 Lili Sheng
Lili Sheng George Bayliss
George Bayliss Shougang Zhuang
Shougang Zhuang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 26 January 2021
Sec. Renal Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.598910
This article is part of the Research Topic Combating Diabetes and Diabetic Kidney Disease View all 13 articles
Diabetic kidney disease (DKD) is a leading cause of end-stage renal disease worldwide and the major cause of renal failure among patients on hemodialysis. Numerous studies have demonstrated that transient activation of epidermal growth factor receptor (EGFR) pathway is required for promoting kidney recovery from acute injury whereas its persistent activation is involved in the progression of various chronic kidney diseases including DKD. EGFR-mediated pathogenesis of DKD is involved in hemodynamic alteration, metabolic disturbance, inflammatory response and parenchymal cellular dysfunction. Therapeutic intervention of this receptor has been available in the oncology setting. Targeting EGFR might also hold a therapeutic potential for DKD. Here we review the functional role of EGFR in the development of DKD, mechanisms involved and the perspective about use of EGFR inhibitors as a treatment for DKD.
Diabetic kidney disease (DKD) is a complication of diabetes mellitus and one of the leading causes of end-stage renal disease (ESRD) worldwide. DKD places a heavy personal burden on the many people world-wide who need hemodialysis and heavy economic burden on health care systems. It is urgent to find ways to slow the progression of DKD. However, its pathogenesis is complex and the mechanism is still poorly understood.
Increasing evidence indicates that various signaling pathways are activated and involved in the pathogenesis of DKD. Among them, the role of epidermal growth factor receptor (EGFR) has been extensively studied (Matrougui 2010; Advani et al., 2011; Zhang et al., 2014; Koya 2015). The EGFR belongs to a family of receptors that harbor tyrosine kinase activity and is composed of four members: EGFR (ErbB1), ErbB2, ErbB3, and ErbB4. They can be activated by several ligands, including epidermal growth factor (EGF), transforming growth factor-α (TGF-α), amphiregulin, heparin-binding EGF-like growth factor (HB-EGF), betacellulin, epigulin and epigen (Higashiyama et al., 2008; Schneider and Wolf 2009; Rayego-Mateos et al., 2018b). Upon ligand binding, the receptors form homodimers or heterodimers, leading to phosphorylation of some specific tyrosine residues in intracellular domains. These residues act as docking sites for initiating activation of multiple intracellular signaling pathways (Forrester et al., 2016).
Activation of EGFR signaling has been implicated in numerous physiological and pathophysiological processes, including embryonic development, cell proliferation, cell survival, and tumorigenesis. In the mammalian kidney, EGFR is widely expressed in glomeruli and proximal tubules, including renal epithelial cells, glomerular endothelial cells, podocytes, tubular cells, mesangial cells and medullary interstitial cells (Gesualdo et al., 1996; Harskamp et al., 2016). In the past decade, many studies have investigated the role of EGFR signaling in the progression of chronic kidney disease (CKD) (Chen et al., 2012; Harskamp et al., 2016; Rayego-Mateos et al., 2018b). In this review, we will discuss the role and the mechanism of EGFR in the development of DKD and consider the potential use of EGFR inhibitors as a treatment of this disease.
Chronic hyperglycemia in diabetes mellitus can induce dysfunction of all types of cells in the kidney. DKD is mainly manifested by proteinuria, which varies in several stages, including the silent stage, the microalbuminuria stage (30–300 mg/day) and the macroalbuminuria stage (>300 mg/day) (Papadopoulou-Marketou et al., 2017). Proteinuria occurs along with morphological changes in the glomerulus and interstitium (Qi H. et al., 2017.). From the onset of diabetes mellitus to nearly 5 years, kidney size increases along with an increase in renal plasma flow and hyperfiltration, with thickened glomerular basement membrane and mild or severe mesangial expansion. After 5–10 years, glomerular damage progresses with the occurrence of microalbuminuria and nodular accumulation of mesangial matrix. As glomerulosclerosis advances, extra-glomerular lesions also form. Proteinuria is irreversible at this stage as the glomerular filtration rate (GRF) drops below 60 ml/min/1.73 m2 and ultimately reaches end-stage levels below 15 ml/min/1.73 m2 (Sulaiman 2019; Han et al., 2017; Kanwar et al., 2011; Papadopoulou-Marketou et al., 2017; Qi C. et al., 2017). During the progression of DKD, mitochondria generate excess reactive oxidative species (ROS) or reactive nitrogen species (RNS), resulting in the activation of several signaling pathways, transcription factors and cytokines, such as TGF-β/smad/MAPK signaling, JAK/STAT signaling, VEGF, EGFR. Activation of these signaling pathways and transcription factors is associated with cell growth, angiogenesis, and apoptosis, leading to DKD ultimately (Kanwar et al., 2011; Magee et al., 2017; Papadopoulou-Marketou et al., 2017; VR et al., 2019).
Renal EGFR phosphorylation levels were significantly increased in animal models of diabetes mellitus and in cultured cells treated with high glucose (Konishi and Berk 2003; Saad et al., 2005; Portik-Dobos et al., 2006; Uttarwar et al., 2011; Li R. et al., 2015). EGFR inhibition slowed the progression of DKD, including the improvement of proteinuria and morphologic changes (Wassef et al., 2004; Chen et al., 2012; Zhang et al., 2014). The concentration of EGFR ligands in plasma and kidneys, such as EGF, TGF-α and HB-EGF, was also increased in DKD (Uttarwar et al., 2011; Miyazawa et al., 2013; Perlman et al., 2015). Some reports suggested that connective tissue growth factor (CTGF) is a novel EGFR ligand and that blocking CTGF-mediated profibrotic effects could also be a potential therapeutic option to treat fibrotic renal diseases (Rayego-Mateos et al., 2013; Rayego-Mateos et al., 2018a).
Besides direct activation by its ligands, EGFR transactivation has been recognized as another important mechanism for signal transduction. The process of EGFR transactivation is not mediated through direct ligand binding, but through other second messengers. Several stimuli known to be involved in the pathogenesis of DKD were found capable of transactivating EGFR, such as ROS, TGF-β and PKC. In streptozotocin-induced diabetes and in cultured cells exposed to high glucose, ROS inhibition with superoxide dismutase (SOD) or an NADPH oxidase inhibitor attenuated the upregulation of EGFR phosphorylation (Chen et al., 2015; Sheng et al., 2016). Endothelin-1 (ET-1) mediated activation of endothelin A (ETA) receptor also contributed to EGFR transactivation in diabetic animals (Portik-Dobos et al., 2006). The mechanism by which EGFR transactivation occurs upon stimulation with these active factors remains unclear. A well-accepted hypothesis is that these substances act on their own receptors and then induce release of EGFR ligands (Konishi and Berk 2003; Higashiyama et al., 2008; Chen et al., 2012). EGFR ligands including EGF, HB-EGF and TNF-α are synthesized as precursors anchored on the cell membrane. Upon stimulation, they are released from the membrane in soluble bioactive forms by specific metalloproteases such as ADAM17 (Ohtsu et al., 2006; Uttarwar et al., 2011) (Uttarwar et al., 2011; Li T. et al., 2015; Morgado-Pascual et al., 2015). In diabetes, several second messengers, such as ROS and protein kinases can induce activation of ADAMs, leading to shedding of EGFR ligands. The shed ligands can bind to EGFR in an autocrine or paracrine-dependent manner (Schreier et al., 2014). In addition, Src, a non-receptor tyrosine kinase, can also mediate EGFR transactivation initiated by activation of G-protein-coupled receptors (GPCRs) (Taniguchi et al., 2013; Forrester et al., 2016).
The pathogenesis of DKD is a complex process involving many factors, including hemodynamic alteration, metabolic disturbance, inflammatory response and parenchymal cellular dysfunction (Tung et al., 2018; VR et al., 2019). EGFR transactivation by high glucose causes multicellular dysfunction, which initiates and accelerates kidney injury. Studies have found that EGFR inhibition can reduce kidney size after in STZ treated diabetic mice, without affecting body weight, blood glucose or blood pressure (Wassef et al., 2004). Inhibition of EGFR with erlotinib also markedly reduces albuminuria and renal expression of CTGF, collagen I, collagen IV in diabetic mice (Zhang et al., 2014).
Hemodynamic alteration plays an important role in the pathogenesis of DKD. Chronic hyperglycemia induces metabolic alteration and dysfunction in endothelial and vascular smooth muscle cells, leading to vascular dysfunction and hemodynamic alteration in kidneys and other organs (Matrougui 2010; Li T. et al., 2015). Glomerular hemodynamic alterations such as hyperfiltration and hyper-perfusion are found in the early stages of DKD. Hyperfiltration is a result of a decrease in glomerular afferent and efferent arteriolar resistance; dilation of the efferent arteriole is relatively less than dilation of the afferent arteriole, causing a relative increase in glomerular transcapillary hydraulic pressure (Hostetter 2003; Wolf and Ziyadeh 2007; Ziyadeh and Wolf 2008). This facilitates the development of albumin leakage from the glomerular capillary compartment to Bowman’s space. Many factors, especially angiotensin Ⅱ (AngⅡ), have been implicated as important biologically active agents that cause hyperperfusion and hyperfiltration. (Cooper 2001; Wolf 2004; Forbes et al., 2007). Since Ang II can induce transactivation of EGFR, it has been suggested that blockade of EGFR can reduce Ang Ⅱ-mediated hemodynamic alteration (Krishna et al., 2007; Akhtar et al., 2012).
In diabetic animal models, treatment with EGFR inhibitors results in a significant normalization of the altered vasoconstrictor and vasodilator response without effecting blood glucose levels (Benter et al., 2005; Yousif et al., 2005; Benter et al., 2009; Akhtar et al., 2012; Schreier et al., 2014). Mechanistic studies showed that EGFR inhibition mediated vascular response to different stimuli occurs through reduction of ROS generation in mesenteric resistance arteries (Kassan et al., 2015). This may involve the correction of diabetes-induced reduction in nitric oxide synthase (eNOS) activity and nitric oxide (NO) generation in vascular smooth muscle cells (VSMC) (Benter et al., 2015). Despite disturbed vascular response, EGFR also mediates vascular remolding in diabetes. Akhtar and others found that inhibition of EGFR activation results in a remarkable reduction in blood vessels thickening both in intima and media, and attenuates vascular hyper-responsiveness via ERK1/2-ROCK pathway (Palen and Matrougui 2008; Akhtar et al., 2019). Thus, EGFR inhibition could help restore some vascular endothelial functions, independent of glucose lowering, providing considerable therapeutic strategy for vascular protection in DKD.
Diabetic kidneys are highly sensitive to metabolic alteration. Patients with diabetes mellitus experience chronic hyperglycemia. Glucose was translocated into cells by various transporters including glucose transporter (GLUT)-1, GLUT-4, and sodium-glucose-linked transporters. Excess glucose influx into cells leads to glucose transport along various metabolic pathways, along with the generation of reactive oxygen species (ROS) and advanced glycation end product (AGEs) (Magee et al., 2017). These metabolic derangements induced activation of several signaling pathways related to proliferation and fibrosis, such as the transforming growth factor-β (TGF-β) and the protein kinase C (PKC) signaling pathways (Cooper 2001; Forbes et al., 2007; Kanwar et al., 2011). In addition, researchers found activation of GLUT1 synthesis itself was associated with growth factor upregulation and extracellular matrix secretion (Heilig et al., 2013).
EGFR has been implied in the regulation of the metabolic pathways. In a diabetes model with eNOS knockout, inhibition of EGFR attenuated albuminuria, glomerulosclerosis and tubulointerstitial fibrosis, along with a decreased urinary excretion of F2-isoprostane, a marker of oxidative stress (Portero-Otin et al., 2002; Li et al., 2018). In addition, inhibition of EGFR tyrosine increased glucose tolerance and ameliorated insulin resistance (Li et al., 2018). In STZ-induced diabetes models, EGFR inhibition markedly reduced renal oxidative stress and endoplasmic reticulum stress (ERS), and attenuated renal fibrosis and apoptosis (Xu et al., 2017). In another study, inhibition of EGFR reversed the accumulation of ROS and superoxide levels, probably by improving p-eNOS expression and inhibiting Nox4 expression (Wang et al., 2020). Advanced glycation end product receptors (AGERs) are receptors that mediate AGEs-induced toxicity to cells. AGER can interact with EGFR and mediate oxidative species generation, as characterized by H2O2 formation in mesangial cells and in human embryonic kidney epithelium-like cells (Cai et al., 2006). AGE product precursors could also impair EGFR signaling (Portero-Otin et al., 2002). EGFR activation along with alteration of these metabolic pathways leads to disturbed signaling and mediates kidney injury.
Low-grade systemic inflammation seems to play a critical role in the pathogenesis of DKD (Rivero et al., 2009; Matoba et al., 2019; Vasanth et al., 2019). Scurt et al. found that serum markers of inflammation such as CXCL-16, MCP-1, ANGP-2 could predict the onset of microalbminuria in patients with diabetes mellitus type 2 (Scurt et al., 2019). Other studies indicate that serum IL-18 and TNF-α levels were increased in diabetic patients, especially in those with kidney impairment (Moriwaki et al., 2003; Mora and Navarro 2004). In the onset of diabetic mellitus, excess AGEs and ROS, and activation of several signaling pathways, induced transcription of various adhesion molecules and pro-inflammatory cytokines, and mediated macrophage infiltration and the progression of DKD (Matoba et al., 2019). EGFR inhibition decreases renal T-cell infiltration and islet macrophage infiltration in diabetic glomeruli and the interstitium (Li et al., 2018). Aldosterone-induced proinflammatory gene (CCL-2 and CCL-5) expression in cultured tubular epithelial cells was also shown to occur through the ADAM-17/TGF-α/EGFR pathway (Morgado-Pascual et al., 2015). Zhang et al. also found that treatment with erlotinib, an EGFR inhibitor, reduced kidney macrophage infiltration and oxidative stress in the tubular interstitium (Zhang et al., 2014). Thus, EGFR may be involved in renal inflammatory responses in DKD.
It has been thought that pathologic changes to mesangial cells represent the central feature of glomerulosclerosis in DKD. However, damage to other cell types, including endothelial cells, podocytes, tubular epithelial cells and fibroblasts, also contributes to progression of DKD (Qian et al., 2008; Magee et al., 2017).
Mesangial proliferation and expansion is considered the hallmark of DKD. Mesangial cells are mesenchymal in origin, and are easily activated to undergo proliferation and matrix secretion. Activation of growth factors promotes different signaling pathways that mediate proliferation of mesangial cells. Research by Wu and colleagues indicated that high glucose induced collagen production in mesangial cells through EGFR-mediated activation of the PI3K-Akt signaling pathway (Wu et al., 2007; Wu et al., 2009). Another study also indicated that high glucose induced collagen accumulation in mesangial cells through the Src/TACE/HB-EGF signaling pathway (Taniguchi et al., 2013). EGFR transactivation by high glucose does not require PKC, ROS, or AngII, but HB-EGF release is essential for transactivation of EGFR in mesangial cells to induce mesangial cell proliferation and matrix secretion (Uttarwar et al., 2011).
Endothelial cells can produce NO and regulate platelet adhesion, immune function, control of volume. Endothelial dysfunction is the inability of vasculature to dilate in response to certain stimuli acting on the endothelium (Goligorsky 2015). It is always associated with a deficiency of eNOS activity and NO release (Rivero et al., 2009; Kolluru et al., 2012; Sharma et al., 2012). Wang et al. applied a novel rhynchophylline analogue, Y396, to study the role of EGFR on endothelial function. They found that Y396 inhibits the tyrosine kinase activity of EGFR by directly targeting EGFR and restores endothelium-dependent vascular relaxation without affecting vascular structure (Wang et al., 2020). This effect of EGFR inhibition may be mediated by downregulation of Nox2 and Nox4 expression and ROS suppression (Galan et al., 2012). Similarly, Belmadani et al. also demonstrated that EGFR activation is elevated and induces resistance artery dysfunction and endothelium-dependent relaxation in diabetic mice without interfering blood pressure (Belmadani et al., 2008).
Another mechanism that regulates vascular dysfunction is the endothelial-to- mesenchymal transition (EndoMT) (Piera-Velazquez et al., 2016; Pardali et al., 2017; Piera-Velazquez and Jimenez 2019). EndoMT is known as endothelial cells transition into a mesenchymal cell type. In 2013, LeBleu et al. employed cell linage tracing and found that approximately 10–15% of myofibroblasts were derived from EndoMT (LeBleu et al., 2013). Since then, much attention has been paid to the EndoMT in DKD, especially the renal interstitial fibrosis (Li et al., 2009; Li and Bertram 2010). The role of EGFR in EndoMT in the kidney was less extensively investigated. In an animal model of cardiac fibrosis, Liu et al. observed that EGFR mediated EndoMT promotes several fibrosis-related events post myocardial infarction, (Liu et al., 2020). These events include acquisition by endothelial cells of a spindle-like shape and the ability to migrate. Although glomerular endothelial mitochondrial dysfunction plays a key role in the pathogenesis of DKD as evidenced by podocyte depletion and proteinuria (Qi H. et al., 2017), the role of EGFR in glomerular endothelial cell pathophysiology has not been well investigated.
Podocyte injury is an early event in DKD and is a hallmark of glomerulopathy. Studies have suggested that podocyte injury is associated with the early stage of proteinuria in patients with diabetes (Wolf and Ziyadeh 2007; Bose et al., 2017; Dai et al., 2017). As terminally differentiated cells, podocytes are vulnerable to injury and may not be able to regenerate or repair themselves after injury. They could undergo hypertrophy, epithelial-mesenchymal transition, detachment and apoptosis under certain stimuli, leading to depletion of these cells within the glomerulus, characterized by foot process effacement on biopsy (Han et al., 2017). In DKD, podocytes are involved in the development of glomerular hypertrophy, proteinuria and glomerulosclerosis (Li et al., 2007; Dai et al., 2017; Maestroni and Zerbini 2018) and promote the development of interstitial fibrosis. High concentrations of glucose induce the production of ROS and initiate podocyte apoptosis and podocyte depletion, which may be an early pathological change in DKD (Susztak et al., 2006). EGFR also plays an important role in podocyte injury in DKD. This is evident by the observations that EGFR inhibition led to less podocyte loss in models of diabetic nephropathy while podocyte-specific deletion of EGFR attenuated albuminuria and podocyte loss induced by hyperglycemia (Taniguchi et al., 2013; Li et al., 2018). This may be mediated by activation of TGF-β-SMAD2/3 signaling pathway and enhanced ability of mitochondrial NADPH oxidase to increase ROS production (Chen et al., 2015).
The tubular epithelial cell has been implicated in interstitial fibrosis. Tubular epithelial cells are also vulnerable to pathologic stress due to high glucose levels and can undergo epithelial-mesenchymal-transition and apoptosis (VR et al., 2019). Upon injury, epithelial cells can secrete growth factors and inflammatory cytokines to induce fibroblast activation and renal fibrosis (Sheng and Zhuang 2020). EGFR is highly expressed in proximal tubules. Its transactivation mediates sodium and water transport by regulation of NHE3 and serum glucocorticoid regulated kinase-1 (sgk1) (Panchapakesan et al., 2011). Erlotinib treatment decreased tubular injury and tubulointerstitial fibrosis in db/db mice (Li et al., 2018). EGFR inhibition also attenuated renal tubular epithelial cell proliferation and apoptosis in diabetic rats (Wassef et al., 2004). In addition, erlotinib treatment decreased ER stress and increased autophagy in tubular cells in diabetes (Zhang et al., 2014). The protective effect of some other interventions may be also partly through EGFR. For example, histone deacetylase inhibition can attenuate tubular cell proliferation and early diabetic renal enlargement in response to high glucose by downregulation of EGFR (Gilbert et al., 2011).
EGFR also participated in epithelial-mesenchymal transition (EMT). EMT, a process by which injured renal tubular cells undergo a phenotype change and acquire mesenchymal characteristics, is widely recognized as a critical mediator of fibrogenesis in chronic kidney diseases. EGFR has been thought to mediate EMT. Sustained EGFR activation in the tubule induces epithelial dedifferentiation and cell cycle arrest with an increase in the mesenchymal marker and decreases in the epithelial marker (Overstreet et al., 2017). Administration of CTGF in cultured tubular epithelial cells caused G2/M cell cycle arrest and EMT via EGFR pathways. The cells lost the typical cobblestone pattern and showed a spindle-shaped pattern, along with the elevation of mesenchymal marker and a decrease in epithelial marker. EGFR inhibition attenuated these changes (Rayego-Mateos et al., 2018a). It is possible that the activation of EGFR in diabetic kidney disease may mediate EMT, thus promoting interstitial fibrosis.
Two groups have reported that patients who suffered from non-small-cell lung cancer (NSCLC) experienced an improvement in diabetes after erlotinib (an EGFR inhibitor) treatment (Portero-Otin et al., 2002; Costa and Huberman 2006; Brooks 2012). This improvement may be due to erlotinib-elicited reduction of insulin resistance by inhibition of TNF-α and the T-cell mediated immune response (Brooks 2012). These interesting clinical reports are supported by a striking finding in animal studies showing that erlotinib-treated mice had a relatively slow increase in body weight, a decrease in fasting blood glucose levels, and improved glucose disposition and insulin sensitivity. EGFR inhibition with erlotinib also decreased islet macrophage infiltration and increased autophagy, leading to preservation of pancreatic β-cell function and subsequent improvement of metabolic status. Moreover, EGFR blockade increases circulating levels of the adipokine adiponectin, an adipocyte-derived hormone that has insulin-sensitizing, anti-inflammatory, and kidney-protective effects (Fang et al., 2015; Ding et al., 2016; Li et al., 2018). In addition, treatment with EGFR inhibitor PD153035 reduces low-grade inflammation, macrophage infiltration in adipocytes and improves glucose tolerance and insulin actions (Prada et al., 2009). These studies suggest that EGFR inhibitors may also ameliorate the progression of DKD through improving insulin sensitivity and pancreatic beta cell functions.
Other EGFR tyrosine kinase family members, such as the ErbB2 and ErbB4, may also contribute to the progression of CKD and the pathogenesis of DKD (Zeng et al., 2018a). It relies on heterodimerization with other EGFR family members for signaling. Akhtar et al. investigated the phosphorylation of ErbB2 in diabetes. They found that high glucose exposure enhanced activation of ErbB2, induced vascular dysfunction in VSMCs (Akhtar et al., 2013). ErbB4 expression was increased in the mild fibrotic kidneys, and decreased as fibrosis progressed (Zeng et al., 2018a). ErbB4 suppression significantly attenuated diabetic glomerular injury and albuminuria. Mesangial expansion and sclerosis were reduced with ErbB4 inhibition, as well as STZ-induced podocyte foot process effacement and podocyte loss. TGF-β1 induced MCP-1 expression in podocytes was also suppressed by ErbB4 inhibition (Lee et al., 2017). ErbB4 may also play an important role in glucose homeostasis and lipogenesis. ErbB4 deficiency-related obesity and adipose tissue inflammation may contribute to the development of metabolic syndrome (Zeng et al., 2018b). Some researchers suggest that increased expression of ErbB4 may actually reflect a compensatory effort to prevent development of tubulointerstitial injury (Zeng et al., 2018a).
In the past decades, much attention has been paid on application of tyrosine kinase inhibitors to treat diabetes, including EGFR inhibitors in animal models (Fountas et al., 2015; Malek and Davis 2016). With EGFR inhibitors being extensively used to treat non-small-cell lung cancer (NSCLC), their efficacy in treating CKD and DKD have also been explored in animal models and culture systems. Numerous animal studies and in vitro studies have provided evidence that EGFR inhibition could attenuate or prevent development and progression of DKD. This effect may associate with improvement in β cell function and insulin resistance (Li et al., 2018).
Although there are no clinical trials designed for treatment of human DKD by targeting EGFR, there are two case reports about the application of EGFR inhibitor erlotinib in diabetes. In 2006, Costa et al. observed that administration of erlotinib to a lung cancer patient improved his type 2 diabetes (Costa and Huberman 2006). When given chemotherapy with erlotinib 100 mg daily, the patient felt frequent episodes of hypoglycemia, and her fasting glucose level was stabilized as well. After 8 months, her HbA1c had dropped to 6.5% from 8.2%. In another case report, a 73-year-old man with history of metabolic syndrome, CKD and insulin-dependent type 2 diabetes received erlotinib 150 mg daily after being with metastatic NSCLC. Four weeks after starting erlotinib, the patient's insulin requirement began to decline from 90 units daily. After 10 weeks he was off insulin completely. His HbA1c decreased from to 6.6% from 7.4% in six months. At the same time, an abrupt increase in his serum creatinine slowed down (Brooks 2012).
The first EGFR tyrosine kinase inhibitor (TKI) was approved for clinical use in 2003 and was mostly used in patients with non-small-cell lung cancer (NSCLC) carrying EGFR-activating mutations and in patients with breast and pancreatic cancers. Nevertheless, EGFR-TKIs may cause adverse effects. Since EGFR plays a role in epithelial maintenance, the most frequent and severe side effects are dermatological reactions and diarrhea. Other adverse effects include hepatotoxicity, stomatitis, interstitial lung disease, ocular toxicity and hypomagnesaemia (Shah and Shah 2019; Xu et al., 2019; Huang et al., 2020). Most of the data come from patients with cancer. In addition, seven patients were reported in the literature to develop anti-EGFR-induced nephrotic/nephritic syndrome after 2–24 weeks of therapy. All the cases of kidney disease associated with EGFR inhibitor treatment were identified in patients with cancers and shown by the variable and often prolonged time course between drug exposure (2 weeks–6 months) and clinical recognition of kidney injury (Izzedine and Perazella 2017). Since DKD treatment needs a long-term application of drugs, it is anticipated that use of EGFR-TKI in DKD patients would have additional safety concerns. As such, future clinical observations and/or clinical trials are needed to determine the benefit and side effect of EGFR-TKI in those population of patients.
Nearly one third of patients with diabetes develop DKD, which in many cases progress to end-stage renal disease and the need for dialysis or kidney transplantation. The underlying mechanisms mediating DKD remain incompletely understood. In vitro and in vivo studies have demonstrated that EGFR activation can initiate multiple pathological processes leading to DKD, such as hemodynamic and metabolic alterations, chronic inflammation, and multicellular dysfunction (Figure 1 and Table 1). Given the importance of EGFR in mediating the pathogenesis of DKD, much work has gone into studying whether EGR inhibition could slow or stop the development of DKD. EGFR inhibitors have been extensively used to treat various tumors, in particular lung carcinoma. This suggests an interesting possibility that EGFR inhibitors may be repurposed as a treatment for DKD and CKD caused by other etiologies. Nevertheless, beside their benefit effects, long-term use of EGFR inhibitors may result in some adverse effects including kidney problems. Most of side effects of EGFR inhibitors in patients with tumor are tolerable. But it is uncertain whether they are also applicable and tolerable in patients with CKD, in particular DKD. Therefore, clinical trials are needed to determine the efficacy and adverse effects of EGFR inhibitors in patients with DKD.
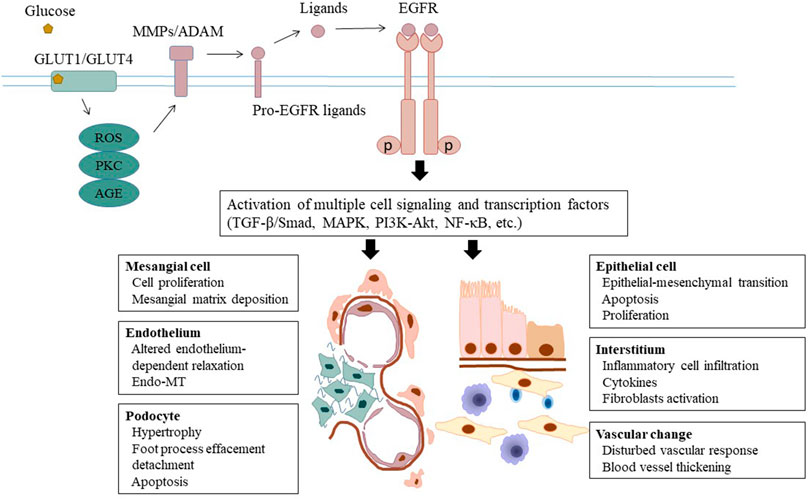
FIGURE 1. Epidermal growth factor receptor and the pathogenesis of DKD. Under the circumstance of diabetes, glucose triggers activation of ADAMs through several second messengers including ROS and protein kinase C. ADAM-mediated shedding of ligands induces phosphorylation of EGFR and subsequent activation of cell several signaling pathways and transcription factors. As a result, multiple cellular and tissue responses are initiated and implicated in the pathogenesis of DKD progression, including hemodynamic alteration, metabolic disturbance, inflammatory response and parenchymal cellular dysfunction. EGFR, epidermal growth factor receptor; DKD, diabetic kidney disease; ADAM, a disintegrin and metalloprotease; ROS, reactive oxidative species.
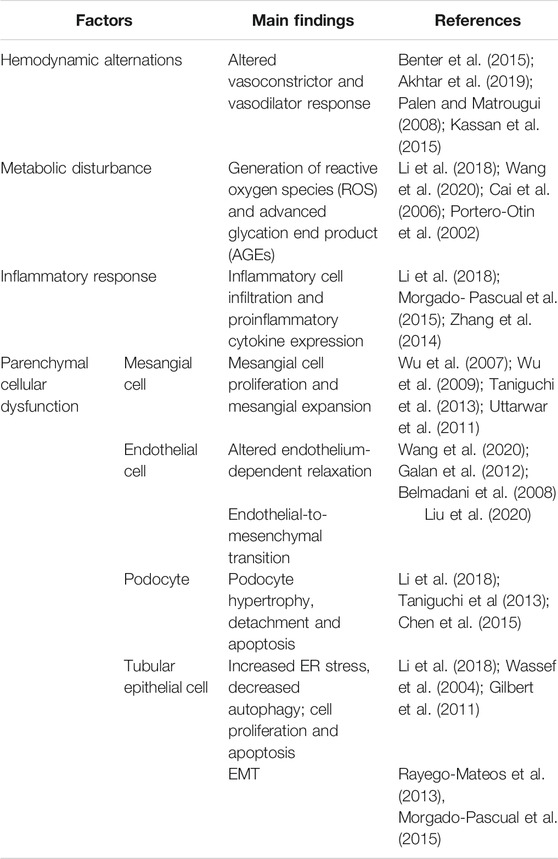
TABLE 1. EGFR and the pathogenesis of DKD.
LS drafted the article, and GB and SZ edited the manuscript. All the authors reviewed the manuscript and approved is for publication.
This work was supported by the National Natural Science Foundation of China (81670623 and 81830021 to SZ, 82000645 to LS), the Branch Grant of National Key Grants of the Ministry of Science and Technology (2018YFA0108802 to SZ) and the US National Institutes of Health (1R01DK113256-01A1 to SZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Advani, A., Wiggins, K. J., Cox, A. J., Zhang, Y., Gilbert, R. E., and Kelly, D. J. (2011). Inhibition of the epidermal growth factor receptor preserves podocytes and attenuates albuminuria in experimental diabetic nephropathy. Nephrology. 16 (6), 573–581. doi:10.1111/j.1440-1797.2011.01451.x
Akhtar, S., Yousif, M. H., Dhaunsi, G. S., Chandrasekhar, B., Al-Farsi, O., and Benter, I. F. (2012). Angiotensin-(1-7) inhibits epidermal growth factor receptor transactivation via a Mas receptor-dependent pathway. Br. J. Pharmacol. 165 (5), 1390–1400. doi:10.1111/j.1476-5381.2011.01613.x
Akhtar, S., Yousif, M. H., Dhaunsi, G. S., Sarkhouh, F., Chandrasekhar, B., Attur, S., and Benter, I. F. (2013). Activation of ErbB2 and downstream signalling via Rho kinases and ERK1/2 contributes to diabetes-induced vascular dysfunction. PloS One. 8 (6), e67813), doi:10.1371/journal.pone.0067813
Akhtar, S., Chandrasekhar, B., Yousif, M. H., Renno, W., Benter, I. F., and El-Hashim, A. Z. (2019). Chronic administration of nano-sized PAMAM dendrimers in vivo inhibits EGFR-ERK1/2-ROCK signaling pathway and attenuates diabetes-induced vascular remodeling and dysfunction. Nanomedicine. 18, 78–89. doi:10.1016/j.nano.2019.02.012
Belmadani, S., Palen, D. I., Gonzalez-Villalobos, R. A., Boulares, H. A., and Matrougui, K. (2008). Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 57 (6), 1629–1637. doi:10.2337/db07-0739
Benter, I. F., Yousif, M. H., Griffiths, S. M., Benboubetra, M., and Akhtar, S. (2005). Epidermal growth factor receptor tyrosine kinase-mediated signalling contributes to diabetes-induced vascular dysfunction in the mesenteric bed. Br. J. Pharmacol. 145 (6), 829–836. doi:10.1038/sj.bjp.0706238
Benter, I. F., Benboubetra, M., Hollins, A. J., Yousif, M. H., Canatan, H., and Akhtar, S. (2009). Early inhibition of EGFR signaling prevents diabetes-induced up-regulation of multiple gene pathways in the mesenteric vasculature. Vasc. Pharmacol. 51 (4), 236–245. doi:10.1016/j.vph.2009.06.008
Benter, I. F., Sarkhou, F., Al-Khaldi, A. T., Chandrasekhar, B., Attur, S., Dhaunsi, G. S., Yousif, M. H., and Akhtar, S. (2015). The dual targeting of EGFR and ErbB2 with the inhibitor Lapatinib corrects high glucose-induced apoptosis and vascular dysfunction by opposing multiple diabetes-induced signaling changes. J. Drug Target. 23 (6), 506–518. doi:10.3109/1061186X.2015.1057150
Bose, M., Almas, S., and Prabhakar, S. (2017). Wnt signaling and podocyte dysfunction in diabetic nephropathy. J. Invest. Med. 65 (8), 1093–1101. doi:10.1136/jim-2017-000456
Brooks, M. B. (2012). Erlotinib appears to produce prolonged remission of insulin-requiring type 2 diabetes associated with metabolic syndrome and chronic kidney disease. Br. J. Diabetes Vasc. Dis. 12 (2), 87–90. doi:10.1177/1474651412442694
Cai, W., He, J. C., Zhu, L., Lu, C., and Vlassara, H. (2006). Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc. Natl. Acad. Sci. USA. 103 (37), 13801–13806. doi:10.1073/pnas.0600362103
Chen, J., Chen, J. K., Nagai, K., Plieth, D., Tan, M., Lee, T. C., Threadgill, D. W., Neilson, E. G., and Harris, R. C. (2012). EGFR signaling promotes TGFβ-dependent renal fibrosis. J. Am. Soc. Nephrol. 23 (2), 215–224. doi:10.1681/ASN.2011070645
Chen, J., Chen, J. K., and Harris, R. C. (2015). EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 26 (5), 1115–1125. doi:10.1681/ASN.2014020192
Cooper, M. E. (2001). Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia. 44 (11), 1957–1972. doi:10.1007/s001250100000
Costa, D. B., and Huberman, M. S. (2006). Improvement of type 2 diabetes in a lung cancer patient treated with Erlotinib. Diabetes Care. 29 (7), 1711. doi:10.2337/dc06-0558
Dai, H., Liu, Q., and Liu, B. (2017). Research progress on mechanism of podocyte depletion in diabetic nephropathy. J Diabetes Res. 2017, 2615286. doi:10.1155/2017/2615286
Ding, W., Cai, Y., Wang, W., Ji, L., Dong, Y., Zhang, X., Su, M., Liu, J., Lu, G., and Zhang, X. (2016). Adiponectin protects the kidney against chronic intermittent hypoxia-induced injury through inhibiting endoplasmic reticulum stress. Sleep Breath. 20 (3), 1069–1074. doi:10.1007/s11325-016-1321-4
Fang, F., Bae, E. H., Hu, A., Liu, G. C., Zhou, X., Williams, V., Maksimowski, N., Lu, C., Konvalinka, A., John, R., and Scholey, J. W. (2015). Deletion of the gene for adiponectin accelerates diabetic nephropathy in the Ins2 (+/C96Y) mouse. Diabetologia. 58 (7), 1668–1678. doi:10.1007/s00125-015-3605-9
Forbes, J. M., Fukami, K., and Cooper, M. E. (2007). Diabetic nephropathy: where hemodynamics meets metabolism. Exp. Clin. Endocrinol. Diabetes. 115 (2), 69–84. doi:10.1055/s-2007-949721
Forrester, S. J., Kawai, T., O'Brien, S., Thomas, W., Harris, R. C., and Eguchi, S. (2016). Epidermal growth factor receptor transactivation: mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu. Rev. Pharmacol. Toxicol. 56, 627–653. doi:10.1146/annurev-pharmtox-070115-095427
Fountas, A., Diamantopoulos, L. N., and Tsatsoulis, A. (2015). Tyrosine kinase inhibitors and diabetes: a novel treatment paradigm?. Trends Endocrinol Metab. 26 (11), 643–656. doi:10.1016/j.tem.2015.09.003
Galán, M., Kassan, M., Choi, S. K., Partyka, M., Trebak, M., Henrion, D., and Matrougui, K. (2012). A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 60 (1), 71–80. doi:10.1161/HYPERTENSIONAHA.112.192500
Gesualdo, L., Di Paolo, S., Calabró, A., Milani, S., Maiorano, E., Ranieri, E., Pannarale, G., and Schena, F. P. (1996). Expression of epidermal growth factor and its receptor in normal and diseased human kidney: an immunohistochemical and in situ hybridization study. Kidney Int. 49 (3), 656–665. doi:10.1038/ki.1996.94
Gilbert, R. E., Huang, Q., Thai, K., Advani, S. L., Lee, K., Yuen, D. A., Connelly, K. A., and Advani, A. (2011). Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 79 (12), 1312–1321. doi:10.1038/ki.2011.39
Goligorsky, M. S. (2015). Pathogenesis of endothelial cell dysfunction in chronic kidney disease: a retrospective and what the future may hold. Kidney Res Clin Pract. 34 (2), 76–82. doi:10.1016/j.krcp.2015.05.003
Han, Q., Zhu, H., Chen, X., and Liu, Z. (2017). Non-genetic mechanisms of diabetic nephropathy. Front. Med. 11 (3), 319–332. doi:10.1007/s11684-017-0569-9
Harskamp, L. R., Gansevoort, R. T., van Goor, H., and Meijer, E. (2016). The epidermal growth factor receptor pathway in chronic kidney diseases. Nat. Rev. Nephrol. 12 (8), 496–506. doi:10.1038/nrneph.2016.91
Heilig, C. W., Deb, D. K., Abdul, A., Riaz, H., James, L. R., Salameh, J., and Nahman, N. S. (2013). GLUT1 regulation of the pro-sclerotic mediators of diabetic nephropathy. Am. J. Nephrol. 38 (1), 39–49. doi:10.1159/000351989
Higashiyama, S., Iwabuki, H., Morimoto, C., Hieda, M., Inoue, H., and Matsushita, N. (2008). Membrane-anchored growth factors, the epidermal growth factor family: beyond receptor ligands. Canc. Sci. 99 (2), 214–220. doi:10.1111/j.1349-7006.2007.00676.x
Hostetter, T. H. (2003). Hyperfiltration and glomerulosclerosis. Semin. Nephrol. 23 (2), 194–199. doi:10.1053/anep.2003.50017
Huang, J., Meng, L., Yang, B., Sun, S., Luo, Z., and Chen, H. (2020). Safety Profile of epidermal growth factor receptor tyrosine kinase inhibitors: a disproportionality analysis of FDA adverse event reporting system. Sci. Rep. 10 (1), 4803. doi:10.1038/s41598-020-61571-5
Izzedine, H, and Perazella, M. A. (2017). Adverse kidney effects of epidermal growth factor receptor inhibitors. Nephrol. Dial. Transplant. 32 (7), 1089–1097. doi:10.1093/ndt/gfw467
Kanwar, Y. S., Sun, L., Xie, P., Liu, F. Y., and Chen, S. (2011). A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 6, 395–423. doi:10.1146/annurev.pathol.4.110807.092150
Kassan, M., Ait-Aissa, K., Ali, M., Trebak, M., and Matrougui, K. (2015). Augmented EGF receptor tyrosine kinase activity impairs vascular function by NADPH oxidase-dependent mechanism in type 2 diabetic mouse. Biochim. Biophys. Acta. 1853 (10 Pt A), 2404–2410. doi:10.1016/j.bbamcr.2015.05.032
Kolluru, G. K., Bir, S. C., and Kevil, C. G. (2012). Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. 2012, 918267. doi:10.1155/2012/918267
Konishi, A, and Berk, B. C. (2003). Epidermal growth factor receptor transactivation is regulated by glucose in vascular smooth muscle cells. J. Biol. Chem. 278 (37), 35049–35056. doi:10.1074/jbc.M304913200
Koya, D. (2015). Epidermal growth factor receptor signaling and the progression of diabetic nephropathy. J. Diabetes Investig. 6 (5), 519–521. doi:10.1111/jdi.12317
Krishna, S. B., Alfonso, L. F., Thekkumkara, T. J., Abbruscato, T. J., and Bhat, G. J. (2007). Angiotensin II induces phosphorylation of glucose-regulated protein-75 in WB rat liver cells. Arch. Biochem. Biophys. 457 (1), 16–28. doi:10.1016/j.abb.2006.10.011
LeBleu, V. S., Taduri, G., O'Connell, J., Teng, Y., Cooke, V. G., Woda, C., Sugimoto, H., and Kalluri, R. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19 (8), 1047–1053. doi:10.1038/nm.3218
Lee, H. W., Khan, S. Q., Khaliqdina, S., Altintas, M. M., Grahammer, F., Zhao, J. L., Koh, K. H., Tardi, N. J., Faridi, M. H., Geraghty, T., Cimbaluk, D. J., Susztak, K., Moita, L. F., Baltimore, D., Tharaux, P. L., Huber, T. B., Kretzler, M., Bitzer, M., Reiser, J., and Gupta, V. (2017). Absence of miR-146a in podocytes increases risk of diabetic glomerulopathy via up-regulation of ErbB4 and Notch-1. J. Biol. Chem. 292 (2), 732–747. doi:10.1074/jbc.M116.753822
Li, J., and Bertram, J. F. (2010). Review: endothelial-myofibroblasts transition, a new player in diabetic renal fibrosis. Nephrology. 15 (5), 507–512. doi:10.1111/j.1440-1797.2010.01319.x
Li, J. J., Kwak, S. J., Jung, D. S., Kim, J. J., Yoo, T. H., et al. (2007). Podocyte biology in diabetic nephropathy. Kidney Int. Suppl. Suppl (106), S36–S42. doi:10.1038/sj.ki.5002384
Li, J., Qu, X., and Bertram, J. F. (2009). Endothelial-myofibroblasts transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 175 (4), 1380–1388. doi:10.2353/ajpath.2009.090096
Li, Z., Li, Y., Overstreet, J. M., Chung, S., Niu, A., Fan, X., Wang, S., Wang, Y., Zhang, M. Z., and Harris, R. C. (2018). Inhibition of epidermal growth factor receptor activation is associated with improved diabetic nephropathy and insulin resistance in type 2 diabetes. Diabetes. 67 (9), 1847–1857. doi:10.2337/db17-1513
Li, R., Uttarwar, L., Gao, B., Charbonneau, M., Shi, Y., Chan, J. S., et al. (2015). High glucose up-regulates ADAM17 through HIF-1α in mesangial cells. J. Biol. Chem. 290 (35), 21603–21614. doi:10.1074/jbc.M115.651604
Li, T., Yang, G. M., Zhu, Y., Wu, Y., Chen, X. Y., et al. (2015). Diabetes and hyperlipidemia induce dysfunction of VSMCs: contribution of the metabolic inflammation/miRNA pathway. Am. J. Physiol. Endocrinol. Metab. 308 (4), E257–E269. doi:10.1152/ajpendo.00348.2014
Liu, L., Song, S., Zhang, Y. P., Wang, D., Zhou, Z., et al. (2020). Amphiregulin promotes cardiac fibrosis post myocardial infarction by inducing the endothelial-mesenchymal transition via the EGFR pathway in endothelial cells. Exp. Cell Res. 390 (2), 111950. doi:10.1016/j.yexcr.2020.111950
Maestroni, S, and Zerbini, G. (2018). Glomerular endothelial cells versus podocytes as the cellular target in diabetic nephropathy. Acta Diabetol. 55 (11), 1105–1111. doi:10.1007/s00592-018-1211-2
Magee, C., Grieve, D. J., Watson, C. J., and Brazil, D. P. (2017). Diabetic nephropathy: a tangled web to unweave. Cardiovasc. Drugs Ther. 31 (5–6), 579–592. doi:10.1007/s10557-017-6755-9
Malek, R., and Davis, S. N. (2016). Tyrosine kinase inhibitors under investigation for the treatment of type II diabetes. Expet Opin. Invest. Drugs. 25 (3), 287–296. doi:10.1517/13543784.2016.1142531
Matoba, K., Takeda, Y., Nagai, Y., Kawanami, D., Utsunomiya, K., and Nishimura, R. (2019). Unraveling the role of inflammation in the pathogenesis of diabetic kidney disease. Int. J. Mol. Sci. 20 (14), 3393. doi:10.3390/ijms20143393
Matrougui, K. (2010). Diabetes and microvascular pathophysiology: role of epidermal growth factor receptor tyrosine kinase. Diabetes Metab. Res. Rev. 26 (1), 13–16. doi:10.1002/dmrr.1050
Miyazawa, T., Zeng, F., Wang, S., Fan, X., Cheng, H., Yang, H., et al. (2013). Low nitric oxide bioavailability upregulates renal heparin binding EGF-like growth factor expression. Kidney Int. 84 (6), 1176–1188. doi:10.1038/ki.2013.214
Mora, C., and Navarro, J. F. (2004). Inflammation and pathogenesis of diabetic nephropathy. Metab. Clin. Exp. 53 (2), 265–267. doi:10.1016/j.metabol.2003.11.005
Morgado-Pascual, J. L., Rayego-Mateos, S., Valdivielso, J. M., Ortiz, A., Egido, J., and Ruiz-Ortega, M. (2015). Paricalcitol inhibits aldosterone-induced proinflammatory factors by modulating epidermal growth factor receptor pathway in cultured tubular epithelial cells. BioMed. Res. Int. 2015, 783538. doi:10.1155/2015/783538
Moriwaki, Y., Yamamoto, T., Shibutani, Y., Aoki, E., Tsutsumi, Z., Takahashi, S., et al. (2003). Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metab. Clin. Exp. 52 (5), 605–608. doi:10.1053/meta.2003.50096
Ohtsu, H., Dempsey, P. J., Frank, G. D., Brailoiu, E., Higuchi, S., Suzuki, H., et al. (2006). ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 26 (9), e133–7. doi:10.1161/01.ATV.0000236203.90331.d0
Overstreet, J. M., Wang, Y., Wang, X., Niu, A., Gewin, L. S., Yao, B., et al. (2017). Selective activation of epidermal growth factor receptor in renal proximal tubule induces tubulointerstitial fibrosis. Faseb. J. 31 (10), 4407–4421. doi:10.1096/fj.201601359RR
Palen, D. I., and Matrougui, K. (2008). Role of elevated EGFR phosphorylation in the induction of structural remodelling and altered mechanical properties of resistance artery from type 2 diabetic mice. Diabetes Metab. Res Rev. 24 (8), 651–656. doi:10.1002/dmrr.905
Panchapakesan, U., Pollock, C., and Saad, S. (2011). Renal epidermal growth factor receptor: its role in sodium and water homeostasis in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 38 (2), 84–88. doi:10.1111/j.1440-1681.2010.05472.x
Papadopoulou-Marketou, N., Chrousos, G. P., and Kanaka-Gantenbein, C. (2017). Diabetic nephropathy in type 1 diabetes: a review of early natural history, pathogenesis, and diagnosis. Diabetes Metab. Res Rev. 33 (2). doi:10.1002/dmrr.2841
Pardali, E., Sanchez-Duffhues, G., Gomez-Puerto, M. C., and Ten Dijke, P. (2017). TGF-β-Induced endothelial-mesenchymal transition in fibrotic diseases. Int. J. Mol. Sci. 18 (10). doi:10.3390/ijms18102157
Perlman, A. S., Chevalier, J. M., Wilkinson, P., Liu, H., Parker, T., Levine, D. M., et al. (2015). Serum inflammatory and immune mediators are elevated in early stage diabetic nephropathy. Ann. Clin. Lab. Sci. 45 (3), 256–263.
Piera-Velazquez, S., and Jimenez, S. A. (2019). Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 99 (2), 1281–1324. doi:10.1152/physrev.00021.2018
Piera-Velazquez, S., Mendoza, F. A., and Jimenez, S. A. (2016). Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J. Clin. Med. 5 (4). doi:10.3390/jcm5040045
Portero-Otín, M., Pamplona, R., Bellmunt, M. J., Ruiz, M. C., Prat, J., Salvayre, R., and Nègre-Salvayre, A. (2002). Advanced glycation end product precursors impair epidermal growth factor receptor signaling. Diabetes. 51 (5), 1535–1542. doi:10.2337/diabetes.51.5.1535
Portik-Dobos, V., Harris, A. K., Song, W., Hutchinson, J., Johnson, M. H., Imig, J. D., et al. (2006). Endothelin antagonism prevents early EGFR transactivation but not increased matrix metalloproteinase activity in diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290 (2), R435–R441. doi:10.1152/ajpregu.00300.2005
Prada, P. O., Ropelle, E. R., Mourão, R. H., de Souza, C. T., Pauli, J. R., Cintra, D. E., et al. (2009). EGFR tyrosine kinase inhibitor (PD153035) improves glucose tolerance and insulin action in high-fat diet-fed mice. Diabetes. 58 (12), 2910–2919. doi:10.2337/db08-0506
Qi, C, Mao, X., Zhang, Z., and Wu, H. (2017). Classification and differential diagnosis of diabetic nephropathy. J. Diabetes Res. 2017, 8637138. doi:10.1155/2017/8637138
Qi, H, Casalena, G., Shi, S., Yu, L., Ebefors, K., Sun, Y., et al. (2017). Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes. 66 (3), 763–778. doi:10.2337/db16-0695
Qian, Y., Feldman, E., Pennathur, S., Kretzler, M., and Brosius, F. C. (2008). From fibrosis to sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes. 57 (6), 1439–1445. doi:10.2337/db08-0061
Rayego-Mateos, S., Rodrigues-Díez, R., Morgado-Pascual, J. L., Rodrigues Díez, R. R., Mas, S., Lavoz, C., et al. (2013). Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J. Mol. Cell Biol. 5 (5), 323–335. doi:10.1093/jmcb/mjt030
Rayego-Mateos, S., Morgado-Pascual, J. L., Rodrigues-Diez, R. R., Rodrigues-Diez, R., Falke, L. L., Mezzano, S., et al. (2018a). Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 244 (2), 227–241. doi:10.1002/path.5007
Rayego-Mateos, S., Rodrigues-Diez, R., Morgado-Pascual, J. L., Valentijn, F., Valdivielso, J. M., Goldschmeding, R., et al. (2018b). Role of epidermal growth factor receptor (EGFR) and its ligands in kidney inflammation and damage. Mediat. Inflamm. 2018, 8739473. doi:10.1155/2018/8739473
Rivero, A., Mora, C., Muros, M., García, J., Herrera, H., and Navarro-González, J. F. (2009). Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin. Sci. 116 (6), 479–492. doi:10.1042/CS20080394
Saad, S., Stevens, V. A., Wassef, L., Poronnik, P., Kelly, D. J., Gilbert, R. E., and Pollock, C. A. (2005). High glucose transactivates the EGF receptor and up-regulates serum glucocorticoid kinase in the proximal tubule. Kidney Int. 68 (3), 985–997. doi:10.1111/j.1523-1755.2005.00492.x
Schneider, M. R., and Wolf, E. (2009). The epidermal growth factor receptor ligands at a glance. J. Cell. Physiol. 218 (3), 460–466. doi:10.1002/jcp.21635
Schreier, B., Gekle, M., and Grossmann, C. (2014). Role of epidermal growth factor receptor in vascular structure and function. Curr. Opin. Nephrol. Hypertens. 23 (2), 113–121. doi:10.1097/01.mnh.0000441152.62943.29
Scurt, F. G., Menne, J., Brandt, S., Bernhardt, A., Mertens, P. R., Haller, H., and Chatzikyrkou, C. (2019). Systemic inflammation precedes microalbuminuria in diabetes. Kidney Int Rep. 4 (10), 1373–1386. doi:10.1016/j.ekir.2019.06.005
Shah, R. R., and Shah, D. R. (2019). Safety and tolerability of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in oncology. Drug Saf. 42 (2), 181–198. doi:10.1007/s40264-018-0772-x
Sharma, A., Bernatchez, P. N., and de Haan, J. B. (2012). Targeting endothelial dysfunction in vascular complications associated with diabetes. Int J Vasc Med, 2012, 750126. doi:10.1155/2012/750126
Sheng, L, and Zhuang, S. (2020). New insights into the role and mechanism of partial epithelial-mesenchymal transition in kidney fibrosis. Front. Physiol. 11, 569322. doi:10.3389/fphys.2020.569322
Sheng, L., Yang, M., Ding, W., Zhang, M., Niu, J., Qiao, Z., et al. (2016). Epidermal growth factor receptor signaling mediates aldosterone-induced profibrotic responses in kidney. Exp. Cell Res. 346 (1), 99–110. doi:10.1016/j.yexcr.2016.06.009
Sulaiman, M. K. (2019). Diabetic nephropathy: recent advances in pathophysiology and challenges in dietary management. Diabetol. Metab. Syndrome. 11, 7. doi:10.1186/s13098-019-0403-4
Susztak, K., Raff, A. C., Schiffer, M., and Böttinger, E. P. (2006). Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 55 (1), 225–233. doi:10.2337/diabetes.55.01.06.db05-0894
Taniguchi, K., Xia, L., Goldberg, H. J., Lee, K. W., Shah, A., Stavar, L., et al. (2013). Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 62 (11), 3874–3886. doi:10.2337/db12-1010
Tung, C. W., Hsu, Y. C., Shih, Y. H., Chang, P. J., and Lin, C. L. (2018). Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology. 23 (Suppl. 4), 32–37. doi:10.1111/nep.13451
Uttarwar, L., Peng, F., Wu, D., Kumar, S., Gao, B., Ingram, A. J., and Krepinsky, J. C. (2011). HB-EGF release mediates glucose-induced activation of the epidermal growth factor receptor in mesangial cells. Am. J. Physiol. Ren. Physiol. 300 (4), F921–F931. doi:10.1152/ajprenal.00436.2010
Vasanth, V. R., Tan, S. H., Candasamy, M., and Bhattamisra, S. K. (2019). Diabetic nephropathy: an update on pathogenesis and drug development. Diabetes Metab. Syndr. 13 (1), 754–762. doi:10.1016/j.dsx.2018.11.054
Wang, Z. J., Chang, L. L., Wu, J., Pan, H. M., Zhang, Q. Y., Wang, M. J., et al. (2020). A novel rhynchophylline analog, Y396, inhibits endothelial dysfunction induced by oxidative stress in diabetes through epidermal growth factor receptor. Antioxidants Redox Signal. 32 (11), 743–765. doi:10.1089/ars.2018.7721
Wassef, L., Kelly, D. J., and Gilbert, R. E. (2004). Epidermal growth factor receptor inhibition attenuates early kidney enlargement in experimental diabetes. Kidney Int. 66 (5), 1805–1814. doi:10.1111/j.1523-1755.2004.00955.x
Wolf, G., and Ziyadeh, F. N. (2007). Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron. Physiol. 106 (2), p26–31. doi:10.1159/000101797
Wolf, G. (2004). New insights into the pathophysiology of diabetic nephropathy: from haemodynamics to molecular pathology. Eur. J. Clin. Invest. 34 (12), 785–796. doi:10.1111/j.1365-2362.2004.01429.x
Wu, D., Peng, F., Zhang, B., Ingram, A. J., Gao, B., and Krepinsky, J. C. (2007). Collagen I induction by high glucose levels is mediated by epidermal growth factor receptor and phosphoinositide 3-kinase/Akt signalling in mesangial cells. Diabetologia. 50 (9), 2008–2018. doi:10.1007/s00125-007-0721-1
Wu, D., Peng, F., Zhang, B., Ingram, A. J., Kelly, D. J., Gilbert, R. E., et al. (2009). EGFR-PLCgamma1 signaling mediates high glucose-induced PKCbeta1-Akt activation and collagen I upregulation in mesangial cells. Am. J. Physiol. Ren. Physiol. 297 (3), F822–F834. doi:10.1152/ajprenal.00054.2009
Xu, Z., Zhao, Y., Zhong, P., Wang, J., Weng, Q., Qian, Y., et al. (2017). EGFR inhibition attenuates diabetic nephropathy through decreasing ROS and endoplasmic reticulum stress. Oncotarget. 8 (20), 32655–32667. doi:10.18632/oncotarget.15948
Xu, R., Shao, H., Zhu, J., Ju, Q., and Shi, H. (2019). Combination strategies based on epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors for cancer patients: pooled analysis and subgroup analysis of efficacy and safety. Medicine. 98 (13), e14135. doi:10.1097/MD.0000000000014135
Yousif, M. H., Benter, I. F., and Akhtar, S. (2005). The role of tyrosine kinase-mediated pathways in diabetes-induced alterations in responsiveness of rat carotid artery. Auton Autacoid Pharmacol. 25 (2), 69–78. doi:10.1111/j.1474-8673.2004.00333.x
Zeng, F., Miyazawa, T., Kloepfer, L. A., and Harris, R. C. (2018a). ErbB4 deletion accelerates renal fibrosis following renal injury. Am. J. Physiol. Ren. Physiol. 314 (5), F773–F787. doi:10.1152/ajprenal.00260.2017
Zeng, F., Wang, Y., Kloepfer, L. A., Wang, S., and Harris, R. C. (2018b). ErbB4 deletion predisposes to development of metabolic syndrome in mice. Am. J. Physiol. Endocrinol. Metab. 315 (4), E583–E593. doi:10.1152/ajpendo.00166.2018
Zhang, M. Z., Wang, Y., Paueksakon, P., and Harris, R. C. (2014). Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 63 (6), 2063–2072. doi:10.2337/db13-1279
Keywords: Epidermal growth factor receptor, diabetic nephropathy, hemodynamic alternation, metabolic disturbance, inflammation, multicellular dysfunction
Citation: Sheng L, Bayliss G and Zhuang S (2021) Epidermal Growth Factor Receptor: A Potential Therapeutic Target for Diabetic Kidney Disease. Front. Pharmacol. 11:598910. doi: 10.3389/fphar.2020.598910
Received: 25 August 2020; Accepted: 30 November 2020;
Published: 26 January 2021.
Edited by:
Valeria Mas, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Onkar Prakash Kulkarni, Birla Institute of Technology and Science, IndiaCopyright © 2021 Sheng, Bayliss and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shougang Zhuang, c3podWFuZ0BsaWZlc3Bhbi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.