dencker, D., Gould, R. W., Grannan, M., Noetzel, M. J., et al. (2014). Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem. Neurosci. 5, 920–942. doi:10.1021/cn500128b
Buckley, N. J., Bonner, T. I., and Brann, M. R. (1988). Localization of a family of muscarinic receptor mRNAs in rat brain. J. Neurosci. 8, 4646–4652. doi:10.1523/jneurosci.08-12-04646.1988
Burford, N. T., and Nahorski, S. R. (1996). Muscarinic m1 receptor-stimulated adenylate cyclase activity in chinese hamster ovary cells is mediated by Gs alpha and is not a consequence of phosphoinositidase C activation. Biochem. J. 315 (Pt 3) (Pt 3), 883–888. doi:10.1042/bj3150883
Bymaster, F. P., Carter, P. A., Zhang, L., Falcone, J. F., Stengel, P. W., Cohen, M. L., et al. (2001). Investigations into the physiological role of muscarinic M2 and M4 muscarinic and M4 receptor subtypes using receptor knockout mice. Life Sci. 68, 2473–2479. doi:10.1016/s0024-3205(01)01041-4
Bymaster, F. P., McKinzie, D. L., Felder, C. C., and Wess, J. (2003). Use of M1-M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem. Res. 28, 437–442. doi:10.1023/a:1022844517200
Byun, N. E., Grannan, M., Bubser, M., Barry, R. L., Thompson, A., Rosanelli, J., et al. (2014). Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology. 39, 1578–1593. doi:10.1038/npp.2014.2
Canals, M., Lane, J. R., Wen, A., Scammells, P. J., Sexton, P. M., and Christopoulos, A. (2012). A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J. Biol. Chem. 287, 650–659. doi:10.1074/jbc.M111.314278
Carpenter, A. C., Saborido, T. P., and Stanwood, G. D. (2012). Development of hyperactivity and anxiety responses in dopamine transporter-deficient mice. Dev. Neurosci. 34, 250–257. doi:10.1159/000336824
Carruthers, S. P., Gurvich, C. T., and Rossell, S. L. (2015). The muscarinic system, cognition and schizophrenia. Neurosci. Biobehav. Rev. 55, 393–402. doi:10.1016/j.neubiorev.2015.05.011
Carver, C. M., and Shapiro, M. S. (2019). Gq-coupled muscarinic receptor enhancement of KCNQ2/3 channels and activation of TRPC channels in multimodal control of excitability in dentate gyrus granule cells. J. Neurosci. 39, 1566–1587. doi:10.1523/jneurosci.1781-18.2018
Chambon, C., Jatzke, C., Wegener, N., Gravius, A., and Danysz, W. (2012). Using cholinergic M1 receptor positive allosteric modulators to improve memory via enhancement of brain cholinergic communication. Eur. J. Pharmacol. 697, 73–80. doi:10.1016/j.ejphar.2012.10.011
Chan, W. Y., McKinzie, D. L., Bose, S., Mitchell, S. N., Witkin, J. M., Thompson, R. C., et al. (2008). Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 105, 10978–10983. doi:10.1073/pnas.0800567105
Chatzidaki, A., and Millar, N. S. (2015). Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 97, 408–417. doi:10.1016/j.bcp.2015.07.028
Chopko, T. C., Han, C., Gregro, A. R., Engers, D. W., Felts, A. S., Poslusney, M. S., et al. (2019). SAR inspired by aldehyde oxidase (AO) metabolism: discovery of novel, CNS penetrant tricyclic M4 PAMs. Bioorg. Med. Chem. Lett. 29, 2224–2228. doi:10.1016/j.bmcl.2019.06.032
Choy, K. H., Shackleford, D. M., Malone, D. T., Mistry, S. N., Patil, R. T., Scammells, P. J., et al. (2016). Positive allosteric modulation of the muscarinic M1 receptor improves efficacy of antipsychotics in mouse glutamatergic deficit models of behavior. J. Pharmacol. Exp. Ther. 359, 354–365. doi:10.1124/jpet.116.235788
Christopoulos, A., Changeux, J. P., Catterall, W. A., Fabbro, D., Burris, T. P., Cidlowski, J. A., et al. (2014). International union of basic and clinical pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol. Rev. 66, 918–947. doi:10.1124/pr.114.008862
Christopoulos, A., and Kenakin, T. (2002). G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 54, 323–374. doi:10.1124/pr.54.2.323
Christopoulos, A., Lanzafame, A., and Mitchelson, F. (1998). Allosteric interactions at muscarinic cholinoceptors. Clin. Exp. Pharmacol. Physiol. 25, 185–194. doi:10.1111/j.1440-1681.1998.t01-4-.x
Christopoulos, A., Lanzafame, A., Ziegler, A., and Mitchelson, F. (1997). Kinetic studies of co-operativity at atrial muscarinic M2 receptors with an "infinite dilution" procedure. Biochem. Pharmacol. 53, 795–800. doi:10.1016/s0006-2952(96)00814-3
Christopoulos, A., and Mitchelson, F. (1995). [3H]N-methylscopolamine dissociation from muscarine receptors affected by low concentrations of allosteric modulators. Eur. J. Pharmacol. 290, 259–262. doi:10.1016/0922-4106(95)90002-0
Christopoulos, A. (2014). Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol. 86, 463–478. doi:10.1124/mol.114.094342
Clark, A. L., and Mitchelson, F. (1976). The inhibitory effect of gallamine on muscarinic receptors. Br. J. Pharmacol. 58, 323–331. doi:10.1111/j.1476-5381.1976.tb07708.x
Croy, C. H., Schober, D. A., Xiao, H., Quets, A., Christopoulos, A., and Felder, C. C. (2014). Characterization of the novel positive allosteric modulator, LY2119620, at the muscarinic M(2) and M(4) receptors. Mol. Pharmacol. 86, 106–115. doi:10.1124/mol.114.091751
Dallagnol, J. C. C., Khajehali, E., van der Westhuizen, E. T., Jörg, M., Valant, C., Gonçalves, A. G., et al. (2018). Synthesis and pharmacological evaluation of heterocyclic carboxamides: positive allosteric modulators of the M1 muscarinic acetylcholine receptor with weak agonist activity and diverse modulatory profiles. J. Med. Chem. 61, 2875–2894. doi:10.1021/acs.jmedchem.7b01812
Davey, A. E., Leach, K., Valant, C., Conigrave, A. D., Sexton, P. M., and Christopoulos, A. (2012). Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology. 153, 1232–1241. doi:10.1210/en.2011-1426
Davie, B. J., Valant, C., White, J. M., Sexton, P. M., Capuano, B., Christopoulos, A., et al. .(2014). Synthesis and pharmacological evaluation of analogues of benzyl quinolone carboxylic acid (BQCA) designed to bind irreversibly to an allosteric site of the M ₁ muscarinic acetylcholine receptor. J. Med. Chem. 57, 5405–5418. doi:10.1021/jm500556a
Davis, A. A., Fritz, J. J., Wess, J., Lah, J. J., and Levey, A. I. (2010a). Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J. Neurosci. 30, 4190–4196. doi:10.1523/JNEUROSCI.6393-09.2010
Davis, A. A., Heilman, C. J., Brady, A. E., Miller, N. R., Fuerstenau-Sharp, M., Hanson, B. J., et al. (2010b). Differential effects of allosteric M(1) muscarinic acetylcholine receptor agonists on receptor activation, arrestin 3 recruitment, and receptor downregulation. ACS Chem. Neurosci. 1, 542–551. doi:10.1021/cn100011e
Davis, C. N., Bradley, S. R., Schiffer, H. H., Friberg, M., Koch, K., Tolf, B. R., et al. (2009). Differential regulation of muscarinic M1 receptors by orthosteric and allosteric ligands. BMC Pharmacol. 9, 14. doi:10.1186/1471-2210-9-14
Davis, G. W., Schuster, C. M., and Goodman, C. S. (1996). Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron. 17, 669–679. doi:10.1016/s0896-6273(00)80199-3
Davoren, J. E., Lee, C. W., Garnsey, M., Brodney, M. A., Cordes, J., Dlugolenski, K., et al. (2016a). Discovery of the potent and selective M1 PAM-agonist N-[(3R,4S)-3-Hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): evaluation of efficacy and cholinergic side effects. J. Med. Chem. 59, 6313–6328. doi:10.1021/acs.jmedchem.6b00544
Davoren, J. E., O'Neil, S. V., Anderson, D. P., Brodney, M. A., Chenard, L., Dlugolenski, K., et al. (2016b). Design and optimization of selective azaindole amide M1 positive allosteric modulators. Bioorg. Med. Chem. Lett. 26, 650–655. doi:10.1016/j.bmcl.2015.11.053
Davoren, J. E., Garnsey, M., Pettersen, B., Brodney, M. A., Edgerton, J. R., Fortin, J. P., et al. (2017). Design and synthesis of gamma- and delta-Lactam M1 positive allosteric modulators (PAMs): Convulsion and cholinergic Toxicity of an M1-selective PAM with weak agonist activity. J. Med. Chem. 60, 6649–6663. doi:10.1021/acs.jmedchem.7b00597.s002
Dean, B., Hopper, S., Conn, P. J., and Scarr, E. (2016). Changes in BQCA allosteric modulation of [(3)H]NMS binding to human cortex within schizophrenia and by divalent cations. Neuropsychopharmacology. 41, 1620–1628. doi:10.1038/npp.2015.330
Degroot, A., and Nomikos, G. G. (2006). Genetic deletion of muscarinic M4 receptors is anxiolytic in the shock-probe burying model. Eur. J. Pharmacol. 531, 183–186. doi:10.1016/j.ejphar.2005.12.036
Digby, G. J., Noetzel, M. J., Bubser, M., Utley, T. J., Walker, A. G., Byun, N. E., et al. (2012). Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J. Neurosci. 32, 8532–8544. doi:10.1523/JNEUROSCI.0337-12.2012
Ehlert, F. J. (1988). Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol. Pharmacol. 33, 187–194
Ehlert, F. J. (2008). On the analysis of ligand-directed signaling at G protein-coupled receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 377, 549–577. doi:10.1007/s00210-008-0260-4
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 36, 131–137. doi:10.1038/ng1296
Emamian, E. S. (2012). AKT/GSK3 signaling pathway and schizophrenia. Front. Mol. Neurosci. 5, 33. doi:10.3389/fnmol.2012.00033
Emmerling, M. R., Moore, C. J., Doyle, P. D., Carroll, R. T., and Davis, R. E. (1993). Phospholipase A2 activation influences the processing and secretion of the amyloid precursor protein. Biochem. Biophys. Res. Commun. 197, 292–297. doi:10.1006/bbrc.1993.2474
Engers, D. W., Melancon, B. J., Gregro, A. R., Bertron, J. L., Bollinger, S. R., Felts, A. S., et al. (2019a). VU6005806/AZN-00016130, an advanced M4 positive allosteric modulator (PAM) profiled as a potential preclinical development candidate. Bioorg. Med. Chem. Lett. 29, 1714–1718. doi:10.1016/j.bmcl.2019.05.026
Engers, J. L., Bender, A. M., Kalbfleisch, J. J., Cho, H. P., Lingenfelter, K. S., Luscombe, V. B., et al. (2019b). Discovery of tricyclic Triazolo- and Imidazopyridine Lactams as M1 positive allosteric modulators. ACS Chem. Neurosci. 10, 1035–1042. doi:10.1021/acschemneuro.8b00311
Enriquez-Barreto, L., and Morales, M. (2016). The PI3K signaling pathway as a pharmacological target in Autism related disorders and Schizophrenia. Mol. Cell Ther. 4, 2. doi:10.1186/s40591-016-0047-9
Felder, C. C., Kanterman, R. Y., Ma, A. L., and Axelrod, J. (1989). A transfected m1 muscarinic acetylcholine receptor stimulates adenylate cyclase via phosphatidylinositol hydrolysis. J. Biol. Chem. 264, 20356–20362
Felder, C. C., Porter, A. C., Skillman, T. L., Zhang, L., Bymaster, F. P., Nathanson, N. M., et al. (2001). Elucidating the role of muscarinic receptors in psychosis. Life Sci. 68, 2605–2613. doi:10.1016/s0024-3205(01)01059-1
Felder, C. C., Williams, H. L., and Axelrod, J. (1991). A transduction pathway associated with receptors coupled to the inhibitory guanine nucleotide binding protein Gi that amplifies ATP-mediated arachidonic acid release. Proc. Natl. Acad. Sci. U.S.A. 88, 6477–6480. doi:10.1073/pnas.88.15.6477
Felder, C. C. (1995). Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. 9, 619–625
Ferguson, G. N., Valant, C., Horne, J., Figler, H., Flynn, B. L., Linden, J., et al. (2008). 2-aminothienopyridazines as novel adenosine A1 receptor allosteric modulators and antagonists. J. Med. Chem. 51, 6165–6172. doi:10.1021/jm800557d
Ferrari-DiLeo, G., and Flynn, D. D. (1993). Diminished muscarinic receptor-stimulated [3H]-PIP2 hydrolysis in alzheimer’s disease. Life Sci. 53, PL439–PL444. doi:10.1016/0024-3205(93)90037-4
Figueroa, K. W., Griffin, M. T., and Ehlert, F. J. (2009). Selectivity of agonists for the active state of M1 to M4 muscarinic receptor subtypes. J. Pharmacol. Exp. Ther. 328, 331–342. doi:10.1124/jpet.108.145219
Fisher, A., Heldman, E., Gurwitz, D., Haring, R., Karton, Y., Meshulam, H., et al. (1996). M1 agonists for the treatment of Alzheimer's disease. Novel properties and clinical update. Ann. N. Y. Acad. Sci. 777, 189–196. doi:10.1111/j.1749-6632.1996.tb34418.x
Fisher, A., Pittel, Z., Haring, R., Bar-Ner, N., Kliger-Spatz, M., Natan, N., et al. (2003). M1 muscarinic agonists can modulate some of the hallmarks in Alzheimer's disease: implications in future therapy. J. Mol. Neurosci. 20, 349–356. doi:10.1385/JMN:20:3:349
Flohr, A., Hutter, R., Mueller, B., Bohnert, C., Pellisson, M., and Schaffhauser, H. (2017). Discovery of the first low-shift positive allosteric modulators for the muscarinic M1 receptor. Bioorg. Med. Chem. Lett. 27, 5415–5419. doi:10.1016/j.bmcl.2017.11.008
Flynn, D. D., Weinstein, D. A., and Mash, D. C. (1991). Loss of high-affinity agonist binding to M1 muscarinic receptors in Alzheimer’s disease: implications for the failure of cholinergic replacement therapies. Ann. Neurol. 29, 256–262. doi:10.1002/ana.410290305
Flynn, D. D., Ferrari-DiLeo, G., Mash, D. C., and Levey, A. I. (1995). Differential regulation of molecular subtypes of muscarinic receptors in Alzheimer’s disease. J. Neurochem. 64, 1888–1891.
Foote, M., and Zhou, Y. (2012). 14-3-3 proteins in neurological disorders. Int. J. Biochem. Mol. Biol. 3, 152–164.
Foster, D. J., Wilson, J. M., Remke, D. H., Mahmood, M. S., Uddin, M. J., Wess, J., et al. (2016). Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release. Neuron. 91, 1244–1252. doi:10.1016/j.neuron.2016.08.017
Funk, A. J., McCullumsmith, R. E., Haroutunian, V., and Meador-Woodruff, J. H. (2012). Abnormal activity of the MAPK- and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology. 37, 896–905. doi:10.1038/npp.2011.267
Gainetdinov, R. R., Wetsel, W. C., Jones, S. R., Levin, E. D., Jaber, M., and Caron, M. G. (1999). Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 283, 397–401. doi:10.1126/science.283.5400.397
Gerber, D. J., Sotnikova, T. D., Gainetdinov, R. R., Huang, S. Y., Caron, M. G., and Tonegawa, S. (2001). Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 15312–15317. doi:10.1073/pnas.261583798
Gomeza, J., Zhang, L., Kostenis, E., Felder, C., Bymaster, F., Brodkin, J., et al. (1999). Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 10483–10488. doi:10.1073/pnas.96.18.10483
Gotti, C., Clementi, F., Fornari, A., Gaimarri, A., Guiducci, S., Manfredi, I., et al. (2009). Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 78, 703–711. doi:10.1016/j.bcp.2009.05.024
Goupil, E., Tassy, D., Bourguet, C., Quiniou, C., Wisehart, V., Pétrin, D., et al. (2010). A novel biased allosteric compound inhibitor of parturition selectively impedes the prostaglandin F2alpha-mediated Rho/ROCK signaling pathway. J. Biol. Chem. 285, 25624–25636. doi:10.1074/jbc.M110.115196
Graham, K., Zhang, J., Qiao, H., Wu, Y., and Zhou, Y. (2019). Region-specific inhibition of 14-3-3 proteins induces psychomotor behaviors in mice. NPJ schizophr. 5, 1. doi:10.1038/s41537-018-0069-1
Gregory, K. J., Herman, E. J., Ramsey, A. J., Hammond, A. S., Byun, N. E., Stauffer, S. R., et al. (2013). N-aryl piperazine metabotropic glutamate receptor 5 positive allosteric modulators possess efficacy in preclinical models of NMDA hypofunction and cognitive enhancement. J. Pharmacol. Exp. Ther. 347, 438–457. doi:10.1124/jpet.113.206623
Griffin, R. J., Moloney, A., Kelliher, M., Johnston, J. A., Ravid, R., Dockery, P., et al. (2005). Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J. Neurochem. 93, 105–117. doi:10.1111/j.1471-4159.2004.02949.x
Gurwitz, D., Haring, R., Heldman, E., Fraser, C. M., Manor, D., and Fisher, A. (1994). Discrete activation of transduction pathways associated with acetylcholine m1 receptor by several muscarinic ligands. Eur. J. Pharmacol. 267, 21–31. doi:10.1016/0922-4106(94)90220-8
Hadar, A., Milanesi, E., Squassina, A., Niola, P., Chillotti, C., Pasmanik-Chor, M., et al. (2016). RGS2 expression predicts amyloid-β sensitivity, MCI and Alzheimer's disease: genome-wide transcriptomic profiling and bioinformatics data mining. Transl. Psychiatry. 6, e909. doi:10.1038/tp.2016.179
Haga, K., Kameyama, K., Haga, T., Kikkawa, U., Shiozaki, K., and Uchiyama, H. (1996). Phosphorylation of human m1 muscarinic acetylcholine receptors by G protein-coupled receptor kinase 2 and protein kinase C. J. Biol. Chem. 271, 2776–2782. doi:10.1074/jbc.271.5.2776
Haga, K., Kruse, A. C., Asada, H., Yurugi-Kobayashi, T., Shiroishi, M., Zhang, C., et al. (2012). Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 482, 547–551. doi:10.1038/nature10753
Hager, M. V., Clydesdale, L., Gellman, S. H., Sexton, P. M., and Wootten, D. (2017). Characterization of signal bias at the GLP-1 receptor induced by backbone modification of GLP-1. Biochem. Pharmacol. 136, 99–108. doi:10.1016/j.bcp.2017.03.018
Hamilton, S. E., Loose, M. D., Qi, M., Levey, A. I., Hille, B., McKnight, G. S., et al. (1997). Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. U.S.A. 94, 13311–13316. doi:10.1073/pnas.94.24.13311
Hamilton, S. E., and Nathanson, N. M. (2001). The M1 receptor is required for muscarinic activation of mitogen-activated protein (MAP) kinase in murine cerebral cortical neurons. J. Biol. Chem. 276, 15850–15853. doi:10.1074/jbc.M011563200
Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., et al. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. 141, 1917–1933. doi:10.1093/brain/awy132
Hangya, B., Ranade, S. P., Lorenc, M., and Kepecs, A. (2015). Central cholinergic neurons are rapidly recruited by reinforcement feedback. Cell. 162, 1155–1168. doi:10.1016/j.cell.2015.07.057
Harkany, T., Lengyel, Z., Soós, K., Penke, B., Luiten, P. G., and Gulya, K. (1995). Cholinotoxic effects of beta-amyloid (1-42) peptide on cortical projections of the rat nucleus basalis magnocellularis. Brain Res. 695, 71–75. doi:10.1016/0006-8993(95)00823-9
Hartl, D., Gu, W., Mayhaus, M., Pichler, S., Schöpe, J., Wagenpfeil, S., et al. (2015). Amyloid-β protein precursor cleavage products in postmortem ventricular cerebrospinal fluid of Alzheimer’s disease patients. J Alzheimers Dis. 47, 365–372. doi:10.3233/JAD-150191
Harvey, R. D. (2012). Muscarinic receptor agonists and antagonists: effects on cardiovascular function. Handb. Exp. Pharmacol. 208, 299–316. doi:10.1007/978-3-642-23274-9_13
Hawes, B. E., van Biesen, T., Koch, W. J., Luttrell, L. M., and Lefkowitz, R. J. (1995). Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. J. Biol. Chem. 270, 17148–17153. doi:10.1074/jbc.270.29.17148
Holroyd, E. W., Szekeres, P. G., Whittaker, R. D., Kelly, E., and Edwardson, J. M. (1999). Effect of G protein-coupled receptor kinase 2 on the sensitivity of M4 muscarinic acetylcholine receptors to agonist-induced internalization and desensitization in NG108-15 cells. J. Neurochem. 73, 1236–1245. doi:10.1046/j.1471-4159.1999.0731236.x
Hopper, S., Pavey, G. M., Gogos, A., and Dean, B. (2019). Widespread changes in positive allosteric modulation of the muscarinic M1 receptor in some participants with schizophrenia. Int. J. Neuropsychopharmacol. 22, 640–650. doi:10.1093/ijnp/pyz045
Hoshi, N., Langeberg, L. K., Gould, C. M., Newton, A. C., and Scott, J. D. (2010). Interaction with AKAP79 modifies the cellular pharmacology of PKC. Mol. Cell. 37, 541–550. doi:10.1016/j.molcel.2010.01.014
Huynh, T., Valant, C., Crosby, I. T., Sexton, P. M., Christopoulos, A., and Capuano, B. (2013). Probing structural requirements of positive allosteric modulators of the M4 muscarinic receptor. J. Med. Chem. 56, 8196–8200. doi:10.1021/jm401032k
Huynh, T., Valant, C., Crosby, I. T., Sexton, P. M., Christopoulos, A., and Capuano, B. (2015). Synthesis and pharmacological evaluation of M4 muscarinic receptor positive allosteric modulators derived from VU10004. ACS Chem. Neurosci. 6, 838–844. doi:10.1021/acschemneuro.5b00035
Ilyaskina, O. S., Lemoine, H., and Bünemann, M. (2018). Lifetime of muscarinic receptor-G-protein complexes determines coupling efficiency and G-protein subtype selectivity. Proc. Natl. Acad. Sci. U. S. A. 115, 5016–5021. doi:10.1073/pnas.1715751115
Itoh, M., Nagatomo, K., Kubo, Y., and Saitoh, O. (2006). Alternative splicing of RGS8 gene changes the binding property to the M1 muscarinic receptor to confer receptor type-specific Gq regulation. J. Neurochem. 99, 1505–1516. doi:10.1111/j.1471-4159.2006.04220.x
Jablensky, A. (2006). Subtyping schizophrenia: implications for genetic research. Mol. Psychiatry. 11, 815–836. doi:10.1038/sj.mp.4001857
Janickova, H., Rudajev, V., Zimcik, P., Jakubik, J., Tanila, H., El-Fakahany, E. E., et al. (2013). Uncoupling of M1 muscarinic receptor/G-protein interaction by amyloid beta(1-42). Neuropharmacol. 67, 272–283. doi:10.1016/j.neuropharm.2012.11.014
Jeon, J., Dencker, D., Wörtwein, G., Woldbye, D. P., Cui, Y., Davis, A. A., et al. (2010). A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J. Neurosci. 30, 2396–2405. doi:10.1523/JNEUROSCI.3843-09.2010
Jiang, T., Yu, J. T., Tan, M. S., Zhu, X. C., and Tan, L. (2013). β-Arrestins as potential therapeutic targets for Alzheimer’s disease. Mol. Neurobiol. 48, 812–818. doi:10.1007/s12035-013-8469-8
Jope, R. S., Song, L., Li, X., and Powers, R. (1994). Impaired phosphoinositide hydrolysis in Alzheimer’s disease brain. Neurobiol. Aging. 15, 221–226. doi:10.1016/0197-4580(94)90116-3
Jörg, M., van der Westhuizen, E. T., Khajehali, E., Burger, W. A. C., White, J. M., Choy, K. H. C., et al. (2019). 6-Phenylpyrimidin-4-ones as positive allosteric modulators at the M1 mAChR: the determinants of allosteric activity. ACS Chem. Neurosci. 10, 1099–1114. doi:10.1021/acschemneuro.8b00613
Jorg, M., Khajehali, E., van der Westhuizen, E. T., Choy, K. H. C., Shackleford, D., Tobin, A. B., et al. (2020). Development of novel 4-Arylpyridin-2-one and 6-Arylpyrimidin-4-one positive allosteric modulators of the M1 muscarinic acetylcholine receptor. ChemMedChem. [Epub ahead of print]. doi:10.1002/cmdc.202000540
Jung, S. R., Kushmerick, C., Seo, J. B., Koh, D. S., and Hille, B. (2017). Muscarinic receptor regulates extracellular signal regulated kinase by two modes of arrestin binding. Proc. Natl. Acad. Sci. U.S.A. 114, E5579–E5588. doi:10.1073/pnas.1700331114
Kandel, E. R. (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain. 5, 14. doi:10.1186/1756-6606-5-14
Kar, S., Slowikowski, S. P., Westaway, D., and Mount, H. T. (2004). Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J. Psychiatry Neurosci. 29, 427–441.
Karam, C. S., Ballon, J. S., Bivens, N. M., Freyberg, Z., Girgis, R. R., Lizardi-Ortiz, J. E., et al. (2010). Signaling pathways in schizophrenia: emerging targets and therapeutic strategies. Trends Pharmacol. Sci. 31, 381–390. doi:10.1016/j.tips.2010.05.004
Kelly, J. F., Furukawa, K., Barger, S. W., Rengen, M. R., Mark, R. J., Blanc, E. M., et al. (1996). Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc. Natl. Acad. Sci. U.S.A. 93, 6753–6758. doi:10.1073/pnas.93.13.6753
Kenakin, T., and Christopoulos, A. (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216. doi:10.1038/nrd3954
Kenakin, T., Watson, C., Muniz-Medina, V., Christopoulos, A., and Novick, S. (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203. doi:10.1021/cn200111m
Kenakin, T. P. (2012). Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br. J. Pharmacol. 165, 1659–1669. doi:10.1111/j.1476-5381.2011.01749.x
Keov, P., López, L., Devine, S. M., Valant, C., Lane, J. R., Scammells, P. J., et al. (2014). Molecular mechanisms of bitopic ligand engagement with the M1 muscarinic acetylcholine receptor. J. Biol. Chem. 289, 23817–23837. doi:10.1074/jbc.M114.582874
Kesby, J. P., Eyles, D. W., McGrath, J. J., and Scott, J. G. (2018). Dopamine, psychosis and schizophrenia: the widening gap between basic and clinical neuroscience. Transl. Psychiatry. 8, 30. doi:10.1038/s41398-017-0071-9
Klawonn, A. M., Wilhelms, D. B., Lindström, S. H., Singh, A. K., Jaarola, M., Wess, J., et al. (2018). Muscarinic M4 receptors on cholinergic and dopamine D1 receptor-expressing neurons have opposing functionality for positive reinforcement and influence impulsivity. Front. Mol. Neurosci. 11, 139. doi:10.3389/fnmol.2018.00139
Koshimizu, H., Leiter, L. M., and Miyakawa, T. (2012). M4 muscarinic receptor knockout mice display abnormal social behavior and decreased prepulse inhibition. Mol. Brain. 5, 10. doi:10.1186/1756-6606-5-10
Kruse, A. C., Ring, A. M., Manglik, A., Hu, J., Hu, K., Eitel, K., et al. (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 504, 101–106. doi:10.1038/nature12735
Kuduk, S. D., Chang, R. K., Di Marco, C. N., Pitts, D. R., Greshock, T. J., Ma, L., et al. (2011). Discovery of a selective allosteric M1 receptor modulator with suitable development properties based on a quinolizidinone carboxylic acid scaffold. J. Med. Chem. 54, 4773–4780. doi:10.1021/jm200400m
Kuduk, S. D., Chang, R. K., Di Marco, C. N., Ray, W. J., Ma, L., Wittmann, M., et al. (2010). Quinolizidinone carboxylic acids as CNS penetrant, selective m1 allosteric muscarinic receptor modulators. ACS Med. Chem. Lett. 1, 263–267. doi:10.1021/ml100095k
Kuduk, S. D., Di Marco, C. N., Saffold, J. R., Ray, W. J., Ma, L., Wittmann, M., et al. (2014). Identification of a methoxynaphthalene scaffold as a core replacement in quinolizidinone amide M(1) positive allosteric modulators. Bioorg. Med. Chem. Lett. 24, 1417–1420. doi:10.1016/j.bmcl.2014.01.012
Lanciego, J. L., Luquin, N., and Obeso, J. A. (2012). Functional neuroanatomy of the basal ganglia. Cold Spring Harb. Perspect. Med. 2, a009621. doi:10.1101/cshperspect.a009621
Langmead, C. J., Watson, J., and Reavill, C. (2008). Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 117, 232–243. doi:10.1016/j.pharmthera.2007.09.009
Lanzafame, A., Christopoulos, A., and Mitchelson, F. (1997). Three allosteric modulators act at a common site, distinct from that of competitive antagonists, at muscarinic acetylcholine M2 receptors. J. Pharmacol. Exp. Ther. 282, 278–285.
Le, U., Melancon, B. J., Bridges, T. M., Vinson, P. N., Utley, T. J., Lamsal, A., et al. (2013). Discovery of a selective M₄ positive allosteric modulator based on the 3-amino-thieno[2,3-b]pyridine-2-carboxamide scaffold: development of ML253, a potent and brain penetrant compound that is active in a preclinical model of schizophrenia. Bioorg. Med. Chem. Lett. 23, 346–350. doi:10.1016/j.bmcl.2012.10.073
Leach, K., Sexton, P. M., and Christopoulos, A. (2007). Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 28, 382–389. doi:10.1016/j.tips.2007.06.004
Leach, K., Loiacono, R. E., Felder, C. C., McKinzie, D. L., Mogg, A., Shaw, D. B., et al. (2010). Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 35, 855–869. doi:10.1038/npp.2009.194
Leach, K., Davey, A. E., Felder, C. C., Sexton, P. M., and Christopoulos, A. (2011). The role of transmembrane domain 3 in the actions of orthosteric, allosteric, and atypical agonists of the M4 muscarinic acetylcholine receptor. Mol. Pharmacol. 79, 855–865. doi:10.1124/mol.111.070938
Levey, A. I., Kitt, C. A., Simonds, W. F., Price, D. L., and Brann, M. R. (1991). Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci. 11, 3218–3226
Levey, A. I. (1996). Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 93, 13541–13546. doi:10.1073/pnas.93.24.13541
Li, H., Eishingdrelo, A., Kongsamut, S., and Eishingdrelo, H. (2016). G-protein-coupled receptors mediate 14-3-3 signal transduction. Signal Transduct. Target. Ther. 1, 16018. doi:10.1038/sigtrans.2016.18
Li, X., Yu, B., Sun, Q., Zhang, Y., Ren, M., Zhang, X., et al. (2018). Generation of a whole-brain atlas for the cholinergic system and mesoscopic projectome analysis of basal forebrain cholinergic neurons. Proc. Natl. Acad. Sci. U. S. A. 115, 415–420. doi:10.1073/pnas.1703601115
Li, Y., Li, H., Liu, X., Bao, G., Tao, Y., Wu, Z., et al. (2009). Regulation of amygdalar PKA by beta-arrestin-2/phosphodiesterase-4 complex is critical for fear conditioning. Proc. Natl. Acad. Sci. U.S.A. 106, 21918–21923. doi:10.1073/pnas.0906941106
Lindsley, M., Crespo, P., Pellici, P. G., Gutkind, J. S., and Wetzker, R. (1997). Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science. 275, 394–397. doi:10.1126/science.275.5298.394
Liu, W., Dou, F., Feng, J., and Yan, Z. (2011). RACK1 is involved in β-amyloid impairment of muscarinic regulation of GABAergic transmission. Neurobiol. Aging. 32, 1818–1826. doi:10.1016/j.neurobiolaging.2009.10.017
Lleó, A. (2007). Current therapeutic options for Alzheimer’s disease. Curr. Genomics. 8, 550–558. doi:10.2174/138920207783769549
Lockhart, I. A., Mitchell, S. A., and Kelly, S. (2009). Safety and tolerability of donepezil, rivastigmine and galantamine for patients with Alzheimer’s disease: systematic review of the ‘real-world’ evidence. Dement. Geriatr. Cogn. Disord. 28, 389–403. doi:10.1159/000255578
Long, M. F., Engers, J. L., Chang, S., Zhan, X., Weiner, R. L., Luscombe, V. B., et al. (2017). Discovery of a novel 2,4-dimethylquinoline-6-carboxamide M4 positive allosteric modulator (PAM) chemotype via scaffold hopping. Bioorg. Med. Chem. Lett. 27, 4999–5001. doi:10.1016/j.bmcl.2017.10.016
Ma, L., Seager, M. A., Seager, M., Wittmann, M., Jacobson, M., Bickel, D., et al. (2009). Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 15950–15955. doi:10.1073/pnas.0900903106
Mandai, T., Kasahara, M., Kurimoto, E., Tanaka, M., Suzuki, M., Nakatani, A., et al. (2019). In Vivo pharmacological comparison of TAK-071, a positive allosteric modulator of muscarinic M1 receptor, and xanomeline, an agonist of muscarinic M1/M4 receptor, in rodents. Neuroscience. 414, 60–76. doi:10.1016/j.neuroscience.2019.07.003
Marlo, J. E., Niswender, C. M., Days, E. L., Bridges, T. M., Xiang, Y., Rodriguez, A. L., et al. (2009). Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol. 75, 577–588. doi:10.1124/mol.108.052886
May, L. T., Leach, K., Sexton, P. M., and Christopoulos, A. (2007). Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51. doi:10.1146/annurev.pharmtox.47.120505.105159
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W., et al. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 34, 807–820. doi:10.1016/s0896-6273(02)00716-x
McGuire, J. L., Depasquale, E. A., Funk, A. J., O'Donnovan, S. M., Hasselfeld, K., Marwaha, S., et al. (2017). Abnormalities of signal transduction networks in chronic schizophrenia. NPJ Schizophr. 3, 30. doi:10.1038/s41537-017-0032-6
McOmish, C. E., Burrows, E., Howard, M., Scarr, E., Kim, D., Shin, H. S., et al. (2008a). Phospholipase C-beta1 knockout mice exhibit endophenotypes modeling schizophrenia which are rescued by environmental enrichment and clozapine administration. Mol. Psychiatry. 13, 661–672. doi:10.1038/sj.mp.4002046
McOmish, C. E., Burrows, E. L., Howard, M., and Hannan, A. J. (2008b). PLC-beta1 knockout mice as a model of disrupted cortical development and plasticity: behavioral endophenotypes and dysregulation of RGS4 gene expression. Hippocampus. 18, 824–834. doi:10.1002/hipo.20443
Medeiros, R., Kitazawa, M., Caccamo, A., Baglietto-Vargas, D., Estrada-Hernandez, T., Cribbs, D. H., et al. (2011). Loss of muscarinic M1 receptor exacerbates Alzheimer's disease-like pathology and cognitive decline. Am. J. Pathol. 179, 980–991. doi:10.1016/j.ajpath.2011.04.041
Migeon, J. C., and Nathanson, N. M. (1994). Differential regulation of cAMP-mediated gene transcription by m1 and m4 muscarinic acetylcholine receptors. Preferential coupling of m4 receptors to Gi alpha-2. J. Biol. Chem. 269, 9767–9773.
Migeon, J. C., Thomas, S. L., and Nathanson, N. M. (1995). Differential coupling of m2 and m4 muscarinic receptors to inhibition of adenylyl cyclase by Gi alpha and G(o)alpha subunits. J. Biol. Chem. 270, 16070–16074. doi:10.1074/jbc.270.27.16070
Migeon, J. C., Thomas, S. L., and Nathanson, N. M. (1994). Regulation of cAMP-mediated gene transcription by wild type and mutated G-protein alpha subunits. Inhibition of adenylyl cyclase activity by muscarinic receptor-activated and constitutively activated G(o) alpha. J. Biol. Chem. 269, 29146–29152
Millar, J. K., Pickard, B. S., Mackie, S., James, R., Christie, S., Buchanan, S. R., et al. (2005). DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 310, 1187–1191. doi:10.1126/science.1112915
Mirnics, K., Middleton, F. A., Stanwood, G. D., Lewis, D. A., and Levitt, P. (2001). Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol. Psychiatry. 6, 293–301. doi:10.1038/sj.mp.4000866
Mistry, R., Dowling, M. R., and Challiss, R. A. (2005). An investigation of whether agonist-selective receptor conformations occur with respect to M2 and M4 muscarinic acetylcholine receptor signalling via Gi/o and Gs proteins. Br. J. Pharmacol. 144, 566–575. doi:10.1038/sj.bjp.0706090
Mistry, S. N., Jörg, M., Lim, H., Vinh, N. B., Sexton, P. M., Capuano, B., et al. (2016a). 4-Phenylpyridin-2-one derivatives: a novel class of positive allosteric modulator of the M1 muscarinic acetylcholine receptor. J. Med. Chem. 59, 388–409. doi:10.1021/acs.jmedchem.5b01562
Mistry, S. N., Lim, H., Jörg, M., Capuano, B., Christopoulos, A., Lane, J. R., et al. (2016b). Novel fused arylpyrimidinone based allosteric modulators of the M1 muscarinic acetylcholine receptor. ACS Chem. Neurosci. 7, 647–661. doi:10.1021/acschemneuro.6b00018
Mistry, S. N., Valant, C., Sexton, P. M., Capuano, B., Christopoulos, A., and Scammells, P. J. (2013). Synthesis and pharmacological profiling of analogues of benzyl quinolone carboxylic acid (BQCA) as allosteric modulators of the M1 muscarinic receptor. J. Med. Chem. 56, 5151–5172. doi:10.1021/jm400540b
Mitchell, R., Robertson, D. N., Holland, P. J., Collins, D., Lutz, E. M., and Johnson, M. S. (2003). ADP-ribosylation factor-dependent phospholipase D activation by the M3 muscarinic receptor. J. Biol. Chem. 278, 33818–33830. doi:10.1074/jbc.M305825200
Miyakawa, T., Yamada, M., Duttaroy, A., and Wess, J. (2001). Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J. Neurosci. 21, 5239–5250. doi:10.1523/jneurosci.21-14-05239.2001
Moehle, M. S., Pancani, T., Byun, N., Yohn, S. E., Wilson, G. H., Dickerson, J. W., et al. (2017). Cholinergic projections to the substantia nigra pars reticulata inhibit dopamine modulation of basal ganglia through the M4 muscarinic receptor. Neuron. 96, 1358–e4. doi:10.1016/j.neuron.2017.12.008
Montminy, M. (1997). Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem. 66, 807–822. doi:10.1146/annurev.biochem.66.1.807
Moran, S. P., Xiang, Z., Doyle, C. A., Maksymetz, J., Lv, X., Faltin, S., et al. (2019). Biased M1 receptor-positive allosteric modulators reveal role of phospholipase D in M1-dependent rodent cortical plasticity. Sci. Signal. 12, eaax2057. doi:10.1126/scisignal.aax2057
Mrzljak, L., Levey, A. I., and Goldman-Rakic, P. S. (1993). Association of m1 and m2 muscarinic receptor proteins with asymmetric synapses in the primate cerebral cortex: morphological evidence for cholinergic modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 90, 5194–5198. doi:10.1073/pnas.90.11.5194
Mueller, R., Dawson, E. S., Meiler, J., Rodriguez, A. L., Chauder, B. A., Bates, B. S., et al. (2012). Discovery of 2-(2-benzoxazoyl amino)-4-aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu₅): from an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem. 7, 406–414. doi:10.1002/cmdc.201100510
Nair, A. G., Castro, L. R. V., El Khoury, M., Gorgievski, V., Giros, B., Tzavara, E. T., et al. (2019). The high efficacy of muscarinic M4 receptor in D1 medium spiny neurons reverses striatal hyperdopaminergia. Neuropharmacol. 146, 74–83. doi:10.1016/j.neuropharm.2018.11.029
Nathanson, N. M. (2000). A multiplicity of muscarinic mechanisms: enough signaling pathways to take your breath away. Proc. Natl. Acad. Sci. U.S.A. 97, 6245–6247. doi:10.1073/pnas.97.12.6245
Nawaratne, V., Leach, K., Felder, C. C., Sexton, P. M., and Christopoulos, A. (2010). Structural determinants of allosteric agonism and modulation at the M4 muscarinic acetylcholine receptor: identification of ligand-specific and global activation mechanisms. J. Biol. Chem. 285, 19012–19021. doi:10.1074/jbc.M110.125096
Nevo, I., Avidor-Reiss, T., Levy, R., Bayewitch, M., Heldman, E., and Vogel, Z. (1998). Regulation of adenylyl cyclase isozymes on acute and chronic activation of inhibitory receptors. Mol. Pharmacol. 54, 419–426. doi:10.1124/mol.54.2.419
Nordberg, A., and Winblad, B. (1986). Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci. Lett. 72, 115–119. doi:10.1016/0304-3940(86)90629-4
Panarese, J. D., Cho, H. P., Adams, J. J., Nance, K. D., Garcia-Barrantes, P. M., Chang, S., et al. (2016). Further optimization of the M1 PAM VU0453595: discovery of novel heterobicyclic core motifs with improved CNS penetration. Bioorg. Med. Chem. Lett. 26, 3822–3825. doi:10.1016/j.bmcl.2016.04.083
Park, S. M., Chen, M., Schmerberg, C. M., Dulman, R. S., Rodriguiz, R. M., Caron, M. G., et al. (2016). Effects of β-arrestin-biased dopamine D2 receptor ligands on schizophrenia-like behavior in hypoglutamatergic mice. Neuropsychopharmacology. 41, 704–715. doi:10.1038/npp.2015.196
Passmore, G. M., Reilly, J. M., Thakur, M., Keasberry, V. N., Marsh, S. J., Dickenson, A. H., et al. (2012). Functional significance of M-type potassium channels in nociceptive cutaneous sensory endings. Front. Mol. Neurosci. 5, 63. doi:10.3389/fnmol.2012.00063
Pei, J. J., Khatoon, S., An, W. L., Nordlinder, M., Tanaka, T., Braak, H., et al. (2003). Role of protein kinase B in Alzheimer's neurofibrillary pathology. Acta Neuropathol. 105, 381–392. doi:10.1007/s00401-002-0657-y
Picciotto, M. R., Higley, M. J., and Mineur, Y. S. (2012). Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 76, 116–129. doi:10.1016/j.neuron.2012.08.036
Pittenger, C., Fasano, S., Mazzocchi-Jones, D., Dunnett, S. B., Kandel, E. R., and Brambilla, R. (2006). Impaired bidirectional synaptic plasticity and procedural memory formation in striatum-specific cAMP response element-binding protein-deficient mice. J. Neurosci. 26, 2808–2813. doi:10.1523/JNEUROSCI.5406-05.2006
Plonk, S. G., Park, S. K., and Exton, J. H. (1998). The alpha-subunit of the heterotrimeric G protein G13 activates a phospholipase D isozyme by a pathway requiring Rho family GTPases. J. Biol. Chem. 273, 4823–4826. doi:10.1074/jbc.273.9.4823
Popiolek, M., Nguyen, D. P., Reinhart, V., Edgerton, J. R., Harms, J., Lotarski, S. M., et al. (2016). Inositol phosphate accumulation in Vivo provides a measure of muscarinic M1 receptor activation. Biochem. 55, 7073–7085. doi:10.1021/acs.biochem.6b00688.s001
Porter, A. C., Bymaster, F. P., DeLapp, N. W., Yamada, M., Wess, J., Hamilton, S. E., et al. (2002). M1 muscarinic receptor signaling in mouse hippocampus and cortex. Brain Res. 944, 82–89. doi:10.1016/s0006-8993(02)02721-x
Poslusney, M. S., Salovich, J. M., Wood, M. R., Melancon, B. J., Bollinger, K. A., Luscombe, V. B., et al. (2019). Novel M4 positive allosteric modulators derived from questioning the role and impact of a presumed intramolecular hydrogen-bonding motif in beta-amino carboxamide-harboring ligands. Bioorg. Med. Chem. Lett. 29, 362–366. doi:10.1016/j.bmcl.2018.12.039
Price, M. R., Baillie, G. L., Thomas, A., Stevenson, L. A., Easson, M., Goodwin, R., et al. (2005). Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 68, 1484–1495. doi:10.1124/mol.105.016162
Raedler, T. J., Bymaster, F. P., Tandon, R., Copolov, D., and Dean, B. (2007). Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry. 12, 232–246. doi:10.1038/sj.mp.4001924
Rai, S. N., Dilnashin, H., Birla, H., Singh, S. S., Zahra, W., Rathore, A. S., et al. (2019). The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 35, 775–795. doi:10.1007/s12640-019-0003-y
Rajagopal, S., Ahn, S., Rominger, D. H., Gowen-MacDonald, W., Lam, C. M., Dewire, S. M., et al. (2011). Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 80, 367–377. doi:10.1124/mol.111.072801
Reiner, C., and Nathanson, N. M. (2008). The internalization of the M2 and M4 muscarinic acetylcholine receptors involves distinct subsets of small G-proteins. Life Sci. 82, 718–727. doi:10.1016/j.lfs.2008.01.013
Ridley, A. J., and Hall, A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70, 389–399. doi:10.1016/0092-8674(92)90163-7
Ridley, A. J., Paterson, H. F., Johnston, C. L., Diekmann, D., and Hall, A. (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 70, 401–410. doi:10.1016/0092-8674(92)90164-8
Ridley, A. J. (2001). Rho proteins: linking signaling with membrane trafficking. Traffic. 2, 303–310. doi:10.1034/j.1600-0854.2001.002005303.x
Rook, J. M., Abe, M., Cho, H. P., Nance, K. D., Luscombe, V. B., Adams, J. J., et al. (2017). Diverse effects on M1 signaling and adverse effect liability within a series of M1 ago-PAMs. ACS Chem. Neurosci. 8, 866–883. doi:10.1021/acschemneuro.6b00429.s001
Rosenblum, K., Futter, M., Jones, M., Hulme, E. C., and Bliss, T. V. (2000). ERKI/II regulation by the muscarinic acetylcholine receptors in neurons. J. Neurosci. 20, 977–985. doi:10.1523/jneurosci.20-03-00977.2000
Roth, B. L. (2016). DREADDs for neuroscientists. Neuron. 89, 683–694. doi:10.1016/j.neuron.2016.01.040
Roy, A. A., Lemberg, K. E., and Chidiac, P. (2003). Recruitment of RGS2 and RGS4 to the plasma membrane by G proteins and receptors reflects functional interactions. Mol. Pharmacol. 64, 587–593. doi:10.1124/mol.64.3.587
Rumenapp, U., Asmus, M., Schablowski, H., Woznicki, M., Han, L., Jakobs, K. H., et al. (2001). The M3 muscarinic acetylcholine receptor expressed in HEK-293 cells signals to phospholipase D via G12 but not Gq-type G proteins: regulators of G proteins as tools to dissect pertussis toxin-resistant G proteins in receptor-effector coupling. J. Biol. Chem. 276, 2474–2479. doi:10.1074/jbc.M004957200
Sabbir, M. G., and Fernyhough, P. (2018). Muscarinic receptor antagonists activate ERK-CREB signaling to augment neurite outgrowth of adult sensory neurons. Neuropharmacol. 143, 268–281. doi:10.1016/j.neuropharm.2018.09.020
Salah-Uddin, H., Scarr, E., Pavey, G., Harris, K., Hagan, J. J., Dean, B., et al. (2009). Altered M(1) muscarinic acetylcholine receptor (CHRM1)-Galpha(q/11) coupling in a schizophrenia endophenotype. Neuropsychopharmacol. 34, 2156–2166. doi:10.1038/npp.2009.41
Salovich, J. M., Vinson, P. N., Sheffler, D. J., Lamsal, A., Utley, T. J., Blobaum, A. L., et al. (2012). Discovery of N-(4-methoxy-7-methylbenzo[d]thiazol-2-yl)isonicatinamide, ML293, as a novel, selective and brain penetrant positive allosteric modulator of the muscarinic 4 (M4) receptor. Bioorg. Med. Chem. Lett. 22, 5084–5088. doi:10.1016/j.bmcl.2012.05.109
Sanchez-Mejia, R. O., Newman, J. W., Toh, S., Yu, G. Q., Zhou, Y., Halabisky, B., et al. (2008). Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nature Neurosci. 11, 1311–1318. doi:10.1038/nn.2213
Santini, F., Penn, R. B., Gagnon, A. W., Benovic, J. L., and Keen, J. H. (2000). Selective recruitment of arrestin-3 to clathrin coated pits upon stimulation of G protein-coupled receptors. J. Cell Sci. 113 (Pt 13), 2463–2470.
Satoh, Y., Endo, S., Ikeda, T., Yamada, K., Ito, M., Kuroki, M., et al. (2007). Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J. Neurosci. 27, 10765–10776. doi:10.1523/JNEUROSCI.0117-07.2007
Scarr, E., Cowie, T. F., Kanellakis, S., Sundram, S., Pantelis, C., and Dean, B. (2009). Decreased cortical muscarinic receptors define a subgroup of subjects with schizophrenia. Mol. Psychiatry. 14, 1017–1023. doi:10.1038/mp.2008.28
Scarr, E., Udawela, M., Thomas, E. A., and Dean, B. (2018). Changed gene expression in subjects with schizophrenia and low cortical muscarinic M1 receptors predicts disrupted upstream pathways interacting with that receptor. Mol. Psychiatry. 23, 295–303. doi:10.1038/mp.2016.195
Schubert, J. W., Harrison, S. T., Mulhearn, J., Gomez, R., Tynebor, R., Jones, K., et al. (2019). Discovery, optimization, and biological characterization of 2,3,6-Trisubstituted pyridine-containing M4 positive allosteric modulators. ChemMedChem. 14, 943–951. doi:10.1002/cmdc.201900088
Shekhar, A., Potter, W. Z., Lightfoot, J., Lienemann, J., Dube, S., Mallinckrodt, C., et al. (2008). Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry. 165, 1033–1039. doi:10.1176/appi.ajp.2008.06091591
Shimohama, S., Homma, Y., Suenaga, T., Fujimoto, S., Taniguchi, T., Araki, W., et al. (1991). Aberrant accumulation of phospholipase C-delta in Alzheimer brains. Am. J. Pathol. 139, 737–742.
Shirey, J. K., Brady, A. E., Jones, P. J., Davis, A. A., Bridges, T. M., Kennedy, J. P., et al. (2009). A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci. 29, 14271–14286. doi:10.1523/JNEUROSCI.3930-09.2009
Smesny, S., Milleit, B., Nenadic, I., Preul, C., Kinder, D., Lasch, J., et al. (2010). Phospholipase A2 activity is associated with structural brain changes in schizophrenia. Neuroimage. 52, 1314–1327. doi:10.1016/j.neuroimage.2010.05.009
Smith, J. S., Lefkowitz, R. J., and Rajagopal, S. (2018). Biased signalling: from simple switches to allosteric microprocessors. Nature Rev. Drug. Discov. 17, 243–260. doi:10.1038/nrd.2017.229
Song, X., Coffa, S., Fu, H., and Gurevich, V. V. (2009). How does arrestin assemble MAPKs into a signaling complex?. J. Biol. Chem. 284, 685–695. doi:10.1074/jbc.M806124200
Stockton, J., Birdsall, N. J., Burgen, A. S., and Hulme, E. C. (1982). The theory, methods and relevance of muscarinic receptor binding studies. Scand. J. Gastroenterol. Suppl. 72, 43–47.
Sunahara, R. K., Dessauer, C. W., and Gilman, A. G. (1996). Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 36, 461–480. doi:10.1146/annurev.pa.36.040196.002333
Suratman, S., Leach, K., Sexton, P., Felder, C., Loiacono, R., and Christopoulos, A. (2011). Impact of species variability and 'probe-dependence' on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br. J. Pharmacol. 162, 1659–1670. doi:10.1111/j.1476-5381.2010.01184.x
Suzuki, N., Hajicek, N., and Kozasa, T. (2009). Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals. 17, 55–70. doi:10.1159/000186690
Tarr, J. C., Wood, M. R., Noetzel, M. J., Bertron, J. L., Weiner, R. L., Rodriguez, A. L., et al. (2017). Challenges in the development of an M4 PAM preclinical candidate: the discovery, SAR, and in vivo characterization of a series of 3-aminoazetidine-derived amides. Bioorg. Med. Chem. Lett. 27, 2990–2995. doi:10.1016/j.bmcl.2017.05.014
Temple, K. J., Engers, J. L., Long, M. F., Gregro, A. R., Watson, K. J., Chang, S., et al. (2019). Discovery of a novel 3,4-dimethylcinnoline carboxamide M4 positive allosteric modulator (PAM) chemotype via scaffold hopping. Bioorg. Med. Chem. Lett. 29, 126678. doi:10.1016/j.bmcl.2019.126678
Temple, K. J., Engers, J. L., Long, M. F., Watson, K. J., Chang, S., Luscombe, V. B., et al. (2020a). Discovery of a novel 2,3-dimethylimidazo[1,2-a]pyrazine-6-carboxamide M4 positive allosteric modulator (PAM) chemotype. Bioorg. Med. Chem. Lett. 30, 126812. doi:10.1016/j.bmcl.2019.126812
Temple, K. J., Long, M. F., Engers, J. L., Watson, K. J., Chang, S., Luscombe, V. B., et al. (2020b). Discovery of structurally distinct tricyclic M4 positive allosteric modulator (PAM) chemotypes. Bioorg. Med. Chem. Lett. 30, 126811. doi:10.1016/j.bmcl.2019.126811
Tence, M., Cordier, J., Premont, J., and Glowinski, J. (1994). Muscarinic cholinergic agonists stimulate arachidonic acid release from mouse striatal neurons in primary culture. J. Pharmacol. Exp. Ther. 269, 646–653.
Thal, D. M., Sun, B., Feng, D., Nawaratne, V., Leach, K., Felder, C. C., et al. (2016). Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature. 531, 335–340. doi:10.1038/nature17188
Thathiah, A., and De Strooper, B. (2011). The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat. Rev. Neurosci. 12, 73–87. doi:10.1038/nrn2977
Thathiah, A., Horre, K., Snellinx, A., Vandewyer, E., Huang, Y., Ciesielska, M., et al. (2013). beta-arrestin 2 regulates Abeta generation and gamma-secretase activity in Alzheimer’s disease. Nat. Med. 19, 43–49. doi:10.1038/nm.3023
Thiele, A. (2013). Muscarinic signaling in the brain. Annu. Rev. Neurosci. 36, 271–294. doi:10.1146/annurev-neuro-062012-170433
Thomas, R. L., Mistry, R., Langmead, C. J., Wood, M. D., and Challiss, R. A. (2008). G protein coupling and signaling pathway activation by m1 muscarinic acetylcholine receptor orthosteric and allosteric agonists. J. Pharmacol. Exp. Ther. 327, 365–374. doi:10.1124/jpet.108.141788
Tsang, S. W., Pomakian, J., Marshall, G. A., Vinters, H. V., Cummings, J. L., Chen, C. P., et al. (2007). Disrupted muscarinic M1 receptor signaling correlates with loss of protein kinase C activity and glutamatergic deficit in Alzheimer’s disease. Neurobiol. Aging. 28, 1381–1387. doi:10.1016/j.neurobiolaging.2006.06.001
Turlington, M., Noetzel, M. J., Chun, A., Zhou, Y., Gogliotti, R. D., Nguyen, E. D., et al. (2013). Exploration of allosteric agonism structure-activity relationships within an acetylene series of metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulators (PAMs): discovery of 5-((3-fluorophenyl)ethynyl)-N-(3-methyloxetan-3-yl)picolinamide (ML254). J. Med. Chem. 56, 7976–7996. doi:10.1021/jm401028t
Turski, L., Ikonomidou, C., Turski, W. A., Bortolotto, Z. A., and Cavalheiro, E. A. (1989). Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 3, 154–171. doi:10.1002/syn.890030207
Valant, C., Aurelio, L., Devine, S. M., Ashton, T. D., White, J. M., Sexton, P. M., et al. (2012a). Synthesis and characterization of novel 2-amino-3-benzoylthiophene derivatives as biased allosteric agonists and modulators of the adenosine A(1) receptor. J. Med. Chem. 55, 2367–2375. doi:10.1021/jm201600e
Valant, C., Felder, C. C., Sexton, P. M., and Christopoulos, A. (2012b). Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol. Pharmacol. 81, 41–52. doi:10.1124/mol.111.074872
van Biesen, T., Hawes, B. E., Raymond, J. R., Luttrell, L. M., Koch, W. J., and Lefkowitz, R. J. (1996). G(o)-protein alpha-subunits activate mitogen-activated protein kinase via a novel protein kinase C-dependent mechanism. J. Biol. Chem. 271, 1266–1269. doi:10.1074/jbc.271.3.1266
van der Westhuizen, E. T., Spathis, A., Khajehali, E., Jorg, M., Mistry, S. N., Capuano, B., et al. (2018). Assessment of the molecular mechanisms of action of novel 4-Phenylpyridine-2-one and 6-Phenylpyrimidin-4-one allosteric modulators at the M1 muscarinic acetylcholine receptors. Mol. Pharmacol. 94, 770–783. doi:10.1124/mol.118.111633
van Enkhuizen, J., Janowsky, D. S., Olivier, B., Minassian, A., Perry, W., Young, J. W., et al. (2015). The catecholaminergic-cholinergic balance hypothesis of bipolar disorder revisited. Eur. J. Pharmacol. 753, 114–126. doi:10.1016/j.ejphar.2014.05.063
Varga, E. V., Stropova, D., Rubenzik, M., Wang, M., Landsman, R. S., Roeske, W. R., et al. (1998). Identification of adenylyl cyclase isoenzymes in CHO and B82 cells. Eur. J. Pharmacol. 348, R1–R2. doi:10.1016/s0014-2999(98)00258-1
Vilaro, M. T., Mengod, G., and Palacios, J. M. (1993). Advances and limitations of the molecular neuroanatomy of cholinergic receptors: the example of multiple muscarinic receptors. Prog. Brain Res. 98, 95–101. doi:10.1016/s0079-6123(08)62385-7
Vogler, O., Nolte, B., Voss, M., Schmidt, M., Jakobs, K. H., and van Koppen, C. J. (1999). Regulation of muscarinic acetylcholine receptor sequestration and function by beta-arrestin. J. Biol. Chem. 274, 12333–12338. doi:10.1074/jbc.274.18.12333
Wang, H., Xu, J., Lazarovici, P., Quirion, R., and Zheng, W. (2018). cAMP response element-binding protein (CREB): a possible signaling molecule link in the pathophysiology of schizophrenia. Front. Mol. Neurosci. 11, 255. doi:10.3389/fnmol.2018.00255
Weinberger, D. R. (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen. Psychiatry. 44, 660–669. doi:10.1001/archpsyc.1987.01800190080012
Wess, J. (2004). Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu. Rev. Pharmacol. Toxicol. 44, 423–450. doi:10.1146/annurev.pharmtox.44.101802.121622
Willets, J. M., Nahorski, S. R., and Challiss, R. A. (2005). Roles of phosphorylation-dependent and -independent mechanisms in the regulation of M1 muscarinic acetylcholine receptors by G protein-coupled receptor kinase 2 in hippocampal neurons. J. Biol. Chem. 280, 18950–18958. doi:10.1074/jbc.M412682200
Wood, M. R., Noetzel, M. J., Engers, J. L., Bollinger, K. A., Melancon, B. J., Tarr, J. C., et al. (2016a). Discovery and optimization of a novel series of highly CNS penetrant M4 PAMs based on a 5,6-dimethyl-4-(piperidin-1-yl)thieno[2,3-d]pyrimidine core. Bioorg. Med. Chem. Lett. 26, 3029–3033. doi:10.1016/j.bmcl.2016.05.010
Wood, M. R., Noetzel, M. J., Tarr, J. C., Rodriguez, A. L., Lamsal, A., Chang, S., et al. (2016b). Discovery and SAR of a novel series of potent, CNS penetrant M4 PAMs based on a non-enolizable ketone core: challenges in disposition. Bioorg. Med. Chem. Lett. 26, 4282–4286. doi:10.1016/j.bmcl.2016.07.042
Wood, M. R., Noetzel, M. J., Melancon, B. J., Poslusney, M. S., Nance, K. D., Hurtado, M. A., et al. (2017a). Discovery of VU0467485/AZ13713945: an M4 PAM Evaluated as a preclinical candidate for the treatment of schizophrenia. ACS Med. Chem. Lett. 8, 233–238. doi:10.1021/acsmedchemlett.6b00461
Wood, M. R., Noetzel, M. J., Poslusney, M. S., Melancon, B. J., Tarr, J. C., Lamsal, A., et al. (2017b). Challenges in the development of an M4 PAM in vivo tool compound: the discovery of VU0467154 and unexpected DMPK profiles of close analogs. Bioorg. Med. Chem. Lett. 27, 171–175. doi:10.1016/j.bmcl.2016.11.086
Wootten, D., Christopoulos, A., Marti-Solano, M., Babu, M. M., and Sexton, P. M. (2018). Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nature Rev. Mol. Cell Biol. 19, 638–653. doi:10.1038/s41580-018-0049-3
Wootten, D., Savage, E. E., Valant, C., May, L. T., Sloop, K. W., Ficorilli, J., et al. (2012). Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol. Pharmacol. 82, 281–290. doi:10.1124/mol.112.079319
Wootten, D., Christopoulos, A., and Sexton, P. M. (2013a). Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov. 12, 630–644. doi:10.1038/nrd4052
Wootten, D., Savage, E. E., Willard, F. S., Bueno, A. B., Sloop, K. W., Christopoulos, A., et al. (2013b). Differential activation and modulation of the glucagon-like peptide-1 receptor by small molecule ligands. Mol. Pharmacol. 83, 822–834. doi:10.1124/mol.112.084525
Wu, E. H., and Wong, Y. H. (2006). Activation of muscarinic M4 receptor augments NGF-induced pro-survival Akt signaling in PC12 cells. Cell. Signal. 18, 285–293. doi:10.1016/j.cellsig.2005.04.009
Xia, Z., and Storm, D. R. (2012). Role of signal transduction crosstalk between adenylyl cyclase and MAP kinase in hippocampus-dependent memory. Learn. Mem. 19, 369–374. doi:10.1101/lm.027128.112
Xue, B., Mao, L. M., Jin, D. Z., and Wang, J. Q. (2015). Regulation of synaptic MAPK/ERK phosphorylation in the rat striatum and medial prefrontal cortex by dopamine and muscarinic acetylcholine receptors. J. Neurosci. Res. 93, 1592–1599. doi:10.1002/jnr.23622
Yeatman, H. R., Lane, J. R., Choy, K. H., Lambert, N. A., Sexton, P. M., Christopoulos, A., et al. (2014). Allosteric modulation of M1 muscarinic acetylcholine receptor internalization and subcellular trafficking. J. Biol. Chem. 289, 15856–15866. doi:10.1074/jbc.M113.536672
Yohn, S. E., Foster, D. J., Covey, D. P., Moehle, M. S., Galbraith, J., Garcia-Barrantes, P. M., et al. (2018). Activation of the mGlu1 metabotropic glutamate receptor has antipsychotic-like effects and is required for efficacy of M4 muscarinic receptor allosteric modulators. Mol. Psychiatry. 25 (11), 2786–2799. doi:10.1038/s41380-018-0206-2
Yuan, L., Barbash, S., Kongsamut, S., Eishingdrelo, A., Sakmar, T. P., and Eishingdrelo, H. (2019). 14-3-3 signal adaptor and scaffold proteins mediate GPCR trafficking. Sci. Rep. 9, 11156. doi:10.1038/s41598-019-47478-w
Yuan, P., Zhou, R., Wang, Y., Li, X., Li, J., Chen, G., et al. (2010). Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J. Affect. Disord. 124, 164–169. doi:10.1016/j.jad.2009.10.017
Keywords: muscarinic receptor, allosteric modulation, Alzheimer’s disease, schizophrenia, biased agonism, biased allostery
Citation: van der Westhuizen ET, Choy KHC, Valant C, McKenzie-Nickson S, Bradley SJ, Tobin AB, Sexton PM and Christopoulos A (2021) Fine Tuning Muscarinic Acetylcholine Receptor Signaling Through Allostery and Bias. Front. Pharmacol. 11:606656. doi: 10.3389/fphar.2020.606656
Received: 15 September 2020; Accepted: 30 November 2020;
Published: 29 January 2021.
Edited by:
Anthony Peter Davenport, University of Cambridge, United KingdomReviewed by:
Nadezhda A. German, Texas Tech University Health Sciences Center, United StatesCopyright © 2021 van der Westhuizen, Choy, Valant, McKenzie-Nickson, Bradley, Tobin, Sexton and Christopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma T. van der Westhuizen, RW1tYS5WYW5EZXJXZXN0aHVpemVuQG1vbmFzaC5lZHU=; Arthur Christopoulos, QXJ0aHVyLkNocmlzdG9wb3Vsb3NAbW9uYXNoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.