
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 23 September 2020
Sec. Neuropharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.588160
 Paola Devoto1,2*
Paola Devoto1,2* Claudia Sagheddu1
Claudia Sagheddu1 Michele Santoni1
Michele Santoni1 Giovanna Flore3Pierluigi Saba1
Giovanna Flore3Pierluigi Saba1 Marco Pistis1,4
Marco Pistis1,4 Gian Luigi Gessa2,4
Gian Luigi Gessa2,4Previous results indicate that dopamine (DA) release in the medial prefrontal cortex (mPFC) is modified by α2 adrenoceptor- but not D2 DA receptor- agonists and antagonists, suggesting that DA measured by microdialysis in the mPFC originates from noradrenergic terminals. Accordingly, noradrenergic denervation was found to prevent α2-receptor-mediated rise and fall of extracellular DA induced by atipamezole and clonidine, respectively, in the mPFC. The present study was aimed to determine whether DA released by dopaminergic terminals in the mPFC is not detected by in vivo microdialysis because is readily taken up by norepinephrine transporter (NET). Accordingly, the D2-antagonist raclopride increased the electrical activity of DA neurons in the ventral tegmental area (VTA) and enhanced extracellular DOPAC but failed to modify DA in the mPFC. However, in rats whose NET was either inactivated by nisoxetine or eliminated by noradrenergic denervation, raclopride still elevated extracellular DOPAC and activated dopaminergic activity, but also increased DA. Conversely, the D2-receptor agonist quinpirole reduced DOPAC but failed to modify DA in the mPFC in control rats. However, in rats whose NET was eliminated by noradrenergic denervation or inhibited by locally perfused nisoxetine, quinpirole maintained its ability to reduce DOPAC but acquired that of reducing DA. Moreover, raclopride and quinpirole, when locally perfused into the mPFC of rats subjected to noradrenergic denervation, were able to increase and decrease, respectively, extracellular DA levels, while being ineffective in control rats. Transient inactivation of noradrenergic neurons by clonidine infusion into the locus coeruleus, a condition where NET is preserved, was found to reduce extracellular NE and DA in the mPFC, whereas noradrenergic denervation, a condition where NET is eliminated, almost totally depleted extracellular NE but increased DA. Both transient inactivation and denervation of noradrenergic neurons were found to reduce the number of spontaneously active DA neurons and their bursting activity in the VTA. The results indicate that DA released in the mPFC by dopaminergic terminals is not detected by microdialysis unless DA clearance from extracellular space is inactivated. They support the hypothesis that noradrenergic terminals are the main source of DA measured by microdialysis in the mPFC during physiologically relevant activities.
Dopaminergic and noradrenergic projections from the ventral tegmental area (VTA) and the locus coeruleus (LC), respectively, converge to the prefrontal cortex (PFC), where they play a key role in the cognitive and motivational functions of this region (Descarries et al., 1987; Seguela et al., 1990; Phillips et al., 2004; Xing et al., 2016). Accordingly, impairments of catecholamine transmission in the PFC have been implicated in the cognitive and emotional deficits in schizophrenia (Bird et al., 1979; van Kammen and Kelley, 1991; Tassin, 1992; Slifstein and Abi-Dargham, 2017; Rao et al., 2019), depressive disorders (Bunney and Davis, 1965; Schildkraut, 1965; Kapur and Mann, 1992; Dunlop and Nemeroff, 2007; El Mansari et al., 2010), substance use disorder (Koob and Nestler, 1997; Wise, 1998; Volkow et al., 2007; Weinshenker and Schroeder, 2007; Aston-Jones and Kalivas, 2008) and attention deficit/hyperactivity disorder (Bymaster et al., 2002; Engert and Pruesner, 2008; Holzer et al., 2013; Gold et al., 2014).
Conversely, the efficacy of atypical antipsychotics, including clozapine and olanzapine, in ameliorating the negative symptoms and cognitive deficits in schizophrenia has been attributed to their ability to increase norepinephrine (NE) and dopamine (DA) release in the PFC (Meltzer and Stahl, 1976; Moghaddam and Bunney, 1990; Lidow et al., 1998; Melis et al., 1999; Gessa et al., 2000; Devoto et al., 2003).
Different pharmacological treatments, such as psychostimulants (Darracq et al., 1998), antipsychotic drugs (Westerink et al., 1998; Devoto et al., 2003), fluoxetine (Bymaster et al., 2002), stress (Finlay et al., 1995; Feenstra, 2000), aversive and rewarding conditioned and unconditioned stimuli (see Fenstra, 2000) have been shown to produce concomitant and parallel changes in extracellular NE and DA in the PFC, suggesting a cross-talk between dopaminergic and noradrenergic systems to modulate the activity of target neurons in the PFC (Guiard et al., 2008; Masana et al., 2011; Chandler et al., 2014; Xing et al., 2016; Ranjbar-Slamloo and Fazlali, 2020).
A key role has been attributed to the NE transporter (NET) and α2-adrenoceptors in governing the cross-talk between dopaminergic and noradrenergic inputs to the PFC. Thus, in view of the low level of DA transporter (Sesack et al., 1998) and abundance of NET in the PFC (Freed et al., 1995; Miner et al., 2003), it has been suggested that DA uptake from extracellular spaces is carried out almost exclusively by NET (Carboni et al., 1990; Pozzi et al., 1994; Gresch et al., 1995; Moron et al., 2002), and that DA competes with NE for the same transporter for which DA has the same affinity as NE (Raiteri et al., 1977; Horn, 1990). Accordingly, an increase of extracellular NE would reduce DA uptake, leading to extracellular DA increase and, vice versa, a reduced NE would facilitate DA clearance leading to a reduction of extracellular DA levels (Carboni et al., 1990).
In addition to the above hypotheses, it has been suggested that presynaptic α2-heteroreceptors control catecholamine release from dopaminergic terminals, so that their blockade and stimulation would respectively result in the increase or decrease of DA in the PFC (Pozzi et al., 1994; Gresch et al., 1995; Hertel et al., 1999).
At variance from these hypotheses, early studies from our laboratory suggest that extracellular DA in the cerebral cortex originates other than from dopaminergic also from noradrenergic terminals, where DA would act both as the precursor and co-transmitter of NE (Devoto et al., 2001; Devoto et al., 2002; Devoto et al., 2003; Devoto et al., 2004; Devoto and Flore, 2006).
Accordingly, we have recently demonstrated that central noradrenergic denervation prevented the elevation of extracellular DA in the mPFC elicited by the α2-adrenoceptor antagonist atipamezole, suggesting that noradrenergic terminals are the primary source of DA released by α2-adrenoceptor antagonists in the mPFC (Devoto et al., 2019).
The present study was aimed to test the hypothesis that DA released by dopaminergic terminals in the mPFC is readily taken up from extracellular fluid by the NET into NE nerve terminals, so that DA cannot be detected by in vivo microdialysis unless its clearance from extracellular space is inactivated.
This hypothesis might provide an answer to several questions, such as why D2-receptor antagonists, including haloperidol, raclopride, amisulpride, and sulpiride do not, or only weakly, increase extracellular DA in the prefrontal cortex (Moghaddam and Bunney, 1990; Di Giovanni et al., 1998; Gessa et al., 2000; Devoto et al., 2001; Westerink et al., 2001; Tanda et al., 2015), why the D2-receptor agonist quinpirole does not reduce dialysate levels of DA in the mPFC (Devoto et al., 2001; Devoto et al., 2003) and why noradrenergic denervation fails to reduce extracellular DA in the mPFC (Carboni et al., 1990; Pozzi et al., 1994; Yamamoto and Novotney, 1998; Valentini et al., 2004; Devoto et al., 2015; Devoto et al., 2019), which should be otherwise expected according to the hypothesis of the noradrenergic source of extracellular DA in the mPFC.
These questions are relevant not so much to the technical limitations of microdialysis as to the interpretation of DA measurements by in vivo microdialysis, in the sense that DA changes attributed to meso-prefrontal dopaminergic transmission may be indeed mediated by noradrenergic one.
Male Sprague-Dawley rats (ENVIGO, Italy) weighing 250–350 g, were group-housed and kept on a regular 12:12 h light/dark cycle, in temperature- and humidity-controlled facilities, with food and water available ad libitum. The experimental protocols were conducted to minimize pain and suffering and to reduce the number of animals used. Experiments were approved by the Animal Ethics Committees of the University of Cagliari and were carried out in accordance with the European Directive on the protection of animals used for scientific purposes (2010/63/EU). In total 126 animals were used.
Central noradrenergic system ablation was achieved as previously described (Devoto et al., 2015; Devoto et al., 2019) by the administration of the selective neurotoxin anti-dopamine-β-hydroxylase saporin (aDBH). Briefly, rats were anesthetized with Equithesin (0.97 g pentobarbital, 2.1 g MgSO4, 4.25 g chloral hydrate, 42.8 ml propylene glycol, 11.5 ml 90% ethanol in 100 ml; 5 ml/kg, IP) and placed in a Kopf stereotaxic apparatus. A hole was drilled in the skull, directed to the lateral ventricle [AP −1.0, L ± 1.5 from the bregma, V −4.3 from skull, coordinates according to Paxinos and Watson (2007)] for administration of 5 µg/5 µl immunotoxin (n = 39, aDBH rats) or 5 µl vehicle (n = 40, control rats), with a 10 µl syringe operated by a CMA/100 microinjection pump (CMA Microdialysis, Stockholm, Sweden) at 1 µl/min during 5 min, followed by 2 min pause before slowly withdrawing the needle. Injections were randomly distributed into either the left or right lateral ventricles. Rats were given antibiotic therapy (enrofloxacin, Bayer HealthCare, Shawnee Mission, KS) for five days and allowed to recover in their home cages for fifteen to eighteen days before the experiment. Some electrophysiology or microdialysis experiments were performed in intact rats (n = 35).
Rats were stereotaxically implanted with vertical microdialysis probes (membrane AN 69-HF, Hospal-Dasco, Bologna, Italy; cut-off 40,000 Daltons, 4 mm active membrane length), in the mPFC (AP +3.0, L ± 0.6, V −6.5 from the bregma, according to the coordinates of Paxinos and Watson, 1997), under Equithesin anesthesia. The day after probe implantation, artificial cerebrospinal fluid (aCSF: 147 mM NaCl, 4 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, pH 6–6.5) was pumped through the dialysis probes at a constant rate of 1.1 µl/min via a CMA/100 microinjection pump (Carnegie Medicine, Stockholm, Sweden) in freely moving animals, and dialysate samples were collected every 20 min. A subgroup of 11 rats was perfused with aCSF containing 10 µM nisoxetine. Drugs were administered after stable extracellular levels were obtained, i.e. three consecutive samples with a variance not exceeding 15%. The average of these three values was considered as baseline and used as 100% for the subsequent calculation of the variations induced by the administration of drugs. NE, DOPAC and DA were simultaneously analyzed by HPLC with electrochemical detection, by HPLC systems equipped with 3.0 × 150 mm C18 (3.5 µ) Symmetry columns (Waters, Milan, Italy), maintained at 40°C by Series 1100 thermostats (Agilent Technologies, Waldbronn, Germany), and ESA Coulochem II detectors (Chelmford, MA, USA). The mobile phase consisted of 80 mM Na2HPO4, 0.27 mM EDTA, 0.6 mM sodium octyl sulfate, 7% methanol, 4% acetonitrile, pH 2.4 with H3PO4, delivered at 0.3 ml/min; the Coulochem analytical cell first electrode was set at +200 mV, the second at −200 mV. Quantification was performed by recording the second electrode signal. Under these conditions, NE and DA detection limits (signal to noise ratio 3:1) were 0.3 pg per injection on column. In most aDBH-lesioned rats, NE concentration fell below the HPLC detection limit, therefore the actual NE value was in the range of 0 to 0.3 pg. To avoid bias in data analysis, according to the suggestion by McKittrick and Abercrombie (2007) the samples whose NE was not detectable were attributed the theoretical value of 0.15 pg, chosen taking into account that the actual value may range between the two extremes.
A sub-group of animals was implanted with a microdialysis probe into the locus coeruleus (LC, AP 11.1 mm, L ± 1.2 mm, V −8.2 mm from cortical surface, with an inclination of 16°, 2 mm dialyzing membrane), homolaterally to mPFC or VTA to be analyzed.
On completion of testing, rats were sacrificed by Equithesin overdose, the brains removed and sectioned by a cryostat (Leica CM3050 S) in 40 µm thick coronal slices to verify locations of dialysis probes. No animal was found with errant location of the device.
To measure tissue contents of NE, DA and DOPAC, rats were sacrificed by decapitation 15 days after aDBH (n = 6) or vehicle (n = 6) i.c.v. injection; brains were rapidly removed and placed on a brain cutting block maintained on ice. The mPFC was dissected out from 2 mm slices, immediately frozen on dry ice, and stored at –80°C until processing for catecholamine content. Briefly, tissues were weighed, homogenized by sonication in 0.1 M HClO4 (1:20 weight tissue per solvent volume), centrifuged at 10,000g, the supernatant filtered using microspin centrifuge tubes (0.22 µm nylon filter), and directly injected into the HPLC in the same analytical conditions described for microdialysis experiments. Data were expressed as pg per mg tissue.
Rats were anaesthetized with urethane 1.3 g/kg, IP. Rats were placed in a stereotaxic apparatus (Kopf, Tujunga, CA, USA) with their body temperature maintained at 37 ± 1°C by a heating pad. The recording electrode was placed above the VTA (5.1–5.7 posterior to bregma, 0.2–0.6 mm lateral to midline, 7.0–8.0 mm from cortical surface), according to the stereotaxic rat brain atlas of Paxinos and Watson (Paxinos and Watson, 2007). Single unit activity of neurons was recorded extracellularly (bandpass filter 0.1–10,000 Hz) with glass micropipettes filled with 2% Pontamine sky blue dissolved in 0.5 M sodium acetate. Individual action potentials were isolated and amplified by means of a window discriminator (Neurolog System, Digitimer, Hertfordshire, UK) and displayed on a digital storage oscilloscope (TDS 3012, Tektronics, Marlow, UK). Experiments were sampled online and offline with Spike2 software by a computer connected to CED1401 interface (Cambridge Electronic Design, Cambridge, UK). Dopamine neurons were isolated and identified according to previously described electrophysiological characteristics (Grace and Bunney, 1983; Ungless et al., 2004). VTA dopamine neurons were recorded only when criteria for identification were fulfilled (firing rate ≤ 10 Hz, duration of action potential ≥ 2.5 ms). Bursts were defined as the occurrence of two spikes at interspike interval < 80 ms and terminated when the interspike interval exceeded 160 ms.
To analyze cell population activity, the electrode was passed within each area in 12 predetermined tracks separated by 200 µm (5.1 to 5.7 mm posterior to bregma; 0.2 to 0.6 mm lateral to midline) and the total number of active cells encountered in each area was divided by the number of tracks.
The immunotoxin aDBH was purchased from Advanced Targeting System (San Diego, CA, USA); nisoxetine hydrochloride and clonidine hydrochloride were from Tocris (Bristol, UK), quinpirole hydrochloride and raclopride tartrate were from Sigma-Aldrich. 2. A single, full effective dose of each drug was chosen, according to literature and our previous experience, to describe a qualitative rather than a quantitative effect (Nisoxetine: Chen and Reith, 1994; Rowley et al., 2000; Devoto et al., 2019. Quinpirole: Devoto et al., 2001; Marinelli et al., 2006. Clonidine: Devoto et al., 2001; Gobbi et al., 2001. Raclopride: Wong et al., 2018; Devoto et al., 2019). Drugs were dissolved in sterile distilled water, and IP or subcutaneously (SC) administered in a volume of 1 ml/kg body weight. Sub-groups of animals were treated by infusion through microdialysis probe with 20 µM clonidine in aCSF into the LC, or with 10 µM nisoxetine in aCSF into the mPFC.
Data were analyzed by unpaired Student’s t test, with or without Welch’s correction, one-way or two-way repeated measures ANOVA, as appropriate and specified in Results. Post-hoc multiple comparisons were made using Sidak’s multiple comparison test. In all cases, P < 0.05 was considered significant. Statistic was performed using GraphPad Prism version 7.00 (GraphPad Software, La Jolla California USA, www.graphpad.com).
As shown in Figure 1, in control rats raclopride (0.5 mg/kg, IP) increased extracellular DOPAC by about 140% in the mPFC (Figure 1A) and activated the firing rate and the bursting activity (measured ad spikes in burst and burst rate) of DA neurons in the VTA (Figure 2). However, consistent with previous results with different D2-receptor antagonists (Moghaddam and Bunney, 1990; Kuroki et al., 1999; Gessa et al., 2000; Devoto et al., 2001; Tanda et al., 2015), raclopride failed to modify extracellular DA in the mPFC (Figure 1A).
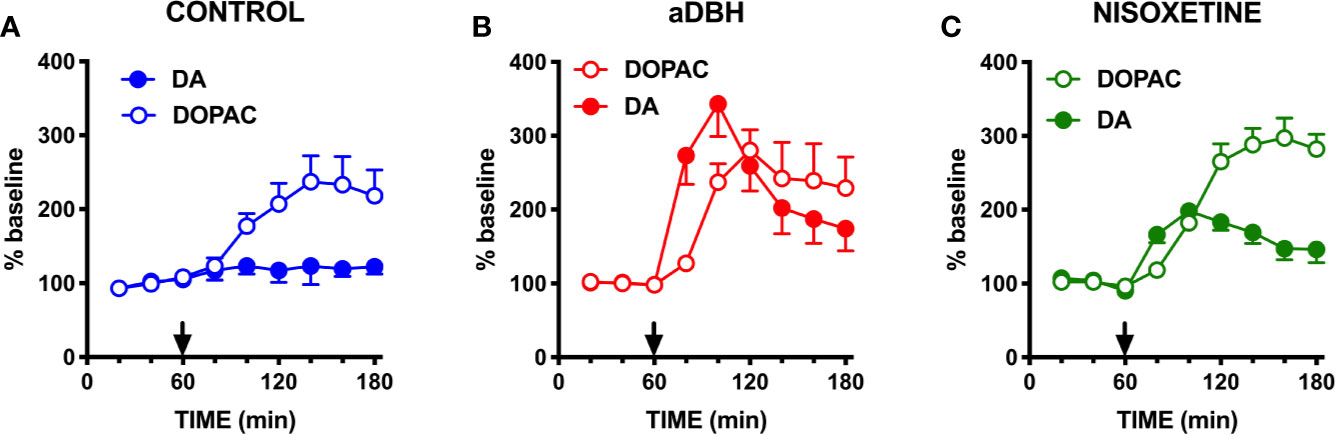
Figure 1 Effect of raclopride on extracellular DA and DOPAC levels in (A) control rats, (B) in aDBH denervated rats and (C) during continuous nisoxetine perfusion into the mPFC. Raclopride was administered at time point 60 min, as indicated by the arrow. Data are expressed as percent of baseline and are the mean ± SEM obtained from 4 control rats (A), 6 aDBH-lesioned rats (B) and 5 nisoxetine-perfused rats (C). One-way repeated measures ANOVA indicated a significant effect of raclopride on DOPAC (F(1.223, 3.668) = 12.54, P = 0.0262) but not DA (F(1.366, 4.097) = 0.9247, P = 0.4255) in control rats. In aDBH lesioned rats one-way repeated measures ANOVA indicated a significant effect of raclopride on for both DOPAC (F(1.306, 6.532) = 10.48, P = 0.0128) and DA (F(1.654, 8.268) = 18.06, P = 0.0013). In nisoxetine perfused rats, one-way repeated measures ANOVA indicated a significant effect of raclopride on DOPAC (F(2.379, 9.515) = 43.01, P < 0.0001) and DA (F(1.905, 7.62) = 18.12, P = 0.0014). Data were further analyzed by two-way repeated measures ANOVA, which indicated a significant effect in time x experimental condition interaction (F(10, 60) = 8.08, P < 0.0001), time (F(2.65, 31.8) = 15.7, P < 0.0001) and experimental condition (F(2, 12) = 5.55, P = 0.0197) for DA and in time × experimental condition interaction (F(10, 60) = 2.13, P = 0.0359), time (F(1.64, 19.7) = 24.2, P < 0.0001) but not in experimental condition (F(2, 12) = 0.508, P = 0.6143) for DOPAC.
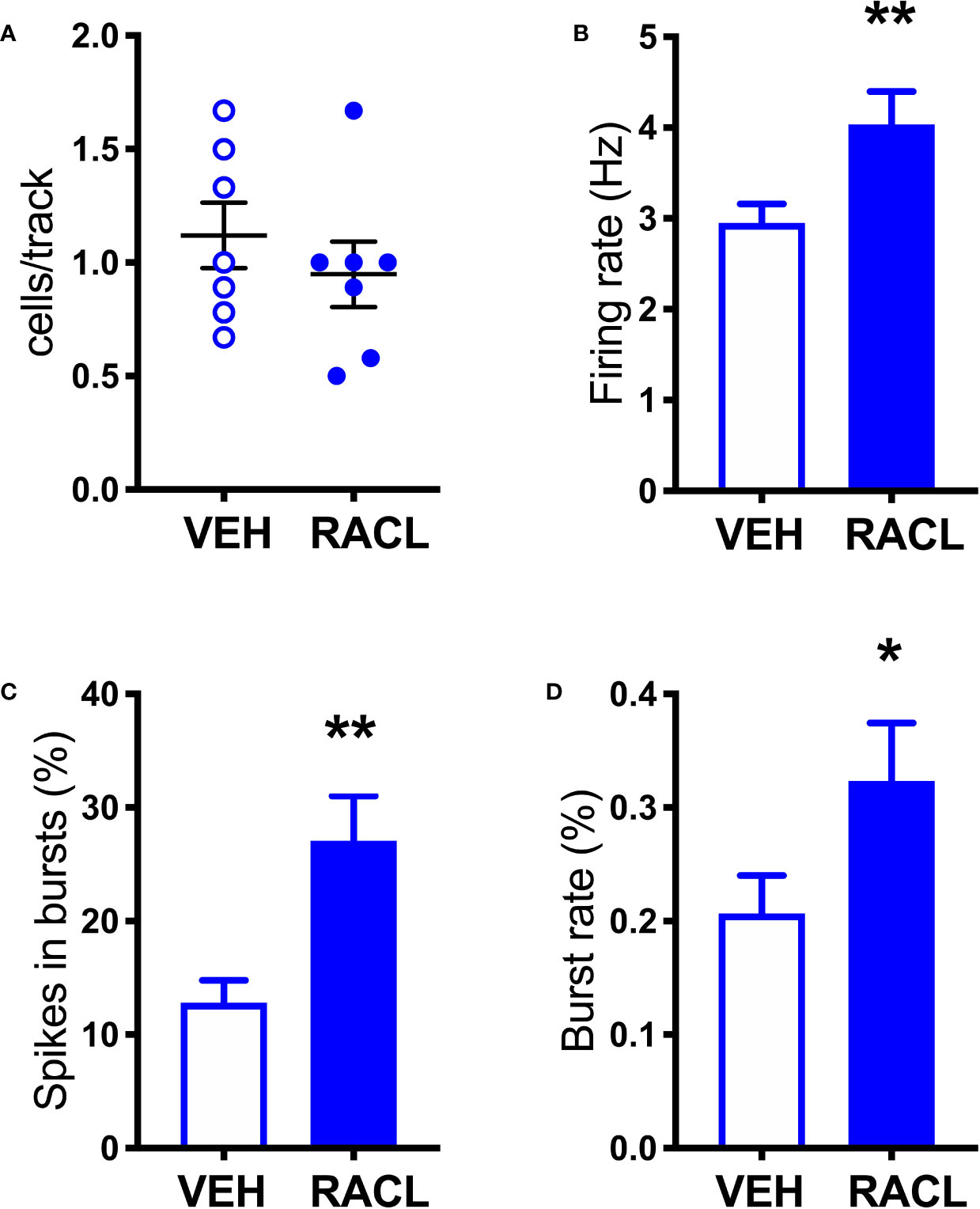
Figure 2 Raclopride increases the electrical activity of DA cells in control rats. (A) Raclopride did not change the number of spontaneously active VTA DA neurons per track (Veh 1.1 ± 0.1, n = 7; Racl: 0.9 ± 0.1, n = 7, t = 0.8410, df = 12, P = 0.41). (B) Raclopride increased DA neuron firing rate (Veh: 2.9 ± 0.2 Hz, n = 61, Racl:4.0 ± 0.4 Hz, n = 31, t = 2.795, df = 90, P = 0.006); (C) the percentage of spikes in bursts (Veh: 12.8 ± 2.0%, n = 61, Racl: 27.1 ± 3.9, n = 31, t = 3.624, df = 90, P = 0.0005), (D) burst rate (Veh: 0.2 ± 0.03, n = 48, Racl: 0.3 ± 0.05, n = 29, t = 2.002, df = 75, P = 0.048). *,P < 0.05, **P < 0.01 (unpaired Student’s t-test with or without Welch’s correction, as appropriate).
To verify whether DA uptake by NET into noradrenergic terminals prevented DA from being detected by microdialysis, raclopride was administered in rats whose NET was either eliminated by noradrenergic denervation with the neurotoxin aDBH or inactivated by nisoxetine.
As shown in Figure 1B, in denervated rats raclopride increased extracellular DOPAC (by 170%) in the mPFC and, as in control rats, activated the firing rate and bursting activity (measured ad spikes in burst and burst rate) of DA neurons in the VTA (Figure 3) but, unlike in control rats, raclopride also increased extracellular DA in the mPFC by 230% (Figure 1B).
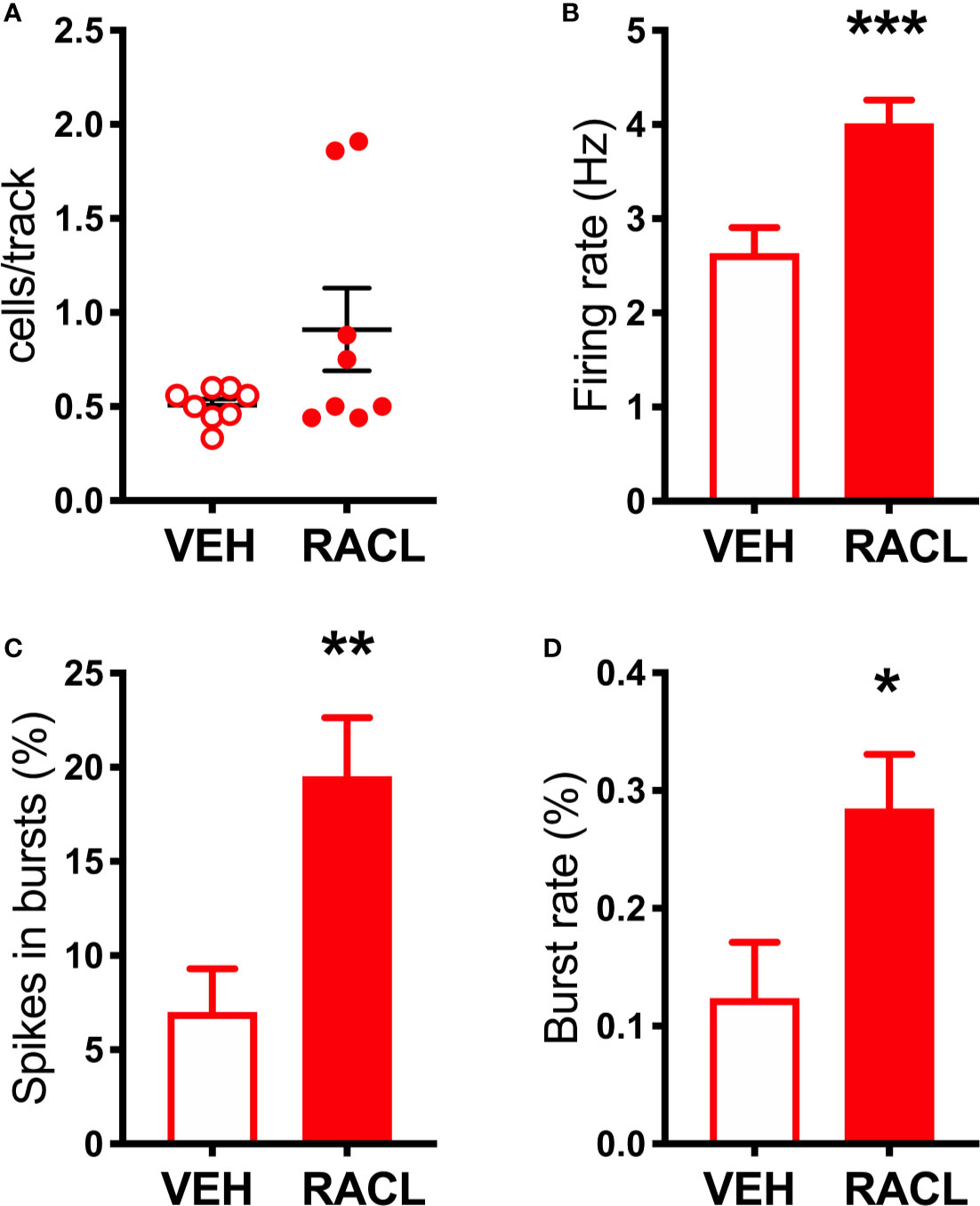
Figure 3 Raclopride increases the electrical activity of DA cells in aDBH-lesioned rats (A) Raclopride did not significantly affect the number of spontaneously active VTA DA neurons per track (Veh 0.5 ± 0.03, n = 8; Racl: 0.9 ± 0.2, n = 8, t = 1.812, df = 14, P = 0.09). (B) Raclopride increased DA neuron firing rates (Veh: 2.6 ± 0.3 Hz, n = 32, Racl:4.0 ± 0.2 Hz, n = 59, t = 3.558, df = 89, P = 0.0006); (C) the percentage of spikes in bursts (Veh: 7.0 ± 2.3%, n = 32, Racl: 19.5 ± 3.0, n = 59, t = 2.765, df = 89, P = 0.0069), (D) burst rate (Veh: 0.12 ± 0.04, n = 20, Racl: 0.28 ± 0.04, n = 49, t = 2.057, df = 67, P = 0.046). *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired Student’s t-test with or without Welch’s correction, as appropriate).
As Table 1 shows, consistent with earlier results (Devoto et al., 2015; Devoto et al., 2019), the ICV injection of aDBH (0.5 µg/5µl) depleted tissue and extracellular NE almost totally and did not modify tissue DA and DOPAC. Notably, in apparent contrast to our hypothesis that noradrenergic terminals supply DA in the mPFC, noradrenergic denervation did not reduce extracellular DA (actually increased it by about 38%) and reduced extracellular DOPAC by 50%.
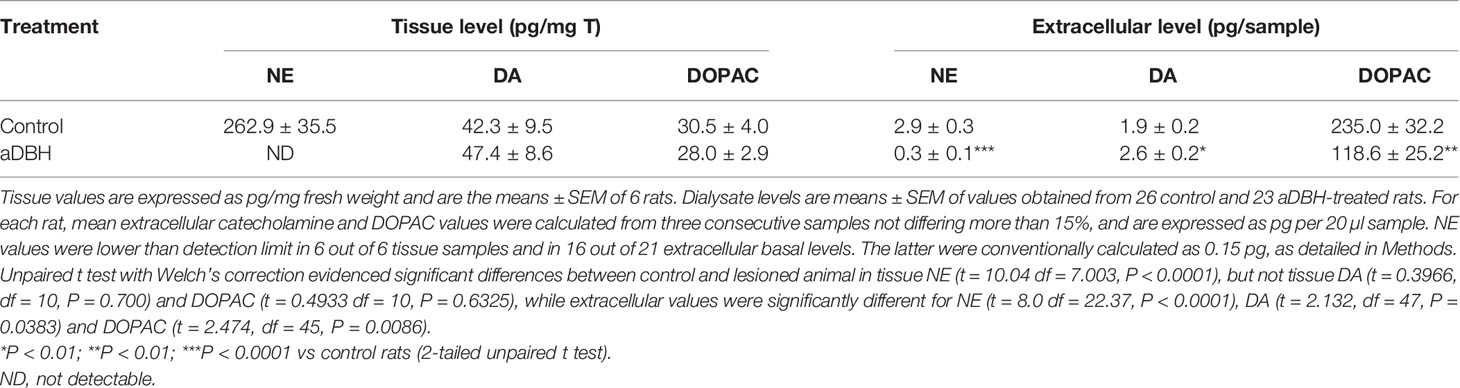
Table 1 Tissue and extracellular catecholamine levels in the medial prefrontal cortex of rats treated with aDBH or its vehicle.
As shown in Figure 1C, raclopride (0.5 mg/kg, IP) administration to rats whose NET was inactivated by nisoxetine (10 µM, locally infused into the mPFC by reverse dialysis), increased both extracellular DOPAC and DA to approximately 200% and 300% above the levels produced by nisoxetine alone (Figure 1C).
As expected from previous results (Carboni et al., 1990; Linnér et al., 2001; Bymaster et al., 2002), NET inhibition by nisoxetine increased extracellular NE and DA by about 600% and 170%, respectively, and did not increase, but actually reduced, extracellular DOPAC in the mPFC (Table 2).

Table 2 Extracellular catecholamine levels in the medial prefrontal cortex of rats locally perfused with nisoxetine (10 µM) and control rats.
Consistent with previous results (Devoto et al., 2001; Devoto et al., 2002; Devoto et al., 2003), quinpirole administration (0.1 mg/kg, SC) to control rats reduced extracellular DOPAC by 30% but failed to modify extracellular DA in the mPFC (Figure 4A), even though this dose is able to fully inhibit the activity of both fast- and slow-firing VTA dopaminergic neurons (Marinelli et al., 2006; Perez and Lodge, 2012). To clarify whether quinpirole-induced changes in extracellular DA remained undetected because of DA being taken-up by NET, the effect of quinpirole (0.1 mg/kg, SC) was tested in rats whose NET was eliminated by noradrenergic denervation or inactivated by nisoxetine. As shown in Figure 4B, after noradrenergic denervation quinpirole reduced extracellular DOPAC (by 40%), like in control rats, but, unlike the latter, also reduced extracellular DA by 70%. In rats whose NET was inactivated by nisoxetine (10 µM, locally infused into the mPFC by reverse dialysis), quinpirole not only reduced extracellular DOPAC (by 30%), like in control rats, but did also reduce extracellular DA by 50% in the mPFC (Figure 4C). Notably, quinpirole and raclopride perfused via reverse microdialysis into the mPFC reproduced the same effects seen after systemic administration, in that quinpirole (10 µM) and raclopride (100 µM) failed to modify extracellular DA in control rats but quinpirole reduced by 60% and raclopride increased by 75% extracellular DA after noradrenergic denervation (Figure 5). These results are consistent with the notion that D2-receptor on presynaptic terminals play a key role in the action of D2-receptor agonists and antagonists (Anzalone et al., 2012; Ford, 2014).
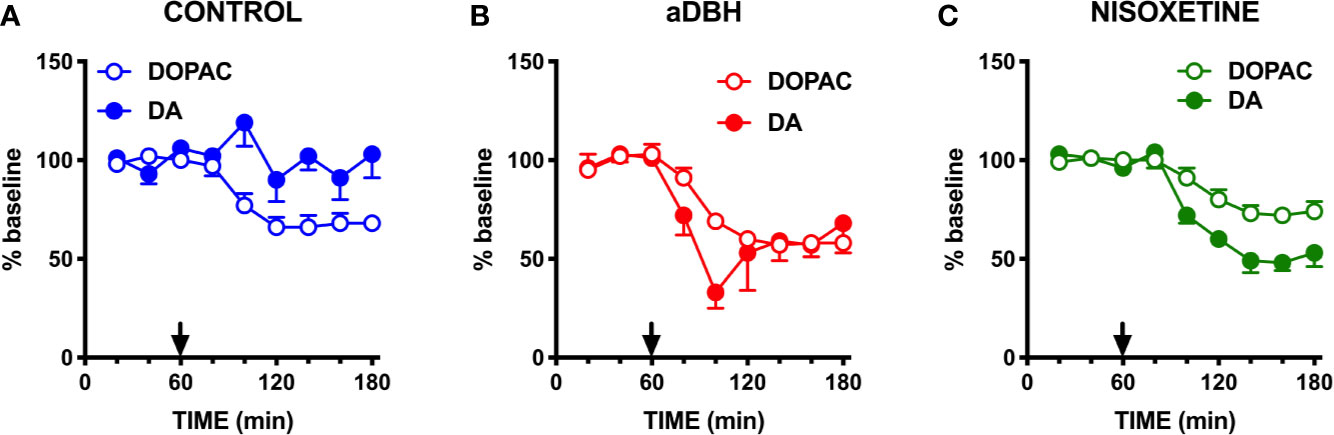
Figure 4 Effect of quinpirole on extracellular DA and DOPAC levels in (A) control rats, (B) in aDBH denervated rats and (C) during continuous nisoxetine perfusion into the mPFC. Quinpirole was administered at time point 60 min, as indicated by the arrow. Data are expressed as percent of mean basal value and are the mean ± SEM obtained from 5 control, 3 (aDBH) and 6 nisoxetine rats. One-way repeated measures ANOVA indicated a significant effect of quinpirole on DOPAC (F(2.14, 8.56) = 24.52, P = 0.0003) but not DA (F(2.538, 10.15) = 1.329, P=0.3143) in control rats; for DA (F(1.958, 9.788) = 28.88, P < 0.0001) and DOPAC (F(1.917, 9.587) = 14.87, P = 0.0012) in nisoxetine-perfused rats and for both DA (F(1.815, 3.631) = 8.861, P = 0.0409) and DOPAC (F(1.245, 2.49) = 28.13, P = 0.0202) in a-DBH-lesioned rats. Data were further analyzed by two-way repeated measures ANOVA, which indicated a significant effect in time x experimental condition interaction (F(10, 55) = 4.54, P = 0.0001), time (F(2.14, 23.5) = 5.52, P = 0.0097) and experimental condition (F(2, 11) = 14.0, P = 0.001) for DA, and in time (F(1.88, 20.6) = 37.7, P = 0.0097) and experimental condition (F(2, 11) = 4.66, P = 0.0342, but not in time x experimental condition interaction (F(10, 60) = 0.752, P = 0.6732) for DOPAC.
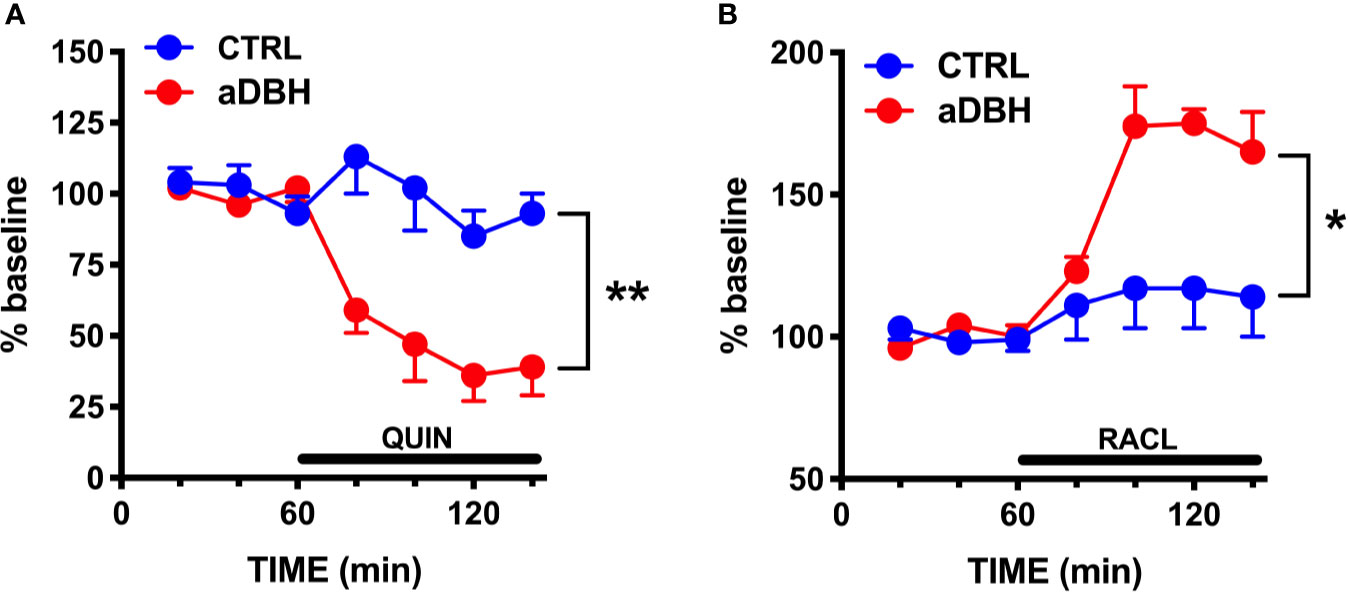
Figure 5 Effect of quinpirole (A) and raclopride (B) infusion into the mPFC on extracellular DA levels of control (CTRL) and aDBH-lesioned (aDBH) rats. Data are expressed as % of baseline and are the mean ± SEM of samples collected from 5 (aDBH) and 4 (CTRL) rats for quinpirole (10 µM) and from 5 (aDBH) and 6 (CTRL) rats for raclopride (100 µM). Drugs were perfused into the mPFC from time point 60 to the end of experiment (horizontal bar). Two-way ANOVA with repeated measures indicated a significant difference in experimental condition (F(1,7) = 22.4; P = 0.0021) but not in time (F(3, 21) = 2.56, P = 0.0824) and time x experimental condition interaction (F(3, 21) = 0.697, P = 0.5645) for quinpirole effect, and in experimental condition (F(1,9) = 9.03, P = 0.0148), time (F(3,27) = 5.93, P = 0.0030) and time x experimental condition interaction (F(3,27) = 3.69, P = 0.0240) for raclopride effect. **P = 0.0148; *P = 0.0021.
To clarify whether the loss of NET prevented noradrenergic denervation from reducing extracellular DA in the mPFC, this condition was compared with the transient inactivation of noradrenergic neurons, produced by clonidine infusion into the locus coeruleus, a condition in which NET activity is preserved.
As shown in Figure 6, clonidine (20 µM) reduced extracellular NE and DA by 80% and 60%, respectively, in the mPFC of intact rats, but did not modify extracellular DA in aDBH-lesioned rats.
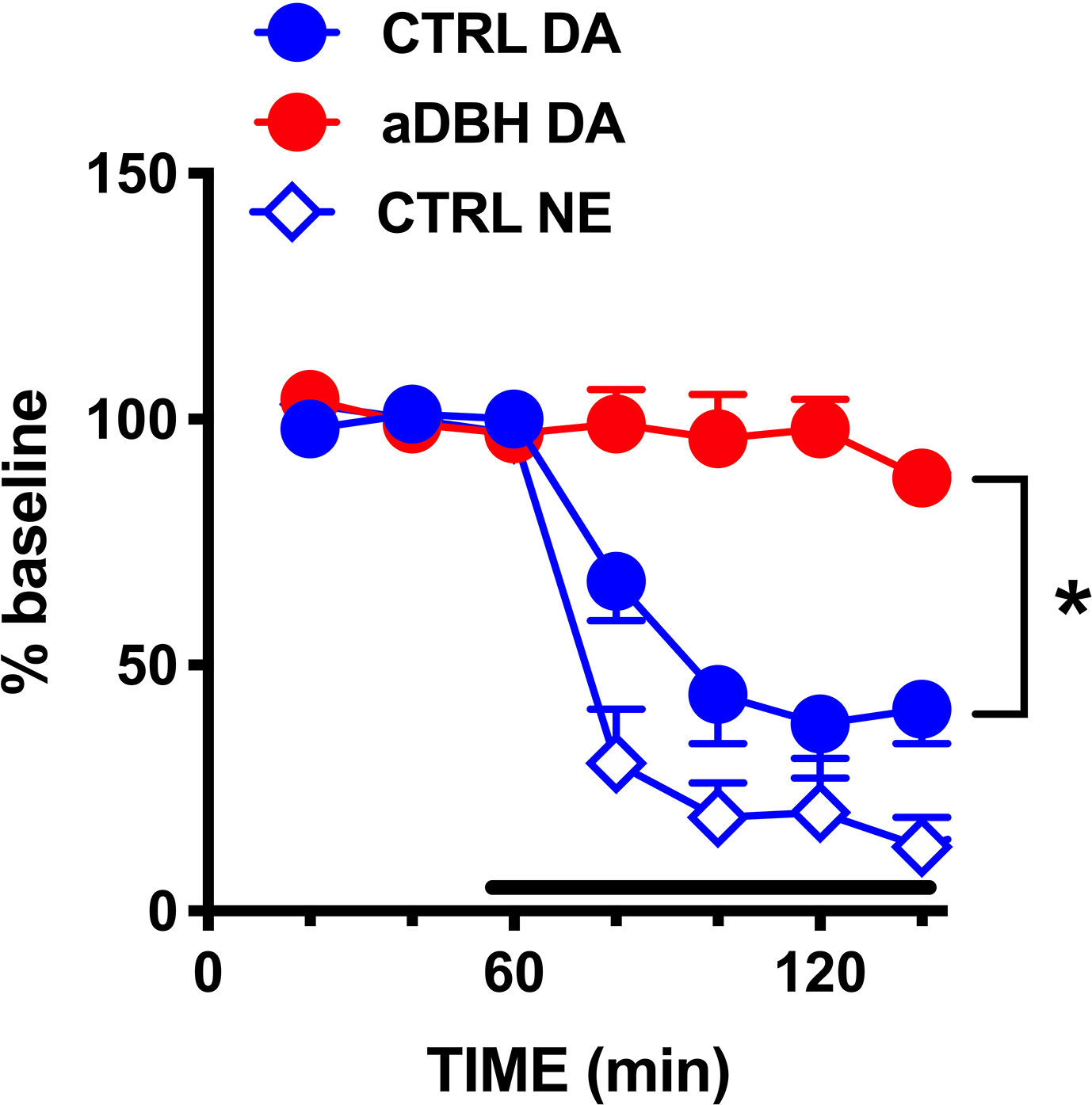
Figure 6 Effect of clonidine infusion into the LC on extracellular DA and NE in the mPFC. Data are expressed as % of baseline and are the mean ± SEM of samples collected from 4 (aDBH) and 7 (CTRL) rats. Clonidine was perfused into the LC homolateral to the mPFC from time point 60 to the end of experiment (horizontal bar). Two-way ANOVA with repeated measures of DA levels indicated a significant effect of aDBH treatment (F(1,9) = 30.5; P = 0.0004), but not in time (F(2, 18) = 1.93, P = 1737, and in time x experimental condition interaction (F(3, 27) = 0.989, P = 0.4126). *P = 0.0004
To rule out the possibility that differences in DA release in the mPFC between denervation and transient inhibition might depend on differences in the activity of dopaminergic neurons, the electrical activity of DA neurons in the VTA in the two conditions was tested. As shown in Figures 7, 8, denervation and transient inactivation of noradrenergic neurons produced the same changes in the activity of DA neurons, both reducing the number of spontaneously active DA neurons and the bursting activity (assessed as the percentage of spikes in burst and burst rate) but both failed to modify the firing rate of DA neurons.
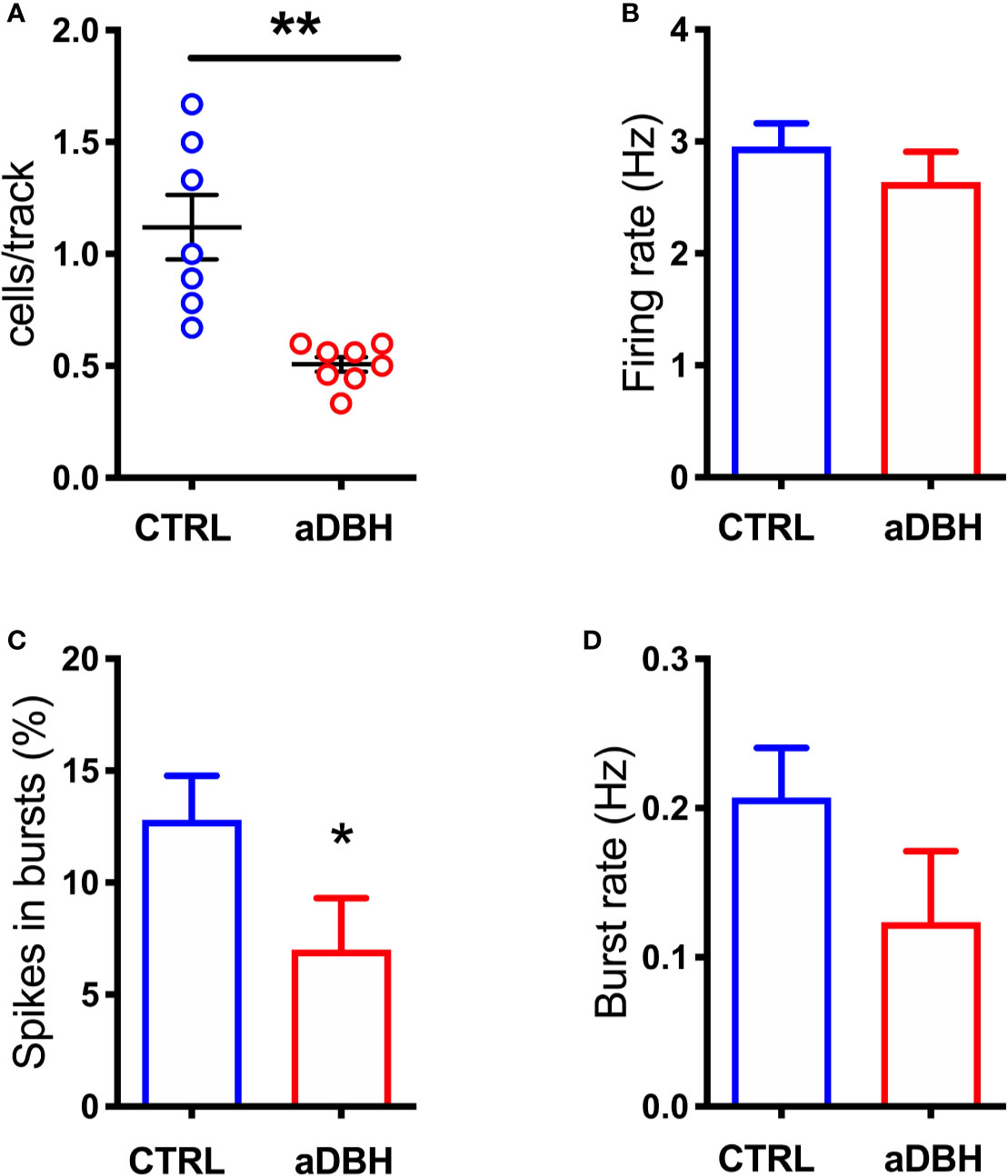
Figure 7 Effect of aDBH-induced noradrenergic denervation on firing of VTA DA neurons. The scatter plot (A) shows the number of spontaneously active VTA DA neurons encountered by the electrode per each track (unpaired Student’s t-test with Welch correction, t = 4.138, df = 6.609, P= 0.0025). Graph histograms represent (B) individual DA neuron firing rates, in CTRL and aDBH-treated rats (control group: 2.954 ± 0.207 Hz, n = 61; aDBH-treated group: 2.637 ± 0.27 Hz, n = 32; t = 0.913 df = 91, P = 0.364); (C) percentage of spikes in bursts (control group: 12.8 ± 1.974%, n = 61; aDBH-treated group: 7.01 ± 2.29%, n = 32; t = 1.814 df = 91, P = 0.0365) and (D) burst rate (control group: 0.207 ± 0.033 Hz, n = 48; aDBH-treated group: 0.124 ± 0.048 Hz, n = 20, t = 1.391 df = 66, P = 0.169). Data are expressed as mean ± SEM and were analyzed with unpaired Student’s t-test. *P < 0.05; **P < 0.01.
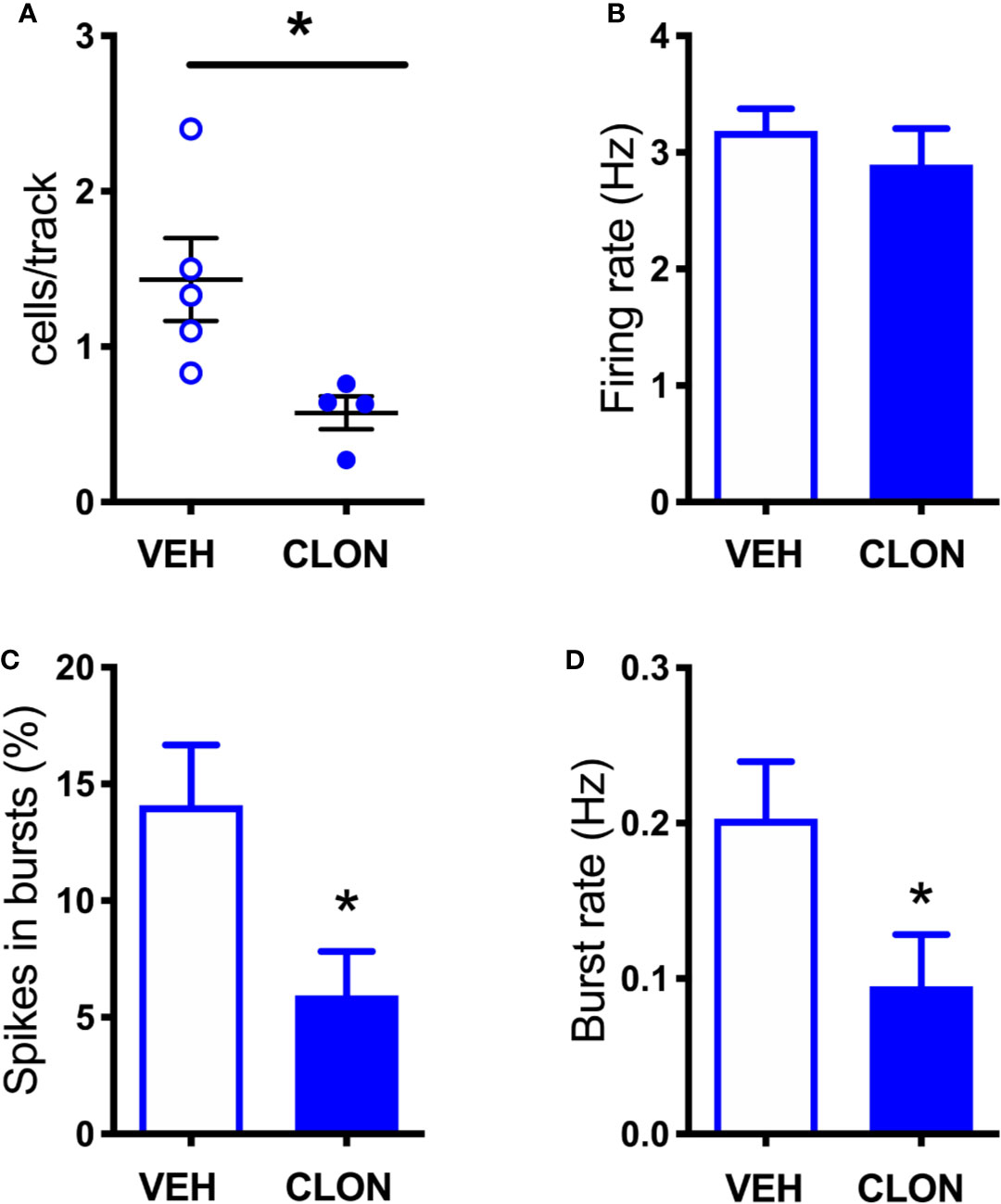
Figure 8 Effect of clonidine continuously infused into the LC on the firing of VTA DA neurons. (A) number of spontaneously active VTA DA neurons per track in vehicle (VEH) and clonidine -treated rats (t = 2.707 df = 7, P = 0.0303). (B) individual DA neuron firing rates (control group: 3.185 ± 0.19 Hz, n = 58; clonidine-treated group: 2.896 ± 0.308 Hz, n = 25; t = 0.816, df = 81, P = 0.4168). (C) percentage of spikes in bursts (control group: 14.08 ± 2.59%, n = 47; clonidine-treated group: 5.94 ± 1.88%, n = 25; t = 2.546, df = 80.06, P = 0.0128). (D) burst rate (control group: 0.203 ± 0.037 Hz, N = 47; clonidine-treated group: 0.095 ± 0.033 Hz, n = 21; t = 2.175 df = 59.89, P = 0.0336). *P < 0.05 (unpaired Student’s t-test with or without Welch’s correction, as appropriate).
The present results support the hypothesis that DA released by dopaminergic terminals in the mPFC is readily taken up from extracellular space into noradrenergic terminals by NET, so that it cannot be detected by in vivo microdialysis unless its clearance from extracellular space is prevented.
Accordingly, as previously reported for other selective D2-antagonists, including sulpiride, haloperidol, amisulpride, (Moghaddam and Bunney, 1990; Kuroki et al., 1999; Gessa et al., 2000; Devoto et al., 2001; Tanda et al., 2015), raclopride failed to increase extracellular DA in the mPFC, even though it stimulated the electrical activity of DA neurons in the VTA and increased extracellular DOPAC, the deaminated DA metabolite, in the mPFC. However, after NET was inactivated by nisoxetine or eliminated by noradrenergic denervation, raclopride uncovered the ability to increase extracellular DA in the mPFC, as measured by in vivo microdialysis, while maintaining that of stimulating the firing of DA neurons in the VTA and increasing extracellular DOPAC in the mPFC.
Importantly, the loss of NET provides a likely explanation for why noradrenergic denervation failed to reduce extracellular DA in the mPFC (Harik, 1984; Carboni et al., 1990; Pozzi et al., 1994; Valentini et al., 2004; Masana et al., 2011; Devoto et al., 2015; Devoto et al., 2019), in apparent contradiction with our hypothesis that noradrenergic terminals supply extracellular DA in the mPFC. Indeed, in accord with this hypothesis, transient inactivation of noradrenergic neurons by clonidine perfusion into the LC, a condition in which NET activity is preserved, produced a profound reduction of extracellular NE and DA in the mPFC. Moreover, consistent with the hypothesis that cortical DA measured by microdialysis originates from noradrenergic terminals, the D2 receptor agonist quinpirole, both given systemically or locally perfused into the mPFC, failed to modify extracellular DA in the mPFC in control rats but, opposite to raclopride, reduced extracellular DA after NET was inactivated by nisoxetine or eliminated by noradrenergic denervation, in line with the hypothesis that extracellular DA from dopaminergic terminals may be detected when DA clearance is impaired.
Notably, as expected from the hypothesis that extracellular DA in the mPFC derives from noradrenergic terminals, in a previous study (Devoto et al., 2019) the α2-adrenoceptor agonist clonidine was found to reduce extracellular DA in the mPFC of control rats but to be ineffective after noradrenergic denervation with aDBH.
Our results are consistent with previous observations using in vivo microdialysis that quinpirole, locally perfused into the mPFC, lowered extracellular DA in the mPFC, after nomifensine was included in the perfusate (Santiago et al., 1993), and with results in in vitro slices of PFC from rats and rabbits showing that different D2-receptor agonists reduced potassium- and electrically stimulated DA release, provided that NET and/or DAT was inhibited by nisoxetine or desmethyl-imipramine (Talmaciu et al., 1986; Wolf and Roth, 1987; Hoffman, 1988).
Our results are not in contrast with the finding that two D3-receptor agonists, (+)-7-OH-DPAT and PD128,907, as well as apomorphine (Ozaki et al., 1989), reduced extracellular DA in the mPFC or frontal cortex (Gobert et al., 1996; Westerink et al., 1998), since the effect of the D3 agonists on DA release in the PFC does not seem to be mediated by D2-autoreceptors (Gobert et al., 1995), while apomorphine is a D2 and D1 receptor agonist (Chipkin et al., 1987) and D1 receptor agonists may reduce extracellular DA in the mPFC (Santiago et al., 1993).
Importantly, results obtained by in vivo microdialysis may not reproduce results obtained with voltammetry or chronoamperometry, because microdialysis can measure changes of extracellular DA level with a time resolution of minutes to hours, related to tonic firing of DA neurons, while voltammetry and chronoamperometry can monitor DA released by phasic activation of DA neurons and also single-spike firing with a sub-second time resolution from a closer proximity to the synaptic site of release, likely before DA is cleared by NET (Gobert et al., 1995; Hauber, 2010).
These considerations may explain why, when using chronoamperometry, quinpirole was found to lower extracellular DA in the mPFC (Weber et al., 2018), unlike what we saw by microdialysis.
It is important to note that the hypothesis that DA released by dopaminergic terminals in the mPFC may not be detectable by microdialysis does not contrast the notion that DA is tonically and phasically released by dopaminergic terminals in the mPFC (see Hauber, 2010), and that noradrenergic and dopaminergic systems interact reciprocally both at the level of the LC and VTA and at nerve terminal level in the mPFC (Grenhoff et al., 1993; Guiard et al., 2008; Hauber, 2010; Mejias-Aponte, 2016; Xing et al., 2016; Park et al., 2017; Kielbinski et al., 2019). Indeed, the finding that both transient inactivation and denervation of noradrenergic neurons reduced the activity of DA neurons in the VTA is consistent with the view that NE exerts a tonic excitatory effect on VTA DA cells via α1-adrenoceptors (Grenhoff et al., 1993; Grenhoff et al., 1993; Park et al., 2017; Kielbinski et al., 2019).
However, the findings that denervation and transient inactivation of noradrenergic neurons similarly reduced the electrical activity of DA neurons in the VTA, but differently modified extracellular DA in the mPFC, while raclopride similarly activated VTA DA neurons in control as in noradrenergic denervated rats, but produced distinct effects on extracellular DA in the mPFC, indicate that changes in extracellular DA, as measured by microdialysis in the mPFC may be dissociated from the electrical activity of DA neurons in the VTA.
Importantly, previous observations (Gessa et al., 2000) indicate that such dissociation may apply to meso-prefrontal DA neurons as well, since haloperidol, clozapine and olanzapine, in doses equally effective in stimulating the electrical activity of meso-prefrontal DA neurons, were found differ in modifying extracellular DA in the mPFC: namely, D2-receptor antagonist haloperidol failed to modify extracellular DA in the mPFC, while clozapine and olanzapine, that inhibit both D2- and α2-receptors (Baldessarini et al., 1992; Bymaster et al., 1996; Devoto et al., 2003; Tauscher et al., 2004), increased extracellular DA, supposedly from dopaminergic and noradrenergic terminals (Gessa et al., 2000).
An important corollary of our results regards the formation and function of extracellular DOPAC in the mPFC. The finding that extracellular DOPAC paralleled the changes of dopaminergic activity supports the view that extracellular DOPAC, unlike DA, is a reliable indicator of DA activity in the mPFC.
Extracellular DOPAC in the mPFC is thought to originate from the oxidation by MAO-A of DA captured by DAT and NET into dopaminergic and noradrenergic terminals, respectively (Mazei et al., 2002; Kaenmaki et al., 2010; Tammimaki et al., 2016).
The finding that extracellular DOPAC was reduced by 50% after noradrenergic denervation and was slightly, although not significantly, reduced after NET inhibition by nisoxetine, in spite of the marked increase of extracellular DA, indicates that noradrenergic terminals substantially contribute to DOPAC formation in the mPFC.
As to the source of DA to form DOPAC by noradrenergic terminals, previous results (Devoto et al., 2003) indicated that DOPAC measured by microdialysis in the mPFC was five times higher than that in the occipital cortex, a region equally innervated by noradrenergic afferences but with scarce dopaminergic terminals. These results suggest that the majority of DA oxidized to DOPAC by noradrenergic terminals originates from dopaminergic terminals.
Our results suggest a mechanism by which atypical antipsychotics rescue the impaired dopaminergic functioning and improve the negative symptoms in schizophrenia. These compounds, which have a considerable affinity for α2-adrenoceptors, would block α2-autoreceptors on noradrenergic neurons, activate noradrenergic activity and eventually produce a co-release of NE and DA in the PFC. This mechanism may also explain the enhanced efficacy of typical antipsychotics by add-on therapy with α2-adrenoceptors antagonists (Wadenberg et al., 2007; Hecht and Landy, 2012).
In conclusion, the finding that DA released by pharmacologically activated dopaminergic neurons may not be detected in the mPFC by in vivo microdialysis raises the important question of whether DA levels measured by microdialysis in the mPFC after stimuli such as feeding, drinking, sex, novelty, handling, foot-shock, etc. (Horvitz, 2000; Phillips et al., 2004) may indeed represent DA released from noradrenergic terminals.
These considerations challenge for further research on the role DA from of noradrenergic source in the physiology and dysfunctions of the PFC and indicate noradrenergic neurons as an important target for interventions aimed at modifying extracellular DA level in the PFC.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Ministero della Salute, Direzione Generale della sanità animale e dei farmaci veterinari, Uff. 6, Roma, Italy - Aut. n. 611/2017-PR.
PD and GLG designed the study and wrote the manuscript. PD supervised and analyzed microdialysis experiments. MP supervised electrophysiology experiments and contributed to the writing of the manuscript. CS and MS performed and analyzed electrophysiology experiments. PS and GF performed noradrenergic lesion and microdialysis experiments. All authors contributed to the article and approved the submitted version.
The research was founded by the “Guy Everett Laboratory”, by “FSC 2014–2020—Patto per lo Sviluppo della Regione Sardegna, Legge Regionale n. 7 del 7 agosto 2007 (Bando 2017)” and by “Progetti di Rilevante Interesse Nazionale” (PRIN) 2017 (2017YH3SXK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank dr. Barbara Tuveri and her team for their dedication and expertise in animal care.
Anzalone, A., Lizardi-Ortiz, J. E., Ramos, M., De Mei, C., Hopf, F. W., Iaccarino, C., et al. (2012). Dual control of dopamine synthesis and release by presynaptic and postsynaptic dopamine D2 receptors. J. Neurosci. 32, 9023–9034. doi: 10.1523/jneurosci.0918-12.2012
Aston-Jones, G., Kalivas, P. W. (2008). Brain norepinephrine rediscovered in addiction research. Biol. Psychiatry 63, 1005–1006. doi: 10.1016/j.biopsych.2008.03.016
Baldessarini, R. J., Huston-Lyons, D., Campbell, A., Marsh, E., Cohen, B. M. (1992). Do central antiadrenergic actions contribute to the atypical properties of clozapine? Br. J. Psychiatry Suppl. 17, 12–16. doi: 10.1192/S0007125000296852
Bird, E. D., Spokes, E. G., Iversen, L. L. (1979). Increased dopamine concentration in limbic areas of brain from patients dying with schizophrenia. Brain 102, 347–360. doi: 10.1093/brain/102.2.347
Bunney, W. E., Jr, Davis, J. M. (1965). Norepinephrine in depressive reactions. A. Rev. Arch. Gen. Psychiatry 13, 483–494. doi: 10.1001/archpsyc.1965.01730060001001
Bymaster, F. P., Calligaro, D. O., Falcone, J. F., Marsh, R. D., Moore, N. A., Tye, N. C., et al. (1996). Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 14, 87–96. doi: 10.1016/0893-133x(94)00129-n
Bymaster, F. P., Katner, J. S., Nelson, D. L., Hemrick-Luecke, S. K., Threlkeld, P. G., Heiligenstein, J. H., et al. (2002). Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27, 699–711. doi: 10.1016/s0893-133x(02)00346-9
Carboni, E., Tanda, G. L., Frau, R., Di Chiara, G. (1990). Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J. Neurochem. 55, 1067–1070. doi: 10.1111/j.1471-4159.1990.tb04599.x
Chandler, D. J., Waterhouse, B. D., Gao, W. J. (2014). New perspectives on catecholaminergic regulation of executive circuits: evidence for independent modulation of prefrontal functions by midbrain dopaminergic and noradrenergic neurons. Front. Neural Circuits 8, 53. doi: 10.3389/fncir.2014.00053
Chen, N. H., Reith, M. E. (1994). Effects of locally applied cocaine, lidocaine, and various uptake blockers on monoamine transmission in the ventral tegmental area of freely moving rats: a microdialysis study on monoamine interrelationships. J. Neurochem. 63 (5), 1701–1713. doi: 10.1046/j.1471-4159.1994.63051701.x
Chipkin, R. E., McQuade, R. D., Iorio, L. C. (1987). D1 and D2 dopamine binding site up-regulation and apomorphine-induced stereotypy. Pharmacol. Biochem. Behav. 28, 477–482. doi: 10.1016/0091-3057(87)90509-0
Darracq, L., Blanc, G., Glowinski, J., Tassin, J. P. (1998). Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. J. Neurosci. 18, 2729–2739. doi: 10.1523/jneurosci.18-07-02729.1998
Descarries, L., Lemay, B., Doucet, G., Berger, B. (1987). Regional and laminar density of the dopamine innervation in adult rat cerebral cortex. Neuroscience 21, 807–824. doi: 10.1016/0306-4522(87)90038-8
Devoto, P., Flore, G. (2006). On the origin of cortical dopamine: Is it a co-transmitter in noradrenergic neurons? Curr. Neuropharmacol. 4, 115–125. doi: 10.2174/157015906776359559
Devoto, P., Flore, G., Pani, L., Gessa, G. L. (2001). Evidence for co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex. Mol. Psychiatry 6, 657–664. doi: 10.1038/sj.mp.4000904
Devoto, P., Flore, G., Pira, L., Diana, M., Gessa, G. L. (2002). Co-release of noradrenaline and dopamine in the prefrontal cortex after acute morphine and during morphine withdrawal. Psychopharmacol. (Berl) 160, 220–224. doi: 10.1007/s00213-001-0985-y
Devoto, P., Flore, G., Vacca, G., Pira, L., Arca, A., Casu, M. A., et al. (2003). Co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex induced by clozapine, the prototype atypical antipsychotic. Psychopharmacology 167, 79–84. doi: 10.1007/s00213-002-1381-y
Devoto, P., Flore, G., Pira, L., Longu, G., Gessa, G. L. (2004). Alpha2-adrenoceptor mediated co-release of dopamine and noradrenaline from noradrenergic neurons in the cerebral cortex. J. Neurochem. 88, 1003–1009. doi: 10.1046/j.1471-4159.2003.02239.x
Devoto, P., Flore, G., Saba, P., Frau, R., Gessa, G. L. (2015). Selective inhibition of dopamine-beta-hydroxylase enhances dopamine release from noradrenergic terminals in the medial prefrontal cortex. Brain Behav. 5, e00393. doi: 10.1002/brb3.393
Devoto, P., Flore, G., Saba, P., Scheggi, S., Mulas, G., Gambarana, C., et al. (2019). Noradrenergic terminals are the primary source of α2-adrenoceptor mediated dopamine release in the medial prefrontal cortex. Progr. Neuropsychopharm. Biol. Psy. 90, 97–103. doi: 10.1016/j.pnpbp.2018.11.015
Di Giovanni, G., Di Mascio, M., Di Matteo, V., Esposito, E. (1998). Effects of acute and repeated administration of amisulpride, a dopamine D2/D3 receptor antagonist, on the electrical activity of midbrain dopaminergic neurons. E. J. Pharmacol. Exp. Ther. 287, 51–57.
Dunlop, B. W., Nemeroff, C. B. (2007). The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry 64, 327–337. doi: 10.1001/archpsyc.64.3.327
El Mansari, M., Guiard, B. P., Chernoloz, O., Ghanbari, R., Katz, N., Blier, P. (2010). Relevance of norepinephrine-dopamine interactions in the treatment of major depressive disorder. Version 2. CNS Neurosci. Ther. 16 (3), e1–17. doi: 10.1111/j.1755-5949.2010.00146.x
Engert, V., Pruessner, J. C. (2008). Dopaminergic and noradrenergic contributions to functionality in ADHD: the role of methylphenidate. Curr. Neuropharmacol. 6 (4), 322–328. doi: 10.2174/157015908787386069
Feenstra, M. G. (2000). Dopamine and noradrenaline release in the prefrontal cortex in relation to unconditioned and conditioned stress and reward. Prog. Brain Res. 126, 133–163. doi: 10.1016/S0079-6123(00)26012-3
Finlay, J. M., Zigmond, M. J., Abercrombie, E. D. (1995). Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: effects of diazepam. Neuroscience 64 (3), 619–628. doi: 10.1016/0306-4522(94)00331-x
Ford, C. P. (2014). The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 282, 13–22. doi: 10.1016/j.neuroscience.2014.01.025
Freed, C., Revay, R., Vaughan, R. A., Kriek, E., Grant, S., Uhl, G. R., et al. (1995). Dopamine transporter immunoreactivity in rat brain. J. Comp. Neurol. 359 (2), 340–349. doi: 10.1002/cne.903590211
Gessa, G. L., Devoto, P., Diana, M., Flore, G., Melis, M., Pistis, M. (2000). Dissociation of haloperidol, clozapine, and olanzapine effects on electrical activity of mesocortical dopamine neurons and dopamine release in the prefrontal cortex. Neuropsychopharmacology 22 (6), 642–649. doi: 10.1016/S0893-133X(00)00087-7
Gobbi, G., Muntoni, A. L., Gessa, G. L., Diana, M. (2001). Clonidine fails to modify dopaminergic neuronal activity during morphine withdrawal. Psychopharmacol. (Berl) 158 (1), 1–6. doi: 10.1007/s002130100832
Gobert, A., Rivet, J. M., Audinot, V., Cistarelli, L., Spedding, M., Vian, J., et al. (1995). Functional correlates of dopamine D3 receptor activation in the rat in vivo and their modulation by the selective antagonist, (+)-S 14297: II. Both D2 and “silent” D3 autoreceptors control synthesis and release in mesolimbic, mesocortical and nigrostriatal pathways. J. Pharmacol. Exp. Ther. 275 (2), 899–913.
Gobert, A., Lejeune, F., Rivet, J. M., Cistarelli, L., Millan, M. J. (1996). Dopamine D3 (Auto)receptors Inhibit Dopamine Release in the Frontal Cortex of Freely Moving Rats In Vivo. J. Neurochem. 66 (5), 2209–2212. doi: 10.1046/j.1471-4159.1996.66052209.x
Gold, M. S., Blum, K., Oscar-Berman, M., Braverman, E. R. (2014). Low dopamine function in attention deficit/hyperactivity disorder: should genotyping signify early diagnosis in children? Postgrad. Med. 126 (1), 153–177. doi: 10.3810/pgm.2014.01.2735
Grace, A. A., Bunney, B. S. (1983). Intracellular and extracellular electrophysiology of nigral dopaminergic neurons–1. Identification and characterization. Neuroscience 10 (2), 301–315. doi: 10.1016/0306-4522(83)90135-5
Grenhoff, J., Nisell, M., Ferré, S., Aston-Jones, G., Svensson, T. H. (1993). Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J. Neural Transm. 93 (1), 11–25. doi: 10.1007/BF01244934
Gresch, P. J., Sved, A. F., Zigmond, M. J., Finlay, J. M. (1995). Local influence of endogenous norepinephrine on extracellular dopamine in rat medial prefrontal cortex. J. Neurochem. 65 (1), 111–116. doi: 10.1046/j.1471-4159.1995.65010111.x
Guiard, B. P., El Mansari, M., Blier, P. (2008). Cross-talk between dopaminergic and noradrenergic systems in the rat ventral tegmental area, locus ceruleus, and dorsal hippocampus. Mol. Pharmacol. 74 (5), 1463–1475. doi: 10.1124/mol.108.048033
Harik, S. I. (1984). Locus coeruleus lesion by local 6-hydroxydopamine infusion causes marked and specific destruction of noradrenergic neurons, long-term depletion of norepinephrine and the enzymes that synthesize it, and enhanced dopaminergic mechanisms in the ipsilateral cerebral cortex. J. Neurosci. 4 (3), 699–707. doi: 10.1523/JNEUROSCI.04-03-00699.1984
Hauber, W. (2010). Dopamine release in the prefrontal cortex and striatum: temporal and behavioral aspects. Pharmacopsychiatry 43 (S1), 32–41. doi: 10.1055/s-0030-1248300
Hecht, E. M., Landy, D. C. (2012). Alpha-2 receptor antagonist add-on therapy in the treatment of schizophrenia; a meta-analysis. Schizophr. Res. 134 (2-3), 202–206. doi: 10.1016/j.schres.2011.11.030
Hertel, P., Fagerqist, M. V., Svensson, T. H. (1999). Enhanced cortical dopamine output and antipsychotic-like effect of raclopride by α2 adrenoceptor blockade. Science 286, 105–107. doi: 10.1126/science.286.5437.105
Hoffmann, I. S., Talmaciu, R. K., Ferro, C. P., Cubeddu, L. X. (1988). Sustained high release at rapid stimulation rates and reduced functional autoreceptors characterize prefrontal cortex dopamine terminals. J. Pharmacol. Exp. Ther. 245 (3), 761–772.
Holzer, B., Lopes, V., Lehman, R. (2013). Combination use of atomoxetine hydrochloride and olanzapine in the treatment of attention-deficit/hyperactivity disorder with comorbid disruptive behavior disorder in children and adolescents 10-18 years of age. J. Child Adolesc. Psychopharmacol. 23 (6), 415–418. doi: 10.1089/cap.2013.0029
Horn, A. S. (1990). Dopamine uptake: a review of progress in the last decade. Prog. Neurobiol. 34 (5), 387–400. doi: 10.1016/0301-0082(90)90033-d
Horvitz, J. C. (2000). Mesolimbocortical and nigrostriatal dopamine responses to salient non-reward events. Neuroscience 96 (4), 651–656. doi: 10.1016/s0306-4522(00)00019-1
Käenmäki, M., Tammimäki, A., Myöhänen, T., Pakarinen, K., Amberg, C., Karayiorgou, M., et al. (2010). Quantitative role of COMT in dopamine clearance in the prefrontal cortex of freely moving mice. J. Neurochem. 114 (6), 1745–1755. doi: 10.1111/j.1471-4159.2010.06889.x
Kapur, S., Mann, J. J. (1992). Role of the dopaminergic system in depression. Biol. Psychiatry 32 (1), 1–17. doi: 10.1016/0006-3223(92)90137-o
Kielbinski, M., Bernacka, J., Solecki, W. B. (2019). Differential regulation of phasic dopamine release in the forebrain by the VTA noradrenergic receptor signaling. J. Neurochem. 149, 747–759. doi: 10.1111/jnc.14706
Koob, G. F., Nestler, E. J. (1997). The neurobiology of drug addiction. J. Neuropsychiat. Clin. Neurosci. 9 (3), 482–497. doi: 10.1176/jnp.9.3.482
Kuroki, T., Meltzer, H. Y., Ichikawa, J. (1999). Effects of antipsychotic drugs on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens. J. Pharmacol. Exp. Ther. 288 (2), 774–781.
Lidow, M. S., Williams, G. V., Goldman-Rakic, P. S. (1998). The cerebral cortex: a case for a common site of action of antipsychotics. Trends Pharmacol. Sci. 19 (4), 136–140. doi: 10.1016/s0165-6147(98)01186-9
Linnér, L., Endersz, H., Ohman, D., Bengtsson, F., Schalling, M., Svensson, T. H. (2001). Reboxetine modulates the firing pattern of dopamine cells in the ventral tegmental area and selectively increases dopamine availability in the prefrontal cortex. J. Pharmacol. Exp. Ther. 297 (2), 540–546.
Marinelli, M., Rudick, C. N., Hu, X. T., White, F. J. (2006). Excitability of dopamine neurons: modulation and physiological consequences. CNS Neurol. Disord. Drug Targ. 5 (1), 79–97. doi: 10.2174/187152706784111542
Masana, M., Bortolozzi, A., Artigas, F. (2011). Selective enhancement of mesocortical dopaminergic transmission by noradrenergic drugs: therapeutic opportunities in schizophrenia. Int. J. Neuropsychopharmacol. 14 (1), 53–68. doi: 10.1017/S1461145710000908
Mazei, M. S., Pluto, C. P., Kirkbride, B., Pehek, A. E. (2002). Effects of Catecholamine Uptake Blockers in the Caudate-Putamen and Subregions of the Medial Prefrontal Cortex of the Rat. Brain Res. 936 (1-2), 58–67. doi: 10.1016/s0006-8993(02)02542-8
McKittrick, C. R., Abercrombie, E. D. (2007). Catecholamine mapping within nucleus accumbens: differences in basal and amphetamine-stimulated efflux of norepinephrine and dopamine in shell and core. J. Neurochem. 100, 1247–1256.
Mejias-Aponte, C. A. (2016). Specificity and impact of adrenergic projections to the midbrain dopamine system. Brain Res. 1641 (Pt B), 258–273. doi: 10.1016/j.brainres.2016.01.036
Melis, M., Diana, M., Gessa, G. L. (1999). Clozapine potently stimulates mesocortical dopamine neurons. Eur. J. Pharmacol. 366 (2-3), R11–R13. doi: 10.1016/s0014-2999(98)00934-0
Meltzer, H. Y., Stahl, S. M. (1976). The dopamine hypothesis of schizophrenia: a review. Schizophr. Bull. 2 (1), 19–76. doi: 10.1093/schbul/2.1.19
Miner, L. H., Schroeter, S., Blakely, R. D., Sesack, S. R. (2003). Ultrastructural localization of the norepinephrine transporter in superficial and deep layers of the rat prelimbic prefrontal cortex and its spatial relationship to probable dopamine terminals. J. Comp. Neurol. 466 (4), 478–494. doi: 10.1002/cne.10898
Moghaddam, B., Bunney, B. S. (1990). Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J. Neurochem. 54 (5), 1755–1760. doi: 10.1111/j.1471-4159.1990.tb01230.x
Morón, J. A., Brockington, A., Wise, R. A., Rocha, B. A., Hope, B. T. (2002). Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J. Neurosci. 22 (2), 389–395. doi: 10.1523/JNEUROSCI.22-02-00389.2002
Ozaki, N., Nakahara, D., Miura, H., Kasahara, Y., Nagatsu, T. (1989). Effects of apomorphine on in vivo release of dopamine and its metabolites in the prefrontal cortex and the striatum, studied by a microdialysis method. J. Neurochem. 53 (6), 1861–1864. doi: 10.1111/j.1471-4159.1989.tb09253.x
Park, J. W., Bhimani, R. V., Park, J. (2017). Noradrenergic Modulation of Dopamine Transmission Evoked by Electrical Stimulation of the Locus Coeruleus in the Rat Brain. ACS Chem. Neurosci. 8 (9), 1913–1924. doi: 10.1021/acschemneuro.7b00078
Paxinos, G., Watson, C. (2007). The rat brain in stereotaxic coordinates. 7th Edition (London: Elsevier Academic Press).
Perez, S. M., Lodge, D. J. (2012). Aberrant dopamine D2-like receptor function in a rodent model of schizophrenia. J. Pharmacol. Exp. Ther. 343 (2), 288–295. doi: 10.1124/jpet.112.193201
Phillips, A. G., Ahn, S., Floresco, S. B. (2004). Magnitude of dopamine release in medial prefrontal cortex predicts accuracy of memory on a delayed response task. J. Neurosci. 24 (2), 547–553. doi: 10.1523/JNEUROSCI.4653-03.2004
Pozzi, L., Invernizzi, R., Cervo, L., Vallebuona, F., Samanin, R. (1994). Evidence that extracellular concentrations of dopamine are regulated by noradrenergic neurons in the frontal cortex of rats. J. Neurochem. 63, 195–200. doi: 10.1046/j.1471-4159.1994.63010195.x
Raiteri, M., Del Carmine, R., Bertollini, A., Levi, G. (1977). Effect of sympathomimetic amines on the synaptosomal transport of noradrenaline, dopamine and 5-hydroxytryptamine. Eur. J. Pharmacol. 41 (2), 133–143. doi: 10.1016/0014-2999(77)90202-3
Ranjbar-Slamloo, Y., Fazlali, Z. (2020). Dopamine and Noradrenaline in the Brain; Overlapping or Dissociate Functions? Front. Mol. Neurosci. 12, 334. doi: 10.3389/fnmol.2019.00334
Rao, N., Northoff, G., Tagore, A., Rusjan, P., Kenk, M., Wilson, A., et al. (2019). Impaired Prefrontal Cortical Dopamine Release in Schizophrenia During a Cognitive Task: A [11C]FLB 457 Positron Emission Tomography Study. Schizophr. Bull. 45 (3), 670–679. doi: 10.1093/schbul/sby076
Rowley, H. L., Needham, P. L., Kilpatrick, I. C., Heal, D. J. (2000). A comparison of the acute effects of zotepine and other antipsychotics on rat cortical dopamine release, in vivo. Naunyn Schmiedebergs Arch. Pharmacol. 361 (2), 187–192. doi: 10.1007/s002109900170
Santiago, M., Machado, A., Cano, J. (1993). Regulation of prefrontal cortical dopamine release by dopamine receptor agonists and antagonists. Eur. J. Pharmacol. 239 (1-3), 83–91. doi: 10.1016/0014-2999(93)90979-r
Schildkraut, J. J. (1965). The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am. J. Psychiatry 122 (5), 509–522. doi: 10.1176/ajp.122.5.509
Séguéla, P., Watkins, K. C., Geffard, M., Descarries, L. (1990). Noradrenaline axon terminals in adult rat neocortex: an immunocytochemical analysis in serial thin sections. Neuroscience 35 (2), 249–264. doi: 10.1016/0306-4522(90)90079-j
Sesack, S. R., Hawrylak, V. A., Matus, C., Guido, M. A., Levey, A. I. (1998). Dopamine axon varicosities in the prelimbic division of the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J. Neurosci. 18 (7), 2697–2708. doi: 10.1523/JNEUROSCI.18-07-02697.1998
Slifstein, M., Abi-Dargham, A. (2017). Recent Developments in Molecular Brain Imaging of Neuropsychiatric Disorders. Semin. Nucl. Med. 47 (1), 54–63. doi: 10.1053/j.semnuclmed.2016.09.002
Talmaciu, R. K., Hoffmann, I. S., Cubeddu, L. X. (1986). Dopamine autoreceptors modulate dopamine release from the prefrontal cortex. J. Neurochem. 47 (3), 865–870. doi: 10.1111/j.1471-4159.1986.tb00691.x
Tammimaki, A., Aonurm-Helm, A., Kaenmaki, M., Mannisto, P. T. (2016). Elimination of extracellular dopamine in the medial prefrontal cortex of conscious mice analysed using selective enzyme and uptake inhibitors. J. Physiol. Pharmacol. 67 (2), 301–309.
Tanda, G., Valentini, V., De Luca, M. A., Perra, V., Serra, G. P., Di Chiara, G. (2015). A systematic microdialysis study of dopamine transmission in the accumbens shell/core and prefrontal cortex after acute antipsychotics. Psychopharmacol. (Berl.) 232 (8), 1427–1440. doi: 10.1007/s00213-014-3780-2
Tassin, J. P. (1992). NE/DA interactions in prefrontal cortex and their possible roles as neuromodulators in schizophrenia. J. Neural Transm. Suppl. 36, 135–162. doi: 10.1007/978-3-7091-9211-5_7
Tauscher, J., Hussain, T., Agid, O., Verhoeff, N. P., Wilson, A. A., Houle, S., et al. (2004). Equivalent occupancy of dopamine D1 and D2 receptors with clozapine: differentiation from other atypical antipsychotics. Am. J. Psychiatry 161 (9), 1620–1625. doi: 10.1176/appi.ajp.161.9.1620
Ungless, M. A., Magill, P. J., Bolam, J. P. (2004). Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science 303 (5666), 2040–2042. doi: 10.1126/science.1093360
Valentini, V., Frau, R., Di Chiara, G. (2004). Noradrenaline transporter blockers raise extracellular dopamine in medial prefrontal but not parietal and occipital cortex: differences with mianserin and clozapine. J. Neurochem. 88 (4), 917–927. doi: 10.1046/j.1471-4159.2003.02238.x
van Kammen, D. P., Kelley, M. (1991). Dopamine and norepinephrine activity in schizophrenia. An integrative perspective. Schizophr. Res. 4 (2), 173–191. doi: 10.1016/0920-9964(91)90032-m
Volkow, N. D., Fowler, J. S., Wang, G. J., Swanson, J. M., Telang, F. (2007). Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch. Neurol. 64 (11), 1575–9. doi: 10.1001/archneur.64.11.1575
Wadenberg, M.-L., Wiker, C., Svensson, T. H. (2007). Enhanced efficacy of both typical and atypical antipsychotic drugs by adjunctive alpha2 adrenoceptor blockade: experimental evidence. Int. J. Neuropsychopharmacol. 10 (2), 191–202. doi: 10.1017/S1461145706006638
Weber, M. A., Graack, E. T., Scholl, J. L., Renner, K. J., Forster, G. L., Watt, M. J. (2018). Enhanced dopamine D2 autoreceptor function in the adult prefrontal cortex contributes to dopamine hypoactivity following adolescent social stress. Eur. J. Neurosci. 48 (2), 1833–1850. doi: 10.1111/ejn.14019
Weinshenker, D., Schroeder, J. P. (2007). There and back again: a tale of norepinephrine and drug addiction. Neuropsychopharmacology 32 (7), 1433–1451. doi: 10.1038/sj.npp.1301263
Westerink, B. H., de Boer, P., de Vries, J. B., Kruse, C. G., Long, S. K. (1998). Antipsychotic drugs induce similar effects on the release of dopamine and noradrenaline in the medial prefrontal cortex of the rat brain. Eur. J. Pharmacol. 361 (1), 27–33. doi: 10.1016/s0014-2999(98)00711-0
Westerink, B. H., Kawahara, Y., De Boer, P., Geels, C., De Vries, J. B., Wikström, H. V., et al. (2001). Antipsychotic Drugs Classified by Their Effects on the Release of Dopamine and Noradrenaline in the Prefrontal Cortex and Striatum. Eur. J. Pharmacol. 412 (2), 127–138. doi: 10.1016/s0014-2999(00)00935-3
Wise, R. A. (1998). Drug-activation of brain reward pathways. Drug Alcohol Depend. 51 (1-2), 13–22. doi: 10.1016/s0376-8716(98)00063-5
Wolf, M. E., Roth, R. H. (1987). Dopamine neurons projecting to the medial prefrontal cortex possess release-modulating autoreceptors. Neuropharmacology 26 (8), 1053–1059. doi: 10.1016/0028-3908(87)90248-6
Wong, Y. C., Ilkova, T., van Wijk, R. C., Hartman, R., de Lange, E. C. M. (2018). Development of a population pharmacokinetic model to predict brain distribution and dopamine D2 receptor occupancy of raclopride in non-anesthetized rat. Eur. J. Pharm. Sci. 111, 514–525. doi: 10.1016/j.ejps.2017.10.031
Xing, B., Li, Y.-C., Gao, W.-Y. (2016). Norepinephrine Versus Dopamine and Their Interaction in Modulating Synaptic Function in the Prefrontal Cortex. Brain Res. 1641 (Pt B), 217–233. doi: 10.1016/j.brainres.2016.01.005
Keywords: dopamine-norepinephrine interaction, noradrenergic denervation, norepinephrine transporter, D2 receptor drug, neuronal activity, ventral tegmental area
Citation: Devoto P, Sagheddu C, Santoni M, Flore G, Saba P, Pistis M and Gessa GL (2020) Noradrenergic Source of Dopamine Assessed by Microdialysis in the Medial Prefrontal Cortex. Front. Pharmacol. 11:588160. doi: 10.3389/fphar.2020.588160
Received: 28 July 2020; Accepted: 07 September 2020;
Published: 23 September 2020.
Edited by:
Philippe De Deurwaerdere, Université de Bordeaux, FranceReviewed by:
Andrea Contini, INRAE Nouvelle-Aquitaine Bordeaux, FranceCopyright © 2020 Devoto, Sagheddu, Santoni, Flore, Saba, Pistis and Gessa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Devoto, cGRldm90b0B1bmljYS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.